Предпосылки создания настоящего изобретения
Область изобретения
В настоящем изобретении предлагается новый класс соединений, фармацевтические композиции, включающие указанные соединения и способы применения указанных соединений для лечения или профилактики заболеваний или нарушений, ассоциированных с аномальной или разрегулированной киназной активностью, прежде всего заболеваний или нарушений, вызванных аномальной активацией киназ Abl, Bcr-Abl, Bmx, ВТК, b-RAF, c-RAF, CSK, cSRC, Fes, FGFR3, Flt3, IKKα, IKKβ, JNK1α1, JNK2α2, Lck, Met, MKK4, MKK6, p70S6K, PAK2, PDGFRα, PKA, PKCα, PKD2, ROCK-II, Ros, Rsk1, SAPK2α, SAPK2β, SAPK3, SAPK4, SGK, Syk, Tie2 и TrkB.
Протеинкиназы представляют собой многочисленное семейство белков, которые играют центральную роль в регуляции широкого круга клеточных процессов и в осуществлении контроля клеточных функций. Перечень таких киназ включает, без ограничения перечисленным, рецепторные тирозинкиназы, такие как рецепторная киназа тромбоцитарного фактора роста (PDGF-R), рецептор фактора роста нервов, trkB, Met, и рецептор фактора роста фибробластов, FGFR3; не-рецепторные тирозин киназы, такие как Abl и гибридная киназа BCR-Abl, Lck, Csk, Fes, Bmx и c-src, а также серин/треонинкиназы, такие как c-RAF, sgk, киназы MAP (например, МКК4, MKK6 и т.п.) и SAPK2α, SAPK2β и SAPK3. Аномальная активность киназ наблюдается при многих патологических состояниях, включая доброкачественные и злокачественные пролиферативные заболевания, а также заболевания, обусловленные аномальной активацией иммунной и нервной систем.
Новые соединения по настоящему изобретению ингибируют активность одной или более протеинкиназ и, следовательно, их можно использовать для лечения заболеваний, ассоциированных с киназами.
Краткое описание сущности изобретения
В одном объекте настоящего изобретения предлагаются соединения формулы I
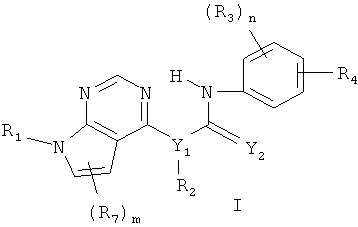
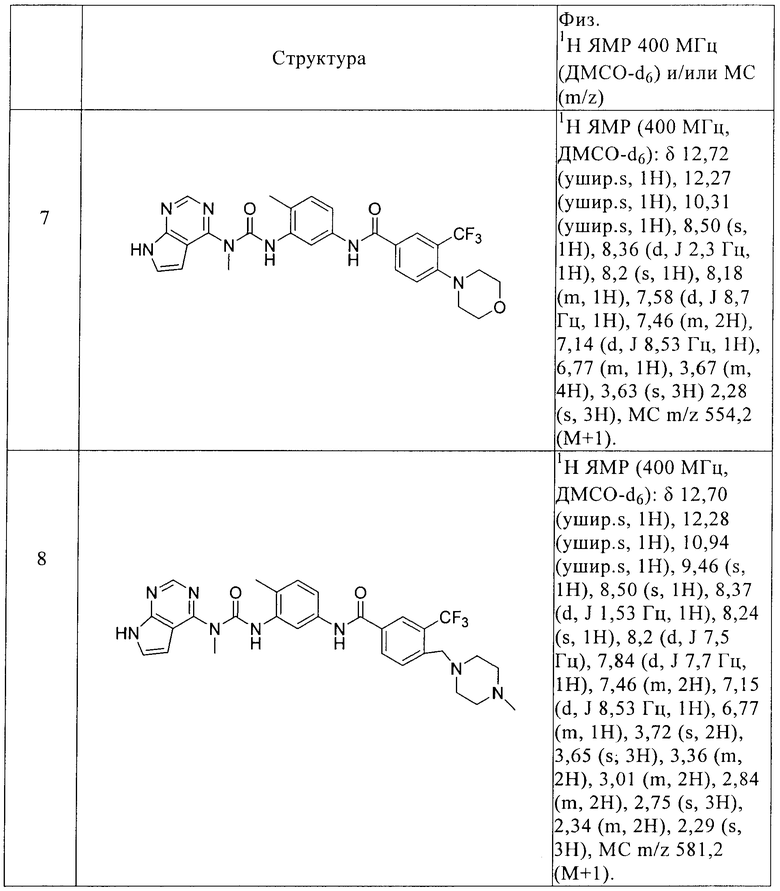
где
n равно 0, 1 и 2,
m равно 0, 1 и 2,
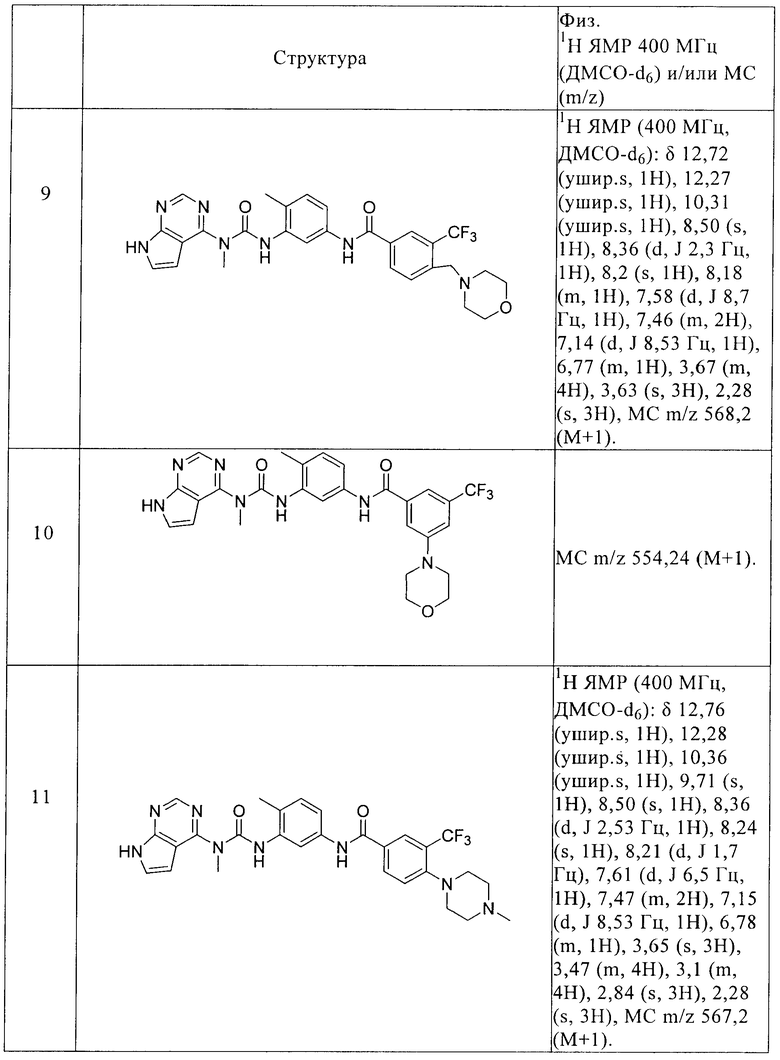
Y1 выбирают из N и CR5, причем R5 выбирают из водорода и С1-С6алкила,
Y2 выбирают из О и S,
R1 выбирают из водорода и С1-C6алкила,
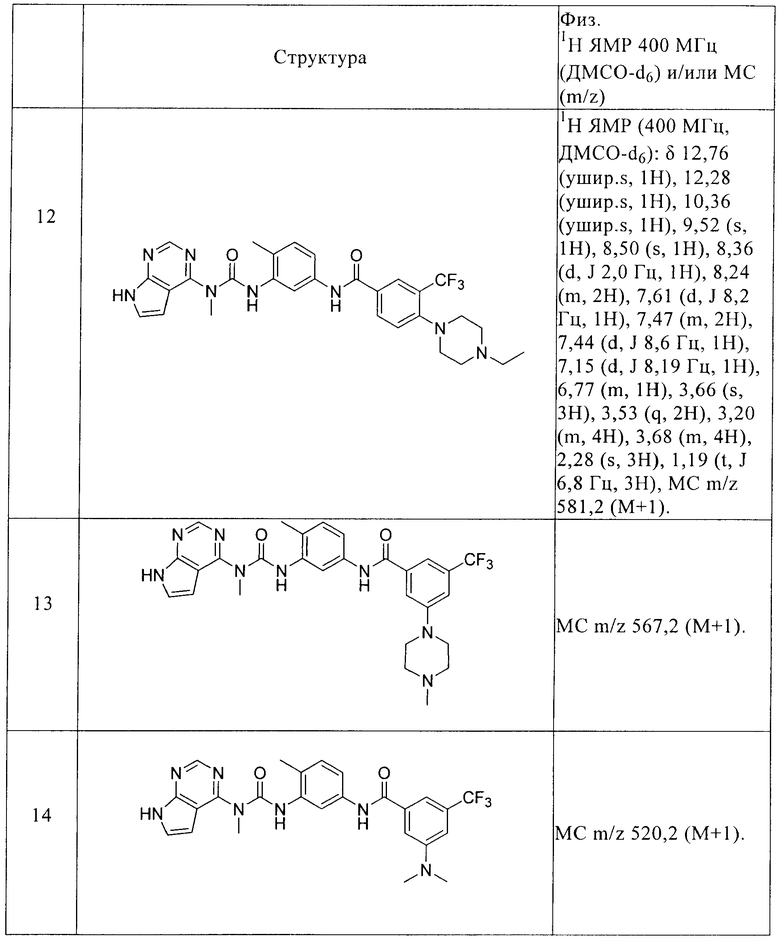
R2 выбирают из водорода и С1-С6алкила,
R3 выбирают из группы, включающей водород, галоген, гидрокси, С1-С6алкил, C1-С6алкокси, галогензамещенный C1-С6алкил и галогензамещенный C1-С6алкокси,
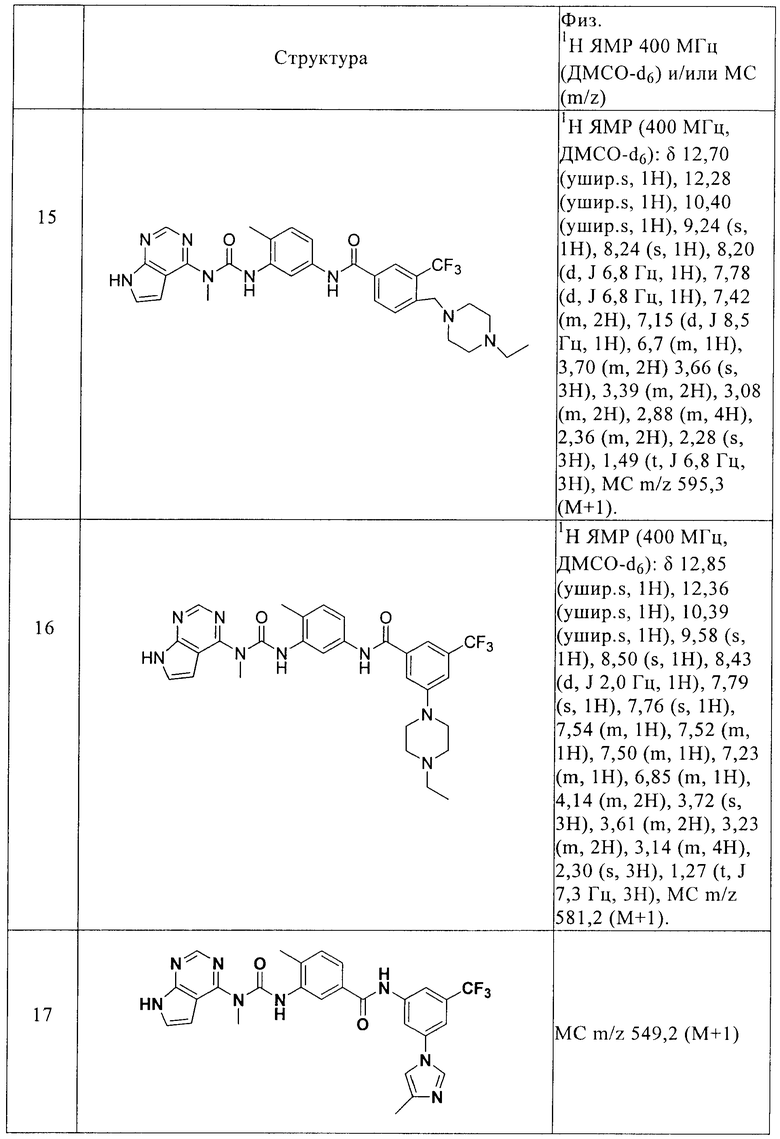
R4 выбирают из -NR5C(О)R6 и -C(О)NR5R6, где R5 выбирают из водорода и C1-С6алкила, а R6 выбирают из группы, включающей С6-С10арил(С0-С4)алкил, С5-С10гетероарил (С0-С4)алкил, С3-С10циклоалкил(С0-С4)алкил и С3-С10гетероциклоалкил(С0-С4)алкил, причем любой арил, гетероарил, циклоалкил или гетероциклоалкил в составе R6 необязательно замещен 1-3 радикалами, выбранными из группы, включающей галоген, гидрокси, -NR5R5, C1-С6алкил, C1-С6алкокси, галогензамещенный C1-С6алкил и галогензамещенный C1-С6алкокси, С5-С10гетероарил (С0-С4)алкил, С3-С8гетероцикло (С0-С4) алкил и С3-С8гетероцикло(С0-С4)алкокси, причем любой гетероарил или гетероциклоалкил в составе R6 необязательно замещен 1-3 радикалами, независимо выбранными из группы, включающей C1-С6 алкил и гидрокси(С1-С6)алкил,
R7 выбирают из группы, включающей водород, галоген, гидрокси, С1-С6алкил, C1-С6алкокси, галогензамещенный C1-С6алкил, галогензамещенный С1-С6алкокси, С6-С10арил(С0-С4)алкил, С3-С10гетероарил(С0-С4)алкил, С3-С10циклоалкил (С0-С4)алкил и С3-С10гетероциклоалкил(С0-С4)алкил, а также N-оксиды, пролекарства, защищенные производные, индивидуальные изомеры и смеси изомеров, а также фармацевтически приемлемые соли и сольваты (например, гидраты) таких соединений.
Во втором объекте настоящего изобретения предлагается фармацевтическая композиция, включающая соединение формулы I или N-оксид, индивидуальные изомеры и смеси изомеров, или их фармацевтически приемлемые соли, в смеси с одним или более пригодных эксципиентов.
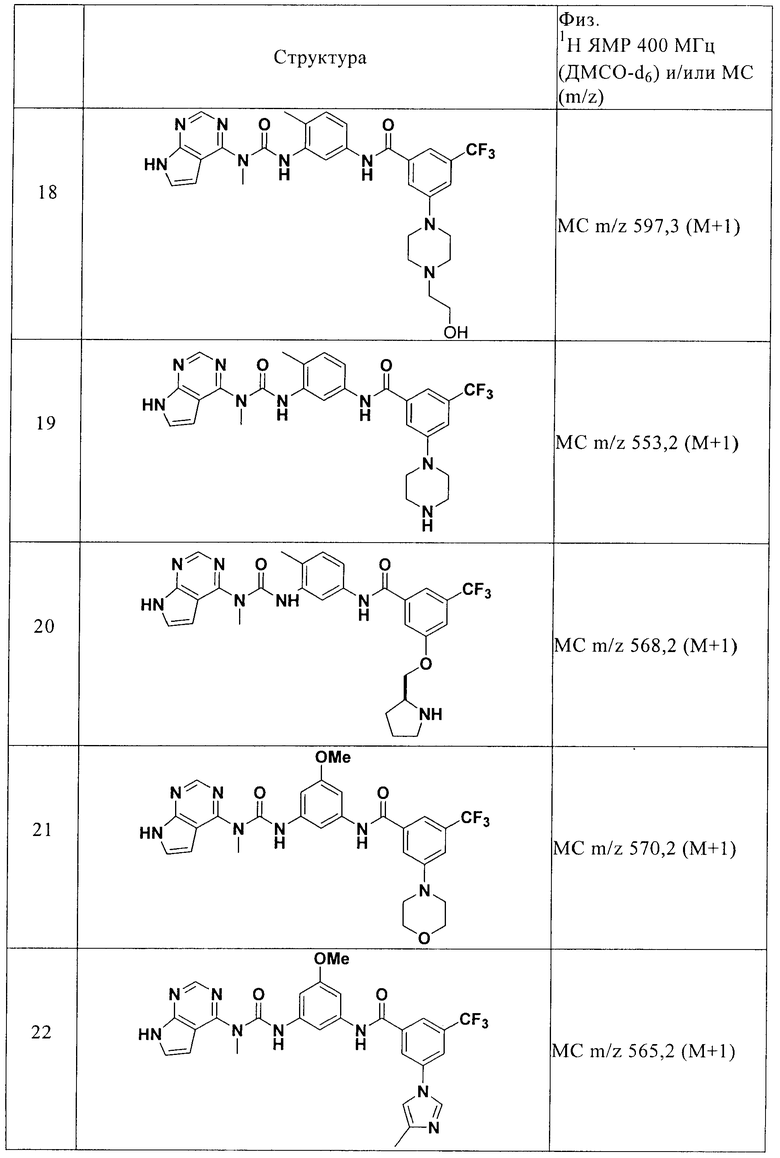
В третьем объекте настоящего изобретения предлагается способ лечения заболевания у животного, который заключается в том, что при ингибировании киназной активности, прежде всего активности Abl, Bcr-Abl, Bmx, ВТК, b-RAF, c-RAF, CSK, cSRC, Fes, FGFR3, Flt3, IKKα, IKKβ, JNK1α1, JNK2α2, Lck, Met, МКК4, МКК6, p70S6K, PAK2, PDGFRα, PKA, PKCα, PKD2, ROCK-II, Ros, Rskl, SAPK2α, SAPK2β, SAPK3, SAPK4, SGK, Syk, Tie2 и/или TrkB, наблюдается предотвращение, подавление или снижение интенсивности заболевания и/или симптомов заболеваний, причем способ включает введение животному терапевтически эффективного количества соединения формулы I или N-оксида, индивидуальных изомеров и смеси изомеров, или их фармацевтически приемлемой соли.
В четвертом объекте настоящего изобретения предлагается применение соединения формулы I для получения лекарственного средства, предназначенного для лечения заболевания у животного, причем киназная активность, прежде всего активность Abl, Bcr-Abl, Bmx, ВТК, b-RAF, c-RAF, CSK, cSRC, Fes, FGFR3, Flt3, IKKα, IKKβ, JNK1α1, JNK2α2, Lck, Met, МКК4, МКК6, p70S6K, PAK2, PDGFRα, PKA, PKCα, PKD2, ROCK-II, Ros, Rsk1, SAPK2α, SAPK2β, SAPK3, SAPK4, SGK, Syk, Tie2 и/или TrkB, связана с развитием заболевания и/или симптомов заболевания.
В пятом объекте настоящего изобретения предлагается способ получения соединения формулы I и N-оксидов, пролекарств, защищенных производных, индивидуальных изомеров и смеси изомеров, а также фармацевтически приемлемых солей.
Подробное описание предпочтительных вариантов осуществления изобретения
Определение терминов
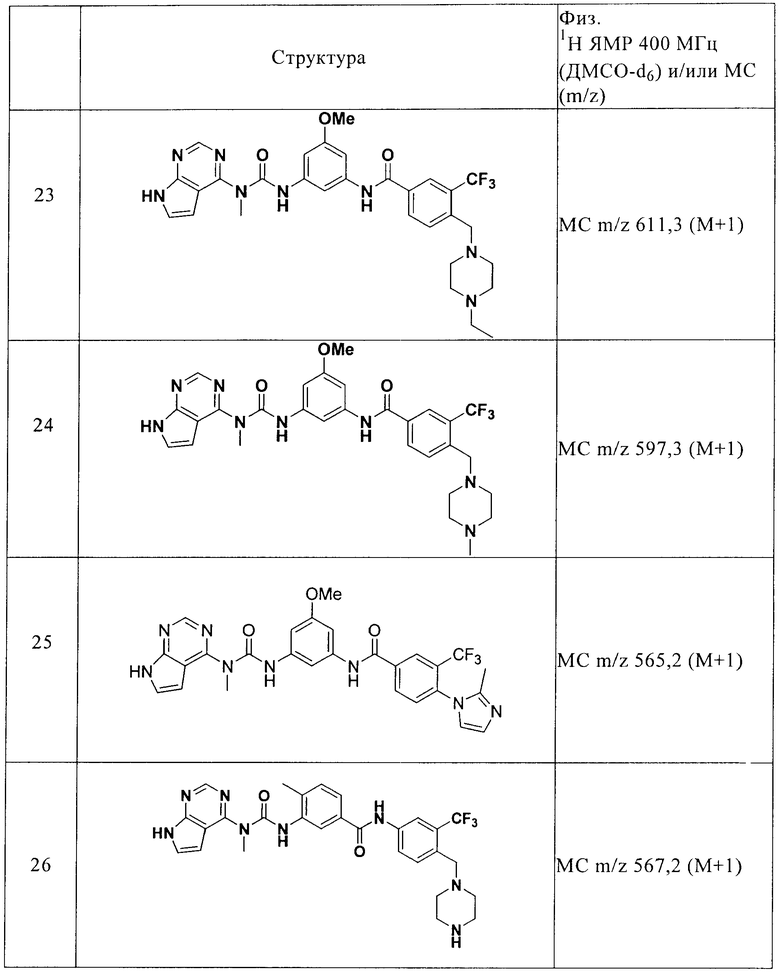
«Алкил» в качестве группы и структурного элемента других групп, например, галогензамещенный алкил и алкокси, является линейным или разветвленным. С1-С4алкокси включает метокси, этокси и т.п. Галогензамещенный алкил включает трифторметил, пентафторэтил и т.п.
«Арил» означает моноциклический или конденсированный бициклический ароматический цикл, содержащий 6-10 атомов углерода в цикле. Например, арил означает фенил или нафтил, предпочтительно фенил. «Арилен» означает двухвалентный радикал, производное арила.
«Гетероарил» имеет значения, определенные выше для арила, причем один или более атомов в цикле являются гетероатомами. Например, гетероарил включает пиридил, индолил, индазолил, хиноксалинил, хинолинил, бензофуранил, бензопиранил, бензотиопиранил, бензо[1,3]диоксол, имидазолил, бензимидазолил, пиримидинил, фуранил, оксазолил, изоксазолил, триазолил, тетразолил, пиразолил, тиенил и т.п.
«Циклоалкил» означает насыщенный или частично ненасыщенный, моноциклический, конденсированный бициклический или мостиковый полициклический цикл, содержащий указанное количество атомов в цикле. Например, С3-С10циклоалкил включает циклопропил, циклобутил, циклопентил, циклогексил и т.п.
«Гетероциклоалкил» означает циклоалкил, как определено в данном контексте, при условии, что один или более указанных атомов углерода в цикле заменены на группу, выбранную из следующих групп: -О-, -N=, -NR-, -С(О)-, -S-, -S(О)- или -S(О)2-, где R означает водород, С1-С4алкил или азотзащитную группу. Например, С3-С8гетероциклоалкил, использованный в данной заявке для описания соединений по настоящему изобретению, включает морфолино, пирролидинил, пирролидинил-2-он, пиперазинил, пиперидинил, пиперидинилон, 1,4-диокса-8-азаспиро[4.5]дец-8-ил и т.п.
«Галоген» предпочтительно означает хлор или фтор, но также может означать бром или иод.
«Панель киназ» включает следующие киназы: Abl (человека), Abl (T315I), JAK2, JAK3, ALK, JNK1α1, ALK4, KDR, Aurora-A, Lck, B1k, MAPK1, Bmx, MAPKAP-K2, BRK, MEK1, CaMKII (крысы). Met, CDK1/циклинВ, p70S6K, CHK2, PAK2, CK1, PDGFRα, CK2, PDK1, c-kit, Pim-2, c-RAF, РКА(человека), CSK, PKBα, cSrc, PKCα, DYRK2, Plk3, EGFR, ROCK-I, Fes, Ron, FGFR3, Ros, Flt3, SAPK2α, Fms, SGK, Fyn, SIK, GSK3β, IGF-1R, Tie-2, IKKβ, TrKB, IR, WNK3, IRAK4, ZAP-70, ITK, AMPK (крысы), LIMK1, Rsk2, Ax1, LKB1, SAPK2β, BrSK2, Lyn (человека), SAPK3, ВТК, МАРКАР-К3, SAPK4, KaMKIV, MARK1, Snk, CDK2/циклинА, MINK, SRPK1, CDK3/циклинЕ, МКК4(мыши), ТАК1, CDK5/p25, МКК6(человека), ТВК1, CDK6/циклинD3, MLCK, TrkA, CDK7/циклинН/МАТ1, MRCKβ, TSSK1, CHK1, MSK1, Yes, CK1d, MST2, ZIPK, c-Kit (D816V), MuSK, DAPK2, NEK2, DDR2, NEK6, DMPK, PAK4, DRAK1, PAR-1Bα, EphA1, PDGFRβ, EphA2, Pim-1, EphA5, PKBβ, EphB2, PKCβI, EphB4, PKCδ, FGFR1, РКСη, FGFR2, PKCθ, FGFR4, PKD2, Fgr, PKG1β, Fit1, PRK2, Hck, PYK2, HIPK2, Ret, IKKα, RIPK2, IRR, ROCK-II (человека), JNK2α2, Rse, JNK3, Rsk1 (человека), PI3 Kγ, PI3 Kδ и PI3-Кβ. Соединения по настоящему изобретению анализировали с использованием указанной панели киназ (дикий тип и/или мутантная форма), и полученные результаты свидетельствуют о том, что соединения ингибируют активность по крайней мере одной киназы из указанной панели.
"Мутантные формы BCR-Abl" означают замену одной или многих аминокислот в последовательности киназы дикого типа. Мутации в киназе BCR-ABL часто приводят к повреждению важных контактных участков между белком и ингибитором (например, ингибитором Gleevec, и т.п.), индуцируя переход из неактивного состояния в активное состояние, т.е. переход в конформацию, при которой BCR-ABL и Gleevec неспособны связываться. Анализ клинических случаев свидетельствует о том, что спектр мутаций, ассоциированных с устойчивым фенотипом, со временем медленно, но неуклонно увеличивается. По-видимому, мутации накапливаются в четырех основных фрагментах. Одна группа мутаций (G250E, Q252R, Y253F/H, E255K/V) включает аминокислоты, которые образуют фосфат-связывающую петлю для АТФ (известную также, как Р-петля). Вторая группа мутаций (V289A, F311L, T315I, F317L) обнаружена в участке связывания с ингибитором Gleevec и непосредственно взаимодействует с ингибитором за счет водородных связей и Вандер-Ваальсовых сил. Третья группа мутаций (М351Т, E355G) расположена вблизи каталитического домена. Четвертая группа мутаций (H396R/P) локализована в петле активации, конформация которой является молекулярным переключателем, регулирующим активацию/инактивацию киназной активности. Точечные мутации в BCR-ABL, ассоциированные с устойчивостью к ингибитору Gleevec, обнаруженные у пациентов с диагнозом CML и ALL, включают M224V, L248V, G250E, G250R, Q252R, Q252H, Y253H, Y253F, Е255К, E255V, D276G, Т277А, V289A, F311L, T315I, T315N, F317L, М343Т, М315Т, E355G, F359V, F359A, V379I, F382L, L387M, L387F, Н396Р, H396R, А397Р, S417Y, Е459К и F486S (положения аминокислот, указанных однобуквенным кодом, приводятся в соответствии с последовательностью, указанной в базе данных GenBank, номер ААВ60394, соответствующей ABL тип la, Martinelli и др., Haematologica/The Hematology Journal, 90-94, (2005, апрель). Если не указано иное, Bcr-Abl означает фермент дикого типа и его мутантные формы.
"Лечение" означает способ снижения или ослабления интенсивности заболевания и/или его симптомов.
Подробное описание предпочтительных вариантов осуществления настоящего изобретения
Гибридный белок BCR-Abl образуется в результате взаимной транслокации, которая гибридизует протоонкоген Abl с геном Bcr. Затем BCR-Abl трансформирует В-клетки благодаря увеличению митогенной активности. Такое увеличение активности приводит к снижению чувствительности к апоптозу, а также к изменению адгезии и хоминга клеток-предшественников CML (хронического миелолейкоза). В настоящем изобретении предлагаются соединения, композиции и способы лечения заболевания, ассоциированного с киназой, прежде всего заболеваний, ассоциированных с киназой Abl, Bcr-Abl, ВМХ, ВТК, b-RAF, c-RAF, CSK, c-SRC, Fes, FGFR3, Flt3, IKKa, IKKβ, JNK1α1, JNK2α2, Lck, Met, MKK4, MKK6, p70S6K, PAK2, PDGFRα, PKA, PKBα, PKD2, ROCK-II, Ros, Rsk1, SAPK2α, SAPK2β, SAPK3, SGK, Syk, Tie2 и TrkB. Например, лейкоз и другие пролиферативные заболевания, ассоциированные с Bcr-Abl, можно лечить при ингибировании киназы Bcr-Abl дикого типа и ее мутантных форм.
В одном варианте осуществления изобретения соединение формулы I означает соединение формулы Ia,
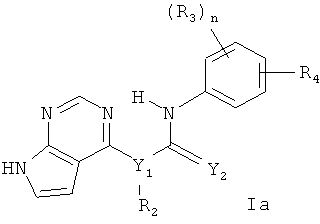
где
n равно 0 и 1,
Y1 выбирают из N и СН,
Y2 выбирают из О и S,
R1 выбирают из водорода и C1-С6алкила,
R2 выбирают из водорода и C1-С6алкила,
R3 выбирают из следующих групп: водород, галоген, гидрокси, С1-С6алкил, C1-С6алкокси, галогензамещенный C1-С6алкил и галогензамещенный C1-С6алкокси,
R4 выбирают из -NR5C(O)R6 и -C(O)NR5R6, где R5 выбирают из водорода и C1-С6алкила, a R6 выбирают из следующих групп: С6-С10арил(С0-С4)алкил, С5-С10гетероарил(С0-С4)алкил, С3-С10циклоалкил(С0-С4)алкил и С3-С10гетероциклоалкил(С0-С4)алкил, причем любой арил, гетероарил, циклоалкил или гетероциклоалкил в составе R6 необязательно замещен 1-3 радикалами, выбранными из группы, включающей галоген, гидрокси, -NR5R5, C1-С6алкил, C1-С6алкокси, галогензамещенный C1-С6алкил, галогензамещенный C1-С6алкокси, С5-С10гетероарил(С0-С4)алкил, С3-С8гетероцикло(С0-С4)алкил и С3-С8гетероцикло(С0-С4)алкокси, причем любой гетероарил или гетероциклоалкил в составе R6 необязательно замещен 1-3 радикалами, независимо выбранными из группы, включающей C1-С6алкил и гидрокси (С1-С6)алкил.
В другом варианте R2 выбирают из водорода и метила, а R3 выбирают из метила и метокси.
В другом варианте R4 выбирают из -NHC(O)R6 и -C(O)NHR6, где R6 выбирают из фенила, необязательно замещенного 1-3 радикалами, выбранными из группы, включающей трифторметил, диметиламино, имидазолил, морфолино, морфолинометил, пиперазинил, пиперазинилметил и пирролидинилметокси, причем указанные имидазолил, пиперазинил, пиперазинилметил необязательно замещены группой, выбранной из метила, этила и гидроксиэтила.
Предпочтительные соединения по настоящему изобретению выбирают из группы, включающей N-[4-(4-этилпиперазин-1-илметил)-3-трифторметилфенил]-4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]бензамид, 4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]-N-(4-пиперазин-1-илметил-3-трифторметилфенил)бензамид, 3-(4-метилимидазол-1-ил)-N-{4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-5-трифторметилбензамид, N-[4-метил-3-(2-7Н-пирроло[2,3-d]пиримидин-4-илацетиламино)фенил]-3-трифторметилбензамид, N-(3-имидазол-1-ил-5-трифторметилфенил)-4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]бензамид, N-{4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-3-трифторметилбензамид, 4-(2-метилимидазол-1-ил)-N-{4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-3-трифторметилбензамид, N-{4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-4-морфолин-4-ил-3-трифторметилбензамид, N-{4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-4-(4-метилпиперазин-1-илметил)-3-трифторметилбензамид, N-{4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-4-морфолин-4-илметил-3-трифторметилбензамид, N-{4-метил-3-[3-метил-3-(7Н-пирроло[2,3-(1]пиримидин-4-ил)уреидо]фенил}-3-морфолин-4-ил-5-трифторметилбензамид, N-{4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-4-(4-метилпиперазин-1-ил)-3-трифторметилбензамид, 4-(4-этилпиперазин-1-ил)-N-{4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-3-трифторметилбензамид, N-{4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-3-(4-метилпиперазин-1-ил)-5-трифторметилбензамид, 3-диметиламино-N-{4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-5-трифторметилбензамид, 4-(4-этилпиперазин-1-илметил)-N-{4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-3-трифторметилбензамид, 3-(4-этилпиперазин-1-ил)-N-{4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-5-трифторметилбензамид, 4-метил-[3-(4-метилимидазол-1-ил)-5-трифторметилфенил]-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]бензамид, 3-[4-(2-гидроксиэтил)пиперазин-1-ил]-{4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-5-трифторметилбензамид, N-{4-метил-3-[3-метил-3-(7H-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-3-пиперазин-l-ил-5-трифторметилбензамид, N-{4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-3-(пирролидин-2-илметокси)-5-трифторметилбензамид, N-{3-метокси-5-[3-метил-3-(7H-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-3-морфолин-4-ил-5-трифторметилбензамид, N-{3-метокси-5-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-3-(4-метилимидазол-1-ил)-5-трифторметилбензамид, 4-(4-этилпиперазин-1-илметил)-N-{3-метокси-5-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-3-трифторметилбензамид, N-{3-метокси-5-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-4-(4-метилпиперазин-1-илметил)-3-трифторметилбензамид и N-{3-метокси-5-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-4-(2-метилимидазол-1-ил)-3-трифторметилбензамид.
Другие предпочтительные соединения по изобретению подробно описаны в примерах и таблице 1.
Фармакология и промышленная применимость
Соединения по изобретению модулируют активность киназ и как таковые используются для лечения заболеваний или нарушений, при которых киназы принимают участие в развитии патологии и/или симптоматике заболевания. Примеры киназ, которые ингибируются соединениями и композициями, описанными в данном контексте, и для подавления которых используются методы, описанные в данном контексте, включают, без ограничения перечисленным, Abl и Bcr-Abl.
Тирозинкиназа Абельсона (т.е. Abl, с-Abl) принимает участие в регуляции клеточного цикла, в клеточном ответе на генотоксический стресс и в передаче информации о клеточном окружении через интегриновый сигнал. Таким образом белок Abl выполняет сложную функцию в качестве клеточного модуля, который интегрирует сигналы от различных межклеточных и внутриклеточных источников, что оказывает влияние на принятие решений относительно клеточного цикла и апоптоза. Тирозинкиназа Абельсона включает подтипы, такие как химерный гибридный белок (онкопротеин) BCR-Abl с разрегулированной тирозинкиназной активностью или v-Abl. BCR-Abl играет решающую роль в патогенезе в 95% случаев хронического миелогенного лейкоза (CML) и в 10% случаев острого лимфолейкоза. STI-571 (Gleevec) является ингибитором онкогенной тирозинкиназы BCR-Abl и используется при лечениии хронического миелоидного лийкоза (CML). Однако у некоторых пациентов на стадии бластного криза CML наблюдается устойчивость к STI-571 из-за мутаций в киназе BCR-Abl. К настоящему времени идентифицировано 22 мутации, наиболее частыми из которых являются G250E, E255V, T315I, F317L и М351Т.
Соединения по настоящему изобретению ингибируют киназу abl, прежде всего киназу v-abl. Соединения по настоящему изобретению также ингибируют дикий тип киназы BCR-Abl и мутированные формы киназы BCR-Abl и, следовательно, пригодны для лечения Bcr-abl-позитивного рака и опухолевых заболеваний, таких как лейкоз (прежде всего хронический миелоидный лейкоз и острый лимфолейкоз, при развитии которых прежде всего установлен апоптотический механизм действия), а также оказывают действие на подгруппу лейкозных стволовых клеток и могут использоваться для очистки этих клеток in vitro после их удаления из организма (например, удаления костного мозга) и реплантации популяции клеток, очищенных от раковых клеток (например, реплантации очищенных клеток костного мозга).
Сигнальный путь Ras-Raf-MEK-ERK опосредует клеточный ответ на ростовые сигналы. Ras мутирован в онкогенную форму в ~15% случаев рака человека. Семейство Raf относится к семейству серин/треонинпротеинкиназ и включает три члена семейства A-Raf, B-Rafn c-Raf(или Raf-1). Исследование Raf как мишени лекарственных средств направлено на изучение действия Raf в качестве подавляющего эффектора на Ras. Однако полученные недавно данные свидетельствуют о том, что B-Raf выполняет важную функцию в образованиия некоторых опухолей без обязательного участия активированной аллели Ras (Nature, 417, 949-954 (01 июля 2002)). Мутации B-Raf прежде всего обнаружены в большинстве злокачественных меланом.
Существующие методы лечения меланомы характеризуются недостаточной эффективностью, прежде всего на поздней стадии заболевания. Соединения по настоящему изобретению ингибируют также клеточные процессы с участием киназы b-Raf и представляют собой новый терапевтически подход к лечению рака человека, прежде всего меланомы.
Соединения по настоящему изобретению кроме того ингибируют клеточные процессы с участием киназы c-Raf. c-Raf активируется онкогеном ras, который мутирован при многих видах рака человека. Следовательно, ингибирование киназной активности c-Raf представляет собой перспективный способ предотвращения роста опухоли, опосредованного геном ras (Campbell S.L., Oncogene, 17, 1395 (1998)).
PDGF (фактор роста тромбоцитов) представляет собой очень распространенный ростовой фактор, который играет важную роль как в процессе нормального роста, так и в патологической клеточной пролиферации, например, которая наблюдается при онкогенезе и при заболеваниях клеток гладкой мускулатуры кровеносных сосудов, например при атеросклерозе и тромбозе. Соединения по изобретению ингибируют активность рецептора PDGF (PDGFR) и, следовательно, могут использоваться при лечении опухолевых заболеваний, таких как глиома, саркома, рак предстательной железы и рак ободочной кишки, молочной железы и яичника.
Соединения по настоящему изобретению можно использовать не только в качестве подавляющих опухоль агентов, например при мелкоклеточном раке легких, но и в качестве агента для лечения незлокачественных пролиферативных нарушений, таких как атеросклероз, тромбоз, псориаз, склеродерма и фиброз, а также для защиты стволовых клеток, например, для борьбы с гемотоксическим действием химиотерапевтических агентов, таких как 5-фторурацил, и для лечения астмы. Соединения по изобретению прежде всего можно использовать для лечения заболеваний, которые чувствительны к ингибированию рецепторной киназы PDGF.
Соединения по настоящему изобретению оказывают лечебное действие при лечении нарушений, возникающих в результате трансплантации, например аллогенной трансплантации, прежде всего при отторжении тканей, таких как прежде всего облитерирующий бронхиолит (OB), т.е. хроническое отторжение аллогенных трансплантатов легкого. В отличие от пациентов, не страдающих от OB, у пациентов с диагнозом OB часто наблюдается повышенная концентрация PDGF в бронхоальвеолярной промывной жидкости.
Соединения по настоящему изобретению также являются эффективными при заболеваниях, ассоциированных с миграцией и пролиферацией клеток гладкой мускулатуры сосудов (при которых PDGF и PDGF-R часто играют заметную роль), таких как рестеноз и атеросклероз. Такое воздействие и его последующее влияние на пролиферацию и миграцию клеток гладкой мускулатуры сосудов in vitro и in vivo можно оценивать при введении соединений по настоящему изобретению, а также при исследовании воздействия на утолщение внутренней оболочки сосудов после механического повреждения in vivo.
Семейство trk рецепторов нейтрофина (trkA, trkB, trkC) стимулирует выживание, рост и дифференциацию нейронных и ненейронных тканей. Белок TrkB экспрессируется в клетках нейроэндокринного типа в тонкой кишке и ободочной кишке, в α-клетках поджелудочной железы, в моноцитах и макрофагах лимфатических узлов и селезенки, и в гранулярных слоях эпидермиса (Shibayama и Koizumi, 1996). Экспрессия белка TrkB ассоциируется с неблагоприятным прогнозом и прогрессированием опухолей Вилмса и нейробластомы. Более того, TkrB экспрессируется в злокачественных клетках предстательной железы, но не обнаруживается в нормальных клетках. Сигнальный путь подавления (экспрессии) рецепторов trk включает каскад активации МАРК с участием Shc, активированного Ras, генов ERK-1 и ERK-2, и пути трансдукции PLC-γ1 (Sugimoto и др., 2001).
Киназа c-Src передает онкогенные сигналы многих рецепторов. Например, сверхэкспрессия EGFR или HER2/neu в опухолях приводит к конститутивной активации c-src, что является характеристикой злокачественных клеток, но не наблюдается в нормальных клетках. С другой стороны, мыши с дефицитом экспрессии c-src представляют собой фенотип «мраморной болезни», что свидетельствует о ключевой роли c-src в функционировании остеокластов и возможном участии в развитии ассоциированных нарушений.
Киназа семейства Tec, Bmx, не-рецепторная протеинтирозинкиназа, контролирует пролиферацию эпителиальных раковых клеток молочной железы.
Установлено, что рецептор 3 фактора роста фибробластов обладает отрицательным регуляторным действием на рост костной ткани и ингибирование пролиферации хондроцитов. Летальная дисплазия вызывается различными мутациями в рецепторе 3 фактора роста фибробластов, а одна мутация, TDII FGFR3, обладает конститутивной тирозинкиназной активностью, которая активирует фактор транскрипции Stal1, что приводит к экспрессии ингибитора клеточного цикла, остановке роста и аномальному развитию костной ткани (Su и др., Nature, 386, 288-292 (1997)). Кроме того, FGFR3 часто экспрессируется в опухолях типа множественной миеломы. Ингибиторы активности FGFR3 можно использовать для лечения воспалительных или аутоиммунных заболеваний, опосредованных Т-клетками, включающих, без ограничения перечисленным, ревматоидный артрит (RA), коллагеновый артрит II, рассеянный склероз (МС:), системную красную вочанку (SLE), псориаз, юношеский диабет, болезнь Шенгрена, заболевание щитовидной железы, саркоидоз, аутоиммунный увеит, воспалительное заболевание кишечника (болезнь Крона и язвенный колит), глютеновую болезнь и тяжелую псевдопаралитическую миастению.
Активность сывороточной и глюкокортикоид-регулируемой киназы (SGK) коррелирует с активностями возмущенных ионных каналов, прежде всего активностью натриевых и/или калиевых каналов, и соединения по изобретению можно использовать для лечения гипертензии.
В работах Lin и др., J. Clin. Invest. 100, 8, 2072-2078 (1997) и Р.Lin, PNAS, 95, 8829-8834 (1998) установлено подавление роста опухоли и васкуляризации, а также снижение метастазирования в легких при аденовирусных инфекциях или при инъекциях внеклеточного домена Tie-2 (Tek) в опухоли молочной железы и модели ксенотрансплантата меланомы. Ингибиторы Tie2 можно использовать в случаях, при которых наблюдается аномальная неоваскуляризация (например, при диабетической ретинопатии, хроническом воспалении, псориазе, саркоме Капоши, хронической неоваскуляризации при дегенерации желтого пятна, ревматоидном артрите, инфантильной гемангиоме и раке).
Lck принимает участие в передаче сигнала Т-клетками. У мышей с отсутствием гена Lck наблюдается низкий уровень тимоцитов. Функция Lck в качестве положительного активатора передачи сигнала Т-клетками свидетельствует о том, что ингибиторы Lck можно использовать для лечения аутоиммунного заболевания, такого как ревматоидный артрит.
Киназы JNK и другие МАРК принимают участие в опосредовании клеточного ответа на рак, тромбин-индуцированную агрегацию тромбоцитов, нарушения типа иммунодефицита, аутоиммунные заболевания, гибель клеток, аллергию, остеопороз и болезнь сердца. Терапевтические мишени, связанные с активацией пути JNK, включают хронический миелогенный лейкоз (CML), ревматоидный артрит, астму, остеоартрит, ишемию, рак и нейродегенеративные заболевания. В связи с активацией JNK, ассоциированной с заболеванием печени или приступами гепатической ишемии, соединения по изобретению можно также использовать для лечения различных нарушений функции печени. Сообщается также об участии JNK в развитии сердечно-сосудистого заболевания, такого как инфаркт миокарда или застойная сердечная недостаточность, а также установлено, что JNK опосредует гипертрофические ответные реакции на различные формы сердечного стресса. Установлено, что каскад JNK принимает участие в активации Т-клеток, включая активацию промотора IL-2. Таким образом, ингибиторы JNK могут оказывать лечебное действие при коррекции патологических ответных иммунных реакций. Установлена также роль активации JNK при развитии различных видов рака, что свидетельствует о возможности эффективного использования ингибиторов JNK при заболевании раком. Например, конститутивно активируемая JNK ассоциируется с онкогенезом, опосредованным HTLV-1 (Oncogene, 13, 135-142 (1996)). JNK принимает участие в развитии саркомы Капоши (KS). Пролиферативное действие других цитокинов, принимающих участие в пролиферации KS, таких как эндотелиальный фактор роста сосудов (VEGF), IL-6 и TNFa, также может оказаться опосредованным JNK. Кроме того, регуляция гена c-jun в клетках р210, трансформированных BCR-ABL, соответствует активности JNK, что свидетельствует о возможности применения ингибиторов JNK при лечении хронического миелогенного лейкоза (CML) (Blood 92, 2450-2460 (1998)).
Принято считать, что некоторые аномальные пролиферативные состояния ассоциированы с экспрессией raf и, следовательно, чувствительны к ингибированию экспресии raf. Аномально высокие уровни экспрессии белка raf также связаны с трансформацией и аномальной пролиферацией клеток. Предполагается также, что указанные аномальные пролиферативные состояния чувствительны к ингибированию экспрессии raf. Например, предполагается, что экспрессия белка c-raf играет роль в аномальной клеточной пролиферации, поскольку сообщается, что 60% всех клеточных линий карциномы легкого экспрессируют необычно высокий уровень мРНК и белка c-raf. Другими примерами аномальных пролиферативных состояний являются гиперпролиферативные нарушения, такие как рак, опухоли, гиперплазия, фиброз легких, ангиогенез, псориаз, атеросклероз и пролиферация клеток гладкой мускулатуры кровеносных сосудов, такие как стеноз или рестеноз после пластической операции на сосудах. Клеточный сигнальный путь, частью которого является raf, также принимает участие в воспалительных нарушениях, характеризующихся пролиферацией Т-клеток (активация и рост Т-клеток), таких, например, как отторжение тканевого трансплантата, эндотоксиновый шок и гломерулярный нефрит.
Стресс-активируемые протеинкиназы (SAPK) представляют собой семейство протеинкиназ, которые принимают участие на предпоследней стадии пути передачи сигнала, при этом происходит активация фактора транскрипции с-jun и экспрессия генов, регулируемых c-jun. Прежде всего c-jun принимает участие в транскрипции генов, которые кодируют белки, связанные с репарацией ДНК, поврежденной вследствие генотоксических эффектов. Следовательно, агенты, которые ингибируют активность SAPK в клетке, предотвращают репарацию ДНК и сенсибилизируют клетку к агентам, которые индуцируют повреждение ДНК или ингибируют синтез ДНК и индуцируют апоптоз клетки, или к агентам, которые ингибируют клеточную пролиферацию.
Митоген-активируемые протеинкиназы (МАРК) являются членами консервативных путей передачи сигнала, которые активируют факторы транскрипции, факторы трансляции и другие молекулы-мишени в ответ на различные внеклеточные сигналы. МАРК активируются митоген-активируемыми протеинкиназами (МКК) за счет двойного фосфорилирования по двум аминокислотам в последовательности Thr-X-Tyr. У высших эукариотов физиологическая роль передачи сигнала МАРК коррелирует с клеточными процессами, такими как пролиферация, онкогенез, развитие и дифференциация. Соответственно, способность регулировать передачу сигнала по указанным путям (прежде всего с участием МКК4 и МКК6) можно использовать при разработке способов лечения и профилактики заболеваний человека, ассоциированных с передачей сигнала МАРК, таких как воспалительные заболевания, аутоиммунные заболевания и рак.
Семейство протенкиназ рибосомального белка S6 человека включает по крайней мере 8 членов (RSK1, RSK2, RSK3, RSK4, MSK1, MSK2, p70S6K и p70S6 Kb). Протеинкиназы рибосомального белка S6 выполняют важные плеотропные функции, в том числе играют ключевую роль в регуляции трансляции мРНК при биосинтезе белка (Eur. J. Biochem., 267(21), 6321-6330 (2000, ноябрь); Exp Cell Res., 253 (1), 100-109 (1999, 25 ноября); Mol Cell Endocr1nol., 151(1-2), 65-77 (1999, 25 мая)). Кроме того, фосфорилирование рибосомального белка S6 киназой p70S6 связано с регуляцией клеточной подвижности (Immunol. Cell B1ol., 78(4), 447-451 (2000, август)) и клеточного роста (Prog. Nucle1c Ac1d Res. Mol. Biol., 65, 101-127 (2000)), и, следовательно, является важным фактором при метастазировании опухоли, иммунном ответе и репарации ткани, а также при других патологических состояниях.
Киназы SAPK (называемые также "jun N-концевые киназы" или "JNK") представляют собой семейство протеинкиназ, которые принимают участие на предпоследней стадии пути передачи сигнала, при этом происходит активация фактора транскрипции c-jun и экспрессия генов, регулируемых c-jun. Прежде всего c-jun принимает участие в транскрипции генов, которые кодируют белки, связанные с репарацией ДНК, поврежденной вследствие генотоксических эффектов. Агенты, которые ингибируют активность SAPK в клетке, предотвращают репарацию ДНК и сенсибилизируют клетку к таким терапевтическим противоопухолевым агентам, которые оказывают действие за счет индуцирования повреждения ДНК.
ВТК играют важную роль в развитии аутоиммунного и/или воспалительного заболевания, такого как системная красная волчанка (SLE), ревматоидный артрит, рассеянный васкулит, идиопатическая тромбоцитопеническая пурпура (ITP), тяжелая псевдопаралитическая миастения и астма. Благодаря участию ВТК в активации В-клеток, ингибиторы ВТК можно использовать для подавления патогенных состояний, опосредованных В-клетками, таких как продуцирование аутоантител, и при лечении лимфомы и лейкоза В-клеток.
СНК2 является киназой контрольных точек семейства серин/треонинпротеинкиназ и принимает участие в механизме контроля повреждений ДНК, таких как повреждения, вызванные мутагенами окружающей среды и эндогенными видами активного кислорода. В результате, ее можно использовать в качестве супрессора опухоли и мишени при онкотерапии.
CSK влияет на метастазирующую активность опухолевых клеток, прежде всего рака ободочной кишки.
Fes является не-рецепторной протеинтирозинкиназой, которая принимает участие в ряде путей передачи сигнала с участием цитокинов, а также дифференциации миелоидных клеток. Fes является также ключевым компонентом в механизме дифференциации гранулоцитов.
Рецепторная тирозинкиназа Flt3 принимает участие в развитии лейкоза и миелодиспластического синдрома. Приблизительно 25% клеток лейкоза AML экспрессируют конститутивно активную форму аутофосфорилированной тирозинкиназы (ф)FLT3 на клеточной поверхности. Активность ф-FLT3 обеспечивает рост и выживание предпочтительно лейкозных клеток. Пациенты с острым лейкозом, у которых лейкозные клетки экспрессируют киназу ф-FLT3, в основном характеризуются неблагоприятным клиническим прогнозом. Ингибирование киназы ф-FLT3 индуцирует апоптоз (программируемую гибель клеток) лейкозных клеток.
Ингибиторы IKKα и IKKβ (1 и 2) являются лекарственными средствами для лечения заболеваний, включающих ревматоидный артрит, отторжение трансплантата, воспалительное заболевание кишечника, остеоартрит, астму, хроническое обструктивное заболевание легких, атеросклероз, псориаз, рассеянный склероз, инсульт, системную красную волчанку, болезнь Альцгеймера, ишемию головного мозга, травматическое повреждение мозга, болезнь Паркинсона, боковой амиотрофический склероз, субарахноидальное кровоизлияние и другие заболевания или нарушения, ассоциированные с избыточным продуцированном воспалительных медиаторов в головном мозге и центральной нервной системе.
Met ассоциируется с большинством типов основных раков человека и экспрессия этого фермента часто коррелирует с неблагоприятным прогнозом и метастазированием. Ингибиторы Met являются лекарственными средствами для лечения заболеваний, включающих различные виды рака, такие как рак легких, NSCLC (немелкоклеточный рак легких), рак костной ткани, рак поджелудочной железы, рак кожи, рак головы и шеи, кожная и внутриглазная меланома, рак матки, рак яичника, ректальный рак, рак анальной области, рак желудка, рак ободочной кишки, рак молочой железы, гинекологические опухоли (например, саркома матки, карцинома маточных труб, карцинома эндометрия, карцинома шейки матки, карцинома влагалища или карцинома вульвы), болезнь Ходжкина, рак пищевода, рак тонкой кишки, рак эндокринной системы (например, рак щитовидной железы, паратиреоидный рак или рак надпочечников), саркомы мягких тканей, рак мочеиспускательного канала, рак пениса, рак предстательной железы, хронический или острый лейкоз, детские солидные опухоли, лимфоцитарные лимфомы, рак мочевого пузыря, рак почек или мочеточника (например, почечноклеточный рак, карцинома почечного пелвиса), детские злокачественные заболевания, опухоли центральной нервой системы (например, первичная лимфома ЦНС, опухоли позвоночника, глиома мозгового ствола или гипофизарная аденома), рак крови, такой как острый миелоидный лейкоз, хронический миелоидный лейкоз и т.п., язва пищевода Баррета (предраковый синдром), неопластическое заболевание кожи, псориаз, грибовидные микозы и доброкачественная гипертрофия предстательной железы, заболевания, ассоциированные с диабетом, такие как диабетическая ретинопатия, ретинальная ишемия и ретинальная неоваскуляризация, цирроз печени, сердечно-сосудистые заболевания, такие как атеросклероз, иммунологические заболевания, такие как аутоиммунное заболевание и почечное заболевание. Предпочтительно заболевание означает рак, такой как острый миелоидный лейкоз и колоректальный рак.
Nima-ассоциированная киназа 2 (Nek2) является протеинкиназой, регулирующей клеточный цикл, причем фермент локализуется в центросоме и обладает максимальной активностью на стадии митоза. Функциональные исследования свидетельствуют об участии Nek2 в регуляции разделения центросомы и образовании веретена. Повышенное содержание белка Nek2 (в 2-5 раз) наблюдается в клеточных линиях, полученных из некоторых опухолей человека, таких как рак шейки матки, яичника, предстательной железы и прежде всего рака молочной железы.
Заболевания или состояния, опосредованные p70S6K, включают, без ограничения перечисленным, пролиферативные нарушения, такие как рак и туберозный склероз.
В настоящее время существует постоянно возрастающее множество данных о надежном и эффективном лечении ингибиторами киназы раков, при которых киназа-мишень лекарственного средства конститутивно активируется мутациями в гене. Опубликовано множество работ о мутациях, выявленных в последовательностях киназ, которые происходят в результате естественного отбора опухоли. Список примеров таких киназ включает, без органичения перечисленным, мутант b-raf V599E в более 60% случае меланом, мутанты Flt3-ITD в 30% случае AML, мутации c-kit у пациентов с GIST, PDGFRα у пациентов с GIST и HES, PDGFRβ у пациентов с CMML, мутанты Pi3K при раке ободочной кишки и желудка, а также при глиобластоме, и мутанты EGFR в 10% случаев рака легких (чувствительных к ирессе) и глиобластомы.
В соответствии с вышеизложенным, в настоящем изобретении предлагается также способ профилактики или лечения любых заболеваний или нарушений, указанных выше, у субъекта, который нуждается в таком лечении, причем указанный способ включает введение указанному субъекту терапевтически эффективного количества (см. ниже в разделе Способы введения и фармацевтические композиции) соединения формулы I или его фармацевтически приемлемой соли. При любом вышеуказанном применении требуемые дозы могут изменяться в зависимости от способа введения, конкретного состояния, подлежащего лечению, и ожидаемого действия.
Способы введения и фармацевтические композиции
В общем случае, соединения по изобретению вводят в терапевтически эффективных количествах любыми обычными и приемлемыми известными способами, отдельно или в комбинации с одним или более терапевтическими агентами. Терапевтически эффективное количество может изменяться в широком диапазоне в зависимости от тяжести заболевания, возраста и относительного состояния здоровья субъекта, активности используемого соединения и других факторов. В общем случае удовлетворительные результаты достигаются при системном введении суточных доз от приблизительно 0,03 до 2,5 мг/кг массы тела. Для более крупного млекопитающего, например, человека, назначаемая суточная доза составляет от приблизительно 0,5 мг до приблизительно 100 мг, которую можно вводить, например, разделенными дозами до четырех раз в сутки или в виде формы с замедленным высвобождением. Пригодные стандартные лекарственные формы для перорального введения включают от приблизительно 1 до 50 мг активного ингредиента.
Соединения по изобретению можно вводить в виде фармацевтических композиций любым приемлемым способом, прежде всего энтеральным способом, например, пероральным способом в форме таблеток или капсул, или парентеральным способом, например, в форме инъекционных растворов или суспензий, местным способом, например, в форме лосьонов, гелей, мазей или кремов, или назальным способом или в форме суппозиториев. Фармацевтические композиции, включающие соединение по настоящему изобретению в свободной форме или в форме фармацевтически приемлемой соли в смеси по крайней мере с одним фармацевтически приемлемым носителем или разбавителем, получают обычным способом с использованием обычных методов смешивания, гранулирования или нанесения покрытия. Например, пероральные композиции могут представлять собой таблетки или желатиновые капсулы, включающие активный ингредиент в смеси с а) разбавителями, например лактозой, декстрозой, сахарозой, маннитом, сорбитом, целлюлозой и/или глицином, б) замасливателями, например диоксидом кремния, тальком, стеариновой кислотой, ее кальциевой или магниевой солью и/или полиэтиленгликолем, а таблетки могут также включать в) связующие агенты, например силикат магния/алюминия, крахмал, желатин, трагакант, метилцеллюлозу, Na-соль карбоксиметилцеллюлозы и/или поливинилпирролидон, и при необходимости г) дезинтегрирующие агенты, например крахмалы, агар, альгиновую кислоту или ее натриевую соль, или шипучие смеси, и/или д) абсорбенты, красители, ароматизаторы и подсластители. Инъекционные композиции могут представлять собой водные изотонические растворы или суспензии, а суппозитории получают из жировых эмульсий или суспензий. Композиции можно стерилизовать или они могут содержать адъюванты, такие как консерванты, стабилизирующие, смачивающие или эмульгирующие агенты, способствующие растворению агенты, соли для регуляции осмотического давления и/или буферные вещества. Кроме того, они могут содержать другие терапевтически ценные соединения. Пригодные составы для чрескожного применения включают эффективное количество соединения по настоящему изобретению в смеси с носителем. Носитель может включать абсорбируемые фармакологически приемлемые растворители для обеспечения проницаемости через кожу пациента. Например, системы для чрескожного введения представляют собой повязку, включающую защитную пленку, резервуар, содержащий соединение необязательно в смеси с носителем, необязательно мембрану, регулирующую скорость доставки соединения на кожу пациента с заданной и контролируемой скоростью в течение продолжительного времени, и устройство, обеспечивающее удерживание повязки на поверхности кожи. Можно также использовать матричные чрескожные составы. Пригодные составы для местного применения, например, для нанесения на кожу и в глаза, предпочтительно представляют собой водные растворы, мази, кремы или гели, известные в данной области. Такие составы могут содержать солюбилизирующие, стабилизирующие, тонизирующие агенты, буферные вещества и консерванты.
Соединения по изобретению можно вводить в терапевтически эффективных количествах в комбинации с одним или более терапевтических агентов (фармацевтические комбинации). Например, синергетическое действие достигается при использовании комбинации с другими иммуномодулирующими или противовоспалительными веществами, например, при использовании комбинации с циклоспорином, рапамицином или аскомицином, или их иммунодепрессантными аналогами, например циклоспорином A (CsA), циклоспорином G, FK-506, рапамицином, или аналогичными соединениями, кортикостероидами, циклофосфамидом, азатиоприном, метотрексатом, бреквинаром, лефлуномидом, мизорибином, микофеноловой кислотой, микофенолатом мофетила, 15-дезоксиспергуалином, иммунодепрессантными антителами, прежде всего моноклональными антелами к рецепторам лейкоцитов, например, МНС, CD2, CD3, CD4, CD7, CD25, CD28, В7, CD45, CD58 или их лигандам, или другими иммонумодулирующими соединениями, такими как CTLA41g. Если соединения по изобретению вводят совместно с другими терапевтически активными агентами, дозы совместно вводимых соединений варьируют в зависимости от типа совместно используемого лекарственного средства, от конкретного лекарственного средства, от состояния, подлежащего лечению, и т.п.
Кроме того, изобретение относится к фармацевтическим комбинациям, например, набору, включащему а) первый агент, который представляет собой соединение по изобретению, указанное выше, в свободной форме или в форме фармацевтически приемлемой соли, и б) по крайней мере один сопутствующий агент. Набор включает инструкции по введению лекарственных средств.
Термины "совместное введение" или "комбинированное введение" или аналогичные термины, используемые в описании, означают введение выбранных терапевтических агентов одному пациенту, а также курс лечения, согласно которому агенты необязательно вводят одновременно и одним и тем же способом.
Термин "фармацевтическая комбинация", используемый в описании заявки, означает продукт, который образуется при смешивании или объединении более одного активного ингредиента и включает фиксированные и нефиксированные комбинации активных ингредиентов. Термин "фиксированная комбинация" означает, что активные ингредиенты, например, соединение формулы I и сопутствующий агент, вводятся пациенту одновременно в форме одного материала или дозы. Термин "нефиксированная комбинация" означает, что активные ингредиенты, например, соединение формулы I и сопутствующий агент, вводят пациенту раздельно, одновременно, совместно или последовательно, без ограничений по времени, причем такое введение обеспечивает достижение терапевтически эффективного уровня двух соединений в организме пациента. Последнее относится также к комбинированному лечению, например к введению трех или более активных ингредиентов.
Способы получения соединений по изобретению
Настоящее изобретение включает также способы получения соединений по изобретению. В описанных реакциях существует необходимость защищать реакционно-способные функциональные группы, например гидрокси, амино, имино, тио или карбоксигруппы, если указанные группы должны присутствовать в конечном продукте, чтобы исключить их нежелательное участие в реакциях. Стандартные защитные группы используют по обычным методикам, как описано, например, в книге T.W.Greene and P.G.M.Wuts «Protective Groups in Organic Chemistry», John Wiley and Sons, (1991).
Соединения формулы I получают, как показано на схеме I.
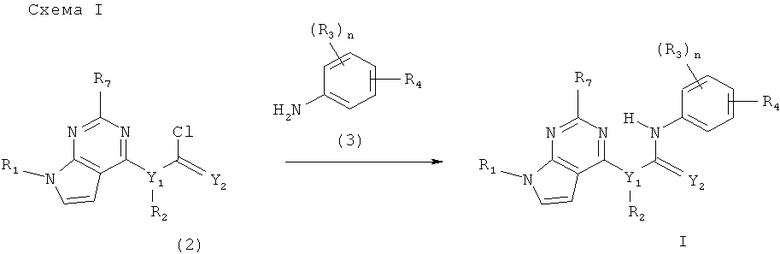
где n, R1, R2, R3, R4, R7, Y1 и Y2 имеют значения, определенные в разделе «Краткое описание сущности изобретения». Соединение формулы I получают при взаимодействии соединения формулы 2 с соединением формулы 3 в присутствии пригодного растворителя (например, дихлорметана и т.п.) и пригодного основания (например, диизопропилэтиламина и т.п.). Реакцию проводят при температуре от приблизительно 0°С до приблизительно 40°С в течение приблизительно 10 ч до завершения реакции. Реакционную смесь затем необязательно обрабатывают для удаления любых защитных групп.
Подробное описание синтеза соединения формулы I приведено ниже в разделе «Примеры».
Дополнительные способы получения соединений по изобретению
Соединение по изобретению получают в виде фармацевтически приемлемой кислотно-аддитивной соли при взаимодействии соединения в форме свободного основания с фармацевтически приемлемой неорганической или органической кислотой. В другом варианте фармацевтически приемлемую основно-аддитивную соль получают при взаимодействии соединения в форме свободной кислоты с фармацевтически приемлемым неорганическим или органическим основанием.
В другом варианте соединения по изобретению в форме соли получают из солей исходных или промежуточных материалов.
Соединения по изобретению в форме свободной кислоты или свободного основания получают из основно-аддитивной соли или кислотно-аддитивной соли соответственно. Например, соединение по изобретению в форме кислотно-аддитивной соли можно превратить в соответствующее свободное основание при обработке пригодным основанием (например, раствором гидроксида аммония, гидроксида натрия и т.п.). Соединение по изобретению в форме основно-аддитивной соли можно превратить в соответствующую свободную кислоту при обработке пригодной кислотой (например, хлористо-водородной кислотой и т.п.).
Соединения по изобретению в неокисленной форме получают из N-оксидов соединений по изобретению при обработке восстанавливающим агентом (например, серой, диоксидом серы, трифенилфосфином, боргидридом лития, боргидридом натрия, трихлоридом фосфора, трибромидом или т.п.) в пригодном инертном органическом растворителе (например, ацетонитриле, этаноле, водном диоксане или т.п.) при температуре от 0 до 80°С.
Пролекарства соединений по изобретению получают известными методами (см. Saulnier и др., Bioorganic and Medicinal Chemistry Letters, т.4, с.1985, (1994)). Например, соответствующие пролекарства получают при взаимодействии немодифицированного соединения по изобретению с пригодным карбамилирующим агентом (например, 1,1-ацилоксиалкилкарбанохлоридатом, пара-нитрофенилкарбонатом или т.п.).
Защищенные производные соединений по изобретению получают известными способами. Более подробно методы введения и удаления защитных групп приведены в книге T.W.Greene «Protective Groups in Organic Chemistry», 3 изд., John Wiley and Sons, Inc., (1999).
Соединения по изобретению получают в виде сольватов (например, гидратов). Гидраты соединений по настоящему изобретению получают перекристаллизацией из водной/органической смеси растворителей, используя такие органические растворители, как диоксан, тетрагидрофуран или метанол.
Соединения по изобретению получают в виде индивидуальных стереоизомеров при взаимодействии рацемической смеси соединения с оптически активным разделяющим агентом с образованием пары диастереоизомерных соединений, при разделении диастереомеров и получении оптически чистых энантиомеров. Так как разделение энантиомеров происходит с использованием ковалентных диастереомерных производных соединений по изобретению, предпочтительными являются диссоциирующие комплексы (например, кристаллические диастереомерные соли). Диастереомеры обладают различными физическими свойствами (например, температура плавления, температура кипения, растворимость, реакционная способность и т.п.) и их разделяют простым методом, используя эти различия. Диастереомеры разделяют хроматографией или, предпочтительно, способами, основанными на их различной растворимости, при этом получают оптически чистые энантиомеры наряду с разделяющим агентом, по любой методике, при которой не происходит рацемизации. Более подробно описание выделения стереоизомеров из их рацемических смесей приведено в книге Jean Jacques, Andre Collet, Samuel H. Wilen, «Enantiomers, Racemates and Resolutions», John Wiley and Sons, Inc., (1981).
Таким образом, соединения формулы I получают способом, который заключается в том, что
(a) проводят реакцию, как указано на схеме I, и
(b) соединение по изобретению необязательно превращают в фармацевтически приемлемую соль,
(c) соль соединения по изобретению необязательно превращают в несолевую форму,
(d) неокисленную форму соединения по изобретению необязательно превращают в фармацевтически приемлемый N-оксид,
(e) N-оксид соединения по изобретению необязательно превращают в неокисленную форму,
(f) из смеси изомеров необязательно выделяют индивидуальный изомер соединения по изобретению,
(g) немодифицированное соединение по изобретению необязательно превращают в фармацевтически приемлемое пролекарство, и
(h) пролекарство необязательно превращают в немодифицированное соединение по изобретению.
Если получение исходных материалов не описано подробно, соединения известны или их получают по известным методикам, или как описано ниже в разделе «Примеры».
Следует понимать, что приведенные выше превращения даны только для иллюстрации способов получения соединений по настоящему изобретению и что можно использовать также другие известные способы.
Примеры
Приведенные примеры иллюстрируют получение соединений формулы I по настоящему изобретению, не ограничивая его объем.
Пример 1
Синтез 3-(4-метилимидазол-1-ил)-N-{4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-5-трифторметилбензамида
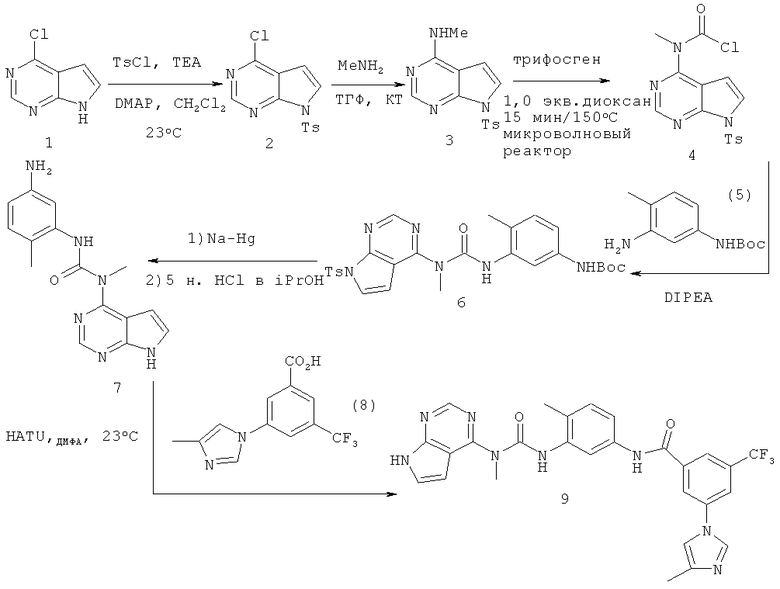
4-Хлор-7-(толуол-4-сульфонил)-7Н-пирроло[2,3-d]пиримидин
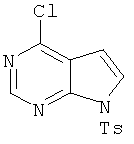
В раствор 6-хлордеазапурина (2,0 г, 13 ммолей, 1,0 экв.) в дихлорметане (65 мл) добавляли триэтиламин (1,98 мл, 14,3 ммоля, 1,1 экв.) и 4-диметиламинопиридин (катализатор). Затем в реакционную смесь порциями добавляли тозилхлорид (2,55 г, 13,4 ммоля, 1,03 экв.) и смесь выдерживали в течение 30 мин. Затем реакционную смесь распределяли между дихлорметаном и водой. Органический слой отделяли, водный слой экстрагировали дихлорметаном. Объединенные органические экстракты промывали водой, сушили над Na2SO4, фильтровали и концентрировали, при этом получали указанный в заголовке продукт 2. Указанное в заголовке соединение использовали на следующей стадии без дополнительной очистки.
Метил[7-(толуол-4-сульфонил)-7H-пирроло[2,3-d]пиримидин-4-ил]амин
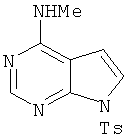
В раствор соединения 2 (1,7 г, 5,5 ммоля, 1,0 экв.) в ТГФ (5 мл) при 23°С добавляли метиламин (6,6 мл 2,0 М раствора в ТГФ, 13,1 ммоля, 2,4 экв.), затем полученную смесь перемешивали при КТ в течение 3 ч, твердый остаток распределяли между этилацетатом и водой. Органические слои отделяли, сушили над Na2SO4, фильтровали и концентрировали, при этом получали указанный в заголовке продукт. Остаток использовали на следующей стадии без дополнительной очистки.
N-Хлоркарбонил-N-метил[7-(толуол-4-сульфонил)-7H-пирроло[2,3-d]пиримидин-4-ил]амин
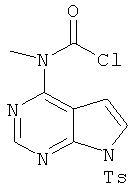
Флакон Смита (объемом 2,5 мл), содержащий соединение 3 (0,250 г, 0,82 ммоля, 1,0 экв.), трифосген (0,24 г, 0,82 ммоля, 1,0 экв.) и диоксан (2 мл), помещали в микроволновый реактор синтезатора Смита и выдерживали при 150°С в течение 20 мин. Затем реакционную смесь распределяли между дихлорметаном и водой. Органический слой отделяли, водный слой экстрагировали дихлорметаном. Объединенные органические экстракты промывали водой, сушили над Na2SO4, фильтровали и концентрировали, при этом получали указанный в заголовке продукт, который использовали на следующей стадии без дополнительной очистки.
Трет-бутиловый эфир (4-метил-3-{3-метил-3-[7-(толуол-4-сульфонил)-7H-пирроло[2,3-d]пиримидин-4-ил]уреидо}фенил)карбаминовой кислоты
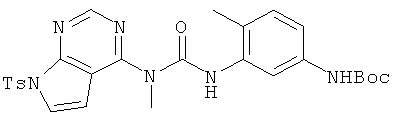
В раствор соединения 5 (0,182 г, 0,82 ммоля, 1,0 экв.) и диизопропилэтиламина (0,14 мл, 0,82 ммоля, 1,0 экв.) в дихлорметане (2,0 мл) при 23°С по каплям добавляли карбамоилхлорид 4 (0,3 г, 0,82 ммоля, 1,0 экв.) в течение 15 мин. Полученную смесь перемешивали при КТ в течение 8 ч, а затем распределяли между дихлорметаном и водой. Объединенные органические экстракты промывали водой, сушили над Na2SO4, фильтровали и концентрировали, при этом получали указанный в заголовке продукт, который использовали на следующей стадии без дополнительной очистки.
3-(5-Амино-2-метилфенил)-1-метил-1-(7H-пирроло[2,3-d]пиримидин-4-ил]мочевина
В неочищенную мочевину 6 (3,2 г, 5,8 ммоля, 1,0 экв.) в дихлорметане (30 мл) добавляли 5-6 н. HCl в изопропаноле (11,6 мл, 58 ммолей, 10,0 экв.), при этом получали прозрачный раствор. Полученную смесь перемешивали при 23°С в течение 30 мин до осаждения продукта. Реакционную массу концентрировали в вакууме и нейтрализовали насыщенным водным раствором бикарбоната натрия. Органические слои экстрагировали этилацетатом, сушили над Na2SO4, фильтровали и концентрировали, при этом получали продукт с удаленной Вос-группой.
В раствор неочищенного первичного амина (2,3 г, 5,1 ммоля, 1,0 экв.) в ТГФ/МеОН (1:1) добавляли Na-Hg (3,5 г, 3,0 экв. в расчете на Na). Реакционную смесь перемешивали в течение 15 мин, затем смесь декантировали для удаления солей ртути и жидкий супернатант концентрировали при пониженном давлении. Твердый осадок распределяли между этилацетатом и водой. Органический слой отделяли, сушили над Na2SO4, фильтровали и концентрировали, при этом получали неочищенный продукт. Неочищенный продукт очищали экспресс-хроматографией на колонке (элюент: CH2Cl2/МеОН, 9:1, об./об.), при этом получали указанное в заголовке соединение 7 в виде твердого вещества.
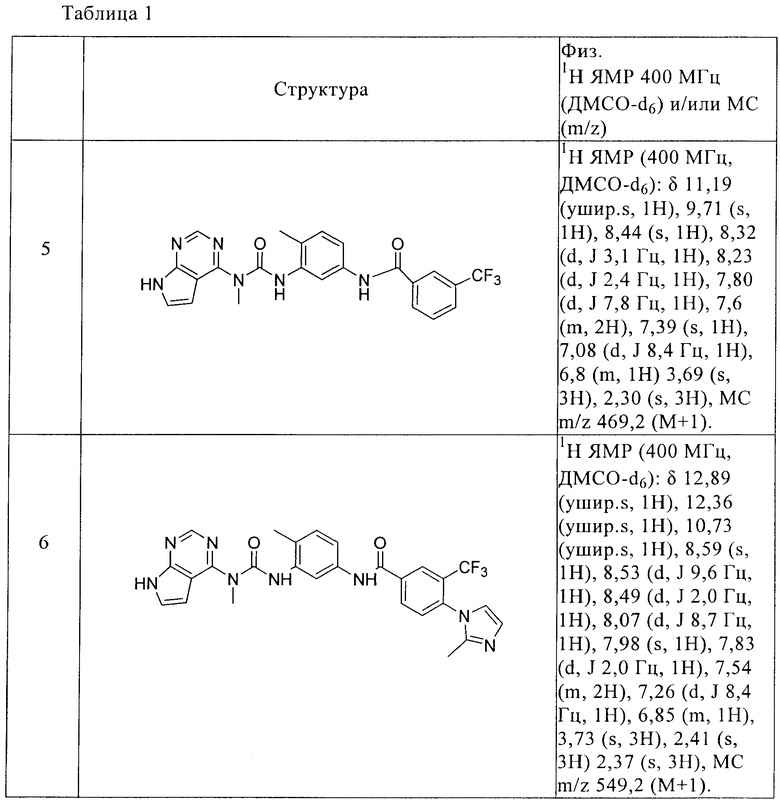
3-(4-Метилимидазол-1-ил)-N-{4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-5-трифторметилбензамид
В раствор соединения 7 (7 мг, 0,23 ммоля, 1,0 экв.), 3-(4-метилимидазол-1-ил)-5-трифторметилбензойной кислоты (7,6 мг, 0,27 ммоля, 1,2 экв.) и DIPEA (40 мкл, 0,27 ммоля, 1,2 экв.) в ДМФА (0,5 мл) добавляли HATU (9,9 мг, 0,25 ммоля, 1,1 экв.), перемешивали при КТ в течение 1 ч, растворитель удаляли в вакууме. Остаток растворяли в ДМСО (1 мл). Полученный раствор очищали обращено-фазовой ЖХ-МС, при этом получали указанное в заголовке соединение в виде трифторацетата. МС m/z: 549,2 (M+1).
1Н ЯМР (400 МГц, ДМСО-d6): δ 12,81 (ушир.s, 1Н), 12,28 (ушир.s, 1H), 10,52 (ушир.s, 1H), 9,40 (s, 1H), 8,52 (s, 1H), 8,51 (s, 1H), 8,40 (d, J 2,0 Гц, 1H), 8,36 (s, 1H), 8,33 (s, 1H), 7,48 (m, 2H), 7,19 (d, J 8,32 Гц, 1H), 6,78 (m, 1H), 3.05 (s, 3Н), 2,30 (s, 3H), 2,27 (s, 3H).
Пример 2
Синтез N-[4-метил-3-(2-7Н-пирроло[2,3-d]пиримидин-4-илацетиламино)фенил]-3-трифторметилбензамида
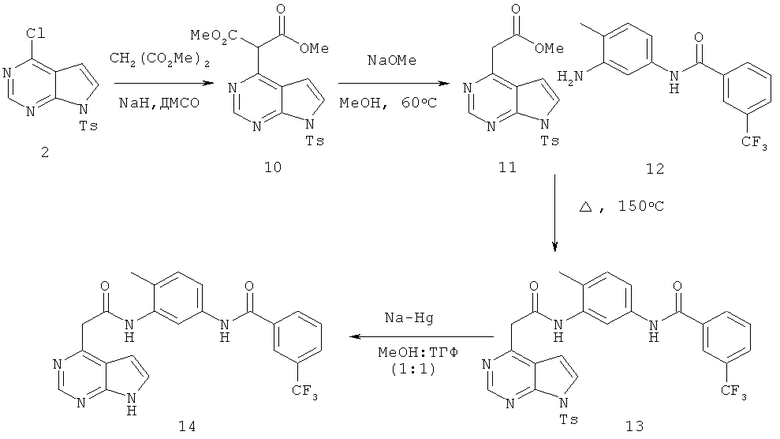
Диметиловый эфир 2-[7-(толуол-4-сульфонил)-7Н-пирроло[2,3-d]пиримидин-4-ил]малоновой кислоты
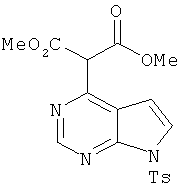
В суспензию NaH (25 мг, 0,62 ммоля, 3,1 экв.) в ДМСО (2,0 мл) при 23°С добавляли диметилмалонат (0,071 мл, 0,62 ммоля, 3,1 экв.). После прекращения выделения водорода добавляли соединение 2 (64 мг, 20 ммолей, 1,0 экв.) и реакционную смесь выдерживали при 80°С в течение 3 ч. Затем реакционную смесь охлаждали до КТ и реакцию останавливали добавлением насыщенного NH4Cl. Органические слои экстрагировали этилацетатом, сушили над Na2SO4, фильтровали и концентрировали, при этом получали требуемый продукт (79 мг, 94%).
Метиловый эфир 7-(толуол-4-сульфонил)-7Н-пирроло[2,3-d]пиримидин-4-ил]уксусной кислоты
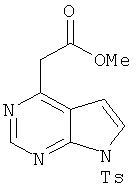
Смесь соединения 10 (0,63 г, 1,56 ммоля, 1,0 экв.) и метоксида натрия (0,038 мл 25% мас./об. раствора, 0,16 ммоля, 0,1 экв.) в МеОН (15 мл) нагревали при 60°С в течение 1 ч. Затем реакционную смесь концентрировали, реакцию останавливали NH4Cl и смесь экстрагировали этилацетатом. Органические слои сушили над Na2SO4, фильтровали и концентрировали. Неочищенный продукт очищали (элюент: гексан/этилацетат, 2:1), при этом получали требуемый продукт.
N-(1-Метилен-3-{2-[7-(толуол-4-сульфонил)-7Н-пирроло[2,3-d]пиримидин-4-ил]ацетиламино}пент-2-енил)-3-трифторметилбензамид
Смесь соединения 11 (60 мг, 0,17 ммоля, 1,0 экв.) и соединения 12 (153 мг, 0,51 ммоля, 3,0 экв.) нагревали при 150°С в течение 4 ч. После завершения реакции реакционную массу охлаждали до КТ, неочищенный продукт очищали (элюент: гексан/этилацетат, 2:1), при этом получали требуемый продукт.
[4-Метил-3-(2-7Н-пирроло[2,3-d]пиримидин-4-илацетиламино)фенил]амид 5,5,5-трифтор-2-метилпент-2-еновой кислоты
В раствор соединения 13 (61 мг, 0,1 ммоля, 1,0 экв.) в ТГФ/МеОН (1:1) добавляли Na-Hg (172 мг, 7,0 экв. в расчете на Na). Реакционную смесь перемешивали в течение 30 мин, затем смесь декантировали для удаления солей ртути и жидкий супернатант концентрировали при пониженном давлении. Твердый осадок распределяли между этилацетатом и водой. Органический слой отделяли, сушили над Na2SO4, фильтровали и концентрировали, при этом получали неочищенный продукт, который перемешивали в течение 1 ч при КТ, а растворитель удаляли в вакууме. Остаток растворяли в ДМСО (1 мл), полученный раствор очищали обращено-фазовой ЖХ-МС, при этом получали указанное в заголовке соединение в виде трифторацетата. МС (m/z): 454,2 (M+1).
Пример 3
Синтез N-(3-имидазол-1-ил-5-трифторметилфенил)-4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]бензамида
4-Метил-N-[3-(4-метилимидазол-1-ил)-5-трифторметилфенил]-3-{3-метил-3[7-(толуол-4-сульфонил)-7H-пирроло[2,3-d]пиримидин-4-ил]уреидо}бензамид
В раствор соединения 15 (15 мг, 0,04 ммоля, 1,0 экв.) и диизопропилэтиламина (6,8 мкл, 0,04 ммоля, 1,0 экв.) в дихлорметане (0,5 мл) при 23°С по каплям добавляли карбамоилхлорид 4 (14,5 мг, 0,04 ммоля, 1,0 экв.) в течение 15 мин. Затем реакционную смесь перемешивали при КТ в течение 8 ч и распределяли между дихлорметаном и водой. Органический слой отделяли, водный слой экстрагировали дихлорметаном. Объединенные органические экстракты промывали водой, сушили над Na2SO4, фильтровали и концентрировали, при этом получали указанный в заголовке продукт 16. Остаток использовали на следующей стадии без дополнительной очистки.
4-Метил-N-[3-(4-метилимидазол-1-ил)-5-трифторметилфенил]-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]бензамид
В раствор соединения 16 (22 мг, 0,031 ммоля, 1,0 экв.) в ТГФ/МеОН (1:1) добавляли Na-Hg (22 мг, 2,0 экв. в расчете на Na). Реакционную смесь перемешивали в течение 30 мин, затем смесь декантировали для удаления солей ртути и жидкий супернатант концентрировали при пониженном давлении. Твердый осадок распределяли между этилацетатом и водой. Органический слой отделяли, сушили над Na2SO4, фильтровали и концентрировали, при этом получали неочищенный продукт, который перемешивали в течение 1 ч при КТ, а растворитель удаляли в вакууме. Остаток растворяли в ДМСО (1 мл), полученный раствор очищали обращено-фазовой ЖХ-МС, при этом получали указанное в заголовке соединение в виде трифторацетата. МС (m/z): 549,2 (M+1).
Пример 4
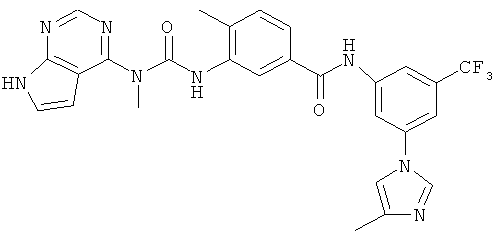
Синтез N-[4-(4-этилпиперазин-1-илметил)-3-трифторметилфенил]-4-метил-3-[3-метил-3-(7Н-пирроло[2.3-d]пиримидин-4-ил)уреидо]бензамида
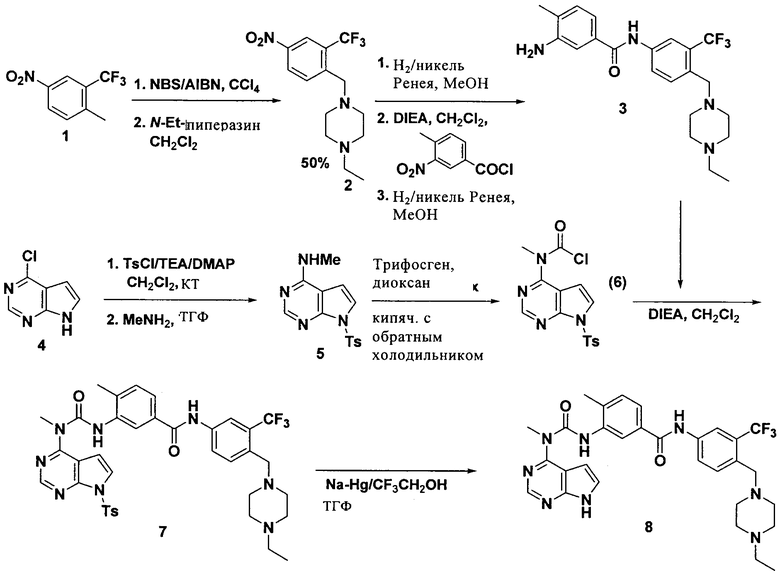
1. 1-Этил-4-(4-нитро-2-трифторбензил)пиперазин
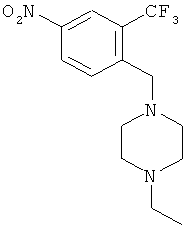
В раствор 1-метил-4-нитро-2-трифторметилбензола (соединение 1, 15 г, 73 ммоля, 1,0 экв.) в четыреххлористом углероде (250 мл) добавляли NBS (13 г, 98 мл, 73 ммоля, 1,0 экв.) и AIBN (1,19 г, 7,3 ммоля, 0,1 экв.) в качестве инициатора. Реакционную смесь кипятили с обратным холодильником в течение ночи, затем распределяли в воде. Органический слой отделяли и водный слой экстрагировали дихлорметаном. Объединенные органические экстракты промывали водой, сушили над Na2SO4, фильтровали и концентрировали, при этом получали твердое вещество, которое растворяли в дихлорметане (300 мл). Прозрачный раствор обрабатывали DIEA (12,55 мл, 73 ммоля, 1,0 экв.) и N-этилпиперазином (8,25 г, 73 ммоля, 1,0 экв.). Реакционную смесь перемешивали при КТ в течение 30 мин (до завершения реакции по данным ЖХ-МС). Реакционную смесь промывали водой, сушили над Na2SO4, фильтровали и концентрировали, при этом получали неочищенный продукт. Неочищенный продукт очищали экспресс-хроматографией на колонке (элюент:гексан/этилацетат, 1:1, об./об.), при этом получали указанное соединение 2 в виде твердого вещества.
2. 4-(4-Этилпиперазин-1-илметил)-3-трифторметилфениламин
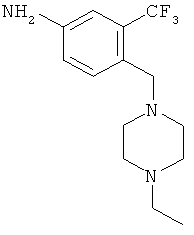
В раствор 4-(4-этилпиперазин-1-илметил)-3-трифторметилфениламина (10 г, 31,54 ммоля, 1,0 экв.) в МеОН (250 мл) добавляли никель Ренея (1,0 г, 10 мас.%), суспензию перемешивали в атмосфере водорода (1 атм) в течение 24 ч. После окончания реакции (по данным ЖХ-МС) реакционную массу фильтровали через целит и фильтрат концентрировали при пониженном давлении, при этом получали требуемый продукт.
3. N-[4-(4-Этилпиперазин-1-илметил)-3-трифторметилфенил]-4-метил-3-нитробензамид
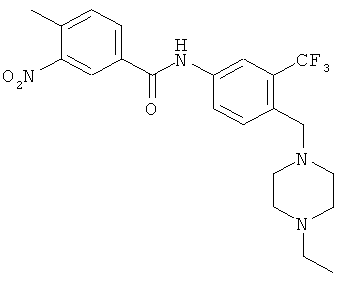
В раствор 4-(4-этилпиперазин-1-илметил)-3-трифторметилфениламина (8,0 г, 27,9 ммоля, 1,0 экв.) в дихлорметане (140 мл) добавляли диизопропилэтиламин (5,27 мл, 30,69 ммоля, 1,1 экв.). Полученный раствор охлаждали до 0°С, затем порциями добавляли 4-метил-3-нитробензоилхлорид (4,18 мл, 28,73 ммоля, 1,03 экв.) и полученную реакционную смесь выдерживали в течение 30 мин. Реакционную смесь распределяли между дихлорметаном и водой. Органический слой отделяли и водный слой экстрагировали дихлорметаном. Объединенные органические экстракты промывали водой, сушили над Na2SO4, фильтровали и концентрировали, при этом получали требуемый продукт, который использовали на следующей стадии без дополнительной очистки.
4. 3-Амино-N-[4-(4-этилпиперазин-1-илметил)-3-трифторметилфенил]-4-метилбензамид
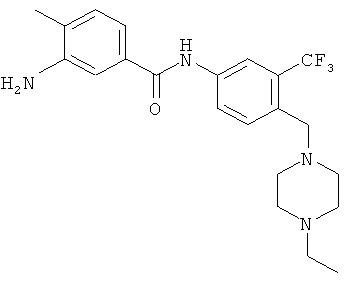
В раствор N-[4-(4-этилпиперазин-1-илметил)-3-трифторметилфенил]-4-метил-3-нитробензамида (10 г, 22,2 ммоля, 1,0 экв.) в МеОН (250 мл) добавляли никель Ренея (1,0 г, 10 мас.%), суспензию перемешивали в атмосфере водорода (1 атм) в течение 24 ч. После завершения реакции (по данным ЖХВР) реакционную массу фильтровали через целит и фильтрат концентрировали при пониженном давлении, при этом получали требуемый продукт 3.
1Н ЯМР (400 МГц, ДМСО-d6): δ 10,24 (s, 1Н), 8,18 (s, 1H), 8,02 (d, J 8,0 Гц, 1H), 7,66 (d, J 8,8 Гц, 1H), 7,16 (s, 1H), 7,13 (m, 2H), 5,1 (s, 2H), 3,55 (s, 2H), 2,37 (m, 10Н), 2,11 (s, 3H), 1,05 (t, 3H).
5. 4-Хлор-7-(толуол-4-сульфонил)-7Н-пирроло[2,3-d]пиримидин
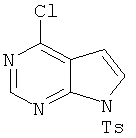
В раствор 6-хлордеазапурина (соединение 4, 10,0 г, 65 ммолей, 1,0 экв.) в дихлорметане (325 мл) добавляли триэтиламин (9,9 мл, 71,5 ммоля, 1.1 экв.) и 4-диметиламинопиридин (катализатор), затем порциями добавляли тозилхлорид (12,76 г, 66,9 ммоля, 1,03 экв.) и реакционную смесь выдерживали в течение 30 мин. Реакционную смесь распределяли между дихлорметаном и водой. Органический слой отделяли и водный слой экстрагировали дихлорметаном. Объединенные органические экстракты промывали водой, сушили над Na2SO4, фильтровали и концентрировали, при этом получали указанный в заголовке продукт, который использовали на следующей стадии без дополнительной очистки.
6. Метил[7-(толуол-4-сульфонил)-7Н-пирроло[2,3-d]пиримидин-4-ил]амин
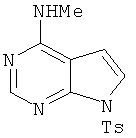
В раствор 4-хлор-7-(толуол-4-сульфонил)-7Н-пирроло[2,3-d]пиримидина (17 г, 55 ммолей, 1,0 экв.) в ТГФ (50 мл) при 23°С добавляли метиламин (66 мл 2,0 М раствора в ТГФ, 13,1 ммоля, 2,4 экв.). Реакционную смесь перемешивали в течение 3 ч при КТ. После завершения реакции ТГФ удаляли при пониженном давлении и твердый остаток распределяли между этилацетатом/ТГФ (1:1) и водой. Органический слой отделяли, сушили над Na2SO4, фильтровали и концентрировали, при этом получали указанный в заголовке продукт 5, который использовали на следующей стадии без дополнительной очистки.
7. N-Хлоркарбонил-N-метил[7-(толуол-4-сульфонил)-7Н-пирроло[2,3-d]пиримидин-4-ил]амин
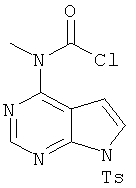
Сосуд Смита (объемом 20 мл), содержащий соединение 5 (2,50 г, 8,2 ммоля, 1,0 экв.), трифосген (2,4 г, 8,2 ммоля, 1,0 экв.) и диоксан (15 мл), помещали в микроволновый нагреватель синтезатора Смита при 150°С в течение 20 мин. Реакционную смесь концентрировали, затем остаток распределяли между дихлорметаном и водой. Органический слой отделяли и водный слой экстрагировали дихлорметаном. Объединенные органические экстракты промывали водой, сушили над Na2SO4, фильтровали и концентрировали, при этом получали указанный в заголовке продукт 6, который использовали на следующей стадии без дополнительной очистки.
8. N-[4-(4-Этилпиперазин-1-илметил)-3-трифторметилфенил]-4-метил-3-{3-метил-3-[7-(толуол-4-сульфонил)-7Н-пирроло[2,3-d]пиримидин-4-ил]уреидо}бензамид
В раствор соединения 3 (3,45 г, 8,2 ммоля, 1,0 экв.) и пиридина (0,66 мл, 0,82 ммоля, 1,0 экв.) в дихлорметане (40 мл) при 23°С по каплям в течение 15 мин добавляли карбамоилхлорид 6 (3 г, 8,2 ммоля, 1,0 экв.). Реакционную смесь перемешивали при КТ в течение 8 ч, затем распределяли между дихлорметаном и водой. Органический слой отделяли и водный слой экстрагировали дихлорметаном. Объединенные органические экстракты промывали водой, сушили над Na2SO4, фильтровали и концентрировали, при этом получали неочищенный продукт 7. Остаток очищали колоночной хроматографией (элюент: МеОН/CH2Cl2, 5:1), при этом получали указанный в заголовке продукт 7.
9. N-[4-(4-Этилпиперазин-1-илметил)-3-трифторметилфенил]-4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]бензамид
В раствор тозилмочевины 7 (1,5 г, 2,0 ммоля, 1,0 экв.) в ТГФ (10 мл) добавляли Na-Hg (0,92 г, 2,0 экв. в расчете на Na), затем трифторэтанол (291 мкл, 4,0 ммоля. 2,0 экв.). Реакционную смесь перемешивали в течение 15 мин, затем смесь декантировали для удаления солей ртути и жидкий супернатант концентрировали при пониженном давлении. Твердый осадок распределяли между этилацетатом и водой. Органический слой отделяли, сушили над Na2SO4, фильтровали и концентрировали, при этом получали неочищенный продукт, который очищали препаративной ЖХВР, при этом получали указанное в заголовке соединение 8. МС (m/z): 595,3 (M+1).
1Н ЯМР (400 МГц, ДМСО-d6): δ 12,98 (ушир.s, 1Н), 12,42 (ушир.s, 1H), 10,52 (ушир.s, 1H), 8,68 (s, 1H), 8,65 (s, 1H), 8,27 (s, 1H), 8,11 (d, J 7,3 Гц, 1H), 7,76 (d, J 7,2 Гц, 1H), 7,72 (d, J 7,0 Гц, 1H), 7,6 (m, 1H), 7,47 (d, J 8,3 Гц, 1H) 6,9 (m, 1H), 3,78 (s, 3Н), 3,62 (s, 2H), 2,51 (s, 3H), 2,43-2,35 (m, 10Н), 1,04 (t, J 6,9 Гц, 3Н).
Соединения формулы I, приведенные в таблице 1, получали по методикам, описанным в приведенных выше примерах, с использованием соответствующих исходных материалов.
Методы анализа
Соединения по настоящему изобретению анализируют по их способности селективно ингибировать пролиферацию клеток 32D, экспрессирующих BCR-Abl (32D-p210), в сравнении с исходными клетками 32D. Соединения, селективно ингибирующие пролиферацию этих клеток, трансформированных BCR-Abl, анализируют на наличие антипролиферативной активности в отношении клеток Ba/F3, экспрессирующих дикий тип или мутантные формы Bcr-abl.
Ингибирование BCR-Abl-зависимой клеточной пролиферации (экспресс-анализ)
Для анализа использовали линию клеток мыши, которые представляли собой гемопоэтические клетки-предшественники линии 32D, трансформированнные кДНК BCR-Abl (32D-p210). Указанные клетки культивировали в среде RPMI/10% эмбриональная телячья сыворотка (RPMI/FCS), содержащей пенициллин 50 мкг/мл, стрептомицин 50 мкг/мл и 200 мМ L-глутамин. Нетрансформированные клетки 32D культивировали при добавлении среды, содержащей 15% WEHI в качестве источника IL3.
Суспензию клеток 32D или 32D-p210 высеивали в 384-луночные микропланшеты Грейнера (черного цвета) с плотностью 5000 клеток в лунке в 50 мкл среды. В каждую лунку добавляли по 50 нл раствора анализируемого соединения (1 мМ раствор в ДМСО) (включая STI571 в качестве положительного контроля). Клетки инкубировали при 37°С в течение 72 ч в атмосфере 5% СО2, в каждую лунку добавляли по 10 мкл 60% раствора реагента Alamar Blue (фирма Tek diagnostics) и клетки инкубировали в течение еще 24 ч. Интенсивность флуоресценции (возбуждение при 530 нм, испускание при 580 нм) измеряли с использованием системы Acquest™ (фирма Molecular Devices).
Ингибирование BCR-Abl-зависимой клеточной пролиферации
Клетки 32D-p210 высеивали в 96-луночные планшеты ТС с плотностью 15000 клеток в лунке. В каждую лунку добавляли по 50 мкл раствора анализируемого соединения при двухкратном серийном разведении (Смакс составляет 40 мкМ) (включая STI571 в качестве положительного контроля). Клетки инкубировали при 37°С в течение 48 ч в атмосфере 5% СО2, в каждую лунку добавляли по 15 мкл раствора МТТ (фирма Promega) и клетки инкубировали в течение еще 5 ч. Оптическую плотность при 570 нм измеряли спектрофотометрией и по графику зависимости доза/ответ определяли величины IC50, т.е. концентрацию соединения, обеспечивающую 50% ингибирование.
Действие на клеточный цикл
Клетки 32D и 32D-p210 высеивали в 6-луночные планшеты ТС с плотностью 2,5×106 клеток в лунке в 5 мл среды и добавляли анализируемое соединение при концентрации 1 или 10 мкМ (включая STI571 в качестве контроля). Затем клетки инкубировали при 37°С в течение 24 ч или 48 ч в атмосфере 5% СО2. Клеточную суспензию (2 мл) промывали ФСБ, фиксировали в 70% EtOH в течение 1 ч и обрабатывали ФБС/ЭДТА/РНКаза А в течение 30 мин. Затем добавляли иодид пропидия (Cf 10 мкг/мл) и измеряли интенсивность флуоресценции проточной цитометрией с использованием системы FACScalibur™ (фирма BD Biosciences). Анализируемые соединения по настоящему изобретению проявляли апоптотическое действие на клетки 32D-p210, но не вызывали апоптоза в исходных клетках 32D.
Действие на аутофосфорилирование клеточного белка BCR-Abl
Аутофосфорилирование BCR-Abl измеряли методом ИФА со связыванием антигена с использованием иммобилизованных антител, специфичных к с-abl, и антител к фосфотирозину. Клетки 32D-p210 высеивали в 96-луночные планшеты ТС с плотностью 2×105 клеток в лунке в 50 мкл среды. В каждую лунку добавляли по 50 мкл раствора анализируемого соединения при двухкратном серийном разведении (Смакс составляет 10 мкМ) (включая STI571 в качестве положительного контроля). Клетки инкубировали при 37°С в течение 90 мин в атмосфере 5% СО2. Затем клетки обрабатывали в течение 1 ч на ледяной бане 150 мкл буферного раствора для лизиса (50 мМ трис-HCl, рН 7,4, 150 мМ NaCI, 5 мМ EDTA, 1 мМ EGTA и 1% NP-40), содержащего ингибиторы протеаз и фосфатаз. 50 мкл Клеточного лизата добавляли в 96-луночные планшеты (optiplates), предварительно покрытые специфичными антителами анти-abl и блокированные. Планшеты инкубировали при 4°С в течение 4 ч, промывали буферным раствором TBS/твин 20, добавляли 50 мкл раствора конъюгата щелочной фосфатазы с антителами к фосфотирозину и планшет инкубировали при 4°С в течение ночи. Планшет промывали буферным раствором TBS/твин 20, добавляли 90 мкл люминесцентного субстрата и измеряли флуоресценцию с использованием системы Acquest™ (фирма Molecular Devices). Анализированные соединения по изобретению, которые ингибируют пролиферацию клеток, экспрессирующих BCR-Abl, ингибируют аутофосфорилирование клеточного BCR-Abl дозозависимым способом.
Действие на пролиферацию клеток, экспрессирующих мутантные формы Bcr-abl
Соединения по изобретению анализировали на наличие антипролиферативного действия на клетки Ba/F3, экспрессирующие BCR-AbI дикого типа или его мутантные формы (G250E, E255V, T315I, F317L, М351Т), которые обладают устойчивостью или пониженной чувствительностью к STI571. Антипролиферативное действие указанных соединений на клетки, экспрессирующие мутантный BCR-Abl, и на нетрансформированные клетки анализировали при концентрациях 10, 3,3, 1,1 и 0,37 мкМ, как описано выше (в среде, не содержащей IL3). Величины IC50 для соединений, не обладающих токсичностью в отношении нетрансформированных клеток, определяли по графику зависимости доза/ответ, как описано выше.
FGFR3 (ферментативный анализ)
Анализ киназной активности очищенного FGFR3 (фирма Upstate) проводили в конечном объеме 10 мкл, содержащем 0,25 мкг/мл фермента в буферном растворе для определения киназы (30 мМ трис-HCl, рН 7,5, 15 мМ MgCl2, 4,5 мМ MnCl2, 15 мкМ Na3VO4 и 50 мкг/мл БСА), субстраты (5 мкг/мл биотин-поли-EY (Glu, Tyr) (фирма CIS-US, Inc.) и 3 мкМ АТФ. Анализ проводили с использованием двух растворов: первый раствор (5 мкл), содержащий фермент FGFR3 в буферном растворе для определения киназы, добавляли в 384-луночные планшеты ProxiPlate® (фирма Perkin-Elmer), в каждую лунку добавляли 50 нл раствора соединений в ДМСО, а затем 5 мкл второго раствора, содержащего субстрат (поли-EY) и АТФ в буферном растворе для определения киназы. Реакционную смесь инкубировали при комнатной температуре в течение 1 ч, реакцию останавливали добавлением 10 мкл смеси для обнаружения HTRF, содержащей 30 мМ трис-HCl, рН 7,5, 0,5 М KF, 50 мМ ETDA, 0,2 мг/мл БСА, 15 мкг/мл стрептавидин-XL665 (фирма CIS-US, Inc.) и 150 нг/мл конъюгата криптата и антифосфотирозиновых антител (фирма CIS-US, Inc.). Смесь инкубировали при комнатной температуре в течение 1 ч для образовния комплекса стрептавидин/биотин, сигналы флуоресценции с разрешением по времени регистрировали на флуориметре Analyst GT (фирма Molecular Devices Corp.). Величины IC50 рассчитывали методом линейной регрессии по проценту ингибирования каждым соединением при 12 концентрациях (разведение 1:3 исходного раствора от 50 мкМ до 0,28 нМ). По данным указанного анализа соединения по изобретению обладают величинами IC50 в интервале от 10 нМ до 2 мкМ.
FGFR3 (анализ в культуре клеток)
Соединения по изобретению анализировали по способности ингибировать пролиферацию трансформированных клеток Ba/F3-TEL-FGFR3, которая является зависимой от активности клеточной киназы FGFR3. Клетки Ba/F3-TEL-FGFR3 культивировали в суспензии в среде RPMI 1640, содержащей 10% эмбриональной телячьей сыворотки, до концентрации 800000 клеток/мл. Клетки высеивали в 384-луночные планшеты с плотностью 5000 клеток/лунку в 50 мкл культуральной среды. Соединения по изобретению растворяли и разбавляли диметилсульфоксидом (ДМСО). Получали 12 растворов в ДМСО при серийном разведении 1:3 обычно от 10 мМ до 0,05 мкМ. В лунки с клетками добавляли по 50 нл разбавленных растворов соединений и инкубировали в течение 48 ч в инкубаторе для клеточных культур. Затем в лунки с клетками добавляли реагент AlamarBlue® (фирма TREK Diagnostic Systems) до конечной концентрации 10%, который используется для регистрации восстанавливающей среды, создаваемой пролиферирующими клетками. Смесь инкубировали при 37°С в инкубаторе для клеточных культур в течение еще 4 ч и определяли интенсивность флуоресценции восстановленного AlamarBlue® (возбуждение при 530 нм, испускание при 580 нм) на флуориметре Analyst GT (фирма Molecular Devices Corp.). Величины IC50 рассчитывали методом линейной регрессии по проценту ингибирования каждым соединением при 12 концентрациях.
FLT3 и PDGFRβ (анализ в культуре клеток)
Действие соединений по изобретению на клеточную активность (активность в клетках) FLT3 и PDGFRβ оценивали с использованием методов, описанных выше для определения клеточной активности FGFR3, при этом вместо клеток Ba/F3-TEL-FGFR3 использовали клетки Ba/F3-FLT3-ITD и Ba/F3-Tel-PDGFRβ соответственно.
b-Raf (ферментативный анализ)
Соединения по изобретению анализировали по способности ингибировать активность b-Raf. Анализ проводили в 384-луночных планшетах MaxiSorp (фирма NUNC) с черными стенками и прозрачным дном. Субстрат IκВα разбавляли в DPBS (1:750) и в каждую лунку добавляли по 15 мкл. Планшеты инкубировали при 4°С в течение ночи и трижды промывали TBST (25 мМ трис, рН 8,0, 150 мМ NaCl и 0,05% твин 20) с использованием устройства для промывки планшетов EMBLA. Планшеты блокировали реагентом Superblock (15 мкл/лунка) в течение 3 ч при комнатной температуре, трижды промывали TBST и планшет сушили постукиванием. Затем в каждую лунку добавляли 10 мкл буферного раствора для анализа, содержащего 20 мкМ АТФ, затем 100 нл или 500 нл раствора соединения. B-Raf разбавляли в буферном растворе для анализа (1 мкл до 25 мкл) и в каждую лунку добавляли по 10 мкл разбавленного раствора b-Raf (0,4 мкг/лунка). Планшеты инкубировали при комнатной температуре в течение 2,5 ч. Киназную реакцию останавливали при шестикратной промывке планшета раствором TBST. Антитела фосф-IκВα (Ser32/36) разбавляли в реагенте Superblock (1:10000) и в каждую лунку добавляли по 15 мкл. Планшеты инкубировали при 4°С в течение ночи и шестикратно промывали раствором TBST. Коньюгат АР и антимышиного, козьего IgG разбавляли в реагенте Superblock (1:1500) и в каждую лунку добавляли по 15 мкл. Планшеты инкубировали при комнатной температуре в течение 1 ч и шестикратно промывали раствором TBST. В каждую лунку добавляли по 15 мкл флуоресцентного субстрата Attophos АР (фирма Promega) и планшеты инкубировали при комнатной температуре в течение 15 мин. Планшеты анализировали на планшет-ридере Acquest или Analyst GT с использованием программы для анализа интенсивности флуоресценции (возбуждение при 455 нм, излучение при 580 нм).
b-Raf (анализ в культуре клеток)
Соединения по изобретению анализировали с использованием клеток A375 по способности ингибировать фосфорилирование МЕК. Клетки линии A375 (АТСС) были получены от пациента с меланомой и содержали мутацию V599E в гене B-Raf. Уровень фосфорилированного МЕК повышался за счет мутации В-Raf. Клетки А375, от субконфлюентных до конфлюентных, инкубировали в присутствии соединений в течение 2 ч при 37°С в бессывороточной среде. Клетки однократно промывали холодным ФСБ и лизировали в буферном расторе для лизиса, содержащем 1% тритона Х100. После центрифугирования супернатанты анализировали электрофорезом в ДСН-ПААГ и переносили на нитроцеллюлозные мембраны. Затем мембраны анализировали методом вестерн-блоттинг с использованием антител анти-фосфо-МЕК (сер217/221, фирма Cell Signaling). Количество фосфорилированного МЕК регистрировали по интенсивности зон фосфо-МЕК на нитроцеллюлозных мембранах.
Радиоферментный анализ при связывании субстрата на фильтре с использованием набора «Upstate KinaseProfiler™»
Соединения по изобретению анализировали по их способности ингибировать активность индивидуальных членов киназной панели. Соединения анализировали при двойном повторе с конечной концентрацией 10 мкМ по следующей общей методике. При этом состав буферного раствора для определения киназы и субстраты варьируют при анализе различных киназ, включенных в набор "Upstate KinaseProfiler™". Буферный раствор для определения киназы (2,5 мкл, 10×, содержащий при необходимости MnCl2), активную киназу (0,001-0,01 ед., 2,5 мкл), специфический пептид или поли(Glu4-Tyr) (5-500 мкМ или 0,01 мг/мл) в буферном растворе для определения киназы и буферный раствор для определения киназы (50 мкМ, 5 мкл) смешивали в пробирке эппендорф на ледяной бане. Затем добавляли смесь Mg/АТФ (10 мкл, 67,5 (или 33,75) мМ MgCl2, 450 (или 225) мкМ АТФ и 1 мкКи/мкл [γ-32P]-АТФ (3000 Ки/ммоль)) и реакционную смесь инкубировали при приблизительно 30°С в течение приблизительно 10 мин. Реакционную смесь (20 мкл) наносили на квадратные кусочки бумаги Р81 (фосфоцеллюлоза, 2 см × 2 см, в случае положительно заряженного пептидного субстрата) или ватман №1 (в случае пептидного субстрата поли(Glu4-Tyr). Квадратики четырехкратно промывали (каждый раз в течение 5 мин) 0,75% фосфорной кислотой, однократно ацетоном (в течение 5 мин), переносили в сцинтилляционный флакон, добавляли 5 мл сцинтилляционной жидкости и измеряли включение фосфора 32Р (расп/мин) в пептидный субстрат на сцинтиляционном счетчике Beckman. Для каждой реакции рассчитывали процент ингибирования.
Соединения формулы I, в свободной форме или в форме фармацевтически приемлемой соли, проявляют ценные фармакологические свойства, например, как показано при испытаниях in vitro, описанных в тексте заявки. Например, соединения формулы I предпочтительно характеризуются величинами IC50 в интервале от 1×10-10 до 1×10-5 М, предпочтительно менее 500 нМ, 250 нМ, 100 нМ и 50 нМ для дикого типа и мутанта киназы BCR-Abl.
Например, N-[4-(4-этилпиперазин-1-илметил)-3-трифторметилфенил]-4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]бензамид (пример 4) характеризуется величинами IC50<5 нМ, <5 нМ, <5 нМ, <5 нМ, <5 нМ и <5 нМ в отношении киназы дикого типа и мутантных форм G250E, E255V, T315I, F317L и М351Т Bcr-abl соответственно.
Например, N-[4-(4-этилпиперазин-1-илметил)-3-трифторметилфенил]-4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]бензамид (пример 4) характеризуется величинами IC50 36 нМ, 15 нМ, 3,8 нМ и 100 нМ для киназ FGFR3, FLT-3, PDGFR-β и B-RAF соответственно.
Соединения по изобретению ингибируют некоторые киназы, указанные в панели киназ, на более 40%, предпочтительно более 50%, наиболее предпочтительно более 60%. Например, N-[4-(4-этилпиперазин-1-илметил)-3-трифторметилфенил]-4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]бензамид (пример 4) характеризуется следующим профилем ингибирования (процент ингибирования каждой киназы заключен в скобки): Abl (T315L) (98%), Abl (мыши) (98%), Bmx (человека) (100%), ВТК (человека) (99%), c-RAF (человека) (97%), CSK (человека) (100%), cSRC (человека) (99%), Fes (человека) (97%), FGFR3 (человека) (98%), Flt3 (человека) (96%), IKKα (человека) (98%), IKKβ (человека) (96%), JNK1α1 (человека) (81%), JNK2α2 (человека) (99%), Lck (человека) (100%), Met (человека) (70%), МКК4 (мыши) (87%), МКК6 (человека) (98%), p70S6K (человека) (94%), РАК2 (человека) (62%), PDGFRα (человека) (93%), РКА (человека) (92%), PKCα (человека) (61%), PKD 2 (человека) (94%), ROCK-II (человека) (78%), Ros (человека) (62%), Rsk1 (человека) (95%), SAPK2α (человека) (92%), SAPK2β (h) (85%), SAPK3 (человека) (92%), SAPK4 (человека) (82%), SGK (h) (81%), Syk (человека) (63%), Tie2 (человека) (98%) и TrkB (человека) (99%).
Следует понимать, что описанные в данном контексте примеры и варианты осуществления изобретения представлены только для иллюстрации и что различные модификации или изменения, очевидные для специалиста в данной области техники, включены в объем и сущность настоящего изобретения и пункты прилагаемой формулы изобретения. Все публикации, патенты и опубликованные заявки, цитированные в данном контексте, включены в настоящее описание в качестве ссылок.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ | 2006 |
|
RU2383545C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗ | 2006 |
|
RU2387653C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗ | 2006 |
|
RU2368602C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ КАК ИНГИБИТОРЫ ПРОТЕИНКИНАЗЫ | 2005 |
|
RU2406725C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ | 2005 |
|
RU2401265C2 |
| ИНГИБИТОРЫ ТИРОЗИНКИНАЗЫ БРУТОНА | 2013 |
|
RU2619465C2 |
| ПРОИЗВОДНЫЕ 4-АМИНО-6-ФЕНИЛПИРРОЛО[2,3] ПИРИМИДИНА, ОБЛАДАЮЩИЕ ИНГИБИРУЮЩИМ ДЕЙСТВИЕМ В ОТНОШЕНИИ ДЕЙСТВИЯ ТИРОЗИНКИНАЗЫ, ИХ ПРИМЕНЕНИЕ И СПОСОБЫ ПОЛУЧЕНИЯ (ВАРИАНТЫ) | 2002 |
|
RU2318826C2 |
| Новое производное пиридо [3,4-d] пиримидин-8-она, обладающее ингибирующей протеинкиназы активностью, и фармацевтическая композиция для предупреждения, облегчения или лечения рака, содержащее указанное выше | 2020 |
|
RU2780168C1 |
| КОМБИНАЦИИ, ВКЛЮЧАЮЩИЕ ИНГИБИТОРЫ Bcr-Abl/c-Kit/PDGF-R TK, ДЛЯ ЛЕЧЕНИЯ РАКА | 2007 |
|
RU2452492C2 |
| ПРОИЗВОДНЫЕ ТИЕНО[3,2-d]ПИРИМИДИНА, ОБЛАДАЮЩИЕ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ ПРОТЕИНКИНАЗЫ | 2011 |
|
RU2524210C2 |
Изобретение относится к новым соединениям формулы I и их фармацевтически приемлемым солям, обладающим ингибирующей активностью в отношении киназ, выбранных из Abl, Bcr-Abl, Bmx, ВТК, b-RAF, c-RAF, CSK, cSRC, Fes, FGFR3, Flt3, IKKα, IKKβ, JNK1α1, JNK2α2, Lck, Met, MKK4, MKK6, p70S6K, PAK2, PDGFRα, PKA, PKCα, PKD2, ROCK-II, Ros, Rsk1, SAPK2α, SAPK2β, SAPK3, SAPK4, SGK, Syk, Tie2 и TrkB. Соединения могут найти применение для лечения или профилактики заболеваний или нарушений, ассоциированных с аномальной или разрегулированной киназной активностью, таких как пролиферативные заболевания, заболевания иммунной и нервной системы. В соединениях формулы I
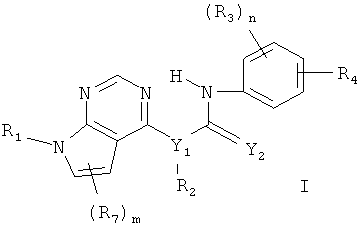
n равно 1, m равно 0, Y1 выбран из N и CR5, причем R5 представляет собой водород, Y2 представляет собой О, R1 представляет собой водород, R2 выбран из водорода и С1-С6алкила, R3 выбран из группы, включающей водород, C1-С6алкил, C1-С6алкокси, R4 выбран из -NR5C(O)R6 и -C(O)NR5R6, где R5 выбран из водорода и C1-C6алкила, а R6 представляет собой фенил, необязательно замещенный 1-3 радикалами, выбранными из группы, включающей NR5R5, галогензамещенный C1-С6алкил, C5-С6гетероарил(С0-С4)алкил, где гетероарил содержит 1-2 гетероатома, выбранных из N и О, C5-C6гетероцикло(С0-С4)алкил, где гетероциклил содержит 1-2 гетероатома N, и C5-С6гетероцикло(С0-С4)алкокси, где гетероциклил содержит 1-2 гетероатома N, причем любой гетероарил или гетероциклил в составе R6 необязательно замещен 1-3 радикалами, независимо выбранными из группы, включающей C1-С6алкил и гидрокси (C1-С6)алкил. 4 н. и 4 з.п. ф-лы.
1. Соединение формулы I
где n равно 1,
m равно 0,
Y1 выбран из N и CR5, причем R5 представляет собой водород,
Y2 представляет собой О,
R1 представляет собой водород,
R2 выбран из водорода и C1-С6алкила,
R3 выбран из группы, включающей водород, С1-С6алкил, C1-С6алкокси,
R4 выбран из -NR5C(O)R6 и -C(O)NR5R6, где R5 выбран из водорода и С1-С6алкила,
a R6 представляет собой фенил, необязательно замещенный 1-3 радикалами, выбранными из группы, включающей NR5R5, галогензамещенный С1-С6алкил, C5-C6гетероарил(С0-С4)алкил, где гетероарил содержит 1-2 гетероатома, выбранных из N и О, C5-С6гетероцикло(С0-С4)алкил, где гетероциклил содержит 1-2 гетероатома N, и C5-С6гетероцикло(С0-С4)алкокси, где гетероциклил содержит 1-2 гетероатома N, причем любой гетероарил или гетероциклил в составе R6 необязательно замещен 1-3 радикалами, независимо выбранными из группы, включающей С1-С6алкил и гидрокси (C1-С6)алкил,
и его фармацевтические приемлемые соли.
2. Соединение по п.1 формулы Ia
где n равно 1,
Y1 выбран из N и СН,
R2 выбран из водорода и C1-C4алкила,
R3 выбран из группы, включающей водород, C1-C6алкил, C1-С6алкокси,
R4 выбран из -NR5C(O)R6 и -C(O)NR5R6, где R5 выбран из водорода и C1-C6алкила,
a R6 представляет собой фенил, необязательно замещенный 1-3 радикалами, выбранными из группы, включающей NR5R5, галогензамещенный C1-C6алкил, С5-С6гетероарил(С0-С4)алкил, где гетероарил содержит 1-2 гетероатома, выбранных из N и О, С5-С6гетероцикло(С0-С4)алкил, где гетероциклил содержит 1-2 гетероатома N, и C5-С6гетероцикло(С0-С4)алкокси, где гетероциклил содержит 1-2 гетероатома N, причем любой гетероарил или гетероциклил в составе R6 необязательно замещен 1-3 радикалами, независимо выбранными из группы, включающей C1-С6алкил и гидрокси (C1-C6)алкил.
3. Соединение по п.2, в котором R2 выбран из водорода и метила, a R3 выбран из метила и метокси.
4. Соединение по п.3, в котором R4 выбран из -NHC(O)R6 и -C(O)NHR6, где R6 представляет собой фенил, необязательно замещенный 1-3 радикалами, выбранными из группы, включающей трифторметил, диметиламино, имидазолил, морфолино, морфолинометил, пиперазинил, пиперазинилметил и пирролидинилметокси, причем указанные имидазолил, пиперазинил, пиперазинилметил необязательно замещены группой, выбранной из метила, этила и гидроксиэтила.
5. Соединение по п. 4, выбранное из группы, включающей
N-[4-(4-этилпиперазин4-илметал)-3-трифторметилфенил]-4-метил-3-[3-метил-3-(7Н-пирроло [2,3-d] пиримидин-4-ил)уреидо]бензамид,
4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]-N-(4-пиперазин-1-илметил-3-трифторметилфенил)бензамид,
3-(4-метилимидазол-1-ил)-N-{4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-5-трифторметилбензамид,
N-4-[4-метил-3-(2-7Н-пирроло[2,3-d]пиримидин-4-илацетиламино)фенил]-3-трифторметилбензамид,
N-(3-имидазол-1-ил-5-трифторметилфенил)-4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]бензамид,
N-{4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-3 -трифторметилбензамид,
4-(2-метилимидазол-1-ил)-N-{4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-3-трифторметилбензамид,
N-{4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-4-морфолин-4-ил-3-трифторметилбензамид,
N-{4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-4-(4-метилпиперазин-1-илметил)-3-трифторметилбензамид,
N-{4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-4-морфолин-4-илметил-3-трифторметилбензамид,
N-{4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-3-морфолин-4-ил-5-трифторметилбензамид,
N-{4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-4-(4-метилпиперазин-1-ил)-3-трифторметилбензамид,
4-(4-этилпиперазин-1-ил)-N-{4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-3-трифторметилбензамид,
N-{4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-3-(4-метилпиперазин-1-ил)-5-трифторметилбензамид,
3-диметиламино-N-{4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-5-трифторметилбензамид,
4-(4-этилпиперазин-1-илметил)-N-{4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-3-трифторметилбензамид,
3-(4-этилпиперазин-1-ил)-N-{4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-5-трифторметилбензамид,
4-метил-N-[3-(4-метилимидазол-1-ил)-5-трифторметилфенил]-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]бензамид,
3-[4-(2-гидроксиэтил)пиперазин-1-ил]-N-{4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-5-трифторметилбензамид,
N-{4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил)-3-пиперазин-1-ил-5-трифторметилбензамид,
N-{4-метил-3-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-3-(пирролидин-2-илметокси)-5-трифторметилбензамид,
N-{3-метокси-5-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-3-морфолин-4-ил-5-трифторметилбензамид,
N-{3-метокси-5-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-3-(4-метилимидазол-1-ил)-5-трифторметилбензамид,
4-(4-этилпиперазин-1-илметил)-N-{3-метокси-5-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-3-трифторметилбензамид,
N-{3-метокси-5-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-4-(4-метилпиперазин-1-илметил)-3-трифторметилбензамид и
N-{3-метокси-5-[3-метил-3-(7Н-пирроло[2,3-d]пиримидин-4-ил)уреидо]фенил}-4-(2-метилимидазол-1-ил)-3-трифторметилбензамид.
6. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении киназ, включающая терапевтически эффективное количество соединения по п.1 в комбинации с фармацевтически приемлемым эксципиентом.
7. Способ ингибирования аномальной активности киназ, выбранных из группы, включающей Abl, Bcr-Abl, Bmx, ВТК, b-RAF, c-RAF, CSK, cSRC, Fes, FGFR3, Flt3, IKKα, IKKβ, JNK1α1, JNK2α2, Lck, Met, MKK4, MKK6, p70S6K, PAK2, PDGFRα, PKA, PKCα, PKD2, ROCK-II, Ros, Rskl, SAPK2α, SAPK2β, SAPK3, SAPK4, SGK, Syk, Tie2 и TrkB киназы, путем введения терапевтически эффективного количества соединения по п.1.
8. Применение соединения по п.1 для получения лекарственного средства, предназначенного для лечения заболевания, при котором активность киназ Abl, Bcr-Abl, Bmx, ВТК, b-RAF, c-RAF, CSK, cSRC, Fes, FGFR3, Flt3, IKKα, IKKβ, JNK1α1, JNK2α2, Lck, Met, MKK4, MKK6, p70S6K, PAK2, PDGFRα, PKA, PKCα, PKD2, ROCK-II, Ros, Rskl, SAPK2α, SAPK2β, SAPK3, SAPK4, SGK, Syk, Tie2 и TrkB принимает участие в развитии заболевания и/или симптомов заболевания.
| WO 2005039486 A2, 06.05.2005 | |||
| WO 2005016528 А2, 24.02.2005. |

