ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к применению новых соединений, которые ингибируют тирозинкиназу Брутона (Btk) и полезны для лечения аутоиммунных и воспалительных заболеваний, вызванных аномальной активацией В-клеток.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Протеинкиназы составляют одно из самых больших семейств ферментов человека и регулируют множество различных сигнальных процессов путем присоединения фосфатных групп к белкам (Т. Hunter, Cell 1987 50: 823-829). В частности, тирозинкиназы фосфорилируют белки по фенольной группе тирозиновых остатков. Семейство тирозинкиназ включает члены, которые контролируют клеточный рост, миграцию и дифференцировку. Аномальная киназная активность вовлечена в патогенез множества заболеваний человека, включая различные виды рака, аутоиммунные и воспалительные заболевания. Поскольку протеинкиназы стоят в ряду ключевых регуляторов клеточной сигнальной системы, они представляют собой мишень для модуляции клеточной функции посредством низкомолекулярных ингибиторов киназ и, таким образом, являются хорошей мишенью для разработки лекарств. Помимо лечения патологических процессов, опосредованных киназами, селективные и эффективные ингибиторы активности киназ полезны также для исследования клеточных сигнальных процессов и выявления других клеточных мишеней, представляющих терапевтический интерес.
Имеются надежные свидетельства тому, что В-клетки играют ключевую роль в патогенезе аутоиммунных и/или воспалительных заболеваний. Лекарственные средства на основе белка, которые истощают популяцию В-клеток, например Rituxan, эффективны против вызванных антителами воспалительных заболеваний, таких как ревматоидный артрит (Rastetter et al. Annu Rev Med 2004 55: 477). Поэтому ингибиторы протеинкиназ, которые участвуют в активации В-клеток, должны представлять собой полезные терапевтические средства для патологических состояний, опосредованных В-клетками, таких как продукция аутоантител.
Сигнальный путь, опосредованный рецептором В-клеток (BCR), контролирует целый ряд В-клеточных реакций, включая пролиферацию и дифференцировку в зрелые клетки, продуцирующие антитела. BCR является ключевым регуляторным пунктом активности В-клеток, и аномалии в сигнальной системе могут стать причиной разрегулированой пролиферации В-клеток и образования патогенных аутоантител, что приводит к различным аутоиммунным и/или воспалительным заболеваниям. Тирозинкиназа Брутона (Btk) представляет собой не ассоциированную с BCR-рецептором киназу, находящуюся вблизи мембраны и расположенную в сигнальном пути сразу после BCR. Было показано, что недостаток киназы Btk блокирует BCR-опосредованный сигнальный путь, и поэтому ингибирование киназы Btk может представлять собой полезный терапевтический подход для блокирования патологических процессов, опосредованных В-клетками.
Киназа Btk является членом Тес-семейства тирозинкиназ, и было показано, что она является ключевым регулятором раннего развития В-клеток, а также активации и выживаемости зрелых В-клеток (Khan et al. Immunity 1995 3: 283; Ellmeier et al. J. Exp. Med. 2000 192: 1611). Мутации киназы Btk у человека приводят к состоянию Х-связанной агаммаглобулинемии (XLA) (см. обзор: Rosen et al. New Eng. J. Med. 1995 333: 431 and Lindvall et al. Immunol. Rev. 2005 203: 200). У таких пациентов ослаблен иммунитет, а также показано, что у них нарушено созревание В-клеток, понижен уровень иммуноглобулинов и периферических В-клеток, снижен Т-клеточный независимый иммунный ответ, а также ослаблена мобилизация кальция после BCR-стимуляции.
Роль, которую играет Btk в аутоиммунных и воспалительных заболеваниях, доказана также с помощью моделей Btk-дефицитных мышей. В преклинических мышиных моделях системной красной волчанки (SLE) Btk-дефицитные мыши демонстрируют существенное улучшение в прогрессировании заболевания. Кроме того, Btk-дефицитные мыши резистентны по отношению к индуцируемому коллагеном артриту (Jansson and Holmdahl Clin. Exp. Immunol. 1993 94: 459). Была показана дозозависимая эффективность селективного ингибитора киназы Btk в мышиной модели артрита (Z. Pan et al., Chem. Med Chem. 2007 2: 58-61).
Киназа Btk экспрессируется также клетками, отличными от В-клеток, которые могут быть вовлечены в прогрессирование заболевания. Например, киназа Btk экспрессируется тучными клетками, и тучные клетки из Btk-дефицитного костного мозга демонстрируют нарушенную антиген-индуцируемую дегрануляцию (Iwaki et al. J. Biol. Chem. 2005 280: 40261). Это говорит о том, что киназа Btk может быть полезной для лечения патологических реакций тучных клеток, таких как аллергия и астма. Кроме того, моноциты от пациентов с XLA, у которых отсутствует активность киназы Btk, демонстрируют сниженную продукцию фактора некроза опухолей альфа (TNF-α) после стимуляции (Horwood et al. J Exp Med 197: 1603, 2003). Таким образом, опосредованное TNF-α воспаление можно модулировать низкомолекулярными ингибиторами киназы Btk. Кроме того, сообщалось, что киназа Btk участвует в апоптозе (Islam and Smith Immunol. Rev. 2000 178: 49) и поэтому ингибиторы Btk могут быть полезными для лечения конкретных типов В-клеточных лимфом и лейкозов (Feldhahn et al. J. Exp. Med. 2005 201: 1837).
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В настоящей заявке предложены соединения формулы I, представляющие собой ингибиторы Btk, и способы их применения, описанные ниже.
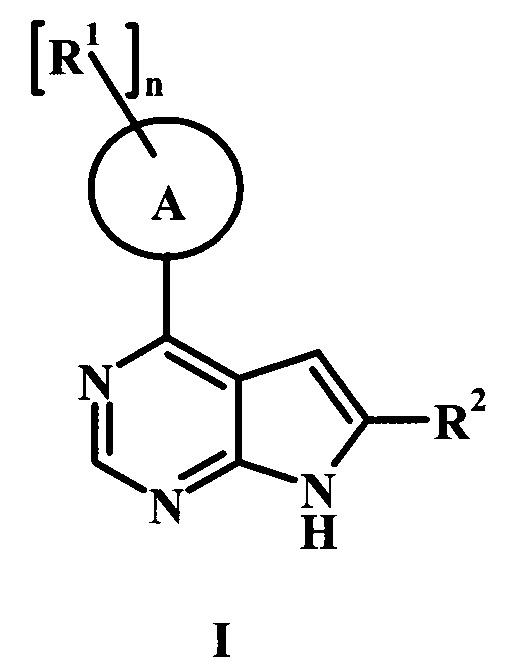
В настоящей заявке предложено соединение формулы I,
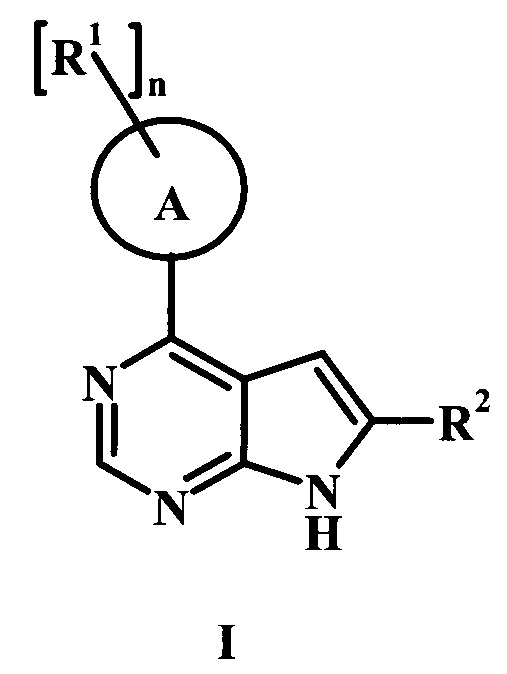
в котором:
А представляет собой фенил или пиперидинил;
каждый радикал R1 представляет собой независимо галоген, низший алкил, CH2NHC(=O)R1', CH2N(СН3)С(=O)R1', CH2NHC(=O)CH2NHR1', CH2R1' или CH2NHR1';
n равно 0, 1, или 2;
R1' представляет собой фенил, ненасыщенный или частично ненасыщенный бициклический или моноциклический гетероарил или гетероциклоалкил, возможно замещенный одним или более R1'';
каждый радикал R1'' представляет собой независимо низший алкил, галоген, циклоалкил, гетероциклоалкил, низший алкил-гетероциклоалкил, оксогруппу, циано-низший алкил, гидрокси-низший алкил или низшую алкоксигруппу;
R2 представляет собой Н, R3 или R4;
R3 представляет собой C(=O)OR3', C(=O)R3' или C(=O)NH(CH2)2R3';
R3' представляет собой Н, низший алкил, гетероциклоалкил, аминогруппу или ОН;
R4 представляет собой низший алкил или гетероарил, возможно замещенный одним или более R4': и
R4' представляет собой гидроксил, аминогруппу, ОС(=O) CH2CH3, или С(=O)ОН;
или его фармацевтически приемлемая соль.
В настоящей Заявке предложено соединение Формулы I,
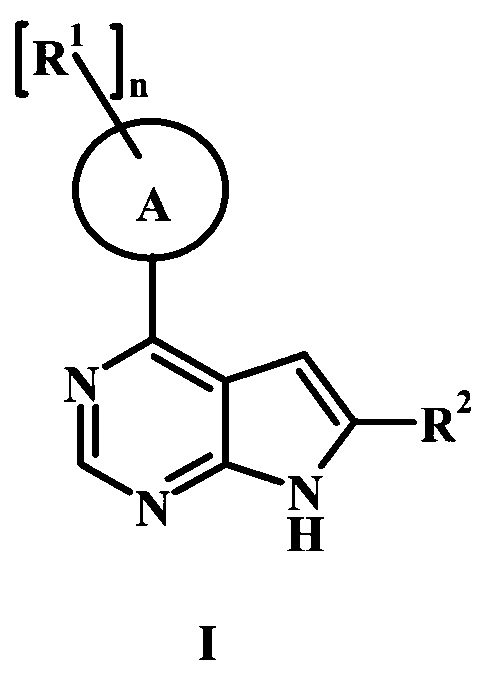
в котором:
А представляет собой фенил или пиперидинил;
каждый радикал R1 представляет собой независимо галоген, низший алкил, CH2NHC(=O)R1', CH2N(СН3)С(=O)R1', CH2NHC(=O)CH2NHR1', CH2R1' или CH2NHR1';
n равно 0, 1, или 2;
R1' представляет собой фенил, ненасыщенный или частично ненасыщенный бициклический или моноциклический гетероарил или гетероциклоалкил, возможно замещенный одним или более R1'';
каждый радикал R1'' представляет собой независимо низший алкил, галоген, циклоалкил, гетероциклоалкил, низший алкил гетероциклоалкил, оксогруппу, циано-низший алкил, гидрокси-низший алкил или низшую алкоксигруппу;
R2 представляет собой Н, R3 или R4;
R3 представляет собой C(=O)OR3', C(=O)R3' или C(=O)NH(CH2)2R3';
R3' представляет собой Н, низший алкил, гетероциклоалкил, аминогруппу или ОН;
R4 представляет собой низший алкил или гетероарил, возможно замещенный одним или более R4': и
R4' представляет собой метил, гидроксил, аминогруппу, СН2-CH2N(СН3)2, ОС(=O) CH2CH3, CH2C(=O)ОН, CH2CH2OH или С(=O)ОН;
или его фармацевтически приемлемая соль.
В настоящей Заявке предложен способ лечения воспалительного и/или аутоиммунного состояния, включающий введение пациенту, который в этом нуждается, терапевтически эффективного количества соединения Формулы I.
В настоящей Заявке предложена фармацевтическая композиция, включающая соединение Формулы I, смешанное по меньшей мере с одним фармацевтически приемлемым носителем, эксципиентом или разбавителем.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Определения
Форма единственного числа по отношению к некоторому объекту обозначает один или несколько таких объектов; например, термин «соединение» относится к одному или более соединений или, по меньшей мере, к одному соединению. Таким же образом, термины "один", "один или более" и "по меньшей мере один" могут быть использованы в данном тексте равнозначно.
Выражение "как определено выше в данном тексте" относится к наиболее широкому определению для каждой группы, приведенной в описании сущности изобретения или в наиболее широком пункте формулы. Во всех других воплощениях, представленных ниже, заместители, которые могут присутствовать в каждом воплощении и которые не определены однозначно, включают наиболее широкое определение, представленное в описании сущности изобретения.
В настоящем описании используемые в промежуточных фразах или в пунктах формулы термины "включает(ют)" и "включающий" следует толковать в открытом значении. Это означает, что эти термины следует интерпретировать как синонимы фраз "имеющий по меньшей мере" или "включающий по меньшей мере". В отношении способа, термин "включающий" означает, что такой способ включает по меньшей мере перечисленные стадии, но может включать и дополнительные стадии. В контексте соединения или композиции термин "включающий" означает, что такое соединение или такая композиция включает по меньшей мере перечисленные признаки или компоненты, но может включать также дополнительные признаки или компоненты.
В данном тексте, если не указано иное, слово "или" употребляется во "включающем" значении союза "и/или", а не в "исключающем" значении союза "или/или".
Термин "независимо" в данном тексте указывает на то, что переменная применяется в каждом случае безотносительно наличия или отсутствия переменной, имеющей такое же или другое значение в одном и том же соединении. Так, в соединении, в котором R'' встречается дважды и определен как "независимо углерод или азот", оба R'' могут представлять собой углерод, оба R'' могут представлять собой азот, или один R'' может представлять собой углерод, а другой - азот.
В тех случаях, когда любая переменная встречается более одного раза в любом фрагменте или любой формуле, обозначающей или описывающей соединения, используемые или заявленные в настоящем изобретении, ее определение в каждом случае независимо от ее определения в любом другом случае. Кроме того, комбинации заместителей и/или переменных допустимы только, если такие соединения соответствуют стабильным соединениям.
Символ "*" в конце связи или символ " ", проведенный через связь, обозначает место присоединения функциональной группы или другой химической группы к остальной части молекулы, фрагментом которой они являются. Так, например:
", проведенный через связь, обозначает место присоединения функциональной группы или другой химической группы к остальной части молекулы, фрагментом которой они являются. Так, например:
MeC(=O)OR4, где 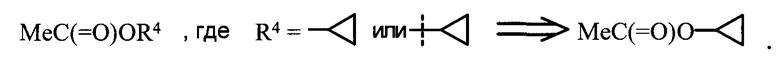 или .
или .
Связь, проведенная внутрь циклической системы (в отличие от связи, проведенной к определенной вершине) указывает на то, что эта связь может находиться при любом подходящем кольцевом атоме.
Термин "возможный" или "возможно" в данном тексте означает, что описанное далее событие или обстоятельство может, но не обязательно, иметь место, и что такое описание включает случаи, в которых указанное событие или обстоятельство имеют место, и случаи, в которых они не имеют место. Например, "возможно замещенный" означает, что такая возможно замещенная группа может включать атом водорода или заместитель.
Выражение "возможная связь" означает, что указанная связь может присутствовать или отсутствовать, и такое описание включает одинарную, двойную и тройную связи. Если указано, что заместитель представляет собой "связь" или "отсутствует", то атомы, присоединенные к таким заместителям, непосредственно соединены между собой.
Термин "примерно" в данном тексте обозначает приблизительно, в районе, грубо или около. В тех случаях, когда термин "примерно" используется вместе с числовым диапазоном, он модифицирует этот диапазон, расширяя его границы выше и ниже указанных числовых значений. В общем случае термин "примерно" используется в данном тексте для модификации численных величин выше и ниже указанного значения с колебанием 20%.
Некоторые соединения формулы I могут проявлять таутомерию. Таутомерные соединения могут существовать в виду двух или более взаимопревращающихся форм. Прототропные таутомеры возникают вследствие миграции ковалентно связанного атома водорода между двух атомов. Таутомеры обычно существуют в равновесии, и попытки выделить индивидуальные таутомеры, как правило, приводят к смеси, химические и физические свойства которой соответствуют смеси соединений. Положение равновесия зависит от химических свойств молекулы. Например, у многих алифатических альдегидов и кетонов, таких как ацетон, преобладает кето-форма, в то время как у фенолов преобладает енольная форма. Подобные прототропные таутомеры включают кето-енольные 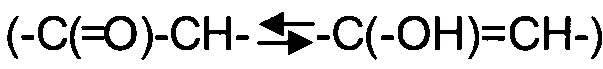 , амидо/имидно-кислотные
, амидо/имидно-кислотные 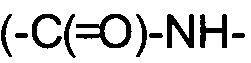
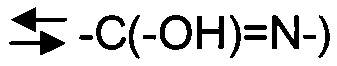 и амидиновые
и амидиновые  таутомеры. Два последних особенно характерны для гетероарильных и гетероциклических колец, и настоящее изобретение включает все таутомерные формы таких соединений.
таутомеры. Два последних особенно характерны для гетероарильных и гетероциклических колец, и настоящее изобретение включает все таутомерные формы таких соединений.
Технические и научные термины, используемые в данном тексте, имеют общепринятое значение, известное специалисту в той области техники, к которой относится настоящее изобретение, если не определено иное. В тексте даны ссылки на многочисленные материалы и методы, известные специалисту в данной области техники. К авторитетным справочным изданиям, где представлены общие принципы фармакологии, относятся следующие: Goodman and Gilman's The Pharmacological Basis of Therapeutics, 10th Ed., McGraw Hill Companies Inc., New York (2001). Любые подходящие материалы и/или методы, известные специалисту в данной области техники, можно применять при воплощении настоящего изобретения. Тем не менее, в настоящей Заявке описаны предпочтительные материалы и методы. Материалы, реагенты и т.п., на которые даны ссылки в ниже следующем описании и примерах, можно получить из коммерческих источников, если не указано иное.
Определения, описанные в данном тексте, можно применять с получением химически релевантных комбинаций, таких как "гетероалкиларил", "галогеналкилгетероарил", "арилалкилгетероциклил", "алкилкарбонил", "алкоксиалкил" и т.п. Если термин "алкил" используется как окончание, следующее за другим термином (например "фенилалкил" или "гидроксиалкил"), он обозначает алкильную группу, раскрытую выше, содержащую один или два заместителя, выбранных из другой указанной группы. Так, например, "фенилалкил" обозначает алкильную группу, содержащую один или два фенильных заместителя, и таким образом включает бензил, фенилэтил и бифенил. "Алкиламиноалкил" представляет собой алкильную группу, содержащую в качестве заместителя одну или две алкиламиногруппы. Термин "гидроксиалкил" включает 2-гидроксиэтил, 2-гидроксипропил, 1-(гидроксиметил)-2-метилпропил, 2-гидроксибутил, 2,3-дигидроксибутил, 2-(гидроксиметил), 3-гидроксипропил и т.д. Таким образом, в данном тексте термин "гидроксиалкил" обозначает подмножество гетероалкильных групп, раскрытых ниже. Термин -(ар)алкил обозначает либо незамещенный алкил, либо аралкильную группу. Термин (гетеро)арил или (гет)арил обозначает либо арильную, либо гетероарильную группу.
Термин "спироциклоалкил", используемый в данном тексте, означает спироциклическую циклоалкильную группу, такую как, например, спиро[3.3]гептан. Термин "спирогетероциклоалкил", используемый в данном тексте, означает спироциклический гетероциклоалкил, такой как, например, 2,6-диаза-спиро[3.3]гептан.
Термин "ацил", используемый в данном тексте, обозначает группу формулы -C(=O)R, в которой R представляет собой водород или низший алкил, раскрытый в данном описании. Термин "алкилкарбонил", используемый в данном тексте, обозначает группу формулы C(=O)R, в которой R представляет собой алкил, раскрытый в данном описании. Термин C1-6-ацил обозначает группу -C(=O)R, содержащую 1-6 атомов углерода. Термин "арилкарбонил", используемый в данном тексте, обозначает группу формулы C(=O)R, в которой R представляет собой арильную группу; термин "бензоил", используемый в данном тексте, обозначает "арилкарбонильную" группу, в которой R представляет собой фенил.
Термин "сложный эфир", используемый в данном тексте, обозначает группу формулы -C(=O)OR, в которой R представляет собой низший алкил, раскрытый в данном описании.
Термин "алкил", используемый в данном тексте, обозначает насыщенный моновалентный углеводородный остаток с неразветвленной или разветвленной цепью, содержащий от 1 до 10 атомов углерода. Термин "низший алкил" обозначает углеводородный остаток с линейной или разветвленной цепью, содержащий от 1 до 6 атомов углерода. Термин "С1-10алкил", используемый в данном тексте, обозначает алкил, состоящий из 1-10 атомов углерода. Примеры алкильных групп включают, без ограничения, низшие алкильные группы, в т.ч. метил, этил, пропил, изопропил, н-бутил, изобутил, трет-бутил или пентил, изопентил, неопентил, гексил, гептил и октил.
Если термин "алкил" используется как окончание, следующее после другого термина (например "фенилалкил" или "гидроксиалкил"), это обозначает алкильную группу, раскрытую выше, содержащую один или два заместителя, выбранных из другой указанной группы. Таким образом, например, "фенилалкил" обозначает радикал R'R''-, в котором R' представляет собой фенильный радикал, а R'' представляет собой алкиленовый радикал, раскрытый в данном описании, при этом подразумевается, что место присоединения фенилалкильной группы находится на алкиленовом радикале. Примеры арилалкиловых радикалов включают, без ограничения, бензил, фенилэтил и 3-фенилпропил. Термины "арилалкил" или "аралкил" понимают аналогичным образом, за исключением того, что R' представляет собой арильный радикал. Термины "(гет)арилалкил" или "(гет)аралкил" понимают аналогичным образом, за исключением того, что R' представляет собой по выбору арильный или гетероарильный радикал.
Термины "галогеналкил" или "галоген-низший алкил" или "низший галогеналкил" обозначает углеводородный остаток с линейной или разветвленной цепью, содержащий от 1 до 6 атомов углерода, при этом один или более атомов углерода замещены одним или более атомами галогена.
Термин "алкилен" или "алкиленил", используемый в данном тексте, обозначает бивалентный насыщенный углеводородный радикал, включающий от 1 до 10 атомов углерода (например (CH2)n), или разветвленный насыщенный бивалентный углеводородный радикал, включающий от 2 до 10 атомов углерода (например -СНМе- или -CH2CH(i-Pr)CH2-), если не указано иное. За исключением метилена, открытые валентности алкиленовой группы не расположены при одном и том же атоме. Примеры алкиленовых радикалов включают, без ограничения, метилен, этилен, пропилен, 2-метилпропилен, 1,1-диметилэтилен, бутилен, 2-этилбутилен.
Термин "алкоксигруппа", используемый в данном тексте, обозначает -O-алкильную группу, в которой алкил раскрыт выше, например метокси, этокси, н-пропилокси, изопропилокси, н-бутилокси, изобутилокси, трет-бутилокси, пентилокси и гексилоксигруппы, включая их изомеры. Термин "низшая алкоксигруппа", используемый в данном тексте, обозначает алкоксигруппу, включающую "низшую алкильную" группу, раскрытую ранее. Термин "C1-10-алкоксигруппа", используемый в данном тексте, обозначает -O-алкил, в котором алкил представляет собой C1-10.
Термин "РСу3" обозначает фосфин, трижды замещенный тремя циклическими радикалами.
Термины "галогеналкоксигруппа", или "галоген-низшая алкоксигруппа", или "низшая галогеналкоксигруппа" обозначает низшую алкоксигруппу группу, в которой один или более атомов углерода содержат в качестве заместителя один или более атомов галогена.
Термин "гидроксиалкил", используемый в данном тексте, обозначает алкильный радикал, раскрытый в данном описании, в котором от одного до трех атомов водорода на различных атомах углерода замещен(ы) гидроксигруппами.
Термины "алкилсульфонил" и "арилсульфонил", используемые в данном тексте, обозначают группу формулы -S(=O)2R, в которой R представляет собой алкил или арил, соответственно, при этом алкил и арил раскрыты в данном описании. Термин "гетероалкилсульфонил", используемый в данном тексте, обозначает группу формулы -S(=O)2R, в которой R представляет собой "гетероалкил", раскрытый в данном описании.
Термины "алкилсульфониламино" и "арилсульфониламино", используемые в данном тексте, обозначают группу формулы -NR'S(=O)2R, в которой R представляет собой алкил или арил, соответственно, R' представляет собой водород или C1-3-алкил, при этом алкил и арил раскрыты в данном описании.
Термин "циклоалкил", используемый в данном тексте, обозначает насыщенное карбоциклическое кольцо, содержащее от 3 до 8 атомов углерода, т.е. циклопропил, циклобутил, циклопентил, циклогексил, циклогептил или циклооктил. Термин "С3-7-циклоалкил", используемый в данном тексте, обозначает циклоалкил, включающий от 3 до 7 атомов углерода в карбоцикле.
Термин "карбоксиалкил", используемый в данном тексте, обозначает алкильный радикал, в которой один атом водорода замещен карбоксильной группой, при этом подразумевается, что точка присоединения гетероалкильного радикала расположена при атоме углерода. Термин "карбоксигруппа" или "карбоксил" обозначает группу -CO2H.
Термин "гетероарил" или "гетероароматический", используемый в данном тексте, обозначает моноциклический или бициклический радикал, включающий от 5 до 12 кольцевых атомов, содержащий по меньшей мере одно ароматическое или частично ненасыщенное кольцо, включающее от четырех до восьми атомов в кольце, в т.ч. один или более гетероатомов N, О, или S, при этом оставшиеся атомы кольца представляют собой атомы углерода, при этом подразумевается, что место присоединения гетероарильного радикала расположено на ароматическом или частично ненасыщенном кольце. Специалисту в данной области техники хорошо известно, что гетероарильные кольца носят менее ароматичный характер, по сравнению с их полностью углеродными аналогами. Поэтому в целях изобретения, гетероарильная группа должна лишь в некоторой степени проявлять ароматический характер. Примеры гетероарильных групп включают моноциклические ароматические гетероциклы, содержащие от 5 до 6 атомов кольца и от 1 до 3 гетероатомов, и представляет собой, без ограничения, пиридинил, пиримидинил, пиразинил, оксазинил, пирролил, пиразолил, имидазолил, оксазолил, 4,5-дигидрооксазолил, 5,6-дигидро-4Н-[1,3]оксазолил, изоксазол, тиазол, изотиазол, триазолин, тиадиазол и оксадиаксолин, возможно содержащие один или более, предпочтительно один или два заместителя, выбранных из группы, включающей гидроксигруппу, цианогруппу, алкил, алкоксигруппу, тиогруппу, низшую галогеналкоксигруппу, алкилтиогруппу, галоген, низший галогеналкил, алкилсульфинил, алкилсульфонил, галоген, аминогруппу, алкиламиногруппу, диалкиламиногруппу, аминоалкил, алкиламиноалкил и диалкиламиноалкил, нитрогруппу, алкоксикарбонил и карбамоил, алкилкарбамоил, диалкилкарбамоил, арилкарбамоил, алкилкарбониламиногруппу и арилкарбониламиногруппу. Примеры бициклических радикалов включают, без ограничения, хинолинил, изохинолинил, бензофурил, бензотиофенил, бензоксазол, бензизоксазол, бензотиазол, нафтиридинил, 5,6,7,8-тетрагидро-[1,6]нафтиридинил и бензизотиазол. Бициклические радикалы возможно могут быть замещены по любому кольцу, при этом место присоединения расположено на кольце, содержащем гетероатом.
Термин "гетероциклил", или "гетероциклоалкил", или "гетероцикл", используемый в данном тексте, обозначает моновалентный насыщенный циклический радикал, состоящий из одного или более колец, предпочтительно от одного до двух колец, в т.ч. спироциклические кольцевые системы, состоящие из трех-восьми атомов в каждом кольце, включающий один или более кольцевых гетероатомов (выбранных из N, O или S(O)0-2), и который, возможно, независимо содержит один или более, предпочтительно один или два заместителя, выбранных из гидроксигруппы, оксогруппы, цианогруппы, низшего алкила, низшей алкоксигруппы, низшей галогеналкоксигруппы, алкилтиогруппы, галогена, низшего галогеналкила, гидроксиалкила, нитрогруппы, алкоксикарбонила, аминогруппы, алкиламиногруппы, алкилсульфонила, арилсульфонила, алкиламиносульфонила, ариламиносульфонила, алкилсульфониламиногруппы, арилсульфониламиногруппы, алкиламинокарбонила, ариламинокарбонила, алкилкарбониламиногруппы, арилкарбониламиногруппы, а также их ионных форм, если не указано иное. Примеры гетероциклических радикалов включают, без ограничения, морфолинил, пиперазинил, пиперидинил, азетидинил, пирролидинил, гексагидроазепинил, оксетанил, тетрагидрофуранил, тетрагидротиофенил, оксазолидинил, тиазолидинил, изоксазолидинил, тетрагидропиранил, тиоморфолинил, хинуклидинил и имидазолинил, а также их ионные формы. Примерами могут быть также бициклические системы, например 3,8-диазабицикло[3.2.1]октан, 2,5-диазабицикло[2.2.2]октан или октагидро-пиразино[2,1-с][1,4]оксазин.
ИНГИБИТОРЫ КИНАЗЫ ВТК
В настоящей Заявке предложено соединение Формулы I,
в котором:
А представляет собой фенил или пиперидинил;
каждый радикал R1 представляет собой независимо галоген, низший алкил, CH2NHC(=O)R1', CH2N(СН3)С(=O)R1', CH2NHC(=O)CH2NHR1', CH2R1' или CH2NHR1';
n равно 0, 1, или 2;
R1' представляет собой фенил, ненасыщенный или частично ненасыщенный бициклический или моноциклический гетероарил или гетероциклоалкил, возможно замещенный одним или более R1'';
каждый радикал R1'' представляет собой независимо низший алкил, галоген, циклоалкил, гетероциклоалкил, низший алкил-гетероциклоалкил, оксогруппу, циано-низший алкил, гидроксил-низший алкил или низшую алкоксигруппу;
R2 представляет собой Н, R3 или R4;
R3 представляет собой C(=O)OR3', C(=O)R3' или C(=O)NH(CH2)2R3';
R3' представляет собой Н, низший алкил, гетероциклоалкил, аминогруппу или ОН;
R4 представляет собой низший алкил или гетероарил, возможно замещенный одним или более R4': и
R4' представляет собой метил, гидроксил, аминогруппу, CH2-CH2N(СН3)2, ОС(=O) CH2CH3, CH2C(=O)ОН, CH2CH2OH или С(=O)ОН;
или его фармацевтически приемлемая соль.
Кроме того, следует понимать, что каждое воплощение, касающееся конкретного остатка A, R1, R1', R1'', R2, R3, R3', R4 и R4', раскрытого в данном тексте, можно комбинировать с любым другим воплощением, касающимся другого остатка A, R1, R1', R1'', R2, R3, R3', R4 и R4', раскрытого в данном тексте.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, R2 представляет собой Н и n равно 1.
В настоящей Заявке предложено соединение Формулы I, в котором R1 представляет собой галоген, R2 представляет собой Н и n равно 1.
В настоящей Заявке предложено соединение Формулы I, в котором R1 представляет собой галоген.
В настоящей Заявке предложено соединение Формулы I, в котором R2 представляет собой Н и n равно 2.
В настоящей Заявке предложено соединение Формулы I, в котором R2 представляет собой Н, n равно 2 и один R1 представляет собой галоген.
В настоящей Заявке предложено соединение Формулы I, в котором R2 представляет собой Н, n равно 2 и один R1 представляет собой низший алкил.
В настоящей Заявке предложено соединение Формулы I, в котором R1 представляет собой CH2NHC(=O)R1'.
В настоящей Заявке предложено соединение Формулы I, в котором R1 представляет собой CH2NHC(=O)CH2NHR1'.
В настоящей Заявке предложено соединение Формулы I, в котором R1 представляет собой CH2NHR1'.
В настоящей Заявке предложено соединение Формулы I, в котором R1 представляет собой CH2NHC(=O)R1', R2 представляет собой Н и n равно 1.
В настоящей Заявке предложено соединение Формулы I, в котором R1 представляет собой CH2NHC(=O)CH2NHR1', R2 представляет собой Н и n равно 1.
В настоящей Заявке предложено соединение Формулы I, в котором R1 представляет собой CH2NHR1', R2 представляет собой Н и n равно 1.
В настоящей Заявке предложено соединение Формулы I, в котором n равно 2, один R1 представляет собой CH2NHC(=O)R1' и R2 представляет собой C(=O)OR3', C(=O)R3' или C(=O)NH(CH2)2R3'.
В настоящей Заявке предложено соединение Формулы I, в котором n равно 2, один R1 представляет собой CH2NHC(=O)R1' и R2 представляет собой низший алкил или гетероарил.
В настоящей Заявке предложено соединение Формулы I, в котором R1 представляет собой трет-бутил или галоген.
В настоящей Заявке предложено соединение Формулы I, в котором R1' представляет собой трет-бутил или галоген, R1 представляет собой CH2NHC(=O)R1', R2 представляет собой Н и n равно 1.
В настоящей Заявке предложено соединение Формулы I, в котором один R1 представляет собой фтор, а R1' представляет собой трет-бутил.
В настоящей Заявке предложено соединение Формулы I, в котором один R1 представляет собой фтор и R1' представляет собой трет-бутил, n равно 2, один R1 представляет собой CH2NHC(=O)R1' и R2 представляет собой C(=O)OR3', C(=O)R3' или C(=O)NH(CH2)2R3'.
В настоящей Заявке предложено соединение Формулы I, в котором один R1 представляет собой фтор и R1' представляет собой трет-бутил, n равно 2, один R1 представляет собой CH2NHC(=O)R1' и R2 представляет собой низший алкил или гетероарил.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой пиперидинил.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой пиперидинил и n=1.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой пиперидинил, n=1 и R1 представляет собой CH2NHC(=O)R1'.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой пиперидинил, n=1, R1 представляет собой CH2NHC(=O)R1' и R1' представляет собой фенил, возможно замещенный одним или более R1''.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой пиперидинил, n=1, R1 представляет собой CH2NHC(=O)R1' и R1' представляет собой фенил, возможно замещенный одним или более низшим алкилом.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой пиперидинил, n=1, R1 представляет собой CH2NHC(=O)R1' и R1' представляет собой фенил, возможно замещенный трет-бутилом.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил и n=1 или 2.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, n=1 или 2 и один R1 представляет собой CH2NHC(=O)R1'.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, n=2, один R1 представляет собой CH2NHC(=O)R1', a другой представляет собой галоген и R1' представляет собой фенил, ненасыщенный или частично ненасыщенный бициклический или моноциклический гетероарил или гетероциклоалкил, возможно замещенный одним или более R1''.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, n=2, один R1 представляет собой CH2NHC(=O)R1', a другой представляет собой F, и R1' представляет собой фенил, возможно замещенный одним или более R1''.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, n=2, один R1 представляет собой CH2NHC(=O)R1', a другой представляет собой галоген и R1' представляет собой фенил, возможно замещенный одним или более низшим алкил, галоген, циклоалкил или гетероциклоалкил.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, n=2, один R1 представляет собой CH2NHC(=O)R1', a другой представляет собой галоген и R1' представляет собой фенил, возможно замещенный одним или более низшим алкилом или галогеном.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, n=2, один R1 представляет собой CH2NHC(=O)R1', a другой представляет собой F и R1' представляет собой фенил, возможно замещенный одним или более низшим алкилом.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, n=2, один R1 представляет собой CH2NHC(=O)R1', a другой представляет собой галоген и R1' представляет собой фенил, возможно замещенный одним или более трет-бутилом.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, n=2, один R1 представляет собой CH2NHC(=O)R1', a другой представляет собой F и R1' представляет собой фенил, возможно замещенный одним или более трет-бутилом.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, n=2, один R1 представляет собой CH2NHC(=O)R1', a другой представляет собой F, R1' представляет собой фенил, возможно замещенный одним или более трет-бутилом и R4 представляет собой гетероарил, возможно замещенный одним или более R4'.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, n=2, один R1 представляет собой CH2NHC(=O)R1', a другой представляет собой F, R1' представляет собой фенил, возможно замещенный одним или более трет-бутилом, и R4 представляет собой гетероарил, возможно замещенный одним или более метилом.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, n=2, один R1 представляет собой CH2NHC(=O)R1', a другой представляет собой F, R1' представляет собой фенил, возможно замещенный одним или более трет-бутилом и R4 представляет собой гетероарил, возможно замещенный метилом.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, n=2, один R1 представляет собой CH2NHC(=O)R1', a другой представляет собой F, R1' представляет собой фенил, возможно замещенный одним или более трет-бутилом и R4 представляет собой пиразолил, возможно замещенный метилом.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, n=1, R1 представляет собой CH2NHC(=O)R1' и R1' представляет собой фенил, возможно замещенный одним или более трет-бутилом.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, n=1, R1 представляет собой CH2NHC(=O)R1', R1' представляет собой фенил, возможно замещенный одним или более трет-бутилом и R4 представляет собой гетероарил, возможно замещенный одним или более R4'.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, n=1, R1 представляет собой CH2NHC(=O)R1', R1' представляет собой фенил, возможно замещенный одним или более трет-бутилом и R4 представляет собой пиразолил, возможно замещенный одним или более R4'.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, n=1, R1 представляет собой CH2NHC(=O)R1', R1' представляет собой фенил, возможно замещенный одним или более трет-бутилом и R4 представляет собой пиразолил, возможно замещенный одним или более метилом.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, n=1, R1 представляет собой CH2NHC(=O)R1', R1' представляет собой фенил, возможно замещенный одним или более трет-бутилом и R4 представляет собой пиразолил, возможно замещенный метилом.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, R2 представляет собой Н, n=2, один R1 представляет собой CH2NHC(=O)R1', а другой представляет собой F и R1' представляет собой фенил, возможно замещенный одним или более галогеном, низшим алкилом или циклоалкилом.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, R2 представляет собой Н, n=2, один R1 представляет собой CH2NHC(=O)R1', а другой представляет собой F и R1' представляет собой фенил, возможно замещенный одним или более Cl, трет-бутилом или циклопропилом.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, n=1 и R4 = гетероарил, возможно замещенный одним или более R4'.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, n=2 и R4 = гетероарил, возможно замещенный одним или более R4'.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, n=1 и R4 = гетероарил, возможно замещенный одним или более метилом.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, n=2 и R4 = гетероарил, возможно замещенный одним или более метилом.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, R2 представляет собой Н, n=2 и R1 представляет собой CH2NHC(=O)R1'.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, R2 представляет собой Н, n=2, один R1 представляет собой CH2NHC(=O)R1', а другой представляет собой галоген.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, R2 представляет собой Н, n=2, один R1 представляет собой CH2NHC(=O)R1', а другой представляет собой F.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, R2 представляет собой Н, n=2, один R1 представляет собой CH2NHC(=O)R1', а другой представляет собой F и R1' представляет собой фенил.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, R2 представляет собой Н, n=2, один R1 представляет собой CH2NHC(=O)R1', а другой представляет собой F, R1' представляет собой фенил и R1'' представляет собой низший алкил или циклоалкил.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, R2 представляет собой Н, n=2, один R1 представляет собой CH2NHC(=O)R1', а другой представляет собой F, R1' представляет собой фенил и R1'' представляет собой трет-бутил или циклопропил.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, R2 представляет собой Н, n=2, один R1 представляет собой CH2NHC(=O)R1', а другой представляет собой F, R1' представляет собой фенил и R1'' представляет собой трет-бутил.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, R2 представляет собой Н, n=2, один R1 представляет собой CH2NHC(=O)R1', а другой представляет собой F, и R1' представляет собой ненасыщенный или частично ненасыщенный моноциклический гетероарил.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, R2 представляет собой Н, n=2, один R1 представляет собой CH2NHC(=O)R1', а другой представляет собой F, и R1' представляет собой ненасыщенный или частично ненасыщенный бициклический гетероарил.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, R2 представляет собой Н, n=2, один R1 представляет собой CH2NHC(=O)R1', а другой представляет собой F, R1' представляет собой ненасыщенный или частично ненасыщенный моноциклический гетероарил и R1'' представляет собой низший алкил.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, R2 представляет собой Н, n=2, один R1 представляет собой CH2NHC(=O)R1', а другой представляет собой F, R1' представляет собой ненасыщенный или частично ненасыщенный бициклический гетероарил и R1'' представляет собой низший алкил.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, R2 представляет собой Н, n=2, один R1 представляет собой CH2NHC(=O)R1', а другой представляет собой F, R1' представляет собой ненасыщенный или частично ненасыщенный моноциклический гетероарил, R1'' представляет собой низший алкил и R4 представляет собой гетероарил, возможно замещенный одним или более R4'.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, R2 представляет собой Н, n=2, один R1 представляет собой CH2NHC(=O)R1', а другой представляет собой F, R1' представляет собой ненасыщенный или частично ненасыщенный бициклический гетероарил, R1'' представляет собой низший алкил и R4 представляет собой гетероарил, возможно замещенный одним или более R4'.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, R2 представляет собой Н, n=2, один R1 представляет собой CH2NHC(=O)R1', а другой представляет собой F, R1' представляет собой ненасыщенный или частично ненасыщенный моноциклический гетероарил, R1'' представляет собой низший алкил и R4 представляет собой гетероарил, возможно замещенный одним или более метилом, гидроксилом, аминогруппой, CH2-CH2N(СН3)2, ОС(=O) CH2CH3, CH2C(=O)ОН, CH2CH2OH или С(=O)ОН.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, R2 представляет собой Н, n=2, один R1 представляет собой CH2NHC(=O)R1', а другой представляет собой F, R1' представляет собой ненасыщенный или частично ненасыщенный бициклический гетероарил, R1'' представляет собой низший алкил и R4 представляет собой гетероарил, возможно замещенный одним или более метилом.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, R2 представляет собой Н, n=2, один R1 представляет собой CH2NHC(=O)R1', а другой представляет собой F, R1' представляет собой ненасыщенный или частично ненасыщенный бициклический гетероарил, R1'' представляет собой низший алкил и R4 представляет собой гетероарил, возможно замещенный метилом.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, R2 представляет собой Н, n=2, один R1 представляет собой CH2NHC(=O)R1', а другой представляет собой F, R1' представляет собой ненасыщенный или частично ненасыщенный бициклический гетероарил, R1'' представляет собой низший алкил и R4 представляет собой пиразолил, возможно замещенный метилом.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, R2 представляет собой Н и n=1.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, R2 представляет собой Н, n=1 и R1 представляет собой галоген или низший алкил.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, R2 представляет собой Н, n=1 и R1 представляет собой Cl, F или метил.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, R2 представляет собой Н и n=2.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, R2 представляет собой Н, n=2 и один R1 представляет собой галоген, а другой представляет собой низший алкил.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, R2 представляет собой Н, n=2 и один R1 представляет собой Cl или F, а другой представляет собой метил.
В настоящей Заявке предложено соединение Формулы I, в котором А представляет собой фенил, R2 представляет собой Н, n=2 и оба радикала R1 представляют собой метил.
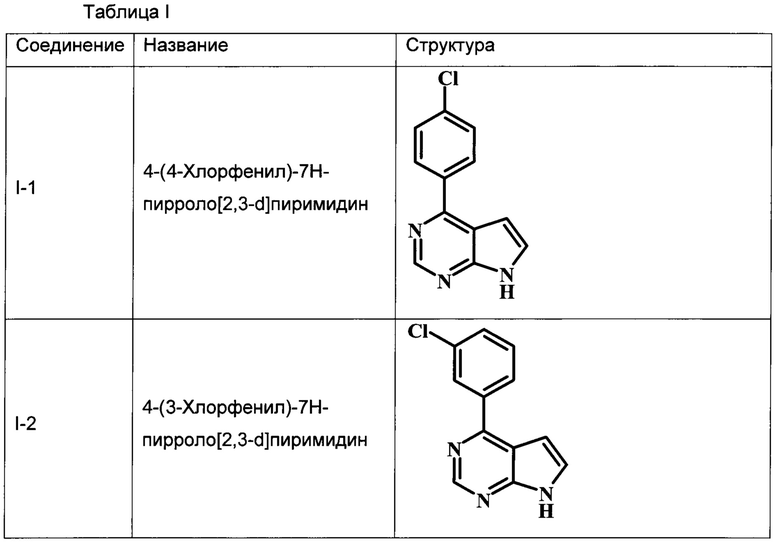
В настоящей Заявке предложено соединение Формулы I, выбранное из группы, состоящей из следующих веществ:
4-(4-хлорфенил)-7Н-пирроло[2,3-d]пиримидин;
4-(3-хлорфенил)-7Н-пирроло[2,3-d]пиримидин;
4-(2-хлорфенил)-7Н-пирроло[2,3-d]пиримидин;
4-(3-фтор-4-метилфенил)-7Н-пирроло[2,3-d]пиримидин;
4-(2,4-диметилфенил)-7Н-пирроло[2,3-d]пиримидин;
4-(3,4-диметилфенил)-7Н-пирроло[2,3-d]пиримидин;
4-п-толил-7Н-пирроло[2,3-d]пиримидин;
4-(3-хлор-4-метилфенил)-7Н-пирроло[2,3-d]пиримидин;
4-трет-бутил-N-[4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-бензил]-бензамид;
3-хлор-N-[4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-бензил]-бензамид;
2-(3-хлорфениламино)-N-[4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-бензил]-ацетамид;
4-трет-бутил-N-[2-фтор-4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-бензил]-бензамид;
трет-бутиловый эфир 4-{4-[(4-трет-бутилбензоиламино)-метил]-3-фторфенил}-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты;
4-(4-((4-трет-бутилбензамидо)метил)-3-фторфенил)-7Н-пирроло[2,3-d]пиримидин-6-карбоновая кислота;
4-трет-бутил-N-(2-фтор-4-(6-(морфолин-4-карбонил)-7Н-пирроло[2,3-d]пиримидин-4-ил)бензил)бензамид;
диметиламид 4-{4-[(4-трет-бутилбензоиламино)-метил]-3-фторфенил}-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты;
метиламид 4-{4-[(4-трет-бутилбензоиламино)-метил]-3-фторфенил}-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты;
(2-гидроксиэтил)-амид 4-{4-[(4-трет-бутилбензоиламино)-метил]-3-фторфенил}-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты;
(2-диметиламиноэтил)-амид 4-{4-[(4-трет-бутилбензоиламино)-метил]-3-фторфенил}-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты;
4-трет-бутил-N-{1-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-пиперидин-4-илметил}-бензамид;
4-трет-бутил-N-{4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-бензамид;
4-циклопропил-N-{4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-бензамид;
4-изопропил-N-{4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-бензамид;
N-{4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-4-оксетан-3-илбензамид;
4-(3-метилоксетан-3-ил)-N-{4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-бензамид;
4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензиламид 4,5,6,7-тетрагидробензо[b]тиофен-2-карбоновой кислоты;
4-трет-бутил-N-{2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-бензамид;
6-трет-бутил-N-{2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-никотинамид;
2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензиламид 5-метилтиофен-2-карбоновой кислоты;
4-трет-бутил-N-(2-фтор-4-{6-[1-(2-гидрокси-этил)-1Н-пиразол-4-ил]-7Н-пирроло[2,3-d]пиримидин-4-ил}-бензил)-бензамид;
4-трет-бутил-N-{2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-N-метилбензамид;
{2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-метиламид 5-метилтиофен-2-карбоновой кислоты;
2-трет-бутил-5-{2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-4,5-дигидротиено[2,3-d]пиррол-6-он;
2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензиламид 5-трет-бутилизоксазол-3-карбоновой кислоты;
N-{2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-4-(3-метилоксетан-3-ил)-бензамид;
4-(цианодиметилметил)-N-{2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-бензамид;
2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензиламид 4,5,6,7-тетрагидробензо[b]тиофен-2-карбоновой кислоты;
N-{2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-4-(1-гидрокси-1-метилэтил)-бензамид;
2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензиламид 3-трет-бутилизоксазол-5-карбоновой кислоты
2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензиламид 3-трет-бутоксиазетидин-1-карбоновой кислоты;
2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензиламид 1,3-дигидроизоиндол-2-карбоновой кислоты;
4-трет-бутил-N-(4-{6-[1-(2-диметиламиноэтил)-1Н-пиразол-4-ил]-7Н-пирроло[2,3-d]пиримидин-4-ил}-2-фторбензил)-бензамид;
4-{6-[1-(2-диметиламиноэтил)-1Н-пиразол-4-ил]-7Н-пирроло[2,3-d]пиримидин-4-ил}-2-фторбензиламид 3-трет-бутоксиазетидин-1-карбоновой кислоты;
4-{6-[1-(2-диметиламиноэтил)-1Н-пиразол-4-ил]-7Н-пирроло[2,3-d]пиримидин-4-ил}-2-фторбензиламид 1,3-дигидроизоиндол-2-карбоновой кислоты;
этиловый эфир [4-(4-{4-[(4-трет-бутилбензоиламино)-метил]-3-фторфенил}-7Н-пирроло[2,3-d]пиримидин-6-ил)-пиразол-1-ил]-уксусной кислоты;
[4-(4-{4-[(4-трет-бутилбензоиламино)-метил]-3-фторфенил}-7Н-пирроло[2,3-d]пиримидин-6-ил)-пиразол-1-ил]-уксусная кислота;
N-(2-фтор-4-(6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил)бензил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиридин-2-карбоксамид;
2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензиламид 5-трет-бутилизоксазол-3-карбоновой кислоты;
2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензиламид 3-трет-бутил-[1,2,4]оксадиазол-5-карбоновой кислоты;
трет-бутиловый эфир {2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-карбаминовой кислоты; и
N-{2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-бензамид.
В настоящей Заявке предложен способ лечения воспалительного и/или аутоиммунного состояния, включающий введение пациенту, который в этом нуждается, терапевтически эффективного количества соединения Формулы I.
В настоящей Заявке предложен способ лечения ревматоидного артрита, включающий введение пациенту, который в этом нуждается, терапевтически эффективного количества соединения Формулы I.
В настоящей Заявке предложен способ лечения астмы, включающий введение пациенту, который в этом нуждается, терапевтически эффективного количества соединения Формулы I.
В настоящей Заявке предложена фармацевтическая композиция, включающая соединение Формулы I.
В настоящей Заявке предложена фармацевтическая композиция, включающая соединение Формулы I, смешанное по меньшей мере с одним фармацевтически приемлемым носителем, эксципиентом или разбавителем.
В настоящей Заявке предложено применение соединения Формулы I в изготовлении лекарственного средства для лечения воспалительного расстройства.
В настоящей Заявке предложено применение соединения Формулы I в изготовлении лекарственного средства для лечения аутоиммунного расстройства.
В настоящей Заявке предложено применение соединения Формулы I в изготовлении лекарственного средства для лечения ревматоидного артрита.
В настоящей Заявке предложено применение соединения Формулы I в изготовлении лекарственного средства для лечения астмы.
В настоящей Заявке предложено применение соединения, описанного выше, для лечения воспалительного и/или аутоиммунного состояния.
В настоящей Заявке предложено применение соединения, описанного выше, для лечения ревматоидного артрита.
В настоящей Заявке предложено применение соединения, описанного выше, для лечения астмы.
В настоящей Заявке предложено соединение, описанное выше, для применения в лечении воспалительного и/или аутоиммунного состояния.
В настоящей Заявке предложено соединение, описанное выше, для применения в лечении ревматоидного артрита.
В настоящей Заявке предложено соединение, описанное выше, для применения в лечении астмы.
В настоящей Заявке предложено соединение, способ или композиция, описанные выше.
В настоящей Заявке предложен способ лечения воспалительного и/или аутоиммунного состояния, включающий введение пациенту, который в этом нуждается, терапевтически эффективного количества соединения Формулы I', представляющего собой ингибитор киназы Btk.
В настоящей Заявке предложен способ лечения артрита, включающий введение пациенту, который в этом нуждается, терапевтически эффективного количества соединения Формулы I', представляющего собой ингибитор киназы Btk.
В настоящей Заявке предложен способ лечения астмы, включающий введение пациенту, который в этом нуждается, терапевтически эффективного количества соединения Формулы I', представляющего собой ингибитор киназы Btk.
В настоящей Заявке предложен способ ингибирования пролиферации В-клеток, включающий введение пациенту, который в этом нуждается, терапевтически эффективного количества соединения Формулы I', представляющего собой ингибитор киназы Btk.
В настоящей Заявке предложен способ ингибирования активности киназы Btk, включающий введение соединение по любой Формуле I', представляющего собой ингибитор киназы Btk, в котором соединение-ингибитор Btk характеризуется величиной константы IC50, составляющей 50 мкмоль или менее в биохимическом анализе активности киназы Btk in vitro.
В одной вариации указанного выше способа соединение-ингибитор Btk характеризуется величиной константы IC50, составляющей 100 наномоль или менее в биохимическом анализе активности киназы Btk in vitro.
В другой вариации указанного выше способа, соединение характеризуется величиной константы IC50, составляющей 10 наномоль или менее в биохимическом анализе активности киназы Btk in vitro.
В настоящей Заявке предложен способ лечения воспалительного состояния, включающий совместное введение пациенту, который в этом нуждается, терапевтически эффективного количества противовоспалительного соединения в комбинации с соединением Формулы I', представляющим собой ингибитор Btk.
В настоящей Заявке предложен способ лечения артрита, включающий совместное введение пациенту, который в этом нуждается, терапевтически эффективного количества противовоспалительного соединения в комбинации соединением Формулы I', представляющим собой ингибитор Btk.
В настоящей Заявке предложен способ лечения лимфомы или лейкемических клеток BCR-ABL1+ путем введения пациенту, который в этом нуждается, терапевтически эффективного количества соединения Формулы I', представляющего собой ингибитор киназы Btk.
В настоящей Заявке предложена фармацевтическая композиция, включающая соединение Формулы I', представляющее собой ингибитор Btk, смешанное по меньшей мере с одним фармацевтически приемлемым носителем, эксципиентом или разбавителем.
В настоящей Заявке предложено применение соединения Формулы I' в изготовлении лекарственного средства для лечения воспалительного расстройства.
В настоящей Заявке предложено применение соединения Формулы I' в изготовлении лекарственного средства для лечения аутоиммунного расстройства.
В настоящей Заявке предложено соединение, способ или композиция, описанные выше.
СОЕДИНЕНИЯ И ИХ ПОЛУЧЕНИЕ
Выбранные примеры соединений, охватываемых настоящим изобретением и включенных в объем настоящего изобретения, представлены в ниже следующей таблице. Эти ниже следующие примеры и способы получения представлены с той целью, чтобы помочь специалисту в данной области техники лучше понять и практически осуществить настоящее изобретение. Следует понимать, что они не ограничивают объем настоящего изобретения изобретение, но лишь являются его примером и наглядным представлением.
В общем случае номенклатура, использованная в настоящей Заявке, получена на основе AUTONOM™ v. 4.0, компьютеризированной системы Института Бельштейна для генерирования систематической номенклатуры IUPAC. В случае несоответствия между изображенной структурой и приведенным для этой структуры названием, большая значимость приписывается изображенной структуре. Кроме того, если стереохимия структуры или части структуры не указана посредством, например жирной или пунктирной линии, следует считать, что такая структура или часть структуры охватывает все ее стереоизомеры.
В Табл. 1 представлены примеры соединений родовой Формулы I.
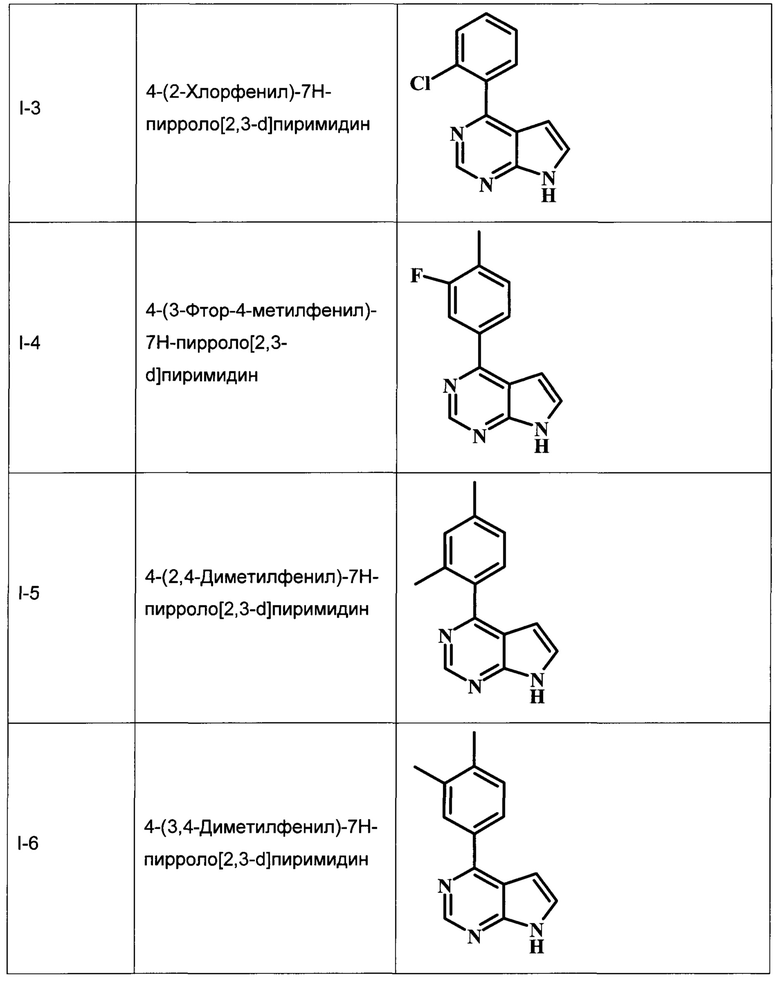
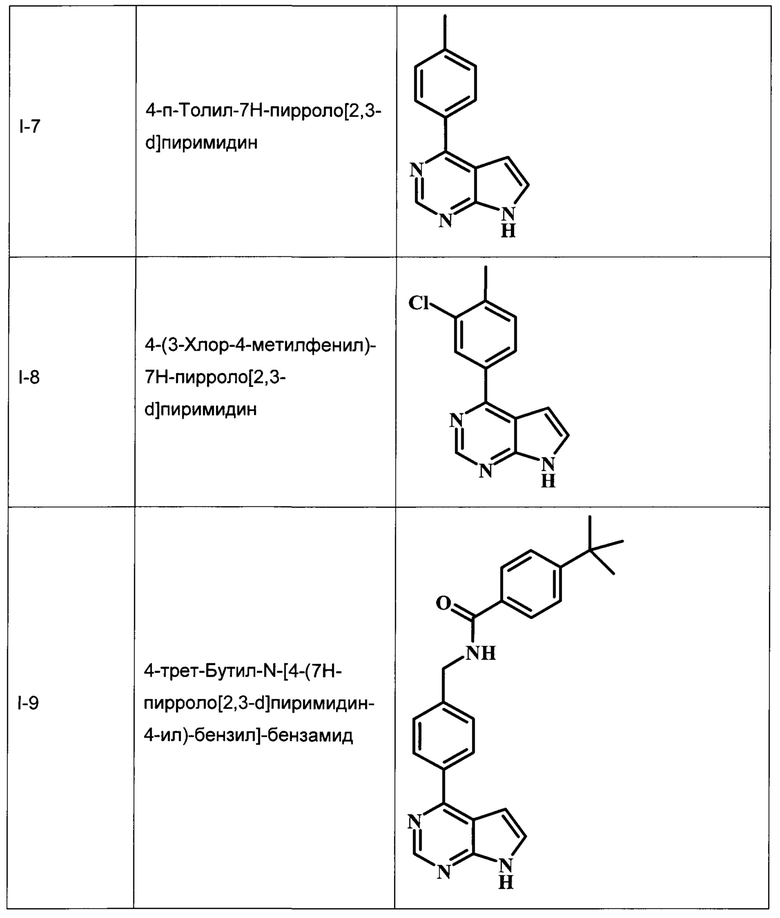
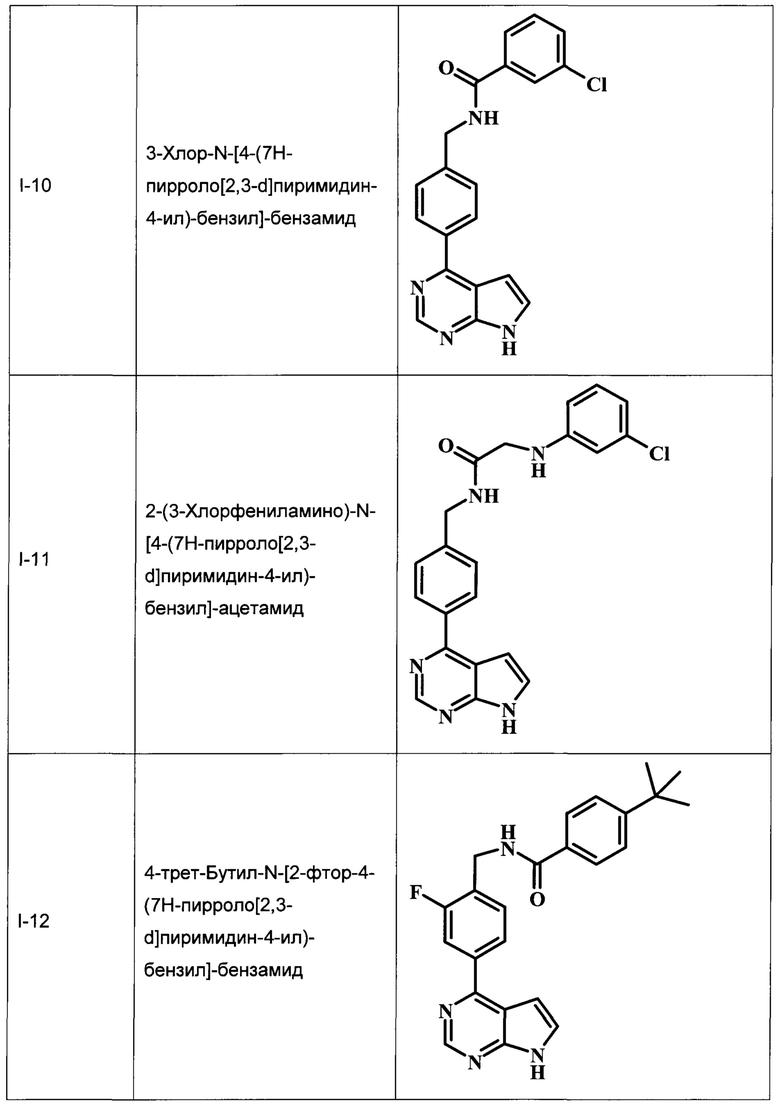
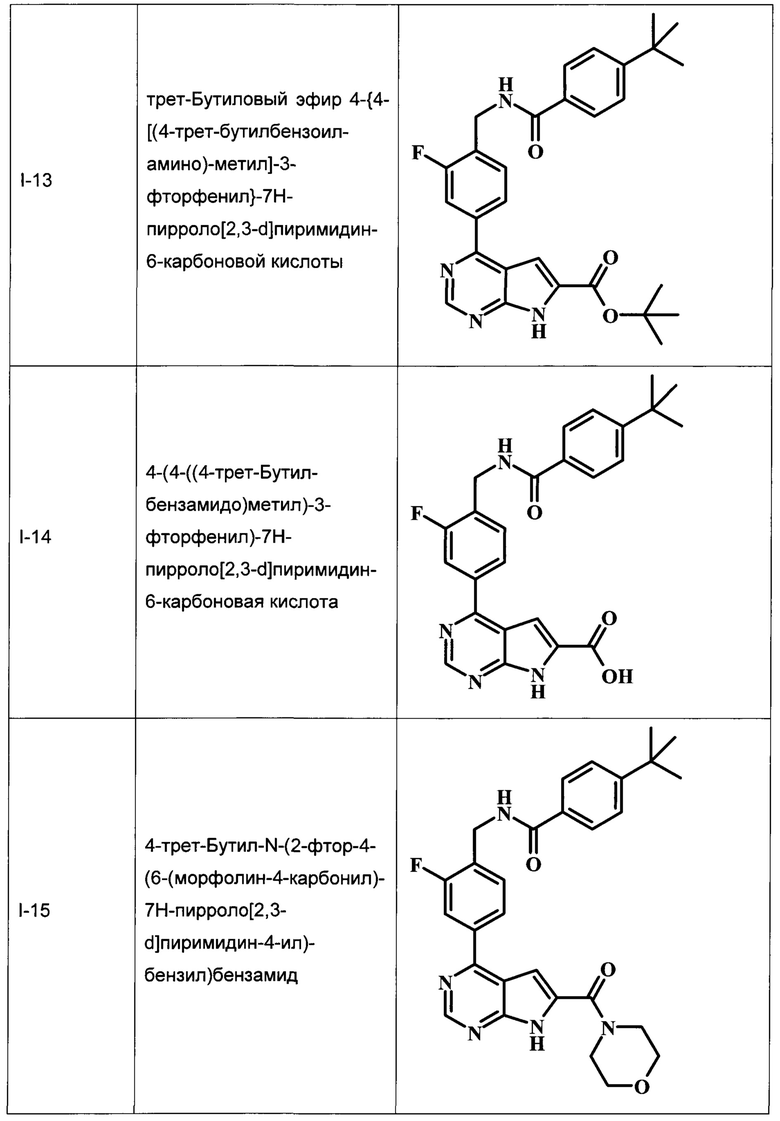
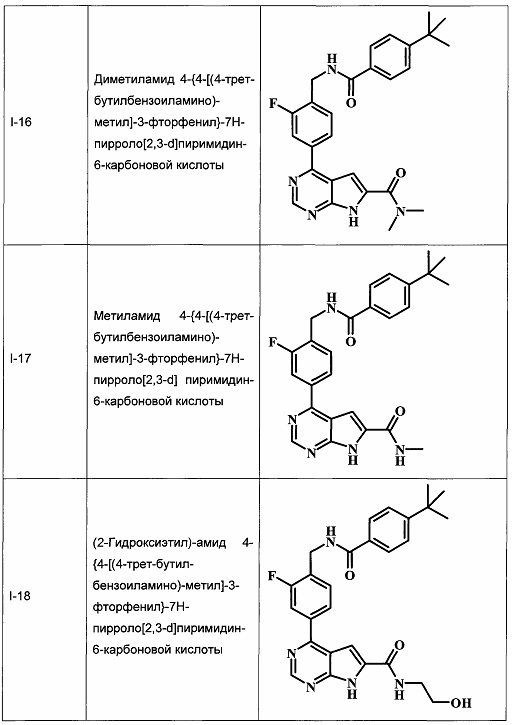
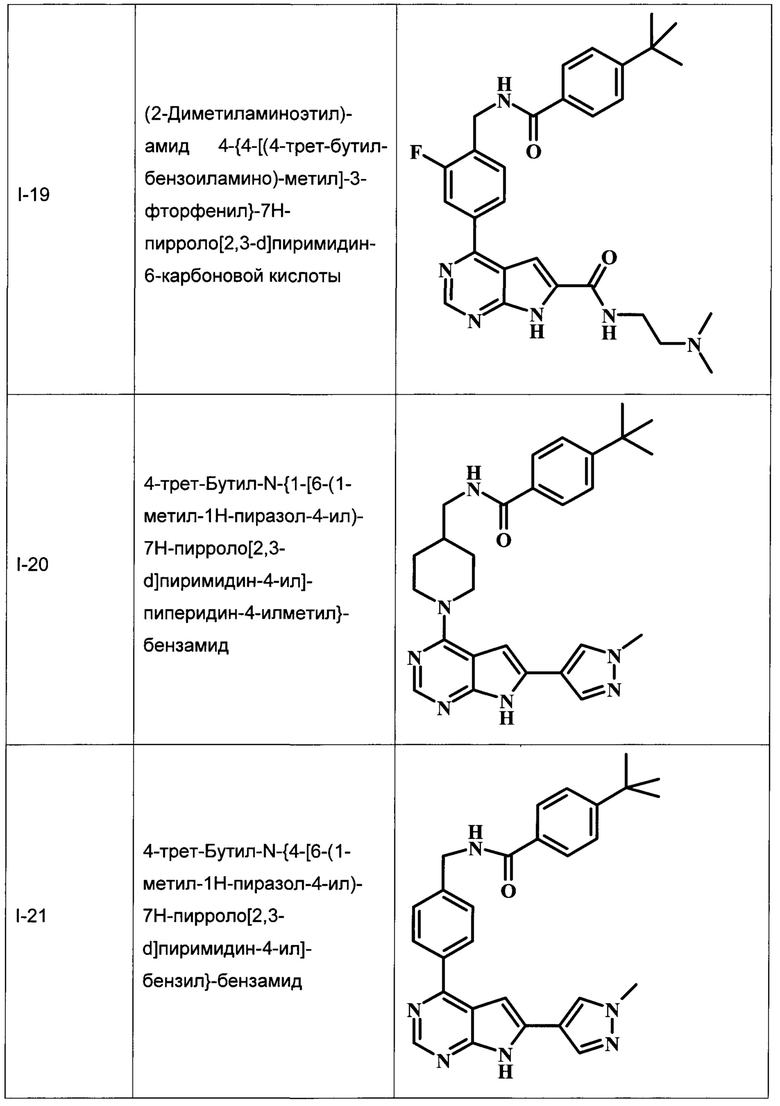

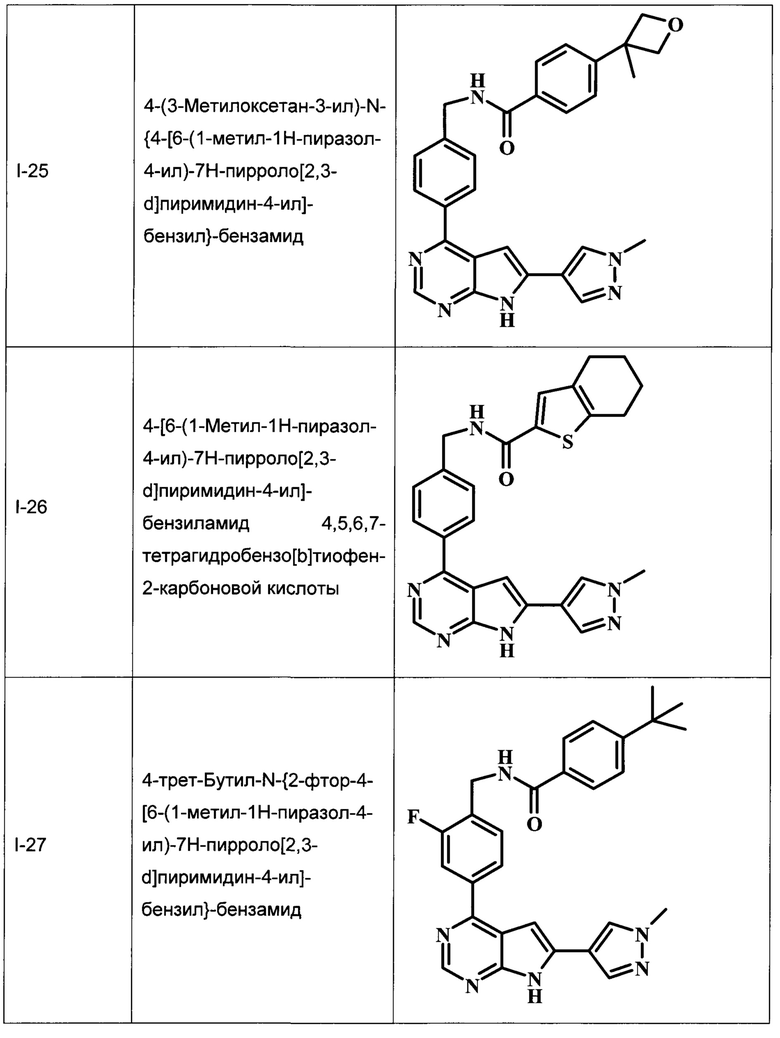
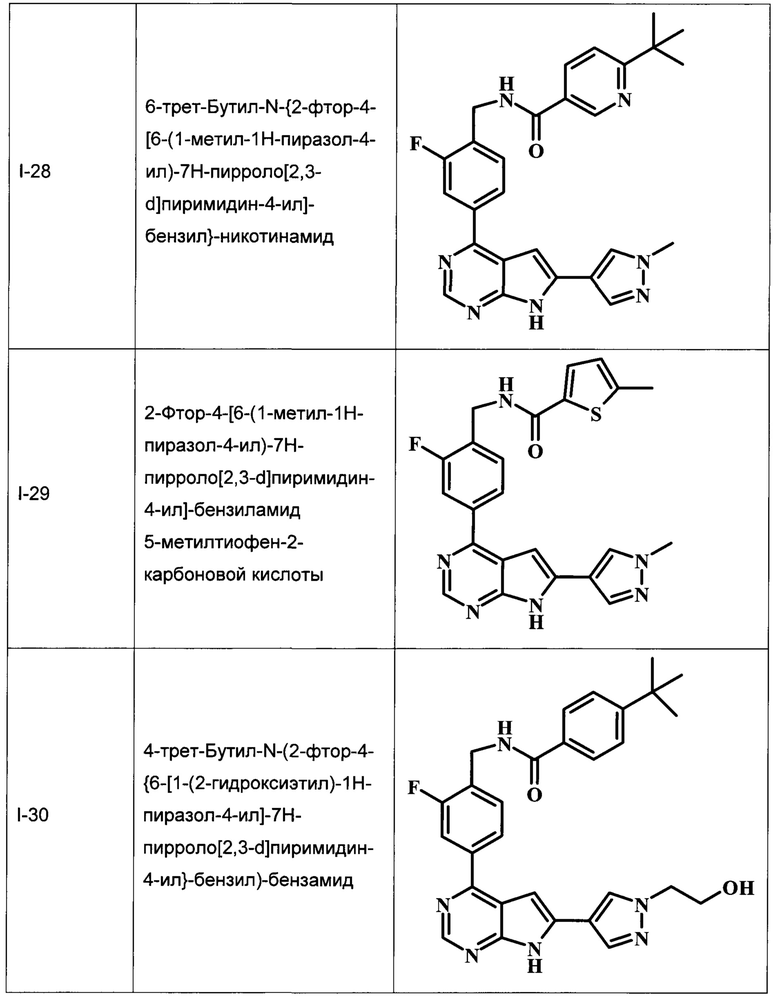
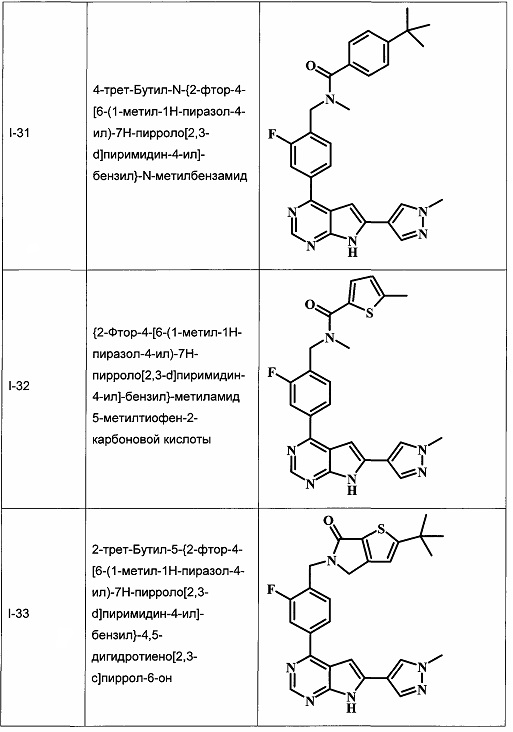
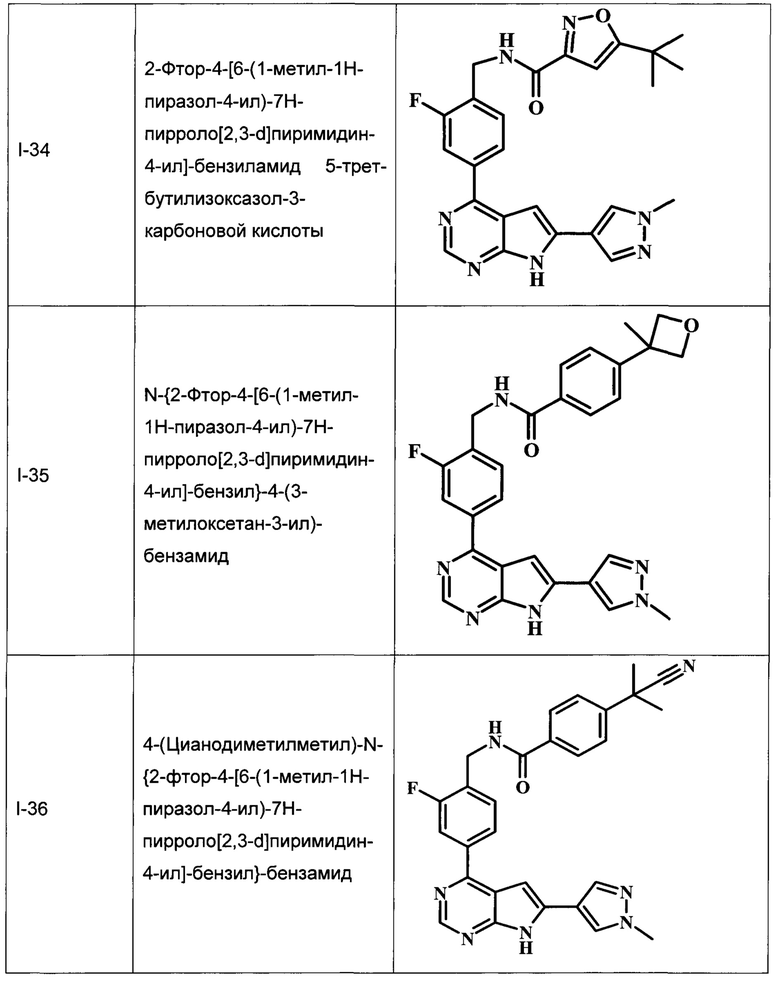
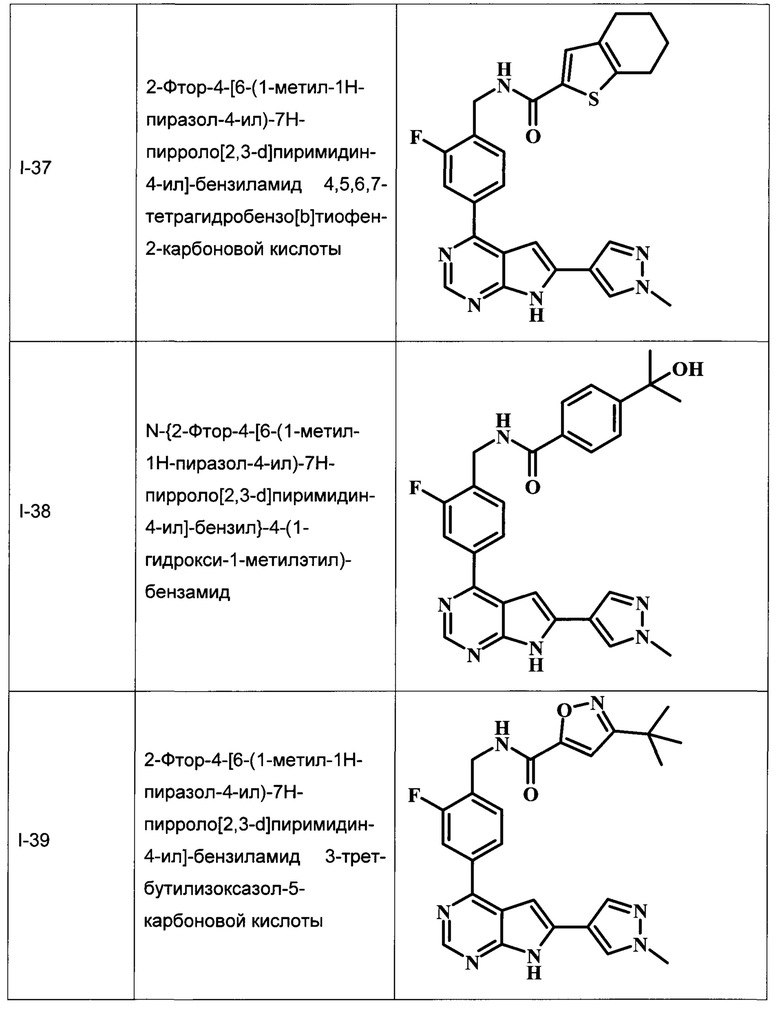
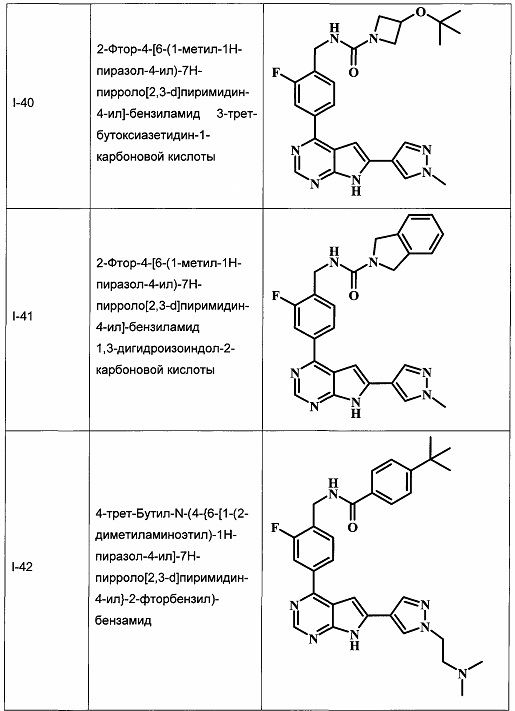
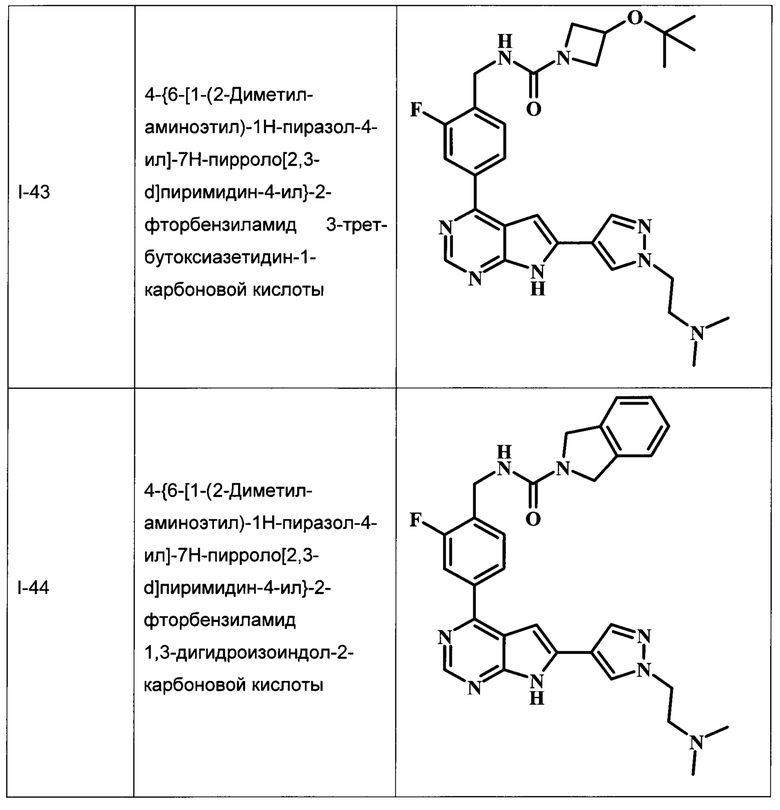
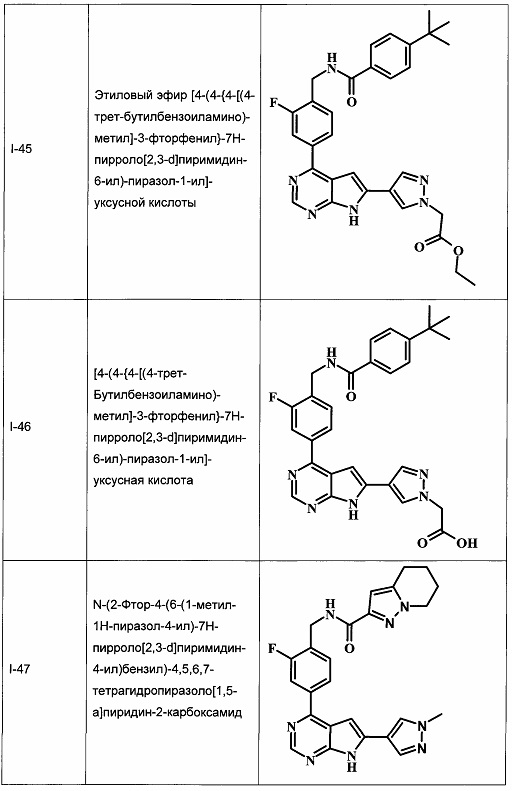
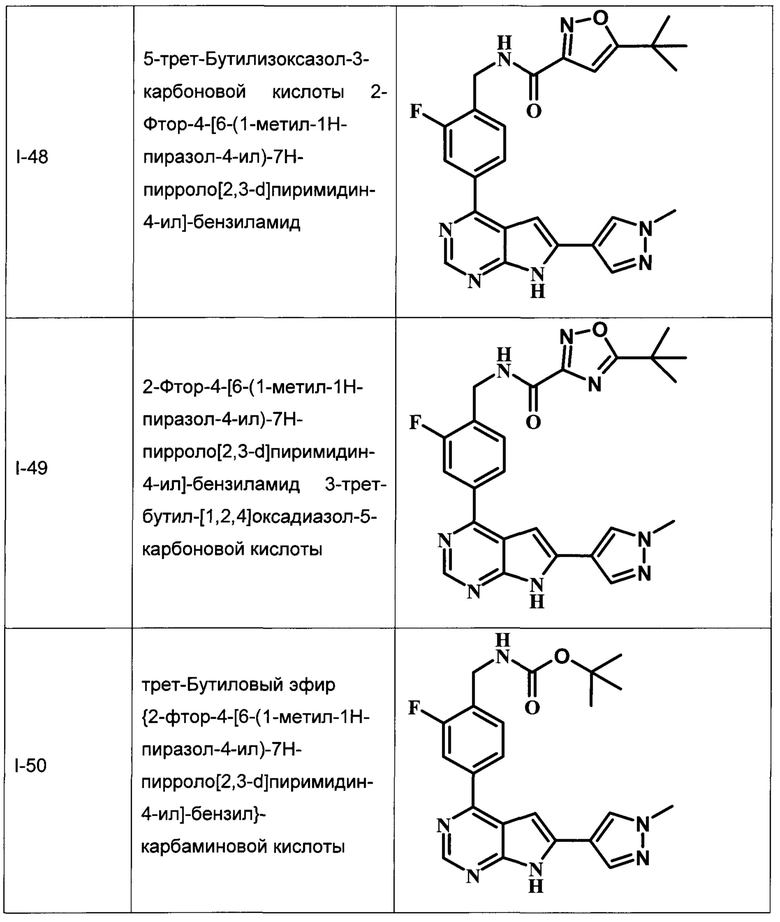
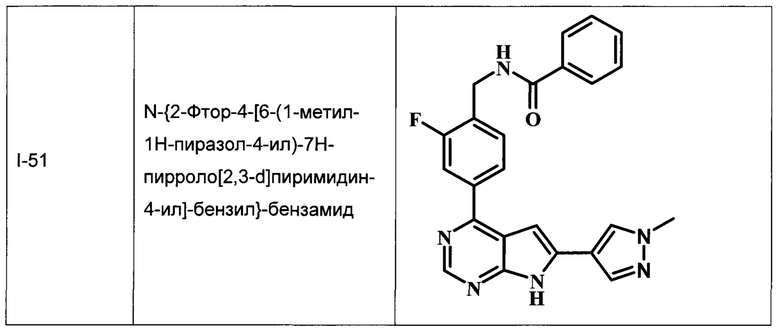
ОБЩИЕ СХЕМЫ СИНТЕЗА
Соединения по настоящему изобретению можно получить способами, известными в данной области техники. Подходящие способы для синтеза этих соединений приведены в примерах. В общем случае соединения по настоящему изобретению можно получить одним из описанных ниже путей синтеза (Схемы 1-5). Исходные вещества либо коммерчески доступны, либо их можно синтезировать способами, известными средним специалистам в данной области техники.
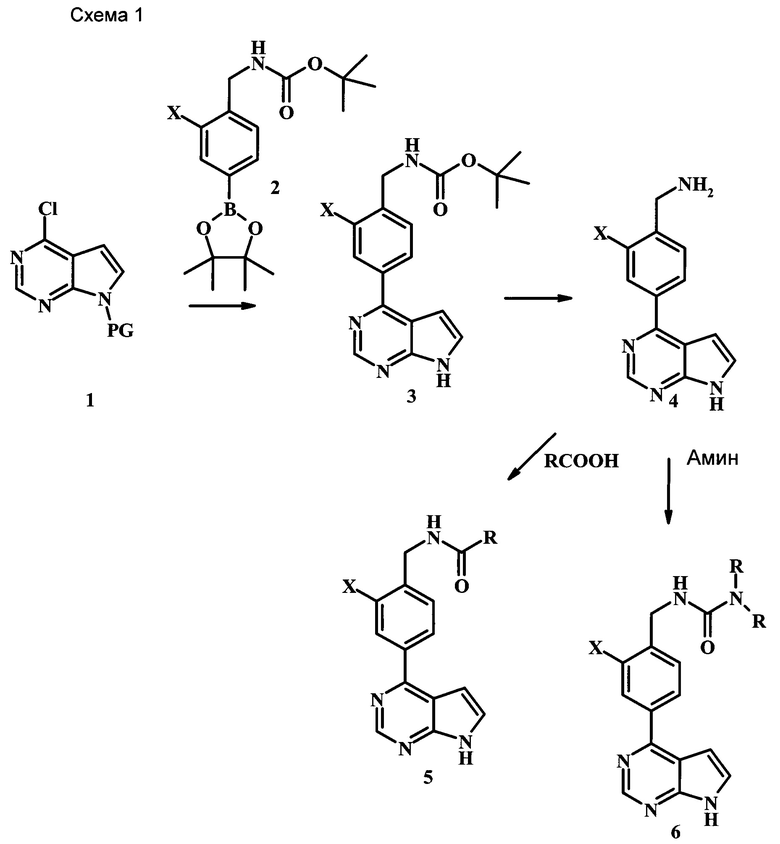
Целевые соединения Формулы 5 и 6, в которых Х представляет собой фтор, водород или метил, a R таков, как описано выше в общей Формуле I, можно получить по схеме 1. Исходя из коммерчески доступного 4-хлор-7-тозил-7Н-пирроло[2,3-d]пиримидина 1, реакция ариларильного сочетания Сузуки с боронатным сложным эфиром 2 дает производное 3. Условия для ариларильного сочетания по Сузуки можно найти в обзоре (Modern Arene Chemistry 2002, 53-106). При проведении реакции можно применять любые условия, обычно используемые в реакции Сузуки. Обычно реакцию сочетания Сузуки проводят в присутствии катализатора переходного металла, такого как тетракис(трифенилфосфин)палладий(0)), стандартного органического растворителя, такого как диметоксиэтан, и слабого неорганического основания, такого как карбонат калия. Эту реакцию проводят температуре между комнатной температурой и примерно 100°С со временем проведения реакции в интервале от 1 часа до нескольких часов, с применением стандартного нагревания. Эту реакцию можно также осуществлять при микроволновом облучении, которое обычно проводят при более высокой температуре (например при 160°С), но в течение более короткого промежутка времени (5-60 мин). В ходе этой реакции наблюдается также удаление тозильной группы. Трет-бутоксикарбонильную (ВОС) защитную группу в производном 3 можно легко удалять в кислых условиях, например в смеси трифторуксусной кислоты (TFA) и дихлорметана (DCM), для получения производного 4 со свободной аминогруппой. Эта реакция может также протекать при комнатной температуре со временем реакции в интервале от 15 минут до 3 часов. Реакцию сочетания между соединением 4 и производными карбоновой кислоты можно осуществлять с помощью стандартных реагентов для пептидной конденсации, таких как O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфат (HATU), стандартного органического растворителя, такого как N,N-диметилформамид (DMF), и основания, такого как диизопропилэтиламин (DIPEA), с получением соединения 5. Перечень сшивающих реагентов, которые можно применять для данного превращения, можно найти в литературе: Chemical Review 2011, 111, 6557. Эта реакция может протекать при комнатной температуре, со временем реакции в интервале от одного до нескольких часов. Иначе свободный амин 4 можно сочетать с другим амином с помощью 1,1'-карбонилдиимидазола в качестве связующего реагента для получения производных мочевины, таких как соединение 6. Эту реакцию можно проводить в DMF при температуре между комнатной и 90°С в течение нескольких часов. Как известно специалистам в данной области техники, в данной схеме можно использовать другие защитные группы, отличные от тозильной группы или ВОС-группы (основной источник: P.G.M. Wuts and Т.W. Greene in Green's Protective Groups in Organic Synthesis, Wiley and Sons, 2007).
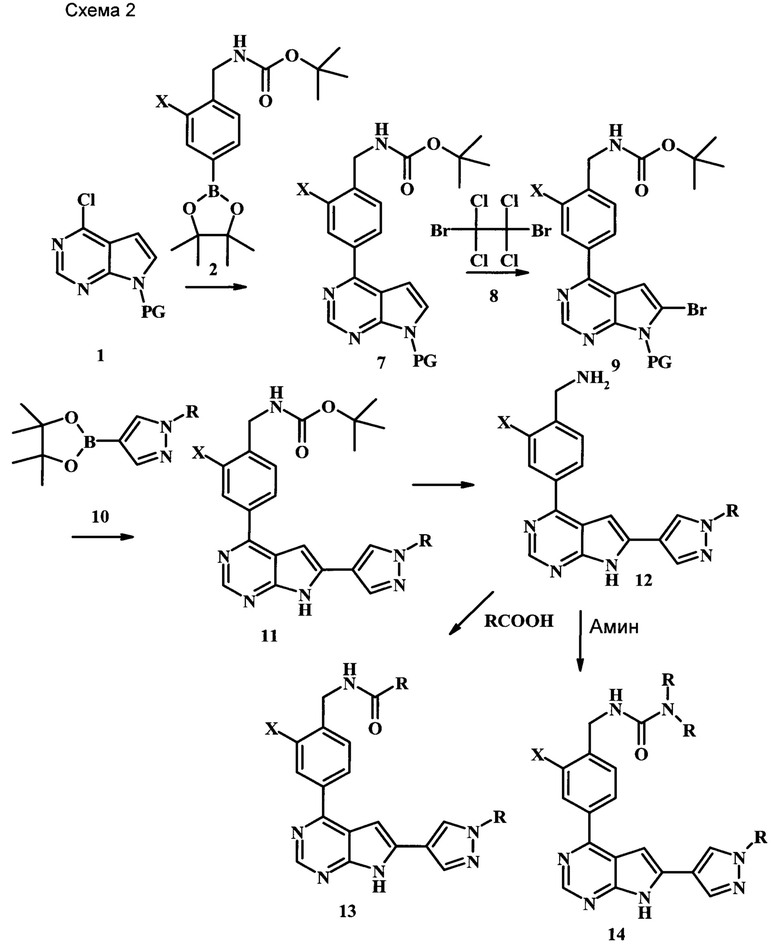
Целевые соединения Формулы 13 и 14, в которых Х представляет собой фтор, водород или метил, и R таков, как описано выше в родовой Формуле I, можно получить по схеме 2. Проводили ариларильное сочетание Сузуки, как описано на схеме 1. При этом за счет более короткого времени реакции (30 минут) и применения нагревания микроволновым облучением при 160°С, тозильную защитную группу можно сохранить в этих условиях. Бромирование по положению С-2 углеродного скелета пирролопиримидина можно осуществлять с помощью 1,2-дибромтетрахлорэтана в присутствии сильного основания, такого как диизопропиламид лития (LDA), с получением производного 9. Эта реакция может протекать в инертном растворителе, таком как тетрагидрофуран (THF), при -78°С со временем реакции в интервале от 2 часов до нескольких часов (WO 2004/093812). Сочетание Сузуки между соединениями 9 и 10 может протекать в стандартных условиях Сузуки. В этой реакции используется более длительное время реакции (60 минут) с микроволновым облучением при 160°С, и в этом случае тозильная защитная группа также удаляется. Последующие стадии для получения производных 13 и 14 были описаны выше.
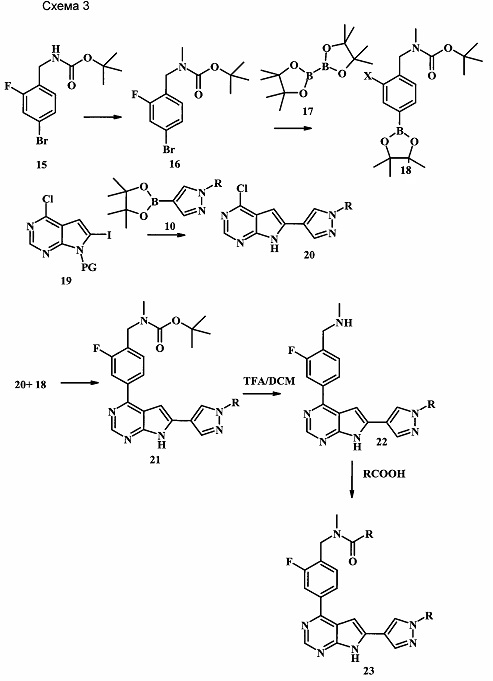
На схеме 3 представлен синтез соединения 23. Метилирование по атому азота карбамата 15 может протекать при участии сильного основания, такого как гидрид натрия (NaH), в присутствии метилиодида и в полярном растворителе, таком как DMF. Эта реакция протекает при температуре от 4°С до комнатной температуры, со временем реакции в интервале от 2 часов до нескольких часов. Катализируемая палладием реакция борилирования карбамата 16 может протекать при участии бис(пинаколато)диборона 17, подходящего источника палладия в качестве катализатора, такого как дихлорид 1,1'-бис(дифенилфосфино)ферроцен-палладия(II), и ацетата калия (Journal of Organic Chemistry 1995, 60, 7508-7510). Эта реакция может протекать в подходящем растворителе, таком как диоксан, DMF или NMP, с использованием либо стандартного нагревания, либо нагревания микроволновым облучением, при температуре в интервале между 90°С и 150°С и со временем реакции в интервале от одного часа до нескольких часов. 4-Хлор-6-иод-7-(фенилсульфонил)-7Н-пирроло[2,3-d]пиримидин 19 можно сочетать с боронатным сложным эфиром 10 в условиях сочетания по Сузуки, описанных выше, с получением производного 20. Аналогичным образом, сочетание соединений 18 и 20 осуществляют в таких же стандартных условиях с получением соединения 21. Последующие стадии для получения производных соединения 23 были описаны выше.

Целевые соединения общей формулы 33, в которых R таков, как раскрыто для родовой Формулы I, можно получить по схеме 4. Вводимый в реакцию сочетания реагент 27 можно получить в две стадии из коммерчески доступных исходных веществ. Как описано выше, образование амидной связи в производном 26 можно осуществлять с помощью стандартных связывающих реагентов. Аналогичным образом, введение боронатной сложноэфирной функциональной группы в соединение 27 также можно проводить в стандартных условиях. Синтез производного 30 уже был описан в литературе (WO 2011/149827). Сочетание Сузуки между соединениями 27 и 30 может проводить в полярных растворителях, таких как DME, диоксан или DMF, при температуре между 60°С и 100°С, стандартными способами нагревания, со временем реакции в интервале от 1 часа до нескольких часов. За счет нагревания микроволновым облучением можно значительно сократить время реакции для сочетания по Сузуки (Current Organic Chemistry, 2010, 14, 1050-1074). Как правило, требуется всего лишь 10-60 минут для завершения реакции. Удаление ВОС-группы в соединении 31 и последующая реакция сочетания с образованием амидных связей, как в соединении 33, были описаны выше.
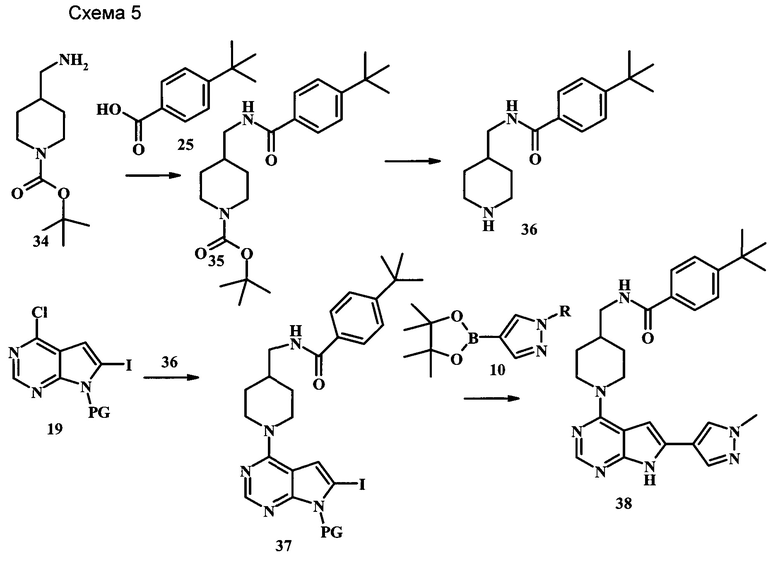
Целевые соединения 38 можно получить по схеме 5. Реакцию между соединениями 19 и 36 можно осуществлять в полярных протонных растворителях, таких как этанол, в присутствии основания, такого как DIPEA или триэтиламин (TEA). Данную реакцию может проводить при 80°С, со временем реакции в интервале от 1 часа до нескольких часов. Последующие стадии, приводящие к получению соединения 38, были описаны выше.
ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И СПОСОБЫ ВВЕДЕНИЯ
Лекарственные средства на основе соединений по настоящему изобретению можно изготавливать с применением большого разнообразия лекарственных форм для орального введения и носителей. Оральное введение можно осуществлять в форме таблеток, таблеток, покрытых оболочкой, драже, твердых и мягких желатиновых капсул, растворов, эмульсий, сиропов или суспензий. Соединения по настоящему изобретению эффективны при их введении другими способами, включая непрерывное (внутривенное вливание), местное, парентеральное, внутримышечное, внутривенное, подкожное, трансдермальное (возможно совместно с агентом, повышающим проницаемость), трансбуккальное, назальное, а также ведение путем ингаляции и суппозиториев, наряду с другими способами введения. Предпочтительным способом введения, как правило, является оральный способ с удобным режимом суточного дозирования, который можно подбирать с учетом тяжести заболевания и реакции пациента на действующее вещество.
Соединение или соединения по настоящему изобретению, а также их фармацевтически применимые соли, совместно с одним или более стандартными эксципиентами, носителями или разбавителями, могут быть изготовлены в виде фармацевтических композиций и форм со стандартной дозой. Такие фармацевтические композиции и стандартные лекарственные формы могут включать обычные компоненты в стандартных пропорциях, при наличии или отсутствии дополнительных активных соединений или компонентов, при этом такие лекарственные формы могут содержать любое подходящее эффективное количество действующего вещества, соответствующее диапазону предполагаемых суточных дозировок. Такие фармацевтические композиции можно применять в виде твердых субстанций, например, таких как таблетки или наполненные капсулы, полутвердые субстанции, порошки, лекарственные формы с пролонгированным высвобождением; или жидкостей, таких как растворы, суспензии, эмульсии, эликсиры или наполненные капсулы для орального применения; или в форме суппозиториев для ректального или вагинального введения; или в форме стерильных растворов для инъекций для парентерального применения. Стандартный лекарственный препарат содержит примерно от 5% до 95% действующего вещества или веществ (вес/вес). Подразумевается, что термин "лекарственный препарат" или "лекарственная форма" включает как твердые, так и жидкие лекарственные формы действующего вещества, и специалисту в данной области техники будет понятно, что активный компонент может присутствовать в различных лекарственных средствах, в зависимости от органа-мишени или ткани-мишени, а также желаемой дозы и фармакокинетических параметров.
Термин "экспципиент", используемый в данном тексте, обозначает соединение, применяемое при изготовлении фармацевтической композиции, в общем случае безопасное, нетоксичное, а также не являющееся нежелательным с биологической или иной точки зрения, и включает эксципиенты, приемлемые для ветеринарного применения, а также для фармацевтического применения у человека. Соединения по настоящему изобретению можно вводить в чистом виде, но как правило, их вводят в смеси с одним или более подходящих фармацевтических эксципиентов, разбавителей или носителей, выбранных в соответствии с предполагаемым способом введения и общепринятой фармацевтической практикой.
Термин "фармацевтически приемлемый" обозначает полезный при изготовлении фармацевтической композиции, которая в общем случае безопасна, нетоксична и не является нежелательной с биологической или иной точки зрения, и которая подходит для фармацевтического применения в ветеринарии, а также у человека.
Форма "фармацевтически приемлемой соли" активного компонента может изначально обеспечивать желаемые фармакокинетические свойства действующего вещества, не характерные для его несолевой формы, и может даже положительно повлиять на фармакодинамику действующего вещества в плане его терапевтического воздействия на организм. Фраза "фармацевтически приемлемая соль" соединения обозначает соль, которая является фармацевтически приемлемой и обладает желаемой фармакологической активностью соединения-предшественника. Такие соли включают: (1) кислотно-аддитивные соли, полученные с помощью неорганических кислот, таких как хлороводородная кислота, бромоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.п.; или полученные с помощью органических кислот, таких как уксусная кислота, пропионовая кислота, гексановая кислота, циклопентанпропионовая кислота, гликолевая кислота, пировиноградная кислота, молочная кислота, малоновая кислота, янтарная кислота, яблочная кислота, малеиновая кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, 3-(4-гидроксибензоил)бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, 1,2-этандисульфоновая кислота, 2-гидроксиэтансульфоновая кислота, бензолсульфоновая кислота, 4-хлорбензолсульфоновая кислота, 2-нафталинсульфоновая кислота, 4-толуолсульфоновая кислота, камфорсульфоновая кислота, 4-метилбицикло[2.2.2]-окт-2-ен-1-карбоновая кислота, глюкогептоновая кислота, 3-фенилпропионовая кислота, триметилуксусная кислота, трет-бутилуксусная кислота, лаурилсерная кислота, глюконовая кислота, глутаминовая кислота, гидроксинафтойная кислота, салициловая кислота, стеариновая кислота, муконовая кислота, и т.п.; или (2) соли, полученные за счет того, что кислотный атом водорода, присутствующий в соединении-предшественнике, либо замещается ионом металла, например, ионом щелочного металла, ионом щелочноземельного металла или ионом алюминия; либо координируется с органическим основанием, таким как этаноламин, диэтаноламин, триэтаноламин, трометамин, N-метилглюкамин, и т.п.
Твердые лекарственные формы включают порошки, таблетки, пилюли, капсулы, саше, суппозитории и измельчаемые гранулы. Твердый носитель может представлять собой одно или более веществ, которые могут выступать также в качестве разбавителей, ароматизаторов, солюбилизаторов, лубрикантов, суспендирующих агентов, связующих веществ, консервантов, веществ, улучшающих распадаемость таблеток или инкапсулирующего материала. В случае порошков носителем обычно является тонкоизмельченное твердое вещество, представляющее собой смесь с тонкоизмельченным активным компонентом. В случае таблеток активный компонент обычно смешивают с носителем, обязательно обладающим связывающей способностью, в приемлемом соотношении, и придают желаемые форму и размер. Приемлемые носители включают, без ограничения, карбонат магния, стеарат магния, тальк, сахар, лактозу, пектин, декстрин, крахмал, желатин, трагакант, метилцеллюлозу, натрий-карбоксиметилцеллюлозу, низкоплавкий воск, какао масло и т.п. Твердые лекарственные формы могут содержать, помимо активного компонента, красители, ароматические добавки, стабилизаторы, буферные агенты, искусственные и натуральные подсластители, диспергирующие вещества, загустители, солюбилизаторы и т.п.
Для орального введения подходят также жидкие лекарственные формы, включая жидкие лекарственные формы, в т.ч. эмульсии, сиропы, эликсиры, водные растворы и водные суспензии. К этим формам относятся твердые лекарственные формы, которые необходимо переводить в жидкие формы непосредственно перед использованием. Эмульсии могут быть приготовлены в растворах, например, в водных растворах пропиленгликоля, или они могут содержать эмульгаторы, например лецитин, сорбитан моноолеат или камедь. Водные растворы можно изготавливать растворением активного компонента в воде и добавлением подходящих красителей, ароматизаторов, стабилизаторов и загустителей. Водные суспензии можно изготавливать диспергированием тонкоизмельченного активного компонента в воде совместно с вязким веществом, таким как натуральные или синтетические каучуки, смолы, метилцеллюлоза, натрий-карбоксиметилцеллюлоза и другие хорошо известные суспендирующие агенты.
Соединения по настоящему изобретению можно включать в лекарственные формы для парентерального введения (например, путем инъекции, например болюсной инъекции или непрерывной инфузии) и могут быть представлены в виде лекарственных форм со стандартной дозой в ампулах, предварительно заполненных шприцах, контейнерах для малообъемной инфузии или многодозовых контейнерах, с добавлением консерванта. Такие композиции могут быть изготовлены в форме, например, суспензий, растворов или эмульсий на масляной или водной основе, например растворов в водном полиэтиленгликоле. Примеры маслянистых или неводных носителей, разбавителей, растворителей или основ включают пропиленгликоль, полиэтиленгликоль, растительные масла (например оливковое масло) и вводимые инъекцией органические эфиры (например этилолеат) и могут включать агенты для создания лекарственной формы, такие как консерванты, увлажняющие агенты, эмульгаторы или суспендирующие агенты, стабилизаторы и/или диспергирующие агенты. Как вариант, активный компонент может присутствовать в форме порошка, полученного путем асептического выделения стерильного твердого вещества или путем лиофилизации из раствора для восстановления перед применением с помощью подходящей основы, например стерильной апирогенной воды.
Соединения по настоящему изобретению можно включать в лекарственные формы для местного применения на коже, такие как мази, крема и лосьоны, или такие как трансдермальный пластырь. Мази или крема могут быть приготовлены, например, на водной или масляной основе с добавлением подходящих загустителей и/или гелеобразующих агентов. Лосьоны можно изготавливать на водной или масляной основе, и в общем случае они содержат один или более эмульгаторов, стабилизаторов, диспергирующих агентов, суспендирующих агентов, загустителей или красителей. Лекарственные формы, которые подходят для местного введения через рот, включают леденцы, содержащие действующее вещество, включенное в ароматизированную основу, обычно сахарозу и камедь или трагакант; пастилки, содержащие действующее вещество, включенное в инертную основу, такую как желатин и глицерин или сахароза и камедь; и ополаскиватели для рта, содержащие действующее вещество в подходящем жидком носителе.
Соединения по настоящему изобретению можно включать в лекарственную форму для введения в виде суппозиториев. Сначала расплавляют низкоплавкий воск, например смесь глицеридов жирной кислоты или какао масло, и гомогенно диспергируют активный компонент, например, путем перемешивания. Затем расплавленную гомогенную смесь разливают в формы подходящего размера и оставляют для охлаждения и отвердевания.
Соединения по настоящему изобретению можно включать в лекарственные формы для вагинального введения: пессарии, тампоны, крема, гели, пасты, пенки или спреи, содержащие помимо, активного компонента, подходящие носители, хорошо известные в данной области техники.
Соединения по настоящему изобретению можно включать в лекарственные формы для интраназального введения. Растворы или суспензии вводят непосредственно в носовую полость удобным способом, например, с помощью капельницы, пипетки или спрея. Такие лекарственные формы могут быть реализованы в виде форм с однократной или многократной дозировкой. В последнем случае, для капельницы или пипетки, этого достигают введением пациенту подходящего, предварительно определенного объема раствора или суспензии. В случае спрея этого достигают, например, с помощью насоса с дозированным распылением.
Соединения по настоящему изобретению можно включать в лекарственные формы для аэрозольного введения, в частности через дыхательные пути, включая интраназальное введение. Такое соединение обычно имеет маленький размер частиц, например, порядка пяти (5) микрон или менее. Такой размер частиц можно обеспечить способами, известными в данной области техники, например путем микронизации. Активный компонент помещают в сосуд под давлением с подходящим пропеллентом, таким как хлорфторуглерод (CFC), например дихлордифторметан, трихлорфторметан или дихлортетрафторэтан, или диоксид углерода или другой подходящий газ. Такой аэрозоль в целях удобства может также содержать поверхностно-активное вещество, например лецитин. Дозу лекарства можно контролировать с помощью дозирующего клапана. Как вариант, активные компоненты можно изготавливать в форме сухого порошка, например порошковой смеси соединений в подходящей порошковой основе, такой как лактоза, крахмал, производные крахмала, например гидроксипропилметилцеллюлоза или поливинилпирролидин (PVP). Порошковый носитель образует гель в носовой полости. Порошковую композицию можно изготавливать в виде стандартной лекарственной формы, например в капсулах или картриджах, в желатиновой или блистерной упаковке, из которой порошок можно вводить путем ингаляции.
По желанию можно изготавливать лекарственные формы с кишечнорастворимой оболочкой, адаптированные для введения активного компонента с замедленным или контролируемым высвобождением. Например, соединения по настоящему изобретению можно включать в лекарственную форму в виде систем для трансдермальной или подкожной доставки лекарственного вещества. Такие системы доставки имеют преимущество в случаях, когда необходимо замедленное высвобождение соединения и когда очень важно строгое соблюдение пациентом схемы лечения. Соединения в трансдермальных системах доставки часто наносят на твердую подложку, которую можно приклеивать к коже. Соединение, представляющее интерес, можно также сочетать с агентом, повышающим проницаемость, например, таким как Azone (1-додецилазациклогептан-2-он). Системы доставки с замедленным высвобождением внедряют подкожно в субдермальный слой хирургическим путем или путем инъекции. В подкожных имплантантах соединение инкапсулировано в липидно-растворимой мембране, например такой, как силиконовый каучук, или биодеградируемом полимере, например в полилактиде.
Подходящие лекарственные формы, а также фармацевтические эксципиенты, носители, разбавители и эксципиенты описаны в литературе (Remington: The Science и Practice of Pharmacy 1995, edited by E.W. Martin, Mack Publishing Company, 19th edition, Easton, Pennsylvania). Опытный специалист по лекарственным формам может вносить модификации в лекарственные формы в пределах сущности настоящего изобретения, с получением множества лекарственных форм для конкретного пути введения, без придания композициям по настоящему изобретению нестабильности и без ограничения их терапевтической активности.
Модификацию соединений по настоящему изобретению с целью улучшения их растворимости в воде или других растворителях, например, можно легко осуществлять путем минимальной модификации (солеобразование, этерификация и т.п.), что доступно среднему специалисту в данной области техники. Средний специалист в данной области техники способен также модифицировать способ введения и режим дозирования конкретного соединения, с тем чтобы привести фармакокинетические характеристики соединения по настоящему изобретению к максимальному благоприятному эффекту у пациентов.
Термин "терапевтически эффективное количество", используемое в данном тексте, обозначает количество, которое необходимо для облегчения симптомов заболевания у индивидуума. Дозировку следует подбирать в соответствии с индивидуальными требованиями в каждом конкретном случае. Дозировку можно варьировать в широких пределах, в зависимости от многочисленных факторов, таких как тяжесть заболевания, на которое направлено лечение, возраст и общее состояние здоровья пациента, другие лекарственные средства, которые принимает пациент, способ и форма введения, а также предпочтения и личный опыт лечащего врача. В случае орального введения приемлемой является суточная доза в интервале между примерно 0,01 и примерно 1000 мг/кг массы тела в сутки при монотерапии и/или комбинированной терапии. Предпочтительная суточная доза находится в пределах примерно от 0,1 до 500 мг/кг массы тела, более предпочтительно примерно от 0,1 до 100 мг/кг массы тела, и наиболее предпочтительно примерно от 1,0 до 10 мг/кг массы тела в сутки. Таким образом, при введении пациенту массой 70 кг, дозировка будет находиться в диапазоне примерно от 7 мг до 0,7 г в сутки. Суточную дозу можно вводить как однократную дозу или разделенными дозами, как правило, от 1 до 5 доз в сутки. В общем случае, лечение начинают с меньших доз, находящихся ниже оптимальной дозы соединения. Затем дозировку постепенно повышают до достижения оптимального эффекта для индивидуального пациента. Средний специалист в лечении заболеваний, описанных в данном тексте, сможет без необходимости нежелательного экспериментирования, основываясь на личных знаниях, опыте и содержании настоящей Заявки, установить терапевтически эффективное количество соединения по настоящему изобретению для конкретного заболевания и пациента.
Фармацевтические препараты предпочтительно представляют собой формы со стандартной дозировкой. В такой форме данный препарат подразделен на стандартные дозы, содержащие подходящие количества активного компонента. Лекарственная форма со стандартной дозировкой может представлять собой упакованный препарат, упаковку, содержащую дискретные количества препарата, например упакованные таблетки, капсулы и порошки во флаконах или ампулах. Кроме того, стандартная лекарственная форма сама по себе может представлять собой капсулу, таблетку, саше или леденец, или она может представлять подходящее количество любого из этих форм в упакованном виде.
НАЗНАЧЕНИЯ И СПОСОБЫ ЛЕЧЕНИЯ
Соединения родовой Формулы I ингибируют тирозинкиназу Брутона (Btk). Активация киназы Btk киназами, расположенными выше по сигнальному, пути приводит к активации фосфолипазы-Сγ, что в свою очередь стимулирует высвобождение провоспалительных медиаторов. Соединения Формулы I полезны в лечении артрита и других противовоспалительных и аутоиммунных заболеваний. Таким образом, соединения Формулы I полезны для лечения артрита. Соединения Формулы I полезны для ингибирования киназы Btk в клетках и для моделирования развития В-клеток. Кроме того, настоящее изобретение включает фармацевтические композиции, содержащие соединения Формулы I, смешанные с фармацевтически приемлемым носителем, эксципиентами или разбавителями.
Соединения, описанные в данном тексте, представляют собой ингибиторы киназ, в частности ингибиторы Btk. Эти ингибиторы могут быть эффективными при лечении одного или более заболеваний, реагирующих на ингибирование киназ, включая заболевания, реагирующих на ингибирование киназы Btk и/или ингибирование пролиферации В-клеток у млекопитающих. Не привязываясь к какой-либо конкретной теории, полагают, что взаимодействие соединений по настоящему изобретению с киназой Btk приводит к ингибированию активности Btk и, соответственно, к реализации фармацевтической эффективности этих соединений. Таким образом, настоящее изобретение включает способ лечения млекопитающего, например человека, страдающего заболеванием, реагирующим на ингибирование активности киназы Btk и/или ингибирование пролиферации В-клеток, включающий введение такому млекопитающему, страдающему таким заболеванием, эффективного количества по меньшей мере одного химического соединения, предложенного в настоящей Заявке. Эффективную концентрацию можно установить экспериментально, например с помощью анализа концентрации такого соединения в крови, или теоретически, путем расчета биодоступности. К другим киназам, на которые может быть оказано воздействие, помимо киназы Btk, относятся, без ограничения, другие тирозинкиназы и сериновые/треониновые киназы.
Киназы играют существенную роль в сигнальных путях, контролирующих основополагающие клеточные процессы, такие как пролиферация, дифференциация и гибель (апоптоз). С патологической киназной активностью связано множество заболеваний, включая различные формы рака, аутоиммунные и/или воспалительные заболевания, а также острые воспалительные реакции. Многоплановая роль киназ в сигнальных клеточных путях создает благоприятные условия для выявления новых лекарственных средств, нацеленных на киназы и сигнальные пути.
Одно из воплощений изобретения включает способ лечения пациента с аутоиммунным и/или воспалительным заболеванием или острой воспалительной реакцией, которые чувствительны к ингибированию активности Btk и/или пролиферации В-клеток.
Аутоиммунные и/или воспалительные заболевания, на которые можно повлиять с помощью соединений и композиций по настоящему изобретению, включают, без ограничения: псориаз, аллергию, болезнь Крона, синдром раздраженного кишечника, болезнь Шегрена, реакцию на тканевый трансплантат и сверхострое отторжение трансплантированных органов, астму, системную красную волчанку (и ассоциированный гломерулонефрит), дерматомиозит, множественный склероз, склеродермию, васкулит (АНЦА-ассоциированный и другие формы васкулита), аутоиммунные гемолитические и тромбоцитопенические состояния, синдром Гудпасчера (и ассоциированный гломерулонефрит и легочное кровотечение), атеросклероз, ревматоидный артрит, хроническую идиопатическую тромбоцитопеническую пурпуру (ITP), Аддисонову болезнь, болезнь Паркинсона, болезнь Альцгеймера, диабет, септический шок и миастению гравис.
Настоящим изобретением охвачены способы лечения, в которых по меньшей мере одно химическое соединение, предложенное в настоящей Заявке, вводится в комбинации с противовоспалительным агентом. Противовоспалительные агенты включают без ограничения нестероидные противовоспалительные препараты (НПВП), неспецифичные и специфичные ко 2 субъединице цитохром С-оксидазы (СОХ-2) ингибиторы фермента циклооксигеназы, соединения золота, кортикостероиды, метотрексат, антагонисты рецепторов фактора некроза опухолей (TNF), иммунодепрессанты и.
Примеры НПВП включают, без ограничения, ибупрофен, флурбипрофен, напроксен и напроксен натрий, диклофенак, комбинации диклофенака натрия и мизопростола, сулиндак, оксапрозин, дифлузинал, пироксикам, индометацин, этодолак, фенопрофен кальций, кетопрофен, набуметон натрий, сульфасалазин, толметин натрий и гидроксихлорохин. Примеры НПВП включают также специфичные к СОХ-2 ингибиторы, такие как целекоксиб, вальдекоксиб, лумиракоксиб и/или эторикоксиб.
В некоторых воплощениях такой противовоспалительный агент представляет собой салицилат. Салицилаты включают, без ограничения, ацетилсалициловую кислоту, или аспирин, салицилат натрия, а также салицилаты холина и магния.
Такой противовоспалительный агент может также представлять собой кортикостероид. Например, указанный кортикостероид может представлять собой кортизон, дексаметазон, метилпреднизолон, преднизолон, преднизолон натрий фосфат или преднизон.
В дополнительных воплощениях противовоспалительный агент представляет собой соединение золота, такое как ауротиомалат натрия или ауранофин.
Настоящее изобретение включают также воплощения, в которых указанный противовоспалительный агент представляет собой метаболический ингибитор, такой как ингибитор дигидрофолатредуктазы, например метотрексат, или ингибитор дигидрооротатдегидрогеназы, например лефлуномид.
Другие воплощения настоящего изобретения относятся к комбинациям, в которых по меньшей мере одно противовоспалительное соединение представляет собой моноклональное антитело против С5-компонента комплемента (например экулизумаб или пекселизумаб), антагонист TNF, например этанерцепт или инфликсимаб, который представляет собой моноклональное антитело против TNFα.
Другие воплощения настоящего изобретения относятся к комбинациям, в которых по меньшей мере одно действующее вещество представляет собой иммунодепрессант, например иммунодепрессант, выбранный из следующих: метотрексат, лефлуномид, циклоспорин, такролимус, азатиоприн и микофенолата мофетил.
В-клетки и предшественники В-клеток, экспрессирующие киназу Btk, вовлечены в патологию В-клеточных опухолей, включая, без ограничения, В-клеточную лимфому, лимфому (включая лимфому Ходжкина и не-ходжки некую лимфому), волосатоклетчатую лимфому, множественную миелому, хронический и острый миелогенный лейкоз, а также хронический и острый лимфоцитарный лейкоз.
Показали, что киназа Btk является ингибитором белка гибели Fas/APO-1 (CD-95), включая индуцирующий смерть сигнальный комплекс (DISC, от англ. Death-Inducing Signaling Complex) в лимфоидных В-клетках. Судьба лейкемических/лимфоидных клеток может зависеть от равновесия между разнонаправленными проапоптотическими воздействиями каспаз, активируемых под действием комплекса DISC, и восходящего анти-апоптозного регулирующего механизма с участием киназы Btk и/или ее субстратов (Vassilev et al., J. Biol. Chem. 1998, 274, 1646-1656).
Также обнаружили, что ингибиторы киназы Btk полезны в качестве хемо-сенсибилизирующих агентов, и таким образом, полезны в комбинации с другими химиотерапевтическими лекарственными веществами, в частности такими лекарственными веществами, которые индуцируют апоптоз. Примеры других химиотерапевтических лекарственных веществ, которые можно применять в комбинации с хемосенсибилизирующими ингибиторами киназы Btk, включают ингибиторы топоизомеразы I (камптотецин или топотекан), ингибиторы топоизомеразы II (например дауномицин и этопозид), алкилирующие агенты (например циклофосфамид, мелфалан и BCNU), агенты, направленные на тубулин (например таксол и винбластин) и биологические агенты (например антитела, такие как антитело против белка CD20, IDEC 8, иммунотоксины и цитокины).
Активность киназы Btk связана также с некоторыми лейкозами, где экспрессируется ген слияния bcr-abl, возникающий за счет транслокации частей хромосомы 9 и 22. Эта аномалия часто наблюдается при хроническом миелогенном лейкозе. Киназа Btk постоянно фосфорилируется под действием bcr-abl-киназы, инициирующей нисходящие сигналы выживания, за счет чего bcr-abl-клетки избегают апоптоза (N. Feldhahn et al. J. Exp. Med. 2005 201(11): 1837-1852).
Способы лечения
В настоящей Заявке предложен способ лечения воспалительного и/или аутоиммунного состояния, включающий введение пациенту, который в этом нуждается, терапевтически эффективного количества соединения Формулы I.
В настоящей Заявке предложен способ лечения воспалительного состояния, включающий введение пациенту, который в этом нуждается, терапевтически эффективного количества соединения Формулы I.
В настоящей Заявке предложен способ лечения ревматоидного артрита, включающий введение пациенту, который в этом нуждается, терапевтически эффективного количества соединения Формулы I.
В настоящей Заявке предложен способ лечения астмы, включающий введение пациенту, который в этом нуждается, терапевтически эффективного количества Формулы I.
В настоящей Заявке предложен способ лечения воспалительного и/или аутоиммунного состояния, включающий введение пациенту, который в этом нуждается, терапевтически эффективного количества соединения Формулы I, представляющего собой ингибитор киназы Btk.
В настоящей Заявке предложен способ лечения артрита, включающий введение пациенту, который в этом нуждается, терапевтически эффективного количества соединения Формулы I, представляющего собой ингибитор киназы Btk.
В настоящей Заявке предложен способ лечения астмы, включающий введение пациенту, который в этом нуждается, терапевтически эффективного количества соединения Формулы I, представляющего собой ингибитор киназы Btk.
В настоящей Заявке предложен способ ингибирования пролиферации В-клеток, включающий введение пациенту, который в этом нуждается, терапевтически эффективного количества соединения Формулы I, представляющего собой ингибитор киназы Btk.
В настоящей Заявке предложен способ ингибирования активности киназы Btk, включающий введение соединения по любой Формуле I, представляющего собой ингибитор Btk, при котором указанное соединение-ингибитор Btk характеризуется величиной IC50, составляющей 50 мкмоль или менее в биохимическом анализе активности киназы Btk in vitro.
В одной вариации указанного выше способа такое соединение-ингибитор Btk характеризуется величиной IC50, составляющей 100 наномоль или менее в биохимическом анализе активности киназы Btk in vitro.
В другой вариации указанного выше способа, соединение характеризуется величиной IC50, составляющей 10 наномоль или менее в биохимическом анализе активности киназы Btk in vitro.
В настоящей Заявке предложен способ лечения воспалительного состояния, включающий совместное введение пациенту, который в этом нуждается, терапевтически эффективного количества противовоспалительного соединения в комбинации с соединением Формулы I, представляющим собой ингибитор киназы Btk.
В настоящей Заявке предложен способ лечения артрита, включающий совместное введение пациенту, который в этом нуждается, терапевтически эффективного количества противовоспалительного соединения в комбинации с соединением Формулы I, представляющим собой ингибитор киназы Btk.
В настоящей Заявке предложен способ лечения лимфомы или лейкемических BCR-ABL1+-клеток путем введения пациенту, который в этом нуждается, терапевтически эффективного количества соединения Формулы I, представляющего собой ингибитор киназы Btk.
ПРИМЕРЫ
Общие условия
Соединения по настоящему изобретению можно получить исходя из коммерчески доступных исходных веществ, с применением стандартных способов и процедур синтеза, известных специалистам в данной области техники. Ниже приведены схемы реакций, подходящие для получения таких соединений. Более подробные детали раскрыты в конкретных примерах.
Примеры получения
Специальная аббревиатура
Общие особенности проведения эксперимента
Реагенты заказывали в Aldrich, Oakwood Matrix и у других поставщиков и использовали без дополнительной очистки. Реакции с применением микроволнового излучения для нагревания проводили либо с помощью системы Personal Chemistry Emrys Optimizer System, либо СЕМ Discovery System. Очистку в масштабах от нескольких миллиграмм до нескольких грамм осуществляли способами, известными специалистам в данной области техники, такими как элюирование с колонки для флэш-хроматографии с силикагелем; в некоторых случаях осуществляли также препаративную очистку на колонке для флэш-хроматографии с применением одноразовых готовых заполненных мультиграммовых колонок с силикагелем (RediSep), элюировали с помощью системы CombiFlash. По настоящему изобретению для очистки промежуточных продуктов, в качестве инструментов для флэш-хроматографии можно использовать также продукцию Biotage™ и ISCO™.
С целью идентификации и оценки чистоты соединений снимали спектры LC/MS (жидкостная хроматография/масс-спектроскопия) с помощью системы, описанной далее. Для записи масс-спектров, данная система состоит из спектрометра Micromass Platform II: электроспрэй-ионизация в режиме определения положительных ионов (диапазон масс: 150-1200). Одновременное хроматографическое разделение осуществляли на следующие системе для ВЭЖХ: колоночный картридж ES Industries Chromegabond WR C-18 3u 120 Å (3,2×30 мм); подвижная фаза А: вода (0,02% TFA) и фаза В: Ацетонитрил (0,02% TFA); градиент от 10% В до 90% B в течение 3 минут; время уравновешивания 1 мин; скорость потока 2 мл/мин.
Многие соединения Формулы I также очищали с помощью обращенной-фазной ВЭЖХ способами, хорошо известными специалистам в данной области техники. В некоторых случаях проводили очистку препаративной ВЭЖХ с использованием масс-спектрометра РЕ Sciex 150 EX, контролирующего коллектор фракций Gilson 215, соединенный с системой для препаративной ВЭЖХ Shimadzu, и автоинжектор Leap. Соединения собирали из элюируемого потока с помощью детекции LC/MS в режиме определения положительных ионов. Элюирование соединений с колонок C-18 (2,0×10 см, элюировали со скоростью 20 мл/мин) осуществляли с помощью подходящего режима линейного градиента в течение 10 минут Растворителя (А) 0,05% TFA/H2O и Растворителя (В) 0,035% TFA/ацетонитрил. Для инжекции в систему ВЭЖХ неочищенные образцы растворяли в смеси метанола, ацетонитрила и DMSO.
Характеристики соединений получали с помощью 1H-ЯМР на ЯМР-спектрометре Bruker при 400 МГц.
Соединения по настоящему изобретению можно синтезировать известными способами. Ниже следующие примеры и ссылки приведены с целью понимания настоящего изобретению. При этом подразумевается, что данные примеры не ограничивают настоящее изобретение, истинный объем которого представлен в приложенной формуле изобретения. Названия конечных продуктов в примерах генерировали с помощью программы Isis AutoNom 2000.
Примеры получения
Пример I-1
4-(4-Хлорфенил)-7Н-пирроло[2,3-d]пиримидин
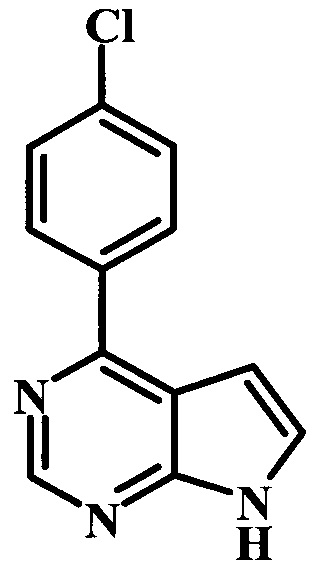
В сосуде для микроволнового облучения на 10 мл 4-хлор-7Н-пирроло[2,3-d]пиримидин (200 мг; 1,3 ммоль; Экв.: 1.00), 4-хлорфенилбороновую кислоту (204 мг; 1,3 ммоль; Экв.: 1.00) и карбонат калия (720 мг; 5,21 ммоль; Экв.: 4.00) в 2 мл воды соединяли с DME (4,00 мл). Добавляли Pd(PPh3)4 (78 мг; 0,068 ммоль). Реакционную смесь облучали микроволнами при 160°С в течение 60 минут. Полученный раствор разбавляли с помощью EtOAc, промывали водой. Объединенные органические фазы высушивали над безводным сульфатом натрия, затем выпаривали. Неочищенное вещество растворяли в DCM и фильтровали. Указанное в заголовке соединение получали в виде зеленого твердого вещества (90 мг; выход 30%). LC/MS: m/z расч. для C12H8ClN3 ([М+Н]+): 230,6 фактич.: 230,1
Пример I-2
4-(3-Хлорфенил)-7Н-пирроло[2,3-d]пиримидин
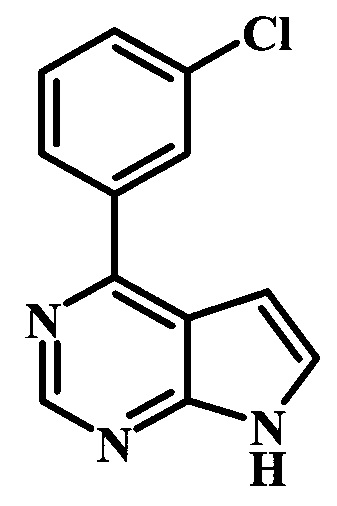
По аналогии со способом, описанным в примере 1, из 3-хлорфенилбороновой кислоты можно получить указанное в заголовке соединение.
Пример I-3
4-(2-Хлорфенил)-7Н-пирроло[2,3-d]пиримидин
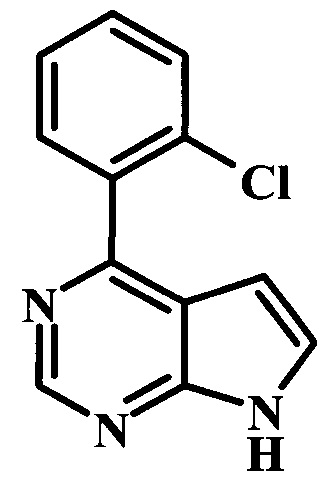
По аналогии со способом, описанным в примере 1, из 2-хлорфенилбороновой кислоты, можно получить указанное в заголовке соединение.
Пример I-4
4-(3-Фтор-4-метилфенил)-7Н-пирроло[2,3-d]пиримидин
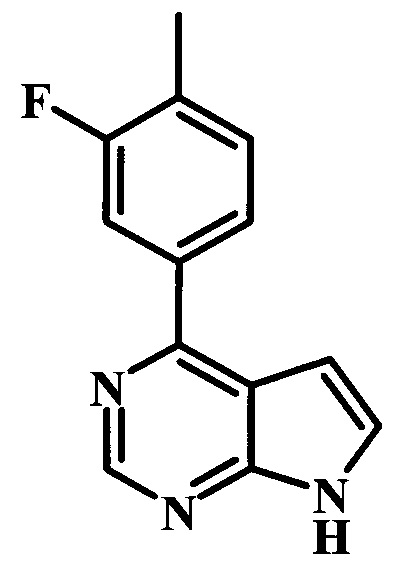
По аналогии со способом, описанным в примере 1, из 3-фтор-4-метилфенилбороновой кислоты можно получить указанное в заголовке соединение.
Пример I-5
4-(2,4-Диметилфенил)-7Н-пирроло[2,3-d]пиримидин
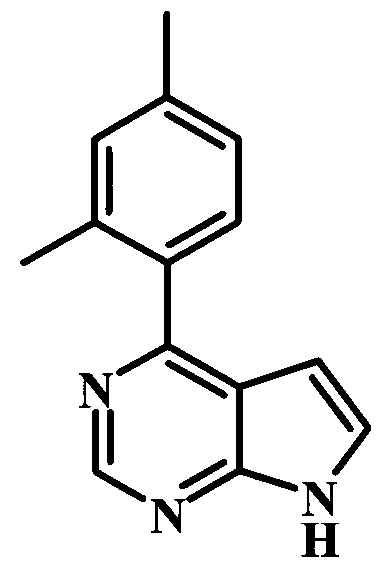
В герметизируемом сосуде для микроволнового облучения на 10 мл 4-хлор-7-тозил-7Н-пирроло[2,3-d]пиримидин (210 мг; 0,681 ммоль; Экв.: 1,00), 2,4-диметилфенилбороновую кислоту (112 мг; 0,749 ммоль; Экв.: 1,1) и карбонат калия (376 мг; 2,72 ммоль; Экв.: 4,00) в 2 мл воды соединяли с DME (4 мл). Вносили Pd(PPh3)4 (79 мг; 0,068 ммоль; Экв.: 0,1). Реакционную смесь нагревали микроволновым облучением при 160°С в течение 60 минут. Полученный раствор разбавляли с помощью EtOAc и промывали солевым раствором. Объединенные органические фазы высушивали над безводным сульфатом натрия, затем удаляли растворитель при пониженном давлении. Неочищенное вещество очищали хроматографией на колонке (силикагель, 5-70% этилацетат в гексане). Указанное в заголовке соединение получали в виде твердого вещества (51 мг; выход 34%). LC/MS: m/z расч. для C14H13N3 ([М+Н]+): 224,2 Фактич.: 224,2
Пример I-6
4-(3,4-Диметилфенил)-7Н-пирроло[2,3-d]пиримидин
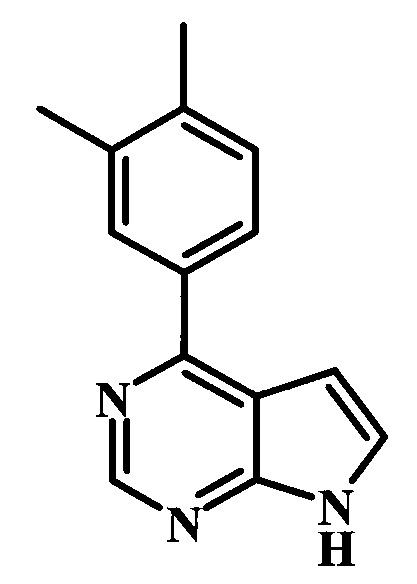
В герметизируемой трубке на 10 мл для микроволнового облучения 4-хлор-7-тозил-7Н-пирроло[2,3-d]пиримидин (210 мг; 0,681 ммоль; Экв.: 1,00), 3,4-диметилфенилбороновую кислоту (112 мг; 0,749 ммоль; Экв.: 1,1) и карбонат калия (376 мг; 2,72 ммоль; Экв.: 4,00) в 2 мл воды соединяли с DME (4 мл). Вносили Pd(PPh3)4 (79 мг; 0,0681 ммоль; Экв.: 0,1). Реакционную смесь нагревали микроволновым облучением при 160°С в течение 60 минут. Полученный раствор разбавляли с помощью EtOAc и промывали солевым раствором. Объединенные органические фазы высушивали над безводным сульфатом натрия, затем растворитель удаляли при пониженном давлении. Неочищенное вещество очищали хроматографией на колонке (силикагель, 5-70% этилацетат в гексане). Указанное в заголовке соединение получали в виде твердого вещества (17 мг; выход 11%). LC/MS: m/z расч. для C14H13N3 ([М+Н]+): 224,2 Фактич.: 224,2
Пример I-7
4-п-толил-7Н-пирроло[2,3-d]пиримидин
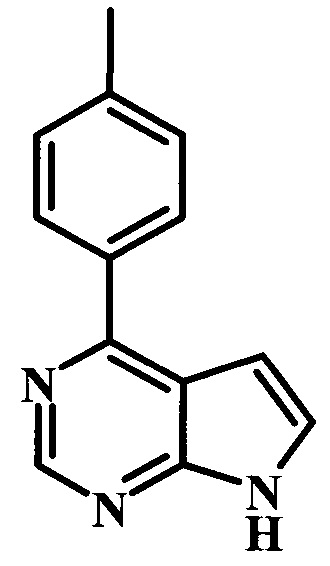
В герметизируемом сосуде на 10 мл для микроволнового облучения 4-хлор-7-тозил-7Н-пирроло[2,3-d]пиримидин (210 мг; 0,681 ммоль; Экв.: 1,00), п-толилбороновую кислоту (102 мг; 0,749 ммоль; Экв.: 1,1) и карбонат калия (376 мг; 2,72 ммоль; Экв.: 4,00) в 2 мл воды соединяли с DME (4 мл). Вносили Pd(PPh3)4 (79 мг; 0,0681 ммоль; Экв.: 0,1). Реакционную смесь нагревали микроволновым облучением при 160°С в течение 60 минут. Полученный раствор разбавляли с помощью EtOAc и промывали солевым раствором. Объединенные органические фазы высушивали над безводным сульфатом натрия, затем растворитель удаляли при пониженном давлении. Неочищенное вещество очищали хроматографией на колонке (силикагель, 5-70% этилацетат в гексане. Указанное в заголовке соединение получали в виде твердого вещества (60 мг; выход 42%). LC/MS: m/z расч. для C13H11N3 ([M+H]+): 210,2 Фактич.: 210,2
Пример I-8
4-(3-Хлор-4-метилфенил)-7Н-пирроло[2,3-d]пиримидин
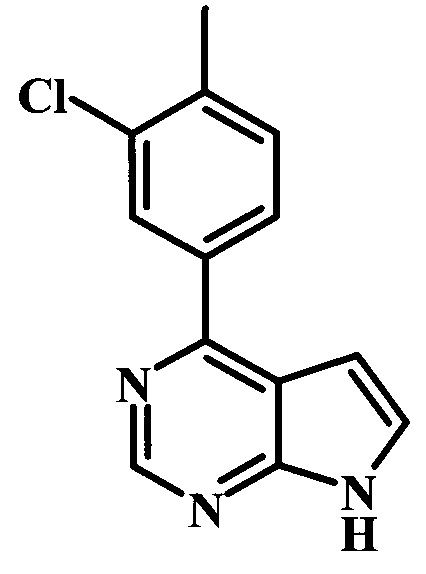
В герметизируемой трубке на 10 мл для микроволнового облучения 4-хлор-7-тозил-7Н-пирроло[2,3-d]пиримидин (210 мг; 0,681 ммоль; Экв.: 1,00), 3-хлор-4-метилфенилбороновую кислоту (128 мг; 0,749 ммоль; Экв.: 1,1) и карбонат калия (376 мг; 2,72 ммоль; Экв.: 4,00) в 2 мл воды соединяли с DME (4 мл). Вносили Pd(PPh3)4 (79 мг; 0,068 ммоль; Экв.: 0,1). Реакционную смесь нагревали микроволновым облучением при 160°С в течение 60 минут. Полученный раствор разбавляли с помощью EtOAc и промывали солевым раствором. Объединенные органические фазы высушивали над безводным сульфатом натрия, затем растворитель удаляли при пониженном давлении. Неочищенное вещество обрабатывали с помощью DCM. Твердое вещество отфильтровывали. Указанное в заголовке соединение получали в виде твердого вещества (14 мг; выход 8%). LC/MS: m/z расч. для C13H10ClN3 ([M+H]+): 244,7 Фактич.: 244,2
Пример I-9
4-трет-Бутил-N-[4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-бензил]-бензамид
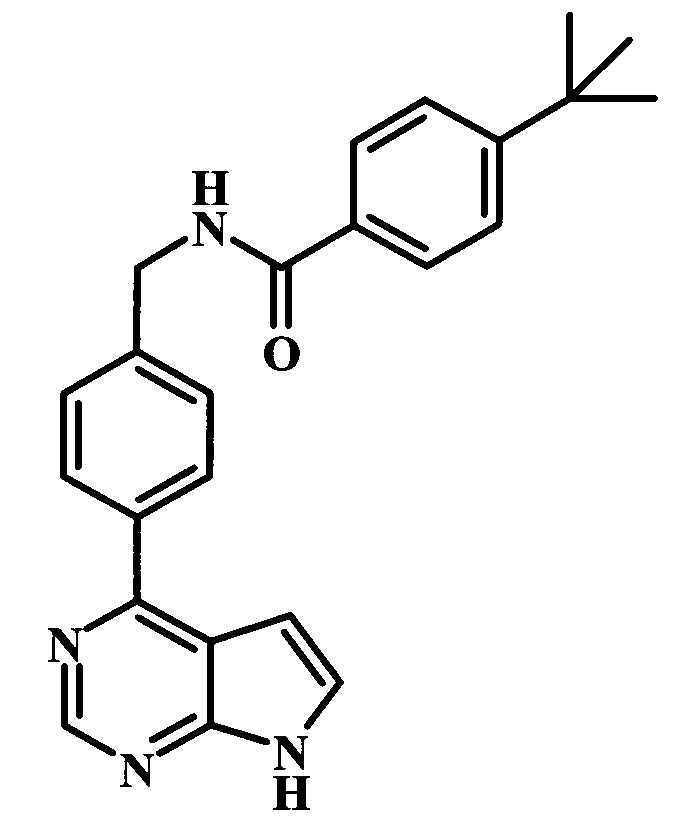
Стадия 1: Трет-бутиловый эфир [4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-бензил]-карбаминовой кислоты
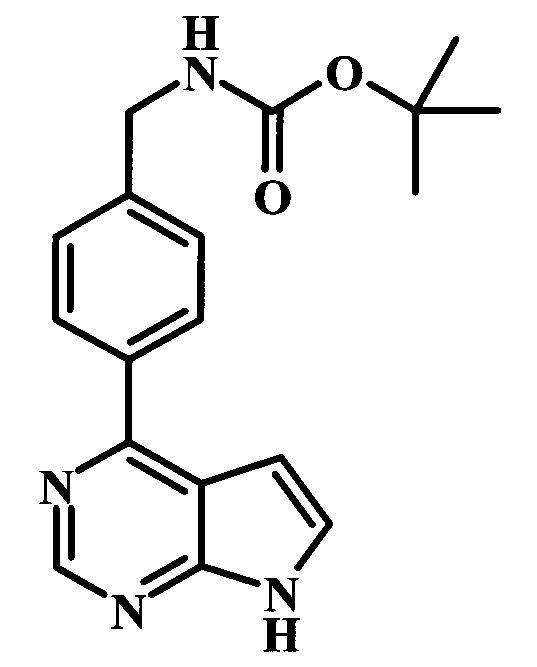
В герметизируемой трубке на 20 мл для микроволнового облучения 4-хлор-7Н-пирроло[2,3-d]пиримидин (500 мг; 3,26 ммоль; Экв.: 1,00), трет-бутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензилкарбамат (1,6 г; 4,8 ммоль; Экв.: 1,47) и карбонат калия (1,8 г; 13,0 ммоль; Экв.: 4,00) соединяли с DME (10 мл) и водой (5 мл). Вносили Pd(PPh3)4 (376 мг; 0,326 ммоль; Экв.: 0,1) и эту реакционную смесь нагревали при 150°С в течение 60 минут. Полученный раствор разбавляли с помощью EtOAc и промывали солевым раствором. Объединенные органические фазы высушивали над безводным сульфатом натрия, затем растворитель удаляли при пониженном давлении. Неочищенное вещество очищали хроматографией на колонке (силикагель, 5-70% этилацетат в гексане). Указанное в заголовке соединение получали в виде твердого вещества (545 мг; выход 52%). LC/MS: m/z расч. для C18H20N4O2 ([M+H]+): 325,3 Фактич.: 325,2
Стадия 2: 4-трет-Бутил-N-[4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-бензил]-бензамид
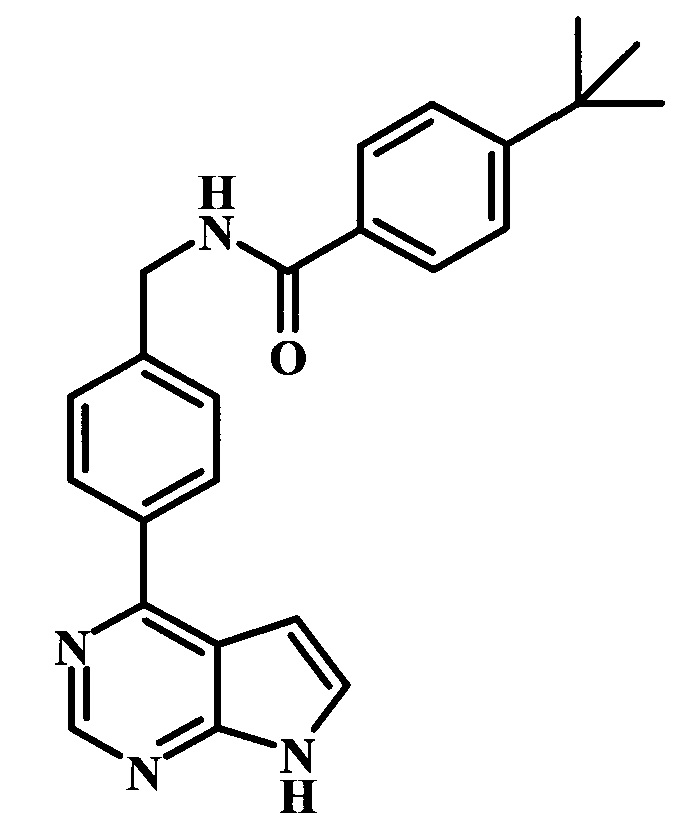
В сцинтилляционном флаконе на 20 мл трет-бутил-4-(7Н-пирроло[2,3-d]пиримидин-4-ил)бензилкарбамат (200 мг; 0,617 ммоль; Экв.: 1,00) растворяли в 2 мл DCM и 2 мл TFA. Реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Растворитель удаляли при пониженном давлении. Неочищенное вещество растворяли в DMF (4 мл). Вносили 4-трет-бутилбензойную кислоту (121 мг; 0,678 ммоль; Экв.: 1,1), DIPEA (0,43 мл, 2,47 ммоль; Экв.: 4,00) и HATU (258 мг; 0,678 ммоль; Экв.: 1,1). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли с помощью EtOAc и воды, затем перемешивали при комнатной температуре в течение 30 минут. Полученный раствор промывали солевым раствором. Объединенные органические фазы высушивали над безводным сульфатом натрия, затем растворитель удаляли при пониженном давлении. Неочищенное вещество обрабатывали с помощью DCM и полученное твердое вещество фильтровали с получением указанного в заголовке соединения в виде твердого вещества (65 мг; выход 27%). LC/MS: m/z расч. для C24H24N4O ([М+Н]+): 385,4 Фактич.: 385,1
Пример I-10
3-Хлор-N-[4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-бензил]-бензамид
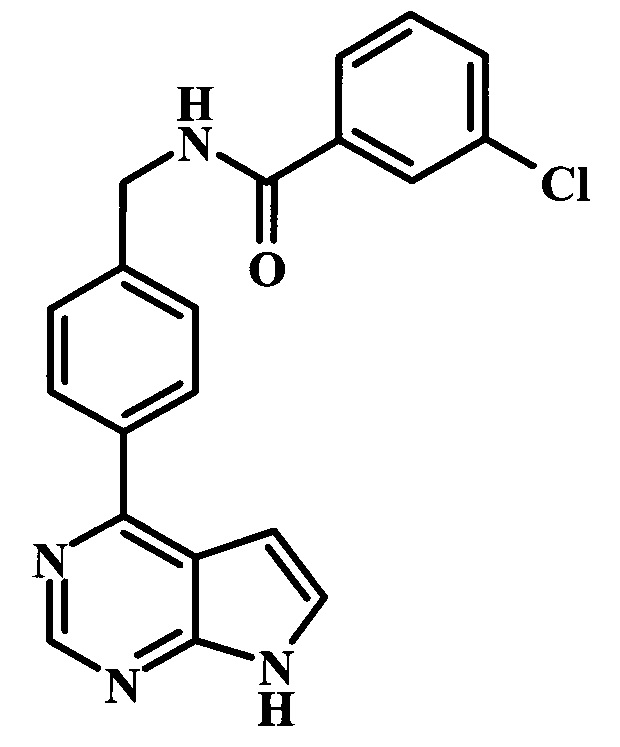
По аналогии со способом, описанным в примере 9, стадия 2, из 3-хлорбензойной кислоты (96.5 мг; 0,617 ммоль; Экв.: 1,00) получали указанное в заголовке соединение в виде твердого вещества (45 мг; 20% выход). LC/MS: m/z расч. для C20H15ClN4O ([M+H]+): 363,8 Фактич.: 363,0
Пример I-11
2-(3-Хлорфениламино)-N-[4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-бензил]-ацетамид
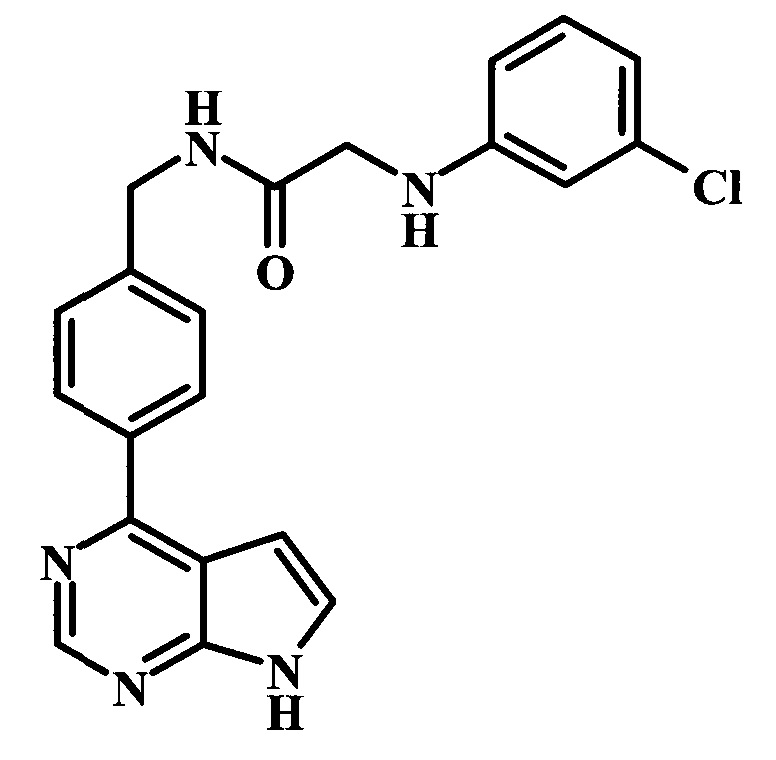
Стадия 1: Метиловый эфир (3-хлорфениламино)-уксусной кислоты
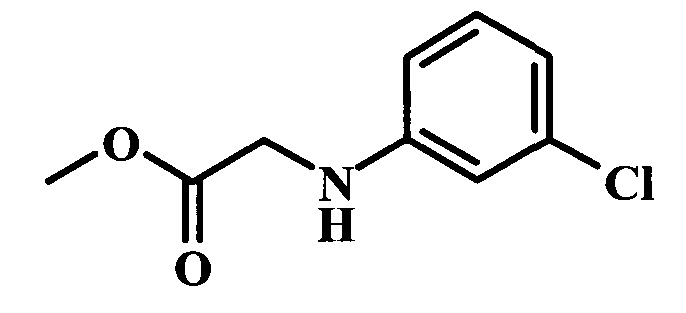
В круглодонной колбе на 250 мл метилбромацетат (1,66 г; 1 мл, 10,9 ммоль; Экв.: 1,00), 3-хлоранилин (1,66 г; 1,4 мл; 13,0 ммоль; Экв.: 1,2) и DIPEA (1,9 мл; 10,9 ммоль; Экв.: 1,00) соединяли с DMF (20 мл) с получением светло-желтого раствора. Реакционную смесь нагревали при 60°С в течение ночи. Реакционную смесь разбавляли с помощью EtOAc, затем промывали солевым раствором. Объединенные органические фазы высушивали над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении с получением указанного в заголовке соединения (2,1 г; выход 97%). LC/MS: m/z расч. для C9H10ClNO2 ([М+Н]+): 200,6 Фактич.: 200,0
Стадия 2: (3-Хлорфениламино)-уксусная кислота
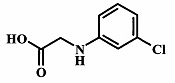
В сцинтилляционном флаконе на 20 мл метиловый эфир (3-хлорфениламино)-уксусной кислоты (500 мг; 2.5 ммоль; Экв.: 1,00) и NaOH (500 мг; 12,5 ммоль; Экв.: 4,99) в 5 мл Н2О соединяли с EtOH (8 мл) с получением светло-желтого раствора. Реакционную смесь нагревали при 60°С в течение 4 часов. Реакционную смесь разбавляли с помощью EtOAc и промывали с помощью 10% водной HCl. Объединенные органические фазы высушивали над безводным сульфатом натрия, затем выпаривали при пониженном давлении с получением указанного в заголовке соединения в виде коричневого твердого вещества. LC/MS: m/z расч. для C8H8ClNO2 ([М+Н]+): 186,0 Фактич.: 186,0
Стадия 3: 2-(3-Хлорфениламино)-N-[4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-бензил]-ацетамид
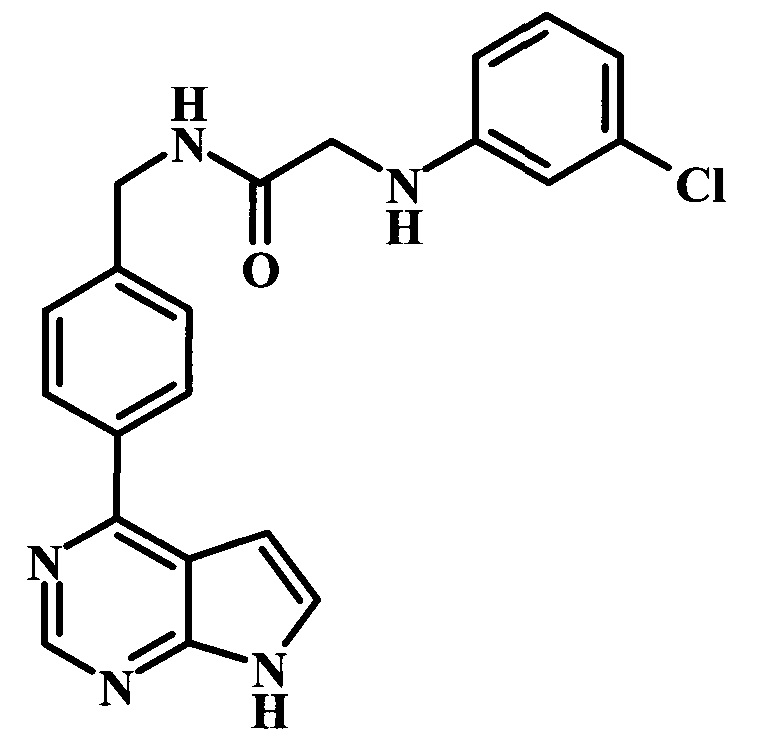
По аналогии со способом, описанным в примере 9, стадия 2, из трет-бутил-4-(7Н-пирроло[2,3-d]пиримидин-4-ил)бензилкарбамата (345 мг; 1,06 ммоль; Экв.: 1,00), 2-(3-хлорфениламино)уксусной кислоты (217 мг; 1,17 ммоль; Экв.: 1,1), DIPEA (740 мг; 1 мл; 5,73 ммоль; Экв.: 5,38) и HATU (445 мг; 1,17 ммоль; Экв.: 1,1) получали указанное в заголовке соединение в виде твердого вещества (71 мг; выход 17%). LC/MS: m/z расч. для C21H18ClN5O ([М+Н]+): 392,8 Фактич.: 392,1
Пример I-12
4-трет-Бутил-N-[2-фтор-4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-бензил]-бензамид
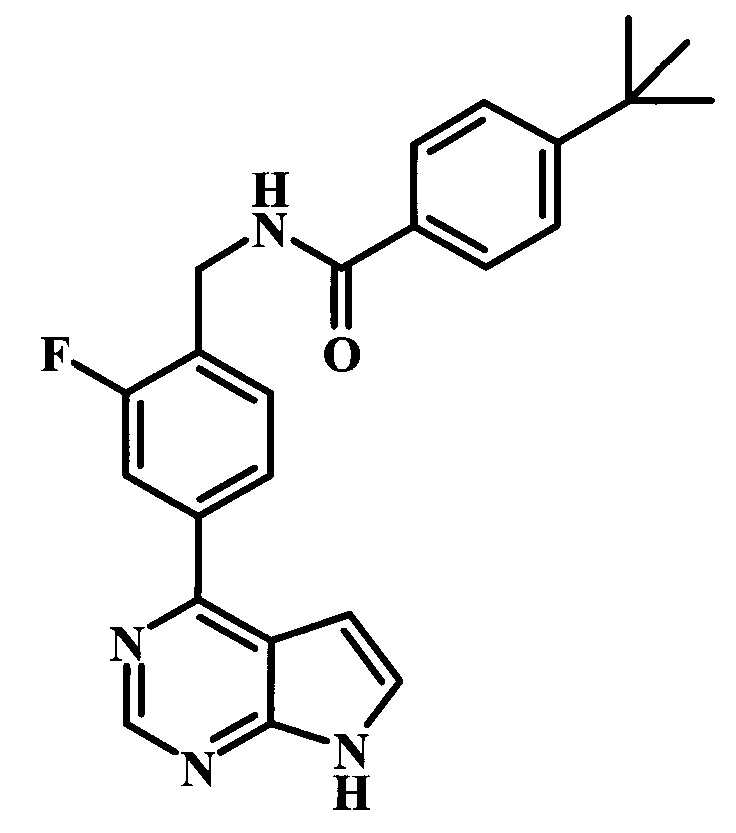
Стадия 1: трет-Бутиловый эфир [2-фтор-4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-бензил]-карбаминовой кислоты
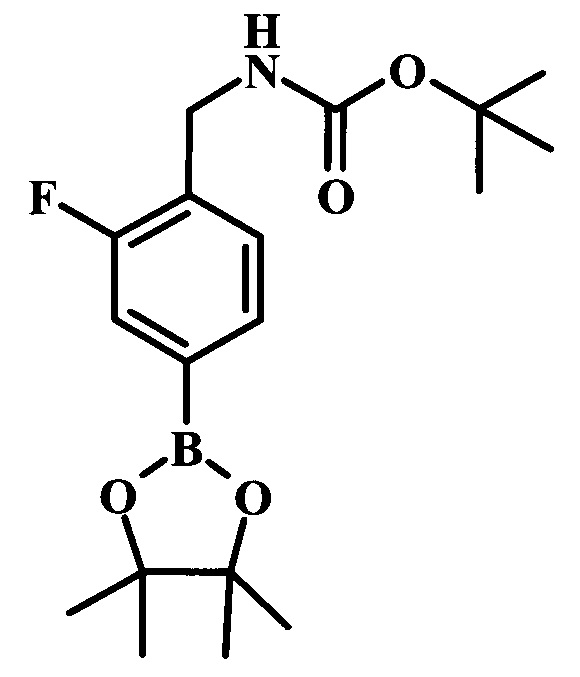
В трубке под давлением трет-бутил-4-бром-2-фторбензилкарбамат (5 г; 16,4 ммоль), бис(пинаколато)диборон (6,26 г; 24,7 ммоль) и ацетат калия (4,84 г; 49,3 ммоль) соединяли с NMP (75,0 мл) с получением светло-желтого раствора. Реакционную смесь дегазировали в атмосфере азота в течение 10 минут. Добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II) (722 мг; 0,986 ммоль). Реакционную смесь нагревали при 100°С в течение 20 часов. Реакционную смесь гасили водой и экстрагировали с помощью DCM (3×100 мл). Объединенные органические слои промывали водой, солевым раствором, высушивали над Na2SO4, фильтровали и концентрировали. Неочищенное вещество очищали флэш-хроматографией (силикагель, 120 г; 0%-30% этилацетат в гексане). трет-Бутиловый эфир [2-фтор-4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-бензил]-карбаминовой кислоты (5,8 г; 100%) получали в виде желтого масла.
Стадия 2: трет-Бутиловый эфир [2-фтор-4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-бензил]-карбаминовой кислоты
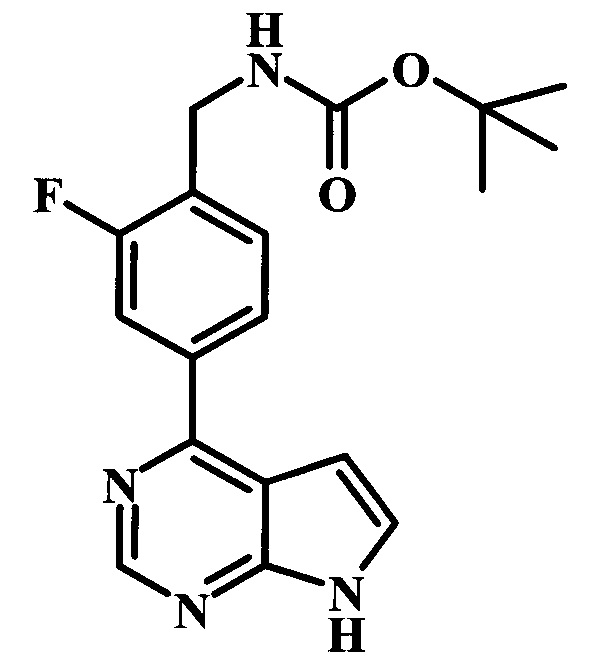
В герметизируемом сосуде на 10 мл для микроволнового облучения 4-хлор-7-тозил-7Н-пирроло[2,3-d]пиримидин (157 мг; 0,511 ммоль; Экв.: 1,00), трет-бутиловый эфир [2-фтор-4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-бензил]-карбаминовой кислоты (197 мг; 0,562 ммоль; Экв.: 1,1) и карбонат калия (282 мг; 2,04 ммоль; Экв.: 4,00) в 2 мл воды соединяли с DME (4 мл). Добавляли Pd(Ph3P)4 (59,0 мг; 0,051 ммоль; Экв.: 0,1) и эту реакционную смесь нагревали при 160°С в течение 60 минут. Полученный раствор разбавляли с помощью EtOAc и промывали солевым раствором. Объединенные органические фазы высушивали над безводным сульфатом натрия, затем растворитель удаляли при пониженном давлении. Неочищенное вещество очищали хроматографией на колонке (силикагель, 5-70% этилацетат в гексане). Указанное в заголовке соединение получали в виде твердого вещества (75 мг; 43% выход). LC/MS: m/z расч. для C18H19FN4O2 ([М+Н]+): 343,3 Фактич.: 343,3
Стадия 3: 4-трет-Бутил-N-[2-фтор-4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-бензил]-бензамид
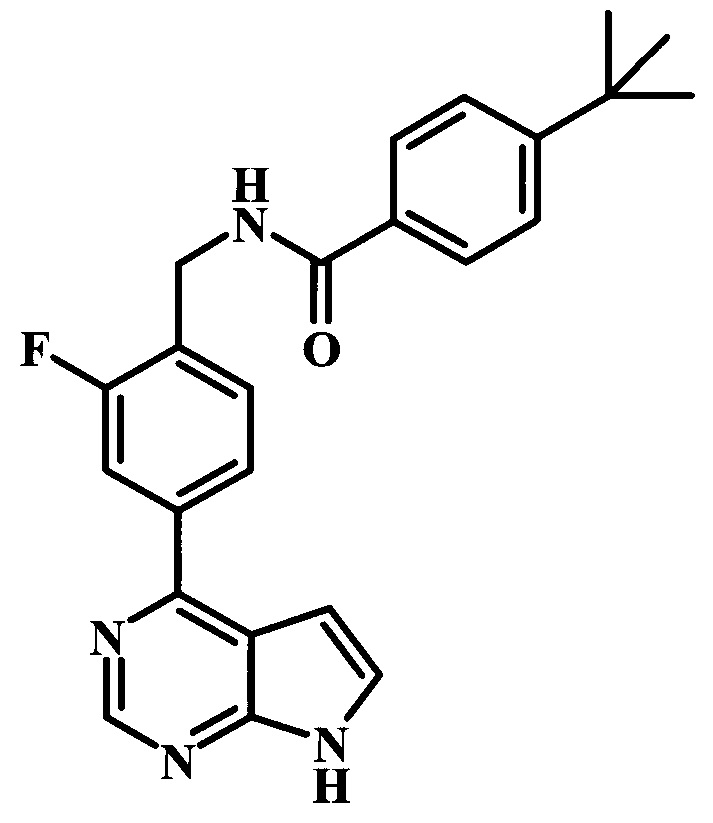
В сцинтилляционном флаконе на 20 мл трет-бутиловый эфир [2-фтор-4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-бензил]-карбаминовой кислоты (70 мг; 0,134 ммоль; Экв.: 1,00) растворяли в 2 мл DCM и 2 мл TFA. Реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Растворитель удаляли при пониженном давлении. Неочищенное вещество растворяли в DMF (4 мл). Вносили 4-трет-бутилбензойную кислоту (26 мг; 0,148 ммоль; Экв.: 1,1), DIPEA (0,047 мл; 0,269 ммоль; Экв.: 4,00) и HATU (56 мг; 0,148 ммоль; Экв.: 1,1). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли с помощью EtOAc и воды, затем перемешивали при комнатной температуре в течение 30 мин. Полученный раствор промывали солевым раствором. Объединенные органические фазы высушивали над безводным сульфатом натрия, затем растворитель удаляли при пониженном давлении. Неочищенное вещество обрабатывали с помощью DCM и это твердое вещество фильтровали с получением указанного в заголовке соединения в виде твердого вещества (32 мг; выход 59%). LC/MS: m/z расч. для C24H23FN4O ([M+H]+): 403,4 Фактич.: 403,2
Пример I-13
трет-Бутиловый эфир 4-{4-[(4-трет-бутилбензоиламино)-метил]-3-фторфенил}-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты
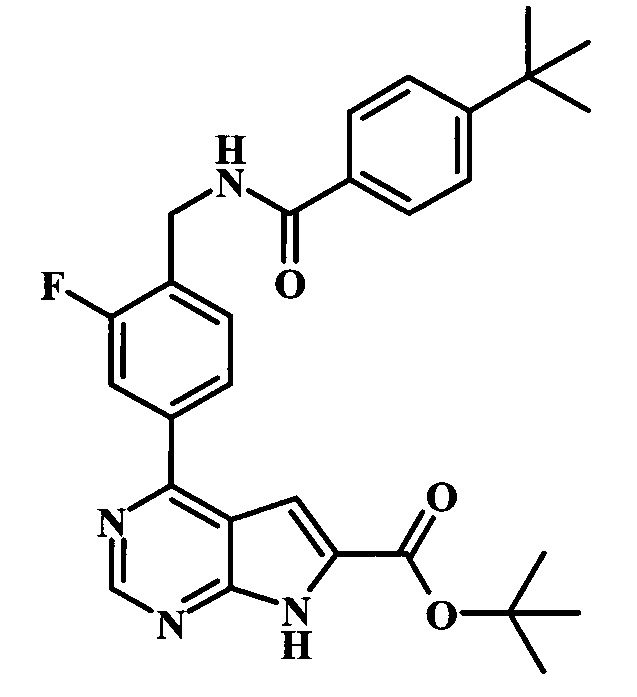
Стадия 1: трет-Бутиловый эфир 4-хлор-5-гидрокси-6,7-дигидро-5Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты
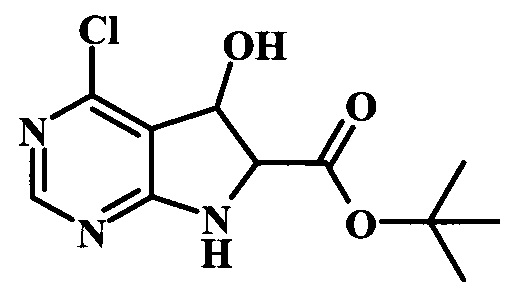
К суспензии 4,6-дихлорпиримидин-6-карбальдегида (2 г; 11,3 ммоль; Экв.: 1,00) в EtOH (50 мл) добавляли трет-бутил-2-аминоацетат (1,48 г; 11,3 ммоль; Экв.: 1,00), а затем триэтиламин (2,86 г; 3,94 мл; 28,3 ммоль; Экв.: 2,5) и перемешивали при КТ в течение 48 ч. Растворитель удаляли при пониженном давлении. Неочищенное вещество разбавляли дихлорметаном и промывали водой. Объединенные органические фазы высушивали над безводным сульфатом натрия и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали хроматографией на колонке (силикагель, 10-90% этилацетат в гексане) с получением указанного в заголовке соединения (634 мг; выход 21%) в виде белого твердого вещества. LC/MS: m/z расч. для C11H14ClN3O ([М+Н]+): 272,7 Фактич.: 272,1
Стадия 2: трет-Бутиловый эфир 4-хлор-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты
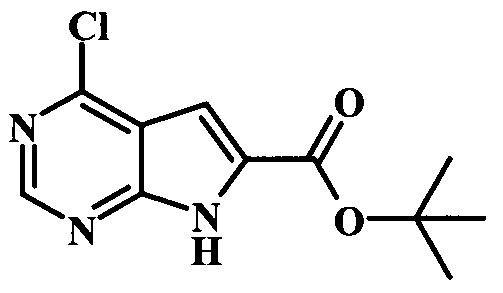
К раствору трет-бутил-4-хлор-5-гидрокси-6,7-дигидро-5Н-пирроло[2,3-d]пиримидин-6-карбоксилата (634 мг; 2,33 ммоль; Экв.: 1,00) в DMF (10 мл) добавляли гидрид натрия (93,3 мг; 2,33 ммоль; Экв.: 1,00) при 0°С и затем перемешивали при КТ в течение 1 ч. Реакционную смесь гасили водой, затем промывали с помощью NH4Cl и солевым раствором. Объединенные органические слои высушивали над безводным сульфатом натрия, затем растворитель удаляли при пониженном давлении. Неочищенное вещество очищали хроматографией на колонке (силикагель, 5-35% этилацетат в гексане) с получением указанного в заголовке соединения (417 мг; выход 70,4%) в виде белого твердого вещества. LC/MS: m/z расч. для C11H12ClN3O ([М+Н]+): 254,6 Фактич.: 254,1
Стадия 3: N-(4-Бром-2-фторбензил)-4-трет-бутил-бензамид
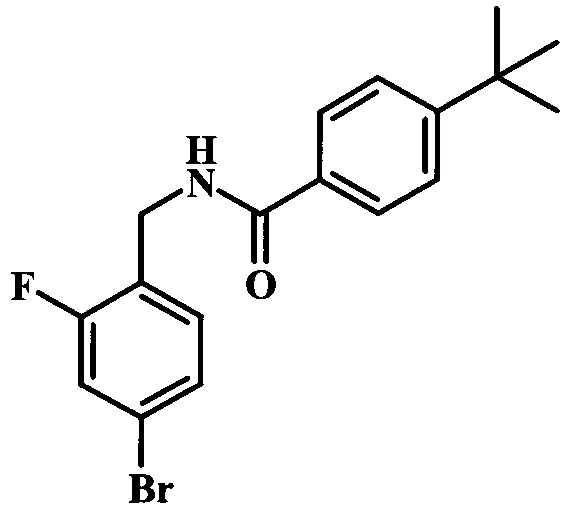
К раствору (4-бром-2-фторфенил)метанамина (1,5 г; 7,35 ммоль; Экв.: 1,00) в DCM (25 мл), охлажденному до 0°С, добавляли раствор 4-трет-бутилбензоилхлорида (1,45 г; 7,35 ммоль; Экв.: 1,00), и триэтиламина (1,49 г; 2,05 мл; 14,7 ммоль; Экв.: 2,00) в DCM (5 мл). Реакционную смесь доводили до КТ в течение 1 ч. Реакционную смесь очищали хроматографией на колонке (силикагель, 5-40% этилацетат в гексане) с получением указанного в заголовке соединения (2,59 г; 7,11 ммоль; выход 96,7%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ ppm 7,74 (d, J=8,0 Гц, 2H), 7,48 (d, J=8,5 Гц, 2H), 7,35 (t, J=7,5 Гц, 1H), 6,48 (s, 1H), 4,67 (d, J=5,7 Гц, 2H), 1,36 (s, 9H).
Стадия 4: 4-трет-Бутил-N-[2-фтор-4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-бензил]-бензамид
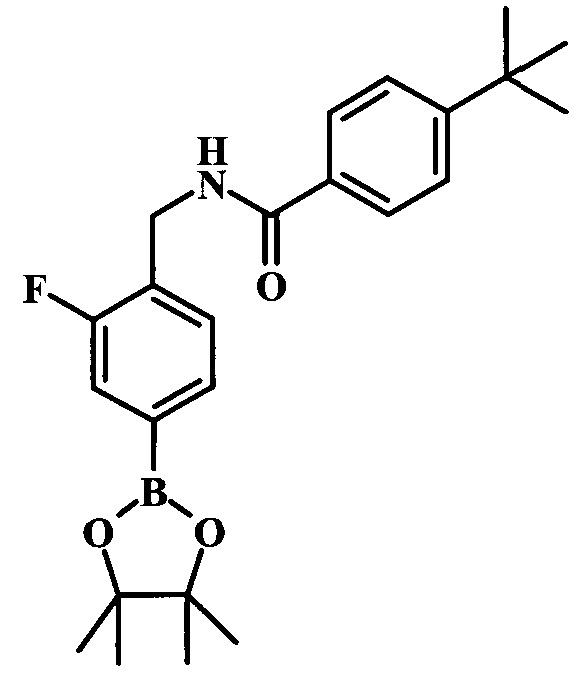
К смеси N-(4-бром-2-фторбензил)-4-трет-бутилбензамида (600 мг; 1,65 ммоль; Экв.: 1,00), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (627 мг; 2,47 ммоль; Экв.: 1,5), ацетата калия (485 мг; 4,94 ммоль; Экв.: 3) и PdCl2(dppf)-CH2Cl2 (121 мг; 165 мкмоль; Экв.: 0,1) при перемешивании в атмосфере N2 добавляли NMP (12 мл) и нагревали до 100°С в течение 16 ч. Реакционную смесь разбавляли с помощью EtOAc и промывали водой и солевым раствором. Объединенные органические слои высушивали над безводным сульфатом натрия и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали хроматографией на колонке (силикагель, 15-60% этилацетат в гексане) с получением указанного в заголовке соединения (545 мг; выход 80%) в виде грязно-белого твердого вещества. 1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ ppm 7,74 (d, J=8,3 Гц, 2H), 7,75 (d, J=7,4 Гц, 1H), 7,51 (d, J=10,4 Гц, 1H), 7,47 (d, J=8,4 Гц, 2H), 7,44 (t, J=7,0 Гц, 1H), 6,46 (s, 1H), 4,74 (d, J=5,9 Гц, 2Н), 1,37 (s, 12H), 1,36 (s, 9H).
Стадия 5: трет-Бутиловый эфир 4-{4-[(4-трет-бутилбензоиламино)-метил]-3-фторфенил}-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты
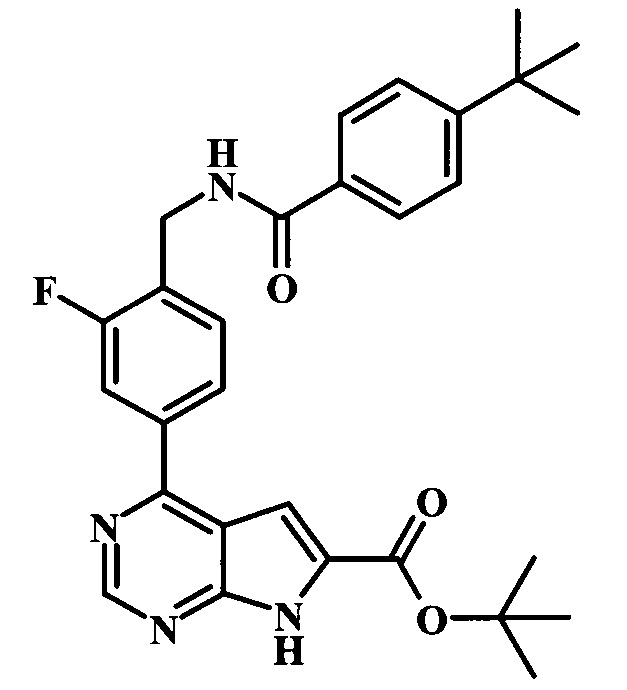
К смеси трет-бутил-4-хлор-7Н-пирроло[2,3-d]пиримидин-6-карбоксилата (100 мг; 0,394 ммоль; Экв.: 1,00), 4-трет-бутил-N-(2-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензил)бензамида (162 мг; 0,394 ммоль; Экв.: 1,00), тетракис(трифенилфосфин)палладия(0) (46 мг; 0,04 ммоль; Экв.: 0,1) и карбоната калия (163 мг; 1,18 ммоль; Экв.: 3,00) добавляли DME (1 мл) и воду (500 мкл) и нагревали микроволновым облучением при 150°С в течение 30 мин. Реакционную смесь фильтровали через слой Celite и разбавляли дихлорметаном. Полученный раствор очищали хроматографией на колонке (силикагель, 15-60% этилацетат в гексане, а затем 0-30% [10% метанол/дихлорметан] в дихлорметане) с получением указанного в заголовке соединения (80 мг; выход 40%) в виде грязно-белого твердого вещества. 1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ ppm 10,93 (s, 1Н), 9,18 (s, 1H), 7,94 (t, J=9,3 Гц, 2H), 7,80 (d, J=8,5 Гц, 2H), 7,68 (t, J=7,8 Гц, 1H), 7,50 (d, J=8,4 Гц, 1H), 7,39 (s, 1H), 6,67 (t, J=5,9 Гц, 1H), 4,83 (d, J=5,8 Гц, 2H), 1,69 (s, 9H), 1,37 (s, 9H); LC/MS: m/z расч. для C29H31FN4O3 ([М+Н]+): 503,5 Фактич.: 503,3
Пример I-14
4-{4-[(4-трет-Бутилбензоиламино)-метил]-3-фторфенил}-7Н-пирроло[2,3-d]пиримидин-6-карбоновая кислота
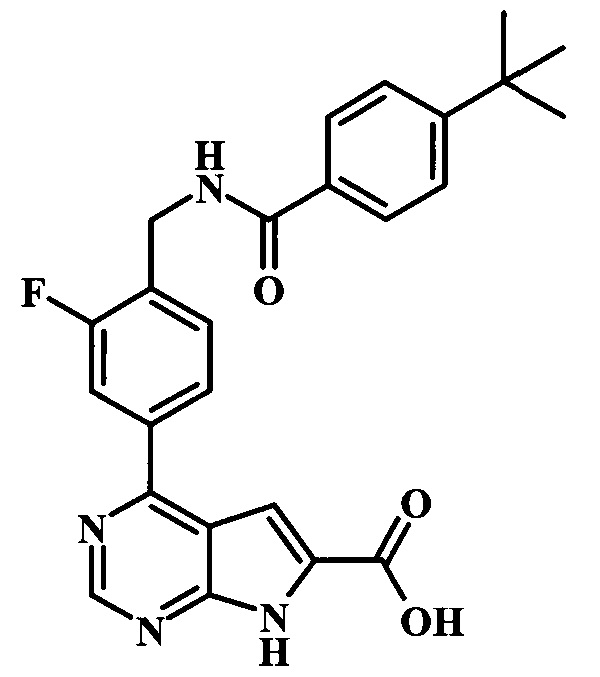
К раствору трет-бутилового эфира 4-{4-[(4-трет-бутилбензоиламино)-метил]-3-фторфенил}-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты (55 мг; 0,109 ммоль; Экв.: 1,00) в дихлорметане (1 мл) добавляли трифторуксусную кислоту (843 мкл, 10,9 ммоль; Экв.: 100) и перемешивали при КТ в течение 2 ч. Растворитель концентрировали в вакууме из метанола (3×) с получением указанного в заголовке соединения (43 мг; выход 88%) в виде светло-коричневого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ ppm 13,05 (s, 1H), 9,10 (t, J=5,9 Гц, 1H), 8,98 (s, 1H), 8,06 (d, J=8,2 Гц, 1H), 7,97 (d, J=11,2 Гц, 1H), 7,89 (d, J=8,4 Гц, 2H), 7,59 (t, J=7,6 Гц, 1H), 7,54 (m, 3H), 4,64 (d, J=5,6 Гц, 2H), 1,33 (s, 9H); LC/MS: m/z расч. для C25H23FN4O3 ([M+H]+): 447,4 Фактич.: 447,2
Пример I-15
4-трет-Бутил-N-(2-фтор-4-(6-(морфолин-4-карбонил)-7Н-пирроло[2,3-d]пиримидин-4-ил)бензил)бензамид
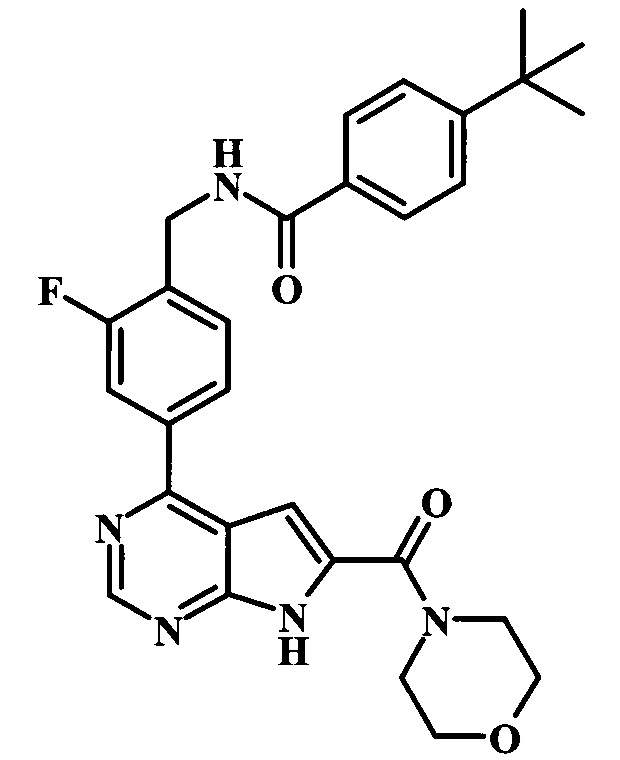
К раствору 4-{4-[(4-трет-бутилбензоиламино)-метил]-3-фторфенил}-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты (50 мг; 0,112 ммоль; Экв.: 1,00), HBTU (42,5 мг; 0,112 ммоль; Экв.: 1,00) и DIPEA (59 мкл; 0,336 ммоль; Экв.: 3) в DMF (1,5 мл) добавляли морфолин (19,5 мг; 0,224 ммоль; Экв.: 2,00) и перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь разбавляли этилацетатом и промывали солевым раствором. Объединенные органические фазы высушивали над безводным сульфатом натрия, затем растворитель удаляли при пониженном давлении. Неочищенное вещество очищали хроматографией на колонке (силикагель, 0-5% метанол в дихлорметане, а затем 50-100% этилацетат в гексане) с получением указанного в заголовке соединения (26 мг; выход 45%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ ppm 8,96 (s, 1H), 7,74 (m, 2H), 7,71 (d, J=8,4 Гц, 2H), 7,58 (t, J=8,2 Гц, 1H), 7,41 (d, J=8,4 Гц, 1H), 6,91 (s, 1H), 4,71 (s, 1H), 3,81 (br. s, 4H), 3,72 (br. s, 4H), 1,27 (s, 9H).
Пример I-16
Диметиламид 4-{4-[(4-трет-бутилбензоиламино)-метил]-3-фторфенил}-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты
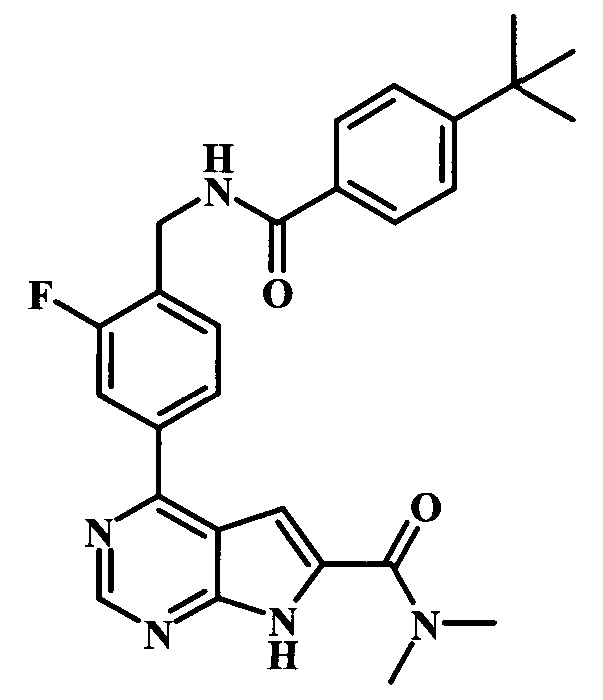
К раствору 4-{4-[(4-трет-бутилбензоиламино)-метил]-3-фторфенил}-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты (50 мг; 0,112 ммоль; Экв.: 1,00), HBTU (42,5 мг; 0,112 ммоль; Экв.: 1,00) и DIPEA (59 мкл; 0,336 ммоль; Экв.: 3,00) в DMF (1 мл) добавляли диметиламин в THF (112 мкл; 0,224 ммоль; Экв.: 2,00) и перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь разбавляли этилацетатом и промывали солевым раствором. Объединенные органические фазы высушивали над безводным сульфатом натрия, затем растворитель удаляли при пониженном давлении. Неочищенное вещество очищали хроматографией на колонке (силикагель, градиент 50-100% этилацетат в гексане) с получением указанного в заголовке соединения (18 мг; выход 34%) в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d) δ ppm 12,78 (s, 1H), 9,10 (t, J=5,8 Гц, 1H), 8,92 (s, 1H), 8,04 (d, J=8,0 Гц, 1H), 7,96 (d, J=11,6 Гц, 1H), 7,89 (d, J=8,6 Гц, 2H), 7,56 (t, J=7,9 Гц, 1H), 7,53 (d, J=8,3 Гц, 2H), 7,23 (s, 1H), 4,62 (s, 2H), 3,26 (br. s, 3H), 3,06 (br. s, 3H), 1,32 (s, 9H); LC/MS: m/z расч. для C27H28FN5O2 ([М+Н]+): 474,5 Фактич.: 474,3
Пример I-17
Метиламид 4-{4-[(4-трет-бутилбензоиламино)-метил]-3-фторфенил}-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты
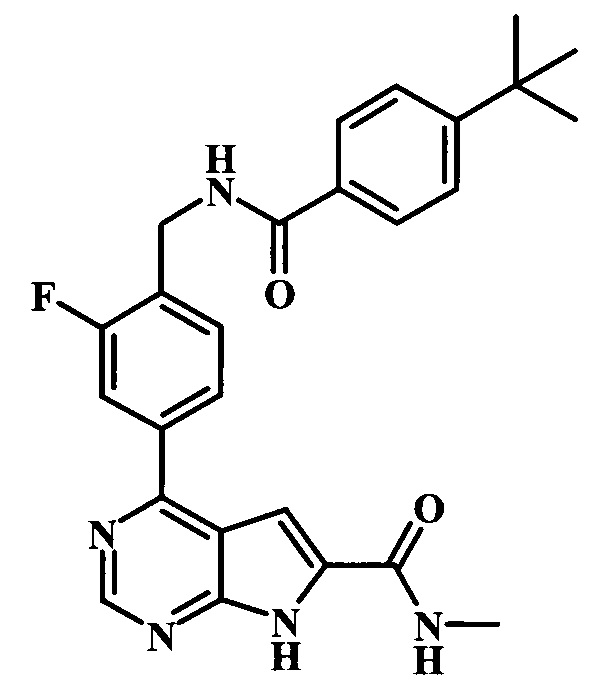
К раствору 4-{4-[(4-трет-бутилбензоиламино)-метил]-3-фторфенил}-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты (50 мг; 0,112 ммоль; Экв.: 1,00), HBTU (42,5 мг; 0,112 ммоль; Экв.: 1,00) и DIPEA (43 мг; 59 мкл; 0,336 ммоль; Экв.: 3,00) в DMF (1,00 мл) добавляли метанамин в THF (112 мкл; 0,224 ммоль; Экв.: 2,00) и перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь разбавляли этилацетатом и промывали солевым раствором. Объединенные органические фазы высушивали над безводным сульфатом натрия, затем растворитель удаляли при пониженном давлении. Неочищенное вещество очищали хроматографией на колонке (силикагель, 50-100% этилацетат в гексане) с получением указанного в заголовке соединения (25 мг; выход 48,6%) в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ ppm 12,82 (s, 1H), 9,11 (t, J=5,7 Гц, 1H), 8,92 (s, 1H), 8,70 (d, J=4,8 Гц, 1H), 8,01 (d, J=7,8 Гц, 1H), 7,93 (d, J=11,6 Гц, 1H), 7,89 (d, J=8,7 Гц, 2H), 7,61 (s, 1H), 7,59 (t, J=8,1 Гц, 1H), 7,53 (d, J=8,4 Гц, 2H), 4,64 (d, J=5,4 Гц, 2H), 2,84 (d, J=4,5 Гц, 3H), 1,32 (s, 9H); LC/MS: m/z расч. для C26H26FN5O2 ([М+Н]+): 460,5 Фактич.: 460,3
Пример I-18
(2-Гидроксиэтил)-амид 4-{4-[(4-трет-бутилбензоиламино)-метил]-3-фторфенил}-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты
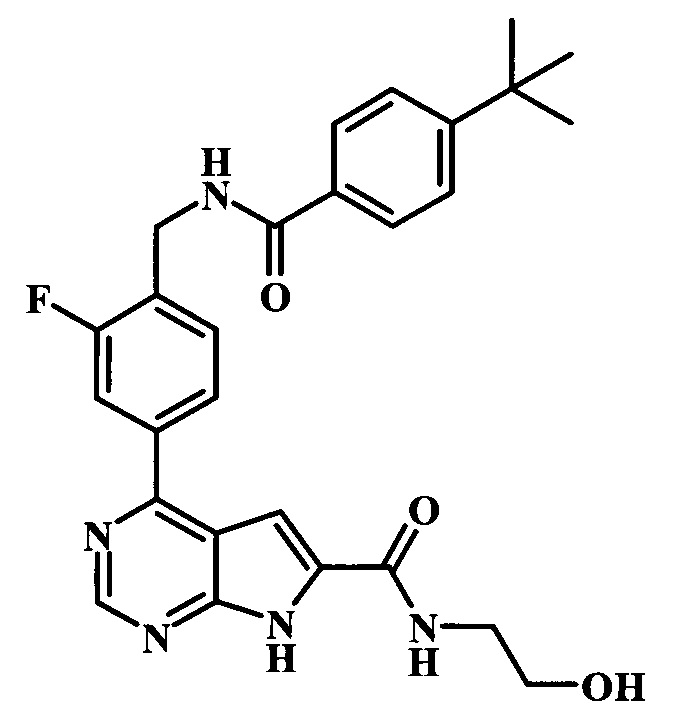
К раствору 4-{4-[(4-трет-бутилбензоиламино)-метил]-3-фторфенил}-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты (50 мг; 0,112 ммоль; Экв.: 1,00), HBTU (42,5 мг; 0,112 ммоль; Экв.: 1,00) и DIPEA (59 мкл; 0,336 ммоль; Экв.: 3,00) в DMF (1,00 мл) добавляли 2-аминоэтанол (14 мг; 0,224 ммоль; Экв.: 2,00) и перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь разбавляли этилацетатом и промывали солевым раствором. Объединенные органические фазы высушивали над безводным сульфатом натрия, затем растворитель удаляли при пониженном давлении. Неочищенное вещество очищали хроматографией на колонке (силикагель, 50-100% этилацетат в гексане, а затем 0-10% метанол в дихлорметане, содержащем NH4OH) с получением указанного в заголовке соединения (16 мг; выход 29%) в виде грязно-белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ ppm 12,80 (s, 1H), 9,10 (t, J=5,6 Гц, 1H), 8,92 (s, 1H), 8,73 (t, J=6,4 Гц, 1H), 8,03 (d, J=8,6 Гц, 1H), 7,95 (d, J=11,0 Гц, 1H), 7,89 (d, J=8,4 Гц, 2H), 7,69 (s, 1H), 7,59 (t, J=7,5 Гц, 1H), 7,53 (d, J=8,2 Гц, 2H), 4,64 (d, J=6,0 Гц, 2H), 3,55 (t, J=5,2 Гц, 2H), 3,38 (m, 2H), 1,32 (s, 9H); LC/MS: m/z расч. для C27H28FN5O3 ([М+Н]+): 490,5 Фактич.: 490,4
Пример I-19
(2-Диметиламиноэтил)-амид 4-{4-[(4-трет-бутилбензоиламино)-метил]-3-фторфенил}-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты
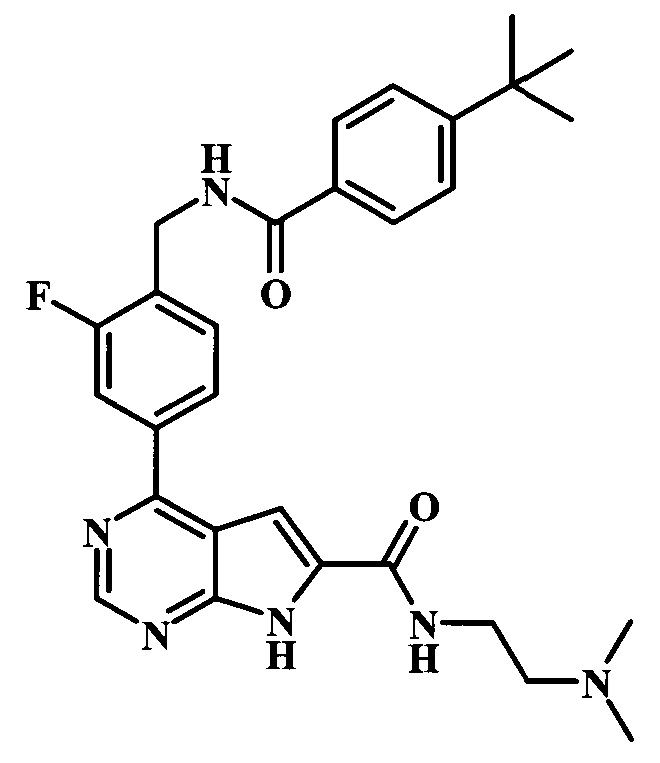
К раствору 4-{4-[(4-трет-бутилбензоиламино)-метил]-3-фторфенил}-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты (50 мг; 0,112 ммоль; Экв.: 1,00), N1,N1-диметилэтан-1,2-диамин (10 мг; 0,112 ммоль; Экв.: 1,00) и DIPEA (49 мкл; 0,280 ммоль; Экв.: 2,5) в DMF (1,00 мл), охлажденному до 0°С, добавляли циклический ангидрид 1-пропанфософоновой кислоты (80 мкл; 0,134 ммоль; Экв.: 1,2) и доводили до КТ в течение 4 ч. Реакционную смесь разбавляли этилацетатом и промывали солевым раствором. Объединенные органические фазы высушивали над безводным сульфатом натрия, затем растворитель удаляли при пониженном давлении. Неочищенное вещество очищали хроматографией на колонке (0-15% метанол в дихлорметане против NH4OH) с получением указанного в заголовке соединения (25 мг; выход 43%) в виде грязно-белого твердого вещества. 1H ЯМР (400 МГц, МЕТАНОЛ-d4) δ ppm 8,93 (s, 1H), 7,97 (d, J=8,1 Гц, 1H), 7,91 (d, J=11,0 Гц, 1H), 7,86 (d, J=8,5 Гц, 2H), 7,66 (t, J=7,8 Гц, 1H), 7,57 (s, 1H), 7,56 (d, J=8,5 Гц, 2H), 4,76 (s, 2H), 3,75 (t, J=6,1 Гц, 2H), 3,22 (m, 2H), 2,84 (s, 6Н), 1,38 (s, 9H); LC/MS: m/z расч. для C29H33FN6O2 ([M+H]+): 517,6 Фактич.: 517,4
Пример I-20
4-трет-Бутил-N-{1-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-пиперидин-4-илметил}-бензамид
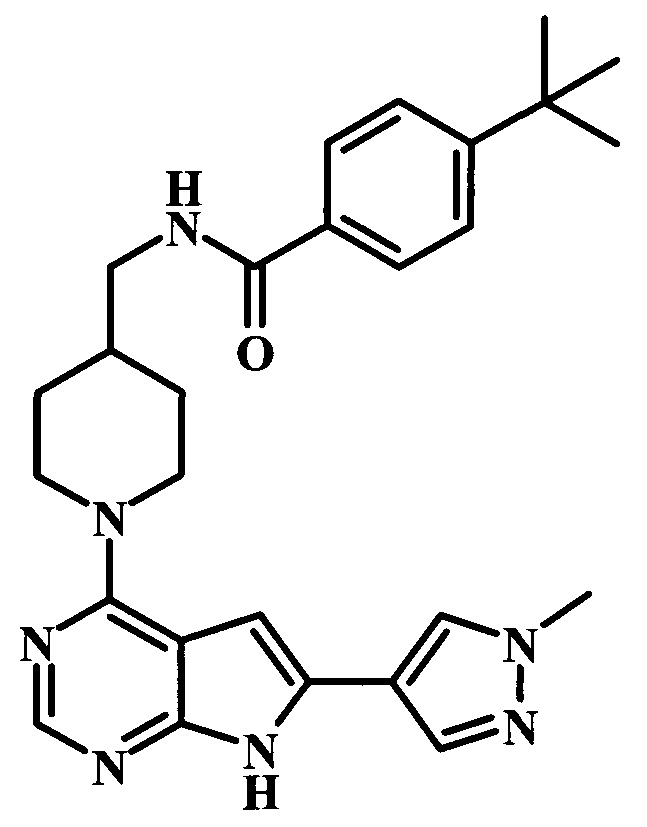
Стадия 1: трет-Бутиловый эфир 4-[(4-трет-бутилбензоиламино)-метил]-пиперидин-1-карбоновой кислоты
К раствору трет-бутил-4-(аминометил)пиперидин-1-карбоксилата (1 г; 4,67 ммоль; Экв.: 1,00) в DCM (16,7 мл), охлажденному до 0°С, добавляли раствор 4-трет-бутилбензоилхлорида (918 мг; 4,67 ммоль; Экв.: 1,00), триэтиламина (361 мкл; 4,67 ммоль; Экв.: 1,00) в DCM (5 мл). Реакционную смесь доводили до КТ в течение 1 ч. Реакционную смесь очищали хроматографией на колонке (30-70% этилацетат в гексане) с получением указанного в заголовке соединения (1,75 г; 4,67 ммоль; 100% выход) в виде бесцветного вязкого масла. 1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ ppm 7,73 (d, J=8,5 Гц, 2H), 7,48 (d, J=8,1 Гц, 2H), 6,21 (s, 1H), 4,15 (d, J=12,9 Гц, 2H), 3,39 (t, J=6,0 Гц, 2H), 2,72 (t, J=14,7 Гц, 2H), 1,83 (m, 1H), 1,76 (d, J=14,3 Гц, 2H), 1,48 (s, 9H), 1,36 (s, 9H).
Стадия 2: 4-трет-Бутил-N-пиперидин-4-илметилбензамид
К раствору трет-бутилового эфира 4-[(4-трет-бутил-бензоиламино)-метил]-пиперидин-1-карбоновой кислоты (1,75 г; 4,67 ммоль; Экв.: 1,00) в DCM (35 мл) добавляли трифторуксусную кислоту (7,2 мл; 93,5 ммоль; Экв.: 20) и перемешивали при КТ в течение 4 ч. Растворитель удаляли при пониженном давлении и высушивали в вакууме с получением указанного в заголовке соединения (2,92 г; выход 124%) в виде вязкого бесцветного масла. 1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ ppm 8,72 (br. s, 3H), 7,71 (d, J=8,5 Гц, 2H), 7,49 (d, J=8,5 Гц, 2H), 6,81 (t, J=6,7 Гц, 1H), 3,56 (d, J=12,8 Гц, 2H), 3,46 (t, J=6,4 Гц, 2H), 3,01 (q, J=11,7 Гц, 2H), 2,02 (d, J=14,3 Гц, 2H), 1,69 (q, J=14,1 Гц, 2H), 1,36 (s, 9Н).
Стадия 3: N-[1-(7-Бензолсульфонил-6-иод-7Н-пирроло[2,3-d]пиримидин-4-ил)-пиперидин-4-илметил]-4-трет-бутил-бензамид
К суспензии 4-хлор-6-иод-7-(фенилсульфонил)-7Н-пирроло[2,3-d]пиримидина (200 мг; 0,477 ммоль; Экв.: 1,00) в EtOH (3,00 мл) добавляли 4-трет-бутил-N-пиперидин-4-илметилбензамид (185 мг; 477 мкмоль; Экв.: 1,00) и триэтиламин (332 мкл; 2,38 ммоль; Экв.: 5,00) и нагревали до 80°С в течение 2 ч. Реакционную смесь охлаждали до КТ. Образовавшейся осадок отфильтровывали. Фильтрат очищали хроматографией на колонке (силикагель, 20-80% этилацетат в гексане) с получением указанного в заголовке соединения (159 мг; выход 51%) в виде белого твердого вещества. 1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ ppm 8,37 (s, 1H), 8,25 (d, J=8,21 Гц, 2H), 7,72 (d, J=8,4 Гц, 2H), 7,63 (t, J=8,1 Гц, 1H), 7,54 (t, J=7,6 Гц, 2H), 7,47 (d, J=8,5 Гц, 2H), 6,96 (s, 1H), 6,28 (s, 1H), 4,61 (d, J=12,4 Гц, 2H), 3,40 (t, J=6,3 Гц, 2H), 3,09 (t, J=13,3 Гц, 2H), 2,03 (m, 1H), 1,91 (d, J=13,3 Гц, 2H), 1,36 (s, 9H).
Стадия 4: N-{1-[7-Бензолсульфонил-6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-пиперидин-4-илметил}-4-трет-бутил-бензамид
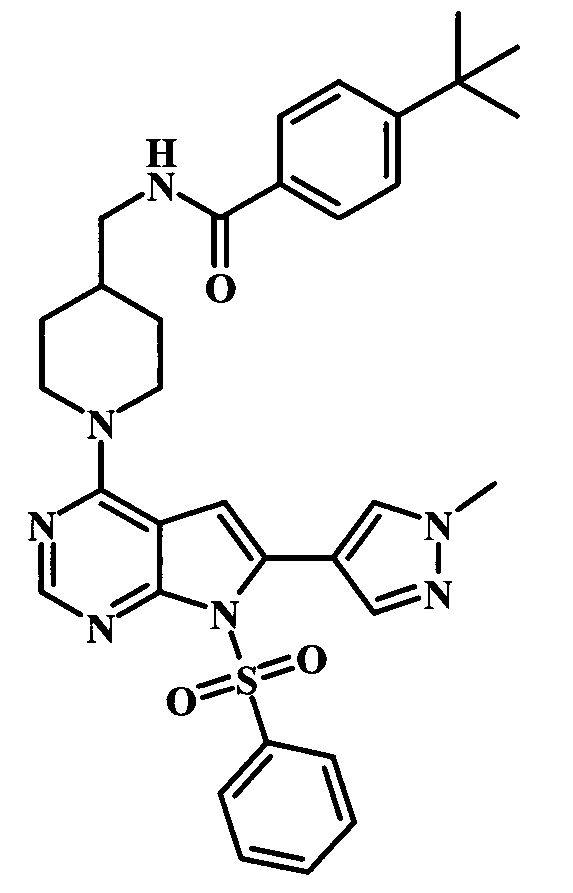
К смеси N-[1-(7-бензолсульфонил-6-иод-7Н-пирроло[2,3-d]пиримидин-4-ил)-пиперидин-4-илметил]-4-трет-бутил-бензамида (159 мг; 0,242 ммоль; Экв.: 1,00), 1-метил-1Н-пиразол-4-илбороновой кислоты (36,5 мг; 0,290 ммоль; Экв.: 1,2), Pd(PPh3)4 (28 мг; 0,024 ммоль; Экв.: 0,1) и карбоната калия (100 мг; 0,725 ммоль; Экв.: 3,00) добавляли смесь DME (1,29 мл)/ вода (322 мкл) и нагревали микроволновым облучением до 150°С в течение 1 ч. Реакционную смесь разбавляли с помощью DCM и промывали водой. Объединенные органические фазы высушивали над безводным сульфатом натрия, затем растворитель удаляли при пониженном давлении. Неочищенное вещество очищали хроматографией на колонке (силикагель, 30-100% этилацетат в гексане) с получением указанного в заголовке соединения (85 мг; выход 58%) в виде белого твердого вещества. LC/MS: m/z расч. для C33H37N7O3S ([М+Н]+): 612,7 Фактич.: 612,4
Стадия 5: 4-трет-Бутил-N-{1-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-пиперидин-4-илметил}-бензамид
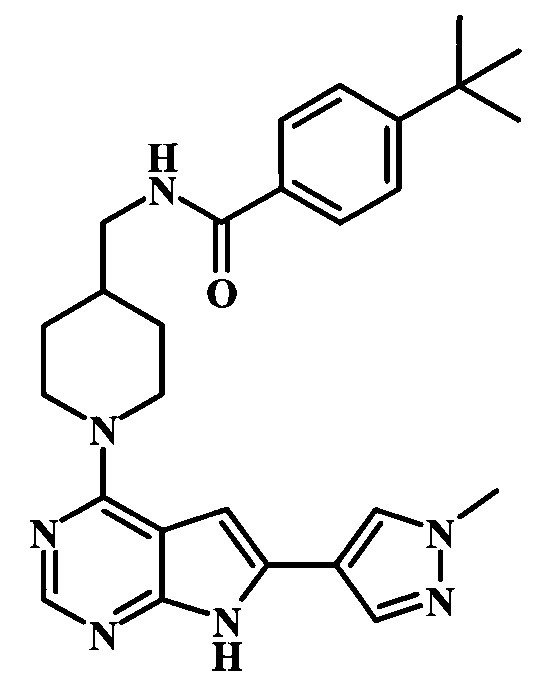
К раствору N-{1-[7-бензолсульфонил-6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-пиперидин-4-илметил}-4-трет-бутил-бензамида (85 мг; 0,139 ммоль; Экв.: 1,00) в THF (926 мкл)/ МеОН (463 мкл) добавляли карбонат цезия (136 мг; 0,417 ммоль; Экв.: 3,00) и перемешивали при КТ в течение 16 ч. Реакционную смесь очищали хроматографией на колонке (силикагель, 1-6% метанол в DCM, содержащем NH4OH) с получением указанного в заголовке соединения (45 мг; выход 68,7%) в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ ppm 11,95 (s, 1H), 8,45 (t, J=5,4 Гц, 1H), 8,11 (s, 1H), 7,93 (s, 1H), 7,80 (d, J=7,9 Гц, 2H), 7,49 (d, J=7,9 Гц, 2H), 6,75 (s, 1H), 4,71 (d, J=13,8 Гц, 2H), 3,88 (s, 3H), 3,20 (t, J=6,2 Гц, 2H), 3,06 (t, J=12,8 Гц, 2H), 1,97 (m, 1H), 1,82 (d, J=13,3 Гц, 2H), 1,31 (s, 9H), 1,23 (d, J=14,3 Гц, 2 H); LC/MS: m/z расч. для C27H33N7O ([М+Н]+): 472,6 Фактич.: 472,4
Пример I-21
4-трет-Бутил-N-{4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-бензамид
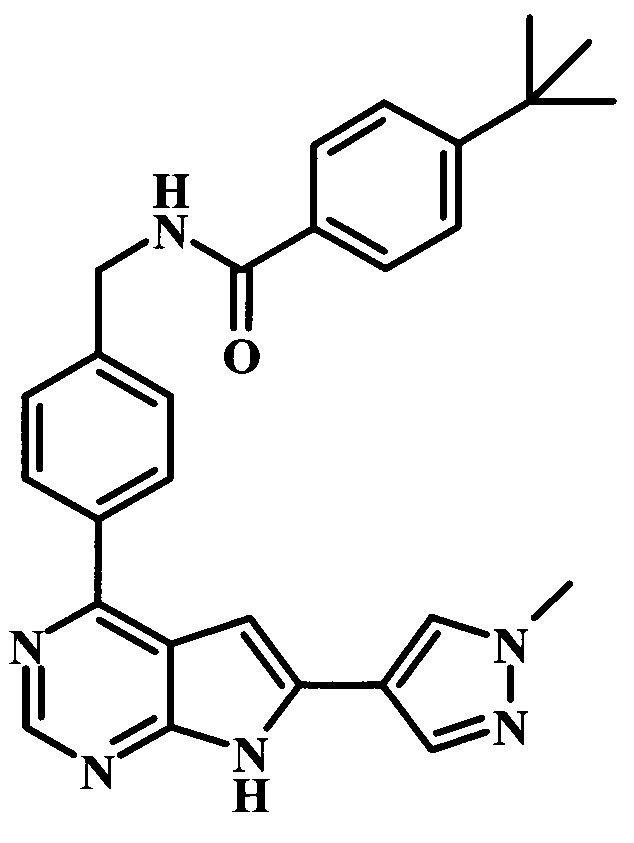
Стадия 1: трет-Бутиловый эфир 4-[7-(толуол-4-сульфонил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-карбаминовой кислоты
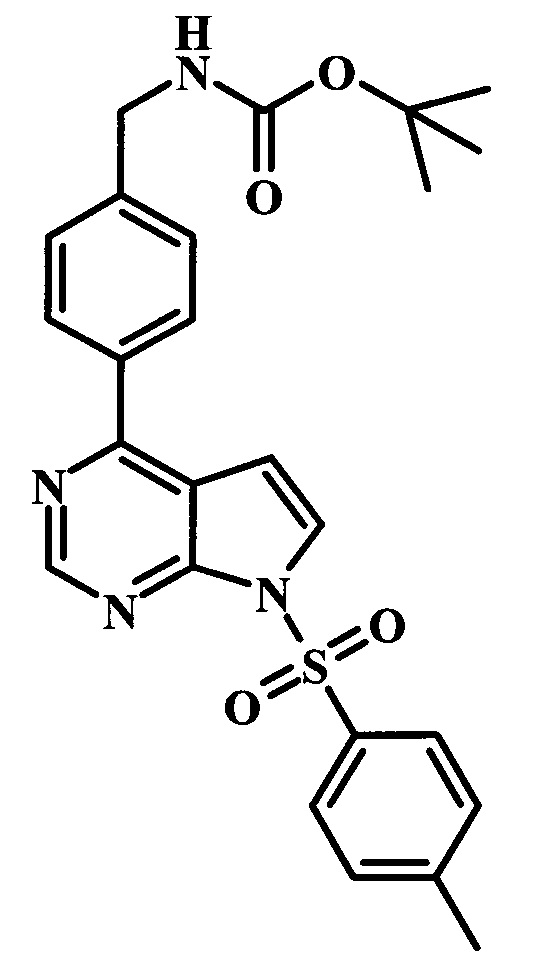
В герметизируемой трубке на 20 мл для микроволнового облучения 4-хлор-7-тозил-7Н-пирроло[2,3-d]пиримидин (1 г; 3,25 ммоль; Экв.: 1,00), 4-((трет-бутоксикарбониламино)метил)фенилбороновую кислоту (1,22 г; 4,87 ммоль; Экв.: 1,5) и карбонат калия (1,8 г; 13,0 ммоль; Экв.: 4,00) в 5 мл воды соединяли с DME (10 мл). Вносили Pd(PPh3)4 (375 мг; 0,325 ммоль; Экв.: 0,1). Реакционную смесь нагревали микроволновым облучением при 150°С в течение 30 мин. Полученный раствор промывали с помощью EtOAc и солевым раствором. Объединенные органические фазы высушивали над безводным сульфатом натрия, затем растворитель удаляли при пониженном давлении. Неочищенное вещество очищали хроматографией на колонке (силикагель, 5-70% этилацетат в гексане). Указанное в заголовке соединение получали в виде белого твердого вещества (700 мг; выход 45%). LC/MS: m/z расч. для C25H26N4O4S ([M+H]+): 479,5 Фактич.: 479,3
Стадия 2: трет-Бутиловый эфир 4-[6-бром-7-(толуол-4-сульфонил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-карбаминовой кислоты
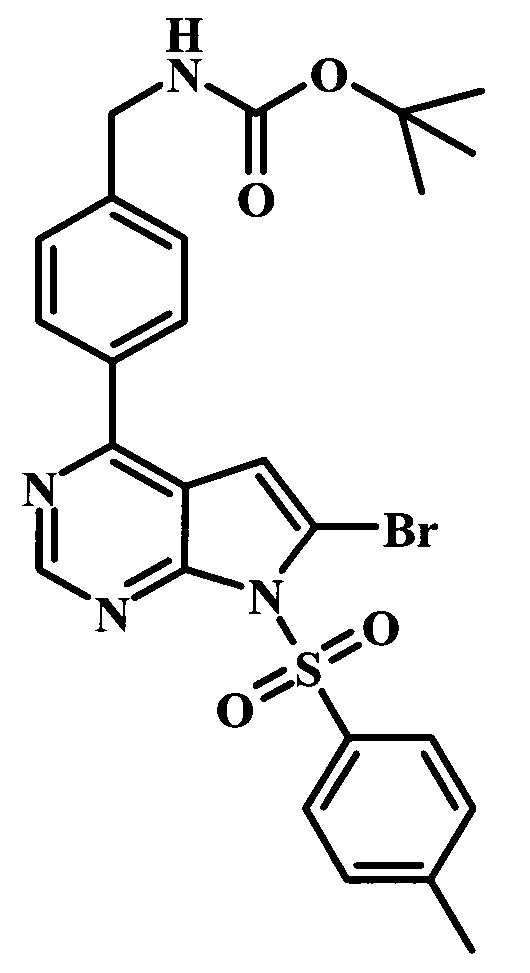
В круглодонной колбе на 100 мл трет-бутиловый эфир 4-[7-(толуол-4-сульфонил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-карбаминовой кислоты (500 мг; 1,04 ммоль; Экв.: 1,00) растворяли в THF (10 мл) и охлаждали до -78°С. Добавляли LDA 2M раствор в смеси гептан/THF/этилбензол (1,31 мл; 2,61 ммоль; Экв.: 2,5) при -78°С в атмосфере азота с получением темно-коричневого раствора. Реакционную смесь перемешивали при -78°С в течение 1 ч 30 мин. Добавляли 1,2-дибром-1,1,2,2-тетрахлорэтан (851 мг; 2,61 ммоль; Экв.: 2,5) в 5 мл THF и эту реакционную смесь перемешивали при -78°С в течение 2 часов. Вносили воду. Реакционную смесь разбавляли с помощью EtOAc и промывали солевым раствором. Объединенные органические фазы высушивали над безводным сульфатом натрия, затем выпаривали. Неочищенное вещество очищали хроматографией на колонке (силикагель, 5-70% этилацетат в гексане). Указанное в заголовке соединение получали в виде белого твердого вещества (450 мг; выход 77%). LC/MS: m/z расч. для C25H25BrN4O4S ([M+H]+): 558,4 Фактич.: 558,8
Стадия 3: трет-Бутиловый эфир {4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-карбаминовой кислоты
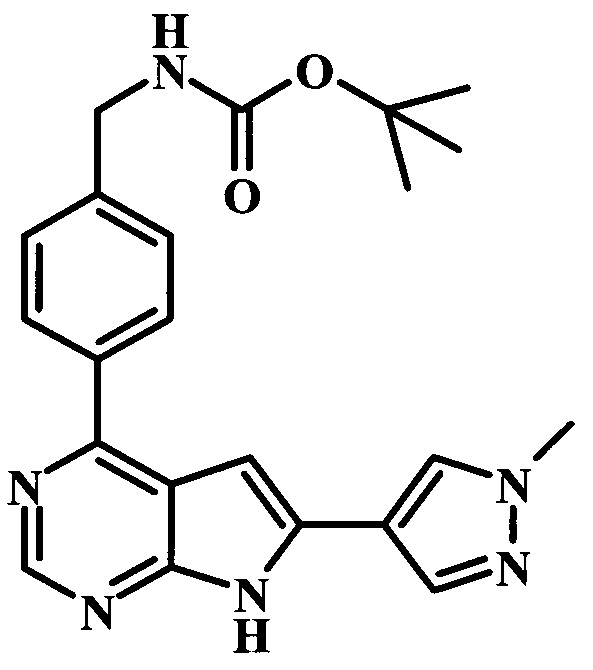
В герметизируемой трубке на 10 мл для микроволнового облучения трет-бутиловый эфир 4-[6-бром-7-(толуол-4-сульфонил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-карбаминовой кислоты (200 мг; 0,359 ммоль; Экв.: 1,00), 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол (192 мг; 0,923 ммоль; Экв.: 2,57) и карбонат калия (198 мг; 1,44 ммоль; Экв.: 4,00) в воде (1 мл) соединяли с DME (4 мл). Вносили Pd(PPh3)4 (42 мг; 0,036 ммоль; Экв.: 0,1), сосуд с реакционной смесью закрывали и нагревали микроволновым облучением при 150°С в течение 60 мин. Реакционную смесь разбавляли с помощью EtOAc затем промывали солевым раствором. Объединенные органические фазы высушивали над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенное вещество обрабатывали с помощью DCM, затем фильтровали с получением указанного в заголовке соединения в виде желтого твердого вещества (32 мг; выход 22%). LC/MS: m/z расч. для C22H24N6O2 ([M+H]+): 405,4 Фактич.: 405,2
Стадия 4: 4-трет-Бутил-N-{4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-бензамид
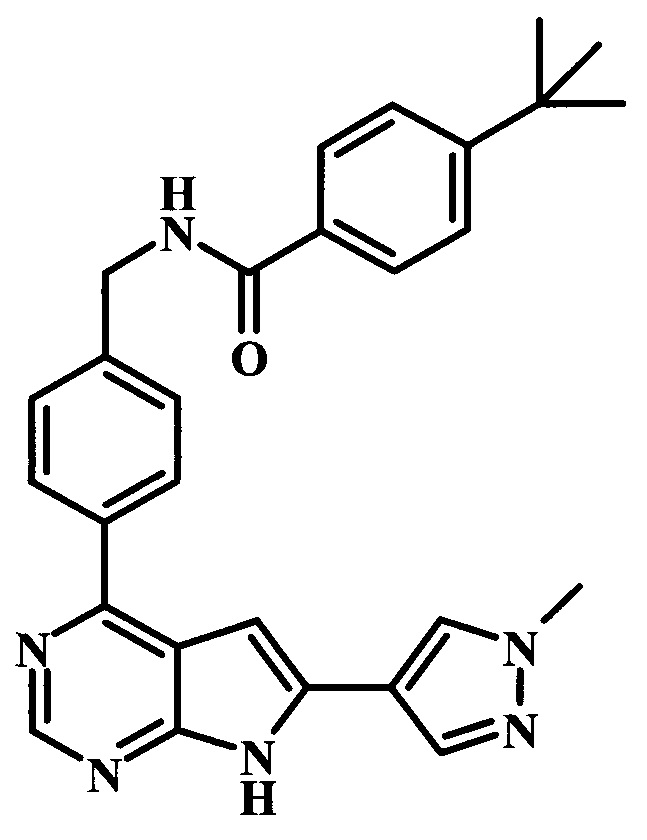
В сцинтилляционном флаконе на 20 мл трет-бутиловый эфир {4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-карбаминовой кислоты (30 мг; 0,074 ммоль; Экв.: 1,00) объединяли с 1 мл DCM и 1 мл TFA. Полученный раствор перемешивали при комнатной температуре в течение 1 час. Растворитель удаляли при пониженном давлении. Неочищенное вещество растворяли в DMF (2 мл). Вносили 4-трет-бутилбензойную кислоту (14,5 мг; 0,082 ммоль; Экв.: 1,1), DIPEA (0,052 мл; 0,297 ммоль; Экв.: 4,00) и HATU (31,0 мг; 0,082 ммоль; Экв.: 1,1). Реакционную смесь перемешивали в течение ночи. Полученный раствор разбавляли с помощью 10 мл воды и 5 мл EtOAc. Полученный раствор перемешивали при комнатной температуре в течение еще 30 мин. Органические фазы экстрагировали и высушивали над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Полученное твердое вещество обрабатывали с помощью DCM и это твердое вещество фильтровали. Указанное в заголовке соединение получали в виде твердого вещества (11 мг; выход 32%). LC/MS: m/z расч. для C28H28N6O ([M+H]+): 465,5 Фактич.: 465,2
Пример I-22
4-Циклопропил-N-{4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-бензамид
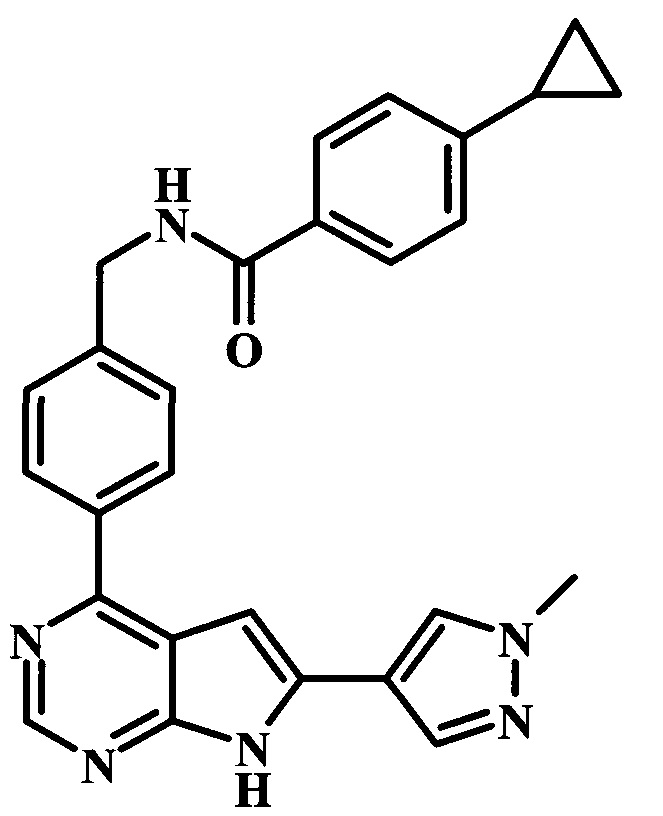
По аналогии со способом, описанным в примере 21, стадия 4, из 4-циклопропилбензойной кислоты (12,0 мг; 0,074 ммоль; Экв.: 1,00) получали указанное в заголовке соединение в виде твердого вещества (16 мг; выход 48%). LC/MS: m/z расч. для C27H24N6O ([M+H]+): 449,5 Фактич.: 449,2
Пример I-23
4-Изопропил-N-{4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-бензамид
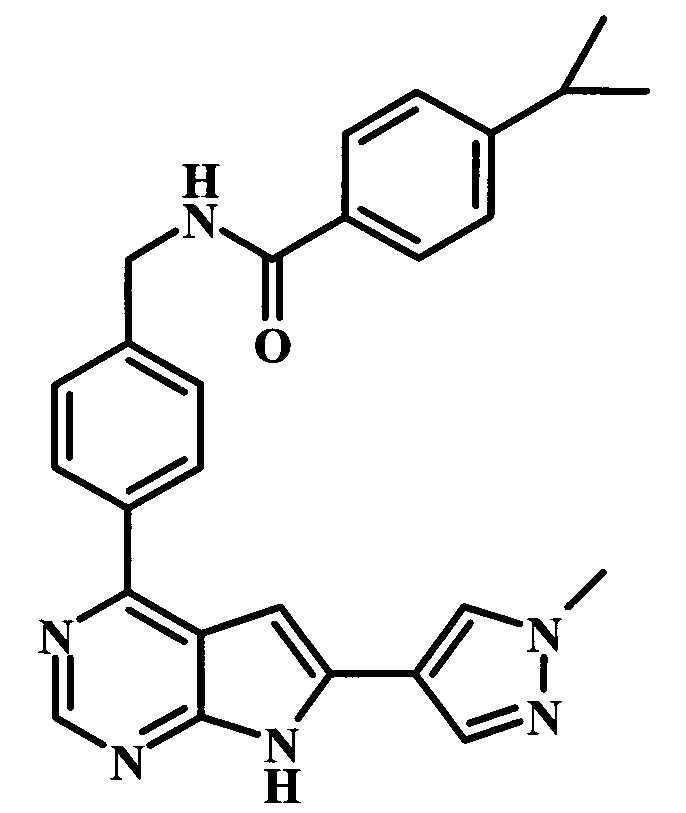
По аналогии со способом, описанным в примере 21, стадия 4, из 4-изопропилбензойной кислоты (13,4 мг; 0,082 ммоль; Экв.: 1,1) получали указанное в заголовке соединение в виде твердого вещества (5 мг; выход 15%). LC/MS: m/z расч. для C27H26N6O ([М+Н]+): 451,5 Фактич.: 451,3
Пример I-24
N-{4-[6-(1-Метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-4-оксетан-3-илбензамид
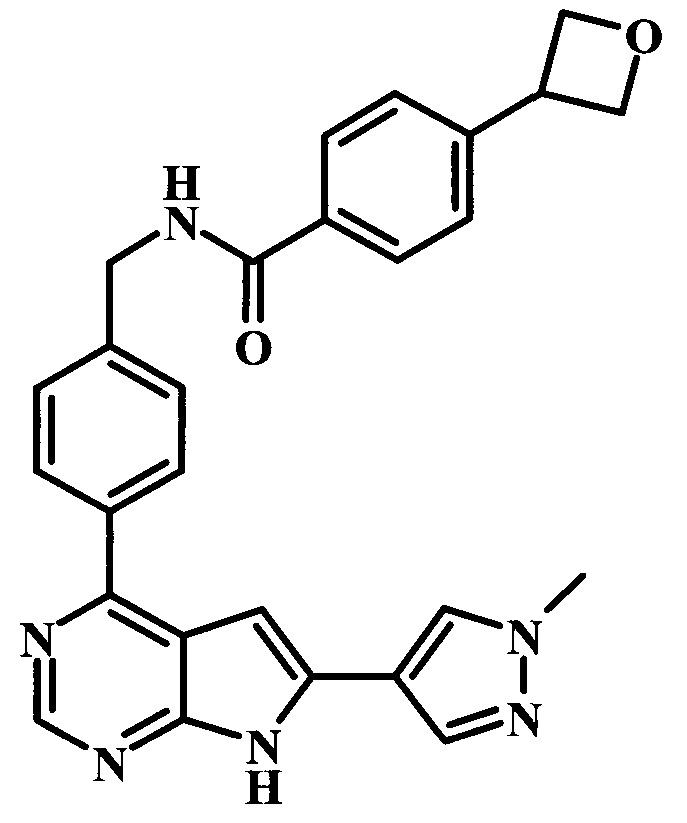
Стадия 1: Метиловый эфир 4-оксетан-3-ил-бензойной кислоты
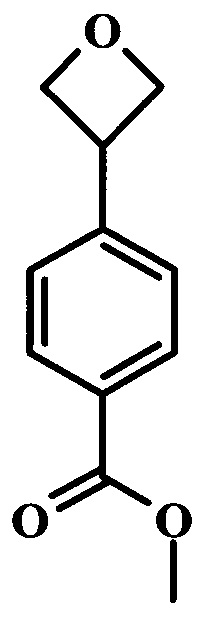
В герметизируемом сосуде для микроволнового облучения на 20 мл 4-(метоксикарбонил)фенилбороновую кислоту (978 мг; 5,44 ммоль; Экв.: 2,0), транс-2-аминоциклогексанол гидрохлорид (50 мг; 0,326 ммоль; Экв.: 0,12) и иодид никеля (102 мг; 0,326 ммоль; Экв.: 0,12) соединяли с изопропиловым спиртом (8 мл) с получением белой суспензии. Добавляли NaHMDS (997 мг; 5,44 ммоль; Экв.: 2,0). Сосуд с реакционной смесью заполняли аргоном и перемешивали в течение 5 мин. Добавляли 3-иодоксетан (0,5 г; 2,72 ммоль; Экв.: 1,00). Реакционный сосуд закрывали и затем нагревали при 80°С в течение 20 мин микроволновым облучением. По истечении этого времени ТСХ показала наличие двух пятен возможных продуктов с близкими параметрами удерживания (Rf). Реакционную смесь разбавляли изопропиловым спиртом, затем фильтровали через фильтровальную бумагу. Растворитель концентрировали с получением желтого масла. Этот продукт растворяли в метиленхлориде и полученный раствор концентрировали на силикагеле. Неочищенный продукт на силикагеле наносили на 40-граммовую силикагелевую колонку. Проводили флэш-хроматографию (переход от 5% этилацетат-гексан до 10% этилацетат-гексан). Указанное в заголовке соединение выделяли в виде масла (118 мг; выход 23%). Другой выделенный побочный продукт соответствовал изопропиловому эфиру 4-оксетан-3-ил-бензойной кислоты (77 мг; выход 13%).
Стадия 2: 4-Оксетан-3-ил-бензойная кислота
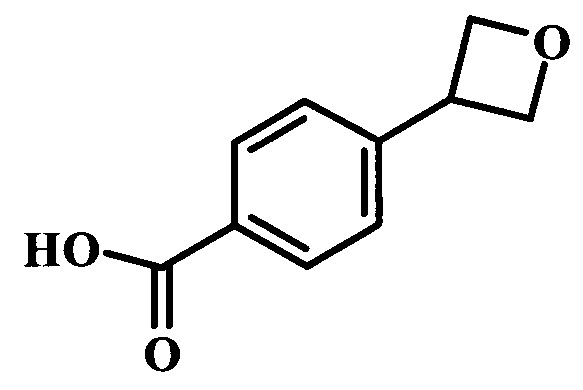
В грушевидной колбе на 100 мл метил-4-(оксетан-3-ил)бензоат (118 мг; 0,614 ммоль; Экв.: 1,00) и гидроксид лития моногидрат (40 мг; 0,953 ммоль; Экв.: 1,55) соединяли с THF (2,5 мл) с получением бесцветного раствора. Добавляли воду (2,5 мл). Реакционную смесь перемешивали в течение ночи при комнатной температуре. Утром с помощью ТСХ было выявлено небольшое количество оставшегося исходного вещества. Вносили еще 40 мг LiOH моногидрата и эту реакционную смесь снова перемешивали в течение ночи при комнатной температуре. Растворитель удаляли при пониженном давлении. Водный остаток затем разбавляли с помощью 10 мл воды и этот раствор экстрагировали с помощью 20 мл смеси 1:1 гексан-этилацетат. Водную фазу затем подкисляли несколькими каплями 4 н водной HCl, что давало белую суспензию. Эту суспензию экстрагировали этилацетатом. Органические экстракты высушивали над безводным сульфатом натрия и растворитель удаляли при пониженном давлении. Указанное в заголовке соединение получали в виде белого твердого вещества (88 мг; выход 80%). Данный продукт использовали в таком виде без дополнительной очистки.
Стадия 3: N-{4-[6-(1-Метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-4-оксетан-3-илбензамид
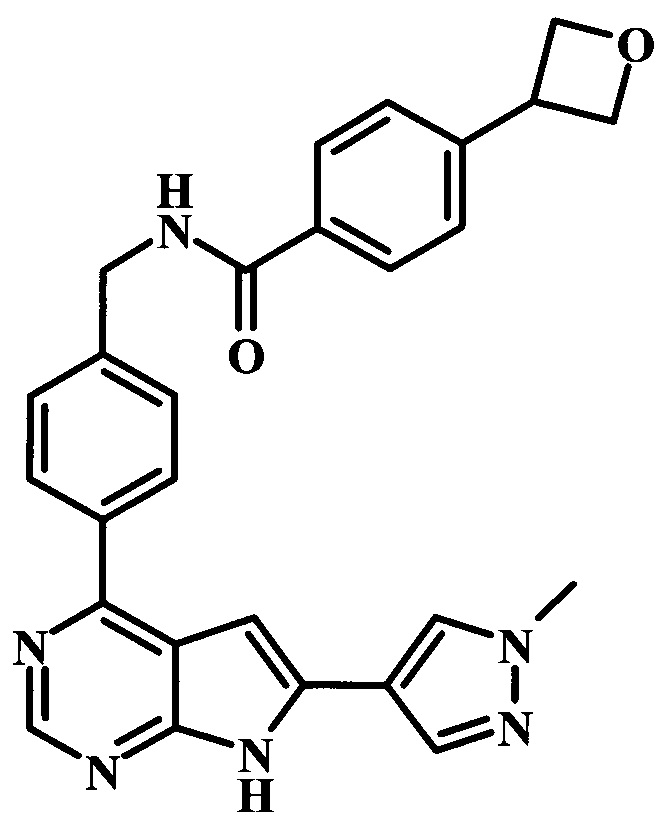
В сцинтилляционном флаконе на 20 мл трет-бутиловый эфир {4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-карбаминовой кислоты (40 мг; 0,099 ммоль; Экв.: 1,00) объединяли с 3 мл DCM и 3 мл TFA. Полученный раствор перемешивали при комнатной температуре в течение 1 час. Растворитель удаляли при пониженном давлении. Неочищенное вещество растворяли в DMF (4 мл). Вносили 4-оксетан-3-ил-бензойную кислоту (18 мг; 0,099 ммоль; Экв.: 1,00), DIPEA (0,069 мл; 0,396 ммоль; Экв.: 4,00) и HATU (38 мг; 0,099 ммоль; Экв.: 1,00). Реакционную смесь перемешивали в течение ночи. Полученный раствор разбавляли с помощью 10 мл воды и 5 мл EtOAc. Полученный раствор перемешивали при комнатной температуре в течение еще 30 мин. Органические фазы экстрагировали и высушивали над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Полученное твердое вещества обрабатывали с помощью DCM и это твердое вещество фильтровали. Указанное в заголовке соединение получали в виде твердого вещества (3 мг; выход 7%). LC/MS: m/z расч. для C27H24N6O2 ([M+H]+): 465,5 Фактич.: 465,2
Пример I-25
4-(3-Метилоксетан-3-ил)-N-{4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-бензамид
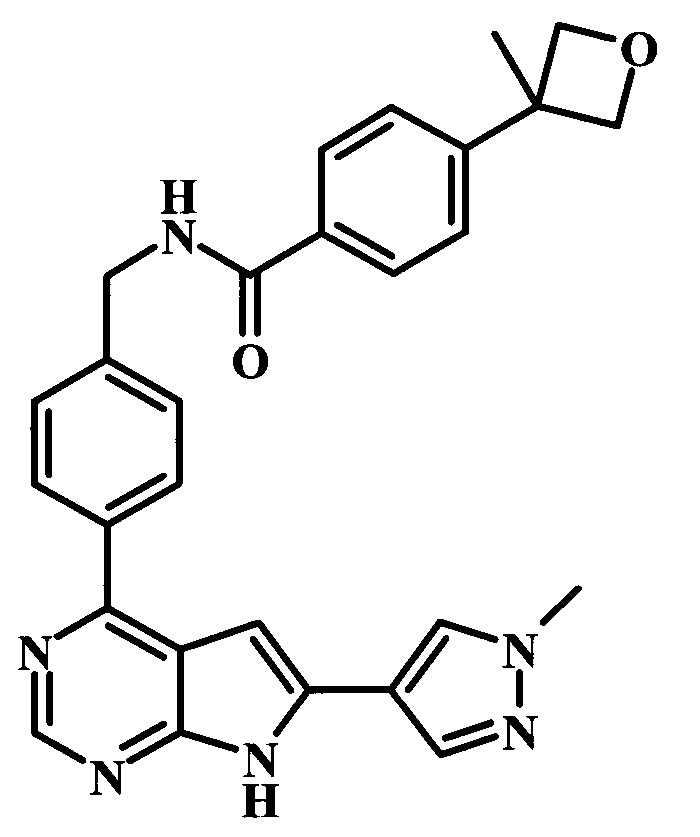
Стадия 1: 4-(3-Метилоксетан-3-ил)-бензойная кислота
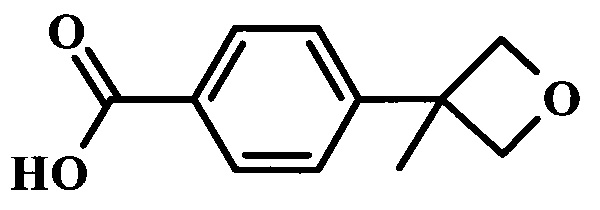
В трехгорловой колбе на 250 мл 3-(4-бромфенил)-3-метилоксетан (1,05 г; 4,62 ммоль) объединяли с THF (35 мл) с получением бесцветного раствора. Этот раствор охлаждали до -78°С на бане со смесью сухого льда и ацетона. К холодному раствору добавляли по каплям 1,6 М раствор nBuLi в гексане (3,32 мл; 5,32 ммоль). Внесение по каплям проводили в течение 10 мин. Реакционную смесь перемешивали при -78°С в течение 1 час. По истечении этого времени в реакционную смесь через длинную иглу вводили газообразный диоксид углерода, который генерировали в отдельной колбе из сухого льда. Реакционная смесь быстро изменяла цвет на светло-желтый. Диоксид углерода пропускали при низкой температуре в течение еще 20 мин. По истечении этого времени реакционная смесь представляла собой белую суспензию. Реакционную смесь нагревали до комнатной температуры, затем медленно гасили водой. Органический растворитель выпаривали. Полученную смесь экстрагировали с помощью раствора 1:1 этилацетата и гексана. Водную фазу затем подкисляли до кислых значений рН добавлением 4 н водной HCl. Полученную белую суспензию фильтровали под вакуумом на воронке Бюхнера. Собранное белое вещество высушивали на вакуумной воронке и затем дополнительно высушивали в вакуумном сушильном шкафу, что давало 4-(3-метилоксетан-3-ил)-бензойную кислоту (456 мг; 51%). 1H ЯМР (300 МГц, DMSO-d6) δ ppm 12,89 (br. s., 1H), 7,93 (d, J=8,48 Гц, 2H), 7,36 (d, J=8,67 Гц, 2H), 4,81 (d, J=5,84 Гц, 2H), 4,56 (d, J=6,03 Гц, 2H), 1,64 (s, 3H).
Стадия 2: 4-(3-Метилоксетан-3-ил)-N-{4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-бензамид
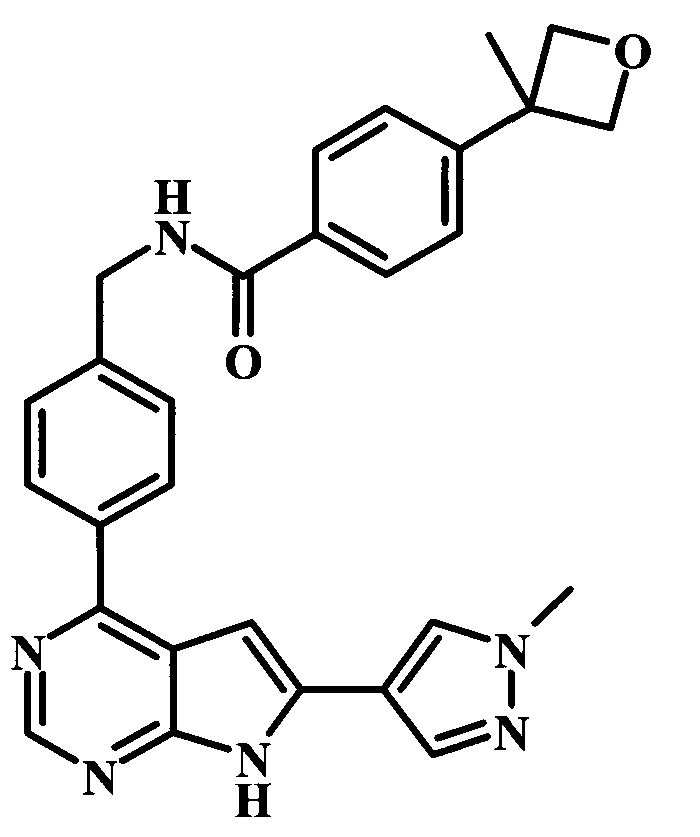
В сцинтилляционном флаконе на 20 мл трет-бутиловый эфир {4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-карбаминовой кислоты (60 мг; 0,148 ммоль; Экв.: 1,00) объединяли с 3 мл DCM и 3 мл TFA. Полученный раствор перемешивали при комнатной температуре в течение 1 час. Растворитель удаляли при пониженном давлении. Неочищенное вещество растворяли в DMF (4 мл). Вносили 4-оксетан-3-ил-бензойную кислоту (31 мг; 0,163 ммоль; Экв.: 1,1), DIPEA (0,104 мл; 0,593 ммоль; Экв.: 4,00) и HATU (62 мг; 0,163 ммоль; Экв.: 1,1). Реакционную смесь перемешивали в течение ночи. Полученный раствор разбавляли с помощью 10 мл воды и 5 мл EtOAc. Полученный раствор перемешивали при комнатной температуре в течение еще 30 мин. Органические фазы экстрагировали и высушивали над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Полученное твердое вещество обрабатывали с помощью DCM и это твердое вещество фильтровали. Указанное в заголовке соединение получали в виде желтого твердого вещества (15 мг; выход 21%). LC/MS: m/z расч. для C28H26N6O2 ([M+H]+): 479,5 Фактич.: 479,2
Пример I-26
4-[6-(1-Метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензиламид4,5,6,7-тетрагидробензо[b]тиофен-2-карбоновой кислоты
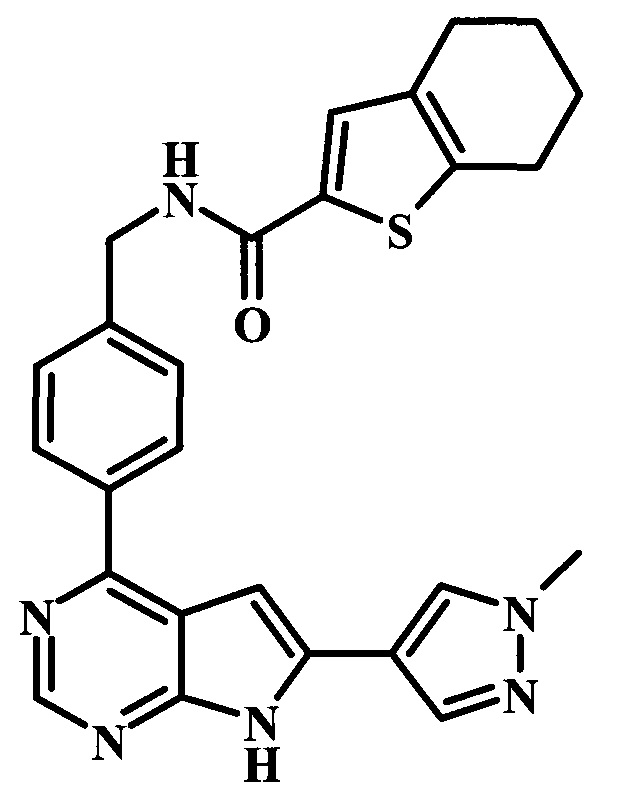
В сцинтилляционном флаконе на 20 мл трет-бутиловый эфир {4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-карбаминовой кислоты (40 мг; 0,099 ммоль; Экв.: 1,00) объединяли с 3 мл DCM и 3 мл TFA. Полученный раствор перемешивали при комнатной температуре в течение 1 час. Растворитель удаляли при пониженном давлении. Неочищенное вещество растворяли в DMF (4 мл). Вносили 4,5,6,7-тетрагидробензо[b]тиофен-2-карбоновую кислоту (20 мг; 0,109 ммоль; Экв.: 1,1), DIPEA (0,069 мл; 0,396 ммоль; Экв.: 4,00) и HATU (41 мг; 0,109 ммоль; Экв.: 1,1). Реакционную смесь перемешивали в течение ночи. Полученный раствор разбавляли с помощью 10 мл воды и 5 мл EtOAc. Раствор перемешивали при комнатной температуре в течение еще 30 мин. Органические фазы экстрагировали и высушивали над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенное вещество очищали хроматографией на колонке (силикагель, 0-10% МеОН в DCM). Указанное в заголовке соединение получали в виде твердого вещества (17 мг; выход 37%). LC/MS: m/z расч. для C26H24N6OS ([M+H]+): 469,5 Фактич.: 469,2
Пример I-27
4-трет-Бутил-N-{2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-бензамид
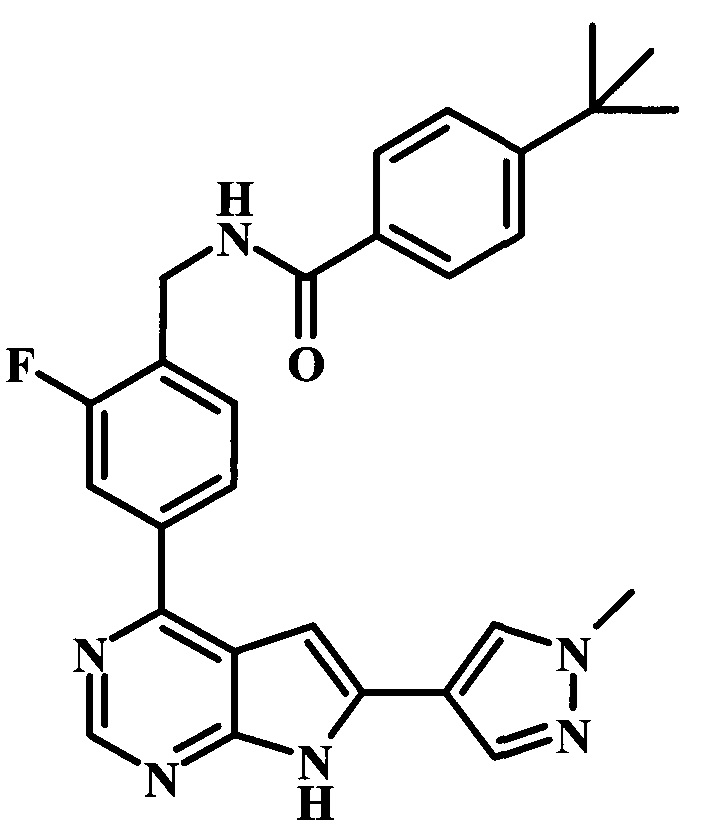
Стадия 1: трет-Бутиловый эфир {2-фтор-4-[7-(толуол-4-сульфонил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-карбаминовой кислоты
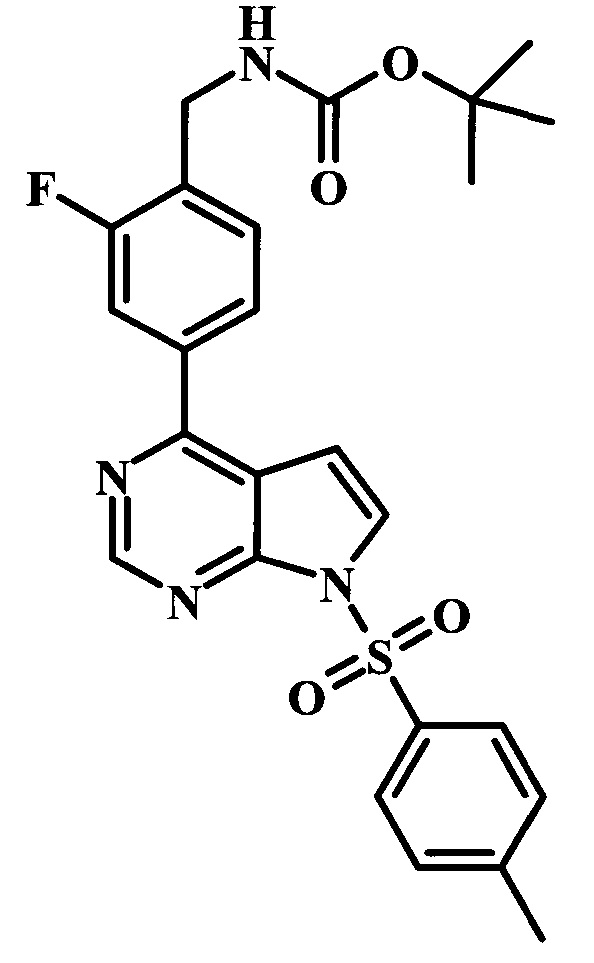
В герметизируемой трубке на 20 мл для микроволнового облучения 4-хлор-7-тозил-7Н-пирроло[2,3-d]пиримидин (1 г; 3,25 ммоль; Экв.: 1,00), трет-бутиловый эфир [2-фтор-4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-бензил]-карбаминовой кислоты (1,14 г; 3,25 ммоль; Экв.: 1,00) и карбонат калия (1,8 г; 13,0 ммоль; Экв.: 4,00) в 5 мл воды соединяли с DME (10 мл). Вносили Pd(PPh3)4 (375 мг; 0,325 ммоль; Экв.: 0,1). Реакционную смесь нагревали микроволновым облучением при 150°С в течение 30 мин. Полученный раствор промывали с помощью EtOAc и солевым раствором. Объединенные органические фазы высушивали над безводным сульфатом натрия, затем растворитель удаляли при пониженном давлении. Неочищенное вещество очищали хроматографией на колонке (силикагель, 5-70% этилацетат в гексане). Указанное в заголовке соединение получали в виде белого твердого вещества (950 мг; выход 59%). LC/MS: m/z расч. для C25H25FN4O4S ([M+H]+): 497,5 Фактич.: 497,2
Стадия 2: трет-Бутиловый эфир {4-[6-бром-7-(толуол-4-сульфонил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-2-фторбензил}-карбаминовой кислоты
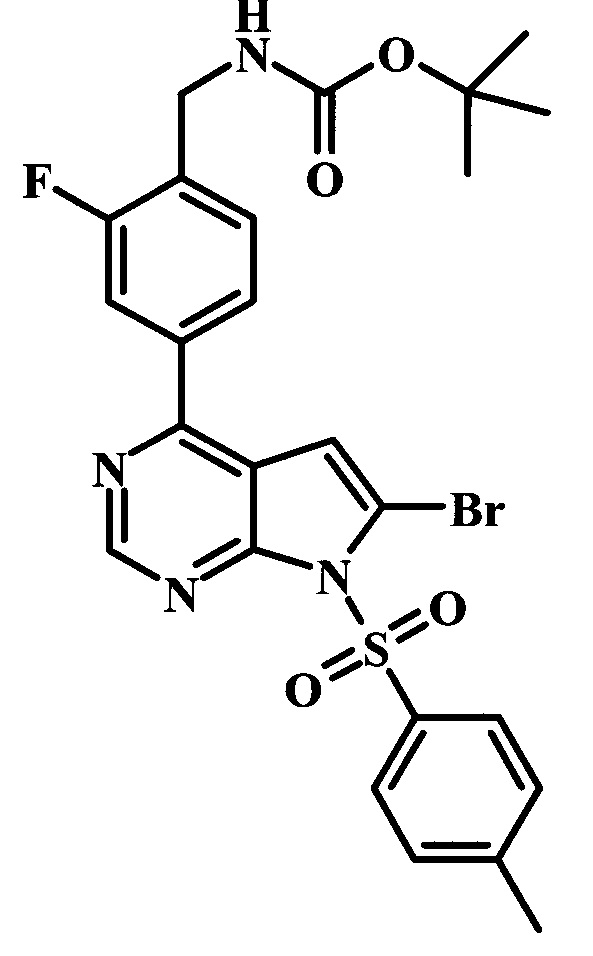
В круглодонной колбе на 100 мл трет-бутиловый эфир {2-фтор-4-[7-(толуол-4-сульфонил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-карбаминовой кислоты (2 г; 4,03 ммоль; Экв.: 1,00) растворяли в THF (40 мл) и охлаждали до -78°С. Добавляли LDA, 2 М раствор в смеси гептан/THF/этилбензол (5,03 мл; 10,1 ммоль; Экв.: 2,5), при -78 С в атмосфере азота с получением темно-коричневого раствора. Реакционную смесь перемешивали при -78°С в течение 1 ч 30 мин. Добавляли 1,2-дибром-1,1,2,2-тетрахлорэтан (3,28 г; 10,1 ммоль; Экв.: 2,5) в 10 мл THF и эту реакционную смесь перемешивали при -78°С в течение 2 часов. Добавляли солевой раствор. Реакционную смесь разбавляли с помощью EtOAc и промывали солевым раствором. Объединенные органические фазы высушивали над безводным сульфатом натрия, затем выпаривали. Неочищенное вещество очищали хроматографией на колонке (силикагель, 5-70% этилацетат в гексане). Указанное в заголовке соединение получали в виде бежевого твердого вещества (1,2 г; выход 52%). LC/MS: m/z расч. для C25H24BrFN4O4S ([M+H]+): 576,4 Фактич.: 577,1
Стадия 3: трет-Бутиловый эфир {2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-карбаминовой кислоты
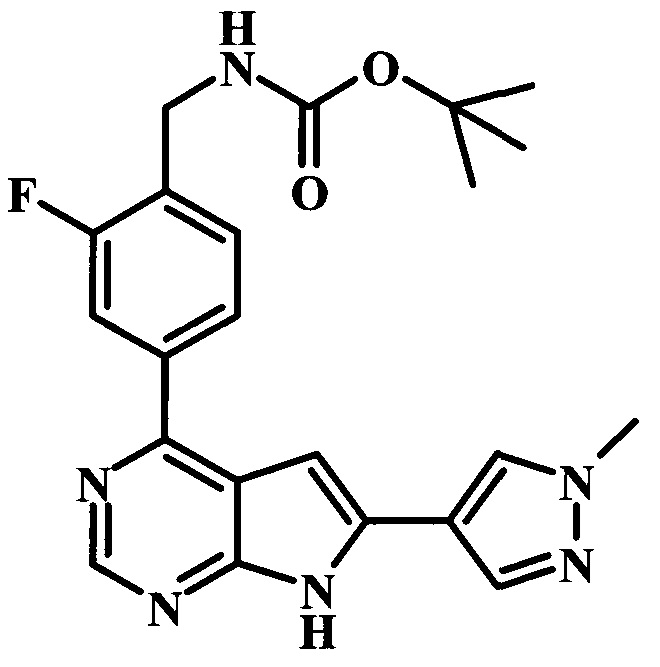
В герметизируемой трубке на 20 мл для микроволнового облучения трет-бутиловый эфир {4-[6-бром-7-(толуол-4-сульфонил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-2-фторбензил}-карбаминовой кислоты (400 мг; 0,695 ммоль; Экв.: 1,00), 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол (226 мг; 1,09 ммоль; Экв.: 1,6) и карбонат калия (384 мг; 2,78 ммоль; Экв.: 4,00) в воде (3 мл) соединяли с DME (6 мл). Вносили Pd(PPh3)4 (80 мг; 0,069 ммоль; Экв.: 0,1) и сосуд с этой реакционной смесью закрывали и нагревали микроволновым облучением при 160°С в течение 60 мин. Реакционную смесь разбавляли с помощью EtOAc затем промывали солевым раствором. Объединенные органические фазы высушивали над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенное вещество обрабатывали с помощью DCM, затем фильтровали с получением указанного в заголовке соединения в виде коричневого твердого вещества (75 мг; выход 26%). LC/MS: m/z расч. для C22H23FN6O2 ([M+H]+): 423,4 Фактич.: 423,3
Стадия 4: 4-трет-Бутил-N-{2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-бензамид
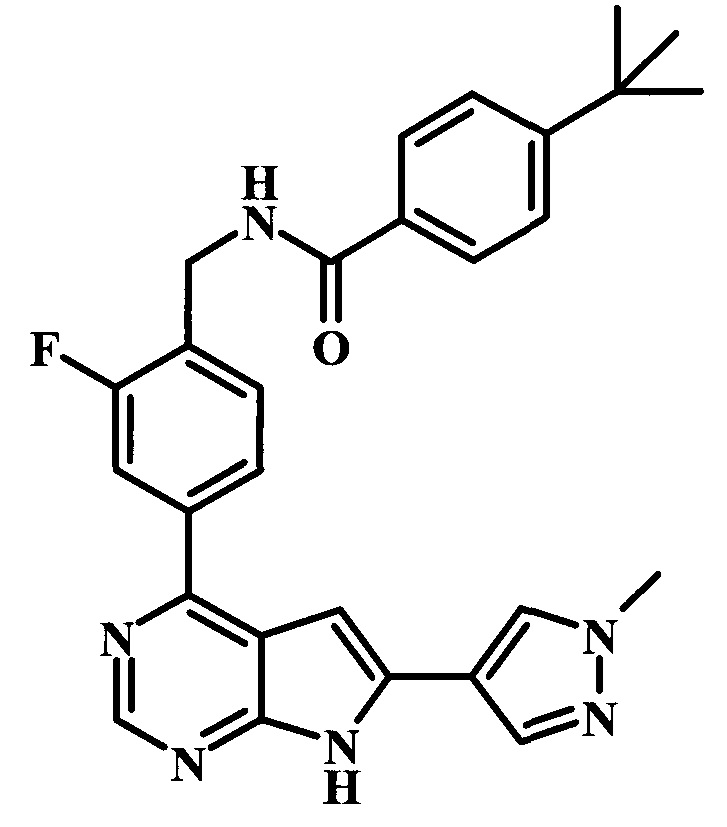
В сцинтилляционном флаконе на 20 мл трет-бутиловый эфир {2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-карбаминовой кислоты (40 мг; 0,094 ммоль; Экв.: 1,00) объединяли с 1 мл DCM и 1 мл TFA. Полученный раствор перемешивали при комнатной температуре в течение 1 час.Растворитель удаляли при пониженном давлении. Неочищенное вещество растворяли в DMF (2 мл). Вносили 4-трет-бутилбензойную кислоту (19 мг; 0,104 ммоль; Экв.: 1,1), DIPEA (0,066 мл; 0,379 ммоль; Экв.: 4,00) и HATU (40 мг; 0,104 ммоль; Экв.: 1,1). Реакционную смесь перемешивали в течение ночи. Полученный раствор разбавляли с помощью 10 мл воды и 5 мл EtOAc. Раствор перемешивали при комнатной температуре в течение еще 30 мин. Органические фазы экстрагировали и высушивали над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Полученное твердое вещества обрабатывали с помощью DCM и твердое вещество фильтровали. Указанное в заголовке соединение получали в виде твердого вещества (17 мг; выход 37%). LC/MS: m/z расч. для C28H27FN6O ([M+H]+): 483,5 Фактич.: 483,2
Пример I-28
6-трет-Бутил-N-{2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-никотинамид
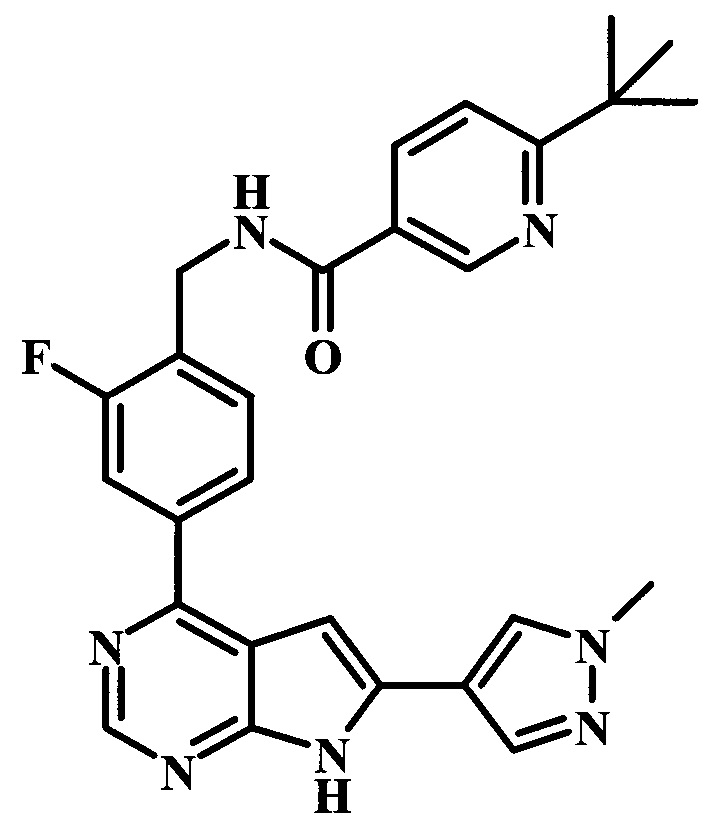
Стадия 1: 6-трет-Бутил-никотиновая кислота
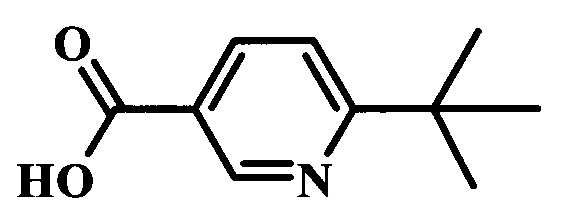
К суспензии никотиновой кислоты (2 г; 16 ммоль; Экв.: 1,00) в воде добавляли концентрированную серную кислоту (1 мл; 18,8 ммоль; Экв.: 1,2) и эту смесь перемешивали в атмосфере азота с образованием прозрачного раствора. Добавляли триметилуксусную кислоту (1,83 г; 17,9 ммоль; Экв.: 1,1) и продолжали перемешивать в атмосфере аргона при комнатной температуре в течение 10 мин. Добавляли нитрат серебра (125 мг; 0,736 ммоль), а затем персульфат аммония (295 мг; 1,29 ммоль; Экв.: 0,08), колбу оборачивали алюминиевой фольгой, чтобы защитить от света, и смесь нагревали при 90°С в атмосфере азота. Через 2 часа реакционную смесь охлаждали до комнатной температуры и выстаивали в течение ночи. Экстракция реакционной смеси этилацетатом не приводила к получению существенного количества ожидаемого продукта. Анализ LC/MS не показывал присутствие продукта в водном слое. Водную смесь концентрировали в вакууме до получения бесцветного твердого вещества. Твердое вещество обрабатывали с помощью THF, фильтровали и фильтрат концентрировали в вакууме. Остаток перерастворяли в метаноле, фильтровали и затем фильтрат концентрировали в вакууме. Концентрированные фильтраты очищали обращенно-фазной хроматографией на колонке С-18, 85 г, элюировали градиентом от 10% ацетонитрила в воде до 100% ацетонитрила. Фракции, содержащие целевой продукт, объединяли и концентрировали с получением бесцветной водной суспензии (объем ~5 мл). Вносили дополнительное количество воды (~20 мл) с получением прозрачного раствора и эту смесь лиофилизовали с получением указанного в заголовке соединения в виде бесцветного, аморфного лиофилизованного твердого вещества (139 мг; выход 5%). LC/MS: m/z расч. для C10H14NO2 ([М+Н]+): 180,2, 483,5 Фактич.: 180,1
Стадия 2: 6-трет-Бутил-N-{2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-никотинамид
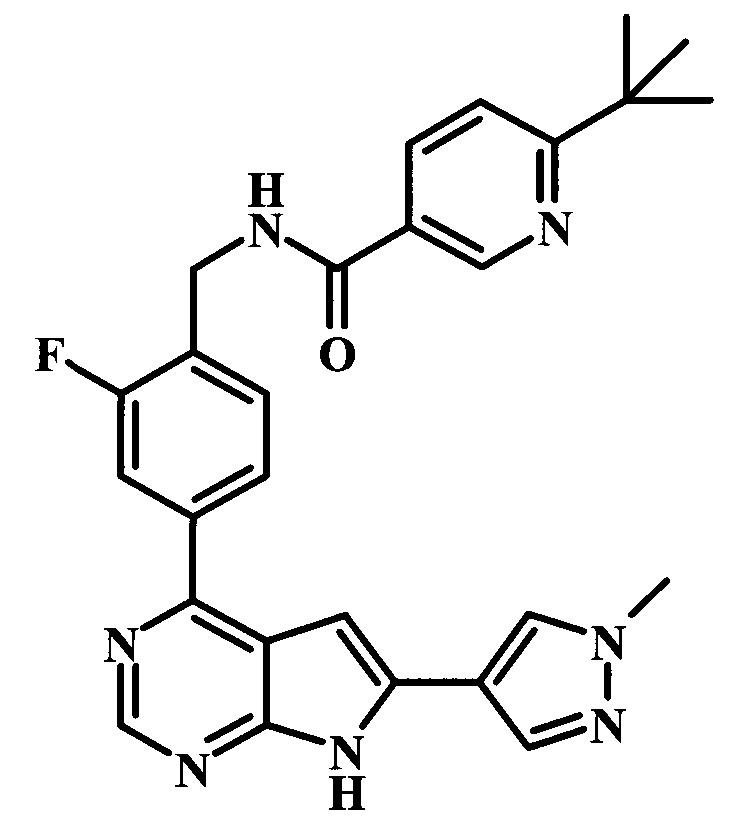
В сцинтилляционном флаконе на 20 мл трет-бутиловый эфир {2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-карбаминовой кислоты (56 мг; 0,133 ммоль; Экв.: 1,00) объединяли с 2 мл DCM и 2 мл TFA. Полученный раствор перемешивали при комнатной температуре в течение 30 мин. Растворитель удаляли при пониженном давлении. Неочищенное вещество растворяли в DMF (2 мл). Вносили 6-трет-бутил-никотиновую кислоту (40 мг; 0,233 ммоль; Экв.: 1,7), DIPEA (0,093 мл; 0,53 ммоль; Экв.: 4,00) и HATU (55 мг; 0,146 ммоль; Экв.: 1,1). Реакционную смесь перемешивали в течение ночи. Полученный раствор разбавляли с помощью 10 мл воды и 5 мл EtOAc. Полученный раствор перемешивали при комнатной температуре в течение еще 30 мин. Органические фазы экстрагировали и высушивали над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Полученное твердое вещество обрабатывали с помощью DCM и фильтровали. Указанное в заголовке соединение получали в виде желтого твердого вещества (40 мг; выход 62%). LC/MS: m/z расч. для C27H26FN7O ([M+H]+): 484,5 Фактич.: 483,3
Пример I-29
2-Фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензиламид 5-метилтиофен-2-карбоновой кислоты
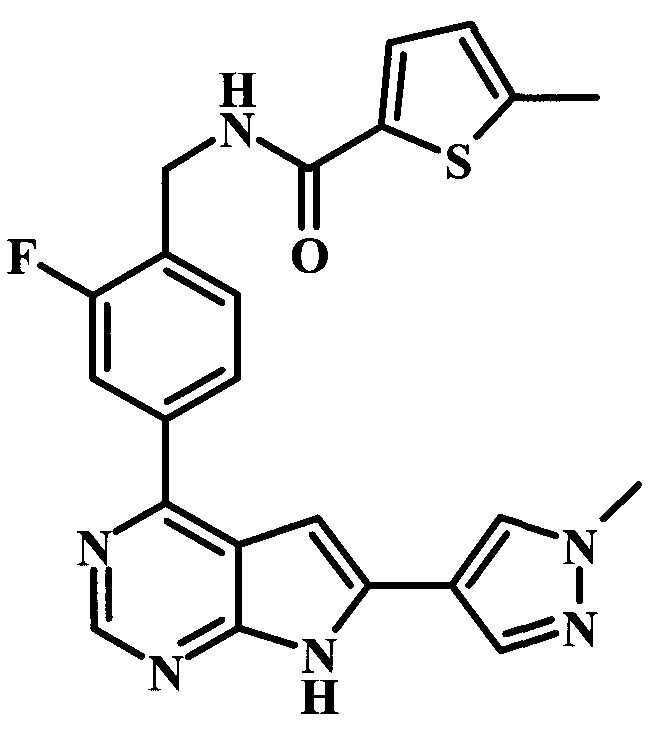
По аналогии со способом, описанным в примере 28, стадия 2, из 5-метилтиофен-2-карбоновой кислоты (22 мг; 0,156 ммоль; Экв.: 1,1) получали указанное в заголовке соединение в виде твердого вещества (42 мг; выход 66%). LC/MS: m/z расч. для C23H19FN6OS ([M+H]+): 447,5 Фактич.: 447,2
Пример I-30
4-трет-Бутил-N-(2-фтор-4-{6-[1-(2-гидроксиэтил)-1Н-пиразол-4-ил]-7Н-пирроло[2,3-d]пиримидин-4-ил}-бензил)-бензамид
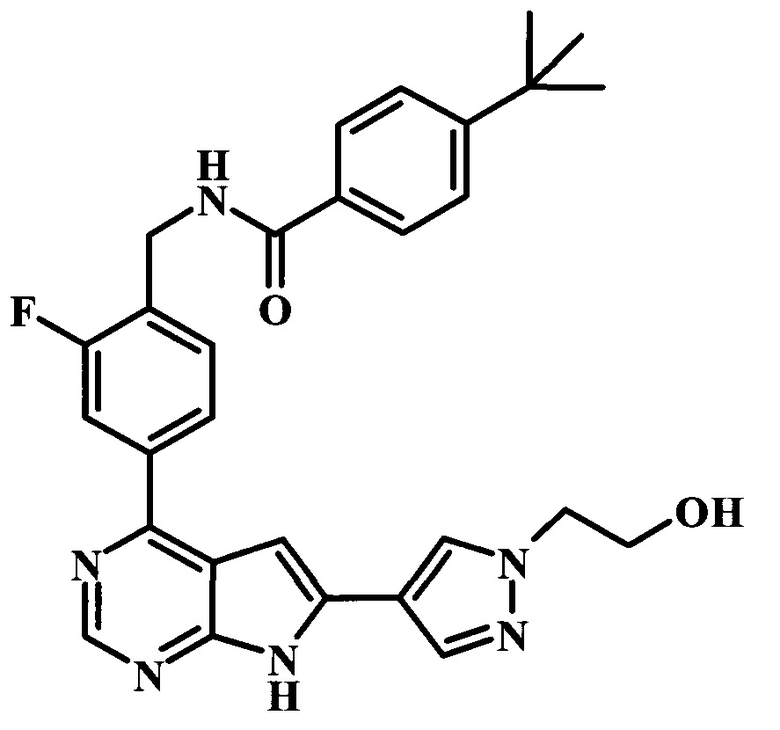
Стадия 1: N-[4-(6-Бром-7Н-пирроло[2,3-d]пиримидин-4-ил)-2-фторбензил]-4-трет-бутил-бензамид

В сцинтилляционном флаконе на 20 мл трет-бутиловый эфир {4-[6-бром-7-(толуол-4-сульфонил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-2-фторбензил}-карбаминовой кислоты (200 мг; 0,348 ммоль; Экв.: 1,00) объединяли с 1 мл DCM и 1 мл TFA. Полученный раствор перемешивали при комнатной температуре в течение 30 мин. Растворитель удаляли при пониженном давлении. Неочищенное вещество растворяли в DMF (2 мл). Вносили 4-трет-бутилбензойную кислоту (68 мг; 0,382 ммоль; Экв.: 1,1), DIPEA (0,243 мл; 1,39 ммоль; Экв.: 4,00) и HATU (145 мг; 0,382 ммоль; Экв.: 1,1). Реакционную смесь перемешивали в течение ночи. Полученный раствор разбавляли с помощью 10 мл воды и 5 мл EtOAc. Полученный раствор перемешивали при комнатной температуре в течение еще 30 мин. Органические фазы экстрагировали и высушивали над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенное вещество выдерживали при комнатной температуре в течение ночи. Полученное твердое вещество обрабатывали с помощью DCM и фильтровали. Указанное в заголовке соединение получали в виде твердого вещества (96 мг; выход 57%). LC/MS: m/z расч. для C24H22BrFN4O ([М+Н]+): 482,3 Фактич.: 483,0
Стадия 2: 4-трет-Бутил-N-(2-фтор-4-{6-[1-(2-гидроксиэтил)-1Н-пиразол-4-ил]-7Н-пирроло[2,3-d]пиримидин-4-ил}-бензил)-бензамид
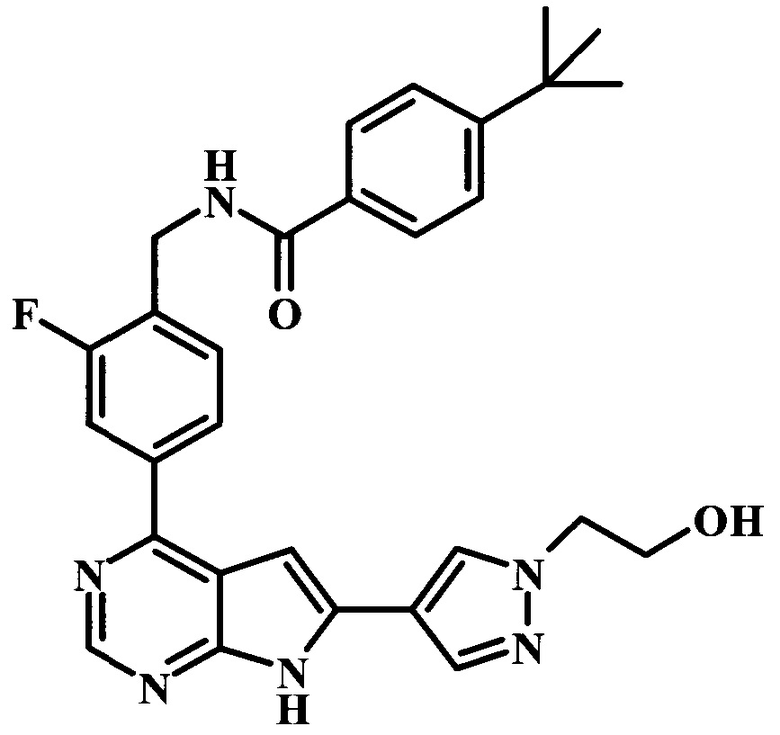
В герметизируемом сосуде на 20 мл для микроволнового облучения, N-[4-(6-Бром-7Н-пирроло[2,3-d]пиримидин-4-ил)-2-фторбензил]-4-трет-бутил-бензамид (90 мг; 0,187 ммоль; Экв.: 1,00), 2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол-1-ил)этанол (49,0 мг; 0,206 ммоль; Экв.: 1,1) и карбонат калия (103 мг; 0,748 ммоль; Экв.: 4,00) в воде (1 мл) соединяли с DME (4 мл). Вносили Pd(PPh3)4 (22 мг; 0,019 ммоль; Экв.: 0,1) и сосуд с этой реакционной смесью закрывали и нагревали микроволновым облучением при 150°С в течение 30 мин. Реакционную смесь разбавляли с помощью EtOAc, затем промывали солевым раствором. Объединенные органические фазы высушивали над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенное вещество обрабатывали с помощью DCM, затем фильтровали с получением указанного в заголовке соединения в виде коричневого твердого вещества (22 мг; выход 23%). LC/MS: m/z расч. для C29H29FN6O2 ([M+H]+): 513,5 Фактич.: 513,2
Пример I-31
4-трет-Бутил-N-{2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-N-метилбензамид
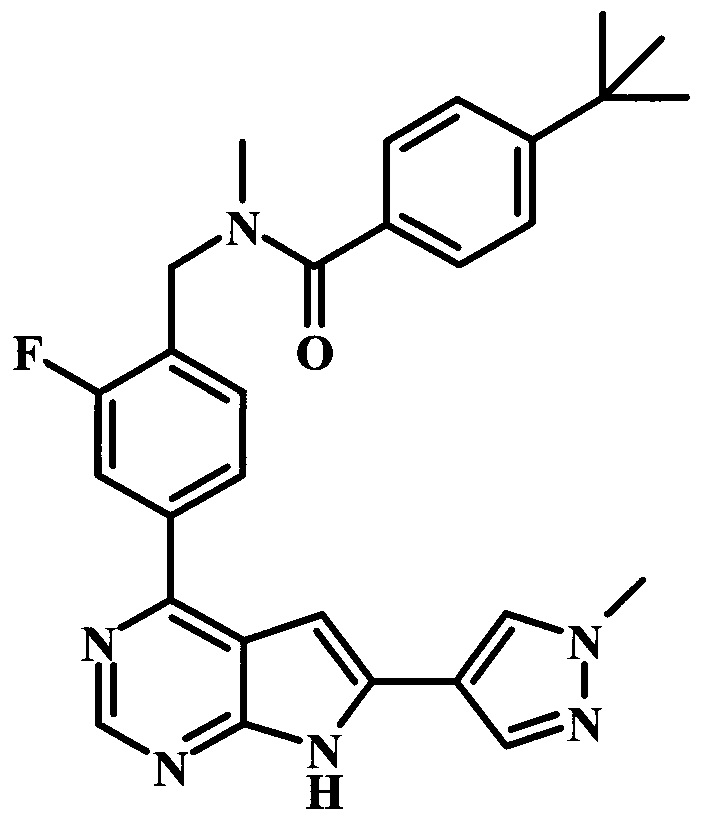
Стадия 1: трет-Бутиловый эфир (4-бром-2-фторбензил)-метилкарбаминовой кислоты

В круглодонной колбе на 100 мл трет-бутил-4-бром-2-фторбензилкарбамат (2 г; 6,58 ммоль; Экв.: 1,00), метилиодид (0,7 мл; 11,2 ммоль; Экв.: 1,7) и NaH в 60% масляной дисперсии (395 мг; 16,5 ммоль; Экв.: 2,5) соединяли с DMF (40 мл) при 0°С. Реакционную смесь перемешивали и оставляли нагреться до комнатной температуры в течение 3 часов. Реакционную смесь гасили с помощью МеОН. Реакционную смесь разбавляли с помощью EtOAc и промывали солевым раствором. Объединенные органические фазы высушивали над безводным сульфатом натрия, затем растворитель удаляли при пониженном давлении. Неочищенное вещество очищали хроматографией на колонке (силикагель, 5-40% этилацетат в гексане). Указанное в заголовке соединение получали в виде масла (1,8 г, выход 86%).
Стадия 2: трет-Бутиловый эфир [2-фтор-4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-бензил]-метил-карбаминовой кислоты
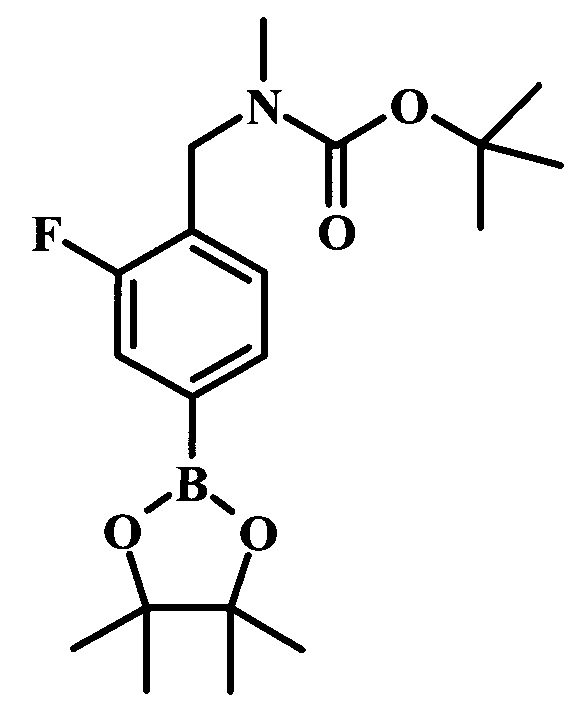
В круглодонной колбе на 250 мл трет-бутиловый эфир (4-бром-2-фторбензил)-метилкарбаминовой кислоты (1,8 г; 5,66 ммоль; Экв.: 1,00), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (2,15 г; 8,49 ммоль; Экв.: 1,5) и ацетат калия (1,67 г; 17,0 ммоль; Экв.: 3,00) соединяли с NMP (40 мл). Полученный раствор дегазировали в атмосфере азота в течение 10 мин. Добавляли 1,1'-бис(дифенилфосфино)ферроцен]-дихлорпалладий(II) (414 мг; 0,566 ммоль; Экв.: 0,1) и эту реакционную смесь нагревали при 100°С в течение 24 часов. Реакционную смесь охлаждали, затем разбавляли водой и EtOAc. Органические фазы объединяли, затем высушивали над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении и полученное неочищенное вещество очищали хроматографией на колонке (силикагель, 5-40% этилацетат в гексане). Указанное в заголовке соединение получали в виде масла (1,01 г, выход 49%).
Стадия 3: 7-Бензолсульфонил-4-хлор-6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин
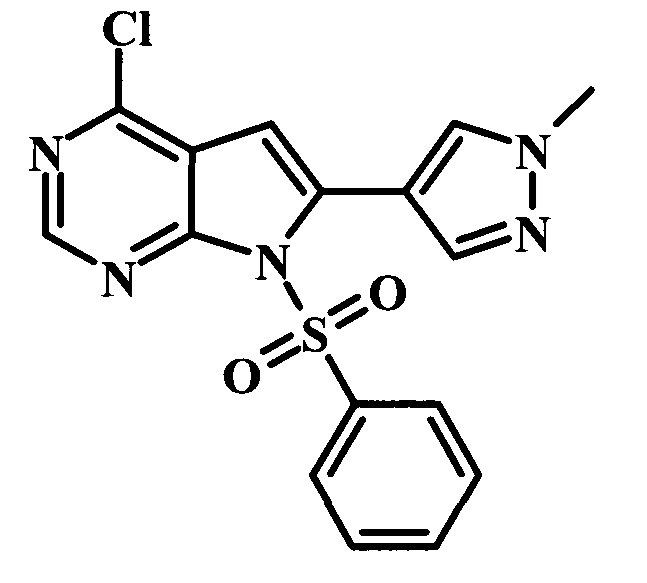
В сцинтилляционном флаконе на 20 мл 4-хлор-6-иод-7-(фенилсульфонил)-7Н-пирроло[2,3-d]пиримидин (1 г; 2,38 ммоль; Экв.: 1,00), карбонат калия (1,32 г; 9,53 ммоль; Экв.: 4,00) в 5 мл воды и 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол (744 мг; 3,57 ммоль; Экв.: 1,5) соединяли с DME (10 мл). Вносили Pd(PPh3)4 (275 мг; 0,238 ммоль; Экв.: 0,1) и эту реакционную смесь нагревали при 90°С в течение 6 часов. Реакционную смесь выстаивали в течение ночи при комнатной температуре. При этом образовывался осадок, который фильтровали в вакууме с получением указанного в заголовке соединения в виде бежевого твердого вещества (194 мг; выход 22%). LC/MS: m/z расч. для C16H12ClN5O2S ([М+Н]+): 374,8 Фактич.: 374,1
Стадия 4: трет-Бутиловый эфир {2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-метилкарбаминовой кислоты
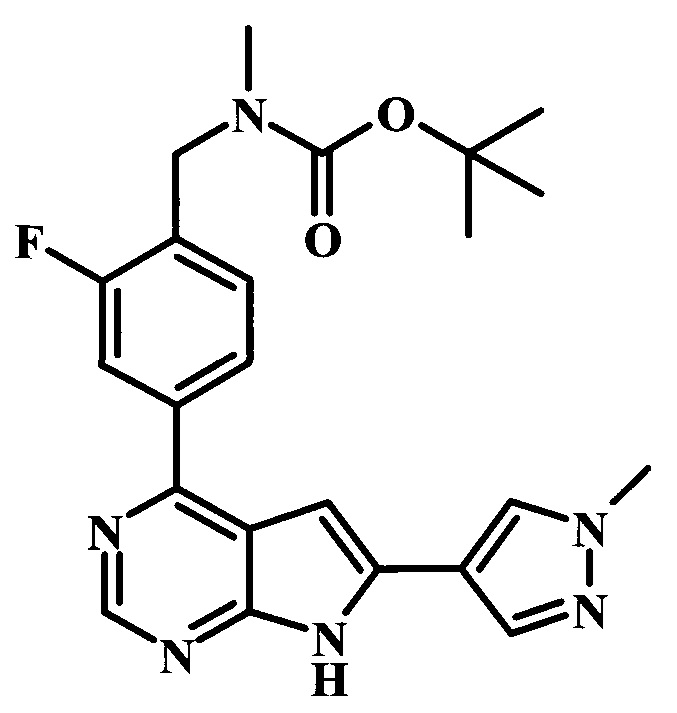
В сцинтилляционном флаконе на 20 мл 7-бензолсульфонил-4-хлор-6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин (150 мг; 0,401 ммоль; Экв.: 1,00), трет-бутиловый эфир [2-фтор-4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-бензил]-метилкарбаминовой кислоты (161 мг; 0,441 ммоль; Экв.: 1,1) и карбонат калия (222 мг; 1,61 ммоль; Экв.: 4,00) в 3 мл воды соединяли с DME (6 мл). Вносили Pd(PPh3)4 (46,4 мг; 0,04 ммоль; Экв.: 0,1) и эту реакционную смесь нагревали при 160°С в течение 60 мин. Реакционную смесь разбавляли с помощью EtOAc и промывали солевым раствором. Объединенные органические фазы высушивали над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении и неочищенное вещество очищали хроматографией на колонке (силикагель, 5-70% этилацетат в гексане). Указанное в заголовке соединение получали в виде твердого вещества (74 мг; выход 42%). LC/MS: m/z расч. для C23H25FN6O2 ([M+H]+): 437,4 Фактич.: 437,3
Стадия 5: 4-трет-Бутил-N-{2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-N-метилбензамид
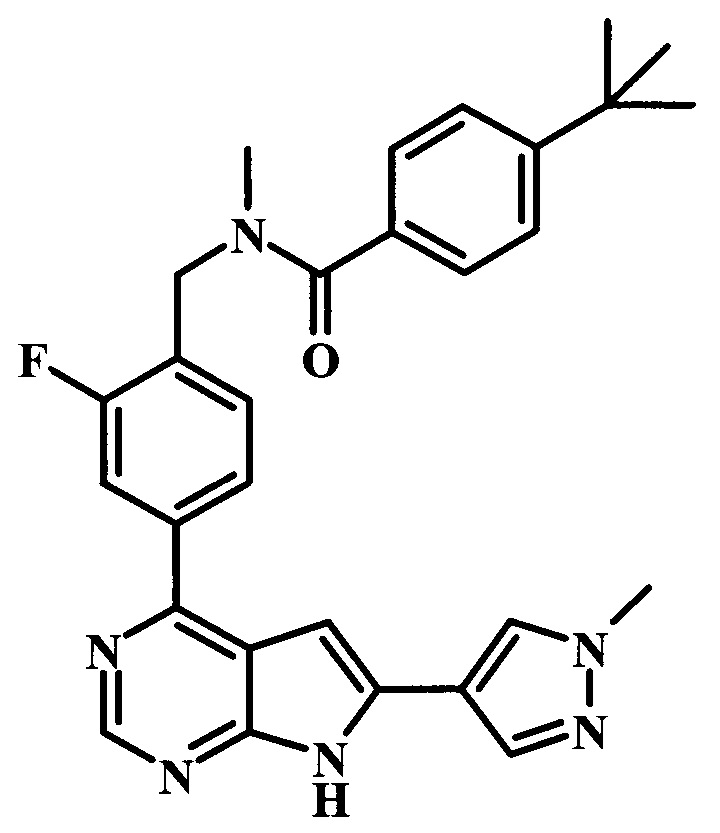
В сцинтилляционном флаконе на 20 мл трет-бутиловый эфир {2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-метил-карбаминовой кислоты (40 мг; 0,092 ммоль; Экв.: 1,00) объединяли с DCM (3 мл) и TFA (3 мл). Полученный раствор перемешивали при комнатной температуре в течение 30 мин. Растворитель удаляли при пониженном давлении. Неочищенное вещество растворяли в DMF (4 мл). Вносили 4-трет-бутилбензойную кислоту (18 мг; 0,101 ммоль; Экв.: 1,1), DIPEA (0,1 мл; 0,573 ммоль; Экв.: 6,25) и HATU (38 мг; 0,101 ммоль; Экв.: 1,1). Реакционную смесь перемешивали в течение ночи. Полученный раствор разбавляли с помощью 10 мл воды и 5 мл EtOAc. Этот раствор перемешивали при комнатной температуре в течение еще 30 мин. Органические фазы экстрагировали и высушивали над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенное вещество очищали хроматографией на колонке (силикагель, 0-10% МеОН в DCM). Указанное в заголовке соединение получали в виде твердого вещества (38 мг; выход 84%). LC/MS: m/z расч. для C29H29FN6O ([M+H]+): 497,5 Фактич.: 497,4
Пример I-32
{2-Фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-метиламид 5-метилтиофен-2-карбоновой кислоты
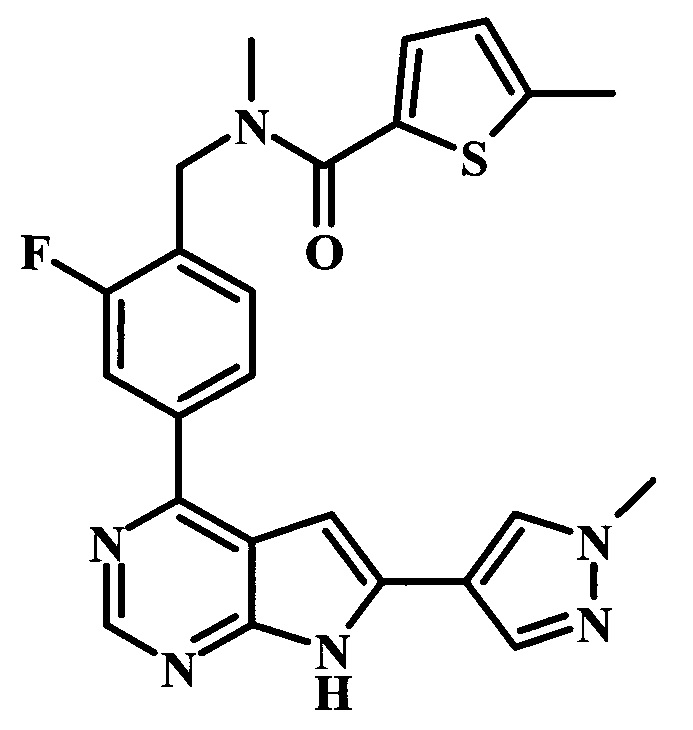
В сцинтилляционном флаконе на 20 мл трет-бутиловый эфир {2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-метил-карбаминовой кислоты (30 мг; 0,069 ммоль; Экв.: 1,00) объединяли с DCM (3 мл) и TFA (3 мл). Полученный раствор перемешивали при комнатной температуре в течение 30 мин. Растворитель удаляли при пониженном давлении. Неочищенное вещество растворяли в DMF (4 мл). Вносили 5-метилтиофен-2-карбоновую кислоту (11 мг; 0,076 ммоль; Экв.: 1,1), DIPEA (0,05 мл; 0,275 ммоль; Экв.: 4,00) и HATU (29 мг; 0,076 ммоль; Экв.: 1,1). Реакционную смесь перемешивали в течение ночи. Полученный раствор разбавляли с помощью 10 мл воды и 5 мл EtOAc. Полученный раствор перемешивали при комнатной температуре в течение еще 30 мин. Органические фазы экстрагировали и высушивали над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенное вещество очищали хроматографией на колонке (силикагель, 0-10% МеОН в DCM). Указанное в заголовке соединение получали в виде твердого вещества (14 мг; выход 44%). LC/MS: m/z расч. для C24H21FN6OS ([M+H]+): 461,5 Фактич.: 461,2
Пример I-33
2-трет-Бутил-5-{2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-4,5-дигидротиено[2,3-с]пиррол-6-он
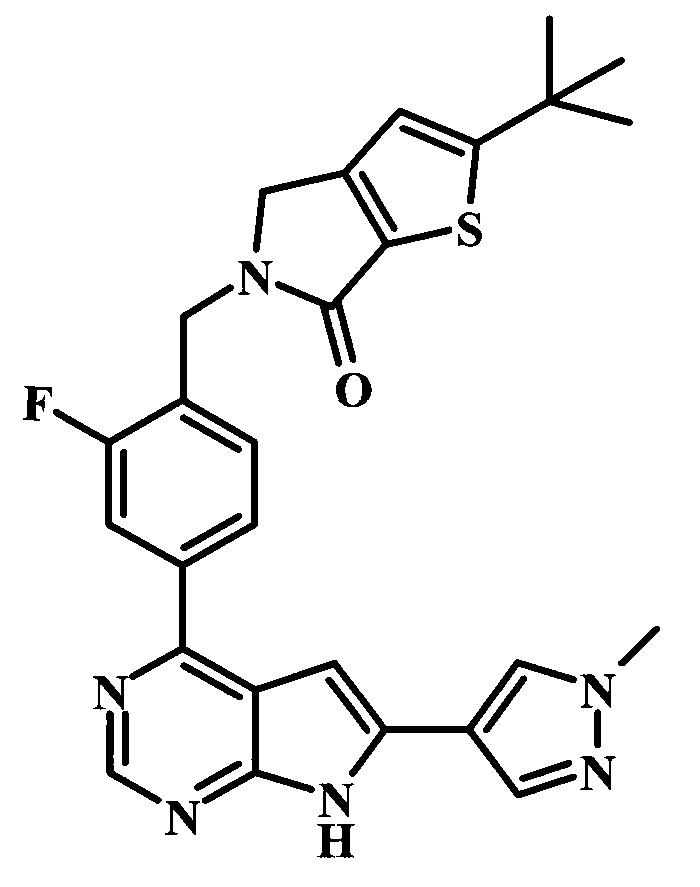
Стадия 1: Метиловый эфир 3-метилтиофен-2-карбоновой кислоты
В круглодонной колбе на 1 л 3-метилтиофен-2-карбоновую кислоту (15 г; 106 ммоль) объединяли с метанолом (211 мл) с получением грязно-белой суспензии. Эту смесь охлаждали до 0°С в воде со льдом. К холодной суспензии добавляли по каплям концентрированную серную кислоту (6 мл; 113 ммоль). Реакционную смесь перемешивали, постепенно доводя до комнатной температуры. Смесь перемешивали при комнатной температуре в течение трех дней. По истечении этого времени ТСХ показала полное превращение исходного вещества в менее полярный продукт. Реакционную смесь концентрировали для удаления метанола. Оставшееся светло-коричневое масло делили между этилацетатом и насыщенным водным бикарбонатом натрия. Органическую фазу высушивали над Na2SO4, фильтровали и концентрировали с получением коричневого масла, которое содержало смесь целевого метилового эфира (84%) и исходного вещества (16%), по данным 1H ЯМР-интегрирования. Неочищенный продукт перерастворяли в этилацетате и полученный раствор промывали 1 М водным NaOH. Органическую фазу высушивали над MgSO4, фильтровали и концентрировали с получением метилового эфира 3-метилтиофен-2-карбоновой кислоты (13,6 г; 82%) в виде светло-коричневого масла. 1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ ppm 7,39 (d, J=5,09 Гц, 1H), 6,92 (d, J=5,20 Гц, 1H), 3,87 (s, 3H), 2,57 (s, 3H).
Стадия 2: Метиловый эфир 5-трет-бутил-3-метилтиофен-2-карбоновой кислоты
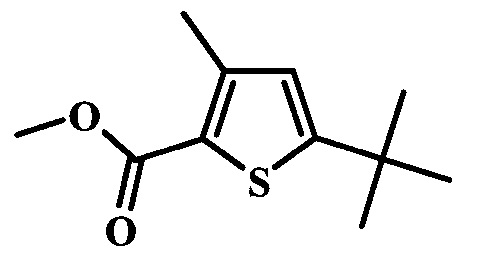
В круглодонной колбе на 500 мл трихлорид алюминия (17,3 г; 130 ммоль) объединяли с DCM (20 мл) с получением грязно-белой суспензии. Сосуд с этой смесью заполняли аргоном и затем охлаждали до -78°С на бане с сухим льдом/ацетоном. Добавляли по каплям раствор метил-3-метилтиофен-2-карбоксилата (13,5 г; 86,4 ммоль) в 10 мл DCM в течение 5 мин. Реакционную смесь перемешивали при -78°С в течение 5 мин. К холодной реакционной смеси добавляли по каплям раствор 2-хлор-2-метилпропана (9,87 мл; 90,7 ммоль) в 10 мл DCM в течение 30 мин. Реакционную смесь перемешивали в течение выходных с обратным холодильником на бане со смесью сухого льда/ацетона, которая постепенно плавилась, и доводили реакционную колбу до комнатной температуры. Реакционную смесь вливали в лед с водой. После того как лед растаял, органическую фазу отделяли и затем высушивали над Na2SO4. Органическую фазу фильтровали, затем концентрировали с получением коричневого масла. Это масло наносили непосредственно на 330-граммовую колонку с силикагелем. С помощью флэш-хроматографии (0-5% EtOAc-гексан) выделяли метиловый эфир 5-трет-бутил-3-метилтиофен-2-карбоновой кислоты (7,05 г; 38%) в виде желтого масла. 1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ ppm 6,68 (s, 1H), 3,84 (s, 3H), 2,50 (s, 3H), 1,38 (s, 9H).
Стадия 3: Метиловый эфир 3-бромметил-5-трет-бутилтиофен-2-карбоновой кислоты
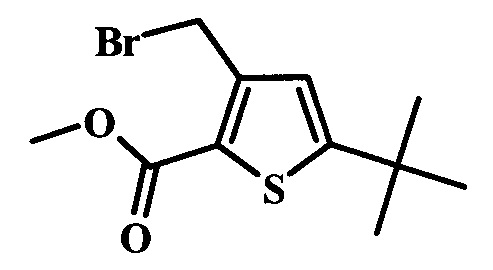
В грушевидной колбе на 1 л метил-5-трет-бутил-3-метилтиофен-2-карбоксилат (6,06 г; 28,5 ммоль), N-бромсукцинимид (6,1 г; 34,3 ммоль) и азобисизобутиронитрил (234 мг; 1,43 ммоль) соединяли с четыреххлористым углеродом (80 мл) с получением оранжевой суспензии. Эту смесь нагревали при 90°С в течение ночи. На утро реакционную смесь охлаждали до комнатной температуры и затем фильтровали для удаления выпавшего в осадок твердого вещества. Фильтрат концентрировали до состояния коричневого масла. Этот продукт наносили непосредственно на 120-граммовую колонку с силикагелем. С помощью флэш-хроматографии (5% EtOAc-гексан) удалось лишь частично очистить этот продукт. Фракции, содержащие чистый продукт, соединяли с получением метилового эфира 3-бромметил-5-трет-бутилтиофен-2-карбоновой кислоты (2,65 г; 32%) в виде желтого масла. Второй прогон хроматографии на колонке проводили с неочищенными фракциями, поученными выше (снова с использованием 120-граммовой колонке силикагель и 5% EtOAc-гексан). Получали еще одну партию очищенного метилового эфира 3-бромметил-5-трет-бутилтиофен-2-карбоновой кислоты (2,54 г; 30%). 1H ЯМР (300 МГц, ХЛОРОФОРМ-d) δ ppm 6,93 (s, 1H), 4,87 (s, 2H), 3,88 (s, 3H), 1,39 (s, 8H).
Стадия 4: Метиловый эфир 3-[(4-бром-2-фторбензиламино)-метил]-5-трет-бутилтиофен-2-карбоновой кислоты
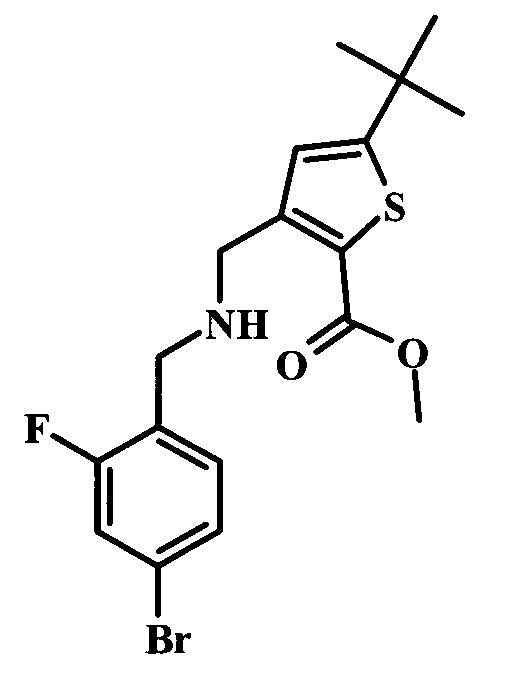
В круглодонной колбе на 250 мл 4-бром-2-фторбензиламин (5,34 г; 26,2 ммоль), метиловый эфир 3-бромметил-5-трет-бутилтиофен-2-карбоновой кислоты (2,54 г; 8,72 ммоль) и карбонат цезия (3,73 г; 11,4 ммоль) соединяли с ацетонитрилом (50 мл) с получением белой суспензии. Реакционную смесь перемешивали в течение выходных при комнатной температуре. Реакционную смесь фильтровали, затем фильтрат концентрировали на роторном испарителе. Неочищенный продукт наносили непосредственно на 120-граммовую колонку с силикагелем. В ходе флэш-хроматографии (5-25% EtOAc-гексан) получали метиловый эфир 3-[(4-бром-2-фторбензиламино)-метил]-5-трет-бутилтиофен-2-карбоновой кислоты (1,88 г; 52%) в виде бледно-желтого масла. LC/MS: m/z расч. для C18H22BrFN ([М+Н]+): 414 и 416 Фактич.: 416,0
Стадия 5: 3-[(4-Бром-2-фторбензиламино)-метил]-5-трет-бутилтиофен-2-карбоновая кислота
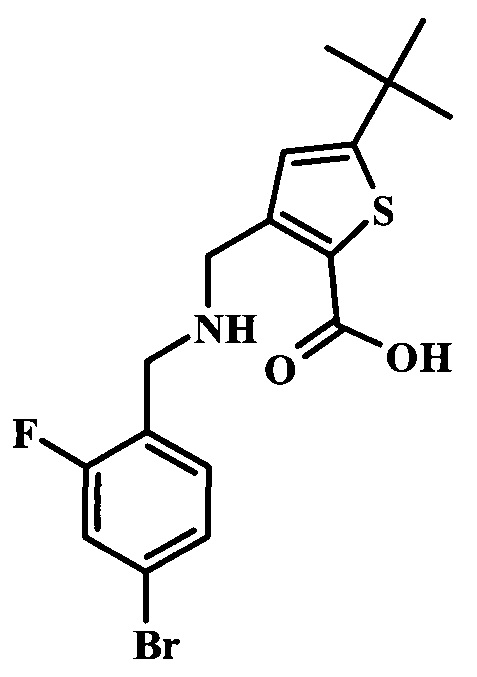
В грушевидной колбе на 1 л метиловый эфир 3-[(4-бром-2-фторбензиламино)-метил]-5-трет-бутилтиофен-2-карбоновой кислоты (1,85 г; 4,47 ммоль) и гидроксид лития моногидрат (1,87 г; 44,7 ммоль) соединяли с THF (12 мл) и водой (12 мл) с получением бесцветного суспензии. Эту смесь перемешивали при комнатной температуре в течение ночи. На утро с помощью LCMS было выявлено преимущественно исходное вещество и небольшое количество продукта. Добавляли метанол (5 мл) и эту реакционную смесь нагревали при 50°С в течение 20 часов. Реакционную смесь охлаждали до комнатной температуры и концентрировали досуха на роторном испарителе. Полученное грязно-белое твердое вещество частично растворяли в воде, затем добавляли 4 н водный HCl до превращения смеси в белую суспензию. Эту суспензию экстрагировали этилацетатом. Органическую фазу высушивали (Na2SO4), фильтровали и затем концентрировали с получением 3-[(4-бром-2-фторбензиламино)-метил]-5-трет-бутилтиофен-2-карбоновой кислоты (1,77 г; 99%) в виде грязно-белой пены. LC/MS: m/z расч. для C17H20BrFNO ([М+Н]+): 400 и 402 Фактич.: 402,0
Стадия 6: 5-(4-Бром-2-фторбензил)-2-трет-бутил-4,5-дигидротиено[2,3-с]пиррол-6-он
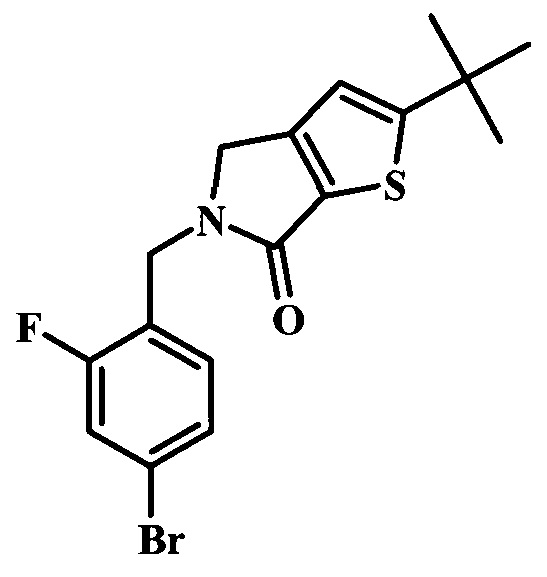
В круглодонной колбе на 1 л 3-((4-бром-2-фторбензиламино)метил)-5-трет-бутилтиофен-2-карбоновую кислоту (1,77 г; 4,42 ммоль) объединяли с метиленхлоридом (80 мл) с получением светло-желтого раствора. Сосуд с реакционной смесью заполняли аргоном, затем добавляли по каплям тионилхлорид (1,96 г; 1,2 мл; 16,4 ммоль) в течение 5 мин. Реакционную смесь перемешивали при комнатной температуре в атмосфере аргона в течение 18 часов. По истечении этого времени LCMS-анализ выявил смесь исходного вещества и продукта. К реакционной смеси добавляли еще 1,5 мл тионилхлорида и эту реакционную смесь перемешивали еще в течение 24 часов при комнатной температуре. По истечении этого времени LCMS-анализ показал завершение реакции. Реакционную смесь концентрировали с получением коричневато-желтого масла. Этот неочищенный продукт растворяли в метиленхлориде и полученный раствор концентрировали над силикагелем. Неочищенный продукт на силикагеле наносили на 120-граммовую колонку с силикагелем. В ходе флэш-хроматографии (5-25% EtOAc-гексан) получали 5-(4-бром-2-фторбензил)-2-трет-бутил-4,5-дигидротиено[2,3-с]пиррол-6-он (1,22 г; 72%) в виде бледно-желтого масла. LC/MS: m/z расч. для C17H18BrFNOS ([М+Н]+): 382 и 384 Фактич.: 384,0
Стадия 7: 2-трет-Бутил-5-[2-фтор-4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-бензил]-4,5-дигидротиено[2,3-с]пиррол-6-он
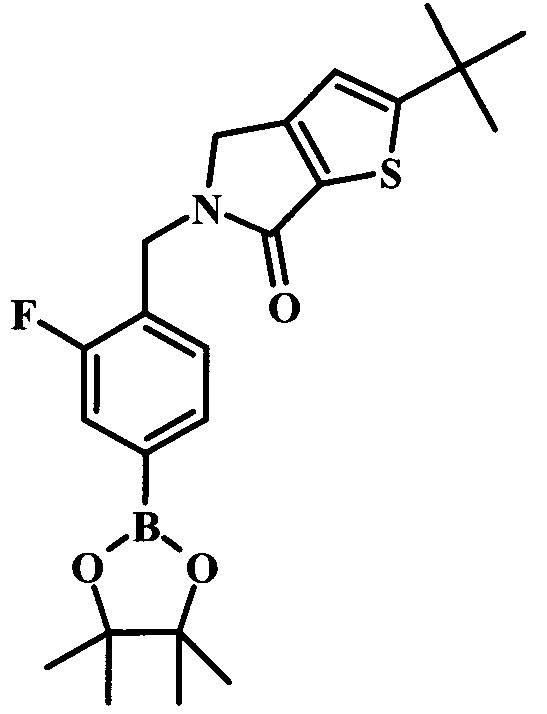
В круглодонной колбе на 250 мл бис(пинаколато)диборон (1,15 г; 4,53 ммоль), 5-(4-бром-2-фторбензил)-2-трет-бутил-4,5-дигидротиено[2,3-с]пиррол-6-он (1,07 г; 2,8 ммоль) и ацетат калия (825 мг; 8,41 ммоль) соединяли с диоксаном (9 мл) с получением темно-коричневой суспензии. К этой смеси добавляли комплекс дихлорида 1,1'-бис(дифенилфосфино)ферроцен-палладия(II) и дихлорметана (185 мг; 227 мкмоль). Реакционную смесь нагревали при 110°С в течение восьми часов. По истечении этого времени реакционную смесь охлаждали до комнатной температуры и диоксан выпаривали. Неочищенный продукт растворяли в метиленхлориде, затем этот раствор вливали в воду (30 мл). Органическую фазу отделяли и затем высушивали над MgSO4, фильтровали и концентрировали над силикагелем. Неочищенный продукт на силикагеле наносили на 120-граммовую колонку с силикагелем. В ходе флэш-хроматографии (5-25% этилацетат в гексане) получали 2-трет-бутил-5-[2-фтор-4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-бензил]-4,5-дигидротиено[2,3-с]пиррол-6-он (0,72 г; 60%) в виде белого порошка. LC/MS: m/z расч. для C23H30BFNO3S ([М+Н]+): 430 Фактич.: 430,2
Стадия 8: 2-трет-Бутил-5-{2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-4,5-дигидротиено[2,3-с]пиррол-6-он
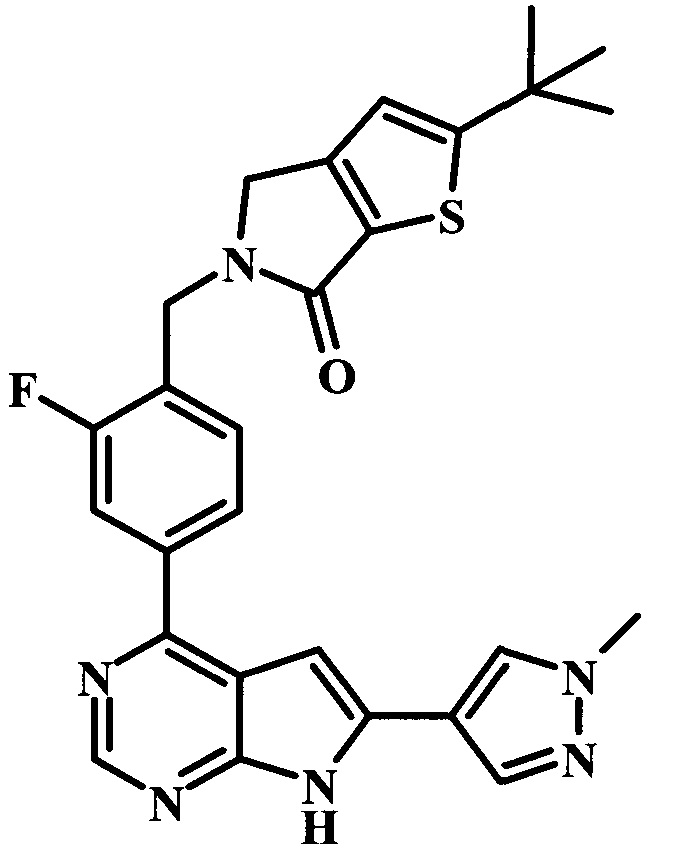
В трубке для микроволнового облучения на 10 мл 7-бензолсульфонил-4-хлор-6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин (150 мг; 0,401 ммоль; Экв.: 1,00), 2-трет-бутил-5-[2-фтор-4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-бензил]-4,5-дигидротиено[2,3-с]пиррол-6-он (190 мг; 0,441 ммоль; Экв.: 1,1) и карбонат калия (222 мг; 1,61 ммоль; Экв.: 4,00) в 2 мл воды соединяли с DME (4 мл). Добавляли Pd(Ph3P)4 (46 мг; 0,04 ммоль; Экв.: 0,1). Реакционную смесь нагревали микроволновым облучением при 160°С в течение 60 мин. Полученный раствор разбавляли с помощью EtOAc, затем промывали солевым раствором. Объединенные органические фазы высушивали над безводным сульфатом натрия и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали хроматографией на колонке (силикагель, 0-10% МеОН в DCM). Указанное в заголовке соединение получали в виде светло-желтого твердого вещества (17 мг; выход 9%). LC/MS: m/z расч. для C27H25FN6OS ([M+H]+): 501,6 Фактич.: 501,3
Пример I-34
2-Фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензиламид 5-трет-бутилизоксазол-3-карбоновой кислоты
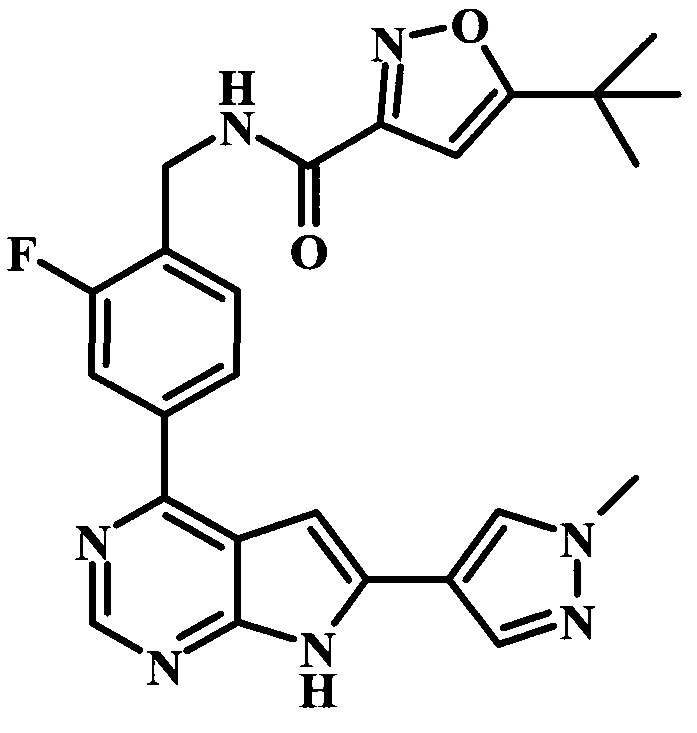
В сцинтилляционном флаконе на 20 мл трет-бутиловый эфир {2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-метил-карбаминовой кислоты (75 мг; 0,178 ммоль; Экв.: 1,00) объединяли с DCM (3 мл) и TFA (3 мл). Полученный раствор перемешивали при комнатной температуре в течение 30 мин. Растворитель удаляли при пониженном давлении. Неочищенное вещество растворяли в DMF (4 мл). Вносили 5-трет-бутилизоксазол-3-карбоновую кислоту (18 мг; 0,195 ммоль; Экв.: 1,1), DIPEA (0,124 мл; 0,710 ммоль; Экв.: 4,00) и HATU (74 мг; 0,195 ммоль; Экв.: 1,1). Реакционную смесь перемешивали в течение ночи. Полученный раствор разбавляли с помощью 10 мл воды и 5 мл EtOAc. Этот раствор перемешивали при комнатной температуре в течение еще 30 мин. Органические фазы объединяли и высушивали над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенное вещество очищали хроматографией на колонке (силикагель, 0-10% МеОН в DCM). Указанное в заголовке соединение получали в виде желтого твердого вещества (35 мг; выход 42%). LC/MS: m/z расч. для C25H24FN7O2 ([М+Н]+): 474,5 Фактич.: 474,3
Пример I-35
N-{2-Фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-4-(3-метилоксетан-3-ил)-бензамид
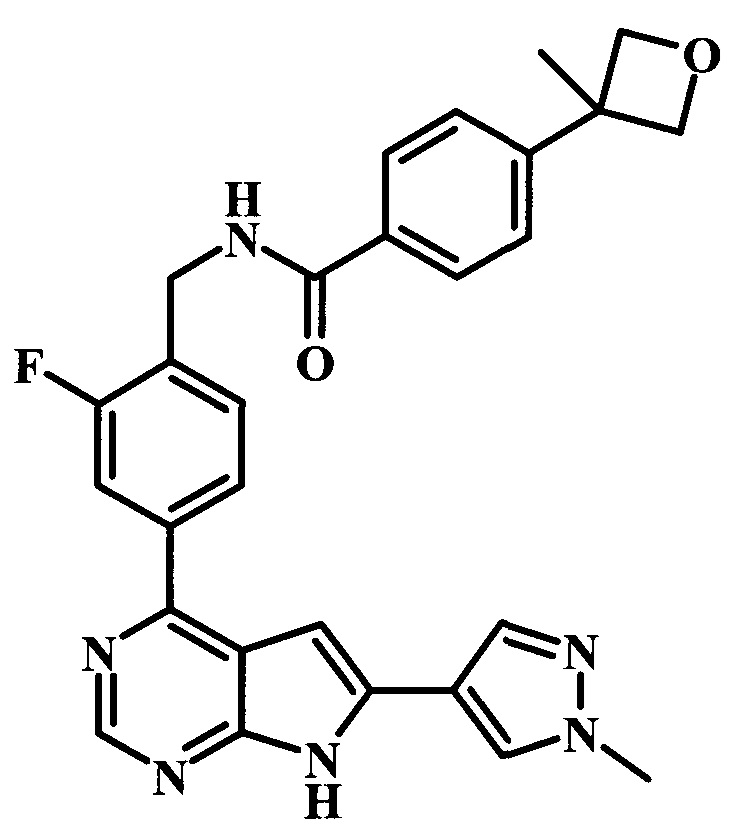
В герметизируемой трубке на 10 мл трет-бутиловый эфир {2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-метилкарбаминовой кислоты (65 мг; 0,155 ммоль; Экв.: 1,00) объединяли с DCM (3 мл) и TFA (3 мл). Полученный раствор перемешивали при комнатной температуре в течение 60 мин. Растворитель удаляли при пониженном давлении и высушивали в вакууме. Неочищенное вещество растворяли в DMF (5 мл). Вносили 4-(3-метилоксетан-3-ил)бензойную кислоту (59,6 мг; 0,31 ммоль; Экв.: 2,00), DIPEA (0,14 мл; 0,78 ммоль; Экв.: 5,00) и HATU (118 мг; 0,31 ммоль; Экв.: 2,00). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Полученный раствор разбавляли с помощью EtOAc, промывали водой и солевым раствором. Объединенные органические фазы высушивали над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенное вещество очищали хроматографией на колонке (силикагель, 0-10% МеОН в DCM). Указанное в заголовке соединение получали в виде твердого вещества (44 мг; выход 57%). LC/MS: m/z Расч. для C28H25FN6O2 ([М+Н]+): 497,5 Фактич.: 497,2
Пример I-36
4-(Цианодиметилметил)-N-{2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-бензамид

По аналогии со способом, описанным в примере 35, из 4-(2-цианопропан-2-ил)бензойной кислоты (58,7 мг; 0,310 ммоль; Экв.: 2,00) получали указанное в заголовке соединение в виде твердого вещества (43 мг; выход 53%). LC/MS: m/z Расч. для C28H24FN7O ([М+Н]+): 494,5 Фактич.: 494,2
Пример I-37
2-Фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензиламид 4,5,6,7-тетрагидробензо[b]тиофен-2-карбоновой кислоты
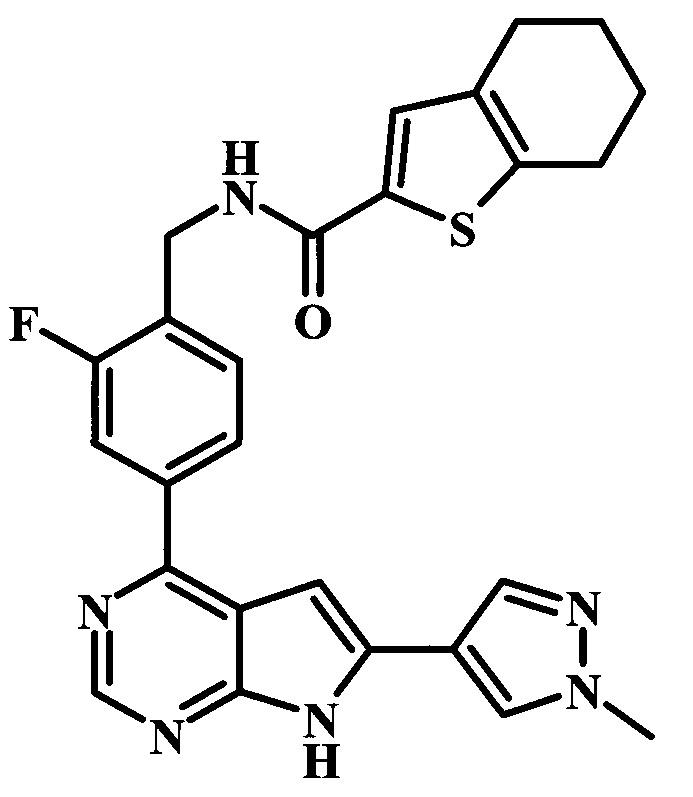
По аналогии со способом, описанным в примере 35, из 4,5,6,7-тетрагидро-бензо[b]тиофен-2-карбоновой кислоты (57 мг; 0,31 ммоль; Экв.: 2,00) получали указанное в заголовке соединение в виде твердого вещества (53 мг; выход 63%). LC/MS: m/z Расч. для C26H23FN6OS ([М+Н]+): 487,5 Фактич.: 487,2
Пример I-38
N-{2-Фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-4-(1-гидрокси-1-метилэтил)-бензамид
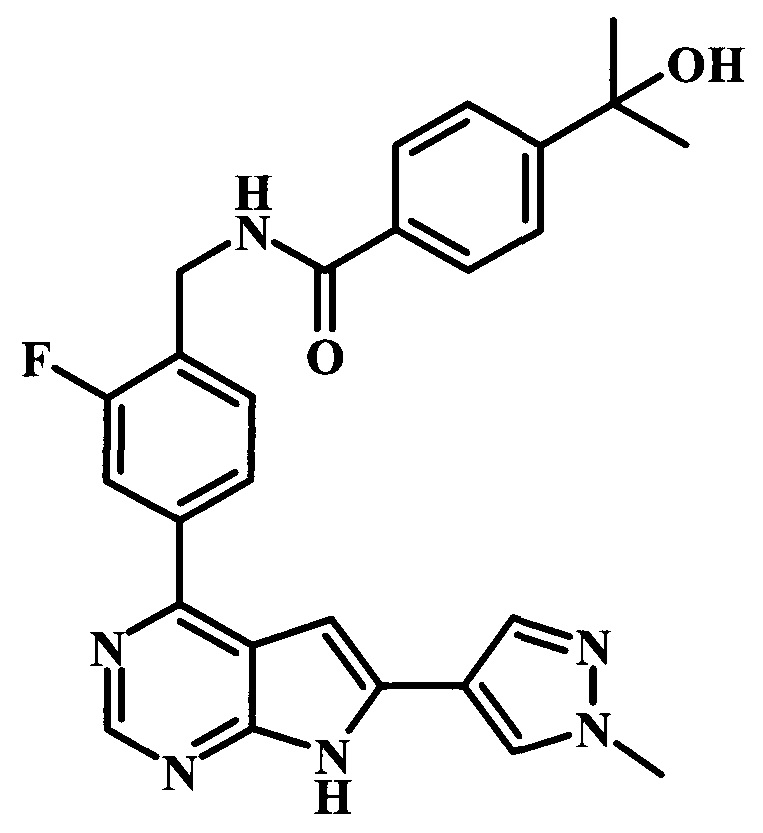
Стадия 1: 4-(1-Гидрокси-1-метилэтил)-бензойная кислота
В круглодонной колбе на 500 мл 4-изопропилбензойную кислоту (1,0 г; 6,09 ммоль) объединяли с 5 мл 10% KOH в воде с получением мутной суспензии. Вносили KOH в воде (96 мл; 19,2 ммоль) и перманганат калия (1,92 г; 12,2 ммоль) в 100 мл воды. Реакционную смесь нагревали при 70°С в течение 1 час. К этой смеси добавляли 5 капель глицерина. Реакционную смесь охлаждали до 0°С. Твердый остаток фильтровали через слой Celite. Фильтрат дважды промывали эфиром. Объединенные органические фазы промывали солевым раствором, высушивали над безводным сульфатом натрия, концентрировали и высушивали в течение ночи. Полученный продукт, 4-(1-гидрокси-1-метилэтил)-бензойную кислоту, собирали в виде белого твердого вещества (870 мг; 79%) и использовали на следующей стадии без дополнительной очистки. 1H ЯМР (400 МГц, DMSO-d6) δ ppm 12,80 (br. s, 1H), 7,83-7,89 (m, 2H), 7,55-7,60 (m, 2H), 5,15 (s, 1H), 1,43 (s, 6H).
Стадия 2: N-{2-Фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-4-(1-гидрокси-1-метилэтил)-бензамид

По аналогии со способом, описанным в примере 35, из 4-(2-гидроксипропан-2-ил)бензойной кислоты (41,4 мг; 0,230 ммоль; Экв.: 2,00) получали указанное в заголовке соединение в виде твердого вещества (46 мг; выход 82%). LC/MS: m/z Расч. для C27H25FN6O2 ([М+Н]+): 485,5 Фактич.: 485,4
Пример I-39
2-Фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензиламид 3-трет-бутилизоксазол-5-карбоновой кислоты
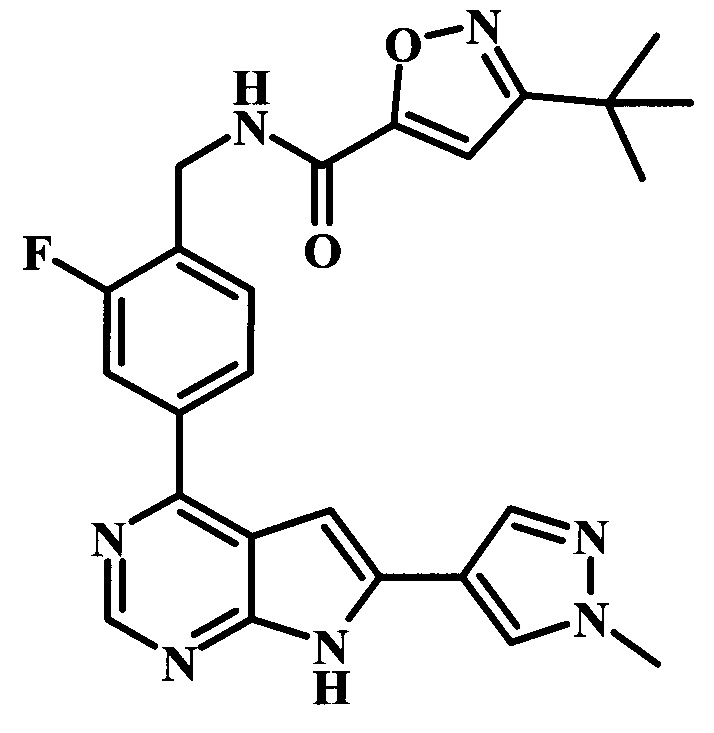
Стадия 1: Метиловый эфир 3-трет-бутилизоксазол-5-карбоновой кислоты
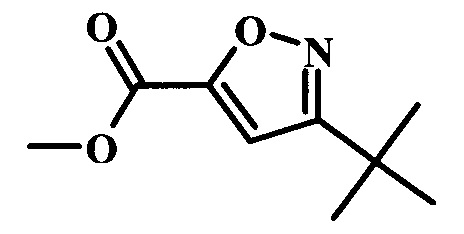
К раствору триметилуксуного альдегида (1,0 г; 11,6 ммоль) в смеси 1:1 трет-бутиловый спирт/вода (40 мл) добавляли гидроксиламин гидрохлорид (807 мг; 11,6 ммоль) и гидроксид натрия (464 мг; 11,6 ммоль). Смесь перемешивали при комнатной температуре в течение 30 мин, после чего добавляли порциями хлорамин-Т (5,49 г; 23,4 ммоль) в течение 5 мин, а затем сульфат меди(II) (327 мг; 1,3 ммоль), порошок меди (73,8 мг; 1,16 ммоль) и метилпропиоанат (976 мг; 11,6 ммоль). Реакционную смесь нагревали с обратным холодильником и выдерживали в таком режиме в течение 2 ч. По истечении этого времени смесь охлаждали до комнатной температуры и вливали в смесь лед/вода (50 г). Добавляли гидроксид аммония (10 мл) и этот раствор экстрагировали с помощью DCM (3×200 мл). Органические слои объединяли, высушивали (Na2SO4), фильтровали и концентрировали при пониженном давлении. Неочищенное вещество очищали флэш-хроматографией (силикагель, 40 г; от 0% до 10% EtOAc в гексане) с получением метилового эфира 3-трет-бутилизоксазол-5-карбоновой кислоты (427 мг; 20%) в виде бесцветного масла. LC/MS: m/z Расч. для C9H13NO3 [(М+Н)]+ 184 Фактич.: 184,1
Стадия 2: 3-трет-Бутилизоксазол-5-карбоновая кислота
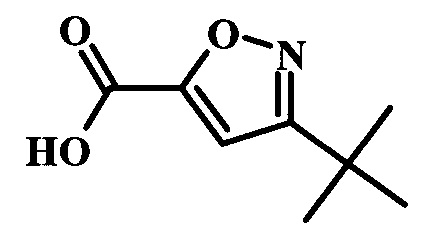
К раствору метилового эфира 3-трет-бутилизоксазол-5-карбоновой кислоты (425 мг; 2,32 ммоль) в метаноле (4 мл) добавляли 1 н водный NaOH (11,6 мл; 11,6 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 час, затем концентрировали и нейтрализовали 1 н хлороводородной кислотой (10 мл). Эту смесь экстрагировали этилацетатом, высушивали над сульфатом натрия, фильтровали и концентрировали с получением 3-трет-бутилизоксазол-5-карбоновой кислоты (318 мг; 81%) в виде белого полутвердого вещества. LC/MS: m/z Расч. для C8H11NO3 [(M+H)+] 170 Фактич.: 170
Стадия 3: 2-Фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензиламид 3-трет-бутилизоксазол-5-карбоновой кислоты
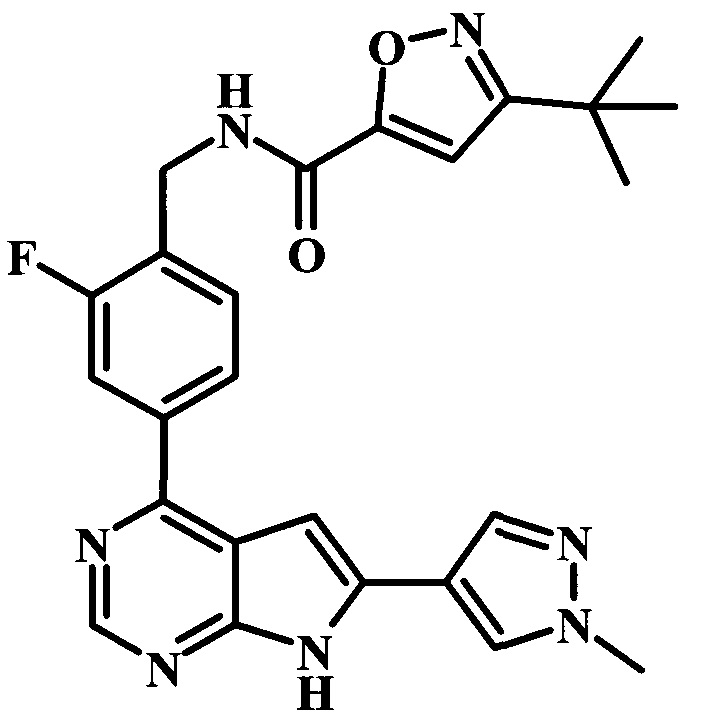
По аналогии со способом, описанным в примере 35, из 3-трет-бутилизоксазол-5-карбоновой кислоты (39 мг; 0,230 ммоль; Экв.: 2,00) получали указанное в заголовке соединение в виде твердого вещества (39 мг; выход 71%). LC/MS: m/z Расч. для C25H24FN7O2 ([М+Н]+): 474,5. Фактич.: 474,3.
Пример I-40
2-Фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензиламид 3-трет-бутоксиазетидин-1-карбоновой кислоты
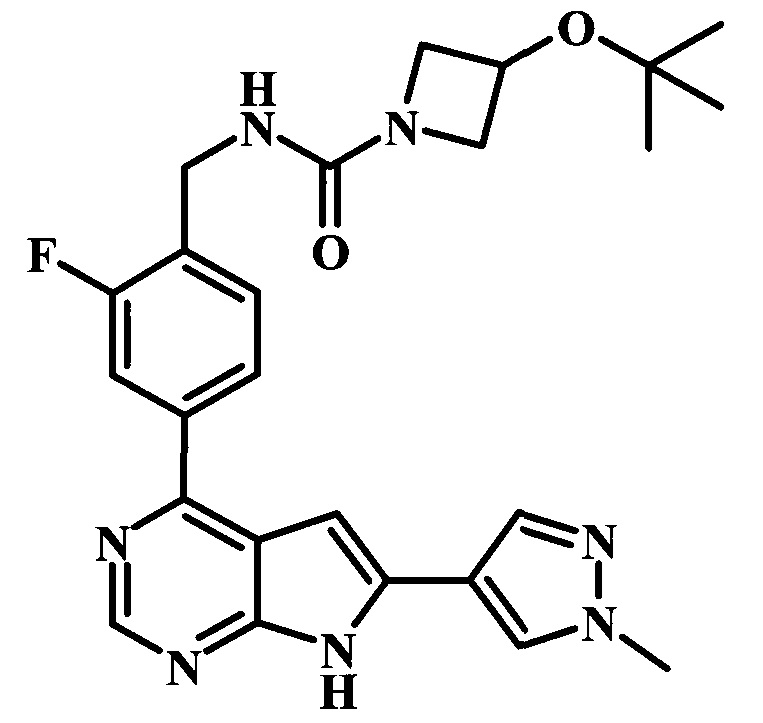
В герметизируемом сосуде на 20 мл для микроволнового облучения трет-бутиловый эфир {2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-метилкарбаминовой кислоты (65 мг; 0,155 ммоль; Экв.: 1,00) объединяли с DCM (3 мл) и TFA (3 мл). Полученный раствор перемешивали при комнатной температуре в течение 60 мин. Растворитель удаляли при пониженном давлении и высушивали в вакууме. Неочищенное вещество растворяли в DMF (4 мл). К реакционной смеси добавляли ди(1Н-имидазол-1-ил)метанон (50,3 мг; 0,31 ммоль; Экв.: 2,00) и DIPEA (0,14 мл; 0,78 ммоль; Экв.: 5,00) с получением светло-желтого раствора. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Добавляли 3-трет-бутоксиазетидин (40 мг; 0,310 ммоль; Экв.: 2,00) и перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли с помощью EtOAc и промывали солевым раствором. Органические фазы объединяли и высушивали над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенное вещество очищали хроматографией на колонке (силикагель, 0-10% МеОН в DCM). Указанное в заголовке соединение получали в виде желтого твердого вещества (32 мг; выход 43%). LC/MS: m/z Расч. для C25H28FN7O2 ([М+Н]+): 478,5 Фактич.: 478,6
Пример I-41
2-Фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензиламид 1,3-дигидроизоиндол-2-карбоновой кислоты
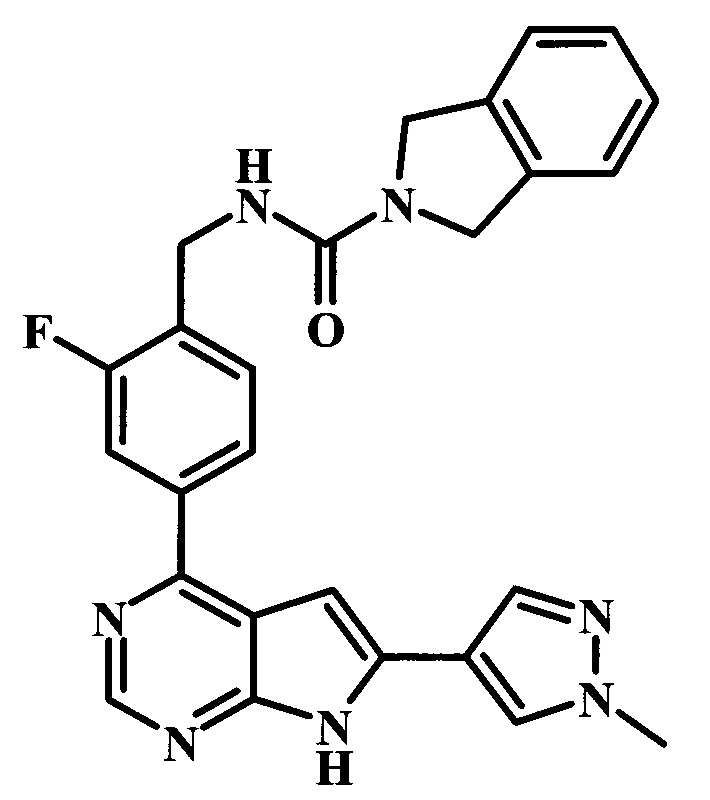
По аналогии со способом, описанным в примере 40, из изоиндолина (37,0 мг; 0,31 ммоль; Экв.: 2,00) получали указанное в заголовке соединение в виде твердого вещества (43 мг; выход 59%). LC/MS: m/z Расч. для C26H22FN7O ([М+Н]+): 468,5 Фактич.: 468,1
Пример 42
4-трет-Бутил-N-(4-{6-[1-(2-диметиламиноэтил)-1Н-пиразол-4-ил]-7Н-пирроло[2,3-d]пиримидин-4-ил}-2-фторбензил)-бензамид
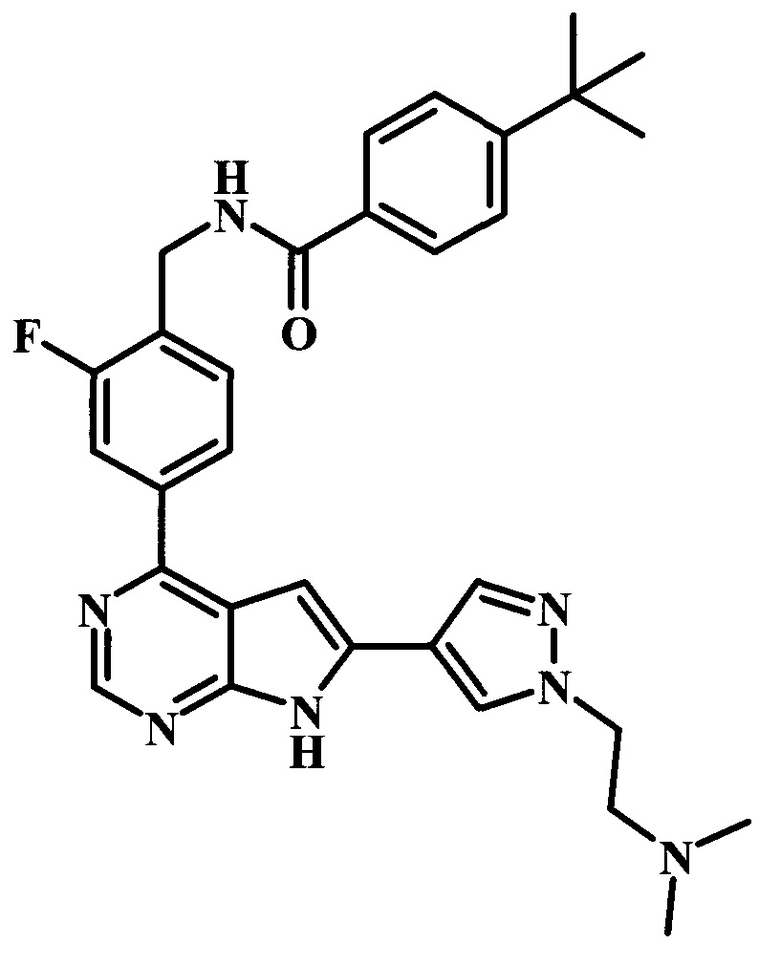
Стадия 1: N,N-Диметил-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол-1-ил)-этанамин
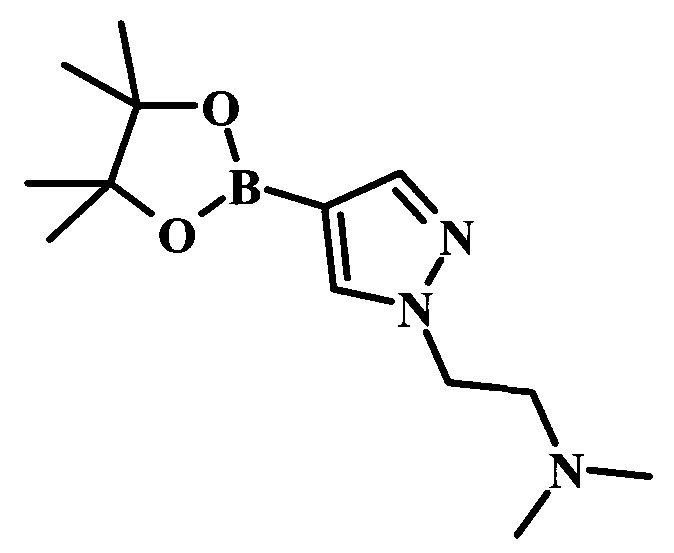
В круглодонной колбе на 250 мл 2-хлор-N,N-диметилэтанамин (998 мг; 9,28 ммоль), 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол (1,2 г; 6,18 ммоль) и карбонат цезия (4,03 г; 12,4 ммоль) соединяли с ацетонитрилом (20 мл) с получением белой суспензии. Реакционную смесь нагревали при 100°С в течение ночи. На утро эту реакционную смесь охлаждали до комнатной температуры, фильтровали и полученный фильтрат концентрировали с получением указанного в заголовке соединения (1,26 г; выход 77%) в виде бесцветного масла. Этот неочищенный продукт использовали в последующих реакциях без очистки.
Стадия 2: трет-Бутиловый эфир (4-{6-[1-(2-диметиламиноэтил)-1Н-пиразол-4-ил]-7Н-пирроло[2,3-d]пиримидин-4-ил}-2-фторбензил)-карбаминовой кислоты
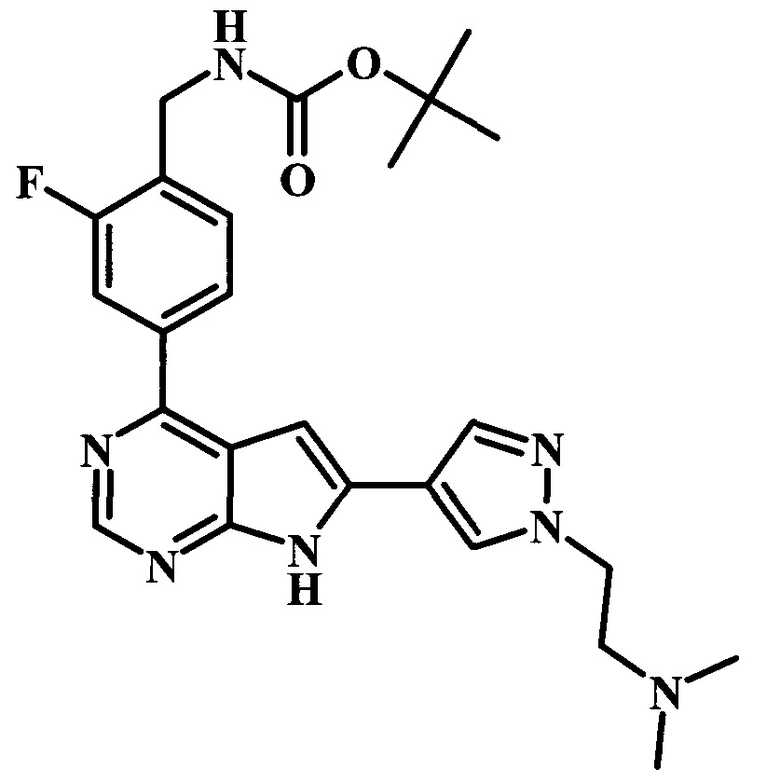
В герметизируемой трубке на 20 мл для микроволнового облучения трет-бутил-4-(6-бром-7-тозил-7Н-пирроло[2,3-d]пиримидин-4-ил)-2-фторбензилкарбамат (250 мг; 0,434 ммоль; Экв.: 1,00), N,N-диметил-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол-1-ил)этанамин (319 мг; 1,2 ммоль; Экв.: 2,77) и Pd(PPh3)4 (50 мг; 0,043 ммоль; Экв.: 0,1) соединяли с DME (4 мл) с получением светло-коричневой суспензии. Добавляли воду (1 мл), а затем карбонат калия (240 мг; 1,74 ммоль; Экв.: 4,00). Реакционную смесь нагревали при 150°С микроволнами в течение 1 ч. Реакционную смесь разбавляли с помощью EtOAc и промывали солевым раствором и вод. Органические фазы объединяли и высушивали над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенное вещество очищали хроматографией на колонке (силикагель, 0-10% МеОН в DCM). Указанное в заголовке соединение получали в виде желтого твердого вещества (62 мг; выход 29%). LC/MS: m/z Расч. для C25H30FN7O2 ([М+Н]+): 480,6 Фактич.: 480,3
Стадия 3: (2-{4-[4-(4-Аминометил-3-фторфенил)-7Н-пирроло[2,3-d]пиримидин-6-ил]-пиразол-1-ил}-этил)-диметиламин
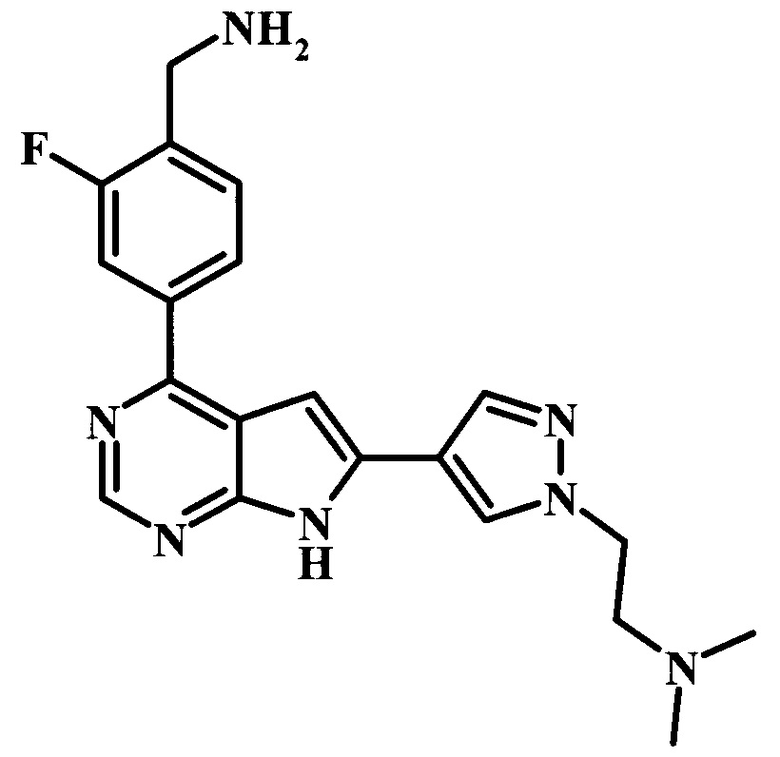
В круглодонной колбе на 100 мл трет-бутил-4-(6-(1-(2-(диметиламино)этил)-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил)-2-фторбензилкарбамат (62 мг; 0,129 ммоль; Экв.: 1,00) объединяли с DCM (8 мл) с получением светло-желтой суспензии. Добавляли TFA (4 мл; 51,9 ммоль; Экв.: 402) и перемешивали при комнатной температуре в течение 1 ч. Растворитель удаляли при пониженном давлении. Неочищенное вещество высушивали в высоком вакууме в течение 2 ч. Этот остаток использовали на следующей стадии без дополнительной очистки. LC/MS: m/z Расч. для C20H22FN7 ([М+Н]+): 380,4 Фактич.: 380,2
Стадия 4: 4-трет-Бутил-N-(4-{6-[1-(2-диметиламиноэтил)-1Н-пиразол-4-ил]-7Н-пирроло[2,3-d]пиримидин-4-ил}-2-фторбензил)-бензамид
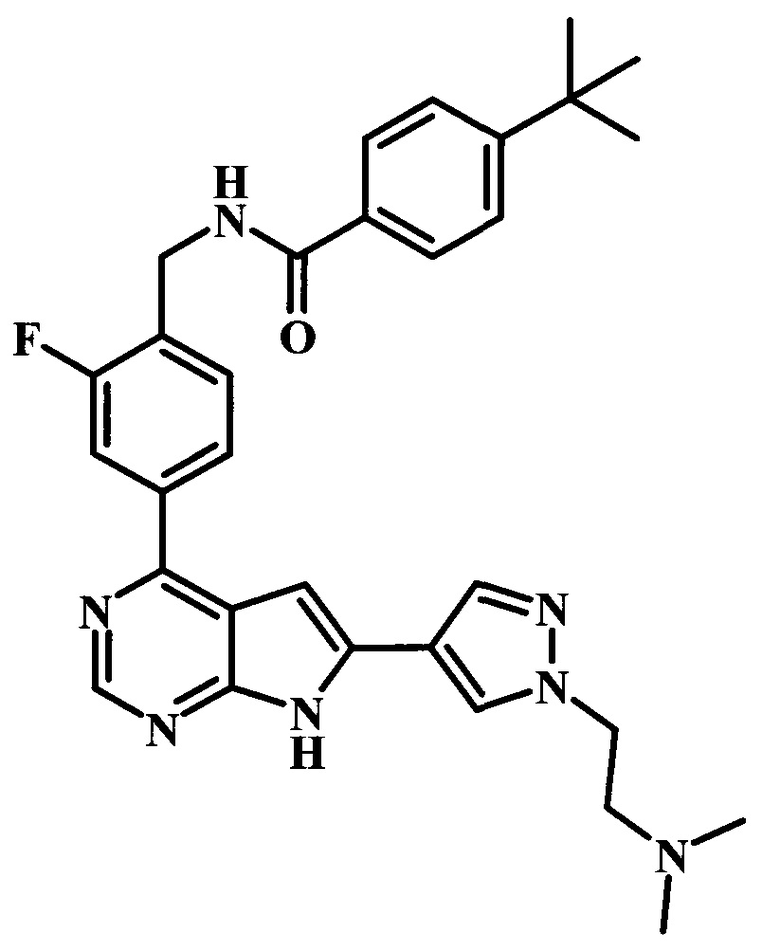
В герметизируемой трубке на 10 мл (2-{4-[4-(4-аминометил-3-фторфенил)-7Н-пирроло[2,3-d]пиримидин-6-ил]-пиразол-1-ил}-этил)-диметиламин (48 мг; 0,127 ммоль; Экв.: 1,00), 4-трет-бутилбензойную кислоту (45 мг; 0,253 ммоль; Экв.: 2,00) и HATU (96 мг; 0,253 ммоль; Экв.: 2,00) соединяли с DMF (4 мл) с получением желтого раствора. Реакционную смесь перемешивали в течение 5 мин, затем добавляли DIPEA (0,110 мл; 0,630 ммоль; Экв.: 5,00) и перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли с помощью EtOAc и промывали солевым раствором. Органические фазы объединяли и высушивали над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенное вещество очищали хроматографией на колонке (силикагель, 0-10% МеОН в DCM). Указанное в заголовке соединение получали в виде твердого вещества (49 мг; выход 71%). LC/MS: m/z Расч. для C31H34FN7O ([М+Н]+): 540,6 Фактич.: 540,3
Пример I-43
4-{6-[1-(2-Диметиламиноэтил)-1Н-пиразол-4-ил]-7Н-пирроло[2,3-d]пиримидин-4-ил}-2-фторбензиламид 3-трет-бутоксиазетидин-1-карбоновой кислоты
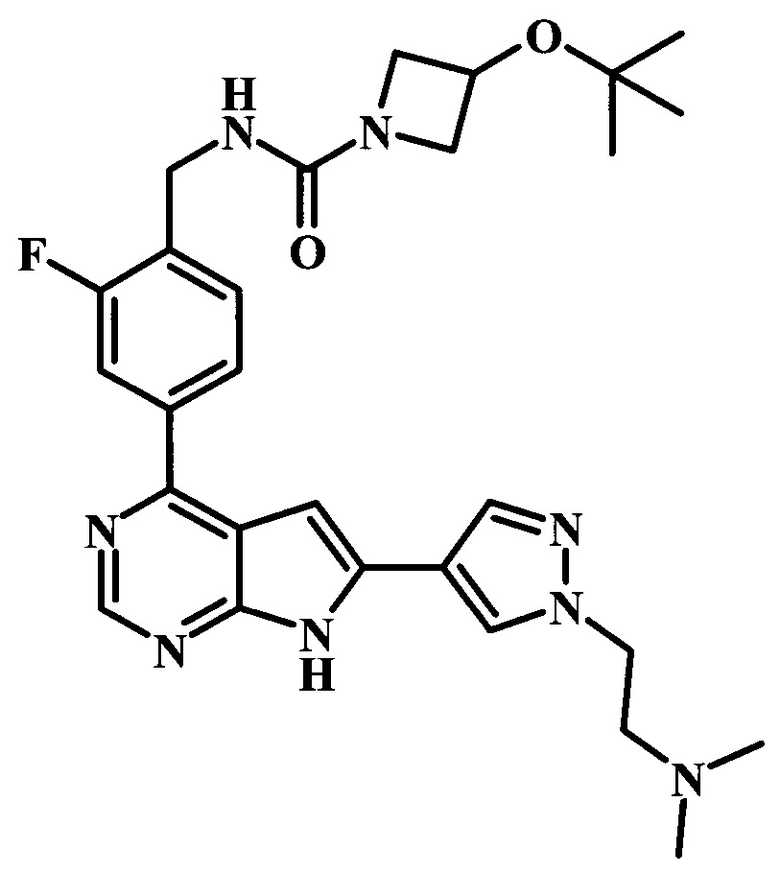
В герметизируемой трубке на 20 мл для микроволнового облучения (2-{4-[4-(4-аминометил-3-фторфенил)-7Н-пирроло[2,3-d]пиримидин-6-ил]-пиразол-1-ил}-этил)-диметиламин (22 мг; 0,056 ммоль; Экв.: 1,00), ди(1Н-имидазол-1-ил)метанон (19 мг; 0,116 ммоль; Экв.: 2,00) и DIPEA (51 мкл; 0,290 ммоль; Экв.: 5,00) соединяли с DMF (2 мл) с получением светло-желтого раствора. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Добавляли 3-трет-бутоксиазетидин (15,0 мг; 0,116 ммоль; Экв.: 2,00) и перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли с помощью EtOAc и промывали солевым раствором и водой. Органические фазы объединяли и высушивали над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенное вещество очищали хроматографией на колонке (силикагель, 0-10% МеОН в DCM). Указанное в заголовке соединение получали в виде желтого твердого вещества (18 мг; выход 58%). LC/MS: m/z Расч. для C28H35FN8O2 ([М+Н]+): 535,6. Фактич.: 535,4.
Пример I-44
4-{6-[1-(2-Диметиламиноэтил)-1Н-пиразол-4-ил]-7Н-пирроло[2,3-d]пиримидин-4-ил}-2-фторбензиламид 1,3-дигидроизоиндол-2-карбоновой кислоты
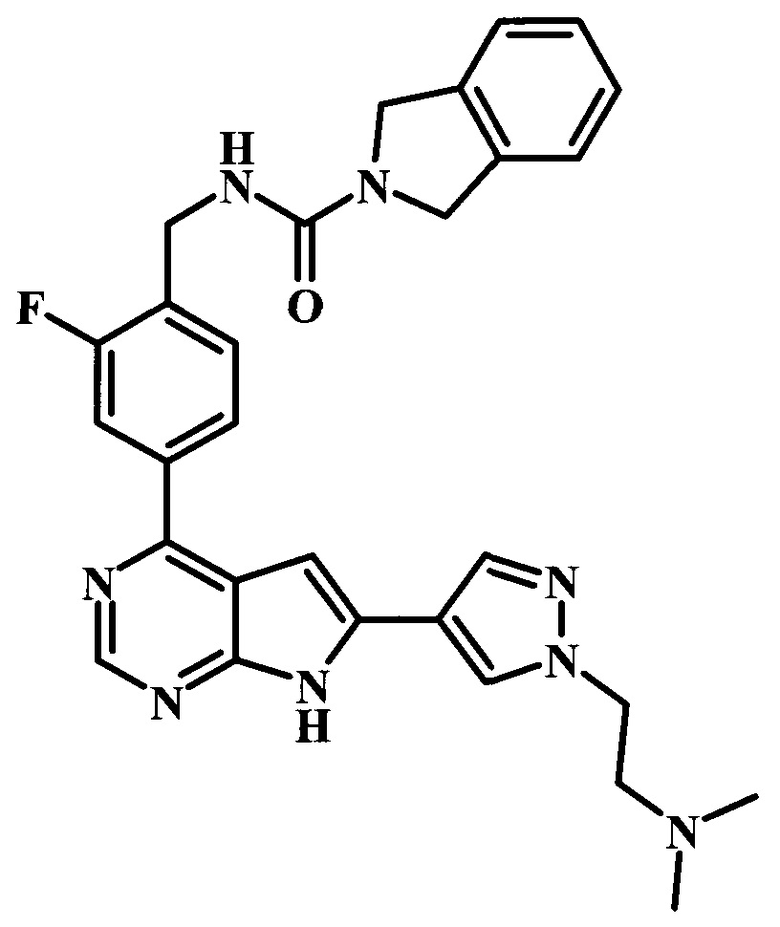
По аналогии со способом, описанным в примере 43, из изоиндолина (14 мг; 0,116 ммоль; Экв.: 2,00) получали указанное в заголовке соединение в виде желтого твердого вещества (20 мг; выход 63%). LC/MS: m/z Расч. для C29H29FN8O ([М+Н]+): 525,6 Фактич.: 525,3
Пример I-45
Этиловый эфир [4-(4-{4-[(4-трет-бутилбензоиламино)-метил]-3-фторфенил}-7Н-пирроло[2,3-d]пиримидин-6-ил)-пиразол-1-ил]-уксусной кислоты
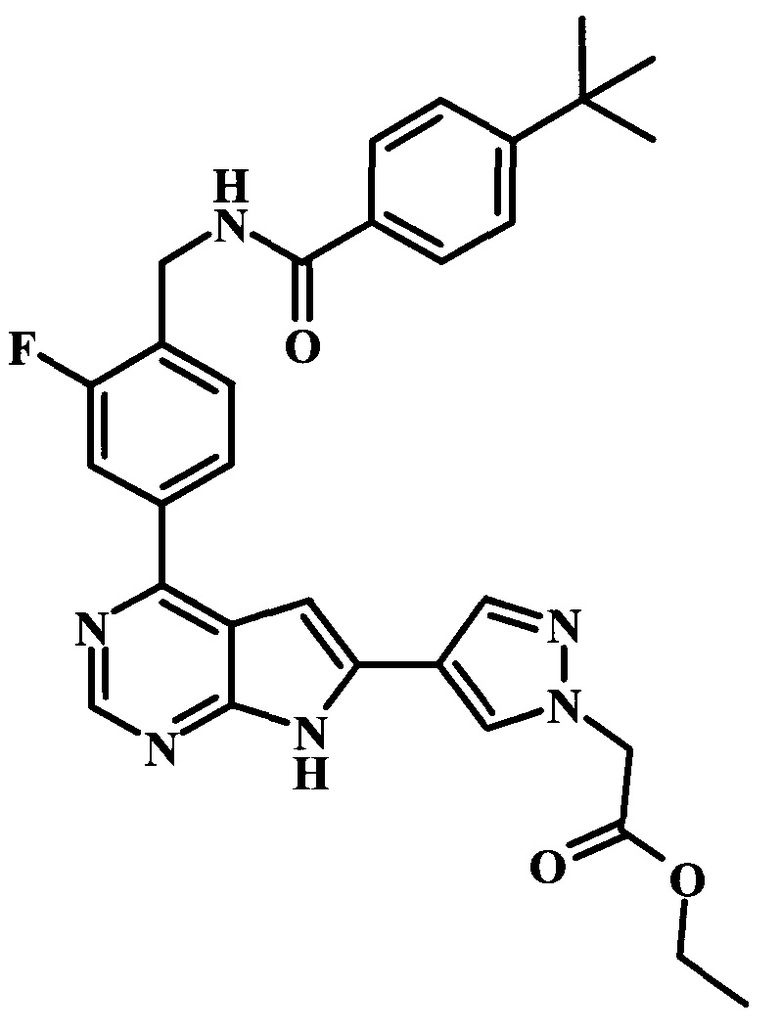
Стадия 1: Этиловый эфир (4-{4-[4-(трет-бутоксикарбониламинометил)-3-фторфенил]-7Н-пирроло[2,3-d]пиримидин-6-ил}-пиразол-1-ил)-уксусной кислоты
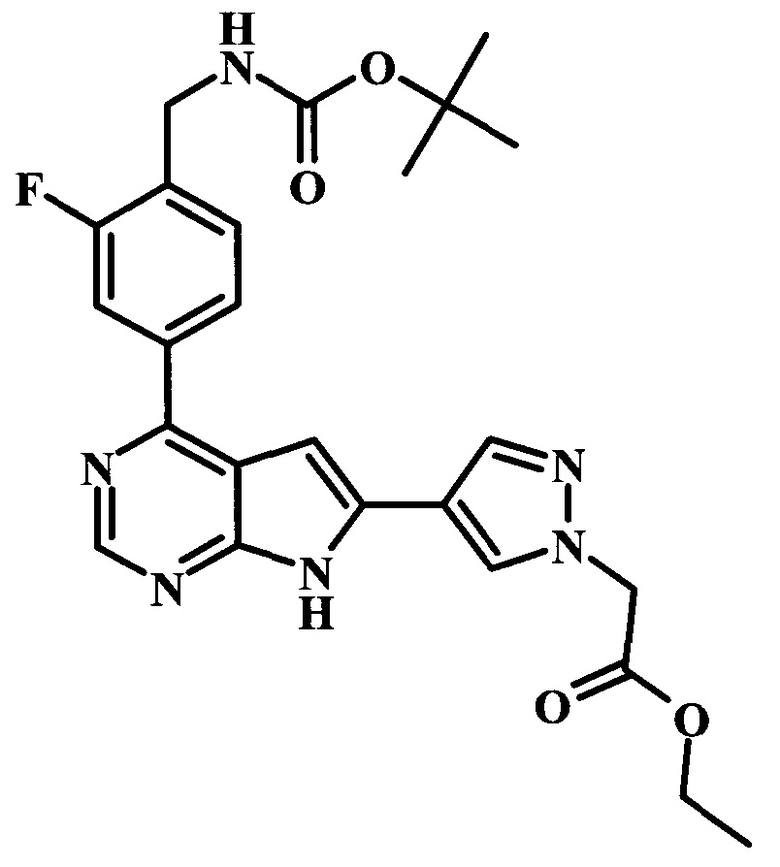
В герметизируемой трубке на 20 мл для микроволнового облучения трет-бутил-4-(6-бром-7-тозил-7Н-пирроло[2,3-d]пиримидин-4-ил)-2-фторбензилкарбамат (250 мг; 0,434 ммоль; Экв.: 1,00), этил-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол-1-ил)ацетат (365 мг; 1,3 ммоль; Экв.: 3) и Pd(PPh3)4 (50 мг; 0,043 ммоль; Экв.: 0,1) соединяли с DMF (10 мл) с получением светло-коричневой суспензии. Добавляли карбонат калия (240 мг; 1,74 ммоль; Экв.: 4,00). Реакционную смесь нагревали при 155°С микроволнами в течение 1 ч. Реакционную смесь разбавляли с помощью EtOAc и промывали солевым раствором и водой. Органические фазы объединяли и высушивали над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенное вещество очищали хроматографией на колонке (силикагель, 5-60% EtOAc в гексане). Указанное в заголовке соединение получали в виде твердого вещества (72 мг; выход 33%). LC/MS: m/z Расч. для C25H27FN6O4 ([М+Н]+): 495,5 Фактич.: 495,3
Стадия 2: Этиловый эфир {4-[4-(4-аминометил-3-фторфенил)-7Н-пирроло[2,3-d]пиримидин-6-ил]-пиразол-1-ил}-уксусной кислоты
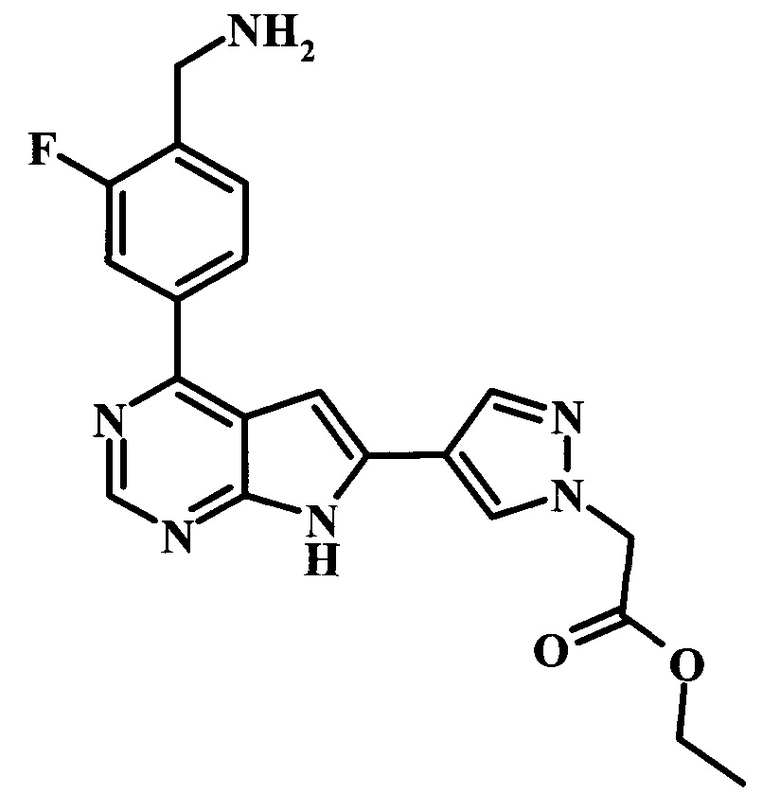
В круглодонной колбе на 50 мл этиловый эфир (4-{4-[4-(трет-бутоксикарбониламинометил)-3-фторфенил]-7Н-пирроло[2,3-d]пиримидин-6-ил}-пиразол-1-ил)-уксусной кислоты (72 мг; 0,146 ммоль; Экв.: 1,00) объединяли с DCM (8 мл) с получением желтого раствора. Добавляли TFA (4 мл; 51,9 ммоль; Экв.: 357) и перемешивали при комнатной температуре в течение 1 ч. Растворитель удаляли при пониженном давлении, затем высушивали в вакууме в течение 3 ч. Этот остаток использовали на следующей стадии без дополнительной очистки. LC/MS: m/z Расч. для C20H19FN6O2 ([М+Н]+): 395,4. Фактич.: 395,2.
Стадия 3: Этиловый эфир [4-(4-{4-[(4-трет-бутилбензоиламино)-метил]-3-фторфенил}-7Н-пирроло[2,3-d]пиримидин-6-ил)-пиразол-1-ил]-уксусной кислоты
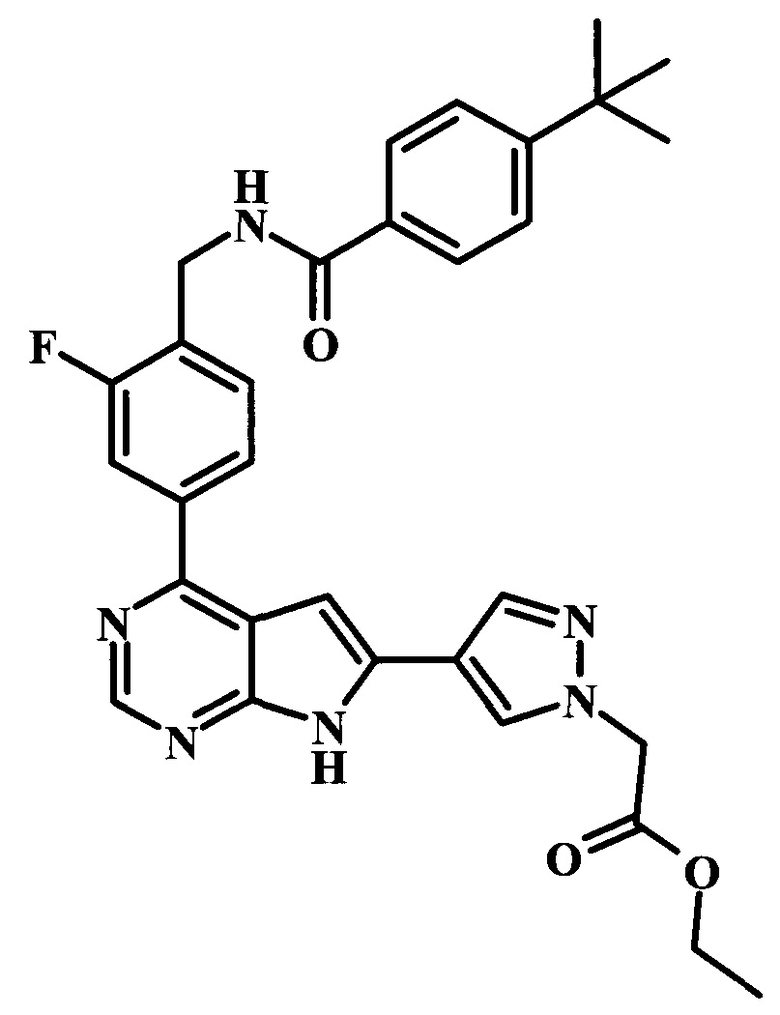
В круглодонной колбе на 50 мл этиловый эфир {4-[4-(4-аминометил-3-фторфенил)-7Н-пирроло[2,3-d]пиримидин-6-ил]-пиразол-1-ил}-уксусной кислоты (52 мг; 0,132 ммоль; Экв.: 1,00), 4-трет-бутилбензойную кислоту (47,0 мг; 0,264 ммоль; Экв.: 2,00) и HATU (100 мг; 0,264 ммоль; Экв.: 2,00) соединяли с DMF (4 мл) с получением желтого раствора. Реакционную смесь перемешивали в течение 5 мин, затем добавляли DIPEA (115 мкл; 659 мкмоль; Экв.: 5,00) и перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли с помощью EtOAc и промывали солевым раствором и водой. Органические фазы объединяли и высушивали над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенное вещество очищали хроматографией на колонке (силикагель, 0-10% МеОН в DCM). Указанное в заголовке соединение получали в виде твердого вещества (52 мг; выход 69%). LC/MS: m/z Расч. для C31H31FN6O3 ([М+Н]+): 555,6 Фактич.: 555,4
Пример 46
[4-(4-{4-[(4-трет-Бутилбензоиламино)-метил]-3-фторфенил}-7Н-пирроло[2,3-d]пиримидин-6-ил)-пиразол-1-ил]-уксусная кислота
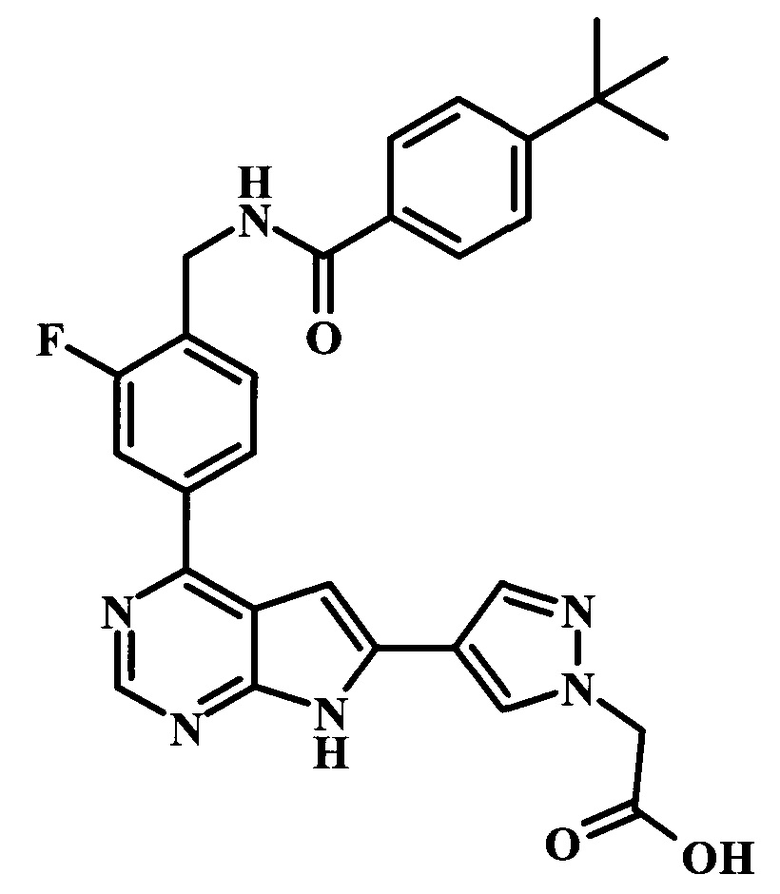
В круглодонной колбе на 50 мл этиловый эфир [4-(4-{4-[(4-трет-бутилбензоиламино)-метил]-3-фторфенил}-7Н-пирроло[2,3-d]пиримидин-6-ил)-пиразол-1-ил]-уксусной кислоты (47 мг; 0,085 ммоль; Экв.: 1,00) соединяли с THF (5 мл) с получением желтой суспензии. Добавляли 1 М раствор NaOH (0,135 мл; 0,135 ммоль; Экв.: 1,59) и перемешивали при комнатной температуре в течение ночи. Реакционную смесь подкисляли добавлением 1 н HCl. Растворитель удаляли при пониженном давлении. Этот остаток очищали с помощью ВЭЖХ с получением указанного в заголовке соединения в виде желтого твердого вещества (34 мг; выход 76%). LC/MS: m/z Расч. для C29H27FN6O3 ([М+Н]+): 527,6 Фактич.: 527,3
Пример 47
N-(2-фтор-4-(6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил)бензил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиридин-2-карбоксамид
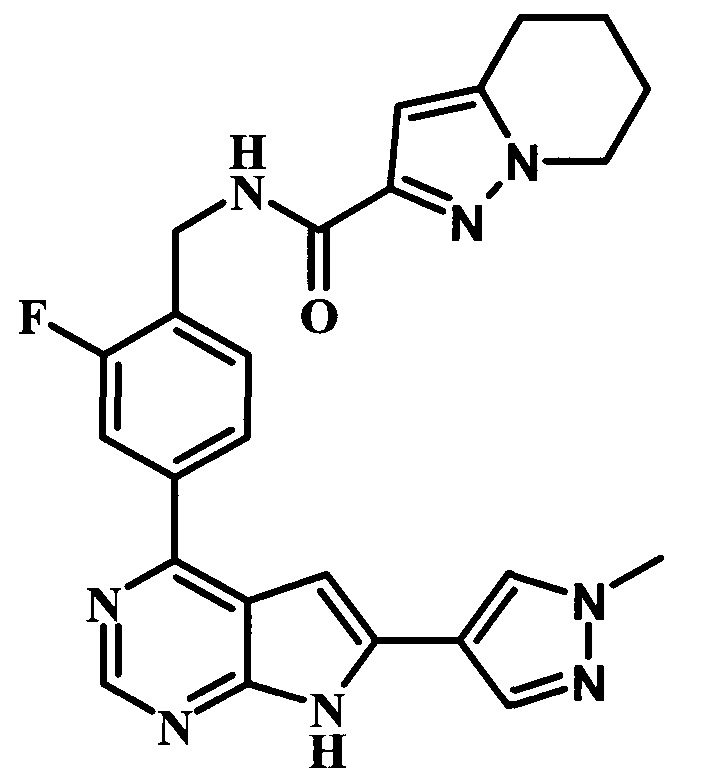
Стадия 1: N-(4-бром-2-фторбензил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиридин-2-карбоксамид
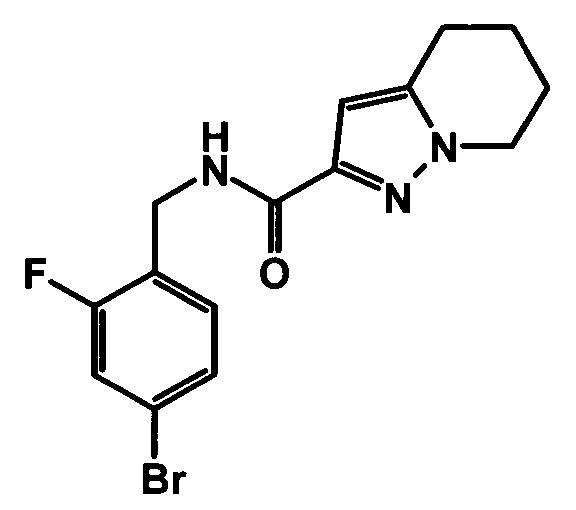
(4-Бром-2-фторфенил)метанамин (193 мг; 945 мкмоль; Экв.: 1,00), 4,5,6,7-тетрагидропиразоло[1,5-а]пиридин-2-карбоновую кислоту (157 мг; 945 мкмоль; Экв.: 1,00), HBTU (358 мг; 945 мкмоль; Экв.: 1,00) и DIPEA (366 мг; 495 мкл; 2,83 ммоль; Экв.: 3) в DMF (3,15 мл) перемешивали при КТ в течение 16 ч. Реакционную смесь разбавляли этилацетатом, промывали водой и солевым раствором. Объединенные органические слои высушивали над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Полученное неочищенное вещество очищали хроматографией на колонке (силикагель, 10-65% этилацетат в гексане) с получением N-(4-бром-2-фторбензил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиридин-2-карбоксамида (178 мг; выход 54%) в виде бесцветного масла. LC/MS: m/z расч. для C15H15BrFN3O ([М+Н]+): 353,2 Фактич.: 354,1
Стадия 2: N-(2-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиридин-2-карбоксамид
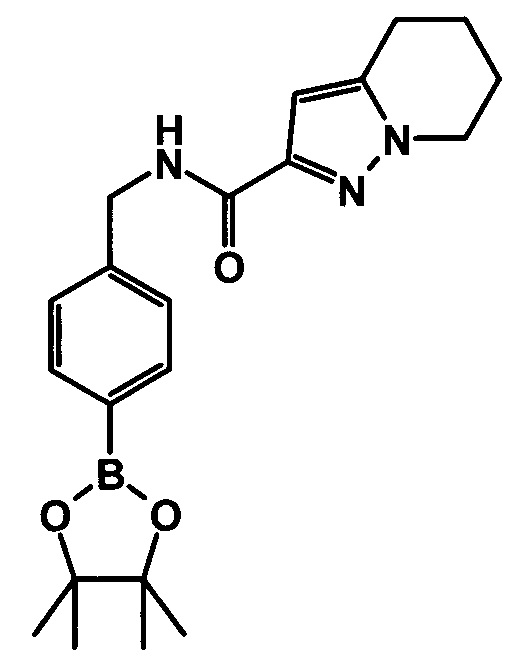
N-(4-бром-2-фторбензил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиридин-2-карбоксамид (178 мг; 505 мкмоль; Экв.: 1,00), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (193 мг; 758 мкмоль; Экв.: 1,5), аддукт PdCl2(dppf)-CH2Cl2 (37,0 мг; 50,5 мкмоль; Экв.: 0,1) и ацетат калия (149 мг; 1,52 ммоль; Экв.: 3) в NMP (3 мл) нагревали до 100°С в течение 16 ч. Реакционную смесь разбавляли этилацетатом, промывали водой и солевым раствором. Объединенные органические слои высушивали над безводным сульфатом натрия и растворитель удаляли при пониженном давлении. Полученное неочищенное вещество очищали хроматографией на колонке (силикагель, 30-100% этилацетат в гексане) с получением N-(2-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиридин-2-карбоксамида (74 мг; выход 37%) в виде белого твердого вещества. LC/MS: m/z расч. для C21H27BFN3O3 ([М+Н]+): 400,2 Фактич.: 400,2
Стадия 3: N-(2-фтор-4-(6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил)бензил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиридин-2-карбоксамид
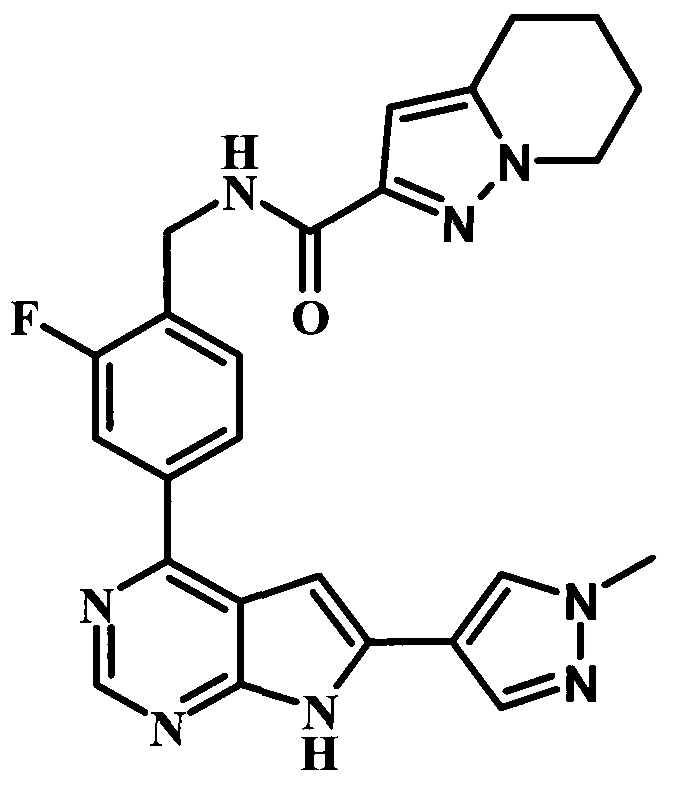
4-Хлор-6-(1-метил-1Н-пиразол-4-ил)-7-(фенилсульфонил)-7Н-пирроло[2,3-d]пиримидин (69,3 мг; 185 мкмоль; Экв.: 1,00), N-(2-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиридин-2-карбоксамид (74 мг; 185 мкмоль; Экв.: 1,00), тетракис(трифенилфосфин)палладий(0) (21,4 мг; 18,5 мкмоль; Экв.: 0,1) и карбонат калия (76,8 мг; 556 мкмоль; Экв.: 3) в смеси DME (1,48 мл)/ вода (371 мкл) нагревали до 150°С микроволновым облучением в течение 45 мин. Очищали хроматографией на колонке (силикагель, 0-100% этилацетат в смеси [10% МеОН/этилацетат]), а затем очищали с помощью ВЭЖХ с получением N-(2-фтор-4-(6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил)бензил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиридин-2-карбоксамида (5,7 мг; выход 7%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, DMSO-d) δ ppm 12,63 (s, 1H), 8,79 (s, 1H), 8,73 (t, J=5,8 Гц, 1H), 8,31 (s, 1H), 8,11 (s, 1H), 8,04 (d, J=7,8 Гц, 1H), 7,94 (d, J=11,5 Гц, 1H), 7,52 (t, J=8,2 Гц, 1H), 7,15 (s, 1H), 6,43 (s, 1H), 4,56 (d, J=6,2 Гц, 2H), 4,15 (t, J=5,3 Гц, 2H), 3,92 (s, 3H), 2,80 (t, J=5,0 Гц, 2H), 2,01 (br. s, 2H), 1,82 (br. s, 2H); LC/MS: m/z расч. для C25H23FN8O ([M+H]+): 471,5 Фактич.: 471,2
Пример 48
2-Фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензиламид 5-трет-бутилизоксазол-3-карбоновой кислоты
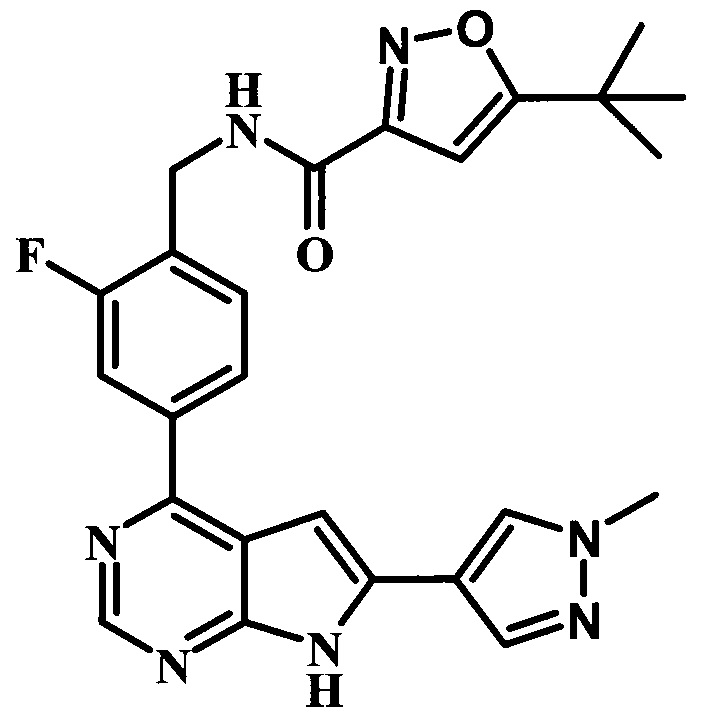
В сцинтилляционном флаконе на 20 мл трет-бутиловый эфир {2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-карбаминовой кислоты (75 мг; 0,178 ммоль; Экв.: 1,00) объединяли с 2 мл DCM и 2 мл TFA. Полученный раствор перемешивали при комнатной температуре в течение 30 мин. Растворитель удаляли при пониженном давлении. Неочищенное вещество растворяли в DMF (2 мл). Вносили 5-трет-бутилизоксазол-3-карбоновую кислоту (33,0 мг; 0,195 ммоль; Экв.: 1,1), DIPEA (0,124 мл; 0,71 ммоль; Экв.: 4,00) и HATU (74 мг; 0,195 ммоль; Экв.: 1,1). Реакционную смесь перемешивали в течение ночи. Полученный раствор разбавляли с помощью 10 мл воды и 5 мл EtOAc. Полученный раствор перемешивали при комнатной температуре в течение еще 30 мин. Органические фазы экстрагировали и высушивали над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Полученное твердое вещество очищали хроматографией на колонке (силикагель, 0-10% МеОН в DCM). Указанное в заголовке соединение получали в виде желтого твердого вещества (35 мг; выход 42%). LC/MS: m/z расч. для C25H24FN7O2 ([M+H]+): 474,5 Фактич.: 474,3
Пример 49
2-Фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензиламид 3-трет-бутил-[1,2,4]оксадиазол-5-карбоновой кислоты
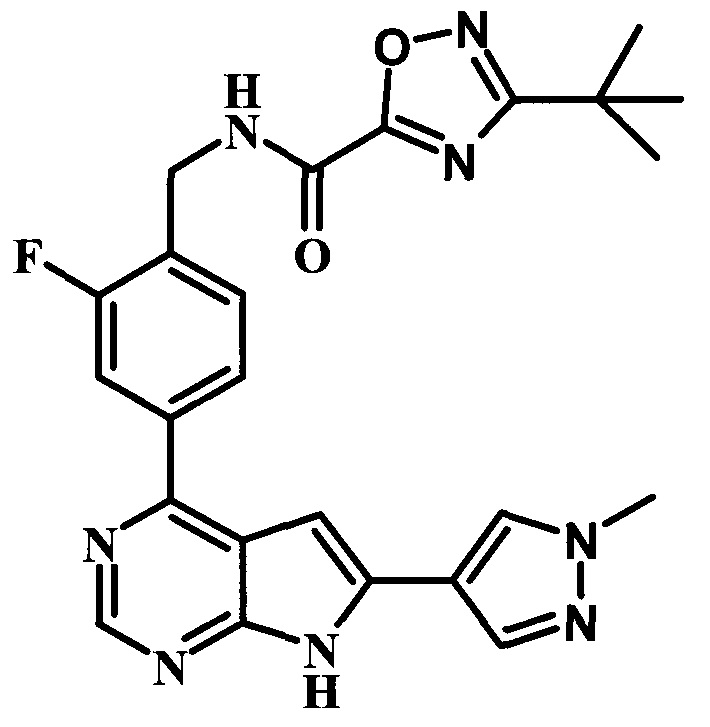
В сцинтилляционном флаконе на 20 мл трет-бутиловый эфир {2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-карбаминовой кислоты (150 мг; 0,355 ммоль; Экв.: 1,00) объединяли с 3 мл DCM и 3 мл TFA. Полученный раствор перемешивали при комнатной температуре в течение 30 мин. Растворитель удаляли при пониженном давлении. Неочищенное вещество растворяли в DMF (4 мл). Вносили 3-трет-бутил-1,2,4-оксадиазол-5-карбоновую кислоту (66,5 мг; 0,391 ммоль; Экв.: 1,1), DIPEA (0,25 мл; 1,42 ммоль; Экв.: 4,00) и бромтрипирролидин-1-илфосфоний (Pybrop) (182 мг; 0,391 ммоль; Экв.: 1,1). Реакционную смесь перемешивали в течение ночи. Полученный раствор разбавляли с помощью 10 мл воды и 5 мл EtOAc. Полученный раствор перемешивали при комнатной температуре в течение еще 30 мин. Органические фазы экстрагировали и высушивали над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Полученное твердое вещество препаративной ВЭЖХ (10-100% ацетонитрил-вода). Указанное в заголовке соединение получали в виде желтого твердого вещества (20 мг; выход 12%). LC/MS: m/z расч. для C24H23FN8O2 ([М+Н]+): 475,5 Фактич.: 475,2
Пример 50
трет-Бутиловый эфир {2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-карбаминовой кислоты
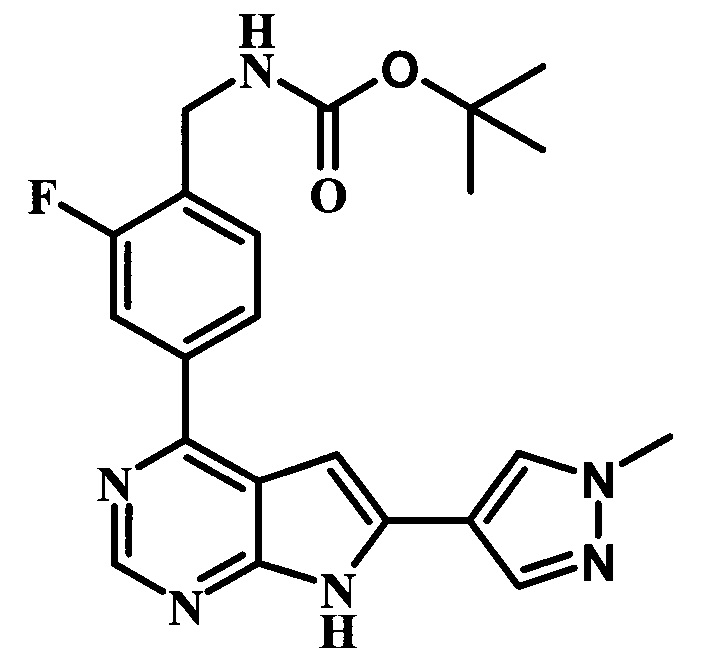
В герметизируемом сосуде на 20 мл для микроволнового облучения 7-бензолсульфонил-4-хлор-6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин (500 мг; 1,34 ммоль; Экв.: 1,00), трет-бутиловый эфир [2-фтор-4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-бензил]-карбаминовой кислоты (541 мг; 1,61 ммоль; Экв.: 1,2) и карбонат калия (739 мг; 5,35 ммоль; Экв.: 4,00) в 5 мл воды соединяли с DME (10 мл). Вносили Pd(PPh3)4 (375 мг; 0,325 ммоль; Экв.: 0,1). Реакционную смесь нагревали микроволновым облучением при 160°С в течение 60 мин. Полученный раствор промывали с помощью EtOAc и солевым раствором. Объединенные органические фазы высушивали над безводным сульфатом натрия, затем растворитель удаляли при пониженном давлении. Неочищенное вещество очищали хроматографией на колонке (силикагель, 0-10% МеОН в DCM). Указанное в заголовке соединение получали в виде желтого твердого вещества (150 мг; выход 27%). LC/MS: m/z расч. для C22H23FN6O2 ([M+H]+): 423,4 Фактич.: 423,2
Пример 51
N-{2-Фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-бензамид
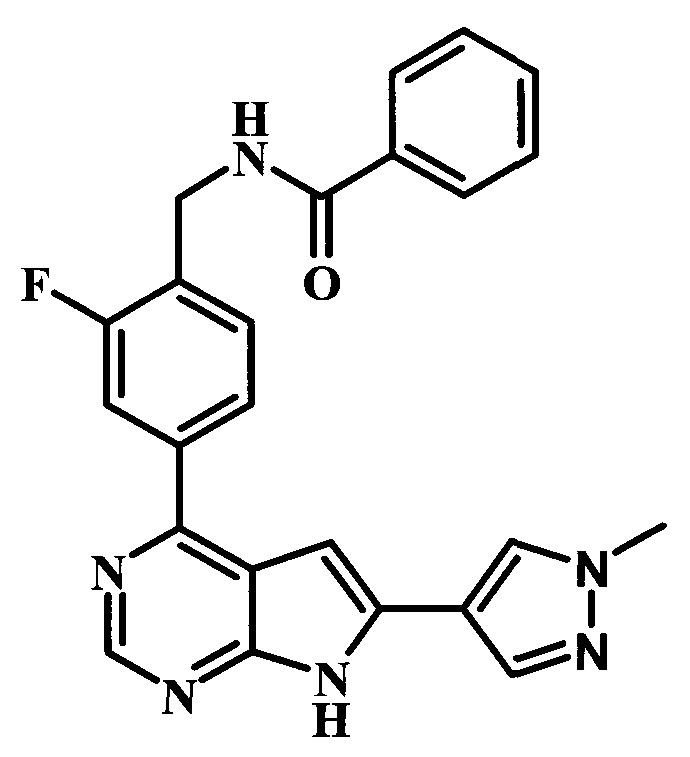
В сцинтилляционном флаконе на 20 мл трет-бутиловый эфир {2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-карбаминовой кислоты (100 мг; 0,237 ммоль; Экв.: 1,00) объединяли с 2 мл DCM и 2 мл TFA. Полученный раствор перемешивали при комнатной температуре в течение 30 мин. Растворитель удаляли при пониженном давлении. Неочищенное вещество растворяли в DMF (2 мл). Вносили бензойную кислоту (32 мг; 0,26 ммоль; Экв.: 1,1), DIPEA (0,165 мл; 0,95 ммоль; Экв.: 4,00) и HATU (99 мг; 0,26 ммоль; Экв.: 1,1). Реакционную смесь перемешивали в течение ночи. Полученный раствор разбавляли с помощью 10 мл воды и 5 мл EtOAc. Раствор перемешивали при комнатной температуре в течение еще 30 мин. Органические фазы экстрагировали и высушивали над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Полученное твердое вещество очищали хроматографией на колонке (силикагель, 0-10% МеОН в DCM). Указанное в заголовке соединение получали в виде желтого твердого вещества (8 мг; выход 8%). LC/MS: m/z расч. для C24H19FN6O ([M+H]+): 427,5 Фактич.: 427,1
Биологические Примеры
Анализ ингибирования тирозинкиназы Брутона (Btk)
Данный анализ представляет собой захват радиоактивного 33P-фосфорилированного продукта при фильтрации. Взаимодействия Btk, биотинилированного SH2 пептидного субстрата (Src гомология) и аденозинтрифосфата (АТФ) приводит к фосфорилированию этого пептидного субстрата. Биотинилированный продукт связывается со стрептавидин-сефарозными гранулами. Все связанные радиоактивно-меченные продукты детектируют с помощью сцинтилляционного счетчика.
Планшеты для анализа представляют собой 96-луночные полипропиленовые (Greiner) и 96-луночные планшеты с гидрофильным поливинилиденфторидным (PVDF) фильтром (1,2 мкм, Millipore). Указанные далее концентрации представляют собой конечные аналитические концентрации: 10-100 мкМ соединения в DMSO (Burdick and Jackson); 5-10 нМ фермент Btk (с гистидиновой меткой, полноразмерный); 30 мкМ пептидный субстрат (Biotin-Aca-AAAEEIYGEI-NH2); 100 мкМ АТФ (Sigma); 8 мМ имидазол (Sigma, pH 7,2); 8 мМ глицерин-2-фосфат (Sigma); 200 мкМ EGTA (Roche Diagnostics); 1 мМ MnCl2 (Sigma); 20 мМ MgCl2 (Sigma); 0,1 мг/мл BSA (Sigma); 2 мМ DTT (Sigma); 1 мкКи 33P АТР (Amersham); 20% стрептавилин-сефарозные гранулы (Amersham); 50 мМ EDTA (Gibco); 2 М NaCl (Gibco); 2 M NaCl / 1%(вес) фосфорная кислота (Gibco); microscint-20 (Perkin Elmer).
Значения IC50 рассчитывали по 10 точкам на каждое соединение с помощью данных, полученных по стандартному шаблону для анализа на 96-луночном планшете. На каждом планшете тестировали одно контрольное соединение и семь неизвестных ингибиторов, и каждый планшет прогоняли дважды. Обычно соединения разбавляли с полулогарифмическим шагом, начиная от 100 мкМ и заканчивая 3 нМ. Контрольное соединение представляло собой стауроспорин. Уровень фона определяли в отсутствии пептидного субстрата. Общую активность определяли в присутствии пептидного субстрата. Для определения ингибирования Btk использовали следующий протокол.
1) Подготовка образцов: тестовые соединения разбавляли с полулогарифмическим шагом в аналитическом буфере (имидазол, глицерин-2-фосфат, EGTA, MnCl2, MgCl2, BSA).
2) Подготовка гранул
a) промыть гранулы центрифугированием при 500 g
b) восстановить гранулы с помощью PBS и EDTA с получением 20% кашицы гранул
3) Пре-инкубировать реакционный микс без субстрата (аналитический буфер, DTT, АТФ, 33P АТФ) и микс с субстратом (аналитический буфер, DTT, АТФ, 33Р АТФ, пептидный субстрат) при 30°С в течение 15 мин.
4) Для начала анализа пре-инкубировать 10 мкл Btk в буфере для фермента (имидазол, глицерин-2-фосфат, BSA) и 10 мкл тестового соединения в течение 10 мин при КТ.
5) Добавить 30 мкл реакционной смеси, не содержащей или содержащей субстрат, к Btk и соединению.
6) Инкубировать 50 мкл общего аналитического микса в течение 30 мин при 30°С.
7) Перенести 40 мкл аналитической смеси к 150 мкл кашицы гранул на планшет с фильтром для остановки реакции.
8) Промыть планшет с фильтром через 30 мин, используя следующие стадии:
а. 3×250 мкл NaCl
b. 3×250 мкл NaCl, содержащего 1% фосфорной кислоты
с. 1×250 мкл H2O
9) Высушить планшет в течение 1 ч при 65°С или в течение ночи при КТ.
10) Добавить 50 мкл реагента microscint-20 и считывать число импульсов в минуту (cpm) от 33P на сцинцилляционном счетчике.
Рассчитать процентную долю активности по исходным данным, выраженным в cpm:
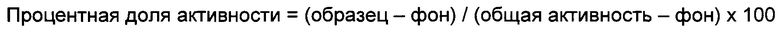
Рассчитать IC50 по процентной доле активности, с использованием односайтовой сигмоидальной модели «доза-отклик»:
y=А+((В-А)/(1+((x/С)D))))
где:
х = конц. соединения, y = % активности, А = минимум, В = максимум, С = IC50, D = 1 (коэффициент Хилла)
Анализ ингибирования тирозинкиназы Брутона (Btk) TR-FRET (анализ резонансного переноса энергии флуоресценции с разрешением по времени)
С помощью этого конкурентного анализа киназы BTK можно измерить активность соединения (IC50) для инактивированного состояния тирозинкиназы Брутона с помощью технологии FRET (резонансного переноса энергии флуоресценции). Комплекс Btk-Eu инкубировали на льду за один час перед использованием при начальной концентрации 50 нМ BTK-bioease™: 10 нМ Eu-стрептавидин (Perkin-Elmer, кат. № AD0062). Использовали аналитический буфер следующего состава: 20 мМ HEPES (рН 7,15); 0,1 мМ DTT; 10 мМ MgCl2; 0,5 мг/мл BSA плюс 3% стабилизатор киназы (Fremont Biosolutions, кат. № STB-K02). Через 1 ч описанную выше реакционную смесь разбавляли в 10 раз аналитическим буфером до достижения 5 нМ BTK:1 нМ комплекс Eu-Стрептавидин (донорный флюорофор). Затем наносили 18 мкл смеси 0,11 нМ BTK-Eu и 0,11 нМ Kinase Tracer 178 (Invitrogen, кат. № PV5593), с использованием только BTK-Eu в качестве отрицательного контроля, на 384-луночный планшет с плоским дном (Greiner, 784076). Тестируемые соединения готовили в 10-кратной концентрации и получали серийные разведения в DMSO с полулогарифмическим шагом для получения 10 точек для кривой. Для инициации FRET-реакции, соединения, приготовленные в виде 10-кратных стоковых растворов в DMSO, наносили на планшеты и инкубировали планшеты 18-24 ч при 14°С.
После инкубации планшеты считывали на ридере BMG Pherastar Fluorescent (или эквивалентном ему) и использовали для измерения энергии испускания от европиевого донороного флюорофора (испускание при 620 нм) и FRET (испускание при 665 нм). Усредняли лунки с отрицательным контролем для получения среднего минимума. Положительные контрольные лунки "без ингибитора" усредняли для получения среднего максимума. Процент от максимального значения FRET рассчитывали по следующему уравнению:
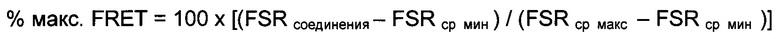
где FSR = отношение сигнала FRE. Кривые % макс. FRET строили с помощью приложения Activity Base (Excel) и IC50 (%), определяли коэффициент Хилла, z' и коэффициент вариации (%CV). Среднее значение IC50 и стандартное отклонение выводили по двум кривым (кривые синглетного ингибирования от двух независимых разведении) с помощью Microsoft Excel.
Результаты этого анализа для выбранных соединений перечислены ниже в Таблице II.
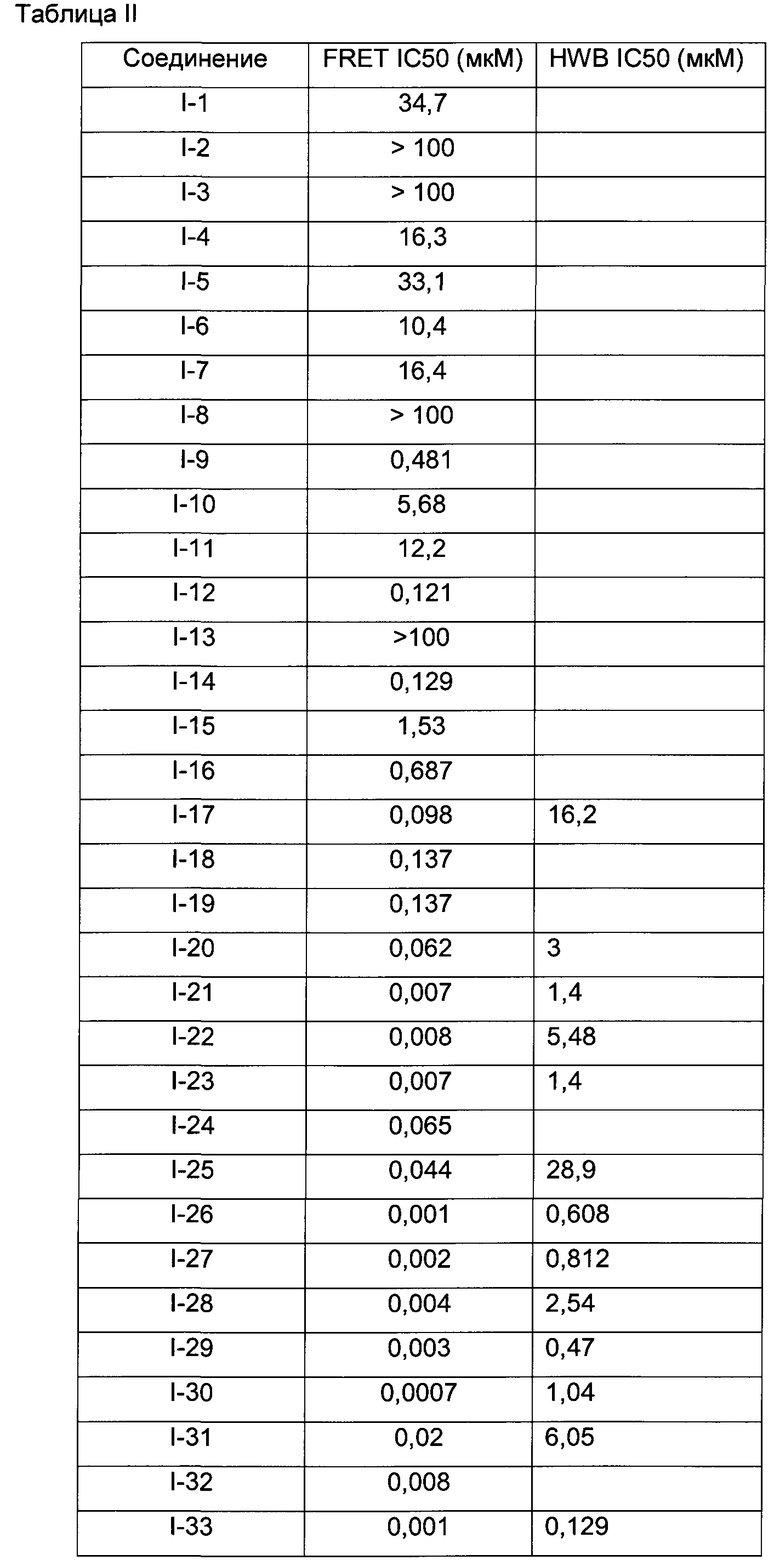
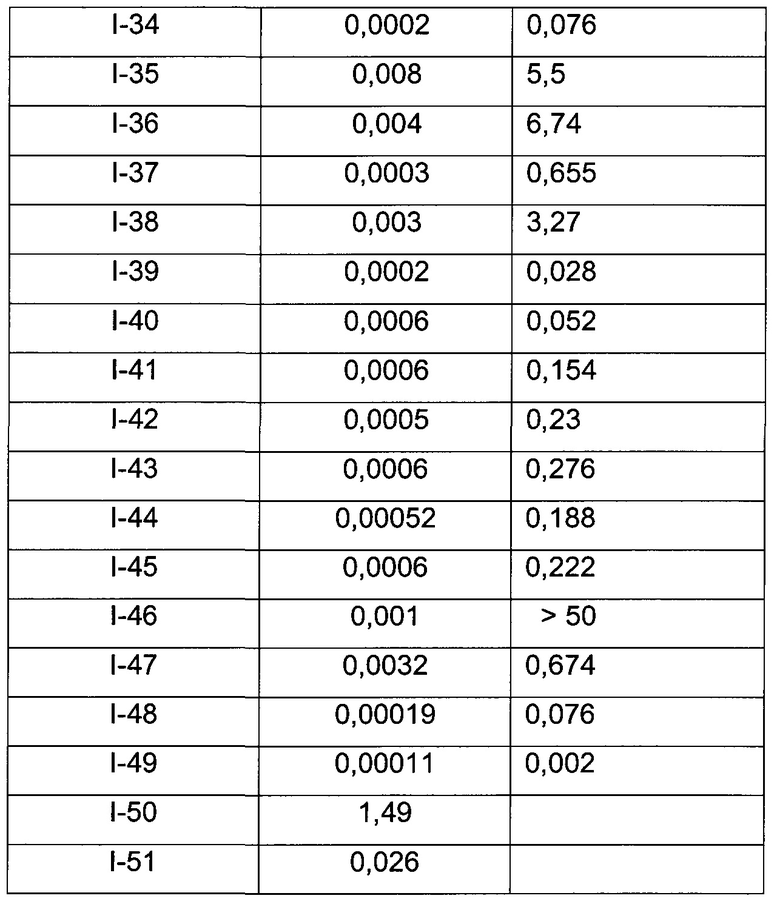
Ингибирование активации В-клеток в цельной крови, измеренное по экспрессии CD69
Методика исследования способности ингибиторов Btk подавлять опосредованную рецептором В-клеток активацию В-клеток в крови человека описана ниже.
Цельную кровь человека (HWB, от англ. Human Whole Blood) получали от здоровых доноров при следующих ограничениях: 24 ч без приема лекарств, некурящие. Кровь собирали путем венопункции в пробирки Vacutainer с гепарином натрия в качестве антикоагулянта. Тестовые соединения разбавляли до 10-кратной желаемой исходной концентрации лекарственного вещества в PBS (20×), а затем готовили трехкратные серийные разведения в 10% DMSO в PBS для получения девяти точек для построения кривой доза-отклик. Вносили по 5,5 мкл каждого разведения соединения в дупликатах на 96-луночный планшет с лунками объемом 2 мл и V-образным дном (Analytical Sales and Services, #59623-23); вносили по 5,5 мкл 10% DMSO в PBS в контрольные лунки и лунки без стимуляции. Вносили HWB (100 мкл) в каждую лунку, перемешивали и инкубировали планшеты при 37С, 5% CO2, 100% влажности в течение 30 минут. Вносили козьи F(ab')2 против IgM человека (Southern Biotech, #2022-14) (10 мкл раствора 500 мкг/мл, конечная концентрация 50 мкг/мл) в каждую лунку (за исключением лунок без стимуляции) при перемешивании, и инкубировали планшеты еще в течение 20 часов.
По окончании 20-часовой инкубации образцы инкубировали с антителами, меченными флуоресцентным зондом (15 мкл, РЕ мыши против CD20 человека, BD Pharmingen, #555623, и/или 20 мкл АРС мыши против CD69 человека, BD Pharmingen #555533) в течение 30 минут, при 37С, 5% CO2, 100% влажности. В анализ включали индуцированный контроль, неокрашенный и отдельные красители для установки компенсации и выставления начального напряжения. Затем образцы лизировали с помощью 1 мл 1-кратного буфера Pharmingen Lyse Buffer (BD Pharmingen # 555899) и планшеты центрифугировали при 1800 об/мин в течение 5 минут. Супернатанты удаляли отсасыванием и оставшиеся гранулы снова лизировали с помощью 1 мл однократного буфера Pharmingen Lyse Buffer, и планшеты откручивали, как ранее. Супернатанты отсасывали и оставшиеся гранулы промывали в буфере FACs (PBS + 1% FBS). После окончательного откручивания Супернатанты удаляли и гранулы ресуспендировали в 180 мкл буфера FACs. Образцы переносили на 96-луночный планшет, подходящий для прогона на 96-луночной системе HTS 96 на проточном цитофлуориметре BD LSR II.
При подходящей длине волны возбуждения и испускания для использованного флюорофора были получены данные и рассчитаны процентные доли позитивных клеток с помощью программы Cell Quest Software. Результаты сначала анализировали с помощью программы FACS analysis (Flow Jo). Величину IC50 для тестового соединения определяли как концентрацию, которая снижает на 50% процентную долю CD69-позитивных клеток, которые также являются CD20-позитивными после стимуляции с помощью анти-IgM (среднее из 8 контрольных лунок, после вычитания среднего из 8 лунок для фонового уровня без стимуляции). Значения IC50 рассчитывали с помощью программы XLfit, версия 3, уравнение 201.
Ингибирование активации В-клеток - анализ FLIPR В-клеток на клетках Ramos
Ингибирование активации В-клеток соединениями по настоящему изобретению можно продемонстрировать при исследовании воздействия тестовых соединений на IgM-стимулированные отклики В-клеток.
В-клеточный FLIPR-анализ представляет собой клеточный функциональный метод определения эффекта потенциальных ингибиторов повышения внутриклеточного кальция при стимуляции с помощью антител против IgM. Клетки Ramos (линия человеческих клеток лимфомы Беркитта, номер в АТСС CRL-1596) культивировали в ростовой среде (описана ниже). За сутки до анализа клетки Ramos ресуспендировали в свежей ростовой среде (такой же, как и выше) и высеивали в концентрации 0,5×106/мл в колбы для тканевых культур. В день анализа клетки считали и высеивали в концентрации 1×106/мл в ростовой среде, содержащей дополнительно 1 мкМ FLUO-ЗАМ (TefLabs кат. №0116, приготовленного в безводном DMSO и 10% плюрониловой кислоте) в колбах для тканевой культуры, и инкубировали при 37°С (4% CO2) в течение 1 ч. Для удаления внеклеточного красителя клетки собирали центрифугированием (5 мин, 1000 об/мин), ресуспендировали в буфере FLIPR (описанном ниже) при концентрации 1×106 клеток/мл и затем наносили на 96-луночные, покрытые поли-D-лизином черно-прозрачные планшеты (BD кат. №356692) в количестве 1×105 клеток/лунку. Добавляли тестовые соединения при различных концентрациях, в интервале от 100 мкМ до 0,03 мкМ (7 концентраций, подробно указаны ниже) и инкубировали с клетками в течение 30 мин при КТ. Са2+-сигнальный путь в клетках Ramos стимулировали добавлением 10 мкг/мл антител против IgM (Southern Biotech, кат. №2020-01) и измеряли на FLIPR (Molecular Devices, осуществляет снимки с 96-луночных планшетов с помощью камеры CCD с аргоновым лазером, возбуждение при 480 нМ).
Состав сред/буферов:
Ростовая среда: RPMI 1640 среда, содержащая L-глутамин (Invitrogen, кат. №61870-010); 10% фетальную сыворотку коровы (FBS, Summit Biotechnology кат. № FP-100-05); 1 мМ пируват натрия (Invitrogen кат. №11360-070).
Буфер FLIPR: HBSS (Invitrogen, кат. №141175-079); 2 мМ CaCl2 (Sigma кат. № С-4901); HEPES (Invitrogen, кат. №15630-080); 2,5 мМ пробенецид (Sigma, кат. № Р-8761); 0,1% BSA (Sigma, кат. № А-7906); 11 мМ глюкоза (Sigma, кат. № G-7528).
Подробное описание разведения соединений:
Для того чтобы получить наибольшую конечную концентрацию в анализе, равную 100 мкМ, 24 мкл 10 мМ стокового раствора соединения (приготовленного в DMSO) непосредственно вносили в 576 мкл буфера FLIPR. Тестовые соединения разбавляли в буфере FLIPR (с помощью автоматического дозатора Biomek 2000) по следующей схеме разведении: растворитель; 1,00×10-4М; 1,00×10-5; 3,16×10-6; 1,00×10-6; 3,16×10-7; 1,00×10-7; 3,16×10-8.
Проведение исследования и анализ данных:
Внутриклеточное возрастание кальция представляли с помощью статистического минимума-максимума (вычитая полученный фоновый уровень из пика, вызванного добавлением стимулирующего антитела, с использованием контроля (Molecular Devices FLIPR) и программного обеспечения с функцией статистической обработки. Значения IC50 определяли с помощью нелинейной аппроксимации (программное обеспечение GraphPad Prism).
Коллаген-индуцированный артрит у мышей (mCIA)
В 0 день мышам делали инъекцию в основание хвоста или в несколько точек на спине эмульсией коллагеном II типа внутрикожно (i.d.) в полном адъюванте Фройнда (CFA, от англ. Complete Freund's Adjuvant). После иммунизации коллагеном у животных развивался артрит примерно за период от 21 до 35 суток. Возникновение артрита синхронизировали (усиливали) систематическим введением коллагена в неполном адъюванте Фройнда (IFA, от англ. Incomplete Freund's Adjuvant; i.d.) на 21 день. Животных обследовали ежедневно после 20-го дня на предмет любых проявлений легкого артрита (1 или 2 балла; см. описание шкалы ниже), которые являются сигналом для усиления. После усиления мышей оценивали по шкале и вводили им дозу потенциальных терапевтических агентов в течение назначенного периода времени (как правило, 2-3 недели) и с частотой дозирования ежедневно (QD) или дважды в день (BID).
Коллаген-индуцированный артрит у крыс (rCIA)
В 0 день крысам делали инъекцию эмульсией бычьего коллагена II типа в неполном адъюванте Фройнда (IFA) интрадермально (i.d.) в нескольких точках на спине. Усиливающую инъекцию эмульсией коллагена делали примерно на 7-ой день (i.d.), в основание хвоста или в других положениях на спине. Артрит обычно наблюдали через 12-14 дней после первой инъекции коллагеном. Животных оценивали на предмет развития артрита, как описано ниже (Оценка артрита) начиная с 14 дня. Животным вводили дозу потенциальных терапевтических агентов в режиме профилактики, начиная с момента повторной иммунизации и в течение предписанного периода времени (как правило, 2-3 недели), и с частотой дозирования ежедневно (QD) или дважды в день (BID).
Оценка артрита:
В обеих моделях развитие воспаления конечностей и суставов конечностей оценивали количественно по системе баллов, включающей оценку четырех конечностей по критериям, описанным ниже:
Баллы:
1 = опухание и/или покраснение конечности или одного пальца.
2 = опухание одного или более суставов.
3 = сильное опухание конечности, с вовлечением более чем двух суставов.
4 = тяжелый артрит всей конечности и пальцев.
Оценку проводили в 0 день для установления базовой линии и начиная снова при первых признаках опухания вплоть до трех раз в неделю до конца эксперимента. Артритический индекс для каждой мыши получали путем сложения баллов по каждой их четырех конечностей, что давало максимальный балл 16 на одно животное.
Крысиная модель астмы In Vivo
Самцов серой крысы сенсибилизировали интраперитонеально с помощью 100 мкг овальбумина (ОА) в 0,2 мл алюмогеля один раз в неделю в течение 3 недель (дни 0, 7 и 14). На 21-й день (через неделю после последней сенсибилизации) крысам вводили дозу q.d. в виде композиции либо с инертным веществом, либо с соединением, подкожно, а через 0,5 часа осуществляли провокацию аэрозолем с помощью ОА (1% ОА в течение 45 минут) и умерщвляли через 4 или 24 ч после провокации. Когда животное забивали, собирали сыворотку и плазму у всех животных для серологического анализа и фармакокинетики (PK), соответственно. Вставляли трахеальную канюлю и легкие трижды промывали с помощью PBS. Жидкость бронхоальвеолярного лаважа (БАЛ) исследовали на общее число лейкоцитов и дифференцированный подсчет лейкоцитов. Общее число лейкоцитов в аликвоте клеток (20-100 мкл) определяли с помощью устройства Coulter Counter. Для дифференцированного подсчета лейкоцитов 50-200 мкл образца центрифугировали в Cytospin и препарат окрашивали с помощью Diff-Quik. Соотношение моноцитов, эозинофилов, нейтрофилов и лимфоцитов подсчитывали с помощью световой микроскопии по стандартным морфологическим критериям и выражали в процентах. Выбранные примеры ингибиторов Btk демонстрируют снижение общего числа лейкоцитов в БАЛ у сенсибилизированных и провоцированных с помощью ОА крыс, по сравнению с контрольным уровнем.
Представленное выше изобретение было детально описано с помощью иллюстраций и примеров, в целях ясности и лучшего понимания. Специалисту в данной области техники очевидно, что можно осуществить изменения и модификации в пределах объема приложенной формулы изобретения. Таким образом, следует понимать, что выше представленное описание является иллюстративным, но не ограничивающим. Таким образом, объем настоящего изобретения следует определять не со ссылкой на представленное выше описание, а со ссылкой на прилагаемую формулу изобретения, в совокупности с полным охватом эквивалентов, на которые дают право пункты указанной формулы.
Все патенты, заявки на патент и публикации, процитированные в данной заявке, включены посредством ссылок полностью во всех смыслах в той же мере, как если бы каждый отдельный патент, заявка на патент или публикация были таким образом индивидуально обозначены.
| название | год | авторы | номер документа |
|---|---|---|---|
| {3-[(7H-ПИРРОЛО[2,3-d]ПИРИМИДИН-4-ИЛ)АЗОЛИЛ]АЗЕТИДИН-3-ИЛ}АЦЕТОНИТРИЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ЯНУС КИНАЗ | 2015 |
|
RU2601410C1 |
| СКОНДЕНСИРОВАННЫЕ С ГЕТЕРОЦИКЛИЧЕСКИМ КОЛЬЦОМ ПРОИЗВОДНЫЕ ПИРИМИДИНА | 1996 |
|
RU2136683C1 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛОПИРИМИДОНА ИЛИ ПИРРОЛОТРИАЗОНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ФАРМАЦЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ | 2014 |
|
RU2674977C2 |
| ПИРРОЛОПИРИМИДИНЫ В КАЧЕСТВЕ ПОТЕНЦИАТОРОВ МВТР | 2017 |
|
RU2757457C2 |
| ПЕРВИЧНЫЕ КАРБОКСАМИДЫ В КАЧЕСТВЕ ИНГИБИТОРОВ BТK | 2014 |
|
RU2708395C2 |
| ПРОИЗВОДНЫЕ АМИНОМЕТИЛПИПЕРИДИНА В КАЧЕСТВЕ ИНГИБИТОРА КИНАЗЫ | 2018 |
|
RU2756505C1 |
| ПРОИЗВОДНЫЕ ОКСИФТОРПИПЕРИДИНА В КАЧЕСТВЕ ИНГИБИТОРА КИНАЗЫ | 2018 |
|
RU2758370C1 |
| НОВЫЕ БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2016 |
|
RU2760554C1 |
| НОВЫЕ (ГЕТЕРО)АРИЛ-ЗАМЕЩЕННЫЕ ПИПЕРИДИНИЛЬНЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2017 |
|
RU2742271C2 |
| НОВЫЕ ПИПЕРИДИНИЛЬНЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2017 |
|
RU2743163C2 |
Изобретение относится к новому соединению формулы I или его фармацевтически приемлемой соли, которые обладают свойствами ингибитора тирозинкиназы Брутона. Соединения могут быть использованы в качестве терапевтически активного вещества для лечения воспалительного и/или аутоиммунного состояния, выбранного из ревматоидного артрита и астмы. В формуле I
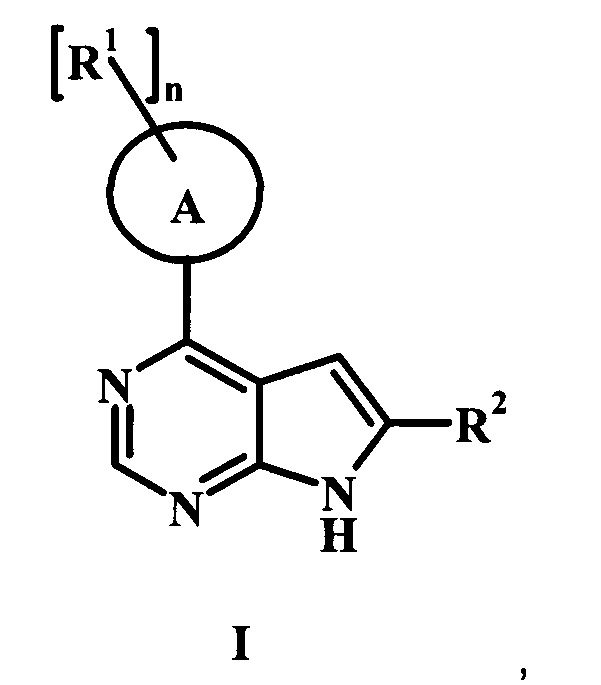
А представляет собой фенил; n равно 1 или 2; R1 представляет собой CH2NHC(=O)R1' или CH2NHC(=O)CH2NHR1', когда n равно 1; и один R1 представляет собой галоген, а второй R1 представляет собой CH2NHC(=O)R1' или CH2N(CH3)C(=O)R1', когда n равно 2; R1' представляет собой низшую алкоксигруппу, фенил, ненасыщенный или частично ненасыщенный 8-9-членный бициклический гетероцикл, содержащий 1-2 гетероатома в бициклической системе, выбранных из азота и серы, или 4-6-членный моноциклический гетероарил с 1-3 гетероатомами, выбранными из азота, кислорода и серы, или 4-5-членный гетероциклоалкил, содержащий атом кислорода или азота в качестве гетероатома, возможно замещенный одним или более R1ʺ; каждый радикал R1ʺ представляет собой независимо низший алкил, галоген, 3-6-членный циклоалкил, 4-членный гетероциклоалкил с атомом кислорода в качестве гетероатома, низший алкил-4-членный гетероциклоалкил с атомом кислорода в качестве гетероатома, оксогруппу, циано-низший алкил, гидроксил-низший алкил или низшую алкоксигруппу; R2 представляет собой Н, R3 или R4; R3 представляет собой C(=O)OR3', C(=O)R3' или C(=O)NH(CH2)2R3'; R3' представляет собой Н, низший алкил, 6-членный гетероциклоалкил с 1-2 гетероатомами, выбранными из азота и кислорода, аминогруппу или ОН; R4' представляет собой пиразолил, возможно замещенный R4'; и R4' представляет собой метил, CH2-CH2N(CH3)2, СН2С(=O)ОСН2СН3, СН2С(=O)ОН или CH2CH2OH. 9 н. и 9 з.п. ф-лы, 2 табл., 51 пр.,
1. Соединение формулы I
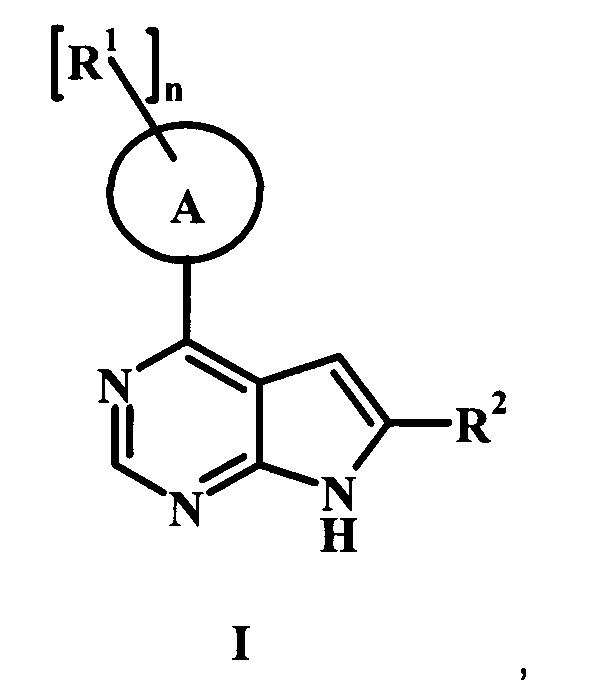
в котором
А представляет собой фенил;
n равно 1 или 2;
R1 представляет собой CH2NHC(=O)R1' или CH2NHC(=O)CH2NHR1', когда n равно 1; и один R1 представляет собой галоген, а второй R1 представляет собой CH2NHC(=O)R1' или CH2N(CH3)C(=O)R1', когда n равно 2;
R1' представляет собой низшую алкоксигруппу, фенил, ненасыщенный или частично ненасыщенный 8-9-членный бициклический гетероцикл, содержащий 1-2 гетероатома в бициклической системе, выбранных из азота и серы, или 4-6-членный моноциклический гетероарил с 1-3 гетероатомами, выбранными из азота, кислорода и серы, или 4-5-членный гетероциклоалкил, содержащий атом кислорода или азота в качестве гетероатома, возможно замещенный одним или более R1ʺ;
каждый радикал R1ʺ представляет собой независимо низший алкил, галоген, 3-6-членный циклоалкил, 4-членный гетероциклоалкил с атомом кислорода в качестве гетероатома, низший алкил-4-членный гетероциклоалкил с атомом кислорода в качестве гетероатома, оксогруппу, циано-низший алкил, гидроксил-низший алкил или низшую алкоксигруппу;
R2 представляет собой Н, R3 или R4;
R3 представляет собой C(=O)OR3', C(=O)R3' или C(=O)NH(CH2)2R3';
R3' представляет собой Н, низший алкил, 6-членный гетероциклоалкил с 1-2 гетероатомами, выбранными из азота и кислорода, аминогруппу или ОН;
R4' представляет собой пиразолил, возможно замещенный R4'; и
R4' представляет собой метил, CH2-CH2N(CH3)2, СН2С(=O)ОСН2СН3, СН2С(=O)ОН или CH2CH2OH;
или его фармацевтически приемлемая соль.
2. Соединение по п. 1, в котором R2 представляет собой Н и n равно 2.
3. Соединение по п. 1, в котором n равно 1 и R1 представляет собой CH2NHC(=O)R1' или CH2NHC(=O)CH2NHR1'.
4. Соединение по п. 1, в котором n равно 2, один R1 представляет собой CH2NHC(=O)R1'.
5. Соединение по п. 4, в котором R2 представляет собой C(=O)OR3', C(=O)R3' или C(=O)NH(CH2)2R3'.
6. Соединение по п. 4, в котором R2 представляет собой пиразолил, замещенный R4'.
7. Соединение по п. 1, в котором R1ʺ представляет собой трет-бутил или галоген.
8. Соединение по п. 1, в котором n равно 2, один R1 представляет собой фтор и R1ʺ представляет собой трет-бутил.
9. Соединение по п. 1, выбранное из группы, состоящей из следующих веществ:
4-трет-бутил-N-[4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-бензил]-бензамид;
3-хлор-N-[4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-бензил]-бензамид;
2-(3-хлорфениламино)-N-[4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-бензил]-ацетамид;
4-трет-бутил-N-[2-фтор-4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-бензил]-бензамид;
трет-бутиловый эфир 4-{4-[(4-трет-бутилбензоиламино)-метил]-3-фторфенил}-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты;
4-(4-((4-трет-бутилбензамидо)метил)-3-фторфенил)-7Н-пирроло[2,3-d]пиримидин-6-карбоновая кислота;
4-трет-бутил-N-(2-фтор-4-(6-(морфолин-4-карбонил)-7Н-пирроло[2,3-d]пиримидин-4-ил)бензил)бензамид;
диметиламид 4-{4-[(4-трет-бутилбензоиламино)-метил]-3-фторфенил}-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты;
метиламид 4-{4-[(4-трет-бутилбензоиламино)-метил]-3-фторфенил}-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты;
(2-гидроксиэтил)-амид 4-{4-[(4-трет-бутилбензоиламино)-метил]-3-фторфенил}-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты;
(2-диметиламиноэтил)-амид 4-{4-[(4-трет-бутилбензоиламино)-метил]-3-фторфенил}-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты;
4-трет-бутил-N-{4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-бензамид;
4-циклопропил-N-{4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-бензамид;
4-изопропил-N-{4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-бензамид;
N-{4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-4-оксетан-3-илбензамид;
4-(3-метилоксетан-3-ил)-N-{4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-бензамид;
4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензиламид 4,5,6,7-тетрагидробензо[b]тиофен-2-карбоновой кислоты;
4-трет-бутил-N-{2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-бензамид;
6-трет-бутил-N-{2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-никотинамид;
2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензиламид 5-метилтиофен-2-карбоновой кислоты;
4-трет-бутил-N-(2-фтор-4-{6-[1-(2-гидроксиэтил)-1Н-пиразол-4-ил]-7Н-пирроло[2,3-d]пиримидин-4-ил}-бензил)-бензамид;
4-трет-бутил-N-{2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-N-метилбензамид;
{2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-метиламид 5-метилтиофен-2-карбоновой кислоты;
2-трет-бутил-5-{2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-4,5-дигидротиено[2,3-с]пиррол-6-он;
2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензиламид 5-трет-бутилизоксазол-3-карбоновой кислоты;
N-{2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-4-(3-метилоксетан-3-ил)-бензамид;
4-(цианодиметилметил)-N-{2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-бензамид;
2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензиламид 4,5,6,7-тетрагидробензо[b]тиофен-2-карбоновой кислоты;
N-{2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-4-(1-гидрокси-1-метилэтил)-бензамид;
2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензиламид 3-трет-бутилизоксазол-5-карбоновой кислоты;
2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензиламид 3-трет-бутоксиазетидин-1-карбоновой кислоты;
2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-с1]пиримидин-4-ил]-бензиламид 1,3-дигидроизоиндол-2-карбоновой кислоты;
4-трет-бутил-N-(4-{6-[1-(2-диметиламиноэтил)-1Н-пиразол-4-ил]-7Н-пирроло[2,3-d]пиримидин-4-ил}-2-фторбензил)-бензамид;
4-{6-[1-(2-диметиламиноэтил)-1Н-пиразол-4-ил]-7Н-пирроло[2,3-d]пиримидин-4-ил}-2-фторбензиламид 3-трет-бутоксиазетидин-1-карбоновой кислоты;
4-{6-[1-(2-диметиламиноэтил)-1Н-пиразол-4-ил]-7Н-пирроло[2,3-d]пиримидин-4-ил}-2-фторбензиламид 1,3-дигидроизоиндол-2-карбоновой кислоты;
этиловый эфир [4-(4-{4-[(4-трет-бутилбензоиламино)-метил]-3-фторфенил}-7Н-пирроло[2,3-d]пиримидин-6-ил)-пиразол-1-ил]-уксусной кислоты;
[4-(4-{4-[(4-трет-бутилбензоиламино)-метил]-3-фторфенил}-7Н-пирроло[2,3-d]пиримидин-6-ил)-пиразол-1-ил]-уксусная кислота;
N-(2-фтор-4-(6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил)бензил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиридин-2-карбоксамид;
2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензиламид 5-трет-бутилизоксазол-3-карбоновой кислоты;
2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензиламид 3-трет-бутил-[1,2,4]оксадиазол-5-карбоновой кислоты;
трет-бутиловый эфир {2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-карбаминовой кислоты и
N-{2-фтор-4-[6-(1-метил-1Н-пиразол-4-ил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензил}-бензамид.
10. Способ лечения воспалительного и/или аутоиммунного состояния, опосредованного активностью тирозинкиназы Брутона, выбранного из ревматоидного артрита и астмы, включающий введение пациенту, который в этом нуждается, терапевтически эффективного количества соединения по любому из пп. 1-9.
11. Способ лечения воспалительного состояния, опосредованного активностью тирозинкиназы Брутона, включающий введение пациенту, который в этом нуждается, терапевтически эффективного количества соединения по любому из пп. 1-9.
12. Способ лечения ревматоидного артрита, опосредованного активностью тирозинкиназы Брутона, включающий введение пациенту, который в этом нуждается, терапевтически эффективного количества соединения по любому из пп. 1-9.
13. Способ лечения астмы, опосредованной активностью тирозинкиназы Брутона, включающий введение пациенту, который в этом нуждается, терапевтически эффективного количества соединения по любому из пп. 1-9.
14. Фармацевтическая композиция, обладающая свойствами ингибитора тирозинкиназы Брутона, включающая терапевтически эффективное количество соединения по любому из пп. 1-9, смешанное по меньшей мере с одним фармацевтически приемлемым носителем, эксципиентом или разбавителем.
15. Применение соединения по любому из пп. 1-9 в качестве терапевтически активного вещества, обладающего свойствами ингибитора тирозинкиназы Брутона, для лечения воспалительного и/или аутоиммунного состояния, выбранного из ревматоидного артрита и астмы.
16. Применение соединения по любому из пп. 1-9 для изготовления лекарственного средства для лечения воспалительного и/или аутоиммунного состояния, опосредованного активностью тирозинкиназы Брутона, выбранного из ревматоидного артрита и астмы.
17. Применение соединения по любому из пп. 1-9 для лечения воспалительного и/или аутоиммунного состояния, опосредованного активностью тирозинкиназы Брутона, выбранного из ревматоидного артрита и астмы.
18. Соединение по любому из пп. 1-9 для применения в лечении воспалительного и/или аутоиммунного состояния, опосредованного активностью тирозинкиназы Брутона.
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| WO 2010036316 A1, 01.04 | |||
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| WO 2009080682 A1, 02.07.2009 | |||
| US 2012157442 A1, 21.06.2012 | |||
| WO 2011029046 A1, 10.03.2011 | |||
| US 2006189638 A1, 24.12 | |||
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Регистратор для дел | 1925 |
|
SU690A1 |
| Способ образования азотной кислоты | 1921 |
|
SU1428A1 |
| Bisagni, Emile; et al., Condensation of hydrazine on 4-chloro-5-acetonylpyrimidines | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |

