Изобретение относится к способам получения низкомолекулярного пектина и может быть использовано в различных отраслях промышленности и медицины, в частности в фармацевтической промышленности для создания новых лечебных и профилактических средств и низкомолекулярных сорбентов.
Пектины представляют собой высокомолекулярные природные полимеры. Они присутствуют во всех высших цветковых растениях, входят в состав первичных клеточных стенок и срединных пластинок. Основой пектиновых молекул является поли-D-галактуроновая кислота. Часть остатков D-галактуроновой кислоты в пектине находится в форме метиловых эфиров. Если доля таких остатков составляет менее 50%, то такой пектин считается низкоэтерифицированным. Полностью деэтерифицированный, то есть не содержащий метиловых эфиров или содержащий незначительное их количество, пектин называется также пектовой кислотой.
В ходе исследований биологической активности пектинов обнаружен ряд фармакологических эффектов у низкомолекулярных производных пектинов - олигогалактуронидов. В этом плане особенный интерес представляют олигогалактурониды со степенью полимеризации от 6 до 25-30 моносахаридных остатков, что соответствует их молекулярной массе от 1 до 4,5-5 кДа. У этой группы олигогалактуронидов обнаружена иммуномодулирующая, интерферон-индуцирующая, макрофаг-активирующая и противоопухолевая активность (Kumazawa Y., Mizunoe K., Otsuka Y. Immunostimulating polysaccharide separated from hot water extract of Angelic acutiloba Kitagawa (Yamato tohki) // Immunology. 1982. Vol.47. P.75-83.). Кроме этого, данные олигосахариды стимулировали апоптоз клеток аденокарциномы кишечника у людей (Olano-Martin Е., Rimbah G.H., Gibson G.R., Rastall R.A. Pectin and pectic-oligosaccharides induce apoptosis in in vitro human colonicadenocarcinoma cell // Anticancer Res. 2003. Vol.23. P.341-346.).
Установлено, что олигогалактурониды со степенью полимеризации не менее 6-9 способны образовывать прочные комплексы с ионами многих тяжелых металлов, таких как свинец, ртуть, кадмий, и рядом других (Kohn R. Binding of divalent cations to oligomeric fragments of pectin // Carbohydr. Res. 1987. Vol.160. P.343-353.). Это открывает перспективу создания на основе олигогалактуронидов эффективных средств для выведения из организма человека и животных депонированных форм указанных токсичных элементов, т.к. благодаря небольшой молекулярной массе олигогалактурониды могут достаточно легко всасываться в желудочно-кишечном тракте, а затем выводиться из организма через почки.
Получение низкомолекулярных пектинов (олигогалактуронидов) обычно осуществляют путем деполимеризации природных высокомолекулярных пектинов. Для этого может применяться ряд методов. Наиболее известным является метод ферментативного гидролиза пектинов с помощью пектиназ. Так, еще в 1944 году был предложен способ получения D-галактуроновой кислоты путем ферментативного гидролиза пектина (пат. США №2338534, опубл. 04.01.1944 г.).
Известен способ получения композиции олигогалактуронидов, обладающей антибактериальным действием, также путем ферментативного гидролиза пектина (IP 10226701 (A), С08 В 37/00, опубл. 25.08.2010 г.). Согласно этому способу, пектин сначала подвергают предварительному гидролизу в водном растворе при 80-100°С, а затем проводят окончательный гидролиз смеси с помощью пектиназы. Полученный продукт содержит смесь насыщенных и ненасыщенных (имеющих двойные углерод-углеродные связи в молекулах в результате окислительной деградации) олигогалактуронидов со степенью полимеризации от 2 до 15 (0,4-2,7 кДа).
Недостатком данного способа является то, что для успешного осуществления ферментативного гидролиза требуется, как правило, определенная предварительная обработка пектина, например, как в данном случае, частичная деполимеризация и деэтерификация в ходе предварительного гидролиза. Другим существенным недостатком ферментативного гидролиза является то, что в силу специфики действия пектинолитических ферментов в полученных гидролизатах всегда преобладают моно-, ди- и трисахариды, а доля молекул с массой 1 кДа (что соответствует степени полимеризации - 6) и более незначительна.
Другим известным способом является способ щелочной деполимеризации пектина (ЕР 0487340 (А1), А23Р 1/08, опубл. 27.05.92). Согласно этому способу суспензию пектина в 60% об. изопропаноле обрабатываю трехкратным (по химическому эквиваленту галактуроновой кислоты) количеством гидроокиси натрия в течение 5 дней при 25°С или 2 дней при 40°С, с последующим отделением продукта на фильтре, нейтрализацией и сушкой. Полученный продукт содержит пектины с молекулярной массой от 2 до 20 кДа и предназначен в качестве вспомогательного вещества в кулинарии.
Недостатком данного способа является большая длительность процесса и большой разброс молекулярных масс пектинов в продукте, а также то, что галактуроновая кислота малоустойчива в щелочной среде и легко распадается с образованием ненасыщенных, сильноокрашенных и сильнопахнущих соединений (Kertesz Z.I. The pectic substances. Interscience Publishers, Inc., N.Y., L., 1951, 576 р.).
Известен также способ гидролиза пектина при высокой температуре (WO 2009/004153, С08В 37/06, опубл. 08.01.2009). Согласно этому способу 1% раствор пектина с рН 4-5 нагревают под давлением при 110°С в течение 30-60 минут, затем раствор охлаждают, осаждают олигогалактурониды изопропанолом и сушат. Полученный продукт на 95% состоит из олигогалактуронидов со степенью полимеризации 4-11 (0,7-2 кДа).
Недостатком данного способа является то, что при температуре более 100°С резко ускоряются процессы термического разложения олигогалактуронидов - разрывы пиранозного цикла, окисление, декарбоксилирование, образование фурфурола и его производных. Это сопровождается снижением выхода олигогалактуронидов и загрязнением гидролизата продуктами их разложения (Kertesz Z.I. The pectic substances. Interscience Publishers, Inc., N.Y., L., 1951, 576 p.). С химической точки зрения олигогалактурониды, имеющие в составе молекул продукты термической деградации галактуроновой кислоты, не могут в полной мере считаться олигогалактуронидами.
Известен способ деполимеризации полиуронидов - альгинатов и пектинов - с помощью окислителя - перекиси водорода (пат. США US 2002/0016453 (А1), С08В 37/06, опубл. 07.02.2002). Согласно этому способу пектовая кислота в виде пектата лития окисляется перекисью водорода концентрацией около 2% в присутствии сульфата железа в течение 4-х часов. Затем олигогалактурониды осаждают уксусной кислотой, промывают пропанолом и сушат. Средняя степень полимеризации олигогалактуронидов - 16 (2,8 кДа). Выход продукта составляет 38%.
К недостаткам данного способа следует отнести, во-первых, низкий выход продукта. Во-вторых, перекись водорода одновременно с деполимеризацией пектина вызывает значительную деструкцию остатков галактуроновой кислоты - декарбоксилирование, разрывы пиранозного цикла, окисление спиртовых групп, и, как отмечалось в предыдущем примере, полученные продукты не могут в полной мере считаться олигогалактуронидами (Kertesz Z.I. The pectic substances. Interscience Publishers, Inc., N.Y., L., 1951, 576 p.).
Наиболее близким по технической сущности к заявляемому способу является способ получения низкомолекулярного пектина (олигогалактуронидов) из пектинов льна путем кислотного гидролиза (Bedouet L., Courtois В., Courtois J. Methods for obtaining neutral and acid oligosaccharides from flax pectins // Biotechnol. Lett. 2005. Vol.27. P.34, 36, табл.1 и 37). Способ осуществляют следующим образом. Из образца пектина массой 50 мг, содержащего 22,9 мг (45,8%) уроновой (галактуроновой) кислоты, готовят водный раствор пектина концентрацией 5 мг/мл (0,5%). Затем добавляют 12 М раствор соляной кислоты до концентрации 100 мМ (0,1 г-экв/л) и проводят гидролиз при 80°С в течение 24 часов Полученный гидролизат охлаждают до комнатной температуры. Жидкую фазу, содержащую низкомолекулярные продукты гидролиза, отделяют от высокомолекулярной полигалактуроновой кислоты центрифугированием при 4°С и относительном центробежном ускорении 20000 g в течение 30 минут. Затем низкомолекулярные продукты гидролиза - олигогалактурониды - осаждают из жидкой фазы изопропанолом и отделяют центрифугированием. Средняя степень полимеризации олигогалактуронидов - 15 (2,6 кДа). Выход олигогалактуронидов составляет 33,9% от первоначального содержания галактуроновой кислоты.
Существенным недостатком данного способа является низкая исходная концентрация пектина в растворе. Результатом этого является, во-первых, низкая скорость гидролиза, так как она при неизменной константе гидролиза пропорциональна концентрации пектина в растворе. Во-вторых, низкая исходная концентрация пектина приводит к соответствующей низкой концентрации конечного продукта в готовом гидролизате - содержание олигогалактуронидов в готовом гидролизате, полученном вышеописанным методом, составляет всего 0,78 мг/мл за 24 часа гидролиза (выход продукта рассчитывается умножением исходной концентрации пектина в растворе на относительное содержание галактуроновой кислоты в пектине и на относительный выход галактуронидов и составляет соответственно 5 мг/мл × 0,458 × 0,339=0,78 мг/мл). А это, в свою очередь, приводит к более высоким удельным затратам на производство единицы массы продукта. В данном случае это, например, затраты, связанные с термостатированием длительное время всего объема гидролизата при 80°С и расходом кислоты на гидролиз.
Препятствием для существенного повышения концентрации пектина в растворе является то обстоятельство, что высокомолекулярные пектины уже при концентрации около 1% образуют достаточно вязкие растворы и использование более концентрированных растворов пектинов может создать определенные технологические трудности. Высоковязкие растворы при низких концентрациях образуют практически все коммерческие пектины, так как именно с этой способностью связано одно из главных направлений их использования - в качестве загустителей и структурирующих агентов в пищевой промышленности. Так, например, вязкость 1,5% растворов наиболее распространенных коммерческих пектинов - яблочного и цитрусового (производитель - Herbstreith & Fox KG, Германия) - в 25-30 раз превышает вязкость воды.
Другим недостатком является низкая технологическая эффективность метода, которая приводит к низкому выходу продукта. Выход продукта - олигогалактуронидов - составляет около 34%, 33% составляет остаток высокомолекулярной полигалактуроновой кислоты и 33% - потери олигогалактуронидов вследствие их структурной деградации. Таким образом, потери продукта составляют практически такую же величину, как и его выход.
Технологическим недостатком метода является то, что для отделения жидкой фазы, содержащей готовый продукт, требуются слишком жесткие условия: величина относительного центробежного ускорения 20000 g для большинства промышленных центрифуг и сепараторов либо недостижима, либо близка к их предельному рабочему режиму, необходимость охлаждения системы до 4°С также создает дополнительные технические проблемы и приводит к удорожанию процесса.
Задачей, поставленной перед изобретением, является получение низкомолекулярного пектина, обогащенного фракцией олигогалактуронидов с молекулярной массой 1-5 кДа, увеличение выхода целевого продукта, упрощение и удешевление способа.
Поставленная цель достигается тем, что в известном способе получения низкомолекулярного пектина (олигогалактуронидов), включающем гидролиз пектина в водном растворе минеральной кислоты при нагревании, с последующим отделением жидкой фазы от нерастворимого остатка пектина, выделением из нее низкомолекулярных продуктов гидролиза пектина путем их осаждения органическим растворителем, смешивающимся с водой, согласно изобретению, в качестве исходного сырья для гидролиза используют низкоэтерифицированный пектин со степенью этерификации не более 30%, гидролиз ведут циклично, при этом время проведения одного цикла рассчитывают по формуле:
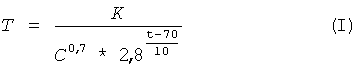
где Т - время проведения одного цикла, ч;
С - концентрация кислоты: 0,1-2,0 г-моль/л;
t - температура процесса: 70-100°С;
К - коэффициент пропорциональности, значения которого находятся в пределах 12-18;
перед осаждением низкомолекулярных продуктов гидролиза пектина органическим растворителем жидкую фазу нейтрализуют до рН не менее 4,0. После осаждения полученные низкомолекулярные продукты гидролиза пектина сушат.
Использование в качестве исходного сырья для гидролиза низкоэтерифицированного пектина со степенью этерификации не более 30%, имеющего ограниченную набухаемость в кислой среде, позволяет проводить процесс кислотного гидролиза в гетерофазной системе - суспензии: гель низкоэтерифицированного пектина (твердая фаза) - раствор минеральной кислоты (жидкая фаза), что, во-первых, позволяет на практике повысить концентрацию пектина в суспензии до 5-15% (в пересчете на сухое вещество пектина). Использование таких высоких, по сравнению с прототипом, концентраций исходного сырья при прочих равных условиях (при одинаковой степени гидролиза пектина) позволяет получать гидролизаты с соответственно большим процентным содержанием конечного продукта - олигогалактуронидов, что значительно уменьшает удельные затраты на производство единицы массы продукта. И, как следствие, ведет к упрощению и удешевлению процесса получения целевого продукта.
Во-вторых, гидролиз пектина в гетерофазной системе позволяет вести процесс циклично, за счет чего удается полностью использовать исходное сырье и предупредить деградацию олигогалактуронидов в жидкой фазе, обеспечивая тем самым повышение выхода целевого продукта и понижение удельных затрат на его получение.
При увеличении степени этерификации более 30% происходит резкое увеличение объема набухшего геля пектина и значительное снижение его механической прочности, что существенно осложняет последующее отделение геля пектина от жидкой фазы, также возможен частичный переход высокомолекулярного пектина в жидкую фазу.
Для осуществления заявленного способа может быть использован практически любой коммерческий пектин. Для этого предварительно проводят снижение его степени этерификации до необходимой величины в пределах 0-30%, используя любой из известных методов деэтерификации: щелочной, кислотный или ферментативный. Наиболее удобным и экономичным, по мнению авторов, может считаться метод щелочной деэтерификации пектина в среде органического растворителя, например этанола или изопропанола. Данный метод приводится ниже, в описании осуществления заявляемого способа.
Использование для расчета времени гидролиза пектина формулы (I), разработанной авторами, позволяет в указанном диапазоне температур (70-100°С), концентрации кислоты (0,1-2,0 г-моль/л) и коэффициенте пропорциональности (К=12-18) с удовлетворительной точностью рассчитать время одного цикла гидролиза, необходимое для получения продукта с максимальным относительным содержанием фракции олигогалактуронидов 1-5 кДа.
В ходе исследования авторами гетерофазного кислотного гидролиза низкоэтерифицированного пектина было установлено, что олигогалактурониды с молекулярной массой 1-5 кДа, образовавшиеся в ходе гидролиза пектина, практически полностью переходят в раствор - жидкую фазу, в то время как доля таких молекул, оставшихся в пектиновом геле -твердой фазе, не превышает 1-2% от их общего количества. Таким образом, важным преимуществом предлагаемого метода гетерофазного кислотного гидролиза низкоэтерифицированного пектина является наличие эффективной и постоянной молекулярной сепарации, происходящей естественным путем непосредственно в процессе самого гидролиза.
Сочетание в данном способе эффекта межфазной молекулярной сепарации и технологической простоты разделения фаз позволяет останавливать процесс гидролиза в нужный момент времени, быстро отделять жидкую фазу, содержащую готовый продукт, и затем проводить следующий цикл гидролиза оставшегося высокомолекулярного пектина.
Указанные ограничения температуры и концентрации кислоты связаны, в первую очередь, с экономическими и технологическими причинами. Так, при температуре среды ниже 70°С и концентрации кислоты ниже 0,1 г-моль/л происходит существенное снижение скорости гидролиза пектина и значительное увеличение времени процесса, это ведет к повышению затрат на производство целевого продукта.
При температуре более 100°С резко ускоряются процессы термического разложения олигогалактуронидов - разрывы пиранозного цикла, окисление, декарбоксилирование, образование фурфурола и его производных. Это сопровождается снижением выхода олигогалактуронидов и загрязнением гидролизата продуктами их разложения. Увеличение концентрации кислоты более 2,0 г-моль/л не дает значительного увеличения скорости гидролиза, но приводит к существенному перерасходу кислоты и дополнительным затратам на ее последующую нейтрализацию и освобождение продукта от образовавшихся солей, что, в конечном результате, ведет к усложнению процесса получения целевого продукта.
Нейтрализация жидкой среды до рН не менее 4,0 перед осаждением низкомолекулярных продуктов гидролиза пектина органическим растворителем обеспечивает получение осадка низкомолекулярного пектина (олигогалактуронидов) с оптимальной консистенцией - плотного, хорошо отделяющегося от жидкой фазы. При рН ниже 4,0 происходит увеличение удельного объема осадка и ослабление его структуры, и, как следствие, затрудняется отделение осадка от жидкой фазы.
В качестве минеральной кислоты могут быть использованы, в частности, соляная, серная, азотная кислоты.
В качестве органического растворителя могут быть использованы известные органические растворители, смешивающиеся с водой, в частности ацетон, пропанол, изопропанол, этанол, метанол.
Отделение жидкой фазы от нерастворимого осадка можно вести либо фильтрацией, либо центрифугированием при сравнительно небольших величинах центробежного ускорения 500-1000 g. Это значительно снижает затраты и упрощает процесс получения целевого продукта по сравнению с прототипом, поскольку в прототипе центрифугирование ведут при ускорении 20000 g и температуре 4°С.
С экономической точки зрения целесообразно для поддержания процесса гидролиза пектина в постоянном технологическом режиме, т.е. при постоянном количестве пектина в реакционной среде, осуществлять добавку новых порций сырья к остатку пектина после отделения жидкой фазы для компенсации его расхода в ходе гидролиза. Это позволяет проводить подряд любое количество однотипных циклов гидролиза и получать любое требуемое количество целевого продукта с нужной молекулярной массой, 1-5 кДа.
Сырье может добавляться к остатку пектина после каждого или после нескольких циклов гидролиза, а его количество может быть рассчитано либо по изменению объема геля пектина, либо по количеству пектина, оставшегося после отделения жидкой фазы.
В первом случае, для определения количества добавляемого сырья по изменению объема геля пектина, берут определенное количество пектина (m1), добавляют минеральную кислоту до образования суспензии и перед проведением гидролиза суспензию пектина в минеральной кислоте центрифугируют при относительном центробежном ускорении 500-1000 g в течение 5-15 минут и фиксируют первоначальный объем геля - пектина, V1, мл. А перед проведением последующих циклов гидролиза остаток пектина после гидролиза центрифугируют в тех же условиях - при той же величине относительного центробежного ускорения и времени, фиксируя V2 - объем остатка пектина после гидролиза, мл.
Для определения количества добавляемого сырья по объему остатка пектина после центрифугирования его рассчитывают по формуле:
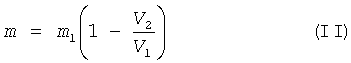
где m - масса добавляемого сырья, г;
m1 - масса исходного сырья, г;
V1 - первоначальный объем набухшего сырья - геля низкоэтерифицированного пектина, мл;
V2 - объем остатка пектина после гидролиза, мл.
Использование для расчета формулы (II), предлагаемой авторами, позволяет с достаточной точностью определить массу добавляемого сырья, обеспечивающего восстановление содержания пектина до исходной концентрации. В конечном результате, это обеспечивает максимально полное использование остатка пектина, повышение выхода целевого продукта и возможность получения его в необходимом количестве.
Во втором случае, для определения количества добавляемого сырья по количеству пектина, оставшегося после отделения жидкой фазы, перед началом процесса гидролиза фиксируют массу исходного сырья, m1, также определяют в нем содержание галактуроновой кислоты, C1. По окончании времени гидролиза, рассчитанного по формуле (I), отделяют остаток пектина от жидкой фазы, фиксируют его массу m2 и определяют содержание в нем галактуроновой кислоты С2.
Количество добавляемого сырья рассчитывают по формуле:
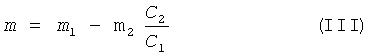
где m - масса добавляемого сырья, г;
m1 - масса исходного сырья, г;
m2 - масса остатка пектина после отделения жидкой фазы, г;
C1 - содержание галактуроновой кислоты в исходном сырье, %;
С2 - содержание галактуроновой кислоты в остатке пектина, %.
Формула (III), предлагаемая авторами, так же как и формула (II), позволяет с необходимой точностью определять количество добавляемого сырья.
Для определения содержания галактуроновой кислоты в исходном сырье и остатке пектина может быть использован любой из известных методов, например титриметрический (Афанасьев С.П., Чирва В.Ю., Кацева Г.Н. и др. Модификация титриметрических методов анализа пектиновых веществ // Химия природ. соединений. 1984. №4. С.428-431.) или фотометрический (Blumenkrantz N., Asboe-Hansen G. New method for quantitative determination of uronic acids // Anal. Biochem. 1973. Vol.54. P.484-489). Учитывая, что в ходе каждого цикла гидролиза расходуется примерно одинаковое количество сырья, то нет необходимости проводить расчеты количества добавляемого сырья после каждого цикла гидролиза - достаточно проводить их через каждые 5-10 циклов.
Способ осуществляется следующим образом.
Пример 1
Берут 30,0 г пектовой кислоты (Serva, №31680) с содержанием галактуроновой кислоты 82,2% и степенью этерификации менее 1% и смешивают с 200 мл раствора соляной кислоты концентрацией 0,1 г-моль/л (С). Полученную 15% суспензию нагревают до 100°С (t) на кипящей водяной бане и выдерживают при этой температуре и постоянном перемешивании в течение времени (Т), рассчитанного по формуле (I) при коэффициенте пропорциональности К=18:
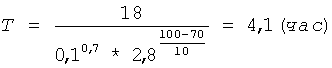
Затем суспензию охлаждают и фильтруют, для фильтрации суспензии используют полипропиленовую фильтрткань PL-30 (LOSMA, Италия). Остаток пектина на фильтре промывают 200 мл соляной кислоты концентрацией 0,1 г-моль/л (С) для более полного отделения олигогалактуронидов. К остатку пектина добавляют раствор соляной кислоты концентрацией 0,1 г-моль/л - до восстановления первоначального объема суспензии. Остаток пектина ресуспендируют в жидкой фазе и проводят следующий цикл гидролиза в прежних условиях - Т=4,1 часов при t=100°C. Всего проводят 5 циклов гидролиза, каждый раз отделяя и промывая остаток пектина, как было описано выше. Общий объем полученного фильтрата составляет 1440 мл, содержание олигогалактуронидов - 8,23 мг/мл.
Жидкую фазу (фильтрат) нейтрализуют до рН 4,0 5,0 М раствором гидроокиси натрия и осаждают олигогалактурониды добавлением 4,3 л этанола. Смесь интенсивно перемешивают в течение минуты, затем, спустя 12 часов, выпавший осадок олигогалактуронидов отделяют центрифугированием при ОЦУ 1000-1500 g в течение 15 минут и сушат при 80°С. Выход продукта - сухого порошка олигогалактуроната натрия - 13,38 г (содержание в олигогалактуронате натрия чистой галактуроновой кислоты составляет 88,5%). Для определения относительного (по сырью) выхода продукта, в пересчете на галактуроновую кислоту, устанавливают расход сырья по разнице между общим количеством пектовой кислоты, использованной для гидролиза (30,0 г), и количеством пектина, оставшимся после гидролиза. Для этого количество пектовой кислоты пересчитывают на содержание в ней галактуроновой кислоты, а в остатке пектина определяют общее содержание галактуроновой кислоты фотометрическим методом. Установленный расход сырья составляет по галактуроновой кислоте 13,30 г. Относительный выход продукта по галактуроновой кислоте составляет, соответственно, 89,1%. По данным эксклюзионной хроматографии полученного продукта в 0,05 М ацетатном буфере с рН 3,0 на колонке с сефадексом G-50 superfine, откалиброванной с помощью стандартного набора пуллуланов, относительное содержание фракции олигогалактуронидов 1-5 кДа в полученном низкомолекулярном пектине составляет 62,5%.
Пример 2
Берут 10,0 г пектовой кислоты (Serva, №31680) (m1) с содержанием галактуроновой кислоты 82,2% и степенью этерификации менее 1% и смешивают с 200 мл раствора соляной кислоты концентрацией 0,1 г-моль/л (С), фиксируют объем полученной 5% суспензии - он составляет 210 мл, затем суспензию центрифугируют 15 минут при относительном центробежном ускорении (ОЦУ) 500 g и фиксируют первоначальный объем геля пектина - он составляет 48 мл (V1). Затем осадок геля пектина ресуспендируют в жидкой фазе, нагревают до 100°С (t) на кипящей водяной бане и выдерживают при этой температуре и постоянном перемешивании в течение времени (Т), рассчитанного по формуле (I) при коэффициенте пропорциональности К=18:
Затем суспензию охлаждают до комнатной температуры и центрифугируют 15 минут при ОЦУ 500 g. Жидкую фазу отделяют от осадка геля пектина, замеряют объем осадка - он составляет 40 мл (V2). Подставляя полученные данные по первоначальному объему низкоэтерифицированного геля пектина (V1) и объему остатка геля пектина после гидролиза (V2), равному 40 мл, в формулу (II), рассчитывают необходимую добавку сырья (m) для восстановления первоначального количества пектовой кислоты:
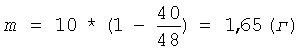
К осадку добавляют указанное количество пектовой кислоты и раствор соляной кислоты концентрацией 0,1 г-моль/л (С) - до восстановления первоначального объема суспензии. Осадок ресуспендируют в жидкой фазе и проводят следующий цикл гидролиза в прежних условиях - 4,1 часа при 100°С. Всего проводят 5 циклов гидролиза. При этом каждый раз после отделения жидкой фазы замеряют объем осадка пектина: если он остается постоянным (равным 40 мл (V2)), то количество сырья, добавляемое к остатку пектина, также остается постоянным - 1,65 г (m), поскольку объем осадка пектина в процессе гидролиза не изменялся (если объем осадка изменяется, то по формуле (II) заново рассчитывают необходимую добавку сырья). Объем полученного фугата - жидкой фазы - составляет 810 мл, содержание олигогалактуронидов - 7,15 мг/мл. Жидкую фазу нейтрализуют до рН 4,0 5,0 М раствором гидроокиси натрия и осаждают олигогалактурониды добавлением 2,4 л ацетона. Смесь интенсивно перемешивают в течение минуты, затем, спустя 12 часов, выпавший осадок олигогалактуронидов отделяют центрифугированием при ОЦУ 1000-1500 g в течение 15 минут и сушат при 80°С. Выход продукта - сухого порошка олигогалактуроната натрия - 6,53 г. Относительный выход продукта по галактуроновой кислоте, определенный как описано в предыдущем примере, составляет 88,3%. Относительное содержание фракции олигогалактуронидов 1-5 кДа в полученном низкомолекулярном пектине составляет 62,0%.
Пример 3
Получают низкоэтерифицированный пектин путем контролируемой щелочной деэтерификации высокоэтерифицированного пектина.
Берут 50 г высокоэтерифицированного цитрусового пектина (Herbstreith & Fox KG, Германия) суспендируют в 0,5 л 60% об. этанола и постепенно, порциями добавляют 5,0 М раствор гидроокиси натрия, контролируя величину степени этерификации пектина в пробах, взятых после добавления каждой порции гидроокиси натрия. После достижения величины степени этерификации менее 30% к суспензии добавляют 20 мл концентрированной соляной кислоты (плотность - d=1,19 г/см3), пектин отделяют фильтрованием, промывают 150 мл 50% этанола и сушат при 80°С. Выход низкоэтерифицированного пектина - 47 г, содержание галактуроновой кислоты - 74,8%, степень этерификации - 30,0%.
Берут 10,0 г полученного низкоэтерифицированного пектина (m1), смешивают с 200 мл раствора соляной кислоты концентрацией 0,5 г-моль/л (С) и фиксируют объем полученной 5% суспензии - он составляет 210 мл, затем суспензию центрифугируют 5 минут при ОЦУ 1000 g и фиксируют первоначальный объем геля пектина - он составляет 112 мл (V1). Затем осадок геля пектина ресуспендируют в жидкой фазе и термостатируют при 80°С (t) и постоянном перемешивании в течение времени (Т), рассчитанного по формуле (I) при коэффициенте пропорциональности К=12:
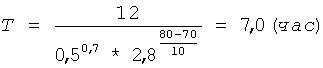
Затем суспензию охлаждают до комнатной температуры и центрифугируют 5 минут при ОЦУ 1000 g. Жидкую фазу отделяют от осадка геля пектина, а осадок дважды промывают раствором кислоты для более полного отделения олигогалактуронидов: осадок суспендируют в 100 мл соляной кислоты концентрацией 0,5 г-моль/л (С), затем центрифугируют 5 минут при ОЦУ 1000 g, жидкую фазу сливают и повторяют промывку осадка. После промывки замеряют объем осадка пектина после гидролиза - он составляет 96 мл (V2). Подставляя полученные значения V1, V2, и m1 в формулу (II), рассчитывают необходимую добавку сырья (m) для восстановления первоначального количества пектина:
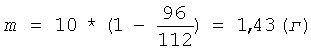
К осадку добавляют указанное количество низкоэтерифицированного пектина (1,43 г) и раствор соляной кислоты концентрацией 0,5 г-моль/л (С) - до восстановления первоначального объема суспензии. Осадок ресуспендируют в жидкой фазе и проводят следующий цикл гидролиза в прежних условиях - Т=7,0 часа при t=80°C. Всего проводят 5 циклов гидролиза. При этом каждый раз после отделения жидкой фазы замеряют объем осадка: если он остается постоянным (равным 96 мл), то количество сырья, добавляемое к остатку пектина, также остается постоянным - 1,43 г, если объем осадка изменяется, то по формуле (II) заново рассчитывают необходимую добавку сырья. Объем полученного фугата - жидкой фазы - составляет 1450 мл, содержание олигогалактуронидов - 3,48 мг/мл. Жидкую фазу нейтрализуют: вначале сухой гидроокисью натрия до рН 1-2, затем 5,0 М раствором гидроокиси натрия до рН 6,0. Олигогалактурониды осаждают добавлением 4,5 л этанола. Выпавший осадок олигогалактуронидов отделяют центрифугированием 15 минут при ОЦУ 1000-1500 g, затем очищают от солей: ресуспендируют в 0,6 л 70% этанола и повторно центрифугируют. Влажный осадок сушат при 80°С. Выход продукта - сухого порошка олигогалактуроната натрия - 5,70 г. Относительный выход продукта по галактуроновой кислоте, определенный как описано в предыдущем примере, составляет 94,2%. Относительное содержание фракции олигогалактуронидов 1-5 кДа в полученном низкомолекулярном пектине составляет 67,0%.
Пример 4
Берут 30 г пектовой кислоты (см. пример 1) (m1) с содержанием галактуроновой кислоты 82,2% (C1), смешивают с 200 мл раствора серной кислоты концентрацией 2,0 моль/л (С) и фиксируют объем полученной 15% суспензии - он составляет 230 мл. Суспензию термостатируют при 70°С (t) и постоянном перемешивании в течение времени (Т), рассчитанного по формуле (I) при коэффициенте К=12:
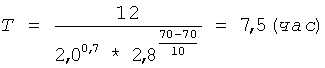
Затем горячую суспензию фильтруют, для фильтрации суспензии используют полипропиленовую фильтрткань PL-30 (LOSMA, Италия). Остаток пектина на фильтре промывают 200 мл серной кислоты концентрацией 2,0 моль/л (С) для более полного отделения олигогалактуронидов. Полученный фильтрат охлаждают до комнатной температуры. Остаток пектина взвешивают и определяют в нем процентное содержание галактуроновой кислоты титриметрическим методом. По полученным значениям массы остатка пектовой кислоты - 127,1 (m2) и содержания в нем галактуроновой кислоты - 16,0% (С2) рассчитывают по формуле (III) необходимую добавку сырья (m) для восстановления первоначального количества пектовой кислоты:
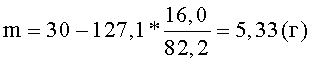
К остатку пектина добавляют рассчитанное количество пектовой кислоты (m) и раствор серной кислоты концентрацией 2,0 моль/л (С) - до восстановления первоначального объема суспензии. Остаток пектина ресуспендируют в жидкой фазе и проводят следующий цикл гидролиза в прежних условиях - T=7,5 часов при t=70°C. Всего проводят 5 циклов гидролиза. При этом каждый раз после отделения жидкой фазы остаток пектина снова взвешивают, определяют в нем процентное содержание галактуроновой кислоты и по формуле (III) рассчитывают необходимую добавку сырья. Общий объем полученного фильтрата составляет 1280 мл, содержание олигогалактуронидов - 16,4 мг/мл. Фильтрат нейтрализуют: вначале сухой гидроокисью натрия до рН 1-2, затем 5,0 М раствором гидроокиси натрия до рН 8. Олигогалактурониды осаждают добавлением 4,0 л изопропанола. Выпавший осадок олигогалактуронидов отделяют центрифугированием 15 минут при ОЦУ 1000-1500 g, затем очищают от солей: ресуспендируют в 2,0 л 70% изопропанола и повторно центрифугируют. Влажный осадок сушат при 80-100°С. Выход продукта - сухого порошка олигогалактуроната натрия - 23,75 г. Относительный выход продукта по галактуроновой кислоте составляет 95,1%. Относительное содержание фракции олигогалактуронидов 1-5 кДа составляет 62,7%.
Пример 5
Получают низкоэтерифицированный пектин. Для этого в 0,5 л 50% этанола растворяют 20,0 г гидроокиси натрия, затем при перемешивании добавляют 100,0 г высокоэтерифицированного цитрусового пектина (Herbstreith & Fox KG, Германия). Смесь перемешивают 50 минут, затем постепенно небольшими порциями добавляют 40,0 мл концентрированной соляной кислоты, пектин отделяют фильтрованием, промывают 300 мл 50% этанола и сушат при 80°С. Выход низкоэтерифицированного пектина - 93,3 г, содержание галактуроновой кислоты - 76,5% (C1), степень этерификации - 0%.
Берут 20,0 г (m1) полученного низкоэтерифицированного пектина, смешивают с 200 мл раствора азотной кислоты концентрацией 0,5 г-моль/л (С) и фиксируют объем полученной 10% суспензии - он составляет 220 мл. Суспензию термостатируют при t=90°C и постоянном перемешивании в течение времени (Т), рассчитанного по формуле (I) при коэффициенте К=14,5:
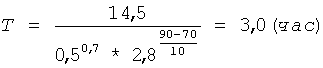
По окончании гидролиза суспензию охлаждают и фильтруют, для фильтрации суспензии используют полипропиленовую фильтрткань PL-30. Остаток пектина на фильтре промывают 150 мл азотной кислоты концентрацией 0,5 г-моль/л (С) для более полного отделения олигогалактуронидов. К остатку пектина добавляют раствор азотной кислоты концентрацией 0,5 г-моль/л (С) - до восстановления первоначального объема суспензии. Остаток пектина ресуспендируют в жидкой фазе и проводят следующий цикл гидролиза в прежних условиях - 3,0 часа (Т) при 90°С (t). Всего проводят 3 цикла гидролиза. Промытый остаток пектина после последнего цикла гидролиза взвешивают и определяют в нем процентное содержание галактуроновой кислоты титриметрическим методом. По полученным значениям массы остатка пектина - 56,1 г (m2) и содержания в нем галактуроновой кислоты - 16,4% (С2) рассчитывают по формуле (III) необходимую добавку сырья (m) для восстановления первоначального количества пектина:

К остатку пектина добавляют указанное количество низкоэтерифицированного пектина и проводят следующие 3 цикла гидролиза в прежних условиях. Общий объем полученного фильтрата составляет 1660 мл, содержание олигогалактуронидов - 6,81 мг/мл. Фильтрат нейтрализуют: вначале сухой гидроокисью натрия до рН 1-2, затем 5,0 М раствором гидроокиси натрия до рН 5. Олигогалактурониды осаждают добавлением 5,0 л этанола. Выпавший осадок олигогалактуронидов отделяют центрифугированием 15 минут при ОЦУ 1000-1500 g, затем очищают от солей: ресуспендируют в 1,3 л 70% об. этанола и повторно центрифугируют. Влажный осадок сушат при 80°С. Выход продукта - сухого порошка олигогалактуроната натрия - 12,77 г. Относительный выход продукта по галактуроновой кислоте составляет 92,5%. Относительное содержание фракции олигогалактуронидов 1-5 кДа в полученном низкомолекулярном пектине составляет 65,0%.
Пример 6
Получают низкоэтерифицированный пектин.
В 0,4 л 50% об. изопропанола растворяют 20 г гидроокиси натрия, затем при перемешивании добавляют 100 г высокоэтерифицированного яблочного пектина (Herbstreith & Fox KG, Германия). Смесь перемешивают 10 мин, затем постепенно небольшими порциями добавляют 40 мл концентрированной соляной кислоты, пектин отделяют фильтрованием, промывают 300 мл 50% изопропанола и сушат при 80-100°С. Выход низкоэтерифицированного пектина - 94,5 г, содержание галактуроновой кислоты - 78,4% (С1), степень этерификации - 13,8%.
Берут 20 г полученного низкоэтерифицированного пектина (m1), смешивают с 200 мл раствора серной кислоты концентрацией 1,0 моль/л (С) и фиксируют объем полученной 10% суспензии - он составляет 220 мл. Суспензию термостатируют при t=90°C и постоянном перемешивании в течение времени (Т), рассчитанного по формуле (I) при коэффициенте К=16:
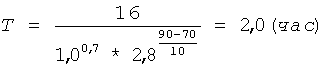
Затем суспензию охлаждают и фильтруют, для фильтрации суспензии используют полипропиленовую фильтрткань PL-30. Остаток пектина на фильтре промывают 150 мл серной кислоты концентрацией 1,0 моль/л (С) для более полного отделения олигогалактуронидов. Остаток пектина взвешивают и определяют в нем процентное содержание галактуроновой кислоты титриметрическим методом. По полученным значениям массы остатка пектина - 79,0 г (m2) и содержания в нем галактуроновой кислоты - 15,5% (С2) рассчитывают по формуле (III) необходимую добавку сырья (m) для восстановления первоначального количества пектина:
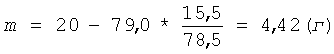
К остатку пектина добавляют указанное количество низкоэтерифицированного пектина и раствор серной кислоты концентрацией 1,0 моль/л (С) - до восстановления первоначального объема суспензии. Остаток пектина ресуспендируют в жидкой фазе и проводят следующий цикл гидролиза в прежних условиях Т=2,0 часа при t=90°C. Всего проводят 5 циклов гидролиза. При этом каждый раз после отделения жидкой фазы к остатку пектина добавляют 4,42 г низкоэтерифицированного пектина (m). Так как в ходе проведенного гидролиза пектина может возникнуть некоторое допустимое расхождение между количеством добавленного сырья и реальной величиной расхода сырья, то для устранения такого возможного расхождения после 5 циклов гидролиза снова проводят расчет необходимой добавки сырья. Для этого промытый остаток пектина после последнего цикла гидролиза взвешивают и определяют в нем процентное содержание галактуроновой кислоты титриметрическим методом. По полученным значениям массы остатка пектина - 82,8 г (m2) и содержания в нем галактуроновой кислоты - 15,1% (С2) рассчитывают по формуле (III) необходимую разовую корректирующую добавку сырья (m) для восстановления первоначального количества пектина:
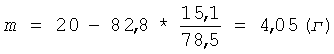
К остатку пектина добавляют указанное количество низкоэтерифицированного пектина. Затем проводят следующие 5 циклов гидролиза в прежних условиях. При этом каждый раз после отделения жидкой фазы к остатку пектина по-прежнему добавляют 4,42 г низкоэтерифицированного пектина. Общий объем полученного фильтрата составляет 2720 мл, содержание олигогалактуронидов - 11,40 мг/мл. Фильтрат нейтрализуют: вначале сухой гидроокисью натрия до рН 1-2, затем 5,0 М раствором гидроокиси натрия до рН 5. Олигогалактурониды осаждают добавлением 8,0 л изопропанола. Выпавший осадок олигогалактуронидов отделяют центрифугированием 15 минут при ОЦУ 1000-1500 g, затем очищают от солей: ресуспендируют в 2,6 л 70% об. изопропанола и повторно центрифугируют. Влажный осадок сушат при 80-100°С. Выход продукта - сухого порошка олигогалактуроната натрия - 35,05 г. Относительный выход продукта по галактуроновой кислоте составляет 90,5%. Относительное содержание фракции олигогалактуронидов 1-5 кДа в полученном низкомолекулярном пектине составляет 67,7%.
Как видно из приведенных примеров, заявляемый способ гетерофазного гидролиза позволяет за счет нерастворимости исходного пектина в кислоте проводить гидролиз целенаправленно - до получения продукта с необходимой молекулярной массой, и при этом использовать исходный пектин практически полностью, т.е. позволяет избегать каких-либо значительных технологических потерь сырья и повысить конечный выход продукта (по галактуроновой кислоте) по сравнению с прототипом в 2,6-2,8 раза до 88-95% от исходного сырья.
Кроме того, в заявляемом способе в отличие от известного способа, гидролиз сырья ведется отдельными, сравнительно короткими циклами, что позволяет существенно ограничить деструкцию олигогалактуронидов в процессе гидролиза и, как следствие, дополнительно повысить выход целевого продукта.
Более того, предлагаемое авторами техническое решение обеспечивает по сравнению с прототипом более высокую скорость процесса и, соответственно, снижение удельных производственных затрат и времени на выпуск одного килограмма продукта при значительной экономии сырья, также позволяет отказаться от использования сложного и дорогостоящего технологического оборудования для отделения жидкой фазы от остатка пектина.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ НИЗКОМОЛЕКУЛЯРНОГО ПЕКТИНА | 2010 |
|
RU2441024C1 |
| СПОСОБ ПОЛУЧЕНИЯ НИЗКОМОЛЕКУЛЯРНОГО ПЕКТИНА | 2011 |
|
RU2478650C1 |
| СПОСОБ ПОЛУЧЕНИЯ НИЗКОМОЛЕКУЛЯРНОГО ПЕКТИНА | 2011 |
|
RU2478649C1 |
| СПОСОБ ПОЛУЧЕНИЯ ОКТАГАЛАКТУРОНИДА | 2014 |
|
RU2576535C1 |
| СПОСОБ ПОЛУЧЕНИЯ ГЕПТАГАЛАКТУРОНИДА | 2014 |
|
RU2570709C1 |
| СПОСОБ ПОЛУЧЕНИЯ ОКТАГАЛАКТУРОНИДА | 2014 |
|
RU2570708C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПЕКТАТА КАЛЬЦИЯ | 2008 |
|
RU2375377C1 |
| СРЕДСТВО, ОБЛАДАЮЩЕЕ ПРЕБИОТИЧЕСКОЙ АКТИВНОСТЬЮ | 2007 |
|
RU2366429C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПЕКТИНА ИЗ РАСТИТЕЛЬНОГО СЫРЬЯ | 2016 |
|
RU2610312C1 |
| СПОСОБ ПОЛУЧЕНИЯ АЛЬГИНАТА КАЛЬЦИЯ | 2008 |
|
RU2395525C1 |
Изобретение относится к способам получения низкомолекулярного пектина. Способ предусматривает гидролиз пектина в водном растворе минеральной кислоты при нагревании. Далее жидкую фазу отделяют от нерастворимого остатка пектина и нейтрализуют до рН не менее 4,0. Выделение низкомолекулярных продуктов гидролиза пектина проводят путем их осаждения органическим растворителем, смешивающимся с водой. После осаждения полученные низкомолекулярные продукты гидролиза пектина сушат. При этом в качестве исходного сырья для гидролиза используют низкоэтерифицированный пектин со степенью этерификации не более 30%. Гидролиз ведут циклично. При этом время проведения одного цикла рассчитывают по определенной формуле. Изобретение позволяет получать низкомолекулярный пектин без технологических потерь сырья, а также существенно обеспечить деструкцию олигогалактуронидов, тем самым повышая конечный выход продукта. 1. з.п. ф-лы.
1. Способ получения низкомолекулярного пектина, включающий гидролиз пектина в водном растворе минеральной кислоты при нагревании с последующим отделением жидкой фазы от нерастворимого остатка пектина, выделением из нее низкомолекулярных продуктов гидролиза пектина путем их осаждения органическим растворителем, смешивающимся с водой, отличающийся тем, что в качестве исходного сырья для гидролиза используют низкоэтерифицированный пектин со степенью этерификации не более 30%, гидролиз ведут циклично, при этом время проведения одного цикла рассчитывают по формуле:

где Т - время проведения одного цикла, ч;
С - концентрация кислоты 0,1-2,0 г-моль/л;
t - температура процесса 70-100°С;
К - коэффициент пропорциональности, значения которого находятся в пределах 12-18;
перед осаждением низкомолекулярных продуктов гидролиза пектина органическим растворителем жидкую фазу нейтрализуют до рН не менее 4,0, а после осаждения низкомолекулярные продукты гидролиза пектина сушат.
2. Способ по п.1, отличающийся тем, что после каждого или нескольких циклов гидролиза к остатку пектина добавляют новую порцию сырья, количество которого рассчитывают по формуле:
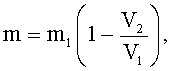
где m - масса добавляемого сырья, г;
m1 - масса исходного сырья, г;
V1 - объем геля пектина до гидролиза, мл;
V2 - объем остатка геля пектина после гидролиза, мл,
при этом V1 и V2 определяют после центрифугирования геля пектина, или по формуле:
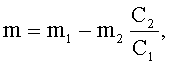
где m - масса добавляемого сырья, г;
m1 - масса исходного сырья, г;
m2 - масса остатка пектина после отделения жидкой фазы, г;
C1 - содержание галактуроновой кислоты в исходном сырье, %;
С2 - содержание галактуроновой кислоты в остатке пектина, %.
| BEDOUET L, COURTOIS В, COURTOIS J | |||
| Methods for obtaining neutral and acid oligosaccharides from flax pectines | |||
| Biotechnology Letters, 2005, v.27, p.34, 36, 37 and table 1 | |||
| ПРЕПАРАТ ДЛЯ УМЕНЬШЕНИЯ И/ИЛИ БЛОКИРОВАНИЯ АДГЕЗИИ ПАТОГЕННЫХ ВЕЩЕСТВ И ОРГАНИЗМОВ, ПРИМЕНЕНИЕ АНТИАДГЕЗИВНЫХ УГЛЕВОДОВ | 2001 |
|
RU2267324C2 |
| KR 20000012173 A, 06.03.2000. | |||

