Область техники, к которой относится изобретение
Настоящее изобретение относится к способу получения органосилсесквиоксанов и к применению композиции, содержащей органосилсесквиоксаны, для создания защитного покрытия на подложке с тем, чтобы придать подложке стойкость к механическому и химическому разрушению при одновременном сохранении превосходных оптических свойств, и в качестве материала в массе.
Предпосылки создания изобретения
Органосилсесквиоксаны представляют собой кремний-кислород-содержащую структуру, имеющую общую формулу (RSiO1,5)n, в которой n представляет собой четное число ≥ 4. Органосилсесквиоксаны, которые имеют очень специфическую структуру (например, соединение, имеющее формулу (RSiO1,5)8, имеет октаэдрическую каркасную структуру), называются в области знаний как органоолигосилсесквиоксаны, или полиэдрические олигомерные силсесквиоксаны (или ПОСС (POSS)).
Органосилсесквиоксаны обладают хорошими механическими свойствами, например, как покрытия с хорошей износостойкостью, и могут быть выражены формулой так, чтобы иметь хорошую химическую стойкость, например, гидролитическую стабильность или стойкость к УФ-деструкции. Указанные и другие свойства превращают органосилсесквиоксаны в используемые в качестве защитных покрытий для широкого ряда подложек, в частности, полимерсодержащих материалов, таких как акриловые полимеры и поликарбонаты, которые рутинно используются в качестве альтернативы стеклу во многих ситуациях, когда масса, тенденция разбиваться или стоимость стекла являются противопоказаниями к его использованию.
Ряд способов получения и модификации органосилсесквиоксанов описан в литературе, включая:
(1) получение частично или полностью конденсированных структур;
(2) функционализацию частично или полностью конденсированных структур;
(3) получение структур, содержащих более одной органической функциональности.
Примеры таких способов рассматриваются в работах Brown et al., J. Am. Chem. Soc., (1965), 87: 4313-4317 и Feher et al., J. Am. Chem. Soc., (1965), 111: 1741-1748. В общих чертах указанные способы используют очень низкие концентрации мономера в растворителе и введение больших количеств воды с обеспечением очень медленного получения (обычно в течение ряда месяцев) специфических и определенных каркасных структур. Как результат получаемые материалы имеют ограниченное применение, например, в области защитных покрытий, благодаря значительным затратам, связанным с их медленным получением.
Введение функциональных органических лигандов в основную органосилсесквиоксановую структуру осуществляется рядом способов. В данном контексте термин «функциональный» используется для описания органических групп, которые действуют с приданием конкретных механических и/или химических свойств конечному материалу. Например, введение органических лигандов достигается:
(1) обменом функциональности в существующей полностью конденсированной структуре;
(2) расщеплением и введением функциональности в полностью конденсированную структуру;
(3) введением функциональности в частично конденсированную структуру; и
(4) введением функциональности в процессе получения структуры.
Примеры указанных способов описываются в US-A-5047492 и US-A-5484867.
Последний из указанных способов - способ (4) применяется редко, но, когда он используется, он включает согидролиз и конденсацию силановых предшественников, содержащих нефункциональные органические группы, и силановых предшественников, содержащих функциональные органические группы, в частности, со способностью к полимеризации. Однако получение указанных функционализированных органосилсесквиоксанов остается медленным.
Поэтому было бы желательно создать способ получения органосилсесквиоксанов и композиций, их содержащих, который не имеет продолжительности реакций, описанных в прототипе, и который делает указанные материалы доступными для промышленности на более экономически эффективной основе.
Краткое описание изобретения
Согласно первому аспекту настоящего изобретения предусматривается способ получения композиции, содержащей органосилсесквиоксаны, включающий следующие стадии:
(1) частичный гидролиз гидролизующихся предшественников неорганических мономеров, содержащих, по меньшей мере, 50 мол.% первых гидролизующихся предшественников неорганического мономера, имеющих формулу RSiY3, в которой R представляет собой органическую группу, R-Si-связь является негидролизующейся связью, каждая Y-группа представляет собой одинаковую или отличающуюся друг от друга Y-группу и выбрана из химически реакционнх групп, так что каждая Si-Y-связь гидролизуется с образованием Si-OH-связи с образованием неорганических мономеров и обеспечением частичной конденсации неорганических мономеров с образованием жидкой композиции, содержащей неорганические олигомеры;
(2) перед завершением конденсации неорганических мономеров охлаждение жидкой композиции количеством воды, которое в комбинации с водой, используемой на стадии (1), и любой водой, отщепляемой при конденсации неорганических мономеров на стадии (1), превышает стехиометрическое количество воды, необходимой для осуществления полного гидролиза всех присутствующих предшественников гидролизующихся неорганических мономеров; и
(3) сушка композиции.
Способ настоящего изобретения обеспечивает контролируемое и быстрое получение широкого ряда органосилсесквиоксансодержащих композиций. Композиции могут содержать органосилсесквиоксаны, имеющие одну или более органических функциональностей и имеющие полностью или неполностью конденсированные структуры. Кроме того, композиции могут содержать или не содержать только органосилсесквиоксаны.
Согласно второму аспекту настоящего изобретения композиция, содержащая органосилсесквиоксаны, получается вышеуказанным способом.
Другие аспекты настоящего изобретения включают нанесение вышеуказанных композиций на ряд подложек, покрытые ими подложки, изделия, выполненные из вышеуказанных композиций, и применение вышеуказанных композиций в качестве связующих агентов.
Композиции настоящего изобретения способны обеспечить покрытия, которые обладают износостойкостью, гидролитической стабильностью и стойкостью к УФ-деструкции, на широком ряду подложек, в частности, на полимерных подложках, и могут быть тонко отрегулированы, чтобы иметь другие свойства в зависимости от природы введенных в них функциональностей и условий, используемых в их получении.
Подробное описание изобретения
В общих чертах способ настоящего изобретения включает двухстадийную реакцию гидролиза/конденсации и последующую сушку и, необязательно, отверждение получаемой композиции. На первой стадии гидролизующиеся предшественники неорганических мономеров, из которых, по меньшей мере, 50 мол.% имеет формулу RSiY3 (называемые «первыми гидролизующимися предшественниками мономеров»), частично гидролизуются и подвергаются частичной, но не полной конденсации, с образованием неорганических олигомеров, которые образуют составные блоки конечных органосилсесквиоксановых молекул.
На второй стадии способа неорганические олигомеры охлаждаются добавлением или введением в них относительно большого количества воды, что имеет эффект вызывать быструю конденсацию неорганических олигомеров. В зависимости от имеющегося времени и условий, используемых для конденсации, степень конденсации может в некоторых пределах варьироваться до по существу полной конденсации указанных олигомеров.
Точная природа конечных структур, образованных при сушке и, необязательно, отверждении полученной композиции является предметом предположения, но несущественно для достигаемых благоприятных механических и химических свойств.
В контексте настоящей заявки под «гидролизующимся предшественником неорганического мономера» понимается любая неорганическая молекула, которая превращается гидролизом в полимеризующийся неорганический мономер, который при поликонденсации с подобными гидролизованными или частично гидролизованными мономерами образует неорганические олигомеры и в конечном счете неорганическую сетку. Термин «неорганический» используется для обозначения присутствия в молекуле предшественника неорганического элемента, обычно обуславливающего оксидный керамический материал, например кремния, алюминия, титана, циркония, иттрия или других переходных металлов.
Рассматривая первую стадию способа более подробно, первые гидролизующиеся предшественники неорганических мономеров RSiY3 могут быть выбраны из ряда материалов. Однако определяющим является то, что R-Si-связь не должна быть гидролизующейся, так как в противном случае органическая группа R может быть утрачена в конечной структуре. Предпочтительно, группа R сама является химически стабильной, и, в частности, она является устойчивой к гидролизу в присутствии воды или влаги. Подходящие примеры группы R включают водород и необязательно замещенные (цикло)алкильные, арильные, алкенильные, амидо-, эпокси-, (мет)акрильные, стирольные, нитрильные, ангидридные, сложноэфирные, фосфино-, галогенидные, амино-, меркапто- и цианатные группы и их смеси. Предпочтительные группы R выбираются из (цикло)алкил-, арил- и алкенил-групп, необязательно замещенных группами, выбранными из эпоксидных, винильных, (мет)акриловых и цианатных групп.
Кроме того, предпочтительно, размер группы R выбран так, что конечная композиция является жидкостью по природе, обеспечивая ее применение в качестве состава покрытия.
Группы Y в первых гидролизующихся предшественниках неорганических мономеров могут быть одинаковыми или отличающимися друг от друга, хотя обычно они являются одинаковыми. Si-Y-связь является высокореакционной связью, которая легко подвергается гидролизу с образованием силанольной группы, т.е. Si-OH. Подходящими примерами групп Y являются алкокси-, ацетокси-, амино- и нитро- группы и атомы галогенов. Будучи образованной, силанольная группа может конденсироваться через реакцию с отщеплением воды с другой силанольной группой в соответствии со следующим уравнением реакции:
RY3-nSi(OH)n + (HO)nSiY3-nR → RY3-nSi(OH)n-1Si-O-Si(OH)n-1Y3-nR
Альтернативно, конденсация может проходить гетерофункциональным образом, когда присутствуют различные гидролизующиеся предшественники неорганических мономеров, как рассмотрено более подробно ниже, так, как показано в следующих двух уравнениях реакции:
-Si-OH + ClSi- → -Si-O-Si- + HCl
-Si-OH + ROSi- → -Si-O-Si- + ROH
Способ настоящего изобретения может включать использование единственного типа гидролизующегося предшественника неорганического мономера, имеющего формулу RSiY3. Альтернативно, могут использоваться различные гидролизующиеся предшественники неорганических мономеров, например, различающиеся по природе и/или числу органических групп R и/или по природе и/или числу органических групп Y.
В качестве примера могут использоваться первые гидролизующиеся предшественники неорганических мономеров, имеющие различные R-группы, например, RSiY3, R1SiY3 и т.д., причем группы R и R1 выбраны с обеспечением конкретных механических и/или химических свойств конечного продукта. В этом отношении настоящее изобретение не ограничивается использованием двух различных типов первых гидролизующихся предшественников мономеров, но может включать использование более двух различных типов предшественников мономеров при условии, что поддерживается адекватный контроль способа с тем, чтобы достигнуть желаемый уровень введения различных R-групп в конечный продукт.
Как указано выше, при введении или, альтернативно, при использовании первых гидролизующихся предшественников неорганических мономеров, имеющих различные R и/или Y группы, способ может включать использование гидролизующихся предшественников неорганических мономеров, различающихся по числу гидролизующихся групп Y. В частности, в дополнение к первым гидролизующимся предшественникам мономеров RSiY3 могут использоваться гидролизующиеся предшественники неорганических мономеров, имеющие общую формулу RnSiY4-n, в которой n составляет 0, 2 или 3, R представляет собой органическую группу, например, выбранную из групп, приведенных для R выше, R-Si-связь представляет собой негидролизующуюся связь, и R-группы являются одинаковыми или отличающимися друг от друга, и каждая Y-группа является одинаковой или отличающейся от каждой другой Y-группы и выбирается из химически реакционных групп, так что каждая Si-Y-связь гидролизуется с образованием Si-OH-связи, и смеси таких предшественников мономеров. В последующем указанные дополнительные предшественники мономеров называются термином «вторые гидролизующиеся предшественники неорганических мономеров».
В дополнение к первым гидролизующимся предшественникам неорганических мономеров могут использоваться гидролизующиеся предшественники неорганических мономеров, имеющие четыре гидролизующиеся связи, т.е. имеющие формулу SiY4 (n=0).
Дополнительно или альтернативно способ может включать использование гидролизующихся предшественников неорганических мономеров, имеющих меньше гидролизующихся связей, чем первые гидролизующиеся предшественники неорганических мономеров, например, имеющих формулу, выбранную из R2SiY2 (n=2) и R3SiY (n=3), и их смеси.
В контексте любого из вторых гидролизующихся предшественников неорганических мономеров, указанных выше, природа групп R и Y является такой, как определено выше для первых предшественников органических мономеров. Для ясности, однако, группы R и/или Y во вторых гидролизующихся предшественниках неорганических мономеров могут быть одинаковыми или отличающимися от групп R и/или Y в первых гидролизующихся предшественниках неорганических мономеров.
Соответствующие примеры первых гидролизующихся предшественников неорганических мономеров и вторых гидролизующихся предшественников неорганических мономеров, имеющих формулу R2SiY2 или R3SiY, включают:
(i) (алкил)алкоксисиланы, такие как триметоксисилан, триэтоксисилан, три-н-пропоксисилан, диметоксисилан, диэтоксисилан, ди-изо-пропоксисилан, монометоксисилан, моноэтоксисилан, монобутоксисилан, метилдиметоксисилан, этилдиэтоксисилан, диметилметоксисилан, ди-изо-пропил-изопропоксисилан, метилтриметоксисилан, этилтриэтоксисилан, н-пропил-три-н-пропоксисилан, бутилтрибутоксисилан, диметилдиметоксисилан, диэтилдиэтоксисилан, ди-изо-пропил-ди-изо-пропоксисилан, дибутилдибутоксисилан, триметилметоксисилан, триэтилэтоксисилан, три-н-пропил-н-пропоксисилан, трибутилбутоксисилан, фенилтриметоксисилан, дифенилдиэтоксисилан и трифенилметоксисилан;
(ii) (алкил)алкоксисиланы, имеющие изоцианатную группу, такие как 3-изоцианатопропилтриметоксисилан, 3-изоцианато-пропилтриэтоксисилан, 3-изоцианатопропилметилдиметоксисилан, 3-изоцианатопропилэтилдиэтоксисилан, 3-изоцианатопропилдиметил-изо-пропоксисилан, 3-изоцианатопропилдиэтилэтоксисилан, 2-изоцианатоэтилдиэтилбутоксисилан, ди-(3-изоцианатопропил)-диэтоксисилан, ди-(3-изоцианатопропил)метилэтоксисилан и этокситриизоцианатосилан;
(iii) (алкил)алкоксисиланы, имеющие эпокси-группу, такие как 3-глицидоксипропилтриметоксисилан, 3-глицид-оксипропилтриэтоксисилан, 3-глицидоксипропилметилдиметоксисилан, 3-глицидоксипропилметилдиэтоксисилан, 3-глицидоксипропилдиметилэтоксисилан, 2-(3,4-эпоксициклогексил)этилтриметоксисилан и 3,4-эпоксибутилтриметоксисилан;
(iv) (алкил)алкоксисиланы, имеющие карбоксильную группу, такие как карбоксиметилтриэтоксисилан и карбоксиметилэтилдиэтоксисилан;
(v) алкоксисиланы, имеющие кислотноангидридную группу, такие как 3-(триэтоксисилил)-2-метилпропилсукциновый ангидрид;
(vi) алкоксисиланы, имеющие галогенангидридную группу, такие как 2-(4-хлоросульфонилфенил)этилтриэтоксисилан;
(vii) (алкил)алкоксисиланы, имеющие аминогруппу, такие как N-2-(аминоэтил)-3-аминопропилтриэтоксисилан, N-2-(аминоэтил)-3-аминопропилметилдиметоксисилан и N-фенил-3-аминопропилтриметоксисилан;
(viii) (алкил)алкоксисиланы, имеющие тиольную группу, такие как 3-меркаптопропилтриметоксисилан, 3-меркаптопропилтриэтоксисилан, 2-меркаптоэтилтриэтоксисилан и 3-меркаптопропилметилдиметоксисилан;
(ix) (алкил)алкоксисиланы, имеющие винильную группу, такие как винилтриметоксисилан, винилтриэтоксисилан и винилметилдиэтоксисилан;
(x) (алкил)алкоксисиланы, имеющие акрилатную или метакрилатную группу, такие как 3-метакрилоксипропилтриметоксисилан, 3-метакрилоксипропилтриэтоксисилан, 3-метакрилоксипропилметилдиметилсилан и 3-акрилоксипропилтриэтоксисилан;
(xi) (алкил)алкоксисиланы, имеющие атом галогена, такие как триэтоксифторосилан, 3-хлоропропилтриметоксисилан, 3-бромоалкилалкоксисилан и 2-хлороэтилметилдиметоксисилан;
(xii) (алкил)алкоксисиланы, имеющие галогенированный алкил-лиганд, такие как (3,3,3-трифторопропил)триметоксисилан и 1Н, 1Н, 2Н, 2Н-перфтородецилтриэтоксисилан; и
(xiii) (алкил)алкоксисиланы, использующие алкокси-группу в качестве функциональной группы, такие как изопропилтриизопропоксисилан и триизопропилизопропоксисилан.
В вышеуказанных соединениях алкил-группа может быть замещена циклоалкил-группой, арил-группой или алкенил-группой и, необязательно, может быть замещена, предпочтительно, (мет)акрилатной группой или эпокси-группой. Когда в конечном продукте желательна гидролитическая стабильность, необходимо избегать гидролизующихся предшественников мономеров, имеющих гидролизующиеся R-группы.
Примеры подходящих вторых гидролизующихся предшественников неорганических мономеров, имеющих формулу SiY4, включают кремнийтетраалкоксиды, такие как тетраметоксисилан, тетраметоксисилан, тетраизопропоксисилан и тетрабутоксисилан.
Способ может также, или альтернативно, включать использование гидролизующихся предшественников неорганических мономеров, содержащих неорганический атом, иной, чем кремний, например, имеющих формулу MYn, в которой М обычно представляет собой металл, n представляет собой валентность металла, и каждая Y-группа является одинаковой или отличающейся друг от друга и выбирается из химически реакционных групп, так что каждая M-Y-связь гидролизуется в М-ОН-связь.
Подходящие примеры таких материалов включают:
i) титантетраалкоголяты, такие как титантетра-н-пропилат, титантетра-изо-пропилат и титантетрабутилат;
ii) алюминийтетраалкоголяты, такие как алюминийтри-втор-бутилат, алюминийтри-н-бутилат, алюминийтри-изо-пропилат;
iii) цирконийтетраалкоголяты, такие как цирконийтетра-н-пропилат, цирконийтетра-изо-пропилат и цирконийтетрабутилат; и
iv) алкоголяты металла, такие как диметилат меди, диэтилат бария, триметилат бора, триэтилат галлия, тетраэтилат германия, тетрабутилат свинца, пента-н-пропилат тантала и гексаэтилат вольфрама.
Способ может также, или альтернативно, включать использование гидролизующихся предшественников неорганических мономеров общей формулы (R2SiO)xY2, в которой х ≥ 1, R-Si-связь является негидролизующейся связью, и R является таким, как определено выше (и каждая R-группа может быть одинаковой или отличающейся от каждой другой R-группы), и Y представляет собой химически реакционную группу с каждой Si-Y-связью, гидролизующейся в Si-ОН-связь. Примеры подходящей группы Y (которая может быть одинаковой или отличающейся от другой) включают хлор, ацетокси, амин, оксим (т.е. R2C=NOSi) и алкокси. Целое число х может варьироваться от 1 до большого числа, например, до или даже больше 100, давая рост до мультикремниевых полимеров.
Когда способ включает использование гидролизующихся предшественников неорганических мономеров, которые различаются по числу присутствующих гидролизующихся групп, существенным является то, что, по меньшей мере, 50 мол.%, предпочтительно, более 60 мол.%, более предпочтительно, по меньшей мере, 70 мол.%, и, наиболее предпочтительно, по меньшей мере, 80 % мол. или выше, например, по меньшей мере, 90 мол.%, всех присутствующих гидролизующихся предшественников неорганических мономеров должно быть первыми гидролизующимися предшественниками неорганических мономеров для того, чтобы получить наиболее желательные свойства для нанесения покрытий.
Гидрофильный/гидрофобный характер R-группы (групп) будет определять поведение конечного продукта при воздействии воды. Это поведение может быть модифицировано подходящим выбором растворителя. Например, тенденция к отталкиванию воды алифатического углеводорода может быть изменена использованием такого протонного растворителя, как спирт, по сравнению с таким апротонным растворителем, как тетрагидрофуран.
Когда способ включает использование различных гидролизующихся предшественников неорганических мономеров, предпочтительно, чтобы гидролиз различных гидролизующихся предшественников неорганических мономеров проводился отдельно с тем, чтобы избежать конкурирования для присутствующих молекул воды, что в противном случае может привести к неоднородному или нежелательному распределению мономеров и, в частности, связанных органических групп, в конечном продукте. После отдельного частичного гидролиза различные образованные неорганические олигомеры смешиваются вместе перед обработкой на стадии охлаждения, которая следует за этим.
Для того чтобы гидролизовать гидролизующиеся предшественники неорганических мономеров, вода либо добавляется к гидролизующимся предшественникам неорганических мономеров, либо образуется на месте. Обычно гидролиз достигается путем образования гомогенной смеси с водой и, необязательно, органическим растворителем. Альтернативно, предшественники могут быть растворены в органическом растворителе, и вода добавляется к получаемому раствору контролируемым образом, чтобы избежать неконтролируемого развития агломерирования частично гидролизованных молекул. Подходящие органические растворители включают низкокипящие органические жидкости, например, имеющие точку кипения ниже 100°C, такие как спирты.
Предпочтительно, смесь воды и гидролизующихся предшественников мономеров смешивается с обеспечением того, чтобы как можно больше молекул предшественника подверглось воздействию воды с достижением в результате как можно более гомогенного гидролиза и конденсации.
Если (хотя это менее предпочтительно) вода образуется на месте в реакционной смеси, это может быть осуществлено, например, при добавлении к гидролизующимся предшественникам неорганических мономеров спирта, а затем слабой кислоты снова контролируемым образом. В данном случае (и как описано выше) предпочтительно, чтобы проводился отдельный частичный гидролиз различных гидролизующихся предшественников неорганических мономеров с последующим смешением вместе различных образовавшихся неорганических олигомеров.
Для инициирования гидролиза гидролизующихся предшественников неорганических мономеров может использоваться катализатор при условии, что он не взаимодействует с предшественниками неорганических мономеров и не влияет на природу частиц, образовавшихся при гидролизе. Подходящие катализаторы включают минеральные кислоты, такие как соляная кислота, серная кислота и азотная кислота. Для этой цели требуется только небольшое количество кислоты. Хотя, в зависимости от природы предшественников неорганических мономеров, гидролиз может проходить самопроизвольно.
Количество воды, используемой для гидролиза, обычно должно быть достаточным для гидролиза, по меньшей мере, одной из гидролизующихся связей, присутствующих в каждой молекуле предшественника неорганического мономера. Однако предпочтительно, количество воды и условия реакции гидролиза выбираются так, чтобы достигнуть гидролиза только одной или самое большое двух из гидролизующихся связей, присутствующих в первых гидролизующихся предшественниках неорганических мономеров, т.к. это определяет типы структуры, которые получаются на второй стадии способа.
Как описано выше, часть первой стадии способа включает, по меньшей мере, частичную конденсацию, но не полную конденсацию мономеров, образованных при гидролизе, с образованием неорганических олигомеров. Степень конденсации, которая имеет место, может быть определена, например, методом ЯМР. Предпочтительно, степень конденсации является такой, что большинство олигомеров, образовавшихся из первых гидролизующихся предшественников неорганических мономеров, имеет одну из следующих формул, в которых каждая или часть из R-групп может быть одинаковой или различной:
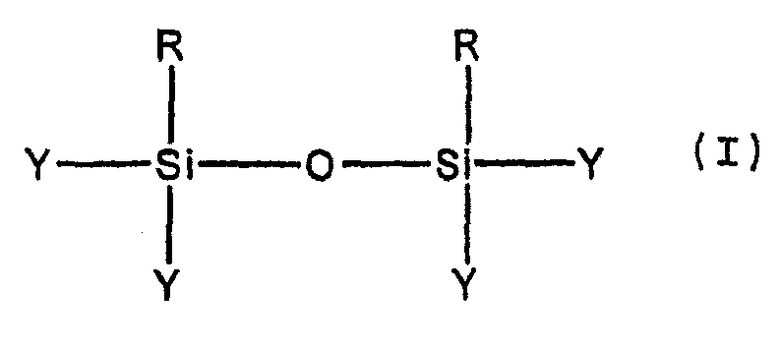
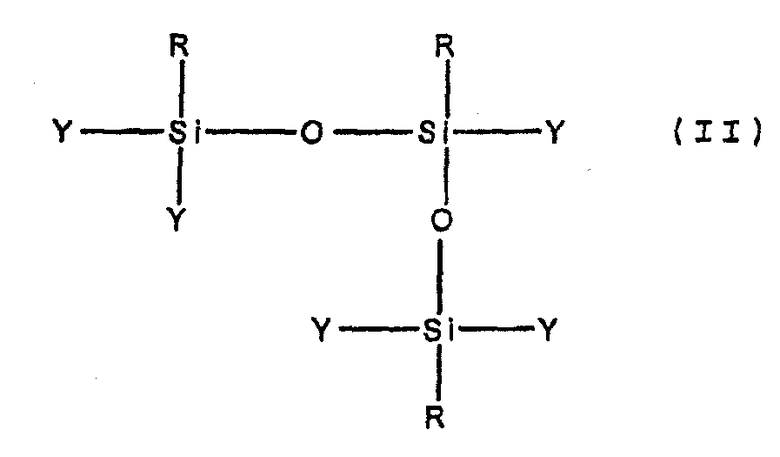
Более предпочтительно, большинство олигомеров имеет формулу (I), и, наиболее предпочтительно, большая часть (например, по меньшей мере, 80 мол.% или даже, по меньшей мере, 90 мол.%) олигомеров имеет формулу (I).
Первая стадия (гидролиз) способа может варьироваться по длительности, например, она может занимать менее часа или много дней в зависимости от свойств, требуемых в конечном продукте. Считается, что большая длительность дает большую связанность получаемой неорганической сетки и, таким образом, более высокую молекулярную массу.
Как только проходит достаточное время для того, чтобы имела место частичная конденсация или желаемая степень конденсации, смесь охлаждается в воде полным гидролизом присутствующих частиц и созданием условий, которые способствуют полному гидролизу и дополнительной конденсации. Конкретная природа созданных структур зависит от природы и концентрации используемых гидролизующихся предшественников неорганических мономеров, растворителя, начальных условий гидролиза и условий соконденсации (время, температура, рН). На практике реакции гидролиза и конденсации первой стадии способа могут протекать в течение периода времени, начиная от минут, обычно не менее 10 мин, до 24 ч или более.
Охлаждение может осуществляться либо введением воды в жидкую композицию, полученную на первой стадии способа, либо введением жидкой композиции в воду, предпочтительно, одностадийно. Объем воды, используемой на стадии охлаждения, в комбинации с любой водой, используемой на первой стадии, и любой водой, отщепившейся в результате конденсации на этой стадии, должен быть больше стехиометрического количества воды, необходимого для осуществления полного гидролиза (т.е. всех гидролизующихся связей) всех гидролизующихся предшественников неорганических мономеров. Предпочтительно, количество воды, по меньшей мере, в два раза и, более предпочтительно, по меньшей мере, в пять раз превышает указанное стехиометрическое количество. Затем позволяют проходить дополнительной конденсации с перемешиванием обычно в течение от часов до нескольких дней в зависимости от свойств, требуемых в конечном продукте, и поэтому желательно, чтобы указанный продукт был только частично конденсирован или по существу полностью конденсирован.
Полученный продукт затем сушат. В контексте данной заявки «сушка» включает удаление любой свободной воды и летучих, например, нагреванием при повышенной температуре, обычно в интервале 40-80°C.
Высушенный продукт готов для использования, например, в качестве композиции покрытия или в форме массы, или он может быть модифицирован перед использованием. Например, продукт может быть сшит (или отвержден) и/или модифицирован с введением дополнительной органической природы с модификацией в результате свойств конечного продукта, и/или он может быть дегидратирован со снижением или предотвращением дополнительной реакции в процессе использования. Для улучшения стабильности при хранении в композицию может быть введен растворитель.
Сшивание, или отверждение продукта может быть осуществлено через зависимые R-группы и/или через остаточные силанолы с образованием ряда 3-димерных структур.
Сшивание органических групп может быть осуществлено любым из известных средств, например, использованием подходящих сшивающих реагентов или условий переработки или того и другого, выбранных, чтобы способствовать межмолекулярному сшиванию в большей степени, чем внутримолекулярному сшиванию. Например, эпоксисодержащие R-группы могут быть сшиты или дополнительно полимеризованы при использовании реагентов, которые действуют как ускорители или отвердители, например, аминов, или при использовании кислот Льюиса.
Хотя высушенный продукт не может содержать много остаточных силанолов, они могут тем не менее влиять на поведение продукта в процессе отверждения и после отверждения. Конденсация части или всех указанных остаточных силанолов может быть особенно желательной, когда дополнительная конденсация в процессе использования продукта будет отрицательно влиять на свойства продукта. Остаточные силанолы могут быть подвергнуты самоконденсации при использовании подходящих катализаторов конденсации, таких как триэтиламин или оловосодержащие катализаторы, такие как олово(II)этилгексаноат, или при выборе подходящих условий реакции, известных в технике.
Альтернативно, конденсация остаточных силанолов может быть осуществлена при введении дополнительных силанолсодержащих соединений или других соединений, конденсирующихся с остаточными силанолами, уже присутствующими в продукте. Подходящими материалами являются материалы, имеющие формулу RnSiY'4-n, в которой n составляет 0-3, R представляет собой органическую группу, как определено выше, причем каждая R-Si-связь является негидролизующейся связью, и каждая R-группа является одинаковой или отличающейся от каждой другой R-группы, и Y' представляет собой группы, которые обеспечивают образование силоксана через конденсацию силанола или другими путями, причем каждая Y'-группа является такой же или отличающейся от каждой другой Y'-группы.
Например, силанолсодержащие соединения для использования в указанной конденсации могут быть получены гидролизом гидролизующихся предшественников неорганических мономеров, имеющих общую формулу RnSiY'4-n, в которой каждая SiY'-связь является гидролизующейся в Si-OH, причем Y' выбран, например, из групп, приведенных для Y выше в контексте первых гидролизующихся предшественников неорганических мономеров. Такой гидролиз дает соединения, такие как (но не ограничиваясь этим) R2Si(OH)2 и R3SiOH.
Обычно указанные силанолсодержащие соединения добавляют к высушенному продукту в присутствии агента конденсации, такого как олово(II)этилгексаноат, для промотирования конденсации.
Другим вариантом, указанным выше, осуществления конденсации остаточных силанолов является введение других соединений, конденсирующихся с указанными силанолами и имеющих общую формулу RnSiY'4-n, в которой SiY'-связь может не быть гидролизующейся прямо, или совсем, в Si-OH. Такие соединения включают материалы, обычно используемые в качестве агентов дериватизации для превращения стеклянных поверхностей в гидрофобные, и примеры таких материалов хорошо известны в технике. Например, для этой цели являются подходящими материалы, имеющие общую формулу R3SiY', в которой R является таким, как определено выше, и Y' представляет собой, например, хлор. Например, одним частным примером такого агента дериватизации является триметилхлоросилан.
Еще другим вариантом осуществления конденсации силанолов является использование материалов, имеющих общую формулу (R2Si)xY2, как определено выше в контексте вторых гидролизующихся предшественников неорганических мономеров, и гидролиз перед добавлением к высушенному продукту.
Дополнительно или в качестве альтернативы снижению содержания остаточного силанола конденсацией это может быть достигнуто, по меньшей мере, частичной дегидратацией и, предпочтительно, по существу полной дегидратацией. Дегидратация осуществляется любым из традиционных способов, известных в технике. Например, высушенный продукт может быть растворен в подходящем растворителе, например тетрагидрофуране ((ТГФ)(THF)), и свободная вода удалена с помощью молекулярного сита. Альтернативно, дегидратация может быть ускорена использованием катализатора конденсации, таким как триэтиламин, растворенный в ТГФ и помещенный на молекулярное сито. После периода дегидратации высушенного продукта летучий растворитель выпаривается, оставляя полностью или частично дегидратированный продукт.
Хотя дополнительная конденсация остаточных силанолов описана выше как проводимая на высушенном продукте, может быть желательно осуществлять такую конденсацию перед сушкой или перед завершением сушки продукта. Например, может быть желательно осуществлять конденсацию на продукте, который был, по меньшей мере, частично дегидратирован после сушки с удалением свободной воды.
Кроме того, хотя дегидратация описана выше как проводимая на высушенном продукте, по меньшей мере, частичная дегидратация может быть проведена на более ранних стадиях способа.
Способ настоящего изобретения способен обеспечить появление широкого ряда структур продукта. Тенденция к «каркасной» или «лестничной» структуре в конечном продукте регулируется параметрами способа, например, типом и/или концентрацией образуемых олигомеров, используемым растворителем, временем и температурой стадии смешения, концентрацией на стадии охлаждения, временем и температурой, при которых проводится охлаждение, и способом извлечения конечного продукта.
Когда плотность сшивки продукта является меньше теоретических 100%, продукт может еще считаться реакционным, что может быть или может не быть приемлемым согласно применению, для которого предназначен материал. Если это неприемлемо, различные варианты осуществления дополнительного сшивания и снижения реакционности описаны выше.
Для нанесения покрытия сшивание через органические группы (т.е. R-группы) может быть инициировано перед нанесением и проводится с завершением либо перед нанесением, либо после нанесения на подложку, например, с использованием известных способов облучения излучением (например, ультрафиолетовым излучением), термического или химического. Если сшивание выполняется с завершением перед нанесением покрытия, может быть желательно растворять композицию в растворителе для того, чтобы нанести композицию на подложку и затем выпарить растворитель для того, чтобы высушить нанесенную композицию.
Перед или после любого сшивания или дополнительной конденсации продукт, полученный при сушке (третья стадия способа), может быть смешан с органическим мономером или олигомером (обычно называемые в последующем как «полимеризующиеся органические соединения»), которые могут быть затем дополнительно полимеризованы, или органическим полимером, таким как латекс.
Природа полимеризующихся органических соединений выбирается в соответствии со свойствами, требуемыми в конечном продукте. Обычно полимеризующиеся органические соединения выбираются с обеспечением прочности и износостойкости и, когда желательно, прозрачности. Кроме того, если требуется химическая стойкость, например, стойкость к набуханию или другому разрушению при контакте с растворителем, желательно использовать полимеризующиеся органические соединения, способные образовывать двухмерные или трехмерные, т.е. сшитые, полимерные сетки. Такие полимеризующиеся органические соединения могут считаться имеющими двухфункциональную или трехфункциональную реакционность тем, что они обладают двумя или более реакционными участками, способными к полимеризации.
Для нанесения покрытий предпочтительными полимеризующимися органическими соединениями являются такие, которые при полимеризации образуют термоотверждающиеся полимеры. Примеры подходящих полимеризующихся органических соединений включают карбонаты, сложные эфиры, такие как терефталаты, эпоксисодержащие материалы, метил(мет)акрилаты, уретаны и другие двухфункциональные и трехфункциональные мономеры, такие как некоторые уретанакрилаты, ненасыщенные алифатические углеводороды и их смеси. Особенно предпочтительными являются уретановые предшественники, такие как изоцианаты и диизоцианаты и полиолы, и уретанакрилаты. Могут также использоваться металлоорганические мономеры, но в таком случае они не будут содержать гидролизующиеся связи.
Предпочтительно, полимеризующиеся органические соединения полимеризуются при относительно низкой температуре, например, ниже 150°C, после введения подходящего инициатора, или при облучении, например, ультрафиолетовым излучением или ИК-излучением, или при обработке рентгеновскими лучами или электронным пучком, с тем, чтобы быть применимыми в качестве покрытия для термопластичных материалов или термоотверждающихся материалов, имеющих низкие температуры плавления.
Полимеризация полимеризующихся органических соединений может быть инициирована любым традиционным способом, который будет определяться природой полимеризующихся органических соединений. Он обычно включает использование инициатора полимеризации.
Для нанесения покрытий значительная органическая полимеризация обычно замедляется до нанесения покрытия на подложку. Поэтому, если для инициирования или ускорения органической полимеризации используется нагрев, температура должна быть выбрана так, чтобы не иметь какое-либо вредное воздействие на подложку, на которую должно наноситься покрытие. В случае термопластичных или термоотверждающихся подложек должны использоваться относительно низкие температуры, обычно ниже 150°C, и, более типично, в интервале 30-80°C. Когда используется химический инициатор полимеризации, это может означать замедление его введения до момента перед операцией нанесения покрытия или возможно в процессе операции нанесения.
Пропорция органического полимера, вводимого в конечный продукт, зависит от свойств, требуемых в конечных продуктах.
Свойства конечного продукта могут дополнительно регулироваться путем использования добавок, традиционно используемых в технике.
При использовании в качестве композиции покрытия композиция может быть нанесена на подложку любым традиционным способом, например моканием, напылением, валиком или кистью. Композиция может быть нанесена на широкий ряд подложек, и особенно подходящим является нанесение покрытий на полимерные материалы, имеющие относительно низкие температуры плавления, например, 150°C или ниже. Примеры таких полимерных материалов включают термопластичные материалы и термоотверждающиеся материалы, такие как поликарбонаты, сложные полиэфиры, такие как полиакрилаты и политерефталаты, полиуретаны и полиакриловые полимеры. Улучшенные стойкость к царапанию/истиранию и химическая стойкость, придаваемые указанным материалам путем нанесения покрытий настоящего изобретения, позволяют им быть значительно более широко используемыми, чем они используются в настоящее время.
Композиция покрытия может также использоваться для нанесения на подложки, выбранные из следующего: стекло, металлы, включая мягкие металлы, такие как алюминий, латунь и серебро, керамические материалы и природные материалы, такие как кожа и древесина, или синтетические заменители указанных материалов. Она находит, в частности, использование в качестве покрытия для стекла и заменителей стекла. Например, она может использоваться в качестве покрытия для зданий или транспортных средств, окон и ветровых стекол, например, для автомобиля, самолета и поезда, стекол очков, объективов фотокамер, защитных колпаков, оптических фильтров и световых футляров, например, осветительных приборов, компакт-дисков, экранов дисплеев, например, в персональных компьютерах и мобильных телефонах, и для защиты белых изделий, например, холодильников и моечных машин, и небелых изделий, например, аудиовизуального оборудования.
Композиция настоящего изобретения также находит использование в качестве покрытия катализатора для ряда подложек. Для данного применения, предпочтительно, чтобы композиция не была сшитой, с тем чтобы сохранить ее жидкую природу.
Композиция настоящего изобретения также находит использование для соединения вместе, по меньшей мере, двух изделий. Например, композиция может быть нанесена на поверхность одного или каждого изделия, поверхности сводятся вместе, и композиция отверждается с образованием надежного соединения.
Способ настоящего изобретения также способен давать материалы, которые находят использование в качестве материалов в массе в большей степени, чем как покрытия. В данном случае материал может быть формован, например, формованием в форме, или иным образом формован в широкий ряд различных изделий.
Настоящее изобретение дополнительно иллюстрируется следующими примерами.
Примеры
Пример 1
Метакрилатсилановая смола
Смесь 111,1 г промышленного метилированного спирта ((ПМС (IMS)), 16,3 г воды и 0,16 г 37% HCl тщательно перемешивают и затем добавляют к 150,0 г 3-триметоксисилилпропилмет-акрилата. Данную смесь интенсивно перемешивают в течение 4 ч и затем выливают (или охлаждают) в 555 г дистиллированной воды. Охлажденную смесь интенсивно перемешивают в течение не менее 18 ч перед выливанием в большой полипропиленовый контейнер и нагревают при приблизительно 50°C в течение приблизительно 6 ч с удалением воды. Затем извлекают оставшуюся вязкую жидкость (смолу).
Пример 2
Метакрилатсилановое покрытие
10,0 г смолы, полученной в примере 1, разбавляют 30,0 г ПМС и добавляют 0,1 г фотоинициатора Irgacure 184. После тщательного смешения данный раствор наносят поливом на пластину из поликарбоната Lexan. Покрытую пластину сушат в воздушной атмосфере в течение 5 мин при 50°C и затем отверждают с использованием ультрафиолетового излучения.
Пример 3
Метакрилатсилановое и уретанакрилатное покрытие
9,0 г смолы, полученной в примере 1, разбавляют 27,0 г промышленного метилированного спирта и перемешивают до получения гомогенного раствора. К данному раствору добавляют 1,0 г алифатического уретанакрилата 260GP25 и 0,1 г фотоинициатора Irgacure 184. После перемешивания до получения гомогенного раствора жидкость наносят поливом на пластину из поликарбоната Lexan. Покрытую пластину сушат в воздушной атмосфере в течение 5 мин при 50°C и затем отверждают с использованием ультрафиолетового излучения.
Пример 4
Акрилатсилановая смола
Смесь 117,8 г промышленного метилированного спирта ((ПМС (IMS)), 17,3 г воды и 0,17 г 37% HCl тщательно перемешивают и затем добавляют к 150,0 г 3-триметоксисилилпропилакрилата. Данную смесь интенсивно перемешивают в течение 4 ч и затем выливают (или охлаждают) в 570 г дистиллированной воды. Охлажденную смесь интенсивно перемешивают в течение не менее 18 ч перед выливанием в большой полипропиленовый контейнер и нагревают при приблизительно 50°C в течение 6 ч с удалением воды. Затем извлекают оставшуюся вязкую жидкость (смолу).
Пример 5
Акрилатсилановое покрытие
10,0 г смолы, полученной в примере 4, разбавляют 30,0 г ПМС и добавляют 0,1 г фотоинициатора Irgacure 184, когда было 0,1 г FC4430 (от фирмы 3M Corporation) в качестве добавки, улучшающей текучесть. После тщательного смешения данный раствор наносят поливом на пластину из поликарбоната Lexan. Покрытую пластину сушат в воздушной атмосфере в течение 5 мин при 50°C и затем отверждают с использованием ультрафиолетового излучения.
Пример 6
Акрилатсилановое и уретанакрилатное покрытие
9,0 г смолы, полученной в примере 4, разбавляют 27,0 г промышленного метилированного спирта и перемешивают до получения гомогенного раствора. К данному раствору добавляют 1,0 г алифатического уретанакрилата 260GP25 и 0,1 г фотоинициатора Irgacure 184. После перемешивания до получения гомогенного раствора жидкость наносят поливом на пластину из поликарбоната Lexan. Покрытую пластину сушат в воздушной атмосфере в течение 5 мин при 50°C и затем отверждают с использованием ультрафиолетового излучения.
Пример 7
Эпоксисилановая смола
Смесь 77,9 г промышленного метилированного спирта ((ПМС (IMS)) и 11,4 г дистиллированной воды тщательно перемешивают и затем добавляют к 100,0 г 3-глицидоксипропилтриметоксисилана. Данную смесь интенсивно перемешивают в течение 4 ч и затем выливают (или охлаждают) в 370 г дистиллированной воды. Охлажденную смесь интенсивно перемешивают в течение не менее 18 ч перед выливанием в большой полипропиленовый контейнер и нагревают при приблизительно 50°C в течение приблизительно 6 ч с удалением воды. Затем извлекают оставшуюся вязкую жидкость (смолу).
Пример 8
Эпоксисилановое и диаминооктановое покрытие
5,0 г смолы, полученной в примере 7, разбавляют в 15,0 г ПМС и перемешивают до получения гомогенной жидкости. К данной жидкости добавляют 1,08 г диаминооктана. Смесь интенсивно перемешивают в течение 5 мин и затем наносят в качестве покрытия на пластину из поликарбоната Lexan. Покрытую пластину затем сушат и отверждают в воздушной атмосфере при 130°C в течение 18 ч.
Пример 9
Эпоксисилановое и ксилилендиаминовое покрытие
===5,0 г смолы, полученной в примере 7, разбавляют в 15,0 г ПМС и перемешивают до получения гомогенной жидкости. К данной жидкости добавляют 1,02 г ксилилендиамина. Смесь интенсивн перемешивают в течение 5 мин и затем наносят в качестве покрытия на пластину из поликарбоната Lexan. Покрытую пластину затем сушат и отверждают в воздушной атмосфере при 130°C в течение 18 ч.
Пример 10
Смешанная эпокси-фенилсилановая смола
Компонент А и компонент В получают раздельно.
Компонент А. 7,0 г 3-глицидоксипропилтриметоксисилана помещают в химический стакан и к нему добавляют однородную смесь 5,5 г ПМС и 0,80 г воды.
Компонент В. 40,0 г фенилтриметоксисилана помещают в химический стакан и к нему добавляют однородную смесь 37,1 г ПМС и 5,45 г воды.
Компоненты А и В перемешивают отдельно в закрытых химических стаканах в течение примерно 1 ч, после чего их объединяют и перемешивают в течение примерно 4 ч снова в закрытом химическом стакане. Данную смесь интенсивно перемешивают в течение 4 ч и затем выливают (или охлаждают) в 192 г дистиллированной воды. Охлажденную смесь интенсивно перемешивают в течение не менее 18 ч перед выливанием в большой полипропиленовый контейнер и нагревают при приблизительно 50°C в течение приблизительно 6 ч с удалением воды. Затем извлекают оставшуюся вязкую жидкость (смолу).
Пример 11
Смешанное эпокси-фенилсиландиаминооктановое покрытие
5,0 г смолы, полученной в примере 10, разбавляют в 15,0 г ПМС и перемешивают до получения гомогенной жидкости. К данной жидкости добавляют 0,17 г диаминооктана. Смесь интенсивно перемешивают в течение 5 мин и затем наносят в качестве покрытия на пластину из поликарбоната Lexan. Покрытую пластину затем сушат и отверждают в воздушной атмосфере при 130°C в течение 18 ч.
Пример 12
Циклогексилэпоксисилановая смола
Смесь 44,9 г промышленного метилированного спирта ((ПМС (IMS)) и 6,59 г дистиллированной воды тщательно перемешивают и затем добавляют к 60,0 г 2-(3,4-эпоксициклогексил)этил-триметоксисилана. Данную смесь интенсивно перемешивают в течение 4 ч и затем выливают (или охлаждают) в 223 г дистиллированной воды. Охлажденную смесь интенсивно перемешивают в течение не менее 18 ч перед выливанием в большой полипропиленовый контейнер и нагревают при приблизительно 50°C в течение приблизительно 6 ч с удалением воды. Затем извлекают оставшуюся вязкую жидкость (смолу).
Пример 13
Циклогексилэтоксисиландиаминооктановое покрытие
5,0 г смолы, полученной в примере 12, разбавляют в 15,0 г ПМС и перемешивают до получения гомогенной жидкости. К данной жидкости добавляют 0,20 г диаминооктана. Смесь интенсивно перемешивают в течение 5 мин и затем наносят в качестве покрытия на пластину из поликарбоната Lexan. Покрытую пластину затем сушат и отверждают в воздушной атмосфере при 130°C в течение 18 ч.
Пример 14
Аминосилановая смола
Смесь 49,9 г промышленного метилированного спирта ((ПМС (IMS)) и 7,32 г дистиллированной воды тщательно перемешивают и затем добавляют к 60,0 г 3-аминопропилтриэтоксисилана. Данную смесь интенсивно перемешивают в течение 4 ч и затем выливают (или охлаждают) в 234 г дистиллированной воды. Охлажденную смесь интенсивно перемешивают в течение не менее 18 ч перед выливанием в большой полипропиленовый контейнер и нагревают при приблизительно 50°C в течение приблизительно 6 ч с удалением воды. Затем извлекают оставшуюся вязкую жидкость (смолу).
Пример 15
Смешанная эпоксисмола
Компонент А и компонент В получают раздельно.
Компонент А. 40,0 г 3-глицидоксипропилтриметоксисилана помещают в химический стакан и к нему добавляют однородную смесь 31,1 г ПМС и 4,57 г воды.
Компонент В. 5,0 г тетраэтоксисилана помещают в химический стакан и к нему добавляют однородную смесь 4,4 г ПМС и 0,65 г воды.
Компоненты А и В перемешивают отдельно в закрытых химических стаканах в течение примерно 1 ч, после чего их объединяют и перемешивают в течение примерно 4 ч снова в закрытом химическом стакане. Данную смесь интенсивно перемешивают в течение 4 ч и затем выливают (или охлаждают) в 172 г дистиллированной воды. Охлажденную смесь интенсивно перемешивают в течение не менее 18 ч перед выливанием в большой полипропиленовый контейнер и нагревают при приблизительно 50°C в течение приблизительно 6 ч с удалением воды. Затем извлекают оставшуюся вязкую жидкость (смолу).
Пример 16
Дегидратирование смолы
1,5 г смолы, полученной в примере 4, растворяют в смеси 0,25 г триэтиламина и 4,5 г тетрагидрофурана ((ТГФ), (THF)). Раствор затем помещают на высушенное молекулярное сито типа 4А. Через 24 ч растворитель выпаривается, дегидратированную смолу растворяют в 4,5 г ПМС и добавляют 0,1 г фотоинициатора Irgacure 184. После тщательного перемешивания раствор затем наносят поливом на пластину из поликарбоната Lexan. Покрытую пластину сушат в воздушной атмосфере в течение 5 мин и затем отверждают с использованием ультрафиолетового излучения.
Пример 17
Блокирование остаточных силанолов
1,5 г смолы, полученной в примере 4, растворяют в смеси 0,25 г триэтиламина, 4,5 г ТГФ и 0,25 г хлоротриметилсилана. Через 24 ч смесь фильтруют через 1 мкм фильтр с удалением твердой хлоридной соли. Растворитель выпаривают, смолу растворяют в 4,5 г ПМС и добавляют 0,1 г фотоинициатора Irgacure 184. После тщательного перемешивания раствор затем наносят поливом на пластину из поликарбоната Lexan. Покрытую пластину сушат в воздушной атмосфере в течение 5 мин и затем отверждают с использованием ультрафиолетового излучения.
Пример 18
Блокирование остаточных силанолов силаном типа R 2 SiY 2
1,5 г смолы, полученной в примере 4, растворяют в смеси 0,25 г триэтиламина, 4,5 г ТГФ и 0,5 г диэтоксидиметилсилана. Раствор затем помещают на высушенное молекулярное сито типа 4А. Через 24 ч растворитель выпаривается, смолу растворяют в 4,5 г ПМС и добавляют 0,1 г фотоинициатора Irgacure 184. После тщательного перемешивания раствор затем наносят поливом на пластину из поликарбоната Lexan. Покрытую пластину сушат в воздушной атмосфере в течение 5 мин и затем отверждают с использованием ультрафиолетового излучения.
Пример 19
Блокирование остаточных силанолов длинноцепочечными силанами
1,5 г смолы, полученной в примере 4, растворяют в смеси 0,25 г триэтиламина, 4,5 г ТГФ и 0,25 г полидиметилсилоксана с гидрокси-окончанием (вязкость = 90-150 сСт). Раствор затем помещают на высушенное молекулярное сито типа 4А. Через 24 ч растворитель выпаривается, смолу растворяют в 4,5 г ПМС и добавляют 0,1 г фотоинициатора Irgacure 184. После тщательного перемешивания раствор затем наносят поливом на пластину из поликарбоната Lexan. Покрытую пластину сушат в воздушной атмосфере в течение 5 мин и затем отверждают с использованием ультрафиолетового излучения.
Пример 20
Введение силана R 2 SiY 2 в золь
Смесь 117,8 г промышленного метилированного спирта ((ПМС (IMS)), 17,3 г дистиллированной воды и 0,17 г 37% HCl тщательно перемешивают и затем добавляют к 150,0 г 3-три-метоксисилилпропилакрилата. Данную смесь перемешивают в течение 240 мин. Отдельно смесь 58,3 г промышленного метилированного спирта ((ПМС (IMS)), 5,46 г дистиллированной воды и 0,1 г 37% HCl тщательно перемешивают и затем добавляют к 45,0 г диэтоксидиметилсилана и перемешивают в течение 5 мин. Два силановых раствора затем смешивают и перемешивают вместе в течение 2 ч и затем выливают (или охлаждают) в 570 г дистиллированной воды. Охлажденную смесь интенсивно перемешивают в течение не менее 18 ч перед выливанием в большой полипропиленовый контейнер и нагревают при приблизительно 50°C в течение приблизительно 6 ч с удалением воды. Затем извлекают оставшуюся вязкую жидкость (смолу).
Пример 21
Дегидратирование смолы. Использование в качестве клея/герметика
1,5 г смолы, полученной в примере 4, растворяют в смеси 0,25 г триэтиламина и 4,5 г ТГФ. Раствор затем помещают на высушенное молекулярное сито типа 4А. Через 24 ч растворитель выпаривается, дегидратированную смолу растворяют в 0,5 г ПМС и добавляют 0,1 г фотоинициатора Irgacure 184 вместе с 0,5 г уретанакрилата 260Gp25. После тщательного смешения раствор затем распыляют на лист из сложного полиэфира. Поверх первого листа помещают второй лист, и два листа спрессовывают вместе с получением линии соединения ~ 50 мкм. Сэндвичевую структуру затем отверждают под ультрафиолетовым излучением в течение 5 мин, и смесь смол дает клеевой шов.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПОКРЫВНАЯ КОМПОЗИЦИЯ, СПОСОБ ЕЕ ПОЛУЧЕНИЯ И ЕЕ ПРИМЕНЕНИЕ | 2013 |
|
RU2674057C2 |
| ПРОЦЕСС ПОЛУЧЕНИЯ МОДИФИЦИРОВАННОГО ИНТЕРПОЛИМЕРА ИЛИ МОДИФИЦИРОВАННОГО ПОЛИМЕРА | 2009 |
|
RU2531821C2 |
| ОРГАНОСИЛАНОВЫЕ КОМПОЗИЦИИ ДЛЯ ПОКРЫТИЙ И ИХ ИСПОЛЬЗОВАНИЕ | 2008 |
|
RU2514962C2 |
| ЭПОКСИДНЫЕ ПОЛИСИЛОКСАНОВЫЕ СОСТАВЫ ДЛЯ ПОКРЫТИЙ И ШПАТЛЕВКИ | 1995 |
|
RU2159260C2 |
| СПОСОБ ПОКРЫТИЯ СУБСТРАТОВ | 2006 |
|
RU2430129C2 |
| ПОРОШКОВАЯ ФЕРРОМАГНИТНАЯ КОМПОЗИЦИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2009 |
|
RU2510993C2 |
| СПОСОБ МОДИФИКАЦИИ НЕОРГАНИЧЕСКОГО КИСЛОРОДСОДЕРЖАЩЕГО ЗЕРНИСТОГО МАТЕРИАЛА, ПОЛУЧЕННЫЙ ИЗ НЕГО ПРОДУКТ И ИХ ПРИМЕНЕНИЕ | 2006 |
|
RU2415893C2 |
| ВИНИЛСИЛАНЫ ДЛЯ ПРИМЕНЕНИЯ В ФУНКЦИОНАЛИЗИРОВАННЫХ ЭЛАСТОМЕРНЫХ ПОЛИМЕРАХ | 2013 |
|
RU2669799C2 |
| АМИНОВЫЕ СОЕДИНЕНИЯ И ПОЛУЧЕННЫЕ ИЗ НИХ ОТВЕРЖДАЕМЫЕ КОМПОЗИЦИИ | 2002 |
|
RU2296129C2 |
| СПОСОБ НАНЕСЕНИЯ ПОКРЫТИЙ НА МЕТАЛЛИЧЕСКИЕ ПОВЕРХНОСТИ МНОГОСТАДИЙНЫМ МЕТОДОМ | 2010 |
|
RU2542184C2 |
Изобретение относится к способу получения композиции, содержащей органосилсесквиоксаны. Предложен способ получения композиции, включающей органосилсесквиоксаны, содержащий следующие стадии: (1) частичный гидролиз гидролизующихся предшественников неорганических мономеров, содержащих, по меньшей мере, 50 мол.% первых гидролизующихся предшественников неорганических мономеров формулы RSiY3, где R выбран из (цикло)алкильных, арильных, алкенильных, эпокси-, (мет)акрильных, сложноэфирных, амино-, меркапто- и цианатных групп и их смесей, R-Si-связь является негидролизующейся связью, каждая Y-группа является одинаковой или отличающейся от каждой другой Y-группы алкокси-группой; (2) охлаждение жидкой композиции перед завершением конденсации неорганических мономеров количеством воды, которое в комбинации с водой, используемой на стадии (1), и любой водой, отщепляемой при конденсации неорганических мономеров на стадии (1), превышает стехиометрическое количество воды, необходимой для осуществления полного гидролиза всех присутствующих предшественников гидролизующихся неорганических мономеров; и (3) сушка композиции; причем суммарное количество используемой воды по меньшей мере в 5 раз больше стехиометрического количества воды, необходимого для полного гидролиза всех присутствующих гидролизующихся предшественников мономеров. Технический результат - контролируемое и быстрое получение широкого ряда органосилсесквилоксансодержащих композиций, пригодных для получения защитных покрытий и в качестве связующих агентов. 3 н. и 14 з.п. ф-лы.
1. Способ получения композиции, содержащей органосилсесквиоксаны, который включает следующие стадии:
(1) частичный гидролиз гидролизующихся предшественников неорганических мономеров, содержащих, по меньшей мере, 50 мол.% первых гидролизующихся предшественников неорганических мономеров, имеющих формулу RSiY3, в которой R-группа представляет собой органическую группу, которая является устойчивой к гидролизу, и которая является одинаковой или отличающейся от каждой другой R-группы, где R-группа в первых гидролизующихся предшественниках неорганических мономеров выбрана из (цикло)алкильных, арильных, алкенильных, эпокси-, (мет)акрильных, сложноэфирных, амино-, меркапто- и цианатных групп и их смесей, R-Si-связь является негидролизующейся связью, каждая Y-группа является алкокси группой и является одинаковой или отличающейся от каждой другой Y-группы и выбирается из химически реакционных групп, так что каждая Si-Y-связь является гидролизующейся с образованием Si-OH-связи, с образованием неорганических мономеров и обеспечением частичной конденсации неорганических мономеров с образованием жидкой композиции, содержащей неорганические олигомеры; где минеральные кислоты используют в качестве катализатора для инициирования гидролиза гидролизующихся предшественников неорганических мономеров;
(2) охлаждение жидкой композиции перед завершением конденсации неорганических олигомеров количеством воды, которое в комбинации с водой, используемой на стадии (1), и любой водой, отщепляемой при конденсации неорганических мономеров на стадии (1), превышает стехиометрическое количество воды, необходимое для осуществления полного гидролиза всех гидролизующихся предшественников неорганических мономеров, присутствующих вначале стадии (1), с тем, чтобы полностью гидролизовать олигомеры и обеспечить их дополнительную конденсацию; и
(3) сушка композиции,
в котором на стадии (1) количество воды добавляется к гидролизующимся предшественникам мономеров так, чтобы обеспечить гидролиз одной или самое большое двух гидролизующихся связей, присутствующих в гидролизующихся предшественниках мономеров,
в котором количество воды, используемой на стадии (2) в комбинации с количеством воды, используемой на стадии гидролиза (1), и любой воды, высвободившейся при конденсации на стадии (1), составляет, по меньшей мере, в 5 раз больше стехиометрического количества воды, необходимого для полного гидролиза всех присутствующих гидролизующихся предшественников мономеров.
2. Способ по п.1, в котором гидролизующиеся предшественники неорганических мономеров содержат более 60 мол.%, предпочтительно, по меньшей мере, 70 мол.%, и, более предпочтительно, по меньшей мере, 80 мол.% первых гидролизующихся предшественников неорганических мономеров.
3. Способ по п.1 или 2, в котором R-группа в первых гидролизующихся предшественниках неорганических мономеров выбрана из (цикло)алкильных, арильных, алкенильных, групп, необязательно замещенных группами выбранными из эпокси, винила и метил(акриловых) групп.
4. Способ по п.1 или 2, где первые гидролизующиеся предшественники неорганических мономеров выбраны из (алкил)алкоксисиланов; (алкил)алкоксисиланов, имеющих эпоксигруппу; (алкил)алкоксисиланов, имеющих карбоксильную группу; (алкил)алкоксисиланов, имеющих аминогруппу; (алкил)алкоксисиланов, имеющих тиольную группу; (алкил)алкоксисиланов, имеющих винильную группу; (алкил)алкоксисиланов, имеющих акрилатную или метакрилатную группу; (алкил)алкоксисиланов, имеющих атом галогена; (алкил)алкоксисиланов, имеющих галогенированный алкил-лиганд; и
где алкил-группа может быть замещена циклоалкил-группой, арил-группой или алкенил-группой и необязательно может быть замещена предпочтительно (мет)акрилатной группой или эпоксигруппой.
5. Способ по любому из пп.1-2, в котором стадия (1) содержит частичный гидролиз различных гидролизующихся предшественников неорганических мономеров, указанные различные гидролизующиеся предшественники неорганических мономеров гидролизуются отдельно друг от друга с последующей частичной конденсацией получаемых различных неорганических мономеров и затем смешением получаемых жидких композиций перед осуществлением стадии (2).
6. Способ по любому из пп.1-2, в котором первые гидролизующиеся предшественники неорганических мономеров содержат молекулы, имеющие различные R-группы, в котором предпочтительно на стадии (1) различные первые гидролизующиеся предшественники мономеров гидролизуются отдельно друг от друга и обеспечивают частичную конденсацию, и получаемые жидкие композиции затем смешиваются вместе перед осуществлением стадии (2).
7. Способ по любому из пп.1-2, в котором гидролизующиеся предшественники неорганических мономеров дополнительно содержат вторые гидролизующиеся предшественники мономеров, имеющие формулу RnSiY4-n, в которой п составляет 0, 2 или 3, каждая R-Si-связь является негидролизующейся связью, R представляет собой органическую группу, и каждая R-группа является одинаковой или отличающейся от каждой другой R-группы, и каждая Y-группа является одинаковой или отличающейся от каждой другой Y-группы и выбирается из химически реакционных групп, так что каждая Si-Y-связь гидролизуется в Si-OH, и в котором предпочтительно на стадии (1) первые и вторые гидролизующиеся предшественники мономеров частично гидролизуются отдельно друг от друга и обеспечивают частичную конденсацию, и получаемые жидкие композиции затем смешиваются вместе перед осуществлением стадии (2).
8. Способ по п.7, в котором вторые гидролизующиеся предшественники неорганических мономеров имеют формулу SiY4.
9. Способ по любому одному из пп.1, 2 или 8, в котором гидролизующиеся предшественники неорганических мономеров дополнительно содержат гидролизующиеся предшественники мономеров, имеющие формулу (R2SiO)xY2, в которой х≥1, каждая R-Si-связь является негидролизующейся связью, R представляет собой органическую группу, и каждая R-группа является одинаковой или отличающейся от каждой другой R-группы, и каждая Y-группа является одинаковой или отличающейся от каждой другой Y-группы и выбирается из химически реакционных групп, так что каждая Si-Y-связь гидролизуется в Si-OH, и в котором предпочтительно на стадии (1) различные гидролизующиеся предшественники мономеров частично гидролизуются отдельно друг от друга и обеспечивают частичную конденсацию, и получаемые жидкие композиции затем смешиваются вместе перед осуществлением стадии (2).
10. Способ по любому одному из пп.1, 2 или 8, в котором на стадии (1), по меньшей мере, 80 мол.%, предпочтительно, по меньшей мере, 90 мол.%, первых гидролизующихся предшественников мономеров подвергаются гидролизу и конденсации через одну из Si-Y-связей с образованием неорганических олигомеров, имеющих формулу RY2-Si-O-SiY2R, в которой каждая R-группа является одинаковой или отличающейся от каждой другой R-группы.
11. Способ по одному из пп.1, 2 или 8, который дополнительно включает в качестве стадии (4) дегидратирование композиции, полученной на стадии (3), с получением дегидратированной композиции.
12. Способ по любому одному из пп.1, 2 или 8, который дополнительно включает смешение композиции, полученной на конечной стадии (3) и дополнительно стадии (4), если присутствует, с органическими мономерами или олигомерами.
13. Способ по п.12, который дополнительно включает полимеризацию органических мономеров или олигомеров.
14. Способ по любому одному из пп.1, 2 или 8, который дополнительно включает смешение композиции, полученной на конечной стадии (3) и дополнительно стадии (4), если присутствует, с органическим жидким полимером.
15. Способ по любому одному из пп.1, 2 или 8, который дополнительно включает сшивание композиции, полученной на конечной стадии (3) и дополнительно стадии (4), если присутствует, через R-группы с получением сшитой композиции.
16. Способ получения покрытия на подложке, включающий выполнение способа по любому из пп.1-15 с получением композиции, содержащей органосилсесквиоксаны, нанесение на подложку и необязательно сшивание композиции.
17. Способ соединения вместе, по меньшей мере, двух изделий, включающий выполнение способа по любому из пп.1-15, с получением композиции, содержащей органосилсесквиоксаны, нанесение на поверхность одного или каждого изделия композиции, приведение поверхностей в контакт друг с другом и затем сшивание композиции с образованием соединения.
| GB 1217082 А, 23.12.1970 | |||
| US 20030212228 A1, 13.11.2003 | |||
| US 6586104 B2, 01.07.2003 | |||
| Коршак В.В., Козырева Н.М., Кирилин А.И | |||
| и др | |||
| Успехи в области синтеза элементоорганических полимеров | |||
| - М.: Наука, 1988, с.180-181 | |||
| US 2003224286 A1, 04.12.2003 | |||
| US 2003191269 A1, 09.10.2003 | |||
| JP 2004284221 A, 14.10.2004 | |||
| Установка для бурения скважин | 1986 |
|
SU1469072A1 |
| Устройство для индикации количества магнитной ленты | 1975 |
|
SU538417A1 |
| JP 2005122815 A, 12.05.2005. | |||

