ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новому и усовершенствованному способу лечения легко передающихся эндобронхиальных инфекций сухими порошковыми препаратами на базе аминогликозидных антибиотиков, таких как тобрамицин.
ПРЕДПОСЫЛКИ ДЛЯ СОЗДАНИЯ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Кистозный фиброз (КФ) - наиболее распространенное генетическое заболевание в США и Северной Европе, ведущее к уменьшению продолжительности жизни, в США им болеет примерно 30000 человек (Cunningham, J.C. et al., "An Introduction to Cystic Fibrosis for Patients and Families" 5th ed., Bethesda: Cystic Fibrosis Foundation (2003)) и примерно столько же человек в Западной Европе. Генным нарушением в этом аутосомном рецессивном заболевании является мутация в гене КФ трансмембранного транспортного регулятора (CFTR), который зашифрован в белке хлорного канала (Collins, F.S., "Cystic Fibrosis Molecular Biology and Therapeutic Implications," Science 256: 774-779 (1992)). Люди с кистозным фиброзом обычно страдают от хронических эндобронхиальных инфекций, синуситов, мальабсорбции, вызванной панкреатической недостаточностью, повышенным выделением солей при потении, обструктивными гепатобилиарными заболеваниями и снижением фертильности. (FitzSimmons, S.С. "The Changing Epidemiology of Cystic Fibrosis," J Pediatr 122: 1-9 (1993)). Респираторные заболевания являются главной причиной заболеваемости и 90% смертей у людей с кистозным фиброзом (Cystic Fibrosis Foundation, Cystic Fibrosis Foundation Patient Registry 2003 Annual Data Report, Bethesda, MD: Cystic Fibrosis Foundation, (2004); Davis, P.B. et al., "Cystic fibrosis," Amer J. Respir Crit Care Med 154(5): 1229-56 (1996)). Легочная функция (измеряемая через объем воздуха при форсированном выдохе за 1 секунду (ОФВ1 должн., в %1)) является значимым показателем выживаемости при кистозном фиброзе. Двухлетняя выживаемость для группы людей с кистозным фиброзом уменьшается вдвое с каждым 10%-ным уменьшением ОФВ1 должн., а у людей с ОФВ1 на 30% ниже, чем ОФВ1 должн., двухлетняя выживаемость составляет меньше 50% (Kerem, E. et al., "Prediction of Mortality in Patients with Cystic Fibrosis," N Engl J Med 326: 1187-1191 (1992)). Коэффициенты снижения легочной функции неодинаковы как у разных людей, так и в течение жизни одного индивида. Ретроспективные лонгитюдные исследования показывают, что скорость ухудшения состояния варьируется в диапазоне от менее 2% от ОФВ1 должн. за год до более 9% от ОФВ1 должн. за год, с абсолютной скоростью ухудшения состояния, строго привязанной к возрасту на момент смерти (Corey, M. et al., "Longitudinal Analysis of Pulmonary Function Decline in Patients with Cystic Fibrosis," J Pediatr 131 (6): 809-1 (1997)).
Пациенты с кистозным фиброзом страдают от истончения слизистой, вызванного нарушением эпителиального транспорта ионов, что ведет к снижению защитной функции легких и приводит к повышению восприимчивости к ранним эндобронхиальным инфекциям с бактериями Staphylococcus aureus (Золотистый Стафилококк), Haemophilus influenzae (Гемофильная инфекция) и Р.Aeruginosa (Синегнойная Палочка). Большинство обследованных пациентов с диагнозом «кистозный фиброз» в период полового созревания уже имеют бактерию Р. Aeruginosa в мокроте. (Cystic Fibrosis Foundation Patient Registry 2003 Annual Data Report (2004)). Хронические эндобронхиальные инфекции, особенно синегнойные, провоцируют стойкие воспалительные реакции дыхательных путей, что ускоряет прогрессирующее обструктивное заболевание, отличительным признаком которого является диффузный бронхоэктаз (Davis, Р.В. et al. (1996), supra; Winnie, G.B. et al., "Respiratory Tract Colonization with Pseudomonas aeruginosa in Cystic Fibrosis: Correlations Between AxAi-P seudomonas aeruginosa Antibody Levels And Pulmonary Function," Pediatr Pulmonol 10:92-100 (1991); Ballman, M. et al. "Long Term Follow Up of Changes in FEV1 and Treatment Intensity During Pseudomonas Aeruginosa Colonisation in Patients with Cystic Fibrosis," Thorax 53:732-737 (1998); Pamukcu, A. et al., "Effects of Pseudomonas aeruginosa Colonization on Lung Function and Anthropometric Variables in Children with Cystic Fibrosis," Pediatr Pulmonol 19:10-15 (1995)). О связи между развитием хронической эндобронхиальной синегнойной инфекции, воспалением легких, снижением функции легких и окончательной смертью можно предположить, судя по значительному снижению выживаемости, сопутствующему хронической синегнойной инфекцией (Henry, R.L. et al., "Mucoid Pseudomonas aeruginosa is a Marker of Poor Survival in Cystic Fibrosis," Pediatr Pulmonol 12(3): 158-61 (1992)) и по значительным корреляциям между ранним развитием хронической синегнойной инфекции и детской смертности (Demko, CA. et al., "Gender Differences in Cystic Fibrosis: Pseudomonas aeruginosa Infection," J Clin Epidemiol 48:1041-1049 (1995)). Было доказано, что продолжительное применение терапии, подавляющее либо бактериальную флору в легких (MacLusky, LB. et al, "Long-term Effects of Inhaled Tobramycin in Patients with Cystic Fibrosis Colonized with Pseudomonas aeruginosa " Pediatr Pulmonol 7(1):42-8 (1989)), либо последующее воспаление легких (Konstan, M. W. et al., "Effect of high-dose Ibuprofen in Patients with Cystic Fibrosis," N Engl J Med 332(13): 848-54 (1995)), приводит к снижению скорости разрушения легких у зараженных пациентов.
Традиционно при эндобронхиальных синегнойных заболеваниях стандартной терапией служил прием парентеральных антисинегнойных антибиотиков, обычно включающих аминогликозиды, в течение 14-21 дня. Однако парентеральные аминогликозиды, будучи сильно полярными веществами, с трудом проникают в полость бронхов. Чтобы достичь адекватной концентрации лекарства в месте локализации инфекции при парентеральном введении, необходимо обеспечить такой уровень его содержания в сыворотке, при котором повышаются нефротоксичность, вестибулярная токсичность и ототоксичность ("American Academy of Otolaryngology. Guide for the evaluation of hearing handicap," JAMA 241(19):2055-9 (1979); Brummett, R.E., "Drug-induced ototoxicity," Drugs. 19:412-28 (1980)).
Ингаляционное введение аминогликозидов представляет собой привлекательную альтернативу, т.к. обеспечивает высокую концентрацию антибиотиков прямо в месте локализации инфекции в полости бронхов и минимизирует при этом систематическое бионакопление. (Touw, DJ. et al., "Inhalation of Antibiotics in' Cystic Fibrosis," Eur Respir J 8:1594-604 (1995); Rosenfeld, M. et al., "Aerosolized Antibiotics for Bacterial Lower Airway Infections: Principles, Efficacy, and Pitfalls," Clinical Pulmonary Medicine 4(2):101-12 (1997)).
На настоящий момент стандартным лекарством, применяемым для лечения синегнойных инфекций у пациентов с кистозным фиброзом, является ингаляционный раствор тобрамицина TOBI®, изготовляемый по удобной и стойкой формуле тобрамицина, не содержащей консервантов (раствор в пропорции 60 мг/мл тобрамицина в 5 миллилитрах 25%-го физиологического раствора), и предназначенный для введения через струйный небулайзер (распылитель), разработанный PathoGenesis Corporation, Seattle, Wash, (сейчас Chiron Corporation, Emeryville, Calif). Сочетание 5 мл дозы ТРИ (300 мг тобрамицина), принимаемой дважды в день, с компрессорной системой PARI LC PLUS/PulmoAide было одобрено федеральным агентством по лекарственным средствам (FDA) на основании заявки на новые средства (NDA 50-753, декабрь 1997) в качестве продолжительной дробной терапии для лечения синегнойной инфекции у пациентов с кистозным фиброзом и остается стандартом в этой области. Процесс ингаляции одной 300 мг дозы ТРИ (в такой дозировке ТРИ представлен в продаже) может занять 20 минут без учета времени, требующегося для установки и прочистки распылителя. Аэрозольное введение 5 мл дозы препарата, содержащего 300 мг тобрамицина в 25%-ном физиологическом растворе, для подавления синегнойной инфекции в полость бронхов больного описано в патенте США №5508269, который включен в полном объеме в описание настоящей заявки в качестве ссылки.
Тобрамицин - это аминогликозидный антибиотик, который продуцируется актиномицетами (Streptomyces tenebrarius). Низкие концентрации тобрамицина (<4 мкг/мл) эффективно подавляют рост многих грамнегативных бактерий и при определенных условиях могут обладать бактерицидным действием. Тобрамицин плохо абсорбируется поверхностями слизистых оболочек. Традиционно для тобрамицина требуется парентеральное введение. Кроме того, активность тобрамицина снижается при наличии гнойной мокроты: высокая концентрации двувалентных катионов, кислая среда, повышенная ионная сила, макромолекулы, обеспечивающие связь с лекарством, - все это вместе взятое препятствует действию тобрамицина. По оценкам исследователей, для преодоления этих сдерживающих эффектов необходимо в пять или десять раз увеличить концентрацию тобрамицина в мокроте (Levy, J. et al., "Bioactivity of Gentamicin in Purulent Sputum from Patients with Cystic Fibrosis or Bronchiectasis: Comparison with Activity in Serum," J Infect Dis 148(6): 1069-76 (1983)).
Эффективность доставки слабоабсорбируемого антибиотика тобрамицина в дыхательные пути пациентов с кистозным фиброзом путем впрыскивания аэрозоля была тщательно задокументирована. В значительной мере целью этой работы было лечение хронических легочных инфекций с Р. Aeruginosa (синегнойной палочкой) у пациентов с кистозным фиброзом. К примеру, многоцентровое, двойное слепое, плацебо-контролируемое, перекрестное исследование применения 600 мг аэрозольного тобрамицина (за три приема) для лечения эндобронхиальных инфекций Р.Aeruginosa (синегнойной палочкой), в котором принял участие 71 пациент с кистозным фиброзом, продемонстрировало значительное снижение в мокроте этого болезнетворного организма и одновременное улучшение результатов спирометрии в лечебной группе. Обнаружение следов Р.Aeruginosa (синегнойной палочки) с высокой устойчивостью к тобрамицину (определенной через минимальную ингибирующую концентрацию МИК>128 мкг/мл) сравнивалось для плацебо и лечебной групп. Присутствие в мокроте грамотрицательных бактерий, отличных от Р.Aeruginosa (синегнойной палочки) и устойчивых к тобрамицину, отмечалось с одинаковой частотой как при введении тобрамицина, так и при введении плацебо. (Ramsey, В. et al., "Response to Letter to the Editor: Aerosolized Tobramycin in Patients with Cystic Fibrosis," N Engl J Med 329: 1660 (1993)).
Хотя такая схема приема лекарственного средства и была признана в равной мере безопасной и эффективной, она является дорогой и неудобной. Анализ МИК для Р.aeruginosa, выделенных из начальных мокротных культур пациентов Children's Hospital CF Center, Seattle, Wash., в 1993, показал, что 90% выделений имеют МИК<16 мкг/мл и 98% всех выделений имеют МИК<128 мкг/мл. Этот осмотр предполагает, что достижение в мокроте концентраций тобрамицина в 128 мкг/мл должно позволить эффективно лечить эндобронхиальные заболевания у пациентов с кистозным фиброзом. (Levy, J. et al., "Bioactivity of Gentamicin in Purulent Sputum from Patients with Cystic Fibrosis or Bronchiectasis: Comparison with Activity in Serum," J Infect Dis 148(6); 1069-76 (1983)).
Рандомизированное перекрестное исследование сравнивало эффективность разных ингаляторов, доставлящих тобрамицин, путем измерения пиков в концентрации тобрамицина в образцах мокроты, полученных спустя десять минут после завершения введения дозы аэрозоля. В исследовании использовался раствор тобрамицина для ингаляции TOBI®, PathoGenesis Corporation, Seattle, Wash, (сейчас Chiron Corporation, Emeryville, Calif), содержащий тобрамицин в пропорции 60 мг/мл в 5 мл 25% физиологического раствора, с использованием струйного небулайзера PARI® LC, PARI Respiratory Equipment, Inc., Richmond, Va. Было показано, что данная система доставки лекарства дает среднее значение концентрации тобрамицина в мокроте при 678.8 мкг/г (s.d. 661.0 мкг/г) и медианное значение концентрации при 433.0 мкг/г. Только у 13% пациентов уровень концентрации в мокроте оказался меньше <128 мкг/г. У 87% больных концентрация составила >128 мкг/г (Eisenberg, J. et al., "A Comparison of Peak Sputum Tobramycin Concentration in Patients With Cystic Fibrosis Using Jet and Ultrasonic Nebulizer Systems. Aerosolized Tobramycin Study Group," Chest III (4):955-962 (1997)). Недавно струйный небулайзер PARI® LC был усовершенствован за счет клапана одностороннего действия и переименован в PARI® LC PLUS. Благодаря одностороннему клапану устройство PARI® LC PLUS характеризуется лучшей способностью доставки лекарства, нежели струйный небулайзер PARI® LC; односоронний клапан снижает вероятность случайной утечки и позволяет использовать фильтр выдоха. Опыт показал, что среднее значение концентрации тобрамицина в мокроте, достигаемое при использовании струйного небулайзера PARI LC PLUS, значительно выше, чем то же значение при использовании струйного небулайзера PARI®LC, что описано у Eisenberg и др. (1997, см. выше).
Далее, имеются два отчета о двух сопровождаемых плацебо-контролем, мультицентровых, рандомизированных, двойных слепых клинических исследованиях продолжительного применения жидкого аэрозоля тобрамицина путем вдыхания пациентами с кистозным фиброзом с синегнойной инфекцией, представленных в статье Ramsey, В.W. et al., "Intermittent Administration of Inhaled Tobramycin in Patients with Cystic Fibrosis. Cystic Fibrosis Inhaled Tobramycin Study Group." N. Engl. J. Med. 340(1):23-30 (1999). В этих исследованиях пятьсот двадцать субъектов случайным образом получали или 300 мг тобрамицина для ингаляции или плацебо дважды в день в течение 28 дней, с последующими 28 днями без изучаемого лекарства. Субъекты продолжали прием лекарства или плацебо в течение трех циклов «принятия/непринятия» в общей сложности 24 недели. Среди эффективных переменных было содержание Р.Aeruginosa в мокроте. Для пациентов, которых лечили тобрамицином, снижение концентрации Р.aeruginosa с нулевой недели по двадцатую составило в среднем 0,8 log 10. Для сравнения, пациенты, получавшие плацебо, имели прирост в 0,3 log 10 (Р<0,001). Для пациентов, которых лечили тобрамицином, снижение концентрации Р.aeruginosa с нулевой недели по четвертую составило в среднем 1,9 log10. Для сравнения пациенты, получавшие плацебо, не показали никаких изменений (Р<0,001).
В патенте США №6890907 и опубликованной в США заявке на патент 2003/0143162 А1 говорится о том, что пациенты, страдающие от эндобронхиальной инфекции, могут быть эффективно вылечены с помощью ингаляции пациентам дозы, составляющей 4,0 мл (или меньше) распыляемого жидкого аэрозоля, содержащего от примерно 60 до примерно 200 мг/мл аминогликозидного антибиотика, такого как тобрамицин, в физиологически приемлемом носителе, за время, меньшее чем примерно 10 минут. Более эффективное введение аминогликозидного препарата позволяет вводить меньшее количество жидкого аминогликозида по сравнению со стандартным режимом введения за меньшее время, что снижает стоимость лечения и расход лекарства. Более того, было показано, что препараты, содержащие минимальное и уже эффективное количество аминогликозида, заключенного в малом объеме физиологически приемлемого раствора, снижают раздражение легких после ингаляции аминогликозидного состава.
Вдобавок к вдыхаемым антибиотикам, таким как имеющийся в продаже продукт TOBI®, существует разнообразное число других терапий хронических заболеваний, регулярно назначаемых для снижения разрушительных циклов закупорки, заражения или воспаления в легких с КФ. Интенсивная Терапия Очистки Дыхательных путей (Reisman, JJ. et al. 5 "Role of conventional physiotherapy in cystic fibrosis," J Pediatr 113(4):632-6 (1988)), вдыхаемые бронхорасширители (Konig P et al., "Short-term and Long-term Effects of Albuterol Aerosol Therapy in Cystic Fibrosis: A Preliminary Report," Pediatr Pulmonol 20(4): 205-14 (1995)) и муколитические средства, такие как дорназа альфа (рекомбинантная человеческая дезоксирибонуклеаза I) (rhDNase; Fuchs, HJ. et al., "Effect of Aerosolized Recombinant Human DNase on Exacerbations of Respiratory Symptoms and on Pulmonary Function in Patients with Cystic Fibrosis. The Pulmozyme Study Group," N Engl J Med 331(10):637-42 (1994)) - их прописывают на постоянной основе, создавая потенциал для успешного лечения тех инфекций, которым подвержены люди с КФ. Было показано, что строгое соблюдение способа лечения является значительной проблемой для людей с КФ (Conway, S.P. et al., "Compliance with treatment in adult patients with cystic fibrosis," Thorax 51(1):29-33 (1996)) и что несоблюдение правил лечения может меняться в зависимости от специфики лечения (Abbott J et al., "Treatment Compliance in Adults with Cystic Fibrosis," Thorax 49(2): 115-20 (1994)).
Как сказано выше, коммерчески доступный жидкий аэрозоль раствора тобрамицин TOBI® для ингаляции доказал высокую эффективность при лечении синегнойной инфекции у пациентов с КФ. Учитывая необходимость соблюдения правил для обеспечения защиты легочной функции у пациентов с КФ, усовершенствования в существующих способах лечения, которые снижают время введения лекарства или делают для пациентов лечение более удобным, будут способствовать добросовестности пациента и выразятся в росте терапевтического эффекта. Согласно сказанному, существует потребность в новых и усовершенствованных способах и приспособлениях для введения пациенту аминогликозидного антибиотика при ингаляции, снижении сложности введения, увеличении добросовестности пациента и, в итоге, росте эффективности терапии в форме ингаляций.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение предлагает способ лечения больных с эндобронхиальной инфекцией, включающий прописывание больному сухого порошкового аэрозольного препарата, содержащего от 90 до 130 мг аминогликозидного антибиотика, для применения от одного до трех раз в день на первом этапе лечения от 20 до 36 дней. В практическом применении изобретения за первым лечебным этапом может следовать второй нелечебный этап, когда больному не вводятся никакие аминогликозидные антибиотики. При лечении вирулентных инфекций больному прописывается цикл, включающий первый лечебный этап с аминогликозидным лечением, и следующий за ним нелечебный этап, когда больному не вводятся никакие аминогликозидные антибиотики. Этот цикл может быть повторен два и большее число раз до тех пор, пока не будет достигнут желаемый бактериальный эффект. В случае хронических инфекций, таких как инфекции, возникающие у больных кистозным фиброзом, первый и второй лечебные этапы могут повторяться множество раз в ходе оказания медицинской помощи пациенту.
Способ лечения полезен для лечения любой эндобронхиальной инфекции, чувствительной к аминогликозидному антибиотику, такой как синегнойная эндобронхиальная инфекция, связанная с кистозным фиброзом.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Дальнейшие аспекты и многие имеющиеся преимущества настоящего изобретения будут оценены и в то же время лучше поняты при обращении к следующему детальному описанию, сопровождаемому чертежами, где изображено следующее.
Фиг.1 показывает среднее значение концентрации тобрамицина в сыворотке у субъектов в различные моменты после введения определенных доз ТПИ и ТРИ.
Фиг.2 показывает зависимости площади под кривой (ППК/AUC) (0,12) от дозировок тобрамицинового порошка для ингаляции (ТПИ) и тобрамицинового раствора для ингаляции (ТРИ).
Фиг.3 показывает зависимости ППК (0,∞) от дозировок ТПИ и ТРИ.
Фиг.4 показывает среднее значение концентрации тобрамицина в мокроте субъектов, принявших определенные дозы ТРИ и ТПИ.
Детальное описание технического результата
Если нет специального указания, все термины, используемые здесь, имеют те же значения, какие они будут иметь для любого профессионала в области настоящего изобретения. Здесь используются следующие сокращения:
Английская аббревиатура - (русская аббревиатура) - значение
АЕ - НЯ - нежелательное явление
ALT - АЛТ - аланиновая аминотрансфераза
AUC - ППК - площадь под кривой
BID - (-) - дважды в день
BUN-АМК азот мочевины крови
CaCl2 - (-) - хлорид кальция
CF - КФ - кистозный фиброз
CFC - ХФУ - хлорофторуглерод
Cmax - (-) - максимальная концентрация
CFTR - (-) - cystic fibrosis transmembrane conductance regulator (трансмембранный транспортный регулятор кистозного фиброза)
DPI - СПИ - сухой порошковый ингалятор
DSPC - (-) - 1,2-дистеароил-сн-глицеро-3-фосфохолин
FDA - (-) - United States Food and Drug Administration (Федеральное управление по лекарственным средствам)
FEV1 - ОФВ1 - объем форсированного выдоха за 1 секунду
FVC - ФЖЕЛ - форсированная жизненная емкость легких
PEV25-75 - СОС25-75 средняя скорость воздушного потока между 25 и 75% ФЖЕЛ
НРМС - ГПМЦ - 2-гидроксилпропилметилцеллюлоза
IRB - (-) - Institutional Review Board (Институтский наблюдательный совет)
IVRS - СИРО - Система Интерактивного Речевого Ответа
MedDRA - (-) - Medical Dictionary for Regulatory Activities (стандартизированная международная терминология)
MIC - МИК - минимальная ингибиторная концентрация
Р.aeruginosa - (-) - Pseudomonas aeruginosa (Синегнойная палочка)
PFOB - (-) - perfluorooctyl bromide (перфтороктил-бромид)
QPIT - (-) - quantitative pilocarpine iontophoresis test (количественный поликарпиновый ионтофорезный тест)
SAE - СНЯ - серьезные нежелательные явления
tmax - (-)- время достижения максимальной концентрации
TOBI® - ТРИ - 300 мг томбрамициновый раствор для ингаляции, корпорация Chiron, Эмервиль, Калифорния
TIP - ТПИ - Томбрамициновый порошок для ингаляции
С одной стороны, настоящее изобретение представляет собой способ лечения больных с эндобронхиальными инфекциями, включающий прописывание больному сухого порошкового аэрозольного препарата, содержащего от 90 до 130 мг аминогликозидного антибиотика, для применения от одного до трех раз в день на первом этапе лечения от 20 до 36 дней. В практическом применении изобретения за первым лечебным этапом может следовать второй нелечебный этап, когда больному не вводятся никакие аминогликозидные антибиотики. При лечении вирулентных инфекций больному прописывается цикл, включающий первый лечебный этап с аминогликозидным лечением, и следующий за ним нелечебный этап, когда больному не вводятся никакие аминогликозидные антибиотики. Этот цикл может быть повторен два и большее число раз до тех пор, пока не будет достигнут желаемый бактериальный эффект. В случае хронических инфекций, таких как инфекции, возникающие у больных кистозным фиброзом, первый и второй лечебные этапы могут повторяться множество раз в ходе оказания медицинской помощи пациенту.
С другой стороны, данное изобретение представляет собой использование аминогликозидного антибиотика в приготовлении лекарства для лечения больных с эндобронхиальными инфекциями, предписывая введение в эндобронхиальную систему пациента на первом лечебном этапе сухого порошкового аэрозольного препарата, содержащего от 90 до 130 мг аминогликозидного антибиотика, - от одного до трех раз в день на первом лечебном этапе от 20 до 36 дней. В практической реализации данной стороны изобретения, первый лечебный этап может быть аналогично продолжен вторым «нелечебным» этапом, когда больному не вводят никакие аминогликозидные антибиотики, кроме того, первый и второй этапы лечения могут быть повторены в соответствии с тем, как уже было описано.
Способ данной стороны изобретения включает этап назначения лекарства субъекту (человеку или животному), в случае необходимости его применения, вводящегося способом ингаляции в виде терапевтически эффективного количества аэрозольного порошка, содержащего от 20% по весу до 90% по весу антибиотика, где порошок имеет физиологически приемлемый состав, то есть содержит частицы, где по крайней мере 50% частиц имеют аэродинамический диаметр в пределах от 1 мкм до 5 мкм.
Термин «эндобронхиальная инфекция» используется в отношении бактериальной инфекции, локализованной в области бронхов. Примерами эндобронхиальных инфекций, с которыми можно бороться, используя настоящее изобретение, являются инфекции грамотрицательных бактерий, таких как Pseudomonas aeruginosa, Staphylococcus aureus, Haemophilus influenzae, Burkholderia cepacia, Stenotrophomonas maltophilia и Alcaligenes xiloxidants. Настоящее изобретение может быть использовано, к примеру, для лечения людей, страдающих от эндобронхиальных инфекций, связанных с кистозным фиброзом, таких как, к примеру, синегнойная инфекция.
Аминогликозидные антибиотики, полезные в применении изобретения, включают, к примеру, гентамицин, амикацин, канамицин, стрептомицин, неомицин, нетилмицин, парамеции и тобрамицин. Предпочтительным на данный момент аминогликозидным антибиотиком для использования в практическом применении данного изобретения является тобрамицин. Аминогликозидный антибиотик обычно прописывается в форме фармацевтически приемлемой соли (то есть сульфата, цитрата, аскорбата, глюконата, карбоната, тартрата, сукцината, ацетата или фосфата) или сложного эфира.
При применении данного препарата аэрозольный порошок вдыхается субъектом (человеком или животным) и таким образом проникает в легкие субъекта (человека или животного). Аэрозольный порошок состоит из частиц, содержащих аминогликозидный антибиотик. Было установлено, что аэрозольные порошки (содержащие аминогликозидный антибиотик), где по крайней мере 50% частиц имеют аэродинамический диаметр в диапазоне от 1 мкм до 5 мкм, эффективно проникают в легкие субъекта (человека или животного), тем самым эффективно доставляя аминогликозидный антибиотик в легкие субъекта. Например, некоторые аэрозольные порошки (содержащие аминогликозидный антибиотик) в практическом применении настоящего изобретения состоят из частиц, где по крайней мере 60% частиц, или по крайней мере 70% частиц, или по крайней мере 80% частиц, или по крайней мере 90% частиц, или по крайней мере 95% частиц имеют аэродинамический диаметр в диапазоне от 1 мкм до 5 мкм.
Термин «аэродинамический диаметр» относится к диаметру сферы единичной плотности, имеющей ту же конечную скорость осаждения, что и рассматриваемая частица (см. "Aerosol Measurement: Principles, Techniques and Applications". Edited by Klaus Willeke and Paul A.Baron. Van Nostrand Reinhold, New York, 1993). Аэродинамический диаметр используется, например, для предположения, где такая частица осядет в респираторном тракте.
«Срединный массовый аэродинамический диаметр» (аббревиатура СМАД) есть мера аэродинамического размера распыляемой частицы. Распределение по аэродинамическим размерам характеризует оседание аэрозоля при ингаляции и равно диаметру сферы единичной плотности, имеющей обычно ту же скорость осаждения в воздухе, что и частица. Когда наблюдается нормальное логарифмическое распределение по аэродинамическим размерам, то может быть определен срединный массовый аэродинамический диаметр (СМАД). В данном контексте СМАД относится к середине диапазона значений или к медиане по распределению размеров аэродинамических частиц в аэрозольном порошке, определяемым каскадными импакциями Андерсона.
Каскадные импакторы включают серии экранов с уменьшающимся размером пор. Экран задерживает частицы, движущиеся внутри струи, которые проходят через импактор. Количество отсеянного материала (частиц, имеющих размеры меньше нужного диапазона), которое задержано на каждом экране, может быть определено при смывании его с экрана и взвешивании смытого материала. Примеры каскадных импакторов и их использование описаны в 601 Главе (Аэрозоли) в Pharmacopoeia of the United States (26th Revision), упомянутая часть из которой приводится здесь со ссылкой.
Порошковые аминогликозидные препараты-антибиотики, полезные в практическом использовании настоящего изобретения, обычно содержат менее чем 15% по весу влаги, обычно ниже примерно 11% по весу влаги и предпочтительно менее примерно 8% по весу влаги.
В практическом применении данного изобретения пациенту, страдающему от эндобронхиальной инфекции, прописывается для ингаляций терапевтически эффективное количество аэрозольного порошка, содержащего аминогликозидный антибиотик. Терапевтически эффективное количество аэрозольного порошка содержит достаточно аминогликозидного антибиотика для полного или частичного препятствия росту болезнетворных бактерий в легких пациента. Например, выведены терапевтически эффективные дозы для аминогликозидного тобрамицина при предписании пациентам употреблять его от одного до трех раз ежедневно, предпочтительно употребление препарата дважды в день. Содержание тобрамицина в аэрозоли порошкового препарата должно составлять от примерно 90 до 130 мг, более предпочтительно от примерно 100 до 120 мг и наиболее предпочтительно от примерно 110 мг до примерно 115 мг (где вес определяется как вес свободного основания без учета возможно присутствующих противоионов).
Дозу принимаемого аминогликозида, такого как тобрамицин, можно вводить с помощью самостоятельного контейнера, содержащего разовую дозу, либо с помощью нескольких отдельных контейнеров, содержащих разовые дозы для последовательного введения в зависимости от типа устройства, используемого для доставки антибиотика. К примеру, вводимая доза аминогликозида может быть разделена на несколько разовых доз в количестве от двух до шести, более предпочтительно от трех до пяти доз и еще предпочтительнее на четыре дозы. В одном из репрезентативных результатов доза лекарства, содержащая 112 мг тобрамицина, (где вес определяется как вес свободного основания без учета возможно присутствующих противоионов) разделена на четыре отдельные №2 ГПМЦ капсулы с тобрамицином в виде свободного основания весом 27 мг на капсулу.
Сухие порошковые аэрозольные составы вводятся пациенту на первом этапе, длящемся от 20 до 36 дней, предпочтительнее от 26 до 30 дней и даже более предпочтительно около 28 дней. За первым лечебным этапом следует второй нелечебный этап, когда больному не вводятся никакие аминогликозидные антибиотики. Согласно одному из аспектов изобретения, второй нелечебный этап длится от примерно 20 до примерно 36 дней, предпочтительнее от примерно 26 до примерно 30 дней и наиболее предпочтительно около 28 дней.
В одном из репрезентативных результатов описанные в изобретении способы применяются для лечения пациентов с КФ, страдающих хронической синегнойной инфекцией. По данному аспекту изобретение предлагает лечение пациентов с КФ, страдающих от эндобронхиальной инфекции, согласно которому пациенту прописывают прием сухого порошкового аэрозоля, содержащего от 110 до 115 мг антибиотика тобрамицина, дважды в день на первом, 28-дневном, этапе лечения, с последующим нелечебным этапом от 26 до 30 дней, когда больному не вводятся никакие аминогликозидные антибиотики, а затем повторение первого и второго этапов лечения. Согласно данному аспекту изобретения, дозировка тобрамицина в 110-115 мг для последующего введения может быть разделена на разовые дозы в количестве от трех до пяти, предпочтительнее на четыре единичные дозы. Поскольку больные КФ склонны к заражению хронической синегнойной инфекцией, цикл лечения из двух этапов - первого лечебного этапа и следующего за ним второго нелечебного этапа - можно повторять множество раз на протяжении неопределенно долгого времени для долгосрочной терапии эндобронхиальных инфекций у пациентов с КФ.
Аэрозольный порошок обычно содержит от 20% (по весу) до 90% (по весу) аминогликозидного антибиотика. Соответственно, в некоторых реализациях данного изобретения, аэрозольный порошок содержит от 30% (по весу) до 80% (по весу) аминогликозидного антибиотика. В некоторых реализациях аэрозольный порошок содержит от 40% (по весу) до 70% (по весу) аминогликозидного антибиотика. В соответствии с этим, процентное содержание (по весу) аминогликозидного антибиотика соответствует количеству свободного антибиотика, без учета веса возможно присутствующих противоионов.
Аэрозольные порошки, обычно, но не обязательно, используемые в изобретении, включают, по крайней мере, один физиологически приемлемый носитель. Для примера, аэрозольный порошок может включать один или большее число добавок-наполнителей и/или любой другой компонент, который увеличивает эффективность аминогликозидного антибиотика. Такие добавки-наполнители могут просто выполнять функцию агентов-наполнителей, когда необходимо снизить концентрацию активного вещества в порошке, принимаемом пациентом. Также добавки могут служить улучшению диспергирования порошка в порошковом распыляющем устройстве с тем, чтобы обеспечить большую эффективность и воспроизводимость доставки активного вещества и улучшить физико-механические характеристики активных веществ (например, сыпучесть и прочность), обеспечивающие легкость изготовления и заправки порошков. В особенности, добавки-наполнители часто могут служить для улучшения физической и химической стабильности аминогликозида, минимизируя остаточную влажность и препятствуя ее росту, и увеличивая размер частицы, степень агрегации, поверхностные свойства (например, шероховатость), простоту ингаляции, и точность попадания нужных частиц в полость легких.
Фармацевтические наполнители и добавки, используемые в аминогликозидных составах, используются и при практическом применении данного изобретения. Они включают (но не ограничиваются только ими) следующие компоненты: протеины, пептиды, аминокислоты, липиды, полимеры и углеводороды (например, сахарозы, включая моносахариды, ди-, три-, терра- и олигосахариды; производные от сахарозы, такие как альдитолы, альдоновые кислоты, этерифицированные сахарозы; и полисахариды или сахарные полимеры), которые могут присутствовать как по отдельности, так и в различных сочетаниях. К примеру, протеиновые добавки включают сывороточный альбумин, такой как человеческий сывороточный альбумин (ЧСА), рекомбинантный человеческий альбумин (рЧА), желатин и казеин. Характерные аминокислотные/пептидные компоненты, которые также могут выполнять функцию буферной емкости, включают аланин, глицин, аргинин, бетаин, гистидин, глутаминовую кислоту, аспарагиновую кислоту, цистеин, лизин, лейцин, пролин, изолейцин, валин, метионин, фенилаланин и аспартам, хотя аргинин менее предпочтителен. Также подходят для использования в данном изобретении такие полиаминокислоты из типичных аминокислот, как ди-лейцин, и три-лейцин.
Углеводородные наполнители (карбогидраты), которые могут быть использованы в изобретении, включают, для примера, моносахариды, такие как фруктоза, мальтоза, галактоза, глюкоза, Д-манноза и сорбоза; дисахариды, такие как лактоза, сахароза, треголоза, целлобиоза; полисахариды, такие как раффиноза, мелезитоза, мальтодекстрины, декстрины и крахмал; и альдитолы, такие как маннитол, ксилитол, мальтитол, лактитол, ксилитол сорбитол (глюкитол) и мионозит.
Аминогликозидный состав может так же включать буфер или вещества, регулирующие рН; обычно в качестве буфера берется соль, приготовленная из органической кислоты или основания. Типичные буферы включают соли органических кислот, таких как соли лимонной кислоты, аскорбиновой кислоты, глюконовой кислоты, угольной кислоты, винной кислоты, янтарной кислоты, уксусной кислоты, фталевой кислоты; трис, трометанин гидрохлорид, и фосфатные буферы.
Кроме того, аминогликозидные смеси, полезные в практическом использовании настоящего изобретения, могут включать полимерные наполнители/добавки, такие как полифенилпирролидоны, гидроксипропилметилцеллюлозу, метилцеллюлозу, этилцеллюлозу, фиколл (полимерный сахар), декстран, декстраты (например, циклодекстрин, такой как дигидроксипропил-β-циклодекстрин, оксиэтиловый крахмал), полиэтиленгликоли, пектин, соли (например, хлорид натрия, т.е. поваренная соль), антиоксиданты, антистатические агенты, сурфактанты (например, полисорбаты, такие как "TWEEN 20" и "TWEEN 80", лецитин, олеиновая кислота, бензалконий хлорид и сложный эфир сорбитана), липиды (например, фосфолипиды, жирные кислоты), стероиды (например, холестерол) и хелатообразующие агенты (например, этилендиаминтетрауксусная кислота, ЭДТК). Другие примеры фармацевтических наполнителей и/или добавок, годящихся к использованию в аминогликозидных смесях, перечислены в статьях "Remington: The Science & Practice of Pharmacy" (Ремингтон: Наука и Практика Фармакологии), 19th ed., Williams & Williams, (1995), "Physician's Desk Reference", 52nd ed., Medical Economics, Montvale, NJ. (1998), данные из которых приведены здесь со ссылкой.
На данный момент наиболее предпочтительна комбинация наполнителей из лецитина и хлористого кальция (хлорида кальция). Лецитин - член фосфатидилхолиновой группы природных фосфолипидов, которые действуют как сурфактанты в легких млекопитающих (включая человека).
Аминогликозидные составы, полезные в практическом использовании настоящего изобретения, могут включать диспергирующие добавки для улучшения внутренних свойств диспергирования аминогликозидных порошков. Подходящие вещества описаны в заявках РСТ WO 95/31479, WO 96/32096 и WO 96/32149, приведенных здесь со ссылкой в полном объеме. Как в них указано, подходящие вещества включают растворимые водой полипептиды и гидрофобные аминокислоты, такие как триптофан, лейцин, фенилаланин и глицин. Согласно данному изобретению, лейцин и три-лейцин наиболее предпочтительны для использования.
Твердотельная форма, образованная аминогликозидом и наполнителем, придает стабильность окружению аминогликозида. Стабилизирующая форма может быть кристаллом, аморфным стеклом или смесью двух этих форм. Для аминогликозидных сухих порошковых препаратов, которые существенно аморфны, предпочтительна температура фазового перехода для стекла (Тс) выше примерно 35°С, предпочтительнее 45°С и еще более предпочтительно свыше 55°С. Желательно, чтобы температура была выше температуры хранения, по крайней мере, на 20°С. Согласно предпочтительному окружению, аминогликозидные смеси включают фосфолипид как твердотельную форму, что описано в WO 99/16419 и WO 01/85136 и приведено здесь со ссылкой в полном объеме.
Как сказано выше, сухие порошковые аминогликозидные смеси могут быть приготовлены путем распылительной сушки при таких условиях, которые ведут к образованию преимущественно аморфных стеклянных или преимущественно кристаллических биоактивных порошков. Распылительная сушка аминогликозидного растворенного препарата полностью разработана и, для примера, в целом описана в "Spray Drying Handbook," 5th ed., К. Masters, John Wiley & Sons, Inc., NY, NY (1991), и в WO 97/41833, данные из которых приведены здесь со ссылкой в полном объеме.
Для приготовления аминогликозидного раствора для распылительной сушки согласно одному из результатов изобретения, аминогликозид обычно растворяют в физиологически приемлемом растворителе, таком как вода. Диапазон значений рН в растворе, предназначенном для распылительной сушки, в основном поддерживается между 3 и 10, предпочтительнее между 5 и 8, желательно с приблизительно нейтральными рН, поскольку такие рН могут обеспечить физиологическую совместимость порошка после его распыления внутрь легких. Водный состав может опционально (необязательно, факультативно) содержать дополнительные легкосмешиваемые растворы, такие как алкоголь, ацетон и аналогичные. Аминогликозидные растворы, как правило, аминогликозид, растворенный при концентрации от 0,05% (вес/объем) до примерно 20% (вес/объем), обычно от 0,4% до 5,0% (вес/объем).
Затем раствор, содержащий аминогликозиды, сушится распылением при помощи обычного сушащего распылителя, например, при помощи распылителя, представленного коммерческими производителями Niro A/S (Дания), Buchi (Швейцария) или аналогичного, становясь стойким аминогликозидным сухим порошком. Оптимальные условия для сушки распылением аминогликозидного раствора изменяются в зависимости от состава препарата и определяются обычно экспериментально. Для сушки распылением, как правило, используют воздух, хотя также подходят инертные газы, такие как азот или аргон. Кроме того, температура газа на входе и выходе, установленная для сушки материала распылением, обычно определяется экспериментально, хотя в основном температура на входе будет меняться от примерно 50°С до 200°С в то время как температура на выходе будет меняться от примерно 30°С до примерно 150°С.
Аминогликозидные сухие порошки могут быть также приготовлены с помощью лиофильной сушки, вакуумной сушки, сублимационной порошковой сушки, процесса разделения с использованием сверхкритических флюидов и с помощью других форм испарительной сушки, а также перемешиванием, перетиранием или струйным перемалыванием до состояния сухого порошка. В некоторых случаях может быть желательно привести сухой порошковый аминогликозидный препарат к составу с улучшенными физико-технологическими характеристиками, например пониженной статикой, лучшей сыпучестью, слабой осаждаемостью и аналогичными, что предполагает создание смеси, состоящей из агрегатов тонких частиц, таких как агрегаты или агломераты частиц описанных выше аминогликозидных сухих порошков, где агрегаты легко разрушаются до нужного порошкового состава для доставки в легкие, как описано, например, в патенте США №5654007, приведенном здесь со ссылкой. С другой стороны, аминогликозидные порошки могут быть приготовлены путем агломерации компонент порошка, просеиванием материала для получения агломератов, сфероидизации для обеспечения сферичности агломератов и отсева по размеру для получения одноразмерного конечного продукта, как это описано в WO 95/09616, приведенном здесь со ссыпкой. Аминогликозидные сухие порошки предпочтительно держать в сухих условиях (то есть при относительно малой влажности) в процессе производства, технологической обработки и при хранении.
Согласно одному из результатов, типовой тобрамициновый препарат в форме порошка, полезный в практическом применении настоящего изобретения, может быть приготовлен с помощью способов эмульсификации/распылительной сушки, описанными в WO 99/16419 и WO 01/85136, процитированных выше. Согласно представленным в этих заявках результатам препараты из частиц, содержащих, по крайней мере, 75% вес/вес тобрамицина, предпочтительнее, по крайней мере, 85% вес/вес тобрамицина, 2-25% вес/вес фосфолипидов, предпочтительнее 8-18% вес/вес, и 0-5% вес/вес ионов металла, таких как хлористый кальций. Частицы в данной реализации обычно имеют СМАД от 1 микрона до 5 микрон и объемную плотность, большую, чем 0,08 г/см3, предпочтительнее больше, чем 0,12 г/см3.
Другой типовой тобрамициновый препарат в форме порошка, полезный в практическом применении данного изобретения, может быть изготовлен путем создания эмульсии, содержащей активный тобрамицин, 1,2-дистеароил-сн-глицеро-3-фосфохолин (DSPC), CaCl2 и перфтороктил-бромид (РFОВ). Затем эта подготовленная эмульсия распыляется с помощью атомизирующей насадки, дающей капли подходящего размера. По мере того как капли высыхают, вода и PFOB испаряются, давая сферические частицы на базе фосфолипидов с пористой структурой. Эти сферы малой плотности и поэтому показывают лучшие аэродинамические характеристики (сферические частицы имеют аэродинамический диаметр в диапазоне от 1 мкм до 5 мкм). Их высокая поверхностная пористость также снижает двучастичное взаимодействие, снижая энергию, необходимую для аэрозольного распыления.
Аэрозольный порошок (содержащий аминогликозидный антибиотик) может быть введен с помощью порошкового ингалятора, с использованием вдоха пациента (человека или животного) для доставки порошкового аминогликозидного препарата антибиотика в легкие. Примером полезного сухого порошкового ингалятора является Т-326 ингалятор (Nektar Therapeutics, 150 Industrial Road, San Carlos, CA 94070, U.S.A.). Другими примерами полезных сухих порошковых ингаляторных устройств являются устройства, описанные в патентах США №№5458135; 5740794; 5775320 и 5785049, каждый из которых приведен здесь со ссылкой. При введении с использованием устройства данного типа порошковое лекарство размещается в контейнере, имеющем прокалываемую крышку или другую поверхность доступа, предпочтительно блистерную упаковку или картридж, где контейнер может содержать одну разовую дозу или несколько разовых доз. Типовые способы по заполнению большого числа емкостей отмеренными дозами сухого порошкового лекарства описаны в патенте США №5826633, приведенном здесь со ссылкой.
Также подходят для доставки аминогликозидных порошков описанные здесь же сухие порошковые ингаляторы, описание которых содержится, к примеру, в патентах США №№3906950, 4013075, 4069819 и 4995385, каждый из которых приведен здесь со ссылкой, где прописанная для введения субъекту доза аминогликозидного сухого порошка содержится в капсуле, такой как твердая желатиновая капсула. Размер капсулы (речь идет о размерах капсул 00, 0, 1, 2 или 3) зависит, кроме прочих факторов, от ингаляционного устройства, используемого для введения порошков.
Другие распылители сухого порошка для введения в легкие аминогликозидных сухих порошков включают описанные выше, например, в европейских патентах №№ЕР 129985, ЕР 472598, ЕР 467172 и в патенте США №5522385, где каждый из патентов приведен здесь со ссылкой. Для доставки аминогликозидных сухих порошков изобретения также подходят ингаляционные устройства, такие как Astra-Draco "TURBUHALER". Этот тип устройства детально описан в патентах США №№4668218, 4667668 и 4805811, каждый из которых приведен здесь со ссылкой.
Также подходят устройства, которые используют пистон для подачи воздуха либо для подсасывания порошкового медикамента, поднятия медикамента по транспортному решету путем продувания воздуха через решето, либо для смешивания воздуха с порошковым медикаментом в смешивающей камере с последующим введением порошка пациенту через горлышко устройства так, как описано в патенте США №5388572, приведенном здесь со ссылкой.
При дальнейшем изложении будет учитываться тот факт, что терапевтически эффективное количество аэрозольного порошка может быть введено из одного контейнера или из контейнеров числом более одного, помещенных в сухой порошковый ингалятор. К примеру, в сухой порошковый ингалятор может быть помещен отдельный контейнер, содержащий терапевтически эффективное количество аэрозольного порошка (включающего аминогликозидный антибиотик);
содержимое контейнера вдыхается субъектом (человеком или животным). Опять же в качестве примера можно отметить, что в сухой порошковый ингалятор могут быть помещены отдельные контейнеры с разовыми дозами (то есть 2, 3 или 4 контейнера), такие как #2 ГПМЦ капсулы, которые по отдельности содержат недостаточно эффективное количество аэрозольного порошка (содержащего аминогликозидный антибиотик), но которые вместе содержат терапевтически эффективное количество аэрозольного порошка. Ингалятор сухого порошка высвобождает содержимое всех контейнеров, содержащееся внутри, и тем самым обеспечивает потребителя терапевтически эффективным количеством аэрозольного порошка.
Аминогликозидный лечебный курс, предусмотренный настоящим изобретением, может быть использован сам по себе или в сочетании с другим или другими дополнительными веществами для лечения эндобронхиальных инфекций, в особенности синегнойных инфекций. В отношении данного аспекта изобретения одно или большее число дополнительных веществ для лечения эндобронхиальных инфекций могут быть введены в течение первого этапа аминогликозидного лечения или в течение второго нелечебного этапа, когда пациенту не вводятся никакие аминогликозидные антибиотики, либо в течение как первого так и второго этапов. При одной из реализации данного подхода в изобретении пациенту в течение второго нелечебного этапа, когда он не должен принимать аминогликозидные антибиотики, выписывалось одно или больше дополнительных веществ для лечения эндобронхиальных инфекций. Подходящие дополнительные вещества для лечения эндобронхиальных инфекций включают, для примера, неаминогликозидные антимикробные вещества, такие как монобактам, бета-лактам, макролид, фторхинолоны и/или гликопептидные антибиотические составы. К примеру, неаминогликозидным антимикробным веществом может быть азтреонам.
Эмитированная доза (ЭД) порошкообразного аминогликозидного препарата-антибиотика обычно должна быть больше, чем 50%. Более желательна полезная в практическом применении данного изобретения ЭД порошкового аминогликозидного препарата антибиотика, большая 70%, а чаще большая 80%. В данном контексте термин «эмитированная доза» или «ЭД» относится к индикации доставки сухого порошка из подходящего ингаляционного устройства после поджигания или распыления из единицы порошка (капсулы или резервуара). ЭД определяется как отношение доставленной ингаляционным устройством дозы к величине номинальной дозы (то есть масса порошка на единицу дозы, размещенной в подходящем ингаляторе, перед использованием). ЭД - экспериментально определяемая величина и обычно определяется в искусственных лабораторных условиях, которые симулируют введение дозы пациенту. Чтобы определить значение ЭД, номинальная доза сухого порошка (определенная как сказано выше) помещается в подходящем сухом порошковом ингаляторе, который затем активизируют для распыления порошка. Затем полученное облако аэрозоля всасывается в вакуум из устройства, где ловится на тарированный фильтр, присоединенный к отверстию. Количество порошка, достигшее фильтра, составляет доставленную дозу. Возьмем, к примеру, капсулу №2 весом 50 мг, содержащую сухой порошок и помещенную в ингалятор. Если после распыления порошка способом, описанным выше, на фильтре получается 50 мг порошка, тогда ЭД для сухого порошковой смеси составляет 80%: 40 мг (доставленная доза) / 50 мг (номинальная доза) × 100=80%.
По другим аспектам предметом изобретения является устройство для использования при лечении эндобронхиальных инфекций, при этом устройство содержат одну или большее число доз от 90 до 130 мг сухого порошкового аминогликозидного антибиотика вместе с инструкцией по введению дозировки больному с помощью сухого порошкового ингалятора от одного до трех раз в день на первом лечебном этапе от 20 до 36 дней. Согласно данному аспекту, изобретения должны содержать инструкции с рекомендацией, где будет указано, что первый лечебный этап может быть продолжен вторым «нелечебным» этапом, когда пациенту не вводятся никакие аминогликозидные антибиотики, и что цикл из первого лечебного этапа с антигликозидным лечением с последующим вторым «нелечебным» этапом, когда пациенту не вводятся никакие аминогликозидные антибиотики, может быть повторен два и большее число раз по достижении желаемого антибактериального эффекта. В устройства по настоящему изобретению одна и большее число доз могут содержаться в одном контейнере, как единичная разовая доза, или они могут быть разделены по нескольким контейнерам с разовыми дозами для последовательного введения, в зависимости от ингаляционного устройства, используемого для подачи антибиотика. К примеру, вводимая доза аминогликозидного антибиотика может быть разделена на несколько разовых доз в количестве от двух до шести, более предпочтительно от трех до пяти единиц доз и даже более предпочтительно на четыре единицы доз. В одном из репрезентативных результатов изобретения в комплект устройства входят дозы для введения от 112 мг тобрамицина (определяемого как чистый вес, без учета веса возможно присутствующих противоионов), помещенного в четыре отдельные #2 ГПМЦ капсулы с весом в 50 мг (27 мг чистого тобрамицина) на капсулу.
Следующие примеры просто иллюстрируют наиболее удачную модель практического применения изобретения, что, однако, не предполагает ограничения настоящего изобретения только такой моделью применения.
Пример 1
Приготовление Порошка Тобрамицина для Ингаляции (ПТИ)
Сухой порошок сульфат тобрамицина приготовляется согласно следующей процедуре. Стерильная вода для орошения (SWFI) нагревается на геле до температуры жидкого кристалла (свыше 80 С) дистеароилфосфатидилхолина (DSPC). Затем в разогретую воду добавляются DSPC и дигидрат хлорида кальция (CaCl2 + 2H2O). Полученная смесь липидов перемешивается в UltraTurrax T-50 (IKA Labortechnik) при 8000 об/мин в течение 5 минут. Затем капельно (15 мл/мин) добавляется перфтороктил-бромид (PFOB) в липидную смесь при перемешивании. После окончания добавления полученная эмульсия PFOB перемешивается в воде дополнительные 10 минут со скоростью вращения 10000 об/мин. Эмульсификация в UltraTurrax дает капли размером в микронном диапазоне. Затем сульфат тобрамицина растворяется в непрерывной фазе эмульсии и полученная смесь используется как материал для распылительной сушки. Материал сушится распылением, давая сухой порошковый препарат, имеющий состав, приведенный далее в Таблице 1.
Порошок помещен в капсульную заправочную станцию при относительной влажности в 10-15% и с возможностью прийти в равновесие в течение 10 минут, а затем упакован в #2 ГПМЦ капсулы с весом в 50 мг (27 мг чистого тобрамицина) на капсулу.
Пример 2
Этот пример описывает клиническое исследование, которое демонстрирует, что однодозовое введение тобрамицина с сухим порошковым препаратом по изобретению приводит к более эффективной доставке тобрамицина, чем введение тобрамицина с раствором, при поддержании одинаковых фармокинетических характеристик тобрамицина.
Цикловый хронометраж и план исследования
Исследование было сформулировано как рандомизированное, открытое, проспективное когортное, активно конролируемое, однодозовое, с увеличением дозы клиническое исследование. В каждой последовательной когорте субъекты исследования были рандомизированы в отношении 3:1 для получения или одной дозы Тобрамицинового Порошка для Ингаляции (ТПИ), вводимой с использованием ингалятора Т-326 (Nektar Therapeutics, San Carlos, CA, USA), согласно графику применения, описанному ниже, или же одной дозы в 300 мг Раствора Тобрамицина для Ингаляции, распыляемой с помощью струйного небулайзера PARI LC PLUS™ с компрессором DeVilbiss PulmoAide™. Каждый субъект исследования мог принадлежать исключительно к одной когорте.
Эскалация до следующего дозового уровня ТПИ производилась после рассмотрения Комитетом Мониторинга Данных (КМД) всех вызванных лечением нежелательных явлений (НЯ) и других данных по безопасности полной когорты или в том случае, если не выполнялся ни один из следующих критериев: один или больше субъектов когорты, подвергнутой лечению с помощью ТПИ, испытали по меньшей мере относительное двадцатипроцентное падение значения ОФВ1 в течение 30 минут после окончания дозирования; хотя бы один субъект, принимавший ТПИ, испытал относящиеся к изучаемому лекарству серьезные нежелательные явления (СНЯ).
Экспериментальное лечение
Разовая доза ТПИ вводилась с помощью Ингалятора Т-326:
Когорта 1: Две капсулы ТПИ (14 мг тобрамицина в чистом виде на капсулу («дозировка»))
Когорта 2: Четыре капсулы ТПИ (дозировка 14 мг)
Когорта 3: Две капсулы ТПИ (дозировка 28 мг)
Когорта 4: Четыре капсулы ТПИ (дозировка 28 мг)
Когорта 5: Три капсулы ТПИ (дозировка 28 мг)
Контрольное лечение
Одна доза ТРИ с 300 мг/5 мл [натуральный тобрамицин в растворе в пропорции 60 мг/мл (с наполнителем 5 мл 25%-ного физиологического раствора с рН 6,0±0,5)] распылялась струйным небулайзером PARI LC PLUS с компрессором DeVilbiss PulmoAide.
В исследовании были рандомизированы и обследованы до 80 субъектов в возрасте не менее шести лет с подтвержденным диагнозом кистозного фиброза (КФ). В каждой когорте, в которую входило всего по 16 субъектов, способом рандомизации определялись 12 субъектов для получения ТПИ и 4 субъекта для получения ТРИ. Субъекты проверялись на соответствие требованиям от 7 до 9 дней до введения исследуемого лекарства.
Оценивались безопасность лечения для субъектов и концентрации тобрамицина в мокроте и сыворотке крови до дозирования, спустя 30 минут и через 1, 2, 4, 8 и 12 часов после того как изучаемое лекарство в количестве одной дозы вводилось под наблюдением. Производился повторный прием субъекта в клинике через 7 (±2) дней.
Обсуждение хода эксперимента, включая выбор контрольной группы
В ходе данного исследования были зафиксированы общая безопасность и приемлемость экспериментального лечения. Для лучшей оценки этого вывода была использована рандомизированная схема 3:1 для увеличения числа участвующих в экспериментальной лечебной группе.
Контрольным вариантом данного исследования было применение 300 мг/5 мл ТРИ, подаваемого с помощью струйного небулайзера PARI LC PLUS с компрессором DeVilbiss PulmoAide. ТРИ использовался для введения пациентам с КФ, болеющим Р.aeruginosa.
Критерии включения при отборе группы исследуемых
Субъекты удовлетворяли требованиям для участия в исследовании, если они удовлетворяли сразу всем следующим критериям включения.
- Предоставление письменного согласия об информированности и подпись об ознакомлении с законом по обеспечению доступности и подотчетности в медицинском страховании (HIPAA authorization) перед проведением каких-либо относящихся к исследованию процедур.
- Мужчины- и женщины-субъекты не моложе 6 лет на момент скрининга.
- Диагноз КФ с содержанием хлорида в поте >60 мэкв/л по ионтофорезному тесту на количество пилокарпина (QPIT) и/или по генотипу с двумя идентифицируемыми мутациями, указывающими на КФ и подтверждаемыми одним или более клиническим признаком, указывающим на КФ.
- Для женщин-субъектов старше 11 лет или достигших первой менструации: отрицательный тест на беременность. Сексуально активные женщины в возрасте, позволяющем иметь детей, должны были согласиться использовать контрацепцию в течение периода исследований.
- Способность отхаркивать образцы мокроты по требованию.
- ОФВ1>40% от должного значения (рассчитанного с использованием уравнений Кнудсона, основанных на учете пола, возраста и роста).
- Способность выполнять все протокольные требования.
- Клиническая устойчивость по мнению исследователя.
События, являющиеся основанием для исключения при отборе исследуемых субъектов
Субъекты исключались из исследования, если имело место одно из следующих исключающих участие событий:
- Ингаляционное или внутривенное введение аминогликозидов за 14 дней перед началом введения изучаемого лекарства или в период проведения исследования.
- Введение любого исследуемого лекарства за 14 дней до введения изучаемого лекарства или в период проведения исследования.
- Введение петлевого диуретика за 7 дней до введения изучаемого лекарства или в период проведения исследования.
- Кровохаркание чаще чем "60 сс" в любое время в течение 30 дней, предшествующих введению исследуемого лекарства.
- Установленная местная или системная гиперчувствительность к аминогликозидам или ингалируемым антибиотикам.
- Сыворотка креатинина 2 мг/дл и более, АМК 40 мг/дл и более или ненормальный анализ мочи, определяемый как 2 + или большая протеинурия.
Отстранение субъектов от лечения или оценок
Субъекты, или их родители, или официальные опекуны могут отозвать свое согласие на участие в исследовании в любое время без ограничений. Исследователь может отстранить субъект на основе субъективного клинического ощущения, что делается в интересах самого субъекта, или если субъект не мог выполнить требования протокола. Когда было возможно, приводились результаты тестов и заключения, составленные на момент последнего визита.
Если субъект не выполнял обязательные визиты, осуществлялась работа для определения причин. Эта информация была указана в соответствующей индивидуальной регистрационной форме (ИРФ).
Рандомизированные субъекты, отказавшиеся от исследования до введения первой дозы, были замещены. Любой рандомизированный субъект, кто отказался от исследования после введения первой дозы, замещен не был. Причина отказа и время отказа указывались в индивидуальной регистрационной форме (ИРФ). Причины отказов классифицировались следующим образом:
- Нежелательное явление;
- Нарушение протокола;
- Невозможность дальнейшего наблюдения;
- Отзыв согласия на участие;
- Смерть;
- Ошибочное включение;
- Административные причины;
- Другие, не указанные выше.
Введенные лечебные средства
До восьмидесяти субъектов были рандомизированы и получили лечение. Каждый субъект получил единичную дозу либо контрольного лекарства, либо экспериментального, как указано ниже.
Экспериментальное лекарственное средство, введенное с помощью ингалятора Т-326
Когорта 1: Две капсулы ТПИ (дозировка 14 мг)
Когорта 2: Четыре капсулы ТПИ (дозировка 14 мг)
Когорта 3: Две капсулы ТПИ (дозировка 28 мг)
Когорта 4: Четыре капсулы ТПИ (дозировка 28 мг)
Когорта 5: Три капсулы ТПИ (дозировка 28 мг)
Контрольное лечение.
ТРИ в 300 мг/5 мл [натуральный тобрамицин в растворе в пропорции 60 мг/мл (наполнитель 5 мл 1/4 физиологического раствора с рН 6.0±0.5)] подавался с помощью струйного небулайзера PARI LC PLUS с компрессором DeVilbiss PulmoAide.
Характеристики исследуемого продукта
ТПИ, используемый в этом исследовании, - это сухой порошковый препарат тобрамицина и двух наполнителей: 1,2-дистеароил-сн-глицеро-3-фосфохолина (DSPC) и хлористого кальция (CaCl2). ТПИ был упакован в капсулы 2-гидроксипропилметилцеллюлозы (ГПМЦ) двух типов, содержащих либо 25 мг, либо 50 мг порошка. В настоящем исследовании использовались две количественные дозы тобрамицинового порошка: 14 мг тобрамицина на капсулу и 28 мг тобрамицина на капсулу.
TOBI® тобрамициновый раствор для ингаляции (300 мг/5 мл) - это стерильный невоспламеняющийся свободный от консервантов антибиотик, приготовленный для введения в виде аэрозоля. Каждый мл исследуемого лекарства содержит 60 мг тобрамицина и 2,25 мг хлорида натрия в стерильной воде для инъекций с рН 6.0±0.5.
Порядок организации процесса
Субъекты, рандомизированные для контрольного лечения, получали по 300 мг тобрамицинового раствора для ингаляции TOBI® в пропорции 60 мг/мл. Тобрамициновый раствор для ингаляции TOBI® был введен субъектам с помощью струйного небулайзера PARI LC PLUS и компрессора DeVilbiss PulmoAide. Доза тобрамицинового раствора для ингаляции TOBI® в 300 мг была исполнена в виде коммерчески доступной ампулы тобрамицинового раствора для ингаляции TOBI®. Две 5-миллилитровые ампулы исследуемого лекарства были упакованы в фольгу. Хотя это было изучение разовой дозы, фольговая упаковка содержала две 5-миллилитровые ампулы предоставляемого субъектам исследуемого лекарства на случай, если произойдет потеря изучаемого медикамента в ходе приготовления и использования распылителя и системы подачи.
Субъекты, рандомизированные для экспериментального лечения, получали одну дозу ТПИ, содержащую две или четыре капсулы с дозировкой 14 мг или две, три или четыре капсулы с дозировкой 28 мг. ТПИ вводился испытуемым с помощью ингалятора Т-326. ТПИ капсулы были герметично упакованы во влагонепроницаемый контейнер, укрытый двухслойной фольгой, и вводились в течение 30 минут сразу после открытия контейнера. Для когорт 1, 2 и 3 для введения одной дозы использовался только один ингалятор Т-326. Для когорт 4 и 5 ингалятор Т-326 заменялся после введения второй капсулы; тем самым для введения одной дозы в этих когортах использовались два ингалятора Т-326.
Подробные инструкции по подготовке и проведению как контрольного, так и экспериментального лечения были предоставлены исследователям клинических исследовательских центров.
Принцип пополнения лечебных групп субъектами
Подходящие субъекты способом рандомизации присоединялись в отношении 3:1 или к экспериментальной, или контрольной лечебным группам. Если исследователь или координатор исследований признал, что испытуемый удовлетворяет критериям отбора, персонал заполнял ведомость рандомизации субъекта, получал номер субъекта и лечебные предписания через Систему интерактивного речевого ответа (СИРО).
В начале исследования предполагалось, что до 80 субъектов в пяти группах случайным образом будут распределены: 60 на экспериментальное лечение (12 субъектов в каждой когорте) и 20 на контрольное лечение (4 субъекта в каждой когорте).
Выбор дозировок, используемых в исследовании
Дозой для контрольного лечения была признанная Федеральным Агенством по лекарственным средствам (FDA) доза ТРИ 300 мг для лечения Р.aeruginosa у пациентов с КФ в возрасте от 6 лет и старше. В соответствии с фармакинетическим моделированием (РК modeling) систематическое накопление 300-миллиграммовой дозы ТРИ, вводимой с помощью жидкого небулайзера PARI LC PLUS с компрессором DeVilbiss PulmoAide, было оценено в 11,7% от распыляемой дозы. Среднее и стандартное отклонение в концентрации 300-миллиграммовой дозы ТРИ в мокроте, спустя час после ингаляции, составили 1,0±0,58 мкг/мл, с учетом возможного широкого диапазона осаждения. Дозой для экспериментального лечения были две или четыре капсулы ТПИ, содержащие 14 мг тобрамицина на капсулу; или две, три или четыре капсулы ТПИ, содержащие 28 мг тобрамицина на капсулу.
Выбор и учет дозы для каждого субъекта
Субъектам давалась либо разовая доза, содержащая 300 мг тобрамицинового раствора для ингаляции TOBI® в пропорции 60 мг/мл, либо одна доза ТПИ, содержащая две или четыре капсулы с 14 мг дозы, или две, три или четыре капсулы 28 мг вещества дозы. Никаких ограничений на время применения дозы относительно времени принятие еды не накладывалось.
Слепое исследование
Исследование было клиническим открытым исследованием. Использование двух различных систем введения препаратов не позволяет проводить слепое исследование.
Предшествующая и сопутствующая терапии.
Дополнительная поддерживающая терапия проводилась согласно стандартной практике на каждом этапе исследования. Лекарства, приведенные в исключающих критериях, не вводились пациентам с момента скрининга до завершающего посещения.
Субъектам разрешалось пользоваться бронхорасширителями перед употреблением исследуемого лекарства. Бронхорасширители были прописаны только тем испытуемым, кто постоянно использовал бронхорасширители для клинической терапии. Постоянное использование определялось как один или большее число раз в день за две недели до наблюдения. Субъектам с кратко действующими бронхорасширителями прописывалось их применение в период от 15 до 60 минут до принятия исследуемого лекарства. Субъекты с длительно действующими бронхорасширителями принимали лекарства, как предписано, по прошествии 24 часов.
Запрещено было использование следующих препаратов: любых форм аминогликозидов или исследуемых лекарств за 14 дней до введения изучаемого лекарства и в течение периода исследования; петлевых диуретиков - за семь дней до введения исследуемого лекарства и в течение всего периода исследования.
Комплаенс лечения (степень соответствия поведения пациента врачебным рекомендациям по лечению болезни)
Субъект самостоятельно вводил разовую дозу изучаемого лекарства в присутствии исследователя или координатора исследований. Причины любых преждевременных прекращений, повреждений или задержек при введении исследуемого лекарства были записаны в первичную документацию и в индивидуальную регистрационную форму. Общее время введения изучаемого лекарства записывалось в первичную документацию и в ИРФ.
Каждый субъект (или родитель/официальный опекун, соответственно) в присутствии исследователя предоставлял письменное согласие об информированности, включающее подтверждение об ознакомлении с законом об отчетности и безопасности медицинского страхования (HIPAA authorization), и давал согласие на участие в исследовании, до того как начинались какие-либо относящиеся к исследованию процедуры. Исследователи наблюдали субъектов при встречах от семи до девяти дней до первого дня (второго визита) с введением исследуемого лекарства для определения их соответствия при внесении в списки. Исследователи просматривали и записывали существенные моменты медицинской истории испытуемого, включая истории текущей болезни и других относящихся к делу респираторных заболеваний, базисные явные симптомы и вероятные симптомы, ингаляции сухим порошком и ингаляции антибиотиков за шесть месяцев до скрининга, и текущие лекарства и терапии, применяемые на момент скрининга.
Исследователи проводили физическое наблюдение субъектов, включающее измерение роста, веса и основных показателей жизнедеятельности. Основные показатели жизнедеятельности включали пульс, дыхание, температуру во рту и артериальное давление в сидячем положении. Пульс и дыхание измерялись в течение одной минуты после того, как испытуемый пробыл в покое не менее 10 минут. Давление крови измерялось, когда испытуемый просидел в покое не менее 10 минут.
Все субъекты, чей регулярный лечебный режим включал постоянное использование кратко действующих бронхорасширителей при клинической терапии один или большее число раз ежедневно в течении двух недель до скрининга, применяли бронхорасширители в период от 15 до 30 минут до контрольного спирометрического теста и в период от 15 до 60 минут до введения исследуемого лекарства. Субъекты, принимающие длительно действующие бронхорасширители, принимали средство в соответствии с предписанием за 24 часа до приема изучаемого лекарства.
Субъекты проходят стандартный спирометрический тест в соответствии с Рекомендациями Американского торакального общества от 1994 г. для того, чтобы определить объем форсированного выдоха в 1 секунду (ОФВ1), форсированную жизненную емкость легких и объемную форсированную скорость выдоха (СОС25-75), для подтверждения их способности надежно пройти спирометрию и для уверенности, что они удовлетворят критерию на включение, по которому ОФВ1 равен или превосходит 40% от должного значения, рассчитанного по уравнениям Кнудсона с учетом пола, возраста и веса.
Субъекты предоставляли образцы крови для скрининг-тестов химии и гематологии и образцы мочи для измерения протеинурии. Женщины-испытуемые от 11 лет и старше или те, кто достиг первой менструации, сдавали мочу для теста на беременность.
Субъекты, удовлетворяющие всем требованиям, были признаны удовлетворившими требованиям на участие в исследовании и были случайно распределены по лечебным когортам, как описано в протоколе. Рандомизированные субъекты вернулись в клинику через 7-9 дней (второй визит) для осуществления исследуемого лечения и сопутствующих процедур.
До проведения первого дня исследуемого лечения при втором визите все рандомизированные субъекты выполнили процедуры, предшествующие принятию дозы и включавшие подтверждения об отсутствии изменений в базисных симптомах, изменений в сопутствующем лечении и в основных жизненных показателях. Исследователи провели повторные физические наблюдения на случай, если какие-то телесные системы были аномальны и клинически значимы при скрининге или если субъект имел какие-либо клинически значимые изменения в самочувствии с момента скрининга. Полосковый тест мочи на протеинурию также повторно проводился перед принятием дозы, если имелся след или 1+ на момент скрининга; в случае отклонений также проведены проверки химического состава сыворотки крови и гематология. Исследователи провели предшествующий приему дозы спирометрический тест (от 15 до 30 минут после использования субъектом кратко действующего бронхорасширителя, если требуется) и собрали образцы сыворотки крови и мокроты для оценки начального уровня тобрамицина.
До проведения исследуемого лечения субъекты ТПИ, использующие кратко действующие бронхорасширители или регулярно, или по случаю, могли воспользоваться их кратко действующими бронхорасширителями по решению исследователя. Субъекты, не использующие кратко действующие бронхорасширители, могли ими воспользоваться, если они имели 10% и большее значение снижения ОФВ1 между скринингом и предшествующими принятию дозы тестами легочной функции. Относительный процент изменения с момента скрининга в ОФВ1 рассчитывался следующим образом:
относительные проценты ОФВ1 изменения при наблюдении
= [(предшествующий принятию дозы ОФВ1 - скрининг ОФВ1)/скрининг ОФВ1]×100
Единичная доза исследуемого средства применялась спустя 60 минут после предшествующего принятию дозы употребления бронхорасширителя, проводимого по необходимости, или спирометрии, и оценки субъектов на возможность применения аэрозоля и протокола безопасности, как описано в последующих разделах. Во время дозирования испытуемым предлагалось сидеть прямо, дышать спокойно и использовать затычки для носа в ходе ингаляции. Координатор исследования записывал время начала и остановки введения исследуемого лечения. Если испытуемый испытывал длительный кашель (дольше 10 секунд), координатор исследования останавливал таймер и перезапускал его, после чего испытуемый продолжал лечение. Для ТПИ испытуемых исследователь и/или координаторы исследования отмечали время, когда при второй ингаляции исследуемого лекарства становился слышен шипящий звук, идущий от ингалятора. Сразу после завершения введения изучаемого лекарства субъекты полоскали рот 30 мл физиологического раствора от 5 до 10 секунд и сплевывали его; данная процедура полоскания производилась три раза.
Субъекты для полностью безопасного окончания исследования оставались в клинике на 12 часов после начала введения исследуемых средств. Затем субъекты отпускались из клиники и назначались на третий визит по прошествии 8 (±2) дней. Вариации во времени последнего визита были связаны с личными обстоятельствами испытуемых (в этом примере последний визит обозначается как «день 8»). Предполагалось, что слабые отклонения от протокольного расписания оказывают минимальный или никакого эффекта на состояние исследуемых показателей.
В ходе визита в «день 8» координатор исследования отмечал любые изменения в медицинской истории субъекта, включая сопутствующие болезни, новые или ухудшившиеся нежелательные явления, связанные с КФ симптомы, текущие лекарства и дозировки, а также лекарства, продаваемые без рецепта. Было продолжено обследование субъектов с клинически значимыми относящимися к изучаемому лекарству побочными реакциями (или по телефону, или при визитах в клинику) вплоть до восстановления их удовлетворительного самочувствия. Исследователь проводил физическое наблюдение исследуемых, измерял рост, вес, основные показатели жизнедеятельности и спирометрические результаты, собирал кровь и образцы мочи для химии, гематологии и полосковых тестов мочи.
Главные переменная(ые) в доставке аэрозоля
Расчеты сравнимых доз ТПИ к ТРИ 300 мг/5 мл были основаны на оценке характеристик подачи аэрозоля при исследуемом и контрольном лечениях. Характеристики подачи аэрозоля тестируемого и контрольного лечения были определены на основе концентраций тобрамицина в сыворотке крови и мокроте во времени, расчете точных фармакокинетических характеристик сыворотки крови и мокроты, как описано в этом Примере, измерении времени введения лекарства и оценки работы ингалятора Т-326.
Концентрации тобрамицина в сыворотке крови
Образцы крови были собраны до дозирования и спустя 0,5, 1, 2, 4, 8 и 12 часов после начала первого вдоха исследуемого средства в ходе ингаляции. Образцы собирались как можно ближе к данному графику, и предполагалось, что отклонение в ±2 минуты несущественно для образцов, собранных через 0,5 часов, и в ±10 минут для образцов, собранных через час и более. Данные, собранные вне этих интервалов, рассматривались в протоколе, как отклонения.
Сыворотка была собрана и до начала анализа хранилась при температуре -20°С и ниже. Концентрация тобрамицина в сыворотке анализировалась по способу флуоресцентного поляризационного иммуноанализа (FPIA) с использованием системы Abbott TDx®/TDxFLx®. Образцы помещались непосредственно в лунки картриджей для образцов. Интенсивность поляризации устанавливалась с помощью аппарата TDx/TDxFLx (иммуноферментного анализатора). Для вычисления концентраций тобрамицина использовалось взвешенное четырехпараметрическое логарифмическое уравнение. Концентрации тобрамицина были получены в единицах независимых базовых эквивалентов.
Для оценки взятых у субъектов исследуемых образцов были приготовлены калибровочные стандарты (0,05, 0,10, 0,40, 0,80 и 0,90 мкг/мл) и образцы для контроля качества (0,10, 0,40 и 0,80 мкг/мл). Оценка производилась при шести замерах.
Нижним пределом количественного анализа было значение 0,05 мкг/мл. Точность оценки, определенная исходя из коэффициентов изменчивости образцов для контроля качества, составила 4,9%, 5,7% и 5,6% для образцов с 0,10, 0,40 и 0,80 мкг/мл, соответственно. Точность способа для образцов, различных по внутренним количественным характеристикам (различным концентрациям), составила 103%, 103% и 101% для образцов с 0,10, 0,40 и 0,80 мкг/мл, соответственно. Таким образом, способ демонстрирует удовлетворительную точность для фармакокинетического анализа.
Расчет фармакокинетических параметров сыворотки и удовлетворительная оценка дозы ТПИ, вызывающий тот же эффект, что и ТРИ, приведены в этом примере.
Концентрации тобрамицина в мокроте
Образцы мокроты были взяты у субъектов при сильном откашливании и собраны до дозирования и через 0,5, 1, 2, 4, 8 и 12 часов после начала первого вдоха в ходе ингаляции исследуемого лечения. Образцы собирались как можно ближе к указанным временам и в одних и тех же интервалах около них. Данные, собранные вне этих интервалов, рассматривались в протоколе как отклонения.
Образец мокроты (минимум 100 мг) собирался до принятия разовой дозы исследуемого лекарства для определения базовой концентрации тобрамицина.
Образцы мокроты хранились при -70°С или ниже вплоть до анализа. Концентрация тобрамицина устанавливалась с помощью жидкой хроматографии высокой производительности (HPLC) с ультрафиолетовым детектированием.
Образцы мокроты субъектов были разбавлены 20%-ным нормальным раствором гидроксида натрия и растворены в трис (гидроксиметиламинометан) буфере (20,0 г трис основания/л). Стандарты образцов мокроты были приготовлены при смешивании тобрамицина с мокротой от доноров с КФ, разведенной и перемешанной до финальных концентраций в 0, 20, 40, 100, 200, 400 и 1000 мкг/г мокроты. Образцы контроля качества были получены при смешивании разведенной и смешанной мокроты от нескольких доноров до концентраций 40, 300 и 800 мкг/г мокроты. К 100 мкл каждого образца-стандарта, образца контроля качества и исследуемого образца добавлялся внешний стандарт сисомицина (100 мкл, 0,15 мг/мл в трис-буфере), затем 400 мкл ацетонитрила и 50 мкл 2,4-динитрофторобензина (0,17 г/мл). Образцы смеси нагревались в сухом цилиндрическом нагревателе в течение 1 часа при температуре 80°С. После добавления 600 мкл ацетонитрила и воды в соотношении 60/40 (объем/объем) 50 мкл от полученной смеси анализировались с помощью HPLC.
Образцы помещались в колонку Waters Nova-Pak® C-18, 3.9×150 мм, 4 мкм присоединенную к Waters HPLC с насосом 600Е и ультрафиолетовым (λmax=360 нм) детектором 486 или 2487 и автоматическим пробоотборником 717 Plus. Подвижная фаза, содержащая 0,2% уксусной кислоты в ацетонитриле (39/61, объем/объем), откачивалась на скорости 1,5 мл/мин в течение 5 минут, 2,0 мл/мин в течение дополнительных 9 или 10 минут, в зависимости от длительности прохождения через колонку. Программное обеспечение Waters Millennium-32 C/S LC Software (версии 3.20) использовалось для работы с Waters HPLC инструментарием для ввода необработанных данных, а также для обработки, расчета и вывода результатов анализа. Было вычислено отношение максимальной концентрации тобрамицина к внутреннему стандарту сисомицина (PHR). Анализ производился на 17 замерах.
Время удерживания варьировалось от 3,8 до 4,1 минут и от 10,0 до 10,6 минут наблюдались для тобрамицина и сисомицина, соответственно. Между PHR и концентраций тобрамицина в мокроте (от 20 до 1000 мкг/г) имела место линейная зависимость. Ее регрессионная модель описывается уравнением PHR=Вх+А (где х= концентрация тобрамицина), взвешенным по 1/х. Нижним пределом количественного анализа было значение 20 мкг/г. Концентрация в образцах стандарта варьировалась в пределах от 97 до 102% номинальной концентрации с коэффициентами изменчивости не выше чем 5,2%. Точность способа для образцов, различных по внутренним количественным характеристикам (различным концентрациям), составила 5,2%, 3,9% и 2,0% для образцов с 40, 300 и 800 мкг/г, соответственно. Таким образом, способ демонстрирует удовлетворительную точность для фармакокинетического анализа.
Время введения препарата
Время ввода препарата было определено как время с момента первой ингаляции субъекта до окончания введения препарата. Введение препарата завершалось, когда тестируемый ингалятор Т-326 начинал шуметь и когда контрольный небулайзер PARI LC PLUS начинал шипеть. Для капсул с ТПИ исследователь отмечал время, когда на втором вдохе субъекта становился слышен шум, сообщающий о том, что капсула пуста.
Если введение лекарства по какой-то причине было прервано, также записывалось время прерывания и старта и остановки при повторном введении. Если дозирование было прервано, время прерывания не учитывалось при фиксации общего время введения. Если дозирование было прервано и не было зафиксировано время остановки или начала повторного дозирования, считалось, что время введения лекарства подсчитано быть не может.
Осмотр и анализ осадка тобрамицина в капсулах с ТПИ и использовавшихся ингаляторах Т-326.
Хотя это и не указано в протоколе исследования, используемый ингалятор Т-326 Inhaler и используемые капсулы с ТПИ анализировались для оценки их эксплуатационных характеристик.
Нежелательные явления
Нежелательное явление определено как любое нежелательное медицинское явление при клиническом обследовании субъекта, принимавшего фармацевтический продукт, вне зависимости от его дозы; такое явление не обязательно должно иметь непременную связь с этим лечением. Тем самым НЯ может быть любой неблагоприятный и непредусмотренный признак (объективные знаки болезни, включая отклонения при лабораторных исследованиях), симптом (субъективные признаки заболевания) или болезнь, которые совпали во времени с использованием медицинского продукта, в независимости от того, связаны они с приемом медицинского продукта или нет. Сюда относятся сопутствующие болезни или травмы, а также обострение заболеваний, уже имевшихся до начала исследования. Неожиданное нежелательное явление (НЯ) - это явление, по своим характеристикам и серьезности не согласующееся с имеющейся информацией о применяемом продукте.
О нежелательных явлениях у субъектов исследователи могли узнавать по их спонтанным добровольным признаниям либо в ходе расспросов, проводимых исследователем или координатором.
Все НЯ контролировались до их окончания или, если НЯ определялось как хроническое, до выяснения причин его возникновения. Если причина НЯ оставалась неустановленной до завершения исследования, исследователь и медицинский контроль должны были дать клиническую оценку о необходимости дальнейшего наблюдения, заслужившего внимание НЯ.
Степень серьезности явлений, указанных в графе НЯ ИРФ, классифицировалась исследователем по следующей схеме.
Легкая: никаких ограничений обычной активности.
Умеренная: некоторые ограничения в обычной активности.
Тяжелая: невозможность обычной активности.
Связь изучаемого лечения с НЯ определялась исследователем на основании следующих определений.
Нет связи: реакции на исследуемый продукт нет, или наличие НЯ не очевидно связано с продуктом во времени, или предполагается, что связь между НЯ и исследуемым продуктом маловероятна.
Возможная связь: введение исследуемого лекарства и НЯ логически связаны во времени, при этом НЯ может быть относительно хорошо объяснено причинами, не связанными с реакцией на исследуемый продукт.
Вероятная связь: исследуемое лечение и НЯ логически связаны во времени, и НЯ наиболее вероятно объясняется реакцией на исследуемый продукт, нежели другими причинами, либо исследуемый продукт является самой вероятной причиной НЯ.
Серьезные нежелательные явления
В этом исследовании СНЯ выявлено не было.
Клиническая лабораторная проверка
Лабораторные проверки базового состава крови, химии мокроты и полосочный тест (с использованием тест-полосок) на белок в моче проводились во время скрининга и в день последнего визита (день 8). В день дозирования (день 1) были измерены азот мочевины крови (АМК) и сыворотка крови через 12 часов после начала введения исследуемого лекарства. Кроме того, полосочный тест на белок в моче был повторен сразу перед дозированием, если при скрининге тест показал след или 1+; химия мокроты и состава крови также были повторены, если при скрининге имелись отклонения.
Гематологические тесты, проведенные в этом исследовании, включали определение уровня лейкоцитов в крови (WBC), содержание эритроцитов в крови (RBC), гемоглобин, гематокрит, определение лейкоцитарной формулы и количества тромбоцитов.
Химический анализ крови выявил содержание натрия, калия, хлора, бикарбоната, азота мочевины крови, креатина, глюкозы, кальция, фосфора, гамма-глутаминтрансферазы, Аланин-трансаминазы (АЛТ), аспартат-трансаминазы (AST or SGOT), алкалин-фосфатазы, лактик-дегидрогеназы(LDH), общего билирубина, прямого билирубина, свободного билирубина, общего протеина, альбумина и сыворотку хорионгонадотропина (HCG).
Бронхоспазм и спирометрия
Спирометрия (объем форсированного выдоха в одну секунду [ОФВ1 в литрах], проверки форсированной жизненной емкости легких [ФЖЕЛ в литрах] и скорости потока форсированного выдоха в течение середины второй четверти форсированной жизненной емкости легких [СОС25-75] проводились за день до дня 1 принятия разовой дозы и через 30 минут после завершения дозирования для оценки чувствительности дыхательных путей к исследуемым лекарствам. Стандартный спирометрический тест также проводился в день 8 при последнем визите.
Протокол исследований предварительно определял бронхоспазм как относительное падение ОФВ1 должн. на 20% или более от начала дозирования и до 30-й минуты после окончания дозирования. Субъекты с относительно малым падением в 10% и более в ОФВ1 должн. отмечались особо.
Через 30 минут после дозирования, если относительный % падения ОФВ1 составил 20% и более, спирометрические измерения (ОФВ1, ФЖЕЛ и СОС25-75) надо было повторять в соответствии с графиком, определенным исследователем, до тех пор, пока падение ОФВ1 не составит менее 10% (для субъекта 12/317, единственного субъекта, у которого падение ОФВ1 составило 20% и более от ОФВ 1 должн., повторные измерительные операции проведены не были). Отдельные случаи падения ОФВ1 должн. на 20% и более были квалифицированы как НЯ (это не было сделано для испытуемого 12/317). Лечение с подходящими медикаментами и дальнейшее обследование назначались по усмотрению исследователя.
Основные показатели жизнедеятельности
Измерение основных показателей жизнедеятельности до дозирования в день 1 и через 30 минут и 1, 4, 8 и 12 часов после начала дозирования включало измерение артериального кровяного давления, пульса, дыхания после 10 минут покоя и температуры тела. Также Основные показатели жизнедеятельности были измерены в день 8 - во время последнего визита.
Исследования состояния здоровья
Исследования состояния здоровья испытуемых проводилось в день 8 - во время последнего визита - и состояло из физического осмотра для определения общего состояния субъекта: кожи, лимфатических узлов, HEENT (головы, глаз, ушей, носа, горла), легких, кардиоваскулярной системы (сердечно-сосудистой), брюшной полости, конечностей, мышечно-скелетной, неврологической и генитоуритарной (мочеполовой) систем тела.
Адекватность измерений
Измерения эффективности способа в настоящем исследовании проводились в соответствии с установленными стандартами измерений, характеризуемых их широкой используемостью и общепризнанностью в качестве надежных, точных, релевантных и селективных в отношении эффективных и неэффективных веществ. Измерения надежности, использовавшиеся в данном исследовании, представляли собой стандартные клинические и лабораторные процедуры.
Фармакокинетика
Для получения фармакокинетических параметров зависимость концентрации (С) от времени (t) для проб сыворотки и мокроты были проанализированы на основании независимых моделей. Были получены величины максимумов концентрации (Cmax) и времени (tmax), при котором наблюдались максимумы концентрации. Окончательная скорость процесса (λz) была определена из расчета логарифмической регрессии на конечной фазе. Период полурастворения был вычислен по формуле t1/2=ln(2)/λz. Концентрации ниже предела количественного анализа хроматографии принимались как равные нулю при всех вычислениях. Площади под кривыми (ППК) концентрация-время для сыворотки и мокроты на временном отрезке от нуля (где ноль соответствовал процедурам, производившимся сразу перед дозированием) и до 12 часов, т.е. ППК (0,12) были вычислены по правилу трапеций. Площадь под кривой при устремлении времени окончания к бесконечности, т.е. ППК (0,∞), определялась по формуле ППК (0,12)+С(12)/λz, где С(12) - это концентрация вещества спустя 12 часов от начала дозирования.
Все фармакокинетические параметры были выражены как среднее значение ± стандартное отклонение. Среднее значение периода полурастворения было вычислено по формуле:
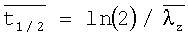 ,
,
где 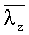 - среднее арифметическое конечных скоростей процессов для каждой дозы. Стандартное отклонение от среднего периода полурастворения
- среднее арифметическое конечных скоростей процессов для каждой дозы. Стандартное отклонение от среднего периода полурастворения 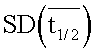 было получено по формуле:
было получено по формуле:
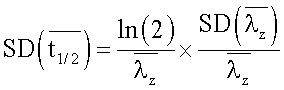
где 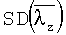 - стандартная ошибка среднего конечного значения скорости для каждой дозы.
- стандартная ошибка среднего конечного значения скорости для каждой дозы.
Оценка сравнимых доз ТПИ и ТРИ
Для оценки сравнимых доз ТПИ и ТРИ кривая логарифмической регрессии сравнивалась с логарифмической кривой ППК (0,12) и логарифмической зависимостью (ТПИ доза) концентрации ТПИ в сыворотке крови для всех когорт. Сравнимые дозы и доверительный интервал в 95% были определены способом инверсии подогнанной линии регрессии и рассмотрением значений, соответствующих верхней и нижней границам 95-процентного доверительного интервала для среднего логарифмической зависимости ППК (0,12) по показателям ТРИ.
Наблюдение
Исследование проводилось согласно принципам надлежащей клинической практики (НКП).
Обработка данных
Информация из индивидуальной регистрационной карты вводилась в базу данных Clintrial®.
Контроль за качеством ввода осуществлялся с использованием процедурного языка/секвенциального языка запросов (PL/SQL) и программного обеспечения SAS® версии 8.2 или выше (Институт SAS, Кери, Северная Каролина). Анализ проводился с использованием Analysis SAS® версии 8.2 или выше и базировался на заранее разработанном плане анализа. Оценка уровня итоговой ошибки базы данных составила 0,022% при верхней границе 95-процентного доверительного интервала 0,157% (округленно). Такое значение нижней границы доверительного интервала ниже допустимого значения в 0,5%.
Статистические способы и определение размера образца
Целями первой фазы исследования были оценка безопасности тестируемого лечения с помощью ТПИ и контрольного лечения с помощью ТРИ, а также оценка дозы ТПИ, которая могла бы дать фармакокинетическую кривую, сравнимую с кривой, получаемой при лечении ТРИ 300 мг/5 мл. В данном исследовании никакого сравнения эффективности лекарственных средств не предусматривалось. Все запланированные заключения и анализы носили диагностирующий характер. Не проводилось никаких статистических сравнений между группами, подвергнутыми лечению с помощью ТПИ и ТРИ, и статистических проверок на различие между двумя способами лечения. План анализа исследования учитывал, что из-за малого числа субъектов в каждом клиническом центре, участвующем в исследовании, данные со всех центров должны быть объединены для анализа.
Все заключения и анализы проводились для всех шести лечебных групп. Если не указано особо, частоты и процентные распределения вычислялись для категорных переменных, а число имеющихся (non-missing) значений, среднее, стандартное отклонение, минимум, максимум и медиана вычислялись для континуальных переменных. Все данные, занесенные в индивидуальные регистрационные формы (ИРФ), были представлены в листингах индивидуальных данных субъекта.
Были определены три группы субъектов:
- группа всех участвующих: субъекты, допущенные к участию и рандомизированные в ходе исследования,
- группа оценки безопасности: субъекты, рандомизированные и принимавшие все или часть исследуемого лекарства,
- группа оценки фармакокинетики: субъекты, рандомизированные и принимавшие все исследуемое лекарство.
Базисные и демографические характеристики были суммированы для всех групп участников. Оценка фармакокинетики тобрамицина проводилась с привлечением группы оценки фармакокинетики. Оценка дозы ТПИ, дающей системное воздействие тобрамицина, сравнимое с ТРИ, проводилась на группе оценки сравнимых дозировок. Анализ всех других переменных проводился с привлечением группы оценки безопасности.
Для проведения анализа использовалось программное обеспечение SAS версии 8.2. Для графического представления полученных результатов использовался Microsoft Excel.
Анализ доставки аэрозоля и оценка сравнимой дозировки ТПИ
Все субъекты, получавшие разовую дозу лекарства, были включены в анализ и оценку характеристик подачи аэрозоля, которые производились на основании концентраций тобрамицина в сыворотке и мокроте, оценки фармокинетических параметров сыворотки и регистрации времени введения лекарства.
Сывороточная ППК (0,12) использовалась для оценки сравнимых дозировок ТПИ и ТРИ. В линейный регрессионный анализ, где зависимой переменной (игрек) был логарифм ППК (0,12), а независимой переменной (икс) был логарифм дозы ТПИ, были включены данные по всем ТПИ группам. Сравнимая доза ТПИ была определена способом инверсии линии регрессии, подогнанной к среднему значению логарифма ППК (0,12) по концентрациям тобрамицина в сыворотке субъектов, принимавших ТРИ, из всех пяти ТРИ когорт.
Вторичный анализ доставки аэрозоля
Протокол исследования определял отсутствие вторичных переменных доставки аэрозоля.
Анализ безопасности
Все субъекты, получившие дозировку изучаемого лекарства, были оценены на безопасность на основании анализа НЯ, изменений функции легких, клинических лабораторных результатов и врачебного осмотра.
Оценка побочных явлений
Симптомы базового уровня и вызванные лечением побочные реакции (НЯ) были закодированы с использованием тезауруса MedDRA. Общее обострение индивидуальных вызванных лечением НЯ (процент субъектов, кто испытал обострение, по крайней мере, один раз в течение или после исследуемого лечения) был описательно оценен для любых заслуживающих внимание различий при лечении с помощью ТПИ и ТРИ. Статистического анализа запланировано не было. НЯ были также разделены по степени серьезности (легкая, умеренная, тяжелая) и отношения к лекарству (не относится, возможно относится) для групп тестируемого ТПИ и контрольного лечения. Для оценок взаимосвязи с лекарством классификация вида «возможно относится» включала индивидуальные НЯ, которые разделялись исследователем на вероятно связанные, возможно связанные и несвязанные с изучаемым лечением.
Изменение функции легких
Нормальные значения были получены для ОФВ1, ФЖЕЛ и СОС25-75 (спирометрических измерений) в тех случаях, когда субъекты не имели болезни легких. Эти нормы обычно используются в изучении испытуемых с болезнью легких. Непосредственные спирометрические измерения были приведены к виду процентных нормативных значений с использованием уравнений Кнудсона, как это описано ниже. Для каждого испытуемого нормативное значение по Кнудсону представляло собой линейную комбинацию возраста субъекта (лет) и его роста (см), выраженную в следующей формуле:
Нормативное Значение Кнудсона = С0+С1×Рост+С2×Возраст,
где коэффициенты С0, C1 и С2 определялись на основании половой и возрастной группы субъекта.
При наличии непосредственных наблюдаемых значений из спирометрических измерений и кнудсоновского нормативного значения процентное должное значение вычисляется по формуле:
% должн. = (непосредственно наблюдаемая величина/кнудсоновское нормативное значение)×100
Изменение и относительное изменение в ОФВ1 должн. (1%) с момента дозирования до 30-й минуты после окончания дозирования были подсчитаны, используя следующие формулы:
Изменение = последозовый ОФВ1% - преддозовый ОФВ1%
Относительное изменение = [(последозовый ОФВ1% - преддозовый ОФВ1%)/Преддозовый ОФВ1%)]×100.
Субъект классифицировался как испытывающий бронхоспазм, если относительное изменение представляло собой снижение ОФВ должн. на 20% и больше.
Изменение и относительное изменение, а также наличие реакции бронхоспазма описательно сравнивались между лечебными группами. Кроме того, следующие сходным образом были выведены переменные:
- реакция субъекта с относительным падением ОФВ1 на 10% и больше должного по данным с момента дозирования до 30-й минуты после окончания дозирования,
- изменение и относительное изменение в ОФВ1 и СОС25-75% от ОФВ1 должн. по данным с момента дозирования до 30-й минуты после окончания дозирования,
- изменения и относительные изменения в ОФВ1, ФЖЕЛ и СОС25-75 должн. в процентах от предсказанных по данным с момента дозирования до 30-й минуты после окончания дозирования.
Другие переменные безопасности
Описательно был произведен учет других переменных безопасности, включающих клинические лабораторные результаты, основные показатели жизнедеятельности, прием сопровождающих лекарств, медицинские процедуры, врачебные осмотры. Кроме того, изменения от базисного (первоначального) уровня к концу исследования были подсчитаны и учтены для лабораторных данных, основных показателей жизнедеятельности и спирометрических измерений; в этих подсчетах базисное значение было последней доступной оценкой до начала исследования. Также были оценены изменения в лабораторных результатах, которые вышли за стандартно допустимый диапазон.
Определение размеров образца
Размер образца и выбор дизайна параллельных групп исследования был сделан скорее из учета клинических и практических, нежели статистических соображений. Все анализы рассматривались как экспираторные (диагностирующие) и проводились с использованием описательных способов. Никаких заключений статистического анализа запланировано в настоящем исследовании не было.
Предварительный анализ
После изучения каждой исследуемой когорты подсчитывались итоговые значения ключевых переменных безопасности и передавались в Комитет Мониторинга Данных (КМД) для рассмотрения и решения об увеличении дозы (эскалации до следующего дозового уровня). Следующие результаты указывались для каждого субъекта и суммировались по окончании лечения каждой когорты для взвешенного рассмотрения КМД:
- демографическая принадлежность субъекта;
- ОФВ1 должн. до и после дозирования и процентное изменение;
- серьезные нежелательные явления;
- НЯ, вызванные лечением;
- НЯ респираторной системы, вызванные лечением.
Этот предварительный анализ был проведен единственно для рассмотрения безопасности и принятия КМД решения об увеличении дозировки. Никакого статистического анализа изучаемых данных запланировано не было и никакой проверки уровня альфа-процентного критического значения не требовалось из-за предварительно обеспеченной безопасности и рассмотрения КМД решений по увеличению дозировки.
Скрининг, участие и рандомизация субъектов
Все 97 субъектов перед исследованием прошли скрининг у 15 исследователей. Девяносто из 97 субъектов удовлетворили проходным критериям и были отобраны (Таблица 2) и случайным образом распределены в одну из шести лечебных групп. Семеро из 97 наблюдавшихся субъектов не удовлетворили критерию включения в исследование, и им было отказано в участии.
Отстранение субъектов от лечения
Трое из 90 участвовавших субъектов из-за предшествующих дозированию НЯ были отстранены от лечения и не получили разовую дозу исследуемого лекарства (Таблица 2). Восемьдесят семь из 90 субъектов приняли исследуемое лекарство.
Завершение исследования
Восемьдесят шесть из 87 субъектов, принявших исследуемое лекарство, успешно прошли исследование. Один из 87 принявших дозу субъектов был исключен из исследования из-за вызванных лечением нежелательных явлений.
Один из 86 завершивших исследование субъектов получил дозу, но осмотр возвращенных капсул после завершения исследования выявил, что капсулы не были проколоты ингалятором Т-326. Концентрация тобрамицина в сыворотке крови данного субъекта была ниже количественно измеримого предела (BQL), что подтверждало, что субъект не получил исследуемое лекарство.
Массив данных, проанализированный для оценки подачи аэрозоля
Восемьдесят шесть из 90 участвовавших субъектов были рандомизированы, приняли разовую дозу изучаемого лекарства, и были оценены с точки зрения безопасности протокола. Трое из 90 субъектов были исключены из-за нежелательных явлений до того, как они получили исследуемое лекарство, и не учитывались при оценке безопасности. Еще один субъект не получил никакого исследуемого лекарства из-за сбоя ингалятора Т-326 при прокалывании лечебной капсулы и также не учитывался при оценке безопасности.
Восемьдесят четыре субъекта, принявших лечение, подходили для определения фармакокинетических параметров и оценки дозы ТПИ, сходной с дозой ТРИ. Двое из лечившихся субъектов не учитывались при оценке фармакокинетических параметров и оценке сходных доз.
Субъекты, не получившие лечение, также не подходили для оценок фармакокинетических параметров.
Демографические характеристики
В исследовании приняли участие сорок три мужчины и сорок семь женщин, от семи до 50 лет, с диагнозом КФ. Средний возраст был сходен между лечебными группами, меняясь от 19,5 в группе ТРИ до 24,1 в группе ТПИ 2×14 мг. Пятнадцать субъектов были в возрасте от 7 до 12 лет, 22 от 13 до 17 и 53 в возрасте от 18 до 50 лет. Семьдесят девять субъектов были представителями белой расы (Caucasian), кроме них пятеро латиноамериканцев, трое субъектов принадлежали черной расе и трое имели другие корни. Половое и расовое распределение было сходным для ТПИ и ТРИ лечебных групп, хотя малое несоответствие в полах было отмечено между группами ТПИ 4×14 мг и ТРИ (ТПИ: 11 женщин, 3 мужчин; ТРИ: 8 женщин, 12 мужчин). Эффект этого дисбаланса для результатов исследования незначителен. При скрининге ТПИ и ТРИ субъекты имели в среднем сравнимые рост и вес.
Другие базисные характеристики
Участвующие субъекты проходили лабораторное (хлорид в поте >60 мэкв/л по количественному поликарпиновому ионтофорезному тесту (QPIT) или/и с двумя различимыми мутациями в гене) и клиническое освидетельствования, диагностирующие наличие клинически стабильного кистозного фиброза. Имеющиеся до начала исследования медицинская история, явная симптоматика и косвенные симптомы для лечебных групп были сходными.
Субъекты, участвующие в исследовании, удовлетворяли включающим и исключающим критериям. Отметим, что у четырех субъектов за день до дозирования имело место слабое или среднее устойчивое кровохаркание. Все субъекты удовлетворяли включающему критерию, по которому их ОФВ1 был меньше ОФВ1 должного, рассчитанного по полу, возрасту и росту, на 40%. Средний ОФВ1 должн. для субъектов в шести группах был сходным (от 58,51% в группе ТПИ 2×14 мг до 82,40% в группе ТПИ 3×28 мг и 67,95% в группе ТРИ). Все субъекты, кроме трех, удовлетворили включающему критерию, по которому результат полоскового теста на протеинурию должен был показать меньше 2+; трое субъектов не предоставили мочу при скрининге.
Тридцать девять из 90 участвовавших субъектов использовали сухой порошок при ингаляции за предшествовавшие шесть месяцев до скрининга, что составило от 25% до 60% человек на лечебную группу. Тридцать семь из 90 субъектов использовали ТРИ (от 40% до 100% субъектов на лечебную группу), ни один из субъектов не получал других аминогликозидов, а 10 из 90 субъектов использовали другие ингаляционные антибиотики за предыдущие шесть месяцев.
Тридцать шесть из 90 участвовавших (то есть субъекты ТПИ и ТРИ групп) использовали кратко действующие бронхорасширители как часть их стандартного лечения КФ в пределах от 15 до 60 минут до начала исследуемого лечения. В двух случаях бронхорасширители использовались более чем за 60 минут до введения исследуемого лекарства.
Измерение комплаенса лечения
Комплаенс с требованиями протокола по однодозовому введению лекарства был признан приемлемым, так как 86 из 90 участвующих субъектов употребили изучаемое лекарство (трое субъектов были исключены до дозирования из-за базисных симптомов и один субъект дозировался, но не получил лекарство из-за того, что устройство Т-326 не прокололо лечебные капсулы). Четверо других субъектов не имели нарушений в приеме лекарства, причины которых указаны в Таблице 6.
Концентрации тобрамицина в сыворотке и фармакокинетические параметры
Фиг.1, на которой изображен график зависимости средней концентрации тобрамицина в сыворотке от времени после введения ТПИ и ТРИ, свидетельствует о том, что лекарство быстро абсорбируется: среднее tmax при любом лечении было 1 час. Распределение лекарства оказалось очень быстрым, и уровень падения имел характерный вид простой экспоненты со средней конечной скоростью полурастворения от 2,8 до 3,5 часа. Значения фармакокинетических параметров тобрамицина после введения ТРИ соответствуют ранее проведенным исследованиям.
Увеличение дозы ТПИ приводило к увеличению накопления тобрамицина, как следствие этого росли значения ППК (0,∞), ППК (0,12) и Cmax (Table 8). Это увеличение было почти пропорционально дозе.
У четырех субъектов начался кашель, который привел к прерыванию приема доз. Прерывания отмечены для доз ТПИ 4×14 мг, 3×28 мг и 4×28 мг. У всех оцениваемых субъектов с кашлем значения ППК (0,∞), ППК (0,12) и Cmax находились в одном диапозоне со значениями у субъектов из их же группы дозирования, выполнивших дозирование без прерываний.
Разница в чувствительности к тобрамицину (ее показывают графики ППК и Cmax) у субъектов, получивших 4×14 мг в капсулах, заполненных вручную, и в капсулах 2×28 мг, заполненных автоматом (Р>0,5), зафиксирована не была. Следовательно, биосовместимости капсул, заполненных вручную и автоматически, были аналогичны, и эти две группы были объединены в одну.
В целом, наблюдалась слабая отрицательная корреляция между процентом порошка, оставшегося в ингаляторе, и ППК (0, ∞) (r=-0,22, Р=0,0771), ППК (0,12) (r=-0,23, Р=0,0707) и Cmax (r=-0,22, Р=0,0838). Меньшая чувствительность связывалась с большим количеством ТПИ, оставшимся в устройстве для ингаляции. Один из субъектов в ТПИ группе 4×28 мг (субъект 04/409) имел больше, чем 45% порошка, оставшегося в устройстве, и имел чувствительность к тобрамицину, которая была одной из наименьших в его лечебной группе.
Не было обнаружено значительных корреляций между изменениями от базисного уровня ОФВ1 и сывороточных ППК и Cmax [для субъектов ТПИ: ППК(0,∞) (Р=0,9838), ППК (0,12) (Р=0,9990) и Cmax (Р=0,9110); для субъектов ТРИ: ППК (0,∞) (Р=0,5216), ППК (0,12) (Р=0,4337) и Cmax (Р=0,3878)].
Сравнимые дозы ТПИ и ТРИ
Воздействие, достигнутое после введения ТРИ 300 мг, было сходным с тем, что наблюдались после введения 4×28 мг капсул ТПИ (фиг.2 и 3). Когда данные ППК (0,12) всех групп были проанализированы как функция от дозы (фиг.2), результаты показали, что доза 115 мг ТПИ вызывает среднее ППК (0,12), сходное с ТРИ. Когда рассматривалась ППК (0, ∞) (фиг.3), было выявлено, что доза 112 мг дает сходное ППК (0, ∞) с ТРИ. На основании этих результатов становится ясно, что четыре капсулы 28 мг ТПИ (общая доза в 112 мг) будут оказывать воздействие, очень близкое к воздействию ТРИ.
Концентрация тобрамицина в мокроте и фармакокинетические параметры
После введения ТПИ и ТРИ максимум концентрации тобрамицина в мокроте был достигнут в среднем через 30 минут (фиг.4) с последующим спадом и средним временем полурастворения от 0,8 до 2,2 часов (Таблица 8).
Вариативность фармакокинетических параметров по мокроте была выше, чем по сыворотке. Помимо того, что фармакокинетические параметры, определяемые по мокроте, сильно варьировались, некоторые субъекты не смогли предоставить мокроту по требованию (недостаток образца мокроты в какой-либо момент времени имелся не более чем у троих субъектов на группу). Это оказало влияние на расчеты величин воздействия: ППК (0, ∞), ППК (0,12) и Cmax. Следовательно, хотя и имеется тенденция к росту накопления веществ в мокроте с увеличением дозы, пропорциональная зависимость фармакокинетических параметров в мокроте от дозы не может быть подтверждена окончательно.
Изучение времени введения
Время введения лекарства составило в среднем около 16 минут у субъектов, принимавших ТРИ (Таблица 9). Для сравнения, время введения для двух капсул ТПИ составило в среднем 1,7 и 2,5 минут для 2×14 мг и 2×28 мг доз, соответственно. Времена введения составили в среднем 4,2, 4,5 и 4,9 минут для ТПИ доз 4×14 мг, 3×28 мг и 4×28 мг, но для этих трех групп второе устройство распаковывалось и использовалось для дозирования третьей и четвертой капсул, соответственно. Следовательно, время введения ТПИ преимущественно возрастает с увеличением количества капсул и с увеличением дозировки.
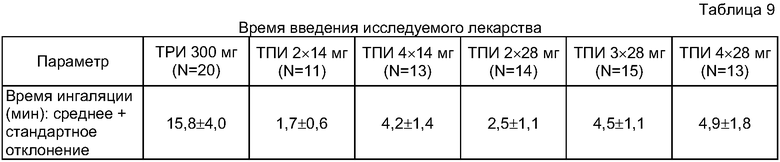
Оценка эксплуатационных показателей ингалятора Т-326 и ТПИ капсул
Из всех капсул только две капсулы ТПИ вводились не так, как требовалось (исключение составляют 3 и 4 капсулы для субъектов из группы ТПИ 4×28 мг). Шум был слышен на втором вдохе при использовании 85% и больше капсул. Ни о каких сбоях ингаляторов не сообщено ни центрами проведения исследования, ни инспекцией аналитиков Nektar.
Общее количество тобрамицина, оставшегося в ингаляторах и капсулах, составило в среднем 10,5% от номинальной дозы. Этот результат включает субъекта 04/409, у которого в устройстве осталось свыше 45% порошка из-за неудачного дозирования, так что более чем вероятно, что действительное количество остающегося в устройствах порошка было еще меньше.
Обработка потерянной или невалидной информации
В ходе исследования, трое рандомизированных субъектов были отстранены от участия в исследовании до того, как они получили исследуемые лекарства из-за предшествующих НЯ. Другой рандомизированный субъект прошел исследование, но позже было обнаружено, что он не принял исследуемое лекарство, поскольку обе капсулы с ТПИ не были проколоты. Скрининг и участие этих субъектов продолжились до тех пор, пока они не были заменены; для замещения были отобраны субъекты со следующими номерами в рандомизационном коде. Один из субъектов предварительно отказался от исследования.
Никакой стратегии переноса данных последнего измерения вперед (LRCF) не применялось для обработки данных по субъектам, отстраненным от исследования на раннем этапе.
Концентрации тобрамицина меньше количественного предела принимались равными нулю при всех расчетах.
Использование субъектами «аэрозольного комплекта доставки»
В протоколе было отмечено, что рандомизированные субъекты, принявшие полную дозу изучаемого лекарства, подходили для оценок фармакокинетики. В ходе проведения исследования непредусмотренное дозирование и вопросы, связанные с состоянием капсул ТПИ, привели к пересмотру оценочного критерия для фармакокинетики. Данные по шести субъектам были исключены из сравнительного анализа фармакокинетических характеристик и доз (четверо не получали исследуемого лекарства и двое не получали полную дозу). Восемьдесят четыре субъекта подходили для фармакокинетических оценок и сравнительного анализа по дозам.
Данные всех 86 участвовавших субъектов, принявших исследуемое лекарство при ингаляции, были включены в оценку времени введения изучаемого лекарства и в оценку количества тобрамицина, остающегося в капсулах и в ингаляторе Т-326.
Активный контроль исследований, призванный показать эквивалентность продуктов
Исследование не было проведено или призвано продемонстрировать клиническую и фармакокинетическую эквивалентность между тестируемыми и контрольными продуктами.
Исследование подгрупп
Следующие подгруппы были проанализированы на основании нормированных на дозу зависимостей (ППК (0,12), ППК (0,∞) 5 и Cmax) - накопления тобрамицина в сыворотке крови после введения ТПИ:
- субъекты младше чем 12 лет и субъекты от 12 и старше (Р>0,4);
- ранее использовавшие сухие порошки и не использовавшие (Р>0,8);
- мужчины и женщины (Р>0,1);
- вес тела (Р>0,2).
Выявлено, что ни одна из этих переменных не влияет на накопление тобрамицина.
При анализе ППК и Cmax было выявлено, что накопление тобрамицина у ТПИ субъектов (N=27), использовавших бронхорасширители через от 15 до 60 минут после дозирования, происходит интенсивнее, чем у тех субъектов, которые не использовали бронхорасширители (N=37), а значения ППК(0,∞), ППК (0,12) и Cmax были выше на 19% (Р=0,0685), 27% (Р=0,0240) и 38% (Р=0,0038) соответственно. Использование бронхорасширителей может увеличить количество тобрамицина, который в итоге попадает в кровеносную систему, вероятно, за счет увеличения количества вещества, оседающего в легких.
Заключение по аэрозольной подаче лекарства
Графики зависимостей средней концентрации в сыворотке от времени после введения ТПИ и ТРИ показывают, что лекарство быстро абсорбируется: среднее tmax составило 1 час при любом лечении. Распределение лекарства оказалось очень быстрым, и уровень снижения концентрации имел вид, характерный для простой экспоненты, со средней конечной скоростью полурастворения от 2,8 до 3,5 часов. Значения фармакокинетических параметров тобрамицина после введения ТРИ соответствовали ранее проведенным исследованиям.
Увеличение дозы ТПИ приводило к увеличению накопления тобрамицина; как свидетельство этому, наблюдался рост значений ППК(0,∞), ППК (0,12) и Cmax. Эта зависимость была несколько слабее, чем она была бы, если бы накопление тобрамицина было пропорционально дозе. Никаких различий в накоплении тобрамицина между субъектами, принявшими 4×14 мг в виде вручную заправленных капсул, и субъектами, принявшими 2×28 капсул, заправленных автоматически, выявлено не было. Вследствие этого, эти две группы были объединены для анализа аналогичных дозировок. В целом, наблюдалась слабая отрицательная корреляция между процентом порошка, оставшегося в ингаляторе, и ППК(0, ∞) (r=-0,22, Р=0,0771), ППК (0,12) (r=-0,23, Р=0,0707) и Cmax (r=-0,22, Р=0,0838). Меньшая чувствительность связывалась с большим оставшимся количеством ТПИ в ингаляторе. Не было обнаружено значительных корреляций между отклонениями от базисного уровня ОФВ1 и сывороточных ППК(0, ∞) (Р=0,9838), ППК (0,12) (Р=0.9990) и Cmax (Р=0,9110).
Концентрации тобрамицина, зафиксированные после введения ТРИ 300 мг, имели значения, попавшие в интервал между значениями концентрации при введении 3×28 мг и 4×28 мг капсул ТПИ. На основании этих результатов видно, что четыре капсулы с 28 мг ТПИ (общая доза весом в 112 мг) обеспечивают наиболее близкое системное накопление к системному накоплению, демонстрируемому при приеме ТРИ.
Анализ подгрупп субъектов, принимавших ТПИ, показал, что возраст, предшествовавшее использование сухих порошков, пол и вес тела не оказывают влияние на накопление тобрамицина из ТПИ, а также, что фармакокинетические характеристики тобрамицина в этих подгруппах ТПИ сходны. Накопление тобрамицина у субъектов, принимавших ТПИ, использовавших бронхорасширители (N=27), было несколько выше, нежели у субъектов, принимавших ТПИ, которые бронхорасширителями не пользовались (N=37).
После введения ТПИ и ТРИ максимум концентрации в мокроте достигался в среднем через 30 минут и среднее время полурастворения варьировалось от 0,8 до 2,2 часов.
Изменения в фармакокинетических характеристиках были значительнее в мокроте, чем в сыворотке крови.
Время введения лекарства составляло в среднем 16 минут для субъектов, принимавших ТРИ. Для сравнения, время введения для двух капсул ТПИ в среднем составило 1,7 и 2,5 минут для 2×14 мг и 2×28 мг доз соответственно. Времена введения составили в среднем 4,2, 4,5 и 4,9 минут для ТПИ доз 4×14 мг, 3×28 мг и 4×28 мг соответственно. Таким образом, время введения ТПИ преимущественно возрастает по мере увеличения количества капсул, а также с увеличением дозировки.
Объем накопленного препарата
Восемьдесят шесть субъектов из 90 участвовавших получили одну дозу изучаемого лекарства, как приведено ниже:
- 12 субъектов получили до 28 мг ТПИ в разовой дозе 2х14 мг
- 13 субъектов получили до 56 мг ТПИ в разовой дозе 4х14 мг
- 14 субъектов получили до 56 мг ТПИ в разовой дозе 2х28 мг
- 15 субъектов получили до 84 мг ТПИ в разовой дозе 3х28 мг
- 13 субъектов получили до 112 мг ТПИ в разовой дозе 4×28 мг
- 20 субъектов получили до 300 мг ТРИ в 5-миллилитровой разовой дозе 5-миллилитрового раствора в пропорции 60 мг/мл.
Краткое заключение по нежелательным явлениям
В ходе или после введения однократной дозы исследуемого лекарства НЯ, вызванные лечением, испытало больше субъектов, принимавших ТПИ (40 из 66 субъектов, 60,6%), чем принимавших ТРИ (6 из 20, 30,0%). Процент субъектов с какой-либо НЯ был сходен для различных уровней дозирования ТПИ (ТПИ 2×14 мг = 45%; 4×14 мг = 54%, 2×28 мг = 64%, 3×28 мг = 67%, 4×28 мг = 69%); размеры образцов были слишком малы, чтобы определить, как тенденция к возникновению любой НЯ связана с увеличением дозы ТПИ. Все вызванные лечением НЯ были слабыми или средними по интенсивностям.
Один из субъектов ТПИ 4×28 мг имел средний по силе кашель и увеличение образуемой мокроты, которые на второй день не прекратились и привели к госпитализации на восьмой день после введения лекарства. Затем, на восьмой день после прохождения исследования субъектом, эти НЯ были классифицированы как СНЯ. Ни одно из этих СНЯ, выявленных у субъектов, не могло быть отнесено к лечению ТПИ. Другой субъект, принимавший ТПИ 4×28 мг, имел несерьезные НЯ средней тяжести, вероятно относящиеся к лечению, а именно усиление кашля, дисгевзию (расстройство вкуса) и слезоточивость, что привело к прерыванию приема лекарства с последующим прекращением; затем субъект отозвал согласие и был исключен из исследования. Четверо субъектов имели несерьезные НЯ в форме кашля при ингаляции ТПИ, что привело к коррекции, прерыванию и откладыванию приема доз (один субъект из группы ТПИ 4×14 мг, другой 4×28 мг и двое из 3×28 мг). Каждая НЯ была рассмотрена исследователем как возможно относящаяся к лечению ТПИ.
Среди НЯ, испытанных большинством субъектов, принимавших ТПИ, были кашель и усиление кашля (13 из 66 субъектов = 19,7%; 12 из 13 субъектов, принимавших ТПИ, сообщали о кашле как о базисном симптоме еще до начала исследуемого лечения); дисгевзия (11 субъектов, 16,7%); фарингит, кровохаркание и ринорея (4 субъекта, 6,1% каждое); рост мокротообразования, крепитация (потрескивание в легких), рост слезоточивости, боли в области живота, головокружение, головные боли (без пояснений) и раздражение в горле (3 субъекта, 4,5% каждое). Случаи кашля, усиления кашля и дигевзии медленно возрастали по мере увеличения дозы ТПИ, но никаких убедительных тенденций к возникновению индивидуальных НЯ по полученным данным не наблюдалось. Для сравнения, не более чем один субъект, принимавший ТРИ, испытывал какую-либо НЯ. Субъекты, принимавшие ТРИ, не испытывали никакого кашля, усиления кашля или дисгевзии. Однако большинство субъектов, принимавших ТРИ (17 из 20, 85%), получали ТРИ как часть их обычной терапии.
Исследователи рассмотрели все случаи кашля и его усиления, все случаи дисгевзии и большинство случаев кровохаркания и раздражения горла как вероятно или возможно относящиеся к лечению ТПИ. Для сравнения, единичные случаи сдавленности в груди, простого герпеса, роста числа эозинофильных клеток, сухость горла, фарингит, рост мокротообразования и вязкости мокроты рассматривались как относящиеся к лечению ТРИ.
Неожиданные НЯ при лечении: все случаи
Сорок из 66 субъектов, принимавших ТПИ, и шестеро из 20 субъектов, принимавших ТРИ, имели неожиданные лечебные НЯ в ходе или после самого лечения. Процент субъектов с какой-либо НЯ был сходен для групп с разным уровнем дозы ТПИ. Все неожиданные лечебные НЯ были слабыми или умеренными по силе. НЯ, перенесенные наибольшим числом субъектов, принимавших ТПИ, были кашель и усиление кашля (13 из 66 субъектов = 19,7%); дисгевзия (11 субъектов, 16,7%); фарингит, кровохаркание и ринорея (4 субъекта, 6,1% каждый); рост мокротообразования, крепитация, рост слезоточивости, боли в области живота, головокружения, головные боли (без пояснений) и раздражение в горле (3 субъекта, 4,5% каждое). Не более одного субъекта, принимавшего ТРИ, имели какие-либо НЯ. Субъекты, принимавшие ТРИ, не испытывали ни кашля, ни усиления кашли, ни дисгевзии.
Неожиданные лечебные НЯ, вероятно вызванные лечением
Двадцать четыре из 66 субъектов (36,4%) и 2 из 20 субъектов, принимавших ТРИ (10%), имели НЯ, которые рассматривались исследователями как вероятно или возможно относящиеся к лечению (Таблица 12). Исследователи рассмотрели большинство случаев кашля и усиления кашля (10 из 66 ТПИ субъектов = 15,2%) и все случае дисгевзии (16,7%) как вероятно или возможно относящиеся к лечению ТПИ (Таблица 11). Кровохаркание и раздражения в горле (обе 4,5%) и рост слезоточивости (3,0%) также рассматривались как относящиеся к лечению ТПИ. Для сравнения, единичные случаи сдавленности в груди, простого герпеса, роста числа эозинофильных клеток, сухости в горле, фарингита, роста мокротообразования и вязкости мокроты рассматривались как относящиеся к лечению ТРИ.
Другие серьезные нежелательные явления
Один из субъектов, принимавших ТПИ 4×28 мг, имел СНЯ на второй день (средней силы кашель и рост мокротообразования), что привело к госпитализации из-за обострения болезни легких КФ на восьмой день после принятия разовой дозы исследуемого лекарства. Ни одно из СНЯ, имевшихся у данного субъекта, не было признано относящимся к лечению ТПИ.
Отказ субъекта от участия в исследовании из-за нежелательных явлений, относящихся лечению
Один из субъектов, принимавших ТПИ 4×28 мг, испытывал среднее усиление кашля, дисгевзию и рост слезоточивости, что привело к прерыванию и остановке введения исследуемого лекарства, а субъект был отстранен от исследования. Каждое НЯ рассматривалось исследователем как возможно относящееся к лечению ТПИ.
Прерывания в дозировании из-за нежелательных явлений, вызванных лечением
У четырех субъектов был кашель, который привел к обострению, прерыванию или задержкам при дозировании (у одного субъекта, принимавшего ТПИ 4×14 мг, другого - 4×28 мг и у двоих - 3×28 мг). Каждое НЯ рассматривалось исследователем как возможно относящееся к лечению ТПИ.
Анализ и обсуждение смертей, других серьезных нежелательных явлений и других значимых нежелательных явлений
Ни одной смерти в ходе данного исследования не произошло. Единственными СНЯ были умеренный кашель и рост мокротообразования, отмеченные на второй день у единственного субъекта, приведшие к госпитализации в восьмой день после прохождения субъектом всего исследования; эти реакции были признаны не относящимися к изучаемому лечению. Пять субъектов, принимавших ТПИ, продемонстрировали умеренный кашель или усиление кашля спустя минуты после ингаляции ТПИ, что привело одного из субъектов (у которого также проявилась дисгевзия и рост слезоточивости) к отказу от исследования и четверых к прерываниям дозирования. Все субъекты сообщали о базовых респираторных симптомах и среди тех, кто сообщил о кашле как НЯ, все кроме одного субъекта также испытали усиление кашля как НЯ. Эти НЯ проходили через 5-35 минут без всякого вмешательства или с минимальным вмешательством. Четверо последних субъектов начали заново и успешно завершили дозирование через пару минут после первоначальной остановки. Эти НЯ возможно относились к изначальной бронхиальной раздражимости, которая часто сопровождает кистозный фиброз, хотя исследователь рассматривал каждую реакцию как возможно относящуюся к введению изучаемого лекарства. Таким образом, в этом исследовании, ТПИ каждого уровня дозировки и ТРИ 300 мг/5 мл продемонстрировали хорошую переносимость.
Состав крови
Никаких значительных отклонений от базисного уровня в результатах последующих гематологических тестов для групп, принимавших ТРИ и ТПИ, не наблюдалось. Для групп, принимавших ТРИ и ТПИ, не было зафиксировано существенных различий в среднем изменении базисного уровня, а также никаких следов роста среднего отклонения по мере увеличения дозы ТПИ на основании любого гематологического теста. Не наблюдалось никакого значимого роста по сравнению с базисным уровнем для последующих тестов групп, принимавших ТРИ или ТПИ, в сторону превышения или уменьшения от нормального уровня гематологических тестов. При увеличении дозы ТПИ также не было обнаружено существенных различий между группами ТРИ и ТПИ и никаких следов увеличения отклонений для гематологических результатов, отстоящих от среднего значения. Один субъект, принимавший ТРИ, имел клинически значимый последующий рост эозинофильных клеток, определенный как НЯ.
Химический состав сыворотки
Никаких значимых средних отклонений от базисного уровня в последующих результатах для групп, принимавших ТРИ и ТПИ, не наблюдалось. Не было никаких важных различий между группами ТРИ и ТПИ в среднем изменении базового уровня и никаких следов роста среднего отклонения по мере увеличения дозы ТПИ на основании любого химического теста. Не наблюдалось никакого значимого роста по сравнению с базисным уровнем для последующих тестов групп, принимавших ТРИ или ТПИ, в сторону превышения или уменьшения от нормального уровня химии сыворотки крови. При увеличении дозы ТПИ также не было обнаружено существенных различий между группами ТРИ и ТПИ и никаких следов увеличения отклонений для химических результатов, отстоящих от среднего значения.
Один субъект имел клинически значимый последующий результат по глюкозе; этот результат был отнесен к КФ-диабетам, не был записан как НЯ и не был отнесен к лечению ТПИ.
Полосковый тест на белок в моче
Поскольку полосковый тест на белок в моче - это качественный тест, количественных изменений от базового уровня для последующих результатов подсчитано не было. Не наблюдалось никакого значительного превышения базового уровня вплоть до окончания исследования для ТРИ или ТПИ групп, на что указывали результаты полоскового теста на белок в моче, не превышающие положительное значение 2+.
Единственный 3 + результат был отмечен у субъекта, который получал дозу ТПИ 4×28 мг.
Индивидуальные отклонения при изучении состава крови
С единственным исключением, никаких клинически значимых результатов изучения состава крови в ходе исследования не наблюдалось. Субъект ТРИ 12/405 имел клинически значимый последующий результат по эозинофилам в 9,0% (нормальный диапазон от 0 до 6%), который был определен как НЯ и рассматривался исследователем как возможно относящийся к лечению ТРИ. Результат базового уровня по эозинофилам субъекта был выше предела нормального (5,9%), но субъект имел повышенный уровень эозинофилов за восемь дней до базового результата. Клиническая важность конечного числа эозинофилов в 9,0% неясна.
Химический состав сыворотки
С единственным исключением, никаких клинически значимых результатов химии сыворотки в ходе исследования не наблюдалось. Единственный субъект (ТПИ 3×28 мг) имел клинически значимый результат по глюкозе; этот результат был отнесен к диабету, связанному с КФ.
Полосковый тест на белок в моче
С единственным исключением, никаких клинических значимых результатов полоскового теста на белок в моче в ходе исследования не наблюдалось. Один субъект (ТПИ 4×28 мг) имел положительный 3 + результат на шестой день; этот субъект имел положительный 1 + результат при скрининге и при повторном тесте вне графика в день дозирования. Координатор исследования предположил, что 3 + белок в моче - результат, связанный с началом СНЯ (кашель и рост мокротообразования) на второй день, приведших к госпитализации субъекта.
Бронхоспазм и острая болезнь легочной функции
Единственный случай бессимптомного бронхоспазма (падение ОФВ1 должн. на 20,9% через 30 минут после ингаляции ТПИ 2×28 мг; (Таблица 11)) наблюдался в ходе спирометрической процедуры, которая не была проведена в соответствии со стандартами Американского торакального общества. Если бы процедура проводилась по стандарту, снижение ОФВ1 могло бы быть менее 11%. Такое же бессимптомное снижение ОФВ1 должн. также наблюдалось у ТРИ субъекта (снижение на 19.1%). Еще один субъект, принимавший ТРИ, и шесть других субъектов, принимавших ТПИ, имели падение больше чем 10%, но меньше чем 20% от ОФВ1 должн. (Таблица 11). У троих из шести субъектов, принимавших ТПИ (субъект 02/507, принимавший дозу ТПИ в 3×28 мг, и субъекты 01/415 и 08/406, принимавшие дозу ТПИ в 4×28 мг), был кашель или усиление кашля через несколько минут после дозирования.
Количественные изменения в легочной функции
Среднее изменения ОФВ1 должн., ФЖЕЛ % должн. и СОС25-75% должн. у ТРИ и всех ТПИ групп было относительно стабильно с начала скрининга до дозирования 1 (не включая) и со дня 1 дозирования и до дня 8 последнего визита. Не было выявлено никаких существенных различий для ТРИ и ТПИ групп и никаких существенных относящихся к дозировке различий для ТПИ групп в среднем изменении предсказанного процента ОФВ1 должн., ФЖЕЛ или СОС25-75 для 30 минут после дозирования.
Основные жизненные показатели
Не было выявлено значительных или сопоставимых различий между ТПИ и ТРИ группами в основных жизненных показателях или в изменениях основных жизненных показателей в ходе всего исследования.
Сопутствующие лекарства
Тридцать восемь из 90 субъектов принимали сопутствующие лекарства и терапии после дозирования. Не было выявлено никаких различий для разных видов лекарств и терапий.
Заключения о безопасности
Вызванные лечением неожиданные НЯ в ходе и после лечения испытало больше субъектов, принимавших ТПИ (60,6%), нежели субъектов, принимавших ТРИ (30,0%). Процент субъектов с НЯ был сходен среди различных уровней дозы ТПИ (от 45% до 69%); размеры образцов были слишком малы, чтобы определить, наблюдается ли тенденция к увеличению вероятности появления какой-либо НЯ при увеличении дозы ТПИ. Все неожиданные лечебные НЯ были слабыми или умеренными по силе.
Один из субъектов, принимавших ТПИ 4×28 мг, имел две СНЯ (умеренный кашель и рост мокротообразования), согласующиеся с обострением легочного заболевания КФ, что привело к госпитализации на восьмой день после применения разовой дозы исследуемого лекарства; ни одно из этих СНЯ не рассматривалось как СНЯ, относящееся к лечению. Другой субъект, принимавший ТПИ 4×28 мг, имел умеренный, возможно связанный с лечением, учащенный кашель, дисгевзию и рост слезоотделения, что привело к прерыванию приема изучаемого лекарства, а затем и к остановке; после этого субъект отозвал согласие и был исключен из исследования. У четырех субъектов был кашель в ходе ингаляции ТПИ, что привело к коррекции, прерыванию и откладыванию введения доз (один из субъектов, принимавший ТПИ 4×14 мг, другой - ТПИ 4×28 мг, и двое субъектов, принимавших ТПИ 3×28 мг); каждая НЯ рассматривалась исследователем как возможно относящаяся к лечению ТПИ. Однако во всех четырех случаях дозирование было только кратковременно прервано и субъекты продолжили и завершили дозирование без срывов. У всех субъектов в данном исследовании имели место базисные респираторные симптомы: приблизительно по 90% субъектов от каждой когорты сообщали о кашле. Надо отметить, что наличие слабого кашля не является неожиданным при первом применении сухого порошкового ингалятора, особенно, если уже имеются нарушения в дыхательных путях.
НЯ, испытанными большинством субъектов, принимавших ТПИ, были кашель или усиление кашля (13 из 66 субъектов = 19,7%; 12 из 13 субъектов первоначально сообщали о кашле как о базисном симптоме до начала лечения); дисгевзия (11 субъектов, 16,7%); фарингит, кровохаркание и ринорея (4 субъекта, 6,1% каждое); рост мокротообразования, крепитация, рост слезоточивости, боли в области живота, головокружение, головные боли (без пояснений) и раздражение в горле (3 субъекта, 4,5% каждое). Наличие кашля, усиление кашля и дисгевзия слабо увеличиваются по мере увеличения дозы ТПИ. Никто, кроме одного из ТРИ субъектов, не испытывали никаких НЯ. У субъектов, принимавших ТРИ, не было кашля, усиления кашля или дисгевзии, но 85% принимавших ТРИ постоянно употребляли ТРИ.
Исследователи рассмотрели все случаи кашля и его усиления, все случаи дисгевзии и большинство случаев кровохаркания и раздражения горла как вероятно или возможно относящихся к лечению с помощью ТПИ. Для сравнения, единичные случаи сдавленности в груди, простого герпеса, роста числа эозинофильных клеток, сухости горла, фарингита, роста мокротообразования и вязкости слюны рассматривались как относящиеся к лечению ТРИ.
Был зафиксирован единственный случай бессимптомного бронхоспазма (снижение на 20,9% ОФВ1 должн.), аналогичный бессимптомному бронхоспазму (снижение ОФВ1 должн. на 19,1%) у испытуемого, принимавшего ТРИ.
Не было зафиксировано значимых изменений по сравнению с базовым уровнем клинических лабораторных результатов, никаких следов усиления изменений по мере увеличения дозы ТПИ, никаких увеличений в частоте результатов выше и ниже нормального значения и никаких явных различий между ТПИ и ТРИ. Только у одного субъекта, принимавшего ТРИ, рост эозинофильных клеток был квалифицирован как НЯ.
Обсуждение и итоговые заключения
Введение одной дозы ТПИ приводит к более эффективной доставке тобрамицина, чем тобрамициновый раствор для ингаляции TOBI®, при поддержании сходной фармакокинетки тобрамицина. Систематическое накопление, достигаемое после введения тобрамицинового раствора для ингаляции TOBI®, было сходно с тем, что наблюдалось при введении 4×28 мг капсул ТПИ с эквивалентно рассчитанной дозой в 115 мг ТПИ. Следовательно, четыре капсулы по 28 мг ТПИ (всего 112 мг) должны производить такое же системное накопление, что и аналогичная доза 300 мг тобрамицинового раствора для ингаляции TOBI®.
Минимальные прерывания дозирования (временные во всех, кроме одного, случаях), вызванные кашлем, подверглись изучению, которое показало, что такие прерывания в приеме доз не исказили фармакокинетические характеристики разовой дозы тобрамицина в сыворотке после введения ТПИ.
Примерно у 20% субъектов, принявших ТПИ, появился кашель, незначительный в большинстве случаев, иногда он приводил к коротким перерывам в приеме доз и обычно быстро проходил. Кашель - это ожидаемое НЯ при любой терапии с ингаляциями сухим порошком, присутствовавшее у большинства субъектов еще на базовом уровне. В то время как наличие НЯ было выше у лечащихся ТПИ, чем у тех, кто лечился ТРИ, большинство субъектов в большинстве групп получали тобрамициновый раствор для ингаляции TOBI® для длительного периодического применения.
В одном случае, молодой субъект забыл воспользоваться своим обычным лекарством (длительно действующим бронхостимулятором) и испытывал умеренный кашель после дозирования, вследствие чего он отказался от дальнейшего приема. Утрата обычной бронхиальной защиты - наиболее вероятная причина такой реакции.
Введение одной дозы ТПИ в дозах от 28 мг до 112 мг хорошо переносилось больными КФ в ходе исследования. Сто двадцать миллиграмм порошка ТПИ дает близкое систематическое накопление тобрамицина, к тому, что имеется при приеме тобрамицинового раствора для ингаляции TOBI®.
Это исследование демонстрирует, что ингаляция с использованием способов изобретения с простым ингаляционным устройством производится намного быстрее и эффективнее, чем ингаляция стандартной дозы тобрамицинового раствора для ингаляции TOBI® через небулайзер. Побочные эффекты, такие как усилившийся кашель и раздражение в горле после ингаляции ТПИ, не были неожиданны и зависели от количества порошка, содержащегося в каждой дозе. Однако, в большинстве случаев эффект был мягким и субъекты высоко оценили скорость и простоту дозирования ТПИ. Недостаток, связанный с сопутствующим кашлем и нарушением вкуса у группы, принимавшей тобрамициновый раствор для ингаляции TOBI®, может быть отнесена к тому факту, что 85% из субъектов использовали тобрамициновый раствор для ингаляции TOBI® на постоянной основе еще до исследования. Эти субъекты могли привыкнуть к эффекту влажной аэрозольной формы тобрамицина.
Таким образом, введение одной дозы сухого порошка тобрамицина приводит к более эффективной доставке тобрамицина, чем тобрамициновый раствор для ингаляции TOBI®, при поддержании сходной кинетики тобрамицина. Системное накопление, достигаемое после введения тобрамицинового раствора для ингаляции TOBI®, было очень сходно с тем, что наблюдалось после введения 4×28 мг капсул ТПИ с эквивалентной дозой, рассчитанной как 115 мг ТПИ. Следовательно, четыре капсулы 28 мг ТПИ (всего 112 мг) должны вызывать то же системное накопление, что и 300 мг тобрамицинового раствора для ингаляции TOBI®.
В то время как технические результаты изобретения были здесь приведены и описаны, было бы ценно, чтобы были произведены различные изменения без ухода от духа и за границы изобретения.
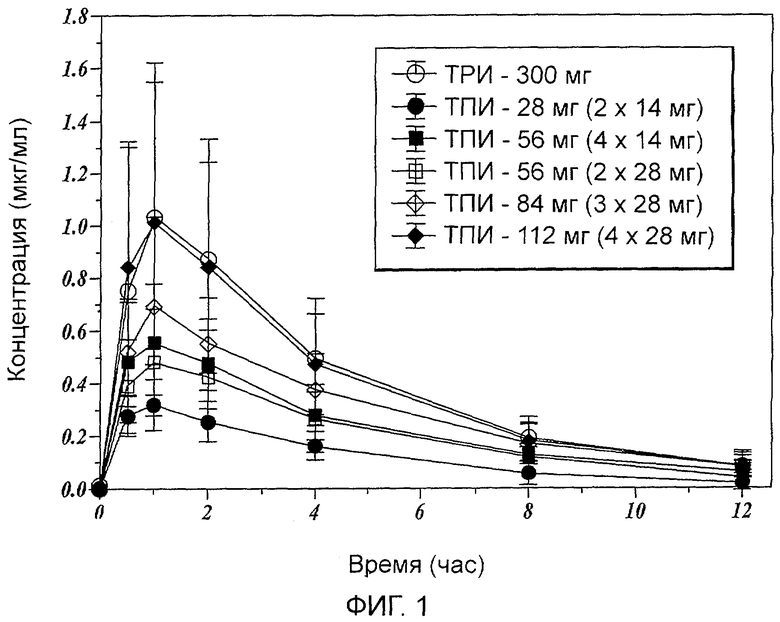
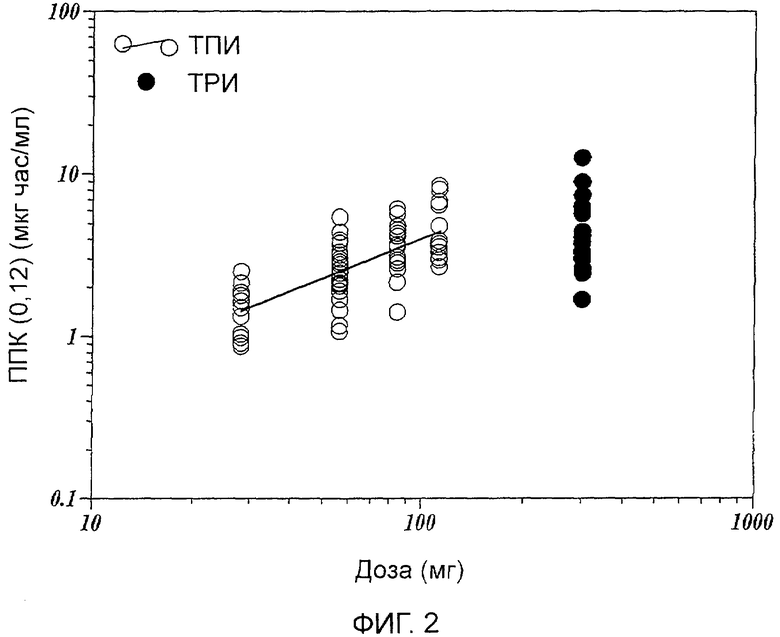
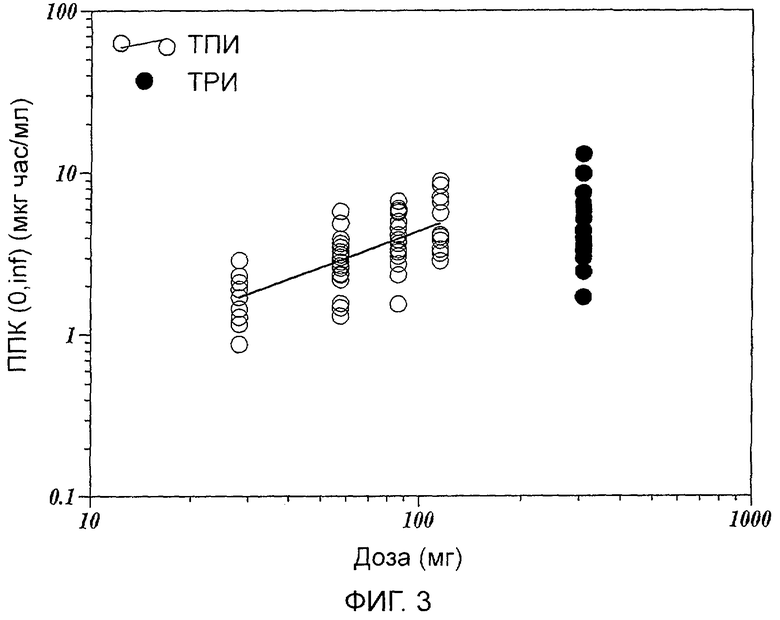
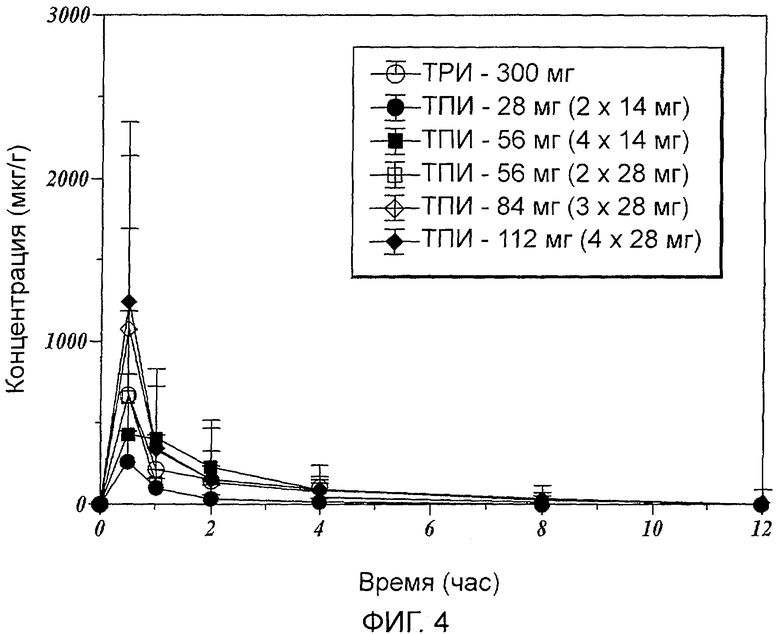
Изобретение относится к медицине, а именно к пульмонологии. Изобретение содержит два варианта осуществления способа. Оба его варианта включают введение в эндобронхиальную систему пациента сухого порошкового аэрозольного препарата тобрамицина. В первом варианте одна доза содержит от 90 до 130 мг тобрамицина или его фармацевтически приемлемой соли. Одну дозу на первом лечебном этапе вводят от одного до трех раз в день. Длительность первого лечебного этапа от 20 до 36 дней. Во втором варианте одна доза содержит от 110 до 115 мг тобрамицина. Эту лечебную дозу на первом лечебном этапе вводят дважды в день. Первый лечебный этап длится 28 дней. За лечебным этапом следует второй нелечебный этап. Нелечебный этап длится от 26 до 30 дней. В этот этап в эндобронхиальную систему больного не вводят никакого антибиотика. Затем первый и второй лечебные этапы повторяют. Способ обеспечивает достижение необходимого фармакологического эффекта введением меньшей дозы сухого порошка тобрамицина в более короткие сроки при лечении синегнойной инфекции у больных кистозным фиброзом. 2 н. и 22 з.п. ф-лы, 11 табл., 4 ил.
1. Способ лечения эндобронхиальной синегнойной инфекции у пациента с кистозным фиброзом, включающий введение в эндобронхиальную систему пациента сухого порошкового аэрозольного препарата, содержащего от 90 до 130 мг тобрамицина или его фармацевтически приемлемой соли на дозу, причем одна доза композиции вводится от одного до трех раз в день на первом лечебном этапе от 20 до 36 дней.
2. Способ по п.1, в котором за первым лечебным этапом следует второй нелечебный этап, когда в эндобронхиальную систему пациента не вводят никаких аминогликозидных антибиотиков.
3. Способ по п.1, в котором одна доза сухого порошкового аэрозольного препарата содержит 100 до 120 мг тобрамицина.
4. Способ по п.3, в котором одна доза сухого порошкового аэрозольного препарата содержит от 110 до 115 мг тобрамицина.
5. Способ по п.1, в котором одна доза сухого порошкового аэрозольного препарата, содержащая от 90 до 130 мг тобрамицина, вводится пациенту разделенной на две-шесть единиц.
6. Способ по п.5, в котором одна доза сухого порошкового аэрозольного препарата вводится пациенту разделенной на три - пять единиц.
7. Способ по п.6, в котором одна доза сухого порошкового аэрозольного препарата вводится пациенту разделенной на четыре единицы.
8. Способ по п.2, в котором второй нелечебный этап длится от 20 до 36 дней.
9. Способ по п.2, в котором первый лечебный этап длится от 26 до 30 дней.
10. Способ по п.9, в котором второй нелечебный этап длится от 26 до 30 дней.
11. Способ по п.2, в котором первый лечебный этап длится 28 дней.
12. Способ по п.11, в котором второй нелечебный этап длится от 26 до 30 дней.
13. Способ по п.2, в котором лечебный курс, включающий первый лечебный этап и следующий за ним второй нелечебный этап, повторяют множество раз.
14. Способ по п.1, в котором сухой порошковый аэрозольный препарат содержит частицы, по крайней мере, 50% которых имеют аэродинамический диаметр в диапазоне от 1 мкм до 5 мкм.
15. Способ по п.14, в котором частицы представляют из себя обладающие пористой структурой сферические частицы на основе фосфолипида.
16. Способ по п.15, в котором частицы содержат, как минимум, 75% тобрамицина, 2-25% фосфолипида и 0-5% иона металла и имеют общую плотность более 0,08 г/см3.
17. Способ по п.15, в котором частицы содержат, как минимум, 85% тобрамицина, 8-18% фосфолипида и 0-5% хлорида кальция и имеют общую плотность более 0,12 г/см3.
18. Способ по п.1, в котором сухой порошковый аэрозольный препарат вводят пациенту с помощью сухого порошкового ингалятора.
19. Способ по п.1, в котором сухой порошковый аэрозольный препарат размещают в отдельном контейнере, размещенном внутри сухого порошкового ингалятора, и аэрозольный порошок доставляют ингалятором из контейнера в легкие при помощи ингалятора.
20. Способ по п.1, в котором сухой порошковый аэрозольный препарат помещают внутрь нескольких контейнеров, размещенных внутри сухого порошкового ингалятора, и аэрозольный порошок доставляется ингалятором из контейнеров в легкие при помощи ингалятора.
21. Способ лечения эндобронхиальной синегнойной инфекции у пациента с кистозным фиброзом, включающий введение в эндобронхиальную систему пациента сухого порошкового аэрозольного препарата, содержащего от 110 до 115 мг антибиотика тобрамицина на дозу, причем одна доза композиции вводится дважды в день на первом лечебном этапе в 28 дней, за которым следует второй нелечебный этап от 26 до 30 дней, когда в эндобронхиальную систему больного не вводят никакого антибиотика тобрамицина, а затем первый и второй лечебные этапы повторяют.
22. Способ по п.21, в котором одна доза сухого порошкового аэрозольного препарата, содержащая от 110 до 115 мг антибиотика тобрамицина, вводится пациенту разделенной на три-пять единиц.
23. Способ по п.22, в котором одна доза сухого порошкового аэрозольного препарата, содержащая от 110 до 115 мг антибиотика тобрамицина, вводится пациенту разделенной на четыре единицы.
24. Способ по п.21, в котором первый и второй лечебные этапы повторяют множество раз.
| WO 2000035461, 22.06.2000 | |||
| Способ лечения больных хроническими заболеваниями легких | 1984 |
|
SU1335297A1 |
| СИСТЕМА ДОСТАВКИ ЛЕКАРСТВА | 1999 |
|
RU2202340C2 |
| Кипятильник для воды | 1921 |
|
SU5A1 |
| СЕМЫКИН С.Ю | |||
| Эффективность и безопасность применения ципрофлоксацина при лечении обострений бронхолегочного процесса у детей с муковисцидозом: Автореферат кандидатской диссертации, М., 2002, с 8-11, 23 | |||
| RAMSEY B.W | |||
| et al | |||
| «Intermittent | |||

