Данное изобретение в общем случае относится к способу жидкофазного каталитического окисления ароматического соединения. Один аспект изобретения относится к неполному окислению диалкилароматического соединения (например, пара-ксилола) до получения сырой неочищенной ароматической дикарбоновой кислоты (например, сырой неочищенной терефталевой кислоты), которую после этого можно будет подвергнуть очистке и разделению. Другой аспект изобретения относится к улучшенной барботажной реакторной колонне, которая позволяет реализовать более эффективный и экономичный способ жидкофазного окисления.
УРОВЕНЬ ТЕХНИКИ
Реакции жидкофазного окисления используют в широком ассортименте существующих коммерческих способов. Например, жидкофазное окисление в настоящее время используют для окисления альдегидов до получения кислот (например, пропионового альдегида до получения пропионовой кислоты), окисления циклогексана до получения адипиновой кислоты и окисления алкилароматики до получения спиртов, кислот или дикислот. Способом коммерческого окисления, имеющим в особенности большое значение и относящимся к последней категории (окисление алкилароматики), является жидкофазное каталитическое неполное окисление пара-ксилола до получения терефталевой кислоты. Терефталевая кислота представляет собой важное соединение, характеризующееся широким ассортиментом сфер применения. Основным вариантом использования терефталевой кислоты является использование в качестве исходного сырья при получении полиэтилентерефталата (ПЭТФ). ПЭТФ представляет собой хорошо известный пластик, используемый в больших количествах по всему миру для получения продукции, такой как бутылки, волокна и упаковка.
В типичном способе жидкофазного окисления, включающем неполное окисление пара-ксилола до получения терефталевой кислоты, поток жидкофазного исходного подаваемого материала и поток газофазного окислителя вводят в реактор, и в реакторе они образуют многофазную реакционную среду. Вводимый в реактор поток жидкофазного исходного подаваемого материала содержит, по меньшей мере, одно окисляемое органическое соединение (например, пара-ксилол), в то время как поток газофазного окислителя содержит молекулярный кислород. По меньшей мере, часть молекулярного кислорода, вводимого в реактор в качестве газа, растворяется в жидкой фазе реакционной среды, что обеспечивает доступность кислорода для жидкофазной реакции. Если жидкая фаза многофазной реакционной среды будет содержать недостаточную концентрацию молекулярного кислорода (то есть если определенные части реакционной среды будут «обеднены кислородом»), то тогда нежелательные побочные реакции могут привести к образованию примесей и/или целевые реакции могут замедлиться по скорости. Если жидкая фаза реакционной среды будет содержать чрезмерно мало окисляемого соединения, то тогда скорость реакции может оказаться нежелательно низкой. Кроме того, если жидкая фаза реакционной среды будет содержать избыточную концентрацию окисляемого соединения, то тогда дополнительные нежелательные побочные реакции могут привести к образованию примесей.
Обычно используемые реакторы жидкофазного окисления оборудуют средствами перемешивания, предназначенными для перемешивания многофазной реакционной среды, содержащейся в них. Перемешивание реакционной среды проводят в целях стимулирования растворения молекулярного кислорода в жидкой фазе реакционной среды, выдерживания относительно однородных концентраций растворенного кислорода в жидкой фазе реакционной среды и выдерживания в жидкой фазе реакционной среды относительно однородных концентраций окисляемого органического соединения.
Перемешивание реакционной среды, подвергающейся жидкофазному окислению, зачастую проводят при использовании механических средств перемешивания в емкостях, таких как, например, корпусные реакторы с непрерывным перемешиванием (CSTR). Несмотря на то что реакторы CSTR могут обеспечить проведение тщательного перемешивания реакционной среды, реакторам CSTR свойственны несколько недостатков. Например, реакторы CSTR характеризуются относительно высоким уровнем капитальных затрат вследствие наличия у них потребности в дорогостоящих двигателях, подшипниках с жидкостным уплотнением и приводных валах и/или сложных перемешивающих механизмах. Кроме того, вращающиеся и/или осциллирующие механические компоненты обычно используемых реакторов CSTR требуют регулярного проведения технического обслуживания. Работы и время остановки, связанные с проведением такого технического обслуживания, увеличивают эксплуатационные расходы для реакторов CSTR. Однако даже при регулярном проведении технического обслуживания механические системы перемешивания, используемые в реакторах CSTR, подвержены отказам механической части и могут потребовать замены по истечении относительно коротких периодов времени.
Барботажные реакторные колонны представляют собой привлекательную альтернативу для реакторов CSTR и других реакторов окисления с механическим перемешиванием. Барботажные реакторные колонны обеспечивают перемешивание реакционной среды без возникновения потребности в дорогостоящем и ненадежном механическом оборудовании. Барботажные реакторные колонны обычно включают удлиненную прямостоячую зону реакции, внутри которой содержится реакционная среда. Перемешивание реакционной среды в зоне реакции обеспечивается главным образом за счет естественного всплывания пузырьков газа, поднимающихся через жидкую фазу реакционной среды. Данное обусловленное естественным всплыванием перемешивание, достигаемое в барботажных реакторных колоннах, приводит к уменьшению капитальных затрат и расходов на техническое обслуживание в сопоставлении с реакторами с механическим перемешиванием. Кроме того, по существу отсутствие подвижных механических деталей, связанное с барботажными реакторными колоннами, обеспечивает получение системы окисления, которая менее подвержена отказам механической части в сопоставлении с реакторами с механическим перемешиванием.
Если жидкофазное неполное окисление пара-ксилола будут проводить в обычно используемом реакторе окисления (CSTR или барботажная колонна), то тогда продуктом, отбираемым из реактора, обычно будет являться суспензия, содержащая сырую неочищенную терефталевую кислоту (СТА) и маточный раствор. СТА характеризуется относительно высокими уровнями содержания примесей (например, 4-карбоксибензальдегида, пара-толуиловой кислоты, флуоренонов и других окрашенных веществ), что делает ее непригодной для использования в качестве исходного сырья при получении ПЭТФ. Таким образом, СТА, полученную в обычно используемых реакторах окисления, обычно подвергают технологическому процессу очистки, который превращает СТА в очищенную терефталевую кислоту (РТА), подходящую для использования при получении ПЭТФ.
Один типичный способ очистки при превращении СТА в РТА включает следующие далее стадии: (1) замена маточного раствора в суспензии, содержащей СТА, на воду, (2) нагревание суспензии СТА/вода для растворения СТА в воде, (3) каталитическое гидрирование раствора СТА/вода для превращения примесей в более желательные и/или легче отделяемые соединения, (4) осаждение полученной в результате РТА из подвергнутого гидрированию раствора при использовании нескольких стадий кристаллизации и (5) отделение закристаллизованной РТА от остающихся жидкостей. Несмотря на свою эффективность, данный тип обычно используемого способа очистки может оказаться очень дорогостоящим. Индивидуальные факторы, вносящие свой вклад в высокую стоимость обычно используемых способов очистки СТА, включают, например, тепловую энергию, необходимую для стимулирования растворения СТА в воде, катализатор, необходимый для проведения гидрирования, поток водорода, необходимый для проведения гидрирования, потери выхода, вызванные гидрированием некоторой части терефталевой кислоты, и наличие нескольких емкостей, необходимых для проведения многоступенчатой кристаллизации. Таким образом, было бы желательным предложение продукта СТА, который можно было бы очищать без возникновения потребности в стимулированном нагреванием растворении в воде, гидрировании и/или многоступенчатой кристаллизации.
ОБЪЕКТЫ ИЗОБРЕТЕНИЯ
Поэтому объект настоящего изобретения заключается в предложении более эффективных и экономичных реактора и способа жидкофазного окисления.
Другой объект изобретения заключается в предложении более эффективных и экономичных реактора и способа для жидкофазного каталитического неполного окисления пара-ксилола до получения терефталевой кислоты.
Еще один объект изобретения заключается в предложении барботажной реакторной колонны, которая облегчает проведение улучшенных реакций жидкофазного окисления при пониженной эффективности образования примесей.
И еще один объект изобретения заключается в предложении более эффективной и экономичной системы, предназначенной для получения чистой терефталевой кислоты (РТА) в результате проведения жидкофазного окисления пара-ксилола до получения сырой неочищенной терефталевой кислоты (СТА), а после этого очистки СТА до получения РТА.
Дополнительный объект изобретения заключается в предложении барботажной реакторной колонны, предназначенной для окисления пара-ксилола и получения продукта СТА, способного подвергнуться очистке без возникновения потребности в стимулированном нагреванием растворении СТА в воде, гидрировании растворенной СТА и/или многоступенчатой кристаллизации гидрированной РТА.
Необходимо отметить, что объем настоящего изобретения, определенный в прилагаемой формуле изобретения, не ограничивается способами или аппаратами, способными обеспечить реализацию всех целей, перечисленных выше. Вместо этого объем заявленного изобретения может включать широкий ассортимент систем, которые не позволяют добиться достижения всех или любых из перечисленных выше целей. Дополнительные цели и преимущества настоящего изобретения станут вполне очевидными для специалиста в соответствующей области техники после ознакомления со следующим далее подробным описанием и сопутствующими чертежами.
КРАТКОЕ ИЗЛОЖЕНИЕ ИЗОБРЕТЕНИЯ
Один вариант реализации настоящего изобретения относится к способу, включающему следующие далее стадии: (а) подачу ароматического соединения в реактор окисления; (b) окисление, по меньшей мере, части упомянутого ароматического соединения в жидкой фазе многофазной реакционной среды, содержащейся в упомянутом реакторе окисления, до получения таким образом сырой неочищенной терефталевой кислоты и одного или нескольких оксидов углерода; и (с) выдерживание во время упомянутого окисления соотношения между молями полученных упомянутых оксидов углерода и молями подаваемого упомянутого ароматического соединения в диапазоне от приблизительно 0,02:1 до приблизительно 0,24:1.
Еще один вариант реализации настоящего изобретения относится к способу, включающему следующие далее стадии: (а) подачу ароматического соединения в реактор окисления; (b) окисление, по меньшей мере, части упомянутого ароматического соединения в жидкой фазе многофазной реакционной среды, содержащейся в упомянутом реакторе окисления, до получения таким образом сырой неочищенной терефталевой кислоты и диоксида углерода; и (с) выдерживание во время упомянутого окисления соотношения между молями полученного упомянутого диоксида углерода и молями подаваемого упомянутого ароматического соединения в диапазоне от приблизительно 0,01:1 до приблизительно 0,21:1.
А еще один вариант реализации настоящего изобретения относится к способу, включающему следующие далее стадии: (а) подачу ароматического соединения в реактор окисления; (b) окисление, по меньшей мере, части упомянутого ароматического соединения в жидкой фазе многофазной реакционной среды, содержащейся в упомянутом реакторе окисления, до получения таким образом сырой неочищенной терефталевой кислоты и монооксида углерода; и (с) выдерживание во время упомянутого окисления соотношения между молями полученного упомянутого монооксида углерода и молями подаваемого упомянутого ароматического соединения в диапазоне от приблизительно 0,005:1 до приблизительно 0,09:1.
И еще один вариант реализации настоящего изобретения относится к способу, включающему следующие далее стадии: (а) подачу ароматического соединения в реактор окисления; (b) окисление, по меньшей мере, части упомянутого ароматического соединения в жидкой фазе многофазной реакционной среды, содержащейся в упомянутом реакторе окисления, до получения таким образом сырой неочищенной терефталевой кислоты и диоксида углерода; и (с) выдерживание во время упомянутого окисления молярной доли выживания для упомянутого ароматического соединения, большей чем приблизительно 98 процентов.
Дополнительный вариант реализации настоящего изобретения относится к способу, включающему следующие далее стадии: (а) подачу ароматического соединения в реактор окисления; (b) окисление, по меньшей мере, части упомянутого ароматического соединения в жидкой фазе многофазной реакционной среды, содержащейся в упомянутом реакторе окисления, до получения таким образом сырой неочищенной терефталевой кислоты и метилацетата; и (с) выдерживание во время упомянутого окисления соотношения между молями полученного упомянутого метилацетата и молями подаваемого упомянутого ароматического соединения в диапазоне от приблизительно 0,005:1 до приблизительно 0,09:1.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Предпочтительные варианты реализации изобретения подробно описываются далее со ссылкой на прилагаемые чертежи, где:
Фиг. 1 представляет собой вид сбоку для реактора окисления, сконструированного в соответствии с одним вариантом реализации настоящего изобретения, в частности, иллюстрирующий введение потоков исходного подаваемого материала, окислителя и среды орошения в реактор, присутствие в реакторе многофазной реакционной среды и отбор газа и суспензии из верха и низа реактора соответственно;
Фиг. 2 представляет собой увеличенный вид сбоку в разрезе для низа барботажной реакторной колонны, полученный вдоль линии 2-2 на Фиг. 3, в частности, иллюстрирующий местоположение и конфигурацию барботера окислителя, используемого для введения в реактор потока окислителя;
Фиг. 3 представляет собой вид сверху для барботера окислителя Фиг. 2, в частности, иллюстрирующий отверстия для окислителя в области верха барботера окислителя;
Фиг. 4 представляет собой вид снизу для барботера окислителя Фиг. 2, в частности, иллюстрирующий отверстия для окислителя в области низа барботера окислителя;
Фиг. 5 представляет собой вид сбоку в разрезе для барботера окислителя, полученный вдоль линии 5-5 на Фиг. 3, в частности, иллюстрирующий ориентацию отверстий для окислителя в областях верха и низа барботера окислителя;
Фиг. 6 представляет собой увеличенный вид сбоку для нижней части барботажной реакторной колонны, в частности, иллюстрирующий систему, предназначенную для введения в реактор потока исходного подаваемого материала в нескольких разнесенных по вертикали позициях;
Фиг. 7 представляет собой вид сверху в разрезе, полученный вдоль линии 7-7 на Фиг. 6, в частности, иллюстрирующий то, как система введения исходного подаваемого материала, продемонстрированная на Фиг. 6, распределяет поток исходного подаваемого материала в зоне предпочтительной радиальной подачи исходного подаваемого материала (FZ) и более чем одном азимутальном квадранте (Q1, Q2, Q3, Q4);
Фиг. 8 представляет собой вид сверху в разрезе, подобный Фиг. 7, но иллюстрирующий альтернативное средство выпуска потока исходного подаваемого материала в реактор, использующее байонетные трубы, каждая из которых имеет множество небольших отверстий для исходного подаваемого материала;
Фиг. 9 представляет собой изометрическое изображение для альтернативной системы, предназначенной для введения потока исходного подаваемого материала в зону реакции в нескольких разнесенных по вертикали позициях без возникновения потребности в наличии нескольких точек проникновения в емкость, в частности, иллюстрирующее то, что система распределения исходного подаваемого материала может, по меньшей мере, отчасти опираться на барботер окислителя;
Фиг. 10 представляет собой вид сбоку для системы распределения исходного подаваемого материала с одной точкой проникновения в емкость и барботера окислителя, проиллюстрированных на Фиг. 9;
Фиг. 11 представляет собой вид сверху в разрезе, полученный вдоль линии 11-11 на Фиг. 10 и дополнительно иллюстрирующий систему распределения исходного подаваемого материала с одной точкой проникновения в емкость, опирающуюся на барботер окислителя;
Фиг. 12 представляет собой изометрическое изображение для альтернативного барботера окислителя, имеющего все отверстия для окислителя расположенными в области низа кольцевого элемента;
Фиг. 13 представляет собой вид сверху для альтернативного барботера окислителя Фиг. 12;
Фиг. 14 представляет собой вид снизу для альтернативного барботера окислителя Фиг. 12, в частности, иллюстрирующий местоположение нижних отверстий, предназначенных для введения потока окислителя в зону реакции;
Фиг. 15 представляет собой вид сбоку в разрезе для барботера окислителя, полученный вдоль линии 15-15 на Фиг. 13, в частности, иллюстрирующий ориентацию нижних отверстий для окислителя;
Фиг. 16 представляет собой вид сбоку для барботажной реакторной колонны, оборудованной внутренней деаэрационной емкостью, расположенной поблизости от нижнего выпускного отверстия реактора;
Фиг. 17 представляет собой увеличенный вид сбоку в разрезе для нижней части барботажной реакторной колонны Фиг. 16, полученный вдоль линии 17-17 на Фиг. 18, в частности, иллюстрирующий конфигурацию внутренней деаэрационной емкости, расположенной у нижнего выпускного отверстия барботажной реакторной колонны;
Фиг. 18 представляет собой вид сверху в разрезе, полученный вдоль линии 18-18 на Фиг. 16, в частности, иллюстрирующий стабилизатор потока, расположенный в деаэрационной емкости;
Фиг. 19 представляет собой вид сбоку для барботажной реакторной колонны, оборудованной внешней деаэрационной емкостью, иллюстрирующий способ, по которому часть деаэрированной суспензии, покидающей низ деаэрационной емкости, можно использовать для промывания линии уменьшения коэффициента заполнения, соединенной с низом реактора;
Фиг. 20 представляет собой вид сбоку для барботажной реакторной колонны, оборудованной гибридной внутренне/внешней деаэрационной емкостью, предназначенной для отделения газовой фазы реакционной среды, отбираемой из боковой позиции повышенного уровня расположения в реакторе;
Фиг. 21 представляет собой вид сбоку для барботажной реакторной колонны, оборудованной альтернативной гибридной деаэрационной емкостью, расположенной поблизости от низа реактора;
Фиг. 22 представляет собой увеличенный вид сбоку в разрезе для нижней части барботажной реакторной колонны Фиг. 21, в частности, иллюстрирующий применение альтернативного барботера окислителя, использующего каналы впускных отверстий, которые принимают поток окислителя через нижнее днище реактора;
Фиг. 23 представляет собой увеличенный вид сбоку в разрезе, подобный Фиг. 22, в частности, иллюстрирующий альтернативное средство введения потока окислителя в реактор через множество отверстий в нижнем днище реактора и необязательно с использованием отбойников для более равномерного распределения потока окислителя в реакторе;
Фиг. 24 представляет собой вид сбоку для барботажной реакторной колонны, использующей внутренний канал для течения в целях содействия улучшению диспергирования окисляемого соединения в результате рециркуляции части реакционной среды с ее переходом из верхней части реактора в нижнюю часть реактора;
Фиг. 25 представляет собой вид сбоку для барботажной реакторной колонны, использующей внешний канал для течения в целях содействия улучшению диспергирования окисляемого соединения в результате рециркуляции части реакционной среды с ее переходом из верхней части реактора в нижнюю часть реактора;
Фиг. 26 представляет собой вид сбоку в разрезе для горизонтального эдуктора, который можно использовать для улучшения диспергирования окисляемого соединения в реакторе окисления, в частности, иллюстрирующий эдуктор, который использует поступающий жидкий исходный подаваемый материал для затягивания реакционной среды в эдуктор и выпускает смесь исходного подаваемого материала и реакционной среды в зону реакции с высокой скоростью;
Фиг. 27 представляет собой вид сбоку в разрезе для вертикального эдуктора, который можно использовать для улучшения диспергирования окисляемого соединения в реакторе окисления, в частности, иллюстрирующий эдуктор, который объединяет жидкий исходный подаваемый материал и газ из впускного отверстия и использует объединенную двухфазную текучую среду для затягивания реакционной среды в эдуктор и выпускает смесь жидкого исходного подаваемого материала, газа из впускного отверстия и реакционной среды в зону реакции с высокой скоростью;
Фиг. 28 представляет собой вид сбоку для барботажной реакторной колонны, содержащей многофазную реакционную среду, в частности, иллюстрирующий реакционную среду, теоретически разделенную на 30 горизонтальных долей с равным объемом для того, чтобы количественно установить определенные градиенты в реакционной среде;
Фиг. 29 представляет собой вид сбоку для барботажной реакторной колонны, содержащей многофазную реакционную среду, в частности, иллюстрирующий первый и второй дискретные 20-процентные сплошные объемы реакционной среды, которые характеризуются существенно различными концентрациями кислорода и/или скоростями расходования кислорода;
Фиг. 30 представляет собой вид сбоку для двух расположенных друг над другом реакционных емкостей с использованием или без использования необязательного механического перемешивания, содержащих многофазную реакционную среду, в частности, иллюстрирующий то, что емкости вмещают дискретные 20-процентные сплошные объемы реакционной среды, характеризующиеся существенно различными концентрациями кислорода и/или скоростями расходования кислорода;
Фиг. 31 представляет собой вид сбоку для трех расположенных друг рядом с другом реакционных емкостей с использованием или без использования необязательного механического перемешивания, содержащих многофазную реакционную среду, в частности, иллюстрирующий то, что емкости вмещают дискретные 20-процентные сплошные объемы реакционной среды, характеризующиеся существенно различными концентрациями кислорода и/или скоростями расходования кислорода;
Фиг. 32А и 32В представляют собой увеличенные изображения для частиц сырой неочищенной терефталевой кислоты (СТА), полученных в соответствии с одним вариантом реализации настоящего изобретения, в частности, иллюстрирующие то, что каждая частица СТА представляет собой частицу, характеризующуюся малой плотностью и большой площадью удельной поверхности и состоящую из множества неплотно связанных субчастиц СТА;
Фиг. 33А и 33В представляют собой увеличенные изображения для обычно получаемой СТА, в частности, иллюстрирующие то, что обычная частица СТА характеризуется большим размером частиц, меньшей плотностью и меньшей площадью удельной поверхности в сопоставлении с частицей СТА изобретения Фиг. 32А и 32В;
Фиг. 34 представляет собой упрощенную схему технологичного процесса для способа получения очищенной терефталевой кислоты (РТА) предшествующего уровня техники; и
Фиг. 35 представляет собой упрощенную схему технологического процесса для способа получения РТА в соответствии с одним вариантом реализации настоящего изобретения.
ПОДРОБНОЕ ОПИСАНИЕ
Один вариант реализации настоящего изобретения относится к жидкофазному неполному окислению окисляемого соединения. Такое окисление предпочтительно проводят в жидкой фазе многофазной реакционной среды, содержащейся в одном или нескольких реакторах с перемешиванием. Подходящие реакторы с перемешиванием включают, например, реакторы с барботажным перемешиванием (например, барботажные реакторные колонны), реакторы с механическим перемешиванием (например, корпусные реакторы с непрерывным перемешиванием) и реакторы с перемешиванием потоком (например, струйные реакторы). В одном варианте реализации изобретения жидкофазное окисление проводят в одной барботажной реакторной колонне.
В соответствии с использованием в настоящем документе термин «барботажная реакторная колонна» должен обозначать реактор, предназначенный для облегчения проведения химических реакций в многофазной реакционной среде, где перемешивание реакционной среды главным образом обеспечивается в результате перемещения пузырьков газа снизу вверх через реакционную среду. В соответствии с использованием в настоящем документе термин «перемешивание» должен обозначать работу, затрачиваемую в реакционной среде, которая приводит к возникновению течения и/или перемешивания текучей среды. В соответствии с использованием в настоящем документе термины «основная часть», «главным образом» и «преимущественно» должны обозначать более чем 50 процентов. В соответствии с использованием в настоящем документе термин «механическое перемешивание» должен обозначать перемешивание реакционной среды, вызываемое физическим перемещением жестких или гибких элементов (элемента) по отношению к реакционной среде или внутри реакционной среды. Например, механическое перемешивание можно обеспечить при использовании вращения, осцилляции и/или вибрации внутренних мешалок, лопастей, вибраторов или акустических диафрагм, расположенных в реакционной среде. В соответствии с использованием в настоящем документе термин «перемешивание потоком» должен обозначать перемешивание реакционной среды, вызванное высокоскоростными инжектированием и/или рециркуляцией одной или нескольких текучих сред в реакционной среде. Например, перемешивание потоком можно обеспечить при использовании сопел, эжекторов и/или эдукторов.
В предпочтительном варианте реализации настоящего изобретения менее чем приблизительно 40 процентов от величины перемешивания реакционной среды в барботажной реакторной колонне во время окисления обеспечивают в результате наличия механического перемешивания и/или перемешивания потоком, более предпочтительно менее чем приблизительно 20 процентов от величины перемешивания обеспечивают в результате наличия механического перемешивания и/или перемешивания потоком, а наиболее предпочтительно менее чем 5 процентов от величины перемешивания обеспечивают в результате наличия механического перемешивания и/или перемешивания потоком. Предпочтительно величина механического перемешивания и/или перемешивания потоком, придаваемая многофазной реакционной среде во время окисления, составляет величину, меньшую чем приблизительно 3 киловатта на один кубический метр реакционной среды, более предпочтительно меньшую чем приблизительно 2 киловатта на один кубический метр, а наиболее предпочтительно меньшую чем 1 киловатт на один кубический метр.
Если обратиться теперь к Фиг. 1, то можно сказать, что на ней проиллюстрирована предпочтительная барботажная реакторная колонна 20 как включающая оболочку емкости 22, включающую секцию реакции 24 и секцию отделения 26. Секция реакции 24 определяет внутреннюю зону реакции 28, в то время как секция отделения 26 определяет внутреннюю зону отделения 30. Поток преимущественно жидкофазного исходного подаваемого материала вводят в зону реакции 28 через впускные отверстия для исходного подаваемого материала 32а, b, c, d. Поток преимущественно газофазного окислителя вводят в зону реакции 28 через барботер окислителя 34, расположенный в нижней части зоны реакции 28. Поток жидкофазного исходного подаваемого материала и поток газофазного окислителя совместно образуют многофазную реакционную среду 36 внутри зоны реакции 28. Многофазная реакционная среда 36 включает жидкую фазу и газовую фазу. Более предпочтительно многофазная реакционная среда 36 включает трехфазную среду, включающую твердофазный, жидкофазный и газофазный компоненты. Твердофазный компонент реакционной среды 36 предпочтительно выпадает в осадок внутри зоны реакции 28 в результате прохождения реакции окисления, проводимой в жидкой фазе реакционной среды 36. Барботажная реакторная колонна 20 включает выпускное отверстие для суспензии 38, расположенное поблизости от низа зоны реакции 28, и выпускное отверстие для газа 40, расположенное поблизости от верха зоны отделения 30. Отходящий поток суспензии, включающий жидкофазный и твердофазный компоненты реакционной среды 36, отбирают из зоны реакции 28 через выпускное отверстие для суспензии 38, в то время как отходящий поток преимущественно газа отбирают из зоны отделения 30 через выпускное отверстие для газа 40.
Поток жидкофазного исходного подаваемого материала, вводимый в барботажную реакторную колонну 20 через впускные отверстия для исходного подаваемого материала 32а, b, c, d, предпочтительно включает окисляемое соединение, растворитель и систему катализатора.
Окисляемое соединение, присутствующее в потоке жидкофазного исходного подаваемого материала, предпочтительно имеет, по меньшей мере, одну гидрокарбильную группу. Более предпочтительно окисляемое соединение представляет собой ароматическое соединение. Еще более предпочтительно окисляемое соединение представляет собой ароматическое соединение, имеющее, по меньшей мере, одну присоединенную гидрокарбильную группу или, по меньшей мере, одну присоединенную замещенную гидрокарбильную группу или содержащее, по меньшей мере, один присоединенный гетероатом или, по меньшей мере, одну присоединенную функциональность карбоновой кислоты (-СООН). Еще более предпочтительно окисляемое соединение представляет собой ароматическое соединение, имеющее, по меньшей мере, одну присоединенную гидрокарбильную группу или, по меньшей мере, одну присоединенную замещенную гидрокарбильную группу, при этом каждая присоединенная группа содержит от 1 до 5 атомов углерода. И еще более предпочтительно окисляемое соединение представляет собой ароматическое соединение, имеющее ни больше, ни меньше чем две присоединенные группы, при этом каждая присоединенная группа содержит ни больше, ни меньше чем один атом углерода и состоит из метильных групп и/или замещенных метильных групп и/или самое большее одной группы карбоновой кислоты. Даже еще более предпочтительно окисляемое соединение представляет собой пара-ксилол, мета-ксилол, пара-толуиловый альдегид, мета-толуиловый альдегид, пара-толуиловую кислоту, мета-толуиловую кислоту и/или ацетальдегид. Наиболее предпочтительно окисляемое соединение представляет собой пара-ксилол.
«Гидрокарбильная группа» в соответствии с определением в настоящем документе представляет собой, по меньшей мере, один атом углерода, который связан только с атомами водорода или с другими атомами углерода. «Замещенная гидрокарбильная группа» в соответствии с определением в настоящем документе представляет собой, по меньшей мере, один атом углерода, связанный, по меньшей мере, с одним гетероатомом и, по меньшей мере, с одним атомом водорода. «Гетероатомы» в соответствии с определением в настоящем документе представляют собой все атомы, отличные от атомов углерода и водорода. Ароматические соединения в соответствии с определением в настоящем документе включают ароматическое кольцо, предпочтительно содержащее, по меньшей мере, 6 атомов углерода, еще более предпочтительно содержащее только атомы углерода в качестве части кольца. Подходящие примеры таких ароматических колец включают нижеследующее, но не ограничиваются только им: бензольное, бифенильное, терфенильное, нафталиновое и другие конденсированные ароматические кольца на углеродной основе.
Подходящие примеры окисляемого соединения включают алифатические углеводороды (например, алканы, разветвленные алканы, циклические алканы, алифатические алкены, разветвленные алкены и циклические алкены); алифатические альдегиды (например, ацетальдегид, пропионовый альдегид, изомасляный альдегид и н-масляный альдегид); алифатические спирты (например, этанол, изопропанол, н-пропанол, н-бутанол и изобутанол); алифатические кетоны (например, диметилкетон, этилметилкетон, диэтилкетон и изопропилметилкетон); алифатические сложные эфиры (например, метилформиат, метилацетат, этилацетат); алифатические пероксиды, перкислоты и гидропероксиды (например, трет-бутилгидропероксид, перуксусная кислота и ди-трет-бутилгидропероксид); алифатические соединения, имеющие группы, которые представляют собой комбинации вышеупомянутых алифатических вариантов плюс другие гетероатомы, (например, алифатические соединения, содержащие один или несколько молекулярных сегментов углеводородов, альдегидов, спиртов, кетонов, сложных эфиров, пероксидов, перкислот и/или гидропероксидов в комбинации с натрием, бромом, кобальтом, марганцем и цирконием); различные бензольные кольца, нафталиновые кольца, бифенилы, терфенилы и другие ароматические группы, имеющие одну или несколько присоединенных гидрокарбильных групп, (например, толуол, этилбензол, изопропилбензол, н-пропилбензол, неопентилбензол, пара-ксилол, мета-ксилол, орто-ксилол, все изомеры триметилбензолов, все изомеры тетраметилбензолов, пентаметилбензол, гексаметилбензол, все изомеры этилметилбензолов, все изомеры диэтилбензолов, все изомеры этилдиметилбензолов, все изомеры диметилнафталинов, все изомеры этилметилнафталинов, все изомеры диэтилнафталинов, все изомеры диметилбифенилов, все изомеры этилметилбифенилов и все изомеры диэтилбифенилов, стильбен и стильбен, имеющий одну или несколько присоединенных гидрокарбильных групп, флуорен и флуорен, имеющий одну или несколько присоединенных гидрокарбильных групп, антрацен и антрацен, имеющий одну или несколько присоединенных гидрокарбильных групп, и дифенилэтан и дифенилэтан, имеющий одну или несколько присоединенных гидрокарбильных групп); различные бензольные кольца, нафталиновые кольца, бифенилы, терфенилы и другие ароматические группы, имеющие одну или несколько присоединенных гидрокарбильных групп и/или содержащие один или несколько присоединенных гетероатомов, которые могут соединяться с другими атомами или группами атомов, (например, фенол, все изомеры метилфенолов, все изомеры диметилфенолов, все изомеры нафтолов, простой бензилметиловый эфир, все изомеры бромфенолов, бромбензол, все изомеры бромтолуолов, включающие альфа-бромтолуол, дибромбензол, нафталенат кобальта и все изомеры бромбифенилов); различные бензольные кольца, нафталиновые кольца, бифенилы, терфенилы и другие ароматические группы, имеющие одну или несколько присоединенных гидрокарбильных групп и/или содержащих один или несколько присоединенных гетероатомов и/или имеющих одну или несколько присоединенных замещенных гидрокарбильных групп (например, бензальдегид, все изомеры бромбензальдегидов, все изомеры бромированных толуиловых альдегидов, в том числе все изомеры альфа-бромтолуиловых альдегидов, все изомеры гидроксибензальдегидов, все изомеры бромгидроксибензальдегидов, все изомеры бензолдикарбоксальдегидов, все изомеры бензолтрикарбоксальдегидов, пара-толуиловый альдегид, мета-толуиловый альдегид, орто-толуиловый альдегид, все изомеры толуолдикарбоксальдегидов, все изомеры толуолтрикарбоксальдегидов, все изомеры толуолтетракарбоксальдегидов, все изомеры диметилбензолдикарбоксальдегидов, все изомеры диметилбензолтрикарбоксальдегидов, все изомеры диметилбензолтетракарбоксальдегидов, все изомеры триметилбензолтрикарбоксальдегидов, все изомеры этилтолуолальдегидов, все изомеры триметилбензолдикарбоксальдегидов, тетраметилбензолдикарбоксальдегид, гидроксиметилбензол, все изомеры гидроксиметилтолуолов, все изомеры гидроксиметилбромтолуолов, все изомеры гидроксиметилтолуиловых альдегидов, все изомеры гидроксиметилбромтолуиловых альдегидов, бензилгидропероксид, бензоилгидропероксид, все изомеры толилметилгидропероксидов и все изомеры метилфенолметилгидропероксидов); различные бензольные кольца, нафталиновые кольца, бифенилы, терфенилы и другие ароматические группы, имеющие одну или несколько присоединенных избранных групп, при этом избранные группы обозначают гидрокарбильные группы, и/или присоединенные гетероатомы, и/или замещенные гидрокарбильные группы, и/или группы карбоновой кислоты, и/или группы пероксикислоты (например, бензойная кислота, пара-толуиловая кислота, мета-толуиловая кислота, орто-толуиловая кислота, все изомеры этилбензойных кислот, все изомеры пропилбензойных кислот, все изомеры бутилбензойных кислот, все изомеры пентилбензойных кислот, все изомеры диметилбензойных кислот, все изомеры этилметилбензойных кислот, все изомеры триметилбензойных кислот, все изомеры тетраметилбензойных кислот, пентаметилбензойная кислота, все изомеры диэтилбензойных кислот, все изомеры бензолдикарбоновых кислот, все изомеры бензолтрикарбоновых кислот, все изомеры метилбензолдикарбоновых кислот, все изомеры диметилбензолдикарбоновых кислот, все изомеры метилбензолтрикарбоновых кислот, все изомеры бромбензойных кислот, все изомеры дибромбензойных кислот, все изомеры бромтолуиловых кислот, в том числе альфа-бромтолуиловые кислоты, толилуксусная кислота, все изомеры гидроксибензойных кислот, все изомеры гидроксиметилбензойных кислот, все изомеры гидрокситолуиловых кислот, все изомеры гидроксиметилтолуиловых кислот, все изомеры гидроксиметилбензолдикарбоновых кислот, все изомеры гидроксибромбензойных кислот, все изомеры гидроксибромтолуиловых кислот, все изомеры гидроксиметилбромбензойных кислот, все изомеры карбоксибензальдегидов, все изомеры дикарбоксибензальдегидов, пербензойная кислота, все изомеры гидропероксиметилбензойных кислот, все изомеры гидропероксиметилгидроксибензойных кислот, все изомеры гидропероксикарбонилбензойных кислот, все изомеры гидропероксикарбонилтолуолов, все изомеры метилбифенилкарбоновых кислот, все изомеры диметилбифенилкарбоновых кислот, все изомеры метилбифенилдикарбоновых кислот, все изомеры бифенилтрикарбоновых кислот, все изомеры стильбена, имеющие одну или несколько присоединенных избранных групп, все изомеры флуоренона, имеющие одну или несколько присоединенных избранных групп, все изомеры нафталина, имеющие одну или несколько присоединенных избранных групп, бензил, все изомеры бензила, имеющие одну или несколько присоединенных избранных групп, бензофенон, все изомеры бензофенона, имеющие одну или несколько присоединенных избранных групп, антрахинон, все изомеры антрахинона, имеющие одну или несколько присоединенных избранных групп, все изомеры дифенилэтана, имеющие одну или несколько присоединенных избранных групп, бензокумарин и все изомеры бензокумарина, имеющие одну или несколько присоединенных избранных групп).
Если окисляемое соединение, присутствующее в потоке жидкофазного исходного подаваемого материала, будет представлять собой соединение, твердое при нормальных условиях (то есть твердое вещество при стандартных температуре и давлении), то тогда предпочитается, чтобы при введении в зону реакции 28 окисляемое соединение было бы по существу растворено в растворителе. Предпочитается, чтобы температура кипения окисляемого соединения при атмосферном давлении была бы равной, по меньшей мере, приблизительно 50°С. Более предпочтительно температура кипения окисляемого соединения находится в диапазоне от приблизительно 80 до приблизительно 400°С, а наиболее предпочтительно в диапазоне от 125 до 155°С. Количество окисляемого соединения, присутствующего в жидкофазном исходном подаваемом материале, предпочтительно находится в диапазоне от приблизительно 2 до приблизительно 40 массовых процентов, более предпочтительно в диапазоне от приблизительно 4 до приблизительно 20 массовых процентов, а наиболее предпочтительно в диапазоне от 6 до 15 массовых процентов.
В данный момент следует отметить то, что окисляемое соединение, присутствующее в жидкофазном исходном подаваемом материале, может включать комбинацию двух или более различных окисляемых реагентов. Подачу данных двух или более различных химических веществ можно проводить при их смешении в потоке жидкофазного исходного подаваемого материала или ее можно проводить при их разделении на несколько потоков исходного подаваемого материала. Например, окисляемое соединение, включающее пара-ксилол, мета-ксилол, пара-толуиловый альдегид, пара-толуиловую кислоту и ацетальдегид, можно подавать в реактор через единственное впускное отверстие или несколько раздельных впускных отверстий.
Растворитель, присутствующий в потоке жидкофазного исходного подаваемого материала, предпочтительно включает кислотный компонент и водный компонент. Растворитель предпочтительно присутствует в потоке жидкофазного исходного подаваемого материала с концентрацией в диапазоне от приблизительно 60 до приблизительно 98 массовых процентов, более предпочтительно в диапазоне от приблизительно 80 до приблизительно 96 массовых процентов, а наиболее предпочтительно в диапазоне от 85 до 94 массовых процентов. Кислотный компонент растворителя предпочтительно представляет собой главным образом органическую низкомолекулярную монокарбоновую кислоту, содержащую 1-6 атомов углерода, более предпочтительно 2 атома углерода. Наиболее предпочтительно кислотный компонент растворителя представляет собой главным образом уксусную кислоту. Предпочтительно кислотный компонент составляет, по меньшей мере, приблизительно 75 массовых процентов растворителя, более предпочтительно, по меньшей мере, приблизительно 80 массовых процентов растворителя, а наиболее предпочтительно от 85 до 98 массовых процентов растворителя, при этом баланс образует главным образом вода. Растворитель, вводимый в барботажную реакторную колонну 20, может содержать небольшие количества примесей, таких как, например, пара-толуиловый альдегид, терефталевый альдегид, 4-карбоксибензальдегид (4-СВА), бензойная кислота, пара-толуиловая кислота, пара-толуиловый альдегид, альфа-бром-пара-толуиловая кислота, изофталевая кислота, фталевая кислота, тримеллитовая кислота, полиароматика и/или суспендированный дисперсный материал. Предпочитается, чтобы совокупное количество примесей в растворителе, вводимом в барботажную реакторную колонну 20, составляло бы величину, меньшую чем приблизительно 3 массовых процента.
Система катализатора, присутствующая в потоке жидкофазного исходного подаваемого материала, предпочтительно является однородной жидкофазной системой катализатора, способной стимулировать прохождение окисления (в том числе неполного окисления) окисляемого соединения. Более предпочтительно система катализатора включает, по меньшей мере, один многовалентный переходный металл. Еще более предпочтительно многовалентный переходный металл включает кобальт. Даже более предпочтительно система катализатора включает кобальт и бром. Наиболее предпочтительно система катализатора включает кобальт, бром и марганец.
В случае присутствия в системе катализатора кобальта предпочитается, чтобы количество кобальта, присутствующего в потоке жидкофазного исходного подаваемого материала, было бы таким, чтобы концентрация кобальта в жидкой фазе реакционной среды 36 выдерживалась бы в диапазоне от приблизительно 300 до приблизительно 6000 массовых частей на миллион частей (ч./млн (мас.)), более предпочтительно в диапазоне от приблизительно 700 до приблизительно 4200 ч./млн (мас.), а наиболее предпочтительно в диапазоне от 1200 до 3000 ч./млн (мас.). В случае присутствия в системе катализатора брома предпочитается, чтобы количество брома, присутствующего в потоке жидкофазного исходного подаваемого материала, было бы таким, чтобы концентрация брома в жидкой фазе реакционной среды 36 выдерживалась бы в диапазоне от приблизительно 300 до приблизительно 5000 ч./млн (мас.), более предпочтительно в диапазоне от приблизительно 600 до приблизительно 4000 ч./млн (мас.), а наиболее предпочтительно в диапазоне от 900 до 3000 ч./млн (мас.). В случае присутствия в системе катализатора марганца предпочитается, чтобы количество марганца, присутствующего в потоке жидкофазного исходного подаваемого материала, было бы таким, чтобы концентрация марганца в жидкой фазе реакционной среды 36 выдерживалась бы в диапазоне от приблизительно 20 до приблизительно 1000 ч./млн (мас.), более предпочтительно в диапазоне от приблизительно 40 до приблизительно 500 ч./млн (мас.), наиболее предпочтительно в диапазоне от 50 до 200 ч./млн (мас.).
Концентрации кобальта, брома и/или марганца в жидкой фазе реакционной среды 36, представленные выше, выражают в виде средневременных и среднеобъемных величин. В соответствии с использованием в настоящем документе термин «средневременной» должен обозначать среднюю величину, по меньшей мере, для 10 измерений, проведенных через равные промежутки времени в течение непрерывного периода времени продолжительностью, по меньшей мере, в 100 секунд. В соответствии с использованием в настоящем документе термин «среднеобъемный» должен обозначать среднюю величину, по меньшей мере, для 10 измерений, проведенных через однородные 3-мерные интервалы по всему определенному объему.
Массовое соотношение между кобальтом и бромом (Со:Br) в системе катализатора, вводимой в зону реакции 28, предпочтительно находится в диапазоне от приблизительно 0,25:1 до приблизительно 4:1, более предпочтительно в диапазоне от приблизительно 0,5:1 до приблизительно 3:1, а наиболее предпочтительно в диапазоне от 0,75:1 до 2:1. Массовое соотношение между кобальтом и марганцем (Со:Mn) в системе катализатора, вводимой в зону реакции 28, предпочтительно находится в диапазоне от приблизительно 0,3:1 до приблизительно 40:1, более предпочтительно в диапазоне от приблизительно 5:1 до приблизительно 30:1, а наиболее предпочтительно в диапазоне от 10:1 до 25:1.
Поток жидкофазного исходного подаваемого материала, вводимый в барботажную реакторную колонну 20, может содержать небольшие количества примесей, таких как, например, толуол, этилбензол, пара-толуиловый альдегид, терефталевый альдегид, 4-карбоксибензальдегид (4-СВА), бензойная кислота, пара-толуиловая кислота, пара-толуиловый альдегид, альфа-бром-пара-толуиловая кислота, изофталевая кислота, фталевая кислота, тримеллитовая кислота, полиароматика и/или суспендированный дисперсный материал. В случае использования барботажной реакторной колонны 20 для получения терефталевой кислоты мета-ксилол и орто-ксилол также рассматриваются в качестве примесей. Предпочитается, чтобы совокупное количество примесей в потоке жидкофазного исходного подаваемого материала, вводимого в барботажную реакторную колонну 20, составляло бы величину, меньшую чем приблизительно 3 массовых процента.
Несмотря на то что Фиг. 1 иллюстрирует вариант реализации, в котором окисляемое соединение, растворитель и систему катализатора перемешивают друг с другом и вводят в барботажную реакторную колонну 20 в виде единственного потока исходного подаваемого материала, в альтернативном варианте реализации настоящего изобретения окисляемое соединение, растворитель и катализатор можно вводить в барботажную реакторную колонну 20 по отдельности. Например, поток чистого пара-ксилола можно подавать в барботажную реакторную колонну 20 через отдельное впускное отверстие, а не через впускное отверстие (отверстия) для растворителя и катализатора.
Поток преимущественно газофазного окислителя, вводимый в барботажную реакторную колонну 20 через барботер окислителя 34, включает молекулярный кислород (О2). Предпочтительно поток окислителя включает молекулярный кислород в количестве в диапазоне от приблизительно 5 до приблизительно 40 мольных процентов, более предпочтительно молекулярный кислород в количестве в диапазоне от приблизительно 15 до приблизительно 30 мольных процентов, а наиболее предпочтительно молекулярный кислород в количестве в диапазоне от 18 до 24 мольных процентов. Предпочитается, чтобы баланс потока окислителя главным образом составлял бы газ или газы, такие как азот, которые являются инертными по отношению к окислению. Более предпочтительно поток окислителя состоит по существу из молекулярного кислорода и азота. Наиболее предпочтительно поток окислителя представляет собой сухой воздух, который содержит приблизительно 21 мольный процент молекулярного кислорода и азот в количестве в диапазоне от приблизительно 78 до приблизительно 81 мольного процента. В альтернативном варианте реализации настоящего изобретения поток окислителя может включать по существу чистый кислород.
Если обратиться опять к Фиг. 1, то можно сказать, что барботажную реакторную колонну 20 предпочтительно оборудуют распределителем среды орошения 42, расположенным выше верхней поверхности 44 реакционной среды 36. Распределитель среды орошения 42 может функционировать таким образом, чтобы вводить капли потока преимущественно жидкофазной среды орошения в зону отделения 30 при использовании любого средства каплеобразования, известного на современном уровне техники. Более предпочтительно распределитель среды орошения 42 производит распыление капель сверху вниз в направлении к верхней поверхности 44 реакционной среды 36. Предпочтительно данное распыление капель в направлении сверху вниз оказывает воздействие (то есть распространяется и влияет), по меньшей мере, приблизительно на 50 процентов от максимальной площади горизонтального поперечного сечения зоны отделения 30. Более предпочтительно распыление капель оказывает воздействие, по меньшей мере, приблизительно на 75 процентов от максимальной площади горизонтального поперечного сечения зоны отделения 30. Наиболее предпочтительно распыление капель оказывает воздействие, по меньшей мере, на 90 процентов от максимальной площади горизонтального поперечного сечения зоны отделения 30. Данное распыление жидкой среды орошения в направлении сверху вниз может способствовать предотвращению вспенивания на или выше верхней поверхности 44 реакционной среды 36, а также может содействовать отделению любых капель жидкости или суспензии, захваченных в двигающемся снизу вверх газе, который перемещается в направлении выпускного отверстия для газа 40. Кроме того, жидкую среду орошения можно использовать для уменьшения количества дисперсного материала и потенциально выпадающих в осадок соединений (например, растворенных бензойной кислоты, пара-толуиловой кислоты, 4-СВА, терефталевой кислоты и металлических солей катализатора), уходящих с отходящим потоком газа, отбираемым из зоны отделения 30 через выпускное отверстие для газа 40. В дополнение к этому в результате проведения перегонки введение капель среды орошения в зону отделения 30 можно использовать для регулирования состава отходящего потока газа, отбираемого через выпускное отверстие для газа 40.
Поток жидкой среды орошения, вводимый в барботажную реакторную колонну 20 через распределитель среды орошения 42, предпочтительно имеет приблизительно тот же самый состав, что и образуемый растворителем компонент потока жидкофазного исходного подаваемого материала, вводимого в барботажную реакторную колонну 20 через впускные отверстия для исходного подаваемого материала 32а, b, c, d. Таким образом, предпочитается, чтобы поток жидкой среды орошения содержал бы кислотный компонент и воду. Кислотный компонент потока среды орошения предпочтительно представляет собой низкомолекулярную органическую монокарбоновую кислоту, содержащую 1-6 атомов углерода, более предпочтительно 2 атома углерода. Наиболее предпочтительно кислотный компонент потока среды орошения представляет собой уксусную кислоту. Предпочтительно кислотный компонент составляет, по меньшей мере, приблизительно 75 массовых процентов потока среды орошения, более предпочтительно, по меньшей мере, приблизительно 80 массовых процентов потока среды орошения, а наиболее предпочтительно от 85 до 98 массовых процентов потока среды орошения, при этом баланс составляет вода. Поскольку поток среды орошения обычно имеет по существу тот же самый состав, что и растворитель в потоке жидкофазного исходного подаваемого материала, то, когда в данном описании будут упоминать «совокупный растворитель», вводимый в реактор, такой «совокупный растворитель» должен будет включать как поток среды орошения, так и образуемую растворителем часть потока исходного подаваемого материала.
Во время жидкофазного окисления в барботажной реакторной колонне 20 предпочитается, чтобы потоки исходного подаваемого материала, окислителя и среды орошения в зону реакции 28 по существу непрерывно бы вводили, в то время как отходящие потоки газа и суспензии из зоны реакции 28 по существу непрерывно бы отбирали. В соответствии с использованием в настоящем документе термин «по существу непрерывно» должен обозначать период продолжительностью, по меньшей мере, в 10 часов с перерывами продолжительностью, меньшей чем 10 минут. Предпочитается, чтобы во время окисления окисляемое соединение (например, пара-ксилол) по существу непрерывно вводили бы в зону реакции 28 при расходе, равном, по меньшей мере, приблизительно 8000 килограммам в час, более предпочтительно при расходе в диапазоне от приблизительно 13000 до приблизительно 80000 килограммов в час, еще более предпочтительно в диапазоне от приблизительно 18000 до приблизительно 50000 килограммов в час, а наиболее предпочтительно в диапазоне от 22000 до 30000 килограммов в час. Несмотря на то что в общем случае предпочитается, чтобы расходы поступающих потоков исходного подаваемого материала, окислителя и среды орошения по существу являлись бы стационарными, в данный момент следует отметить то, что один вариант реализации настоящего изобретения предусматривает пульсацию поступающего потока исходного подаваемого материала, окислителя и/или среды орошения для того, чтобы улучшить прохождение перемешивания и массопереноса. В случае введения поступающего потока исходного подаваемого материала, окислителя и/или среды орошения в пульсирующем режиме предпочитается, чтобы их расходы варьировались бы в пределах от приблизительно 0 до приблизительно 500 процентов от упоминаемых в настоящем документе стационарных расходов, более предпочтительно в пределах от приблизительно 30 до приблизительно 200 процентов от упоминаемых в настоящем документе стационарных расходов, а наиболее предпочтительно в пределах от 80 до 120 процентов от упоминаемых в настоящем документе стационарных расходов.
Среднюю скорость реакции за один проход в единицу времени (STR) в барботажной реакторной колонне окисления 20 определяют как массу окисляемого соединения, подаваемого на единицу объема реакционной среды 36 в единицу времени (например, килограммы пара-ксилола, подаваемые на один кубический метр в час). При обычном использовании количество окисляемого соединения, не превращенного в продукт, обычно вычитали бы из количества окисляемого соединения в потоке исходного подаваемого материала перед вычислением значения STR. Однако для многих окисляемых соединений, предпочитаемых в настоящем документе (например, пара-ксилола), степени превращения и выходы обычно являются высокими, и в настоящем документе данный термин удобно определить так, как это заявляется выше. Помимо прочего, по причинам, связанным с капитальными затратами и рабочим коэффициентом заполнения, в общем случае предпочитается, чтобы реакцию проводили бы при высоком значении STR. Однако проведение реакции при значительно повышенных значениях STR может оказывать влияние на качество или выход неполного окисления. Барботажная реакторная колонна 20 является в особенности хорошо подходящей для использования тогда, когда значение STR по окисляемому соединению (например, по пара-ксилолу) находится в диапазоне от приблизительно 25 килограммов на один кубический метр в час до приблизительно 400 килограммов на один кубический метр в час, более предпочтительно в диапазоне от приблизительно 30 килограммов на один кубический метр в час до приблизительно 250 килограммов на один кубический метр в час, еще более предпочтительно от приблизительно 35 килограммов на один кубический метр в час до приблизительно 150 килограммов на один кубический метр в час, а наиболее предпочтительно в диапазоне от 40 килограммов на один кубический метр в час до 100 килограммов на один кубический метр в час.
Значение STR по кислороду в барботажной реакторной колонне окисления 20 определяют как массу молекулярного кислорода, расходуемого на единицу объема реакционной среды 36 в единицу времени (например, килограммы молекулярного кислорода, расходуемого на один кубический метр в час). Помимо прочего, по причинам, связанным с капитальными затратами и расходованием растворителя при окислении, в общем случае предпочитается, чтобы реакцию проводили бы при высоком значении STR по кислороду. Однако проведение реакции при значительно повышенных значениях STR по кислороду в конечном счете приводит к ухудшению качества или выхода неполного окисления. Не в порядке связывания себя теорией представляется, что это возможно связано со скоростью переноса молекулярного кислорода из газовой фазы в жидкость на площади межфазной поверхности и, следовательно, в объем жидкости. Чрезмерно высокое значение STR по кислороду возможно будет приводить к чрезмерно низкому уровню содержания растворенного кислорода в объеме жидкой фазы реакционной среды.
Значение глобальной средней STR по кислороду в настоящем документе определяют как массу всего кислорода, расходуемого во всем объеме реакционной среды 36 в единицу времени (например, килограммы молекулярного кислорода, расходуемого на один кубический метр в час). Барботажная реакторная колонна 20 является в особенности хорошо подходящей для использования тогда, когда значение глобальной средней STR по кислороду находится в диапазоне от приблизительно 25 килограммов на один кубический метр в час до приблизительно 400 килограммов на один кубический метр в час, более предпочтительно в диапазоне от приблизительно 30 килограммов на один кубический метр в час до приблизительно 250 килограммов на один кубический метр в час, еще более предпочтительно от приблизительно 35 килограммов на один кубический метр в час до приблизительно 150 килограммов на один кубический метр в час, а наиболее предпочтительно в диапазоне от 40 килограммов на один кубический метр в час до 100 килограммов на один кубический метр в час.
Предпочитается, чтобы во время окисления в барботажной реакторной колонне 20 соотношение между массовым расходом совокупного растворителя (из потоков как исходного подаваемого материала, так и среды орошения) и массовым расходом окисляемого соединения, поступающего в зону реакции 28, выдерживалось бы в диапазоне от приблизительно 2:1 до приблизительно 50:1, более предпочтительно в диапазоне от приблизительно 5:1 до приблизительно 40:1, а наиболее предпочтительно в диапазоне от 7,5:1 до 25:1. Предпочтительно соотношение между массовым расходом растворителя, введенного в виде части потока исходного подаваемого материала, и массовым расходом растворителя, введенного в виде части потока среды орошения, выдерживают в диапазоне от приблизительно 0,5:1 до уровня вообще нулевого расхода потока среды орошения, более предпочтительно в диапазоне от приблизительно 0,5:1 до приблизительно 4:1, еще более предпочтительно в диапазоне от приблизительно 1:1 до приблизительно 2:1, а наиболее предпочтительно в диапазоне от 1,25:1 до 1,5:1.
Предпочитается, чтобы во время жидкофазного окисления в барботажной реакторной колонне 20 поток окислителя вводили бы в барботажную реакторную колонну 20 в количестве, которое обеспечивает некоторое превышение количества молекулярного кислорода в сопоставлении со стехиометрической потребностью в кислороде. Количество избыточного молекулярного кислорода, необходимого при достижении наилучших результатов для конкретного окисляемого соединения, оказывает влияние на общие экономические показатели жидкофазного окисления. Предпочитается, чтобы во время жидкофазного окисления в барботажной реакторной колонне 20 соотношение между массовым расходом потока окислителя и массовым расходом окисляемого органического соединения (например, пара-ксилола), поступающего в реактор 20, выдерживалось бы в диапазоне от приблизительно 0,5:1 до приблизительно 20:1, более предпочтительно в диапазоне от приблизительно 1:1 до приблизительно 10:1, а наиболее предпочтительно в диапазоне от 2:1 до 6:1.
Если обратиться опять к Фиг. 1, то можно сказать, что потоки исходного подаваемого материала, окислителя и среды орошения, вводимые в барботажную реакторную колонну 20, совместно образуют, по меньшей мере, часть многофазной реакционной среды 36. Реакционная среда 36 предпочтительно представляет собой трехфазную среду, включающую твердую фазу, жидкую фазу и газовую фазу. Как упоминалось выше, окисление окисляемого соединения (например, пара-ксилола) происходит преимущественно в жидкой фазе реакционной среды 36. Таким образом, жидкая фаза реакционной среды 36 содержит растворенный кислород и окисляемое соединение. Экзотермическая природа реакции окисления, которая протекает в барботажной реакторной колонне 20, вызывает вскипание/испарение части растворителя (например, уксусной кислоты и воды), введенного через впускные отверстия для исходного подаваемого материала 32а, b, c, d. Таким образом, газовая фаза реакционной среды 36 в реакторе 20 образуется главным образом из испарившегося растворителя и не растворившейся и не вступившей в реакцию части потока окислителя. Определенные реакторы окисления предшествующего уровня техники используют теплообменные трубки/ребра для нагревания или охлаждения реакционной среды. Однако такие теплообменные конструкции могут оказаться нежелательными в реакторе и способе изобретения, описанных в настоящем документе. Таким образом, предпочитается, чтобы барботажная реакторная колонна 20 по существу не включала бы поверхностей, которые находятся в контакте с реакционной средой 36 и демонстрируют средневременную плотность теплового потока, превышающую 30000 ватт на один квадратный метр. В дополнение к этому предпочитается, чтобы при помощи поверхностей теплообмена отводили бы менее, чем приблизительно 50 процентов от средневременной теплоты реакции в реакционной среде 36, более предпочтительно при помощи поверхностей теплообмена отводили бы менее чем приблизительно 30 процентов от теплоты реакции, а наиболее предпочтительно при помощи поверхностей теплообмена отводили бы менее чем 10 процентов от теплоты реакции.
Концентрацию растворенного кислорода в жидкой фазе реакционной среды 36 формирует динамическое равновесие между скоростью массопереноса из газовой фазы и скоростью расходования за счет реакции в жидкой фазе (то есть она не просто устанавливается парциальным давлением молекулярного кислорода в поступающей газовой фазе, хотя это - один фактор в скорости подачи растворенного кислорода, и он на самом деле оказывает влияние на предельную верхнюю концентрацию растворенного кислорода). Количество растворенного кислорода локально варьируется, при этом оно будет выше поблизости от межфазных поверхностей пузырьков. В целом количество растворенного кислорода зависит от баланса между факторами подачи и потребности в различных частях реакционной среды 36. Во времени количество растворенного кислорода зависит от однородности перемешивания газа и жидкости по отношению к скоростям химического расходования. При проектировании, обеспечивающем достижение надлежащего соответствия между подачей и потребностью в отношении растворенного кислорода в жидкой фазе реакционной среды 36, предпочитается, чтобы средневременная и среднеобъемная концентрация кислорода в жидкой фазе реакционной среды 36 выдерживалась бы на уровне, превышающем приблизительно 1 ч./млн (моль.), более предпочтительно находящемся в диапазоне от приблизительно 4 до приблизительно 1000 ч./млн (моль.), еще более предпочтительно в диапазоне от приблизительно 8 до приблизительно 500 ч./млн (моль.), а наиболее предпочтительно в диапазоне от 12 до 120 ч./млн (моль.).
Реакция жидкофазного окисления, проводимая в барботажной реакторной колонне 20, предпочтительно представляет собой реакцию осаждения, в ходе которой образуется твердая фаза. Более предпочтительно жидкофазное окисление, проводимое в барботажной реакторной колонне 20, приводит к тому, что твердое соединение (например, частицы сырой неочищенной терефталевой кислоты) в реакционной среде 36 будут образовывать, по меньшей мере, приблизительно 10 массовых процентов окисляемого соединения (например, пара-ксилола), вводимого в зону реакции 28. Еще более предпочтительно жидкофазное окисление приводит к тому, что твердое соединение в реакционной среде 36 будут образовывать, по меньшей мере, приблизительно 50 массовых процентов окисляемого соединения. Наиболее предпочтительно жидкофазное окисление приводит к тому, что твердое соединение в реакционной среде 36 будут образовывать, по меньшей мере, 90 массовых процентов окисляемого соединения. Предпочитается, чтобы совокупное количество твердой фазы в реакционной среде 36 превышало бы приблизительно 3 массовых процента при выражении через средневременные и среднеобъемные величины. Более предпочтительно совокупное количество твердой фазы в реакционной среде 36 выдерживают в диапазоне от приблизительно 5 до приблизительно 40 массовых процентов, еще более предпочтительно в диапазоне от приблизительно 10 до приблизительно 35 массовых процентов, а наиболее предпочтительно в диапазоне от 15 до 30 массовых процентов. Предпочитается, чтобы существенная часть продукта окисления (например, терефталевой кислоты), полученного в барботажной реакторной колонне 20, присутствовала бы в реакционной среде 36 в виде твердой фазы в противоположность веществу, остающемуся растворенным в жидкой фазе реакционной среды 36. Количество твердофазного продукта окисления, присутствующего в реакционной среде 36, предпочтительно составляет, по меньшей мере, приблизительно 25 массовых процентов от совокупного (твердо- и жидкофазного) продукта окисления в реакционной среде 36, более предпочтительно, по меньшей мере, приблизительно 75 массовых процентов от совокупного продукта окисления в реакционной среде 36, а наиболее предпочтительно, по меньшей мере, 95 массовых процентов от совокупного продукта окисления в реакционной среде 36. Численные диапазоны, приведенные выше для количества твердой фазы в реакционной среде 36, используются для по существу стационарного режима функционирования барботажной колонны 20 в течение по существу непрерывного периода времени, а не для запуска, остановки или не вполне оптимального режима функционирования барботажной реакторной колонны 20. Количество твердой фазы в реакционной среде 36 определяют при использовании гравиметрического метода. В данном гравиметрическом методе представительную часть суспензии отбирают из реакционной среды и взвешивают. В условиях, которые эффективно обеспечивают выдерживание общего разделения твердой и жидкой фаз, протекающего в реакционной среде, свободную жидкость из части, образуемой твердой фазой, эффективно удаляют при использовании седиментации или фильтрования, без потерь осажденной твердой фазы и при сохранении в части, образуемой твердой фазой, менее чем приблизительно 10 процентов от первоначальной массы жидкости. Остающуюся жидкость в твердой фазе эффективно выпаривают до сухости без сублимации твердой фазы. Оставшуюся часть, образуемую твердой фазой, взвешивают. Соотношение между массой части, образуемой твердой фазой, и массой первоначальной части, образуемой суспензией, представляет собой долю твердой фазы, обычно выражаемую через процентное содержание.
Реакция осаждения, проводимая в барботажной реакторной колонне 20, может приводить к возникновению обрастания (то есть накоплению отложений твердой фазы) на поверхности определенных жестких конструкций, которые находятся в контакте с реакционной средой 36. Таким образом, в одном варианте реализации настоящего изобретения предпочитается, чтобы барботажная реакторная колонна 20 в зоне реакции 28 по существу не включала бы каких-либо внутренних теплообменных, перемешивающих или перегораживающих конструкций, поскольку такие конструкции были бы подвержены возникновению обрастания. Если в зоне реакции 28 будут присутствовать внутренние конструкции, то тогда желательно было бы избежать наличия внутренних конструкций, имеющих внешние поверхности, которые характеризуются значительной величиной площади обращенных кверху плоскостных поверхностей, поскольку такие обращенные кверху плоскостные поверхности были бы в исключительно высокой степени подвержены возникновению обрастания. Таким образом, если в зоне реакции 28 будут присутствовать какие-либо внутренние конструкции, то тогда предпочитается, чтобы по существу плоскостными поверхностями, наклоненными под углом, меньшим чем приблизительно на 15 градусов от горизонтали, были бы образованы менее чем приблизительно 20 процентов от совокупной площади обращенных кверху обнаженных внешних поверхностей таких внутренних конструкций.
Если обратиться опять к Фиг. 1, то можно сказать, что физическая конфигурация барботажной реакторной колонны 20 способствует обеспечению проведения оптимизированного окисления окисляемого соединения (например, пара-ксилола) при минимальном образовании примесей. Предпочитается, чтобы удлиненная секция реакции 24 оболочки емкости 22 включала бы по существу цилиндрическое основное тело 46 и нижнее днище 48. Верхний край зоны реакции 28 определяется горизонтальной плоскостью 50, проходящей через верх цилиндрического основного тела 46. Нижний край 52 зоны реакции 28 определяется самой нижней внутренней поверхностью нижнего днища 48. Обычно нижний край 52 зоны реакции 28 располагается поблизости от устья выпускного отверстия для суспензии 38. Таким образом, удлиненная зона реакции 28, определяемая внутри барботажной реакторной колонны 20, имеет максимальную длину «L», измеряемую от верхнего края 50 до нижнего края 52 зоны реакции 28 вдоль продольной оси цилиндрического основного тела 46. Длина «L» зоны реакции 28 предпочтительно находится в диапазоне от приблизительно 10 до приблизительно 100 метров, более предпочтительно в диапазоне от приблизительно 20 до приблизительно 75 метров, а наиболее предпочтительно в диапазоне от 25 до 50 метров. Зона реакции 28 имеет максимальный диаметр (ширину) «D», который обычно равен максимальному внутреннему диаметру цилиндрического основного тела 46. Максимальный диаметр «D» зоны реакции 28 предпочтительно находится в диапазоне от приблизительно 1 до приблизительно 12 метров, более предпочтительно в диапазоне от приблизительно 2 до приблизительно 10 метров, еще более предпочтительно в диапазоне от приблизительно 3,1 до приблизительно 9 метров, а наиболее предпочтительно в диапазоне от 4 до 8 метров. В предпочтительном варианте реализации настоящего изобретения зона реакции 28 характеризуется соотношением длины к диаметру «L:D» в диапазоне от приблизительно 6:1 до приблизительно 30:1. Еще более предпочтительно зона реакции 28 характеризуется соотношением L:D в диапазоне от приблизительно 8:1 до приблизительно 20:1. Наиболее предпочтительно зона реакции 28 характеризуется соотношением L:D в диапазоне от 9:1 до 15:1.
Как это обсуждается выше, зона реакции 28 барботажной реакторной колонны 20 вмещает многофазную реакционную среду 36. Реакционная среда 36 имеет нижний край, совпадающий с нижним краем 52 зоны реакции 28, и верхний край, расположенный на уровне верхней поверхности 44. Верхняя поверхность 44 реакционной среды 36 определяется положением вдоль горизонтальной плоскости, которая проходит через зону реакции 28 в позиции по вертикали, где содержимое зоны реакции 28 переходит из газофазного сплошного состояния в жидкофазное сплошное состояние. Верхняя поверхность 44 предпочтительно располагается в позиции по вертикали, где локальная средневременная величина удерживания газа в тонкой горизонтальной доле содержимого зоны реакции 28 составляет 0,9.
Реакционная среда 36 имеет максимальную высоту «Н», измеренную между ее верхним и нижним краями. Максимальная ширина «W» реакционной среды 36 обычно равна максимальному диаметру «D» цилиндрического основного тела 46. Предпочитается, чтобы во время жидкофазного окисления в барботажной реакторной колонне 20 величину Н выдерживали бы в диапазоне от приблизительно 60 до приблизительно 120 процентов от L, более предпочтительно от приблизительно 80 до приблизительно 110 процентов от L, а наиболее предпочтительно от 85 до 100 процентов от L. В предпочтительном варианте реализации настоящего изобретения реакционная среда 36 характеризуется соотношением высоты к ширине «Н:W», превышающим приблизительно 3:1. Более предпочтительно реакционная среда 36 характеризуется соотношением Н:W в диапазоне от приблизительно 7:1 до приблизительно 25:1. Еще более предпочтительно реакционная среда 36 характеризуется соотношением Н:W в диапазоне от приблизительно 8:1 до приблизительно 20:1. Наиболее предпочтительно реакционная среда 36 характеризуется соотношением Н:W в диапазоне от 9:1 до 15:1. В одном варианте реализации изобретения L=H и D=W, так что различные размеры или соотношения, представленные в настоящем документе для L и D, также относятся к Н и W и наоборот.
Относительно высокие соотношения L:D и Н:W, предусматриваемые в соответствии с одним вариантом реализации изобретения, могут вносить свой вклад в некоторые существенные преимущества системы изобретения. Как обсуждается более подробно далее, было обнаружено то, что более высокие соотношения L:D и H:W, а также определенные другие признаки, обсуждающиеся далее, могут способствовать созданию выгодных вертикальных градиентов в концентрациях молекулярного кислорода и/или окисляемого соединения (например, пара-ксилола) в реакционной среде 36. В противоположность общепринятой точке зрения, согласно которой считалось бы выгодным иметь хорошо перемешанную реакционную среду при относительно однородных концентрациях по всему объему, было обнаружено то, что разбиение по вертикали на ступени в отношении концентраций кислорода и/или окисляемого соединения облегчает более эффективное и экономичное прохождение реакции окисления. Сведение к минимуму концентраций кислорода и окисляемого соединения поблизости от верха реакционной среды 36 может способствовать предотвращению потерь не вступившего в реакцию кислорода и не вступившего в реакцию окисляемого соединения в результате уноса через верхнее выпускное отверстие для газа 40. Однако если концентрации окисляемого соединения и не вступившего в реакцию кислорода будут низкими по всему объему реакционной среды 36, то тогда скорость и/или селективность окисления уменьшатся. Таким образом, предпочитается, чтобы концентрации молекулярного кислорода и/или окисляемого соединения были бы значительно выше поблизости от низа реакционной среды 36 в сопоставлении с тем, что имеет место поблизости от верха реакционной среды 36.
В дополнение к этому высокие соотношения L:D и H:W приводят к тому, что давление низа реакционной среды 36 будет существенно превышать давление верха реакционной среды 36. Данный градиент давления по вертикали представляет собой следствие высоты и плотности реакционной среды 36. Одно преимущество наличия данного градиента давления по вертикали заключается в том, что повышенное давление низа емкости становится движущей силой, обеспечивающей достижение более значительных растворимости и массопереноса кислорода в сопоставлении с тем, чего можно было бы добиться в других случаях при сопоставимых температурах и давлениях верха реактора в мелких реакторах. Таким образом, реакцию окисления можно проводить при более низких температурах в сопоставлении с тем, что потребовалось бы в более мелкой емкости. В случае использования барботажной реакторной колонны 20 для неполного окисления пара-ксилола до получения сырой неочищенной терефталевой кислоты (СТА) способность функционирования при пониженных температурах реакции при тех же самых или лучших скоростях массопереноса кислорода позволяет воспользоваться несколькими преимуществами. Например, низкотемпературное окисление пара-ксилола приводит к уменьшению количества растворителя, сжигаемого во время прохождения реакции. Как обсуждается более подробно далее, низкотемпературное окисление также благоприятствует образованию небольших, характеризующихся большой площадью удельной поверхности, неплотно связанных, легко растворимых частиц СТА, которые могут быть подвергнуты воздействию более экономичных методик очистки в сопоставлении с крупными, характеризующимися малой площадью удельной поверхности, плотными частицами СТА, получаемыми в соответствии с обычно используемыми способами высокотемпературного окисления.
Предпочитается, чтобы во время окисления в реакторе 20 средневременную и среднеобъемную температуру реакционной среды 36 выдерживали бы в диапазоне от приблизительно 125 до приблизительно 200°С, более предпочтительно в диапазоне от приблизительно 140 до приблизительно 180°С, а наиболее предпочтительно в диапазоне от 150 до 170°С. Давление верха реактора над реакционной средой 36 предпочтительно выдерживают в диапазоне от приблизительно 1 до приблизительно 20 бар избыточного давления (бар (изб.)), более предпочтительно в диапазоне от приблизительно 2 до приблизительно 12 бар (изб.), а наиболее предпочтительно в диапазоне от 4 до 8 бар (изб.). Предпочтительно разность давлений между верхом реакционной среды 36 и низом реакционной среды 36 находится в диапазоне от приблизительно 0,4 до приблизительно 5 бар, более предпочтительно разность давлений находится в диапазоне от приблизительно 0,7 до приблизительно 3 бар, а наиболее предпочтительно разность давлений находится в диапазоне от 1 до 2 бар. Несмотря на то что в общем случае предпочитается, чтобы давление верха реактора над реакционной средой 36 выдерживалось бы на уровне относительно постоянного значения, один вариант реализации настоящего изобретения предусматривает наличие пульсации давления верха реактора для облегчения улучшенного прохождения перемешивания и/или массопереноса в реакционной среде 36. В случае наличия пульсаций давления верха реактора предпочитается, чтобы пульсации давления находились бы в диапазоне от приблизительно 60 до приблизительно 140 процентов от упоминавшегося в настоящем документе стационарного давления верха реактора, более предпочтительно от приблизительно 85 до приблизительно 115 процентов от упоминавшегося в настоящем документе стационарного давления верха реактора, а наиболее предпочтительно от 95 до 105 процентов от упоминавшегося в настоящем документе стационарного давления верха реактора.
Дополнительное преимущество высокого соотношения L:D для зоны реакции 28 заключается в том, что оно может вносить свой вклад в увеличение среднего расхода на единицу сечения потока для реакционной среды 36. Термины «расход на единицу сечения потока» и «расход газа на единицу сечения потока» в соответствии с использованием в настоящем документе в отношении реакционной среды 36 должны обозначать объемный расход газовой фазы реакционной среды 36 на некотором уровне по высоте в реакторе, поделенный на площадь горизонтального поперечного сечения реактора на данном уровне по высоте. Повышенный расход на единицу сечения потока, достигаемый благодаря высокому соотношению L:D для зоны реакции 28, может способствовать прохождению локального перемешивания и увеличению удерживания газа в реакционной среде 36. Средневременные расходы на единицу сечения потока для реакционной среды 36 на одной четверти высоты, половине высоты и/или трех четвертях высоты реакционной среды 36 предпочтительно превышают приблизительно 0,3 метра в секунду, более предпочтительно находятся в диапазоне от приблизительно 0,8 до приблизительно 5 метров в секунду, еще более предпочтительно в диапазоне от приблизительно 0,9 до приблизительно 4 метров в секунду, а наиболее предпочтительно в диапазоне от 1 до 3 метров секунду.
Если обратиться опять к Фиг. 1, то можно сказать, что секция отделения 26 барботажной реакторной колонны 20 представляет собой просто уширенную часть оболочки емкости 22, расположенную непосредственно над секцией реакции 24. Секция отделения 26 обеспечивает уменьшение скорости перемещающейся снизу вверх газовой фазы в барботажной реакторной колонне 20 тогда, когда газовая фаза поднимется выше верхней поверхности 44 реакционной среды 36 и приблизится к выпускному отверстию для газа 40. Данное уменьшение скорости перемещения газовой фазы снизу вверх способствует облегчению удаления жидкой и/или твердой фаз, захваченных в перетекающей снизу вверх газовой фазе, и, таким образом, приводит к уменьшению нежелательных потерь определенных компонентов, присутствующих в жидкой фазе реакционной среды 36.
Секция отделения 26 предпочтительно включает переходную стенку, в общем случае образующую поверхность усеченного конуса, 54, в общем случае цилиндрическую широкую боковую стенку 56 и верхнее днище 58. Узкий нижний край переходной стенки 54 соединяется с верхом цилиндрического основного тела 46 секции реакции 24. Широкий верхний край переходной стенки 54 соединяется с низом широкой боковой стенки 56. Предпочитается, чтобы переходная стенка 54 проходила бы снизу вверх и изнутри наружу от ее узкого нижнего края под углом в диапазоне от приблизительно 10 до приблизительно 70 градусов от вертикали, более предпочтительно в диапазоне от приблизительно 15 до приблизительно 50 градусов от вертикали, а наиболее предпочтительно в диапазоне от 15 до 45 градусов от вертикали. Широкая боковая стенка 56 имеет максимальный диаметр «Х», который в общем случае превышает максимальный диаметр «D» секции реакции 24, хотя если верхняя часть секции реакции 24 будет иметь меньший диаметр в сопоставлении с совокупным максимальным диаметром секции реакции 24, то тогда Х фактически может составить величину, меньшую чем D. В предпочтительном варианте реализации настоящего изобретения соотношение между диаметром широкой боковой стенки 56 и максимальным диаметром секции реакции 24«Х:D» находится в диапазоне от приблизительно 0,8:1 до приблизительно 4:1, наиболее предпочтительно в диапазоне от 1,1:1 до 2:1. Верхнее днище 58 соединяется с верхом широкой боковой стенки 56. Верхнее днище 58 предпочтительно представляет собой элемент в общем случае в виде эллиптического днища, определяющий центральное отверстие, которое делает возможным уход газа из зоны отделения 30 через выпускное отверстие для газа 40. В альтернативном варианте реализации верхнее днище 58 может иметь любую форму, в том числе коническую. Зона отделения 30 имеет максимальную высоту «Y», измеренную от верха 50 зоны реакции 28 самой верхней части зоны отделения 30. Соотношение между длиной зоны реакции 28 и высотой зоны отделения 30 «L:Y» предпочтительно находится в диапазоне от приблизительно 2:1 до приблизительно 24:1, более предпочтительно в диапазоне от приблизительно 3:1 до приблизительно 20:1, а наиболее предпочтительно в диапазоне от 4:1 до 16:1.
Если обратиться теперь к Фиг. 1-5, то можно сказать, что теперь будет более подробно обсуждаться местоположение и конфигурация барботера окислителя 34. Фиг. 2 и 3 демонстрируют то, что барботер окислителя 34 может включать кольцевой элемент 60, поперечный элемент 62 и пару каналов для ввода окислителя 64а, b. В удобном варианте данные каналы для ввода окислителя 64а, b могут входить в емкость на уровне по высоте, находящемся выше кольцевого элемента 60, а после этого поворачиваться книзу так, как это продемонстрировано на Фиг. 2 и 3. В альтернативном варианте канал для ввода окислителя 64а, b может входить в емкость ниже кольцевого элемента 60 или приблизительно на той же самой горизонтальной плоскости, что и кольцевой элемент 60. Каждый канал для ввода окислителя 64а, b включает первый край, соединенный с соответствующим впускным отверстием для окислителя 66а, b, сформированным в оболочке емкости 22, и второй край, через текучую среду соединяющийся с кольцевым элементом 60. Кольцевой элемент 60 предпочтительно образован из каналов, более предпочтительно из множества прямых секций каналов, а наиболее предпочтительно множества прямых трубных секций, жестко соединенных друг с другом до получения трубчатого многоугольного кольца. Предпочтительно кольцевой элемент 60 образован, по меньшей мере, из 3 прямых трубных секций, более предпочтительно из 6-10 трубных секций, а наиболее предпочтительно из 8 трубных секций. В соответствии с этим если кольцевой элемент 60 будет образован из 8 трубных секций, то тогда он будет иметь в общем случае восьмиугольную конфигурацию. Поперечный элемент 62 предпочтительно получают из по существу прямой трубной секции, которая через текучую среду соединяется с противоположными трубными секциями кольцевого элемента 60 и проходит по диагонали между ними. Трубная секция, используемая для поперечного элемента 62, предпочтительно имеет по существу тот же самый диаметр, что и трубные секции, используемые для получения кольцевого элемента 60. Предпочитается, чтобы трубные секции, которые составляют каналы для ввода окислителя 64а, b, кольцевой элемент 60 и поперечный элемент 62, имели бы номинальный диаметр, больший чем приблизительно 0,1 метра, более предпочтительно находящийся в диапазоне от приблизительно 0,2 до приблизительно 2 метров, а наиболее предпочтительно в диапазоне от 0,25 до 1 метра. Как, может быть, наилучшим образом проиллюстрировано на Фиг. 3, каждый элемент, выбираемый из кольцевого элемента 60 и поперечного элемента 62, характеризуется наличием множества верхних отверстий для окислителя 68, предназначенных для выпуска потока окислителя снизу вверх в зону реакции 28. Как, может быть, лучше всего проиллюстрировано на Фиг. 4, кольцевой элемент 60 и/или поперечный элемент 62 могут характеризоваться наличием одного или нескольких нижних отверстий для окислителя 70, предназначенных для выпуска потока окислителя сверху вниз в зону реакции 28. Нижние отверстия для окислителя 70 также могут быть использованы для выпуска жидкой и/или твердой фаз, которые могут проникать внутрь кольцевого элемента 60 и/или поперечного элемента 62. Для того чтобы предотвратить накопление твердой фазы внутри барботера окислителя 34, через барботер 34 можно непрерывно или периодически перепускать поток жидкости для вымывания любых количеств накопившейся твердой фазы.
Если обратиться опять к Фиг. 1-4, то можно сказать, что во время окисления в барботажной реакторной колонне 20 потоки окислителя перепускают через впускные отверстия для окислителя 66а, b в каналы для ввода окислителя 64а, b соответственно. После этого потоки окислителя транспортируют через каналы для ввода окислителя 64а, b в кольцевой элемент 60. Как только поток окислителя поступит в кольцевой элемент 60, поток окислителя будет распределяться по всем внутренним объемам кольцевого элемента 60 и поперечного элемента 62. После этого поток окислителя вытесняется из барботера окислителя 34 в зону реакции 28 через верхние и нижние отверстия для окислителя 68, 70 кольцевого элемента 60 и поперечного элемента 62.
Устья верхних отверстий для окислителя 68 разнесены одно от другого в боковом направлении и располагаются по существу на одном и том же уровне по высоте в зоне реакции 28. Таким образом, устья верхних отверстий для окислителя 68 в общем случае располагаются на по существу горизонтальной плоскости, определенной верхом барботера окислителя 34. Устья нижних отверстий для окислителя 70 разнесены в боковом направлении одно от другого и располагаются по существу на одном и том же уровне по высоте в зоне реакции 28. Таким образом, устья нижних отверстий для окислителя 70 в общем случае располагаются на по существу горизонтальной плоскости, определенной низом барботера окислителя 34.
В одном варианте реализации настоящего изобретения барботер окислителя 34 имеет, по меньшей мере, приблизительно 20 верхних отверстий для окислителя 68, сформированных в нем. Более предпочтительно барботер окислителя 34 имеет сформированные в нем верхние отверстия для окислителя в количестве в диапазоне от приблизительно 40 до приблизительно 800. Наиболее предпочтительно барботер окислителя 34 имеет сформированные в нем верхние отверстия для окислителя 68 в количестве в диапазоне от 60 до 400. Барботер окислителя 34 предпочтительно имеет, по меньшей мере, приблизительно 1 нижнее отверстие для окислителя 70, сформированное в нем. Более предпочтительно барботер окислителя 34 имеет сформированные в нем нижние отверстия для окислителя 70 в количестве в диапазоне от приблизительно 2 до приблизительно 40. Наиболее предпочтительно барботер окислителя 34 имеет сформированные в нем нижние отверстия для окислителя 70 в количестве в диапазоне от 8 до 20. Соотношение между количествами верхних отверстий для окислителя 68 и нижних отверстий для окислителя 70 в барботере окислителя 34 предпочтительно находится в диапазоне от приблизительно 2:1 до приблизительно 100:1, более предпочтительно в диапазоне от приблизительно 5:1 до приблизительно 25:1, а наиболее предпочтительно в диапазоне от 8:1 до 15:1. Диаметры по существу всех верхних и нижних отверстий для окислителя 68, 70 предпочтительно являются по существу одинаковыми, так что соотношение между объемными расходами потока окислителя из верхних и нижних отверстий 68, 70 является по существу тем же самым, что и приведенные выше соотношения для относительных количеств верхних и нижних отверстий для окислителя 68, 70.
Фиг. 5 иллюстрирует направление выпуска окислителя из верхних и нижних отверстий для окислителя 68, 70. Что касается верхних отверстий для окислителя 68, то предпочитается, чтобы, по меньшей мере, часть верхних отверстий для окислителя 68 производила бы выпуск потока окислителя под углом «А» к вертикали. Предпочитается, чтобы процентная доля верхних отверстий для окислителя 68, которые ориентированы под углом «А» к вертикали, находилась бы в диапазоне от приблизительно 30 до приблизительно 90 процентов, более предпочтительно в диапазоне от приблизительно 50 до приблизительно 80 процентов, еще более предпочтительно в диапазоне от 60 до 75 процентов, а наиболее предпочтительно была бы равна приблизительно 67 процентам. Угол «А» предпочтительно находится в диапазоне от приблизительно 5 до приблизительно 60 градусов, более предпочтительно в диапазоне от приблизительно 10 до приблизительно 45 градусов, а наиболее предпочтительно в диапазоне от 15 до 30 градусов. Что касается нижних отверстий для окислителя 70, то предпочитается, чтобы по существу все нижние отверстия для окислителя 70 располагались бы поблизости от самой нижней части кольцевого элемента 60 и/или поперечного элемента 62. Таким образом, любые количества жидкой и/или твердой фаз, которые могут ненамеренно попасть в барботер окислителя 34, можно будет легко выпустить из барботера окислителя 34 через нижние отверстия для окислителя 70. Предпочтительно нижние отверстия для окислителя 70 обеспечивают выпуск потока окислителя сверху вниз по существу под углом, соответствующим вертикали. Для целей данного описания верхним отверстием для окислителя может быть любое отверстие, которое обеспечивает выпуск потока окислителя в общем случае в направлении снизу вверх (то есть под углом, отсчитываемым вверх от горизонтали), а нижним отверстием для окислителя может быть любое отверстие, которое обеспечивает выпуск потока окислителя в общем случае в направлении сверху вниз (то есть под углом, отсчитываемым вниз от горизонтали).
Во многих обычно используемых барботажных реакторных колоннах, содержащих многофазную реакционную среду, по существу вся реакционная среда, расположенная ниже барботера окислителя (или другого механизма, предназначенного для введения потока окислителя в зону реакции), характеризуется очень низкой величиной удерживания газа. Как известно на современном уровне техники, «удерживание газа» представляет собой просто объемную долю многофазной среды, которая находится в газообразном состоянии. Зоны низкой величины удерживания газа в среде также можно называть «неаэрированными» зонами. Во многих обычно используемых суспензионных барботажных реакторных колоннах значительная доля от совокупного объема реакционной среды располагается ниже барботера окислителя (или другого механизма, предназначенного для введения потока окислителя в зону реакции). Таким образом, значительная часть реакционной среды, присутствующей в области низа обычно используемых барботажных реакторных колонн, является неаэрированной.
Было обнаружено то, что сведение к минимуму количества неаэрированных зон в реакционной среде, подвергаемой окислению в барботажной реакторной колонне, может обеспечить сведение к минимуму образования определенных типов нежелательных примесей. Неаэрированные зоны реакционной среды содержат относительно малое количество пузырьков окислителя. Данный малый объем пузырьков окислителя приводит к уменьшению количества молекулярного кислорода, доступного для растворения в жидкой фазе реакционной среды. Таким образом, жидкая фаза в неаэрированной зоне реакционной среды характеризуется относительно низкой концентрацией молекулярного кислорода. Данные обедненные кислородом неаэрированные зоны реакционной среды имеют тенденцию к стимулированию прохождения нежелательных побочных реакций, вместо желательной реакции окисления. Например, в случае неполного окисления пара-ксилола до получения терефталевой кислоты недостаточная доступность кислорода в жидкой фазе реакционной среды может привести к образованию нежелательно больших количеств бензойной кислоты и сопряженных ароматических колец, в частности, в том числе в высшей степени нежелательных молекул окрашенных веществ, известных как флуореноны и антрахиноны.
В соответствии с одним вариантом реализации настоящего изобретения жидкофазное окисление проводят в барботажной реакторной колонне, сконфигурированной и функционирующей таким образом, чтобы свести к минимуму объемную долю реакционной среды, характеризующейся низкими величинами удерживания газа. Данное сведение к минимуму количества неаэрированных зон можно количественно охарактеризовать в результате теоретического разделения совокупного объема реакционной среды на 2000 дискретных горизонтальных долей с одинаковым объемом. За исключением самой верхней и самой нижней горизонтальных долей, каждая горизонтальная доля представляет собой дискретный объем, ограниченный по его боковым сторонам боковой стенкой реактора и ограниченный по его верхней и нижней сторонам воображаемыми горизонтальными плоскостями. Самая верхняя горизонтальная доля ограничена по ее нижней стороне воображаемой горизонтальной плоскостью, а по ее верхней стороне - верхней поверхностью реакционной среды. Самая нижняя горизонтальная доля ограничена по ее верхней стороне воображаемой горизонтальной плоскостью, а по ее нижней стороне - нижним краем емкости. Как только реакционная среда будет теоретически разделена на 2000 дискретных горизонтальных долей с равным объемом, можно будет определить средневременную и среднеобъемную величину удерживания газа для каждой горизонтальной доли. В случае использования данного способа определения количества неаэрированных зон предпочитается, чтобы количество горизонтальных долей, характеризующихся средневременной и среднеобъемной величиной удерживания газа, меньшей 0,1, составляло бы величину, меньшую 30, более предпочтительно меньшую 15, еще более предпочтительно меньшую 6, даже более предпочтительно меньшую 4, а наиболее предпочтительно меньшую 2. Предпочитается, чтобы количество горизонтальных долей, характеризующихся величиной удерживания газа, меньшей 0,2, составляло бы величину, меньшую 80, более предпочтительно меньшую 40, еще более предпочтительно меньшую 20, даже более предпочтительно меньшую 12, а наиболее предпочтительно меньшую 5. Предпочитается, чтобы количество горизонтальных долей, характеризующихся величиной удерживания газа, меньшей 0,3, составляло бы величину, меньшую 120, более предпочтительно меньшую 80, еще более предпочтительно меньшую 40, даже более предпочтительно меньшую 20, а наиболее предпочтительно меньшую 15.
Если обратиться опять к Фиг. 1 и 2, то было обнаружено то, что более низкое расположение барботера окислителя 34 в зоне реакции 28 обеспечивает достижение нескольких преимуществ, включающих уменьшение количества неаэрированных зон в реакционной среде 36. При данных высоте «Н» реакционной среды 36, длине «L» зоны реакции 28 и максимальном диаметре «D» зоны реакции 28 предпочитается, чтобы основную часть (то есть>50 массовых процентов) потока окислителя вводили бы в зону реакции 28 в пределах приблизительно 0,025Н, 0,022L и/или 0,25D от нижнего края 52 зоны реакции 28. Более предпочтительно основную часть потока окислителя вводят в зону реакции 28 в пределах приблизительно 0,02Н, 0,018L и/или 0,2D от нижнего края 52 зоны реакции 28. Наиболее предпочтительно основную часть потока окислителя вводят в зону реакции 28 в пределах 0,015Н, 0,013L и/или 0,15D от нижнего края 52 зоны реакции 28.
В варианте реализации, проиллюстрированном на Фиг. 2, расстояние по вертикали «Y1» между нижним краем 52 зоны реакции 28 и устьями верхних отверстий для окислителя 68 барботера окислителя 34 составляет величину, меньшую приблизительно 0,25Н, 0,022L и/или 0,25D, так что по существу весь поток окислителя поступает в зону реакции 28 в пределах приблизительно 0,25Н, 0,022L и/или 0,25D от нижнего края 52 зоны реакции 28. Более предпочтительно Y1 составляет величину, меньшую чем приблизительно 0,02Н, 0,018L и/или 0,2D. Наиболее предпочтительно Y1 составляет величину, меньшую чем 0,015Н, 0,013L и/или 0,15D, но большую чем 0,005Н, 0,004L и/или 0,06D. Фиг. 2 иллюстрирует линию начала изгиба 72 в позиции, в которой нижний край цилиндрического основного тела 46 оболочки емкости 22 соединяется с верхним краем эллиптического нижнего днища 48 оболочки емкости 22. В альтернативном варианте нижнее днище 48 может иметь любую форму, в том числе коническую, а линию начала изгиба все еще определяют как нижний край цилиндрического основного тела 46. Расстояние по вертикали «Y2» между линией начала изгиба 72 и верхом барботера окислителя 34 предпочтительно составляет, по меньшей мере, приблизительно 0,0012Н, 0,001L и/или 0,01D; более предпочтительно, по меньшей мере, приблизительно 0,005Н, 0,004L и/или 0,05D; а наиболее предпочтительно, по меньшей мере, 0,01Н, 0,008L и/или 0,1D. Расстояние по вертикали «Y3» между нижним краем 52 зоны реакции 28 и устьями нижних отверстий для окислителя 70 барботера окислителя 34 предпочтительно составляет величину, меньшую чем приблизительно 0,015Н, 0,013L и/или 0,15D; более предпочтительно меньшую чем приблизительно 0,012Н, 0,01L и/или 0,1D; а наиболее предпочтительно меньшую чем 0,01Н, 0,008L и/или 0,075D, но большую чем 0,003Н, 0,002L и/или 0,025D.
В предпочтительном варианте реализации настоящего изобретения отверстия, которые обеспечивают выпуск потока окислителя и потока исходного подаваемого материала в зону реакции, имеют такую конфигурацию, чтобы (массовая) величина потока окислителя или исходного подаваемого материала, выпускаемого из отверстия, была бы прямо пропорциональна площади живого сечения отверстия. Таким образом, например, если 50 процентов от совокупной площади живого сечения, определенной всеми отверстиями для окислителя, будут располагаться в пределах 0,15D от низа зоны реакции, то тогда в зону реакции в пределах 0,15D от низа зоны реакции будут поступать 50 массовых процентов потока окислителя и наоборот.
В дополнение к преимуществам, обеспечиваемым в результате сведения к минимуму количества неаэрированных зон (то есть зон, характеризующихся низкой величиной удерживания газа) в реакционной среде 36, было обнаружено то, что прохождение окисления можно улучшить в результате доведения до максимума величины удерживания газа для совокупной реакционной среды 36. Реакционная среда 36 предпочтительно характеризуется средневременной и среднеобъемной величиной удерживания газа, равной, по меньшей мере, приблизительно 0,4, более предпочтительно находящейся в диапазоне от приблизительно 0,6 до приблизительно 0,9, а наиболее предпочтительно в диапазоне от 0,65 до 0,85. Некоторые физические и эксплуатационные признаки барботажной реакторной колонны 20 вносят свой вклад в обсуждавшуюся выше высокую величину удерживания газа. Например, для данных размера реактора и расхода для потока окислителя высокое соотношение L:D для зоны реакции 28 дает в результате меньший диаметр, что вызывает увеличение расхода на единицу сечения потока в реакционной среде 36, что, в свою очередь, приводит к увеличению удерживания газа. В дополнение к этому на среднюю величину удерживания газа даже для данного постоянного расхода на единицу сечения потока, как известно, оказывают влияние фактический диаметр барботажной колонны и соотношение L:D. В дополнение к этому свой вклад в повышенную величину удерживания газа вносит сведение к минимуму количества неаэрированных зон, в частности, в области низа зона реакции 28. Кроме того, на стабильность функционирования при высоких расходах на единицу сечения потока и величинах удерживания газа, описанных в настоящем документе, могут оказывать влияние давление верха реактора и механическая конфигурация барботажной реакторной колонны.
Кроме того, изобретатели выявили важность функционирования при оптимизированном давлении верха реактора для получения повышенного удерживания газа и повышенного массопереноса. Может показаться, что функционирование при пониженном давлении верха реактора, что приводит к уменьшению растворимости молекулярного кислорода в соответствии с действием закона Генри, привело бы к уменьшению скорости массопереноса молекулярного кислорода из газа в жидкость. В емкости с механическим перемешиванием данный вариант обычно и имеет место, поскольку уровни аэрации и скорости массопереноса преимущественным образом определяются конструкцией перемешивающего устройства и давлением верха реактора. Однако в случае барботажной реакторной колонны, соответствующей предпочтительному варианту реализации настоящего изобретения, было выявлено то, как использовать пониженное давление верха реактора для того, чтобы стимулировать занятие заданной массой потока газофазного окислителя большего объема, что вызывает увеличение расхода на единицу сечения потока в реакционной среде 36 и, в свою очередь, приводит к увеличению удерживания газа и скорости переноса молекулярного кислорода.
Установление равновесия между коалесценцией и дроблением пузырьков представляет собой чрезвычайно сложное явление, приводящее, с одной стороны, к возникновению тенденции к вспениванию, что вызывает уменьшение скоростей внутренней циркуляции жидкой фазы и что может потребовать наличия очень, очень больших зон отделения, а с другой стороны, к возникновению тенденции к наличию меньшего количества очень крупных пузырьков, что приводит к пониженному удерживанию газа и пониженной скорости массопереноса из потока окислителя в жидкую фазу. Что касается жидкой фазы, то, как известно, ее состав, плотность, вязкость и поверхностное натяжение, помимо прочих факторов, взаимодействуют очень сложным образом, приводя к получению очень сложных результатов даже в отсутствие твердой фазы. Например, в лабораторных исследованиях исследователи обнаружили то, что при составлении отчетов и оценке результатов по наблюдениям даже для простых водно-воздушных барботажных колонн полезно устанавливать качество «воды» - будет ли это водопроводная вода, дистиллированная вода или деионизованная вода. В случае сложных смесей в жидкой фазе и в случае добавления твердой фазы уровень сложности увеличивается дополнительно. Важными для взаимодействия твердой фазы с жидкой фазой и потоком окислителя при определении того, какими в результате будут получаться характеристики барботирования и схемы течения при естественной конвекции, помимо прочего, являются все параметры, выбираемые из поверхностных неоднородностей индивидуальных частиц твердой фазы, среднего размера твердых частиц, распределения частиц по размерам, количества твердой фазы в сопоставлении с количеством жидкой фазы и способности жидкости смачивать поверхность твердой фазы.
Таким образом, способность барботажной реакторной колонны функционировать подходящим образом при высоких расходах на единицу сечения потока и высоких величинах удерживания газа, описываемых в настоящем документе, зависит, например, от надлежащего выбора: (1) состава жидкой фазы реакционной среды; (2) количества и типа осажденной твердой фазы, где оба параметра можно отрегулировать при помощи условий проведения реакции; (3) величины потока окислителя, подаваемого в реактор; (4) давления верха реактора, которое оказывает влияние на объемный расход потока окислителя, стабильность пузырьков и через энергетический баланс на температуру реакции; (5) самой температуры реакции, которая оказывает влияние на характеристики текучести, свойства осажденной твердой фазы и удельный объем потока окислителя; и (6) геометрии и механических деталей реакционной емкости, в том числе соотношения L:D.
Если обратиться опять к Фиг. 1, то можно сказать, что было обнаружено то, что получения улучшенного распределения окисляемого соединения (например, пара-ксилола) в реакционной среде 36 можно было добиться в результате введения потока жидкофазного исходного подаваемого материала в зону реакции 28 в нескольких позициях, разнесенных по вертикали. Предпочтительно поток жидкофазного исходного подаваемого материала вводят в зону реакции 28 при использовании, по меньшей мере, 3 отверстий для исходного подаваемого материала, более предпочтительно, по меньшей мере, 4 отверстий для исходного подаваемого материала. В соответствии с использованием в настоящем документе термин «отверстия для исходного подаваемого материала» должны обозначать отверстия, через которые поток жидкофазного исходного подаваемого материала выпускают в зону реакции 28 для перемешивания с реакционной средой 36. Предпочитается, чтобы, по меньшей мере, 2 отверстия для исходного подаваемого материала были бы разнесены по вертикали относительно друг друга, по меньшей мере, приблизительно на 0,5D, более предпочтительно, по меньшей мере, приблизительно на 1,5D, а наиболее предпочтительно, по меньшей мере, на 3D. Однако предпочитается, чтобы самое высокое отверстие для исходного подаваемого материала отстояло бы по вертикали от самого нижнего отверстия для окислителя не более чем приблизительно на 0,75Н, 0,65L и/или 8D; более предпочтительно не более чем приблизительно на 0,5Н, 0,4L и/или 5D; а наиболее предпочтительно не более чем на 0,4Н, 0,35L и/или 4D.
Несмотря на то что введение потока жидкофазного исходного подаваемого материала желательно проводить в нескольких позициях по вертикали, также было обнаружено и то, что улучшенное распределение окисляемого соединения в реакционной среде 36 будет достигаться, если основную часть потока жидкофазного исходного подаваемого материала будут вводить в нижнюю половину реакционной среды 36 и/или зоны реакции 28. Предпочтительно в нижнюю половину реакционной среды 36 и/или зоны реакции 28 вводят, по меньшей мере, приблизительно 75 массовых процентов от потока жидкофазного исходного подаваемого материала. Наиболее предпочтительно в нижнюю половину реакционной среды 36 и/или зоны реакции 28 вводят, по меньшей мере, 90 массовых процентов от потока жидкофазного исходного подаваемого материала. В дополнение к этому предпочитается, чтобы, по меньшей мере, приблизительно 30 массовых процентов от потока жидкофазного исходного подаваемого материала вводили бы в зону реакции 28 в пределах приблизительно 1,5D от самой нижней позиции по вертикали, где в зону реакции 28 вводят поток окислителя. Данная самая нижняя позиция по вертикали, где поток окислителя вводят в зону реакции 28, обычно находится в области низа барботера окислителя; однако в одном предпочтительном варианте реализации настоящего изобретения предусматривается широкий ассортимент альтернативных конфигураций, предназначенных для введения потока окислителя в зону реакции 28. Предпочтительно, по меньшей мере, приблизительно 50 массовых процентов от жидкофазного исходного подаваемого материала вводят в пределах приблизительно 2,5D от самой нижней позиции по вертикали, где в зону реакции 28 вводят поток окислителя. Предпочтительно, по меньшей мере, приблизительно 75 массовых процентов от потока жидкофазного исходного подаваемого материала вводят в пределах приблизительно 5D от самой нижней позиции по вертикали, где в зону реакции 28 вводят поток окислителя.
Каждое отверстие для исходного подаваемого материала определяет живое сечение, через которое выпускают исходный подаваемый материал. Предпочитается, чтобы, по меньшей мере, приблизительно 30 процентов от совокупной площади живого сечения всех впускных отверстий для исходного подаваемого материала располагались бы в пределах приблизительно 1,5D от самой нижней позиции по вертикали, где в зону реакции 28 вводят поток окислителя. Предпочтительно, по меньшей мере, приблизительно 50 процентов от совокупной площади живого сечения всех впускных отверстий для исходного подаваемого материала располагаются в пределах приблизительно 2,5D от самой нижней позиции по вертикали, где в зону реакции 28 вводят поток окислителя. Предпочтительно, по меньшей мере, приблизительно 75 процентов от совокупной площади живого сечения всех впускных отверстий для исходного подаваемого материала располагаются в пределах приблизительно 5D от самой нижней позиции по вертикали, где в зону реакции 28 вводят поток окислителя.
Если обратиться опять к Фиг. 1, то можно сказать, что в одном варианте реализации настоящего изобретения впускные отверстия для исходного подаваемого материала 32а, b, c, d представляют собой просто последовательность из отверстий, выровненных по вертикали по одной стороне оболочки емкости 22. Данные отверстия для исходного подаваемого материала предпочтительно имеют по существу подобные диаметры, меньшие чем приблизительно 7 сантиметров, более предпочтительно находящиеся в диапазоне от приблизительно 0,25 до приблизительно 5 сантиметров, а наиболее предпочтительно в диапазоне от 0,4 до 2 сантиметров. Барботажную реакторную колонну 20 предпочтительно оборудуют системой регулирования расхода для потока жидкофазного исходного подаваемого материала из каждого отверстия для исходного подаваемого материала. Такая система регулирования расхода предпочтительно включает индивидуальный клапан регулирования расхода 74а, b, c, d для каждого соответствующего впускного отверстия для исходного подаваемого материала 32а, b, c, d. В дополнение к этому предпочитается, чтобы барботажная реакторная колонна 20 была бы оборудована системой регулирования расхода, которая позволяла бы, по меньшей мере, часть потока жидкофазного исходного подаваемого материала вводить в зону реакции 28 при повышенном расходе на единицу сечения потока во впускном отверстии, равном, по меньшей мере, приблизительно 2 метрам в секунду, более предпочтительно, по меньшей мере, приблизительно 5 метрам в секунду, еще более предпочтительно, по меньшей мере, приблизительно 6 метрам в секунду, а наиболее предпочтительно находящемся в диапазоне от 8 до 20 метров в секунду. В соответствии с использованием в настоящем документе термин «расход на единицу сечения потока во впускном отверстии» обозначает средневременной объемный расход для потока исходного подаваемого материала из отверстия для исходного подаваемого материала, поделенный на площадь отверстия для исходного подаваемого материала. Предпочтительно, по меньшей мере, приблизительно 50 массовых процентов от потока исходного подаваемого материала вводят в зону реакции 28 при повышенном расходе на единицу сечения потока во впускном отверстии. Наиболее предпочтительно по существу весь поток исходного подаваемого материала вводят в зону реакции 28 при повышенном расходе на единицу сечения потока во впускном отверстии.
Если обратиться теперь к Фиг. 6-7, то можно сказать, что на них проиллюстрирована альтернативная система введения в зону реакции 28 потока жидкофазного исходного подаваемого материала. В данном варианте реализации поток исходного подаваемого материала вводят в зону реакции 28 на четырех различных уровнях по высоте. На каждом уровне по высоте оборудуют соответствующую систему распределения исходного подаваемого материала 76а, b, c, d. Каждая система распределения исходного подаваемого материала 76 включает основной канал для исходного подаваемого материала 78 и коллектор 80. Каждый коллектор 80 снабжают, по меньшей мере, двумя выпускными отверстиями 82, 84, соединенными с соответствующими вставными каналами 86, 88, которые проходят в зону реакции 28 оболочки емкости 22. Каждый вставной канал 86, 88 характеризуется наличием соответствующего отверстия для исходного подаваемого материала 87, 89, предназначенного для выпуска потока исходного подаваемого материала в зону реакции 28. Отверстия для исходного подаваемого материала 87, 89 предпочтительно имеют по существу подобные диаметры, меньшие чем приблизительно 7 сантиметров, более предпочтительно находящиеся в диапазоне от приблизительно 0,25 до приблизительно 5 сантиметров, а наиболее предпочтительно в диапазоне от 0,4 до 2 сантиметров. Предпочитается, чтобы отверстия для исходного подаваемого материала 87, 89 в каждой системе распределения исходного подаваемого материала 76а, b, c, d были бы расположены по диаметру напротив друг друга таким образом, чтобы поток исходного подаваемого материала вводить в зону реакции 28 в противоположных направлениях. Кроме того, предпочитается, чтобы расположенные по диаметру напротив друг друга отверстия для исходного подаваемого материала 86, 88 соседних систем распределения исходного подаваемого материала 76 были бы ориентированы с поворотом на угол 90 градусов по отношению друг к другу. В ходе функционирования поток жидкофазного исходного подаваемого материала загружают в основной канал для исходного подаваемого материала 78, а после этого он поступает в коллектор 80. Коллектор 80 равномерно распределяет поток исходного подаваемого материала для одновременного введения с противоположных сторон реактора 20 через отверстия для исходного подаваемого материала 87, 89.
Фиг. 8 иллюстрирует альтернативную конфигурацию, где каждую систему распределения исходного подаваемого материала 76 оборудуют байонетными трубами 90, 92, а не вставными каналами 86, 88 (продемонстрированными на Фиг. 7). Байонетные трубы 90, 92 проходят в зону реакции 28 и включают множество небольших отверстий для исходного подаваемого материала 94, 96, предназначенных для выпуска жидкофазного исходного подаваемого материала в зону реакции 28. Предпочитается, чтобы небольшие отверстия для исходного подаваемого материала 94, 96 у байонетных труб 90, 92 имели бы по существу одни и те же диаметры, меньшие чем приблизительно 50 миллиметров, более предпочтительно находящиеся в диапазоне от приблизительно 2 до приблизительно 25 миллиметров, а наиболее предпочтительно от 4 до 15 миллиметров.
Фиг. 9-11 иллюстрируют альтернативную систему распределения исходного подаваемого материала 100. Система распределения исходного подаваемого материала 100 обеспечивает введение потока жидкофазного исходного подаваемого материала через множество разнесенных по вертикали и разнесенных в боковом направлении позиций без возникновения потребности в наличии нескольких позиций проникновения сквозь боковую стенку барботажной реакторной колонны 20. Система введения исходного подаваемого материала 100 в общем случае включает единственный канал впускного отверстия 102, магистраль 104, множество прямостоячих распределительных труб 106, механизм, создающий боковую опору, 108 и механизм, создающий вертикальную опору, 110. Канал впускного отверстия 102 проникает через боковую стенку основного тела 46 оболочки емкости 22. Канал впускного отверстия 102 через текучую среду соединяется с магистралью 104. Магистраль 104 равномерно распределяет поток исходного подаваемого материала, поступающий из канала впускного отверстия 102, между прямостоячими распределительными трубами 106. Каждая распределительная труба 106 имеет множество разнесенных по вертикали отверстий для исходного подаваемого материала 112а, b, c, d, предназначенных для выпуска потока исходного подаваемого материала в зону реакции 28. Механизм, создающий боковую опору, 108 соединяется с каждой распределительной трубой 106 и предотвращает возникновение относительного бокового перемещения распределительных труб 106. Механизм, создающий вертикальную опору, 110 предпочтительно соединяется с механизмом, создающим боковую опору, 108 и верхом барботера окислителя 34. Механизм, создающий вертикальную опору, 110 по существу предотвращает возникновение вертикального перемещения распределительных труб 106 в зоне реакции 28. Предпочитается, чтобы отверстия для исходного подаваемого материала 112 имели бы по существу одни и те же диаметры, меньшие чем приблизительно 50 миллиметров, более предпочтительно находящиеся в диапазоне от приблизительно 2 до приблизительно 25 миллиметров, а наиболее предпочтительно от 4 до 15 миллиметров. Разнесение по вертикали отверстий для исходного подаваемого материала 112 в системе распределения исходного подаваемого материала 100, проиллюстрированной на Фиг. 9-11, может быть по существу тем же самым, что и описанное выше, в том, что касается системы распределения исходного подаваемого материала Фиг. 1.
Было обнаружено то, что схемы течения реакционной среды во многих барботажных реакторных колоннах могут позволить осуществить неравномерное азимутальное распределение окисляемого соединения в реакционной среде, в особенности тогда, когда окисляемое соединение главным образом вводят по одной стороне реакционной среды. В соответствии с использованием в настоящем документе термин «азимутальный» должен обозначать угол или разнесение по окружности относительно прямостоячей продольной оси зоны реакции. В соответствии с использованием в настоящем документе «прямостоячий» должен обозначать нахождение в пределах 45° от вертикали. В одном варианте реализации настоящего изобретения поток исходного подаваемого материала, содержащий окисляемое соединение (например, пара-ксилол), в зону реакции вводят через множество разнесенных по азимуту отверстий для исходного подаваемого материала. Данные разнесенные по азимуту отверстия для исходного подаваемого материала могут способствовать предотвращению возникновения в реакционной среде областей избыточно высоких и избыточно низких концентраций окисляемого соединения. Различные системы введения исходного подаваемого материала, проиллюстрированные на Фиг. 6-11, представляют собой примеры систем, которые обеспечивают наличие надлежащего разнесения по азимуту для отверстий для исходного подаваемого материала.
Если обратиться опять к Фиг. 7, то можно сказать, что для того, чтобы получить количественные характеристики для разнесенного по азимуту введения потока жидкофазного исходного подаваемого материала в реакционную среду, реакционную среду можно теоретически разделить на четыре прямостоячих азимутальных квадранта «Q1, Q2, Q3, Q4» с приблизительно равным объемом. Данные азимутальные квадранты «Q1, Q2, Q3, Q4» определяются парой воображаемых пересекающихся перпендикулярных вертикальных плоскостей «Р1, Р2», выходящих за пределы максимального вертикального размера и максимального радиального размера реакционной среды. Если реакционная среда будет содержаться в цилиндрической емкости, то тогда линия пересечения воображаемых пересекающихся вертикальных плоскостей Р1, Р2 будет приблизительно совпадать с вертикальной центральной линией цилиндра, а каждый азимутальный квадрант Q1, Q2, Q3, Q4 будет представлять собой в общем случае клиновидный вертикальный объем, имеющий высоту, равную высоте реакционной среды. Предпочитается, чтобы существенную часть окисляемого соединения выпускали бы в реакционную среду через отверстия для исходного подаваемого материала, расположенные, по меньшей мере, в двух различных азимутальных квадрантах.
В предпочтительном варианте реализации настоящего изобретения через отверстия для исходного подаваемого материала, которые могут располагаться в одном азимутальном квадранте, в реакционную среду выпускают не более чем приблизительно 80 массовых процентов от окисляемого соединения. Более предпочтительно через отверстия для исходного подаваемого материала, которые могут располагаться в одном азимутальном квадранте, в реакционную среду выпускают не более чем приблизительно 60 массовых процентов от окисляемого соединения. Наиболее предпочтительно через отверстия для исходного подаваемого материала, которые могут располагаться в одном азимутальном квадранте, в реакционную среду выпускают не более чем 40 массовых процентов от окисляемого соединения. Данные параметры для азимутального распределения окисляемого соединения измеряют тогда, когда азимутальные квадранты будут азимутально ориентированы таким образом, чтобы в один из азимутальных квадрантов обеспечить выпуск максимально возможного количества окисляемого соединения. Например, если поток совокупного исходного подаваемого материала будут выпускать в реакционную среду через два отверстия для исходного подаваемого материала, которые будут азимутально разнесены друг от друга на 89 градусов, то тогда для целей определения азимутального распределения в четырех азимутальных квадрантах 100 массовых процентов потока исходного подаваемого материала будут выпускать в реакционную среду в одном азимутальном квадранте, поскольку азимутальные квадранты могут быть азимутально ориентированы таким образом, чтобы оба отверстия для исходного подаваемого материала были бы расположены в одном азимутальном квадранте.
В дополнение к преимуществам, связанным с надлежащим азимутальным разнесением отверстий для исходного подаваемого материала, также было обнаружено и то, что в барботажной реакторной колонне существенным может являться также и надлежащее радиальное разнесение отверстий для исходного подаваемого материала. Предпочитается, чтобы существенную часть окисляемого соединения, вводимого в реакционную среду, выпускали бы через отверстия для исходного подаваемого материала, которые радиально разнесены в направлении извне вовнутрь от боковой стенки емкости. Таким образом, в одном варианте реализации настоящего изобретения существенная часть окисляемого соединения поступает в зону реакции через отверстия для исходного подаваемого материала, расположенные в «предпочтительной радиальной зоне для исходного подаваемого материала», которая простирается в направлении извне вовнутрь от прямостоячих боковых стенок, определяющих зону реакции.
Если обратиться опять к Фиг. 7, то можно сказать, что предпочтительная радиальная зона для исходного подаваемого материала «FZ» может принимать форму теоретического прямостоячего цилиндра, центрированного в зоне реакции 28 и имеющего внешний диаметр «DO», равный 0,9D, где «D» представляет собой диаметр зоны реакции 28. Таким образом, внешнее кольцевое пространство «ОА», имеющее толщину 0,05D, определяется областью в промежутке между предпочтительной радиальной зоной для исходного подаваемого материала FZ и внутренней стороной боковой стенки, определяющей зону реакции 28. Предпочитается, чтобы через отверстия для исходного подаваемого материала, расположенные в данном внешнем кольцевом пространстве ОА, в зону реакции 28 вводили бы незначительное количество окисляемого соединения или его не вводили бы вообще.
В еще одном варианте реализации предпочитается, чтобы незначительное количество окисляемого соединения вводили бы в центр зоны реакции 28 или туда не вводили бы его вообще. Таким образом, как проиллюстрировано на Фиг. 8, предпочтительная радиальная зона для исходного подаваемого материала FZ может принимать форму теоретического прямостоячего кольцевого пространства, центрированного в зоне реакции 28, имеющего внешний диаметр DO, равный 0,9D, и имеющего внутренний диаметр DI, равный 0,2D. Таким образом, в данном варианте реализации из центра предпочтительной радиальной зоны для исходного подаваемого материала FZ «вырезают» внутренний цилиндр IC, имеющий диаметр 0,2D. Предпочитается, чтобы через отверстия для исходного подаваемого материала, расположенные в данном внутреннем цилиндре IC, в зону реакции 28 вводили бы незначительное количество окисляемого соединения или его не вводили бы вообще.
В предпочтительном варианте реализации настоящего изобретения существенную часть окисляемого соединения в реакционную среду 36 вводят через отверстия для исходного подаваемого материала, расположенные в предпочтительной радиальной зоне для исходного подаваемого материала, вне зависимости от того, будет ли предпочтительная радиальная зона для исходного подаваемого материала иметь цилиндрическую или кольцевую формы, описанные выше. Более предпочтительно через отверстия для исходного подаваемого материала, расположенные в предпочтительной радиальной зоне для исходного подаваемого материала, в реакционную среду 36 выпускают, по меньшей мере, приблизительно 25 массовых процентов от окисляемого соединения. Еще более предпочтительно через отверстия для исходного подаваемого материала, расположенные в предпочтительной радиальной зоне для исходного подаваемого материала, в реакционную среду 36 выпускают, по меньшей мере, приблизительно 50 массовых процентов от окисляемого соединения. Наиболее предпочтительно через отверстия для исходного подаваемого материала, расположенные в предпочтительной радиальной зоне для исходного подаваемого материала, в реакционную среду 36 выпускают, по меньшей мере, 75 массовых процентов от окисляемого соединения.
Несмотря на то что теоретические азимутальные квадранты и теоретическая предпочтительная радиальная зона для исходного подаваемого материала, проиллюстрированные на фиг.7 и 8, описываются со ссылкой на распределение потока жидкофазного исходного подаваемого материала, было обнаружено то, что надлежащее азимутальное и радиальное распределение потока газофазного окислителя также может обеспечить достижение определенных преимуществ. Таким образом, в одном варианте реализации настоящего изобретения описание азимутального и радиального распределения потока жидкофазного исходного подаваемого материала, представленное выше, относится также и к способу, по которому в реакционную среду 36 вводят поток газофазного окислителя.
Если обратиться теперь к Фиг. 12-15, то можно сказать, что на них проиллюстрирован альтернативный барботер окислителя 200 как в общем случае включающий кольцевой элемент 202 и пару каналов для ввода окислителя 204, 206. Барботер окислителя 200 с Фиг.12-15 подобен барботеру окислителя 34 с Фиг. 1-11 при следующих далее трех основных различиях: (1) барботер окислителя 200 не включает диагонального поперечного элемента; (2) верхняя часть кольцевого элемента 202 не имеет отверстий для выпуска окислителя в направлении снизу вверх; и (3) барботер окислителя 200 имеет намного больше отверстий в нижней части кольцевого элемента 202.
Как, может быть, наилучшим образом проиллюстрировано на Фиг. 14 и 15, нижняя часть кольца барботера окислителя 202 характеризуется наличием множества отверстий для окислителя 208. Отверстия для окислителя 208 предпочтительно имеют такую конфигурацию, что, по меньшей мере, приблизительно 1 процент от совокупной площади живого сечения, определенного отверстиями для окислителя 208, располагается ниже центральной линии 210 (Фиг. 15) кольцевого элемента 202, где центральная линия 210 располагается на уровне по высоте для средней точки объема кольцевого элемента 202. Более предпочтительно, по меньшей мере, приблизительно 5 процентов от совокупной площади живого сечения, определенного всеми отверстиями для окислителя 208, располагаются ниже центральной линии 210, при этом, по меньшей мере, приблизительно 2 процента от совокупной площади живого сечения определяются отверстиями 208, которые обеспечивают выпуск потока окислителя в общем случае в направлении сверху вниз в пределах приблизительно 30 градусов от вертикали. Еще более предпочтительно, по меньшей мере, приблизительно 20 процентов от совокупной площади живого сечения, определенного всеми отверстиями для окислителя 208, располагаются ниже центральной линии 210, при этом, по меньшей мере, приблизительно 10 процентов от совокупной площади живого сечения определяются отверстиями 208, которые обеспечивают выпуск потока окислителя в общем случае в направлении сверху вниз в пределах 30 градусов от вертикали. Наиболее предпочтительно, по меньшей мере, приблизительно 75 процентов от совокупной площади живого сечения, определенного всеми отверстиями для окислителя 208, располагаются ниже центральной линии 210, при этом, по меньшей мере, приблизительно 40 процентов от совокупной площади живого сечения определяются отверстиями 208, которые обеспечивают выпуск потока окислителя в общем случае в направлении сверху вниз в пределах 30 градусов от вертикали. Доля от совокупной площади живого сечения, определенной всеми отверстиями для окислителя 208, которые располагаются выше центральной линии 210, предпочтительно составляет величину, меньшую чем приблизительно 75 процентов, более предпочтительно меньшую чем приблизительно 50 процентов, еще более предпочтительно меньшую чем приблизительно 25 процентов, а наиболее предпочтительно меньшую чем 5 процентов.
Как проиллюстрировано на Фиг. 14 и 15, отверстия для окислителя 208 включают отверстия, обращенные книзу, 208а и отверстия, ориентированные под углом, 208b. Отверстия, обращенные книзу, 208а имеют конфигурацию, обеспечивающую выпуск потока окислителя в общем случае в направлении сверху вниз под углом в пределах приблизительно 30 градусов от вертикали, более предпочтительно в пределах приблизительно 15 градусов от вертикали, а наиболее предпочтительно в пределах 5 градусов от вертикали. Отверстия, ориентированные под углом, 208b имеют конфигурацию, обеспечивающую выпуск потока окислителя в общем случае в направлении изнутри наружу и сверху вниз под углом «А», который находится в диапазоне от приблизительно 15 до приблизительно 75 градусов от вертикали, более предпочтительно угол А находится в диапазоне от приблизительно 30 до приблизительно 60 градусов от вертикали, а наиболее предпочтительно угол А находится в диапазоне от 40 до 50 градусов от вертикали.
Предпочитается, чтобы по существу все отверстия для окислителя 208 имели бы приблизительно один и тот же диаметр. Диаметр отверстий для окислителя 208 предпочтительно находится в диапазоне от приблизительно 2 до приблизительно 300 миллиметров, более предпочтительно в диапазоне от приблизительно 4 до приблизительно 120 миллиметров, а наиболее предпочтительно в диапазоне от 8 до 60 миллиметров. Совокупное количество отверстий для окислителя 208 в кольцевом элементе 202 выбирают в соответствии с критериями низкого падения давления, детально описанными далее. Предпочтительно совокупное количество отверстий для окислителя 208, сформированных в кольцевом элементе 202, составляет, по меньшей мере, приблизительно 10, более предпочтительно совокупное количество отверстий для окислителя 208 составляет величину в диапазоне от приблизительно 20 до приблизительно 200, а наиболее предпочтительно совокупное количество отверстий для окислителя 208 составляет величину в диапазоне от 40 до 100.
Несмотря на то что Фиг. 12-15 иллюстрируют очень конкретную конфигурацию барботера окислителя 200, в настоящий момент следует отметить то, что для достижения преимуществ, описанных в настоящем документе, может быть использован широкий ассортимент конфигураций барботеров окислителя. Например, барботер окислителя необязательно должен иметь конфигурацию восьмиугольного кольцевого элемента, проиллюстрированную на Фиг. 12-13. Вместо этого возможно, чтобы барботер окислителя был бы получен из любой конфигурации канала (каналов) для течения, который использует множество пространственно разнесенных отверстий, предназначенных для выпуска потока окислителя. Размер, количество и направление выпуска, характеризующие отверстия для окислителя в канале для течения, предпочтительно находятся в пределах приведенных выше диапазонов. Кроме того, барботер окислителя предпочтительно имеет конфигурацию, обеспечивающую азимутальное и радиальное распределение молекулярного кислорода, описанное выше.
Вне зависимости от конкретной конфигурации барботера окислителя предпочитается, чтобы барботер окислителя имел бы физическую конфигурацию и функционировал бы в соответствии со способом, который обеспечивает сведение к минимуму падения давления, связанного с выпуском потока окислителя из канала (каналов) для течения через отверстия для окислителя в зону реакции. Такое падение давления рассчитывают в виде средневременного статического давления потока окислителя внутри канала для течения на впускных отверстиях для окислителя 66а, b барботера окислителя минус средневременное статическое давление в зоне реакции на уровне по высоте, где половину потока окислителя вводят выше данной позиции по вертикали, а половину потока окислителя вводят ниже данной позиции по вертикали. В предпочтительном варианте реализации настоящего изобретения средневременное падение давления, связанное с выпуском потока окислителя из барботера окислителя, составляет величину, меньшую чем приблизительно 0,3 мегапаскаля (МПа), более предпочтительно меньшую чем приблизительно 0,2 МПа, еще более предпочтительно меньшую, чем приблизительно 0,1 МПа, а наиболее предпочтительно меньшую чем 0,05 МПа. В предпочтительных условиях функционирования барботажной реакторной колонны, описанной в настоящем документе, давление потока окислителя внутри канала (каналов) для течения барботера окислителя предпочтительно находится в диапазоне от приблизительно 0,35 до приблизительно 1 МПа, более предпочтительно в диапазоне от приблизительно 0,45 до приблизительно 0,85 МПа, а наиболее предпочтительно в диапазоне от 0,5 до 0,7 МПа.
Как упоминалось ранее в том, что касается конфигурации барботера окислителя, проиллюстрированной на Фиг. 2-5, может оказаться желательным непрерывное или периодическое промывание барботера окислителя жидкостью (например, уксусной кислотой, водой и/или пара-ксилолом) для предотвращения возникновения обрастания барботера окислителя отложениями твердой фазы. В случае использования такого промывания жидкостью предпочитается, чтобы в течение, по меньшей мере, одного периода продолжительностью, большей чем одна минута каждый день через барботер окислителя и из отверстий для окислителя перепускали бы эффективное количество жидкости (то есть не просто незначительное количество капель жидкости, которые естественным образом могут присутствовать в потоке окислителя). Если из барботера окислителя непрерывно или периодически будут выпускать жидкость, то тогда предпочитается, чтобы средневременное соотношение между массовым расходом жидкости через барботер окислителя и массовым расходом молекулярного кислорода через барботер окислителя находилось бы в диапазоне от приблизительно 0,05:1 до приблизительно 30:1 или в диапазоне от приблизительно 0,1:1 до приблизительно 2:1 или даже в диапазоне от 0,2:1 до 1:1.
В одном варианте реализации настоящего изобретения значительную часть окисляемого соединения (например, пара-ксилола) можно вводить в зону реакции через барботер окислителя. В такой конфигурации предпочитается, чтобы выпуск окисляемого соединения и молекулярного кислорода из барботера окислителя происходил бы через одни и те же отверстия в барботере окислителя. Как отмечалось выше, окисляемое соединение обычно представляет собой жидкость при STP (стандартные температура и давление). Поэтому в данном варианте реализации из барботера окислителя можно выпускать двухфазный поток, при этом жидкая фаза будет содержать окисляемое соединение, а газовая фаза будет содержать молекулярный кислород. Однако необходимо понимать, что во время выпуска из барботера окислителя, по меньшей мере, часть окисляемого соединения может находиться в газообразном состоянии. В одном варианте реализации жидкую фазу, выпускаемую из барботера окислителя, преимущественно образует окисляемое соединение. В еще одном варианте реализации жидкая фаза, выпускаемая из барботера окислителя, имеет по существу тот же самый состав, что и поток исходного подаваемого материала, описанный выше. Если жидкая фаза, выпускаемая из барботера окислителя, будет иметь по существу тот же самый состав, что и поток исходного подаваемого материала, то такая жидкая фаза может содержать растворитель и/или систему катализатора в количествах и при соотношениях, описанных выше в том, что касается состава потока исходного подаваемого материала.
В одном варианте реализации настоящего изобретения предпочитается, чтобы через барботер окислителя вводили бы, по меньшей мере, приблизительно 10 массовых процентов от всего количества окисляемого соединения, вводимого в зону реакции, более предпочтительно через барботер окислителя в зону реакции вводят, по меньшей мере, приблизительно 40 массовых процентов окисляемого соединения, а наиболее предпочтительно через барботер окислителя в зону реакции вводят, по меньшей мере, 80 массовых процентов окисляемого соединения. Если все количество или часть окисляемого соединения будут вводить в зону реакции через барботер окислителя, то предпочитается, чтобы, по меньшей мере, приблизительно 10 массовых процентов от всего количества молекулярного кислорода, вводимого в зону реакции, вводили бы через тот же самый барботер окислителя, более предпочтительно, по меньшей мере, приблизительно 40 массовых процентов окисляемого соединения в зону реакции вводят через один и тот же барботер окислителя, а наиболее предпочтительно, по меньшей мере, 80 массовых процентов окисляемого соединения в зону реакции вводят через один и тот же барботер окислителя. Если в зону реакции через барботер окислителя будут вводить значительную долю окисляемого соединения, то тогда предпочитается, чтобы в барботере окислителя были бы установлены одно или несколько устройств, воспринимающих температуру (например, термопар). Данные температурные датчики можно использовать в качестве вспомогательных средств, помогающих удостовериться в том, что температура в барботере окислителя не становится опасно высокой.
Если обратиться теперь к Фиг. 16-18, то можно сказать, что на них проиллюстрирована барботажная реакторная колонна 20 как включающая внутреннюю деаэрационную емкость 300, расположенную в области низа зоны реакции 28 поблизости от выпускного отверстия для суспензии 38. Было обнаружено то, что во время деаэрации реакционной среды 36 с относительно высокой скоростью протекают побочные реакции, приводящие к образованию примесей. В соответствии с использованием в настоящем документе термин «деаэрация» должен обозначать отделение газовой фазы от многофазной реакционной среды. Если реакционная среда 36 будет высокоаэрированной (величина удерживания газа >0,3), то тогда образование примесей будет минимальным. Если реакционная среда 36 будет высоконеаэрированной (величина удерживания газа <0,01), то тогда образование примесей также будет минимальным. Однако если реакционная среда будет частично аэрирована (величина удерживания газа 0,01-0,3), то тогда будет стимулироваться прохождение нежелательных побочных реакций, и будет образовываться повышенное количество примесей. Деаэрационная емкость 300 имеет целью устранение данной и других проблем в результате сведения к минимуму объема реакционной среды 36 в частично аэрированном состоянии и в результате сведения к минимуму времени, которое потребуется для деаэрации реакционной среды 36. Из низа деаэрационной емкости 300 получают по существу деаэрированную суспензию, и она выходит из реактора 20 через выпускное отверстие для суспензии 38. По существу деаэрированная суспензия предпочтительно содержит менее чем приблизительно 5 объемных процентов газовой фазы, более предпочтительно менее чем приблизительно 2 объемных процента газовой фазы, а наиболее предпочтительно менее чем 1 объемный процент газовой фазы.
На Фиг. 16 барботажная реакторная колонна 20 проиллюстрирована как включающая регулятор уровня 302 и клапан регулирования расхода 304. Регулятор уровня 302 и клапан регулирования расхода 304 взаимодействуют, обеспечивая выдерживание реакционной среды 36 в зоне реакции 28 по существу на постоянном уровне по высоте. Регулятор уровня 302 может функционировать в качестве средства восприятия уровня по высоте (например, при использовании восприятия уровня по разности давлений или восприятия уровня при помощи радиоактивного датчика) для верхней поверхности 44 реакционной среды 36 и генерации управляющего сигнала 306, образующего отклик на уровень по высоте для реакционной среды 36. Клапан регулирования расхода 304 воспринимает управляющий сигнал 306 и регулирует расход суспензии через канал выпускного отверстия для суспензии 308. Таким образом, расход суспензии из выпускного отверстия для суспензии 38 может варьироваться в диапазоне от максимального объемного расхода суспензии (Fмакс) тогда, когда уровень по высоте для реакционной среды 36 будет чрезмерно высоким, до минимального объемного расхода суспензии (Fмин) тогда, когда уровень по высоте для реакционной среды 36 будет чрезмерно низким.
Для того чтобы удалить твердофазный продукт окисления из зоны реакции 28, часть сначала необходимо перепустить через деаэрационную емкость 300. Деаэрационная емкость 300 обеспечивает наличие низкотурбулентного внутреннего объема, который позволяет газовой фазе реакционной среды 36 естественным образом, поднимаясь, выходить из жидкой и твердой фаз реакционной среды 36 по мере того, как жидкая и твердая фазы будут перетекать сверху вниз в направлении к выпускному отверстию для суспензии 38. Подъем газовой фазы из жидкой и твердой фаз вызывается естественной направленной вверх подъемной силой, действующей на газовую фазу в жидкой и твердой фазах. В случае использования деаэрационной емкости 300 переход реакционной среды 36 из состояния полностью аэрированной трехфазной среды в состояние полностью деаэрированной двухфазной суспензии происходит быстро и эффективно.
Если обратиться теперь к Фиг. 17 и 18, то можно сказать, что деаэрационная емкости 300 включает в общем случае прямостоячую боковую стенку 308, определяющую зону деаэрации 312, ограничиваемую ей. Предпочтительно боковая стенка 308 проходит снизу вверх в пределах приблизительно 30 градусов от вертикали, более предпочтительно в пределах приблизительно 10 градусов от вертикали. Наиболее предпочтительно боковая стенка 308 является по существу вертикальной. Зона деаэрации 312 отделена от зоны реакции 28 и имеет высоту «h» и диаметр «d». Верхний край 310 боковой стенки 308 является незамкнутым для того, чтобы принимать реакционную среду из зоны реакции 28 во внутренний объем 312. Нижний край боковой стенки 308 через текучую среду соединяется с выпускным отверстием для суспензии 38 через переходную секцию 314. В определенных случаях, таких как, когда устье выпускного отверстия для суспензии 38 будет большим или когда диаметр «d» боковой стенки 308 будет маленьким, переходную секцию 314 можно исключить. Как, может быть, лучше всего проиллюстрировано на Фиг. 18, деаэрационная емкость 300 также может включать стабилизатор потока 316, расположенный в зоне деаэрации 312. Стабилизатор потока 316 может представлять собой любую конструкцию, функционирующую в целях подавления образования вихрей по мере того, как твердая и жидкая фазы будут перетекать сверху вниз в направлении выпускного отверстия для суспензии 38.
Для того чтобы добиться надлежащего отделения газовой фазы от твердой и жидкой фаз в деаэрационной емкости 300, производят тщательный отбор высоты «h» и площади горизонтального поперечного сечения внутренней зоны деаэрации 312. Высота «h» и площадь горизонтального поперечного сечения внутренней зоны деаэрации 312 должны обеспечивать достижение достаточных расстояния и времени таким образом, чтобы при отборе даже максимального количества суспензии (то есть при отборе суспензии на уровне Fмакс) по существу весь объем пузырьков газа мог бы, поднимаясь, выйти из твердой и жидкой фаз до того, как пузырьки газа достигнут нижнего выпускного отверстия в деаэрационной емкости 300. Таким образом, предпочитается, чтобы площадь поперечного сечения зоны деаэрации 312 была бы такой, чтобы максимальная скорость в направлении сверху вниз (Vdмакс) для жидкой и твердой фаз при прохождении зоны деаэрации 312 была бы существенно меньшей, чем скорость естественного подъема (Vu) для пузырьков газовой фазы при прохождении жидкой и твердой фаз. Максимальная скорость в направлении сверху вниз (Vdмакс) для жидкой и твердой фаз при прохождении зоны деаэрации 312 имеет место при максимальном объемном расходе суспензии (Fмакс), обсуждавшемся выше. Скорость естественного подъема (Vu) для пузырьков газа при прохождении жидкой и твердой фаз варьируется в зависимости от размера пузырьков; однако скорость естественного подъема (Vu0,5) для пузырьков газа с диаметром 0,5 сантиметра при прохождении жидкой и твердой фаз может быть использована в качестве значения отсечки, поскольку по существу весь объем пузырьков, первоначально присутствующий в реакционной среде 36, будет соответствовать величине, превышающей 0,5 сантиметра. Предпочтительно площадь поперечного сечения зоны деаэрации 312 такова, что Vdмакс составляет величину, меньшую чем приблизительно 75 процентов от Vu0,5, более предпочтительно Vdмакс составляет величину, меньшую чем приблизительно 40 процентов от Vu0,5, наиболее предпочтительно Vdмакс составляет величину, меньшую чем 20 процентов от Vu0,5.
Скорость при движении сверху вниз для жидкой и твердой фаз в зоне деаэрации 312 деаэрационной емкости 300 рассчитывают в виде объемного расхода деаэрированной суспензии через выпускное отверстие для суспензии 38, поделенного на минимальную площадь поперечного сечения зоны деаэрации 312. Скорость при движении сверху вниз для жидкой и твердой фаз в зоне деаэрации 312 деаэрационной емкости 300 предпочтительно составляет величину, меньшую чем приблизительно 50 сантиметров в секунду, более предпочтительно меньшую чем приблизительно 30 сантиметров в секунду, а наиболее предпочтительно меньшую чем 10 сантиметров в секунду.
В данный момент следует отметить то, что несмотря на то, что прямостоячая боковая стенка 308 деаэрационной емкости 300 проиллюстрирована как имеющая цилиндрическую конфигурацию, боковая стенка 308 может включать множество боковых стенок, которые образуют широкий спектр конфигураций (например, треугольную, квадратную или овальную), до тех пор, пока стенки будут определять внутренний объем, характеризующийся надлежащими объемом, площадью поперечного сечения, шириной «d» и высотой «h». В предпочтительном варианте реализации настоящего изобретения «d» находится в диапазоне от приблизительно 0,2 до приблизительно 2 метров, более предпочтительно в диапазоне от приблизительно 0,3 до приблизительно 1,5 метров, а наиболее предпочтительно в диапазоне от 0,4 до 1,2 метра. В предпочтительном варианте реализации настоящего изобретения «h» находится в диапазоне от приблизительно 0,3 метра до приблизительно 5 метров, более предпочтительно в диапазоне от приблизительно 0,5 до приблизительно 3 метров, а наиболее предпочтительно в диапазоне от 0,75 до 2 метров.
В предпочтительном варианте реализации настоящего изобретения боковая стенка 308 является по существу вертикальной, так что площадь горизонтального поперечного сечения зоны деаэрации 312 является по существу постоянной по всей высоте «h» зоны деаэрации 312. Предпочтительно максимальная площадь горизонтального поперечного сечения зоны деаэрации 312 составляет величину, меньшую чем приблизительно 25 процентов от максимальной площади горизонтального поперечного сечения зоны реакции 28. Более предпочтительно максимальная площадь горизонтального поперечного сечения зоны деаэрации 312 находится в диапазоне от приблизительно 0,1 до приблизительно 10 процентов от максимальной площади горизонтального поперечного сечения зоны реакции 28. Наиболее предпочтительно максимальная площадь горизонтального поперечного сечения зоны деаэрации 312 находится в диапазоне от 0,25 до 4 процентов от максимальной площади горизонтального поперечного сечения зоны реакции 28. Предпочтительно максимальная площадь горизонтального поперечного сечения зоны деаэрации 312 находится в диапазоне от приблизительно 0,02 до приблизительно 3 квадратных метров, более предпочтительно в диапазоне от приблизительно 0,05 до приблизительно 2 квадратных метров, а наиболее предпочтительно в диапазоне от 0,1 до 1,2 квадратного метра. Объем зоны деаэрации 312 предпочтительно составляет величину, меньшую чем приблизительно 5 процентов от совокупного объема реакционной среды 36 или зоны реакции 28. Более предпочтительно объем зоны деаэрации 312 находится в диапазоне от приблизительно 0,01 до приблизительно 2 процентов от совокупного объема реакционной среды 36 или зоны реакции 28. Наиболее предпочтительно объем зоны деаэрации 312 находится в диапазоне от 0,05 до приблизительно 1 процента от совокупного объема реакционной среды 36 или зоны реакции 28. Объем зоны деаэрации 312 предпочтительно составляет величину, меньшую чем приблизительно 2 кубических метра, более предпочтительно находящуюся в диапазоне от приблизительно 0,01 до приблизительно 1 кубического метра, а наиболее предпочтительно в диапазоне от 0,05 до 0,5 кубического метра.
Если обратиться теперь к Фиг. 19, то можно сказать, что на ней проиллюстрирована барботажная реакторная колонна 20 как включающая внешнюю деаэрационную емкость 400. В данной конфигурации аэрированную реакционную среду 36 отбирают из зоны реакции 28 через отверстие в боковой стороне оболочки емкости 22 повышенного уровня расположения. Отобранную аэрированную среду транспортируют до внешней деаэрационной емкости 400 через канал выпускного отверстия 402 для отделения газовой фазы от твердой и жидкой фаз. Отделенная газовая фаза покидает деаэрационную емкость 400 через канал 404, в то время как по существу деаэрированная суспензия покидает деаэрационную емкость 400 через канал 406.
На Фиг. 19 канал выпускного отверстия 402 продемонстрирован как являющийся приблизительно прямым, горизонтальным и ортогональным по отношению к оболочке емкости 22. Это просто одна удобная конфигурация; и канал выпускного отверстия 402 может быть другим в любом отношении при том условии, что он обеспечивает подходящее соединение барботажной реакторной колонны 20 и внешней деаэрационной емкости 400. Если обратиться к каналу 404, то можно сказать, что в подходящем случае данный канал присоединяется к верху или поблизости от верха деаэрационной емкости 400 для того, чтобы устранить вопросы по технике безопасности, связанные с карманом застаивающегося газа, содержащим окисляемое соединение и окислитель. Кроме того, каналы 402 и 404 в подходящем случае могут включать средства отсечения течения, такие как клапаны.
Если реакционную среду 36 будут отбирать из реактора 20 через выпускное отверстие повышенного уровня расположения, как это продемонстрировано на Фиг. 19, то тогда предпочитается, чтобы барботажная реакторная колонна 20 была бы оборудована нижним выпускным отверстием 408 поблизости от низа 52 зоны реакции 28. Нижнее выпускное отверстие 408 и нижний канал 410, присоединенный к нему, можно использовать для уменьшения коэффициента заполнения (то есть опорожнения) реактора 20 во время остановок. Предпочтительно одно или несколько нижних выпускных отверстий 408 предусматриваются в нижней одной трети высоты реакционной среды 36, более предпочтительно в нижней одной четверти реакционной среды 36, а наиболее предпочтительно в самой нижней точке зоны реакции 28.
В случае отбора суспензии в позиции повышенного уровня расположения и деаэрационной системы, продемонстрированной на Фиг. 19, нижний канал 410 и выпускное отверстие 408 не используются для отбора суспензии из зоны реакции 28 во время окисления. На современном уровне техники известно то, что твердая фаза имеет тенденцию к отстаиванию под действием силы тяжести в неаэрируемых и неперемешиваемых другим образом частях суспензии, в том числе в каналах с застаивающимся течением. Кроме того, отстоявшаяся твердая фаза (например, терефталевая кислота) может иметь тенденцию к затвердеванию в виде больших агломератов вследствие продолжения осаждения и/или перестройки кристаллов. Таким образом, для того чтобы предотвратить закупоривание нижнего канала для течения 410, часть деаэрированной суспензии из низа деаэрационной емкости 400 можно использовать для непрерывного или периодического промывания нижнего канала 410 во время нормального функционирования реактора 20. Предпочтительный способ, обеспечивающий такое промывание канала 410 суспензией, заключается в периодическом открывании клапана 412 в канале 410 и предоставлении возможности части деаэрированной суспензии перетекать через канал 410 в зону реакции 28 через нижнее отверстие 408. Даже тогда, когда клапан 412 будет полностью или частично открыт, только часть деаэрированной суспензии будет перетекать через нижний канал 410 и возвращаться обратно в зону реакции 28. Оставшуюся часть деаэрированной суспензии, не используемую для промывания нижнего канала 410, выводят через канал 414 из реактора 20 для проведения дополнительной переработки на последующих стадиях технологической схемы (например, очистки).
Во время обычного функционирования барботажной реакторной колонны 20 в течение существенного промежутка времени (например, > 100 часов) предпочитается, чтобы количество деаэрированной суспензии, используемой для промывания нижнего канала 410, составляло бы величину, меньшую чем 50 массовых процентов от совокупной деаэрированной суспензии, полученной из низа деаэрационной емкости 400, более предпочтительно меньшую чем приблизительно 20 массовых процентов, а наиболее предпочтительно меньшую чем 5 массовых процентов. Кроме того, предпочитается, чтобы в течение существенного промежутка времени средний массовый расход деаэрированной суспензии, используемой для промывания нижнего канала 410, составлял бы величину, меньшую чем приблизительно 4-кратный средний массовый расход окисляемого соединения, вводимого в зону реакции 28, более предпочтительно меньшую чем приблизительно 2-кратный средний массовый расход окисляемого соединения, вводимого в зону реакции 28, еще более предпочтительно меньшую чем средний массовый расход окисляемого соединения, вводимого в зону реакции 28, а наиболее предпочтительно меньшую чем 0,5-кратный средний массовый расход окисляемого соединения, вводимого в зону реакции 28.
Если обратиться опять к Фиг. 19, то можно сказать, что деаэрационная емкость 400 включает по существу прямостоячую предпочтительно цилиндрическую боковую стенку 416, определяющую зону деаэрации 418. Зона деаэрации 418 имеет диаметр «d» и высоту «h». Высоту «h» измеряют в виде расстояния по вертикали между позицией, в которой аэрированная реакционная среда поступает в деаэрационную емкость 400, и низом боковой стенки 416. Высота «h», диаметр «d», площадь поверхности и объем зоны деаэрации 418 предпочтительно представляют собой по существу то же самое, что и то, что описывается выше в том, что касается зоны деаэрации 312 деаэрационной емкости 300, проиллюстрированной на Фиг. 16-18. В дополнение к этому деаэрационная емкость 400 включает верхнюю секцию 420, полученную в результате продолжения боковой стенки 416 выше зоны деаэрации 418. Верхняя секция 420 деаэрационной емкости 400 может иметь любую высоту, хотя предпочтительно она проходит снизу вверх до положения уровня или положения выше уровня реакционной среды 36 в зоне реакции 28. Верхняя секция 420 обеспечивает то, чтобы газовая фаза имела бы пространство для надлежащего отделения от жидкой и твердой фаз перед тем, как покинуть деаэрационную емкость 400 через канал 404. В данный момент следует отметить то, что несмотря на то, что канал 404 проиллюстрирован как возвращающий отделенную газовую фазу в зону отделения реактора 20, в альтернативном варианте канал 404 может быть соединен с оболочкой емкости 22 на любом уровне по высоте выше канала выпускного отверстия 402. Необязательно канал 404 может быть соединен с каналом выпускного отверстия для газа 40 таким образом, чтобы отделенная газовая фаза из деаэрационной емкости 400 объединялась бы с удаленным потоком паров верха реактора в канале 40 и отправлялась бы на последующие стадии технологической схемы для дополнительной переработки.
Если обратиться теперь к Фиг. 20, то можно сказать, что на ней проиллюстрирована барботажная реакторная колонна 20 как включающая гибридную внутренне-внешнюю деаэрационную емкость 500. В данной конфигурации часть реакционной среды 36 отбирают из зоны реакции 28 через относительно большое отверстие повышенного уровня расположения 502 в боковой стенке оболочки емкости 22. После этого отобранную реакционную среду 36 транспортируют через коленный канал 504 относительно большого диаметра, и она поступает в верх деаэрационной емкости 500. На Фиг. 20 коленный канал 504 продемонстрирован как ортогонально соединяющийся с боковой стенкой оболочки емкости 22 и включающий плавный поворот на угол, равный приблизительно 90 градусам. Это просто одна удобная конфигурация; и коленный канал 504 может быть другим в любом отношении при том условии, что он обеспечивает подходящее соединение барботажной реакторной колонны 20 и внешней деаэрационной емкости 500, как это описано. Кроме того, коленный канал 504 в подходящем случае может включать средства отсечения течения, такие как клапаны.
В деаэрационной емкости 500 газовая фаза перемещается снизу вверх, в то время как твердая и жидкая фазы перемещаются сверху вниз. Перемещающаяся снизу вверх газовая фаза может повторно поступать в коленный канал 504, а после этого уходить через отверстие 502 обратно в зону реакции 28. Таким образом, в отверстии 502 может иметь место противоточное течение поступающей реакционной среды 36 и отходящего отделенного газа. Деаэрированная суспензия покидает деаэрационную емкость 500 через канал 506. Деаэрационная емкость 500 включает по существу прямостоячую, предпочтительно цилиндрическую, боковую стенку 508, определяющую зону деаэрации 510. Зона деаэрации 510 имеет высоту «h» и диаметр «d». Предпочитается, чтобы отверстие повышенного уровня расположения 502 и коленный канал 504 имели бы диаметр, идентичный диаметру «d» зоны деаэрации 510 или превышающий его. Высота «h», диаметр «d», площадь поверхности и объем зоны деаэрации 510 предпочтительно представляют собой по существу то же самое, что и то, что описывалось выше, в том, что касается зоны деаэрации 312 деаэрационной емкости 300, проиллюстрированной на Фиг. 16-18.
Фиг. 19 и 20 иллюстрируют вариант реализации барботажной реакторной колонны 20, в которой твердый продукт (например, сырую неочищенную терефталевую кислоту), полученный в зоне реакции 28, отбирают из зоны реакции 28 через выпускное отверстие повышенного уровня расположения. Отбор аэрированной реакционной среды 36 из позиции повышенного уровня расположения выше низа барботажной реакторной колонны 20 может способствовать предотвращению накопления и застаивания плохо аэрированной реакционной среды 36 в области низа 52 зоны реакции 28. В соответствии с другими аспектами настоящего изобретения концентрации кислорода и окисляемого соединения (например, пара-ксилола) в реакционной среде 36 поблизости от верха реакционной среды 36 предпочтительно составляют величины, меньшие, чем соответствующие концентрации поблизости от низа. Таким образом, отбор реакционной среды 36 в позиции повышенного уровня расположения может обеспечить увеличение выхода в результате уменьшения количества не вступивших в реакцию реагентов, отбираемых из реактора 20. Кроме этого, тогда, когда барботажная реакторная колонна 20 будет функционировать при высоком значении STR и наличии градиентов химического состава, описанных в настоящем документе, температура реакционной среды 36 будет значительно варьироваться в вертикальном направлении. В таких условиях температура реакционной среды 36 обычно будет иметь локальные минимумы поблизости от нижнего края и верхнего края зоны реакции 28. Поблизости от нижнего края минимум относится к испарению растворителя поблизости от того места, где производят введение всего количества или части окислителя. Поблизости от верхнего края минимум опять-таки обуславливается испарением растворителя, хотя в данном случае это объясняется уменьшением давления внутри реакционной среды. В дополнение к этому в промежутке между верхним и нижним краями могут иметь место и другие локальные минимумы каждый раз тогда, когда в реакционную среду будут вводить дополнительные количества исходного подаваемого материала или окислителя. Таким образом, в промежутке между нижним краем и верхним краем зоны реакции 28 существуют один или несколько температурных максимумов, движущей силой возникновения которых является экзотермическая теплота реакций окисления. Отбор реакционной среды 36 в позиции повышенного уровня расположения при повышенной температуре может оказаться в особенности выгодным тогда, когда при повышенных температурах будет происходить переработка на последующих стадиях технологической схемы, поскольку уменьшаются затраты энергии, связанные с нагреванием отобранной среды, предназначенной для переработки на последующих стадиях технологической схемы.
Таким образом, в предпочтительном варианте реализации настоящего изобретения и в особенности тогда, когда переработку на последующих стадиях технологической схемы будут проводить при повышенных температурах, реакционную среду 36 из барботажной реакторной колонны 20 отбирают через выпускное отверстие (отверстия) повышенного уровня расположения, размещенное выше позиции (позиций), в которой в зону реакции 28 поступают, по меньшей мере, 50 массовых процентов от потока жидкофазного исходного подаваемого материала и/или потока газофазного окислителя. Более предпочтительно реакционную среду 36 из барботажной реакторной колонны 20 отбирают через выпускное отверстие (отверстия) повышенного уровня расположения, размещенное выше позиции (позиций), в которой в зону реакции 28 поступает по существу весь поток жидкофазного исходного подаваемого материала и/или поток газофазного окислителя. Предпочтительно через выпускное отверстие (отверстия) повышенного уровня расположения отбирают, по меньшей мере, 50 массовых процентов твердофазных и жидкофазных компонентов, отбираемых из барботажной реакторной колонны 20. Более предпочтительно через выпускное отверстие (отверстия) повышенного уровня расположения отбирают по существу все количество твердофазных и жидкофазных компонентов, отбираемых из барботажной реакторной колонны 20. Предпочтительно выпускное отверстие (отверстия) повышенного уровня расположения размещают, по меньшей мере, приблизительно на 1D выше нижнего края 52 зоны реакции 28. Более предпочтительно выпускное отверстие (отверстия) повышенного уровня расположения размещают, по меньшей мере, приблизительно на 2D выше нижнего края 52 зоны реакции 28. Наиболее предпочтительно выпускное отверстие (отверстия) повышенного уровня расположения размещают, по меньшей мере, на 3D выше нижнего края 52 зоны реакции 28. При наличии высоты «Н» реакционной среды 36 предпочитается, чтобы выпускное отверстие (отверстия) повышенного уровня расположения были бы размещены по вертикали в промежутке между приблизительно 0,2Н и приблизительно 0,8Н, более предпочтительно между приблизительно 0,3Н и приблизительно 0,7Н, а наиболее предпочтительно между 0,4Н и 0,6Н. Кроме того, предпочитается, чтобы температура реакционной среды 36 на выпускном отверстии повышенного уровня расположения, выходящем из зоны реакции 28, по меньшей мере, на 1°С превышала бы температуру реакционной среды 36 на нижнем крае 52 зоны реакции 28. Более предпочтительно температура реакционной среды 36 на выпускном отверстии повышенного уровня расположения, выходящем из зоны реакции 28, превышает температуру реакционной среды 36 на нижнем крае 52 зоны реакции 28 на величину в диапазоне от приблизительно 1,5 до приблизительно 16°С. Наиболее предпочтительно температура реакционной среды 36 на выпускном отверстии повышенного уровня расположения, выходящем из зоны реакции 28, превышает температуру реакционной среды 36 на нижнем крае 52 зоны реакции 28 на величину в диапазоне от 2 до 12°С.
Если обратиться теперь к Фиг. 21, то можно сказать, что на ней проиллюстрирована барботажная реакторная колонна 20 как включающая альтернативную гибридную деаэрационную емкость 600, расположенную в области низа реактора 20. В данной конфигурации аэрированную реакционную среду 36 отбирают из зоны реакции 28 через относительно большое отверстие 602 на нижнем крае 52 оболочки емкости 22. Отверстие 602 определяет незамкнутый верхний край деаэрационной емкости 600. В деаэрационной емкости 600 газовая фаза перемещается снизу вверх в то время, как твердая и жидкая фазы перемещаются сверху вниз. Перемещающаяся снизу вверх газовая фаза может повторно поступать в зону реакции 28 через отверстие 602. Таким образом, в отверстии 602 может иметь место противоточное течение поступающей реакционной среды 36 и отходящего отделенного газа. Деаэрированная суспензия покидает деаэрационную емкость 600 через канал 604. Деаэрационная емкость 600 включает по существу прямостоячую, предпочтительно цилиндрическую, боковую стенку 606, определяющую зону деаэрации 608. Зона деаэрации 608 имеет высоту «h» и диаметр «d». Предпочитается, чтобы отверстие 602 имело бы диаметр, идентичный диаметру «d» зоны деаэрации 608 или превышающий его. Высота «h», диаметр «d», площадь поверхности и объем зоны деаэрации 608 предпочтительно представляют собой по существу то же самое, что и то, что описывалось выше, в том, что касается зоны деаэрации 312 деаэрационной емкости 300, проиллюстрированной на Фиг. 16-18.
Если обратиться теперь к Фиг. 22, то можно сказать, что на ней проиллюстрирована барботажная реакторная колонна 20 Фиг. 21 как включающая альтернативный барботер окислителя 620. Барботер окислителя 620 включает кольцевой элемент 622 и пару каналов впускных отверстий 624, 626. Кольцевой элемент 622 предпочтительно имеет по существу ту же самую конфигурацию, что и кольцевой элемент 202, описанный выше, в том, что касается Фиг. 12-15. Каналы впускных отверстий 624, 626 проходят снизу вверх через отверстия в нижнем днище 48 оболочки емкости 22 и обеспечивают подачу потока окислителя в кольцевой элемент 622.
Если обратиться теперь к Фиг. 23, то можно сказать, что на ней проиллюстрирована барботажная реакторная колонна 20 Фиг. 21 как включающая не использующее барботер средство введения потока окислителя в зону реакции 28. В конфигурации Фиг. 23 подачу потока окислителя в реактор 20 обеспечивают через каналы для окислителя 630, 632. Каналы для окислителя 630, 632 соединяют с соответствующими отверстиями для окислителя 634, 636 в нижнем днище 48 оболочки емкости 22. Поток окислителя вводят непосредственно в зону реакции 28 через отверстия для окислителя 634, 636. Для отклонения течения потока окислителя сразу после его первоначального введения в зону реакции 28 можно предусмотреть наличие необязательных отбойников 638, 640.
Как упоминалось выше, предпочитается, чтобы реактор окисления имел бы конфигурацию и функционировал бы в соответствии со способом, который позволяет избегать возникновения зон высокой концентрации окисляемого соединения в реакционной среде, поскольку такие зоны могут привести к образованию примесей. Один способ улучшения первоначального диспергирования окисляемого соединения (например, пара-ксилола) в реакционной среде заключается в разбавлении окисляемого соединения жидкостью. Жидкость, используемая для разбавления окисляемого соединения, своим источником может иметь часть реакционной среды, расположенную на существенном расстоянии от позиции (позиций), в которой в зону реакции подают окисляемое соединение. Для данной жидкости из удаленной части реакционной среды в результате циркуляции можно обеспечить подачу к позиции, расположенной поблизости от позиции введения окисляемого соединения через канал для течения, который располагается внутри и/или вне основной реакционной емкости.
Фиг. 24 и 25 иллюстрируют два предпочтительных способа организации циркуляции жидкости из удаленной части реакционной среды с подачей в позицию, расположенную поблизости от впускного отверстия для окисляемого соединения, при использовании внутреннего (Фиг. 24) или внешнего (Фиг. 25) канала. Предпочтительно длина канала для течения от его впускного отверстия (то есть отверстия (отверстий), где жидкость поступает в канал) до его выпускного отверстия (то есть отверстия (отверстий), где жидкость выпускают из канала) превышает приблизительно 1 метр, более предпочтительно превышает приблизительно 3 метра, еще более предпочтительно превышает приблизительно 6 метров, а наиболее предпочтительно превышает 9 метров. Однако фактическая длина канала становится менее существенным параметром, если жидкость получают из отдельной емкости, может быть, расположенной непосредственно над или рядом с емкостью, в которую первоначально выпускают исходный подаваемый материал, образуемый окисляемым соединением. Жидкость из любой отдельной емкости, содержащей, по меньшей мере, некоторое количество реакционной среды, представляет собой предпочтительный источник для первоначального разбавления окисляемого соединения.
Предпочитается, чтобы жидкость, перепускаемая через канал, вне зависимости от источника имела бы пониженную стационарную концентрацию окисляемого соединения в сопоставлении с реакционной средой, непосредственно примыкающей, по меньшей мере, к одному выпускному отверстию канала. Кроме того, предпочитается, чтобы жидкость, перепускаемая через канал, имела бы концентрацию окисляемого соединения в жидкой фазе, меньшую приблизительно 100000 ч./млн (мас.), более предпочтительно меньшую приблизительно 10000 ч./млн (мас.), еще более предпочтительно меньшую приблизительно 1000 ч./млн (мас.), а наиболее предпочтительно меньшую 100 ч./млн (мас.), где концентрации измеряют перед добавлением в канал порции образуемого окисляемым соединением исходного подаваемого материала и любого необязательного отдельного исходного подаваемого материала, образуемого растворителем. При проведении измерения после добавления порции образуемого окисляемым соединением исходного подаваемого материала и необязательного исходного подаваемого материала, образуемого растворителем, предпочитается, чтобы объединенный поток жидкости, поступающий в реакционную среду, имел бы концентрацию окисляемого соединения в жидкой фазе, меньшую приблизительно 300000 ч./млн (мас.), более предпочтительно меньшую приблизительно 50000 ч./млн (мас.), а наиболее предпочтительно меньшую 10000 ч./млн (мас.).
Желательно выдерживать течение через канал на уровне достаточно низкого расхода таким образом, чтобы циркулирующая жидкость действительно подавляла бы желательный совокупный градиент окисляемого соединения в пределах реакционной среды. В данном отношении предпочитается, чтобы соотношение между массой жидкой фазы в зоне реакции, в которую первоначально выпускают порцию окисляемого соединения, и массовым расходом жидкости, перепускаемой через канал, превышало бы приблизительно 0,3 минуты, более предпочтительно превышало бы приблизительно 1 минуту, еще более предпочтительно находилось бы в диапазоне от приблизительно 2 минут до приблизительно 120 минут, а наиболее предпочтительно от 3 минут до 60 минут.
Существует множество способов, стимулирующих течение жидкости через канал. Предпочтительные способы включают применение силы тяжести, эдукторов всех типов, использующих либо газ, либо жидкость, либо и газ, и жидкость в качестве движущей текучей среды, и механических насосов всех типов. В случае применения эдуктора в одном варианте реализации изобретения в качестве движущей текучей среды используют, по меньшей мере, одну текучую среду, выбираемую из группы, состоящей из: исходного подаваемого материала, образуемого окисляемым соединением, (жидкости или газа), исходного подаваемого материала, образуемого окислителем, (газа), исходного подаваемого материала, образуемого растворителем, (жидкости) и снабженного насосом источника реакционной среды (суспензии). В еще одном варианте реализации в качестве движущей текучей среды используют, по меньшей мере, две текучие среды, выбираемые из группы, состоящей из: исходного подаваемого материала, образуемого окисляемым соединением, исходного подаваемого материала, образуемого окислителем, и исходного подаваемого материала, образуемого растворителем. В еще одном варианте реализации в качестве движущей текучей среды используют комбинацию исходного подаваемого материала, образуемого окисляемым соединением, исходного подаваемого материала, образуемого окислителем, и исходного подаваемого материала, образуемого растворителем.
Подходящие диаметр или диаметры канала циркуляции могут варьироваться в соответствии с количеством и свойствами транспортируемого материала, энергией, доступной для стимулирования движения потока, и соображениями, касающимися капитальных затрат. Предпочитается, чтобы минимальный диаметр такого канала превышал бы приблизительно 0,02 метра, более предпочтительно находился бы в диапазоне от приблизительно 0,06 метра до приблизительно 2 метров, а наиболее предпочтительно от 0,12 до 0,8 метра.
Как отмечалось выше, течение через канал желательно регулировать, выдерживая в определенных предпочтительных диапазонах. Существует множество известных на современном уровне техники способов оказания воздействия на данное управление в результате задания подходящей фиксированной геометрии во время изготовления канала для течения. Еще один предпочтительный вариант реализации заключается в использовании геометрий, которые могут меняться во время функционирования, а именно включающих клапаны всех сортов и описаний, в том числе как с ручным управлением, так и с механическим управлением при использовании любых средств, включающих контуры управления с обратной связью от воспринимающего элемента или без этого. Еще один предпочтительный способ управления течением разбавленной жидкости заключается в варьировании подвода энергии в промежутке между впускным отверстием и выпускным отверстием канала. Предпочтительные способы включают изменение расхода для одной или нескольких движущих текучих сред, подаваемых в эдуктор, изменение подвода энергии к приводу насоса и изменение разницы плотностей или разницы уровней по высоте при использовании силы тяжести. Данные предпочтительные способы также можно использовать и во всех комбинациях.
Канал, используемый для циркуляции жидкости из реакционной среды, может относиться к любому типу, известному на современном уровне техники. В одном варианте реализации используют канал, сконструированный полностью или частично при использовании обычных материалов для изготовления трубопровода. В еще одном варианте реализации используют канал, сконструированный полностью или частично при использовании стенки реакционной емкости в качестве одной части канала. Канал можно сконструировать полностью включенным в границы реакционной емкости (Фиг. 24), или его можно сконструировать расположенным полностью вне реакционной емкости (Фиг. 25), или он может включать секции, располагаемые как внутри, так и вне реакционной емкости.
Изобретатели предусматривают то, что, в особенности в более крупных реакторах, может оказаться желательным наличие множества каналов и различных вариантов перемещения жидкости через канал. Кроме того, может оказаться желательным наличие множества выпускных отверстий во множестве позиций на одном из каналов или на всех каналах. Детали конструкции будут обеспечивать наличие баланса между желательным совокупным градиентом стационарных концентраций окисляемого соединения и желательным первоначальным разбавлением образуемого окисляемым соединением исходного подаваемого материала в соответствии с другими аспектами настоящего изобретения.
Фиг. как 24, так и 25 иллюстрируют конструкции, которые используют деаэрационную емкость, соединенную с каналом. Данная деаэрационная емкость обеспечивает то, что часть реакционной среды, используемой для разбавления поступающего окисляемого соединения, представляет собой по существу деаэрированную суспензию. Однако в данный момент следует отметить то, что жидкость или суспензия, используемые для разбавления поступающего окисляемого соединения, могут находиться в аэрированной форме, а также в деаэрированной форме.
Использование жидкости, перетекающей через канал, для обеспечения разбавления образуемого окисляемым соединением исходного подаваемого материала является в особенности хорошо подходящим для использования в барботажных реакторных колоннах. Кроме того, в барботажных реакторных колоннах получения больших преимуществ от первоначального разбавления образуемого окисляемым соединением исходного подаваемого материала можно добиться даже без добавления образуемого окисляемым соединением исходного подаваемого материала непосредственно в канал при том условии, что выпускное отверстие канала будет располагаться достаточно близко к позиции добавления окисляемого соединения. В таком варианте реализации предпочитается, чтобы выпускное отверстие канала располагалось бы в пределах приблизительно 27 диаметров выпускного отверстия канала от наиболее близкой позиции добавления окисляемого соединения, более предпочтительно в пределах приблизительно 9 диаметров выпускного отверстия канала, еще более предпочтительно в пределах приблизительно 3 диаметров выпускного отверстия канала, а наиболее предпочтительно в пределах 1 диаметра выпускного отверстия канала.
Также было обнаружено то, что даже без применения каналов для получения жидкости разбавления из удаленной части реакционной среды подходящими для использования при первоначальном разбавлении образуемого окисляемым соединением исходного подаваемого материала в барботажных колоннах окисления, соответствующих одному варианту реализации настоящего изобретения, могут оказаться эдукторы потока. В таких случаях эдуктор располагают внутри реакционной среды, и он имеет свободный проход от реакционной среды до горловины эдуктора, где низкое давление будет затягивать примыкающую реакционную среду. Примеры двух возможных конфигураций эдукторов проиллюстрированы на Фиг. 26 и 27. В предпочтительном варианте реализации данных эдукторов самая близкая позиция подачи окисляемого соединения располагается в пределах приблизительно 4 метров, более предпочтительно в пределах приблизительно 1 метра, а наиболее предпочтительно 0,3 метра от горловины эдуктора. В другом варианте реализации в качестве движущей текучей среды под давлением подают окисляемое соединение. В еще одном варианте реализации в качестве дополнительной движущей текучей среды совместно с окисляемым соединением под давлением подают либо растворитель, либо окислитель. В еще одном варианте реализации в качестве дополнительной движущей текучей среды совместно с окисляемым соединением под давлением подают как растворитель, так и окислитель.
Изобретатели предусматривают то, что, в особенности в более крупных реакторах, может оказаться желательным наличие множества эдукторов различных конструкций, расположенных в различных позициях в пределах реакционной среды. Детали конструкции будут обеспечивать наличие баланса между желательным совокупным градиентом стационарных концентраций окисляемого соединения и желательным первоначальным разбавлением образуемого окисляемым соединением исходного подаваемого материала в соответствии с другими аспектами настоящего изобретения. В дополнение к этому изобретатели предусматривают то, что струи истечения из выпускного отверстия эдуктора могут быть ориентированы в любом направлении. В случае использования множества эдукторов каждый эдуктор может быть ориентирован индивидуально, опять-таки в любом направлении.
Как упоминалось выше, определенные физические и эксплуатационные признаки барботажной реакторной колонны 20, описанные выше в том, что касается Фиг. 1-27, обеспечивают наличие вертикальных градиентов давления, температуры и концентраций реагента (то есть кислорода и окисляемого соединения) в реакционной среде 36. Как обсуждалось выше, данные вертикальные градиенты могут обеспечивать более эффективную и экономичную реализацию способа окисления в сопоставлении с обычно используемыми способами окисления, которые благоприятствуют получению хорошо перемешанной реакционной среды, характеризующейся относительно однородными давлением, температурой и концентрацией реагента во всех ее частях. Далее более подробно будут обсуждаться вертикальные градиенты для кислорода, окисляемого соединения (например, пара-ксилола) и температуры, которые делает возможными использование системы окисления в соответствии с вариантом реализации настоящего изобретения.
Если обратиться теперь к Фиг. 28, то можно сказать, что для того, чтобы получить количественные характеристики градиентов концентраций реагентов, существующих в реакционной среде 36 во время окисления в барботажной реакторной колонне 20, совокупный объем реакционной среды 36 можно теоретически разделить на 30 дискретных горизонтальных долей с равным объемом. Фиг. 28 иллюстрирует концепцию разделения реакционной среды 36 на 30 дискретных горизонтальных долей с равным объемом. За исключением самой верхней и самой нижней горизонтальных долей каждая горизонтальная доля представляет собой дискретный объем, ограниченный по его верхней и нижней сторонам воображаемыми горизонтальными плоскостями и ограниченный по его боковым сторонам стенкой реактора 20. Самая верхняя горизонтальная доля ограничена по ее нижней стороне воображаемой горизонтальной плоскостью, а по ее верхней стороне - верхней поверхностью реакционной среды 36. Самая нижняя горизонтальная доля ограничена по ее верхней стороне воображаемой горизонтальной плоскостью, а по ее нижней стороне - низом оболочки емкости. Как только реакционная среда 36 будет теоретически разделена на 30 дискретных горизонтальных долей с равным объемом, то после этого можно будет определить средневременную и среднеобъемную концентрацию для каждой горизонтальной доли. Индивидуальную горизонтальную долю, имеющую максимальную концентрацию в числе всех 30 горизонтальных долей, можно идентифицировать как «горизонтальную долю С-макс». Индивидуальную горизонтальную долю, расположенную выше горизонтальной доли С-макс и имеющую минимальную концентрацию в числе всех горизонтальных долей, расположенных выше горизонтальной доли С-макс, можно идентифицировать как «горизонтальную долю С-мин». После этого вертикальный градиент концентраций можно рассчитать в виде соотношения между концентрацией в горизонтальной доле С-макс и концентрацией в горизонтальной доле С-мин.
Что касается получения количественных характеристик градиента концентрации кислорода, то тогда, когда реакционная среда 36 будет теоретически разделена на 30 дискретных горизонтальных долей с равным объемом, горизонтальную долю О2-макс идентифицируют как имеющую максимальную концентрацию кислорода в числе всех 30 горизонтальных долей, а горизонтальную долю О2-мин идентифицируют как имеющую минимальную концентрацию кислорода в числе горизонтальных долей, расположенных выше горизонтальной доли О2-макс. Концентрации кислорода в горизонтальных долях измеряют в газовой фазе реакционной среды 36 в виде средневременных и среднеобъемных молярных величин при расчете для влажного состояния. Предпочитается, чтобы соотношение между концентрацией кислорода в горизонтальной доле О2-макс и концентрацией кислорода в горизонтальной доле О2-мин находилось бы в диапазоне от приблизительно 2:1 до приблизительно 25:1, более предпочтительно в диапазоне от приблизительно 3:1 до приблизительно 15:1, а наиболее предпочтительно в диапазоне от 4:1 до 10:1.
Обычно горизонтальная доля О2-макс будет располагаться поблизости от низа реакционной среды 36, в то время как горизонтальная доля О2-мин будет располагаться поблизости от верха реакционной среды 36. Предпочтительно горизонтальная доля О2-мин будет представлять собой одну из 5 самых верхних горизонтальных долей в числе 30 дискретных горизонтальных долей. Наиболее предпочтительно горизонтальная доля О2-мин представляет собой самую верхнюю долю в числе 30 дискретных горизонтальных долей, как это проиллюстрировано на Фиг. 28. Предпочтительно горизонтальная доля О2-макс представляет собой одну из 10 самых нижних горизонтальных долей в числе 30 дискретных горизонтальных долей. Наиболее предпочтительно горизонтальная доля О2-макс представляет собой одну из 5 самых нижних горизонтальных долей в числе 30 дискретных горизонтальных долей. Например, Фиг. 28 иллюстрирует горизонтальную долю О2-макс как третью горизонтальную долю от низа реактора 20. Предпочитается, чтобы разнесение по вертикали между горизонтальными долями О2-мин и О2-макс составляло бы, по меньшей мере, приблизительно 2W, более предпочтительно, по меньшей мере, приблизительно 4W, а наиболее предпочтительно, по меньшей мере, 6W. Предпочитается, чтобы разнесение по вертикали между горизонтальными долями О2-мин и О2-макс составляло бы, по меньшей мере, приблизительно 0,2Н, более предпочтительно, по меньшей мере, приблизительно 0,4Н, а наиболее предпочтительно, по меньшей мере, 0,6Н.
Средневременная и среднеобъемная концентрация кислорода в горизонтальной доле О2-мин при расчете для влажного состояния предпочтительно находится в диапазоне от приблизительно 0,1 до приблизительно 3 мольных процентов, более предпочтительно в диапазоне от приблизительно 0,3 до приблизительно 2 мольных процентов, а наиболее предпочтительно в диапазоне от 0,5 до 1,5 мольных процентов. Средневременная и среднеобъемная концентрация кислорода в горизонтальной доле О2-макс предпочтительно находится в диапазоне от приблизительно 4 до приблизительно 20 мольных процентов, более предпочтительно в диапазоне от приблизительно 5 до приблизительно 15 мольных процентов, а наиболее предпочтительно в диапазоне от 6 до 12 мольных процентов. Средневременная концентрация кислорода в газообразном отходящем потоке, выпускаемом из реактора 20 через выпускное отверстие для газа 40, при расчете для сухого состояния предпочтительно находится в диапазоне от приблизительно 0,5 до приблизительно 9 мольных процентов, более предпочтительно в диапазоне от приблизительно 1 до приблизительно 7 мольных процентов, а наиболее предпочтительно в диапазоне от 1,5 до 5 мольных процентов.
Поскольку концентрация кислорода очень заметно уменьшается по направлению к верху реакционной среды 36, желательно, чтобы потребность в кислороде в области верха реакционной среды 36 была бы пониженной. Получения данной пониженной потребности в кислороде поблизости от верха реакционной среды 36 можно добиться в результате создания вертикального градиента концентрации окисляемого соединения (например, пара-ксилола), когда минимальная концентрация окисляемого соединения будет находиться поблизости от верха реакционной среды 36.
Что касается получения количественных характеристик градиента концентраций окисляемого соединения (например, пара-ксилола), то тогда, когда реакционная среда 36 будет теоретически разделена на 30 дискретных горизонтальных долей с равным объемом, горизонтальную долю ОС-макс будут идентифицировать как имеющую максимальную концентрацию окисляемого соединения в числе всех 30 горизонтальных долей, а горизонтальную долю ОС-мин будут идентифицировать как имеющую минимальную концентрацию окисляемого соединения в числе горизонтальных долей, расположенных выше горизонтальной доли ОС-макс. Концентрации окисляемого соединения в горизонтальных долях измеряют в жидкой фазе при расчете на величины средневременной и среднеобъемной массовой доли. Предпочитается, чтобы соотношение между концентрацией окисляемого соединения в горизонтальной доле ОС-макс и концентрацией окисляемого соединения в горизонтальной доле ОС-мин превышало бы приблизительно 5:1, более предпочтительно превышало бы приблизительно 10:1, еще более предпочтительно превышало бы приблизительно 20:1, а наиболее предпочтительно находилось бы в диапазоне от 40:1 до 1000:1.
Обычно горизонтальная доля ОС-макс будет располагаться поблизости от низа реакционной среды 36, в то время как горизонтальная доля ОС-мин будет располагаться поблизости от верха реакционной среды 36. Предпочтительно горизонтальная доля ОС-мин представляет собой одну из 5 самых верхних горизонтальных долей в числе 30 дискретных горизонтальных долей. Наиболее предпочтительно горизонтальная доля ОС-мин представляет собой самую верхнюю долю в числе 30 дискретных горизонтальных долей, как это проиллюстрировано на Фиг. 28. Предпочтительно горизонтальная доля ОС-макс представляет собой одну из 10 самых нижних горизонтальных долей в числе 30 дискретных горизонтальных долей. Наиболее предпочтительно горизонтальная доля ОС-макс представляет собой одну из 5 самых нижних горизонтальных долей в числе 30 дискретных горизонтальных долей. Например, Фиг. 28 иллюстрирует горизонтальную долю ОС-макс как пятую горизонтальную долю от низа реактора 20. Предпочитается, чтобы разнесение по вертикали между горизонтальными долями ОС-мин и ОС-макс составляло бы, по меньшей мере, приблизительно 2W, где «W» представляет собой максимальную ширину реакционной среды 36. Более предпочтительно разнесение по вертикали между горизонтальными долями ОС-мин и ОС-макс составляет, по меньшей мере, приблизительно 4W, а наиболее предпочтительно, по меньшей мере, 6W. При наличии у реакционной среды 36 высоты «Н» предпочитается, чтобы разнесение по вертикали между горизонтальными долями ОС-мин и ОС-макс составляло бы, по меньшей мере, приблизительно 0,2Н, более предпочтительно, по меньшей мере, приблизительно 0,4Н, а наиболее предпочтительно, по меньшей мере, 0,6Н.
Средневременная и среднеобъемная концентрация окисляемого соединения (например, пара-ксилола) в жидкой фазе в горизонтальной доле ОС-мин предпочтительно составляет величину, меньшую чем приблизительно 5000 ч./млн (мас.), более предпочтительно меньшую чем приблизительно 2000 ч./млн (мас.), еще более предпочтительно меньшую чем приблизительно 400 ч./млн (мас.), а наиболее предпочтительно находящуюся в диапазоне от 1 ч./млн (мас.) до 100 ч./млн (мас.). Средневременная и среднеобъемная концентрация окисляемого соединения в жидкой фазе в горизонтальной доле ОС-макс предпочтительно находится в диапазоне от приблизительно 100 ч./млн (мас.) до приблизительно 10000 ч./млн (мас.), более предпочтительно в диапазоне от приблизительно 200 ч./млн (мас.) до приблизительно 5000 ч./млн (мас.), а наиболее предпочтительно в диапазоне от 500 ч./млн (мас.) до 3000 ч./млн (мас.).
Несмотря на то что предпочитается, чтобы барботажная реакторная колонна 20 обеспечивала бы наличие для концентрации окисляемого соединения градиентов по вертикали, предпочитается также и то, чтобы объемный процент реакционной среды 36, имеющей концентрацию окисляемого соединения в жидкой фазе, превышающую 1000 ч./млн (мас.), был бы сведен к минимуму. Предпочтительно средневременный объемный процент реакционной среды 36, имеющей концентрацию окисляемого соединения в жидкой фазе, превышающую 1000 ч./млн (мас.), составляет величину, меньшую чем приблизительно 9 процентов, более предпочтительно меньшую чем приблизительно 6 процентов, а наиболее предпочтительно меньшую чем 3 процента. Предпочтительно средневременный объемный процент реакционной среды 36, имеющей концентрацию окисляемого соединения в жидкой фазе, превышающую 2500 ч./млн (мас.), составляет величину, меньшую чем приблизительно 1,5 процента, более предпочтительно меньшую чем приблизительно 1 процент, а наиболее предпочтительно меньшую чем 0,5 процента. Предпочтительно средневременный объемный процент реакционной среды 36, имеющей концентрацию окисляемого соединения в жидкой фазе, превышающую 10000 ч./млн (мас.), составляет величину, меньшую чем приблизительно 0,3 процента, более предпочтительно меньшую чем приблизительно 0,1 процента, а наиболее предпочтительно меньшую чем 0,03 процента. Предпочтительно средневременный объемный процент реакционной среды 36, имеющей концентрацию окисляемого соединения в жидкой фазе, превышающую 25000 ч./млн (мас.), составляет величину, меньшую чем приблизительно 0,03 процента, более предпочтительно меньшую чем приблизительно 0,015 процента, а наиболее предпочтительно меньшую чем 0,007 процента. Изобретатели отмечают то, что объем реакционной среды 36, характеризующейся повышенными уровнями содержания окисляемого соединения, не обязательно должен совпадать с одним объемом, образованным примыкающими друг к другу его частями. В различные моменты времени хаотические схемы течения в реакционной емкости барботажной колонны приводят к образованию одновременно двух или более сплошных, но сегрегированных частей реакционной среды 36, характеризующейся повышенными уровнями содержания окисляемого соединения. В каждый момент времени, используемый при усреднении по времени, все такие сплошные, но сегрегированные объемы, превышающие 0,0001 объемного процента от совокупной реакционной среды, складывают друг с другом для определения совокупного объема, характеризующегося повышенными уровнями концентрации окисляемого соединения в жидкой фазе.
В дополнение к градиентам концентраций кислорода и окисляемого соединения, обсуждавшимся выше, предпочитается, чтобы в реакционной среде 36 существовал бы градиент температуры. Если обратиться опять к Фиг. 28, то можно сказать, что количественные характеристики данного градиента температуры можно получить по способу, подобному получению количественных характеристик градиентов концентраций, в результате теоретического разделения реакционной среды 36 на 30 дискретных горизонтальных долей с равным объемом и измерения средневременной и среднеобъемной температуры в каждой доле. Тогда горизонтальную долю, характеризующуюся наименьшей температурой в числе самых нижних 15 горизонтальных долей, можно идентифицировать как горизонтальную долю Т-мин, а горизонтальную долю, расположенную выше горизонтальной доли Т-мин и имеющую максимальную температуру в числе всех долей, расположенных выше горизонтальной доли Т-мин, после этого можно идентифицировать как «горизонтальную долю Т-макс». Предпочитается, чтобы температура горизонтальной доли Т-макс была бы, по меньшей мере, приблизительно на 1°С большей, чем температура горизонтальной доли Т-мин. Более предпочтительно температура горизонтальной доли Т-макс находится в диапазоне температур, превышающих температуру горизонтальной доли Т-мин на величину в диапазоне от приблизительно 1,25 до приблизительно 12°С. Наиболее предпочтительно температура горизонтальной доли Т-макс находится в диапазоне температур, превышающих температуру горизонтальной доли Т-мин на величину в диапазоне от 2 до 8°С. Температура горизонтальной доли Т-макс предпочтительно находится в диапазоне от приблизительно 125 до приблизительно 200°С, более предпочтительно в диапазоне от приблизительно 140 до приблизительно 180°С, а наиболее предпочтительно в диапазоне от 150 до 170°C.
Обычно горизонтальная доля Т-макс будет располагаться поблизости от центра реакционной среды 36, в то время как горизонтальная доля Т-мин будет располагаться поблизости от низа реакционной среды 36. Предпочтительно горизонтальная доля Т-мин представляет собой одну из 10 самых нижних горизонтальных долей в числе 15 самых нижних горизонтальных долей. Наиболее предпочтительно горизонтальная доля Т-мин представляет собой одну из 5 самых нижних горизонтальных долей в числе 15 самых нижних горизонтальных долей. Например, Фиг. 28 иллюстрирует горизонтальную долю Т-мин как вторую горизонтальную долю от низа реактора 20. Предпочтительно горизонтальная доля Т-макс представляет собой одну из 20 средних горизонтальных долей в числе 30 дискретных горизонтальных долей. Наиболее предпочтительно горизонтальная доля Т-мин представляет собой одну из 14 средних горизонтальных долей в числе 30 дискретных горизонтальных долей. Например, Фиг. 28 иллюстрирует горизонтальную долю Т-макс как двенадцатую горизонтальную долю от низа реактора 20 (то есть одну из средних 10 горизонтальных долей). Предпочитается, чтобы разнесение по вертикали между горизонтальными долями Т-мин и Т-макс составляло бы, по меньшей мере, приблизительно 2W, более предпочтительно, по меньшей мере, приблизительно 4W, а наиболее предпочтительно, по меньшей мере, 6W. Предпочитается, чтобы разнесение по вертикали между горизонтальными долями T-мин и T-макс составляло бы, по меньшей мере, приблизительно 0,2Н, более предпочтительно, по меньшей мере, приблизительно 0,4Н, а наиболее предпочтительно, по меньшей мере, 0,6Н.
Как обсуждалось выше, в случае существования в реакционной среде 36 градиента температуры по вертикали может оказаться выгодным отбор реакционной среды 36 в позиции повышенного уровня расположения, где температура реакционной среды является максимальной, в особенности тогда, когда на последующих стадиях технологической схемы отобранный продукт будут подвергать дополнительной переработке при повышенных температурах. Таким образом, если реакционную среду 36 отбирают из зоны реакции 28 через одно или несколько выпускных отверстий повышенного уровня расположения, как это проиллюстрировано на Фиг. 19 и 20, то предпочитается, чтобы выпускное отверстие (отверстия) повышенного уровня расположения располагалось бы поблизости от горизонтальной доли Т-макс. Предпочтительно выпускное отверстие повышенного уровня расположения располагается в пределах 10 горизонтальных долей от горизонтальной доли Т-макс, более предпочтительно в пределах 5 горизонтальных долей от горизонтальной доли Т-макс, а наиболее предпочтительно в пределах 2 горизонтальных долей от горизонтальной доли Т-макс.
В данный момент следует отметить то, что многие из признаков изобретения, описанных в настоящем документе, могут быть использованы в системах с несколькими реакторами окисления, а не просто в системах, использующих единственный реактор окисления. В дополнение к этому определенные признаки изобретения, описанные в настоящем документе, могут быть использованы в реакторах окисления с механическим перемешиванием и/или с перемешиванием потоком, а не просто в реакторах с барботажным перемешиванием (то есть барботажных реакторных колоннах). Например, изобретатели выявили определенные преимущества, связанные с разбиением на ступени/варьированием в отношении концентрации кислорода и/или скорости расходования кислорода по всему объему реакционной среды. Преимущества, реализуемые в результате разбиения на ступени в отношении концентрации/расходования кислорода в реакционной среде, можно реализовать вне зависимости от того, будет ли совокупный объем реакционной среды содержаться в единственной емкости или в нескольких емкостях. Кроме того, преимущества, реализуемые в результате разбиения на ступени в отношении концентрации/расходования кислорода в реакционной среде, можно реализовать вне зависимости от того, будет ли реакционная емкость (емкости) иметь механическое перемешивание, перемешивание потоком и/или барботажное перемешивание.
Один способ получения количественных характеристик для степени разбиения на ступени в отношении концентрации и/или скорости расходования кислорода в реакционной среде заключается в сопоставлении двух или более обособленных 20-процентных сплошных объемов реакционной среды. Данные 20-процентные сплошные объемы не обязательно должны определяться какой-либо конкретной формой. Однако каждый 20-процентный сплошной объем должен быть сформирован из объема реакционной среды, образованного примыкающими друг к другу его частями (то есть каждый объем является «сплошным»), и 20-процентные сплошные объемы не должны перекрываться друг с другом (то есть объемы являются «обособленными»). Фиг. 29-31 иллюстрируют то, что данные обособленные 20-процентные сплошные объемы могут располагаться в одном и том же реакторе (Фиг. 29) или в нескольких реакторах (Фиг. 30 и 31). Необходимо отметить то, что реакторы, проиллюстрированные на Фиг. 29-31, могут представлять собой реакторы с механическим перемешиванием, перемешиванием потоком и/или барботажным перемешиванием. В одном варианте реализации предпочитается, чтобы реакторы, проиллюстрированные на Фиг. 29-31, представляли бы собой реакторы с барботажным перемешиванием (то есть барботажные реакторные колонны).
Если обратиться теперь к Фиг. 29, то можно сказать, что на ней проиллюстрирован реактор 20 как вмещающий реакционную среду 36. Реакционная среда 36 включает первый обособленный 20-процентный сплошной объем 37 и второй обособленный 20-процентный сплошной объем 39.
Если обратиться теперь к Фиг. 30, то можно сказать, что на ней проиллюстрирована система с несколькими реакторами как включающая первый реактор 720а и второй реактор 720b. Реакторы 720а, b совместно вмещают совокупный объем реакционной среды 736. Первый реактор 720а вмещает первую часть реакционной среды 736а, в то время как второй реактор 720b вмещает вторую часть реакционной среды 736b. Первый обособленный 20-процентный сплошной объем 737 реакционной среды 736 продемонстрирован как определенный в пределах первого реактора 720а, в то время как второй обособленный 20-процентный сплошной объем 739 реакционной среды 736 продемонстрирован как определенный в пределах второго реактора 720b.
Если обратиться теперь к Фиг. 31, то можно сказать, что на ней проиллюстрирована система с несколькими реакторами как включающая первый реактор 820а, второй реактор 820b и третий реактор 820с. Реакторы 820а, b, c совместно вмещают совокупный объем реакционной среды 836. Первый реактор 820а вмещает первую часть реакционной среды 836а; второй реактор 820b вмещает вторую часть реакционной среды 836b; а третий реактор вмещает 820с вмещает третью часть реакционной среды 836с. Первый обособленный 20-процентный сплошной объем 837 реакционной среды 836 продемонстрирован как определенный в пределах первого реактора 820а; второй обособленный 20-процентный сплошной объем 839 реакционной среды 836 продемонстрирован как определенный в пределах второго реактора 820b; а третий обособленный 20-процентный сплошной объем 841 реакционной среды 836 продемонстрирован как определенный в пределах третьего реактора 820с.
Разбиение на ступени в отношении доступности кислорода в реакционной среде можно количественно охарактеризовать при отнесении к 20-процентному сплошному объему реакционной среды, содержащей наиболее обогащенную мольную долю кислорода в газовой фазе, и при отнесении к 20-процентному сплошному объему реакционной среды, содержащей наиболее обедненную мольную долю кислорода в газовой фазе. В газовой фазе обособленного 20-процентного сплошного объема реакционной среды, имеющей наивысшую концентрацию кислорода в газовой фазе, средневременная и среднеобъемная концентрация кислорода, при расчете для влажного состояния, предпочтительно находится в диапазоне от приблизительно 3 до приблизительно 18 мольных процентов, более предпочтительно в диапазоне от приблизительно 3,5 до приблизительно 14 мольных процентов, а наиболее предпочтительно в диапазоне от 4 до 10 мольных процентов. В газовой фазе обособленного 20-процентного сплошного объема реакционной среды, имеющей наименьшую концентрацию кислорода в газовой фазе, средневременная и среднеобъемная концентрация кислорода, при расчете для влажного состояния, предпочтительно находится в диапазоне от приблизительно 0,3 до приблизительно 5 мольных процентов, более предпочтительно в диапазоне от приблизительно 0,6 до приблизительно 4 мольных процентов, а наиболее предпочтительно в диапазоне от 0,9 до 3 мольных процентов. Кроме того, соотношение между средневременными и среднеобъемными концентрациями кислорода, при расчете для влажного состояния, в наиболее обогащенном 20-процентном сплошном объеме реакционной среды и в наиболее обедненном 20-процентном сплошном объеме реакционной среды предпочтительно находится в диапазоне от приблизительно 1,5:1 до приблизительно 20:1, более предпочтительно в диапазоне от приблизительно 2:1 до приблизительно 12:1, а наиболее предпочтительно в диапазоне от 3:1 до 9:1.
Количественные характеристики для разбиения на ступени в отношении скорости расходования кислорода в реакционной среде могут быть получены при выражении через значение STR по кислороду, что первоначально описали выше. Значение STR по кислороду было ранее описано в глобальном смысле (то есть из перспективы среднего значения STR по кислороду для совокупной реакционной среды); однако значение STR по кислороду также можно рассматривать и в локальном смысле (то есть часть реакционной среды) для того, чтобы получить количественные характеристики для разбиения на ступени в отношении скорости расходования кислорода по всему объему реакционной среды.
Изобретатели обнаружили то, что очень полезным является стимулирование варьирования значения STR по кислороду по всему объему реакционной среды в общем соответствии с желательными градиентами, описанными в настоящем документе в отношении давления в реакционной среде и мольной доли молекулярного кислорода в газовой фазе реакционной среды. Таким образом, предпочитается, чтобы соотношение между значением STR по кислороду для первого обособленного 20-процентного сплошного объема реакционной среды и значением STR по кислороду для второго обособленного 20-процентного сплошного объема реакционной среды находилось бы в диапазоне от приблизительно 1,5:1 до приблизительно 20:1, более предпочтительно в диапазоне от приблизительно 2:1 до приблизительно 12:1, а наиболее предпочтительно в диапазоне от 3:1 до 9:1. В одном варианте реализации в сопоставлении со «вторым обособленным 20-процентным сплошным объемом» «первый обособленный 20-процентный сплошной объем» располагается ближе к позиции, в которой в реакционную среду первоначально вводят молекулярный кислород. Данные большие градиенты значения STR по кислороду являются желательными вне зависимости от того, будет ли реакционная среда неполного окисления вмещаться в барботажной реакторной колонне окисления или в любом другом типе реакционной емкости, в которой создаются градиенты давления и/или мольной доли молекулярного кислорода в газовой фазе реакционной среды, (например, в емкости с механическим перемешиванием, имеющей несколько расположенных по вертикали зон перемешивания, что достигается в результате использования нескольких крыльчаток, характеризующейся наличием сильного радиального течения, при возможном усилении результата в результате наличия сборных модулей в виде в общем случае горизонтальных перегородок, при этом поток окислителя поднимается в общем случае снизу вверх от позиции подачи поблизости от нижней части реакционной емкости, несмотря на то, что в пределах каждой расположенной по вертикали зоны перемешивания может иметь место значительный уровень обратного смешения потока окислителя и что определенный уровень обратного смешения потока окислителя может иметь место и между примыкающими расположенными по вертикали зонами перемешивания). То есть, изобретатели выявили то, что в случае существования градиента давления и/или мольной доли молекулярного кислорода в газовой фазе реакционной среды желательным является создание подобного градиента химической потребности в растворенном кислороде при использовании способов, описанных в настоящем документе.
Предпочтительные способы стимулирования варьирования локальных значений STR по кислороду заключаются в управлении позициями подачи окисляемого соединения и в управлении перемешиванием жидкой фазы реакционной среды в целях регулирования градиентов концентрации окисляемого соединения в соответствии с другими моментами из описания настоящего изобретения. Другие подходящие способы стимулирования варьирования локальных значений STR по кислороду включают стимулирование варьирования активности в реакции в результате стимулирования варьирования локальной температуры и в результате изменения локальной смеси компонентов катализатора и растворителя (например, в результате введения дополнительного количества газа для стимулирования охлаждения испарением в конкретной части реакционной среды и в результате добавления потока растворителя, содержащего повышенное количество воды для уменьшения активности в конкретной части реакционной среды).
Как обсуждалось выше в том, что касается Фиг. 30 и 31, реакцию неполного окисления в подходящем случае можно проводить в нескольких реакционных емкостях, где, по меньшей мере, часть, предпочтительно, по меньшей мере, 25 процентов, более предпочтительно, по меньшей мере, 50 процентов, а наиболее предпочтительно, по меньшей мере, 75 процентов, от молекулярного кислорода, покидающего первую реакционную емкость, перепускают в одну или несколько последующих реакционных емкостей для расходования дополнительной порции, предпочтительно более чем 10 процентов, более предпочтительно более чем 20 процентов, а наиболее предпочтительно более чем 40 процентов, от молекулярного кислорода, покидающего первую/расположенную раньше на технологической схеме реакционную емкость. При использовании такого последовательного течения молекулярного кислорода из одного реактора в другие желательно, чтобы первая реакционная емкость функционировала бы при интенсивности реакции, более высокой в сопоставлении с тем, что имеет место в, по меньшей мере, одной из последующих реакционных емкостей, предпочтительно при соотношении между средним по емкости значением STR по кислороду в пределах первой реакционной емкости и средним по емкости значением STR по кислороду в пределах последующей реакционной емкости в диапазоне от приблизительно 1,5:1 до приблизительно 20:1, более предпочтительно в диапазоне от приблизительно 2:1 до приблизительно 12:1, а наиболее предпочтительно в диапазоне от 3:1 до 9:1.
Как обсуждалось выше, подходящими для последовательного течения молекулярного кислорода в последующие реакционные емкости в соответствии с настоящим изобретением являются все типы первой реакционной емкости (например, барботажная колонна, ,аппараты с механическим перемешиванием, с обратным перемешиванием, с внутренним разбиением на ступени, с течением в режиме идеального вытеснения и тому подобное) и все типы последующих реакционных емкостей, которые могут относиться, а могут и не относиться к типу, отличному от первой реакционной емкости. Способы стимулирования уменьшения среднего по емкости значения STR по кислороду в пределах последующих реакционных емкостей в подходящем случае включают уменьшение температуры, уменьшение концентраций окисляемого соединения и уменьшение активности в реакции для конкретной смеси каталитических компонентов и растворителя (например, уменьшение концентрации кобальта, увеличение концентрации воды и добавление замедлителя катализатора, такого как небольшие количества ионной меди).
При течении потока из первой реакционной емкости в последующую реакционную емкость поток окислителя можно подвергнуть переработке при использовании любых способов, известных на современном уровне техники, таких как сжатие или уменьшение давления, охлаждение или нагревание и удаление массы или добавление массы в любом количестве или любого типа. Однако использование уменьшения среднего по емкости значения STR по кислороду в последующих реакционных емкостях является в особенности полезным тогда, когда абсолютное давление в верхней части первой реакционной емкости составляет величину, меньшую чем приблизительно 2,0 мегапаскаль, более предпочтительно меньшую чем приблизительно 1,6 мегапаскаль, а наиболее предпочтительно меньшую чем 1,2 мегапаскаль. Кроме того, использование уменьшения среднего по емкости значения STR по кислороду в последующих реакционных емкостях является в особенности полезным тогда, когда соотношение между абсолютным давлением в верхней части первой реакционной емкости и абсолютным давлением в верхней части, по меньшей мере, одной последующей реакционной емкости находится в диапазоне от приблизительно 0,5:1 до 6:1, более предпочтительно в диапазоне от приблизительно 0,6:1 до приблизительно 4:1, а наиболее предпочтительно в диапазоне от 0,7:1 до 2:1. Уменьшение давления в последующих емкостях до уровня, меньшего данных нижних пределов, накладывается на уменьшение доступности молекулярного кислорода, а увеличение давления выше данных верхних пределов требует использования значительных затрат в сопоставлении с применением свежей подачи окислителя.
При использовании последовательного течения молекулярного кислорода в последующие реакционные емкости, характеризующиеся уменьшением средних по емкости значений STR по кислороду, свежие потоки исходного подаваемого материала, образуемые окисляемым соединением, растворителем и окислителем, могут перетекать в последующие реакционные емкости и/или в первую реакционную емкость. Потоки жидкой фазы и твердой фазы, в случае наличия, реакционной среды могут перемещаться в любом направлении между реакционными емкостями. Все количество или часть газовой фазы, покидающей первую реакционную емкость и поступающей в последующую реакционную емкость, могут перетекать отделенными от частей жидкой фазы или твердой фазы, в случае наличия, реакционной среды из первой реакционной емкости или могут перетекать смешанными с ними. Течение потока продукта, содержащего жидкую фазу и твердую фазу, в случае наличия, может направляться на отбор из реакционной среды в любой реакционной емкости в системе.
Если обратиться опять к Фиг. 1-29, то можно сказать, что окисление предпочтительно проводят в барботажной реакторной колонне 20 в условиях, которые в соответствии с предпочтительными вариантами реализации, описанными в настоящем документе, значительно отличаются от того, что имеет место в случае обычно используемых реакторов окисления. В случае использования барботажной реакторной колонны 20 для проведения жидкофазного неполного окисления пара-ксилола до получения сырой неочищенной терефталевой кислоты (СТА) в соответствии с предпочтительными вариантами реализации, описанными в настоящем документе, в образование частиц СТА, обладающих уникальными и выгодными свойствами, вносят свой вклад пространственные профили локальной интенсивности реакции, локальной интенсивности испарения и локальной температуры в комбинации со схемами течения жидкости в реакционной среде и предпочтительными относительно низкими температурами окисления.
Фиг. 32А и 32В иллюстрируют базовые частицы СТА, полученные в соответствии с одним вариантом реализации настоящего изобретения. Фиг. 32А демонстрирует базовые частицы СТА при 500-кратном увеличении, в то время как Фиг. 32В в увеличенном масштабе представляет одну из базовых частиц СТА и демонстрирует данную частицу при 2000-кратном увеличении. Как, может быть, наилучшим образом проиллюстрировано на Фиг. 32В, каждая базовая частица СТА обычно образована из большого количества мелких агломерированных субчастиц СТА, что, таким образом, приводит к получению базовой частицы СТА, характеризующейся относительно большой площадью удельной поверхности, высокой пористостью, низкой плотностью и хорошей растворимостью. Если только не будет указано другого, то различные свойства СТА изобретения, описанные далее, измеряют при использовании представительного образца СТА, где представительный образец весит, по меньшей мере, 1 грамм и/или образован, по меньшей мере, из 10000 индивидуальных частиц СТА. Базовые частицы СТА обычно характеризуются средним размером частиц в диапазоне от приблизительно 20 до приблизительно 150 микронов, более предпочтительно в диапазоне от приблизительно 30 до приблизительно 120 микронов, а наиболее предпочтительно в диапазоне от 40 до 90 микронов. Субчастицы СТА обычно характеризуются средним размером частиц в диапазоне от приблизительно 0,5 до приблизительно 30 микронов, более предпочтительно от приблизительно 1 до приблизительно 15 микронов, а наиболее предпочтительно в диапазоне от 2 до 5 микронов. Относительно большую площадь удельной поверхности базовых частиц СТА, проиллюстрированных на Фиг. 32А и 32В, можно количественно охарактеризовать при использовании метода измерения площади удельной поверхности Браунауэра-Эмметта-Теллера (БЭТ). Предпочтительно базовые частицы СТА характеризуются средней площадью удельной поверхности согласно методу БЭТ, равной, по меньшей мере, приблизительно 0,6 квадратного метра на один грамм (м2/г). Более предпочтительно базовые частицы СТА характеризуются средней площадью удельной поверхности согласно методу БЭТ в диапазоне от приблизительно 0,8 до приблизительно 4 м2/г. Наиболее предпочтительно базовые частицы СТА характеризуются средней площадью удельной поверхности согласно методу БЭТ в диапазоне от 0,9 до 2 м2/г. Физические свойства (например, размер частиц, площадь удельной поверхности согласно методу БЭТ, пористость и растворимость) базовых частиц СТА, полученных в соответствии с оптимизированным способом окисления предпочтительного варианта реализации настоящего изобретения, делают возможной очистку частиц СТА при использовании более эффективных и/или экономичных способов, описанных более подробно далее в связи с Фиг. 35.
Значения средних размеров частиц, представленные выше, определяли при использовании микроскопии в поляризованном свете и методики анализа изображений. Оборудование, использованное при анализе размеров частиц, включало оптический микроскоп Nikon E800 с объективом 4х Plan Flour N. A. 0.13, цифровую камеру Spot RT™ и персональный компьютер с установленным программным обеспечением для анализа изображений Image Pro Plus™ V4.5.0.19. Метод анализа размеров частиц включал следующие основные стадии: (1) диспергирование порошка СТА в минеральном масле; (2) получение препарата для микроскопии в виде дисперсии между предметным и покровным стеклами микроскопа; (3) рассматривание препарата для микроскопии при использовании микроскопии в поляризованном свете (состояние скрещенных поляризаторов - частицы наблюдаются в виде светлых предметов на черном фоне); (4) фиксация различных изображений для каждого случая приготовления образцов (размер поля изображения =3х2,25 мм; размер элемента изображения =1,84 микрон/элемент изображения); (5) проведение анализа изображений при использовании программного обеспечения Image Pro Plus™; (6) перенос результатов измерений для частиц в электронную таблицу; и (7) проведение операции по статистическому обсчету характеристик в электронной таблице. Стадия (5) «проведение анализа изображений при использовании программного обеспечения Image Pro Plus™» включала подстадии: (а) установки порогового значения для изображения в целях детектирования белых частиц на темном фоне; (b) создания черно-белого изображения; (с) приведения в действие однопроходного открытого фильтра для отфильтровывания шума элементов изображения; (d) проведения измерения для всех частиц на изображении; и (е) выдачи данных о среднем диаметре, измеренном для каждой частицы. Программное обеспечение Image Pro Plus™ определяет средний диаметр для индивидуальных частиц в виде среднечисленной длины диаметров частицы, измеренных с интервалами в 2 градуса и проходящих через центр тяжести частицы. Стадия 7 «проведение операции по статистическому обсчету характеристик в электронной таблице» включает вычисление объемно-взвешенного среднего размера частиц следующим образом. Объем каждой из n частиц в образце рассчитывают так, как если бы она была сферической, в результате использования формулы π/6*di^3; умножения объема каждой частицы на ее диаметр до получения π/6*di^4; суммирования для всех частиц в образце значений π/6*di^4; суммирования объемов всех частиц в образце; и вычисления объемно-взвешенного диаметра частиц в виде суммы величин (π/6*di^4) для всех n частиц в образце, поделенной на сумму величин (π/6*di^3) для всех n частиц в образце. В соответствии с использованием в настоящем документе «средний размер частиц» обозначает объемно-взвешенный средний размер частиц, определенный в соответствии с описанным выше методом испытаний; и его также обозначают как D(4,3)

В дополнение к этому стадия 7 включает установление размеров частиц, для которых различные доли от совокупного объема образца характеризуются меньшими размерами частиц. Например, D(v,0,1) представляет собой размер частиц, для которого 10 процентов от совокупного объема образца характеризуются меньшим размером частиц, а 90 процентов - характеризуются большим размером частиц; D(v,0,5) представляет собой размер частиц, для которого половина объема образца характеризуется большим размером частиц, а половина - характеризуется меньшим размером частиц; D(v,0,9) представляет собой размер частиц, для которого 90 процентов от совокупного объема образца характеризуются меньшим размером частиц; и так далее. В дополнение к этому стадия 7 включает вычисление значения D(v,0,9) минус D(v,0,1), которое в настоящем документе определяют как «разброс размеров частиц»; и стадия 7 включает вычисление величины разброса размеров частиц, поделенного на значение D(4,3), что в настоящем документе определяют как «относительный разброс размеров частиц».
Кроме того, предпочитается, чтобы величина D(v,0,1) для частиц СТА, измеренная выше, находилась бы в диапазоне от приблизительно 5 до приблизительно 65 микронов, более предпочтительно в диапазоне от приблизительно 15 до приблизительно 55 микронов, а наиболее предпочтительно в диапазоне от 25 до 45 микронов. Предпочитается, чтобы величина D(v,0,5) для частиц СТА, измеренная выше, находилась бы в диапазоне от приблизительно 10 до приблизительно 90 микронов, более предпочтительно в диапазоне от приблизительно 20 до приблизительно 80 микронов, а наиболее предпочтительно в диапазоне от 30 до 70 микронов. Предпочитается, чтобы величина D(v,0,9) для частиц СТА, измеренная выше, находилась бы в диапазоне от приблизительно 30 до приблизительно 150 микронов, более предпочтительно в диапазоне от приблизительно 40 до приблизительно 130 микронов, а наиболее предпочтительно в диапазоне от 50 до 110 микронов. Предпочитается, чтобы относительный разброс размеров частиц находился бы в диапазоне от приблизительно 0,5 до приблизительно 2,0, более предпочтительно в диапазоне от приблизительно 0,6 до приблизительно 1,5, а наиболее предпочтительно в диапазоне от 0,7 до 1,3.
Значения площади удельной поверхности согласно методу БЭТ, приведенные выше, измеряли при использовании прибора Micromeritics ASAP2000 (доступного в компании Micromeritics Instrument Corporation из Норкросса, Джорджия). На первой стадии метода измерения от 2 до 4 граммов образца частиц отвешивали и высушивали в вакууме при 50°С. После этого образец помещали в газовый коллектор для проведения анализа и охлаждали до 77К. Изотерму адсорбции азота измеряли, как минимум, при 5 равновесных давлениях в результате воздействия на образец известных объемов газообразного азота и измерения падения давления. Равновесные давления в подходящем случае находились в диапазоне Р/Р0=0,01-0,20, где Р представляет собой равновесное давление, а Р0 представляет собой давление паров жидкого азота при 77К. После этого проводили графическое построение для получающейся в результате изотермы в соответствии со следующим далее уравнением метода БЭТ:

где Va представляет собой объем газа, адсорбированного образцом при Р, Vm представляет собой объем газа, необходимый для покрытия совокупной поверхности образца монослоем газа, а С представляет собой константу. Из данного графика определяли значения Vm и С. После этого Vm пересчитывали в площадь удельной поверхности при использовании площади поперечного сечения азота при 77К по формуле

где σ представляет собой площадь поперечного сечения азота при 77К, Т равна 77К, а R представляет собой газовую постоянную.
Как упоминалось выше, СТА, полученная в соответствии с одним вариантом реализации настоящего изобретения, демонстрирует превосходные характеристики растворения в сопоставлении с обычной СТА, полученной при использовании других способов. Данная улучшенная скорость растворения делает возможной очистку СТА изобретения при использовании более эффективных и/или более действенных способов очистки. Следующее далее описание относится к способу, по которому можно получить количественные характеристики для скорости растворения СТА.
Скорость растворения известного количества твердой фазы в известном количестве растворителя в перемешиваемой смеси можно измерять в соответствии с различными протоколами. В соответствии с использованием в настоящем изобретении метод измерения, называемый «испытанием на растворение во времени», определяют следующим образом. В ходе всего испытания на растворение во времени используют давление окружающей среды, равное приблизительно 0,1 мегапаскаля. Температура окружающей среды, используемая в ходе всего испытания на растворение во времени, составляет приблизительно 22°С. Кроме того, перед началом проведения испытаний для твердой фазы, растворителя и всей аппаратуры для растворения добивались достижения полного термического равновесия при данной температуре, и в течение периода времени растворения какого-либо ощутимого нагревания или охлаждения химического стакана или его содержимого не отмечалось. Образуемую растворителем часть в виде свежего тетрагидрофурана марки «чистый для анализа по методу ЖХВД» (степень чистоты >99,9 процента), здесь и далее в настоящем документе обозначаемого как ТГФ, в количестве 250 граммов помещают в очищенный стеклянный химический стакан высокой формы KIMAX объемом 400 миллилитров (номер детали Kimble® 14020, компания Kimble/Kontes, Вайнлэнд, Нью-Джерси), который является нетеплоизолированным, имеющим гладкие стенки и в общем случае цилиндрическую форму. В химический стакан помещают магнитную мешалку с тефлоновым покрытием (номер детали VWR 58948-230, длиной приблизительно 1 дюйм и диаметром 3/8 дюйма, с восьмиугольным поперечным сечением, компания VWR International, Уэст-Честер, Пенсильвания 19380), где она естественным образом опускается на дно. Образец перемешивают при использовании магнитной мешалки Variomag® multipoint 15 (компания H&P Labortechnik AG, Обершляйссхайм, Германия) при ее установке на вращение при 800 оборотах в минуту. Данное перемешивание начинается не более чем за 5 минут до добавления твердой фазы и стационарно продолжается в течение, по меньшей мере, 30 минут после добавления твердой фазы. В нелипкую кювету для отвешивания образцов отвешивают твердый образец в виде частиц сырой неочищенной или очищенной ТРА в количестве 250 миллиграммов. В начальный момент, обозначаемый как t=0, всю отвешенную твердую фазу сразу высыпают в перемешиваемый ТГФ и одновременно запускают секундомер. При правильном проведении операции ТГФ очень быстро смачивает твердую фазу и образует разбавленную хорошо перемешанную суспензию в течение 5 секунд. После этого образцы данной смеси получают в последующие моменты времени, отсчитываемые в минутах от t=0:0,08, 0,25, 0,50, 0,75, 1,00, 1,50, 2,00, 2,50, 3,00, 4,00, 5,00, 6,00, 8,00, 10,00, 15,00 и 30,00. Каждый небольшой образец отбирают из разбавленной хорошо перемешанной смеси при использовании нового одноразового шприца (компания Becton, Dickinson and Co, 5 миллилитров, REF 30163, Франклин-Лэйкс, Нью-Джерси 07417). Сразу же после проведения отбора из химического стакана в новую маркированную стеклянную ампулу для образцов при использовании нового неиспользованного фильтрующего шприца (диаметр 25 мм, 0,45 микрона, Gelman GHP Acrodisc GF®, компания Pall Corporation, Ист-Хиллс, Нью-Йорк 11548) быстро выпускали приблизительно 2 миллилитра прозрачного жидкого образца. Продолжительность каждого заполнения шприца, размещения фильтра и выпускания в ампулу для образцов в надлежащем случае составляет величину, меньшую чем приблизительно 5 секунд, и данный интервал в подходящем случае начинается и заканчивается в течение приблизительно 3 секунд в ту или иную сторону от каждой целевой продолжительности времени отбора образца. В течение приблизительно пяти минут от каждого заполнения ампулы для образцов закрывают крышкой и выдерживают приблизительно при постоянной температуре вплоть до проведения последующего химического анализа. После отбора конечного образца в момент времени, соответствующий прохождению 30 минут после t=0, все шестнадцать образцов анализируют для установления количества растворенной ТРА при использовании метода ЖХВД-ДМД, в общем случае охарактеризованного в другом месте данного описания. Однако в настоящем испытании как калибровочные стандарты, так и приведенные результаты рассчитываются как миллиграммы растворенной ТРА на один грамм растворителя ТГФ (здесь и далее в настоящем документе «ч./млн в ТГФ»). Например, если все 250 миллиграммов твердой фазы представляли бы собой очень чистую ТРА и если данное совокупное количество было бы полностью растворено в 250 граммах растворителя ТГФ до того, как был отобран конкретный образец, то тогда правильно измеренная концентрация составила бы приблизительно 1000 ч./млн в ТГФ.
Если СТА, соответствующую настоящему изобретению, будут подвергать описанному выше испытанию на растворение во времени, то тогда предпочитается, чтобы образец, отобранный по истечении одной минуты после t=0, растворялся бы до концентрации, равной, по меньшей мере, приблизительно 500 ч./млн в ТГФ, более предпочтительно, по меньшей мере, 600 ч./млн в ТГФ. В случае образца, отбираемого по истечении двух минут после t=0, предпочитается, чтобы СТА, соответствующая настоящему изобретению, растворялась бы до концентрации, равной, по меньшей мере, приблизительно 700 ч./млн в ТГФ, более предпочтительно, по меньшей мере, 750 ч./млн в ТГФ. В случае образца, отбираемого по истечении четырех минут после t=0, предпочитается, чтобы СТА, соответствующая настоящему изобретению, растворялась бы до концентрации, равной, по меньшей мере, приблизительно 840 ч./млн в ТГФ, более предпочтительно, по меньшей мере, 880 ч./млн в ТГФ.
Изобретатели обнаружили то, что при описании временной зависимости для полного набора данных из всего испытания на растворение во времени подходящей является относительно простая модель отрицательного экспоненциального роста вне зависимости от сложности частиц образцов и способа растворения. Форма уравнения, здесь и далее в настоящем документе называемого «моделью растворения во времени», представляет собой нижеследующее:
S=A+B*(1-exp(-C*t)),
где t= время с единицей измерения минута;
S= растворимость с единицей измерения ч./млн в ТГФ в момент времени t;
Ехр= показательная функция в основании натурального логарифма 2;
А, В= константы регрессии с единицей измерения ч./млн в ТГФ, где А в основном относится к быстрому растворению более мелких частиц за очень короткие промежутки времени и где сумма А+В в основном относится к совокупному уровню растворения незадолго до завершения указанного периода испытания; и
С= константа времени регрессии с единицей измерения обратная минута.
Константы регрессии подстраивают, добиваясь сведения к минимуму суммы квадратов отклонений фактических экспериментальных данных от соответствующих величин в модели, при этом данный способ обычно называют аппроксимацией по методу «наименьших квадратов». Предпочтительным комплектом программного обеспечения, предназначенным для проведения данной регрессии данных, является JMP Release 5.1.2 (компания SAS Institute Inc., JMP Software, САС-Кэмпус-Драйв, Кэри, Северная Каролина 27513).
В случае проведения для СТА, соответствующей настоящему изобретению, испытания по методу испытания на растворение во времени и аппроксимации в соответствии с описанной выше моделью растворения во времени предпочитается, чтобы СТА характеризовалась бы константой времени «С», превышающей приблизительно 0,5 обратной минуты, более предпочтительно превышающей приблизительно 0,6 обратной минуты, а наиболее предпочтительно превышающей 0,7 обратной минуты.
Фиг. 33А и 33В иллюстрируют обычную частицу СТА, полученную при использовании обычного способа высокотемпературного окисления в корпусном реакторе с непрерывным перемешиванием (CSTR). Фиг. 33А демонстрирует обычную частицу СТА при 500-кратном увеличении, в то время как Фиг. 33В представляет собой изображение в увеличенном масштабе и демонстрирует частицу СТА при 2000-кратном увеличении. Визуальное сопоставление частиц СТА изобретения, проиллюстрированных на Фиг. 32А и 32В, и обычной частицы СТА, проиллюстрированной на Фиг. 33А и 33В, свидетельствует о том, что обычная частица СТА характеризуется большей плотностью, меньшей площадью удельной поверхности, меньшей пористостью и большим размером частиц в сопоставлении с частицами СТА изобретения. Собственно говоря, обычная СТА, представленная на Фиг. 33А и 33В, характеризуется средним размером частиц, равным приблизительно 205 микронам, и площадью удельной поверхности согласно методу БЭТ, равной приблизительно 0,57 м2/г.
Фиг. 34 иллюстрирует обычно используемый способ получения очищенной терефталевой кислоты (РТА). В обычно используемом способе получения РТА пара-ксилол подвергают неполному окислению в высокотемпературном реакторе окисления с механическим перемешиванием 700. Суспензию, содержащую СТА, из реактора 700 отбирают, а после этого очищают в системе очистки 702. Продукт РТА системы очистки 702 вводят в систему разделения 706 для отделения и высушивания частиц РТА. Система очистки 702 составляет большую долю расходов, связанных до получения частиц РТА при использовании обычных способов. Система очистки 702 в общем случае включает систему добавления воды/обмена 708, систему растворения 710, систему гидрирования 712 и три отдельные кристаллизационные емкости 704а, b, c. В системе добавления воды/обмена 708 существенную часть маточного раствора вытесняют водой. После добавления воды суспензию вода/СТА вводят в систему растворения 710, где смесь вода/СТА нагревают до тех пор, пока частицы СТА в воде полностью не растворятся. После растворения СТА раствор СТА в воде подвергают гидрированию в системе гидрирования 712. После этого для отходящего потока продукта гидрирования из системы гидрирования 712 проводят три стадии кристаллизации в кристаллизационных емкостях 704а, b, c с последующим отделением РТА в системе разделения 706.
Фиг. 35 иллюстрирует улучшенный способ получения РТА, использующий барботажную реакторную колонну окисления 800, сконфигурированную в соответствии с вариантом реализации настоящего изобретения. Из реактора 800 отбирают первоначальную суспензию, содержащую твердые частицы СТА и жидкий маточный раствор. Обычно первоначальная суспензия может содержать твердые частицы СТА в количестве в диапазоне от приблизительно 10 до приблизительно 50 массовых процентов, при этом баланс составляет жидкий маточный раствор. Твердые частицы СТА, присутствующие в первоначальной суспензии, обычно содержат, по меньшей мере, приблизительно 400 ч./млн (мас.) 4-карбоксибензальдегида (4-СВА), более часто, по меньшей мере, приблизительно 800 ч./млн (мас.) 4-СВА, а наиболее часто 4-СВА в количестве в диапазоне от 1000 до 15000 ч./млн (мас.). Первоначальную суспензию, отобранную из реактора 800, вводят в систему очистки 802 для уменьшения концентрации 4-СВА и других примесей, присутствующих в СТА. В системе очистки 802 получают более чистую/очищенную суспензию и ее подвергают разделению и высушиванию в системе разделения 804, тем самым получая частицы более чистой твердой терефталевой кислоты, содержащие менее чем приблизительно 400 ч./млн (мас.) 4-СВА, более предпочтительно менее, чем приблизительно 250 ч./млн (мас.) 4-СВА, а наиболее предпочтительно 4-СВА в количестве в диапазоне от 10 до 200 ч./млн (мас.).
Система очистки 802 из системы получения РТА, проиллюстрированной на Фиг. 35, обеспечивает достижение нескольких преимуществ в сопоставлении с системой очистки 802 из системы предшествующего уровня техники, проиллюстрированной на Фиг. 34. Предпочтительно система очистки 802 в общем случае включает систему обмена раствора 806, утилизатор 808 и один кристаллизатор 810. В системе обмена раствора 806, по меньшей мере, приблизительно 50 массовых процентов маточного раствора, присутствующего в первоначальной суспензии, обменивают на свежий растворитель замены до получения, таким образом, суспензии после обмена растворителя, содержащей частицы СТА и растворитель замены. Суспензию после обмена растворителя, покидающую систему обмена раствора 806, вводят в утилизатор (или реактор вторичного окисления) 808. В утилизаторе 808 реакцию вторичного окисления проводят при несколько более высоких температурах в сопоставлении с теми, которые использовали во время реакции первоначального/первичного окисления, проводимой в барботажной реакторной колонне 800. Как обсуждалось выше, большая площадь удельной поверхности, небольшой размер частиц и малая плотность частиц СТА, полученных в реакторе 800, приводят к тому, что определенные примеси, захваченные в частицах СТА, становятся доступными для окисления в утилизаторе 808 без возникновения необходимости в проведении полного растворения частиц СТА в утилизаторе 808. Таким образом, температура в утилизаторе 808 может составлять величину, меньшую, чем во многих подобных способах предшествующего уровня техники. Вторичное окисление, проводимое в утилизаторе 808, предпочтительно приводит к уменьшению концентрации 4-СВА в СТА, по меньшей мере, на 200 ч./млн (мас.), более предпочтительно, по меньшей мере, приблизительно на 400 ч./млн (мас.), а наиболее предпочтительно на величину в диапазоне от 600 до 6000 ч./млн (мас.). Предпочтительно температура вторичного окисления в утилизаторе 808, по меньшей мере, приблизительно на 10°С превышает температуру первичного окисления в барботажной реакторной колоне 800, более предпочтительно превышает температуру первичного окисления в реакторе 800 на величину в диапазоне от приблизительно 20 до приблизительно 80°С, а наиболее предпочтительно превышает температуру первичного окисления в реакторе 800 на величину в диапазоне от 30 до 50°С. Температура вторичного окисления предпочтительно находится в диапазоне от приблизительно 160 до приблизительно 240°С, более предпочтительно в диапазоне от приблизительно 180 до приблизительно 220°С, а наиболее предпочтительно в диапазоне от 190 до 210°С. Очищенный продукт из утилизатора 808 требует наличия только одной стадии кристаллизации в кристаллизаторе 810 перед проведением разделения в системе разделения 804. Подходящие методики вторичного окисления/утилизации обсуждаются более подробно в публикации патентной заявки США № 2005/0065373, описание которой во всей своей полноте посредством ссылки недвусмысленно включается в настоящий документ.
Терефталевую кислоту (например, РТА), полученную при использовании системы, проиллюстрированной на Фиг. 35, предпочтительно образуют частицы РТА, характеризующиеся средним размером частиц, равным, по меньшей мере, приблизительно 40 микронам, более предпочтительно находящимся в диапазоне от приблизительно 50 до приблизительно 2000 микронов, а наиболее предпочтительно в диапазоне от 60 до 200 микронов. Частицы РТА предпочтительно характеризуются средней площадью удельной поверхности согласно методу БЭТ, меньшей чем приблизительно 0,25 м2/г, более предпочтительно находящейся в диапазоне от приблизительно 0,005 до приблизительно 0,2 м2/г, а наиболее предпочтительно в диапазоне от 0,01 до 0,18 м2/г. РТА, полученная при использовании системы, проиллюстрированной на Фиг. 35, является подходящей для использования в качестве исходного сырья при получения ПЭТФ. Обычно ПЭТФ получают в результате проведения этерификации терефталевой кислоты под действием этиленгликоля с последующей поликонденсацией. Предпочтительно терефталевую кислоту, полученную в соответствии с одним вариантом реализации настоящего изобретения, используют в качестве исходного подаваемого материала в способе получения ПЭТФ в трубчатом реакторе, описанном в патентной заявке США с регистрационным номером 10/013318, поданной 7 декабря 2001 года, описание которой во всей своей полноте посредством ссылки включается в настоящий документ.
Частицы СТА, характеризующиеся предпочтительной морфологией, описанной в настоящем документе, являются в особенности хорошо подходящими для использования в описанном выше способе окислительной утилизации, предназначенном для уменьшения уровня содержания 4-СВА. В дополнение к этому данные предпочтительные частицы СТА обеспечивают достижение преимуществ в широком ассортименте других способов последующей переработки, включающих растворение и/или химическую реакцию частиц. Данные дополнительные способы последующей переработки включают нижеследующее, но также и не ограничиваются только этим: реакция, по меньшей мере, с одним гидроксилсодержащим соединением с образованием производных сложных эфиров, в особенности реакция СТА с метанолом с образованием диметилтерефталата и примесных сложных эфиров; реакция, по меньшей мере, с одним диолом с образованием мономерного сложного эфира и/или полимерных производных сложного эфира, в особенности реакция СТА с этиленгликолем с образованием полиэтилентерефталата (ПЭТФ); и полное или частичное растворение в растворителях, включающих нижеследующее, но также и не ограничивающихся только этим: вода, уксусная кислота и N-метил-2-пирролидон, что может включать дополнительную переработку, включающую нижеследующее, но также и не ограничивающуюся только этим: переосаждение более чистой терефталевой кислоты и/или селективное химическое восстановление карбонильных групп, отличных от групп карбоновой кислоты. В частности, включается по существу растворение СТА в растворителе, включающем воду, в сочетании с неполным гидрированием, которое приводит к уменьшению количества альдегидов, в особенности 4-СВА, флуоренонов, фенонов и/или антрахинонов.
Изобретатели также предусматривают возможность получения частиц СТА, обладающих предпочтительными свойствами, описанными в настоящем документе, из частиц СТА, не соответствующих требованиям предпочтительных свойств, описанных в настоящем документе, (частиц СТА, не соответствующих требованиям) при использовании способов, включающих нижеследующее, но также и не ограничивающихся только этим: механическое измельчение частиц СТА, не соответствующих требованиям, и полное или неполное растворение частиц СТА, не соответствующих требованиям, с последующим полным или неполным переосаждением.
В соответствии с одним вариантом реализации настоящего изобретения предлагается способ неполного окисления окисляемого ароматического соединения до получения одного или нескольких типов ароматической карбоновой кислоты, где степень чистоты образуемой растворителем части исходного подаваемого материала (то есть «образуемого растворителем исходного подаваемого материала») и степень чистоты образуемой окисляемым соединением части исходного подаваемого материала (то есть «образуемого окисляемым соединением исходного подаваемого материала») регулируют в определенных диапазонах, указанных далее. Совместно с другими вариантами реализации настоящего изобретения это делает возможным регулирование степени чистоты жидкой фазы и, в случае наличия, твердой фазы и объединенной фазы суспензии (то есть твердой фазы плюс жидкой фазы) реакционной среды в определенных предпочтительных диапазонах, обозначенных далее.
Что касается образуемого растворителем исходного подаваемого материала, то известно окисление окисляемого ароматического соединения (соединений) до получения ароматической карбоновой кислоты, где образуемый растворителем исходный подаваемый материал, вводимый в реакционную среду, представляет собой смесь уксусной кислоты и воды со степенями чистоты «чистый для анализа», и его зачастую используют в лабораторном масштабе и полупромышленном масштабе. Подобным же образом известно проведение окисления окисляемого ароматического соединения до получения ароматической карбоновой кислоты, где растворитель, покидающий реакционную среду, отделяют от полученной ароматической карбоновой кислоты, а после этого отправляют на рецикл обратно в реакционную среду в качестве растворителя в исходном подаваемом материале главным образом по причинам, связанным с производственной себестоимостью. Данное отправление растворителя на рецикл с течением времени приводит к накоплению в отправляемом на рецикл растворителе определенных примесей из исходного подаваемого материала и побочных продуктов технологического процесса. На современном уровне техники известны различные способы содействия очистке отправляемого на рецикл растворителя перед повторным введением в реакционную среду. В общем случае повышенная степень очистки отправляемого на рецикл растворителя приводит к получению значительно более высокой производственной себестоимости в сопоставлении с тем, чего достигают при пониженной степени очистки при использовании подобных способов. Один вариант реализации настоящего изобретения относится к осознанию и определению предпочтительных диапазонов для большого количества примесей в образуемом растворителем исходном подаваемом материале, многие из которых до настоящего времени воспринимались как в основном безвредные, для того, чтобы отыскать оптимальный баланс между общей производственной себестоимостью и общей степенью чистоты продукта.
«Отправляемый на рецикл образуемый растворителем исходный подаваемый материал» в настоящем документе определяют как образуемый растворителем исходный подаваемый материал, включающий, по меньшей мере, приблизительно 5 массовых процентов массы, которую ранее перепускали через реакционную среду, содержащую одно или несколько окисляемых ароматических соединений, претерпевающих неполное окисление. По причинам, связанным с коэффициентом заполнения растворителем и продолжительностью рабочего цикла для производственной установки, предпочитается, чтобы части отправляемого на рецикл растворителя перепускали бы через реакционную среду, по меньшей мере, один раз в день функционирования, более предпочтительно, по меньшей мере, один раз в день в течение, по меньшей мере, семи последовательных дней функционирования, а наиболее предпочтительно, по меньшей мере, один раз в день в течение, по меньшей мере, 30 последовательных дней функционирования. По экономическим причинам предпочитается, чтобы растворитель, отправляемый на рецикл, представлял бы собой, по меньшей мере, приблизительно 20 массовых процентов от образуемого растворителем исходного подаваемого материала, направляемого в реакционную среду настоящего изобретения, более предпочтительно, по меньшей мере, приблизительно 40 массовых процентов, еще более предпочтительно, по меньшей мере, приблизительно 80 массовых процентов, а наиболее предпочтительно, по меньшей мере, 90 массовых процентов.
Изобретатели обнаружили то, что по причинам, связанным с активностью в реакции, и для учета наличия металлсодержащих примесей, остающихся в продукте окисления, концентрации избранных многовалентных металлов в отправляемом на рецикл образуемом растворителем исходном подаваемом материале предпочтительно находятся в диапазонах, приведенных непосредственно далее. Концентрация железа в отправляемом на рецикл растворителе предпочтительно составляет величину, меньшую приблизительно 150 ч./млн (мас.), более предпочтительно меньшую приблизительно 40 ч./млн (мас.), а наиболее предпочтительно находящуюся в диапазоне от 0 до 8 ч./млн (мас.). Концентрация никеля в отправляемом на рецикл растворителе предпочтительно составляет величину, меньшую приблизительно 150 ч./млн (мас.), более предпочтительно меньшую приблизительно 40 ч./млн (мас.), а наиболее предпочтительно находящуюся в диапазоне от 0 до 8 ч./млн (мас.). Концентрация хрома в отправляемом на рецикл растворителе предпочтительно составляет величину, меньшую приблизительно 150 ч./млн (мас.), более предпочтительно меньшую приблизительно 40 ч./млн (мас.), а наиболее предпочтительно находящуюся в диапазоне от 0 до 8 ч./млн (мас.). Концентрация молибдена в отправляемом на рецикл растворителе предпочтительно составляет величину, меньшую приблизительно 75 ч./млн (мас.), более предпочтительно меньшую приблизительно 20 ч./млн (мас.), а наиболее предпочтительно находящуюся в диапазоне от 0 до 4 ч./млн (мас.). Концентрация титана в отправляемом на рецикл растворителе предпочтительно составляет величину, меньшую приблизительно 75 ч./млн (мас.), более предпочтительно меньшую приблизительно 20 ч./млн (мас.), а наиболее предпочтительно находящуюся в диапазоне от 0 до 4 ч./млн (мас.). Концентрация меди в отправляемом на рецикл растворителе предпочтительно составляет величину, меньшую приблизительно 20 ч./млн (мас.), более предпочтительно меньшую приблизительно 4 ч./млн (мас.), а наиболее предпочтительно находящуюся в диапазоне от 0 до 1 ч./млн (мас.). В отправляемом на рецикл растворителе обычно также присутствуют и другие металлсодержащие примеси в общем случае при варьировании содержания на еще более низких уровнях, пропорциональных содержанию одного или нескольких из перечисленных ранее металлов. Регулирование содержания перечисленных выше металлов при выдерживании в предпочтительных диапазонах будет обеспечивать выдерживание содержания других металлсодержащих примесей на подходящих уровнях.
Данные металлы могут выявляться в качестве примесей в любом из поступающих в технологический процесс исходных подаваемых материалов (например, в поступающих окисляемом соединении, соединениях растворителя, окислителя и катализатора). В альтернативном варианте металлы могут возникать в качестве продуктов коррозии в любой из технологических установок, находящихся в контакте с реакционной средой и/или находящихся в контакте с отправляемым на рецикл растворителем. Способы регулирования содержания металлов при выдерживании в описанных диапазонах концентрации включают обеспечение достижения надлежащих технических характеристик и отслеживания степени чистоты для различных исходных подаваемых материалов и надлежащее использование конструкционных материалов, включающих нижеследующее, но не ограничивающихся только этим: многие коммерческие марки титана и нержавеющих сталей, включающие те марки, которые известны под наименованием двухфазных нержавеющих сталей и высокомолибденистых нержавеющих сталей.
Изобретатели также выявили предпочтительные диапазоны для избранных ароматических соединений в отправляемом на рецикл растворителе. Данные соединения включают как осажденные, так и растворенные ароматические соединения в отправляемом на рецикл растворителе.
Как это ни удивительно, но каждый осажденный продукт (например, ТРА), получаемый в результате неполного окисления пара-ксилола, является загрязнителем, в отношении которого необходимо принимать меры в случае растворителя, отправляемого на рецикл. Поскольку, как это ни удивительно, для уровней содержания твердой фазы в реакционной среде существуют предпочтительные диапазоны, любой осажденной продукт в образуемом растворителем исходном подаваемом материале непосредственно уменьшает количество окисляемого соединения, которое можно будет подавать вместе с ним. Кроме того, как было обнаружено, подача осажденной твердой фазы, образуемой ТРА, в отправляемом на рецикл растворителе при повышенных уровнях содержания оказывает неблагоприятное влияние на характер частиц, образуемых в среде осадительного окисления, что приводит к приданию нежелательного характера операциям на последующих стадиях технологической схемы (например, фильтрации продукта, промыванию растворителя, окислительной утилизации для сырого неочищенного продукта, растворению сырого неочищенного продукта для последующей переработки и тому подобному). Еще одна нежелательная характеристика осажденной твердой фазы в отправляемом на рецикл образуемом растворителем исходном подаваемом материале заключается в том, что она зачастую отличается очень высокими уровнями содержания осажденных примесей в сопоставлении с концентрациями примесей в объеме твердой фазы в суспензиях ТРА, из которых получают большое количество отправляемого на рецикл растворителя. Возможно повышенные уровни содержания примесей, наблюдаемые в твердой фазе, суспендированной в отправляемом на рецикл фильтрате, могут быть связаны с временами зародышеобразования, подходящими для осаждения определенных примесей из отправляемого на рецикл растворителя, и/или с охлаждением отправляемого на рецикл растворителя, либо намеренным, либо обусловленным потерями в окружающую среду. Например, концентрации высокоокрашенного и нежелательного 2,6-дикарбоксифлуоренона наблюдались при намного более высоких уровнях содержания в твердой фазе, присутствующей в отправляемом на рецикл растворителе при 80°С, в сопоставлении с тем, что наблюдали в твердой фазе ТРА, отделенной от отправляемого на рецикл растворителя при 160°С. Подобным же образом, концентрации изофталевой кислоты наблюдались при намного более высоких уровнях в твердой фазе, присутствующей в отправляемом на рецикл растворителе, в сопоставлении с уровнями, наблюдаемыми в твердой фазе ТРА из реакционной среды. То, как в точности будут себя вести конкретные осажденные примеси, захваченные в отправляемом на рецикл растворителе, при повторном введении в реакционную среду, по-видимому, представляет собой непостоянное свойство. Это зависит, может быть, от относительной растворимости примеси в жидкой фазе реакционной среды, может быть, от того, как осажденная примесь образует слои в осажденной твердой фазе, а может быть, от локальной скорости осаждения ТРА там, где твердая фаза будет впервые повторно введена в реакционную среду. Таким образом, изобретатели обнаружили то, что регулирование уровня содержания определенных примесей в отправляемом на рецикл растворителе так, как это описывается далее, является полезным вне зависимости от того, будут ли данные примеси присутствовать в отправляемом на рецикл растворителе в растворенной форме, или они будут захватываться в нем в частицах.
Количество осажденной твердой фазы, присутствующей в отправляемом на рецикл фильтрате, определяют при использовании гравиметрического метода следующим образом. Из растворителя, подаваемого в реакционную среду, отбирают представительный образец в то время, как растворитель перепускают в канал в направлении реакционной среды. Подходящий размер образца составляет приблизительно 100 граммов, помещенных в стеклянный контейнер, вмещающий приблизительно 250 миллилитров внутреннего объема. Перед выпуском в условия действия атмосферного давления, но при одновременном непрерывном перепускании в направлении контейнера для образца отправляемый на рецикл фильтрат охлаждают до менее чем 100°С; данное охлаждение производят для того, чтобы ограничить испарение растворителя в течение короткого промежутка времени перед герметичным закрытием в стеклянном контейнере. После помещения образца в условия действия атмосферного давления стеклянный контейнер незамедлительно герметично закрывают. После этого образцу дают возможность охлаждаться до приблизительно 20°С в окружении воздуха с температурой, равной приблизительно 20°С, и в отсутствие принудительной конвекции. После достижения приблизительно 20°С образец выдерживают в данном состоянии в течение, по меньшей мере, приблизительно 2 часов. После этого герметично закрытый контейнер интенсивно встряхивают до тех пор, пока не получают визуально однородное распределение твердой фазы. Непосредственно после этого в контейнер для образца помещают магнитную мешалку и обеспечивают ее вращение при скорости, достаточной для эффективного выдерживания однородного распределения твердой фазы. Отбирают пипеткой и взвешивают аликвоту в количестве 10 миллилитров смешанной жидкости, содержащей суспендированную твердую фазу. После этого объем жидкой фазы из данной аликвоты отделяют в результате проведения перегонки под вакуумом все еще при приблизительно 20°С и при эффективности, позволяющей избежать потерь твердой фазы. Влажную твердую фазу, отфильтрованную из данной аликвоты, после этого высушивают при эффективности, позволяющей избежать сублимации твердой фазы, а данную высушенную твердую фазу взвешивают. Соотношение между массой высушенной твердой фазы и массой первоначальной аликвоты суспензии представляет собой долю твердой фазы, обычно выражаемую в виде процентного содержания и называемую в настоящем документе уровнем содержания «осажденной твердой фазы при 20°С» в образуемом растворителем исходном подаваемом материале.
Изобретатели обнаружили то, что ароматические соединения, растворенные в жидкой фазе реакционной среды и включающие ароматические карбоновые кислоты, не имеющие неароматических гидрокарбильных групп (например, изофталевую кислоту, бензойную кислоту, фталевую кислоту, 2,5,4'-трикарбоксибифенил), как это ни удивительно, являются вредными компонентами. Несмотря на то что данные соединения обладают в значительной мере пониженной химической активностью в рассматриваемой реакционной среде в сопоставлении с окисляемыми соединениями, имеющими неароматические гидрокарбильные группы, изобретатели обнаружили то, что данные соединения, тем не менее, принимают участие во множестве неблагоприятных реакций. Таким образом, выгодно регулировать уровень содержания данных соединений в жидкой фазе реакционной среды при выдерживании в предпочтительных диапазонах. Это приводит к определению предпочтительных диапазонов избранных соединений в отправляемом на рецикл образуемом растворителем исходном подаваемом материале, а также предпочтительных диапазонов избранных предшественников в образуемом окисляемым ароматическим соединением исходном подаваемом материале.
Например, в случае жидкофазного неполного окисления пара-ксилола до получения терефталевой кислоты (ТРА) изобретатели обнаружили то, что высокоокрашенный и нежелательный примесный 2,7-дикарбоксифлуоренон (2,7-DCF) практически не обнаруживается в реакционной среде и отобранном продукте в случае присутствия мета-замещенных ароматических соединений в реакционной среде при очень низких уровнях содержания. Изобретатели обнаружили то, что в случае присутствия в образуемом растворителем исходном подаваемом материале примесной изофталевой кислоты при все более возрастающих уровнях содержания эффективность образования 2,7-DCF увеличивается почти что прямопропорционально. Изобретатели также обнаружили то, что в случае присутствия в исходном подаваемом материале, образуемом пара-ксилолом, примесного мета-ксилола эффективность образования 2,7-DCF опять-таки увеличивается почти что в прямой пропорции. Кроме того, даже в случае отсутствия в образуемом растворителем исходном подаваемом материале и образуемом окисляемым соединением исходном подаваемом материале мета-замещенных ароматических соединений изобретатели обнаружили то, что во время обычного неполного окисления очень чистого пара-ксилола образуется определенное количество изофталевой кислоты, в особенности тогда, когда в жидкой фазе реакционной среды присутствует бензойная кислота. Данная самообразующаяся изофталевая кислота благодаря своей более высокой растворимости, в сопоставлении с ТРА, в растворителе, содержащем уксусную кислоту и воду, с течением времени может накапливаться в коммерческих установках, использующих отправляемый на рецикл растворитель. Таким образом, все характеристики, выбираемые из количества изофталевой кислоты в образуемом растворителем исходном подаваемом материале, количества мета-ксилола в образуемом окисляемым ароматическим соединением исходном подаваемом материале и скорости самообразования изофталевой кислоты в реакционной среде, в надлежащем случае рассматриваются в равновесии друг с другом и в равновесии с любыми реакциями, которые обеспечивают расходование изофталевой кислоты. Как было обнаружено, в дополнение к образованию 2,7-DCF изофталевая кислота претерпевает и другие расходующие ее реакции, как это описывается далее. В дополнение к этому изобретатели обнаружили то, что существуют и другие вопросы, требующие внимания в случае задания подходящих диапазонов для мета-замещенных ароматических соединений при неполном окислении пара-ксилола до получения ТРА. Другие высокоокрашенные и нежелательные примеси, такие как 2,6-дикарбоксифлуоренон (2,6-DCF), по-видимому, очень тесно связаны с растворенными пара-замещенными ароматическими соединениями, которые всегда присутствуют в образуемом пара-ксилолом исходном подаваемом материале, направляемом на жидкофазное окисление. Таким образом, подавление образования 2,7-DCF лучше всего рассматривать в перспективе связи с уровнем содержания других образующихся окрашенных примесей.
Например, в случае жидкофазного неполного окисления пара-ксилола до получения ТРА изобретатели обнаружили то, что по мере увеличения уровней содержания изофталевой кислоты и фталевой кислоты в реакционной среде возрастает эффективность образования тримеллитовой кислоты. Тримеллитовая кислота представляет собой трехфункциональную карбоновую кислоту, присутствие которой приводит к разветвлению полимерных цепей во время получения ПЭТФ из ТРА. Во многих сферах применения ПЭТФ уровни разветвления необходимо регулировать при выдерживании низких уровней, и, таким образом, регулирование в отношении тримеллитовой кислоты должно обеспечить выдерживание низких уровней ее содержания в очищенной ТРА. Присутствие мета-замещенных и орто-замещенных соединений в реакционной среде, помимо приведения к образованию тримеллитовой кислоты, также становится причиной образования и других трикарбоновых кислот (например, 1,3,5-трикарбоксибензола). Кроме того, увеличение присутствия в реакционной среде трикарбоновых кислот приводит к увеличению эффективности образования тетракарбоновых кислот (например, 1,2,4,5-тетракарбоксибензола). Одним фактором при задании предпочтительных уровней содержания мета-замещенных и орто-замещенных соединений в отправляемом на рецикл образуемом растворителем исходном подаваемом материале, в образуемом окисляемым соединением исходном подаваемом материале и в реакционной среде, соответствующей настоящему изобретению, является регулирование эффективности совокупного образования всех ароматических карбоновых кислот, имеющих более чем две группы карбоновой кислоты.
Например, в случае жидкофазного неполного окисления пара-ксилола до получения ТРА изобретатели обнаружили то, что повышенные уровни содержания в жидкой фазе реакционной среды нескольких растворенных ароматических карбоновых кислот, не имеющих неароматических гидрокарбильных групп, непосредственно приводят к повышенной эффективности образования монооксида углерода и диоксида углерода. Данная повышенная эффективность образования оксидов углерода представляет собой потерю выхода как в отношении окислителя, так и в отношении окисляемого соединения, последнее - в связи с тем, что многие из попутно получаемых ароматических карбоновых кислот, которые, с одной стороны, можно рассматривать в качестве примесей, с другой стороны, также имеют коммерческую ценность. Таким образом, надлежащее удаление относительно растворимых карбоновых кислот, не имеющих неароматических гидрокарбильных групп, из отправляемого на рецикл растворителя имеет экономическое значение при предотвращении потерь выхода в отношении как окисляемого ароматического соединения, так и окислителя, в дополнение к подавлению образования в высшей степени нежелательных примесей, таких как различные флуореноны и тримеллитовая кислота.
Например, в случае жидкофазного неполного окисления пара-ксилола до получения ТРА изобретатели обнаружили то, что образование 2,5,4'-трикарбоксибифенила, по-видимому, является неизбежным. 2,5,4'-трикарбоксибифенил представляет собой ароматическую трикарбоновую кислоту, образованную в результате сопряжения двух ароматических колец, может быть, в результате сопряжения растворенных пара-замещенных ароматических соединений с арильным радикалом, может быть, с арильным радикалом, образованным в результате декарбоксилирования или декарбонилирования пара-замещенных ароматических соединений. К счастью, образование 2,5,4'-трикарбоксибифенила обычно происходит до меньших уровней содержания в сопоставлении с тримеллитовой кислотой, и это обычно не становится причиной значительного увеличения трудностей в связи с разветвлением молекул полимера во время получения ПЭТФ. Однако изобретатели обнаружили то, что повышенные уровни содержания 2,5,4'-трикарбоксибифенила в реакционной среде, включающей окисление алкилароматики в соответствии с предпочтительными вариантами реализации настоящего изобретения, приводят к повышению уровней содержания высокоокрашенного и нежелательного 2,6-DCF. Повышенные уровни содержания 2,6-DCF возможно формируются при участии 2,5,4'-трикарбоксибифенила в результате замыкания цикла с потерей молекулы воды, хотя точный механизм реакции достоверно неизвестен. Если 2,5,4'-трикарбоксибифенил, который в большей мере растворим в растворителе, содержащем уксусную кислоту и воду, в сопоставлении с ТРА, получит возможность накапливаться в отправляемом на рецикл растворителе до чрезмерно высоких уровней содержания, то тогда степени превращения в 2,6-DCF могут стать неприемлемо большими.
Например, в случае жидкофазного неполного окисления пара-ксилола до получения ТРА изобретатели обнаружили то, что ароматические карбоновые кислоты, не имеющие неароматических гидрокарбильных групп (например, изофталевая кислота), в общем случае приводят к мягкому подавлению химической активности реакционной среды в случае присутствия в жидкой фазе при достаточной концентрации.
Например, в случае жидкофазного неполного окисления пара-ксилола до получения ТРА изобретатели обнаружили то, что осаждение очень часто является неидеальным (то есть неравновесным) в том, что касается относительных концентраций различных химических соединений в твердой фазе и в жидкой фазе. Может быть, это обуславливается тем, что скорость осаждения очень велика при скоростях реакции за один проход в единицу времени, предпочтительных в настоящем документе, что приводит к неидеальному соосаждению примесей или даже к окклюдированию. Таким образом, в случае желательности ограничения концентрации определенных примесей (например, тримеллитовой кислоты и 2,6-DCF) в сырой неочищенной ТРА в связи с конфигурацией операций на последующих стадиях технологической схемы предпочитается регулировать их концентрацию в образуемом растворителем исходном подаваемом материале, а также скорость их образования в реакционной среде.
Например, изобретатели обнаружили то, что бензофеноновые соединения (например, 4,4'-дикарбоксибензофенон и 2,5,4'-трикарбоксибензофенон), полученные во время неполного окисления пара-ксилола, проявляют нежелательное действие в реакционной среде получения ПЭТФ даже несмотря на то, что бензофеноновые соединения сами по себе не являются настолько высокоокрашенными в ТРА, насколько такими являются флуореноны и антрахиноны. В соответствии с этим желательно ограничить присутствие бензофенонов и избранных предшественников в отправляемом на рецикл растворителе и в образуемом окисляемым соединением исходном подаваемом материале. Кроме того, изобретатели обнаружили то, что наличие повышенных уровней содержания бензойной кислоты вне зависимости от того, будет ли она введена в отправляемый на рецикл растворитель или образована в реакционной среде, приводит к повышенным скоростям получения 4,4'-дикарбоксибензофенона.
Говоря в целом, изобретатели обнаружили и в достаточной степени количественно охарактеризовали удивительный массив реакций для ароматических соединений, не имеющих неароматических гидрокарбильных групп, которые имеют место в случае жидкофазного неполного окисления пара-ксилола до получения ТРА. Резюмируя просто результаты для индивидуального случая, связанного с бензойной кислотой, изобретатели обнаружили то, что повышенные уровни содержания бензойной кислоты в реакционной среде определенных вариантов реализации настоящего изобретения приводят к значительному увеличению эффективности образования высокоокрашенной и нежелательной 9-флуоренон-2-карбоновой кислоты, к получению значительно повышенных уровней содержания 4,4'-дикарбоксибифенила, к получению повышенных уровней содержания 4,4'-дикарбоксибензофенона, к мягкому подавлению химической активности при целевом окислении пара-ксилола и к получению повышенных уровней содержания оксидов углерода и сопутствующим потерям выхода. Изобретатели обнаружили то, что повышенные уровни содержания бензойной кислоты в реакционной среде также приводят к повышенной эффективности образования изофталевой кислоты и фталевой кислоты, уровни содержания которых в желательном случае контролируют, выдерживая в низких диапазонах в соответствии с подобными аспектами настоящего изобретения. Количество и важность реакций, включающих участие бензойной кислоты, может быть, являются даже более удивительными, поскольку некоторые изобретатели последнего времени предусматривают использование бензойной кислоты вместо уксусной кислоты в качестве основного компонента растворителя (смотрите, например, патент США № 6562997). В дополнение к этому изобретатели настоящего изобретения наблюдали то, что во время окисления пара-ксилола бензойная кислота самообразуется со скоростями, которые являются вполне существенными в сопоставлении с ее образованием из примесей, таких как толуол и этилбензол, обычно обнаруживаемых в образуемом окисляемым соединением исходном подаваемом материале, содержащем пара-ксилол технической чистоты.
С другой стороны, изобретатели обнаружили малую значимость дополнительного регулирования состава отправляемого на рецикл растворителя в том, что касается присутствия окисляемого ароматического соединения, и в том, что касается промежуточных соединений в реакции ароматики, которые как сохраняют неароматические гидрокарбильные группы, так и являются также относительно растворимыми в растворителе, отправляемом на рецикл. В общем случае данные соединения либо подают в реакционную среду, либо они образуются в реакционной среде со скоростями, соответствующими значительному превышению уровня их присутствия в отправляемом на рецикл растворителе; а скорость расходования данных соединений в реакционной среде достаточно велика, при сохранения одной или нескольких неароматических гидрокарбильных групп, что позволяет надлежащим образом ограничивать их накопление в отправляемом на рецикл растворителе. Например, во время неполного окисления пара-ксилола в многофазной реакционной среде вместе с большими количествами растворителя в ограниченной степени испаряется и пара-ксилол. В случае покидания данным испарившимся растворителем реактора в виде части отходящего газа и его конденсации для извлечения в качестве растворителя, отправляемого на рецикл, здесь же конденсируется также и существенная часть испарившегося пара-ксилола. Нет никакой необходимости ограничивать концентрацию данного пара-ксилола в растворителе, отправляемом на рецикл. Например, если при покидании суспензией реакционной среды окисления пара-ксилола растворитель от твердой фазы отделяют, то тогда данный извлеченный растворитель будет содержать концентрацию растворенной пара-толуиловой кислоты, подобную концентрации, имеющей место на момент удаления из реакционной среды. Несмотря на то что ограничение стационарной концентрации пара-толуиловой кислоты в жидкой фазе реакционной среды, смотрите далее, может быть важным, смотрите далее, нет никакой необходимости в отдельном регулировании содержания пара-толуиловой кислоты в данной части отправляемого на рецикл растворителя благодаря ее относительно хорошей растворимости и ее малому массовому расходу в сопоставлении с образованием пара-толуиловой кислоты в реакционной среде. Подобным же образом, изобретатели выявили мало оснований для ограничения концентраций в отправляемом на рецикл растворителе ароматических соединений, имеющих метильные заместители (например, толуиловых кислот), ароматических альдегидов (например, терефталевого альдегида), ароматических соединений, имеющих гидроксиметильных заместителей (например, 4-гидроксиметилбензойной кислоты), и бромированных ароматических соединений, сохраняющих, по меньшей мере, одну неароматическую гидрокарбильную группу (например, альфа-бром-пара-толуиловой кислоты), уровнями, более низкими в сопоставлении с теми, которые внутренне присущи жидкой фазе, покидающей реакционную среду, существующую при неполном окислении ксилола в соответствии с предпочтительными вариантами реализации настоящего изобретения. Как это ни удивительно, но изобретатели также обнаружили и то, что нет никакой необходимости также и в регулировании в отправляемом на рецикл растворителе концентрации избранных фенолов, образующихся во время неполного окисления ксилола в соответствии с самой его природой, поскольку данные соединения образуются и разрушаются в реакционной среде со скоростями, соответствующими значительному превышению уровня их присутствия в растворителе, отправляемом на рецикл. Например, изобретатели обнаружили то, что 4-гидроксибензойная кислота оказывает относительно небольшое влияние на химическую активность в предпочтительных вариантах реализации настоящего изобретения в случае совместной подачи с расходами, соответствующими более чем 2 граммам 4-гидроксибензойной кислоты на 1 килограмм пара-ксилола, что намного превышает естественное присутствие в отправляемом на рецикл растворителе, несмотря на то, что другие исследователи сообщали, что она является существенным ядом в подобной реакционной среде (смотрите, например, работу W. Partenheimer, Catalysis Today 23 (1995) p. 81).
Таким образом, существуют многочисленные реакции и многочисленные соображения по заданию предпочтительных диапазонов для различных ароматических примесей в образуемом растворителем исходном подаваемом материале, как в данный момент было описано. Данные открытия сформулированы при выражении через совокупный среднемассовый состав для всех потоков растворителей, подаваемых в реакционную среду в течение заданного периода времени, предпочтительно одного дня, более предпочтительно одного часа, а наиболее предпочтительно одной минуты. Например, если один образуемый растворителем исходный подаваемый материал будут перепускать по существу непрерывно с составом, соответствующим 40 ч./млн (мас.) изофталевой кислоты, при расходе 7 килограммов в минуту, второй образуемый растворителем исходный подаваемый материал будут перепускать по существу непрерывно с составом, соответствующим 2000 ч./млн (мас.) изофталевой кислоты, при расходе 10 килограммов в минуту, и никаких других потоков образуемого растворителем исходного подаваемого материала в реакционную среду поступать не будет, то тогда совокупный среднемассовый состав для образуемого растворителем исходного подаваемого материала будут рассчитывать в виде (40*7+2000*10)/(7+10)=1193 ч./млн (мас.) изофталевой кислоты. Следует отметить то, что масса любого образуемого окисляемым соединением исходного подаваемого материала или любого образуемого окислителем исходного подаваемого материала, которые, может быть, являются перемешанными с образуемым растворителем исходным подаваемым материалом перед их поступлением в реакционную среду, не принимается во внимание при вычислении совокупного среднемассового состава образуемого растворителем исходного подаваемого материала.
Приведенная далее Таблица 1 представляет предпочтительные значения для определенных компонентов в образуемом растворителем исходном подаваемом материале, вводимом в реакционную среду. Компоненты образуемого растворителем исходного подаваемого материала, приведенные в Таблице 1, представляют собой нижеследующее: 4-карбоксибензальдегид (4-СВА), 4,4'-дикарбоксистильбен (4,4'-DCS), 2,6-дикарбоксиантрахинон (2,6-DCA), 2,6-дикарбоксифлуоренон (2,6-DCF), 2,7-дикарбоксифлуоренон (2,7-DCF), 3,5-дикарбоксифлуоренон (3,5-DCF), 9-флуоренон-2-карбоновая кислота (9F-2CA), 9-флуоренон-4-карбоновая кислота (9F-4CA), совокупный ассортимент флуоренонов, включающий другие флуореноны, не перечисленные индивидуально, (совокупный ассортимент флуоренонов), 4,4'-дикарбоксибифенил (4,4'-DCB), 2,5,4'-трикарбоксибифенил (2,5,4'-TCB), фталевая кислота (РА), изофталевая кислота (IPA), бензойная кислота (ВА), тримеллитовая кислота (ТМА), 2,6-дикарбоксибензокумарин (2,6-DCBC), 4,4'-дикарбоксибензил (4,4'-DCBZ), 4,4'-дикарбоксибензофенон (4,4'-DCBP), 2,5,4'-трикарбоксибензофенон (2,5,4'-TCBP), терефталевая кислота (ТРА), осажденная твердая фаза при 20°С и совокупный ассортимент ароматических карбоновых кислот, не имеющих неароматических гидрокарбильных групп. Приведенная далее Таблица 1 представляет предпочтительные количества данных примесей в СТА, полученной в соответствии с вариантом реализации настоящего изобретения.

В отправляемом на рецикл растворителе обычно присутствует также и множество других ароматических примесей, в общем случае при варьировании содержания на еще более низких уровнях и/или в пропорции от содержания одного или нескольких из описанных ароматических соединений. Способы регулирования уровней содержания описанных ароматических соединений при выдерживании в предпочтительных диапазонах обычно будут обеспечивать выдерживание на подходящих уровнях содержания и других ароматических примесей.
В случае использования в реакционной среде брома, как известно, большое количество ионных и органических форм брома будет существовать в динамическом равновесии. Данные различные формы брома характеризуются различными характеристиками стабильности сразу после покидания реакционной среды и прохождения через операции в различных установках, относящиеся к отправляемому на рецикл растворителю. Например, альфа-бром-пара-толуиловая кислота в определенных условиях может присутствовать как таковая или в других условиях может быстро подвергаться гидролизу с образованием 4-гидроксиметилбензойной кислоты и бромида водорода. В настоящем изобретении предпочитается, чтобы, по меньшей мере, приблизительно 40 массовых процентов, более предпочтительно, чтобы, по меньшей мере, приблизительно 60 массовых процентов, а наиболее предпочтительно, чтобы, по меньшей мере, приблизительно 80 массовых процентов от совокупной массы брома, присутствующего в совокупном образуемом растворителем исходном подаваемом материале, вводимом в реакционную среду, имели бы вид одной или нескольких из следующих далее химических форм: ионный бром, альфа-бром-пара-толуиловая кислота и бромуксусная кислота.
Несмотря на то что важность и значение регулирования совокупной среднемассовой степени чистоты образуемого растворителем исходного подаваемого материала при выдерживании в описанных желательных диапазонах настоящего изобретения до настоящего момента не были раскрыты и/или описаны, подходящие способы регулирования степени чистоты образуемого растворителем исходного подаваемого материала могут быть скомпонованы из различных способов, уже известных на современном уровне техники. Во-первых, любой растворитель, испарившийся из реакционной среды, обычно характеризуется подходящей степенью чистоты при том условии, что жидкая или твердая фаза из реакционной среды не будут захватываться испарившимся растворителем. В соответствии с описанием в настоящем документе такой захват надлежащим образом ограничивает подача капель растворителя среды орошения в пространство отделения отходящего газа над реакционной средой; и из такого отходящего газа может быть сконденсирован отправляемый на рецикл растворитель с подходящей степенью чистоты в отношении ароматического соединения. Во-вторых, более трудная и дорогостоящая очистка отправляемого на рецикл образуемого растворителем исходного подаваемого материала обычно относится к растворителю, отбираемому из реакционной среды в жидкой форме, и к растворителю, который впоследствии вступает в контакт с жидкой и/или твердой фазами реакционной среды, отбираемой из реакционной емкости (то есть отправляемому на рецикл растворителю, получаемому из фильтра, в котором твердую фазу концентрируют и/или промывают, отправляемому на рецикл растворителю, получаемому из центрифуги, в которой твердую фазу концентрируют и/или промывают, отправляемому на рецикл растворителю, отбираемому с операции кристаллизации, и тому подобному). Однако на современном уровне техники также известны и способы проведения необходимой очистки данных отправляемых на рецикл потоков растворителя при использовании одного или нескольких описаний предшествующего уровня техники. Что касается регулирования уровня содержания осажденной твердой фазы в отправляемом на рецикл растворителе при выдерживании в пределах указанных диапазонов, то подходящие способы регулирования включают нижеследующее, но не ограничиваются только этим: гравиметрическая седиментация, механическое фильтрование при использовании фильтровального полотна на вращающихся ленточных фильтрах и вращающихся барабанных фильтрах, механическое фильтрование при использовании стационарной фильтрующей среды внутри емкостей, работающих под давлением, гидроциклонов и центрифуг. Что касается регулирования уровня содержания растворенных ароматических соединений в отправляемом на рецикл растворителе при выдерживании в пределах указанных диапазонов, то способы регулирования включают нижеследующее, но не ограничиваются только этим: способы, описанные в патенте США № 4939297 и публикации патентной заявки США № 2005-0038288, посредством ссылки включаемых в настоящий документ. Однако ни одно из данных изобретений предшествующего уровня техники не раскрывает и не описывает предпочтительные уровни степени чистоты в совокупном образуемом растворителем исходном подаваемом материале, как это описывается в настоящем документе. Вместо этого данные изобретения предшествующего уровня техники просто предлагают способы очистки избранных и частных потоков отправляемого на рецикл растворителя без выведения оптимальных значений настоящего изобретения для состава совокупного среднемассового образуемого растворителем исходного подаваемого материала, вводимого в реакционную среду.
Обращаясь теперь к степени чистоты исходного подаваемого материала, образуемого окисляемым соединением, можно сказать, что известно то, что в данном случае имеют место определенные уровни содержания изофталевой кислоты, фталевой кислоты и бензойной кислоты, и при низких уровнях содержания присутствие данных соединений в очищенной ТРА, используемой для получения полимера, является допустимым. Кроме того, известно, что данные соединения являются относительно более растворимыми во многих растворителях, и в выгодном случае их можно удалить из очищенной ТРА при использовании способов кристаллизации. Однако из варианта реализации изобретения, описанного в настоящем документе, на данный момент известно то, что регулирование уровня содержания нескольких относительно растворимых ароматических соединений, а именно соединений, включающих изофталевую кислоту, фталевую кислоту и бензойную кислоту, в жидкой фазе реакционной среды, как это ни удивительно, является важным при регулировании уровня содержания полициклических и окрашенных ароматических соединений, образующихся в реакционной среде, при регулировании уровня содержания соединений, имеющих в молекуле более чем 2 функциональности карбоновой кислоты, при регулировании активности в реакции в реакционной среде для неполного окисления и при регулировании потерь выхода в отношении окислителя и ароматического соединения.
На современном уровне техники известно то, что изофталевая кислота, фталевая кислота и бензойная кислота образуются в реакционной среде следующим образом. Примесный мета-ксилол из исходного подаваемого материала при хороших степени превращения и выходе окисляется до получения IPA. Примесный орто-ксилол из исходного подаваемого материала при хороших степени превращения и выходе окисляется до получения фталевой кислоты. Примесные этилбензол и толуол из исходного подаваемого материала при хороших степени превращения и выходе окисляются до получения бензойной кислоты. Однако изобретатели обнаружили то, что в реакционной среде, содержащей пара-ксилол, значительные количества изофталевой кислоты, фталевой кислоты и бензойной кислоты образуются также и по механизмам, отличным от окисления мета-ксилола, орто-ксилола, этилбензола и толуола. Данные другие присущие природе системы химические маршруты возможно включают декарбонилирование, декарбоксилирование, перегруппировку переходных состояний и присоединение метильных и карбонильных радикалов к ароматическим кольцам.
При определении предпочтительных диапазонов уровней содержания примесей в исходном подаваемом материале, образуемом окисляемым соединением, значение имеет множество факторов. Любая примесь в исходном подаваемом материале, вероятно, будет связана с прямой потерей выхода и затратами на очистку продукта, если требования по степени чистоты к продукту окисления будут достаточно жесткими (например, в случае реакционной среды для неполного окисления пара-ксилола толуол и этилбензол, обычно обнаруживаемые в пара-ксилоле технической чистоты, приводят к образованию бензойной кислоты, и данную бензойную кислоту в основном удаляют из большинства коммерческих вариантов ТРА). В случае участия продукта неполного окисления примеси из исходного подаваемого материала в дополнительных реакциях при рассмотрении вопроса величины необходимых затрат на очистку исходного подаваемого материала существенными становятся факторы, отличные от простой потери выхода и удаления (например, в случае реакционной среды для неполного окисления пара-ксилола, помимо прочего, этилбензол приводит к получению бензойной кислоты, а бензойная кислота после этого приводит к получению высокоокрашенной 9-флуоренон-2-карбоновой кислоты, к получению изофталевой кислоты, к получению фталевой кислоты и к получению повышенных количеств оксидов углерода). В случае самообразования в реакционной среде дополнительных количеств примеси при помощи химических механизмов, не связанных непосредственно с примесями в исходном подаваемом материале, анализ становится еще более сложным (например, в случае реакционной среды для неполного окисления пара-ксилола бензойная кислота также самообразуется из самого пара-ксилола). В дополнение к этому переработка сырого неочищенного продукта окисления на последующих стадиях технологической схемы может оказать влияние на соображения в отношении предпочтительной степени чистоты исходного подаваемого материала. Например, затраты, необходимые для удаления прямой примеси (бензойной кислоты) и последующих примесей (изофталевой кислоты, фталевой кислоты, 9-флуоренон-2-карбоновой кислоты и тому подобного) до подходящих уровней содержания, могут быть одними и теми же, могут быть отличными друг от друга и могут быть отличными от требований по удалению в основном неродственной примеси (например, продукт неполного окисления 4-СВА при окислении пара-ксилола до получения ТРА).
Следующие далее описанные диапазоны степени чистоты для исходного подаваемого материала в случае пара-ксилола предпочитаются тогда, когда пара-ксилол подают в реакционную среду совместно с растворителем и окислителем в целях неполного окисления до получения ТРА. Данные диапазоны являются более предпочтительными в способе получения ТРА, включающем стадии последующего окисления в целях удаления из реакционной среды примесей, отличных от окислителя и растворителя (например, металлов катализатора). Данные диапазоны являются еще более предпочтительными в способах получения ТРА, которые обеспечивают удаление из СТА дополнительных количеств 4-СВА (например, в результате превращения СТА в диметилтерефталат плюс примесные сложные эфиры и последующего отделения сложного метилового сложного эфира 4-СВА при использовании перегонки, в результате реализации способов окислительной утилизации, предназначенных для превращения 4-СВА в ТРА, в результате реализации способов гидрирования, предназначенных для превращения 4-СВА в пара-толуиловую кислоту, которую после этого отделяют при использовании способов неполной кристаллизации). Данные диапазоны являются наиболее предпочтительными в способах получения ТРА, которые обеспечивают удаление дополнительных количеств 4-СВА из СТА в результате реализации способов окислительной утилизации, предназначенных для превращения 4-СВА в ТРА.
При использовании новых знаний о предпочтительных диапазонах для ароматических соединений, отправляемых на рецикл, и об относительных количествах ароматических соединений, образующихся непосредственно в результате окисления примесей из исходного подаваемого материала в сопоставлении с другими присущими природе системы химическими маршрутами, были выявлены улучшенные диапазоны для уровней содержания примесей, соответствующих загрязненному пара-ксилолу, подаваемому в технологический процесс неполного окисления для получения ТРА. Приведенная далее Таблица 2 представляет предпочтительные значения для количества мета-ксилола, орто-ксилола и этилбензола + толуола в образуемом пара-ксилолом исходном подаваемом материале при выражении в массовых частях на миллион частей пара-ксилола.
Компоненты загрязненного образуемого пара-ксилолом исходного подаваемого материала
Специалисты в соответствующей области техники на данный момент должны осознать то, что вышеупомянутые примеси в загрязненном пара-ксилоле могут оказывать свое наибольшее воздействие на реакционную среду после того, как продукты их неполного окисления накопятся в отправляемом на рецикл растворителе. Например, подача верхнего количества из наиболее предпочтительного диапазона для мета-ксилола - 400 ч./млн (мас.) - в жидкой фазе реакционной среды будет незамедлительно приводить к получению приблизительно 200 ч./млн (мас.) изофталевой кислоты при функционировании при уровне содержания твердой фазы в реакционной среде, равном приблизительно 33 массовым процентам. Это сопоставимо с введением верхнего количества из наиболее предпочтительного диапазона для уровня содержания изофталевой кислоты в отправляемом на рецикл растворителе, равного 400 ч./млн (мас.), когда после обеспечения охлаждения реакционной среды в результате испарения типичного растворителя достигают приблизительно 1200 ч./млн (мас.) изофталевой кислоты в жидкой фазе реакционной среды. Таким образом, именно накопление с течением времени продуктов неполного окисления в отправляемом на рецикл растворителе обнаруживает наибольшее из возможных влияние примесных мета-ксилола, орто-ксилола, этилбензола и толуола из исходного подаваемого материала, образуемого загрязненным пара-ксилолом. В соответствии с этим предпочитается, чтобы вышеупомянутые диапазоны примесей в образуемом загрязненным пара-ксилолом исходном подаваемом материале выдерживались бы в течение, по меньшей мере, половины каждого дня функционирования при любой реакционной среде для неполного окисления в конкретной производственной установке, более предпочтительно в течение, по меньшей мере, трех четвертей каждого дня в течение, по меньшей мере, семи последовательных дней функционирования, а наиболее предпочтительным является случай, когда массово-взвешенные средние величины для состава образуемого загрязненным пара-ксилолом исходного подаваемого материала находятся в пределах предпочтительных диапазонов в течение, по меньшей мере, 30 последовательных дней функционирования.
Способы получения загрязненного пара-ксилола предпочтительной степени чистоты уже известны на современном уровне техники, и они включают нижеследующее, но не ограничиваются только этим: перегонка, способы неполной кристаллизации при температурах, меньших температуры окружающей среды, и способы применения молекулярных сит с использованием селективной адсорбции, определяемой размером пор. Однако предпочтительные диапазоны степени чистоты, указанные в настоящем документе, на своем верхнем крае требуют приложения больших усилий и затрат в сопоставлении с тем, что является характерным при использовании на практике коммерческими поставщиками пара-ксилола; и вместе с тем на нижнем крае предпочтительные диапазоны избегают вариантов чрезмерно дорогостоящей очистки пара-ксилола, предназначенного для подачи в реакционную среду для неполного окисления, вследствие раскрытия и описания того, что объединенное действие самообразования примесей из самого пара-ксилола и реакций расходования примесей в реакционной среде становится более существенным параметром в сопоставлении со скоростями подачи примесей совместно с загрязненным пара-ксилолом.
В случае содержания в потоке ксилолсодержащего исходного подаваемого материала избранных примесей, таких как этилбензол и/или толуол, окисление данных примесей может привести к образованию бензойной кислоты. В соответствии с использованием в настоящем документе термин «бензойная кислота, образованная из примесей» должен обозначать бензойную кислоту, получаемую во время окисления ксилола из любого источника, отличного от ксилола.
Как описывается в настоящем документе, часть бензойной кислоты, получаемой во время окисления ксилола, образуется из самого ксилола. Данное получение бензойной кислоты из ксилола явно происходит в дополнение к получению любой части бензойной кислоты, которая может представлять собой бензойную кислоту, образованную из примесей. Не в порядке связывания себя теорией представляется, что бензойная кислота образуется в реакционной среде из ксилола тогда, когда различные промежуточные продукты окисления ксилола самопроизвольно претерпевают декарбонилирование (потерю монооксида углерода) или декарбоксилирование (потерю диоксида углерода) с образованием, таким образом, арильных радикалов. После этого данные арильные радикалы могут отщеплять атом водорода от одного из множества доступных источников в реакционной среде и приводить к получению самообразующейся бензойной кислоты. Вне зависимости от химического механизма термин «самообразующаяся бензойная кислота» в соответствии с использованием в настоящем документе должен обозначать бензойную кислоту, во время окисления ксилола полученную из ксилола.
Как также описывается в настоящем документе, в случае окисления пара-ксилола до получения терефталевой кислоты (ТРА) получение самообразующейся бензойной кислоты приводит к потере выхода в отношении пара-ксилола и потере выхода в отношении окислителя. В дополнение к этому присутствие самообразующейся бензойной кислоты в жидкой фазе реакционной среды коррелирует с увеличением роли многих нежелательных побочных реакций, а именно реакций, включающих образование высокоокрашенных соединений, называемых монокарбоксифлуоренонами. Самообразующаяся бензойная кислота также вносит свой вклад в нежелательное накопление бензойной кислоты в отправляемом на рецикл фильтрате, что дополнительно увеличивает концентрацию бензойной кислоты в жидкой фазе реакционной среды. Таким образом, образование самообразующейся бензойной кислоты желательно свести к минимуму, но данную ситуацию надлежащим образом также рассматривают одновременно и в связи с бензойной кислотой, образованной из примесей, с факторами, оказывающими влияние на расходование бензойной кислоты, с факторами, связанными с другими моментами, определяющими селективность реакции, и с общими экономическими показателями.
Изобретатели обнаружили то, что самообразование бензойной кислоты можно регулировать при выдерживании на низких уровнях в результате надлежащего выбора, например, температуры, распределения ксилола и доступности кислорода в реакционной среде во время окисления. Без желания связывать себя теорией можно сказать, что пониженные температуры и улучшенная доступность кислорода, по-видимому, приводят к уменьшению скоростей декарбонилирования и/или декарбоксилирования, что, таким образом, устраняет аспект потери выхода в связи с самообразующейся бензойной кислотой. Достаточная доступность кислорода, по-видимому, направляет реакцию арильных радикалов в направлении образования других более безопасных продуктов, в частности гидроксибензойных кислот. На баланс между степенью превращения арильных радикалов в бензойную кислоту или в гидроксибензойную кислоту также может оказывать влияние и распределение ксилола в реакционной среде. Вне зависимости от химических механизмов изобретатели выявили условия проведения реакции, которые, несмотря на свою достаточную мягкость в отношении уменьшения эффективности образования бензойной кислоты, являются достаточно жесткими в отношении окисления большой доли полученной гидроксибензойной кислоты до получения монооксида углерода и/или диоксида углерода, которые легко удаляются из продукта окисления.
В предпочтительном варианте реализации настоящего изобретения реактор окисления сконфигурирован и функционирует таким образом, чтобы свести к минимуму образование самообразующейся бензойной кислоты и довести до максимума окисление гидроксибензойных кислот до получения монооксида углерода и/или диоксида углерода. В случае использования реактора окисления для окисления пара-ксилола до получения терефталевой кислоты предпочитается, чтобы пара-ксилол составлял бы, по меньшей мере, приблизительно 50 массовых процентов от совокупного ксилола в потоке исходного подаваемого материала, вводимого в реактор. Более предпочтительно пара-ксилол составляет, по меньшей мере, приблизительно 75 массовых процентов от совокупного ксилола в потоке исходного подаваемого материала. Еще более предпочтительно пара-ксилол составляет, по меньшей мере, 95 массовых процентов от совокупного ксилола в потоке исходного подаваемого материала. Наиболее предпочтительно пара-ксилол составляет по существу все количество совокупного ксилола в потоке исходного подаваемого материала.
В случае использования реактора для окисления пара-ксилола до получения терефталевой кислоты предпочитается, чтобы скорость образования терефталевой кислоты была бы доведена до максимума, в то время как скорость образования самообразующейся бензойной кислоты была бы сведена к минимуму. Предпочтительно соотношение между скоростью образования (при расчете на массу) терефталевой кислоты и скоростью образования (при расчете на массу) самообразующейся бензойной кислоты составляет, по меньшей мере, приблизительно 500:1, более предпочтительно, по меньшей мере, приблизительно 1000:1, а наиболее предпочтительно, по меньшей мере, 1500:1. Как будет продемонстрировано далее, скорость образования самообразующейся бензойной кислоты предпочтительно измеряют тогда, когда концентрация бензойной кислоты в жидкой фазе реакционной среды составляет величину, меньшую 2000 ч./млн (мас.), более предпочтительно меньшую 1000 ч./млн (мас.), а наиболее предпочтительно меньшую 500 ч./млн (мас.), поскольку данные низкие концентрации обеспечивают подавление реакций, которые превращают бензойную кислоту в другие соединения, до подходящих низких скоростей.
Если объединить самообразующуюся бензойную кислоту и бензойную кислоту, образованную из примесей, то соотношение между скоростью образования (при расчете на массу) терефталевой кислоты и скоростью образования (при расчете на массу) совокупной (самообразующейся и образованной из примесей) бензойной кислоты предпочтительно составляет, по меньшей мере, приблизительно 400:1, более предпочтительно, по меньшей мере, приблизительно 700:1, а наиболее предпочтительно, по меньшей мере, 1100:1. Как будет продемонстрировано далее, совокупную скорость образования самообразующейся бензойной кислоты плюс бензойной кислоты, образованной из примесей, предпочтительно измеряют тогда, когда концентрация бензойной кислоты в жидкой фазе реакционной среды составляет величину, меньшую 500 ч./млн (мас.), поскольку данные низкие концентрации обеспечивают подавление реакций, которые превращают бензойную кислоту в другие соединения, до подходящих низких скоростей.
Как описывается в настоящем документе, повышенные концентрации бензойной кислоты в жидкой фазе реакционной среды приводят к увеличению эффективности образования множества других ароматических соединений, некоторые из которых являются вредными примесями в ТРА; и, как описывается в настоящем документе, повышенные концентрации бензойной кислоты в жидкой фазе реакционной среды приводят к увеличению эффективности образования газообразных оксидов углерода, образование которых соответствует потере выхода в отношении окислителя и в отношении ароматических соединений и/или растворителя. Кроме того, в данный момент следует раскрыть то, что изобретатели обнаружили то, что значительная доля данной увеличенной эффективности образования других ароматических соединений и оксидов углерода своим источником имеет реакции, которые обеспечивают превращение некоторого количества самих молекул бензойной кислоты, в противоположность случаю катализирования бензойной кислотой других реакций без расходования ее самой. В соответствии с этим «результирующее образование бензойной кислоты» определяют в настоящем документе как средневременную массу всего количества бензойной кислоты, покидающей реакционную среду, минус средневременную массу всего количества бензойной кислоты, поступающей в реакционную среду в течение того же самого промежутка времени. Данное результирующее образование бензойной кислоты зачастую является положительным, движущей силой для чего являются скорости образования бензойной кислоты, образованной из примесей, и самообразующейся бензойной кислоты. Однако изобретатели обнаружили то, что скорость превращения бензойной кислоты в оксиды углерода и в несколько других соединений, по-видимому, приблизительно линейно увеличивается по мере того, как в жидкой фазе реакционной среды будет увеличиваться концентрация бензойной кислоты, при проведении измерения тогда, когда другие условия проведения реакции, включающие температуру, доступность кислорода, значение STR и активность в реакции, надлежащим образом выдерживают постоянными. Таким образом, если концентрация бензойной кислоты в жидкой фазе реакционной среды будет достаточно большой, может быть, в связи с повышенной концентрацией бензойной кислоты в отправляемом на рецикл растворителе, то тогда степень превращения молекул бензойной кислоты в другие соединения, в том числе оксиды углерода, может стать равной или большей величины, соответствующей химическому образованию новых молекул бензойной кислоты. В данном случае для результирующего образования бензойной кислоты баланс может быть достигнут в области нуля или даже отрицательных величин. Изобретатели обнаружили то, что, если результирующее образование бензойной кислоты является положительным, то тогда соотношение между скоростью образования (при расчете на массу) терефталевой кислоты в реакционной среде и скоростью результирующего образования бензойной кислоты в реакционной среде предпочтительно превышает приблизительно 700:1, более предпочтительно превышает приблизительно 1100:1, а наиболее предпочтительно превышает 4000:1. Изобретатели обнаружили то, что, если результирующее образование бензойной кислоты является отрицательным, тогда соотношение между скоростью образования (при расчете на массу) терефталевой кислоты в реакционной среде и скоростью результирующего образования бензойной кислоты в реакционной среде предпочтительно превышает приблизительно 200:(-1), более предпочтительно превышает приблизительно 1000:(-1), а наиболее предпочтительно превышает 5000:(-1).
Изобретатели также выявили предпочтительные диапазоны для состава суспензии (жидкая фаза + твердая фаза), отбираемой из реакционной среды, и образуемой твердой СТА части суспензии. Предпочтительные составы суспензии и предпочтительные составы СТА, как это ни удивительно, являются превосходными и подходящими для использования. Например, очищенная ТРА, полученная из данной предпочтительной СТА в результате проведения окислительной утилизации, характеризуется достаточно низким уровнем содержания совокупных примесей и окрашенных примесей, так что очищенная ТРА является подходящей для использования, без гидрирования дополнительных количеств 4-СВА и/или окрашенных примесей, для широкого диапазона сфер применения, связанных с волокнами из ПЭТФ, и сфер применения, связанных с упаковкой из ПЭТФ. Например, предпочтительный состав суспензии обеспечивает получение жидкой фазы реакционной среды, которая характеризуется относительно низкой концентрацией существенных примесей, и это значащим образом приводит к уменьшению эффективности образования других даже более нежелательных примесей, описанных в настоящем документе. В дополнение к этому в соответствии с другими вариантами реализации настоящего изобретения предпочтительный состав суспензии существенным образом способствует проведению последующей переработки жидкости из суспензии, которая становится отправляемым на рецикл растворителем с подходящей степенью чистоты.
СТА, получаемая в соответствии с одним вариантом реализации настоящего изобретения, содержит меньше примесей, относящихся к избранным типам, в сопоставлении с СТА, получаемой при использовании обычных способов и аппаратуры, а именно тех, которые используют растворитель, отправляемый на рецикл. Примеси, которые могут присутствовать в СТА, включают нижеследующее: 4-карбоксибензальдегид (4-СВА), 4,4'-дикарбоксистильбен (4,4'-DCS), 2,6-дикарбоксиантрахинон (2,6-DCA), 2,6-дикарбоксифлуоренон (2,6-DCF), 2,7-дикарбоксифлуоренон (2,7-DCF), 3,5-дикарбоксифлуоренон (3,5-DCF), 9-флуоренон-2-карбоновая кислота (9F-2CA), 9-флуоренон-4-карбоновая кислота (9F-4CA), совокупный ассортимент флуоренонов, включающий другие флуореноны, не перечисленные индивидуально, (совокупный ассортимент флуоренонов), 4,4'-дикарбоксибифенил (4,4'-DCB), 2,5,4'-трикарбоксибифенил (2,5,4'-TCB), фталевая кислота (РА), изофталевая кислота (IPA), бензойная кислота (ВА), тримеллитовая кислота (ТМА), пара-толуиловая кислота (РТАС), 2,6-дикарбоксибензокумарин (2,6-DCBC), 4,4'-дикарбоксибензил (4,4'-DCBZ), 4,4'-дикарбоксибензофенон (4,4'-DCBP), 2,5,4'-трикарбоксибензофенон (2,5,4'-TCBP). Приведенная далее Таблица 3 представляет предпочтительные величины для уровней содержания данных примесей в СТА, полученной в соответствии с вариантом реализации настоящего изобретения.
Примеси в СТА
В дополнение к этому предпочитается, чтобы СТА, получаемая в соответствии с вариантом реализации настоящего изобретения, характеризовалась бы пониженным уровнем содержания окрашенных соединений в сопоставлении с СТА, получаемой при использовании обычных способов и аппаратуры, а именно тех, которые используют растворитель, отправляемый на рецикл. Таким образом, предпочитается, чтобы СТА, получаемая в соответствии с одним вариантом реализации настоящего изобретения, характеризовалась бы процентным пропусканием в области 340 нанометров (нм), равным, по меньшей мере, приблизительно 25 процентам, более предпочтительно, по меньшей мере, приблизительно 50 процентам, а наиболее предпочтительно, по меньшей мере, 60 процентам. Кроме того, предпочитается, чтобы СТА, получаемая в соответствии с одним вариантом реализации настоящего изобретения, характеризовалась бы процентным пропусканием в области 400 нанометров (нм), равным, по меньшей мере, приблизительно 88 процентам, более предпочтительно, по меньшей мере, приблизительно 90 процентам, а наиболее предпочтительно, по меньшей мере, 92 процентам.
Испытание с определением процентного пропускания представляет собой способ измерения количества окрашенных светопоглощающих примесей, присутствующих в ТРА или СТА. В соответствии с использованием в настоящем документе испытание относится к измерениям, проводимым для части раствора, полученного в результате растворения 2,00 граммов сухих твердых ТРА или СТА в 20,0 миллилитрах диметилсульфоксида (ДМСО) аналитической или еще более высокой чистоты. После этого часть данного раствора помещают в полумикропроточную кювету Hellma, PN 176.700, которую изготавливают из кварца и которая характеризуется оптическим путем 1,0 см и объемом 0,39 миллилитра (компания Hellma USA, 80 Скайлайн-Драйв, Плейнвью, Нью-Йорк 11803). Для измерения пропускания при различных длинах волн света для данной заполненной проточной кюветы используют спектрофотометр Agilent 8453 Diode Array Spectrophotometer (компания Agilent Technologies, 395 Пэйдж-Милл-Роуд, Пало-Альто, Калифорния 94303). После надлежащей коррекции оптической плотности для учета поглощения фона, в том числе для использованных кюветы и растворителя, но не ограничиваясь только этим, результаты по процентному пропусканию, характеризующему долю падающего света, который проходит через раствор, машина выдает непосредственно. Значения процентного пропускания при длинах волн света в области 340 нанометров и 400 нанометров являются в особенности хорошо подходящими для использования при отделении вклада чистой ТРА от вклада множества примесей, обычно обнаруживаемых в ней.
Предпочтительные диапазоны для уровней содержания различных ароматических примесей в фазе суспензии (твердая фаза + жидкая фаза) реакционной среды представлены далее в Таблице 4.
Примеси в суспензии
Данные предпочтительные составы для суспензии представляют собой вариант реализации предпочтительного состава жидкой фазы реакционной среды при одновременных подходящих устранении экспериментальных трудностей, относящихся к осаждению дополнительных компонентов жидкой фазы из реакционной среды до получения компонентов твердой фазы во время отбора образцов из реакционной среды, разделению жидкой фазы и твердой фазы, и смещении условий к условиям анализа.
В фазе суспензии реакционной среды и в СТА реакционной среды обычно также присутствует и множество других ароматических примесей в общем случае при варьировании содержания на еще более низких уровнях и/или в пропорции от содержания одного или нескольких из описанных ароматических соединений. Регулирование уровней содержания описанных ароматических соединений при выдерживании в предпочтительных диапазонах будет обеспечивать выдерживание на подходящих уровнях содержания и других ароматических примесей. Данные выгодные составы для фазы суспензии в реакционной среде и для твердой СТА, отбираемой непосредственно из суспензии, становятся возможными в результате проведения операций при использовании вариантов реализации изобретения, описанных в настоящем документе для неполного окисления пара-ксилола до получения ТРА.
Измерение концентрации компонентов низкого уровня содержания в растворителе, растворителе, отправляемом на рецикл, СТА, суспензии из реакционной среды и РТА проводят при использовании методов жидкостной хроматографии. В данный момент будут описываться два взаимозаменяемых варианта реализации.
Метод, обозначаемый в настоящем документе как ЖХВД-ДМД, включает жидкостную хроматографию высокого давления (ЖХВД) в сочетании с диодно-матричным детектором (ДМД), что обеспечивает разделение и получение количественных характеристик для различных молекулярных соединений в заданном образце. Прибор, используемый при данном измерении, представляет собой модель 1100 HPLC, оборудованную устройством ДМД, поставляемую компанией Agilent Technologies (Пало-Альто, Калифорния), хотя коммерчески доступными являются также и другие подходящие приборы, приобретаемые и у других поставщиков. Как известно на современном уровне техники, как время элюирования, так и отклик детектора калибруют при использовании известных соединений, присутствующих в известных количествах, при этом соединения и количества являются соответствующими соединениям и количествам, присутствующим в фактических неизвестных образцах.
Метод, обозначаемый в настоящем документе как ЖХВД-MС, включает жидкостную хроматографию высокого давления (ЖХВД) в сочетании с масс-спектрометрией (МС), что обеспечивает разделение, идентификацию и получение количественных характеристик для различных молекулярных соединений в заданном образце. Приборы, используемые при данном измерении, представляют собой Alliance HPLC и ZQ MS, поставляемые компанией Waters Corp. (Милфорд, Массачусетс), хотя коммерчески доступными являются также и другие подходящие приборы, приобретаемые и у других поставщиков. Как известно на современном уровне техники, как время элюирования, так и отклик масс-спектрометра калибруют при использовании известных соединений, присутствующих в известных количествах, при этом соединения и количества являются соответствующими соединениям и количествам, присутствующим в фактических неизвестных образцах.
Еще один вариант реализации настоящего изобретения относится к неполному окислению ароматического окисляемого соединения при надлежащем установлении баланса между подавлением образования вредных ароматических примесей, с одной стороны, и получением диоксида углерода и монооксида углерода, совокупно называемых оксидами углерода (СОх), с другой. Данные оксиды углерода обычно покидают реакционную емкость в отходящем газе, и они соответствуют обусловленной деструкцией потере растворителя и окисляемого соединения, в том числе в конечном счете предпочтительных производных окисленных соединений (например, уксусной кислоты, пара-ксилола и ТРА). Изобретатели выявили нижние границы для образования оксидов углерода, ниже которых, как представляется, высокая эффективность образования вредных ароматических примесей, описанных далее, и низкий уровень совокупной степени превращения неизбежно становятся слишком неудовлетворительными для того, чтобы иметь привлекательность с точки зрения экономики. Изобретатели также выявили верхние границы для оксидов углерода, выше которых образование оксидов углерода продолжает нарастать при малой дополнительной ценности, обеспечиваемой уменьшением эффективности образования вредных ароматических примесей.
Изобретатели обнаружили то, что уменьшение концентраций в жидкой фазе реакционной среды для образуемого ароматическим окисляемым соединением исходного подаваемого материала и ароматических промежуточных соединений приводит к уменьшению скоростей образования вредных примесей во время неполного окисления ароматического окисляемого соединения. Данные вредные примеси включают сопряженные ароматические кольца и/или ароматические молекулы, имеющие большее, чем желательное, количество групп карбоновой кислоты (например, в случае окисления пара-ксилола вредные примеси включают 2,6-дикарбоксиантрахинон, 2,6-дикарбоксифлуоренон, тримеллитовую кислоту, 2,5,4'-трикарбоксибифенил и 2,5,4'-бензофенон). Ароматические промежуточные соединения включают ароматические соединения, происходящие из исходного подаваемого материала, образуемого окисляемым ароматическим соединением, и все еще сохраняющие неароматические гидрокарбильные группы (например, в случае окисления пара-ксилола ароматические промежуточные соединения включают пара-толуиловый альдегид, терефталевый альдегид, пара-толуиловую кислоту, 4-СВА, 4-гидроксиметилбензойную кислоту и альфа-бром-пара-толуиловую кислоту). Образуемый ароматическим окисляемым соединением исходный подаваемый материал и ароматические промежуточные соединения, сохраняющие неароматические гидрокарбильные группы, в случае их присутствия в жидкой фазе реакционной среды, по-видимому, приводят к получению вредных примесей по способу, подобному тому, что уже был описан в настоящем документе для растворенных ароматических соединений, не имеющих неароматических гидрокарбильных групп (например, изофталевой кислоты).
Будучи поставленными перед фактом данной потребности в повышенной активности в реакции для подавления образования вредных ароматических примесей во время неполного окисления окисляемого ароматического соединения, изобретатели обнаружили то, что нежелательным сопутствующим результатом является увеличение эффективности образования оксидов углерода. Важно осознавать то, что данные оксиды углерода представляют собой потерю выхода в отношении окисляемого соединения и окислителя, а не только растворителя. Очевидно, что существенная, а иногда и основная доля оксидов углерода своим происхождением имеет окисляемое соединение и его производные, а не растворитель; и зачастую на единицу углерода окисляемое соединение стоит больше, чем растворитель. Кроме того, важно осознавать то, что желательный продукт в виде карбоновой кислоты (например, ТРА) также подвержен избыточному окислению до получения оксидов углерода в случае его присутствия в жидкой фазе реакционной среды.
Также важно осознавать и то, что настоящее изобретение относится к реакциям в жидкой фазе реакционной среды и к концентрациям реагентов в ней. Это отличает его от некоторых изобретений предшествующего уровня техники, которые непосредственно относятся к получению ароматического соединения, сохраняющего неароматические гидрокарбильные группы, в форме осажденной твердой фазы. Говоря конкретно, в случае неполного окисления пара-ксилола до получения ТРА определенные изобретения предшествующего уровня техники относятся к количеству 4-СВА, осажденного в твердой фазе СТА. Однако для соотношения между количествами 4-СВА в твердой фазе и 4-СВА в жидкой фазе изобретатели настоящего изобретения обнаружили вариативность порядка более двух к одному при использовании тех же самых технических условий в отношении температуры, давления, катализа, состава растворителя и скорости реакции за один проход в единицу времени для пара-ксилола в зависимости от того, будут ли неполное окисление проводить в хорошо перемешиваемом автоклаве или в реакционной среде с разбиением на ступени в отношении кислорода и пара-ксилола в соответствии с настоящим изобретением. Кроме того, изобретатели наблюдали то, что соотношение между количествами 4-СВА в твердой фазе и 4-СВА в жидкой фазе также может варьироваться в области более двух к одному либо в хорошо перемешанной реакционной среде, либо в реакционной среде с разбиением на ступени в зависимости от скорости реакции за один проход в единицу времени для пара-ксилола при прочих подобных технических условиях в отношении температуры, давления, катализа и состава растворителя. В дополнение к этому 4-СВА в твердой фазе СТА, по-видимому, не вносит свой вклад в образование вредных примесей, и 4-СВА в твердой фазе можно без проблем и с высоким выходом извлечь и подвергнуть окислению до получения ТРА (например, в результате проведения окислительной утилизации для суспензии СТА, как это описывается в настоящем документе); в то время как удаление вредных примесей является намного более трудным и дорогостоящим в сопоставлении с удалением 4-СВА твердой фазы, а получение оксидов углерода соответствует необратимой потере выхода. Таким образом, важно осознавать то, что данный аспект настоящего изобретения относится к жидкофазным композициям в реакционной среде.
Изобретатели обнаружили то, что вне зависимости от того, будет ли источником являться растворитель или окисляемое соединение, при степенях превращения, обеспечивающих достижение привлекательности с точки зрения экономики, получение оксидов углерода тесно связано с уровнем общей активности в реакции, несмотря на наличие широкой вариативности для конкретной комбинации температуры, содержания металлов, галогенов, температуры, кислотности реакционной среды согласно измерению показателя рН, концентрации воды, используемых для достижения уровня общей активности в реакции. Изобретатели обнаружили то, что в случае неполного окисления ксилола полезно провести оценку уровня общей активности в реакции при использовании концентрации в жидкой фазе толуиловых кислот на середине высоты реакционной среды, в области низа реакционной среды и в области верха реакционной среды.
Таким образом, формируется важный одновременный баланс, позволяющий свести к минимуму образование вредных примесей в результате увеличения активности в реакции и вместе с тем свести к минимуму образование оксидов углерода в результате уменьшения активности в реакции. То есть, если общее образование оксидов углерода будет подавляться слишком мало, тогда будут формироваться избыточные уровни содержания вредных примесей, и наоборот.
Кроме того, изобретатели обнаружили то, что растворимость и относительная реакционная способность желательной карбоновой кислоты (например, ТРА) и присутствие других растворенных ароматических соединений, не имеющих неароматических гидрокарбильных групп, обеспечивают введение очень важной точки опоры рычага при достижении данного баланса между оксидами углерода и вредными примесями. Желательный продукт в виде карбоновой кислоты обычно растворяется в жидкой фазе реакционной среды даже и при наличии его также и в твердой форме. Например, при температурах в предпочтительных диапазонах ТРА является растворимой в реакционной среде, содержащей уксусную кислоту и воду, при уровнях содержания в диапазоне от приблизительно одной тысячи ч./млн (мас.) до более 1 массового процента, при этом по мере увеличения температуры растворимость увеличивается. Несмотря на существование различий в скоростях реакций, приводящих к образованию различных вредных примесей из образуемого окисляемым ароматическим соединением исходного подаваемого материала (например, пара-ксилола), из ароматических промежуточных соединений в реакции (например, пара-толуиловой кислоты), из желательного продукта в виде ароматической карбоновой кислоты (например, ТРА) и из ароматических соединений, не имеющих неароматических гидрокарбильных групп (например, изофталевой кислоты), наличие и реакционная способность двух последних групп определяет область понижения эффекта в том, что касается дополнительного подавления для первых двух групп - образуемого окисляемым ароматическим соединением исходного подаваемого материала и ароматических промежуточных соединений в реакции. Например, если в случае неполного окисления пара-ксилола до получения ТРА количество растворенной ТРА будет составлять 7000 ч./млн (мас.) в жидкой фазе реакционной среды при заданных условиях, количество растворенной бензойной кислоты будет составлять 8000 ч./млн (мас.), количество растворенной изофталевой кислоты будет составлять 6000 ч./млн (мас.), а количество растворенной фталевой кислоты будет составлять 2000 ч./млн (мас.), то тогда тенденция в направлении дополнительного уменьшения содержания совокупных вредных соединений начнет ослабляться по мере того, как активность в реакции будет увеличиваться, обеспечивая подавление концентрации в жидкой фазе для пара-толуиловой кислоты и 4-СВА до значений, меньших подобных уровней. То есть, присутствие и концентрация в жидкой фазе реакционной среды ароматических соединений, не имеющих неароматических гидрокарбильных групп, подвергаются очень незначительным изменениям в результате увеличения активности в реакции, и их присутствие служит расширению вверх области понижения эффекта при уменьшении концентрации промежуточных соединений в реакции в целях подавления образования вредных примесей.
Таким образом, один вариант реализации настоящего изобретения обеспечивает достижение предпочтительных диапазонов оксидов углерода (монооксида углерода и диоксида углерода), ограниченных с нижнего края низкой активностью в реакции и избыточным образованием вредных примесей, а с верхнего края - избыточными степенями потери углерода, но на уровнях, меньших в сопоставлении с ранее раскрытыми и описанными как коммерчески подходящие для использования. В соответствии с этим образование оксидов углерода предпочтительно регулируют следующим образом. Соотношение между количествами молей полученных совокупных оксидов углерода и количествами молей подаваемого окисляемого ароматического соединения предпочтительно составляет величину в диапазоне от приблизительно 0,02:1 до приблизительно 0,25:1, более предпочтительно в диапазоне от приблизительно 0,04:1 до приблизительно 0,22:1, еще более предпочтительно в диапазоне от приблизительно 0,05:1 до приблизительно 0,19:1, а наиболее предпочтительно в диапазоне от 0,06:1 до 0,15:1. Соотношение между количествами молей полученного диоксида углерода и количествами молей подаваемого окисляемого ароматического соединения предпочтительно составляет величину в диапазоне от приблизительно 0,01:1 до приблизительно 0,21:1, более предпочтительно в диапазоне от приблизительно 0,03:1 до приблизительно 0,19:1, еще более предпочтительно в диапазоне от приблизительно 0,04:1 до приблизительно 0,16:1, а наиболее предпочтительно в диапазоне от 0,05:1 до 0,11:1. Соотношение между количествами молей полученного монооксида углерода и количествами молей подаваемого окисляемого ароматического соединения предпочтительно составляет величину в диапазоне от приблизительно 0,005:1 до приблизительно 0,09:1, более предпочтительно в диапазоне от приблизительно 0,01:1 до приблизительно 0,07:1, еще более предпочтительно в диапазоне от приблизительно 0,015:1 до приблизительно 0,05:1, а наиболее предпочтительно в диапазоне от 0,02:1 до 0,04.
Уровень содержания диоксида углерода в сухом отходящем газе из реактора окисления предпочтительно составляет величину в диапазоне от приблизительно 0,1 до приблизительно 1,5 мольного процента, более предпочтительно в диапазоне от приблизительно 0,20 до приблизительно 1,2 мольного процента, еще более предпочтительно в диапазоне от приблизительно 0,25 до приблизительно 0,9 мольного процента, а наиболее предпочтительно в диапазоне от 0,30 до 0,8 мольного процента. Уровень содержания монооксида углерода в сухом отходящем газе из реактора окисления предпочтительно составляет величину в диапазоне от приблизительно 0,05 до приблизительно 0,6 мольного процента, более предпочтительно в диапазоне от приблизительно 0,10 до приблизительно 0,5 мольного процента, еще более предпочтительно в диапазоне от 0,15 до приблизительно 0,35 мольного процента, а наиболее предпочтительно в диапазоне от 0,18 до 0,28 мольного процента.
Изобретатели обнаружили то, что важным фактором при уменьшении эффективности образования оксидов углерода до данных предпочтительных диапазонов является улучшение степени чистоты отправляемого на рецикл фильтрата и исходного подаваемого материала, образуемого окисляемым соединением, за счет уменьшения концентрации ароматических соединений, не имеющих неароматических гидрокарбильных групп, в соответствии с описанием настоящего изобретения - это одновременно приводит к уменьшению эффективности образования оксидов углерода и вредных примесей. Еще одним фактором является улучшение распределения пара-ксилола и окислителя в реакционной емкости в соответствии с описанием настоящего изобретения. Другие факторы, делающие возможным достижение вышеупомянутых предпочтительных уровней содержания оксидов углерода, заключаются в функционировании при наличии градиентов в реакционной среде, описанных в настоящем документе в отношении давления, в отношении температуры, в отношении концентрации окисляемого соединения в жидкой фазе и в отношении окислителя в газовой фазе. Другие факторы, делающие возможным достижение вышеупомянутых предпочтительных уровней содержания оксидов углерода, заключаются в функционировании в рамках описания настоящего документа в отношении параметров, предпочтительных для скорости реакции за один проход в единицу времени, давления, температуры, состава растворителя, состава катализатора и геометрии механики реакционной емкости.
Одно важное преимущество, вытекающее из функционирования в пределах предпочтительных диапазонов образования оксидов углерода, заключается в том, что может быть уменьшено использование молекулярного кислорода, хотя и не до стехиометрических значений. Несмотря на хорошее разбиение на ступени в отношении окислителя и окисляемого соединения в соответствии с настоящим изобретением, избыток кислорода необходимо сохранять на уровне, превышающем стехиометрическое значение, рассчитываемое для одного только исходного подаваемого материала, образуемого окисляемым соединением, что делает возможными определенные потери в связи с оксидами углерода и обеспечивает наличие избыточного молекулярного кислорода, позволяющего регулировать образование вредных примесей. Говоря конкретно для случая, в котором исходный подаваемый материал, образуемый окисляемым соединением, представляет собой ксилол, соотношение в исходном подаваемом материале между массой молекулярного кислорода и массой ксилола предпочтительно составляет величину в диапазоне от приблизительно 0,9:1 до приблизительно 1,5:1, более предпочтительно в диапазоне от приблизительно 0,95:1 до приблизительно 1,3:1, а наиболее предпочтительно в диапазоне от 1:1 до 1,15:1. Говоря конкретно для исходного подаваемого материала, образуемого ксилолом, средневременный уровень содержания молекулярного кислорода в сухом отходящем газе из реактора окисления предпочтительно составляет величину в диапазоне от приблизительно 0,1 до приблизительно 6 мольных процентов, более предпочтительно в диапазоне от приблизительно 1 до приблизительно 2 мольных процентов, а наиболее предпочтительно в диапазоне от 1,5 до 3 мольных процентов.
Еще одно важное преимущество, вытекающее из функционирования в пределах предпочтительных диапазонов образования оксидов углерода, заключается в том, что в оксиды углерода и другие менее ценные формы превращается меньшее количество ароматического соединения. Данное преимущество оценивают при использовании суммы молей всех ароматических соединений, покидающих реакционную среду, поделенной на сумму молей всех ароматических соединений, поступающих в реакционную среду, в течение непрерывного периода времени продолжительностью предпочтительно в один час, более предпочтительно в один день, а наиболее предпочтительно в 30 последовательных дней. Данное соотношение здесь и далее в настоящем документе называют «молярной долей выживания» для ароматических соединений при прохождении через реакционную среду и выражают через численное процентное соотношение. Если все поступающие ароматические соединения будут покидать реакционную среду в виде ароматических соединений, хотя бы в основном и в окисленных формах поступающих ароматических соединений, то тогда молярная доля выживания будет иметь свое максимальное значение, равное 100 процентам. Если в точности 1 из каждых 100 поступающих ароматических молекул при прохождении через реакционную среду будет превращаться в оксиды углерода и/или другие неароматические молекулы (например, уксусную кислоту), то тогда молярная доля выживания будет равна 99 процентам. Говоря конкретно для случая, в котором основным исходным подаваемым материалом, образуемым окисляемым ароматическим соединением, является ксилол, молярная доля выживания для ароматических соединений при прохождении через реакционную среду предпочтительно составляет величину в диапазоне от приблизительно 98 до приблизительно 99,9 процента, более предпочтительно в диапазоне от приблизительно 98,5 до приблизительно 99,8 процента, а наиболее предпочтительно в диапазоне от 99,0 до 99,7 процента.
Еще один аспект настоящего изобретения включает получение метилацетата в реакционной среде, содержащей уксусную кислоту и одно или несколько окисляемых ароматических соединений. Данный метилацетат является относительно летучим соединением в сопоставлении с водой и уксусной кислотой, и, таким образом, он имеет тенденцию к следованию за отходящим газом, если только для его извлечения и/или деструкции перед высвобождением отходящего газа обратно в окружающую среду не будут использованы дополнительные операции в охлаждающей или другой установке. Таким образом, получение метилацетата осуществляют при наличии эксплуатационных расходов, а также капитальных затрат. Может оказаться так, что метилацетат будут получать в результате сначала объединения метильного радикала, может быть, образующегося в результате разложения уксусной кислоты, с кислородом до получения метилгидропероксида, в результате последующего разложения до получения метанола и, в конце концов, в результате проведения реакции между полученным метанолом и остаточной уксусной кислотой до получения метилацетата. Вне зависимости от пути химической реакции изобретатели обнаружили то, что всякий раз, когда получение метилацетата происходит при чрезмерно малой скорости, эффективность образования оксидов углерода будет также чрезмерно малой, а эффективность образования вредных ароматических примесей будет чрезмерно большой. Если получение метилацетата будет происходить при чрезмерно большой скорости, то тогда также излишне высоким будет и эффективность образования оксидов углерода, что приведет к потерям выхода в отношении растворителя, окисляемого соединения и окислителя. В случае использования предпочтительных вариантов реализации, описанных в настоящем документе, соотношение при получении между количествами молей полученного метилацетата и количествами молей подаваемого окисляемого ароматического соединения предпочтительно составляет величину в диапазоне от приблизительно 0,005:1 до приблизительно 0,09:1, более предпочтительно в диапазоне от приблизительно 0,01:1 до приблизительно 0,07:1, а наиболее предпочтительно в диапазоне от 0,02:1 до приблизительно 0,04:1.
Если образование диоксида углерода, монооксида углерода, их суммы и/или метилацетата будет соответствовать величинам, меньшим предпочтительных диапазонов, описанных в настоящем документе, или если молярная доля выживания для ароматических соединений будет соответствовать величинам, большим предпочтительных диапазонов, описанных в настоящем документе, то тогда необходимо будет увеличить активность в реакции, или необходимо будет уменьшить значение STR. Одним ускорителем для увеличения активности является повышенная температура в пределах предпочтительных диапазонов, описанных в настоящем документе. Другим ускорителем для увеличения активности является повышенная каталитическая активность, обеспечиваемая присутствием смеси каталитических реагентов и растворителя. В общем случае увеличение концентраций кобальта и/или брома будет играть роль ускорителя, увеличивающего активность в реакции, если их будут использовать в пределах диапазонов, предпочтительных в настоящем изобретении. Для ускорения при увеличении активности в реакции также можно использовать и регулирование в реакционной среде концентрации других компонентов катализатора и воды. Значение STR уменьшают в результате уменьшения скорости подачи окисляемого соединения и/или в результате увеличения объема реакционной среды.
Если образование диоксида углерода, монооксида углерода, их суммы и/или метилацетата будет соответствовать величинам, большим предпочтительных диапазонов, описанных в настоящем документе, и/или если молярная доля выживания для ароматических соединений будет соответствовать величинам, меньшим предпочтительных диапазонов, описанных в настоящем документе, то тогда предпочтительные действия по регулированию будут включать порядок действий, обратный по отношению к вышеописанному, опять-таки в пределах предпочтительных диапазонов, описанных в настоящем документе. Изобретатели отмечают то, что в особенности полезным является увеличение значения STR насколько возможно с вхождением в диапазоны настоящего документа при одновременном выдерживании хорошего качества окисления согласно измерению уровня содержания вредных примесей в СТА и в реакционной среде. Изобретатели опять-таки отмечают то, что трудно выдерживать данное качество окисления при таком высоком значении STR и что очень пристальное внимание следует уделить нижеследующему: диспергированию исходного подаваемого материала при поступлении в реакционную среду, качеству аэрирования во всем объеме реакционной среды, деаэрированию при покидании реакционной среды, значению STR по кислороду и количеству растворенного кислорода во всем объеме реакционной среды, избыточному количеству окислителя, покидающего реакционную смесь, желательному пространственному градиенту значения STR по кислороду, желательному пространственному градиенту концентрации окисляемого соединения, желательному пространственному градиенту концентрации окислителя, давлению верха реактора, желательному пространственному градиенту давления и предпочтительной температуре на середине высоты реакционной среды, в соответствии с тем, как все это описывается в настоящем документе. Кроме того, в дополнение к этому и с целью достижения пониженных количеств диоксида углерода, монооксида углерода и/или их суммы и/или с целью увеличения молярной доли выживания для ароматических соединений изобретатели обнаружили то, что подходящим для использования было бы подавление в реакционной среде концентрации растворимых ароматических соединений, не имеющих неароматических гидрокарбильных групп (например, изофталевой кислоты, фталевой кислоты и бензойной кислоты); данное подавление может быть осуществлено в результате использования более чистого исходного подаваемого материала, образуемого окисляемым соединением, и/или более чистого растворителя, в особенности в пределах предпочтительных диапазонов для каждого из них, что описывается в настоящем документе.
Предпочитается, чтобы в реакционной среде при непрерывном окислении пара-ксилола до получения терефталевой кислоты при предпочтительном значении STR, описанном в настоящем документе, количество пара-толуиловой кислоты в жидкой фазе реакционной среды выдерживали бы в диапазоне от приблизительно 200 до приблизительно 10000 ч./млн (мас.), более предпочтительно от приблизительно 800 до приблизительно 8000 ч./млн (мас.), а наиболее предпочтительно от 1600 до 6000 ч./млн (мас.). Кроме того, степень превращения пара-ксилола в терефталевую кислоту в реакционной среде предпочтительно выдерживают превышающей приблизительно 50 мольных процентов, более предпочтительно превышающей приблизительно 90 мольных процентов, еще более предпочтительно превышающей приблизительно 95 мольных процентов, а наиболее предпочтительно превышающей 97 мольных процентов.
В одном варианте реализации настоящего изобретения предпочитается, чтобы значения одного или нескольких рабочих параметров, описанных в настоящем документе (в том числе рабочих параметров, для которых приведены численные количественные характеристики), выдерживали бы в течение коммерчески значащего периода времени. Предпочтительно режим функционирования выдерживают в соответствии с одним или несколькими описанными выше рабочими параметрами в течение, по меньшей мере, приблизительно 1 часа, более предпочтительно, по меньшей мере, приблизительно 12 часов, еще более предпочтительно, по меньшей мере, приблизительно 36 часов, а наиболее предпочтительно, по меньшей мере, 96 часов. Таким образом, если только в настоящем документе не будет указано другого, то тогда рабочие параметры, описанные в настоящем документе, предполагаются для использования при стационарном, оптимальном/коммерческом режиме функционирования - не при запуске, остановке или не вполне оптимальном режиме функционирования.
Изобретатели отмечают то, что для всех численных диапазонов, приведенных в настоящем документе, верхние и нижние края диапазонов могут быть независимы друг от друга. Например, численный диапазон от 10 до 100 обозначает величины, большие чем 10 и/или меньшие чем 100. Таким образом, диапазон от 10 до 100 обеспечивает обоснование для ограничения пункта формулы изобретения величинами, большими чем 10 (не включая верхней границы), ограничения пункта формулы изобретения величинами, меньшими чем 100 (не включая нижнюю границу), а также величинами из всего диапазона от 10 до 100 (включая как верхнюю, так и нижнюю границы).
Определенные варианты реализации данного изобретения могут быть дополнительно проиллюстрированы при использовании следующих далее примеров, хотя необходимо понимать то, что данные примеры включаются просто для целей иллюстрации и не подразумевают ограничения объема изобретения, если только обратное не будет указано конкретно.
ПРИМЕРЫ
Примеры 1-4 относятся к полупромышленным окислениям пара-ксилола до получения терефталевой кислоты, проводимым в полупромышленной системе, скомпонованной вокруг реакционной емкости из титана объемом 2 галлона, снабженной механическим перемешивающим устройством и рубашкой с горячим маслом. Перемешивающее устройство, относящееся к газодиспергирующему типу, в реакционной емкости вращалось при скорости, равной приблизительно 1500 оборотов в минуту (об/мин), а приводная мощность на перемешивающем устройстве составляла приблизительно 220 Ватт. Полупромышленная система была оборудована средствами регулирования давления и температуры в реакционной емкости и регулирования расходов газа и жидкости, поступающих в реакционную емкость. Исходный подаваемый материал, образуемый пара-ксилолом, подавали с использованием шприцевого насоса при результирующем стационарном расходе, равном приблизительно 0,28 килограмма в час. Раствор исходного подаваемого материала, образуемого катализатором, закачивали из резервуара для исходного подаваемого материала, образуемого катализатором, в реакционную емкость при результирующем стационарном расходе, равном приблизительно 3,2 килограмма в час. Как пара-ксилол, так и раствор исходного подаваемого материала, образуемого катализатором, выпускали в реакционную среду через погружную трубу с концом, расположенным ниже уровня аэрированной суспензии в реакционной емкости. При использовании системы измерения уровня с применением радиоактивного датчика количество реакционной массы в реакторе выдерживали соответствующим указанному значению, составляющему приблизительно 40 процентов, в результате автоматического функционирования сливного клапана, расположенного поблизости от низа реактора. В результате калибрования установили, что данный указанный уровень соответствовал приблизительно 3 килограммам реакционной среды. Через трубу с концом, расположенным ниже уровня газодиспергирующей крыльчатки в реакционной емкости, по существу непрерывно подавали сжатый воздух. Линию отходящего газа из реакционной емкости оборудовали системой конденсатора, предназначенной для конденсации большей части органических паров из отходящего газа. Конденсат, полученный из отходящего газа, удаляли из технологического процесса при расходе, равном приблизительно 1,3 килограмма в час, а оставшееся количество конденсата, полученного из отходящего газа, возвращали в реакционную емкость. Расход при подаче воздуха регулировали для выдерживания концентрации кислорода в газе, покидающем реактор, в диапазоне приблизительно от 3 до 4 мольных процентов при расчете на сухое вещество после конденсатора для отходящего газа. Для газов, покидающих реактор, непрерывно проводили мониторинг по кислороду, диоксиду углерода и монооксиду углерода при использовании встроенных в технологическую линию газоанализаторов. Суспензию продукта, содержащую твердую фазу сырой неочищенной терефталевой кислоты (СТА), собирали в неаэрируемом перемешиваемом приемном резервуаре, из которого периодически проводили слив каждые четыре часа во второй неаэрируемый резервуар, в котором суспензию охлаждали до приблизительно 40°С для кристаллизации дополнительного количества растворенного продукта. Получающуюся в результате охлажденную суспензию фильтровали. Фильтрат собирали, взвешивали и анализировали по методу ЖХВД-МС на предмет выявления низких уровней содержания органических соединений, по методу рентгеновского анализа на предмет выявления металлов, по методу газовой хроматографии на предмет выявления метилацетата и ксилола и по методу спектроскопии в близком инфракрасном диапазоне на предмет выявления воды. Влажную твердую фазу взвешивали, а образец анализировали для определения уровня содержания влаги. Часть оставшейся твердой фазы промывали уксусной кислотой, высушивали и анализировали по методу ЖХВД-МС на предмет выявления конкретных анализируемых веществ. Конденсат реакционной смеси взвешивали и анализировали по методу газовой хроматографии, откалиброванному при использовании известных концентраций конкретных анализируемых веществ.
В каждом эксперименте по окислению раствор исходного подаваемого материала, образуемого катализатором, получали в перемешиваемом резервуаре для исходного подаваемого материала, образуемого катализатором. Раствор исходного подаваемого материала, образуемого катализатором, содержал ледяную уксусную кислоту и деионизованную воду. Кобальт в раствор исходного подаваемого материала, образуемого катализатором, добавляли в виде тетрагидрата ацетата двухвалентного кобальта, марганец добавляли в виде тетрагидрата ацетата двухвалентного марганца, а бром добавляли в виде водной 48-процентной бромисто-водородной кислоты. Количества каждого компонента в растворе исходного подаваемого материала, образуемого катализатором, выбирали в целях получения составов реакционных суспензий, продемонстрированных далее. Для запуска в реакционную емкость загружали раствор исходного подаваемого материала, образуемого катализатором, и ксилол, смесь доводили до температуры реакции при использовании нагревающей масляной рубашки и давления и концентрировали, проводя упаривание до приблизительно половины объема. В смесь вводили воздух, разбавленный азотом, до тех пор, пока не наблюдали возникновения тепловыделения. Как только начиналась реакция, в реакционную смесь подавали пара-ксилол и раствор исходного подаваемого материала, образуемого катализатором, при расходах, приведенных выше. Реакционную смесь выдерживали в данных условиях в течение приблизительно 8 часов до фиксации продукта. После этого продукт в виде суспензии собирали через интервалы продолжительностью приблизительно в 4 часа в течение всего эксперимента и проводили анализы, указанные выше. Величины для расходов, температуры и давления газа регистрировали каждые десять минут и усредняли для каждого эксперимента.
Для некоторых реакций, описанных далее, пара-ксилол марки «чистый для хроматографии (ЖХВД)» непосредственно после получения подвергали дополнительной очистке, проводя два цикла неполной кристаллизации при замораживании-оттаивании. Определенные по методу анализа при помощи газовой хроматографии уровни содержания нескольких примесей в исходном подаваемом материале, образуемом пара-ксилолом, продемонстрированы в Таблице 5.
Степень чистоты исходного подаваемого материала, образуемого пара-ксилолом
Примеры 1 и 2 демонстрируют то, что определенная часть бензойной кислоты (ВА) имеет своим происхождением примеси, обычно встречающиеся в загрязненном пара-ксилоле, и что определенная часть бензойной кислоты образуется из самого пара-ксилола в ходе неполного окисления даже в высокоперемешиваемой и высокоаэрированной реакционной среде. Данные для Примеров 1 и 2 представлены в Таблице 6.
Образование бензойной кислоты
Как можно видеть из Таблицы 6, соотношение между эффективностью образования ТРА и эффективностью результирующего образования бензойной кислоты в случае функционирования с использованием загрязненного пара-ксилола марки «чистый для ЖХВД» составляло только 720. Данное соотношение увеличивалось до 1180 для Примера 2 при использовании пара-ксилола, подвергнутого дополнительной очистке. Однако важно отметить, что определенное количество бензойной кислоты образуется при данных условиях проведения реакции, исходя из самой природы системы, даже в случае подачи пара-ксилола, подвергнутого дополнительной очистке, который по существу не содержит монозамещенных бензольных колец. То есть, образовывалась самообразующаяся бензойная кислота, может быть, в результате потери одного алкильного или ацильного заместителя в ходе окисления самого пара-ксилола. Кроме того, количество самообразующейся бензойной кислоты составляло приблизительно одну и ту же величину с точностью до ошибки эксперимента вне зависимости от того, использовали ли пара-ксилол марки «чистый для ЖХВД» или пара-ксилол, подвергнутый дополнительной очистке. То есть, данные в Таблице 2 демонстрируют то, что совокупное количество бензойной кислоты, полученной в случае пара-ксилола марки «чистый для ЖХВД», с точностью до ошибки эксперимента представляло собой сумму количеств самообразующейся бензойной кислоты плюс бензойной кислоты, образованной в результате окисления соответствующих нексилольных примесей, а именно толуола и этилбензола.
Если теперь продолжить обсуждение Примеров 3 и 4, то можно сказать, что они демонстрируют то, что увеличение концентрации бензойной кислоты в жидкой фазе для жидкой фазы реакционной среды приводит к увеличению скоростей нескольких нежелательных реакций в сопоставлении со скоростью получения ТРА. В Примере 4 концентрацию бензойной кислоты в жидкой фазе увеличивали в результате намеренного растворения желательного количества бензойной кислоты в растворе исходного подаваемого материала, образуемого катализатором. Данный раствор для Примера 4, содержащий уксусную кислоту, воду, кобальт, бром, марганец и бензойную кислоту марки «чистый для анализа», подвергали анализу по методу ЖХВД-МС на предмет выявления низких уровней содержания ароматических примесей, которые наиболее вероятно поступали в смесь совместно с приобретенной бензойной кислотой марки «чистый для анализа». Как для Примера 3, так и для Примера 4 результирующее образование ароматических примесей, приведенных в Таблице 7, рассчитывали в результате сложения массового расхода для каждой примеси во всех потоках, покидающих реакционную среду, а после этого вычитания массового расхода той же самой примеси во всех потоках, поступающих в реакционную среду. Для удобства скорости результирующего образования для примесей выражены через массу полученной примеси, поделенную на теоретическую массу полученной ТРА, рассчитанную в результате умножения массы исходного подаваемого материала, образуемого пара-ксилолом, на 166/106 - соотношение молекулярных масс ТРА и пара-ксилола. После этого данные значения умножают на один миллион для получения массового соотношения, выраженного через ч./млн (мас.).
Образование примесей
При сопоставлении результатов из Примеров 3 и 4 обратите внимание на то, что эффективность образования фталевой кислоты в присутствии бензойной кислоты в жидкой фазе реакционной среды отчетливо увеличивалась. Скорость образования фталевой кислоты в Примере 4 составляла приблизительно 189 ч./млн (мас.), при этом данное количество сопоставимо с количеством, образованным из примесного орто-ксилола, присутствующего в пара-ксилоле в пределах предпочтительных диапазонов, описанных в настоящем документе.
При дальнейшем сопоставлении результатов из Примеров 3 и 4 обратите внимание на то, что присутствие бензойной кислоты в жидкой фазе реакционной среды оказывало значительное влияние на образование одной окрашенной примеси - 9-флуоренон-2-карбоновой кислоты, но не оказывало влияния на образование другого окрашенного вещества - 2,7-дикарбоксифлуоренона. Обратите внимание также и на то, что эффективность образования 4,4'-дикарбоксибифенила увеличивалась на порядок величины в Примере 4 и что значительно увеличивался уровень содержания 4,4'-дикарбоксибензофенона.
Изобретатели также описывают то, что аналогичные результаты наблюдали при введении в жидкую фазу реакционной среды изофталевой кислоты или фталевой кислоты в других подобных экспериментах. Например, скорость образования тримеллитовой кислоты увеличивалась при увеличении концентрации либо изофталевой кислоты, либо фталевой кислоты. Например, уровни содержания конкретных типов полиароматических соединений, в том числе высокоокрашенного 2,7-дикарбоксифлуоренона, согласованно и пропорционально увеличивались при добавлении к жидкой фазе реакционной среды изофталевой кислоты.
При дальнейшем сопоставлении результатов из Примеров 3 и 4 в Примере 4 обратите внимание на существенно повышенное количество диоксида углерода, небольшое уменьшение количества монооксида углерода и значительный сдвиг соотношения между количествами данных двух газов, которые своим происхождением имеют как ароматические источники, так и уксусную кислоту. Из упрощенного рассмотрения следует, что общая потеря углерода на оксиды углерода в Примере 4 на 6 процентов превышала соответствующую потерю в Примере 3. Изобретатели также описывают то, что подобный результат в виде повышенной потери на оксиды углерода наблюдали и в других экспериментах, где в жидкую фазу реакционной среды вводили концентрации изофталевой кислоты или фталевой кислоты, а также в циклических экспериментах при использовании бензойной кислоты. Кроме того, обратите внимание на то, что концентрация пара-толуиловой кислоты в жидкой фазе реакционной среды в Примере 4 является более высокой, чем в Примере 3, что свидетельствует о несколько более низком уровне реакционной способности. Источник данного подавления реакционной способности неочевиден, исходя из концентраций компонентов катализатора или воды, не очевиден он и, исходя из температуры реакции или количества избыточного кислорода. Изобретатели также описывают то, что подобный результат в виде пониженной реакционной способности наблюдали и в других экспериментах, в которых в жидкую фазу реакционной среды вводили концентрации изофталевой кислоты или фталевой кислоты, а также в циклических экспериментах при использовании бензойной кислоты.
Таким образом, повышенная эффективность образования примесей, нежелательных в ТРА, потеря на оксиды углерода и подавление реакционной способности, по-видимому, являются результатом того, что в жидкой фазе реакционной среды при повышенных уровнях содержания присутствует множество ароматических соединений, не имеющих неароматических гидрокарбильных групп. Поэтому желательно регулировать количество указанных примесей в исходном подаваемом материале, образуемом пара-ксилолом, указанных примесей в растворителе, отправляемом на рецикл, и уровень самообразования указанных примесей.
Изобретение было подробно описано с конкретной ссылкой на его предпочтительные варианты реализации, но необходимо понимать то, что в рамках объема и сущности изобретения возможна реализация и его вариаций и модификации.
| название | год | авторы | номер документа |
|---|---|---|---|
| СИСТЕМА ОКИСЛЕНИЯ С ВНУТРЕННИМ ВТОРИЧНЫМ РЕАКТОРОМ | 2006 |
|
RU2448766C2 |
| СИСТЕМА ОКИСЛЕНИЯ, ИСПОЛЬЗУЮЩАЯ ВНУТРЕННЮЮ КОНСТРУКЦИЮ ДЛЯ УЛУЧШЕНИЯ ГИДРОДИНАМИКИ | 2006 |
|
RU2418629C2 |
| ОПТИМИЗИРОВАННОЕ ЖИДКОФАЗНОЕ ОКИСЛЕНИЕ | 2005 |
|
RU2388745C2 |
| ОПТИМИЗИРОВАННОЕ ЖИДКОФАЗНОЕ ОКИСЛЕНИЕ В БАРБОТАЖНОЙ КОЛОННЕ РЕАКТОРНОГО ТИПА | 2005 |
|
RU2363534C2 |
| ОПТИМИЗИРОВАННОЕ ЖИДКОФАЗНОЕ ОКИСЛЕНИЕ | 2005 |
|
RU2382759C2 |
| ОПТИМИЗИРОВАННОЕ ЖИДКОФАЗНОЕ ОКИСЛЕНИЕ | 2005 |
|
RU2382758C2 |
| ОПТИМИЗИРОВАННОЕ ЖИДКОФАЗНОЕ ОКИСЛЕНИЕ | 2005 |
|
RU2363535C2 |
| СОСТАВ СЫРОЙ ТЕРЕФТАЛЕВОЙ КИСЛОТЫ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2005 |
|
RU2388744C2 |
| ОПТИМИЗИРОВАННОЕ ЖИДКОФАЗНОЕ ОКИСЛЕНИЕ | 2005 |
|
RU2381212C2 |
| ОПТИМИЗИРОВАННОЕ ЖИДКОФАЗНОЕ ОКИСЛЕНИЕ | 2005 |
|
RU2388738C2 |



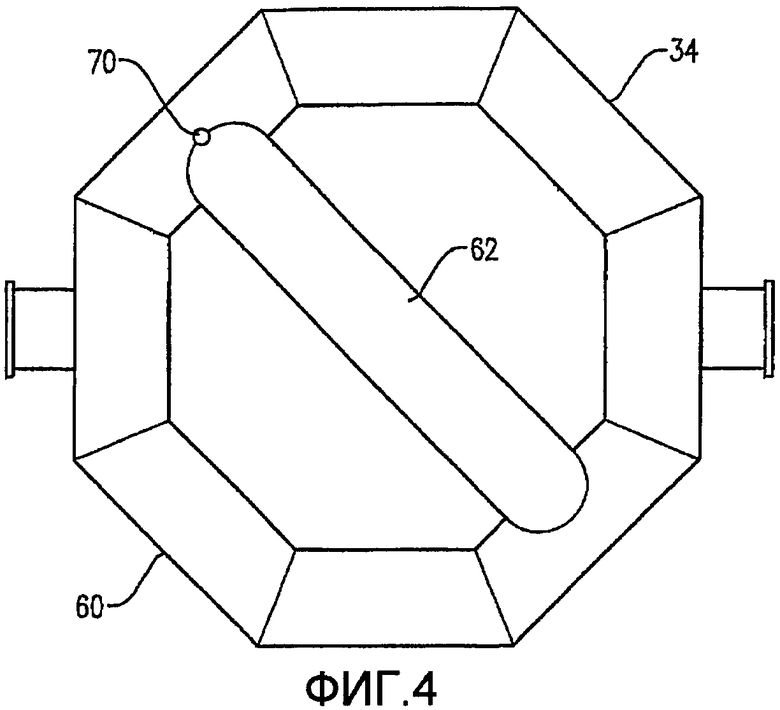


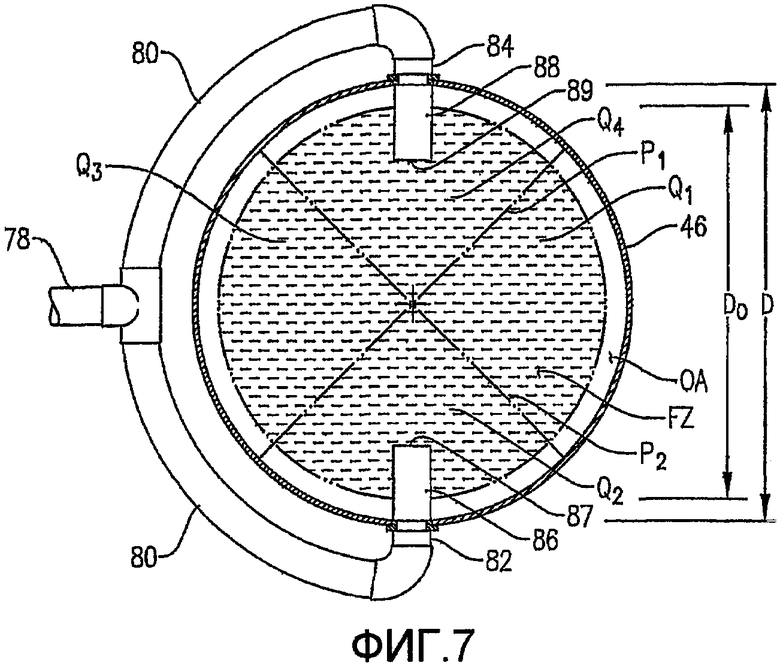





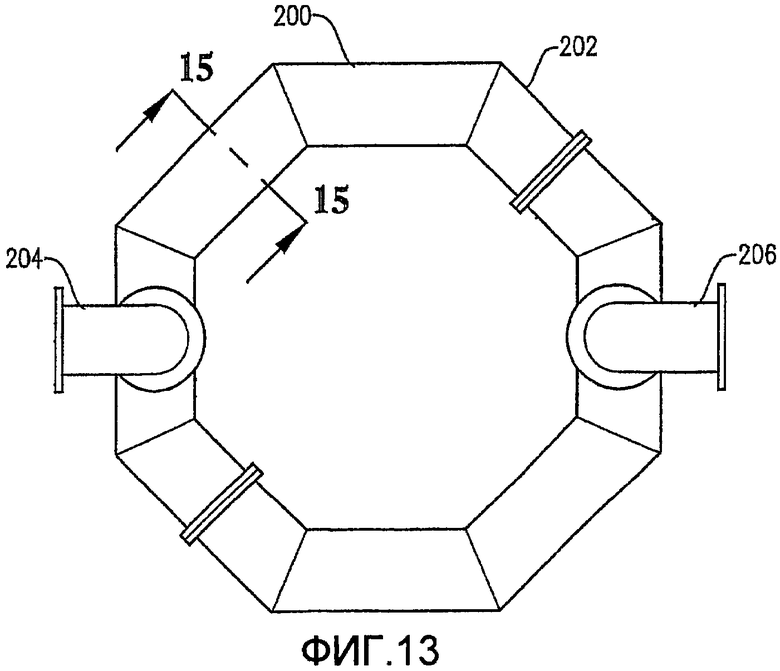


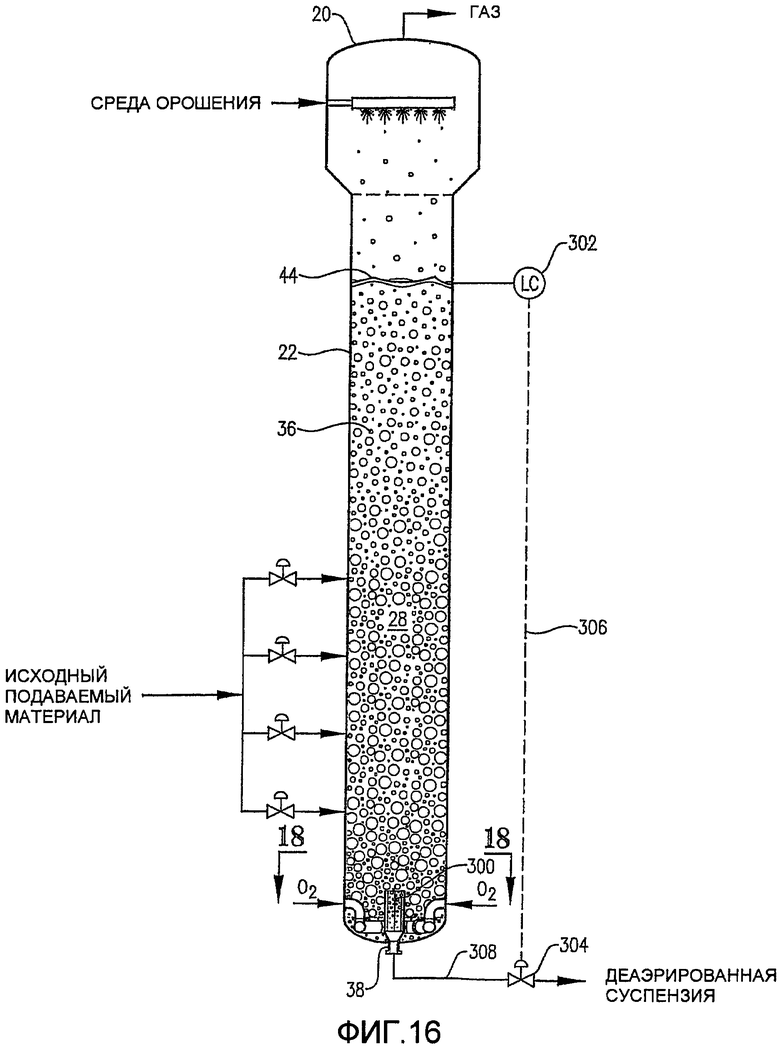

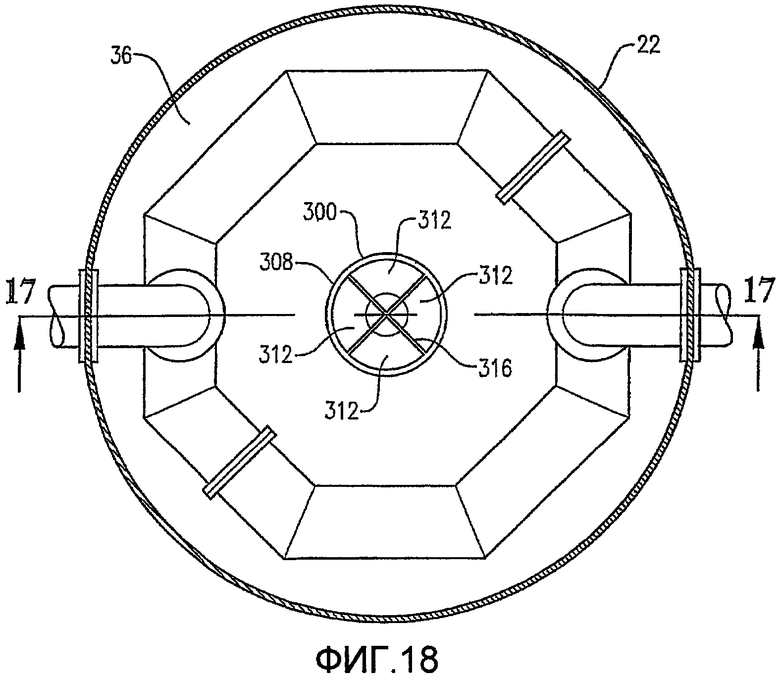




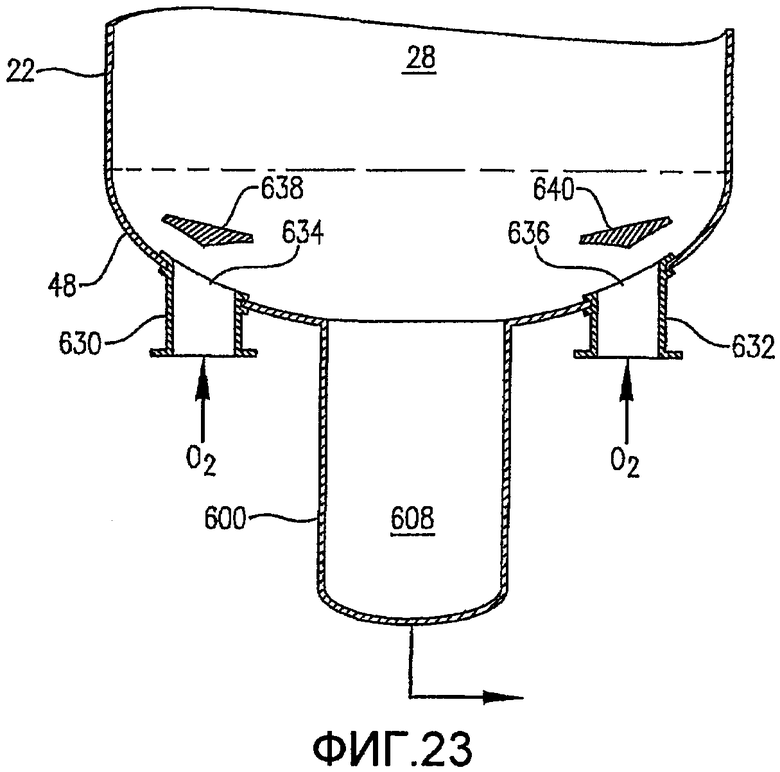

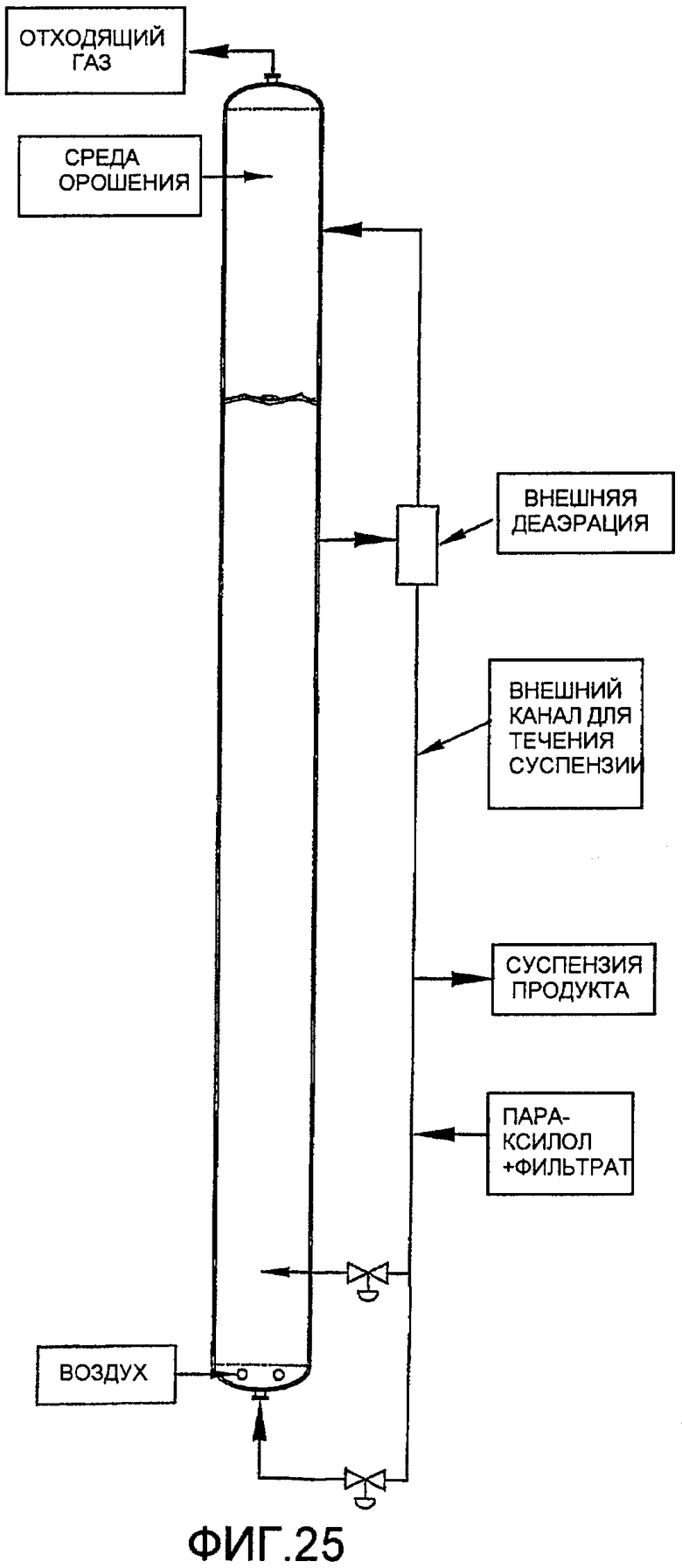












Изобретение относится к усовершенствованному непрерывному способу получения терефталевой кислоты, включающему (а) подачу пара-ксилола в реактор окисления; (b) окисление, по меньшей мере, части упомянутого пара-ксилола в жидкой фазе многофазной реакционной среды, содержащейся в упомянутом реакторе окисления, до получения таким образом сырой неочищенной терефталевой кислоты, где упомянутое окисление приводит к получению диоксида углерода, монооксида углерода и/или метилацетата; и выдерживание во время упомянутого окисления соотношения между молями полученных оксидов углерода и молями подаваемого упомянутого пара-ксилола в диапазоне от 0,02:1 до 0,24:1. Изобретение относится также к непрерывному способу получения терефталевой кислоты, включающему (а) подачу пара-ксилола в реактор окисления; (b) окисление, по меньшей мере, части упомянутого пара-ксилола в жидкой фазе многофазной реакционной среды, содержащейся в упомянутом реакторе окисления, до получения таким образом сырой неочищенной терефталевой кислоты; и (с) выдерживание во время упомянутого окисления молярной доли выживания упомянутого пара-ксилола в диапазоне от 99,0 до 99,7 процента. Способы предназначены для более эффективного и экономичного проведения жидкофазного окисления окисляемого соединения. 2 н. и 31 з.п. ф-лы, 35 ил., 7 табл.
1. Непрерывный способ получения терефталевой кислоты, включающий:
(a) подачу параксилола в реактор окисления;
(b) окисление, по меньшей мере, части упомянутого параксилола в жидкой фазе многофазной реакционной среды, содержащейся в упомянутом реакторе окисления, до получения, таким образом, сырой неочищенной терефталевой кислоты, где упомянутое окисление приводит к получению диоксида углерода, монооксида углерода и/или метилацетата; и выдерживание во время упомянутого окисления соотношения между молями полученных оксидов углерода и молями подаваемого упомянутого параксилола в диапазоне от 0,02:1 до 0,24:1.
2. Способ по п.1, где упомянутое соотношение выдерживают в течение, по меньшей мере, 12 ч.
3. Способ по п.1, где упомянутое соотношение между молями полученных оксидов углерода и молями подаваемого упомянутого ароматического соединения выдерживают в диапазоне от 0,05:1 до 0,19:1.
4. Способ по п.1, где упомянутое соотношение выдерживают в течение, по меньшей мере, 36 ч.
5. Способ по п.1, где упомянутое окисление приводит к превращению, по меньшей мере, 50 мол.% упомянутого ароматического соединения в терефталевую кислоту.
6. Способ по п.1, где упомянутое окисление приводит к образованию твердой фазы в упомянутой реакционной среде, по меньшей мере, из 10 мас.% упомянутого ароматического соединения.
7. Способ по п.1, где упомянутое окисление приводит к тепловыделению, где при помощи поверхностей теплообмена из упомянутой реакционной среды отводят менее чем 50% от теплоты, выделяющейся в результате прохождения упомянутого окисления.
8. Способ по п.1, где упомянутый способ включает подачу в упомянутый реактор окисления потока окислителя, где упомянутый поток окислителя содержит менее чем 40 мол.% молекулярного кислорода.
9. Способ по п.1, где упомянутым реактором окисления является барботажная реакторная колонна.
10. Способ по п.1, где упомянутая реакционная среда включает твердую, жидкую и газовую фазы.
11. Способ по п.1, где упомянутое окисление приводит к превращению, по меньшей мере, 90 мол.% упомянутого параксилола в терефталевую кислоту, где упомянутое окисление приводит к образованию твердой терефталевой кислоты в упомянутой реакционной среде, по меньшей мере, из 50 мас.% упомянутого параксилола, где упомянутое окисление приводит к тепловыделению, где при помощи поверхностей теплообмена из упомянутой реакционной среды отводят менее чем 30% от теплоты, выделяющейся в результате прохождения упомянутого окисления, где упомянутый способ включает подачу в упомянутый реактор потока окислителя, где упомянутый поток окислителя содержит молекулярный кислород в количестве в диапазоне от 15 до 30 мол.%.
12. Способ по п.1, где в жидкой фазе упомянутой реакционной среды концентрация паратолуиловой кислоты составляет величину, меньшую чем 8000 ч./млн (мас.).
13. Способ по п.1, где упомянутый способ дополнительно включает отбор суспензии из упомянутого реактора окисления, где упомянутая суспензия содержит жидкий маточный раствор и твердые частицы упомянутой сырой неочищенной терефталевой кислоты, где представительный образец упомянутой суспензии демонстрирует наличие одной или нескольких из следующих далее характеристик при расчете на объединенное количество твердых и жидких компонентов суспензии:
(v) содержание менее чем 1500 ч./млн (мас.) изофталевой кислоты (IPA),
(vi) содержание менее чем 500 ч./млн (мас.) фталевой кислоты (РА),
(vii) содержание менее чем 500 ч./млн (мас.) тримеллитовой кислоты (ТМА),
(viii) содержание менее чем 2000 ч./млн (мас.) бензойной кислоты (ВА).
14. Способ по п.1, где уровень содержания упомянутого диоксида углерода в сухом отходящем газе из упомянутого реактора окисления находится в диапазоне от 0,1 до 1,5 мол.%, где уровень содержания упомянутого монооксида углерода в сухом отходящем газе из упомянутого реактора окисления находится в диапазоне от 0,05 до 0,6 мол.%, где уровень содержания молекулярного кислорода в сухом отходящем газе из упомянутого реактора окисления находится в диапазоне от 0,1 до 6 мол.%.
15. Способ по п.1, где молярная доля выживания для параксилола при прохождении через упомянутую реакционную среду составляет величину в диапазоне от 98 до 99,9%.
16. Способ по п.1, где упомянутое окисление проводят в присутствии системы катализатора, содержащей кобальт.
17. Способ по п.7, где упомянутая система катализатора дополнительно содержит бром и марганец.
18. Способ по п.1, где существенная часть упомянутой сырой неочищенной терефталевой кислоты существует в виде твердых частиц, где представительный образец упомянутых частиц сырой неочищенной терефталевой кислоты демонстрирует наличие одной или нескольких из следующих далее характеристик:
(ix) содержание менее чем 12 ч./млн (мас.) 4,4-дикарбоксистильбена (4,4-DCS),
(х) содержание менее чем 800 ч./млн (мас.) изофталевой кислоты (IPA),
(xi) содержание менее чем 100 ч./млн (мас.) 2,6-дикарбоксифлуоренона (2,6-DCF),
(xii) процентное пропускание при 340 нм (%Т340) большее чем 25%.
19. Способ по п.1, где существенная часть упомянутой сырой неочищенной терефталевой кислоты существует в виде твердых частиц, где представительный образец упомянутых частиц сырой неочищенной терефталевой кислоты в течение одной минуты растворяется до достижения концентрации, равной, по меньшей мере, 500 ч./млн в ТГФ при проведении описанного в настоящем документе испытания на растворение во времени.
20. Способ по п.1, где существенная часть упомянутой сырой неочищенной терефталевой кислоты существует в виде твердых частиц, где представительный образец упомянутых частиц сырой неочищенной терефталевой кислоты характеризуется константой времени «С», большей чем 0,5 обратной минуты, согласно определению в описанной в настоящем документе модели растворения во времени.
21. Способ по п.1, где упомянутый способ дополнительно включает проведение, по меньшей мере, для части упомянутой сырой неочищенной терефталевой кислоты окисления в реакторе вторичного окисления.
22. Непрерывный способ получения терефталевой кислоты, включающий:
(a) подачу параксилола в реактор окисления;
(b) окисление, по меньшей мере, части упомянутого параксилола в жидкой фазе многофазной реакционной среды, содержащейся в упомянутом реакторе окисления, до получения, таким образом, сырой неочищенной терефталевой кислоты; и
(с) выдерживание во время упомянутого окисления молярной доли выживания упомянутого параксилола в диапазоне от 99,0 до 99,7%.
23. Способ по п.22, где упомянутое окисление приводит к превращению, по меньшей мере, 50 мол.% упомянутого ароматического соединения в терефталевую кислоту.
24. Способ по п.22, где упомянутое окисление приводит к образованию твердой фазы в упомянутой реакционной среде, по меньшей мере, из 10 мас.% упомянутого ароматического соединения.
25. Способ по п.22, где упомянутое окисление приводит к тепловыделению, где при помощи поверхностей теплообмена из упомянутой реакционной среды отводят менее чем 50% от теплоты, выделяющейся в результате прохождения упомянутого окисления.
26. Способ по п.22, где упомянутый способ включает подачу в упомянутый реактор окисления потока окислителя, где упомянутый поток окислителя содержит менее чем 40 мол.% молекулярного кислорода.
27. Способ по п.22, где упомянутую молярную долю выживания выдерживают в течение, по меньшей мере, 12 ч.
28. Способ по п.22, где упомянутым реактором окисления является барботажная реакторная колонна.
29. Способ по п.22, где упомянутая реакционная среда включает твердую, жидкую и газовую фазы.
30. Способ по п.22, где упомянутое окисление приводит к превращению, по меньшей мере, 90 мол.% упомянутого параксилола в терефталевую кислоту, где упомянутое окисление приводит к образованию твердой терефталевой кислоты в упомянутой реакционной среде, по меньшей мере, из 50 мас.% упомянутого параксилола, где упомянутое окисление приводит к тепловыделению, где при помощи поверхностей теплообмена из упомянутой реакционной среды отводят менее чем 30% от теплоты, выделяющейся в результате прохождения упомянутого окисления, где упомянутый способ включает подачу в упомянутый реактор потока окислителя, где упомянутый поток окислителя содержит молекулярный кислород в количестве в диапазоне от 15 до 30 мол.%, где упомянутую молярную долю выживания выдерживают в течение, по меньшей мере, 36 ч.
31. Способ по п.22, где в жидкой фазе упомянутой реакционной среды концентрация паратолуиловой кислоты составляет величину, меньшую чем 8000 ч./млн (мас.).
32. Способ по п.22, где упомянутое окисление проводят в присутствии системы катализатора, содержащей кобальт, бром и марганец.
33. Способ по п.22, где упомянутый способ дополнительно включает проведение, по меньшей мере, для части упомянутой сырой неочищенной терефталевой кислоты окисления в реакторе вторичного окисления.
| Штамп для горячего формирования металлов | 1983 |
|
SU1237298A1 |
| US 4158738 А, 19.06.1979 | |||
| US 5994567 А, 30.11.1999 | |||
| Способ очистки терефталевой кислоты | 1980 |
|
SU929627A1 |

