Область изобретения
Настоящее изобретение относится к смешанным соединениям металлов, обладающим фармацевтической активностью, прежде всего к их применению в качестве соединений, связывающих фосфаты. Изобретение относится также к способам получения указанных соединений, а также к фармацевтическим композициям, содержащим указанные соединения, и к их применению в фармацевтике.
Предпосылки создания настоящего изобретения
При гемодиализе у пациентов с почечной недостаточностью концентрация фосфата в плазме крови может значительно повыситься, и при указанном состоянии, известном как гиперфосфатемия, наблюдается отложение фосфата кальция в мягких тканях. Уровень фосфата в плазме можно снизить благодаря пероральному введению неорганических и органических соединений, связывающих фосфаты. Один из наиболее распространенных способов лечения включает введение дозы геля гидроксида алюминия, который образует нерастворимый фосфат алюминия. Однако при этом наблюдается токсическое действие вследствие накопления алюминия, например снижение скорости продуцирования гемоглобина, нарушение природной репарации и продуцирования костной ткани и возможное нарушение неврологической/познавательной функции. В связи с этим предложены другие соединения алюминия, такие как микрокристаллический оксид/гидроксид алюминия (бемит) и некоторые гидротальциты, описанные в статье Ookubo и др. (Journal Pharmaceutical Sciences, 81(11), 1139-1140, (ноябрь, 1992)). Однако указанные соединения также характеризуются аналогичными недостатками.
Многие известные неорганические препараты для лечения гиперфосфатемии являются эффективными связывающими фосфаты соединениями только в ограниченном интервале рН, прежде всего в кислотной области рН приблизительно 3-5. Указанные соединения, эффективно связывающие фосфаты при рН 3, не обязательно проявляют эффективность при связывании фосфатов при более высоких рН, например >7, в нижних отделах кишечника, т.е. в двенадцатиперстной кишке и ниже, причем в таких отделах происходит по меньшей мере частичное связывание фосфата. Более того, прежде всего щелочные связывающие соединения основного характера могут изменить значение рН в желудке до более высоких значений, при которых снижается фосфат - связывающая активность.
Чтобы исключить указанные недостатки, связанные с применением алюминия, а также проблемы, связанные с эффективностью в ограниченном интервале рН, в заявке WO-A-99/15189 предлагается использовать смешанные соединения металлов, не содержащие алюминий и обладающие по меньшей мере 30 мас.% фосфатсвязывающей способностью в расчете на общую массу фосфата, в интервале рН 2-8.
Как правило, указанные смешанные соединения металлов содержат железо (III) и по меньшей мере один металл из группы, включающей магний, кальций, лантан и церий. Предпочтительно такие соединения также содержат по меньшей мере один анион, выбранный из группы, включающей гидроксил и карбонат, и необязательно дополнительно по меньшей мере один анион, выбранный из группы, включающей сульфат, нитрат, хлорид и оксид. Однако авторами неожиданно было установлено, что из соединений смешанных металлов, описанных в заявке WO-A-99/15189, при их применении высвобождаются некоторые входящие в их состав двухвалентные металлы в растворимой форме.
В заявке JP-A-2004-89760 описано повышение дефосфатирующей активности отдельных соединений смешанных металлов, которые используют для удаления фосфора из бытовых или промышленных сточных вод после термической обработки кристаллов указанных соединений общей формулы
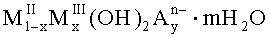
где MII означает по меньшей мере один двухвалентный металл, MIII означает по меньшей мере один трехвалентный металл, An- означает n-валентный анион, а х, у и m находятся в диапазоне 0<х≤0,67, 0<у≤1, 0≤m≤2.
Предполагается, что коэффициент селективности указанных соединений в отношении "фосфорсульфатных ионов", растворенных в воде, приблизительно составляет по меньшей мере 5.
Предпочтительный способ получения указанных термически обработанных соединений заключается в том, что используют смешанный водный раствор водорастворимой соли неорганической или органической кислоты и щелочного гидроксида, который добавляют по каплям к водному раствору, содержащему водорастворимое соединение двухвалентного металла и водорастворимое соединение трехвалентного металла или двухвалентного марганца, и проводят реакцию при температуре 0~90°С, при этом получают кристаллический осадок соединения гидроксида металла общей формулы, приведенной выше. Указанный осадок отделяют и подвергают термической обработке при 200-500°С.
Высвобождение (трехвалентного) алюминия из комплекса MgAlLDH в ходе десорбции фосфата, а также термическая обработка соединения MgMnLDH описана в статье Tezuka S., Bull. Chem. Soc. Jpn., 77, 2101-2107 (2004).
Авторами установлено, что высвобождение двухвалентного металла, например магния, которое происходит при использовании соединений, описанных в заявке WO-A-99/15189, в фармацевтике, можно существенно снизить при термической обработке пригодного смешанного соединения металлов, например слоистого двойного гидроксида или соединения типа гидротальцита. Аналогичным образом можно снизить высвобождение других и двухвалентных металлов, если MII не означает магний.
Термин «смешанное соединение металлов» означает единое соединение, содержащее два или более разного типа металлов. Единое соединение обычно не удается разделить на составляющие компоненты методами физического разделения, для этого требуется проведение химической реакции.
Термин "слоистый двойной гидроксид" (LDH), используемый в описании заявки, означает синтетические или природные слоистые гидроксиды, содержащие два вида катионов металлов, расположенных в основных слоях, а межслойные домены содержат анионы. Указанное многочисленное семейство соединений иногда называют также анионными глинами в отличие от более распространенных катионных глин, содержащих в межслойных доменах катионы. LDH принято называть также гидротальцитоподобными соединениями со ссылкой на одну из разновидностей соответствующего минерала на основе [Mg-Al] (см. "Layered Double Hydroxides: Present and Future", под ред. V.Rives, Nova Science (2001)).
Краткое описание сущности изобретения
В качестве первого объекта в настоящем изобретении предлагается субстанция, предназначенная для применения в качестве лекарственного средства, включающего твердое смешанное соединение металлов формулы (I):

где MII означает по меньшей мере один двухвалентный металл (т.е. металл, имеющий два положительных заряда), М означает по меньшей мере один трехвалентный металл (т.е. металл, имеющий три положительных заряда), An- означает по меньшей мере один n-валентный анион, 2+a=2b+Σcn, а равно числу молей М'''/(число молей М" + число молей М'''), a Σcn<0,9a.
В приведенной выше формуле (I) если А означает более одного аниона, то валентность (то есть заряд аниона n) каждого из них может изменяться.
В приведенной выше формуле (I) "Σcn" означает сумму количества молей каждого аниона в моле соединения формулы (I), умноженную на соответствующую валентность.
Величина z равна предпочтительно 2 или менее, более предпочтительно 1,8 или менее, наиболее предпочтительно 1,5 или менее. Величина z равна 1 или менее.
Величина а составляет прежде всего от 0,1 до 0,5, предпочтительно от 0,2 до 0,4.
Величина b составляет прежде всего 1,5 или менее, предпочтительно 1,2 или менее. Величина b предпочтительно составляет более 0,2, более предпочтительно более 0,4, наиболее предпочтительно более 0,6, наиболее предпочтительно более 0,9.
Если а≥0,3, предпочтительно Σcn<0,5а. Если а≤0,3 предпочтительно Σcn<0,7a.
Величина с для каждого аниона определяется необходимостью обеспечить нейтральность заряда соединения по формуле 2+a=2b+Σcn.
Субстанция согласно первому объекту настоящего изобретения предпочтительно включает более 30 мас.%, более предпочтительно более 50 мас.% соединения или соединений формулы (I), например вплоть до 95 мас.% или 90 мас.% вещества.
Способ получения соединений формулы (I) заключается в изменении некоторых структурных признаков исходного соединения. Формула (I) приведена только для описания его элементного состава и не включает все структурные особенности соединения.
Если соединение формулы (I) включает магний MII и железо MIII в виде катионов и карбонат в качестве аниона, по данным рентгеноструктурного анализа величина угла 2θ составляет 34°. При более низких температурах (≤250°С) на рентгенограмме появляются пики, соответствующие слоистому двойному гидроксиду, тогда как при повышении температуры появляется пик оксида MIIO, но указанные пики можно разрешить методом деконволюции.
Указанные предпочтительные количества субстанции и соединения по первому объекту изобретения можно использовать в других объектах изобретения, как указано в описании заявки.
Вторым объектом настоящего изобретения является субстанция для применения в качестве лекарственного средства, включающая твердое смешанное соединение металлов, полученное или получаемое при нагревании при температуре от 200°С до 600°С, предпочтительно от 225°С до 550°С, более предпочтительно от 250°С до 500°С, соединения формулы (II):
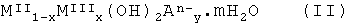
где MII означает по меньшей мере один двухвалентный металл (то есть имеющий 2 положительных заряда), MIII означает по меньшей мере один трехвалентный металл (то есть имеющий три положительных заряда), А означает по меньшей мере один n-валентный анион, x=∑ny, х и m находятся в диапазоне 0<х≤0,5, 0≤m≤10.
Следует отметить, что формула (II) должна обеспечивать суммарный нейтральный заряд соединения. В формуле (I) и/или формуле (II) подклассы соединений каждой формулы могут включать, соответственно, соединения, в которых а или х меньше, чем любое из следующих значений, и соединения, в которых а или х больше или равно любому из следующих значений, причем указанные значения равны 0,1, 0,15, 0,2, 0,25, 0,3, 0,35, 0,4, 0,45. Один такой пример включает подклассы, в которых а более или равно 0,3 и, соответственно, менее 0,3. Величина х обычно составляет от 0,1 до 0,5, предпочтительно от 0,2 до 0,4.
В формуле (II) ∑ny равна сумме индексов при каждом анионе, умноженных на соответствующую валентность аниона.
При нагревании соединения формулы (II) при температуре от 200°С до 600°С, предпочтительно от 225°С до 550°С, более предпочтительно от 250°С до 500°С, предпочтительно происходит снижение количества металла М, переходящего в раствор, по меньшей мере на 50 мас.% по сравнению с потерей металла из ненагретого соединения формулы (II) в условиях, описанных более подробно ниже. Указанное преимущество относится к любому объекту настоящего изобретения формулы (II).
Нагревание прежде всего проводят в атмосфере при температуре от 200°С до 600°С, предпочтительно от 225°С до 550°С, более предпочтительно от 250°С до 500°С, в течение 1 мин или более, более предпочтительно в течение 5 мин или более, более предпочтительно в течение 1 ч или более. Соединение предпочтительно находится в нагретой атмосфере в течение 10 ч или менее, более предпочтительно в течение 5 ч или менее, наиболее предпочтительно в течение 3 ч или менее.
При указанном нагревании происходит прокаливание соединения формулы (II). Предполагается, что при прокаливании образуется субстанция по первому объекту настоящего изобретения. При этом величина а для соединения формулы (I) меньше или равна величине х для соответствующего необработанного соединения формулы (I).
Условия прокаливания ограничены по температуре и/или по времени, т.е. температура прокаливания не превышает 600°С в течение не более 3 ч, иначе снижается активность в отношении связывания фосфатов.
При прокаливании за пределами указанного диапазона происходит снижение величины ∑cn/a в формуле (I) до менее 0,03. Следовательно, предпочтительно, чтобы величина ∑cn/a составляла более 0,03, более предпочтительно более 0,05, наиболее предпочтительно более 0,09, наиболее предпочтительно более 0,10. При прокаливании за пределами указанного диапазона может также образоваться кристаллическая структура типа шпинеля, поэтому предпочтительно отсутствие структуры типа шпинеля по данным рентгеноструктурного анализа. Значение а для структуры типа шпинеля составляет 0,67, в связи с чем предпочтительно значение а для соединения формулы (I) составляет 0,66 или менее, предпочтительно 0,5 или менее, более предпочтительно 0,5 или менее.
Предпочтительно при прокаливании соединения формулы (II) получают субстанцию, обладающую фосфатсвязывающей способностью по меньшей мере на 10% выше по сравнению с соединением формулы (II), из которого при прокаливании получают субстанцию. Предпочтительные значения, описанные выше, также относятся к другому объекту настоящего изобретения, описанному ниже.
Пригодным способом определения степени прокаливания является определение в процентах потери воды, связанной с поверхностью кристаллов, при нагревании при 105°С. Для измерения указанной величины образец выдерживают в течение нескольких суток в определенных условиях окружающей среды (25°С, относительная влажность 20%), после достижения равновесной влажности образец взвешивают, затем нагревают при 105°С в течение 4 ч и повторно взвешивают для определения уменьшения массы, которое выражают в процентах. При нагревании до 105°С удаляется вода, связанная с поверхностью (то есть химически несвязанная вода или вода на поверхности кристаллов).
Обычно смешанные соединения металлов после прокаливания содержат менее 2 мас.%, предпочтительно менее 1,5 мас.%, более предпочтительно менее 1 мас.% по массе воды, связанной с поверхностью кристаллитов.
Для краткости субстанцию для применения в качестве лекарственного средства по первому или второму объектам настоящего изобретения далее по тексту принято называть "субстанцией по изобретению".
Указанные предпочтительные значения для субстанции и соединения по первому и второму объектам по настоящему изобретению относятся к другим объектам настоящего изобретения, как описано в тексте заявки.
Третьим объектом настоящего изобретения является применению в способе получения лекарственного средства, предназначенного для связывания фосфата в организме животных, нуждающихся в указанном лечении, предпочтительно в организме человека, предпочтительно для профилактики или лечения гиперфосфатемии, субстанции, включающей твердое смешанное соединение металлов формулы (I):
где MII означает по меньшей мере один двухвалентный металл, MIII означает по меньшей мере один трехвалентный металл, An- означает по меньшей мере один n-валентный анион, 2+a=2b+∑cn, a=M'''/(M"+M'") и ∑cn<0,9a.
Лекарственное средство используют для лечения животных, предпочтительно человека.
Четвертым объектом настоящего изобретения является применение в способе получения лекарственного средства, предназначенного для связывания фосфата в организме животных, нуждающихся в указанном лечении, предпочтительно в организме человека, предпочтительно для профилактики или лечения гиперфосфатемии, субстанции полученной или получаемой при нагревании при температуре от 200°С до 600°С, предпочтительно от 225°С до 550°С, более предпочтительно от 250°С до 500°С, соединения формулы (II):
где MII означает по меньшей мере один двухвалентный металл, MIII означает по меньшей мере один трехвалентный металл, An- означает по меньшей мере один n-валентный анион, x=Σny, х и m находятся в диапазоне 0<х≤0,5, 0≤m≤10. Величина х предпочтительно составляет от 0,1 до 0,5, более предпочтительно от 0,2 до 0,4.
Подробное описание вариантов осуществления изобретения
Получение активных субстанций
Субстанции по изобретению предпочтительно получают при термической обработке пригодного исходного материала формулы (II), как указано выше. Для получения субстанции по настоящему изобретению необязательно можно использовать другие способы, такие как твердотельный синтез, твердофазные реакции или тонкодисперсное измельчение оксидов или гидроксидов одного или нескольких металлов с использованием гидротермального синтеза или синтеза при низкой температуре.
Субстанции по настоящему изобретению, полученные при термической обработке пригодного исходного материала формулы (II), как описано выше, получают из первого раствора растворимого в воде соединения металла MII и растворимого в воде соединения металла MIII, причем анионы выбирают таким образом, чтобы не происходило образования осадка в первом растворе. Второй раствор также получают из растворимого в воде гидроксида (например, NaOH) и растворимой в воде соли аниона An- (причем катионы выбирают таким образом, чтобы не происходило образования осадка гидроксида металла или осадка аниона и металла из гидроксида). Затем два раствора смешивают и за счет совместного осаждения получают исходный материал, представляющий собой смешанное соединение металлов. Указанный исходный материал включает твердый кристаллический материал, обычно также содержащий небольшое количество твердого аморфного материала. Предпочтительно, чтобы по меньшей мере небольшое количество полученного таким способом материала характеризовалось структурой слоистого двойного гидроксида и/или гидротальцита, обычно также содержащего небольшое количество аморфного и/или слабо кристаллизованного материала. После совместного осаждения материал отделяют фильтрованием или центрифугированием, промывают, а затем высушивают при нагревании.
Материал промывают для удаления водорастворимых солей, являющихся побочными продуктами реакции осаждения. Если твердый осадок содержит значительное количество указанных растворимых солей, то при последующем нагревании растворимые соли могут включатся в полученное твердое вещество, что отрицательно сказывается на фосфатсвязывающих свойствах материала. Материал предпочтительно промывают таким образом, чтобы остаточное содержание водорастворимых солей (имеющих растворимость в воде 1 г/л или более) после высушивания, как описано ниже, составляло менее 15 мас.%, предпочтительно менее 10 мас.%, наиболее предпочтительно менее 5 мас.% от массы твердого смешанного соединения металлов.
После фильтрования или центрифугирования и промывания, высушивание предпочтительно проводят при низкой температуре (до 120°С), например, в печи, в распылительной сушилке или в сушилке с псевдосжиженным слоем.
Перед термической обработкой сухого материала слишком крупные частицы необязательно удаляют за счет размола и/или просеивания и/или при использовании любого другого пригодного способа, например, для получения материала, предназначенного для термической обработки, содержащего частицы диаметром практически не более 100 мкм. Предпочтительно, по данным ситового анализа, менее 10 мас.% частиц характеризуется диаметром более 106 мкм, более предпочтительно менее 5 мас.%. Наиболее предпочтительно материал не содержит частицы диаметром более 106 мкм. Затем полученный сухой материал подвергают необходимой термической обработке, предпочтительно при температуре от 200°С или предпочтительно от 225°С до 550°С, более предпочтительно от 250°С до 500°С, например, в сушильном шкафу, в печи для обжига барабанного типа или в сушилке с псевдоожиженым слоем.
Влажный осадок необязательно непосредственно нагревают при температуре более 200°С и не используют низкотемпературную сушку (например, до 120°С) и измельчение.
Таким образом, пятым объектом настоящего изобретения является способ получения субстанции для применения в качестве лекарственного средства, причем способ заключается в том, что субстанцию, включающую соединение формулы (II):
где MII означает по меньшей мере один двухвалентный металл, MIII означает по меньшей мере один трехвалентный металл, An- означает по меньшей мере один n-валентный анион, x=∑ny, х и m находятся в диапазоне 0<х≤0,5, 0≤m≤10, причем значения х предпочтительно находятся в диапазоне от 0,1 до 0,5, более предпочтительно от 0,2 до 0,4, нагревают при температуре от 200°С до 600°С, предпочтительно от 225°С до 550°С, более предпочтительно от 250°С до 500°С.
Предпочтительно, при нагревании происходит снижение потери металла MII в составе термически обработанного соединения (за счет перехода в раствор) по меньшей мере на 50 мас.% по сравнению с потерей в составе необработанного соединения по данным определения потерь металла MII методом, описанным ниже.
Субстанции по изобретению содержат по меньшей мере одно соединение формулы (I), но при использовании способа, описанного выше для получения исходного материала, в субстанции могут присутствовать также другие материалы, например в промежуточном продукте соединение формулы (II), а в конечном продукте, например, соединения одного металла (в отличие от смешанных соединений), которые также образуются при совместной осаждении.
Твердые смешанные соединения металлов
В формуле (I) и формуле (II) MII предпочтительно выбирают из группы, включающей Mg, Zn, Fe (II), Cu (II) и Ni (II), прежде всего предпочтителен Mg. М предпочтительно выбирают из группы, включающей Mn (III), Fe (III), La (III) и Се (III), прежде всего предпочтителен Fe (III), прежде всего в случае, если MII означает Mg. MII и MIII означают различные металлы или означают один и тот же металл, но с различной валентностью. Например, MII может означать Fe (II), а MIII может означать Fe (III). Однако более предпочтительно MII и MIII означают разные металлы. M (III) может также означать Al (III) в тех случаях, когда накопление алюминия и токсические осложнения являются несущественными.
An- предпочтительно включает по меньшей мере один анион, выбранный из группы, включающей карбонат, гидрокарбонат, сульфат, нитрат, галогенид и гидроксид, прежде всего предпочтителен карбонат.
Любая субстанция по изобретению предпочтительно практически не содержит или совсем не содержит алюминия.
Определение фосфатсвязывающей способности
Специфичный метод определения фосфатсвязывающей способности более подробно описан ниже в разделе Примеры. Однако в общем случае, в том числе в описании заявки, если не указано иное, любая ссылка на фосфатсвязывающую способность в процентах означает, что эта величина определена указанным способом. Субстанцию по изобретению (0,4 г) добавляют в 10 мл 40 мМ раствора натрий-фосфатного буферного раствора с определенным значением рН. Предпочтительно, любое значение фосфатсвязывающей способности в процентах, указанное в данной заявке, определяют при рН от 3 до 7, более предпочтительно от 2 до 8. Образцы гомогенизируют и осторожно встряхивают при комнатной температуре в течение 30 мин. Затем образец центрифугируют в течение 5 мин при 3000 об/мин, супернатант отделяют фильтрованием через фильтры фирмы Millipore с размером пор 0,22 мкм. В супернатанте измеряют содержание растворимого фосфата. Затем рассчитывают содержание фосфата, связанного с фосфатсвязывающим веществом, в процентах относительно необработанного исходного раствора фосфата.
Композиции
Изобретение также относится к фармацевтической композиции, которая включает в качестве активного ингредиента по меньшей мере одну субстанцию по изобретению в смеси с фармацевтически приемлемым носителем.
Таким образом, шестым объектом настоящего изобретения является фармацевтическая композиция, включающая субстанцию по первому объекту изобретения, то есть твердое смешанное соединение металлов формулы (I):
где MII означает по меньшей мере один двухвалентный металл, MIII означает по меньшей мере один трехвалентный металл, An- означает по меньшей мере один n-валентный анион, 2+а=2b+∑cn, а равно числу молей М'"/(число молей М"+число молей М'") и ∑cn<0,9a.
Седьмым объектом настоящего изобретения является фармацевтическая композиция, включающая субстанцию по второму объекту изобретения, то есть твердое смешанное соединение металлов, которое получают при нагревании при температуре от 200°С до 600°С, предпочтительно от 225°С до 550°С, более предпочтительно от 250°С до 600°С, соединения формулы (II):
где MII означает по меньшей мере один двухвалентный металл, MIII означает по меньшей мере один трехвалентный металл, Аn- означает по меньшей мере один n-валентный анион, х=∑ny, а х и m находятся в диапазоне 0<х≤0,5, 0≤m≤10. Значения х предпочтительно находятся в диапазоне от 0,1 до 0,5, более предпочтительно от 0,2 до 0,4.
Предлагается также способ получения фармацевтической композиции, указанной выше, который заключается в том, что по меньшей мере одну субстанцию по изобретению смешивают с фармацевтически приемлемым носителем и необязательно с любыми другими ингредиентами, включая побочные продукты, образующиеся при получении активного ингредиента.
Фармацевтически приемлемым носителем является любой материал, с которым смешивают субстанцию по изобретению для облегчения введения. Можно использовать твердый или жидкий носитель, включая материал, который в нормальных условиях является газообразным, но под давлением образует жидкость, и любой носитель, который обычно используют при получении фармацевтических композиций. Предпочтительно, композиции по изобретению содержат от 0,5 мас.% до 95 мас.% активного ингредиента. Термин фармацевтически приемлемый носитель включает разбавители, эксципиенты и адъюванты.
Другим объектом изобретения является способ связывания избытка фосфата в организме животных, прежде всего человека. Прежде всего указанный объект относится к способу профилактики или лечения гиперфосфатемии у животных, прежде всего человека. Способ включает стадию введения субстанции по изобретению, предпочтительно пероральным способом.
Еще одним объектом изобретения является применение субстанции по изобретению для получения лекарственного средства, предназначенного для связывания фосфата у животных, предпочтительно человека, нуждающегося в указанном лечении, предпочтительно для профилактики или лечения гиперфосфатемии в организме животных, предпочтительно человека.
Субстанции по изобретению можно включать в фармацевтическую композицию в любой пригодной форме, но прежде всего в форме, пригодной для перорального введения, например в твердой стандартной лекарственной форме, такой как таблетки, капсулы, или в жидкой форме, такой как жидкие суспензии, прежде всего водные суспензии. Однако можно использовать также лекарственные формы для экстракорпорального или внутривенного введения. Пригодные составы можно получать известным методом с использованием стандартных твердых носителей, таких, например, как лактоза, крахмал или тальк, или жидких носителей, таких, например, как вода, жирные масла или вазелиновые масла. Другие пригодные носители включают материалы, полученные из животных или растительных белков, такие как желатины, декстрины и белки из семян сои, пшеницы и других растений, смолы, такие как аравийская камедь, гуар, агар и ксантан, полисахариды, альгинаты, карбоксиметилцеллюлозы, каррагинаны, декстраны, пектины, синтетические полимеры, такие как поливинилпирролидон, комплексы полипептид/белок или полисахарид, такие как комплексы желатин/аравийская камедь; сахара, такие как маннит, декстроза, галактоза и трегалоза, циклические сахара, такие как циклодекстрин, неорганические соли, такие как фосфат натрия, хлорид натрия и силикаты алюминия; аминокислоты, включающие от 2 до 12 атомов углерода, такие как глицин, L-аланин, L-аспарагиновая кислота, L-глутаминовая кислота, L-гидроксипролин, L-изолейцин, L-лейцин и L-фенилаланин.
Композиция может также включать вспомогательные компоненты, такие как дезинтегрирующие агенты для таблеток, солюбилизаторы, консерванты, антиоксиданты, ПАВ, загустители, красители, ароматизаторы, агенты для регуляции рН, подсластители или агенты, маскирующие вкус. Пригодные красители включают красный, черный и желтый оксиды железа и красители FD&C, такие как FD&C синий №2 и FD&C красный №40, выпускаемые фирмой Ellis&Everard. Пригодные ароматизирующие агенты включают ароматизаторы со вкусом мяты, малины, лакрицы, апельсина, лимона, грейпфрута, карамели, ванили, вишни и винограда и их комбинации. Пригодные агенты для регуляции рН включают гидрокарбонат натрия, лимонную кислоту, винную кислоту, хлористоводородную кислоту и малеиновую кислоту. Пригодные подсластители включают аспартам, ацесульфам К и тауматин. Пригодные маскирующие агенты включают гидрокарбонат натрия, ионообменные смолы, соединения циклодекстрина, адсорбаты или микроинкапсулированные активные вещества.
Для получения требуемых результатов при лечении и профилактике гиперфосфатемии субстанцию по изобретению в качестве активного соединения предпочтительно вводят в дозе от 0,1 до 500 мг, более предпочтительно от 1 до 200 мг/кг массы тела, один раз в сутки. Тем не менее, при необходимости указанное количество может изменяться в зависимости от массы тела пациента, способа введения, вида животного и его индивидуальной реакции на лекарственный препарат, типа композиции или времени или интервалов, через которое вводят лекарственное средство. В особых случаях достаточной является доза ниже указанного минимального значения, в других случаях можно использовать дозы, превышающие максимальную указанную дозу. При использовании высоких доз рекомендуется разделить дозу на несколько более низких однократных доз. В конечном счете, дозу определяет лечащий врач. Обычно предпочтительным является введение незадолго перед едой, например в течение часа перед едой, или предпочтительно во время еды.
Типичная однократная твердая стандартная лекарственная форма для введения взрослому человеку включает от 1 мг до 1 г, предпочтительно от 10 мг до 800 мг субстанции по изобретению.
Твердая стандартная лекарственная форма может также включать добавку, регулирующую скорость высвобождения активного соединения. Например, субстанция по изобретению удерживается внутри гидрофобного полимерного матрикса и постепенно высвобождается из матрикса при контакте с жидкостями организма. В другом варианте субстанция по изобретению удерживается внутри гидрофильного матрикса, который постепенно или быстро растворяется в присутствии биологических жидкостей организма. Таблетка может включать два или более слоев с разными скоростями высвобождения. Используют гидрофильные, гидрофобные слои или слой из смеси гидрофильного и гидрофобного материалов. Соседние слои в многослойной таблетке разделены нерастворимым барьерным слоем или гидрофильным разделяющим слоем. Нерастворимый барьерный слой формируют из материалов, используемых для получения нерастворимой оболочки. Гидрофильный разделяющий слой получают из материала, более растворимого по сравнению с другими слоями ядра таблетки, и таким образом при растворении разделяющего слоя становятся доступными высвобождающие слои ядра таблетки.
Пригодные полимеры, регулирующие скорость высвобождения, включают полиметакрилаты, этилцеллюлозу, гидроксипропилметилцеллюлозу, метилцеллюлозу, гидроксиэтилцеллюлозу, гидроксипропилцеллюлозу, натриевую соль карбоксиметилцеллюлозы, кальциевую соль карбоксиметилцеллюлозы, полимер акриловой кислоты, полиэтиленгликоль, полиэтиленоксид, каррагенан, ацетат целлюлозы, зеин и т.п.
Пригодные материалы, которые набухают с высокой скоростью при контактировании с водными жидкостями, включают полимерные материалы, такие как натриевая соль сшитой карбоксиметилцеллюлозы, сшитая гидроксипропилцеллюлоза, высокомолекулярная гидроксипропилцеллюлоза, карбоксиметиламид, калиевая соль сополимера метакрилатдивинилбензола, полиметилметакрилат, сшитый поливинилпирролидон и высокомолекулярные поливиниловые спирты. Твердые стандартные лекарственные формы, включающие субстанцию по изобретению, упаковывают в один контейнер или получают в виде блистеров различного типа или т.п., например, с рекомендациями пациенту, где указаны дни недели для введения соответствующих доз.
Таким образом, объектами настоящего изобретения являются:
1. Соединение для применения в качестве лекарственного средства, причем соединение характеризуется формулой (I):
где MII означает по меньшей мере один двухвалентный металл,
MIII означает по меньшей мере один трехвалентный металл,
An- означает по меньшей мере один n-валентный анион,
2+a=2b+∑cn и ∑cn<0,9а.
1а. Соединение для применения в качестве лекарственного средства, причем соединение характеризуется формулой (I):
где MII означает по меньшей мере один двухвалентный металл,
MIII означает по меньшей мере один трехвалентный металл,
An- означает по меньшей мере один n-валентный анион,
2+a=2b+∑cn и ∑cn<0,9a,
z равно 2 или менее, более предпочтительно 1,8 или менее, наиболее предпочтительно 1,5 или менее.
1b. Соединение для применения в качестве лекарственного средства, причем соединение характеризуется формулой (I):
где MII означает по меньшей мере один двухвалентный металл,
MIII означает по меньшей мере один трехвалентный металл,
An- означает по меньшей мере один n-валентный анион,
а равно от 0,1 до 0,5, предпочтительно от 0,2 до 0,4,
b равно 1,5 или менее, предпочтительно 1,2 или менее,
величина с для каждого аниона определяется необходимостью обеспечить нейтральность заряда соединения по формуле 2+a=2b+∑cn,
z равно 2 или менее, более предпочтительно 1,8 или менее, наиболее предпочтительно 1,5 или менее.
2. Соединение по п.п.1, 1a или 1b, в котором в формуле (I) а>0,3.
3. Соединение по п.п.1, 1a или 1b, в котором в формуле (I) а<0,3.
4. Соединение по п.2, в котором в формуле (I) 0,03а<∑cn<0,5а.
5. Соединение по п.3, в котором в формуле (I) 03a<∑cn<0,7a.
6. Соединение по любому предшествующему п., содержащее менее 2 мас.%, предпочтительно менее 1,5 мас.%, более предпочтительно менее 1 мас.% воды, связанной с поверхностью кристалла.
7. Соединение для применения в качестве лекарственного средства, причем соединение получают нагреванием исходного материала, включающего структуру многослойного двойного гидроксида, при температуре от 200°С до 600°С, предпочтительно от 250°С до 500°С.
7а. Соединение для применения в качестве лекарственного средства, причем соединение получают нагреванием исходного материала, включающего структуру многослойного двойного гидроксида, при температуре от 200°С до 600°С, предпочтительно от 250°С до 500°С.
8. Соединение по п.7 или 7а, причем исходный материал включает соединение формулы (II):
где MII означает по меньшей мере один двухвалентный металл,
MIII означает по меньшей мере один трехвалентный металл,
An- означает по меньшей мере один n-валентный анион,
0<x≤0,5,
0<у≤1 и
0<m≤10
и где предпочтительно x=∑yn.
9. Соединение по п.8, причем в формуле (II) х>0,3.
10. Соединение по п.8, причем в формуле (II) х<0,3.
11. Соединение по любому из п.п.7-10, причем субстанция обладает на 10% более высокой фосфатсвязывающей способностью по сравнению с соединением формулы (II), из которого ее получают.
12. Применение соединения в способе получения лекарственного средства для связывания фосфата, причем соединение характеризуется формулой (I):
Где MII означает по меньшей мере один двухвалентный металл,
MIII означает по меньшей мере один трехвалентный металл,
An- означает по меньшей мере один n-валентный анион,
2+a=2b+∑cn и ∑cn<0,9a.
12а. Применение соединения в способе получения лекарственного средства для связывания фосфата, причем соединение характеризуется формулой (I):
Где MII означает по меньшей мере один двухвалентный металл,
MIII означает по меньшей мере один трехвалентный металл,
An- означает по меньшей мере один n-валентный анион,
2+a=2b+∑cn и ∑cn<0,9а,
z равно 2 или менее, более предпочтительно 1,8 или менее, наиболее предпочтительно 1,5 или менее.
12b. Применение соединения в способе получения лекарственного средства для связывания фосфата, причем соединение характеризуется формулой (I):
Где MII означает по меньшей мере один двухвалентный металл,
MIII означает по меньшей мере один трехвалентный металл,
An- означает по меньшей мере один n-валентный анион,
а равно от 0,1 до 0,5, предпочтительно от 0,2 до 0,4,
b равно 1,5 или менее, предпочтительно 1,2 или менее,
величина с для каждого аниона определяется необходимостью обеспечения нейтральности заряда молекулы по формуле 2+а=2b+∑cn,
z равно 2 или менее, более предпочтительно 1,8 или менее, наиболее предпочтительно 1,5 или менее.
13. Применение соединения в способе получения лекарственного средства для профилактики или лечения гиперфосфатемии, причем соединение характеризуется формулой (I):
где MII означает по меньшей мере один двухвалентный металл,
MIII означает по меньшей мере один трехвалентный металл,
An- означает по меньшей мере один n-валентный анион,
2+a=2b+∑cn и ∑cn<0,9a.
13 а. Применение соединения в способе получения лекарственного средства для профилактики или лечения гиперфосфатемии, причем соединение характеризуется формулой (I):
где MII означает по меньшей мере один двухвалентный металл,
MIII означает по меньшей мере один трехвалентный металл,
An- означает по меньшей мере один n-валентный анион,
2+а=2b+∑cn и ∑cn<0,9а,
z равно 2 или менее, более предпочтительно 1,8 или менее, наиболее предпочтительно 1,5 или менее.
13b. Применение соединения в способе получения лекарственного средства для профилактики или лечения гиперфосфатемии, причем соединение характеризуется формулой (I):
Где MII означает по меньшей мере один двухвалентный металл,
MIII означает по меньшей мере один трехвалентный металл,
An- означает по меньшей мере один n-валентный анион,
а равно от 0,1 до 0,5, предпочтительно от 0,2 до 0,4,
b равно 1,5 или менее, предпочтительно 1,2 или менее,
величина с для каждого аниона определяется необходимостью обеспечения нейтральности заряда молекулы по формуле 2+a=2b+∑cn,
z равно 2 или менее, более предпочтительно 1,8 или менее, наиболее предпочтительно 1,5 или менее.
14. Применение по любому из п.п.12-13b, причем в формуле (I) а>0,3 и предпочтительно 0,03a<∑cn<0,5a.
15. Применение по любому из п.п.12-13b, причем в формуле (I) а<0,3 и предпочтительно 0,03а<∑cn<0,7а.
16. Применение соединения в способе получения лекарственного средства для связывания фосфата, причем соединение получают нагреванием исходного материала, включающего структуру многослойного двойного гидроксида, при температуре от 200°С до 600°С, предпочтительно от 250°С до 500°С.
16а. Применение соединения в способе получения лекарственного средства для связывания фосфата, причем соединение получают нагреванием исходного материала, включающего структуру многослойного двойного гидроксида, при температуре от 200°С до 600°С, предпочтительно от 250°С до 500°С.
17. Применение соединения в способе получения лекарственного средства для профилактики или лечения гиперфосфатемии, причем соединение получают нагреванием исходного материала, включающего структуру многослойного двойного гидроксида, при температуре от 200°С до 600°С, предпочтительно от 250°С до 500°С.
17а. Применение соединения в способе получения лекарственного средства для профилактики или лечения гиперфосфатемии, причем соединение получают нагреванием исходного материала, включающего структуру многослойного двойного гидроксида, при температуре от 200°С до 600°С, предпочтительно от 250°С до 500°С.
18. Применение по п.п.16, 16а, 17 или 17а, причем исходный материал включает соединение формулы (II):
где MII означает по меньшей мере один двухвалентный металл,
MIII означает по меньшей мере один трехвалентный металл,
An- означает по меньшей мере один n-валентный анион,
0<x≤0,5,
0<у≤1 и
0<m≤10
и где предпочтительно x=∑yn.
19. Применение по п.18, причем в формуле (II) х>0,3.
20. Применение по п.18, причем в формуле (II) х<0,3.
21. Фармацевтическая композиция, включающая (1) соединение формулы (I):
где MII означает по меньшей мере один двухвалентный металл,
MIII означает по меньшей мере один трехвалентный металл,
An- означает по меньшей мере один n-валентный анион,
2+a=2b+∑cn и ∑cn<0,9а,
z равно 2 или менее, более предпочтительно 1,8 или менее, наиболее предпочтительно 1,5 или менее, и
(2) фармацевтически приемлемый носитель, разбавитель, эксципиент или адъювант.
22. Фармацевтическая композиция, включающая
(1) соединение формулы (I):
где MII означает по меньшей мере один двухвалентный металл,
MIII означает по меньшей мере один трехвалентный металл,
An- означает по меньшей мере один n-валентный анион,
а равно от 0,1 до 0,5, предпочтительно от 0,2 до 0,4,
b равно 1,5 или менее, предпочтительно 1,2 или менее,
величина с для каждого аниона определяется необходимостью обеспечения нейтральности заряда молекулы по формуле 2+a=2b+∑cn,
z равно 2 или менее, более предпочтительно 1,8 или менее, наиболее предпочтительно 1,5 или менее, и
(2) фармацевтически приемлемый носитель, разбавитель, эксципиент или адъювант.
23. Фармацевтическая композиция по п.21 или п.22, причем в формуле (I) а>0,3 и предпочтительно 0,03а<∑cn<0,5а.
24. Фармацевтическая композиция по п.21 или п.22, причем в формуле (I) а<0,3 и предпочтительно 0,03a<∑cn<0,7a.
25. Фармацевтическая композиция, включающая
(1) соединение, которое получают нагреванием исходного материала, включающего структуру многослойного двойного гидроксида, при температуре от 200°С до 600°С, предпочтительно от 250°С до 500°С, и
(2) фармацевтически приемлемый носитель, разбавитель, эксципиент или адъювант.
26. Фармацевтическая композиция, включающая
(1) соединение, полученное нагреванием исходного материала, включающего структуру многослойного двойного гидроксида, при температуре от 200°С до 600°С, предпочтительно от 250°С до 500°С, и
(2) фармацевтически приемлемый носитель, разбавитель, эксципиент или адъювант.
27. Фармацевтическая композиция по п.26, причем исходный материал включает соединение формулы (II):
где MII означает по меньшей мере один двухвалентный металл,
MIII означает по меньшей мере один трехвалентный металл,
An- означает по меньшей мере один n-валентный анион,
0<х≤0,5,
0<у≤1 и
0<m≤10
и где предпочтительно x=∑yn.
28. Фармацевтическая композиция по п.27, причем в формуле (II) х>0,3.
29. Фармацевтическая композиция по п.27, причем в формуле (II) х<0,3.
Все публикации, указанные в приведенном выше описании, включены в настоящую заявку в виде ссылок. Различные модификации и варианты описанных методов и систем по настоящему изобретению представляются очевидными для специалиста в данной области и не выходят за пределы объема и сущности изобретения. Настоящее изобретение описано в виде определенных предпочтительных вариантов осуществления изобретения и не ограничивается указанными конкретными вариантами. Предполагается, что различные модификации указанных способов применения настоящего изобретения, очевидные специалисту в данной области техники, включены в объем перечисленных ниже пунктов формулы изобретения.
Настоящее изобретение иллюстрируется следующими примерами, не ограничивающими его объем.
Примеры
Метод 1 (M1)
(а) Получение исходных материалов
Два исходных материала (партия А и партия В) получали методом, описанным ниже.
Твердый карбонат натрия (21 кг) и твердый гидроксид натрия (25 кг) растворяли в 158 кг деионизированной воды (DIW). В отдельном сосуде смешивали 129 кг деионизированной воды, 52 кг твердого сульфата магния (MgSO4·7H2O) и 50 кг 43 мас.% раствора сульфата железа (фирма Kemira), при этом получали партию А (соотношение Mg/Fe, 2:1). В другом варианте в 143 кг деионизированной воды растворяли 62 кг сульфата магния и 30 кг сульфата железа, при этом получали партию В (соотношение Mg/Fe, 4:1). Затем в течение 60 мин растворы одновременно добавляли в 68 кг холодной воды при перемешивании, поддерживая в реакционной смеси рН 10,3 (±0,2 рН) за счет регуляции скорости подачи растворов и поддерживая температуру реакционной смеси не более 30°С. После завершения добавления реагентов реакционную смесь перемешивали в течение еще 30 мин, затем смесь фильтровали на фильтр-прессе (на рамном фильтр-прессе). Продукт отделяли фильтрованием, трижды порциями по 39 л промывали холодной деионизированной водой (DIW) и твердое вещество продували сжатым воздухом для удаления воды. Затем продукт высушивали в лотковой печи при 100-120°С.
Растворы готовили в чистых пластиковых бочках объемом 200 л с использованием мешалки с большим сдвигом для растворения твердых веществ в DIW.
Для получения раствора гидроксида натрия перед включением смесителя головку полностью погружали в воду. Затем одной порцией добавляли гранулированный гидроксид натрия и добавляли карбонат натрия. Раствор перемешивали до полного растворения твердых веществ.
Для получения раствора сульфата магния/железа перед включением смесителя мешалку Unishear полностью погружали в воду. Сульфат магния добавляли в воду после добавления раствора сульфата железа. Раствор перемешивали до полного растворения сульфата магния.
Для проведения осаждения использовали достаточное количество DIW (деионизированной воды), чтобы покрыть лопасти смесителя в реакционном сосуде (примерно 68 кг воды). Скорости потоков щелочного карбоната и раствора сульфата металла контролировали по показаниям расходомеров.
После включения пропеллерной мешалки в реакционном сосуде объемом 100 галлонов (прибл. 455 л) включали насосы для подачи в реакционный сосуд растворов щелочного карбоната и сульфата металла. В ходе осаждения рН реакционной смеси измеряли на рН-метре. Требуемое значение рН 10,3 (±0,2 рН) достигалось в течение 5-10 мин. При добавлении указанных растворов (в течение 1 ч) значение рН контролировали через определенные интервалы времени, а скорости потоков карбоната или сульфата регулировали при необходимости в соответствии с величиной рН. Температуру также контролировали через равные промежутки времени. Температура реакционной смеси медленно возрастала частично за счет теплоты реакции, а также за счет нагревания растворов при перемешивании в смесителе с высоким сдвигом и при растворении твердых веществ. Если температура достигала 30°С, включали охлаждающую воду. Общее время добавления растворов составляло 60 мин. Затем смесь перемешивали в течение 30 мин при 25-30°С.
Полученную партию фильтровали на фильтр-прессе, продукт на фильтре продували сжатым воздухом в течение примерно 30-60 мин, твердую массу выгружали и высушивали при 100-120°С в течение 16 ч в лотковой печи.
Для анализа образцов перед прокаливанием небольшое количество продукта измельчали пестиком в ступке. Более значительные количества образцов, предназначенных для прокаливания, измельчали в коммерческом блендере (фирма Waring Blender 800 Е (фирма Waring)) в течение не более 8 мин.
Образцы стандартизовали с использованием сита из нержавеющей стали диаметром 200 мм (сито/крышка/приемник) с диаметром отверстий 106 мкм. Образец просеивали вручную, слишком крупную фракцию смешивали с остальной частью высушенного образца для повторного измельчения. Операцию повторяли до тех пор, пока размер всех частиц образца не составлял <106 мкм. Измельченный продукт переносили в запечатанный пластиковый пакет и встряхивали для тщательного его перемешивания.
При этом получали партию А и партию В, в которых соотношение Mg/Fe составляло 2:1 и 4:1 соответственно, и по данным анализа определяли фактические молекулярные формулы:
партия A: [Mg0,67Fe0,33(OH)2][(СО3)0,17(SO4)0,01,0,43H2O][Na2SO4]0,03
партия В: [Mg0,80Fe0,20(OH)2][(СО3)0,16(SO4)0,01,0,60Н2О][Na2SO4]0,03
(b) Примеры получения субстанций по настоящему изобретению
Образцы прокаливали (термическая обработка образцов при температуре до 1000°С) различными способами, а именно в печи для отжига барабанного типа, в сушилке с псевдоожиженным слоем и в печи в стандартном статическом режиме. Измельченные и просеянные образцы (от 10 г до 100 г), полученные по методу 4а и 4b, помещали в печь. Образцы с соотношением 2:1 и 4:1 прокаливали различными методами при 500°С в течение 3 ч, в результате получали образцы с более высокой фосфатсвязывающей способностью и более низким содержанием растворимого Mg по сравнению с непрокаленным материалом, хотя указанное улучшение зависит от способа и условий обработки. Обработка в псевдоожиженном слое, в ходе которой образец перегревали при 500°С в течение 15 ч, приводит к снижению фосфатсвязывающей способности по сравнению с непрокаленным образцом. И наоборот, все образцы, прокаленные при 500°С в течение 3 ч, характеризуются возрастанием фосфатсвязывающей способности и более низким содержанием растворимого магния по сравнению с непрокаленным материалом. Метод прокаливания в печи в стационарном режиме использовали для обжига образцов при различных температурах в течение только 3 ч.
Образцы смешанных соединений металлов массой 10-30 г помещали в фарфоровые чашки (для температуры менее 500°С) или кварцевые чашки (для температуры более 500°С). При этом использовали чашки или тигли диаметром от 7 до 16 см. Во всех экспериментах слой образцов помещали на глубину 1 см. Использовали четыре типа стандартных печей, а именно для температуры до 500°С печь Oven300 plus (фирма Gallenkamp) и печь ML016 (фирма Vectstar), a для температуры выше 500°С печи HTL3 и SP14 (фирма Vecstar). Образцы помещали в предварительно нагретую печь и выдерживали в течение 3 ч. Через 3 ч прокаленные образцы помещали в эксикатор для охлаждения. Затем образцы хранили в сухих и прохладных условиях.
Затем в термически обработанных материалах определяли фосфатсвязывающую способность, количество растворимого магния, содержание поверхностно-абсорбированной воды, а также проводили рентгеноструктурный анализ, как описано ниже.
Метод анализа 1
Определение потери воды (в %), связанной с поверхностью кристаллов, при 105°С
Образцы уравновешивали в течение нескольких суток при температуре 20°С и относительной влажности 20%, высушивали при 105°С в течение 4 ч и определяли потерю массы в процентах. При высушивании при 105°С удаляли поверхностно-абсорбированную воду (то есть химически не связанную воду).
Чистую сухую чашку (тигель), доведенную до постоянной массы высушиванием, точно взвешивали и массу регистрировали (W1). Тигель заполняли образцом (от 1,0 до 3,0 г) и регистрировали общую массу (W2). Затем тигель помещали в печь и выдерживали при 105°С в течение 4 ч. Через 4 ч тигель удаляли из печи, охлаждали до комнатной температуры в эксикаторе, проводили повторное взвешивание и регистрировали конечную массу (W3).
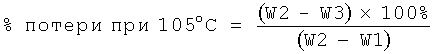
Метод анализа 2
Определение фосфатсвязывающей способности и количества растворимого магния
В 40 мМ натрий-фосфатный буферный раствор (рН 4) добавляли фосфатсвязывающее соединение, полученную смесь центрифугировали, супернатант разбавляли и определяли содержание Fe, Mg и Р методом ICP-OES (оптической эмиссионной спектроскопии индуктивно сопряженной плазмы), известным специалисту в данной области.
Для анализа использовали следующие реактивы: моногидрат монозамещенного фосфата натрия (фирма Aldrich), 1 M соляная кислота, вода AnalaR™, стандартный раствор фосфора (10000 мкг/мл, фирма Romil Ltd), стандартный раствор магния (10000 мкг/мл, фирма Romil Ltd), стандартный раствор железа (1000 мкг/мл), хлорид натрия (фирма BDH).
Для анализа использовали следующие приборы: центрифуга (фирма Metler 2000Е), роторная качалка для анализа крови (фирма Stuart Scientific), миникачалка (MS1), система ICP-OES, пробирки для анализа крови.
Фосфатный буферный раствор (рН 4) получали растворением 5520 г (±0,001 г) дигидрофосфата натрия сначала в небольшом количестве воды AnalaR™, затем переносили в мерную колбу объемом 1 л. В мерную колбу объемом 1 л при постоянном перемешивании добавляли по каплям 1 M HCl до рН 4 (±0,1), затем добавляли воду AnalaR™ до 1 л и тщательно перемешивали.
Навеску каждого образца (0,4 г±0,005 г) помещали в пробирку для анализа крови и пробирки устанавливали в штатив. Все образцы дублировали. В каждую пробирку для анализа крови, содержащую навеску исследуемого материала, добавляли пипеткой 10 мл фосфатного буферного раствора и пробирки закрывали завинчивающимися крышками. Пробирки встряхивали на миникачалке в течение приблизительно 10 с, переносили в роторную качалку для анализа крови и перемешивали в течение 30 мин (±2 мин). Затем пробирки центрифугировали при 3000 об/мин и 20°С в течение 5 мин. Образцы удаляли из центрифуги, пипеткой отбирали аликвотные части (по 2,5 мл) супернатанта и переносили в новые пробирки для анализа крови. В каждую аликвотную часть (2,5 мл) пипеткой добавляли по 7,5 мл воды AnalaR™, закрывали завинчивающейся крышкой и тщательно перемешивали. Затем растворы анализировали в калиброванной системе ICP-OES.
Фосфатсвязывающую способность определяли по следующему уравнению: связывание фосфата (%) = 100-(TP/SP×100)
Высвобождение магния определяли по следующему уравнению:
высвобождение магния (ммоль/1) = TMg-SMg
где
TP означает содержание фосфата в растворе фосфата после реакции с фосфатсвязывающим соединением.
SP означает содержание фосфата в растворе фосфата до реакции с фосфатсвязывающим соединением.
TMg означает содержание магния в растворе фосфата после реакции с фосфатсвязывающим соединением.
SMg означает содержание магния в растворе фосфата до реакции с фосфатсвязывающим соединением.
Метод анализа 3
Рентгеноструктурный анализ
Анализировали порошкообразные образцы при угле 2θ 2-70° с использованием автоматического дифрактометра для рентгеновской порошковой дифрактометрии (излучение CuKα при 40 кВ и 55 мА) (фирма Philips).
Результаты анализа
Результаты вышеуказанного анализа термически обработанных материалов, полученных из исходных материалов партии А и партии В, приведены ниже в таблице 1. Температура, при которой проводили термическую обработку, указана в каждом случае для образцов а-m в таблице 1. Пример "а" является контрольным примером, в каждом случае представляющим собой термически не обработанный исходный материал.
Соединения, описанные в других примерах, получали, как описано ниже, с использованием других способов.
Метод анализа 2 (М2)
Два исходных материала (раствор 1 и раствор 2) получали, как описано ниже.
Сульфат магния и сульфат железа растворяли в воде AnalaR™, при этом получали раствор 1. В отдельном сосуде растворяли карбонат натрия и гидроксид натрия в воде AnalaR™, при этом получали раствор 2. Количество реагентов рассчитывали в соответствии с требуемым соотношением катионов металлов. Затем растворы одновременно добавляли в воду при перемешивании в течение 50 мин, поддерживая в смеси рН 10,3 (±0,2 рН) и температуры не выше 30°С за счет регуляции скорости подачи реагентов. После завершения добавления реакционную смесь перемешивали в течение еще 30 мин и фильтровали на воронке Бюхнера. Продукт отделяли фильтрованием, промывали порциями по 220 мл холодной водой AnalaR™ и высушивали в предварительно нагретой печи.
Для получения раствора 1 воду AnalaR™ взвешивали в отдельном сосуде и при перемешивании с использованием механической мешалки с верхним приводом добавляли требуемое количество раствора сульфата железа (фирма Ferripure, ранее Kemira). В полученный раствор сульфата железа при перемешивании переносили количественно раствор сульфата магния (фирма Epsom Salt, ранее William Biythe) и перемешивали до растворения.
Для получения раствора 2 воду AnalaR™ взвешивали в отдельном сосуде и при перемешивании с использованием механической мешалки с верхним приводом растворяли требуемое количество раствора карбоната натрия (фирма Pharmakarb, ранее Brunner Mond). В полученный раствор карбоната натрия при перемешивании переносили количественно раствор гидроксида натрия (фирма Pearl Caustic Soda, ранее INEOS Chlor) и перемешивали до растворения.
Рассчитанную массу воды AnalaR™ помещали в сосуд. Скорости потоков растворов щелочного карбоната и сульфатов металлов контролировали по показаниям расходомеров.
В сосуде с водой включали пропеллерную мешалку, а затем насосы для подачи растворов щелочного карбоната и сульфата металла. В ходе осаждения величину рН реакционной смеси контролировали на рН-метре. Требуемое значение рН составляло 10,3 (±0,2 единицы рН). Требуемое значение рН устанавливалось в течение приблизительно 5-10 мин. Во время добавления растворов реагентов (50 мин) величину рН измеряли с интервалом 1 мин и поддерживали на требуемом уровне за счет регуляции скорости подачи раствора карбоната. Температуру также измеряли с интервалом 1 мин. Общее время добавления реагентов составляло 50 мин, после чего смесь перемешивали в течение еще 30 мин.
Затем суспензию продукта фильтровали в вакууме на воронке Бюхнера, причем в качестве фильтра использовали уплотненную беззольную фильтровальную бумагу (№541, фирма Whatman™). Осадок на фильтре промывали порциями воды AnalaR™. Полученный продукт переносили в сосуд и высушивали в предварительно нагретой печи при 120°С.
Для анализа образец продукта измельчали с использованием шаровой мельницы (Retsch PM 100) и просеивали через сито из нержавеющей стали диаметром 200 мм (с диаметром отверстий 106 мкм) на качалке для сит (Retsch AS-200). Слишком крупные частицы материала смешивали с исходным материалом высушенного образца для повторного измельчения до размера частиц <106 мкм.
Растворы 1 и 2, использованные для получения соединений, как описано в примерах 1 и 3, получали по методу М2, состав указанных растворов представлен ниже:
Реакция, описанная в примере 1, соответствует следующему уравнению:
4MgSO4+Fe2(SO4)3+12NaOH+Na2CO3→Mg4Fe2(OH)12.CO3.nH2O+7Na2SO4
При изменении соотношения катионов MII/MIII до 2:1, 3:1, 4:1 получали другие композиции.
Метод 3 (М3)
Продукт получали аналогично тому, как описано в методе М2, но два раствора подавали в сосуд с избытком приблизительно до 2 л при постоянном перемешивании.
Первый литр отбрасывали и собирали 3-4 литра суспензии.
Метод 4 (М4)
Продукт получали аналогично тому, как описано в методе М2, вплоть до добавления растворов 1 и 2. При получении суспензии и последующем фильтровании продукт не промывали. Непромытый продукт переносили в сосуд и высушивали в предварительно нагретой печи при 120°С.
Метод 5 (М5)
Материал при соотношении 4:1 получали аналогично тому, как описано в методе М2, вплоть до добавления растворов А и В. При получении суспензии партию разделяли на две равные части. Одну часть суспензии обрабатывали методом М2, т.е. фильтровали через воронку Бюхнера, промывали требуемым количеством воды AnalaR™ и высушивали в предварительно нагретой печи при 120°С. Другую часть суспензии выдерживали, т.е. переносили в аналитический стакан и нагревали на колбонагревателе при 60°С в течение 4 ч. Продукт отделяли фильтрованием, промывали и высушивали, как описано в примере М2. Полученный продукт использовали в примере 5.
Метод 6 (М6)
Соединение, полученное в примере 6, является коммерческой смесью металлов MgAl (Macrosorb CT-100, фирма Ineos Silicas Ltd UK).
Метод (М7)
Указанный способ включает стадию анионообмена (на ионы хлора).
В суспензию материала (соотношение 3:1, получен методом М3) в течение 20 мин добавляли соляную кислоту со скоростью подачи, обеспечивающей поддержание в реакционной смеси рН от 9,5 до 10,5. Указанное добавление проводили в атмосфере азота, т.е. суспензию продували током азота. После добавления кислоты реакционную смесь перемешивали в течение еще 5 мин в потоке азота, а затем смесь фильтровали через воронку Бюхнера. Продукт на фильтре промывали холодной водой AnalaR™ (9×220 мл). Полученный продукт высушивали в предварительно нагретой печи.
Для получения исходной суспензии 22 г материала (соотношение 3:1) взвешивали и при перемешивании механической мешалкой с верхним приводом количественно переносили в сосуд, содержащий 200 мл воды AnalaR™. Через полученную суспензию постоянно пропускали азот.
Для получения раствора соляной кислоты исходный 1 М раствор разбавляли до получения 0,05 М раствора. Перед использованием через раствор в течение 30 мин пропускали азот.
В сосуде, содержащем исходную суспензию, включали механическую мешалку с верхним приводом, а затем насос для подачи соляной кислоты. При добавлении кислоты величину рН и температуру реакционной смеси контролировали на рН-метре. Общее время добавления кислоты составляло 20 мин, после чего смесь перемешивали в течение еще 5 мин при постоянном пропускании азота.
Затем суспензию фильтровали в вакууме на воронке Бюхнера с использованием в качестве фильтра уплотненной беззольной фильтровальной бумаги (№541, фирма Whatman™). Слой твердого вещества промывали 9 порциями по 220 мл воды AnalaR™. Промытый продукт переносили в сосуд и высушивали в предварительно нагретой печи при 120°С.
Образец продукта для анализа измельчали с использованием шаровой мельницы (Retsch PM 100).
Для анализа образец продукта просеивали через сито из нержавеющей стали диаметром 200 мм (с диаметром отверстий 106 мкм) на качалке для сит (Retsch AS-200). Слишком крупные частицы смешивали с исходным материалом для повторного измельчения до размера <106 мкм.
Метод анализа 4
Определение содержания карбоната
Образец известной массы сжигали при 1350°С в печи в атмосфере чистого кислорода. При этом весь углерод в составе образца окислялся до СО2, который пропускали через увлажненную ловушку, продукт анализировали с использованием инфракрасного детектора и содержание углерода в образце определяли при сравнении со стандартом известной концентрации.
Для анализов использовали анализатор углерода и серы Leco SC-144DR с источником кислорода, снабженный керамическими лодочками для сжигания, захватным устройством и трубкой для вдувания.
Образец (0,2±0,01 г) взвешивали в лодочке для сжигания. Затем лодочку помещали в печь Leco и определяли содержание углерода. Анализ повторяли дважды.
Содержание в % карбоната определяли по формуле:
%С (образец) = (%С1+%С2)/2
где С1 и С2 означают содержание углерода, определенное в двух повторах.
%СО3 = %С×60/12.
Метод анализа 5
Рентгеновская флуоресценция (XRF)
Анализ продукта методом рентгеновской флуоресценции проводили на спектрометре PW2400 Wavelength Dispersive XRF (фирма Philips). Образец сплавляли с тетра/метаборатом лития (высокой чистоты, 50:50) и помещали в прибор в виде стекловидного шарика.
Если не указано иное, для анализа использовали реактивы аналитической или эквивалентной чистоты.
Вода AnalaR™, флюс тетрабората/метабората лития (50%:50%) (высокой чистоты, фирма ICPH Fluore-X 50),
Для анализа использовали муфельную печь (фирма Furnace), температура 1025°С, удлиненные щипцы, ручные щипцы, поднос для литья из Pt/5%Au и чашку из Pt/5%/Au.
Образец (1,5±0,0002 г) и тетра/метаборат (7,5000±0,0002 г) в чашке из Pt/5%/Au взвешивали на аналитических весах. Два компонента в чашке перемешивали шпателем, помещали в печь при 1025°С и выдерживали в течение 12 мин. Чашку встряхивали в течение 6 мин и в течение 9 мин для получения гомогенного образца. В печь помещали поднос для литья и выдерживали в течение 9 мин для достижения температурного равновесия. Через 12 мин расплавленный образец выливали на поднос для литья, который вынимали из печи и охлаждали. Состав шарика определяли на спектрофотометре.
Соотношения MII/MIII в исходных образцах А и В, а также в образцах 1-7 и методы получения указаны в таблице 2
В таблице 3 приведены результаты анализов состава (в виде содержания оксидов в %) непрокаленных и прокаленных материалов.
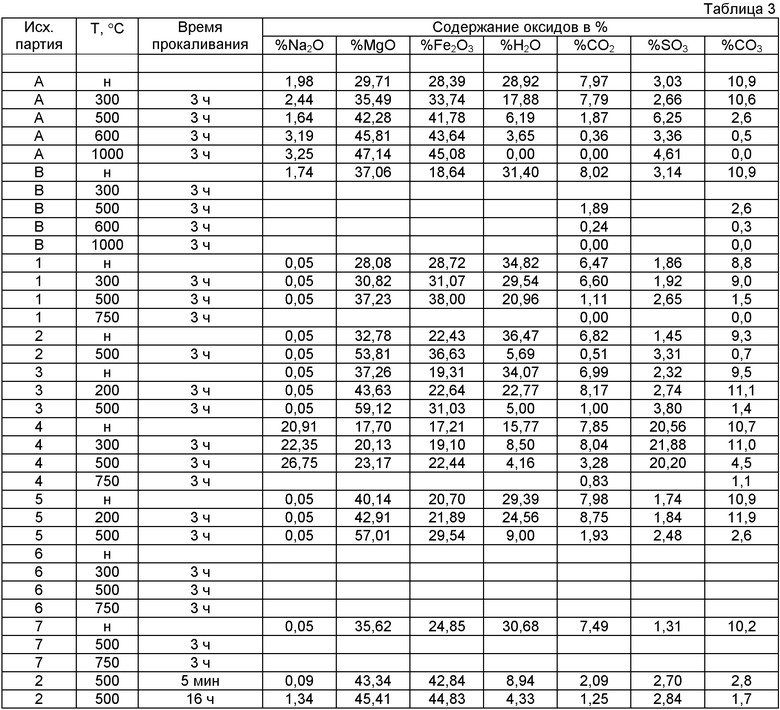
н означает непрокаленный материал.
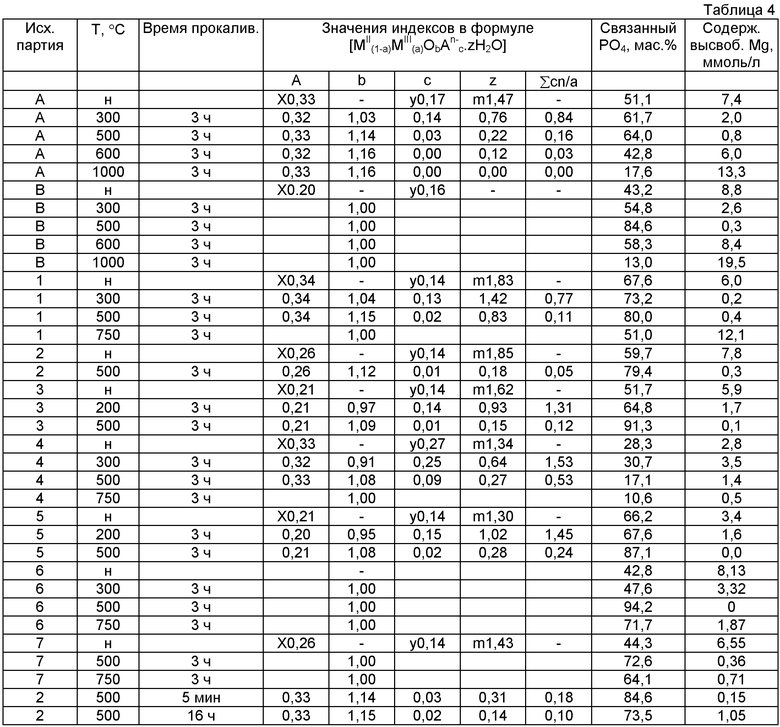
н означает непрокаленный материал.
Для непрокаленного материала, присутствующего в форме формулы (II), вместо индексов а, с и z в таблице использованы индексы х, у и m.
В Таблице 4 содержание Н2О в мас.% рассчитывали по следующему уравнению:
% H2O=100%-(%Na2O+%MgO+%Fe2O3+%SO3+%CO2)
Содержание %Na2O+%MgO+%Fe2O3+%SO3 определяли методом рентгеновской флуоресценции.
Содержание %CO2 определяли на анализаторе углерода LECO.
Содержание %CO2=(ММ CO2/ММ СО3)×(%СО3)=(44/60)×(%СО3).
Значения индексов а, b, с и z в таблице 4 рассчитывали с использованием значений, приведенных в таблице 3, как указано ниже (в расчетах ММ означает молекулярная масса).
Расчет значения а
а=моли MIII/(моли MII+моли MII)
При подстановке молекулярных долей в указанной формуле получают
а=(2×%Fe2O3/ММ Fe2O3)/((%MgO/MM MgO)+(2×%Fe2O3/MM Fe2O3))
ММ Fe2O3=159,6
MM MgO=40,3
Значения %Fe2O3 и %MgO указаны в таблице 4.
Расчет значения с
Молярная доля трехвалентного металла (FeIII), деленная на молярную долю аниона (СО3) = а, деленное на с.
При подстановке значения а, молекулярных масс и мас.% (таблица 3) для Fe2O3 и СО3 в приведенное выше уравнение получают значение с.
с=0,33(%СО3/ММ СО3)/(2×%Fe2O3/ММ Fe2O3)
Молярную долю SO4, присутствующего в качестве аниона промежуточного слоя, рассчитывали вычитанием из общего количества SO4 количества SO4, связанного с Na2O в форме Na2SO4. В указанном выше уравнении количество обменного SO4 суммировали с молярной долей для аниона карбоната.
Расчет значения b
b рассчитывают при условии, что формула [MII (1-a)MIII (a)ObAn- c.zH2O] характеризуется нейтральным зарядом.
Таким образом, сумма произведений зарядов каждого элемента на индексы а, b, с в приведенном ниже уравнении равняется 0:
2(1-a)+3a+(-2b)+(-nc)=0
После преобразования уравнение имеет следующий вид:
b=(2+a-nc)/2
при замене n=2 (заряд карбонатного аниона)
и значения для а и с в приведенной выше формуле получают значение b.
Расчет значения z
Молярная доля трехвалентного металла (FeIII), деленная на молярную долю H2O=а, деленному на z.
(2×%Fe2O3/ММ Fe2O3)/(%H2O/ММ H2O)=a/z
При подстановке а, молекулярной массы и мас.% (таблица 4) для H2O и Fe2O3 в вышеприведенном уравнении получают значение z.
z=0,33(%H2O/ММ H2O)/(2×%Fe2O3/ММ Fe2O3)
Расчет значения ∑cn/a
Указанную величину рассчитывают при подстановке значений с, n и а.
Влияние рН
В приведенной ниже таблице 5 показано влияние рН фосфатного раствора на связывание и высвобождение магния.
ющее соединение
Результаты, приведенные в таблице 5, получали по данным определения связывания фосфата, как описано в методе анализа 2, но со следующими изменениями: 0,5 г фосфатсвязывающего соединения растворяли в 125 мл 4 мМ фосфатного раствора, затем образцы инкубировали в плотно закрытых полипропиленовых конических флаконах на водяной бане при 37°С при встряхивании со скоростью 200 об/мин в течение 30 мин.
| название | год | авторы | номер документа |
|---|---|---|---|
| СМЕШАННЫЕ СОЕДИНЕНИЯ МЕТАЛЛОВ ДЛЯ ПРИМЕНЕНИЯ В КАЧЕСТВЕ АНТАЦИДОВ | 2008 |
|
RU2596489C2 |
| СМЕШАННЫЕ СОЕДИНЕНИЯ МЕТАЛЛОВ ДЛЯ ПРИМЕНЕНИЯ В КАЧЕСТВЕ АНТАЦИДОВ | 2008 |
|
RU2510265C2 |
| МАТЕРИАЛ | 2007 |
|
RU2437650C2 |
| ФОСФАТНЫЙ АДСОРБЕНТ | 2010 |
|
RU2527682C2 |
| КАТАЛИЗАТОРЫ ДЛЯ КОНВЕРСИИ ГИДРОКСИПРОПИОНОВОЙ КИСЛОТЫ ИЛИ ЕЕ ПРОИЗВОДНЫХ В АКРИЛОВУЮ КИСЛОТУ ИЛИ ЕЕ ПРОИЗВОДНЫЕ | 2013 |
|
RU2591192C2 |
| КАТАЛИЗАТОРЫ ДЛЯ ПОЛУЧЕНИЯ АКРИЛОВОЙ КИСЛОТЫ ИЛИ ЕЕ ПРОИЗВОДНЫХ | 2013 |
|
RU2586329C2 |
| СПОСОБ ПОЛУЧЕНИЯ КОМПЛЕКСНОГО МЕТАЛЛОФОСФАТНОГО ПРОДУКТА (ВАРИАНТЫ) | 2014 |
|
RU2579378C2 |
| ГИДРОКСИД КАРБОНАТ ЛАНТАНА, ОКСИКАРБОНАТ ЛАНТАНА И СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2011 |
|
RU2570007C2 |
| КАТАЛИТИЧЕСКАЯ КОНВЕРСИЯ ГИДРОКСИПРОПИОНОВОЙ КИСЛОТЫ ИЛИ ЕЕ ПРОИЗВОДНЫХ В АКРИЛОВУЮ КИСЛОТУ И ЕЕ ПРОИЗВОДНЫЕ | 2013 |
|
RU2586327C2 |
| ПОЛИМЕТАЛЛИЧЕСКИЕ МАТЕРИАЛЫ С ИСПОЛЬЗОВАНИЕМ СОЕДИНЕНИЙ ЧЕТВЕРТИЧНОГО АЛКИЛАММОНИЯ | 2018 |
|
RU2757484C1 |
Субстанция для применения в качестве лекарственного средства, включающая твердое смешанное соединение металлов формулы (I):
где MII означает по меньшей мере один двухвалентный металл, MIII означает Fe3+, An- означает по меньшей мере один n-валентный анион, z равно 2 или менее, а = число молей MIII / (число молей MII + число молей MIII), 2+a=2b+Σcn и Σcn<0,9а. Изобретение также относится к способу получения указанной субстанции, который включает нагревание при температуре от 225°С до 550°С в течение 5 часов соединения, включающего структуру слоистого двойного гидроксида. Изобретение относится также к применению указанной субстанции для получения лекарственного средства и фармацевтической композиции для связывания фосфата и для профилактики или лечения гиперфосфатемии. Изобретение обеспечивает снижение потерь бивалентных металлов в связывающих фосфат субстанциях. 10 н. и 23 з.п. ф-лы, 5 табл.
1. Субстанция для применения в качестве лекарственного средства, включающая твердое смешанное соединение металлов формулы (I):
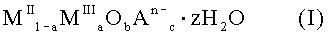
где MII означает по меньшей мере один двухвалентный металл, MIII означает Fe3+, An- означает по меньшей мере один n-валентный анион, z означает 2 или менее; а = число молей MIII / (число молей MII + число молей MIII); 2+a=2b+Σcn и Σcn<0,9a.
2. Субстанция по п.1, причем в формуле (I) а>0,3.
3. Субстанция по п.1, причем в формуле (I) а<0,3.
4. Субстанция по п.2, причем в формуле (I) 0,03a<Σcn<0,5a.
5. Субстанция по п.3, причем в формуле (I) 0,03a<Σcn<0,7a.
6. Субстанция по любому из предшествующих пунктов, в которой твердое смешанное соединение металлов содержит менее 2 мас.%, предпочтительно менее 1,5 мас.%, более предпочтительно менее 1 мас.% поверхностно-поглощенной воды.
7. Субстанция для применения в качестве лекарственного средства, включающая твердое смешанное соединение металлов, определенное в п.1, полученное или получаемое при нагревании исходного материала, включающего структуру слоистого двойного гидроксида, при температуре от 225 до 550°С в течение периода времени около 5 ч или менее.
8. Субстанция по п.7, причем исходный материал включает соединение формулы (II)
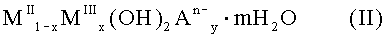
где MII означает по меньшей мере один двухвалентный металл, MIII означает Fe3+, An- означает по меньшей мере один n-валентный анион, х=Σyn, 0<х≤0,5, 0<у≤1 и 0<m≤10.
9. Субстанция по п.8, причем в формуле (II) х≥0,3.
10. Субстанция по п.8, причем в формуле (II) х<0,3.
11. Субстанция по любому из пп.7-10, причем субстанция обладает на 10% более высокой фосфатсвязывающей способностью по сравнению с соединением формулы (II), из которого ее получают.
12. Применение для получения лекарственного средства для связывания фосфата субстанции, включающей твердое смешанное соединение металлов формулы (I):
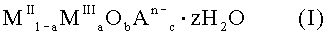
где MII означает по меньшей мере один двухвалентный металл, MIII означает Fe3+, An- означает по меньшей мере один n-валентный анион; z означает 2 или менее; а = число молей MIII / (число молей MII + число молей MIII); 2+а=2b+Σcn и Σcn<0,9а.
13. Применение для получения лекарственного средства для профилактики или лечения гиперфосфатемии субстанции, включающей твердое смешанное соединение металлов формулы (I):
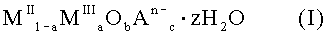
где MII означает по меньшей мере один двухвалентный металл, MIII означает Fe3+, An- означает по меньшей мере один n-валентный анион, z означает 2 или менее; а = число молей MIII / (число молей MII + число молей MIII); 2+а=2b+Σcn и Σcn<0,9a, a z равно 2 или менее.
14. Применение по п.12 или 13, причем в формуле (I) а≥0,3 и предпочтительно 0,03а<Σcn<0,5а.
15. Применение по п.12 или 13, причем в формуле (I) а<0,3 и предпочтительно 0,03а<Σcn<0,7а.
16. Применение для получения лекарственного средства для связывания фосфата в организме животного, нуждающегося в указанном лечении, субстанции, как она определена в п.1, полученной при нагревании исходного материала, включающего структуру слоистого двойного гидроксида, при температуре от 225 до 550°С в течение периода времени около 5 ч или менее.
17. Применение для получения лекарственного средства для профилактики или лечения гиперфосфатемии субстанции по п.1, полученной при нагревании исходного материала, включающего структуру слоистого двойного гидроксида, при температуре от 225 до 550°С в течение периода времени около 5 ч или менее.
18. Применение по п.16 или 17, причем исходный материал включает соединение формулы (II):
где MII означает по меньшей мере один двухвалентный металл, MIII означает Fe3+, An- означает по меньшей мере один n-валентный анион, x=Σny, 0<х≤0,5, 0≤m≤10.
19. Применение по п.18, причем в формуле (II) х≥0,3.
20. Применение по п.18, причем в формуле (II) х<0,3.
21. Способ получения субстанции по п.1, используемой в качестве лекарственного средства, который заключается в том, что исходный материал, включающий структуру слоистого двойного гидроксида, нагревают при температуре от 225 до 550°С в течение периода времени около 5 ч или менее.
22. Способ по п.21, причем исходный материал включает соединение формулы (II):
где MII означает по меньшей мере один двухвалентный металл, MIII означает Fe3+, An- означает по меньшей мере один n-валентный анион, х=Σny, 0<х≤0,5, 0≤m≤10.
23. Способ по п.22, причем в формуле (II) х≥0,3.
24. Способ по п.22, причем в формуле (II) х<0,3.
25. Способ по любому из пп.21-24, причем исходный материал практически не содержит частиц размером более 100 мкм.
26. Фармацевтическая композиция для связывания фосфата и лечения гиперфосфатемии, включающая твердое смешанное соединение металлов формулы (I):
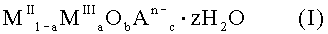
где MII означает по меньшей мере один двухвалентный металл, MIII означает Fe3+, An- означает по меньшей мере один n-валентный анион, z означает 2 или менее; а = число молей MIII / (число молей MII + число молей MIII); 2+а=2b+Σcn и Σcn<0,9a, и фармацевтически приемлемый носитель.
27. Фармацевтическая композиция по п.26, причем в формуле (I) а≥0,3 и предпочтительно 0,03а<Σcn<0,5а.
28. Фармацевтическая композиция по п.26, причем в формуле (I) а<0,3 и предпочтительно 0,03a<Σcn<0,7a.
29. Фармацевтическая композиция для связывания фосфата и лечения гиперфосфатемии, включающая твердое смешанное соединение металлов, как оно определено в п.1, которое получают при нагревании исходного материала, включающего структуру слоистого двойного гидроксида, при температуре от 225 до 550°С в течение периода времени около 5 ч или менее, и фармацевтически приемлемый носитель.
30. Фармацевтическая композиция по п.29, причем исходный материал включает соединение формулы (II):
где MII означает по меньшей мере один двухвалентный металл, MIII означает Fe3+, An- означает по меньшей мере один n-валентный анион, x=Σny, 0<x≤0,5, 0≤m≤10.
31. Фармацевтическая композиция по п.30, причем в формуле (II) х≥0,3.
32. Фармацевтическая композиция по п.30, причем в формуле (II) х<0,3.
33. Способ получения фармацевтической композиции по любому из пп.26-32, заключающийся в том, что твердое соединение смешанных металлов по п.1 смешивают с фармацевтически приемлемым носителем и необязательно любыми другими ингредиентами.
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| TSUGIO SATO «Adsorption of various anions by magnesium aluminium oxide» Ind | |||
| Eng | |||
| Chem | |||
| Prod | |||
| Res | |||
| Dev | |||
| Видоизменение пишущей машины для тюркско-арабского шрифта | 1923 |
|
SU25A1 |
| Способ получения гидроксоалюминатов металлов | 1991 |
|
SU1816738A1 |
| RU 97104121 A, 10.03.1999 | |||
| СПОСОБ ПОЛУЧЕНИЯ НАНОКОМПОЗИТНЫХ МАТЕРИАЛОВ | 2001 |
|
RU2224713C2 |

