Предпосылки изобретения
Область техники
Настоящее изобретение относится к области медицины и химии. Более конкретно настоящее изобретение относится к N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамиду, его тартрату, а также его полиморфам, их синтезу и применениям.
Предшествующий уровень техники
В WO 01/66521 описаны N-азациклоалкил-N-аралкилкарбамиды и амиды карбоновых кислот, которые являются новым классом соединений, эффективных при ингибировании активности моноаминовых рецепторов, включая рецептор серотонина подкласса 5-HT2A. WO 01/66521. Примеры болезненных состояний, при которых могут применяться такие соединения, включают, не ограничиваясь перечисленными, нейропсихиатрические заболевания, такие как шизофрения и родственные идиопатические психозы, депрессию, тревожные состояния, расстройства сна, расстройства аппетита, аффективные расстройства, такие как глубокая депрессия, биполярное расстройство, депрессию с психотическими признаками и синдром Туретта. Другими примерами полезного применения могут являться лечение психозов, вызванных лекарственными средствами, и побочных эффектов болезни Паркинсона, а также психозов, являющихся следствием нейродегенеративных расстройств, таких как болезни Альцгеймера и Хантингтона, гипертензии, мигрени, спазма сосудов, ишемии, и, во-первых, лечение, а, во-вторых, профилактика различных тромботических состояний, включая инфаркт миокарда, тромботический или ишемический удар, идиопатическую и тромботическую тромбоцитопеническую пурпуру и заболевание периферических сосудов.
Сущность изобретения
Один из вариантов осуществления, раскрытый в настоящем описании, включает способ получения соединения формулы (I):

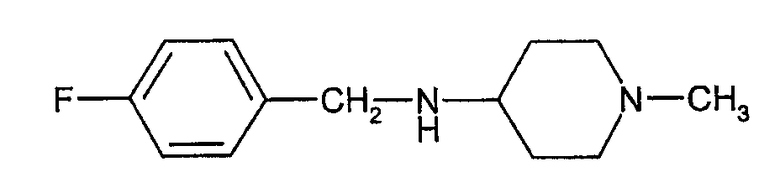
который включает взаимодействие (4-фторбензил)-(1-метилпиперидин-4-ил)амина формулы (II)
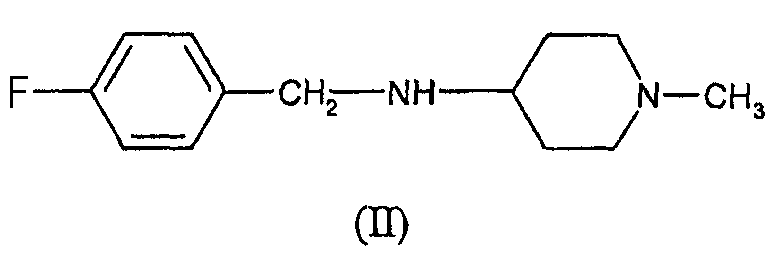
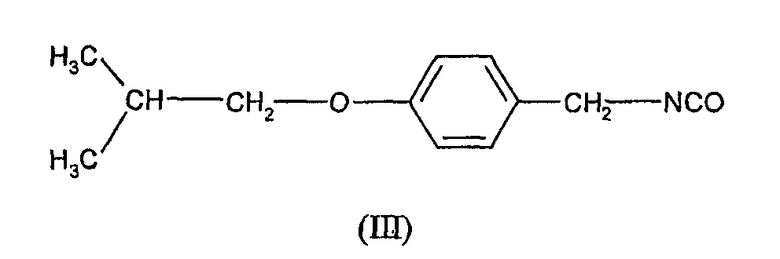
с 4-(2-метилпропилокси)фенилметилизоцианатом формулы (III)
В некоторых вариантах осуществления применяется от приблизительно 0,9 до приблизительно 1,1 эквивалента (4-фторбензил)-(1-метилпиперидин-4-ил)амина на эквивалент 4-(2-метилпропилокси)фенилметилизоцианата. Некоторые варианты осуществления, кроме того, включают выделение соединения формулы (I) после реакции. В некоторых вариантах осуществления выделение включает добавление солеобразующей кислоты после завершения реакции, выделение полученной соли с помощью удаления растворителя, осаждения или как удаления растворителя, так и осаждения, добавление выделенной соли к двухфазной системе, включающей фазу органического растворителя и фазу водного раствора щелочи, и выделение соединения формулы (I) из фазы органического растворителя. В некоторых вариантах осуществления солеобразующая кислота выбрана из группы, состоящей из одной или нескольких из следующих кислот: минеральных кислот, моно- или дикарбоновых кислот и сульфоновых кислот. В некоторых вариантах осуществления pH водной фазы выше чем приблизительно 8,5. В одном из вариантов осуществления это значение pH получают добавлением водного раствора гидроксида щелочного металла. В некоторых вариантах осуществления взаимодействие проводят в присутствии инертного органического растворителя. В некоторых вариантах осуществления растворитель выбран из группы, состоящей из одного или нескольких растворителей из числа следующих: алифатические простые эфиры, сложные эфиры алифатических карбоновых кислот, спирты, лактоны, галогенированные углеводороды и алифатические C3-C8 кетоны. В некоторых вариантах осуществления реакцию проводят при температуре от приблизительно -30 до приблизительно 60°C.
Другой вариант осуществления, раскрытый в настоящем описании, относится к кристаллической форме N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида, которая имеет температуру плавления около 124°C, определенную с помощью дифференциальной сканирующей калориметрии (DSC) при скорости нагревания 10°C/минута.
Другой вариант осуществления, раскрытый в настоящем описании, относится к кристаллической форме N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида, рентгенограмма на порошке которой включает пики, соответствующие величинам d в ангстремах, равным приблизительно 13,0, приблизительно 10,9, приблизительно 6,5, приблизительно 4,7, приблизительно 4,3, приблизительно 4,22 и приблизительно 4,00. В одном из вариантов осуществления рентгенограмма кристаллической формы включает пики, соответствующие значениям d в ангстремах, равным приблизительно 13,0, приблизительно 10,9, приблизительно 6,8, приблизительно 6,5, приблизительно 6,2, приблизительно 5,2, приблизительно 4,7, приблизительно 4,5, приблизительно 4,3, приблизительно 4,22, приблизительно 4,00, приблизительно 3,53, приблизительно 3,40, приблизительно 3,28, приблизительно 3,24, приблизительно 3,19, приблизительно 3,08, приблизительно 2,91 и приблизительно 2,72.
Другой вариант осуществления, раскрытый в настоящем описании, относится к способу получения описанной выше кристаллической формы, включающему растворение соли соединения формулы (I) в воде:
добавление к водному раствору соли органического апротонного растворителя в количестве, достаточном для растворения соединения формулы (I);
доведение значения pH водного раствора соли до величины, как минимум, приблизительно 8,5 путем добавления основания;
удаление части органического апротонного растворителя;
охлаждение оставшегося раствора в органическом апротонном растворителе до температуры ниже 15°C и
выделение образовавшегося осадка.
В некоторых вариантах осуществления соль соединения формулы (I) является гемитартратом. Некоторые варианты осуществления дополнительно включают экстракцию водного раствора органическим растворителем и сбор всех органических фаз до удаления части органического растворителя. В одном из вариантов осуществления органический растворитель выбирают из группы, состоящей из одного или нескольких растворителей из числа следующих: углеводороды, галогенированные углеводороды, сложные эфиры алифатических карбоновых кислот, спирты, лактоны, простые эфиры и алифатические C4-C8 кетоны.
Другой вариант осуществления, раскрытый в настоящем описании, относится к кристаллической форме N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида, полученного способом, который включает растворение гемитартрата соединения формулы (I) в воде:
добавление к водному раствору соли органического апротонного растворителя в количестве, достаточном для растворения соединения формулы (I), доведение значения pH водного раствора соли до величины, как минимум, приблизительно 8,5 добавлением основания, экстракцию водного раствора органическим растворителем и сбор всех органических фаз, удаление части органического апротонного растворителя, охлаждение оставшегося органического апротонного растворителя до температуры ниже 15°C и выделение образовавшегося осадка.
Другим вариантом осуществления, раскрытым в настоящем описании, является гемитартрат N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида формулы (IV)
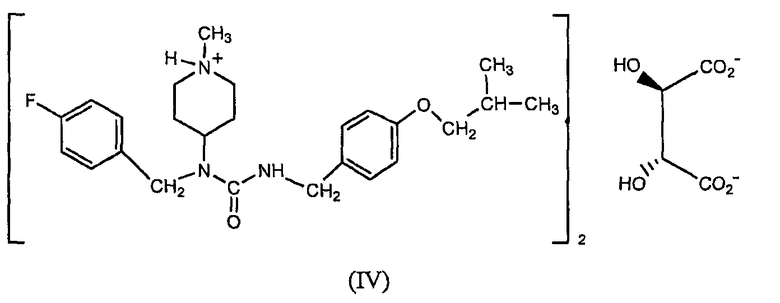
Другой вариант осуществления, раскрытый в настоящем описании, относится к способу получения гемитартрата N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида, включающему проведение реакции синтеза соединения формулы (I), как описано выше, добавление винной кислоты после этой реакции и выделение образовавшегося гемитартрата. В одном из вариантов осуществления выделение включает осаждение гемитартрата с помощью охлаждения, удаления растворителя, добавления жидкости, не являющейся растворителем, или выделение проводится путем комбинации этих способов.
Другой вариант осуществления, раскрытый в настоящем описании, относится к кристаллической форме гемитартрата N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида, рентгенограмма на порошке которой включает пики, соответствующие величинам d в ангстремах, равным приблизительно 18,6, приблизительно 16,7, приблизительно 10,2, приблизительно 6,2, приблизительно 6,1, приблизительно 4,63, приблизительно 4,49, приблизительно 4,44 и приблизительно 3,96. В одном из вариантов осуществления рентгенограмма включает пики, соответствующие величинам d в ангстремах приблизительно 18,6, приблизительно 16,7, приблизительно 10,2, приблизительно 8,2, приблизительно 7,7, приблизительно 7,4, приблизительно 6,5, приблизительно 6,2, приблизительно 6,1, приблизительно 5,86, приблизительно 5,14, приблизительно 5,03, приблизительно 4,78, приблизительно 4,69, приблизительно 4,63, приблизительно 4,49, приблизительно 4,44, приблизительно 4,35, приблизительно 4,10, приблизительно 3,96 и приблизительно 3,66.
В одном из вариантов осуществления указанную выше кристаллическую форму получают растворением соединения формулы IV в этаноле или смеси этанола и изопропанола:
охлаждением раствора до температуры менее чем приблизительно 20°C и выделением образовавшегося твердого осадка. В одном из вариантов осуществления скорость охлаждения на стадии охлаждения составляет от приблизительно 0,1 до приблизительно 3°C/минуту.
Другой вариант осуществления, раскрытый в настоящем описании, относится к кристаллической форме тартрата N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида, полученной способом, который включает растворение соединения формулы (IV) в этаноле или смеси этанола и изопропанола при температуре от приблизительно 55 до приблизительно 90°C:
охлаждение полученного раствора до температуры ниже приблизительно 20°C со скоростью от приблизительно 0,1 до приблизительно 3°C/минуту и выделение полученного твердого вещества.
Другой вариант осуществления, раскрытый в настоящем описании, относится к кристаллической форме тартрата N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида, рентгенограмма на порошке которой включает пики, соответствующие величинам d в ангстремах, равным приблизительно 17,4, приблизительно 10,2, приблизительно 5,91, приблизительно 4,50, приблизительно 4,37 и приблизительно 3,87. В одном из вариантов осуществления рентгенограмма на порошке включает пики, соответствующие величинам d в ангстремах приблизительно 17,4, приблизительно 10,2, приблизительно 8,8, приблизительно 6,4, приблизительно 5,91, приблизительно 5,46, приблизительно 4,99, приблизительно 4,90, приблизительно 4,62, приблизительно 4,50, приблизительно 4,37, приблизительно 4,20, приблизительно 3,87, приблизительно 3,73, приблизительно 3,58, приблизительно 3,42 и приблизительно 2,90.
Другой вариант осуществления, раскрытый в настоящем описании, относится к кристаллической форме тартрата N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида, рентгенограмма на порошке которой включает пики, соответствующие величинам d в ангстремах, равным приблизительно 12,0, приблизительно 10,7, приблизительно 5,86, приблизительно 4,84, приблизительно 4,70, приблизительно 4,57 и приблизительно 3,77, далее по тексту именуемой формой C. В одном из вариантов осуществления рентгенограмма на порошке включает пики, соответствующие величинам d в ангстремах приблизительно 12,0, приблизительно 10,7, приблизительно 7,4, приблизительно 6,9, приблизительно 6,6, приблизительно 6,2, приблизительно 5,86, приблизительно 5,53, приблизительно 5,28, приблизительно 5,16, приблизительно 4,84, приблизительно 4,70, приблизительно 4,57, приблизительно 4,38, приблизительно 4,09, приблизительно 3,94, приблизительно 3,77, приблизительно 3,71, приблизительно 3,49, приблизительно 3,46, приблизительно 3,25, приблизительно 3,08 и приблизительно 2,93.
Другой вариант осуществления, раскрытый в настоящем описании, относится к способу получения описанных выше кристаллических форм, который включает суспендирование твердой формы соединения формулы (IV) в апротонном растворителе:
и перемешивание суспензии в ходе добавления затравок кристаллов кристаллической формы C, описанной в настоящей заявке. В одном из вариантов осуществления температура растворителя на стадии суспендирования находится в пределах от приблизительно 30 до приблизительно 100°C. В одном из вариантов осуществления апротонный растворитель выбран из группы, включающей один или несколько растворителей из числа следующих: алифатические или циклические простые эфиры, сложные эфиры карбоновых кислот, лактоны, алканы и алифатические C3-C8 кетоны. В одном из вариантов осуществления внесение затравок проводят при температуре от приблизительно 40 до приблизительно 80°C. Один из вариантов осуществления, кроме того, включает охлаждение суспензии со скоростью от приблизительно 0,1 до приблизительно 1°C/минуту. В одном из вариантов осуществления суспензию охлаждают приблизительно до комнатной температуры.
Другой вариант осуществления, раскрытый в настоящем описании, относится к способу получения описанных выше кристаллических форм, который включает суспендирование кристаллической формы тартрата N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида или смесей кристаллических форм тартрата N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида в полярном и апротонном растворителе при температурах от приблизительно 30 до приблизительно 70°C, перемешивание полученной суспензии при внесении затравок кристаллов кристаллической формы C, описанной в настоящей заявке, и выделение кристаллического твердого вещества из суспензии.
Еще один вариант осуществления, раскрытый в настоящем описании, относится к способу получения описанных выше кристаллических форм, который включает растворение тартрата N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида в растворителе при температуре от приблизительно 0 до приблизительно 70°C, перемешивание полученного раствора при температуре от приблизительно 50 до приблизительно 70°C при добавлении затравок кристаллов кристаллической формы C, охлаждение полученной суспензии со скоростью от приблизительно 5 до приблизительно 15°C в час до температуры от приблизительно -20°C до приблизительно комнатной температуры и выделение твердых кристаллов из суспензии. В одном из вариантов осуществления растворителем является тетрагидрофуран. В других вариантах осуществления растворитель выбирают из группы, состоящей из одного или нескольких растворителей из числа ацетона, этанола, изопропанола, дихлорметана, 1,4-диоксана и ацетонитрила.
Другой вариант осуществления, раскрытый в настоящем описании, относится к кристаллической форме тартрата N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида, полученной способом, который включает суспендирование кристаллической формы тартрата N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида или смесей кристаллических форм тартрата N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида в полярном и апротонном растворителе при температурах от приблизительно 30 до приблизительно 70°C, перемешивание полученной суспензии при внесении затравок кристаллов кристаллической формы C и выделение кристаллического твердого вещества из суспензии.
Еще один вариант осуществления, раскрытый в настоящем описании, относится к кристаллической форме тартрата N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида, полученной способом, который включает растворение тартрата N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида в тетрагидрофуране или ацетоне при температурах от приблизительно 0 до приблизительно 70°C, перемешивание полученного раствора при температуре от приблизительно 50 до приблизительно 70°C при внесении затравок кристаллов кристаллической формы C, охлаждение полученной суспензии со скоростью от приблизительно 5 до приблизительно 15°C в час до температуры от приблизительно -20°C до приблизительно комнатной температуры и выделение твердого кристаллического вещества из суспензии.
Другой вариант осуществления, раскрытый в настоящем описании, относится к кристаллической форме тартрата N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида, включающей от приблизительно 0% до приблизительно 6,6% изопропанола или этанола, рентгенограмма на порошке которой содержит пики, соответствующие величинам d в ангстремах, равным приблизительно 17,2, приблизительно 16,0, приблизительно 6,1, приблизительно 4,64, приблизительно 4,54 и приблизительно 4,37. В одном из вариантов осуществления рентгенограмма на порошке включает пики, соответствующие величинам d в ангстремах, равным приблизительно 17,2, приблизительно 16,0, приблизительно 10,7, приблизительно 9,8, приблизительно 6,6, приблизительно 6,1, приблизительно 6,00, приблизительно 5,73, приблизительно 5,33, приблизительно 5,17, приблизительно 4,91, приблизительно 4,64, приблизительно 4,54, приблизительно 4,37, приблизительно 4,10, приблизительно 3,91, приблизительно 3,84, приблизительно 3,67, приблизительно 3,55, приблизительно 3,42, приблизительно 3,32, приблизительно 3,13 и приблизительно 3,06.
Еще один вариант осуществления, раскрытый в настоящем описании, относится к кристаллической форме тартрата N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида, включающей приблизительно 5% трет-бутилметилового эфира, рентгенограмма на порошке которой содержит пики, соответствующие величинам d в ангстремах, равным приблизительно 17,3, приблизительно 16,2, приблизительно 10,6, приблизительно 9,8, приблизительно 8,1, приблизительно 7,5, приблизительно 6,6, приблизительно 6,0, приблизительно 5,28, приблизительно 5,09, приблизительно 4,90, приблизительно 4,72, приблизительно 4,51, приблизительно 4,39, приблизительно 4,26, приблизительно 4,04, приблизительно 3,86, приблизительно 3,70, приблизительно 3,54, приблизительно 3,48 и приблизительно 3,02.
Другой вариант осуществления, раскрытый в настоящем описании, относится к кристаллической форме тартрата N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида, включающей приблизительно 3% тетрагидрофурана, рентгенограмма на порошке которой содержит пики, соответствующие величинам d в ангстремах, равным приблизительно 19,0, приблизительно 16,0, приблизительно 13,0, приблизительно 7,8, приблизительно 6,4, приблизительно 6,2, приблизительно 5,74, приблизительно 5,29, приблизительно 5,04, приблизительно 4,83, приблизительно 4,62, приблизительно 4,50, приблизительно 4,34, приблизительно 4,24, приблизительно 4,05, приблизительно 3,89, приблизительно 3,76, приблизительно 3,58 и приблизительно 3,27.
Другой вариант осуществления, раскрытый в настоящем описании, относится к фармацевтической композиции, содержащей соединение формулы (IV), а также фармацевтически приемлемый носитель или носитель или разбавитель:
Еще один вариант осуществления, раскрытый в настоящем описании, относится к фармацевтическим композициям, которые содержат любую из кристаллических форм, описанных выше, и фармацевтически приемлемый носитель или разбавитель.
Другой вариант осуществления, раскрытый в настоящем описании, относится к способу доставки соединения формулы (I) реципиенту, включающему введение субъекту соединения формулы (IV):
Еще один вариант осуществления, раскрытый в настоящем описании, относится к способу ингибирования активности моноаминового рецептора, включающему введение субъекту соединения формулы (IV):
Другой вариант осуществления, раскрытый в настоящем описании, относится к способу лечения нейропсихиатрических заболеваний, включающему введение субъекту соединения формулы (IV):
В некоторых вариантах осуществления нейропсихиатрическое заболевание выбрано из группы, состоящей из психоза, шизофрении, шизоаффективных расстройств, мании, психотической депрессии, аффективных расстройств, деменции, тревожного состояния, расстройств сна, расстройств аппетита, биполярного расстройства, психоза, вызванного гипертензией, мигрени, спазма сосудов и ишемии, двигательных тиков, тремора, психомоторной заторможенности, брадикинезии и невропатической боли.
Еще один вариант осуществления, раскрытый в настоящей заявке, относится к способу лечения нейродегенеративных заболеваний, включающему введение субъекту соединения формулы (IV). В некоторых вариантах осуществления нейродегенеративное заболевание выбрано из группы, состоящей из болезни Паркинсона, болезни Хантингтона, болезни Альцгеймера, спиноцеребеллярной атрофии, синдрома Туретта, атаксии Фридрайха, болезни Мачадо-Джозефа, деменции с тельцами Леви, дистонии, прогрессирующего супрануклеарного паралича и лобно-височной деменции.
Другой вариант осуществления, раскрытый в настоящей заявке, относится к способу лечения дискинезии, связанной с допаминергической терапией, включающему введение субъекту соединения формулы (IV).
Другой вариант осуществления, раскрытый в настоящей заявке, относится к способу лечения дистонии, миоклонуса или тремора, связанных с допаминергической терапией, включающему введение субъекту соединения формулы (IV).
Другой вариант осуществления, раскрытый в настоящей заявке, относится к способу лечения тромботического состояния, включающему введение субъекту соединения формулы (IV). В некоторых вариантах осуществления тромботическое состояние выбрано из группы, состоящей из инфаркта миокарда, тромботического или ишемического удара, идиопатической и тромботической тромбоцитопенической пурпуры, заболеваний периферических сосудов и болезни Рейно.
Краткое описание чертежей
Фиг.1 представляет собой рентгенограмму на порошке кристаллической формы Y соединения формулы I в виде свободного основания.
Фиг.2 представляет собой рентгенограмму на порошке кристаллической формы A соединения формулы IV.
Фиг.3 представляет собой рентгенограмму на порошке кристаллической формы B соединения формулы IV.
Фиг.4 представляет собой рентгенограмму на порошке кристаллической формы C соединения формулы IV.
Фиг.5 представляет собой рентгенограмму на порошке кристаллической формы D соединения формулы IV.
Фиг.6 представляет собой рентгенограмму на порошке кристаллической формы E соединения формулы IV.
Фиг.7 представляет собой рентгенограмму на порошке кристаллической формы F соединения формулы IV.
Подробное описание предпочтительного варианта осуществления изобретения
Одним полезным N-азациклоалкил-N-аралкилкарбамидом является N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамид формулы (I):
Синтез N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида
Один из вариантов осуществления представляет собой способ синтеза соединения формулы (I), включающий взаимодействие соединения формулы (II), т.е. (4-фторбензил)-(1-метилпиперидин-4-ил)амина
с соединением формулы (III) т.е. 4-(2-метилпропилокси)фенилметилизоцианатом
В одном из вариантов осуществления применяют от приблизительно 0,9 до приблизительно 1,1 эквивалента (4-фторбензил)-(1-метилпиперидин-4-ил)амина на эквивалент 4-(2-метилпропилокси)фенилметилизоцианата. В некоторых вариантах осуществления образовавшееся соединение формулы (I) выделяют из реакционной смеси. В одном из вариантов осуществления после завершения реакции добавляют солеобразующую кислоту. Полученную соль можно выделить с помощью удаления растворителя, осаждения или путем как удаления растворителя, так и осаждения с последующим получением соединения формулы (I) в форме свободного основания при действии водного раствора щелочи, путем растворения в органическом растворителе двухфазной системы и выделения соединения формулы (I) из органического раствора. В предпочтительном варианте осуществления в этой реакции применяют 1,0 эквивалент (4-фторбензил)-(1-метилпиперидин-4-ил)амина на эквивалент 4-(2-метилпропилокси)фенилметилизоцианата. Реакцию можно проводить в присутствии кислот Льюиса в качестве катализаторов, таких как соли металлов или более предпочтительно алкоголяты металлов. Отдельными примерами являются MgCl2, FeCl2, FeCl3, FeBr2, Fe(SO4)2, NiCl2, BCl3, AlCl3, BBr3, TiCl4, TiBr4, ZrCl4, BCl3, Al(O-C1-C4-алкил)3 и Ti(O-C1-C4-алкил)3. Количество катализатора может составлять от приблизительно 0,0001 до приблизительно 5 мас.% и предпочтительно от приблизительно 0,01 до приблизительно 3 мас.% по отношению к массе соединения формулы (II).
Взаимодействие предпочтительно проводят в инертных органических растворителях, таких как простые алифатические эфиры (например, диэтиловый эфир, метилпропиловый эфир, дибутиловый эфир, диметиловый эфир этиленгликоля, тетрагидрофуран или диоксан), сложные эфиры алифатических карбоновых кислот или спиртов (например, C2-C4 алкиловые эфиры уксусной кислоты), лактоны (например, валеролактон), галогенированные углеводороды (например, ди- или трихлорметан, тетрахлорэтан) или алифатические C3-C8 кетоны (например, ацетон, метилпропилкетон, диэтилкетон или метил изо- или трет-бутилкетон).
Температура реакции предпочтительно находится в пределах от приблизительно -30 до приблизительно 60°C и более предпочтительно от приблизительно 5 до приблизительно 30°C. Временем реакции можно управлять путем наблюдения за расходованием соединений формулы (II) или формулы (III), либо оперативно анализируя ход процесса, либо отбирая образцы и анализируя их автономно.
Выделение соединения формулы (I) может осуществляться любым подходящим способом, включая удаление растворителя отгонкой из реакционной смеси при пониженном давлении и более низких температурах, как, например, приблизительно до 100°C, предпочтительно приблизительно до 80°C. Выделение также может проходить путем частичного удаления растворителя для повышения концентрации, отделения загрязнений фильтрованием, осаждения твердого соединения формулы (I) либо за счет дальнейшего концентрирования, либо за счет добавления жидкости, не являющейся растворителем, как, например, алифатических углеводородов (например, пентана, гексана, гептана, октана, циклогексана, метилциклогексана) или воды, фильтрования твердого вещества и высушивания. Выделенное соединение формулы (I) может быть очищено известными способами, такими как перегонка или хроматографические методики.
Было обнаружено, что удаление загрязнений, таких как образовавшиеся побочные продукты, до выделения является удобным путем получения соединения формулы (I), обладающего высокой чистотой. Далее было обнаружено, что очистка может быть значительно улучшена путем получения солей карбамида, которые можно осаждать в виде кристаллических соединений и перекристаллизовывать из растворителей для удаления загрязнений. Затем карбамид формулы (I) получают в виде свободного основания с помощью растворения этой соли в воде, добавления основания и экстракции карбамида органическим растворителем. Перед удалением растворителя путем отгонки, необязательно при пониженном давлении, органические растворы могут быть промыты водой и водным раствором хлорида натрия. В этом способе загрязнения могут быть удалены с помощью осаждения или растворения в воде в случае применения двухфазных систем. Если желательно выпадение соли в осадок для более легкого отделения фильтрацией или центрифугированием, может быть осуществлено частичное удаление органического растворителя и добавление нового растворителя. Подходящими растворителями с низкой способностью растворять соли являются апротонные органические растворители, такие как углеводороды, галогенированные углеводороды, простые эфиры, кетоны, эфиры и лактоны карбоновых кислот, ацетонитрил и спирты, включающие не менее 3 атомов углерода.
Солеобразующие кислоты могут быть выбраны из неорганических и органических кислот, таких как минеральные кислоты (HCl, HBr, HI, H2SO4), моно- или дикарбоновые кислоты (муравьиная кислота, уксусная кислота, щавелевая кислота, малоновая кислота, малеиновая кислота, фумаровая кислота, янтарная кислота, винная кислота) или сульфоновые кислоты (метилсульфоновая кислота). Эти кислоты можно добавлять в виде водных растворов в количествах, достаточных для образования твердого или кристаллического осадка. Указанные количества могут меняться от приблизительно 0,5 до приблизительно 2 эквивалентов по отношению к соединению формулы I, причем это количество зависит главным образом от основности кислоты и ее желаемого избытка для полного и быстрого образования соли.
Соли могут быть растворены в системе, состоящей из воды и неводного смешивающегося органического растворителя, растворяющего соединения формулы (I), который добавлен для того, чтобы растворить высвобождающееся при добавлении основания соединение формулы (I). Подходящие основания включают, не ограничиваясь перечисленным, гидроксиды щелочных металлов, такие как LiOH, NaOH или KOH. В одном из вариантов осуществления значение pH водной фазы превышает приблизительно 8,5. Реакция может завершаться за период времени от нескольких минут до 1 часа. Предпочтительно реакцию останавливают через 5-30 минут. Затем органическую фазу отделяют, необязательно промывают водой и насыщенным раствором соли и/или фильтруют. Желаемый продукт может быть получен с помощью удаления растворителя и высушивания или осаждением с помощью не растворяющей жидкости, фильтрования и высушивания твердого остатка. Соединения формулы (I) получают с высокой чистотой и высокими выходами.
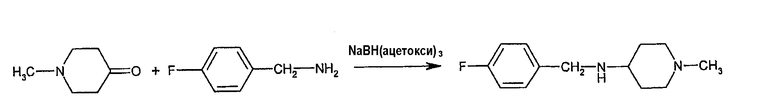
Исходные вещества для описанной выше реакции могут быть получены известными и аналогичными им способами. Конкретно, соединение формулы (II) может быть получено взаимодействием N-метилпиперидин-4-она с 4-фторбензиламином в присутствии гидрида металла, например, согласно следующей схеме:
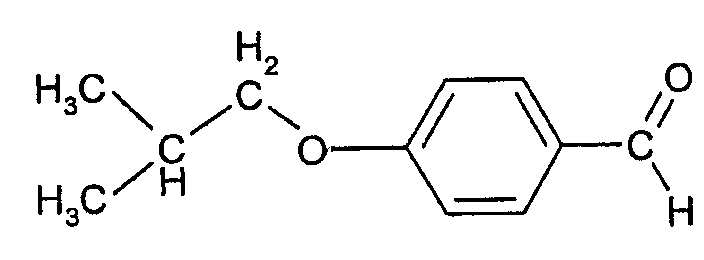
Соединения формулы (III) могут быть получены взаимодействием 4-гидроксибензальдегида с изобутилгалогенидом (например, изобутилбромидом) с образованием 4-изобутоксибензальдегида, который при действии гидроксиламина может быть превращен в форму альдоксима:
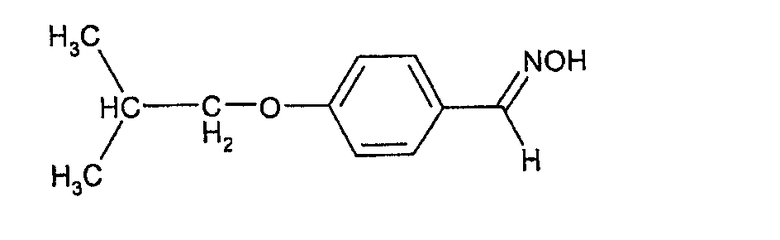
Этот оксим может быть подвергнут каталитическому гидрированию с палладиевым катализатором с образованием соответствующего 4-изобутоксибензиламина, из которого взаимодействием с фосгеном может быть получен изоцианат формулы (III).
Кристаллическая форма N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида (форма Y)
При применении описанного выше способа соединение формулы (I), как правило, образуется в виде в основном аморфного твердого вещества, которое может быть смешано с небольшими количествами кристаллической формы. Неожиданно было обнаружено, что чистая кристаллическая форма может быть получена из солевой формы, такой как гемитартрат, при высвобождении основания в определенных условиях. Эта кристаллизация может быть даже применена для очистки основания путем перекристаллизации солей или перекристаллизации самого основания.
Соответственно, в одном из вариантов осуществления разработана кристаллическая форма N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида, которая обладает характерной температурой плавления, равной приблизительно 124°C (температура пика), определенной с помощью дифференциальной сканирующей калориметрии (DSC), при скорости нагревания, равной 10°C/минуту, называемая далее по тексту формой Y. Энтальпия плавления формы Y составляет приблизительно 99 Дж/г.
Рентгенограмма на порошке формы Y отражена на фиг.1. Конкретно рентгенограмма на порошке данной формы включает следующие характеристические пики, выраженные в величинах d (Е): 13 (vs), 10,9 (vs), 6,8 (vw), 6,5 (s), 6,2 (w), 5,2 (w), 4,7 (m), 4,5 (w), 4,3 (s), 4,22 (vs), 4,00 (m), 3,53 (vw), 3,40 (vw), 3,28 (w), 3,24 (w), 3,19 (w), 3,08 (w), 2,91 (w) и 2,72 (w). Использованные сокращения в круглых скобках означают следующее: (vs) = очень большая интенсивность, (s) = большая интенсивность, (m) = средняя интенсивность, (w) = слабая интенсивность и (vw) = очень слабая интенсивность. В различных вариантах осуществления форма Y присутствует в твердой форме соединения формулы (I) в количествах не менее приблизительно 50%, 70%, 80%, 90%, 95% или 98%, причем остальное количество составляют другие кристаллические формы (включая гидраты и сольваты) и/или аморфные формы.
Форма Y является очень стабильной формой соединения формулы (I) с термодинамической точки зрения. Рентгенограмма и DSC показывают кристаллический характер формы Y, и анализ элементного состава соответствует соединению формулы (I). Кристаллическая форма Y соединения формулы (I) образуется в виде белого порошка.
Соединение формулы (I) растворимо в различных органических растворителях и демонстрирует незначительную растворимость в воде. В противоположность этому соли соединения формулы (I) хорошо растворимы в воде. Эти свойства могут применяться для получения формы Y соединения формулы (I). Например, один способ получения формы Y включает
a) растворение в воде при перемешивании солевой формы соединения формулы (I), предпочтительно гемитартрата;
b) добавление достаточного количества органического апротонного растворителя для растворения образующегося соединения формулы (I);
c) доведение pH водного раствора соли до величины не менее 8,5 добавлением основания;
d) необязательно экстракцию водной фазы органическим растворителем и сбор всех органических фаз;
e) удаление части растворителя и охлаждение оставшегося органического раствора до температуры ниже 15°C;
f) выдерживание при этой температуре с необязательным перемешиванием и
g) отделение осадка фильтрованием, промывание твердого остатка и высушивание его.
Для увеличения выхода маточный раствор можно опять сконцентрировать и охладить. Солеобразующие кислоты могут быть выбраны из неорганических или органических кислот, таких как минеральные кислоты (например, HCl, HBr, HI, H2SO4, H3PO4), моно- и дикарбоновые кислоты (например, муравьиная кислота, уксусная кислота, щавелевая кислота, малоновая кислота, винная кислота, малеиновая кислота, фумаровая кислота, янтарная кислота), сульфоновые кислоты (например, метилсульфоновая кислота), лимонная кислота, глюкуроновая кислота, яблочная кислота, памовая кислота или этан-1,2-дисульфоновая кислота.
Подходящими растворителями являются углеводороды, такие как толуол, галогенированные углеводороды, такие как ди- или трихлорметан, тетрахлорэтан, сложные эфиры алифатических карбоновых кислот и спиртов (C2-C4 алкиловые эфиры уксусной кислоты) (этилацетат), лактоны (валеролактон), простые эфиры (диэтиловый эфир, метилпропиловый эфир, трет-бутилметиловый эфир, дибутиловый эфир, диметиловый эфир), алифатические C4-C8 кетоны (метилпропилкетон, диэтилкетон или метил изо- или трет-бутилкетон). Значение pH на стадии c) может быть успешно доведено, по крайней мере, до 9,5. Подходящие основания включают, не ограничиваясь перечисленными, водные растворы гидроксидов щелочных или щелочноземельных металлов, таких как LiOH, NaOH, KOH или Ca(OH)2.
Удаление части растворителя в основном служит для увеличения концентрации органического раствора, так чтобы он содержал от приблизительно 5 до приблизительно 30 мас.% соединения формулы (I). Температура охлаждения предпочтительно находится в пределах от приблизительно -10 до приблизительно 10°C и наиболее предпочтительно от приблизительно 0 до приблизительно 10°C. Время выдерживания при этой температуре, необязательно при перемешивании, предпочтительно составляет от приблизительно 30 минут до приблизительно 12 часов. Удаление остатка растворителя может осуществляться обычным способом, т.е. в вакууме, в токе инертного газа или обоими способами.
Получение гемитартрата N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида
Соединение формулы (I) обладает низкой растворимостью в воде. Соответственно, в некоторых вариантах осуществления разработаны формы этого соединения, которые растворимы в воде и, следовательно, имеют лучшую биодоступность и улучшенные технологические характеристики для создания препаратов и составов лекарственных композиций. Было обнаружено, что особенно подходящим является гемитартрат N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида. Соответственно, один из вариантов осуществления относится к гемитартрату N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида, представленному формулой (IV),
Получение соединения формулы (IV) может являться составной частью способа синтеза соединения формулы (I), описанного выше, при применении винной кислоты в качестве солеобразующей кислоты. С другой стороны, тартрат может быть получен взаимодействием выделенного соединения формулы (I) с винной кислотой.
В одном из вариантов осуществления гемитартрат N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида получают согласно следующему способу:
a) вводят во взаимодействие от приблизительно 0,9 до приблизительно 1,1 эквивалента (4-фторбензил)-(1-метилпиперидин-4-ил)амина формулы (II)
с 1 эквивалентом 4-(2-метилпропилокси)фенилметилизоцианата формулы (III)
b) добавляют винную кислоту и
c) выделяют гемитартрат соединения формулы (I) из полученной суспензии.
Кроме этого гемитартрат может быть получен путем осаждения при охлаждении, удалении растворителя, добавлении нерастворяющей жидкости или с помощью комбинации перечисленных способов. В одном из вариантов осуществления на стадии b) добавляют один или несколько растворителей, которые обладают низкой растворяющей способностью в отношении гемитартрата, как, например, изопропилацетат, кетон (такой как ацетон или 2-бутанон) и/или тетрагидрофуран. Температура на стадии b) предпочтительно составляет от приблизительно 15 до приблизительно 30°C. Гемитартрат осаждается и образует суспензию, которую можно перемешивать вплоть до 3 дней перед отделением осадка фильтрованием от реакционной смеси, предпочтительно при температуре окружающей среды. Твердый остаток может быть промыт и затем высушен при температуре до 50°C, если желательно, в вакууме.
Гемитартрат формулы (IV) получают с высокими выходами и чистотой. Можно использовать маточный раствор для выделения дополнительного количества гемитартрата формулы (IV) обычным способом. Гемитартрат может быть дополнительно очищен превращением в свободное основание формулы (I) и отделением раствора основания, который затем используют для повторного осаждения гемитартрата добавлением винной кислоты.
Кристаллические формы гемитартрата N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида (формы A-C)
Неожиданно было обнаружено, что соединение формулы (IV) может быть получено в виде нескольких кристаллических форм. Одна из таких твердых кристаллических форм, полученная описанным выше способом, именуется далее по тексту кристаллической формой A. Кристаллическая форма A, как правило, содержит некоторое количество воды, что показано при ее нагревании в ходе термогравиметрического анализа в сочетании с FT инфракрасной спектроскопией или титрованием по Карлу Фишеру. Содержание воды может достигать приблизительно от 2 до 3 мас.%, что, в общем, могло бы соответствовать полугидрату. Однако вода связана достаточно слабо, поэтому потеря массы начинается при температурах несколько выше температуры окружающей среды и завершается приблизительно при 150°C. Кроме того, имеющаяся вода может легко удаляться при действии сухого азота в течение длительного периода времени (приблизительно до 20 часов), и форма A также может существовать в безводном состоянии. DSC показывает, что температура плавления безводной формы A составляет приблизительно 133-135°C (температура пика), при энтальпии плавления приблизительно 70 Дж/г. При действии влаги форма A демонстрирует заметное расплывание, особенно при относительной влажности выше 75%. Если относительная влажность понижается до 50% или менее, вода выделяется обратно. Такое поведение является типичным для твердых веществ, поглощающих влагу. Кристаллическая форма A соединения формулы (IV) хорошо растворима в метаноле, воде или органических растворителях, смешиваемых с водой. Соединение формулы (IV) обладает невысокой растворимостью в других органических растворителях. Кристаллическая форма A при получении описанным выше способом может содержать незначительные количества кристаллической формы C (описанной ниже).
Рентгенограмма на порошке формы A отображена на фиг.2. Конкретно, рентгенограмма содержит следующие характеристические пики, выраженные в величинах d (Е): 18,6 (сильный), 16,7 (очень сильный), 10,2 (сильный), 8,2 (средний), 7,7 (слабый), 7,4 (слабый), 6,5 (слабый), 6,2 (средний), 6,1 (очень сильный), 5,86 (слабый), 5,14 (средний), 5,03 (средний), 4,78 (средний), 4,69 (средний), 4,63 (сильный), 4,49 (сильный), 4,44 (очень сильный), 4,35 (средний), 4,10 (средний), 3,96 (сильный) и 3,66 (средний). В различных вариантах осуществления форма A присутствует в твердой форме соединения формулы (IV) в количествах не менее чем приблизительно 50%, 70%, 80%, 90%, 95% или 98%, причем остаток составляют другие кристаллические формы (включая гидраты и сольваты) и/или аморфные формы.
Кристаллическая форма A может быть получена при управляемой кристаллизации из этанола, необязательно смешанного с изопропанолом. Соответственно, одним из вариантов осуществления является способ получения кристаллической формы A, который включает
a) растворение соединения формулы (IV) в этаноле или смеси этанола и изопропанола при повышенной температуре;
b) медленное охлаждение раствора до температуры менее 20°C и
c) отделение образовавшегося твердого вещества фильтрованием и его высушивание.
В некоторых вариантах осуществления смеси этанола и изопропанола могут содержать до приблизительно 15 и более предпочтительно до 10 объемных процентов изопропанола. Этанол является предпочтительным растворителем. Кроме того, предпочтительно применять высушенный этанол, необязательно в смеси с сухим изопропанолом. В некоторых вариантах осуществления повышенная температура составляет от приблизительно 55 до приблизительно 90°C и предпочтительно от приблизительно 55 до приблизительно 65°C. Смесь перемешивают при повышенной температуре до полного растворения соединения формулы (IV). Медленное охлаждение может означать скорость охлаждения, равную от приблизительно 0,1 до приблизительно 3°C/минуту, предпочтительно от приблизительно 0,2 до приблизительно 2°C/минуту и, в частности, от приблизительно 0,2 до приблизительно 1°C/минуту. Кристаллизация начинается ниже приблизительно 50°C, причем наблюдалось, что в случае перемешивания при этой температуре в течение приблизительно 1 часа может наблюдаться образование густой пасты. Повторное нагревание до более высокой температуры с последующим повторным охлаждением, как правило, приводит к образованию суспензии, которую можно перемешивать при температуре от приблизительно 40 до приблизительно 50°C и также охлаждать далее до температуры менее приблизительно 20°C, предпочтительно от приблизительно 5 до приблизительно 15°C. Скорость охлаждения после перемешивания может быть от приблизительно 0,1 до приблизительно 3°C/минуту и предпочтительно от приблизительно 0,3 до приблизительно 1°C/минуту. Полученное кристаллическое твердое вещество затем отфильтровывают и высушивают просасыванием сухого воздуха через лепешку вещества на фильтре при температурах от приблизительно 25 до менее чем 40°C, предпочтительно приблизительно при 30°C. Для завершения сушки предварительно высушенное твердое вещество можно выдержать в вакууме в течение некоторого времени при температуре окружающей среды или повышенной температуре.
Соединение формулы (IV) может быть превращено полностью в аморфную форму растворением соединения в растворителе, таком как, например, вода и лиофилизацией полученного раствора. Аморфная форма затем может быть использована для получения других полиморфных или псевдополиморфных форм.
В одном из вариантов осуществления другую кристаллическую форму соединения формулы (IV) получают, используя способ установления фазового равновесия, осуществляемого воспроизводимым образом с применением в качестве растворителя этилацетата, ацетона, метилэтилкетона или ацетонитрила. Данная твердая кристаллическая форма далее по тексту именуется кристаллической формой B. Кристаллическая форма B может содержать определенное количество воды, что показано при ее нагревании в ходе термогравиметрического анализа в сочетании с FT инфракрасной спектроскопией или титрованием по Карлу Фишеру. Содержание воды может достигать приблизительно 3,4 мас.%. Это количество указывает на образование в основном моногидрата, стабильного при условиях окружающей среды (теоретическое содержание должно было бы быть 3,5%). Однако вода связана достаточно слабо, поэтому потеря массы наблюдается при температуре окружающей среды и низкой относительной влажности менее чем приблизительно 20%, и форма B также может существовать в безводном состоянии. Температура плавления безводной формы B составляет приблизительно 135°C, и энтальпия плавления составляет около 71 Дж/г. Форма B демонстрирует значительное поглощение воды при действии высокой влажности, особенно при относительной влажности более 80%. Однако ее гигроскопичность является менее выраженной, чем у формы A, причем не было обнаружено расплывания при высокой относительной влажности, составляющей приблизительно 90%.
Рентгенограмма на порошке формы B отображена на фиг.3. Конкретно, рентгенограмма содержит следующие характеристические пики, выраженные в величинах d (Е): 7,4 (очень сильный), 10,2 (сильный), 8,8 (слабый), 6,4 (слабый), 5,91 (очень сильный), 5,46 (слабый), 4,99 (средний), 4,90 (средний), 4,62 (средний), 4,50 (очень сильный), 4,37 (очень сильный), 4,20 (слабый), 3,87 (очень сильный), 3,73 (слабый), 3,58 (средний), 3,42 (слабый) и 2,90 (слабый). В различных вариантах осуществления форма B присутствует в твердой форме соединения формулы (IV) в количествах не менее чем приблизительно 50%, 70%, 80%, 90%, 95% или 98%, причем остаток составляют другие кристаллические формы (включая гидраты и сольваты) и/или аморфные формы.
Кристаллическая форма B может быть получена управляемым образом с помощью различных способов. В одном из вариантов осуществления ее осаждают из растворов в полярных растворителях, таких вода или хлористый метилен, применяя нерастворяющие жидкости, такие как метилэтилкетон, гептан, толуол, ацетонитрил или этилацетат при температурах, равных 0-40°C, с последующим достижением фазового равновесия в основном при комнатной температуре. Другой способ заключается в установлении равновесия суспензий других кристаллических форм, таких как кристаллические формы A и C или их смесей, в таких растворителях, как ацетонитрил, этилацетат, этанол/метилэтилкетон, этанол/ацетон, этилацетат, насыщенный водой, ацетонитрил или этилацетат, содержащий приблизительно 1 объемный процент воды, при температуре от комнатной до приблизительно 40°C, необязательно с циклическим изменением температуры. Установление равновесия суспензий с аморфным веществом соединения формулы (I) при температурах от 0 до приблизительно 45°C, необязательно с применением циклических изменений температуры, является еще одним способом получения формы B. Подходящими растворителями являются гептан, этилацетат, ацетонитрил, метилэтилкетон, этилацетат или трет-бутилметиловый эфир, насыщенные водой, или смесь этилацетат/этанол, содержащая 1 объемный процент воды.
Было обнаружено, что кристаллическая форма A, образующаяся при получении гемитартрата, может содержать определенное количество другой полиморфной формы, причем в ходе дополнительных исследований обнаружилось, что эта полиморфная форма не является ни гидратом, ни сольватом. Эта твердая кристаллическая форма именуется в дальнейшем формой C. Кристаллическая форма C может быть получена при установлении равновесия суспензий кристаллических форм A или B, предпочтительно с добавлением затравок кристаллов формы C. Кристаллическая форма C более стабильна термодинамически и химически, чем формы A или B. Кристаллическая форма C поглощает значительно меньше воды, чем форма A. Поглощение воды при относительной влажности 95% составляет лишь 1%, причем не наблюдается расплывания или гигроскопичности. Действие влажности не приводит к изменению кристаллической формы. Кристаллическая форма C стабильна при относительной влажности 75% в открытой емкости и не поглощает воду вплоть до приблизительно 60°C. Термогравиметрический анализ дает потерю массы ниже 150°C, равную приблизительно 0,9%, которую можно отнести к абсорбированной воде. Исследования способом DSC при скорости нагревания 20°C показывают эндотермический сигнал при 177°C с энтальпией плавления приблизительно 129 Дж/г. Этот сигнал относится к температуре (пиковой) плавления, тогда как первые признаки разложения вещества наблюдаются выше 170°C. Растворимость кристаллической формы C в воде является очень высокой. Кристаллическая форма C в высшей степени подходит в качестве действующего вещества в производстве и рецептурах лекарственных средств.
Рентгенограмма на порошке формы C отображена на фиг.4. Конкретно, рентгенограмма содержит следующие характеристические пики, выраженные в величинах d (Е): 12,0 (слабый), 10,7 (очень сильный), 7,4 (очень слабый), 6,9 (очень слабый), 6,6 (очень слабый), 6,2 (слабый), 5,86 (средний), 5,53 (слабый), 5,28 (средний), 5,16 (средний), 4,84 (очень сильный), 4,70 (средний), 4,57 (сильный), 4,38 (средний), 4,09 (слабый), 3,94 (слабый), 3,77 (сильный), 3,71 (средний), 3,49 (слабый), 3,46 (слабый), 3,25 (слабый), 3,08 (слабый) и 2,93 (слабый). В различных вариантах осуществления форма C присутствует в твердой форме соединения формулы (IV) в количествах не менее чем приблизительно 50%, 70%, 80%, 90%, 95% или 98%, причем остаток составляют другие кристаллические формы (включая гидраты и сольваты) и/или аморфные формы.
В одном из вариантов осуществления разработан способ получения чистой кристаллической формы C в значительных масштабах для промышленного производства этого фармацевтически активного соединения. Было обнаружено, что кристаллизация из нагретых и затем охлажденных растворов не приводит к легкому получению формы C. Далее было обнаружено, что форма C может быть получена управляемым способом, при достижении равновесия суспензиями кристаллических форм A или B в присутствии полярного и апротонного растворителя и добавлении затравок кристаллов формы C. В качестве альтернативы применению затравки кристаллов может применяться исходное вещество, которое после получения соединения формулы (IV) содержит некоторое количество кристаллической формы C. Твердое вещество в суспензии может состоять из кристаллов с размером в диапазоне от 1 до приблизительно 200 и предпочтительно от 2 до 100 мкм, которые могут быть отфильтрованы, промыты и высушены в относительно мягких условиях: например, при 60°C в вакууме. Размер полученных частиц может зависеть от масштаба производства, от примененного растворителя или смеси растворителей, от скорости охлаждения и количества добавленных затравочных кристаллов.
Один из способов получения кристаллической формы C включает образование суспензии твердого соединения формулы (IV) в апротонном растворителе при повышенной температуре и перемешивании этой суспензии необязательно с добавлением затравочных кристаллов формы C, до завершения превращения основного количества вещества в чистую форму C.
Температура, при которой осуществляется описанный выше способ, может быть от 20 до 100°C и предпочтительно от 40 до 80°C. Подходящие растворители для превращения в форму C могут быть выбраны из группы, включающей алифатические или циклические простые эфиры, сложные эфиры карбоновых кислот, лактоны, алканы и алифатические C3-C8 кетоны. Внесение кристаллов формы C предпочтительно выполняют, когда твердая форма частично растворена и образовался насыщенный раствор, в котором суспендирована твердая форма. Внесение затравок проводят предпочтительно в диапазоне температур от 40 до 80°C и более предпочтительно от 55 до 65°C. Время перемешивания суспензии может составлять от 30 минут до нескольких дней и наиболее предпочтительно от 30 минут до 6 часов. Суспензию медленно охлаждают перед выделением твердого осадка фильтрованием или центрифугированием, и скорость охлаждения может составлять от 0,1 до 1°C/минуту. Охлаждение можно проводить до конечной температуры, приблизительно равной комнатной или ниже.
Одним из вариантов осуществления является способ получения кристаллической формы C тартрата N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида формулы (IV), включающий
a) суспендирование аморфной формы или кристаллических форм A, B, D, E или F, или их смеси при перемешивании в полярном и апротонном растворителе при температурах от 30 до 70°C;
b) продолжение перемешивания при температурах от 30 до 70°C и добавление затравочных кристаллов кристаллической формы C, если кристаллическая форма C не присутствует в исходном веществе;
c) продолжение перемешивания при температурах от 30 до 70°C до завершения образования кристаллической формы C;
d) охлаждение до температуры завершения процесса;
e) выделение твердого кристаллического вещества из суспензии и
f) необязательно промывание и затем высушивание твердого кристаллического вещества.
В качестве исходного вещества может применяться кристаллическая форма A, но описанный способ также может реализовываться с формами B, D, E и F или с аморфной формой. Исходное вещество может быть преимущественно высушено перед использованием. Высушивание при 40°C в вакууме является, как правило, достаточным для удаления нежелательных остаточных растворителей (например, спиртов, воды или их смесей), которые оказывают нежелательное влияние на образование формы C. Подходящие растворители для кристаллизации формы C могут быть выбраны из группы, включающей простые эфиры, сложные эфиры карбоновых кислот, лактоны и алифатические кетоны. Отдельными конкретными примерами и предпочтительными растворителями являются диэтиловый эфир, пропилметиловый эфир, трет-бутилметиловый эфир, тетрагидрофуран, этилацетат, трет-бутилметилкетон, ацетон и метилэтилкетон. Наиболее предпочтительными растворителями являются кетоны, причем особенно предпочтителен метилэтилкетон, а также тетрагидрофуран. Количества кристаллических форм A или B в суспензии, которая применяется в качестве исходного вещества, не имеют решающего значения, причем количество исходного вещества выбирается таким образом, чтобы суспензию можно было перемешивать при используемой температуре. Температура на стадии a) предпочтительно приблизительно равна комнатной.
Температура на стадиях b) и c) может изменяться в пределах от 10 до 60°C. Может оказаться предпочтительным применение циклического изменения температуры между более высоким и более низким значением температуры. Количество добавленных затравочных кристаллов может быть от 0,01 до 10 и предпочтительно от 0,1 до 5 мас.% в пересчете на количество кристаллической формы A и/или B. Добавление затравочных кристаллов, как правило, является предпочтительным для ускорения превращения кристаллических форм.
Перемешивание на стадии c) может продолжаться от нескольких часов до нескольких дней, например, от 0,5 часов до 3 дней и предпочтительно от 2 часов до приблизительно 2 дней. Время превращения/конверсии в основном зависит от масштаба синтеза, температуры, примененного растворителя, интенсивности перемешивания и количества затравочных кристаллов, добавленных в суспензию. Время превращения можно контролировать, наблюдая за соотношением исчезающей кристаллической формы и образующейся формы C, либо с помощью оперативного способа анализа, либо путем отбора образцов и анализа в автономном режиме.
Выделение твердого кристаллического вещества можно проводить центрифугированием или фильтрованием. Полученный продукт может быть промыт, например, растворителем и затем высушен в атмосфере сухого инертного газа, который можно прокачивать через лепешку отфильтрованного вещества, необязательно в вакууме, или с применением вакуума в течение времени, достаточного для удаления растворителей. Может применяться дополнительная сушка, или в вакууме и/или при умеренно повышенных температурах вплоть до приблизительно 80°C. Можно отметить, что форма C проявляет отличные свойства с точки зрения фильтрования и высушивания, и образуется твердое вещество, которое в основном не содержит остаточного растворителя; т.е. его количество менее 1000 частей на миллион, предпочтительно менее 200 частей на миллион.
Неожиданно было обнаружено, что кристаллическая форма C также может быть получена путем кристаллизации соединения формулы (IV) из раствора в подобранном растворителе и внесения затравочных кристаллов формы C при повышенной температуре. Соответственно, в одном из вариантов осуществления разработан способ получения кристаллической формы C, включающий
a) растворение аморфной формы или кристаллических форм A, B, D, E или F или их смесей при перемешивании в подходящем растворителе при температурах от 0 до 70°C;
b) продолжение перемешивания и добавление затравочных кристаллов кристаллической формы C к раствору при повышенной температуре; предпочтительно от приблизительно 50 до 70°C и наиболее предпочтительно от 55 до 65°C;
c) продолжение перемешивания образующейся суспензии при той же температуре в течение периода времени, достаточного для превращения соединения формулы (IV) в кристаллическую форму C;
d) охлаждение полученной суспензии со скоростью от 5 до 15°C в час до температуры от -20°C до комнатной и предпочтительно от 0 до 25°C;
e) выделение твердого кристаллического вещества из суспензии и
f) необязательно промывание и затем высушивание твердого кристаллического вещества.
Количество соединения формулы IV на стадии a) выбирают таким образом, чтобы получить концентрированный раствор. Концентрация, которая может быть достигнута, зависит от примененного растворителя или смеси растворителей, а также от растворимости исходного вещества. В качестве растворителей предпочтительны тетрагидрофуран и смеси, содержащие тетрагидрофуран, поскольку обычно при температуре кипения с обратным холодильником может быть растворено приблизительно 200 мг/мл формы A. Однако при растворении исходного вещества может быть применен любой подходящий растворитель. Неограничивающие примеры включают тетрагидрофуран, ацетон, этанол, изопропанол, дихлорметан, 1,4-диоксан и ацетонитрил. Температура на стадии a) предпочтительно составляет от 40 до 70°C. Количество добавленной затравки кристаллов на стадии b) может составлять от 0,1 до 15 мас.% и предпочтительно от 2 до 10 мас.% по отношению к количеству растворенного соединения формулы (IV). Время перемешивания на стадии c) зависит от масштаба синтеза и может меняться от приблизительно 20 минут до приблизительно 24 часов, более предпочтительно от 25 минут до 12 часов и наиболее предпочтительно от 3 минут до 6 часов. Скорость охлаждения на стадии d) предпочтительно составляет от 8 до 12°C в час. Перемешивание может быть продолжено после охлаждения в диапазоне охлаждающих температур в течение периода до 24, предпочтительно до 18 часов и более предпочтительно 14 часов.
Кристаллическая форма C может быть получена с высокой полиморфной чистотой. Вещество, полученное описанными выше способами, может содержать остаточное исходное вещество, например, в количествах до 20 или до 10 мас.% по отношению к кристаллической форме C. Эти смеси также очень хорошо подходят для рецептур лекарственных средств.
Сольваты гемитартрата N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида (Формы D-F)
В некоторых вариантах осуществления соединение формулы (IV) может образовывать различные сольваты с некоторыми растворителями. Псевдополиморфные формы могут применяться в составах лекарственных средств или для получения других полиморфных форм. В некоторых вариантах осуществления эти сольваты могут существовать либо в виде сольватированных форм, т.е. содержащих значительное количество соответствующего растворителя, либо в виде соответствующих несольватированных форм, т.е. форм, не содержащих растворителя, в которых в основном сохранена кристаллическая структура.
Один из таких сольватов образуется при достижении равновесия суспензии кристаллической формы или аморфной формы соединения формулы (IV) в изопропаноле. После высушивания в атмосфере азота в течение приблизительно 30 минут полученный сольват содержит приблизительно 6,0-6,6 мас.% изопропанола. Теоретическое значение для гемисольвата изопропанола составляет 5,6% содержания изопропанола, и был сделан вывод об образовании гемисольвата. Гемисольват с изопропанолом является стабильным при действии относительной влажности 53% в открытой емкости. Эта форма называется в тексте заявки кристаллической формой D.
Рентгенограмма на порошке формы D отображена на фиг.5. Конкретно, рентгенограмма содержит следующие характеристические пики, выраженные в величинах d (Е): 17,2 (сильный), 16,0 (средний), 10,7 (очень слабый), 9,8 (слабый), 6,6 (средний), 6,1 (сильный), 6,00 (средний), 5,73 (слабый), 5,33 (слабый), 5,17 (средний), 4,91 (средний), 4,64 (сильный), 4,54 (очень сильный), 4,37 (очень сильный), 4,10 (средний), 3,91 (средний), 3,84 (средний), 3,67 (слабый), 3,55 (средний), 3,42 (средний), 3,32 (слабый), 3,13 (слабый) и 3,06 (средний). В различных вариантах осуществления форма D присутствует в твердой форме соединения формулы (IV) в количествах не менее чем приблизительно 50%, 70%, 80%, 90%, 95% или 98%, причем остаток составляют другие кристаллические формы (включая гидраты и сольваты) и/или аморфные формы.
Далее было обнаружено, что может быть получен сольват с трет-бутилметиловым эфиром (TBME), если аморфную форму соединения формулы (IV) довести до фазового равновесия в TBME при температуре окружающей среды. Содержание TBME составляет приблизительно 5 мас.% по отношению к соединению формулы (IV) согласно результатам термогравиметрии при скорости нагревания 10°C. Эта форма именуется в тексте заявки кристаллической формой E.
Рентгенограмма на порошке формы E отображена на фиг.6. Конкретно, рентгенограмма содержит следующие характеристические пики, выраженные в величинах d (Е): 17,3 (очень сильный), 16,2 (средний), 10,6 (средний), 9,8 (средний), 8,1 (слабый), 7,5 (слабый), 6,6 (средний), 6,0 (очень сильный), 5,28 (средний), 5,09 (сильный), 4,90 (средний), 4,72 (очень сильный), 4,51 (средний), 4,39 (сильный), 4,26 (сильный), 4,04 (средний), 3,86 (слабый), 3,70 (слабый), 3,54 (средний), 3,48 (средний) и 3,02 (слабый). В различных вариантах осуществления форма E присутствует в твердой форме соединения формулы (IV) в количествах не менее чем приблизительно 50%, 70%, 80%, 90%, 95% или 98%, причем остаток составляют другие кристаллические формы (включая гидраты и сольваты) и/или аморфные формы.
Кроме этого было обнаружено, что кристаллизация соединения формулы (IV) из раствора в тетрагидрофуране (ТГФ) приводит к нестехиометрическому сольвату с ТГФ, который содержит от 0 до приблизительно 3% ТГФ по отношению к соединению формулы (IV), что было установлено по данным термогравиметрии при скорости нагревания 10°C. Выделение растворителя начинается при температуре выше температуры окружающей среды и заканчивается приблизительно 130°C. Эта форма называется в настоящей заявке кристаллической формой F.
Рентгенограмма на порошке формы F отображена на фиг.7. Конкретно, рентгенограмма содержит следующие характеристические пики, выраженные в величинах d (Е): 19,0 (слабый), 16,0 (средний), 13,0 (средний), 7,8 (слабый), 6,4 (средний), 6,2 (средний), 5,74 (слабый), 5,29 (слабый), 5,04 (средний), 4,83 (средний), 4,62 (средний), 4,50 (средний), 4,34 (средний), 4,24 (очень сильный), 4,05 (средний), 3,89 (средний), 3,76 (средний), 3,58 (слабый) и 3,27 (средний). В различных вариантах осуществления форма F присутствует в твердой форме соединения формулы (IV) в количествах не менее чем приблизительно 50%, 70%, 80%, 90%, 95% или 98%, причем остаток составляют другие кристаллические формы (включая гидраты и сольваты) и/или аморфные формы.
Стабильность и фармацевтические композиции
Как упоминалось выше, соединение формулы (IV) особенно хорошо подходит в качестве действующего вещества или пролекарства в фармацевтических композициях для ингибирования активности моноаминового рецептора, предпочтительно рецептора серотонина подкласса 5-HT2A. Соединение формулы (IV) обладает очень хорошей растворимостью в водных системах, и свободное основание высвобождается при физиологических значениях pH, обеспечивая высокую биодоступность. Кроме этого соединение формулы (IV) обладает высокой устойчивостью при хранении.
Было найдено, что кристаллическая форма C является наиболее стабильной из всех обнаруженных кристаллических форм. Кроме этого было найдено, что кристаллические формы A и B стабильны при температурах окружающей среды, стабильны в присутствии формы C и способны сосуществовать с кристаллической формой C. Кристаллические формы A, B и особенно C подходят для широкого круга композиций различных типов, даже в присутствии влагосодержащих компонентов. Эти новые кристаллические формы A, B и особенно C обеспечивают некоторые преимущества для производства, например, они хорошо перерабатываются благодаря удобному размеру и морфологии кристаллов, обладают очень хорошей стабильностью в условиях производства композиций различных типов, устойчивостью при хранении, высокой растворимостью и высокой биодоступностью. Кристаллические формы D, E и F также могут применяться для фармацевтических композиций.
Форма C является химически очень стабильной и может быть легко включена в состав таблеток или любой другой фармацевтически приемлемой дозированной формы. Несмотря на высокую термическую устойчивость, она, тем не менее, проявляет благоприятные свойства с точки зрения растворимости, так что ее растворимость в воде составляет более чем 50-100 мг/мл. Формы A, B, D и F демонстрируют высокую растворимость в воде, составляющую более 200 мг/мл. Растворимость всех форм будет зависеть от pH водной среды.
Формы A и B демонстрируют значительную устойчивость при парциальных давлениях паров воды, соответствующих условиям окружающей среды (т.е. при относительной влажности от 20% до 75%). Кроме того, формы A и B очень удобны для фармацевтической переработки в водных средах, например, для гранулирования с водой или со смесями вода-растворитель.
Соответственно, некоторые варианты осуществления относятся к фармацевтическим композициям, содержащим соединение формулы (IV) и фармацевтически приемлемый носитель или разбавитель. В некоторых вариантах осуществления соединение формулы (IV) выбрано из группы кристаллических форм A, B и C.
Количество требуемого соединения формулы (IV) существенно зависит от типа композиций и желаемых дозировок за период введения. Количество в композиции для орального введения может быть от 0,1 до 500 мг, предпочтительно от 0,5 до 300 мг и более предпочтительно от 1 до 100 мг. Пероральные композиции могут быть твердыми композициями, такими как капсулы, таблетки, пилюли и пастилки, или жидкими, такими как водные суспензии, эликсиры и сиропы. Под твердыми и жидкими композициями понимается также включение соединения формулы (IV) в жидкую или твердую пищу. Кроме того, жидкости включают растворы соединения формулы (IV) для парентеральных применений, таких как инфузия или инъекция.
Описанные выше кристаллические формы могут непосредственно применяться как порошки (например, тонкоизмельченные частицы), гранулы, суспензии или растворы, или они могут быть объединены совместно с другими фармацевтически приемлемыми ингредиентами в смеси компонентов необязательно тонкодисперсных компонентов, которая затем служит для наполнения капсул, изготовленных, например, из твердого или мягкого желатина, прессования таблеток, пилюль или пастилок, или же суспендирования или растворения в носителях для суспензий, эликсиров и сиропов. После прессования могут наноситься покрытия для формирования пилюль.
Фармацевтически приемлемые ингредиенты хорошо известны для различных типов композиций, и они могут представлять собой, например, связующие вещества, такие как природные или синтетические полимеры, эксципиенты, лубриканты, ПАВ, подсластители и вкусоароматические средства, материалы для покрытия, консерванты, красители, загустители, вспомогательные вещества, противомикробные средства, антиоксиданты и носители для различных типов композиций.
Примерами связующих веществ являются трагакантовая камедь, аравийская камедь, крахмал, желатин и биоразрушаемые полимеры, такие как гомо- и сополиэфиры дикарбоновых кислот, алкиленгликолей, полиалкиленгликолей и/или алифатических гидроксикарбоновых кислот; гомо- и сополиамиды дикарбоновых кислот, алкилендиаминов и/или алифатических аминокарбоновых кислот; соответствующие полиэфир-полиамидные сополимеры, полиангидриды, полиортоэфиры, полифосфазены и поликарбонаты. Биоразрушаемые полимеры могут быть линейными, разветвленными или поперечно-сшитыми. Конкретными примерами являются полигликолевая кислота, полимолочная кислота и поли-d,l-лактид/гликолид. Другими примерами полимеров являются водорастворимые полимеры, такие как полиоксаалкилены (например, полиоксаэтилен, полиоксапропилен и их смешанные полимеры), полиакриламиды и гидроксиалкилированные полиакриламиды, полияблочная кислота и ее эфиры или амиды, полиакриловая кислота и ее эфиры или амиды, поливиниловый спирт и его сложные или простые эфиры, поливинилимидазол, поливинилпирролидон, а также природные полимеры, как, например, хитозан.
Примерами эксципиентов являются фосфаты, такие как дикальцийфосфат.
Примерами лубрикантов являются природные или синтетические масла, жиры, воски или соли жирных кислот, как, например, стеарат магния.
Поверхностно-активные вещества могут быть анионными, катионными, амфотерными или нейтральными. Примерами ПАВ являются лецитин, фосфолипиды, октилсульфат, децилсульфат, додецилсульфат, тетрадецилсульфат, гексадецилсульфат и октадецилсульфат, олеат Na или капрат Na, 1-ациламиноэтан-2-сульфоновые кислоты, такие как 1-октаноиламиноэтан-2-сульфоновая кислота, 1-деканоиламиноэтан-2-сульфоновая кислота, 1-додеканоиламиноэтан-2-сульфоновая кислота, 1-тетрадеканоиламиноэтан-2-сульфоновая кислота, 1-гексадеканоиламиноэтан-2-сульфоновая кислота и 1-октадеканоиламиноэтан-2-сульфоновая кислота, а также таурохолевая кислота и тауродезоксихолевая кислота, желчные кислоты и их соли, такие как холевая кислота, дезоксихолевая кислота и гликохолаты натрия, капрат натрия или лаурат натрия, олеат натрия, лаурилсульфат натрия, цетилсульфат натрия, сульфатированное касторовое масло и диоктилсульфосукцинат натрия, кокамидопропилбетаин и лаурилбетаин, жирные спирты, холестерины, моно- или дистеараты глицерина, моно- или диолеаты глицерина, моно- или дипальмитаты глицерина и полиоксиэтиленстеарат.
Примерами подслащивающих средств являются сахароза, фруктоза, лактоза или аспартам.
Примерами вкусоароматических средств являются мята перечная, масло винтергрина или фруктовые ароматизаторы, такие как вишневые или апельсиновые ароматизаторы.
Примерами материалов для покрытия являются желатин, воск, шеллак, сахар или биоразрушаемые полимеры.
Примерами консервантов являются метил или пропилпарабены, сорбиновая кислота, хлорбутанол, фенол и тимеросал.
Примерами вспомогательных веществ являются ароматические добавки.
Примерами загустителей являются синтетические полимеры, жирные кислоты и соли жирных кислот, а также сложные эфиры и жирные спирты.
Примерами антиоксидантов являются витамины, такие как витамин A, витамин C, витамин D или витамин E, экстракты овощей или рыбий жир.
Примерами жидких носителей является вода, спирты, такие как этанол, глицерин, пропиленгликоль, жидкие полиэтиленгликоли, триацетин и масла. Примерами твердых носителей являются тальк, глина, микрокристаллическая целлюлоза, оксид кремния, оксид алюминия и т.п.
Фармацевтическая композиция по настоящему изобретению также может содержать изотонические средства, такие как сахара, буферные составы или хлорид натрия.
Кроме того, соединение формулы (IV) по настоящему изобретению может быть включено в состав шипучих таблеток или порошков, которые разлагаются в водной среде, образуя раствор, пригодный для питья.
Сироп или эликсир могут содержать соединение формулы (IV), сахарозу или фруктозу в качестве подсластителя, консервант, как, например, метилпарабен, краситель и вкусоароматическое средство.
Из соединений формулы (IV) по настоящему изобретению также могут быть получены композиции с медленным высвобождением, для достижения контролируемого высвобождения действующего агента при соприкосновении с жидкими средами организма в желудочно-кишечном тракте, и для обеспечения значительного, постоянного и эффективного уровня действующего агента в плазме крови. С этой целью соединение формулы (IV) может быть внедрено в полимерную матрицу, состоящую из биологически разрушаемого полимера, водорастворимого полимера или полимеров обоих типов и, необязательно, подходящих ПАВ. В данном контексте внедрение может означать включение микрочастиц в матрицу полимеров. Композиции с контролируемым высвобождением, кроме того, получают путем инкапсуляции диспергированных микрочастиц или эмульгированных микрокапель, с помощью известных методик покрытия частиц дисперсий или эмульсий.
Соединение формулы (IV) по настоящему изобретению также применимо для введения комбинации терапевтически активных средств животным. Такая комбинированная терапия может быть проведена при использовании, как минимум, одного дополнительного терапевтического средства, которое может быть дополнительно диспергировано или растворено в композиции. Соединение формулы (IV) по настоящему изобретению и, соответственно, композиции на его основе также могут вводиться в комбинации с другими терапевтическими средствами, которые эффективны для лечения данного состояния с целью осуществления комбинированной терапии.
Кристаллические формы и фармацевтические композиции, описанные в настоящей заявке, хорошо подходят для эффективного лечения нейропсихиатрических заболеваний, включая психоз, аффективные расстройства, деменцию, невропатическую боль и гипертензию.
Один из вариантов осуществления представляет собой способ доставки соединения формулы (I) реципиенту, включающий введение субъекту эффективного количества соединения формулы (IV), как, например, кристаллических форм A, B и C. Еще один вариант осуществления представляет собой применение соединения формулы (IV) для производства лекарственного средства, применимого для ингибирования активности моноаминового рецептора, предпочтительно рецептора серотонина подкласса 5-HT2A.
Один из вариантов осуществления представляет собой способ лечения нейропсихиатрических заболеваний, выбранных из группы, состоящей из психоза, шизофрении, шизоаффективных расстройств, мании, психотической депрессии, аффективных расстройств, деменции, тревожного состояния, расстройств сна, расстройств аппетита, биполярного расстройства, психоза, вызванного гипертензией, мигрени, спазма сосудов и ишемии, двигательных тиков, тремора, психомоторной заторможенности, брадикинезии и невропатической боли, путем введения соединения формулы (IV).
Другой вариант осуществления представляет собой способ лечения нейродегенеративных заболеваний, включая болезнь Паркинсона, болезнь Хантингтона, болезнь Альцгеймера, спиноцеребеллярную атрофию, синдром Туретта, атаксию Фридрайха, болезнь Мачадо-Джозефа, деменцию с тельцами Леви, дистонию, прогрессирующий супрануклеарный паралич и лобно-височную деменцию, путем введения соединения формулы (IV).
Еще один вариант осуществления представляет собой способ лечения дискинезии, связанной с допаминергической терапией, путем введения соединения формулы (IV).
Другой вариант осуществления представляет собой способ лечения дистонии, миоклонуса или тремора, связанных с допаминергической терапией, путем введения соединения формулы (IV).
Еще один вариант осуществления представляет собой способ лечения тромботических состояний, включая инфаркт миокарда, тромботический или ишемический удар, идиопатическую и тромботическую тромбоцитопеническую пурпуру, заболевание периферических сосудов и болезнь Рейно, путем введения соединения формулы (IV).
Другим вариантом осуществления является способ лечения зависимостей, включая алкогольную зависимость, опиоидную наркоманию, никотиновую зависимость, путем введения соединения формулы (IV).
Еще одним вариантом осуществления является способ лечения снижения либидо или проблем с эякуляцией, путем введения соединения формулы (IV).
ПРИМЕРЫ
Экспериментальные методики
Рентгенограмма на порошках (PXRD): PXRD проводили на порошковом рентгеновском дифрактометре Philips 1710, используя излучение CuKα. Межплоскостные расстояния d рассчитывали из значений 2θ, используя длину волны 1,54060 Е. Как правило, значения 2θ получали с ошибкой ±0,1-0,2°. Следовательно, экспериментальная ошибка в межплоскостных расстояниях d зависела от положения пика.
Дифференциальная сканирующая калориметрия (DSC): прибор Perkin Elmer DSC 7 в закрытом золотом резервуаре для образцов в атмосфере азота для определения характеристик формы A и закрытом резервуаре при относительной влажности 50% для определения характеристик формы B. Скорость нагревания 10 K/мин. Все температуры плавления получали из пиковых температур измерений DSC, а не температур начала плавления.
FT-спектроскопия комбинационного рассеяния: прибор Bruker RFS 100. Nd:YAG возбуждение 1064 нм, мощность лазера 100 мВт, Ge-детектор, 64 прохождения в диапазоне 25-3500 см-1, разрешение 2 см-1.
TG-FTIR: термогравиметрические измерения проводили с помощью прибора Netzsch Thermo-Microbalance TG 209, связанного с FTIR спектрометром Brucker Vector 22 (резервуары для образцов с микроотверстием, атмосфера азота, скорость нагревания 10 K/мин).
ВЭЖХ: измерения ВЭЖХ проводили с помощью прибора HP LC1090M, колонка Symmetry C18, 3,0·150 мм.
Пример 1
Получение N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида
a) Получение
К раствору N-метилпиперид-4-она (3,17 кг) и 4-фторбензиламина (3,50 кг) в метаноле (30 л) в течение 1,5 ч добавляли триацетоксиборгидрид (6,5 кг), поддерживая температуру ниже 27°C. Реакционную смесь перемешивали в течение 15 ч при 22°C. Количество не вступившего в реакцию амина контролировали гель-хроматографией (4-фторбензиламин: <5%). В течение 75 минут (мин) добавляли раствор 30% гидроксида натрия (12,1 кг) в воде (13,6 кг), поддерживая температуру ниже 20°C. Отгоняли метанол до объема остатка 26 литров. Добавляли этилацетат (26 л), раствор перемешивали в течение 15 мин, давали фазам разделиться в течение 15 минут и удаляли нижнюю водную фазу. Из органической фазы при пониженном давлении при 73-127°C отгоняли этилацетат. На этой стадии остаток смешивали со второй неочищенной порцией, полученной по описанному способу. Затем объединенные продукты перегоняли при 139-140°C/20 мбар, получая 11,2 кг продукта (>82%).
b) Получение
4-гидроксибензальдегид (4,0 кг) и этанол (20 л) добавляли к раствору изобутилбромида (9,0 кг) в этаноле (15 л). Добавляли карбонат калия (13,6 кг) и суспензию кипятили с обратным холодильником (74-78°C) в течение 5 дней. Количество не вступившего в реакцию 4-гидроксибензальдегида контролировали с помощью ВЭЖХ (<10%). Суспензию охлаждали до 20°C и использовали в следующей стадии.
c) Получение
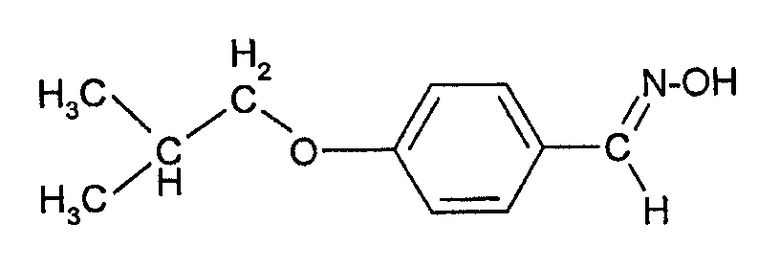
К продукту предыдущей стадии b) (174 л, 176 кг) добавляли гидроксиламин (50% раствор в воде, 8,7 кг) и этанол (54 л). Суспензию кипятили с обратным холодильником (77°C) в течение 3 ч. Не вступившие в реакцию остатки исходных веществ контролировали с помощью ВЭЖХ (<5%). Суспензию охлаждали до 30°C, фильтровали и промывали фильтр этанолом (54 л). Раствор концентрировали отгонкой растворителя при пониженном давлении при 30°C до остаточного объема 67 литров. Раствор охлаждали до 25°C и добавляли воду (110 л). Суспензию концентрировали отгонкой жидкости при пониженном давлении при 30°C до остаточного объема 102 литра. Добавляли петролейный эфир (фракция 60-90, 96 л) и смесь нагревали до кипения с обратным холодильником (70°C). Раствор охлаждали до 40°C и инициировали кристаллизацию внесением затравок кристаллов. Суспензию охлаждали до 5°C и перемешивали в течение 4 ч. Продукт центрифугировали и промывали полученную лепешку осадка петролейным эфиром (фракция 60-90, 32 л). Влажную лепешку высушивали при приблизительно 40°C, получая 16 кг продукта (63%).
d) Получение
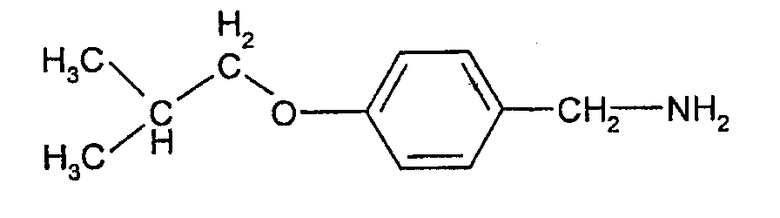
Продукт предыдущей стадии c) (15,7 кг) растворяли в этаноле (123 л). Добавляли уксусную кислоту (8,2 кг) и влажный 5% палладий-на-угле (1,1 кг). Оксим гидрировали при 22°C и давлении 1,5 бар в течение 4 ч. Полноту вступления оксима в реакцию контролировали с помощью ВЭЖХ. Катализатор отделяли фильтрованием и отгоняли растворитель при пониженном давлении при 36°C до конечного объема 31 л. Добавляли этилацетат (63 л) и смесь кипятили с обратным холодильником (75°C) до растворения. Раствор охлаждали до 45°C и инициировали кристаллизацию внесением затравок. Суспензию охлаждали до 6-10°C и перемешивали 2,5 часа. Продукт центрифугировали и лепешку осадка промывали 2 порциями этилацетата (2×0,8 л). Влажную лепешку высушивали при температуре приблизительно 40°C, получая 8 кг (41%) продукта.
e) Получение
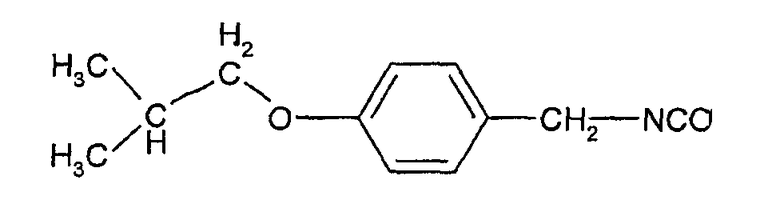
Водный раствор гидроксида натрия (30%, 5,0 кг) добавляли к суспензии продукта предыдущей стадии d) (7,9 кг) в гептане (41 л). Раствор нагревали до 47°C, перемешивали в течение 15 минут и давали расслоиться в течение 15 минут. Проверяли значение pH (pH>12) и отделяли водную фазу. Растворитель удаляли отгонкой при пониженном давлении при 47-65°C. Добавляли гептан (15 л) и затем удаляли отгонкой при пониженном давлении при 58-65°C. Вновь добавляли гептан (7 л), раствор фильтровали и фильтр промывали гептаном (7 л). Растворитель удаляли отгонкой при пониженном давлении при 28-60°C. Добавляли тетрагидрофуран (ТГФ, 107 л) и триэтиламин (ТЭА, 6,8 кг) и фиксировали температуру на уровне 22°C. В другом реакторе в тетрагидрофуран (88 л), предварительно охлажденный до -3°C, вводили фосген (5,0 кг). Раствор, содержащий ТГФ и ТЭА, добавляли к раствору фосгена в течение 3 ч 50 мин, поддерживая температуру на уровне -3°C. Реактор промывали тетрагидрофураном (22 л). Смесь перемешивали в течение 45 мин при 20°C и затем в течение 90 мин при кипячении с обратным холодильником (65°C). Растворитель отгоняли при пониженном давлении при 25-30°C до остаточного объема 149 л. Проверяли отсутствие фосгена. На этой стадии фосген по-прежнему присутствовал, и суспензию дегазировали, барботируя через нее азот. После этой операции содержание фосгена над раствором становилось менее 0,075 частей на миллион. Суспензию фильтровали и промывали тетрагидрофураном (30 л). Растворитель отгоняли при пониженном давлении при 20-25°C до остаточного объема 40 л. Добавляли тетрагидрофуран (51 л) и отгоняли растворитель при пониженном давлении при 20-25°C до остаточного объема 40 л. Конечный объем доводили до приблизительно 52 литров добавлением тетрагидрофурана (11 л). Раствор анализировали и использовали в следующей стадии.
f) Получение указанного в заглавии соединения формулы (I)
Продукт предыдущей стадии e) (51 л) в течение 1 ч добавляли к раствору продукта стадии a) (7,3 кг) в тетрагидрофуране (132 л) при 17°C. Установку промывали тетрагидрофураном (12 л) и смесь перемешивали в течение 15 ч. Остаток не вступившего в реакцию продукта первой стадии контролировали с помощью ВЭЖХ. Растворитель удаляли отгонкой при пониженном давлении при 20-38°C до остаточного объема 165 л. Добавляли древесный уголь (Norit SX1-G, 0,7 кг), смесь перемешивали в течение 15 мин и фильтровали. Установку промывали тетрагидрофураном (7 л), и удаляли растворитель отгонкой при пониженном давлении при 20-25°C до остаточного объема 30 л. Для получения раствора указанного в заглавии соединения формулы (I) добавляли изопропилацетат (96 л), который содержал небольшое количество примесей (в основном, побочных продуктов предыдущих стадий). Удаление растворителя из образца приводило к получению в основном аморфного твердого вещества.
Этот раствор, содержащий неочищенный продукт, применяли для непосредственного получения гемитартрата и одновременно для очистки свободного основания через стадию образования гемитартрата путем перекристаллизации из подходящих растворителей.
Пример 2
Получение чистой кристаллической формы Y соединения формулы I
15,78 г тартрата, полученного по методике примера 10, описанной ниже, растворяли в 130 мл воды. Добавляли 500 мл TBME и доводили pH до 9,8, добавляя 2н раствор NaOH. После осаждения белого твердого вещества водную фазу 5 раз экстрагировали 500 мл TBME. Органические фазы концентрировали до остаточного объема приблизительно 400 мл. Раствор выдерживали при 6°C. Осадок отфильтровывали, промывали TBME и, наконец, высушивали в вакууме в течение 5 часов. Выход: 8,24 г белого порошка. Материнский раствор концентрировали до четвертой части объема и выдерживали при 6°C. Осадок отфильтровывали и высушивали в вакууме в течение 18 часов. Выход: 1,6 г белого порошка.
PXRD выявил кристаллическую структуру образца. Рентгенограмма на порошке показана на фиг.1, и характеристические пики в 2 тета с соответствующими величинами межплоскостных расстояний d в Е показаны в таблице 1. Спектроскопия комбинационного рассеяния также указала на кристаллический характер образца. В спектре комбинационного рассеяния пики винной кислоты обнаружены не были. Анализ TG-FITR выявил потерю массы около 0,4% от 60 до 150°C, вызванную, вероятно, выделением TBME. Выше приблизительно 190°C начиналось разложение образца. DSC (-50°C до 210°C 10°C/мин) выявила поглощение тепла при плавлении при 124°C.
Межплоскостные расстояния для кристаллической формы Y (свободное основание)
Была определена приблизительная растворимость свободного основания при комнатной температуре для 11 растворителей, причем значения приведены в таблице 2.
Приблизительная растворимость кристаллического свободного основания формулы (I) в различных растворителях
Пример 3
Получение гемитартрата формулы (IV) из раствора,
полученного в примере 1(f)
a) Получение неочищенной соли
К раствору соединения формулы (I) в изопропилацетате (96 л) по примеру 1(f) при 23°C добавляли предварительно полученный раствор винной кислоты (1,7 кг) в воде (1,7 л) и тетрагидрофуран (23 л). Полученную суспензию перемешивали в течение 2,5 дней при 22°C. Неочищенный тартрат центрифугировали и промывали лепешку осадка 4 порциями изопропилацетата (4×23 л). Маточные растворы общей массой 107 кг сохраняли для дальнейшего использования при получении тартрата. Влажную лепешку высушивали приблизительно при 40°C, получая 8,3 кг (50%) продукта.
b) Очистка
Неочищенный тартрат, полученный на предыдущей стадии a) (8,1 кг), растворяли в деминерализованной воде (41 л) при 22°C. Добавляли изопропилацетат (40 л), 30% водный раствор гидроксида натрия (4,3 кг) и хлорид натрия (2 кг). Проверяли величину pH (>12) и перемешивали раствор в течение 15 мин. Смеси давали отстояться в течение 15 мин и отделяли водную фазу. Водную фазу повторно экстрагировали изопропилацетатом (12 л). К объединенным органическим фазам добавляли деминерализованную воду (20 л) и хлорид натрия (2,0 кг), раствор перемешивали в течение 15 минут, давали отстояться в течение 15 мин и удаляли водную фазу. Добавляли древесный уголь (0,4 кг), смесь перемешивали 20 мин и фильтровали. После промывки установки изопропилацетатом (12 л), удаляли растворитель при пониженном давлении при 20-25°C. Добавляли гептан (49 л) и суспензию перемешивали в течение 15 мин при 40°C. Затем 8 л растворителя удаляли отгонкой при пониженном давлении при 38-41°C. Суспензию охлаждали до 20°C и перемешивали 1 ч. Продукт центрифугировали и лепешку осадка промывали гептаном (5 л). Влажное соединение формулы (I) (5,5 кг) растворяли в этаноле (28 л) при 45°C. Добавляли раствор винной кислоты (0,72 кг) в этаноле (11 л) при 45°C и установку промывали этанолом (9 л). Раствор охлаждали до 43°C, вносили затравку кристаллов тартрата соединения формулы (I), затем охлаждали суспензию до 35°C в течение 30 мин, перемешивали при этой температуре в течение 1 ч и охлаждали до -5°C. После выдерживания при этой температуре в течение 14 ч продукт центрифугировали и промывали двумя порциями этанола (2×6 л). Маточные растворы общей массой 42 кг сохраняли для последующего использования при получении тартрата. Влажную лепешку осадка высушивали приблизительно при 45°C в течение 76 ч, получая 4 кг продукта.
c) Выделение дополнительного продукта из маточных растворов
Получали дополнительный продукт из сохраненных маточных растворов следующим образом. Растворитель удаляли отгонкой при пониженном давлении при 24-26°C из маточных растворов неочищенного тартрата (107 кг), полученных на стадии a), и маточных растворов тартрата соединения формулы I (42 кг), полученных на стадии b), до остаточного объема 27 л. Добавляли деминерализованную воду (25 л) и смесь концентрировали до остаточного объема 32 л путем отгонки при пониженном давлении при 24-26°C. Добавляли изопропилацетат (30 л) и 30% водный раствор гидроксида натрия (2,7 кг). Проверяли pH (>12) и раствор перемешивали в течение 15 мин. Давали смеси расслоиться в течение 15 минут и отделяли водную фазу. Водную фазу повторно экстрагировали изопропилацетатом (6 л). К объединенным органическим фазам добавляли деминерализованную воду (9 л) и хлорид натрия (0,9 кг), раствор перемешивали в течение 15 мин, давали расслоиться в течение 15 мин и удаляли водную фазу. Добавляли древесный уголь (0,3 кг), смесь перемешивали 20 мин и фильтровали. После промывки установки изопропилацетатом (8 л) растворитель удаляли отгонкой при пониженном давлении при 20-25°C до остаточного объема 12 л, но не досуха. При 30°C добавляли гептан (25 л), суспензию охлаждали до 20°C и перемешивали в течение 1,5 ч. Продукт центрифугировали и лепешку осадка промывали гептаном (2×5 л). Влажную лепешку (4,3 кг) растворяли в этаноле (23 л) при 45°C. При 45°C добавляли раствор винной кислоты (0,58 кг) в этаноле (7,5 л) и промывали установку этанолом (6 л). Раствор перемешивали в течение 20 мин (кристаллизация продукта) и охлаждали суспензию до 35°C в течение 30 мин, перемешивали при этой температуре в течение 1 ч и охлаждали до -5°C. После 14 ч выдерживания при этой температуре продукт центрифугировали и промывали двумя порциями этанола (2×4 л). Влажную лепешку высушивали приблизительно при 45°C в течение 80 ч, получая 3,3 кг продукта.
PXRD обоих продуктов выявил кристаллическую структуру образцов и высокий уровень базовой линии, указывающей на наличие аморфных компонентов и, возможно, небольших количеств кристаллической формы C. PXRD показал, что твердый продукт содержит в основном кристаллическую форму A гемитартрата N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида формулы (IV). Кристаллическая форма A содержала некоторое количество воды, как было показано при нагревании в ходе термогравиметрического анализа (TG-FTIR, потеря 2,2%, соответствовавшая воде и небольшому количеству растворителя). Эти количества показали, что кристаллическая форма A являлась гемигидратом (теоретическое значение содержания воды 1,8%). Однако вода связана весьма слабо, поскольку потеря массы начиналась при температуре несколько выше температуры окружающей среды и заканчивалась приблизительно при 150°C. Кроме этого, вода могла быть легко удалена обработкой сухим азотом в течение продолжительного периода времени (приблизительно до 20 часов). Температура плавления высушенной формы A составляла приблизительно 133-135°C, при энтальпии плавления приблизительно 70 Дж/г (температура пика, измеренного DSC). Форма A продемонстрировала значительное поглощение воды при действии относительной влажности выше 75%. Вода выделялась обратно при снижении относительной влажности до 50% или менее. Это поведение было типичным для твердых веществ, склонных к расплыванию.
Приблизительно измеряли растворимость путем приготовления насыщенных растворов в различных растворителях и гравиметрического определения количества растворенного вещества после удаления растворителя. Результаты приведены в таблице 3.
Приблизительная растворимость кристаллической формы A соединения формулы (IV)
Пример 4
Получение гемитартрата формулы (IV) из неочищенного свободного основания формулы (I)
Неочищенный продукт из примера 1(f) (5,5 кг) растворяли при 45°C в этаноле (28 л). При 45°C добавляли раствор (+)-L-винной кислоты (0,72 кг) в этаноле и промывали установку 9 л этанола. Охлаждали раствор до 43°C и добавляли затравку кристаллов гемитартрата формулы (IV). Затем суспензию охлаждали до 35°C в течение 30 мин, перемешивали при этой температуре в течение 1 часа и охлаждали при перемешивании до -5°C. После перемешивания в течение 14 часов при этой температуре продукт центрифугировали и промывали 2 порциями этанола (2×6 л). Влажную лепешку осадка высушивали при 45°C в течение 76 часов, получая 4,0 кг продукта (выход 83% в пересчете на винную кислоту). PXRD продукта выявил образование полиморфной формы A.
Пример 5
Получение гемитартрата формулы (IV) из неочищенного свободного основания формулы (I)
Неочищенный продукт, полученный по методике примера 1(f) (4,3 кг), растворяли в этаноле (23 л) при 45°C. При 45°C добавляли раствор (+)-L-винной кислоты (0,58 кг) в этаноле и промывали установку 6 л этанола. Раствор перемешивали в течение 20 мин (образование твердого осадка) и суспензию охлаждали до 35°C в течение 30 мин. Суспензию перемешивали при этой температуре в течение 1 часа и затем охлаждали до -5°C. После 14-часового перемешивания при этой температуре продукт центрифугировали и промывали 2 порциями этанола (2×4 л). Влажную лепешку осадка высушивали при 45°C в течение 80 часов, получая 3,3 кг продукта (85% в пересчете на винную кислоту). PXRD продукта выявил образование полиморфной формы A.
Пример 6
Получение аморфной формы соединения формулы (IV) лиофилизацией водного раствора
2,02 г неочищенного свободного основания формулы (I) растворяли при комнатной температуре в 8,0 мл воды (Fluka №95306) при 23±2°C. Полученный раствор фильтровали через 0,22 мкм фильтрующую ячейку Millipore и отфильтрованный раствор переносили в 100-мл круглодонную колбу. Прозрачный раствор замораживали на слое сухого льда (твердый CO2) при -78°C и затем стеклянную колбу с замороженным раствором соединяли с лиофилизатором. Тип лиофилизатора: CHRIST, BETA 2-8 LD-2. Исходное давление составляло около 0,10 мбар, температура холодной ловушки составляла -82°C и конечное давление равнялось 0,007 мбар. Через 15 часов лиофилизация завершалась и колбу отсоединяли. Полученный белый твердый порошок характеризовали дифференциальной сканирующей калориметрией и рентгенограммой на порошке. PXRD полученного продукта показало полностью аморфное состояние и аналогично измерения DSC выявили полную аморфность вещества с температурой стеклования приблизительно 54°C и ΔCp приблизительно 0,5 Дж/г/°C.
Пример 7
Получение чистой кристаллической формы A перекристаллизацией
142,5 г продукта из примера 5 суспендировали в абсолютном этаноле (750 мл). Белую суспензию нагревали при перемешивании в течение 30 минут до 70°C. Начиная с 60°C, раствор приобретал желтоватый цвет и становился прозрачным. Раствор медленно охлаждали, и начало кристаллизации продукта наблюдалось приблизительно при 48°C. Охлаждение от 48°C до 15°C проводили в течение 4 ч. Суспензию перемешивали в течение 1,5 ч при 15°C. После этого образовывался густой осадок. Осадок отфильтровывали в вакууме, дважды промывали 70 мл абсолютного этанола и затем высушивали в вакууме при 40°C. Сухой вес составлял 135,2 г (выход 95%).
Полученный продукт вновь суспендировали при перемешивании в 850 мл абсолютного этанола и нагревали в течение 30 минут до 75°C. Растворение завершалось, и раствор становился в основном бесцветным, начиная с 58-60°C. Раствор фильтровали при 75°C, установку промывали 50 мл абсолютного этанола и затем давали раствору остыть при перемешивании. Кристаллизация начиналась при 48°C. Продукт кристаллизовался при приблизительно 42-44°C и образовывался объемистый осадок. Суспензию оставляли остывать в течение ночи до комнатной температуры. Суспензию фильтровали при 20-22°C и дважды промывали 50 мл абсолютного этанола. Полученный белый и твердый продукт высушивали в течение 48 часов в вакууме при 42°C. Сухой вес составлял 123,6 г (выход 92%).
Рентгенограмма на порошке продукта показана на фиг.2 и характеристические пики, выраженные в величинах 2 тета и соответствующих величинах межплоскостных расстояний d в Е, приведены в таблице 4.
Межплоскостные расстояния d кристаллической формы А соединения формулы (IV)
Пример 8
Получение чистой кристаллической формы A перекристаллизацией
105,0 г соединения формулы (IV), полученного в примере 5, растворяли при перемешивании при 65°C в 560 мл абсолютного этанола и затем при перемешивании охлаждали до 48°C со скоростью 1°C/мин. При этой температуре кристаллизация начиналась через несколько минут, и суспензия превращалась в густую пасту в течение 1 часа. Суспензию вновь нагревали до 60°C и затем охлаждали до 48°C со скоростью 1°C/мин. Полученную суспензию перемешивали и охлаждали до 15°C со скоростью 3°C/ч. Кристаллический осадок отделяли фильтрованием и емкость промывали 50 мл абсолютного этанола, охлажденного до 5°C. Затем кристаллический осадок высушивали на воздухе при 30°C в течение 18 ч, и после этого в вакууме при комнатной температуре в течение 40 ч, получая 98,1 г кристаллического продукта. По данным PXRD продукт оказался кристаллической полиморфной формой A. TG-FTIR показал потерю массы порядка 2,5%, которая была отнесена к выделению воды и небольшого количества этанола.
Пример 9
Получение чистой кристаллической формы A перекристаллизацией
21,0 г соединения формулы (IV), полученного в примере 3(b), растворяли при перемешивании при 65°C в 112 мл абсолютного этанола и затем охлаждали при перемешивании до 48°C со скоростью 1°C/мин. При этой температуре кристаллизация начиналась через несколько минут и суспензия превращалась в густую пасту в течение 1 ч. Суспензию вновь нагревали до 60°C и затем охлаждали до 48°C со скоростью 1°C/мин. Полученную суспензию перемешивали и охлаждали до 15°C со скоростью 3°C/ч. Кристаллический осадок отделяли фильтрованием и емкость промывали 10 мл абсолютного изопропанола, охлажденного до 5°C. Кристаллический осадок на первом этапе высушивали в атмосфере азота при 25°C в течение 18 ч и затем в вакууме при комнатной температуре в течение 20 часов, получая 19,9 г кристаллического продукта. По данным PXRD продукт являлся полиморфной формой A, с чертами сходства с формой D. Анализ TG-FTIR показал потерю массы приблизительно 7,7%, которую отнесли к выделению изопропанола и воды. Продукт был вновь высушен при 30°C на воздухе в течение 20 ч, что дало продукт с потерей массы приблизительно 5% за счет изопропанола и воды.
Пример 10
Получение чистой кристаллической формы A перекристаллизацией
150,0 г соединения формулы (IV), полученного в примере 3(b), растворяли при перемешивании при 65°C в 112 мл абсолютного этанола и затем охлаждали при перемешивании до 48°C при скорости охлаждения 1°C/мин. При этой температуре кристаллизация начиналась через несколько минут и суспензия превращалась в густую пасту в течение 1 ч. Суспензию вновь нагревали до 60°C и затем охлаждали до 48°C со скоростью 1°C/мин. Полученную суспензию перемешивали и охлаждали до 15°C со скоростью 3°C/ч. Кристаллический осадок отделяли фильтрованием и емкость промывали 10 мл абсолютного этанола, охлажденного до 5°C. Кристаллический осадок на первом этапе высушивали в вакууме при 40°C в течение 50 ч, получая 146 г кристаллического продукта, который по данным PXRD являлся чистой полиморфной формой A.
Пример 11
Получение чистой кристаллической формы A
путем приведения суспензии в равновесное состояние
20 мг соединения формулы (IV) из примера 3(b) суспендировали в растворителе и перемешивали в течение 4 дней при переменной температуре, которая циклически изменялась от 18 до 40°C. Продукт идентифицировали как кристаллическую форму A с помощью PXRD или спектроскопии комбинационного рассеяния, при применении следующих растворителей: этанол, изопропанол, гептан, метилэтиловый эфир, трет-бутилметиловый эфир (TBME), этанол и TBME, этанол/гептан, TBME, насыщенный водой.
Пример 12
Получение чистой кристаллической формы A из аморфной формы
путем приведения суспензии в равновесное состояние
64 мг аморфного соединения из примера 6 суспендировали в 1,0 мл тетрагидрофурана и перемешивали при 5°C в течение 18 часов. Твердое вещество отфильтровывали и высушивали в атмосфере азота при комнатной температуре в течение 2 часов. Кристаллическую форму A идентифицировали с помощью PXRD или спектроскопии комбинационного рассеяния.
Пример 13
Получение чистой кристаллической формы A из аморфной формы
путем приведения суспензии в равновесное состояние
20 мг аморфного соединения из примера 6 суспендировали в 500 мкл смеси этанол/ацетон (1:1) и затем перемешивали в течение 3 дней при циклическом изменении температуры от комнатной до 40°C. Кристаллическую форму A идентифицировали с помощью спектроскопии комбинационного рассеяния.
Пример 14
Получение чистой кристаллической формы A из аморфной формы
путем приведения суспензии в равновесное состояние
20 мг аморфного соединения из примера 6 суспендировали в 500 мкл тетрагидрофурана и затем перемешивали в течение 3 дней при циклическом изменении температуры от комнатной до 40°C. Кристаллическую форму A идентифицировали с помощью спектроскопии комбинационного рассеяния.
Пример 15
Получение кристаллической формы B осаждением метилэтилкетоном, не являющимся растворителем
600 мкл водного раствора, содержащего около 160 мг соединения формулы (IV) из примера 3(b), добавляли к 10 мл метилэтилкетона (MEK) при 5°C. Суспензию перемешивали в течение 3 дней. Добавляли 5 мл MEK и продолжали перемешивание в течение 5 часов. Твердое вещество отфильтровывали и высушивали на воздухе при комнатной температуре в течение 12 ч. Кристаллическую форму B идентифицировали XPRD или спектроскопией комбинационного рассеяния. Анализ TG-FITR показал потерю массы приблизительно 2,5%, которую отнесли к выделению воды. Рентгенограмма на порошке продукта показана на фиг.3, и характеристические пики, выраженные в величинах 2 тета с соответствующими величинами межплоскостных расстояний d в Е, приведены в таблице 5.
Межплоскостные расстояния d кристаллической формы B соединения формулы (IV)
Пример 16
Получение кристаллической формы B осаждением гептаном,
не являющимся растворителем
2,0 мл раствора, содержащего 135 мг соединения формулы (IV) из примера 3(b), в хлористом метилене добавляли при комнатной температуре к 3,0 мл гептана. Образовавшуюся суспензию перемешивали в течение 24 ч, затем отфильтровывали осадок и высушивали его на воздухе при комнатной температуре в течение 8 ч. Кристаллическую форму B идентифицировали PXRD или спектроскопией комбинационного рассеяния. Измерения способом DSC позволили определить температуру плавления, равную приблизительно 131°C при энтальпии плавления, равной приблизительно 63 Дж/г.
Пример 17
Получение кристаллической формы B осаждением толуолом,
не являющимся растворителем
2,0 мл раствора, содержащего 135 мг соединения формулы (IV) из примера 3(b), в хлористом метилене добавляли при комнатной температуре к 3,0 мл толуола. Образовавшуюся суспензию перемешивали в течение 24 ч, затем отфильтровывали осадок и высушивали его на воздухе при комнатной температуре в течение 14 ч. Кристаллическую форму B идентифицировали PXRD или спектроскопией комбинационного рассеяния. Измерения способом DSC позволили определить температуру плавления, равную приблизительно 129°C при энтальпии плавления, равной приблизительно 71 Дж/г.
Пример 18
Получение кристаллической формы B осаждением ацетонитрилом,
не являющимся растворителем
2,0 мл раствора, содержащего 135 мг соединения формулы (IV) из примера 3(b), в хлористом метилене добавляли при комнатной температуре к 3,0 мл ацетонитрила. Образовавшуюся суспензию перемешивали в течение 24 ч, затем отфильтровывали осадок и высушивали его на воздухе при комнатной температуре в течение 18 ч. Кристаллическую форму B идентифицировали спектроскопией комбинационного рассеяния.
Пример 19
Получение кристаллической формы B осаждением этилацетатом,
не являющимся растворителем
1,5 мл раствора, содержащего 210 мг соединения формулы (IV) из примера 3(b), в метаноле добавляли при комнатной температуре к 10 мл этилацетата. Осаждения продукта не наблюдалось до того, как приблизительно 50% смеси растворителей этилацетат/метанол не было выпарено при комнатной температуре. Образовавшуюся суспензию перемешивали при 15°C в течение 18 ч, затем отфильтровывали осадок и высушивали его на воздухе при комнатной температуре в течение 12 ч. Кристаллическую форму B идентифицировали спектроскопией комбинационного рассеяния.
Пример 20
Получение кристаллической формы B путем приведения в равновесное состояние суспензии с полиморфной формой A в ацетонитриле
20 мг соединения формулы (IV) из примера 3(b) суспендировали в ацетонитриле и перемешивали в течение 4 дней при температуре, циклически изменявшейся от 18 до 40°C, затем отфильтровывали и высушивали на воздухе при комнатной температуре в течение 18 ч. Кристаллическую форму B идентифицировали спектроскопией комбинационного рассеяния.
Пример 21
Получение кристаллической формы B путем приведения в равновесное состояние суспензии с полиморфной формой A в этилацетате
20 мг соединения формулы (IV) из примера 3(b) суспендировали в 6 мл этилацетата и перемешивали в течение 4 дней при температуре, циклически изменявшейся от 18 до 40°C, затем отфильтровывали и высушивали на воздухе при комнатной температуре в течение 18 ч. Кристаллическую форму B идентифицировали спектроскопией комбинационного рассеяния.
Пример 22
Получение кристаллической формы B путем приведения в равновесное состояние суспензии с полиморфной формой A в этаноле/MEK
20 мг соединения формулы (IV) из примера 3(b) суспендировали в 5 мл смеси этанол/MEK (1:1) и перемешивали в течение 4 дней при температуре, циклически изменявшейся от 18 до 40°C, затем отфильтровывали и высушивали на воздухе при комнатной температуре в течение 18 ч. Кристаллическую форму B идентифицировали спектроскопией комбинационного рассеяния.
Пример 23
Получение кристаллической формы B путем приведения в равновесное состояние суспензии с полиморфной формой A в этилацетате, насыщенном водой
20 мг вещества из примера 6 суспендировали в 500 мкл этилацетата, насыщенного водой, и перемешивали в течение 3 дней при температуре, циклически изменявшейся от комнатной до 40°C, затем отфильтровывали и высушивали на воздухе при комнатной температуре в течение 8 ч. Кристаллическую форму B идентифицировали спектроскопией комбинационного рассеяния.
Пример 24
Получение кристаллической формы B путем приведения в равновесное состояние суспензии с полиморфной формой A в ацетонитриле, содержащем 1% воды
20 мг вещества из примера 6 суспендировали в 500 мкл ацетонитрила, содержащего 1% воды, и перемешивали в течение 3 дней при температуре, циклически изменявшейся от комнатной до 40°C, затем отфильтровывали и высушивали на воздухе при комнатной температуре в течение 16 ч. Кристаллическую форму B идентифицировали спектроскопией комбинационного рассеяния.
Пример 25
Получение кристаллической формы B путем приведения в равновесное состояние суспензии с полиморфной формой A в смеси этилацетат/вода
1,0 г вещества из примера 6 суспендировали в смеси 10 мл этилацетата и 100 мкл воды и перемешивали в течение 100 ч при комнатной температуре, затем отфильтровывали и высушивали на воздухе при комнатной температуре в течение 18 ч. Получали 750 мг кристаллической формы B, что было определено с помощью спектроскопии комбинационного рассеяния и рентгенограммой на порошке.
Пример 26
Получение кристаллической формы B путем приведения в равновесное состояние суспензии с полиморфной формой A в смеси этанол/MEK
20 мг соединения формулы (I) из примера 3(b) суспендировали в 7 мл смеси этанол/MEK (1:1) и перемешивали в течение 4 дней при температуре, циклически изменявшейся от 18 до 40°C, затем отфильтровывали и высушивали на воздухе при комнатной температуре в течение 18 ч. Кристаллическую форму B идентифицировали спектроскопией комбинационного рассеяния.
Пример 27
Получение кристаллической формы B путем приведения в равновесное состояние суспензии с аморфной формой в гептане
60 мг вещества из примера 6 суспендировали в 1,0 мл гептана и перемешивали при 40°C в течение 18 ч. Твердое вещество отфильтровывали и высушивали на воздухе при 40°C в течение 1 ч. Кристаллическую форму B идентифицировали спектроскопией комбинационного рассеяния.
Пример 28
Получение кристаллической формы B путем приведения в равновесное состояние суспензии с аморфной формой в этилацетате
62 мг вещества из примера 6 суспендировали в 1,0 мл этилацетата и перемешивали при 40°C в течение 18 ч. Твердое вещество отфильтровывали и высушивали на воздухе при 40°C в течение 1 ч. Кристаллическую форму B идентифицировали спектроскопией комбинационного рассеяния.
Пример 29
Получение кристаллической формы B путем приведения в равновесное состояние суспензии с аморфной формой в ацетонитриле
62 мг вещества из примера 6 суспендировали в 1,0 мл ацетонитрила и перемешивали при 5°C в течение 18 ч. Твердое вещество отфильтровывали и высушивали в азоте при 22°C в течение 2 ч. Кристаллическую форму B идентифицировали спектроскопией комбинационного рассеяния.
Пример 30
Получение кристаллической формы B путем приведения в равновесное состояние суспензии с аморфной формой в MEK
149 мг вещества из примера 6 суспендировали в 3,0 мл MEK и перемешивали при комнатной температуре в течение 16 ч. Твердое вещество отфильтровывали и высушивали в азоте при 22°C в течение 30 мин. Кристаллическую форму B идентифицировали спектроскопией комбинационного рассеяния.
Пример 31
Получение кристаллической формы B путем приведения в равновесное состояние суспензии с аморфной формой в этилацетате, насыщенном водой
20 мг вещества из примера 6 суспендировали в 500 мкл этилацетата, насыщенного водой, и перемешивали в течение 3 дней при температуре, циклически изменявшейся от комнатной до 40°C, затем отфильтровывали и высушивали на воздухе при комнатной температуре в течение 6 ч. Кристаллическую форму B идентифицировали спектроскопией комбинационного рассеяния.
Пример 32
Получение кристаллической формы B путем приведения в равновесное состояние суспензии с аморфной формой в смеси растворителей, содержащей воду
70 мг вещества из примера 6 суспендировали в 2,0 мл смеси этилацетат/этанол, содержащей 1% воды, перемешивали в течение 1 дня при температуре, циклически изменявшейся от 5°C до комнатной. Затем продолжали перемешивание при 10°C в течение 5 дней. Твердое вещество отфильтровывали и высушивали на воздухе при комнатной температуре в течение 15 мин. Кристаллическую форму B идентифицировали спектроскопией комбинационного рассеяния.
Пример 33
Получение кристаллической формы C путем приведения в равновесное состояние суспензии полиморфной формы A в ацетоне
20 мг соединения формулы (IV) из примера 3(b) суспендировали в 1 мл ацетона, добавляли 2 мг затравочных кристаллов формы C и суспензию перемешивали в течение 4 дней при температуре, циклически изменявшейся от 18 до 40°C, затем отфильтровывали осадок и высушивали на воздухе при комнатной температуре в течение 1 ч. Кристаллическую форму C идентифицировали спектроскопией комбинационного рассеяния.
Пример 34
Получение кристаллической формы C путем приведения в равновесное состояние суспензии полиморфной формы A в тетрагидрофуране (ТГФ)
20 мг соединения формулы (IV) из примера 3(b) суспендировали в 500 мкл ТГФ, добавляли 2 мг затравочных кристаллов формы C и суспензию перемешивали в течение 3 дней при температуре, циклически изменявшейся от 18 до 40°C, отфильтровывали осадок и высушивали его на воздухе при комнатной температуре в течение 3 ч. Кристаллическую форму C идентифицировали спектроскопией комбинационного рассеяния.
Пример 35
Получение кристаллической формы C путем приведения в равновесное состояние суспензии полиморфной формы A в тетрагидрофуране (ТГФ)
255 мг соединения формулы (I) из примера 3(b) суспендировали в 5,0 мл ТГФ, добавляли 25 мг формы C в качестве затравочных кристаллов, и суспензию перемешивали в течение 40 часов при температуре 40°C, отфильтровывали осадок и высушивали в атмосфере азота при комнатной температуре в течение 15 мин. Кристаллическую форму C идентифицировали с помощью PXRD и спектроскопии комбинационного рассеяния.
Пример 36
Получение кристаллической формы C путем приведения в равновесное состояние суспензии полиморфной формы A в тетрагидрофуране (ТГФ)
1,0 г соединения формулы (I) из примера 3(b) суспендировали в 6,0 мл ТГФ, добавляли 50 мг формы C в качестве затравочных кристаллов и полученную суспензию перемешивали в течение 50 часов при комнатной температуре, отфильтровывали осадок и высушивали на воздухе при комнатной температуре в течение 45 минут. Кристаллическую форму C идентифицировали с помощью PXRD и спектроскопии комбинационного рассеяния. Анализ TG-FTIR показал потерю массы менее 0,9% ниже 150°C, которая была отнесена к выделению воды. Эксперименты по динамической абсорбции при добавлении воды показали, что полиморфная форма C не абсорбирует воду, не образует гидрат и не проявляет гигроскопичности. Эксперименты DSC позволили определить температуру плавления около 177°C и энтальпию плавления приблизительно 129 Дж/г.
Рентгенограмма на порошке продукта показана на фиг.4, и характеристические пики, выраженные в величинах 2 тета с соответствующими величинами межплоскостных расстояний d в Е, приведены в таблице 6.
Межплоскостные расстояния d кристаллической формы C соединения формулы (IV)
Пример 37
Получение затравочного материала полиморфной формы C
25 г соединения формулы (IV) из примера 3(b) суспендировали в 100 мл ТГФ и суспензию перемешивали в течение 3 дней при 30°C. Твердое вещество отделяли фильтрованием и высушивали при пониженном давлении при 40°C в течение 2 ч. Получали 23,3 г чистой полиморфной формы C, что было подтверждено данными PXRD и спектроскопии комбинационного рассеяния. Полученное вещество применяли в качестве затравочных кристаллов в последующих экспериментах.
Пример 38
Получение полиморфной формы C
6,0 г кристаллического вещества из примера 9 суспендировали в 30 мл MEK и перемешивали при 50°C. Через 2 часа добавляли 100 мг затравочных кристаллов из примера 37 и продолжали перемешивание в течение 80 часов при комнатной температуре. Твердое кристаллическое вещество отфильтровывали и высушивали в течение 18 часов при 45°C. Получали полиморфную форму C в количестве 4,7 г, содержащую незначительное количество полиморфной формы A, что было подтверждено PXRD. Анализ TG-FTIR показал отсутствие потери массы ниже 170°C.
Пример 39
Получение полиморфной формы C
6,0 г кристаллического вещества из примера 9 суспендировали в 30 мл ТГФ и перемешивали при 50°C. Через 2 часа добавляли 100 мг затравочных кристаллов из примера 37 и продолжали перемешивание в течение 80 часов при комнатной температуре. Твердое кристаллическое вещество отделяли фильтрованием и высушивали в течение 18 часов при 45°C. Получали выход полиморфной формы C, содержавшей небольшое количество полиморфной формы A, составлявший 4,7 г, что было подтверждено PXRD. Анализ TG-FTIR показал потерю массы приблизительно 0,5% ниже 170°C, которая была отнесена к выделению ТГФ.
Пример 40
Получение полиморфной формы C
6,0 г кристаллического вещества из примера 8 суспендировали в 40 мл ТГФ и перемешивали при 50°C. Через 2 часа добавляли 150 мг затравочных кристаллов из примера 37 и продолжали перемешивание в течение 104 часов при 40°C. Вторую порцию затравочных кристаллов из примера 37, равную 200 мг, добавляли через 30 часов. Твердое кристаллическое вещество отделяли фильтрованием и высушивали в течение 18 часов при 45°C. Получали 5,0 г полиморфной формы C, содержавшей небольшое количество полиморфной формы A, что было подтверждено данными PXRD. Анализ TG-FTIR показал потерю массы приблизительно от 0,5 до 0,8% ниже 170°C, которая была отнесена к выделению ТГФ.
Пример 41
Получение полиморфной формы C
6,0 г кристаллического вещества из примера 8 суспендировали в 40 мл MEK и перемешивали при 50°C. Через 2 часа добавляли 150 мг затравочных кристаллов из примера 37 и продолжали перемешивание в течение 104 часов при 40°C. Вторую порцию затравочных кристаллов из примера 37, равную 200 мг, добавляли через 30 часов. Твердое кристаллическое вещество отделяли фильтрованием и высушивали в течение 18 часов при 45°C. Получали 5,4 г полиморфной формы C, содержавшей небольшое количество полиморфной формы A, что было подтверждено данными PXRD. Анализ TG-FTIR показал отсутствие потери массы ниже 170°C.
Пример 42
Получение чистой полиморфной формы C
7,0 г кристаллического вещества из примера 8 суспендировали в 50 мл ацетона и перемешивали при 50°C. Через 2 часа добавляли 200 мг затравочных кристаллов из примера 37. Образовывалась густая паста, к которой добавляли 10 мл ацетона. Перемешивание продолжали в течение 29 часов при 50°C. Затем полученную суспензию охлаждали до 10°C и перемешивали при этой температуре в течение 14 ч. Твердое кристаллическое вещество отделяли фильтрованием и высушивали на воздухе в течение 4,5 часов при 45°C, получая 6,3 г чистой полиморфной формы C, что было подтверждено данными PXRD.
Пример 42
Получение чистой полиморфной формы C
7,0 г кристаллического вещества из примера 8 суспендировали в 50 мл MEK и перемешивали при 60°C. Через 2 часа добавляли 200 мг затравочных кристаллов из примера 37 и продолжали перемешивание в течение 29 ч при 60°C. Затем полученную суспензию охлаждали до 10°C и перемешивали при этой температуре в течение 14 ч. Твердое кристаллическое вещество отделяли фильтрованием и высушивали на воздухе в течение 4,5 часов при 45°C. Получали 6,0 г чистой полиморфной формы C, что было подтверждено данными PXRD.
Пример 43
Получение чистой полиморфной формы C
50,0 г кристаллического вещества из примера 10 суспендировали в 310 мл MEK и перемешивали (600 об/мин) при 50°C. Через 2 часа добавляли 1,5 г затравочных кристаллов из примера 37 (суспензия в 10 мл MEK). Перемешивание продолжали в течение 52 ч при 50°C. Затем полученную суспензию охлаждали до 15°C и перемешивали при этой температуре в течение 2 ч. Твердое кристаллическое вещество отделяли фильтрованием и высушивали в вакууме в течение 16 часов при 50°C. Получали 44,2 г чистой полиморфной формы C, что было подтверждено данными PXRD. Анализ TG-FTIR показал отсутствие потери массы ниже 170°C (продукт не содержал растворителя).
Пример 44
Получение чистой полиморфной формы C
50,0 г кристаллического вещества из примера 10 суспендировали в 360 мл MEK и перемешивали (600 об/мин) при 50°C. Через 2 часа добавляли 1,5 г затравочных кристаллов из примера 37 (суспензия в 10 мл MEK). Перемешивание продолжали в течение 35,5 ч при 50°C. Затем полученную суспензию охлаждали до 15°C и перемешивали при этой температуре в течение 2 ч. Твердое кристаллическое вещество отделяли фильтрованием и высушивали в вакууме в течение 16 часов при 50°C. Получали 41,5 г чистой полиморфной формы C, что было подтверждено данными PXRD. Анализ TG-FTIR показал отсутствие потери массы ниже 170°C (продукт не содержал растворителя).
Пример 45
Получение чистой полиморфной формы C из раствора в ТГФ
7,0 г кристаллического вещества из примера 10 суспендировали в 35 мл ТГФ и нагревали до 65°C. Кристаллическая форма A полностью растворялась, и раствор охлаждали до 60°C. Затем добавляли 0,35 г затравочных кристаллов из примера 37 (суспензия в 1,0 мл ТГФ) и продолжали перемешивание в течение приблизительно 30 мин при 60°C. После этого суспензию охлаждали до 10°C со скоростью 0,15°C в минуту и продолжали перемешивание при этой температуре в течение 2 ч. Твердое кристаллическое вещество отделяли фильтрованием и высушивали в вакууме в течение 16 часов при 50°C. Получали 4,5 г чистой полиморфной формы C, что было подтверждено данными PXRD и спектроскопии комбинационного рассеяния.
Пример 46
Получение формы C непосредственно из раствора
2,0 г кристаллического вещества из примера 10 суспендировали в 10 мл ТГФ при комнатной температуре. Нагревание суспензии до 65°C приводило к образованию прозрачного раствора. Этот раствор охлаждали до 60°C и к раствору добавляли 100 мг затравочных кристаллов формы C из примера 37. При этой температуре суспензия медленно густела, и после перемешивания суспензии в течение 1 часа при 60°C ее охлаждали до 10°C со скоростью 10°C в час. Через 5 часов достигали температуры 10°C и продолжали перемешивание в течение ночи, т.е. приблизительно 14 ч, до отделения твердого вещества фильтрованием и высушивания при 50°C в течение приблизительно 2 часов в вакууме, в результате чего получали чистую кристаллическую форму C.
Пример 47
Тесты на стабильность полиморфной формы C
a) Термическая обработка
Соединения из примеров 3(b) (полиморфная форма A), 25 (полиморфная форма B) и 36 (полиморфная форма C) помещали в запаянные ампулы и выдерживали в течение 1 недели при 100°C. Полиморфные формы A и B образовали компактное расплывшееся вещество, тогда как полиморфная форма C в целом не изменилась и осталась в виде кристаллического сыпучего порошка. Полученные продукты анализировали с помощью ВЭЖХ и определяли чистоту для выявления химической устойчивости к разложению. Чистота полиморфной формы A оказалась равной 25,9%, полиморфной формы B 28,3% и полиморфной формы C 99,7%, что продемонстрировало высокую устойчивость полиморфной формы C.
Пример 48
Воздействие влажности
Соединения из примеров 3(b) (полиморфная форма A), 25 (полиморфная форма B) и 36 (полиморфная форма C) помещали в открытые емкости и выдерживали в течение 1 недели и 2 недель при 60°C и относительной влажности 75%. В полиморфной форме A было зафиксировано содержание воды 2,8% и чистота по данным ВЭЖХ составляла 80%. Полиморфная форма B превратилась в полиморфную форму C, было зафиксировано содержание воды 1,9%, и чистота по данным ВЭЖХ составляла 94,6%. Полиморфная форма C осталась без изменений, и чистота по данным ВЭЖХ составляла 99,7%.
Пример 49
Получение кристаллической формы D с использованием
полиморфной формы A в качестве исходного вещества
600 мкл раствора, содержащего 160 мг соединения формулы (IV) согласно примеру 3(b) в воде, при 5°C добавляли к 10 мл изопропанола. Осаждалось твердое кристаллическое вещество, и суспензию перемешивали в течение 5 часов при 5°C. Твердое кристаллическое вещество отделяли фильтрованием и высушивали в атмосфере азота в течение 1 часа при комнатной температуре. Было получено 164 мг кристаллической формы D, что было подтверждено данными PXRD и спектроскопии комбинационного рассеяния. Анализ TG-FTIR показал потерю массы около 8% до 170°C, что было отнесено к выделению изопропанола и воды.
Пример 50
Получение кристаллической формы D из аморфной формы
в качестве исходного вещества
200 мг вещества из примера 6 суспендировали в 16,0 мл изопропанола. Суспензию перемешивали в течение 18 ч при 40°C и в течение 14 ч при 20°C. Твердое кристаллическое вещество отделяли фильтрованием и высушивали в атмосфере азота в течение 1 часа при комнатной температуре. Получали 178 мг кристаллической формы D, что было подтверждено данными PXRD и спектроскопии комбинационного рассеяния. Анализ TG-FTIR показал потерю массы приблизительно 6,6% ниже 170°C, которое относили к выделению изопропанола. Количество изопропанола показало существование гемисольвата изопропанола (теоретическое содержание изопропанола составляло 5,6%; растворитель трудно удалить высушиванием).
Рентгенограмма на порошке продукта показана на фиг.5, и характеристические пики, выраженные в величинах 2 тета, с соответствующими величинами межплоскостных расстояний d в Е приведены в таблице 7.
Межплоскостные расстояния d кристаллической формы D соединения формулы (IV)
Пример 51
Получение кристаллической формы E с использованием
аморфной формы в качестве исходного вещества
70 мг вещества из примера 6 суспендировали в 1,0 мл трет-бутилметилового эфира (TBME). Суспензию перемешивали в течение 18 ч при 40°C. Твердое кристаллическое вещество отделяли фильтрованием и высушивали на воздухе в течение 1 ч при 40°C. Получали 58 мг кристаллической формы E, что было подтверждено данными PXRD и спектроскопии комбинационного рассеяния.
Пример 52
Получение кристаллической формы E из аморфной формы, использованной в качестве исходного вещества
150 мг вещества из примера 6 суспендировали в 4,0 мл TBME. Суспензию перемешивали в течение 26 ч при комнатной температуре. Твердое кристаллическое вещество отделяли фильтрованием и высушивали на воздухе в течение 5 мин при комнатной температуре. Получали 121 мг кристаллической формы E, что было подтверждено данными PXRD и спектроскопии комбинационного рассеяния. Анализ TG-FTIR (10°C/мин) показал потерю массы около 5,1%, которая начиналась выше комнатной температуры и завершалась ниже 150°C, причем эта потеря была отнесена к выделению TBME. Количество TBME указывало на существование TBME-сольвата.
Рентгенограмма на порошке продукта показана на фиг.6, и характеристические пики, выраженные в величинах 2 тета, с соответствующими величинами межплоскостных расстояний d в Е приведены в таблице 8.
Межплоскостные расстояния d кристаллической формы E соединения формулы (IV)
Пример 53
Получение кристаллической формы F из аморфной формы, использованной в качестве исходного вещества
250 мг вещества из примера 6 растворяли при перемешивании при 65°C в 5,5 мл тетрагидрофурана (ТГФ). Раствор охлаждали до 20°C, в результате чего образовывалась густая паста. Добавляли 3 мл ТГФ и продолжали перемешивание в течение 1 часа при 40°C. Затем суспензию охлаждали до 20°C и перемешивали еще 3 ч. Твердое кристаллическое вещество отделяли фильтрованием и высушивали на воздухе в течение 30 мин при комнатной температуре. Получали 214 мг кристаллической формы F, что было подтверждено данными PXRD и спектроскопии комбинационного рассеяния. Анализ TG-FTIR (10°C/мин) показал потерю массы около 3,0%, которая начиналась выше температуры окружающей среды и завершалась ниже 130°C, причем эта потеря была отнесена к выделению ТГФ. Количество ТГФ указывало на существование нестехиометрического ТГФ-сольвата (теоретическое содержание ТГФ для моно-ТГФ сольвата составляло 12,5%).
Рентгенограмма на порошке продукта показана на фиг.7, и характеристические пики, выраженные в величинах 2 тета, с соответствующими величинами межплоскостных расстояний d в Е приведены в таблице 9.
Межплоскостные расстояния d кристаллической формы F соединения формулы (IV)
Пример 54
Получение гемитартрата N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида
а) Получение:

N-метил-4-пиперидон (исходное вещество, 16,0 кг) и 4-фторбензиламин (17,7 кг, 1,00 экв.) растворяли в метаноле (110,2 кг, 8,70 - объем/масса исходное вещество) при T=15-19°C, затем в атмосфере азота добавляли 5% палладий/C (0,59 кг, 3,68% - масса/масса исходное вещество). Полученную смесь нагревали до T=23-27°C и гидрировали при этой температуре и P=~5 бар до завершения поглощения водорода (~11 часов). Наличие не вступившего в реакцию исходного вещества проверяли ГХ (имин <5%). После этого реакционную смесь очищали от осадка (1575+ фильтровальная бумага GF92) и промывали установку метанолом (5,1 кг, 0,40 - объем/масса исходное вещество). Растворитель отгоняли при пониженном давлении (P=265-60 мбар, T=35-40°C) и маслянистый остаток очищали фракционной перегонкой в вакууме при T=135-140°C, P=8-0,5 мбар, получая 22,15 кг (70%) продукта.
b) Получение
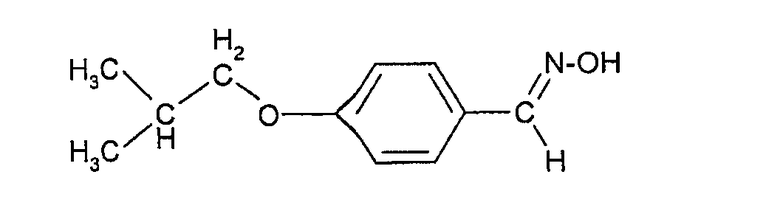
4-гидроксибензальдегид (исходное вещество, 60,0 кг) растворяли в диметилформамиде (142,5 кг, 2,50 - объем/масса исходное вещество) при T=15-25°C, затем порциями добавляли твердый карбонат калия (137,2 кг, 2,02 экв.) и йодид калия (8,1 кг, 0,10 экв.) при T<30°C и суспензию нагревали до T=78-82°C. Температуру холодильника фиксировали на уровне 15°C, к суспензии в течение 4-5 часов при T=78-82°C добавляли изобутилбромид (134,8 кг, 2,00 экв.) и не вступившее в реакцию исходное вещество проверяли ВЭЖХ (исходное вещество <5%). Суспензию охлаждали до T=20-30°C, разбавляли 100% этанолом (213,1 кг, 4,50 - объем/масса исходное вещество), перемешивали 15 мин при T=20-30°C и, наконец, центрифугировали для удаления избытка карбоната и бромида калия. Установку и лепешку осадка промывали 100% этанолом (82,4 кг, 1,74 - объем/масса исходное вещество), затем к фильтрату при комнатной температуре добавляли 50% раствор гидроксиламина в воде (48,8 кг, 1,5 экв.), после чего реакционную смесь нагревали до T=73-77°C и перемешивали при этой температуре в течение 2 часов. Отбирали образец для IPC (альдегид Aca-11<5%), затем реакционную смесь концентрировали при пониженном давлении (270-150 мбар, 45-55°C) до ~1/6 объема, остаток гасили водой (404,5 кг, 6,74 - объем/масса исходное вещество) при T=45-55°C и остаток этанола отгоняли в вакууме (270-150 мбар, 45-55°C, остаточный объем = ~10,4). Реакционную смесь разбавляли бензолом 60-90 (236,9 кг, 5,64 - объем/масса исходное вещество) и нагревали до кипения с обратным холодильником (T=~60°C) до достижения полного растворения (~15 мин, визуальный контроль). Раствор охлаждали до 8-12°C (кристаллизация наблюдалась при T=~17°C, затравки кристаллов вносили при ~12°C в случае необходимости), затем до 0-5°C. Через 2 часа перемешивания при T=0-5°C, реакционную смесь центрифугировали и промывали лепешку осадка двумя порциями бензола 60-90 (59,4 кг, 1,41 - объем/масса исходное вещество), затем высушивали при пониженном давлении при T=40°C, получая 86,7 кг (91,3%) продукта.
c) Получение:
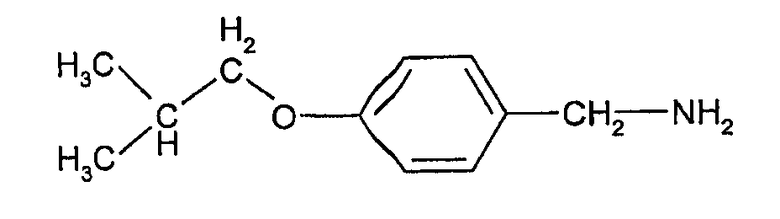
Продукт стадии b) (исходное вещество, 40,0 кг) растворяли в 100% этаноле (229,5 кг, 7,26 - объем/масса исходное вещество) при T=20-25°C, затем в атмосфере азота добавляли безводный никель Ренея (5,8 кг, 14,6% - масса/масса исходное вещество) (катализатор промывали 100% этанолом до достижения содержания KF<300 частей на миллион) и охлаждали суспензию до T=-8 - -12°C. В вакууме через трубку в течение ~8 часов вводили газообразный аммиак (45,8 кг, 13 экв.), затем нагревали суспензию до T=48-50°C (внутреннее давление повышалось до ~2,5 бар). Реакционную массу гидрировали при T=48-50°C и P=4 бар до прекращения поглощения водорода (~9 часов) и контролировали содержание исходного вещества ВЭЖХ (исходное вещество <0,5%). Суспензию охлаждали до T=10-15°C, удаляли избыток аммиака, удаляли твердые частицы из раствора (1575 + фильтровальная бумага GF92 + слой celtrox на фильтре) и промывали установку 100% этанолом (63,4 кг, 2,00 -объем/масса исходное вещество). Растворитель отгоняли при пониженном давлении (P=870-13 мбар, T=42-50°C) и зеленый маслянистый осадок разбавляли 100% этанолом (50,7 кг, 1,60 -объем/масса исходное вещество) и этилацетатом (150,1 кг, 4,17 -объем/масса исходное вещество) и, наконец, охлаждали до T=20-25°C. Медленно добавляли уксусную кислоту (19,9 кг, 1,60 экв.), давая вырасти температуре в процессе добавления (+~14°C), затем нагревали реакционную массу до кипения с обратным холодильником (T=~70°C) для достижения полного растворения. Раствор охлаждали до 40-42°C и вносили затравку кристаллов, после чего суспензию перемешивали при температуре кристаллизации (T=~41°C) в течение 30 мин, охлаждали до T=0-5°C и перемешивали при этой температуре 5 часов. Реакционную массу центрифугировали, лепешку осадка промывали холодным этилацетатом (2×9,4 кг, 2×0,26 - объем/масса исходное вещество) и, наконец, высушивали в вакууме при T=50°C, получая 33,6 кг (67,9%) уксуснокислой соли амина.
Раствор уксуснокислой соли амина (26,4 кг) в питьевой воде (42,2 кг, 1,60 объема) подщелачивали 30% гидроксидом натрия (35,4 кг, ~2,41 экв.) до pH=14 при T=10-25°C, затем продукт экстрагировали толуолом (91,4 кг, 4,00 объема) при T=43-47°C. Реакционной массе давали отстояться при T=43-47°C, доводили pH до 14 с помощью дополнительного количества 30% NaOH при необходимости, затем разделяли фазы. Органическую фазу промывали питьевой водой (35,1 кг, 1,33 объема), затем концентрировали досуха в вакууме (P=170-20 мбар, приблизительно) при T=48-50°C, получая продукт в виде маслянистого остатка.
d) Получение:

Продукт стадии c) растворяли в безводном толуоле (68,5 кг, KF<300 ч/млн, 3,00 объема), раствор переносили в реактор фосгенирования, оборудованный газоочистителем, и установку промывали безводным толуолом (10,3 кг, 0,45 объема). Раствор в толуоле охлаждали до T=0-5°C и медленно по трубке вводили хлористый водород (газообразный, 4,0 кг, 1,00 экв.) в течение ~3 часов при максимальной T=10°C. В конце введения реакционную массу нагревали до 97-103°C и медленно (~3 часов) вводили по трубке фосген (16,6 кг, 1,5 экв.). После окончания введения реакционную смесь перемешивали еще 30 минут при T=97-103°C, прохождение реакции контролировали IPC (ТСХ, исходное вещество <1%) и реакционную массу охлаждали до T=80-85°C. Раствор концентрировали в вакууме (P=500 мбар, приблизительно) при той же температуре до ~2,1 объема, реакционную массу исследовали для подтверждения отсутствия остаточного фосгена, и неочищенный раствор изоцианата охлаждали до T=20-25°C, выливали в цилиндрическую емкость и анализировали.
e) Получение указанного в заглавии соединения формулы (IV):
Продукт стадии d) (~30% раствор в толуоле, 1 экв.) добавляли в течение ~40 мин при T=38-42°C к раствору продукта стадии a) (исходное вещество, 21,8 кг) в ТГФ (189,5 кг, 9,80 - объем/масса исходное вещество). По завершении добавления установку промывали ТГФ (9,0 кг, 0,50 - объем/масса исходное вещество), реакционную массу перемешивали при T=38-42°C до получения прозрачного раствора (~3 часа) и отбирали образец для IPC (ТСХ, Aca-11-фторамин <1%) для проверки полноты образования мочевины. Растворитель отгоняли при пониженном давлении (P=170-70 мбар, T=22-25°C) и твердый остаток растворяли в 100% этаноле (132,5 кг, 7,69 - объем/масса исходное вещество) при T=40-45°C. При T=40-45°C добавляли предварительно полученный раствор L-(+)-винной кислоты (8,1 кг, 1,10 экв.) в 100% этаноле (96,0 кг, 5,57 - объем/масса исходное вещество) и установку промывали 100% этанолом (3,3 кг, 0,19 - объем/масса исходное вещество). Раствор охлаждали до 35-38°C и вносили затравку кристаллов, суспензию перемешивали при температуре кристаллизации (T=~37°C) в течение 30 мин, охлаждали до T=0-5°C в течение ~2 часов и в завершении перемешивали при этой температуре еще 2 часа. Реакционную массу центрифугировали, лепешку осадка промывали холодным 100% этанолом (2×18,9 кг, 2×0,65 - объем/масса исходное вещество) и рассчитывали сухую массу неочищенного продукта на основе LOD (~46%). (LOD - потеря при высушивании).
Неочищенную виннокислую соль (36,7 кг, исходное вещество, масса в сухом состоянии рассчитана на основе измеренной LOD) растворяли при кипячении с обратным холодильником (T=~75°C) в 100% этаноле (205,4 кг, 7,08 - объем/масса исходное вещество, включая спирт, содержащийся во влажном продукте), после чего раствор фильтровали при температуре кипения с обратным холодильником через абсолютный 0,3 мк картридж и установку промывали горячим 100% этанолом (5,9 кг, 0,21 - объем/масса исходное вещество). Раствор охлаждали до 48-50°C и вносили затравку кристаллов, суспензию перемешивали при температуре кристаллизации (T=~49°C) в течение 30 мин, охлаждали в течение ~2 часов до T=20-22°C и, наконец, перемешивали при этой температуре еще 2 часа. Реакционную массу центрифугировали, лепешку осадка промывали предварительно фильтрованным холодным 100% этанолом (2×18,9 кг, 2×0,65 - объем/масса исходное вещество) и продукт высушивали в вакууме при T=45°C не менее 60 часов.
Суспензию соединения формулы (IV) (исходное вещество, 26,5 кг) в предварительно фильтрованном и дегазированном метилэтилкетоне (149,3 кг, 7,00 объемов) нагревали до T=58-63°C и перемешивали при этой температуре в течение 8 часов в атмосфере азота. Образцы для IPC (рентгенограмма порошка, DCS, ИК) отбирали через каждые 2 часа перемешивания. Смесь охлаждали до T=12-17°C в течение ~4,5 часов и перемешивали при этой температуре в течение ~2 часов, затем продукт центрифугировали и лепешку осадка промывали холодным (15°C) предварительно фильтрованным и дегазированным метилэтилкетоном (2×10,7 кг, 2×0,50 объемов). Влажный продукт высушивали ~15 часов в вакууме при T=45°C, выгружали и упаковывали в атмосфере азота, получая 25,2 кг (51,1%) формы C указанного в заглавии соединения формулы (IV).

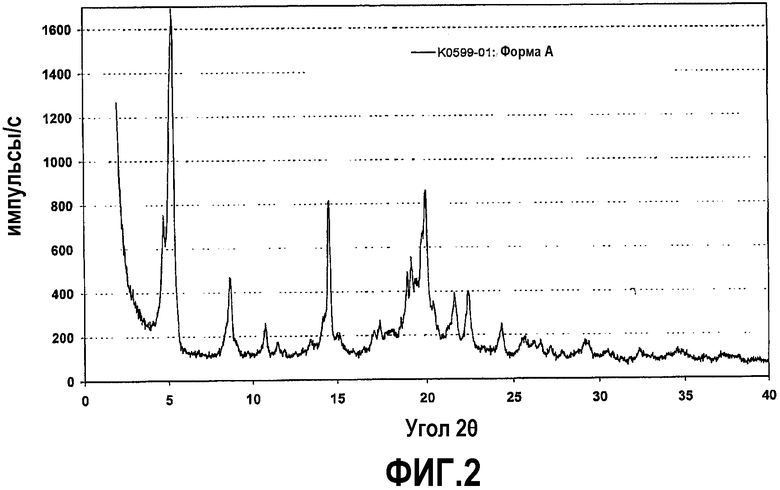
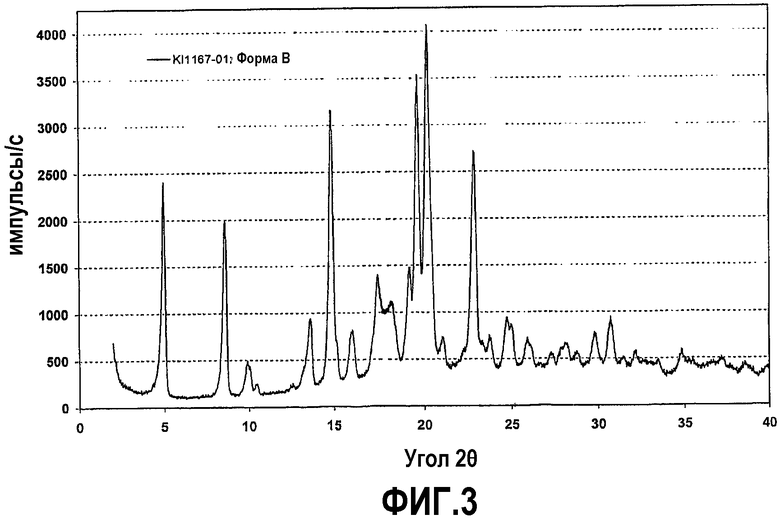
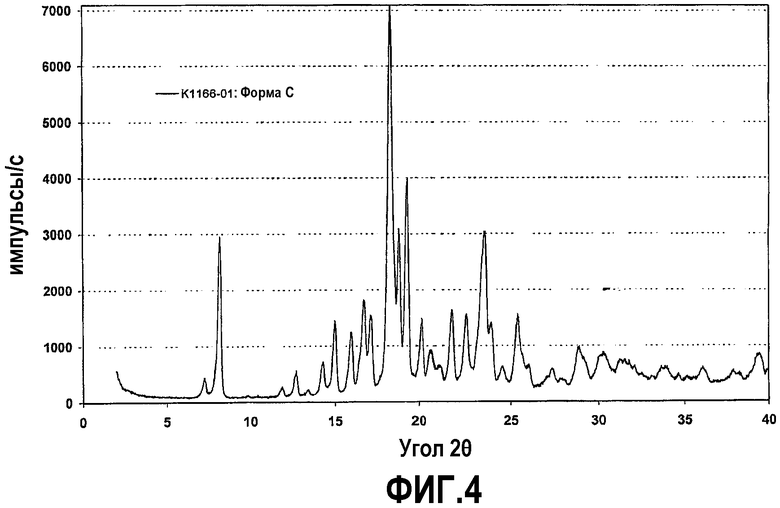
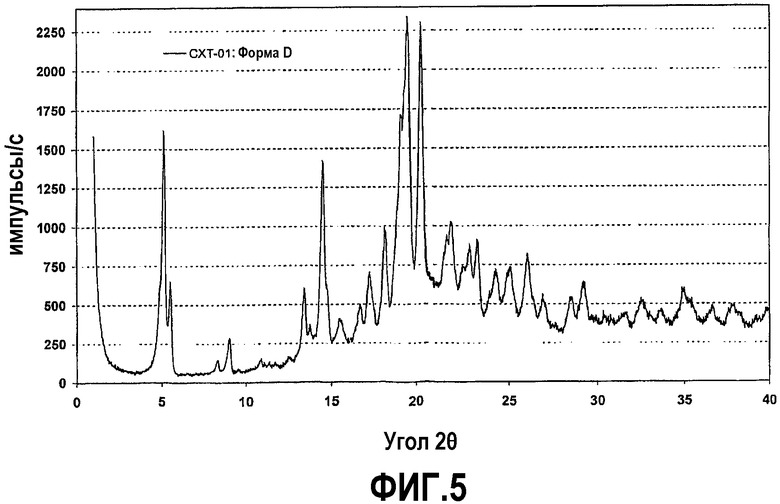
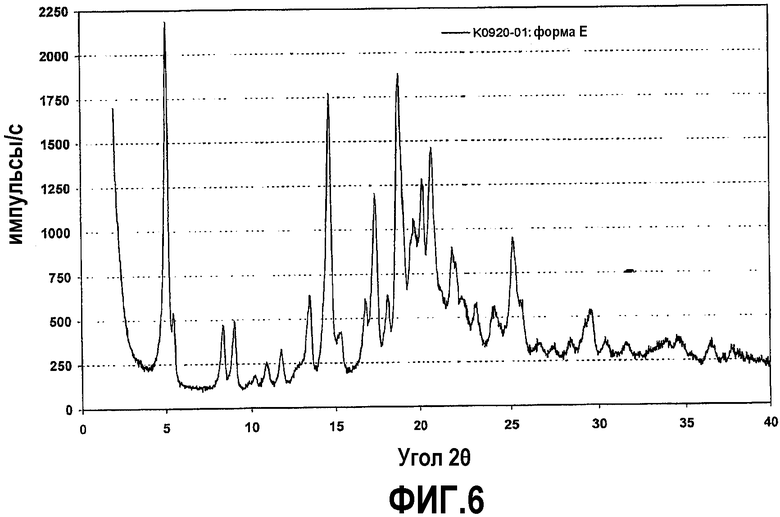
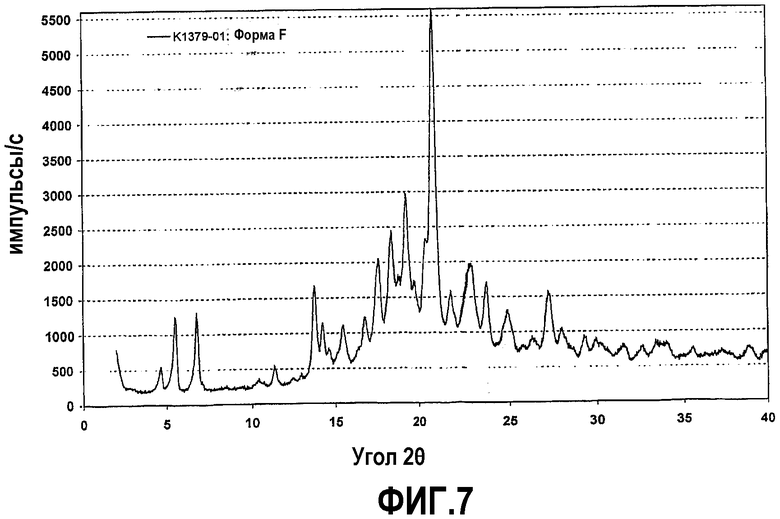
Изобретение относится к кристаллической форме тартрата N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида формулы (IV)
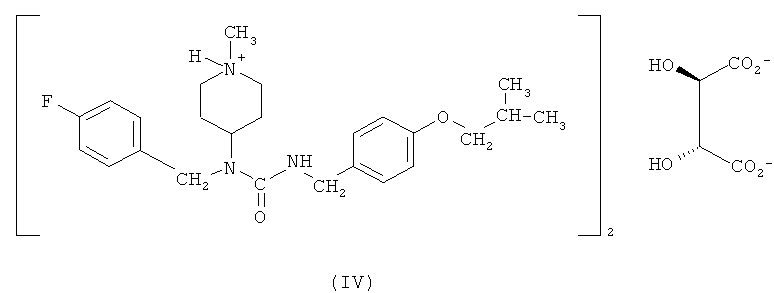 ,
,
где кристаллическая форма является по крайней мере 80% чистой кристаллической формой С, рентгенограмма на порошке которой включает пики, соответствующие величинам d в ангстремах - приблизительно 10,7, приблизительно 4,84, приблизительно 4,57 и приблизительно 3,77.
Изобретение также относится к способам получения кристаллической формы по п.1, к фармацевтической композиции, к способу доставки соединения формулы (I) реципиенту, к способу ингибирования активности моноаминового рецептора, а также к применению соединений по п.1.
Технический результат - получение новой кристаллической формы тартрата N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида формулы (IV), обладающей активностью ингибитора или обратного агониста моноаминового рецептора. 16 н. 23 з.п. ф-лы, 9 табл., 7 ил.
1. Кристаллическая форма тартрата N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида формулы (IV)
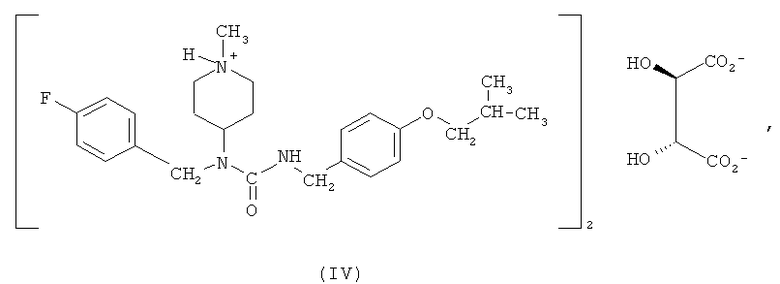
где кристаллическая форма является по крайней мере 80% чистой кристаллической формой С, рентгенограмма на порошке которой включает пики, соответствующие величинам d в ангстремах приблизительно 10,7, приблизительно 4,84, приблизительно 4,57 и приблизительно 3,77.
2. Кристаллическая форма по п.1, рентгенограмма на порошке которой включает пики, соответствующие величинам d в ангстремах приблизительно 10,7, приблизительно 4,84, приблизительно 4,57 и приблизительно 3,77.
3. Способ получения кристаллической формы по п.1, включающий суспендирование твердой формы соединения формулы (IV) в апротонном растворителе:
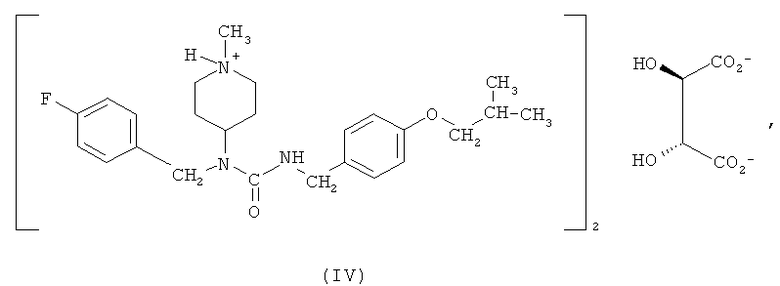
перемешивание полученной суспензии при добавлении затравки кристаллов кристаллической формы С и выделение кристаллической формы С из суспензии.
4. Способ по п.3, в котором температура растворителя на стадии суспендирования составляет от приблизительно 30 до приблизительно 100°С.
5. Способ по п.3, в котором апротонный растворитель выбран из группы, включающей один или несколько растворителей из числа следующих: алифатические или циклические простые эфиры, сложные эфиры карбоновых кислот, лактоны, алканы и алифатические С3-C8 кетоны.
6. Способ по п.3, в котором внесение затравочных кристаллов выполняют при температуре от приблизительно 40 до приблизительно 80°С.
7. Способ по п.3, дополнительно включающий охлаждение суспензии со скоростью от приблизительно 0,1 до приблизительно 1°С/мин.
8. Способ по п.7, в котором суспензию охлаждают приблизительно до комнатной температуры.
9. Способ получения кристаллической формы по п.1, включающий суспендирование кристаллической формы тартрата N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида или смесей кристаллических форм тартрата N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида в полярном и апротонном растворителе при температурах от приблизительно 30 до приблизительно 70°С;
перемешивание полученной суспензии при добавлении затравочных кристаллов кристаллической формы С и
выделение твердого кристаллического вещества из суспензии.
10. Способ получения кристаллической формы по п.1, включающий растворение тартрата N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида в растворителе при температурах от приблизительно 0 до приблизительно 70°С;
перемешивание полученного раствора при температуре от приблизительно 50 до приблизительно 70°С при добавлении затравочных кристаллов кристаллической формы С;
охлаждение полученной суспензии со скоростью от приблизительно 5 до приблизительно 15°С/ч до температуры от приблизительно -20°С до приблизительно комнатной температуры и выделение твердого кристаллического вещества из суспензии.
11. Способ по п.10, в котором растворителем является тетрагидрофуран.
12. Способ по п.10, в котором растворитель выбран из группы, состоящей из ацетона, дихлорметана, 1,4-диоксана, этанола, изопропанола и ацетонитрила.
13. Кристаллическая форма тартрата N-(4-фторбензил)-N-(1-метилпиперидин-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида, полученная способом, включающим
суспендирование кристаллической формы тартрата N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида или смесей кристаллических форм тартрата N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида в полярном и апротонном растворителе при температурах от приблизительно 30 до приблизительно 70°С;
перемешивание суспензии при добавлении затравки кристаллов кристаллической формы по п.1 и
выделение твердого кристаллического вещества из суспензии.
14. Кристаллическая форма тартрата N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида, полученная способом, включающим
растворение тартрата N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида в тетрагидрофуране или ацетоне при температурах от приблизительно 0 до приблизительно 70°С;
перемешивание полученного раствора при температуре от приблизительно 50 до приблизительно 70°С при добавлении затравочных кристаллов кристаллической формы по п.1;
охлаждение полученной суспензии со скоростью от приблизительно 5 до приблизительно 15°С/ч до температуры от приблизительно -20°С до приблизительно комнатной температуры и выделение твердого кристаллического вещества из суспензии.
15. Фармацевтическая композиция, обладающая активностью ингибитора или обратного агониста моноаминового рецептора, содержащая кристаллическую форму по п.1, а также фармацевтически приемлемый носитель или разбавитель.
16. Способ доставки соединения формулы (I) реципиенту:
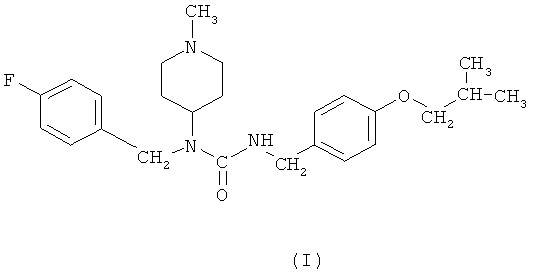
включающий введение субъекту соединения по п.1.
17. Способ ингибирования активности моноаминового рецептора, включающий введение субъекту соединения по п.1.
18. Применение соединения по п.1 для получения лекарственного средства для лечения нейропсихиатрических заболеваний.
19. Применение по п.18, в котором нейропсихиатрическое заболевание выбрано из группы, состоящей из психоза, шизофрении, шизоаффективных расстройств, мании, психотической депрессии, аффективных расстройств, деменции, тревожного состояния, расстройств сна, расстройств аппетита, биполярного расстройства, психоза, вызванного гипертензией, мигрени, спазма сосудов, ишемии, двигательных тиков, тремора, психомоторной заторможенности, брадикинезии и невропатической боли.
20. Применение соединения по п.1 для получения лекарственного средства для лечения нейродегенеративных заболеваний.
21. Применение по п.20, где нейродегенеративное заболевание выбрано из группы, состоящей из болезни Паркинсона, болезни Хантингтона, болезни Альцгеймера, спиноцеребеллярной атрофии, синдрома Туретта, атаксии Фридрайха, болезни Мачадо-Джозефа, деменции с тельцами Леви, дистонии, прогрессирующего супрануклеарного паралича и лобно-височной деменции.
22. Применение соединения по п.1 для получения лекарственного средства для лечения дискинезии, связанной с допаминергической терапией.
23. Применение соединения по п.1 для получения лекарственного средства для лечения дистонии, миоклонуса или тремора, связанных с допаминергической терапией.
24. Применение соединения по п.1 для получения лекарственного средства для лечения тромботического состояния.
25. Применение по п.24, в котором тромботическое состояние выбрано из группы, состоящей из инфаркта миокарда, тромботического удара, ишемического удара, идиопатической тромбоцитопенической пурпуры, тромботической тромбоцитопенической пурпуры, заболеваний периферических сосудов и болезни Рейно.
26. Применение соединения по п.1 для получения лекарственного средства для лечения психоза при болезни Паркинсона, психоза при болезни Альцгеймера, шизофрении или для ингибирования активности моноаминового рецептора.
27. Кристаллическая форма по п.1, где кристаллическая форма является по крайней мере 90% или более чистой кристаллической формой.
28. Кристаллическая форма по п.1, где кристаллическая форма является по крайней мере 95% или более чистой кристаллической формой.
29. Кристаллическая форма по п.1, где кристаллическая форма является по крайней мере 98% или более чистой кристаллической формой.
30. Кристаллическая форма по п.1, которая приводит к потере массы менее 0,9% ниже 150°С, как измерено термогравиметрическим анализом.
31. Кристаллическая форма тартрата N-(4-фторбензил)-N-(1-метилпиперидин-4-ил)-N'-(4-(2-метилпропилокси)фенилметил)карбамида формулы (IV)
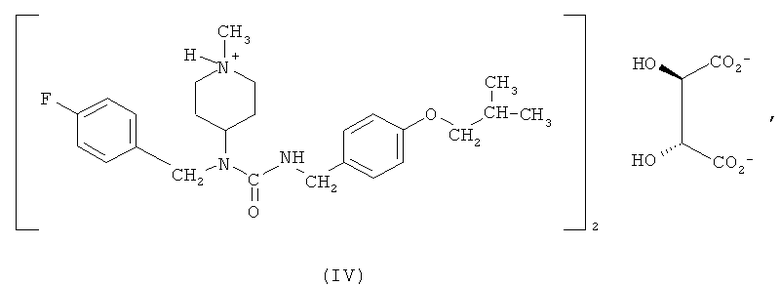
которая показывает эндотермический сигнал при 177°С при измерении дифференциальной сканирующей калориметрией.
32. Кристаллическая форма по п.31, где эндотермический сигнал при 177°С имеет энтальпию плавления приблизительно 129 Дж/г.
33. Кристаллическая форма по п.1, которая содержит менее 1000 ч./млн остаточного растворителя.
34. Кристаллическая форма по п.1, которая имеет кристаллы с размером частиц в диапазоне от 1 мкм до приблизительно 200 мкм.
35. Кристаллическая форма по п.1, где кристаллическая форма показывает эндотермический сигнал приблизительно при 133-135°С при энтальпии плавления приблизительно 70 Дж/г, как определено дифференциальной сканирующей калориметрией.
36. Кристаллическая форма по п.1, где кристаллическая форма имеет температуру плавления, равную приблизительно 131°С при энтальпии плавления, равной приблизительно 63 Дж/г, как определено дифференциальной сканирующей калориметрией.
37. Кристаллическая форма по п.1, где кристаллическая форма показывает эндотермический сигнал при 177°С при энтальпии плавления приблизительно 129 Дж/г, как определено дифференциальной сканирующей калориметрией.
38. Кристаллическая форма по п.1, где остаток 80% чистой кристаллической формы С включает смесь других кристаллических или аморфных форм соединения формулы (IV).
39. Композиция по п.15, где остаток 80% чистой кристаллической формы С включает смесь других кристаллических или аморфных форм соединения формулы (IV).
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| RU 2002126554 A, 20.03.2004. | |||

