Область техники
Изобретение относится к медицине и фармакологии, конкретно к синтетическим биологически активным веществам гетероциклического ряда, обладающим противотуберкулезной активностью: производным 1-(1-бензилиндол-3-ил)2,3,4,9-тетрагидро-1-H-β-карболина, а также к их фармакологически приемлемым солям. Вещества предназначены для использования в медицинской практике для лечения заболеваний, вызванных бактериальными агентами, в том числе микобактериями туберкулеза, а также для аналогичных целей в ветеринарии.
Уровень техники
Среди инфекционных заболеваний, в том числе одну из наиболее серьезных проблем представляет туберкулез, лечение которого затруднено из-за высокой устойчивости туберкулезных микобактерий к воздействию химиотерапевтических агентов [Хоменко А.Г. Химиотерапия туберкулеза - история и современность. // Пробл. туб. - 1996. - №3. - С.2-6]. Кроме того, в настоящее время все больше проблем создает распространение устойчивых форм бактериальных штаммов и микобактерий, малочувствительных к существующим лекарственным препаратам [Carole D. Mitnick et al. Comprehensive Treatment of Extensively Drug-Resistant Tuberculosis. // New Engl. J. Med. 2008. - Vol.359. - P.563-574.; Медников Б.Л. Лекарственная устойчивость у Mycobacterium tuberculosis // Пульмонология. - 2005. - №2. - с.5-9].
В 20-м веке были достигнуты серьезные успехи в химиотерапии туберкулеза, особенно в связи с открытием таких высокоэффективных противотуберкулезных средств, как производные изоникотиновой кислоты - изониазид и его аналоги [Преображенский Н.А., Генкин Э.И. Химия органических лекарственных веществ. - М. - Л.: НТИ Хим. Лит., 1953 г.]. В последующие годы в ассортимент противомикробных и противотуберкулезных агентов вошли другие активные препараты, особенно рифампицин, а также этионамид, этамбутол, препараты аминогликозидного ряда и фторхинолоны [Машковский М.Д. Лекарственные средства. - 15-е изд., перераб., испр. и доп. - М.: Новая Волна, 2006. - 1206 с.]. Некоторое время казалось, что наличие таких эффективных средств, как изониазид и рифампицин, в сочетании с другими препаратами, позволит полностью решить проблему лечения туберкулеза. Но в последней четверти 20-го века стало очевидно, что быстрое нарастание частоты лекарственной устойчивости микобактерий становится главной причиной недостаточной эффективности существующих режимов химиотерапии туберкулеза. [Хоменко А.Г. Химиотерапия туберкулеза - история и современность. // Пробл. туб. - 1996. - №3. - С.2-6]. Резистентный к лечению туберкулез стал серьезной проблемой во многих странах. Например, как сообщили финские исследователи, в Мурманске (Россия) 114 (26%) из 439 образцов оказались устойчивы как минимум к двум препаратам - изониазиду и рифампицину. Фактически, 93 образца (82% от всех мультирезистентных) были устойчивы ко всем 4 препаратам первой линии [Soini H, Marjamaki M, Endourova L, et al. Drug resistance surveillance of tuberculosis in Murmansk, Russia. Program and abstracts of the 45th Interscience Conference on Antimicrobial Agents and Chemotherapy; December 16-19, 2005; Washington, DC. Abstract C2-1443]. С 2006 г. зарегистрирована новая форма туберкулеза с резистентностью еще и к фторхинолонам и другим препаратам второго ряда - это уже туберкулез с широкой лекарственной устойчивостью [Carole D. Mitnick et al. Comprehensive Treatment of Extensively Drug-Resistant Tuberculosis. // New Engl. J. Med. 2008. - Vol.359. - P.563-574].
В настоящее время препараты для лечения туберкулеза делят на две группы - основные и резервные [Машковский М.Д. Лекарственные средства. - 15-е изд., перераб., испр. и доп. - М.: Новая Волна, 2006. - 1206 с.]:
- препараты основной группы - изониазид и его производные (фтивазид, салюзид, метазид), рифампицин, пиразинамид, этамбутол, стрептомицин и его аналоги;
- резервные препараты - этионамид, протионамид, циклосерин, канамицин, флоримицин, парааминосалициловая кислота (ПАСК) и ее производные, а также фторхинолоны - офлоксацин, ципрофлоксацин и др.
Одним из недостатков препаратов основной группы является развитие к ним лекарственной устойчивости.
Резервные препараты для самостоятельного применения практически не применимы, так как основное их действие это профилактика развития множественной лекарственной устойчивости (МЛУ).
В этой связи особо остро стоит проблема изыскания новых противотуберкулезных препаратов, обладающих достаточной эффективностью, особенно по отношению к резистентным штаммам микобактерий. Однако добиться прорыва в данном направлении пока не удалось, так как не было создано новых препаратов, сопоставимых по своей противотуберкулезной активности с изониазидом или рифампицином [М.А.Карачунский. Туберкулез в наши дни. - http://med-lib.ru/speclit/ftiz/15.php]. Именно долгое применение противотуберкулезных препаратов привело к широкому развитию множественной лекарственной устойчивости. Данную проблему пытаются решить с помощью одновременного применения нескольких препаратов.
Нами было обнаружено, что заявляемые вещества по своему химическому строению принадлежат к группе β-карболинов, конкретно к замещенным 1-(1-бензилиндол-3-ил)-2,3,4,9-тетрагидро-1-H-β-|3-карболинам. Среди производных β-карболинового ряда известно немало лекарственных препаратов психотропного, нейротропного, противосудорожного действия и др. [Дуленко В.И., Комиссаров И.В., Долженко А.Т., Николюкин Ю.А. β-Карболины. Химия и нейробиология. - Киев: Наукова думка. - 1992. - 216 с.], но противотуберкулезная активность β-карболинов рассмотрена гораздо меньше. Так, например, слабым антимикобактериальным действием обладают гармин, гармалин и родственные алкалоиды [http://www.taacf.org/downloads/TAACF%20-%20public%20data%20-%20no%20structures.pdf - Antibacterial Activity of a Library of Compounds Against Mycobacterium tuberculosis H37Rv]; имеются сообщения о перспективной антимикобактериальной активности каркасных гетероциклических систем, содержащих β-карболиновый фрагмент [Noren-Muller A., Wilk W., Saxena К., Schwalbe H., Kaiser M., Waldmann H. // Angew. Chem. Intern. Ed. - 2009. - V.42. - N.47. - P.5973], но противотуберкулезных препаратов, используемых в медицинской практике, на основе производных β-карболина до настоящего времени не было создано. При этом следует отметить, что не только уровень противотуберкулезной активности отличает заявляемое вещество от известных производных β-карболина, но и характер заместителей в гетероциклической системе делает его в химическом отношении принципиально непохожим на другие производные β-карболина, описанные в литературе.
Также предполагается, что полезными при туберкулезе могут быть ингибиторы lkB киназы, например описываемые общей формулой производные β-карболина:
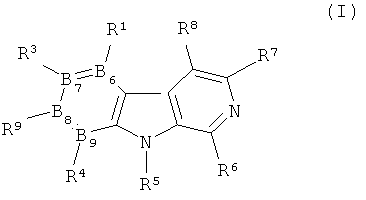
(патенты США 6627637, 7026331, 7348336). Однако в указанных публикациях данные испытаний отсутствуют.
Таким образом, в качестве прототипа выбран гидразид изоникотиновой кислоты (изониазид) [Boyd M.R. The future of new drug development. // Current therapy in oncology. - 1992. - P.11-22]. Выбор основан на его совпадении с заявляемым веществом по наиболее важному назначению - противотуберкулезному действию/клиническому эффекту. Изониазид является одним из наиболее эффективных противотуберкулезных препаратов, может использоваться в любых лекарственных формах (таблетки, инъекции, ингаляции), при этом по своему химическому строению объект изобретения и прототип принадлежат к классу азотистых гетероциклов.
Задача изобретения
Задачей изобретения является получение структурно новых химических соединений, обладающих высокой противотуберкулезной активностью, особенно в отношении штаммов микобактерий, резистентных к веществам, указанным в прототипе - изониазиду и не только. Другими словами, задача изобретения сводится к химическому синтезу нового биологически активного вещества, превосходящего прототип по противотуберкулезной активности и по широте действия.
Существует большая медицинская потребность в новых противотуберкулезных лекарствах с более коротким, чем существующие, курсом лечения и требующие меньшего надзора со стороны медицинского персонала. Кроме того, требуются лекарства, которые смогут (возможно, в комплексе с другими лекарствами) полностью уничтожить микобактерии и убрать риск рецидива.
Очевидно, что наибольшую пользу принесет лекарство, которое снизит срок лечения и количество приемов лекарства пациентом, а также будет активно к лекарственно резистентным штаммам микобактерий туберкулеза.
Сущность изобретения
Поставленная задача решается путем синтеза производных 1-(1-бензилиндол-3-ил)2,3,4,9-тетрагидро-1-H-β-карболина общей формулы 1:

где R1 и R3 - одинаковые или различные, выбраны из группы: H, галоген, метоксигруппа, бензилоксигруппа; R2 - выбран из группы: H, галоген, нитрогруппа, метоксигруппа; R4 - H, или C1-С6 алкил или их солей с фармакологически приемлемыми кислотами.
Предпочтительно R4 представляет собой метил или этил.
В качестве фармакологически приемлемых кислот могут быть выбраны, например, соляная, малеиновая, бензойная, серная, лимонная и т.п.
Галоген означает хлор, фтор, бром или иод.
В качестве предпочтительных соединений могут быть указаны:
- соединение формулы 1, где R1=R2=R3=R4=Н (I)
- соединение формулы 1, где R1=F, R2=R3=R4=Н (II)
- соединение формулы 1, где R1=Cl, R2=R3=R4=Н (III)
- соединение формулы 1, где R1=ОСН3, R2=R3=R4=Н (IV)
- соединение формулы 1, где R1=ОСН2С6Н5, R2=R3=R4=Н (V)
- соединение формулы 1, где R1=R3=R4=Н, R2=F (VI)
- соединение формулы 1, где R1=R3=R4=Н, R2=Cl (VII).
- соединение формулы 1, где R1=R3=R4=Н, R2=ОСН3
- соединение формулы 1, где R1=R3=R4=Н, R2=NO2 (IX).
- соединение формулы 1, где R1=R2=R4=Н, R3=F (X).
- соединение формулы 1, где R1=R2=R4=Н, R3=Cl (XI).
- соединение формулы 1, где R1=R2=R4=Н, R3=ОСН3 (XII).
- соединение формулы 1, где R1=R2=R4=Н, R3=ОСН2С6Н5 (XIII).
- соединение формулы 1, где R1=R2=R3=Н, R4=СН3 (XIV).
- соединение формулы 1, где R1=R2=R3=Н, R4=СН2СН3 (XV).
- соединение формулы 1, где R1=R2=R3=R4=Н в виде солянокислой соли (XVI).
- соединение формулы 1, где R1=R2=R3=R4=Н в виде сульфата (XVII).
- соединение формулы 1, где R1=R2=R3=R4=H в виде соли бензойной кислоты (XVIII).
- соединение формулы 1, где R1=R2=R3=R4=Н в виде соли малеиновой кислоты (XIX).
Заявляемое вещество является новым, поскольку оно не известно из доступных источников информации.
Заявляемое решение является неочевидным, поскольку оно никаким образом не вытекает из современного уровня техники. Заявляемые вещества относятся к группе β-карболинов, среди которых до настоящего времени не создано ни одного противотуберкулезного препарата, использующегося в медицине. Тем более нельзя заранее ожидать высокой противотуберкулезной активности у новых производных β-карболина, замещенных по атому C(1) бензилиндольным фрагментом.
Раскрытие изобретения
Предлагаемый синтез заявляемых веществ состоит из четырех основных этапов.
1) Синтезируют производное 1-бензилиндола (XXII) из производного индола (XX) и производного бензилхлорида (XXI) в присутствии щелочи и межфазного катализатора по общему методу [Жунгиету Г.И., Суворов Н.Н., Кост А.Н. Новые препаративные синтезы в индольном ряду. - Кишинев, «Штиинца». 1983. - 117 с.].
2) Синтезируют производное 1-бензил-индол-3-альдегида (XXIII) из полученного на первом этапе производного 1-бензилиндола (XXII) по общему методу [Жунгиету Г.И., Будылин В.А., Кост А.Н. Препаративная химия индола. - Кишинев, «Штиинца», 1975. - 274 с.].
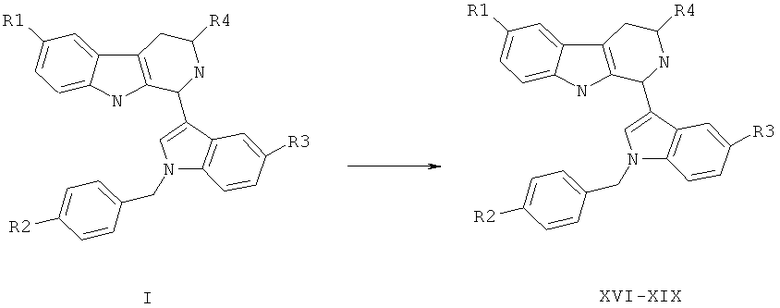
3) Синтезируют целевое вещество из полученного на втором этапе производного 1-бензил-индол-3-альдегида (XXIII) и соответствующего производного триптамина (XXIV).
4) Синтезируют целевое вещество в виде соли из полученного на третьем этапе вещества (I) и кислоты.
Сущность изобретения поясняется приведенными ниже примерами синтеза промежуточных веществ, примерами синтеза заявленного вещества, таблицами выходов и характеристик целевых веществ, экспериментами по изучению биологических свойств и таблицами результатов экспериментов по определению биологических свойств заявляемых веществ,
где примеры 1 и 2 - конкретные варианты выполнения 1-го и 2-го этапов синтеза заявляемого вещества (получение промежуточных производных 1-бензилиндола (XXII) и 1-бензил-индол-3-альдегида (XXIII));
примеры 3 и 4 - конкретные варианты выполнения 3-го и 4-го этапов синтеза заявляемого вещества (получение целевых веществ (I-XV) и (XVI-XIX));
эксперимент 1 - определение антимикобактериальной активности;
эксперимент 2 - определение активности заявляемых веществ против резистентных микобактериальных штаммов;
эксперимент 3 - определение терапевтической эффективности заявляемых веществ при экспериментальном туберкулезе in vivo;
эксперимент 4 - определение острой токсичности;
таблица 1 - выход и температуры плавления целевых веществ (I-XIX);
таблица 2 - данные спектров 1H ЯМР целевых веществ (I-XIX);
таблица 3 - данные элементного анализа целевых веществ (I-XIX);
таблица 4 - результаты определения антимикобактериальной активности заявляемых веществ in vitro в сравнении со стандартами;
таблица 5 - результаты определения активности заявляемых веществ против резистентных микобактериальных штаммов;
таблица 6 - результаты определения терапевтической эффективности заявляемых веществ при экспериментальном туберкулезе in vivo;
таблица 7 - результаты определения определение острой токсичности заявляемых веществ.
Пример 1. Вариант выполнения 1-го этапа синтеза заявляемого вещества (получение промежуточных производных 1-бензилиндола (XXII)).
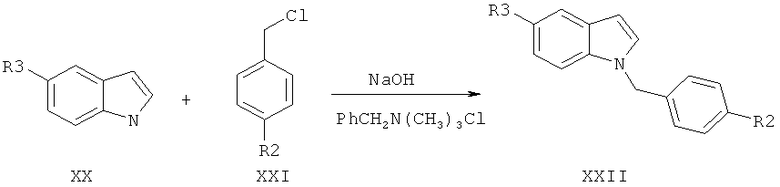
К смеси 15,2 г (0,12 моль) хлористого бензила (XXI, R2=H) и 0,2 г хлористого триметилбензиламмония прибавляют 11,7 г (0.1 моль) индола (XX, R3=H) и 40 мл 50%-ного раствора едкого натра. Перемешивают 5 часов при 70°C, разбавляют 100 мл воды и извлекают эфиром 3×100 мл. Эфирные вытяжки промывают водой и сушат безводным сульфатом магния. Эфир отгоняют и получают 19,2 г масла, которое при стоянии кристаллизуется. Получают 1-бензилиндол (XXII, где R2=R3=H) с выходом 96% от теоретического.
По аналогичной методике получают производные 1-бензилиндола (XXII), где R2=Н, R3=F, Cl, ОСН3 или ОСН2С6Н5; R3=Н, R2=F, Cl, ОСН3 или NO2.
Пример 2. Вариант выполнения 3-го этапа синтеза заявляемого вещества (получение промежуточных производных 1-бензилиндол-3-альдегида (XXIII)).

Растворяют 20,7 г (0,1 моль) 1-бензилиндола (XXII, R2=R3=Н) в 100 мл сухого диметилформамида (DMF). К этому раствору по каплям, при охлаждении приливают раствор 10,7 г (0,1 моль) хлорокиси фосфора в 30 мл DMF, поддерживая температуру смеси не выше 10°C. После этого выдерживают смесь при комнатной температуре 1 ч и выливают в 400 г измельченного льда. Выделившийся осадок отделяют и промывают 3 раза по 100 мл воды, затем растворяют в 70 мл спирта и высаживают 300 мл воды. После сушки получают 1-бензилиндол-3-альдегид (XXIII, где R2=R3=H) в виде светло-коричневых кристаллов с Тпл. 113°C, выход 95% от теоретического.
По аналогичной методике получают производные 1-бензилиндол-3-альдегида (XXIII), где R2=Н, R3=F, Cl, ОСН3 или ОСН2С6Н5; R3=Н, R2=F, Cl, OCH3 или NO2.
Пример 3. Вариант выполнения 3-го этапа синтеза заявляемого вещества (получение целевых веществ (I-XV).

Растворяют 23,5 г (0,1 моль) 1-бензилиндол-3-альдегида (XXIII, R2=R3=Н) и 16,0 г триптамина (XXIV, R1=R4=Н) в 200 мл ледяной уксусной кислоты и нагревают 72 ч при 70°C. Затем реакционную смесь охлаждают до комнатной температуры и выливают в 1 л воды. Выделившийся смолистый продукт отделяют фильтрованием через угольный фильтр, а фильтрат подщелачивают водным аммиаком до pH 9. Сформировавшийся осадок отфильтровывают, промывают водой и обрабатывают 100 мл 5%-ного водного раствора уксусной кислоты. Через 12 ч водно-уксуснокислый раствор фильтруют от не растворившегося осадка и фильтрат подщелачивают аммиаком до рН 9. Осадок отфильтровывают, промывают 1%-ным раствором аммиака и сушат в вакуум-эксикаторе над KOH. Получают 5.65 г целевого продукта (I) в виде светло-желтых кристаллов.
По аналогичной методике получают целевые вещества (II-XV), выход и Тпл. которых приведены в Таблице 1, данные спектров 1H ЯМР - в Таблице 2, данные элементного анализа - в Таблице 3.
Пример 4. Вариант выполнения 4-го этапа синтеза заявляемого вещества (получение целевых веществ (XVI-XIX).
Растворяют 3,77 г (0,01 моль) вещества (I) в 50 мл абсолютного эфира и приливают к этому раствору по каплям, при перемешивании, насыщенный эфирный раствор HCl. Добавляют гексан, выделившийся осадок отфильтровывают, промывают гексаном и сушат в вакуум-эксикаторе. Получают 3,8 г целевого продукта (XVI) в виде бесцветных кристаллов.
По аналогичной методике получают целевые вещества (XVII-XIX), выход и Тпл. которых приведены в Таблице 1, данные спектров 1H ЯМР - в Таблице 2, данные элементного анализа - в Таблице 3.
Температура плавления (Тпл.) и выход целевых веществ (I-XIX) приведены в таблице 1.
Данные спектров 1Н ЯМР целевых веществ (в CDCCl3 (I-XV) и в ДМСО-d6 (XVI-XIX)) приведены в таблице 2.
3H); 1.64 м (СН2, 2Н)
Данные элементного анализа целевых веществ (I-XIX) приведены в таблице 3.
Экспериментальная оценка биологической активности заявляемых веществ.
Эксперимент 1
Тестирование антимикробных свойств заявляемых веществ в in vitro опытах.
Для тестирования антимикробных свойств заявляемых веществ используют стандартный штамм Mycobacterium tuberculosis H37Rv. Вышеуказанный штамм чувствителен ко всем известным антимикробным препаратам.
Для выращивания указанного штамма использована среда Сотона, которая содержит 10% лошадиной сыворотки.
С использованием методики серийных разведений была произведена оценка антимикобактериальных свойств заявляемых веществ.
Для этого тестируемые вещества были растворены в диметилсульфоксиде (ДМСО). Тестируемые вещества были титрованы в среде N-1 так, что каждое из исследуемых веществ содержалось в пробирках со средой в концентрациях 20-0.025 мг/л.
Концентрация тестируемых веществ в соседних пробирках отличалась в 2 раза.
Для контроля был использован ДМСО. ДМСО был титрован так же, как и тестируемые вещества. Тест-штамм микобактерий добавляли в среду в количестве 50×106 на 1 мл.
В качестве препаратов сравнения использовали известные противотуберкулезные препараты. Результаты тестирований веществ приведены в Таблице 4.
Данные, приведенные в Таблице 4, показывают, что активность у ряда заявляемых веществ in vitro не уступает, а порой превосходит прототип и другие препараты сравнения.
Минимальная ингибирующая концентрация (МИК) по отношению к M.tuberculosis H37Rv приведена в таблице 4.
Эксперимент 2.
Проверка in vitro активности заявляемых веществ против резистентных штаммов микобактерий.
На основе результатов Эксперимента 1 были отобраны самые активные из заявляемых веществ
При проведении опыта были использованы следующие модели:
- штамм Mycobacterium avium 84.
- штамм Mycobacterium tuberculosis 61(S), чувствительный к действию известных антимикробных препаратов;
- штамм Mycobacterium tuberculosis 16(R), устойчивый к основным противотуберкулезным препаратам;
Описываемый опыт был проведен на среде Левенштейна-Йенсена. Проверяемые вещества были растворены в ДМСО и погружены в пробирки со средой в конечной концентрации 10 мг/л. Тест-штаммы были засеяны на поверхность среды, после чего пробирки инкубировали в течение 30 суток. В качестве контроля использовали ДМСО и стандартные противотуберкулезные препараты (Таблица 5).
Полученные результаты, приведенные в Таблице 5, показывают, что изученные вещества in vitro подавляют рост всех использованных в эксперименте штаммов микобактерий, в том числе и штамм M.tuberculosis 16(R), устойчивый к основным противотуберкулезным препаратам. Кроме того, представленные вещества подавляют рост M.avium 84. M.avium 84 вызывает заболевания и гибель ВИЧ-инфицированных лиц.
Выявленная антибактериальная активность заявляемых веществ в концентрации 10,0 г/л по отношению к различным микобактериям приведена ниже в таблице 5.
Эксперимент 3.
Определение действия заявляемых веществ на модели экспериментального туберкулеза in vivo.
Определение специфической противотуберкулезной активности в системе in vivo проводили на самцах инбредных мышей линии A/Sn весом 22-23 г. Мышей инфицировали M.tuberculosis штамма H37Rv с использованием камеры аэрогенного заражения "GlasCol" в дозе 100 КОЕ на легкое.
Начало лечения животных - через 1 месяц после заражения, курс лечения - 45 дней. С момента начала лечения ежедневно вводили мышам исследуемые вещества внутрижелудочно, в дозе 15 мг/кг. Для этого вещества растворяли в воде с добавкой 0.05% TWEEN-80, объем вводимого раствора составлял 0.5 мл на мышь.
В качестве препарата сравнения использовали изониазид в той же дозе, что и исследуемые вещества. В каждой экспериментальной группе животных, включая группу, получавшую изониазид, и контрольную группу, не получавшую никакого лечения, насчитывалось по 5 мышей. В течение эксперимента гибели мышей не наблюдалось ни в одной подопытной группе, включая контроль. Для микробиологических исследований мышей выводили из эксперимента методом цервикальной дислокации.
Для определения количества микобактерий (КОЭ Мтб) в легких зараженных мышей легкие мышей гомогенизировали в 2 мл физиологического раствора, готовили серию десятикратных разведений в физиологическом растворе и 50 мкл каждого разведения помещали в чашку Петри, покрытую агаром Дюбо. Чашки Петри с нанесенными разведениями инкубировали в течение 21 суток при 37°C, после чего подсчитывали число колоний на чашке и определяли количество КОЕ микобактерий в легких. Полученные результаты приведены в таблице 6.
Количество KOE M.tuberculosis штамма H37Rv из легких зараженных мышей после 45 дней лечения приведено в таблице 6.
Как видно из полученных результатов, все тестированные вещества обладают противотуберкулезной активностью in vivo. Наиболее активными являются вещества (I, IV и XVI), которые по своей эффективности сопоставимы с изониазидом.
Эксперимент 4.
Определение острой токсичности заявляемых веществ
Острую токсичность (LD 50) заявляемых веществ определяли на беспородных белых мышах массой 18-20 г при пероральном введении. Препараты вводили с помощью желудочного зонда в различных концентрациях в виде эмульсии на ТВИН-80. Для исследования каждой концентрации использовали по 5 мышей.
Острая токсичность заявляемых веществ (LD 50, перорально, мыши) приведена в таблице 7.
Таким образом при терапевтических концентрациях оказывающих МИК (минимально ингибирующая концентрация), варьирующих в пределах 0,1-10 мг/л, для достижения LD 50, мг/кг, требуется увеличение дозировки от 40 до 80-кратного размера. Эти данные свидетельствуют о низкой токсичности и высоком интервале терапевтической широты (диапазон терапевтических доз, т.е. интервал между пороговой и высшей терапевтическими дозами обозначают термином «терапевтическая широта»).
Промышленная применимость
Приведенные выше примеры 1-4, а также описанные в таблицах 1-4 результаты практического синтеза и анализа подтверждают:
а) объективную возможность промышленного синтеза всех заявляемых веществ методами, на настоящее время освоенными современной фармацевтической промышленностью,
б) возможность контроля подлинности и качества таких веществ общепринятыми методами. Физико-химические свойства заявляемых веществ (стабильность и растворимость) позволяют предполагать возможность их использования в любых лекарственных формах - таблетках, инъекциях и ингаляционных формах, в том числе и в комбинации с другими лекарственными препаратами.
Эксперименты 1-4 по определению биологической активности показали, что заявляемые вещества проявляют противотуберкулезное действие как in vitro, так и in vivo, обладая при этом умеренной токсичностью. Это указывает на возможность их использования для лечения туберкулеза и микобактериозов в широком диапазоне лекарственных дозировок.
Таким образом, полученные результаты свидетельствуют о достижении целей, поставленных изобретением: синтезирован новый класс гетероциклических веществ, обладающих высокой противотуберкулезной активностью, которые новы, неочевидны и промышленно применимы.
Любое из веществ, описанных в настоящей заявке, может быть использовано в любой лекарственной форме. Как примеры подходящих композиций и лекарственных форм могут быть приведены любые примеры композиций или лекарственных форм, обычно применяемые для систематически применяемых лекарств. Возможно создание пероральных, инъекционных и ингаляционных форм заявляемых веществ.
Для приготовления фармацевтической композиции настоящего изобретения, может быть взято эффективное количество конкретного вещества, описанного или покрываемого настоящей заявкой, или его соли, кроме того, возможно в смеси (композиции) с другими веществами. Более удобно, чтобы эти фармацевтические композиции были в стандартизированных дозах, например для перорального применения или парентерального введения.
Из-за удобства применения таблетки и капсулы обладают преимуществом, в них, как правило, применяются твердые фармацевтические носители (транспорт).
Для парентеральных композиций, содержащих в себе любое из описанных в настоящей заявке веществ, носителем может быть стерильная вода, а также и другие ингредиенты, например вещества, улучшающие растворимость. В растворах для инъекций, например, носитель может состоять из физиологического или солевого раствора, раствора глюкозы или смеси из солевого раствора и раствора глюкозы. Суспензии для инъекций могут также изготавливаться в подходящем для каждого конкретного случая или ряда случаев представителя или представителей жидких носителей, суспендирующих средств и подобных веществ и составов.
Также в настоящее изобретение входит твердое вещество, описанное в настоящей заявке или покрываемое ей, которое предполагается перевести незадолго до приема в жидкую форму.
Фармацевтическая композиция может также содержать различные другие ингредиенты, известные из уровня техники, такие как стабилизирующие агенты, смазочный материал, буферизирующий агент, эмульгирующий агент, агент, регулирующий вязкость или липкость, сурфактант или поверхностно-активное вещество, консервант, предохраняющее средство, краситель, вкусовая добавка и тому подобное. Некоторые аспекты настоящего изобретения могут содержать сложный эфир, содержащий, по крайней мере, одно из веществ перечисленных выше или покрываемых настоящим изобретением.
Особенное удобство предоставляет создание описанной выше фармацевтической композиции в виде дозировки (дозированной готовой лекарственной формы) для простоты употребления и единообразия.
Дозировка в данном контексте употребляется как физически отдельная часть применимая для унифицированного употребления. Каждая подобная часть содержит заранее определенное количество активного ингредиента, подсчитанного для получения желаемого терапевтического эффекта вместе с приемлемым фармацевтическим носителем.
Примерами таких дозировок являются таблетки (включая рифленые, отламываемые от большей части таблетки, таблетки в оболочке и т.д.), капсулы, пакетики с порошком, пастилки, пластинки, свечи, суппозитории, инъекционные растворы, а также их различные комбинации.
Некоторые аспекты настоящего изобретения включают в себя фармацевтически приемлемую соль, соль с фармацевтически приемлемыми кислотами, некоторых или любого из соединений, описанного в настоящей заявке как изобретение.
Некоторые аспекты настоящего изобретения включают в себя как минимум, одно из соединений, описанных в настоящей заявке как изобретение, для использования в качестве лекарственного средства.
Некоторые аспекты настоящего изобретения включают в себя композицию, включающую в себя, как активное вещество, терапевтически эффективное количество, как минимум, в одно из соединений, описанных в настоящей заявке как изобретение.
Некоторые аспекты настоящего изобретения включают фармацевтическую композицию для предотвращения или лечения туберкулеза, включающая в себя, как минимум, активное вещество, которым может являться соединение, описанное в настоящей заявке как изобретение, в количестве, эффективном для его антитуберкулезного действия, или фармацевтически приемлемую соль такого вещества в комбинации с фармацевтически приемлемым носителем.
Некоторые аспекты настоящего изобретения включают в себя фармацевтическую композицию для предотвращения или лечения туберкулеза у теплокровного животного, которое включает в себя употребление таким животным терапевтически эффективного количества соединения, описанного в настоящей заявке как изобретение.
Некоторые аспекты настоящего изобретения включают в себя соединение, описанное в настоящей заявке как изобретение, или фармацевтическую композицию, описанную в настоящей заявке как изобретение в качестве материала для производства терапевтического или лекарственного средства.
Некоторые аспекты настоящего изобретения включают в себя способ использования соединения, описанного в настоящей заявке как изобретение, в качестве активного соединения в составе лекарственного средства для лечения туберкулеза у млекопитающего.
Некоторые аспекты настоящего изобретения включают в себя метод лечения млекопитающего, больного туберкулезом, или подвергнувшегося риску заболевания туберкулезом, который состоит во введении такому млекопитающему терапевтически эффективного количества соединения или фармацевтической композиции, описанной в настоящей заявке как изобретение.
Некоторые аспекты настоящего изобретения включают в себя применение соединения или фармацевтической композиции, описанных в настоящей заявке как изобретение, в качестве средства, проявляющего противотуберкулезную активность.
Некоторые аспекты настоящего изобретения включают в себя применение соединения или фармацевтической композиции, описанных в настоящей заявке как изобретение, в качестве средства для лечения туберкулеза.
Некоторые аспекты настоящего изобретения включают в себя метод лечения туберкулеза у теплокровного животного или млекопитающего, который включает в себя употребление таким животным или млекопитающим терапевтически эффективного количества соединения или фармацевтической композиции, описанных в настоящей заявке как изобретение.
Некоторые аспекты настоящего изобретения подразумевают под словом «туберкулез» и производными от него словами - фразу «туберкулезная инфекция» и соответствующие производные от нее слова.
Некоторые аспекты настоящего изобретения подразумевают под словом «туберкулез» и производными от него словами лекарственно резистентный туберкулез.
Некоторые аспекты настоящего изобретения включают в себя процесс приготовления соединения или фармацевтической композиции, описанных в настоящей заявке как изобретение.
Некоторые аспекты настоящего изобретения могут включать в себя соединение, описанное в настоящей заявке для использования в качестве лекарственного или противомикробного средства.
Некоторые аспекты настоящего изобретения могут включать в себя использование фразы «терапевтически эффективное количество», «количество, эффективное для его антитуберкулезного действия», а также любые производные от них.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ПРОИЗВОДНЫЕ ХИНОЛИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ МИКОБАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2008 |
|
RU2404971C2 |
| НИКОТИНОИЛГИДРАЗОН ДИМЕФОСФОНА, ОБЛАДАЮЩИЙ ПРОТИВОТУБЕРКУЛЕЗНОЙ АКТИВНОСТЬЮ | 2011 |
|
RU2471787C1 |
| ПРОИЗВОДНЫЕ ХИНОЛИНА, В ЧАСТНОСТИ 5,6,7-ЗАМЕЩЕННЫЕ 1-(2-ХЛОРХИНОЛИН-3-ИЛ)-4-ДИМЕТИЛАМИНО-2-(НАФТАЛИН-1-ИЛ)-1-ФЕНИЛБУТАН-2-ОЛЫ, СПОСОБ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ СОЕДИНЕНИЙ | 2011 |
|
RU2486175C2 |
| ИЗОНИКОТИНОИЛГИДРАЗОН ДИМЕФОСФОНА, ОБЛАДАЮЩИЙ ПРОТИВОТУБЕРКУЛЕЗНОЙ АКТИВНОСТЬЮ | 2011 |
|
RU2457212C1 |
| ЧЕТВЕРТИЧНЫЕ АММОНИЙНЫЕ ПРОИЗВОДНЫЕ 2-АМИНОТИОФЕН-3-КАРБОКСИЛАТОВ, ОБЛАДАЮЩИЕ ПРОТИВОТУБЕРКУЛЕЗНОЙ АКТИВНОСТЬЮ | 2016 |
|
RU2629369C1 |
| ПРОИЗВОДНЫЕ 5-ОКСО-5Н-[1]-БЕНЗОПИРАНО-[5,6-B]4-ОКСО-4Н-[1,2]-ПИРИМИДО-1,4,5,6-ТЕТРАГИД РО-1,3-ТИАЗИНА | 1997 |
|
RU2169732C1 |
| ПРОИЗВОДНЫЕ ХИНОЛИНА, В ЧАСТНОСТИ 6,7-ЗАМЕЩЕННЫЕ 1-(2-ХЛОРХИНОЛИН-3-ИЛ)-4-ДИМЕТИЛАМИНО-2-(НАФТАЛИН-1-ИЛ)-1-ФЕНИЛБУТАН-2-ОЛЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ СОЕДИНЕНИЙ ДЛЯ ЛЕЧЕНИЯ ИНФЕКЦИОННЫХ ЗАБОЛЕВАНИЙ МИКОБАКТЕРИАЛЬНОЙ ПРИРОДЫ, В ЧАСТНОСТИ ТУБЕРКУЛЕЗА | 2013 |
|
RU2530493C1 |
| НАФТОХИНОНОВЫЕ ПРОИЗВОДНЫЕ И ИХ ИСПОЛЬЗОВАНИЕ ДЛЯ ЛЕЧЕНИЯ И БОРЬБЫ С ТУБЕРКУЛЕЗОМ | 2000 |
|
RU2246299C2 |
| СУЛЬФОННЫЕ ПРОИЗВОДНЫЕ 2-НИТРО-2-(3-АРИЛ-1,2,4-ОКСАДИАЗОЛ-5-ИЛ)ЭТАНА, ОБЛАДАЮЩИЕ ПРОТИВОЛЕПРОЗНОЙ И ПРОТИВОТУБЕРКУЛЕЗНОЙ АКТИВНОСТЬЮ | 2009 |
|
RU2415845C1 |
| ПИРИДИНОИЛГИДРАЗОНЫ ДИАЛКИЛ(2-МЕТИЛ-4-ОКСОПЕНТ-2-ИЛ) ФОСФИНОКСИДОВ, ОБЛАДАЮЩИЕ ПРОТИВОТУБЕРКУЛЕЗНОЙ АКТИВНОСТЬЮ | 2012 |
|
RU2498990C1 |
Изобретение относится к медицине и фармакологии, конкретно к применению производных 1-(1-бензилиндол-3-ил)2,3,4,9-тетрагидро-1-Н-β-карболина общей формулы (1), а также их фармакологически приемлемых солей для производств лекарственного средства для лечения или профилактики туберкулеза, к новым производным β-карболина формулы (1), способу их получения, лекарственным средствам на их основе и способу лечения туберкулеза. 5 н. и 39 з.п. ф-лы, 7 табл.
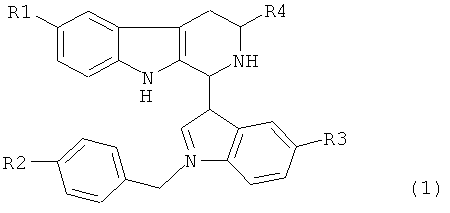
1. Применение, по меньшей мере, одного соединения, представляющего собой производное 1-(1-бензилиндол-3-ил)2,3,4,9-тетрагидро-1-Н-β-карболина общей формулы 1:

где R1 и R3 - одинаковые или различные, выбраны из группы: Н, галоген, метоксигруппа, бензилоксигруппа; R2 - выбран из группы: Н, галоген, нитрогруппа, метоксигруппа; R4 - Н, или С1-С6 алкил или их солей с фармакологически приемлемыми кислотами для производства лекарственного средства для лечения или профилактики туберкулеза.
2. Применение соединения формулы 1 по п.1, где R1=R2=R3=R4=H.
3. Применение соединения формулы 1 по п.1, где R1=F, R2=R3=R4=H.
4. Применение соединения формулы 1 по п.1, где R1=Cl, R2=R3=R4=H.
5. Применение соединения формулы 1 по п.1, где R1=ОСН3, R2=R3=R4=H.
6. Применение соединения формулы 1 по п.1, где R1=ОСН2С6Н5, R2=R3=R4=H.
7. Применение соединения формулы 1 по п.1, где R1=R3=R4=H, R2=F.
8. Применение соединения формулы 1 по п.1, где R1=R3=R4=H, R2=Cl.
9. Применение соединения формулы 1 по п.1, где R1=R3=R4=H, R2=ОСН3.
10. Применение соединения формулы 1 по п.1, где R1=R3=R4=H, R2=NO2.
11. Применение соединения формулы 1 по п.1, где R1=R2=R4=H, R3=F.
12. Применение соединения формулы 1 по п.1, где R1=R2=R4=H, R3=Cl.
13. Применение соединения формулы 1 по п.1, где R1=R2=R4=H, R3=ОСН3.
14. Применение соединения формулы 1 по п.1, где R1=R2=R4=H, R3=ОСН2С6Н5.
15. Применение соединения формулы 1 по п.1, где R1=R2=R3=H, Р4=СН3.
16. Применение соединения формулы 1 по п.1, где R1=R2=R3=H, R4=CH2CH3.
17. Применение соединения формулы 1 по п.1, где R1=R2=R3=R4=H, в виде солянокислой соли.
18. Применение соединения формулы 1 по п.1, где R1=R2=R3=R4=H, в виде сульфата.
19. Применение соединения формулы 1 по п.1, где R1=R2=R3=R4=H, в виде соли бензойной кислоты.
20. Применение соединения формулы 1 по п.1, где R1=R2=R3=R4=H, в виде соли малеиновой кислоты.
21. Лекарственное средство для профилактики и/или лечения туберкулеза у млекопитающего, включающее в себя, по меньшей мере, одно соединение формулы 1 по любому из пп.1-20 в эффективном количестве.
22. Соединение, представляющее собой производное 1-(1-бензилиндол-3-ил)2,3,4,9-тетрагидро-1-Н-β-карболина общей формулы 1:
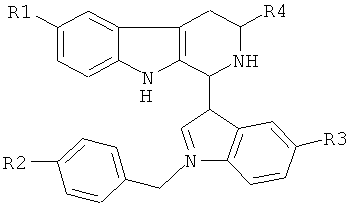
где R1 и R3 - одинаковые или различные, выбраны из группы: Н, галоген, метоксигруппа, бензилоксигруппа; R2 - выбран из группы: Н, галоген, нитрогруппа, метоксигруппа; R4 - Н, или С1-С6 алкил или его соль с фармакологически приемлемыми кислотами.
23. Соединение формулы 1 по п.22, где R1=R2=R3=R4=H.
24. Соединение формулы 1 по п.22, где R1=F, R2=R3=R4=H.
25. Соединение формулы 1 по п.22, где R1=Cl, R2=R3=R4=H.
26. Соединение формулы 1 по п.22, где R1=ОСН3, R2=R3=R4=H.
27. Соединение формулы 1 по п.22, где R1=ОСН2С6Н5, R2=R3=R4=H.
28. Соединение формулы 1 по п.22, где R1=R3=R4=H, R2=F.
29. Соединение формулы 1 по п.22, где R1=R3=R4=Н, R2=Cl.
30. Соединение формулы 1 по п.22, где R1=R3=R4=H, R2=OCH3.
31. Соединение формулы 1 по п.22, где R1=R3=R4=H, R2=NO2.
32. Соединение формулы 1 по п.22, где R1=R2=R4=H, R3=F.
33. Соединение формулы 1 по п.22, где R1=R2=R4=H, R3=Cl.
34. Соединение формулы 1 по п.22, где R1=R2=R4=H, R3=ОСН3.
35. Соединение формулы 1 по п.22, где R1=R2=R4=H, R3=ОСН2С6Н5.
36. Соединение формулы 1 по п.22, где R1=R2=R3=H, R4=СН3.
37. Соединение формулы 1 по п.22, где R1=R2=R3=H, R4=СН2СН3.
38. Соединение формулы 1 по п.22, где R1=R2=R3=R4=H, в виде солянокислой соли.
39. Соединение формулы 1 по п.22, где R1=R2=R3=R4=H, в виде сульфата.
40. Соединение формулы 1 по п.22, где R1=R2=R3=R4=H, в виде соли бензойной кислоты.
41. Соединение формулы 1 по п.22, где R1=R2=R3=R4=H, в виде соли малеиновой кислоты.
42. Соединение по пп.22-42, обладающее антимикобактериальной активностью.
43. Способ синтеза соединения, представляющего собой производное 1-(1-бензилиндол-3-ил)2,3,4,9-тетрагидро-1-Н-β-карболина общей формулы 1:
где R1, R2, R3, R4 - как указано в любом из пп.22-42 или их солей с фармакологически приемлемыми кислотами, в том числе соединений, описанных в любом из пп.22-42, характеризующийся тем, что бензилхлорид
, 
подвергают взаимодействию с производным индола
 ,
,
где R2, R3, как указано выше, при нагревании в присутствии водного раствора щелочи и межфазного катализатора, с последующей обработкой полученного 1-бензилиндола хлорокисью фосфора в среде сухого диметилформамида при охлаждении с получением соответствующего производного 1-бензилиндол-3-альдегида, которое подвергают взаимодействию с производным триптамина
 ,
,
где R1 и R4, как указано выше, при нагревании в ледяной уксусной кислоте с получением целевого производного 1-(1-бензилиндол-3-ил)2,3,4,9-тетрагидро-1-Н-β-карболина общей формулы 1 с последующей обработкой, в случае необходимости, фармацевтически приемлемой кислотой с получением соответствующей солевой формы.
44. Способ лечения туберкулеза у млекопитающего, который включает в себя употребление таким млекопитающим терапевтически эффективного количества соединения, описанного в любом из пп.22-42 или лекарственного средства по п.21.
| ЗАМЕЩЕННЫЕ БЕТА-КАРБОЛИНЫ | 2001 |
|
RU2277095C2 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| US 7348336, B2, 25.03.2008. | |||

