Настоящее изобретение относится к способу получения бетамиметиков формулы 1
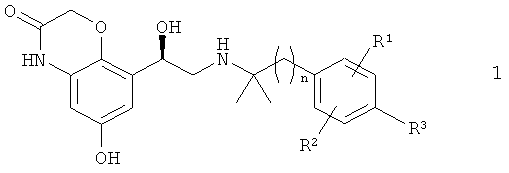
в которой n обозначает 1 или 2,
R1 обозначает водород, галоген, С1-С4алкил или O-С1-С4алкил,
R2 обозначает водород, галоген, С1-С4алкил или O-С1-С4алкил и
R3 обозначает водород, С1-С4алкил, ОН, галоген, O-С1-С4алкил, O-С1-С4алкилен-СООН или O-С1-С4алкилен-СОО-С1-С4алкил.
Предпосылки создания изобретения
Бетамиметики (β-адренергические вещества) известны из уровня техники. В этом отношении можно сослаться, например, на патент US 4460581, где предложены бетамиметики и их применение для терапии различных заболеваний.
Для медикаментозной терапии различных заболеваний часто желательно располагать лекарственными средствами, обладающими продолжительным действием. За счет этого, как правило, удается в течение длительного промежутка времени обеспечить поддержание в организме необходимой для достижения терапевтического эффекта концентрации действующего вещества без необходимости излишне частого повторного введения лекарственного средства в организм. В остальном же введение действующего вещества в организм с меньшей периодичностью в значительной степени способствует хорошему самочувствию пациента. Наиболее желательно при этом располагать лекарственным средством, которое позволяло бы достигать требуемого терапевтического эффекта при его однократном в день введении в организм (т.е. лекарственным средством, рассчитанным на однократный прием в сутки). Преимущество, связанное с применением лекарственного средства только один раз в сутки, состоит в удобстве подобной схемы лечения для пациента благодаря сравнительно быстрому его привыканию к регулярному приему медикамента в определенное время суток.
Исходя из вышеизложенного в основу настоящего изобретения была положена задача разработать способ получения бетамиметиков, которые, с одной стороны, проявляли бы терапевтический эффект при лечении заболеваний дыхательных путей, а с другой стороны, обладали бы более продолжительным действием и тем самым могли бы использоваться для приготовления лекарственных средств с более длительным действием. Задача настоящего изобретения состояла прежде всего в том, чтобы предложить бетамиметики, которые благодаря их длительному действию можно было бы использовать для приготовления лекарственного средства, которое для терапии хронического обструктивного заболевания легких (ХОЗЛ) или астмы требовалось бы вводить в организм один раз в сутки. Наряду с указанными выше задачами еще одна задача настоящего изобретения состояла в том, чтобы предложить бетамиметики, которые обладали бы не только особо высокой эффективностью (сильным действием), но и высокой степенью избирательности по отношению к β2-адренорецепторам.
Подробное описание изобретения
В настоящем изобретении предлагается способ получения соединения формулы 1
в которой n обозначает 1 или 2,
R1 обозначает водород, галоген, С1-С4алкил или O-С1-С4алкил,
R2 обозначает водород, галоген, С1-С4алкил или O-С1-С4алкил и
R3 обозначает водород, С1-С4алкил, ОН, галоген, O-С1-С4алкил, O-С1-С4алкилен-СООН или O-С1-С4алкилен-СОО-С1-С4алкил,
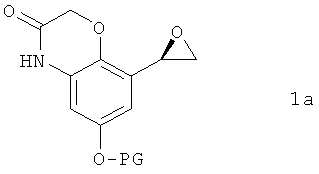
отличающийся тем, что соединение формулы 1а
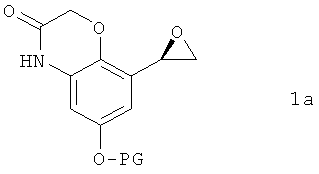
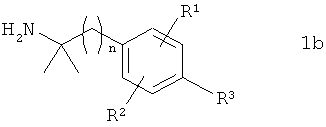
в которой PG обозначает защитную группу, подвергают в органическом растворителе взаимодействию с соединением формулы 1b
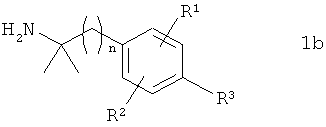
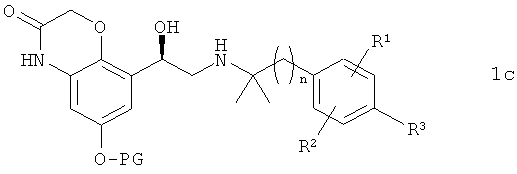
в которой R1, R2, R3 и n имеют указанные выше значения, с получением соединения формулы 1с
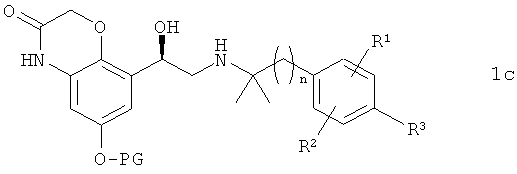
в которой R1, R2, R3, n и PG имеют указанные выше значения, и из этого соединения отщеплением защитной группы PG получают соединение формулы 1.
Описанным выше способом предпочтительно получать соединения формулы 1, в которой
n обозначает 1 или 2,
R1 обозначает водород, галоген или С1-С4алкил,
R2 обозначает водород, галоген или С1-С4алкил и
R3 обозначает водород, С1-С4алкил, ОН, галоген, О-С1-С4алкил, O-С1-С4алкилен-СООН или O-С1-С4алкилен-СОО-С1-С4алкил.
Более предпочтительно получать описанным выше способом соединения формулы 1, в которой
n обозначает 1 или 2,
R1 обозначает водород, фтор, хлор, метил или этил,
R2 обозначает водород, фтор, хлор, метил или этил и
R3 обозначает водород, С1-С4алкил, ОН, фтор, хлор, бром, O-С1-С4алкил, O-С1-С4алкилен-СООН или O-С1-С4алкилен-СОО-С1-С4алкил.
Особенно предпочтительно получать описанным выше способом соединения формулы 1, в которой
n обозначает 1 или 2,
R1 обозначает водород, метил или этил,
R2 обозначает водород, метил или этил и
R3 обозначает водород, метил, этил, ОН, метоксигруппу, этоксигруппу, О-СН2-СООН, O-СН2-СОО-метил или O-СН2-СОО-этил.
Наиболее предпочтительно получать описанным выше способом соединения формулы 1, в которой
n обозначает 1 или 2,
R1 обозначает водород или метил,
R2 обозначает водород или метил и
R3 обозначает водород, метил, ОН, метоксигруппу, О-СН2-СООН или O-СН2-СОО-этил.
При осуществлении предлагаемого в изобретении способа соединение формулы 1а подвергают взаимодействию с соединением формулы 1b в приемлемом растворителе. В качестве растворителей для применения в этих целях пригодны органические растворители, особенно предпочтительно растворители, выбранные из группы, включающей тетрагидрофуран, толуол, этанол, н-пропанол, н-бутанол, н-бутилацетат, диметилформамид, метоксиэтанол, этиленгликоль и диоксан. Наиболее же предпочтительно согласно изобретению использовать в качестве подобных растворителей н-пропанол, тетрагидрофуран и диоксан, среди которых особое значение придается диоксану и н-пропанолу.
Соединение формулы 1b предпочтительно согласно изобретению использовать в по меньшей мере стехиометрическом количестве в пересчете на количество используемого соединения формулы 1а. При необходимости соединение формулы 1b можно также использовать в избытке, например в избытке до 3 эквивалентов, предпочтительно до 2,5 эквивалента, особенно предпочтительно примерно от 1 до 2 эквивалентов, в частности от 1 до 1,5 эквивалента, в пересчете на количество используемого соединения формулы 1а.
Указанное взаимодействие предпочтительно проводить при повышенной температуре, предпочтительно при температуре выше 40°С, особенно предпочтительно при температуре выше 50°С. Наиболее же предпочтительно нагревать реакционную смесь до температуры кипения используемого растворителя.
При такой температуре указанное взаимодействие затем проводят в течение примерно 1-72 ч, предпочтительно в течение 10-60 ч, особенно предпочтительно в течение 20-50 ч.
По завершении реакции растворитель удаляют и остаток растворяют в органическом полярном растворителе, предпочтительно C1-C8спирте или сложном С3-С8эфире, особенно предпочтительно в этаноле или этилацетате, и полученный раствор фильтруют. Фильтрат затем подкисляют, предпочтительно минеральной кислотой, особенно предпочтительно соляной кислотой, и после выдержки продолжительностью от примерно 10 мин до 12 ч, предпочтительно от 20 мин до 6 ч, особенно предпочтительно от 30 мин до 3 ч, продукт отфильтровывают.
Защитную группу предпочтительно отщеплять от соединений формулы 1а путем гидрирования в пригодном для этого растворителе. В качестве растворителей для применения в этих целях пригодны органические растворители, предпочтительно органические полярные растворители, особенно предпочтительно растворители, выбранные из группы, включающей тетрагидрофуран, различные сложные С3-С8эфиры и С1-С8спирты. Предпочтительно же согласно изобретению использовать в качестве подобных растворителей тетрагидрофуран, этанол и метанол, среди которых особое значение придается этанолу и метанолу.
Для гидрирования при осуществлении предлагаемого в изобретении способа предпочтительно использовать катализаторы в присутствии водорода. К предпочтительным для применения при таком гидрировании катализаторам относятся приемлемые катализаторы на основе переходных металлов, предпочтительно гетерогенные катализаторы на основе переходных металлов, особенно предпочтительно палладийсодержащие катализаторы, прежде всего смесь палладия и угля (палладий на угле).
Гидрирование предпочтительно проводить в присутствии избытка водорода. Подобный избыток водорода обеспечивают согласно изобретению путем создания давления водорода в пределах от 1 до 10 бар, предпочтительно от 2 до 7 бар, особенно предпочтительно от 2,5 до 4,5 бара.
Предпочтительно далее проводить гидрирование при повышенной температуре, предпочтительно при температуре в пределах от 25 до 70°С, особенно предпочтительно от 30 до 60°С, прежде всего от 35 до 50°С. По завершении реакции катализатор удаляют предпочтительно путем фильтрации.
После этого удаляют растворитель и продукт перекристаллизовывают из пригодного для этой цели органического растворителя, предпочтительно из C1-C8спирта или смеси C1-C8спиртов, особенно предпочтительно из смеси метанола со спиртом, выбранным из группы, включающей изопропанол, н-пропанол и этанол.
В предпочтительном варианте осуществления предлагаемого в изобретении способа соединение формулы 1а получают путем химического превращения в него соединения формулы 2а
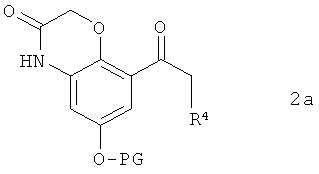
в которой PG имеет указанные выше значения, a R4 обозначает галоген, предпочтительно бром или хлор.
В этом варианте осуществления предлагаемого в изобретении способа соединение формулы 2а подвергают в пригодном для этой цели растворителе взаимодействию с ДИП-хлоридом (диизопинокамфеилхлорборан). В качестве растворителей для применения в этих целях пригодны преимущественно органические растворители. Предпочтительно при этом использовать растворители, выбранные из группы, включающей диэтиловый эфир, трет-бутилметиловый эфир, 2-метилтетрагидрофуран, тетрагидрофуран, толуол и диоксан. Особенно же предпочтительно использовать в качестве растворителей трет-бутилметиловый эфир, тетрагидрофуран и диоксан, среди которых особое значение придается диоксану и тетрагидрофурану.
ДИП-хлорид можно использовать в чистом виде либо в растворе, предпочтительно в инертном органическом растворителе, особенно предпочтительно алифатическом растворителе, прежде всего пентане, гексане, гептане или октане.
ДИП-хлорид добавляют в реакционную среду при пониженной температуре, предпочтительно при температуре ниже 0°С, особенно предпочтительно ниже -10°С, прежде всего при температуре в пределах от -20 до -40°С.
Продолжительность добавления ДИП-хлорида в реакционную среду составляет от 10 мин до 6 ч, предпочтительно от 30 мин до 4 ч, особенно предпочтительно от 1 до 3 ч. Особенно же предпочтительно добавлять ДИП-хлорид в реакционную среду в течение 70-110 мин.
ДИП-хлорид предпочтительно согласно изобретению использовать в по меньшей мере стехиометрическом количестве в пересчете на количество используемого соединения формулы 2а. При необходимости ДИП-хлорид можно также использовать в избытке, например в избытке до 3 эквивалентов, предпочтительно до 2,5 эквивалента, особенно предпочтительно примерно от 1,5 до 2,5 эквивалента, в пересчете на количество используемого соединения формулы 2а.
По завершении добавления ДИП-хлорида реакционную смесь подвергают перемешиванию, которое длится от 10 мин до 4 ч, предпочтительно от 30 мин до 3 ч, особенно предпочтительно от 40 до 80 мин, прежде всего от 50 до 70 мин. В течение этого периода времени температуру реакционной смеси устанавливают на значение в пределах от -20 до 20°С, особенно предпочтительно -10 до 10°С, прежде всего от -5 до 5°С.
По достижении требуемой температуры в реакционную смесь добавляют растворенный в воде гидроксид натрия (NaOH) в по меньшей мере стехиометрическом количестве в пересчете на используемое количество ДИП-хлорида. При необходимости NaOH можно также использовать в избытке, например в избытке до 3 эквивалентов, предпочтительно до 2,5 эквивалента, особенно предпочтительно примерно от 1,5 до 2,5 эквивалента, в пересчете на используемое количество ДИП-хлорида. После добавления NaOH значение pH реакционной смеси предпочтительно должно составлять от 12 до 14, особенно предпочтительно от 12,5 до 13,5, прежде всего от 12,7 до 13,3.
По достижении требуемого значения pH реакционную смесь подвергают перемешиванию, которое длится от 10 мин до 4 ч, предпочтительно от 30 мин до 3 ч, особенно предпочтительно от 40 до 80 мин, прежде всего от 50 до 70 мин. В течение этого периода времени температуру реакционной смеси устанавливают на значение в пределах от 0 до 40°С, особенно предпочтительно от 10 до 30°С, прежде всего от 15 до 25°С. Затем значение pH реакционной смеси добавлением кислоты, предпочтительно минеральной кислоты, особенно предпочтительно соляной кислоты, устанавливают на величину в пределах от 7 до 10, особенно предпочтительно от 8 до 9, прежде всего от 8,2 до 8,8.
В завершение продукт можно выделять из реакционной смеси путем экстракции органическим растворителем и затем путем осаждения другим пригодным для этой цели органическим растворителем получать в виде твердого вещества.
В предпочтительном варианте осуществления предлагаемого в изобретении способа соединение формулы 2а получают путем химического превращения в него соединения формулы 3а
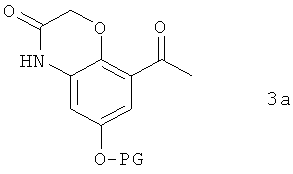
в которой PG имеет указанные выше значения.
В этом варианте осуществления предлагаемого в изобретении способа соединение формулы 3а подвергают в пригодном для этой цели растворителе взаимодействию с галогенирующим агентом. В качестве растворителей для применения в этих целях пригодны преимущественно органические растворители. Предпочтительно при этом использовать растворители, выбранные из группы, включающей уксусную кислоту, бутилацетат, метиленхлорид, тетрагидрофуран, толуол и диоксан. К особенно предпочтительным согласно изобретению растворителям относятся тетрагидрофуран и диоксан.
В качестве галогенирующего агента в предпочтительном варианте осуществления изобретения используют бромирующий агент, особенно предпочтительно бром, N-бромсукцинимид, бензилтриметиламмонийтрибромид и тетрабутиламмонийтрибромид. Галогенирующий агент предпочтительно согласно изобретению использовать в по меньшей мере стехиометрическом количестве в пересчете на используемое количество соединения формулы 3а. При необходимости галогенирующий агент можно также использовать в избытке, например в избытке до 3 эквивалентов, предпочтительно до 2 эквивалентов, особенно предпочтительно примерно от 1 до 1,5 эквивалента, в пересчете на используемое количество соединения формулы 3а. Галогенирующий агент можно при этом добавлять к реакционной смеси в растворителе, предпочтительно в органическом полярном растворителе, особенно предпочтительно в метаноле, этаноле или диоксане, прежде всего в метаноле или диоксане, либо в смеси таких растворителей, прежде всего в смеси метанола с диоксаном.
Указанное взаимодействие предпочтительно проводить при температуре в пределах от 0 до 40°С, предпочтительно от 10 до 30°С, особенно предпочтительно от 15 до 25°С.
По завершении добавления галогенирующего агента реакционную смесь подвергают перемешиванию, которое длится от 10 мин до 6 ч, предпочтительно от 30 мин до 4 ч, особенно предпочтительно от 90 до 150 мин.
Для выделения продукта в реакционную смесь добавляют воду, охлаждая при этом смесь до температуры в пределах от -10 до 10°С, предпочтительно от 0 до 10°С, особенно предпочтительно от 0 до 5°С, и подвергая смесь по завершении добавления к ней воды перемешиванию, которое длится от 10 мин до 4 ч, предпочтительно от 30 мин до 2 ч, особенно предпочтительно от 50 до 70 мин. Целевой продукт можно выделять фильтрацией или центрифугированием с последующей сушкой.
В предпочтительном варианте осуществления предлагаемого в изобретении способа соединение формулы 3а получают путем химического превращения в него соединения формулы 4а
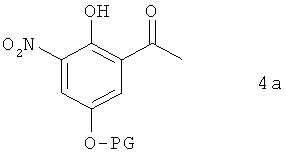
в которой PG имеет указанные выше значения.
В этом варианте осуществления предлагаемого в изобретении способа соединение формулы 4а гидрируют в пригодном для этой цели растворителе. В качестве растворителей для применения в этих целях пригодны органические растворители, предпочтительно органические полярные растворители. Особенно же предпочтительно использовать растворители, выбранные из группы, включающей диметилформамид, N-метилпирролидинон, тетрагидрофуран, 2-метилтетрагидрофуран, толуол и диоксан. К особенно предпочтительным согласно изобретению растворителям при этом относятся диметилформамид, тетрагидрофуран, 2-метилтетрагидрофуран и диоксан, среди которых особое значение придается диметилформамиду и 2-метилтетрагидрофурану.
Для гидрирования при осуществлении предлагаемого в изобретении способа предпочтительно использовать катализаторы в присутствии водорода. К предпочтительным для применения при таком гидрировании катализаторам относятся приемлемые катализаторы на основе переходных металлов, предпочтительно гетерогенные катализаторы на основе переходных металлов, особенно предпочтительно никель- или платиносодержащие катализаторы, прежде всего оксид платины.
Гидрирование предпочтительно проводить в присутствии избытка водорода. Подобный избыток водорода обеспечивают согласно изобретению путем создания давления водорода в пределах от 1 до 10 бар, предпочтительно от 2 до 7 бар, особенно предпочтительно от 2,5 до 4,5 бара.
Предпочтительно далее проводить гидрирование при температуре в пределах от 0 до 50°С, предпочтительно от 10 до 40°С, прежде всего от 20 до 30°С. По завершении реакции катализатор удаляют из жидкой фазы предпочтительно путем фильтрации.
Находящийся в растворе промежуточный продукт формулы 4а#
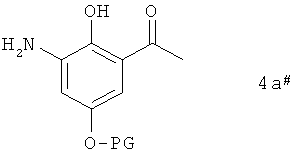
в которой PG имеет указанные выше значения, можно выделять или же без выделения непосредственно превращать далее в соединение формулы 3а.
В соответствии с предлагаемым в изобретении способом соединение формулы 4а# в чистом виде или в виде раствора, прежде всего в виде раствора, от которого на предшествующей стадии был отфильтрован катализатор гидрирования, добавляют к основанию, предпочтительно слабому основанию, особенно предпочтительно карбонату, прежде всего карбонату калия.
Подобное основание предпочтительно использовать в количестве, по меньшей мере в два раза превышающем стехиометрическое в пересчете на используемое количество соединения формулы 4а. При необходимости основание можно также использовать в избытке, например в избытке до 6 эквивалентов, предпочтительно до 4 эквивалентов, особенно предпочтительно примерно от 3 до 3,5 эквивалента, в пересчете на используемое количество соединения формулы 4а.
После этого в реакционную смесь добавляют хлорацетилхлорид. Продолжительность добавления хлорацетилхлорида в реакционную смесь составляет от 10 мин до 2 ч, предпочтительно от 15 мин до 1 ч, особенно предпочтительно от 25 до 35 мин.
Хлорацетилхлорид предпочтительно согласно изобретению использовать в по меньшей мере стехиометрическом количестве в пересчете на используемое количество соединения формулы 4а. При необходимости хлорацетилхлорид можно также использовать в избытке, например в избытке до 4 эквивалентов, предпочтительно до 3 эквивалентов, особенно предпочтительно примерно от 1,5 до 2 эквивалентов, в пересчете на используемое количество соединения формулы 4а.
По завершении добавления хлорацетилхлорида реакционную смесь подвергают перемешиванию, которое длится от 10 мин до 6 ч, предпочтительно от 1 до 4 ч, особенно предпочтительно от 140 до 160 мин.
Указанное взаимодействие предпочтительно проводить при повышенной температуре, предпочтительно при температуре выше 40°С, особенно предпочтительно выше 50°С, наиболее предпочтительно при температуре в пределах от 60 до 70°С.
Реакцию прекращают добавлением воды. Соединение формулы 3а можно очищать и выделять путем экстракционной обработки реакционной смеси водой и последующей перекристаллизации из пригодного для этой цели органического растворителя. Для кристаллизации предпочтительно использовать алифатический углеводород, особенно предпочтительно алифатический циклический углеводород, прежде всего циклогексан или метилциклогексан.
В предпочтительном варианте осуществления предлагаемого в изобретении способа соединение формулы 4а получают путем химического превращения в него соединения формулы 5а
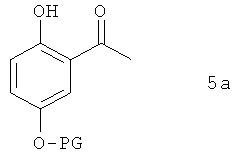
в которой PG имеет указанные выше значения.
В этом варианте осуществления предлагаемого в изобретении способа соединение формулы 5а подвергают в пригодном для этой цели растворителе взаимодействию с нитрующим агентом. В качестве растворителей для применения в этих целях пригодны органические растворители и кислоты, предпочтительно органические протонные растворители и кислоты. К особенно предпочтительным растворителям при этом относятся уксусная кислота и серная кислота, прежде всего уксусная кислота.
Для нитрования при осуществлении предлагаемого в изобретении способа предпочтительно использовать 6-65%-ную азотную кислоту, а также тетрафторборат нитрония или ацетилнитрат. Особенно предпочтительна при этом азотная кислота, прежде всего 65%-ная азотная кислота.
Нитрующий агент предпочтительно согласно изобретению использовать в по меньшей мере стехиометрическом количестве в пересчете на используемое количество соединения формулы 5а. При необходимости нитрующий агент можно также использовать в избытке, например в избытке до 2 эквивалентов, предпочтительно до 1,5 эквивалента, особенно предпочтительно примерно от 1 до 1,1 эквивалента, в пересчете на используемое количество соединения формулы 5а.
По завершении добавления нитрующего агента реакционную смесь подвергают перемешиванию, которое длится от 10 мин до 4 ч, предпочтительно от 20 мин до 3 ч, особенно предпочтительно от 40 до 80 мин.
После этого реакционную смесь разбавляют таким количеством воды, что соединение формулы 4а выпадает из раствора в осадок. Для полного завершения кристаллизации смесь дополнительно подвергают перемешиванию, которое длится от 20 мин до 3 ч, предпочтительно от 30 мин до 2 ч, особенно предпочтительно от 40 до 80 мин, и которое проводят при температуре в пределах от 0 до 20°С, предпочтительно от 5 до 15°С, особенно предпочтительно от 8 до 12°С. Соединение формулы 4а можно выделять путем его отделения от жидкой фазы предпочтительно фильтрацией или центрифугированием.
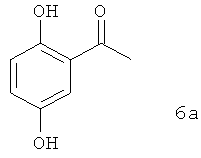
В предпочтительном варианте осуществления предлагаемого в изобретении способа соединение формулы 5а получают путем химического превращения в него соединения формулы 6а
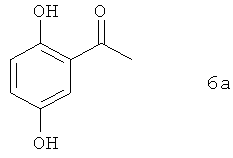
В этом варианте осуществления предлагаемого в изобретении способа соединение формулы 6а подвергают в пригодном для этой цели растворителе взаимодействию с защитной группой PG-A, где А представляет собой приемлемую уходящую группу, такую, например, как хлор, бром, иод, метансульфонил, трифторметансульфонил или пара-толуолсульфонил. Предпочтительно при этом использовать защитную группу, допускающую ее удаление аналогично отщеплению защитной группы PG от соединений формулы 1а. Наиболее предпочтительно использовать необязательно замещенную бензильную защитную группу.
В предпочтительном варианте осуществления предлагаемого в изобретении способа соединение формулы 1b получают путем химического превращения в него соединения формулы 2b
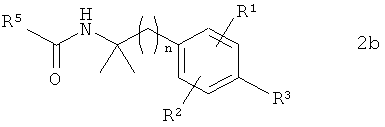
в которой R1, R2, R3 и n имеют указанные выше значения, а R5 обозначает Me.
В этом варианте осуществления предлагаемого в изобретении способа соединение формулы 2b подвергают в пригодном для этой цели растворителе взаимодействию с сильным основанием. В качестве растворителей для применения в этих целях пригодны органические растворители, а особенно предпочтительны растворители, выбранные из группы, включающей этанол, 2-этоксиэтанол и этиленгликоль, а также их смеси. Наиболее же предпочтительно согласно изобретению использовать в качестве растворителя 2-этоксиэтанол или этиленгликоль либо их смесь. В предпочтительном варианте такая смесь состоит из равных объемных частей 2-этоксиэтанола и этиленгликоля, при этом один или другой растворитель может также присутствовать в небольшом избытке.
В качестве сильного основания используют прежде всего неорганические гидроксиды, предпочтительно гидроксиды щелочных либо щелочноземельных металлов, главным образом гидроксид натрия или гидроксид калия. Наиболее предпочтительно при этом использовать гидроксид калия.
Сильное основание предпочтительно согласно изобретению использовать в по меньшей мере стехиометрическом количестве в пересчете на используемое количество соединения формулы 2b. При необходимости сильное основание можно также использовать в избытке, например в избытке до 8 эквивалентов, предпочтительно до 6 эквивалентов, предпочтительно примерно от 2 до 4 эквивалентов, особенно предпочтительно от 3,5 до 4,5 эквивалента, в пересчете на используемое количество соединения формулы 2b.
Указанное взаимодействие предпочтительно проводить при повышенной температуре, предпочтительно при температуре выше 100°С, особенно предпочтительно выше 120°С. Наиболее же предпочтительно нагревать реакционную смесь до температуры в пределах от 140 до 160°С, прежде всего от 145 до 155°С.
После этого реакционную смесь разбавляют для экстракции растворителем и водой. В качестве растворителя при этом используют толуол, ксилол, гептан, метилциклогексан или трет-бутилметиловый эфир, предпочтительно толуол или ксилол. Затем водную фазу удаляют, а органическую фазу на последующих стадиях очистки подвергают экстракционной обработке водой. Воде при этом можно введением традиционных добавок придавать кислое, нейтральное или щелочное значение pH. В предпочтительном варианте органическую фазу подвергают экстракционной обработке сначала подкисленной водой, а затем подщелаченной водой. Продукт можно выделять из органической фазы удалением растворителя.
В предпочтительном варианте осуществления предлагаемого в изобретении способа соединение формулы 2b получают путем химического превращения в него соединения формулы 3b
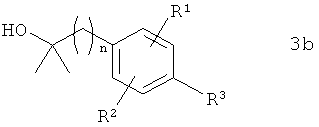
в которой R1, R2, R3 и n имеют указанные выше значения.
В этом варианте осуществления предлагаемого в изобретении способа соединение формулы 3b подвергают в пригодном для этой цели растворителе взаимодействию с ацетонитрилом в присутствии кислоты. В качестве растворителей для применения в этих целях пригодны кислоты, предпочтительно органические кислоты, а особенно предпочтительным растворителем является уксусная кислота.
Ацетонитрил предпочтительно согласно изобретению использовать в по меньшей мере стехиометрическом количестве в пересчете на используемое количество соединения формулы 3b. Предпочтительно при этом использовать ацетонитрил в избытке, например в избытке до 6 эквивалентов, предпочтительно до 5 эквивалентов, особенно предпочтительно примерно от 2 до 4 эквивалентов, прежде всего от 2,5 до 3,5 эквивалента, в пересчете на используемое количество соединения формулы 3b.
В качестве кислоты, в присутствии которой проводят реакцию, предпочтительно использовать серную кислоту, муравьиную кислоту, пара-толуолсульфоновую кислоту, метансульфоновую кислоту, перхлорную кислоту или полифосфорную кислоту, особенно предпочтительно серную кислоту.
Кислоту предпочтительно согласно изобретению использовать в по меньшей мере стехиометрическом количестве в пересчете на используемое количество соединения формулы 3b. При необходимости кислоту можно также использовать в избытке, например в избытке до 2 эквивалентов, предпочтительно до 1,5 эквивалента, особенно предпочтительно примерно от 1 до 1,1 эквивалента, в пересчете на используемое количество соединения формулы 5а. По завершении добавления кислоты реакционную смесь подвергают перемешиванию, которое длится от 1 до 5 ч, предпочтительно от 2 до 4 ч, особенно предпочтительно от 170 до 190 мин.
Указанное взаимодействие предпочтительно проводить при повышенной температуре, предпочтительно при температуре выше 30°С, особенно предпочтительно выше 40°С, особенно предпочтительно при температуре в пределах от 45 до 60°С. При создании изобретения неожиданно было установлено, что при осуществлении подобного способа не происходит нежелательного отщепления метилового эфира в качестве функциональной группы, которого можно было бы ожидать исходя из известных из литературы данных (Can. J. Chem., 56, 1978, cc.3054-3058).
После этого реакционную смесь переносят во второй реактор, содержащий охлажденную смесь растворителей. Для применения в качестве таких растворителей пригодны смеси полярных и неполярных растворителей, предпочтительно водные, органические, полярные и неполярные растворители. К особенно предпочтительным растворителям в качестве компонентов их смеси относятся таковые из группы, включающей воду, трет-бутилметиловый эфир, тетрагидрофуран, толуол, диоксан, гексан, циклогексан и метилциклогексан. Наиболее же предпочтительно согласно изобретению использовать в качестве компонента смеси растворителей воду, трет-бутилметиловый эфир, тетрагидрофуран, толуол, циклогексан и метилциклогексан, при этом особое значение придается смеси из воды, трет-бутилметилового эфира и метилциклогексана.
Смесь растворителей предпочтительно должна иметь пониженную температуру, предпочтительно температуру ниже 20°С, особенно предпочтительно ниже 15°С, наиболее предпочтительно температуру в пределах от 0 до 15°С.
Для осаждения продукта из растворителя повышают значение pH реакционной смеси, которое предпочтительно при этом смещать в область щелочных значений, особенно предпочтительно повышать до 8-12, прежде всего до 9-10. Для повышения значения pH реакционной смеси предпочтительно использовать раствор аммиака.
По завершении процесса добавления подщелачивающего агента и повышения значения pH реакционную смесь подвергают перемешиванию, которое длится от 10 мин до 3 ч, предпочтительно от 20 мин до 2 ч, особенно предпочтительно от 50 до 70 мин.
Затем продукт отделяют центрифугированием и промывают описанными выше, использовавшимися для реакции растворителями. Для дальнейшего повышения чистоты продукта его можно в последующем перекристаллизовывать, соответственно осаждать, например, С1-С8спиртами и водой.
В предпочтительном варианте осуществления предлагаемого в изобретении способа соединение формулы 3b получают путем химического превращения в него соединения формулы 4b
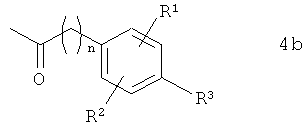
в которой R1, R2, R3 и n имеют указанные выше значения.
В этом варианте осуществления предлагаемого в изобретении способа соединение формулы 4b подвергают в пригодном для этой цели растворителе взаимодействию с метилмагнийбромидом по реакции Гриньяра. В качестве растворителей для применения в этих целях пригодны преимущественно органические растворители. Предпочтительно при этом использовать растворители, выбранные из группы, включающей диэтиловый эфир, трет-бутилметиловый эфир, тетрагидрофуран, толуол и диоксан. Особенно же предпочтительно согласно изобретению использовать в качестве растворителей трет-бутилметиловый эфир, тетрагидрофуран и толуол.
Указанную реакцию предпочтительно проводить при комнатной температуре, предпочтительно при температуре в пределах 10 до 20°С, особенно предпочтительно от 15 до 25°С.
После объединения эдуктов реакционную смесь подвергают перемешиванию, которое длится от 10 мин до 3 ч, предпочтительно от 20 мин до 2 ч, особенно предпочтительно от 50 до 70 мин.
Для прекращения реакции к реакционной смеси добавляют воду и кислоту, предпочтительно серную кислоту. После экстракционной обработки органической фазы стандартными методами продукт можно выделять удалением или отгонкой растворителя. Чистоту продукта можно повысить его перекристаллизацией из органического неполярного растворителя, предпочтительно н-гептана.
Следующим объектом изобретения являются промежуточные продукты формулы 3а
в которой PG имеет указанные выше значения.
Следующим объектом изобретения являются промежуточные продукты формулы 4а
в которой PG имеет указанные выше значения.
Следующим объектом изобретения являются промежуточные продукты формулы 4a#
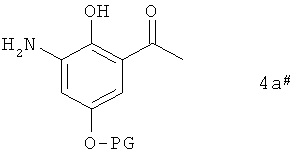
в которой PG имеет указанные выше значения.
Следующим объектом изобретения являются промежуточные продукты формулы 2b
в которой R1, R2, R3 и n имеют указанные выше значения, а R5 обозначает Me.
Объектом изобретения является также способ получения соединений формулы 2а
в которой PG имеет указанные выше значения, а R4 обозначает галоген, предпочтительно бром или хлор, отличающийся тем, что соединение формулы 3а
в которой PG имеет указанные выше значения, подвергают взаимодействию с галогенирующим агентом, выбранным из группы, включающей тетрабутиламмонийтрибромид, бензилтриметиламмонийдихлориодид, N-бромсукцинимид, N-хлорсукцинимид, сульфурилхлорид и бром/диоксан, предпочтительно тетрабутиламмонийтрибромид и N-бромсукцинимид.
Объектом изобретения является также способ получения соединений формулы 3а
в которой PG имеет указанные выше значения, отличающийся тем, что соединение формулы 4а
в которой PG имеет указанные выше значения, подвергают каталитическому гидрированию и затем подвергают взаимодействию с хлорацетилхлоридом.
В предпочтительном варианте осуществления описанного выше способа в качестве промежуточного продукта гидрирования получают соединение формулы 4а#
в которой PG имеет указанные выше значения.
Объектом изобретения является также способ получения соединений формулы 4а
в которой PG имеет указанные выше значения, отличающийся тем, что соединение формулы 5а
в которой PG имеет указанные выше значения, подвергают взаимодействию с нитрующим агентом, выбранным из группы, включающей 65%-ную азотную кислоту, нитрат калия/серную кислоту и тетрафторборат нитрония, предпочтительно взаимодействию с 65%-ной азотной кислотой.
Объектом изобретения является также способ получения соединений формулы 1b
в которой R1, R2, R3 и n имеют указанные выше значения, отличающийся тем, что соединение формулы 2b
в которой R1, R2, R3 и n имеют указанные выше значения, а R5 обозначает Me, подвергают взаимодействию с основанием, выбранным из группы, включающей гидроксид калия, гидроксид натрия, гидроксид лития и гидроксид цезия, предпочтительно взаимодействию с гидроксидом калия или гидроксидом натрия.
Объектом изобретения является также способ получения соединений формулы 2b
в которой R1, R2, R3 и n имеют указанные выше значения, а R5 обозначает Me, отличающийся тем, что соединение формулы 3b
в которой R1, R2, R3 и n имеют указанные выше значения, подвергают в присутствии гигроскопичного реагента, выбранного из группы, включающей серную кислоту, муравьиную кислоту, пара-толуолсульфоновую кислоту, метансульфоновую кислоту, перхлорную кислоту и полифосфорную кислоту, предпочтительно в присутствии серной кислоты, взаимодействию с соединением формулы
в которой R5 имеет указанное выше значение, и затем подвергают взаимодействию с основанием, выбранным из группы, включающей водные растворы аммиака, гидроксид натрия, гидроксид калия, карбонат натрия и карбонат калия.
Используемые термины и понятия
Под "органическим растворителем" согласно изобретению подразумевается органическое низкомолекулярное вещество, способное физическим путем переводить в раствор другие органические вещества. Необходимой предпосылкой для использования того или иного вещества в качестве растворителя является наличие у него свойства химически не изменять в процессе растворения ни растворяющееся, ни растворенное вещество, т.е. свойства, при котором компоненты раствора можно вновь получить из него в их исходном виде методами физического разделения, такими как дистилляция, кристаллизация, сублимация, испарение, адсорбция. По различным причинам возможно использование не только индивидуальных растворителей, но и их смесей, объединяющих в себе растворяющие свойства их компонентов. В качестве примера органических растворителей можно назвать следующие:
- спирты, предпочтительно метанол, этанол, пропанол, бутанол, октанол, циклогексанол,
- гликоли, предпочтительно этиленгликоль, диэтиленгликоль,
- простые эфиры/простые эфиры гликолей, предпочтительно диэтиловый эфир, трет-бутилметиловый эфир, дибутиловый эфир, анизол, диоксан, тетрагидрофуран, простые эфиры моно-, ди-, три-, полиэтиленгликоля,
- кетоны, предпочтительно ацетон, бутанон, циклогексанон,
- сложные эфиры, предпочтительно этилацетат, сложные эфиры гликолей,
- амиды, в частности азотсодержащие соединения, предпочтительно диметилформамид, пиридин, N-метилпирролидон, ацетонитрил,
- серосодержащие соединения, предпочтительно сероуглерод, диметилсульфоксид, сульфолан,
- нитросоединения, предпочтительно нитробензол,
- галогенированные углеводороды, предпочтительно дихлорметан, хлороформ, тетрахлорметан, три- и тетрахлорэтен, 1,2-дихлорэтан, хлорфторуглеводороды,
- алифатические или алициклические углеводороды, предпочтительно бензины, петролейный эфир, циклогексан, метилциклогексан, декалин, терпен-L., или
- ароматические углеводороды, предпочтительно бензол, толуол, о-ксилол, м-ксилол, n-ксилол,
либо их соответствующие смеси.
Под термином "С1-С4алкил" (в том числе и когда он является фрагментом других остатков) подразумеваются разветвленные и неразветвленные алкильные группы с 1-4 атомами углерода. В качестве примера таких групп можно назвать метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил или трет-бутил. Для обозначения подобных групп в некоторых случаях используются также соответствующие им сокращенные названия Me, Et, н-Pr, изо-Pr, н-Bu, изо-Bu, трет-Bu или tBu и т.д. Если не указано иное, то в понятия "пропил" и "бутил" включены также все возможные изомерные формы каждого из таких остатков. Так, например, в понятие "пропил" включены н-пропил и изопропил, в понятие "бутил" включены изобутил, втор-бутил и трет-бутил и т.д.
Под термином "С1-С4алкилен" (в том числе и когда он является фрагментом других остатков) подразумеваются разветвленные и неразветвленные алкиленовые группы с 1-4 атомами углерода. В качестве примера таких групп можно назвать метилен, этилен, пропилен, 1-метилэтилен, бутилен, 1-метилпропилен, 1,1-диметилэтилен или 1,2-диметилэтилен. Если не указано иное, то в понятия "пропилен" и "бутилен" включены также все возможные изомерные формы каждого из таких остатков с таким же числом атомов углерода. Так, например, в понятие "пропилен" включен также 1-метилэтилен, а в понятие "бутилен" включены 1-метилпропилен, 1,1-диметилэтилен и 1,2-диметилэтилен.
Под термином "C1-C8спирт" подразумеваются разветвленные и неразветвленные спирты с 1-8 атомами углерода и одной или двумя гидроксигруппами. Предпочтительны спирты с 1-4 атомами углерода. В качестве примера подобных спиртов можно назвать метанол, этанол, н-пропанол, изопропанол, н-бутанол, изобутанол, втор-бутанол или трет-бутанол. Для обозначения вышеуказанных молекул в некоторых случаях используются также соответствующие им сокращенные названия МеОН, EtOH, н-PrOH, изо-PrOH, н-BuOH, изо-BuOH, трет-BuOH и т.д. Если не указано иное, то в понятия "пропанол", "бутанол", "пентанол" и "гексанол" включены также все возможные изомерные формы каждого из таких спиртов. Так, например, в понятие "пропанол" включены н-пропанол и изопропанол, в понятие "бутанол" включены изобутанол, втор-бутанол и трет-бутанол и т.д.
Под термином "сложный С3-С8эфир" подразумеваются разветвленные и неразветвленные сложные эфиры с в общей сложности 3-8 атомами углерода. Предпочтительны эфиры уксусной кислоты (ацетаты) с 3-6 атомами углерода. В качестве их примеров можно назвать метилацетат, этилацетат, н-пропилацетат, изопропилацетат и н-бутилацетат, предпочтителен среди которых этилацетат.
Под "галогеном" согласно настоящему изобретению подразумевается фтор, хлор, бром или иод. Если не указано иное, то фтор, хлор и бром являются предпочтительными галогенами.
Термин "защитная группа" является согласно настоящему изобретению собирательным понятием для обозначения тех органических остатков, которыми можно временно защищать определенные функциональные группы в молекуле с несколькими активными центрами от воздействия (атаки) реагентов на такие функциональные группы, чтобы реакции протекали только по требуемым (незащищенным) центрам (атомам или группам). Защитные группы должны допускать возможность их избирательного введения в защищаемое ими соединение в мягких условиях. Подобные защитные группы должны оставаться стабильными на протяжении всего времени защиты при любых условиях проведения реакций и операций очистки и не должны допускать рацемизацию и эпимеризацию. Защитные группы должны далее допускать возможность их повторного избирательного отщепления в мягких условиях, в идеальном случае с высоким выходом. Методика выбора пригодной защитной группы, условия реакции по ее введению (растворитель, температура, продолжительность и другие параметры), а также возможные методы повторного удаления защитной группы известны из уровня техники (см. например, Philip Kocienski, Protecting Groups, 3-е изд., изд-во THIEME, Stuttgart, 2004, ISBN 3131370033). К предпочтительным защитным группам относятся необязательно замещенный бензил, дифенилметил, тритил, тозил, мезил и трифлат, среди которых особенно предпочтителен необязательно замещенный бензил.
Экспериментальная часть
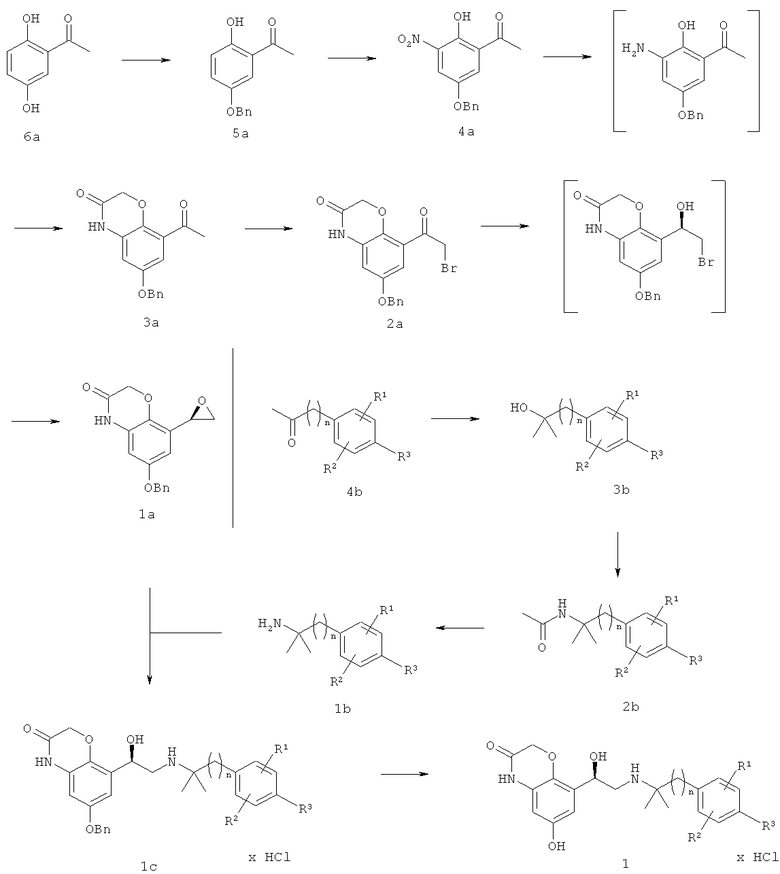
На приведенной выше схеме Bn обозначает бензил, а n может обозначать 1 или 2, R1 может обозначать водород, галоген, С1-С4алкил или O-С1-С4алкил, R2 может обозначать водород, галоген, С1-С4алкил или O-С1-С4алкил и R3 может обозначать водород, С1-C4алкил, ОН, галоген, O-С1-С4алкил, O-С1-С4алкилен-СООН или O-С1-С4алкилен-СОО-С1-С4алкил.
Получение гидрохлорида 8-[(1R)-1-гидрокси-2-[[2-арил-1,1-диметилэтил]амино]этил]-6-(фенилметокси)-2Н-1,4-бензоксазин-3(4Н)-она формулы 1с
7,00 кг (23,54 моля) 8-(2R)-оксиранил-6-(фенилметокси)-2Н-1,4-бензоксазин-3(4Н)-она формулы 1а и 34,70 моля арил-1,1-диметилэтиламина формулы 1b добавляют в 70 л 1,4-диоксана. После этого содержимое реактора нагревают до 97°С и перемешивают при этой температуре в течение 48 ч. Затем смесь охлаждают до 40°С и в вакууме отгоняют 56 л 1,4-диоксана. К остатку добавляют 70 л этанола, охлаждают до 25°С и при 25°С в течение 15 мин добавляют 4,15 кг (34,14 моля) соляной кислоты (30%-ной). Далее вводят затравку и перемешивают до кристаллизации. Образовавшуюся суспензию охлаждают до 20°С и перемешивают в течение 2 ч. Продукт отделяют центрифугированием, промывают 21 л этанола и сушат в вакууме при 50°С. Выход (1с) 84-90%, энантиомерная чистота по данным ЖХВД-анализа 89,5-99,5%.
Получение гидрохлорида 6-гидрокси-8-[(1R)-1-гидрокси-2-[[2-арил-1,1-диметилэтил]амино]этил]-2Н-1,4-бензоксазин-3(4Н)-она формулы 1
19,49 моля гидрохлорида 8-[(1R)-1-гидрокси-2-[[2-(4-метоксифенил)-1,1-диметилэтил]амино]этил]-6-(фенилметокси)-2Н-1,4-бензоксазин-3(4Н)-она формулы 1с загружают в реактор гидрирования и суспендируют в 40 л метанола. 500 г 10%-ного палладия на угле (50% воды) суспендируют в 17 л метанола и путем всасывания подают в реактор гидрирования. Смесь гидрируют при внутренней температуре 40°С и давлении водорода 3 бара до прекращения обнаруживаемого поглощения водорода. Затем катализатор отфильтровывают и промывают 13,3 л метанола. После этого при неглубоком вакууме отгоняют 60 л метанола. При отсутствии кристалообразования в остаток от перегонки вводят затравку. Далее при 50°С дозируют 30 л изопропанола и в течение 1 ч охлаждают до 0°С. Затем смесь перемешивают при 0°С в течение 1 ч, после чего твердое вещество отделяют вакуум-фильтрацией и промывают 15 л холодного изопропанола. Влажный продукт растворяют в 50 л метанола. Полученный раствор фильтруют до прозрачности и напорный фильтр промывают 10 л метанола. После этого при неглубоком вакууме (около 500 мбар) отгоняют 52 л метанола. При отсутствии кристалообразования в остаток от перегонки вводят затравку. Далее дозируют 22,6 л изопропанола. Затем суспензию охлаждают до 0°С и перемешивают в течение 1 ч при 0°С. После этого твердое вещество отделяют от суспензии вакуум-фильтрацией, промывают 15 л холодного изопропанола и сушат в вакууме при 50°С. Выход (1) 63-70%.
Получение 1-[2-гидрокси-5-(фенилметокси)фенил]этанона
20 кг (131,4 моля) 2-ацетилгидрохинона формулы 6а растворяют в 150 л метилизобутилкетона и смешивают с 19,98 кг (144,6 моля) карбоната калия. Далее при 60°С добавляют 22,48 кг (131,5 моля) бензилбромида. После этого реакционную смесь перемешивают в течение 20 ч при 60°С. Затем реакционную смесь охлаждают до 25°С и отфильтровывают твердое вещество. Фильтрат дважды промывают при 25°С раствором, приготовленным из 0,96 кг (11,8 моля) раствора гидроксида натрия (50%-ного) в 60 л воды. Затем метилизобутилкетон практически полностью отгоняют в вакууме и остаток при 60°С растворяют в 80 л метанола. Полученный раствор охлаждают до 0°С и для полного завершения кристаллизации перемешивают в течение 1 ч при этой температуре. Выход (5а) 24,07 кг (75,6%), химическая чистота по данным ЖХВД-анализа 99,2%.
Получение 1-[2-гидрокси-3-нитро-5-(фенилметокси)фенил]этанона
10,00 кг (41,27 моля) 1-[2-гидрокси-5-(фенилметокси)фенил]этанона формулы 5а растворяют в 50 л уксусной кислоты. В полученный раствор при температуре в пределах от 15 до 20°С дозируют 4,40 кг (45,40 моля) 65%-ной азотной кислоты. Питающую емкость промывают 4 л уксусной кислоты. Затем реакционную смесь перемешивают в течение 1 ч. После введения затравки смешивают с 50 л воды. Образовавшуюся суспензию для полного завершения кристаллизации перемешивают в течение 1 ч при 10°С. Продукт отделяют центрифугированием и сушат при 50°С. Выход (4а) 10,34 кг (87,2%), химическая чистота по данным ЖХВД-анализа 99,0%.
Получение 8-ацетил-6-(фенилметокси)-2Н-1,4-бензоксазин-3(4Н)-она
15,00 кг (52,22 моля) 1-[2-гидрокси-3-нитро-5-(фенилметокси)фенил]этанона формулы 4а, 0,165 кг оксида платины(IV) и 45 л 2-метилтетрагидрофурана гидрируют при давлении водорода 3 бара и при внутренней температуре 25°С до прекращения обнаруживаемого поглощения водорода. После этого катализатор отфильтровывают и промывают 20 л 2-метилтетрагидрофурана. В другой реактор загружают 23,09 кг (167,09 моля) карбоната калия и добавляют реакционную смесь из первого реактора. Его дополнительно промывают 22 л 2-метилтетрагидрофурана. После этого к суспензии в течение 30 мин дозируют 9,44 кг (83,55 моля) хлорацетилхлорида. После проведения реакции в течение 2,5 ч при 65°С добавляют 101 л воды. Водную фазу отделяют при 55°С. Затем от органической фазы в вакууме отгоняют 34 л 2-метилтетрагидрофурана. После нагрева до температуры перегонки в течение 30 мин при кипячении с обратным холодильником дозируют 180 л метилциклогексана. Образовавшуюся суспензию охлаждают до 20°С и для полного завершения кристаллизации перемешивают еще в течение 1 ч при этой температуре. Затем осадок отделяют центрифугированием, промывают 113 л метилциклогексана и сушат при 50°С. Выход (3а) 12,70 кг (81,8%), химическая чистота по данным ЖХВД-анализа 98,4%.
Получение 8-(бромацетил)-6-(фенилметокси)-2Н-1,4-бензоксазин-3(4Н)-она
12,00 кг (40,36 моля) 8-ацетил-6-(фенилметокси)-2Н-1,4-бензоксазин-3(4Н)-она формулы 3а растворяют в 108 л 1,4-диоксана. После этого к суспензии при 20°С дозируют раствор 24,33 кг (50,45 моля) тетрабутиламмонийтрибромида в 48 л 1,4-диоксана и 12 л метанола. Содержимое реактора перемешивают в течение 2 ч при 20°С. Затем при 20°С в течение 15 мин добавляют 72 л воды. После охлаждения до 3°С перемешивают в течение 1 ч, продукт отделяют центрифугированием и промывают смесью из 9 л 1,4-диоксана и 4,5 л воды. Далее продукт промывают 60 л воды и сушат в вакууме при 50°С. Выход (2а) 11,29 кг (74,4%), химическая чистота по данным ЖХВД-анализа 98,0%.
Получение 8-(2R)-оксиранил-6-(фенилметокси)-2Н-1,4-бензоксазин-3(4Н)-она
12,00 кг (31,90 моля) 8-(бромацетил)-6-(фенилметокси)-2Н-1,4-бензоксазин-3(4Н)-она формулы 2а растворяют в 180 л тетрагидрофурана и охлаждают до -30°С. После этого в течение 1,5 ч дозируют 34,63 кг (70,18 моля) 65%-ного (-)-ДИП-хлорида в гексане. Далее реакционную смесь перемешивают в течение 1 ч и нагревают до 0°С. При этой температуре дозируют 11,48 кг (143,54 моля) раствора гидроксида натрия (50%-ного) в смеси с 36 л воды. После этого питающую емкость промывают 9 л воды. К концу добавления значение pH должно составлять 13. Смесь нагревают до 20°С и перемешивают в течение 1 ч. Далее дозируют смесь из 4,5 л (42,11 моля) технической соляной кислоты (30%-ной) и 18,6 л воды до тех пор, пока значение рН не достигнет 8,5. После добавления 84 л этилацетата нагревают до 30°С. После разделения фаз от органической фазы отгоняют часть растворителя, остаток смешивают с 120 л трет-бутилметилового эфира, охлаждают до 0°С и перемешивают в течение 1 ч. Продукт выделяют, промывают трет-бутилметиловым эфиром и сушат в вакууме при 50°С. Выход (1а) 8,06 кг (85,0%), энантиомерная чистота по данным ЖХВД-анализа 98,3%.
Получение соединений формулы 3b
24,68 кг (72,6 моля) метилмагнийхлорида (в виде 22%-ного раствора в ТГФ) растворяют в 35 л толуола и охлаждают до 16°С. Далее при температуре в пределах от 16 до 22°С дозируют раствор 60,9 моля арилацетона формулы 4b и 10 л толуола и перемешивают в течение 1 ч при 22°С. Затем реакционный раствор при температуре в пределах от 2 до 17°С дозируют к смеси из 45 л воды и 5,22 кг (51,1 моля) серной кислоты. После этого двухфазную смесь перемешивают и отделяют водную фазу. Органическую фазу промывают раствором 1,00 кг (11,9 моля) гидрокарбоната натрия и 11 л воды. Далее растворитель полностью отгоняют в вакууме. Остаток растворяют в 65,5 л н-гептана. После охлаждения до 2°С реакционную смесь перемешивают при этой температуре в течение 3 ч. Затем продукт выделяют, промывают 17,5 л н-гептана и сушат в вакууме при 25°С. Выход (3b) 75-80%, химическая чистота по данным ЖХВД-анализа 98,9-99,9%.
Получение соединений формулы 2b
55,48 моля 1-арил-2-метилпропан-2-ола формулы 3b добавляют в 6,83 кг (166,44 моля) ацетонитрила и 13 л уксусной кислоты и нагревают до 40°С. Далее при температуре в пределах от 50 до 55°С дозируют 5,66 кг (55,48 моля) серной кислоты. После этого смесь перемешивают в течение 3 ч при 50°С. Во втором реакторе 160 л воды, 20 л трет-бутилметилового эфира и 21 л метилциклогексана охлаждают до 10°С. Затем содержимое первого реактора переносят во второй реактор. После этого значение pH содержимого реактора устанавливают на 9,5 добавлением примерно 40 л раствора аммиака (25%-ного). Суспензию охлаждают до 5°С и перемешивают в течение 1 ч при этой температуре. Продукт отделяют центрифугированием и промывают 30 л воды, а также смесью из 7,5 л трет-бутилметилового эфира и 7,5 л метилциклогексана. Влажный продукт нагревают в 25 л этанола (96%-ного) до 75°С и смешивают при этой температуре с 30 л воды. Раствор перемешивают в течение 15 мин при 85°С, после чего охлаждают до 2°С и перемешивают в течение 1 ч при этой температуре. Затем продукт выделяют, промывают смесью из 5 л воды и 5 л этанола (96%-ного) и сушат. Выход (2b) 65-71%, химическая чистота по данным ЖХВД-анализа 98,6-99,8%.
Получение соединений формулы 1b
Смесь из 45,2 моля N-[2-арил-1,1-диметилэтил]ацетамида формулы 2b, 12,07 кг КОН (180,8 моля), 15 л этоксиэтанола и 15 л этиленгликоля нагревают до 150°С с выдержкой при этой температуре в течение 12 ч. После охлаждения до комнатной температуры смесь разбавляют 61 л воды и 31 л толуола. Фазы разделяют и органическую фазу еще раз промывают 30 л воды. Затем органическую фазу смешивают с 52 л воды. После этого подкисляют добавлением 8,91 кг соляной кислоты (90,4 моля). После разделения фаз содержащую продукт водную фазу смешивают с 30 л толуола и подщелачивают добавлением 9,04 кг 50%-ного раствора NaOH (113,0 молей). После разделения фаз содержащую продукт органическую фазу концентрируют в вакууме до получения маслянистого остатка. Выход (1b) 69-75%, химическая чистота по данным ЖХВД-анализа 94-96%.
В соединениях формул 3b, 2b и 1b при их получении описанными выше методами синтеза остатки R1, R2 и R3 могут иметь, например, следующие значения:
Аналогично описанным выше методам можно тем самым получать следующие соединения формулы 1 в виде их R-энантиомеров:
6-гидрокси-8-{1-гидрокси-2-[2-(4-метоксифенил)-1,1-диметилэтиламино]этил}-4Н-бензо[1,4]оксазин-3-он,
8-{2-[2-(2,4-дифторфенил)-1,1-диметилэтиламино]-1-гидроксиэтил}-6-гидрокси-4Н-бензо[1,4]оксазин-3-он,
8-{2-[2-(3,5-дифторфенил)-1,1-диметилэтиламино]-1-гидроксиэтил}-6-гидрокси-4Н-бензо[1,4]оксазин-3-он,
8-{2-[2-(4-этоксифенил)-1,1-диметилэтиламино]-1-гидроксиэтил}-6-гидрокси-4Н-бензо[1,4]оксазин-3-он,
8-{2-[2-(4-фторфенил)-1,1-диметилэтиламино]-1-гидроксиэтил}-6-гидрокси-4Н-бензо[1,4]оксазин-3-он.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ БЕТАМИМЕТИКОВ | 2008 |
|
RU2462460C2 |
| СПОСОБ ПОЛУЧЕНИЯ ТЕТРАЗАМЕЩЕННЫХ ПРОИЗВОДНЫХ ИМИДАЗОЛА И ИХ НОВЫЕ КРИСТАЛЛИЧЕСКИЕ СТРУКТУРЫ | 2002 |
|
RU2286342C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЦИКЛИЧЕСКИХ ДИКЕТОНОВ | 2005 |
|
RU2384562C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ О-ХЛОРМЕТИЛФЕНИЛГЛИОКСИЛОВОЙ КИСЛОТЫ | 1997 |
|
RU2177472C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ | 2003 |
|
RU2374227C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЦИКЛИЧЕСКИХ ДИКЕТОНОВ | 2003 |
|
RU2316544C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ФЕНИЛПРОПАРГИЛОВОГО ЭФИРА | 2001 |
|
RU2259353C2 |
| НОВЫЕ АНТИХОЛИНЕРГИЧЕСКИЕ СРЕДСТВА, СПОСОБ ИХ ПОЛУЧЕНИЯ, А ТАКЖЕ ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ | 2003 |
|
RU2325388C2 |
| ЭНАНТИОСЕЛЕКТИВНЫЙ СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ХИНОЛИНА | 2005 |
|
RU2383534C2 |
| СПОСОБ ПОЛУЧЕНИЯ СОЛЕЙ БИЦИКЛИЧЕСКИХ ДИКЕТОНОВ И ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ | 2003 |
|
RU2315743C2 |
Описывается способ получения соединений формулы 1
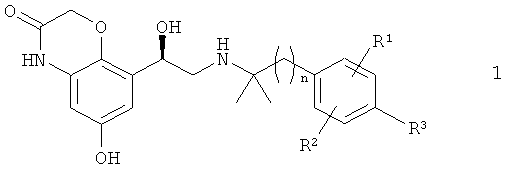
в которой n обозначает 1 или 2, R1 обозначает водород или галоген, R2 обозначает водород или галоген и R3 обозначает водород, галоген или O-C1-С4алкил, заключающийся в том, что соединение формулы 1а
в которой PG обозначает защитную группу, подвергают в органическом растворителе взаимодействию с соединением формулы 1b
с получением соединения формулы 1 с
и из этого соединения отщеплением защитной группы PG получают соединение формулы 1. 11 з.п. ф-лы.
1. Способ получения соединений формулы 1
,
в которой n обозначает 1 или 2,
R1 обозначает водород или галоген,
R2 обозначает водород или галоген, и
R3 обозначает водород, галоген или O-С1-С4алкил,
отличающийся тем, что соединение формулы 1а
,
в которой PG обозначает защитную группу, подвергают в органическом растворителе взаимодействию с соединением формулы 1b
,
в которой R1, R2, R3 и n имеют указанные выше значения, с получением соединения формулы 1с
,
в которой R1, R2, R3, n и PG имеют указанные выше значения, и из этого соединения отщеплением защитной группы PG получают соединение формулы 1.
2. Способ по п.1, которым получают соединения формулы 1, в которой
n обозначает 1 или 2,
R1 обозначает водород, фтор или хлор,
R2 обозначает водород, фтор или хлор, и
R3 обозначает водород, фтор, хлор, бром или O-С1-С4алкил.
3. Способ по п.1, которым получают соединения формулы 1, в которой
n обозначает 1 или 2,
R1 обозначает водород,
R2 обозначает водород, и
R3 обозначает водород, метоксигруппу, этоксигруппу.
4. Способ по п.1, которым получают соединения формулы 1, в которой
n обозначает 1 или 2,
R1 обозначает водород,
R2 обозначает водород, и
R3 обозначает водород или метоксигруппу.
5. Способ по одному из пп.1-4, при осуществлении которого соединение формулы 1а получают путем химического превращения в него соединения формулы 2а
,
в которой PG имеет указанные в п.1 значения, a R4 обозначает галоген.
6. Способ по п.5, при осуществлении которого соединение формулы 2а получают путем химического превращения в него соединения формулы 3а
,
в которой PG имеет указанные в п.1 значения.
7. Способ по п.6, при осуществлении которого соединение формулы 3а получают путем химического превращения в него соединения формулы 4а
,
в которой PG имеет указанные в п.1 значения.
8. Способ по п.7, при осуществлении которого соединение формулы 4а получают путем химического превращения в него соединения формулы 5а
,
в которой PG имеет указанные в п.1 значения.
9. Способ по п.8, при осуществлении которого соединение формулы 5а получают путем химического превращения в него соединения формулы 6а
 ,
,
10. Способ по одному из пп.1-4, при осуществлении которого соединение формулы 1b получают путем химического превращения в него соединения формулы 2b
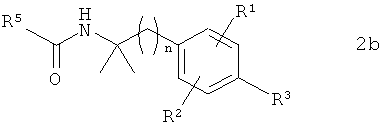 ,
,
в которой R1, R2, R3 и n имеют указанные в пп.1-5 значения, a R5 обозначает Me.
11. Способ по п.10, при осуществлении которого соединение формулы 2b получают путем химического превращения в него соединения формулы 3b
,
в которой R1, R2, R3 и n имеют указанные в пп.1-4 значения.
12. Способ по п.11, при осуществлении которого соединение формулы 3b получают путем химического превращения в него соединения формулы 4b
,
в которой R1, R2, R3 и n имеют указанные в пп.1-4 значения.
| US 4460581, 17.07.1984 | |||
| US 4656168, 07.04.1987 | |||
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| SU 1149876 A, 07.04.1985. | |||

