Настоящее изобретение относится к способу получения солей бициклических 1,3-дикетонов и к новым бициклическим енольным лактоновым промежуточным продуктам для применения в этом способе.
Бициклические 1,3-дикетоны, такие как, например, бицикло[3.2.1]октан-2,4-дион, являются ценными промежуточными продуктами при получении гербицидов, таких как описанные, в частности, в WO 00/15615, WO 00/37437, WO 01/66522 и WO 01/94339.
Известен ряд способов получения таких 1,3-дикетонов. Так, например, бициклические 1,3-дикетоны могут быть получены в соответствии с известными способами из соответствующих соединений в солевой форме.
Такой способ получения бициклических 1,3-дикетонов из соответствующих солей описан, например, в JP 10-265441. Применение алифатических эфиров 3-ацетилциклопентанкарбоновых кислот, которые получают из 3-метиленбицикло[2.2.1]гептан-2-она, в качестве исходных материалов для промышленного получения бицикло[3.2.1]октан-2,4-диона через соответствующую натриевую соль делают такой способ слишком неэкономичным, поскольку окислительное размыкание цикла в присутствии кислот и спиртов, например с использованием сернистой пероксокислоты в присутствии метанола, может привести к образованию не только целевых алифатических эфиров, но также свободной 3-ацетилциклопентанкарбоновой кислоты, которую необходимо вновь превратить в соответствующий алифатический эфир осуществлением перед циклизацией дополнительной реакционной стадии.
Следовательно, целью настоящего изобретения является разработка нового способа получения солей бициклических 1,3-дикетонов, осуществление которого дает возможность экономично получать эти соли с высоким выходом продукта хорошего качества.
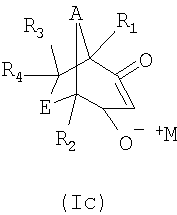
Таким образом, объектом настоящего изобретения является способ получения соединений формулы I
, ,
,
в которой каждый из R1, R2, R3 и R4 независимо друг от друга обозначает водородный атом, С1-С4алкил, атом галогена, гидрокси, С1-С4алкокси, С1-С4алкоксикарбонил, гидроксикарбонил или цианогруппу; каждый из А и Е независимо друг от друга обозначает С1-С2алкилен, который может быть замещен один раз или до четырех раз С1-С4алкильной группой или атомом галогена, гидрокси, С1-С4алкокси, С1-С4алкоксикарбонил или цианогруппу; а М+ обозначает ион щелочного металла, ион щелочно-земельного металла или аммониевый ион, причем этот способ включает следующие стадии:
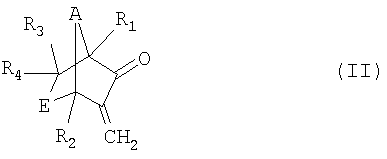
а) реакция соединения формулы II
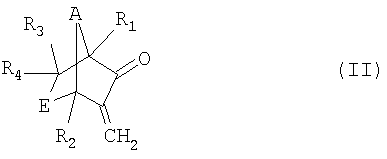 ,
,
в которой R1, R2, R3, R4, А и Е имеют значения, указанные для формулы I, в присутствии окислителя с образованием соединения формулы III
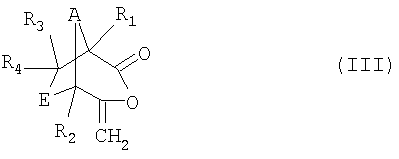 ,
,
в которой R1, R2, R3, R4, А и Е имеют значения, указанные для формулы I, a
б) затем превращение этого соединения в соль формулы I либо в присутствии основания и каталитически эффективного количества цианида, либо в присутствии алкоголята щелочного металла или алкоголята или гидроксида щелочно-земельного металла.
В вышеприведенных определениях заместителей алкильные группы могут быть прямоцепочечными или разветвленными, примером такой группы может служить метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил или трет-бутил. Алкокси представляет собой, например, метокси, этокси, пропокси, изопропокси, н-бутокси, изобутокси, втор-бутокси или трет-бутокси. Алкоксикарбонил представляет собой, например, метоксикарбонил, этоксикарбонил, пропоксикарбонил, изопропоксикарбонил, н-бутоксикарбонил, изобутоксикарбонил, втор-бутоксикарбонил или трет-бутоксикарбонил, предпочтительно метоксикарбонил или этоксикарбонил.
М+ как ион щелочного металла, ион щелочно-земельного металла или аммониевый ион представляет собой, например, натриевый, калиевый, кальциевый, магниевый, триэтиламмониевый или диизопропилэтиламмониевый катион.
Для улучшенной иллюстрации точек присоединения у бициклического соединения можно также представить изображения соединений формулы I следующим образом
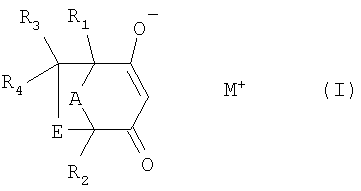 .
.
Поскольку соединения формулы III, которые могут быть получены из хиральных соединений формулы II, и они также могут находиться в хиральных формах, в частности в таких как
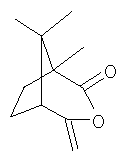 ,
, 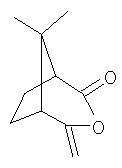 ,
, 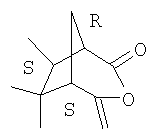
объем настоящего изобретения охватывает все такие хиральные формы, способы их получения и их применение при получении хиральных соединений формулы I.
Соли формулы I могут также встречаться в таутомерных формах, как проиллюстрировано ниже
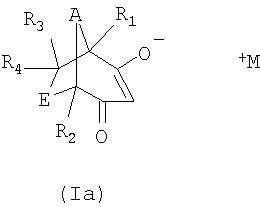
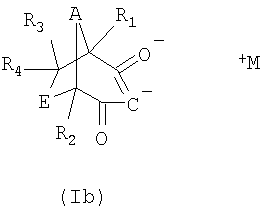
Соединения формулы II известны или могут быть получены в соответствии с известными методами. Получение соединений формулы II, в которой каждый из R1, R2, R3 и R4, обозначает водородный атом, А обозначает метилен и Е обозначает метилен, описаны, например, в JP 10-265415.
Способ в соответствии с изобретением особенно приемлем для получения соединений формулы I, в которой
а) каждый из R1, R2, R3 и R4, независимо друг от друга обозначает водородный атом или С1-С4алкил, каждый из А и Е независимо друг от друга обозначает С1-С2алкилен, который может быть замещен один раз или до четырех раз С1-С4алкильной группой, а М обозначает ион щелочного металла, ион щелочно-земельного металла или аммониевый ион;
б) каждый из R1 и R2, независимо друг от друга обозначает водородный атом или метил;
в) каждый из R3 и R4 независимо друг от друга обозначает водородный атом или метил;
г) А обозначает метилен, который может быть однократно или двукратно замещенным метильной группой, или этилен;
д) Е обозначает метилен, который может быть однократно или двукратно замещенным метильной группой; и/или
е) М+ обозначает натриевый, триэтиламмониевый или диизопропилэтиламмониевый катион.
Способ в соответствии с изобретением еще больше приемлем для получения соединений формулы I, в которой каждый из R1, R2, R3 и R4 обозначает водородный атом, А обозначает метилен и Е обозначает метилен, а М+ обозначает натриевый, триэтиламмониевый или диизопропилэтиламмониевый катион.
Реакционная стадия а).
Кетоны можно окислять до алифатических эфиров в присутствии окислителей, таких как перкислоты, например перуксусная кислота, м-хлорпербензойная кислота и трифторперуксусная кислота, пероксид водорода или пероксид водорода в присутствии каталитически эффективных количеств диоксида селена, причем в только что введенную кислородную группу мигрирует углеродный атом. Такая реакция общеизвестна как перегруппировка Байера-Виллигера. Из специальной химической литературы известно также, что различные стерические, конформационные и электронные эффекты, а также эффекты, вызванные деформацией кольца, определяют положение, в котором вицинально по отношению к карбонильной группе вводят кислородный атом. Следовательно, остается неожиданным то, что в соответствии с изобретением в бициклических экзометиленкетонах с деформированным кольцом формулы II
,
в которой R1, R2, R3, R4, А и Е имеют значения, указанные для формулы I, с высоким уровнем селективности возможно введение между карбонильной группой и экзометиленовой группой кислородной группы и что благодаря этому возможно получение бициклических енольных лактонов формулы III
,
которые можно выделить, которые стабильны и, что является большим преимуществом для промышленной технологии, пригодны для дистилляции.
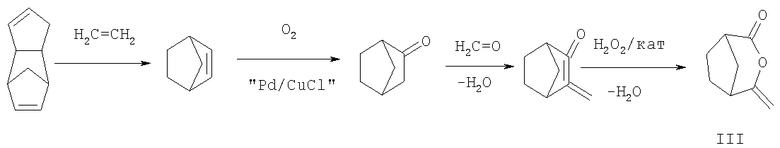
Что касается некоторых соединений формулы III, например бицикло[3.2.1]октан-2,4-диона, то предлагаемый способ обладает особыми экономическими и экологическими преимуществами, поскольку используемые исходные материалы представляют собой нефтехимическое сырье, из которого в результате реакций присоединения, реакций конденсации с удалением воды и в принципе с использованием пероксида водорода как окислителя эффективно и без образования вредоносных сточных вод получают продукт формулы III
На реакционной стадии а) предпочтительными окислителями для превращения соединений формулы II в соединения формулы III являются органические перкислоты, такие как перуксусная кислота, трифторперуксусная кислота, пермуравьиная кислота, перпропионовая кислота, пербензойная кислота, м-хлорпербензойная кислота или монопероксифталевая кислота, пероксид водорода или пероксид водорода в присутствии каталитически эффективных количеств диоксида селена и, когда это уместно, в присутствии дополнительного количества основания.
В предпочтительном варианте реакцию в соответствии с реакционной стадией а) проводят в присутствии основания в инертном растворителе при температурах от -20 до 50°С, преимущественно от -15 до +15°С. Приемлемые растворители включают, например, дихлорметан, дихлорэтан, уксусную кислоту, уксусный ангидрид и их смеси, например дихлорметан и уксусную кислоту или уксусную кислоту и уксусный ангидрид. Подходящие основания включают, например, ацетат натрия, ацетат калия, карбонат натрия, бикарбонат натрия, карбонат калия, карбонат кальция, оксид бария, вторичный кислый фосфат калия и первичный кислый фосфат калия, преимущественно тригидрат ацетата натрия, когда используют пероксид водорода в уксусной кислоте. Основание применяют в количестве от 0,1 до примерно 6 экв., предпочтительно от 1 до 3 экв. Когда используют каталитически эффективное количество диоксида селена, в предпочтительном варианте диоксид селена используют лишь в очень небольших количествах, приблизительно от 0,0001 до 1%.
На реакционной стадии а) окислитель можно использовать в меньше чем стехиометрическом количестве или в эквимолярных количествах, или вплоть до небольшого избытка, до 1,4 экв. В предпочтительном варианте окислитель используют в количестве, которое меньше стехиометрического. Для того чтобы избежать потери селективности вследствие последующего окисления соединения формулы III, предпочтительно окисление до степени превращения от 40 до 85%, преимущественно от 50 до 70%, причем непрореагировавший исходный материал возвращают в процесс. После разрушения избытка окислителя и экстракционной обработки в соответствии с обычными методами исходный материал формулы II можно эффективно выделять в форме низкокипящего дистиллята. Такой технический прием выгоден, в особенности для получения соединений формулы I в промышленном масштабе и их последующего использования при получении бициклических 1,3-дикетонов, поскольку получаемые продукты характеризуются высокой степенью чистоты, в очень значительной мере свободны от остатков и благодаря тому, что они находятся в жидком состоянии, обладают хорошими транспортировочными свойствами (например, пригодны для транспортировки по трубам). Остаток после перегонки можно либо непосредственно использовать для получения солей формулы I, либо, если требуется, концентрировать дистилляцией до содержания от 90 до 99%, например для получения чистых бициклических 1,3-дикетоновых производных прямой реакцией с солями формулы I.
Стадия б) способа
Известно, что в присутствии оснований, таких как, например, метилат натрия, нагреванием в безводном бензоле некоторые 6-метилентетрагидропиран-2-оны можно превращать непосредственно в 1,3-циклогександионы. Такой способ для случаев получения 4,4-диметилциклогексан-1,3-диона и 4-фенилциклогексан-1,3-диона описан в J.Gen.Chem. USSR, 1964, 34, 3509.
Было установлено, что этот способ можно очень успешно применять для конверсии енольных лактонов формулы III в соли бициклических 1,3-дикетонов с деформированным кольцом формулы I в соответствии со стадией б) способа.
С этой целью проводят реакцию соединения формулы III в присутствии по меньшей мере каталитически эффективных количеств ионов алкоголята щелочного металла и алкоголята щелочно-земельного металла в растворителе. Для проведения этой реакции алкоголяты щелочных металлов и щелочно-земельных металлов можно использовать в каталитически эффективных или стехиометрических количествах. Когда применяют каталитически эффективные количества, необходимо добавлять дополнительное основание. Дополнительное основание можно добавлять в стехиометрических количествах или в избытке. Более целесообразно его использовать в количестве от стехиометрического до небольшого избытка. При этом в качестве дополнительных оснований можно применять, например, неорганические основания, такие как карбонаты, например карбонат калия, гидроксиды, например гидроксид натрия или гидроксид калия, оксиды, например оксид бария, и гидриды, например гидрид натрия. Под каталитически эффективными количествами алкоголятов щелочных металлов и щелочно-земельных металлов следует понимать количества от 0,0001 до 25%, предпочтительно от 1 до 10%.
В предпочтительном варианте осуществления способа в соответствии с изобретением алкоголяты щелочных металлов и щелочно-земельных металлов, в особенности алкоголяты лития, натрия и калия, используют без дополнительного основания в стехиометрических количествах или в избытке, но особенно предпочтительно в стехиометрических количествах.
Предпочтительными алкоголятами щелочных металлов и щелочно-земельных металлов являются алкоголяты лития, натрия и калия, преимущественно метилаты и этилаты. Алкоголяты щелочных металлов и щелочно-земельных металлов, которые особенно предпочтительны, представляют собой метилат натрия, этилат натрия, изопропилат натрия, н-бутилат натрия, трет-бутилат калия, пентилат натрия, трет-пентилат натрия, амилат натрия и 2-метоксиэтилат натрия; еще более предпочтителен метилат натрия. Столь же приемлемо применение безводных гидроксидов, например гидроксида лития, гидроксида натрия или гидроксида калия.
Растворителями, подходящими для такого превращения, являются толуол, ксилол, хлорбензол, метилнафталин или спирты, такие как метанол, этанол, изопропанол и амиловый спирт, тетрагидрофуран, диоксан, апротонные растворители, такие как пропионитрил, диметилформамид, N-метилпирролидон и диметилсульфоксид, 2-метил-5-этилпиридин и т.п., а также смеси таких растворителей, например толуола и диметилформамида или толуола с N-метилпирролидоном.
На реакционной стадии б) особое предпочтение отдают применению толуола, а в качестве дополнительного растворителя диметилформамиду или N-метилпирролидону, поскольку в этих обстоятельствах можно особенно успешно осаждать из реакционной смеси соединения формулы I и, следовательно, практически избегать последующих катализируемых основанием вторичных реакций.
На реакционной стадии б) растворитель или растворители используют в количестве, в котором соль, предпочтительно натриевая соль, осаждается из реакционной среды в легкокристаллизующейся форме, а реакционная смесь остается, тем не менее, легкоперемешиваемой. Во время превращения соединений формулы III в соединения формулы I, в которой М обозначает катион щелочного металла, предпочтительно натриевый катион, целесообразно использовать особые смеси растворителей, включающие толуол и приблизительно от 1 до 15% диметилформамида или приблизительно от 1 до 15% N-метилпирролидона, причем особое предпочтение отдают смеси приблизительно от 3 до 8% диметилформамида в толуоле.
В зависимости от растворителя процессы превращения проводят при температурах приблизительно от 0°С до точки кипения и более целесообразно в безводных условиях. В особенно выгодном варианте конверсию проводят в толуоле с использованием в качестве основания метилата натрия в метаноле при температуре от 80°С до точки кипения, причем с целью избежать побочных реакций выделяющийся во время процесса метанол непрерывно отгоняют.
В качестве первоначально загружаемого материала используют преимущественно метилат натрия в форме его приблизительно 30%-ного метанольного раствора в смеси толуола и приблизительно от 1 до 15% диметилформамида, благодаря чему при нагревании прежде всего отгоняют метанол до температуры в головке колонны приблизительно от 105 до 110°С, и только потом соединение формулы III, растворенное в небольшом количестве толуола, добавляют по каплям таким образом, чтобы последующей дистилляцией из реакционной смеси непрерывно отгонять выделяющийся метанол и, следовательно, давать возможность соли формулы I осаждаться из реакционной смеси в форме чистых и легко перемешиваемых мелких кристаллов.
Когда процесс превращения проводят с использованием алкоголятных анионов как катализатора, в качестве основания для осаждения енолята формулы I также целесообразно применять катион, образующий соответствующий алкоголят. Приемлемые количества алкоголятов щелочных металлов составляют от 1,0 до 2,5 экв., преимущественно от 1,0 до приблизительно 1,5 экв. Особое предпочтение отдают применению в качестве основания от 1,0001 до 1,1 экв. метилата натрия.
Соединения формулы I можно либо непосредственно использовать в реакционной смеси для последующих превращений, либо, по-другому, выделять. Соединения формулы I можно выделять из реакционной смеси фильтрованием в соответствии с обычными методами. Другой возможностью является последующее превращение соединений формулы I в соответствующие нейтральные бициклические 1,3-дикетоны, которые, как уже сказано выше, являются промежуточными продуктами в получении гербицидов.
Когда используют метилат натрия в смеси толуола с небольшим количеством диметилформамида или N-метилпирролидона, с этой целью либо соединения формулы I, преимущественно их натриевые соли, можно отфильтровывать, а затем нейтрализовать в водном растворе с применением кислоты, например соляной кислоты, серной кислоты или уксусной кислоты, после чего выделять с помощью экстрагирующего вещества, например этилацетата, трет-бутилметилового эфира, дихлорметана, дихлорэтана или хлорбензола, либо содержащую натриевую соль реакционную смесь можно нейтрализовать непосредственно, введением с перемешиванием водной кислоты, например соляной кислоты крепостью от 2 до 10 н., а затем экстрагировать добавлением, когда это уместно, другого разбавителя, в частности этилацетата, с целью получить нейтральные бициклические 1,3-дикетоны. Нейтрализацию целесообразно проводить с регулированием рН, а полученные 1,3-дикетоны экстрагируют в интервале значений рН от 2 до 7, преимущественно в пределах от 4 до 6.
В другом варианте осуществления способа в соответствии с изобретением на реакционной стадии б) используют каталитически эффективные количества цианидных ионов в присутствии дополнительного основания. Подходящими основаниями служат преимущественно третичные амины, такие как триалкиламины, например триметиламин, триэтиламин, диизопропилэтиламин (основание Хюнига), три-н-бутиламин, N,N-диметиланилин и N-метилморфолин. Приемлемы также такие основания, как безводный гидроксид натрия, бикарбонат натрия и карбонат калия. В качестве предпочтительного источника цианидных ионов используют цианиды щелочных металлов, например цианид натрия или цианид калия, цианид меди (1), органические циангидрины, такие как ацетонциангидрин, триалкилсилилцианиды, такие как триметилсилилцианид, или третичных аммониевых оснований, такие как цианид тетраэтиламмония. В этом варианте осуществления способа в соответствии с изобретением содержание используемого цианида щелочного металла находится в интервале от малого количества до небольшого избытка. Цианиды используют в количествах от 0,1 до приблизительно 25%, предпочтительно от 1 до приблизительно 15%, в присутствии дополнительного основания, такого как преимущественно триэтиламин и основание Хюнига, причем количество основания составляет от 1 до 6 экв., преимущественно от 1,1 до приблизительно 2,5 экв.
Этот предпочтительный вариант способа в соответствии с изобретением осуществляют в инертном растворителе, таком как н-гептан, толуол, ксилол, дихлорметан, дихлорэтан, диметоксиэтан, тетрагидрофуран, диоксан, трет-бутилметиловый эфир, этилацетат, ацетон, 2-бутанон, ацетонитрил, пропионитрил, диметилформамид или N-метилпирролидон, при температуре от -5 до приблизительно +80°С, особенно предпочтительно в ацетонитриле или дихлорметане при температуре от приблизительно 10 до приблизительно 60°С.
Для процессов превращения в зависимости от применяемых растворителей на реакционной стадии б) можно использовать необязательные добавки, например, такие как хлорид лития и бромид лития, или межфазные катализаторы, например, такие как тетрабутиламмонийбромид или преимущественно тетраэтиламмонийцианид, или осушители, такие как сульфат магния, или подходящие молекулярные сита, но обычно такие добавки не требуются.
В данном варианте осуществления способа получения соединений формулы I также существует возможность либо выделять эти последние, либо использовать непосредственно в реакционной смеси для проведения последующих реакций, например с получением, как сказано выше, соединений с гербицидным действием. Получаемые таким образом аммониевые соли формулы I можно выделять, например, после отфильтровывания небольших количеств твердых веществ, таких как калиевая соль формулы I, когда в качестве катализатора используют цианид калия, путем простого концентрирования реакционной смеси выпариванием.
Можно также проводить последующую реакцию соединений формулы I с получением соответствующих нейтральных бициклических 1,3-дикетонов, которые, как сказано выше, служат в качестве промежуточных продуктов при получении гербицидов. С этой целью нейтральные бициклические 1,3-дикетоны можно выделить добавлением воды и кислоты как нейтрализующего агента, например соляной кислоты или серной кислоты, а затем с регулированием рН в интервале приблизительно от 2 до 7, преимущественно приблизительно от 4 до 6, для их выделения с помощью экстрагирующего вещества, например этилацетата, трет-бутилметилового эфира, дихлорметана, дихлорэтана или хлорбензола.
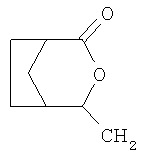
Соединения формулы III
,
в которой R1, R2, R3 R4, А и Е имеют значения, указанные для формулы I, представляют собой ценные промежуточные продукты при получении соединений формулы I, которые были созданы преимущественно для способа, предлагаемого в соответствии с изобретением. Таким образом, эти соединения охватывается объемом настоящего изобретения.
Соединениями формулы III, особенно ценными для получения соединений формулы I, являются преимущественно те, у которых
а) каждый из R1 и R2, независимо друг от друга обозначает водородный атом или метил;
б) каждый из R3 и R4 независимо друг от друга обозначает водородный атом или метил;
в) А обозначает метилен, который может быть однократно или двукратно замещенным метильной группой, или этилен; и/или
г) Е обозначает метилен, который может быть однократно или двукратно замещенным метильной группой.
В качестве промежуточного продукта при получении соединений формулы I особенно приемлемо соединение формулы III, в которой каждый из R1, R2, R3 и R4 обозначает водородный атом, А обозначает метилен и Е обозначает метилен. Предпочтительные соединения формулы III перечислены в следующей таблице.
Таблица 1: соединения формулы III
,
Способ в соответствии с изобретением более подробно проиллюстрирован в следующих примерах получения.
Пример 1: получение 4-метилен-3-оксабицикло[3.2.1 ]октан-2-она из 3-метиленбицикло[2.2.1]гептан-2-она (соединение № 1.001)
а) 98,7 г (0,81 моля) 3-метиленбицикло[2.2.1]гептан-2-она и 32,9 г (0,24 моля) тригидрата ацетата натрия в 400 мл дихлорметана используют в качестве начальной порции, загружаемой в реакционный сосуд. Далее при одновременном регулировании температуры (баня из СО2/ацетона) в пределах от -8 до -10°С с перемешиванием по каплям в течение 2,5 ч вводят 230 г 32%-ной перуксусной кислоты в уксусной кислоте (0,97 моля). После этого реакционную смесь перемешивают при температуре -8°С в течение дополнительного часа. Затем добавляют 200 г льда, а потом 20 г (0,16 моля) сульфита натрия в 100 мл воды. Органическую фазу отделяют и промывают водой, сушат над сульфатом магния и концентрируют с получением в виде жидкого остатка 81,9 г 4-метилен-3-оксабицикло[3.2.1]октан-2-она с его содержанием 93% и выходом продукта 68,2%.
1H-ЯМР (CDCl3): 4,42 част./млн, d, 1Н; 4,18 част./млн, d, 1H; 3,08 част./млн, 2Н; 1,95-2,08 част./млн, 4Н; 1,84 част./млн, m, 1H; 1,67 част./млн, m, 1H.
б) В реакционном сосуде 95,2 г 3-метиленбицикло[2.2.1]гептан-2-она растворяют в 400 мл метиленхлорида, добавляют 32,6 г тригидрата ацетата натрия и смесь охлаждают до температуры -10°С. Далее с перемешиванием при температуре от -8 до -10°С в течение 2,40 ч вводят 199 мл 36-40%-ной перуксусной кислоты и реакционную смесь перемешивают в течение последующих 2 ч при -10°С. В дальнейшем реакционную смесь добавляют в 400 г смеси воды со льдом и органическую фазу отделяют и обрабатывают смесью 100 г льда со 100 мл 15%-ного раствора сульфита натрия. Затем органическую фазу промывают 100 мл 25%-ного раствора карбоната натрия, после чего 100 мл воды. Объединенные водные фазы промывают 200 мл метиленхлорида. Далее объединенные органические фазы с помощью роторного испарителя концентрируют при температуре бани 50°С. Оставшуюся жидкость подвергают фракционной перегонке в колонке под остаточным давлением 53 Па с получением при температуре от 40 до 45°С 27 г 3-метиленбицикло[2.2.1]гептан-2-она (исходный материал) и при температуре от 55 до 60°С 60 г 4-метилен-3-оксабицикло[3.2.1]октан-2-она, что соответствует выходу продукта 55,7% в пересчете на исходный материал и селективности 77,8% (в пересчете на прореагировавший материал).
Пример 2: получение 4-метилен-3-оксабицикло[3.2.2]нонан-2-она из 3-метиленбицикло[2.2.2]октан-2-она (соединение № 1.070)
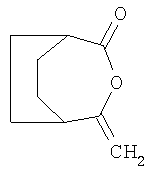
Аналогично примеру 1 проводят взаимодействие 955 мг (7 ммолей) 3-метиленбицикло[2.2.2]октан-2-она с 1,64 г (8,4 ммоля) 32%-ной перуксусной кислоты в присутствии 286 мг (21 ммоль) тригидрата ацетата натрия. Выделяют 1 г 4-метилен-3-оксабицикло[3.2.2]нонан-2-она. После очистки хроматографией в колонке с использованием 10%-ного этилацетата в гексане в виде масла получают чистый 4-метилен-3-оксабицикло[3.2.2]нонан-2-он.
1Н-ЯМР (CDCl3): 4,62 част./млн, "s", 1H; 4,25 част./млн, "s", 1H; 2,9-3,0 част./млн, 2Н; 1,9-2,1 част./млн, 2Н; 1,7-1,9 част./млн, 6Н.
Пример 3: получение триэтиламмониевой соли 4-гидроксибицикло[3.2.1]окт-3-ен-2-она из 4-метилен-3-оксабицикло[3.2.1]октан-2-она
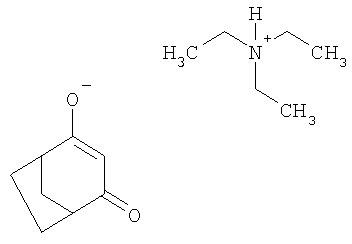
2,76 г (20 ммолей) 4-метилен-3-оксабицикло[3.2.1]октан-2-она при температуре 55°С выдерживают в течение 2,5 ч в присутствии 2,23 г (20 ммолей) триэтиламина и 0,13 г (2 ммоля) цианида калия. Мутную реакционную смесь фильтруют через вспомогательное вещество для фильтрования HyfloR и выпаривают досуха. В виде смолистого гигроскопического продукта получают триэтиламмониевую соль 4-гидроксибицикло[3.2.1]окт-3-ен-2-она.
Пример 4: получение этилдиизопропиламмониевой соли 4-гидроксибицикло[3.2.1]окт-3-ен-2-она из 4-метилен-3-оксабицикло [3.2.1 ]октан-2-она
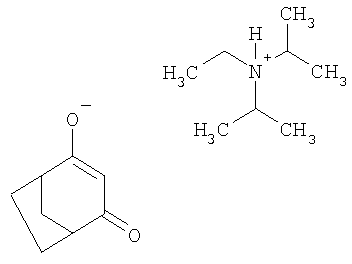
Аналогично примеру 3 1,38 г (10 ммолей) 4-метилен-3-оксабицикло[3.2.1]октан-2-она перемешивают в течение 12 ч в присутствии 1,29 г (10 ммолей) основания Хюнига и 0,13 г цианида калия в 10 мл ацетонитрила. Твердые компоненты (калиевые соли) отфильтровывают и фильтрат выпаривают досуха с получением в виде смолы этилдиизопропиламмониевой соли 4-гидроксибицикло[3.2.1]окт-3-ен-2-она.
Пример 5: получение натриевой соли 4-гидроксибицикло[3.2.1]окт-3-ен-2-она
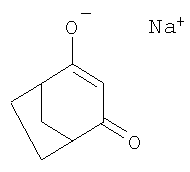
При температуре 110°С 30%-ный раствор 12,1 г (0,22 моля) метилата натрия в метаноле по каплям вводят в раствор 190 мл толуола и 10 мл диметилформамида, причем дистилляцией непрерывно удаляют метанол. Затем в образовавшуюся суспензию в течение 30-минутного периода, продолжая удаление метанола дистилляцией, по каплям добавляют 20,7 г (0,15 моля) 4-метилен-3-оксабицикло[3.2.1]октан-2-она в 20 мл толуола. После перемешивания в течение последующих 2 ч при температуре кипения реакционной смеси дают остыть и выпавший в осадок продукт отфильтровывают и промывают толуолом.
Пример 6: превращение натриевой соли из примера 5 в бицикло[3.2.1]октан-2.4-дион
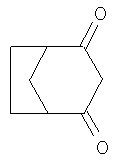
Натриевую соль 4-гидроксибицикло[3.2.1]окт-3-ен-2-она, полученную по изложенному выше, вводят в 300 мл смеси воды со льдом и с помощью концентрированной соляной кислоты рН доводят до 3, осаждая в виде твердого вещества нейтральный бицикло[3.2.1]октан-2,4-дион, который экстрагируют этилацетатом, промывают водой, сушат над сульфатом натрия и концентрируют выпариванием до объема приблизительно 50 мл. Выпавший в осадок продукт (15,2 г, выход: 73,3%) представляет собой чистый бицикло[3.2.1]октан-2,4-дион, температура плавления которого составляет 128-129°С.
Пример 7: прямое превращение в бицикло[3.2.1]октан-2,4-дион без выделения натриевой соли 4-гидроксибицикло[3.2.1]окт-3-ен-2-она
В качестве начальной порции, загружаемой в реакционный сосуд, используют 4,27 г (79 ммолей) метилата натрия в 40 мл диметилсульфоксида. В этот раствор в течение 2,5 ч при температуре от 25 до 35°С с перемешиванием добавляют раствор 7,2 г (52 ммоля) 4-метилен-3-оксабицикло[3.2.1]октан-2-она в 20 мл диметилсульфоксида. По прошествии дополнительно 0,5 ч реакционную смесь разбавляют 200 мл воды и дважды экстрагируют 100 мл этилацетата. Объединенные органические фазы промывают 100 мл воды. Далее водные фазы объединяют, с использованием приблизительно 35 мл 2 н. соляной кислоты рН доводят до 3 и четыре раза экстрагируют, используя каждый раз 400 мл этилацетата. Объединенные органические фазы промывают водой, сушат над сульфатом магния, фильтруют и с помощью роторного испарителя концентрируют. Остающееся коричневое твердое вещество отделяют фильтрованием через силикагель и получают 6,3 г (46 ммолей) бицикло[3.2.1]октан-2,4-диона с содержанием основного вещества 93%, что соответствует выходу продукта 81,4%, и температурой плавления от 129 до 130°С.
Пример 8: прямое превращение в бицикло[3.2.1]октан-2.4-дион без выделения триэтиламмониевой соли 4-гидроксибицикло[3.2.1]окт-3-ен-2-она
Аналогично примеру 3 2,76 г (20 ммолей) 4-метилен-3-оксабицикло[3.2.1]октан-2-она при комнатной температуре перемешивают в течение 15 ч в присутствии 2,23 г (22 ммоля) триэтиламина и 0,13 г (2 ммоля) цианида калия в 10 мл ацетонитрила. Смесь выдерживают при 55°С в течение последующих 30 мин, а затем растворяют в воде и с использованием этилацетата при рН 10 удаляют нейтральные компоненты. Водную фазу, подкисленную до рН 2, экстрагируют этилацетатом, сушат над сульфатом натрия и концентрируют выпариванием, получая 2,05 г выход: 74,3%) чистого бицикло[3.2.1]октан-2,4-диона, температура плавления которого составляет от 129 до 130°С.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ЦИКЛИЧЕСКИХ ДИКЕТОНОВ | 2003 |
|
RU2316544C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЦИКЛИЧЕСКИХ ДИКЕТОНОВ | 2005 |
|
RU2384562C2 |
| ДИФТОРМЕТИЛАМИНОПИРИДИНЫ И ДИФТОРМЕТИЛАМИНОПИРИМИДИНЫ | 2015 |
|
RU2712091C2 |
| СПОСОБ ПОЛУЧЕНИЯ 4-АМИНОПИРИДАЗИНОВ | 2016 |
|
RU2742663C2 |
| СПОСОБ ПОЛУЧЕНИЯ 4-АМИНОПИРИДАЗИНОВ | 2016 |
|
RU2778306C1 |
| ПРОИЗВОДНЫЕ БИФЕНИЛПИРИДОНА И ИХ СОЛИ | 1992 |
|
RU2100350C1 |
| СПОСОБЫ ПОЛУЧЕНИЯ БИЦИКЛИЧЕСКИХ АМИДИНОГИДРАЗОНОВ (ВАРИАНТЫ) И БИЦИКЛИЧЕСКИЕ ГИДРОКСИАМИДИНЫ | 1993 |
|
RU2126381C1 |
| НОВЫЕ КОМПОЗИЦИИ И СПОСОБЫ ИХ СТАБИЛИЗАЦИИ | 2000 |
|
RU2275368C2 |
| СУЛЬФОНАМИДЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ | 1999 |
|
RU2197479C2 |
| ФАРМАЦЕВТИЧЕСКИЕ СОЕДИНЕНИЯ | 2019 |
|
RU2821941C2 |
Изобретение относится к способу получения соединения формулы I
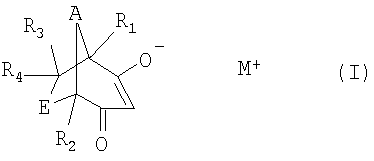 ,
,
в которой каждый из R1, R2, R3 и R4 независимо друг от друга обозначает водородный атом или С1-С4алкил, каждый из А и Е независимо друг от друга обозначает С1-С2алкилен, который может быть замещен один раз или до четырех раз С1-С4алкильной группой, и М+ обозначает ион щелочного металла, ион щелочно-земельного металла или аммониевый ион. Способ включает взаимодействие соединения формулы II
 ,
,
в которой R1, R2, R3, R4, А и Е имеют значения, указанные для формулы I, с окислителем, выбранным из перкислот или пероксида водорода, с образованием соединения формулы III
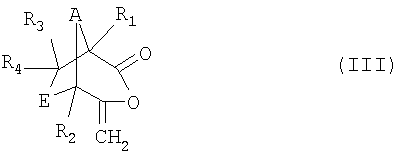 ,
,
в которой R1, R2, R3, R4, А и Е имеют значения, указанные для формулы I, и последующее превращение соединения формулы III в соль формулы I либо в присутствии основания, выбранного из триалкиламинов, безводного гидроксида натрия, бикарбоната натрия или бикарбоната калия, и каталитически эффективного количества цианида, либо в присутствии алкоголята щелочного металла, или алкоголята или гидроксида щелочно-земельного металла. Изобретение также относится к соединению формулы III и к его применению для получения соединения формулы I. Изобретение позволяет получить соли бициклических 1,3-дикетонов экономичным способом с высоким выходом и хорошего качества. 3 н. и 2 з.п. ф-лы, 1 табл.

в которой каждый из R1, R2, R3 и R4 независимо друг от друга обозначает водородный атом, С1-С4алкил; каждый из А и Е независимо друг от друга обозначает С1-С2алкилен, который может быть замещен один раз или до четырех раз С1-С4алкильной группой; и М+ обозначает ион щелочного металла, ион щелочно-земельного металла или аммониевый ион, причем этот способ включает следующие стадии:
а) взаимодействие соединения формулы II
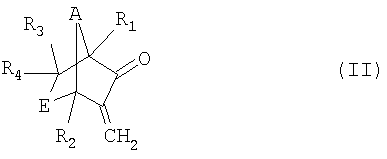
в которой R1, R2, R3, R4, А и Е имеют значения, указанные для формулы I, в присутствии окислителя, выбранного из перкислот или пероксида водорода, с образованием соединения формулы III
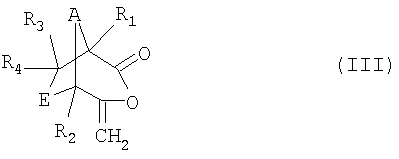
в которой R1, R2, R3, R4, А и Е имеют значения, указанные для формулы I;
б) затем превращение этого соединения в соль формулы I либо в присутствии основания, выбранного из триалкиламинов, безводного гидроксида натрия, бикарбоната натрия или бикарбоната калия, и каталитически эффективного количества цианида, либо в присутствии алкоголята щелочного металла, или алкоголята или гидроксида щелочно-земельного металла.
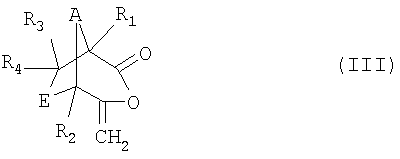
в которой R1, R2, R3, R4, А и Е имеют значения, указанные для формулы I в п.1.
в которой R1, R2, R3, R4, А и Е имеют значения, указанные для формулы I в п.1, для получения соединения формулы I по п.1.
| Способ получения производных 3-окси-2-циклогексен-1-она | 1984 |
|
SU1450733A3 |
| Способ контроля частотных характеристик аппарата магнитной записи | 1977 |
|
SU680044A1 |
| US 3932510 A, 13.01.1976 | |||
| WO 00/15615 A1, 23.03.2000. | |||

