Настоящее изобретение относится к новым определенным бензамидным соединениям или их фармацевтически приемлемым солям, которые являются эффективными ингибиторами фермента деацетилаза гистонов (HDAC). Изобретение также относится к способам приготовления этих новых бензамидных соединений, к фармацевтическим композициям, которые их содержат, и к их применению в терапевтических способах, например, для приготовления лекарственных средств для ингибирования HDAC у теплокровного животного, такого как человек.
Активность HDAC связана с многими болезненными состояниями, такими как злокачественное новообразование (Marks и др., Nature Reviews, 1, 194-202, (2001)), фиброзно-кистозная дегенерация (Li, S. и др., J. Biol. Chem., 274, 7803-7815, (1999)), хорея Хантингдона (Steffan, J.S. и др., Nature, 413, 739-743, (2001)) и серповидно-клеточная анемия (Gabbianelli, M. и др., Blood, 95, 3555-3561, (2000)). Следовательно, настоящее изобретение также относится к способам лечения любого из вышеперечисленных заболеваний с помощью бензамидных соединений согласно настоящему изобретению, а также к применению этих бензамидных соединений для приготовления лекарственного средства для лечения указанных болезненных состояний.
В эукариотических клетках ДНК обычно упакована для предотвращения ее доступности для транскрипционного фактора. При активации клетки эта упакованная ДНК становится доступной для ДНК-связывающих белков, таким образом предоставляется возможность для индукции транскрипции генов (Beato, М., J. Med. Chem., 74, 711-724 (1996); Wolffe, A.P., Nature, 387, 16-17 (1997)). Ядерная ДНК связана с ядерными белками, известными как гистоны, с образованием комплекса, который называют хроматином. Коровые гистоны, которые обозначаются как Н2А, Н2В, Н3 и Н4, окружены 146 парами оснований ДНК с образованием основной единицы хроматина, которая известна как нуклеосома. N-концевые хвосты коровых гистонов содержат остатки лизина, которые являются сайтами для посттранскрипционного ацетилирования. Ацетилирование концевой аминогруппы в боковой цепи лизина нейтрализует потенциал боковой цепи, образует при этом положительный заряд, что, предположительно, сказывается на укладке структуры хроматина.
Деацетилазы гистонов (HDAC) представляют собой цинксодержащие ферменты, катализирующие удаление ацетильных групп из ε-амино конца остатков лизина, которые кластеризуются возле аминоконцов нуклеосомальных гистонов. HDAC можно разделить на два класса, первый из них (HDAC 1, 2, 3 и 8) представляет собой Rpd3-подобные белки дрожжей, а второй (HDAC 4, 5, 6, 7, 9 и 10) представляет собой Hda1-подобные белки дрожжей. Известно, что обратимый процесс ацетилирования является важным для регуляции транскрипции и осуществления клеточного цикла. Дополнительно, нарушение регуляции HDAC связано с некоторыми видами злокачественных новообразований и было показано, что ингибиторы HDAC, такие как трихостатин А (естественный продукт, выделенный из Streptomyces hygroscopicus), проявляют существенное ингибирующее действие на рост клеток и противоопухолевое действие (Meinke, P.Т., Current Medicinal Chemistry, 8, 211-235 (2001)). Yoshida и др., (Exper. Cell Res., 177, 122-131 (1988)) обнаружили, что трихостатин А вызывает задержку клеточного цикла фибробластов крыс в фазах G1 и G2, что свидетельствует о вовлечении HDAC в регуляцию клеточного цикла. Кроме того, было показано, что трихостатин А индуцирует конечную дифференциацию, ингибирует рост клеток и предотвращает образование опухолей у мышей (Finnin и др., Nature, 401, 188-193 (1999)).
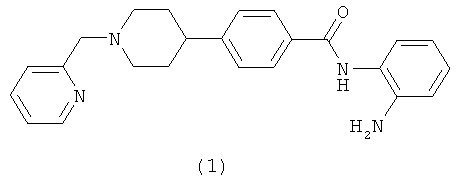
Из опубликованных международных патентных заявок WO 03/087057 и WO 03/092686 известно, что определенные производные бензамида являются ингибиторами HDAC. Одним из предпочтительных соединений, описанных в WO 03/087057, является N-(2-аминофенил)-4-[1-(пирид-2-илметил)пиперидин-4-ил]бензамид [1] (его структура приведена ниже).
Сейчас нами было обнаружено, что определенные производные бензамида, которые несут необязательно замещенную пиразольную группу вместо пиридильной группы, являются эффективными ингибиторами HDAC. Дополнительно, также было обнаружено, что предпочтительные соединения согласно настоящему изобретению обладают другими благоприятными фармакологическими свойствами, включая благоприятную эффективность по отношению к клеткам или в условиях in-vivo, благоприятные DMPK свойства (например, благоприятный профиль биодоступности, и/или благоприятные свободные уровни в плазме крови, и/или благоприятный период полужизни, и/или благоприятный объем распределения), а также хорошую или улучшенную растворимость. Дополнительно, производные бензамида согласно настоящему изобретению обычно проявляют чрезвычайно низкую активность в исследовании ингибирования калиевого канала, кодируемого hERG, которое является индикатором неблагоприятных и тяжелых побочных сердечно-сосудистых действий в клинических условиях.
В соответствии с настоящим изобретением, обеспечивается соединение формулы (I):
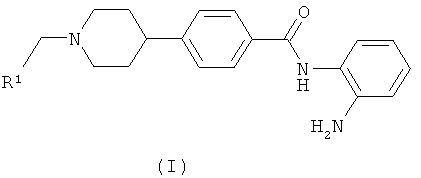
в которой R1 представляет собой углеродсвязанное пиразольное кольцо, которое необязательно замещено одной или несколькими группами, выбранными из С1-4алкила, С3-4циклоалкила, С1-4алкокси и С3-4циклоалкокси;
или его фармацевтически приемлемая соль.
Подразумевается, что определенные соединения формулы (I), указанные выше, могут проявлять явление таутомерии. Подразумевается, что настоящее изобретение охватывает любую такую таутомерную форму или их смесь, которые обладают вышеописанной активностью, и не ограничивается только любой такой одной таутомерной формой, используемой в приведенных формулах или указанную в примерах.
Если необязательные заместители выбирают из "одной или несколько" замещающих групп, то подразумевается, что такое определение охватывает всех таких заместителей, которые выбраны из одной из указанных групп, или заместителей, которые выбраны из двух или более указанных групп.
Подходящие необязательные заместители для R1 могут присутствовать на любых доступных атомах углерода или азота в пиразольном кольце.
R1 подходяще несет от 1 до 3 замещающих групп. Альтернативно, R1 является незамещенным.
Как используется в настоящем изобретении, термин "алкил" относится к неразветвленным или разветвленным цепям. Термин "циклоалкил" охватывает кольцевые структуры, но могут дополнительно включать цепи в виде циклоалкил-алкильных групп. Аналогично, термины "алкокси" и "циклоалкокси" включают алкильные, циклоалкильные или циклоалкил-алкильные группы, связанные с помощью атома кислорода.
Подходящими С1-4алкильными или С3-4циклоалкильными заместителями для R1 являются метил, этил, пропил, циклопропил, циклобутил или циклопропилметил.
Подходящими С1-4алкокси и С3-4циклоалкокси заместителями для R1 являются метокси, этокси, пропокси, циклопропокси, циклобутокси или метилциклопропокси.
В предпочтительном варианте осуществления изобретения, R1 представляет собой углеродсвязанное пиразольное кольцо, которое необязательно замещено 1, 2 или 3 группами, выбранными из С1-4алкила или С1-4алкокси.
В дальнейшем варианте осуществления изобретения, R1 представляет собой углеродсвязанное пиразольное кольцо, которое необязательно замещено 1, 2 или 3 группами, выбранными из С1-2алкила или С1-2алкокси.
Примерами R1 групп являются пиразол-3-ил, пиразол-4-ил, 1-метилпиразол-4-ил, 3-этилпиразол-4-ил, 1,3-диметилпиразол-5-ил, 1,3-диметилпиразол-4-ил, 1,3,5-триметилпиразол-4-ил, 1,3-диметил-5-метоксипиразол-4-ил, 1,5-диметилпиразол-4-ил, 1-этил-5-метилпиразол-4-ил, 1-этилпиразол-4-ил и 1-этил-3-метилпиразол-4-ил (подвержен таутомерии, если это является возможным).
В дальнейшем варианте осуществления изобретения, соединения формулы (I) включают соединения формулы (IA)
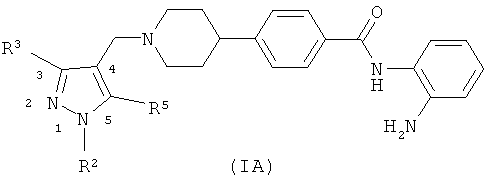
где R2 представляет собой водород, С1-4алкил или С3-4циклоалкил, и R3 и R5 независимо выбирают из водорода, С1-4алкила, С3-4циклоалкила, С1-4алкокси или С3-4циклоалкокси.
Следует принять во внимание, что кольцевые атомы пиразольной части молекулы формулы (IA) обычно нумеруются, как показано на диаграмме выше. Тем не менее, молекула подвержена таутомерии в случае, если R2 представляет собой водород, где переключение групп водорода с одного атома азота пиразольного кольца на другой обозначает, что замещенные пиразолы, где по меньшей мере один из R3 или R5 отличается от водорода, неизбежно представляют смеси каждого таутомера и, следовательно, где R3 и R5 считаются взаимозаменяемыми.
В альтернативном варианте осуществления изобретения, изобретение обеспечивает соединение формулы (IB)
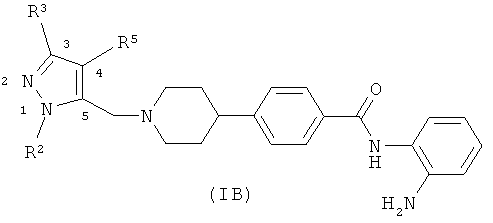
где R2, R3 и R5 имеют значения, определенные выше для формулы (IA).
С другой стороны, следует принять во внимание, что пиразольная часть молекулы формулы (IB) обычно пронумерована в соответствии со схематическим изображением на диаграмме выше. Тем не менее, как обсуждалось выше для соединений формулы (IA), молекула также подвергается таутомерии, если R2 представляет собой водород.
Предпочтительными примерами R2 являются водород, метил, этил, пропил, циклопропил, метилциклопропил или циклобутил.
Например, R2 представляет собой водород, метил, этил, пропил или циклопропил.
В предпочтительном варианте осуществления изобретения, R2 представляет собой водород, метил или этил.
В дальнейшем варианте осуществления изобретения, R2 представляет собой водород или метил.
Предпочтительными примерами групп R3 и R5 являются водород, метил, этил, пропил, циклопропил, циклопропилметил, метокси, этокси, пропокси или циклопропокси.
Предпочтительными примерами групп R3 или R5 являются водород, метил, этил или метокси.
Подходяще, не больше чем одна группа R3 или R5 представляет собой С1-4алкокси.
В предпочтительном варианте осуществления изобретения, по меньшей мере одна и предпочтительно две группы R2, R3 и R5 отличаются от водорода.
В предпочтительном варианте осуществления изобретения, соединения имеют структурную формулу (IA), приведенную выше, в которой R2 представляет собой водород или С1-4алкил, и R3 и R5 каждый независимо выбирают из водорода, С1-4алкила или С1-4алкокси.
В дальнейшем варианте осуществления изобретения, соединения имеют структурную формулу (IA), приведенную выше, в которой R2 представляет собой водород, метил или этил, и R3 и R5 каждый независимо выбирают из водорода, метила, этила или метокси.
В дальнейшем варианте осуществления изобретения, соединения имеют структурную формулу (IA), приведенную выше, в которой R2 представляет собой водород или метил, и R3 и R5 каждый независимо выбирают из водорода, метила, этила или метокси.
В дальнейшем варианте осуществления изобретения, соединения имеют структурную формулу (IA), приведенную выше, в которой R2 представляет собой водород или метил, и R3 и R5 каждый независимо выбирают из водорода, метила или метокси.
В дальнейшем варианте осуществления изобретения, соединения имеют структурную формулу (IA), приведенную выше, в которой R2 представляет собой водород или метил, и R3 и R5 каждый независимо выбирают из водорода или метила.
В предпочтительном варианте осуществления изобретения, соединения имеют структурную формулу (IB), приведенную выше, в которой R2 представляет собой водород или С1-4алкил, и R3 и R5 каждый независимо выбирают из водорода, С1-4алкила, или С1-4алкокси.
В дальнейшем варианте осуществления изобретения, соединения имеют структурную формулу (IB), приведенную выше, в которой R2 представляет собой водород, метил или этил, и R3 и R5 каждый независимо выбирают из водорода, метила, этила или метокси.
В дальнейшем варианте осуществления изобретения, соединения имеют структурную формулу (IB), приведенную выше, в которой R2 представляет собой водород или метил, и R3 и R5 каждый независимо выбирают из водорода, метила, этила или метокси.
В дальнейшем варианте осуществления изобретения, соединения имеют структурную формулу (IB), приведенную выше, в которой R2 представляет собой водород или метил, и R3 и R5 каждый независимо выбирают из водорода, метила или метокси.
Предпочтительные соединения по изобретению включаю любое из следующих соединений:
N-(2-аминофенил)-4-[1-(1H-пиразол-3-илметил)пиперидин-4-ил]бензамид;
N-(2-аминофенил)-4-{1-[(5-метокси-1,3-диметил-1H-пиразол-4-ил)метил]пиперидин-4-ил}бензамид;
N-(2-аминофенил)-4-{1-[(3-этил-1H-пиразол-4-ил)метил]пиперидин-4-ил}бензамид;
N-(2-аминофенил)-4-{1-[(1-метил-1H-пиразол-4-ил)метил]пиперидин-4-ил}бензамид;
N-(2-аминофенил)-4-{1-[(1,3-диметил-1H-пиразол-5-ил)метил]пиперидин-4-ил}бензамид;
N-(2-аминофенил)-4-{1-[(1,3-диметил-1H-пиразол-4-ил)метил]пиперидин-4-ил}бензамид;
N-(2-аминофенил)-4-{1-[(1,3,5-триметил-1H-пиразол-4-ил)метил]пиперидин-4-ил}бензамид;
N-(2-аминофенил)-4-{1-[(1,5-диметил-1H-пиразол-4-ил)метил]пиперидин-4-ил}бензамид;
N-(2-аминофенил)-4-{1-[(1-этил-5-метил-1H-пиразол-4-ил)метил]пиперидин-4-ил}бензамид;
N-(2-аминофенил)-4-{1-[(1-этил-1H-пиразол-4-ил)метил]пиперидин-4-ил}бензамид;
N-(2-аминофенил)-4-{1-[(1-этил-3-метил-1H-пиразол-4-ил)метил]пиперидин-4-ил}бензамид;
или его фармацевтически приемлемая соль.
Подразумевается, что определенные соединения формулы I, указанные выше, могут существовать в виде несольватированных форм, а также в виде сольватов, таких как, например, гидратированные формы. Подразумевается, что изобретение охватывает все такие сольватированные формы, которые обладают антипролиферативной активностью.
Также подразумевается, что определенные соединения формулы I могут обладать полиморфизмом и что настоящее изобретение охватывает все такие формы, которые обладают антипролиферативной активностью.
Подходящая фармацевтически приемлемая соль соединения формулы I представляет собой, например, кислото-аддитивную соль соединения формулы I, например, кислото-аддитивную соль с неорганической или органической кислотой, такой как соляная, бромистоводородная, серная, трифторуксусная, лимонная или малеиновая кислота; или, например, соль соединения формулы I, которое является достаточно кислотным, например, соль щелочного металла или щелочно-земельного металла, такую как кальциевая или магниевая соль, или соль аммония. Дальнейшей подходящей фармацевтически приемлемой солью соединения формулы I является, например, соль, образованная в организме человека или животного после введения соединения формулы I.
Соединения по изобретению могут вводиться в виде пролекарства, представляющего собой соединение, которое распадается в организме человека или животного с высвобождением соединения по изобретению. Пролекарство может применяться для изменения физических свойств и/или фармакокинетических свойств соединения по изобретению. Пролекарство может образовываться, если соединение по изобретению содержит подходящую группу или заместитель, к которой (которому) может присоединяться группа, модифицирующая свойство. Примерами пролекарств являются производные сложных амидов, способные к расщеплению в условиях in vivo, которые могут образовываться у аминогруппы в соединении формулы I.
Таким образом, настоящее изобретение охватывает те соединения формулы I, как определено в настоящем изобретении выше, которые становятся доступными путем органического синтеза или становятся доступными в организме человека или животного при расщеплении их пролекарства. Следовательно, настоящее изобретение охватывает те соединения формулы I, которые образуются с помощью способов органического синтеза, а также те соединения, которые образуются в организме человека или животного при осуществлении метаболизма предшественника соединения, таким образом, соединение формулы I может являться синтетически полученным соединением или метаболически полученным соединением.
Подходящим фармацевтически приемлемым пролекарством соединения формулы I является пролекарство, которое согласно приемлемому медицинскому усмотрению считается пригодным для введения в организм человека или животного без нежелательных фармакологических активностей и без чрезмерной токсичности.
Различные формы пролекарств описаны, например, в следующих документах:
а) Methods in Enzymology, том 42, с.309-396, под ред. K.Widder, и др. (Academic Press, 1985);
б) Design of Pro-drugs, под ред. Н.Bundgaard, (Elsevier, 1985);
в) A Textbook of Drug Design and Development, под ред. Krogsgaard-Larsen и Н.Bundgaard, раздел 5 "Design and Application of Pro-drugs", ред. Н.Bundgaard c. 113-191 (1991);
г) Н.Bundgaard, Advanced Drug Delivery Reviews, 8, 1-38 (1992);
д) Н.Bundgaard, и др., Journal of Pharmaceutical Sciences, 77, 285 (1988);
e) N.Kakeya, и др., Chem. Pharm. Bull., 32, 692 (1984);
ж) Т.Higuchi и V.Stella, "Pro-Drugs as Novel Delivery Systems", A.C.S. Symposium Series, том 14; и
з) E.Roche (ред.), "Bioreversible Carriers in Drug Design", Pergamon Press, 1987.
Подходящим фармацевтически приемлемым пролекарством соединения формулы I является его производное амида, способное к расщеплению в условиях in vivo. Подходящие фармацевтически приемлемые амиды, образующиеся из аминогруппы, включают, например, амид, образованный с (1-10С)алканоильными группами, такими как ацетильная, бензоильная, фенилацетильная и замещенные бензоильная и фенилацетильная группы. Примерами кольцевых заместителей на фенилацетильной и бензоильной группах являются аминометил, N-алкиламинометил, N,N-диалкиламинометил, морфолинометил, пиперазин-1-илметил и 4-(1-4С)алкилпиперазин-1-илметил.
На действия соединения формулы I в условиях in vivo могут оказывать частичные влияния один или несколько метаболитов, которые образуются в организме человека или животного после введения соединения формулы I. Как указывалось в настоящем изобретении ранее, также на действия соединения формулы I в условиях in vivo может оказывать влияние метаболизм предшественника соединения (пролекарства).
Получение соединений формулы I
Специалисту в данной области техники следует принять во внимание, что при осуществлении некоторых процессов/реакций, описанных в настоящем изобретении, может являться необходимым/желательным защищать любые чувствительные группы в соединениях. Примеры, когда такая защита необходима или желательна и подходящие способы защиты известны специалисту в данной области химии. Могут применяться обычные защитные группы в соответствии со стандартными способами (см., например, T.W.Green & P.G.M.Wuts, Protective Groups in Organic Synthesis, 3-е изд., John Wiley and Sons, 1999). Таким образом, если реагирующие вещества содержат, например, такие группы, как аминогруппа, карбоксильная группа или гидроксильная группа, может являться желательным защищать такую группу в некоторых реакциях, описанных в настоящем изобретении.
Любые защитные группы, используемые в способах, описанных в настоящем изобретении, как правило, могут быть выбраны из любых групп, описанных в литературе, или известных специалисту в данной области химии, если это является приемлемым для защиты данной группы, и могут вводиться при помощи обычных способов. Защитные группы могут быть удалены при помощи любого обычного способа, описанного в литературе или известного специалисту в области химии, если это является пригодным для удаления данной защитной группы, эти способы выбирают таким образом, чтобы удаление защитной группы сопровождалось минимальным повреждением групп, находящихся в другой части молекулы.
Конкретные примеры защитных групп приведены ниже, в которых для удобства "низший", как, например, низший алкил, означает, что такая применяемая группа предпочтительно имеет 1-4 атома углерода. Также имеется в виду, что эти примеры не являются исчерпывающими. Если в дальнейшем описываются конкретные примеры способов удаление защитных групп, то они также не являются исчерпывающими. Применение защитных групп и способы снятия защиты, которые конкретно здесь не описаны, несомненно, подпадают под объем изобретения.
Подходящей защитной группой для аминогруппы или алкиламиногруппы является, например, ацильная группа, например, алканоильная группа, такая как ацетил, алкоксикарбонильная группа, например, метоксикарбонильная, этоксикарбонильная или трет-бутоксикарбонильная группа, арилметоксикарбонильная группа, например, бензилоксикарбонил, или ароильная группа, например, бензоил. Условия снятия защиты для вышеупомянутых защитных групп главным образом зависят от выбора защитной группы. Так, например, ацильная группа, такая как алканоильная или алкоксикарбонильная группа или ароильная группа, может быть отщеплена, например, путем гидролиза с подходящим основанием, таким как гидроокись щелочного металла, например, гидроокись лития или натрия. Альтернативно, ацильная группа, такая как трет-бутоксикарбонильная группа может быть отщеплена, например, путем обработки подходящей кислотой, такой как соляная, серная или фосфорная кислота, или трифторуксусная кислота, и арилметоксикарбонильная группа, такая как бензилоксикарбонильная группа, может быть отщеплена, например, путем гидрирования в присутствии катализатора, такого как палладий на угле, или путем обработки кислотой Льюиса, например, трис(трифторацетатом) бора. Подходящей альтернативной защитной группой для первичной аминогруппы является, например, фталоильная группа, которая может быть отщеплена путем обработки алкиламином, например, диметиламинопропиламином, или гидразином.
Подходящей защитной группой для гидроксильной группы является, например, ацильная группа, например, алканоильная группа, такая как ацетил, ароильная группа, например, бензоил, или арилметильная группа, например, бензил. Условия снятия защиты для вышеупомянутых защитных групп главным образом будут зависеть от выбора защитной группы. Так, например, ацильная группа, такая как алканоильная или ароильная группа, может быть отщеплена, например, при помощи гидролиза с подходящим основанием, таким как гидроокись щелочного металла, например гидроокись лития или натрия. Альтернативно, арилметильная группа, такая как бензильная группа, может быть отщеплена, например, путем гидрирования в присутствии катализатора, такого как палладий на угле.
Подходящей защитной группой для карбоксильной группы является, например, эстерифицированная группа, например, метильная или этильная группа, которая может быть отщеплена, например, путем гидролиза с основанием, таким как гидроокись натрия, или, например, трет-бутильная группа, которая может быть отщеплена, например, путем обработки кислотой, например, органической кислотой, такой как трифторуксусная кислота, или, например, бензильная группа, которая может быть отщеплена, например, путем гидрирования в присутствии катализатора, такого как палладий на угле.
Защитные группы могут быть отщеплены на любой подходящей стадии синтеза при помощи обычных методик, хорошо известных в области химии.
В дальнейшем варианте осуществления, настоящее изобретение обеспечивает способ получения соединения формулы (I) или его фармацевтически приемлемой соли, где способ включает:
(а) взаимодействие соединения формулы (II)
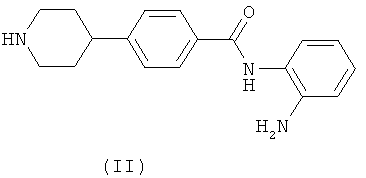
в котором анилиновая часть может быть подходяще защищена;
с соединением формулы (III)
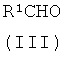
где R1 имеет значения, указанные в данной заявке, в присутствии восстановителя,
и затем, при необходимости, удаления любых оставшихся защитных групп, которые могут присутствовать.
Подходящим восстановителем для способа (а) является, например, неорганическая борогидридная соль, такая как борогидрид натрия, триацетоксиборогидрид натрия или цианоборогидрид натрия и водород. Восстановительное аминирование с помощью водорода необязательно осуществляют в присутствии подходящего катализатора, такого как, например, Pd/C, Pd(OH)2/C, Pt/C, PtO2 или Rh на окиси алюминия, и также можно осуществлять под давлением, например 1-10 бар, при температурах, например 0-150°С.
Способ (а) можно осуществлять в присутствии подходящей кислоты. Подходящей кислотой для способа (а) является кислота Бренстеда, такая как, например, муравьиная кислота, уксусная кислота, трифторуксусная кислота, соляная кислота, серная кислота, паратолуолсульфоновая кислота или камфорсульфоновая кислота; или кислота Льюиса формулы MQz, где М представляет собой металл, Q представляет собой реакционно-способную группу, такую как, например, галоген или сульфонилокси группа, например, хлор, бром, йод, метансульфонилокси, трифторметансульфонилокси или толуол-4-сульфонилокси группа, и z находится в интервале 1-6 и значение z будет зависеть от металла М. Типичными примерами подходящих кислот Льюиса являются трифторид бора, трифторметансульфонат скандия (III), хлорид олова (VI), изопропоксид титана (IV) или хлорид цинка (II).
Альтернативно, соединения формулы (I) могут быть получены путем (б) взаимодействия соединения формулы (II),
где анилин может быть подходяще защищен;
с соединением формулы (IV)
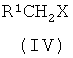
в присутствии подходящего основания;
где Х представляет собой реакционно-способную группу;
и затем, при необходимости, удаления любых оставшихся защитных групп, которые могут присутствовать.
Подходящей реакционно-способной группой Х является, например, галоген или сульфонилокси группа, например, хлор, бром, йод, метансульфонилокси, трифторметансульфонилокси или толуол-4-сульфонилокси группа.
Подходящим основанием для применения в вышеописанном способе (б) является, например, органическое аминовое основание, такое как, например, пиридин, 2,6-лутидин, коллидин, 4-диметиламинопиридин, триэтиламин, морфолин, диизопропилэтиламин (DIPEA), N-метилморфолин или диазабицикло[5,4,0]ундец-7-ен, или, например, карбонат или гидроксид щелочного или щелочно-земельного металла, например, карбонат натрия, карбонат калия, карбонат кальция, гидроксид натрия или гидроксид калия, или, например, гидрид щелочного металла, например, гидрид натрия, гидрокарбонат щелочно-земельного металла, такой как гидрокарбонат натрия, или алкоголят металла, такой как этоксид натрия.
Подходящей защитной группой для анилиновой части или пиперидинового кольца может быть карбамат, такой как трет-бутоксикарбонил или бензилоксикарбонил.
Предпочтительными примерами групп R1 являются группы, приведенные выше.
Реакции, описанные в способах (а) и (б), подходяще осуществляют в присутствии подходящего инертного растворителя или разбавителя, например, алканола или сложного эфира, такого как метанол, этанол, изопропанол или этилацетат, галогенированного растворителя, такого как метиленхлорид, хлороформ или четыреххлористый углерод, простого эфира, такого как тетрагидрофуран, 1,2-диметоксиэтан или 1,4-диоксан, ароматического растворителя, такого как толуол, или диполярного апротонного растворителя, такого как N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидин-2-он или диметилсульфоксид.
Получение исходных веществ
Получения соединения формулы II
Соединение формулы II может быть получено с помощью любого из следующих способов:
(в) Взаимодействие соединения формулы (V), где анилин может быть подходяще защищен,
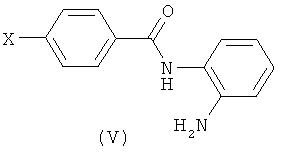
где Х представляет собой реакционно-способную группу, как определено выше, с соединением формулы (VI) в присутствии подходящего основания
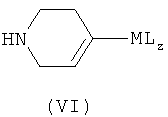
где М представляет собой металл, L представляет собой лиганд, целое число z равно от 0 до 3, и тетрагидропиридиновое кольцо может быть защищено; или взаимодействие соединения формулы (VII), где анилин и тетрагидропиридин могут быть подходяще защищены и М, L и z имеют значения, указанные выше,
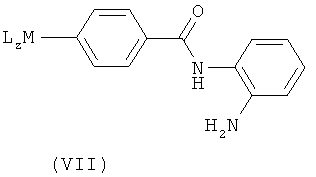
с соединением формулы (VIII):
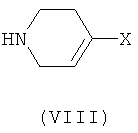
в присутствии подходящего основания;
где Х представляет собой реакционно-способную группу, как определено выше, и затем, при необходимости, и в любом подходящем порядке или комбинации: удаление любых защитных групп из тетрагидропиридина, и/или восстановление тетрагидропиридина до пиперидина и/или удаление любых оставшихся из присутствующих защитных групп.
Подходящей защитной группой для тетрагидропиридинового кольца является такая группа, как трет-бутоксикарбонил (также в настоящем изобретении обозначается как "ВОС") или бензилоксикарбонил. Подходящей защитной группой для анилиновой части также может быть карбамат, такой как ВОС или бензилоксикарбонил.
Подходящим основанием для способа (в) является, например, органическое аминовое основание, такое как, например, пиридин, 2,6-лутидин, коллидин, 4-диметиламинопиридин, триэтиламин, морфолин, N-метилморфолин или диазабицикло[5,4,0]ундец-7-ен, или, например, карбонат или гидроксид щелочного или щелочно-земельного металла, например карбонат натрия, карбонат калия, карбонат кальция, карбонат цезия, гидроксид натрия или гидроксид калия, или, например, гидрид щелочного металла, например гидрид натрия, или гидрокарбонат щелочного металла, такой как гидрокарбонат натрия, или алкоголят металла, такой как этоксид натрия.
Реакцию, определенную выше в способе (в), удобно осуществляют в присутствии подходящего инертного растворителя или разбавителя, например, алканола или сложного эфира, такого как метанол, этанол, изопропанол или этилацетат, галогенированного растворителя, такого как метиленхлорид, хлороформ или четыреххлористый углерод, простого эфира, такого как тетрагидрофуран, 1,2-диметоксиэтан или 1,4-диоксан, ароматического растворителя, такого как толуол, или диполярного апротонного растворителя, такого как N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидин-2-он или диметилсульфоксид. Реакции подходяще осуществляют при температуре в интервале, например, от 10 до 250°С, предпочтительно в интервале от 40 до 80°С.
Металлом М может быть любой металл, который известен в литературе для образования металлоорганических соединений, которые подвергаются каталитическим реакциям перекрестного связывания. Примерами подходящих металлов являются бор, олово, цинк и магний.
Подходящее значение для целого числа z зависит от металла М, но обычно находится в интервале 0-3.
Подходящими значениями для лиганда L, если он присутствует, являются, например, гидрокси, галогеновый, (1-4С)алкокси или (1-6С)алкильный лиганд, например гидрокси, бром, хлор, фтор, йод, метокси, этокси, пропокси, изопропокси, бутокси, метальный, этильный, пропильный, изопропильный или бутильный лиганд или, где целое число z равно 2 и М представляет собой бор, два присутствующих лиганда могут быть связаны таким образом, что вместе с атомом бора, к которому они присоединены, они образуют кольцо. Подходяще, группа MLz представляет собой группу формулы -BL1L2, где В представляет собой бор и L1 и L2 имеют значения, указанные для лиганда L выше. В частности, лиганды L1 и L2 могут быть связаны таким образом, что вместе с атомом бора, к которому они присоединены, они образуют кольцо. Например, L1 и L2 могут вместе образовывать окси-(2-4С)алкиленокси группу, например оксиэтиленокси, пинаколато (-O-С(СН3)2С(СН3)2-O-) или оксипропиленокси группу таким образом, что вместе с атомом бора, к которому они присоединены, они образуют циклическую сложноэфирную группу бороновой кислоты.
Подходящим катализатором для способа (в) являются, например, металлический катализатор, такой как катализатор на основе палладия (0), палладия (II), никеля (0) или никеля (II), например, тетракис(трифенилфосфин)палладий (0), хлорид палладия (II), бромид палладия (II), хлорид бис(трифенилфосфин)палладия (II), тетракис(трифенилфосфин)никель (0), хлорид никеля (II), бромид никеля (II), хлорид бис(трифенилфосфин)никеля (II) или дихлор[1-1'-бис(дифенилфосфино)ферроцен]палладий (II). Дополнительно, подходяще можно добавлять инициатор свободных радикалов, например, азосоединение, такое как азо(бисизобутиронитрил).
Подходяще, тетрагидропиридиновое кольцо восстанавливают до пиперидинового кольца в вышеописанном способе (в) путем гидрирования. Гидрирование необязательно осуществляют в присутствии подходящего катализатора, такого как, например, Pd/C, Pd(OH)2/C, Pt/C, PtO2 или Rh на окиси алюминия, и также можно осуществлять под давлением, например, 1-10 бар. Подходяще также гидрирование осуществлять в подходящей кислоте, например, бромистоводородной кислоте, соляной кислоте, лимонной кислоте, уксусной кислоте и метансульфоновой кислоте, и в подходящем растворителе или смеси растворителей, таких как, например, вода, этанол, тетрагидрофуран (ТГФ), метанол, ацетонитрил или пропан-2-ол.
г) Взаимодействия соединения формулы (IX), где Q1 представляет собой -ОН, -Cl, или -О- 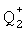 (где
(где  представляет собой катион)
представляет собой катион)
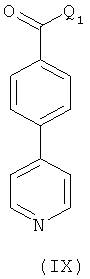
с соединением формулы (X) в присутствии подходящего растворителя и где одна из аминогрупп соединении формулы (X) может быть защищена;
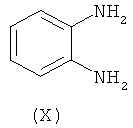
получая соединение формулы (XI)
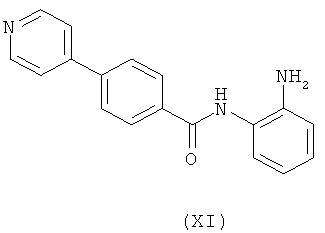
где анилин может быть защищен;
и затем:
превращение соединения формулы (XI) в соединение формулы (II) путем восстановления пириндин-4-ильного кольца до пиперидин-4-ильного кольца с помощью подходящего восстановителя и/или подходящих восстанавливающих условий; и
необязательно удаление любых оставшихся из присутствующих защитных групп.
Подходящим значением для Q1 является -О- Na+ (то есть -О- 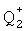 , где
, где 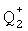 представляет собой Na+).
представляет собой Na+).
Подходяще, одна из аминогрупп соединения формулы (X) защищена с помощью подходящей аминозащитной группы, как определено в данной заявке выше, такой как ВОС группа.
Подходяще, анилин защищают с помощью аминозащитной группы, как определено в данной заявке выше, такой как ВОС группа, в соединении формулы (XI).
Для реакции соединений IX и Х можно использовать любой подходящий растворитель, такой как растворители, описанные ранее.
Соединение формулы (XI) превращают в соединение формулы (II) с помощью подходящего восстановителя и/или подходящих восстанавливающих условий. Подходящим способом является гидрирование. Гидрирование необязательно осуществляют в присутствии подходящего катализатора, такого как, например, Pd/C, Pd(ОН)2/С, Pt/C, PtO2 или Rh на окиси алюминия, и его можно осуществлять под давлением, например, 1-10 бар. Гидрирование подходяще также осуществлять в присутствии подходящей кислоты, например, бромистоводородной кислоты, соляной кислоты, лимонной кислоты, уксусной кислоты и метансульфоновой кислоты, и в подходящем растворителе или смеси растворителей, таких как, например, вода, этанол, тетрагидрофуран (ТГФ), метанол, ацетонитрил или пропан-2-ол.
Подходящий способ получения соединения формулы (XI) включает превращение соединения (IX) в реакционно-способное производное карбоновой кислоты (которое может быть получено in situ и его выделение не является обязательным), с последующим взаимодействием с соединением формулы (X).
Подходящим реакционно-способным производным карбоновой кислоты является, например, ацилгалогенид, например, ацилхлорид, образованный путем взаимодействия кислоты и хлорангидрида кислоты, например, тионилхлорид; смешанный ангидрид, например, образованный путем взаимодействия кислоты и хлороформиата, такого как изобутилхлороформиат; активированный сложный эфир; продукт реакции кислоты и карбодиимида, такой как дициклогексилкарбодиимид; или продукт реакции кислоты с хлоридом 4-(4,6-диметокси-1,3,5-триазинил-2-ил)-4-метилморфолиния (DMTMM), или продукт реакции кислоты с 1,1'-карбонилдиимидазолом (CDI).
Соединения формул (III) и (IV) могут быть получены из коммерческих источников, например, Flurochem Ltd, Old Glossop, Derbyshire SK13 7RY, UK, или они могут быть синтезированы с помощью методов, известных специалисту в данной области техники и/или описанных в литературе, например, Makino, K.; Kim, H.S и Kurasawa Y; J. Heterocyclic Chem. 1998, 35, 489-497 и ссылках, приведенных в этих источниках.
Исследования
Следующие исследования (а)-(в) можно использовать для оценки действия одного или более соединений согласно настоящему изобретению в качестве ингибиторов HDAC, в качестве ингибиторов in vitro рекомбинантной HDAC1 человека, продуцируемой в клетках насекомых Hi5, и в качестве индукторов в условиях in vitro & in vivo ацетилирования гистона Н3 в цельных клетках опухолей. С их помощью также оценивают способность таких соединений ингибировать пролиферацию опухолевых клеток человека.
(а) Ферментативное исследование рекомбинантной HDAC1 в условиях in vitro
Ингибиторы HDAC исследовали по отношению к рекомбинатной HDAC1 человека, продуцируемой Hi5 клетками насекомых. Фермент клонировали с «FLAG» меткой на С-конце гена и аффинно очищали, используя анти-«FLAG» М2 агарозу от SIGMA (A2220).
Исследование деацетилаз осуществляли в реакциях объемом 50 мкл. HDAC1 (75 нг фермента), разведенного в 15 мкл буфера реакции (25 мМ ТрисНСl (рН 8), 137 мМ NaCl, 2,7 мМ KCl, 1 мМ MgCl2) смешивали или с одним буфером (10 мкл) или с буфером, содержащим соединение (10 мкл), в течение 30 минут при температуре окружающей среды. Затем к реакционной смеси добавляли 25 мкМ ацетилированного пептида гистон Н4 (KI 174 Biomol), разведенного в 25 мкл буфера, и инкубировали в течение одного часа при температуре окружающей среды. Реакцию останавливали путем добавления одинакового объема (50 мкл) Fluor de Lys проявителя (Biomol), содержащего трихостатин А в концентрации 2 мкМ. Реакцию оставляли для обнаружения в течение 30 минут при температуре окружающей среды и затем измеряли флуоресценцию при длине возбуждающей волны 360 нм и длине волны эмиссии 465 нм. Значения IC50 для ингибиторов HDAC ферментов измеряли путем анализа кривых зависимости действия от дозы для конкретных соединений и определения концентрации ингибитора, обеспечивающей снижение максимального значения на 50% (контроль с разбавителем).
(б) Исследование ингибирования пролиферации в цельных клетках в условиях in vitro
Ингибирование пролиферации в цельных клетках исследовали с помощью титра клеток Promega 96 водного исследования пролиферации (Promega #G5421). Клетки НСТ116 высевали в 96-луночные планшеты при плотности 1×103 клеток/лунку, и оставляли для прилипания в течение ночи. Их обрабатывали ингибиторами в течение 72 часов. В каждую лунку добавляли 20 мкл тетразолиевого красителя MTS и планшеты повторно инкубировали в течение 3 часов. После этого измеряли абсорбцию на спектрофотометре для анализа: планшет на 96 лунок при 490 нМ. Значения IC50 для ингибиторов HDAC определяли путем анализа кривых зависимости дозы от концентрации для индивидуальных соединений и определения концентрации ингибитора, обеспечивающей снижения максимального сигнала на 50% (контроль с разбавителем).
(в) Исследование активности фермента деацетилазы гистонов в цельных клетках в условиях in vitro
Ацетилирование гистона Н3 в цельных клетках исследовали при помощи иммуногистохимии и анализировали при помощи матричного сканирования Cellomics. Клетки А549 или НСТ116 высевали в 96-луночные планшеты при плотности 1×104 клеток/лунку, и оставляли для прилипания всю ночь. Их обрабатывали ингибиторами в течение 24 часов и затем фиксировали в 1,8% формальдегиде в трис-солевом буферном растворе (TBS) в течение одного часа. Нарушали проницаемость мембран клеток при помощи ледяного метанола в течение 5 минут, промывали в TBS и затем блокировали в TBS 3% высушенного молока со сниженным содержанием жира в течение 90 минут. Затем клетки инкубировали с поликлональными антителами, которые специфичны к ацетилированному гистону Н3 (Upstate #06-599), разведенными 1 к 500 в TBS 3% молока в течение одного часа. Клетки трижды промывали в TBS и затем инкубировали с вторичными антителами, конъюгированными с флуоресцеином (Molecular Probes #A11008) & Hoechst 333542 (1 мкг/мг) (Molecular Probes #H3570) в TBS плюс 1% бычий сывороточный альбумин (Sigma #B6917) в течение одного часа. Несвязанное антитело удаляли путем трехкратного промывания с TBS и после завершающего промывания 100 мкл TBS добавляли к клеткам, и планшеты запечатывали и анализировали при помощи матричного сканирования Cellomics.
Значения ЕС50 для ингибиторов HDAC измеряли путем анализа кривых зависимости действия от дозы для конкретных соединений и затем определяли концентрацию ингибитора, обеспечивающую 50% максимального значения (ссылочное контрольное соединение - трихостатин А (Sigma)).
hERG активность и растворимость соединений по изобретению также может быть оценена с помощью исследований (г)-(е), описанных ниже:
(г) Исследование ингибирования калиевого канала, кодируемого hERG
Культура клеток
Клетки яичника китайского хомячка (СНО), экспрессирующие hERG-кодируемый канал, выращивали до полуслияния при 37°С в увлажненной среде (5% CO2) в питательной среде F-12 Ham, содержащей L-глутамин, 10% бычью телячью сыворотку (FCS) и 0,6 мг/мл гигромицина (все производства Sigma). Перед использованием, монослой промывали с помощью предварительно нагретой (37°С) аликвоты 3 мл Versene 1:5,000 (Invitrogen). После отсасывания этого раствора колбу инкубировали при 37°С в термостате с дополнительным количеством 2 мл Versene 1:5,000 в течение 6 минут. После этого клетки отсоединяли от дна колбы путем осторожного отцеживания и затем в колбу добавляли 10 мл Дульбекко-PBS, содержащей кальций (0,9 мМ) и магний (0,5 мМ) (PBS) (Invitrogen), и отсасывали в центрифужную пробирку объемом 15 мл перед центрифугированием (50 g, в течение 4 минут).
Полученный супернатант отбрасывали и осадок после центрифугирования осторожно повторно ресуспендировали в 3 мл аликвоте PBS. 0,5 мл аликвоты клеточной суспензии удаляли для автоматизированного подсчета клеток (Innovatis Cedex) и конечный объем клеточной суспензии доводили с помощью PBS для получения желательной конечной концентрации клеток.
Электрофизиология
Принципы и эксплуатация этого устройства были описаны ранее (Schroeder и др., Journal of Biomolecular Screening (2003) 8(1), 50-64). Вкратце, технология основывается на планшете на 384 лунок (PatchPlate™), в котором запись осуществляется в каждой лунке с помощью всасывания для установления и поддержания клетки в небольшом отверстии, разделенном двумя изолированными камерами с жидкостями. После запечатывания, раствор в нижней части PatchPlate™ загружали в камеру, содержащую амфотерицин В (Sigma). Это пермеализирует участок клеточной мембраны, покрывающий отверстие, в каждой лунке и в действительности позволяет записывать перфорированный, фиксированный потенциал (пэтч-кламп) в цельной клетке в каждой лунке.
Для каждой серии измерений IonWorks™ HT функционирует следующим образом при комнатной температуре (~21°С). В "лодочку" в положении "Буфер" загружают 4 мл PBS, а в положение "Клетки" вносят клеточную суспензию СНО-hERG, описанную выше. Планшет на 96 лунок (с V-образным дном, Greiner Bio-one), содержащий тестируемые соединения (в 3Х их конечной тестируемой концентрации) помещали в положение "Планшет 1" и PatchPlate™ помещали в устройство и устанавливали в положение с помощью PatchPlate™ крышки.
Планшет с каждым соединением выставляли таким образом, чтобы обеспечить построение десяти, 8-точечных кривых зависимости дозы от концентрации, оставшиеся две колонки на планшете заполняли наполнителем (0,33% ДМСО), для определения исходного уровня исследования, и цизапридом в сверхмаксимальной блокирующей концентрации (10 мкМ), для определения 100% уровня ингибирования. Затем добавляли Fluidics-головную фракцию (F-головная фракция) lonWorks™ HT 3,5 мкл PBS в каждую лунку PatchPlate™ и ее с нижней стороны промывали "Внутренним" раствором следующего состава (в мМ): K-глюконат 100, KCl 40, MgCl2 3,2, EGTA 3 и HEPES 5 (все производства Sigma) (pH 7,25-7,30 с помощью 10 М КОН). После примирования и дебарботирования, Electronics-головную фракцию (Е-головную фракцию) перемещали вокруг PatchPlate™ для осуществления дырочного теста (то есть прикладывали импульс напряжения для определения того, является ли отверстие в каждой клетке открытым). После этого F-головную фракцию диспергировали 3,5 мкл клеточной суспензии, описанной выше, в каждую лунку PatchPlate™ и клеткам предоставляли 200 секунд для достижения и закрытия отверстия в каждой лунке. Затем Е-головную фракцию прогоняли вокруг PatchPlate™ для определения закрытого сопротивления, полученного в каждой лунке.
После этого раствор на нижней стороне PatchPlate™ загружали к раствору "Доступа" следующего состава (в мМ): KCl 140, EGTA I, MgCl2 1 и HEPES 20 (все производства Sigma) (pH 7,25-7,30 с помощью 10 М КОН) плюс 100 мкг/мл амфотерицина В. Через 9 минут для предоставления возможности осуществления перфорации участка, Е-головную фракцию прогоняли вокруг всех 384 лунок пэтч-планшета для получения измерений тока hERG перед действием соединения. Затем 3,5 мкл раствора F-головной фракции из каждой лунки планшета с соединением добавляли к 4 лункам на PatchPlate™. После этого запускали программированное исследование от наиболее разведенной лунки на планшете с соединением и анализ осуществляли в направлении наиболее концентрированной лунки для сведения к минимуму воздействия любых побочных факторов.
После инкубирования приблизительно в течение 3,5 минут, Е-головную фракцию прогоняли вокруг всех 384 лунок PatchPlate™ для получения измерений тока hERG после действия соединений. Таким образом получали некумулятивные дозозависимые кривые при условии, что критерий исходного контроля достигали в достаточном проценте лунок (см. ниже), действие каждой концентрации тестируемого соединения рассчитывали на основе записи от 1 до 4 клеток.
Критерием исходного контроля для каждой лунки являлись: закрытое сопротивление перед сканированием > 60 MΩ, амплитуда хвостового тока hERG перед сканированием >0,15 нА; закрытое сопротивление после сканирования >60 MΩ, Токи hERG перед и после введения соединения вызывали с помощью импульса напряжения, состоящего из 20 с периода, фиксируемого при - 70 мВ, 160 мс шаг к - 60 мВ, 100 мс шаг к - 70 мВ, 1 с шаг к + 40 мВ, 2 с шаг к - 30 мВ и в заключение 500 мс шаг к - 70 мВ. В интервале импульсов напряжения до-соединения и после-соединения потенциал мембраны не фиксировали.
(д) Оценка водной растворимости
Тестируемое соединение (от 1 до 1,6 мг) взвешивали в сосуде и добавляли 1 мл 0,1 М фосфатного буфера (рН 7,4). Тестируемое соединение в интервале между 1,0 и 1,6 мг одновременно растворяли в 1,8 мл ДМСО в сосуде, для применения в качестве калибровочного раствора. Оба раствора перемешивали в течение 24 часов при 25°С. После этого насыщенный водный раствор и ДМСО-калибровочный раствор переносили в глубокие планшеты на 96 лунок. Планшет с насыщенным буферным раствором центрифугировали при относительной центробежной силе 4310 g и после этого водный супернатант переносили во второй глубокий луночный планшет и центрифугировали. После дальнейшего переноса водного супернатанта и 50% разведения буфером, конечный планшет с образцом и ДМСО-калибровочный планшет анализировали с помощью ВЭЖХ-УФ-МС. Количественное определение растворимости образца осуществляли путем сравнения площадей УФ-пиков образца и калибровки при 250 им (выбирали альтернативную длину волны, если 250 нм была неподходящей) с МС-подтверждением идентификации соединения.
(е) Оценка водной растворимости в буферах и стимулированной кишечной жидкости
Растворимость тестировали в следующей среде при указанных температурах:
Стимулированная кишечная жидкость (Fasted) FaSSIF (Galia и Dressman и др., Pharms Res, 15(5), 1998, с.698).
Таурохолат натрия (3 мМ); яичный лецитин (0,75 мМ); KH2PO4 (0,03 М); KCl (0,1 М); NaOH (для доведения до рН 6,5). Измеряли при 37°С.
Фосфатный буфер Соренсена (Handbook of Biochemistry, cc.234-237).
Раствор А 0,067 М монокалийфосфат.
Раствор В 0,067 М Динатрийфосфат.
Измеряли при 25°С и 37°С.
Подходящие количества (определенные с помощью вышеописанного исследования растворимости (д) и/или расчетной кривой рН растворимости) исследуемого соединения точно взвешивали в двух повторах в стеклянных сосудах на 2 драхмы.
К каждому набору параллельных сосудов, минимум 1,50 мл подходящей среды, в которую добавляли рН 6,8 фосфатный буфер Соренсена или FaSSIF. Все взвешивания в каждом случае должны быть достаточными для насыщения среды.
В каждую лунку добавляли покрытый PTFE магнитный повторитель, затем их запечатывали и помещали в магнитный реакционный перемешиваемый блок Variomag (CamLab). Перемешиваемые блоки выдерживали при подходящих температурах (см. выше), покрывали алюминиевой фольгой для уменьшения воздействия света и перемешивали в изменяемых направлениях при 800 об/мин. Из каждого сосуда отбирали пробы в заранее установленные временные промежутки для исследования среды. В каждом образце сначала определяли значение рН, а затем содержание активного компонента в каждый исследуемый момент времени следующим образом.
pH
С помощью подходящего рН-метра (Hydrus 400 - Fisher), электрода и стандартных рН-буферов, прибор калибровали при рН 4,01 и 7,00 при температуре окружающей среды.
Помещая электрод в каждый параллельный образец, определяли значение рН при температуре окружающей среды и результаты выражали в виде десятичной дроби. Электрод промывали деионизированной водой и вытирали насухо перед исследованиями.
Определение содержания активного компонента с помощью ВЭЖХ
Для каждого образца аликвоту 0,4 мл переносили в поликарбонатную ультрацентрифужную пробирку (Beckman). Образцы центрифугировали при 40 тыс. об/мин в течение 15 минут при подходящей температуре для тестирования среды с помощью ультрацентрифуги TL Optima (Beckman). Супернатант из каждой ультрацентрифужной пробирки переносили во вторую ультрацентрифужную пробирку и еще один раз центрифугировали при тех же условиях.
Супернатант из каждого образца исследовали с помощью оптимизированного метода ВЭЖХ для определения тестируемого соединения и содержания активного компонента относительно внешнего стандарта. Может понадобиться разведение супернатанта в подходящем растворителе для доведения концентраций до линейного диапазона для метода ВЭЖХ. Это обычно может быть рассчитано на основе предсказанной кривой водной рН растворимости и, в случае сорастворителей, исходя из добавленного количества соединения
Несмотря на то что фармакологические свойства соединений формулы I различаются в связи со структурными отличиями, как ожидалось, в целом, соединения формулы I могут проявлять активность в следующих концентрациях или дозах в одном или нескольких из вышеприведенных исследований (а), (б), (в) или (г):
Например, при осуществлении исследования (а) ингибирования HDAC1 и исследования (б) ингибирования пролиферации в цельных клетках, соединение, описанное в примере 4 настоящей заявки, проявляет IC50 результаты, приведенные ниже в таблице А. В таблице также приведен соответствующий результат для N-(2-аминофенил)-4-(1-(пирид-2-ил метил)пиперидин-4-ил)бензамида (соединение [1] выше):
Тестируемые соединения согласно настоящему изобретению не обнаруживали физиологически неприемлемого токсического действия в исследовании (г) при введении в эффективных дозах. Таким образом, не ожидается проявления неблагоприятных токсических действий при введении соединения формулы I или его фармацевтически приемлемой соли, как определено выше, в интервале доз, указанных далее.
Дополнительно, несмотря на то что растворимость соединений формулы I неизбежно будут различаться вследствие структурных изменений, как ожидалось, в целом соединения формулы I обладают растворимостью при исследовании (д), описанном выше, например, больше чем 100 мкМ.
В соответствии с дальнейшим вариантом осуществления изобретения, обеспечивается фармацевтическая композиция, которая содержит соединение формулы (I) или его фармацевтически приемлемую соль или его пролекарство, как определено выше, в сочетании с фармацевтически приемлемым разбавителем или носителем.
Композиции по изобретению могут находиться в формах, подходящих для перорального применения (например, в виде таблеток, лепешек, твердых или мягких капсул, водных или масляных суспензий, эмульсий, диспергируемых порошков или гранул, сиропов или эликсиров), для местного введения (например, в виде паст, мазей, гелей, водных или масляных растворов или суспензий), для введения путем ингаляции (например, в виде тонкоизмельченного порошка или жидкого аэрозоля), для введения путем вдувания (например, в виде тонкоизмельченного порошка), или для парентерального введения (например, в виде стерильного водного или масляного раствора для внутривенного, подкожного, внутримышечного введения, или в виде суппозитория для ректального введения).
Композиции по изобретению могут быть получены обычными способами при использовании обычных фармацевтических наполнителей, хорошо известных в данной области. Таким образом, композиции, предназначенные для перорального введения, могут содержать, например, один или несколько красителей, подсластителей, ароматизаторов и/или консервантов.
Подходящие фармацевтически приемлемые наполнители для таблетки включают, например, инертные разбавители, такие как лактоза, карбонат натрия, фосфат кальция или карбонат кальция, гранулирующие и дезинтегрирующие средства, такие как кукурузный крахмал или альгиновая кислота; связующие вещества, такие как крахмал; замасливатели, такие как стеарат магния, стеариновая кислота или тальк; консерванты, такие как этил или пропил п-гидроксибензоат, и антиоксиданты, такие как аскорбиновая кислота. Лекарственные препараты в виде таблеток могут быть без покрытия или могут иметь покрытие или для модификации их распада и последующего всасывания активного вещества в желудочно-кишечном тракте, или для улучшения их стабильности и/или внешнего вида, для этого используются обычные средства для покрытия и методики, которые хорошо известны в данной области.
Композиции для перорального применения могут находиться в виде твердых желатиновых капсул, в которых активный компонент смешан с инертным твердым разбавителем, например, карбонатом кальция, фосфатом кальция или каолином, или в виде мягких желатиновых капсул, в которых активный компонент смешан с водой или маслом, таким как арахисовое масло, жидкий парафин, соевое масло, кокосовое масло или предпочтительно оливковое масло, или другим подходящим наполнителем.
Водные суспензии, как правило, содержат активный компонент в форме тонкоизмельченного порошка совместно с одним или более суспендирующими средствами, такими как карбоксиметилцеллюлоза натрия, метилцеллюлоза, гидроксипропилметилцеллюлоза, альгинат натрия, поливинил-пирролидон, трагакантовая камедь и аравийская камедь; диспергирующими или смачивающими средствами, такими как лецитин или продукты конденсации алкиленоксида с жирными кислотами (например, полиоксиэтиленстеарат), или продукты конденсации этиленоксида с длинноцепочечными алифатическими спиртами, например гептадекаэтиленоксицетанол, или продукты конденсации этиленоксида с производными неполных сложных эфиров жирных кислот и гексита, такие как полиоксиэтиленсорбитмоноолеат, или продукты конденсации этиленоксида с длинноцепочечными алифатическими спиртами, например гептадекаэтиленоксицетанол, или продукты конденсации этиленоксида с производными неполных сложных эфиров жирных кислот и гексита, такие как полиоксиэтиленсорбитмоноолеат, или продукты конденсации этиленоксида с производными неполных сложных эфиров жирных кислот с ангидридами гексита, например полиэтиленсорбитмоноолеат. Водные суспензии также могут содержать один или более консервантов (таких как этил или пропил п-гидроксибензоат, антиоксидантов (таких как аскорбиновая кислота), красителей, ароматизаторов и/или подсластителей (таких как сахароза, сахарин или аспартам).
Масляные суспензии могут быть приготовлены путем суспендирования активного компонента в растительном масле (таком как арахисовое масло, оливковое масло, кунжутное масло или кокосовое масло) или в минеральном масле (таком как жидкий парафин). Масляные суспензии также могут содержать загустители, такие как пчелиный воск, твердый парафин или цетиловый спирт. Также могут добавляться подсластители, такие как перечисленные выше, и ароматизаторы для получения вкусного препарата для перорального введения. Эти композиции также могут быть защищены от разложения путем прибавления антиоксиданта, такого как аскорбиновая кислота.
Диспергируемые или лиофилизированные порошки и гранулы, подходящие для получения водной суспензии или раствора путем добавления воды, как правило, содержат активный компонент совместно с диспергирующим или смачивающим средством, суспендирующим средством и одним или более консервантами. Примерами подходящих диспергирующих или смачивающих средств и суспендирующих средств являются средства, упомянутые выше. Также могут включаться дополнительные наполнители, такие как посластители, ароматизаторы и красители.
Фармацевтические композиции по изобретению также могут находиться в виде эмульсий масло-в-воде. Масляной фазой может быть растительное масло, такое как оливковое масло или арахисовое масло, или минеральное масло, такое как, например, жидкий парафин или смесь любых перечисленных средств. Подходящими эмульгирующими средствами могут быть, например, природные смолы, такие как аравийская камедь или трагакантовая камедь, природные фосфатиды, такие как соя, лецитин, сложные эфиры или производные неполных сложных эфиров жирных кислот с ангидридами гексита (например, сорбитмоноолеат) и продукты конденсации указанных неполных сложных эфиров с этиленоксидом, такие как полиоксиэтиленсорбитмоноолеат. Эмульсии также могут содержать подсластители, ароматизаторы и консерванты.
Сиропы и эликсиры также могут быть приготовлены с подсластителями, такими как глицерин, пропиленгликоль, сорбит, аспартам или сахароза, а также могут содержать средство, уменьшающее раздражение, консервант, ароматизатор и/или краситель.
Фармацевтические композиции также могут находиться в форме стерильного раствора для инъекций или масляной суспензии, растворов, эмульсий или особых систем, которые могут быть приготовлены в соответствии с известными методиками, используя одно или более подходящих диспергирующих или смачивающих средств и суспендирующих средств, описанных выше. Стерильный препарат для инъекций также может представлять собой стерильный раствор для инъекции или суспензию в нетоксичном разбавителе или растворителе, который является приемлемым для парентерального введения, например, раствор в полиэтиленгликоле.
Лекарственные препараты в виде суппозиториев могут быть приготовлены путем смешивания активного компонента с подходящим нераздражающим наполнителем, который является твердым при обычных температурах, но жидким при ректальной температуре, и поэтому он расплавляется в прямой кишке, высвобождая лекарственное средство. Подходящими наполнителями являются, например, масло какао и полиэтиленгликоли.
Лекарственные препараты для местного применения, такие как кремы, мази, гели, водные или масляные растворы или суспензии, как правило, могут быть получены путем приготовления активного компонента с обычным наполнителем или разбавителем, который является подходящим для местного введения, используя обычную методику, известную в данной области техники.
Композиция для введения путем вдувания может находиться в виде тонкоизмельченного порошка, содержащего частицы со средним диаметром, например, 30 мкм или меньше, предпочтительно 5 мкм или меньше, и более предпочтительно в интервале 5 мкм-1 мкм и порошок содержит или сам активный компонент или разведенный одним или несколькими физиологически приемлемыми носителями, такими как лактоза. Затем порошок для вдувания обычно помещают в капсулу, содержащую, например, 1-50 мг активного компонента для применения в устройстве для турбоингаляции, таком как применяется для вдувания известного вещества кромогликата натрия.
Композиции для введения путем ингаляции могут находиться в виде обычного аэрозоля под повышенным давлением, приспособленного для распределения активного компонента или в виде аэрозоля, содержащего тонкоизмельченное твердое вещество или жидкие капли. Могут применяться обычные пропелленты для аэрозоля, такие как летучие фторированные углеводороды или углеводороды, и аэрозольное устройство представляет собой обычное устройство для распределения дозированного количества активного компонента.
Дополнительная информация относительно приготовления лекарственных препаратов содержится в главе 25.2, том 5 «Общей медицинской химии» (Comprehensive Medicinal Chemistry) (Corwin Hansch; председатель редакционной коллегии), Pergamon Press 1990.
В целом, вышеприведенные композиции могут быть приготовлены обычным способом при использовании обычных наполнителей.
Соединение формулы (I) обычно вводится теплокровному животному в стандартной дозе в диапазоне 5-5000 мг/м2 поверхности тела животного, то есть приблизительно 0,1-100 мг/кг и обычно она является терапевтически эффективной дозой. Стандартная дозированная форма, такая как таблетка или капсула, обычно включает, например, 1-250 мг активного компонента. Предпочтительно применяемая суточная доза находится в интервале 1-50 мг/кг. Однако суточная доза обязательно зависит от организма, который подвергается лечению, особенности пути введения и тяжести заболевания, которое поддается лечению. Следовательно, оптимальная доза может быть определена лечащим врачом для каждого пациента.
Нами было обнаружено, что соединения, раскрытые в настоящем изобретении или их фармацевтически приемлемая соль являются эффективными ингибиторами клеточного цикла (средствами, которые подавляют пролиферацию клеток), эти свойства, как полагают, являются результатом их ингибирующего действия по отношению к HDAC. Авторы изобретения также полагают, что соединения согласно настоящему изобретению могут быть вовлечены в ингибирование ангиогенеза, активацию апоптоза и дифференциацию. Таким образом, соединения согласно настоящему изобретению, как ожидается, являются пригодными для лечения заболеваний или болезненных состояний, опосредуемых только ферментами HDAC либо частично опосредуемых этими ферментами, то есть соединения могут применяться для получения ингибирующего действия по отношению к HDAC у теплокровного животного, которое нуждается в таком лечении. Таким образом, соединения согласно настоящему изобретению обеспечивают способ лечения пролиферации озлокачественных клеток, который характеризуется ингибированием HDAC ферментов, то есть соединения могут применяться для получения антипролиферативного действия, опосредованного только ингибированием HDAC, или частично опосредованного ингибированием этого фермента.
В соответствии с другим вариантом осуществления настоящего изобретения, обеспечивается соединение формулы (I), или его фармацевтически приемлемая соль или пролекарство, как определено выше, для применения в способе терапевтического лечения человека или животного.
Таким образом, в соответствии с дальнейшим вариантом осуществления изобретения, обеспечивается соединение формулы (I), или его фармацевтически приемлемая соль, или пролекарство, как определено выше, для применения в качестве лекарственного средства.
В соответствии с дальнейшим вариантом осуществления изобретения, обеспечивается применение соединения формулы (I), или его фармацевтически приемлемой соли, или пролекарства, как определено выше, для приготовления лекарственного средства для применения для получения ингибирующего действия по отношению к HDAC у теплокровного животного, такого как человек.
В соответствии с дальнейшим вариантом осуществления изобретения, обеспечивается способ ингибирования HDAC у теплокровного животного, такого как человек, которое нуждается в таком лечении, который предусматривает введение указанному животному эффективного количества соединения формулы (I), или его фармацевтически приемлемой соли, или пролекарства, как определено выше.
В соответствии с дальнейшим вариантом осуществления изобретения, обеспечивается применение соединения формулы (I), или его фармацевтически приемлемой соли, или пролекарства, как определено выше, для приготовления лекарственного средства для применения для обеспечения ингибирования клеточного цикла (действия, подавляющего пролиферацию клеток) у теплокровного животного, такого как человек.
В соответствии с дальнейшим вариантом осуществления изобретения, обеспечивается способ ингибирования клеточного цикла (действие, подавляющее пролиферацию клеток) у теплокровного животного, такого как человек, которое нуждается в таком лечении, который предусматривает введение указанному животному эффективного количества соединения формулы (I), или его фармацевтически приемлемой соли, или пролекарства, как определено выше.
В соответствии с дальнейшим вариантом осуществления изобретения, обеспечивается способ лечения злокачественного новообразования у теплокровного животного, такого как человек, которое нуждается в таком лечении, который предусматривает введение указанному животному эффективного количества соединения формулы (I), или его фармацевтически приемлемой соли, или пролекарства, как определено выше.
В соответствии с дальнейшим вариантом осуществления изобретения, обеспечивается соединение формулы (I), или его фармацевтически приемлемая соль или пролекарство, как определено выше, для приготовления лекарственного средства для применения для лечения злокачественного новообразования.
В соответствии с дальнейшим вариантом осуществления изобретения, обеспечивается соединение формулы (I), или его фармацевтически приемлемая соль или пролекарство, как определено выше, для применения для лечения злокачественного новообразования.
В соответствии с дальнейшим вариантом осуществления изобретения, обеспечивается применение соединения формулы (I), или его фармацевтически приемлемой соли или пролекарства, как определено выше, для применения для приготовления лекарственного средства для лечения злокачественного новообразования.
В дальнейшем варианте осуществления настоящего изобретения обеспечивается применение соединения формулы (I), или его фармацевтически приемлемой соли или пролекарства, как определено выше, для приготовления лекарственного средства для применения для лечения рака легких, рака прямой кишки, рака молочной железы, рака предстательной железы, лимфомы и/или лейкоза.
В дальнейшем варианте осуществления настоящего изобретения обеспечивается способ лечения рака легких, рака прямой кишки, рака молочной железы, рака предстательной железы, лимфомы или лейкоза, у теплокровного животного, такого как человек, которое нуждается в таком лечении, который предусматривает введение указанному животному эффективного количества соединения формулы (I), или его фармацевтически приемлемой соли или пролекарства, как определено выше.
Злокачественными новообразованиями, которые поддаются лечению согласно настоящему изобретению, являются рак пищевода, миелома, печеночно-клеточный рак, рак поджелудочной железы и рак шейки матки, диффузная эндотелиома костей, нейробластома, саркома Капоши, рак яичников, рак молочной железы, рак прямой кишки, рак предстательной железы, рак мочевого пузыря, меланома, рак легкого [включая немелкоклеточный рак легкого (NSCLC) и мелкоклеточный рак легкого (SCLC)], рак желудка, рак головы и шеи, рак головного мозга, рак почки, лимфома и лейкоз.
Ингибирущее действие по отношению к HDAC, описанное выше, может применяться в виде монотерапии, или, дополнительно к соединению по изобретению, можно применять одно или несколько других веществ и/или лечений. Такое совместное лечение может осуществляться путем одновременного, последовательного или раздельного введения отдельных компонентов для лечения. В медицинской онкологии является обычной практикой применять комбинацию разных форм лечения для лечения каждого пациента со злокачественным новообразованием. В медицинской онкологии другим(и) компонентом(ами) для такого совместного лечения дополнительно к лечению, обеспечивающему ингибирование клеточного цикла, описанному выше, могут быть: хирургия, лучевая терапия или химиотерапия. Такая химиотерапия может охватывать один или несколько следующих классов противоопухолевых средств:
(i) антипролиферативные/противоопухолевые лекарственные средства и их сочетания, которые применяются в онкологии, такие как алкилирующие средства (например, цис-платин, карбоплатин, циклофосфамид, азотный иприт, мельфалан, хлорамбуцил, бусульфан и нитрозомочевины); антиметаболиты (например, антифолаты, такие как фторпиримидины, такие как 5-фторурацил и тегафур, ралтитрексед, метотрексат, арабинозид цитозина и гидроксимочевина; противоопухолевые антибиотики (например, антрациклины, такие как адриамицин, блеомицин, доксорубицин, дауномицин, эпирубицин, идарубицин, митомицин-С, дактиномицин и митрамицин); антимитотические средства (например, алкалоиды барвинка, такие как винкристин, винбластин, виндезин и винорелбин и таксоиды, такие как таксол и таксотер); и ингибиторы топоизомеразы (например, эпиподофиллотоксины, такие этопозид и тенипозид, амсакрин, топотекан и камфотецин);
(ii) цитостатические средства, такие как антиэстрогены (например, тамоксифен, торемифен, ралоксифен, дролоксифен и йодоксифен), ингибиторы рецептора эстрогена (например, фульвестрант), антиандрогены (например, бикалутамид, флутамид, нилутамид и ципротерон ацетат), антагонисты LHRH или агонисты LHRH (например, гозерелин, лейпрорелин и бузерелин), прогестогены (например, мегестрол ацетат), ингибиторы ароматазы (например, анастрозол, летрозол, воразол и эксеместан) и ингибиторы 5α-редуктазы, такие как финастерид;
(iii) средства, которые ингибируют инвазию злокачественных клеток (например, ингибиторы металлопротеиназы, такие как маримастат и ингибиторы функции рецептора урокиназного активатора плазминогена);
(iv) ингибиторы действия фактора роста, например, такие ингибиторы включают антитела к фактору роста, антитела к рецептору фактора роста (например, анти-erbb2 антитело трастузумаб [Herceptin™] и анти-erbb1 антитело цетуксимаб [С225]), ингибиторы фарнезилтрансферазы, ингибиторы МЕК, ингибиторы тирозинкиназы и ингибиторы серин/треонин киназы, например, ингибиторы семейства эпидермального фактора роста (например, ингибиторы тирозинкиназ EGFR семейства, такие как N-(3-хлор-4-фторфенил)-7-метокси-6-(3-морфолинопропокси)хиназолин-4-амин (гефитиниб), N-(3-этинилфенил)-6,7-бис(2-метоксиэтокси)хиназолин-4-амин (эрлотиниб, OSI-774) и 6-акриламидо-N-(3-хлор-4-фторфенил)-7-(3-морфолинопропокси)хиназолин-4-амин (CI 1033)), например, ингибиторы семейства фактора роста производных тромбоцитов и, например, ингибиторы семейства фактора роста гепатоцитов;
(v) антиангиогенные вещества, такие как те, которые ингибируют действие фактора роста эндотелия сосудов (например, антитело к фактору роста клеток эндотелия сосудов бевацизумаб [Avastin™], соединения, которые описаны в международных заявках на патент WO 97/22596, WO 97/30035, WO 97/32856 и WO 98/13354) и соединения, которые действуют по другому механизму (например, линомид, ингибиторы действия интегрина αvβ3 и ангиостатин);
(vi) вещества, которые повреждают сосуды, такие как комбретастатин А4 и соединения, описанные в международных заявках на патент WO 99/02166, WO 00/40529, WO 00/41669, WO 01/92224, WO 02/04434 и WO 02/08213;
(vii) антисмысловая терапия, например, такая, которая направлена на мишени, перечисленные выше, такие как ISIS 2503, антисмысловая терапия на основе гена ras;
(viii) способы генной терапии, включая, например, способы замены аберрантных генов, такие как способы аберрации р53 или аберрации BRCA1 или BRCA2, GDEPT (пролекарственная терапия, направленная на ген фермента), такие как способы с использованием деаминазы цитозина, тимидинкиназы или бактериальной нитроредуктазы и способы повышения устойчивости пациента к химиотерапии или радиотерапии, такие как генная терапия резистентности ко многим лекарственным средствам;
(ix) способы иммунотерапии, включая, например, способы повышения иммуногенности опухолевых клеток пациента в условиях ex vivo и in vivo, такие как трансфекция цитокинами, такими как интерлейкин 2, интерлейкин 4 или фактор стимуляции колоний гранулоцитов-макрофагов, способы снижения активности Т-клеток, способы с использованием трансфектированных иммунных клеток, таких как цитокин-трансфектированные дендритные клетки, способы с использованием цитокин-трансфектированных линий опухолевых клеток и способы с использованием анти-идиотипичных антител;
(х) ингибиторы клеточного цикла, включая, например ингибиторы CDK (например, флавопиридол) и другие ингибиторы контрольных точек клеточного цикла (например, киназа контрольной точки); ингибиторы аурора-киназы и других киназ, вовлеченных в митоз и регуляцию клеточного деления (например, митотические кинезины); и другие ингибиторы деацетилазы гистонов; и
(xi) дифференцирующие средства (например, ретиноевая кислота и витамин D).
В соответствии с этим вариантом осуществления изобретения, обеспечивается фармацевтическая композиция, содержащая соединение формулы (I), как определено выше, и дополнительное противоопухолевое вещество, как определено выше, для совместного лечения злокачественного новообразования.
Также обеспечивается соединение формулы (I), или его фармацевтически приемлемая соль или пролекарство, как определено выше, для применения в способе лечения воспалительных заболеваний, аутоиммунных заболеваний, и аллергических/атопических заболеваний.
В частности, соединение формулы (I), или его фармацевтически приемлемая соль или пролекарство, как определено выше, предусматривается для применения в способе лечения воспаления суставов (особенно ревматоидного артрита, остеоартрита и подагры), воспаления желудочно-кишечного тракта (особенно воспалительных заболеваний кишечника, неспецифического язвенного колита и гастрита), воспаления кожи (особенно псориаза, экземы, дерматитов), рассеянного склероза, атеросклероза, спондилоартропатий (анкилозирующего спондилоартрита, псориатического артрита, артрита, связанного с неспецифическим язвенным колитом), невропатий, связанных со СПИДом, системной красной волчанки, астмы, хронического обструктивного заболевания легких, бронхита, плеврита, респираторного дистресс-синдрома взрослых, сепсиса, и острого и хронического гепатита (вирусного, бактериального или токсического).
Также обеспечивается соединение формулы (I), или его фармацевтически приемлемая соль или пролекарство, как определено выше, для применения в качестве лекарственного средства для лечения воспалительных заболеваний, аутоиммунных заболеваний, и аллергических/атопических заболеваний у теплокровного животного, такого как человек.
В частности, соединение формулы (I), или его фармацевтически приемлемая соль или пролекарство, как определено выше, предусматривается для применения в качестве лекарственного средства для лечения воспаления суставов (особенно ревматоидного артрита, остеоартрита и подагры), воспаления желудочно-кишечного тракта (особенно воспалительных заболеваний кишечника, неспецифического язвенного колита и гастрита), воспаления кожи (особенно псориаза, экземы, дерматитов), рассеянного склероза, атеросклероза, спондилоартропатий (анкилозирующего спондилоартрита, псориатического артрита, артрита, связанного с неспецифическим язвенным колитом), невропатий, связанных со СПИДом, системной красной волчанки, астмы, хронического обструктивного заболевания легких, бронхита, плеврита, респираторного дистресс-синдрома взрослых, сепсиса, и острого и хронического гепатита (вирусного, бактериального или токсического).
Также обеспечивается применение соединения формулы (I), или его фармацевтически приемлемой соли или пролекарства, как определено выше, для приготовления лекарственного средства для применения для лечения воспалительных заболеваний, аутоиммунных заболеваний, и аллергических/атопических заболеваний у теплокровного животного, такого как человек.
Как было указано выше, доза, необходимая для лечения или профилактики конкретного заболевания, связанного с пролиферацией клеток, обязательно зависит от организма, который подвергается лечению, пути введения и тяжести заболевания, которое поддается лечению. Применяемая стандартная доза находится в диапазоне, например, 1-100 мг/кг, предпочтительно 1-50 мг/кг.
Дополнительно к их применению в терапевтической медицине, соединения формулы (I) и их фармацевтически приемлемые соли также полезны в качестве фармацевтических средств для развития и стандартизации тестируемых систем в условиях in vitro и in vivo для оценки действий ингибиторов активности клеточного цикла у лабораторных животных, таких как коты, собаки, кролики, обезьяны, крысы и мыши, как компонент поиска новых терапевтических средств.
Далее изобретение иллюстрируется следующими примерами, в которых, в целом:
(i) действия осуществляли при температуре окружающей среды, то есть в интервале 17-25°С, и в атмосфере инертного газа, такого как аргон, если специально не указано иначе;
(ii) испарение осуществляли путем роторного испарения в вакууме и процедуру обычной обработки осуществляли после удаления остатков твердых веществ путем фильтрации;
(iii) колоночную хроматографию (при помощи флэш-методики) и жидкостную хроматографию среднего давления (MLPC) осуществляли на диоксиде кремния Merck Kieselgel (Art. 9385) или Merck Lichroprep RP-18 (Art. 9303) диоксиде кремния с обращенной фазой, полученного от Е.Merck, Darmstadt, Германия, или используя собственные предварительно упакованные картриджи диоксида кремния с нормальной фазой, например Redisep(TM) доступные картриджи для хроматографии получали от Presearch Ltd., Hitchin, UK, или жидкостную хроматографию высокого давления (ВЭЖХ) осуществляли на диоксиде кремния С18 с обращенной фазой, например, на препаративной колонке с обращенной фазой Dynamax С-18 60Ǻ;
(iv) для выходов, если они представлены, необязательно могут быть достигнуты максимальные значения;
(v) структуру конечных продуктов формулы (I), как правило, подтверждали при помощи ядерного магнитного резонанса (ЯМР) и/или масс-спектральных методов; масс-спектральные данные обычно получали при бомбардировке быстрыми атомами (FAB), используя спектрометр Platform, и, если было целесообразно, данные положительных ионов или данные отрицательных ионов собирали; ЯМР значения химических сдвигов измеряли на дельта-шкале спектры протонного магнитного резонанса измеряли, используя спектрометр Jeol JNM EX 400, работая при напряженности поля 400 МГц, спектрометр Varian Gemini 2000, работая при напряженности поля 300 МГц, Bruker DPX-400, работая при 400 МГц или Bruker AM300, работая при напряженности поля 300 МГц - измерения проводили при температуре окружающей среды, если специально не указано иначе;
(vi) промежуточные продукты, как правило, не были полностью охарактеризованы и чистоту определяли при помощи тонкослойной хроматографии, HPLC, инфракрасного (ИК) и/или ЯМР анализов;
(vii) точки плавления являются несвязанными и они определялись при помощи автоматического прибора для определения точек плавления Mettler SP62 или прибора с масляной баней; точки плавления для конечных продуктов формулы (I) определяли после кристаллизации из обычного органического растворителя, такого как этанол, метанол, ацетон, простой эфир или гексан, отдельно или в добавке;
(viii) использованы следующие сокращения:
Пример 1
N-(2-аминофенил)-4-{1-[(1-метил-1H-пиразол-4-ил)метил]пиперидин-4-ил}бензамид
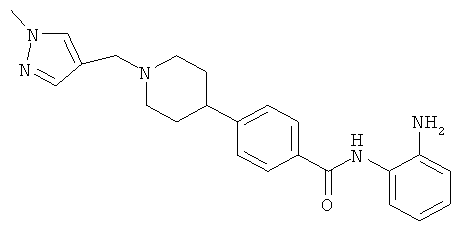
В реакционный сосуд, загруженный 1-метил-1H-пиразол-4-карбоксальдегидом (84,5 мг, 0,77 ммоль), добавляли раствор трет-бутил 2-[(4-пиперидин-4-илбензоил)амино]фенилкарбамата (полученный, как описано в методе 1 ниже; 300 мг, 0,76 ммоль) в дихлорметане (7 мл) и N,N-диметилформамиде (0,5 мл). трет-Бутил 2-[(4-пиперидин-4-илбензоил)амино]фенилкарбамат также может быть приготовлен в соответствии со способом, описанным в методе 4 ниже. Добавляли уксусную кислоту (50 мкл, 0,87 ммоль) и реакционной смеси позволяли перемешиваться при температуре окружающей среды в течение 45 минут. Затем добавляли триацетоксиборогидрид натрия (250 мг, 1,18 ммоль) и реакциям позволяли перемешиваться дополнительно в течение 76 часов, после этого разводили метанолом и наносили непосредственно на SCX-2 картридж (10 г). Картридж промывали метанолом (80 мл), затем продукты элюировали 2М раствором аммиака в метаноле (50 мл). Релевантные фракции упаривали насухо и полученный остаток повторно растворяли в дихлорметане (3 мл) и обрабатывали трифторуксусной кислотой (1 мл). Эту смесь перемешивали при температуре окружающей среды в течение 2 часов, затем разводили дихлорметаном и наносили на SCX-2 картридж (5 г). Картридж промывали метанолом (20 мл), после этого продукты элюировали 2М раствором аммиака в метаноле (20 мл). Аммиачную фракцию упаривали насухо и полученный остаток очищали с помощью флэш-хроматографии на диоксиде кремния, элюируя 10% метанолом в дихлорметане, получая указанное в заглавии соединение (117 мг, 40%); ЯМР-спектр: (ДМСО d6) δ 1,70 (m, 4Н), 2,01 (t, 2H), 2,57 (m, 1H), 2,95 (m, 2H), 3,38 (s, 2H), 3,81 (s, 3H), 4,86 (s, 2H), 6,60 (m, 1H), 6,78 (m, 1H), 6,97 (m, 1H), 7,18 (m, 1H), 7,31 (s, 1H), 7,37 (d, 2H), 7,57 (s, 1H), 7,91 (d, 2H), 9,56 (s, 1H); Масс-спектр: М+Н+ 390.
Пример 2
Используя методику, аналогичную описанной в примере 1, трет-бутил 2-[(4-пиперидин-4-илбензоил)амино]фенилкарбамат подвергали реакции с подходящим пиразолкарбальдегидным исходным веществом (ИВ), получая соединения, приведенные в таблице 1
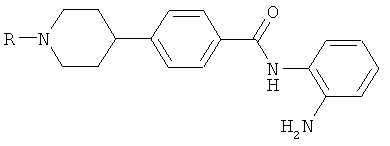
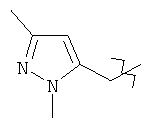
Пример 3
N-(2-аминофенил)-4-[1-(1H-пиразол-3-илметил)пиперидин-4-ил]бензамид
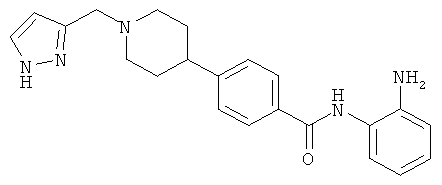
трет-Бутил 2-[(4-пиперидин-4-илбензоил)амино]фенилкарбамат (полученный, как описано в методе 1 ниже; 200 мг, 0,51 ммоль) и 1H-пиразол-3-карбальдегид (50,7 мг, 0,53 ммоль) перемешивали при температуре окружающей среды в дихлорметане (5 мл) в течение 1 часа. Добавляли триацетоксиборогидрид натрия (150 мг, 0,71 ммоль) и смесь перемешивали при температуре окружающей среды в течение 48 часов. Полученный раствор абсорбировали на SCX-2 колонку, которую промывали метанолом (2 объема колонки) и затем продукт элюировали 2М раствором аммиака в метаноле (2 объема колонки), получая пену. Ее растворяли в 1,4-диоксане (2 мл), добавляли 4М раствор хлористого водорода в 1,4-диоксане (2 мл) и раствор перемешивали при температуре окружающей среды в течение 48 часов. Продукт фильтровали и промывали простым диэтиловым эфиром и высушивали на воздухе. Полученное твердое вещество растворяли в воде, подщелачивали 2 н. гидроксидом натрия и полученное твердое вещество фильтровали, промывали водой и высушивали в вакууме, получая указанное в заглавии соединение (61 мг, 44%). ЯМР-спектр: 1Н NMR (ДМСО d6) δ 1,71 (m, 4Н), 2,07 (m, 2H), 2,56 (m, 1H), 2,95 (m, 2H), 3,53 (s, 2H), 4,86 (s, 2H), 6,16 (s, 1H), 6,60 (m, 1H), 6,78 (d, 1H), 6,97 (t, 1H), 7,18 (d, 1H), 7,37 (d, 2H), 7,64 (m, 1H), 7,91 (d, 2H), 9,55 (s, 1H), 12,59 (m, 1H); Масс-спектр: М+Н+ 376.
Пример 4
N-(2-Аминофенил)-4-{1-[(5-метокси-1,3-диметил-1H-пиразол-4-ил)метил]пиперидин-4-ил}бензамид
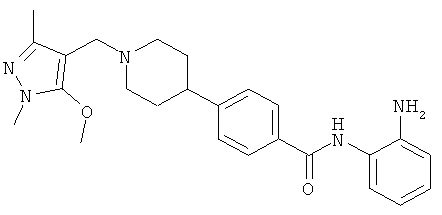
Используя методику, аналогичную описанной в примере 3, трет-бутил 2-[(4-пиперидин-4-илбензоил)амино]фенилкарбамат (полученный, как описано в методе 1 ниже; 200 мг, 0,51 ммоль) подвергали реакции с 5-метокси-1,3-диметил-1H-пиразол-4-карбальдегидом (92,6 мг, 0,60 ммоль), получая указанное в заглавии соединение (68 мг, 36%); ЯМР-спектр: (ДМСО d6) δ 1,63 (m, 2H), 1,78 (m, 2H), 2,00 (m, 2H), 2,06 (s, 3Н), 2,57 (m, 1H), 2,94 (m, 2H), 3,25 (s, 2H), 3,50 (s, 3Н), 3,99 (s, 3Н), 4,86 (br s, 2H), 6,60 (m, 1H), 6,78 (d, 1H), 6,97 (m, 1H), 7,17 (d, 1H), 7,37 (d, 2H), 7,91 (d, 2H), 9,55 (s, 1H); Масс-спектр: М+H+ 434.
Пример 5
N-(2-аминофенил)-4-{1-[(3-этил-1H-пиразол-4-ил)метил]пиперидин-4-ил}бензамид
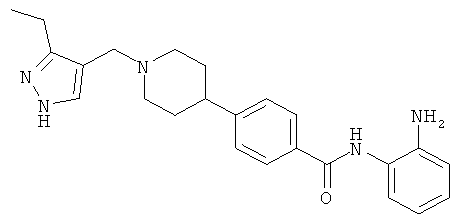
трет-Бутил 2-[(4-пиперидин-4-илбензоил)амино]фенилкарбамат (полученный, как описано в методе 1 ниже; 395 мг, 1,0 ммоль) и 3-этил-1H-пиразол-4-карбальдегид (149 мг, 1,2 ммоль) перемешивали при температуре окружающей среды в дихлорметане (10 мл) в течение 1 часа. Добавляли триацетоксиборогидрид натрия (297 мг, 1,4 ммоль) и смесь перемешивали при температуре окружающей среды в течение 24 часов. Полученный раствор абсорбировали на SCX-2 колонке, которую промывали метанолом (2 объема колонки) и затем продукт элюировали 2М раствором аммиака в метаноле (2 объема колонки), получая продукт в виде белой пены. Его очищали с помощью хроматографии на диоксиде кремния, элюируя 10% метанолом в дихлорметане. Остаток растворяли в дихлорметане (4 мл) и добавляли трифторуксусную кислоту (1 мл) и смесь перемешивали в течение 3 часов при температуре окружающей среды. Полученный раствор абсорбировали на SCX-2 колонке, которую промывали метанолом (2 объема колонки) и затем продукт элюировали 2М раствором аммиака в метаноле (2 объема колонки), получая указанное в заглавии соединение (232 мг, 75%). ЯМР-спектр: (ДМСО d6) δ 1,18 (t, 3H), 1,65 (m, 2H), 1,77 (m, 2H), 2,00 (m, 2H), 2,57 (m, 3H), 2,95 (m, 2H), 3,34 (s, 2H), 4,86 (br s, 2H), 6,60 (m, 1H), 6,78 (d, 1H), 6,97 (m, 1H), 7,17 (d, 1H), 7,29 (br s, 1H), 7,37 (d, 2H), 7,91 (d, 2H), 9,55 (s, 1H), 12,39 (s, 1H); Масс-спектр: М+Н+ 404.
Пример 6
N-(2-аминофенил)-4-{1-[(1,3,5-триметил-1H-пиразол-4-ил)метил]пиперидин-4-ил}бензамид
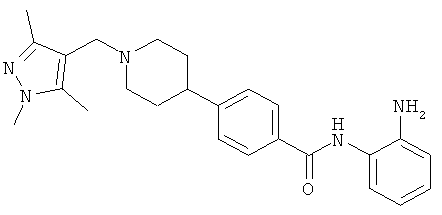
Используя методику, аналогичную описанной в примере 5, трет-бутил 2-[(4-пиперидин-4-илбензоил)амино]фенилкарбамат (полученный, как описано в методе 1 ниже; 200 мг, 0,51 ммоль) подвергали реакции с 1,3,5-триметил-1H-пиразол-4-карбальдегидом (83,5 мг, 0,60 ммоль), получая указанное в заглавии соединение (70 мг, 56%); ЯМР-спектр: (ДМСО d6) δ 1,65 (m, 2H), 1,77 (m, 2H), 1,98 (m, 2H), 2,09 (s, 3H), 2,18 (s, 3H), 2,57 (m, 1H), 2,92 (m, 2H), 3,24 (s, 2H), 3,63 (s, 3H), 4,86 (br s, 2H), 6,60 (m, 1H), 6,78 (d, 1H), 6,97 (m, 1H), 7,17 (d, 1H), 7,37 (d, 2H), 7,91 (d, 2H), 9,55 (s, 1H); Масс-спектр: М+Н+ 418.
Пример 7А
N-(2-аминофенил)-4-{1-[(1,3-диметил-1H-пиразол-4-ил)метил]пиперидин-4-ил}бензамид
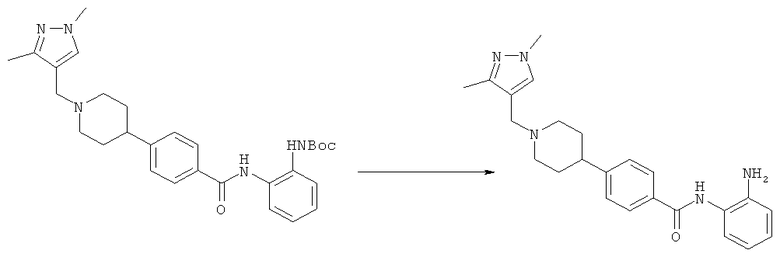
трет-Бутил (2-{[4-(1-{1,3-диметил-1H-пиразол-4-илметил}пиперидин-4-ил)бензоил]амино}фенил)карбамат (полученный, как описано в методе 6 ниже; 7,61 г, 15,11 ммоль) растворяли в 1,4 диоксане (70 мл) и охлаждали до 0°С, используя ледяно-водяную баню. Затем медленно добавляли 4М раствор хлористого водорода в 1,4 диоксане (150 мл, 600 ммоль). Полученной суспензии позволяли нагреться до комнатной температуры и комки разрушали путем перемешивания стеклянным стержнем. Реакционную смесь перемешивали при комнатной температуре в течение 18 часов. Смесь фильтровали с отсасыванием. Полученное твердое вещество растворяли в воде (200 мл), и значение рН раствора доводили до 12 путем медленного добавления 2М водного раствора гидроксида натрия. Полученную смесь экстрагировали дихлорметаном (300 мл) и органические вещества отделяли. Затем водную фазу дополнительно экстрагировали дихлорметаном (200 мл) и объединенные экстракты промывали соляным раствором, высушивали над сульфатом магния, фильтровали и упаривали, получая прозрачную смолу. Эту смолу ресуспендировали в простом диэтиловом эфире и повторно упаривали насухо, получая указанное в заглавии соединение (5,69 г, 93%); ЯМР-спектр (CDCl3) δ 1,81 (m, 4H), 2,07 (m, 2H), 2,25 (s, 3Н), 2,56 (m, 1H), 3,04 (m, 2H), 3,39 (s, 2H), 3,81 (s, 3Н), 3,85 (s, 2H), 6,84 (m, 2H), 7,08 (m, 1H), 7,24 (s, 1H), 7,33 (m, 3Н), 7,82 (m, 3Н). Масс-спектр: М+Н+ 404.
Пример 7В
N-(2-аминофенил)-4-{1-[(1,3-диметил-1H-пиразол-4-ил)метил]пиперидин-4-ил}бензамид
трет-Бутил (2-{[4-(1-{1,3-диметил-1H-пиразол-4-илметил}пиперидин-4-ил)бензоил]амино}фенил)карбамат (Метод 6 ниже; 92,3 г, 183,3 ммоль) суспендировали в метаноле (754 мл) и воде (141 мл) и охлаждали до 0-5°С. Добавляли концентрированную соляную кислоту, поддерживая температуру ниже 20°С. Реакционную смесь перемешивали в течение 20 часов при температуре окружающей среды. Реакцию охлаждали до 0-5°С и добавляли водный раствор гидроксида натрия, поддерживая температуру ниже 20°С до получения значения рН, равного 12-14. Реакционную смесь нагревали до температуры флегмы в течение 30 минут, затем охлаждали до 20°С приблизительно в течение 4 часов. Продукт собирали путем фильтрации и промывали водным метанолом, затем высушивали в вакууме при 45°С до постоянного веса, получая указанное в заглавии соединение (63,3 г 86%). ЯМР-спектр (ДМСО d6), 1,65 (m, 2H), 1,73 (m, 2H), 1,96 (t, 2H), 2,08 (s, 3Н), 2,55 (m, 1H), 2,92 (d, 2H), 3,28 (s, 2H), 3,75 (s, 3Н), 4,87 (s, 2H), 6,59 (m, 1H), 6,77(m, 1H), 6,96 (m, 1H), 7,2 (d, 1H), 7,35 (d, 2H), 7,44 (s, 1H), 7,90 (d, 2H), 9,56 (s, 1H). Масс-спектр: М+Н+ 404.
Пример 8
N-(2-аминофенил)-4-{1-[(1,5-диметил-1H-пиразол-4-ил)метил]пиперидин-4-ил}бензамид
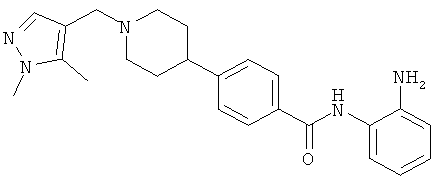
трет-Бутил {2-[(4-{1-[(1,5-диметил-1H-пиразол-4-ил)метил]пиперидин-4-ил}бензоил)амино]фенил}карбамат (полученный, как описано в методе 7; 308 мг, 0,61 ммоль) ресуспендировали в дихлорметане (2 мл) и добавляли трифторуксусную кислоту (1 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 1 часа, затем заливали в SCX-3 картридж (5 г). Картридж промывали дихлорметаном (50 мл) и метанолом (50 мл), после этого продукт элюировали 2М раствором аммиака в метаноле (50 мл). Аммиачную фракцию упаривали, получая белое твердое вещество (182 мг), которое очищали с помощью препаративной ВЭЖХ с обращенной фазой, получая указанное в заглавии соединение (134 мг, 55%); ЯМР-спектр: (ДМСО d6) δ 1,70 (m, 4Н), 2,01 (m, 2Н), 2,22 (s, 3Н), 2,57 (m, 1H), 2,95 (m, 2H), 3,28 (s, 2H), 3,71 (s, 3Н), 4,86 (s, 2H), 6,60 (m, 1H), 6,78 (m, 1H), 6,97 (m, 1H), 7,17 (m, 1H), 7,23 (s, 1H), 7,37 (d, 2H), 7,90 (d, 2H), 9,55 (s, 1H); Масс-спектр: M+H+ 404.
Пример 9
N-(2-аминофенил)-4-{1-[(1-этил-5-метил-1H-пиразол-4-ил)метил]пиперидин-4-ил}бензамид
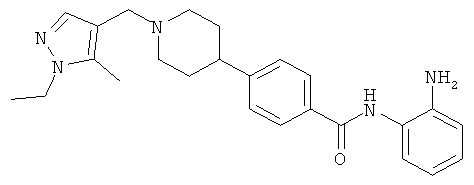
Раствор 1-этил-5-метил-1Н-пиразол-4-карбальдегида (141 мг, 1,02 ммоль) в дихлорметане (1 мл) добавляли к раствору трет-бутил 2-[(4-пиперидин-4-илбензоил)амино]фенилкарбамата (полученного, как описано в методе 1 ниже; 300 мг, 0,76 ммоль) в дихлорметане (6,5 мл). Добавляли уксусную кислоту (45 мкл, 0,79 ммоль) и реакционную смесь перемешивали в течение 4 часов. Затем добавляли N,N-диметилформамид (1 мл) и продолжали перемешивать дополнительно в течение 30 минут, затем добавляли триацетоксиборогидрид натрия (245 мг, 1,16 ммоль) в виде твердого вещества. После этого реакционной смеси позволяли перемешиваться при температуре окружающей среды в течение 18 часов (всю ночь). Реакционную смесь разводили до двойного объема путем добавления метанола и сразу загружали на предварительно промытый (с помощью метанола) SCX-2 картридж (10 г). Картридж промывали метанолом (60 мл), после этого продукты элюировали с помощью 2М раствора аммиака в метаноле (50 мл). Аммиачный элюант упаривали, получая бесцветную пену (420 мг), которую ресуспендировали в дихлорметане (5 мл) и обрабатывали трифторуксусную кислотой (2 мл). Смеси позволяли перемешиваться в течение 2 часов, затем разводили дихлорметаном (10 мл) и заливали на предварительно промытый (с помощью метанола) SCX-2 картридж (10 г). Катридж промывали дихлорметаном (40 мл), метанолом (50 мл) и после этого продукты элюировали 2М раствором аммиака в метаноле (50 мл). При упаривании аммиачной фракции получали светло-желтую смолу (300 мг), которую очищали с помощью препаративной ВЭЖХ с обращенной фазой, получая указанное в заглавии соединение (172 мг, 54%); ЯМР-спектр: (ДМСО d6) δ 1,27 (t, 3Н), 1,66 (m, 4H), 1,98 (m, 2H), 2,21 (s, 3Н), 2,56 (m, 1H), 2,92 (m, 2H), 3,29 (s, 2H), 4,02 (q, 2H), 4,85 (s, 2H), 6,59 (m, 1H), 6,77 (m, 1H), 6,95 (m, 1H), 7,16 (m, 1H), 7,23 (s, 1H), 7,35 (d, 2H), 7,89 (d, 2H), 9,54 (s, 1H); Масс-спектр: М+Н+ 418.
Примеры 10 и 11
Используя методику, аналогичную описанной в примере 9, трет-бутил 2-[(4-пиперидин-4-илбензоил)амино]фенилкарбамат (полученный, как описано в методе 1 ниже) подвергали реакции с подходящим пиразолкарбальдегидным исходным веществом, получая соединения, приведенные в таблице 2.
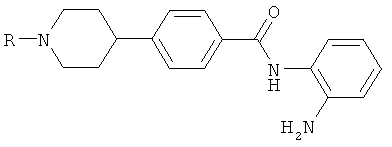
Методы получения исходных веществ
Метод 1
трет-Бутил {2-[(4-пиперидин-4-илбензоил)амино]фенил}карбамат
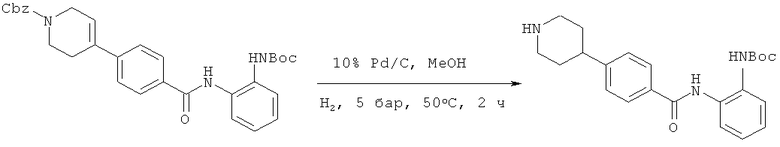
К раствору бензил 4-{4-[({2-[(трет-бутоксикарбонил)амино]фенил}амино)карбонил]фенил}-3,6-дигидропиридин-1(2H)-карбоксилата (269 г, 524 ммоль; полученного, как описано в методе 2 ниже) в метаноле (3000 мл) добавляли 10% палладия на угле (10 г). Реакционную смесь помещали в газообразный водород под давлением 5 бар и нагревали до 50°С в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры, фильтровали через слой целита, и растворитель упаривали при пониженном давлении. Полученную пену растирали в порошок в простом диэтиловом эфире и фильтровали, получая белое твердое вещества. Этот продукт тонко измельчали и перемешивали с 95:5 простой диэтиловый эфир/этилацетат, затем собирали путем фильтрации с отсасыванием. Это твердое вещество промывали простым диэтиловым эфиром, изогексаном и высушивали в вакууме, получая указанное в заглавии соединение (167 г, 81%); ЯМР-спектр: (ДМСО-d6) 1,45 (s, 9H), 1,57 (m, 2Н), 1,72 (m, 2H), 2,61 (t, 2H), 2,69 (m, 1H), 3,07 (m, 2H), 7,18 (m, 2H), 7,40 (d, 2H), 7,53 (d, 2H), 7,91 (d, 2H), 8,70 (br s, 1H), 9,82 (br s, 1H); Масс-спектр: М+Н+ 396.
Метод 2
Бензил 4-{4-[({2-[(трет-бутоксикарбонил)амино]фенил}амино)карбонил]фенил}-3,6-дигидропиридин-1(2H)-карбоксилат
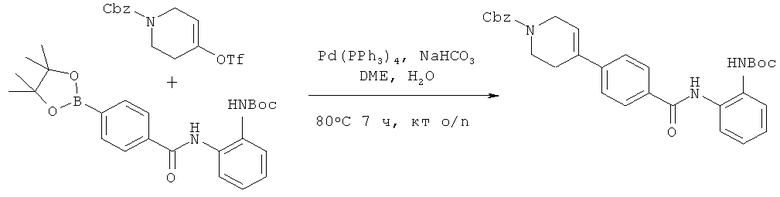
Тетракис(трифенилфосфин)палладий (0) (8,0 г, 6,92 ммоль) добавляли к перемешиваемой суспензии N-(2-трет-бутоксикарбониламинофенил)-4-(4,4,5,5-тетраметил-1,3,2,-диоксаборолан-2-ил) бензамида (288 г, 657 ммоль; полученного, как описано в международной патентной заявке WO 03/087057, Метод 13, страница 60) и бензил 4-{[(трифторметил)сульфонил]окси}-3,6-дигидропиридин-1(2H)-карбоксилата (240 г, 657 ммоль; полученного, как описано в методе 3 ниже) в 1,2-диметоксиэтане (3000 мл) и насыщенного водного раствора бикарбоната натрия (3000 мл). Реакционную смесь нагревали до 80°С в течение 7 часов, затем позволяли охладиться до температуры окружающей среды, при перемешивании. Затем реакционную смесь выливали в воду (2000 мл) и экстрагировали этилацетатом. После этого органические экстракты высушивали над сульфатом магния, фильтровали и упаривали насухо, получая неочищенный продукт в виде серого твердого вещества. Его очищали с помощью колоночной флэш-хроматографии на диоксиде кремния, элюируя этилацетатом/гексаном (30:70 об./об.) и получали указанное в заглавии соединение (279 г. 82%); ЯМР-спектр: (ДМСО-d6) δ 1,44 (s, 9H), 2,56 (m, 2H), 3,66 (m, 2H), 4,14 (m, 2H), 5,13 (s, 2H), 6,34 (m, 1H), 7,18 (m, 2H), 7,33 (m, 1H), 7,40 (m, 4H), 7,54 (m, 2H), 7,62 (d, 2H), 7,94 (d, 2H), 8,64 (br s, 1H), 9,81 (br s, 1H); Масс-спектр: MNa+ 550.
Метод 3
Бензил 4-{[(трифторметил)сульфонил]окси}-3,6-дигидропиридин-1(2H)-карбоксилат.

Бензил 4-оксопиперидин-1-карбоксилат (147 г, 630 ммоль) растворяли в тетрагидрофуране (500 мл), в атмосфере азота. Этот раствор по каплям добавляли в течение 2 часов, к перемешиваемому раствору гексаметилдисилазида лития (20% раствор в тетрагидрофуране, 556 мл, 662 ммоль) в атмосфере азота, поддерживая температуру реакции ниже -70°С. Реакционной смеси позволяли перемешиваться при -75°С дополнительно в течение 1 часа, затем по каплям снова добавляли в течение 2 часов, раствор N-фенил-бис(трифторметансульфонимида) (236 г, 661 ммоль) в тетрагидрофуране (950 мл), поддерживая температуру реакции ниже -70°С. После этого реакции позволяли нагреться до комнатной температуры в течение ночи, затем порциями добавляли 2М водный раствор гидроксида натрия (800 мл). Слои разделяли и органический слой промывали дополнительно 2М водным раствором гидроксида натрия (600 мл), затем упаривали насухо. Полученное твердое вещество растворяли в простом диэтиловом эфире и промывали водой. Затем органический слой фильтровали через целит, высушивали над сульфатом натрия и упаривали насухо, получая указанное в заглавии соединение (140 г, 61%), которое использовали на следующей стадии без дополнительной очистки.
Метод 4
трет-Бутил {2-[(4-пиперидин-4-илбензоил)амино]фенил}карбамат (альтернативный метод)

К трет-бутил {2-[4-пирид-4-илбензоил)амино]фенил}карбамату (20 г, 51,35 ммоль; полученному, как описано в методе 5А или 5В ниже), 10% палладия на угле (3,17 г) и лимонной кислоте (4,75 г, 24,65 ммоль) добавляли воду (80 мл) и IPA (80 мл). Реакционную смесь помещали в газообразный водород под давлением 4 бар и нагревали до 70°С в течение 5 часов. Реакционную смесь охлаждали до 50°С и фильтровали через слой целита. Смесь нагревали до 70°С, затем добавляли 20% мас./мас. водный раствор гидроксида натрия (15 мл) в течение 10 минут до рН 10-11. Дополнительно добавляли воду (30 мл), затем смесь охлаждали до 40°С в течение 1 часа, после этого повторно нагревали до 60°С в течение 30 минут, затем снова охлаждали до температуры окружающей среды. Полученный осадок собирали путем фильтрации, промывали водой (2×20 мл) и высушивали в вакууме при 50°С, получая указанное в заглавии соединение (17,6 г, 84%);
ЯМР-спектр: (ДМСО-d6) δ 1,45 (s, 9Н), 1,53 (m, 2H), 1,70 (m, 2H), 2,58 (m, 1H), 2,66 (m, 2H), 3,03 (m, 2H), 3,31 (br s, 1H), 7,17 (m, 2H), 7,35 (d, 2H), 7,54 (m, 2H), 7,89 (d, 2H), 8,65 (br s, 1H), 9,75 (br s, 1H). Масс-спектр: M+H+ 396.
Метод 5А
трет-Бутил {2-[4-пирид-4-илбензоил)амино]фенил}карбамат
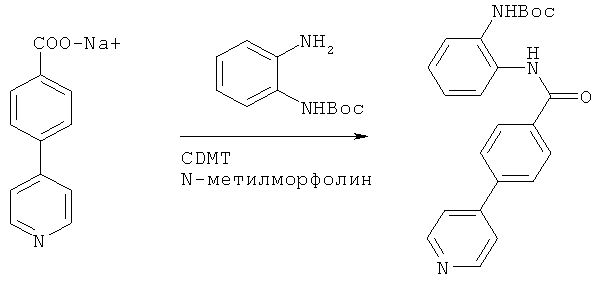
К 4-(4-пиридил)бензоату натрия (55,2 г, 236,2 ммоль), boc о-фенилен диамину (45,7 г.217,6 ммоль) и N-метилморфолину (24 мл) в ацетонитриле (300 мл) добавляли экранированный раствор 2-хлор-4,6-диметокси-1,3,5-триазина (48,5 г, 270,5 ммоль) в ацетонитриле (152 мл) в течение 3 часов. Смесь перемешивали в течение 22 часов, затем добавляли воду (460 мл). Полученный осадок собирали путем фильтрации, промывали 50% водным ацетонитрилом (3×100 мл) и высушивали в вакууме при 50°С, получая указанное в заглавии соединение (75,6 г, 90%);
ЯМР-спектр: (ДМСО-d6): δ 1,45 (s, 9Н), 7,17 (m, 2Н), 7,56 (m, 2H), 7,81 (d, 2H), 7,99 (d, 2H), 8,11 (d, 2H), 8,69 (d, 2H), 9,94 (br s, 1H). Масс-спектр: М+Н+ 390.
Метод 5В
трет-Бутил {2-[4-пирид-4-илбензоил)амино]фенил}карбамат
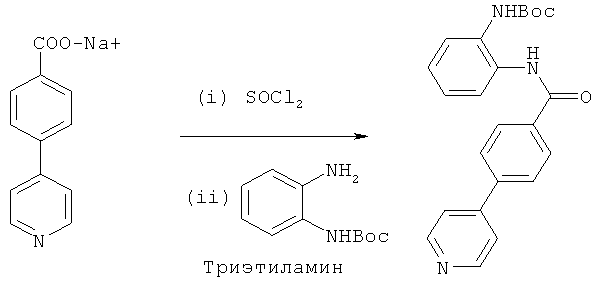
4-(4-Пиридил)бензоат натрия (10 г, 45,2 ммоль) в ацетонитриле (60 мл) нагревали до 70°С, затем добавляли тионилхлорид (6,6 мл, 90,4 ммоль). Реакцию нагревали при температуре флегмы в течение 5 часов, затем охлаждали до температуры окружающей среды. Осторожно добавляли триэтиламин (12,6 мл, 90,4 ммоль), затем нагретый раствор Boc о-фенилен диамина (9,42 г, 45,2 ммоль) в ацетонитриле (15 мл) в течение 10 минут. Добавляли раствор гидроксида натрия (8,6 г, 109 ммоль) в воде (60 мл) и полученное твердое вещество собирали путем фильтрации, промывали водой (20 мл) и высушивали в вакууме при 50°С, получая указанное в заглавии соединение (12,6 г, 68%). ЯМР-спектр: (ДМСО-d6): δ 1,45 (s, 9Н), (7,17 (m, 2H), 7,56 (m, 2H), 7,81 (d, 2H), 7,99 (d, 2H), 8,11 (d, 2H) 8,69 (d, 2H), 9,94 (br s, 1H).
Масс-спектр: M+H+ 390.
Метод 6А
трет-Бутил (2-{[4-(1-{1,3-диметил-1H-пиразол-4-илметил}пиперидин-4-ил)бензоил]амино}фенил)карбамат
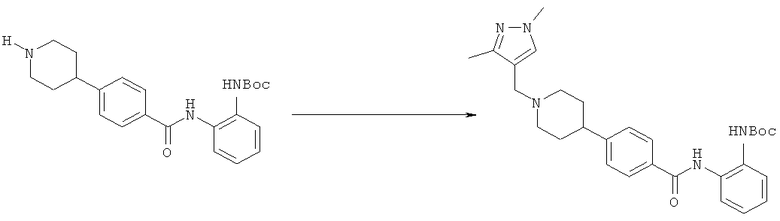
трет-Бутил 2-[(4-пиперидин-4-илбензоил)амино]фенилкарбамат (6,83 г, 17,3 ммоль) и 1,3-диметил-1Н-пиразол-4-карбальдегид (3,0 г, 24,2 ммоль) растворяли в дихлорметане (150 мл). Затем добавляли уксусную кислоту (996 мкл, 17,3 ммоль) и реакционной смеси позволяли перемешиваться при комнатной температуре в течение четырех часов. После этого добавляли триацетоксиборогидрид натрия (5,49 г, 25,9 ммоль) и реакционную смесь перемешивали дополнительно в течение 18 часов. Затем осторожно добавляли насыщенный водный раствор бикарбоната натрия (300 мл), после этого дихлорметан (100 мл). Органический слой отделяли и водный слой повторно экстрагировали дополнительным количеством дихлорметана (150 мл). Объединенные органические экстракты высушивали над сульфатом магния, фильтровали и упаривали насухо. Полученный остаток очищали с помощью флэш-хроматографии на диоксиде кремния, элюируя 5% (об./об.) раствором метанола в дихлорметане, затем повышающимся градиентом 5-10% (об./об.) метанола в дихлорметане, получая прозрачную смолу, которую ресуспендировали в простом диэтиловом эфире и упаривали насухо, получая указанное в заглавии соединение (7,61 г, 87%); ЯМР-спектр: (ДМСО d6) δ 1,43 (s, 9Н), 1,69 (m, 4Н), 1,98 (m, 2H), 2,10 (s, 3Н), 2,56 (m, 1H), 2,92 (m, 2H), 3,26 (s, 2H), 3,70 (s, 3Н), 7,15 (m, 2H), 7,40 (m, 3Н), 7,52 (m, 2H), 7,87 (d, 2H), 8,60 (s, 1H), 9,73 (s, 1H); Масс-спектр: M+H+ 504.
Метод 6В
трет-Бутил (2-{[4-(1-{1,3-диметил-1H-пиразол-4-илметил}пиперидин-4-ил)бензоил]амино}фенил)карбамат
трет-Бутил 2-[(4-пиперидин-4-илбензоил)амино]фенилкарбамат (полученный, как описано в методе 4 выше;108,1 г, 273,3 ммоль), 1,3-диметил-1Н-пиразол-4-карбальдегид (35,6 г, 287 ммоль) и палладия на угле (3,09 г, 1,37 ммоль) загружали в подходящий сосуд высокого давления. Загружали тетрагидрофуран (920 мл), воду (54 мл) и уксусную кислоту (32,8 г, 546,7 ммоль) и перемешиваемую смесь нагревали до 60°С под давлением водорода 3 бара до предположительного завершения реакции. Затем смесь охлаждали до 40°С и добавляли 2 М раствор гидроксида натрия (410 мл, 820 ммоль). После охлаждения до 25°С, смесь фильтровали для удаления катализатора, затем добавляли тетрагидрофуран (650 мл) и органическую фазу отделяли. Органическую фазу частично концентрировали путем перегонки, после этого добавляли толуол (575 мл). Продолжали осуществлять перегонку, в то время как объем реакции поддерживали путем дальнейшего добавления толуола (690 мл). Реакционной смеси позволяли охладиться до температуры окружающей среды приблизительно в течение 3 часов, при этом продукт кристаллизовался. Твердое вещество собирали путем фильтрации, промывали толуолом (460 мл), затем этилацетатом (230 мл), после этого высушивали в вакууме, при 45°С до постоянного веса, получая указанное в заглавии соединение (114,9 г, 83%). Спектр: (ДМСО d6), δ 1,434 (s, 9Н), 1,70 (m, 4Н), 1,98 (m, 2H), 2,11 (s, 3H), 2,56 (m, 1H), 2,93 (m, 2H), 3,28 (s, 2H), 3,71 (s, 3H), 7,16 (m, 2H), 7,40 (m, 3H), 7,52 (m, 2H), 7,87 (d, 2H), 8,60 (s, 1H), 9,73 (s, 1H); Масс-спектр: M+H+ 504.
Метод 7
трет-Бутил {2-[(4-{1-[(1,5-диметил-1H-пиразол-4-ил)метил]пиперидин-4-ил}бензоил)амино]фенил}карбамат
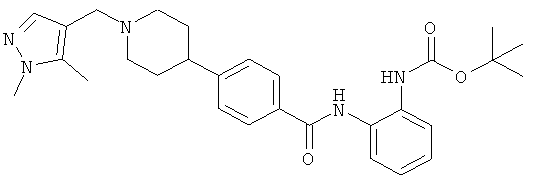
1,5-Диметил-1Н-пиразол-4-карбальдегид (114 мг, 0,92 ммоль) добавляли к раствору трет-бутил 2-[(4-пиперидин-4-илбензоил)амино]фенилкарбамата (289 мг, 0,73 ммоль) в дихлорметане (6 мл), затем добавляли уксусную кислоту (50 мкл, 0,87 ммоль). Реакционной смеси позволяли перемешиваться в атмосфере азота в течение 2,5 часов. Добавляли триацетоксиборогидрид натрия (233 мг, 1,10 ммоль), и реакционной смеси позволяли перемешиваться при температуре окружающей среды в течение 18 часов (в течение ночи). После этого к реакции добавляли насыщенный водный раствор бикарбоната натрия (10 мл) и позволяли перемешиваться в течение 15 минут. Органическую фазу отделяли и водную фазу повторно экстрагировали дихлорметаном (10 мл). Объединенные органические вещества промывали водой, высушивали над сульфатом магния и упаривали насухо, получая продукт в виде бесцветной смолы (308 мг, 84%), который использовали без дополнительной очистки. Масс-спектр: М+Н+ 504.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ ГИСТОНДЕАЦЕТИЛАЗЫ | 2014 |
|
RU2673819C2 |
| ТРИАЗОЛОПИРИДИНОВЫЕ СОЕДИНЕНИЯ-ИНГИБИТОРЫ JAK И СПОСОБЫ | 2009 |
|
RU2561104C2 |
| БЕНЗОИЛАМИНОГЕТЕРОЦИКЛИЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ АКТИВАТОРОВ ГЛЮКОКИНАЗЫ (GLK) | 2007 |
|
RU2440992C2 |
| ИНГИБИТОРЫ ГИСТОНДЕАЦЕТИЛАЗЫ | 2016 |
|
RU2720678C2 |
| ИНГИБИТОРЫ ГИСТОНДЕЗАЦЕТИЛАЗЫ | 2008 |
|
RU2453536C2 |
| АМИНОПИРИМИДИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2006 |
|
RU2427578C2 |
| ПЕРВИЧНЫЕ КАРБОКСАМИДЫ В КАЧЕСТВЕ ИНГИБИТОРОВ BТK | 2014 |
|
RU2708395C2 |
| ТРИАЗОЛПИРИДИНОВЫЕ СОЕДИНЕНИЯ, ИНГИБИРУЮЩИЕ JAK, И СПОСОБЫ | 2009 |
|
RU2560153C2 |
| ПИРИДИЛПИПЕРИДИНОВЫЕ АНТАГОНИСТЫ РЕЦЕПТОРА ОРЕКСИНОВ | 2008 |
|
RU2470021C2 |
| ПРОИЗВОДНЫЕ 3-ЗАМЕЩЕННОГО 1,5-ДИФЕНИЛПИРАЗОЛА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ СВ1 МОДУЛЯТОРОВ | 2005 |
|
RU2375349C2 |
Изобретение относится к бензамидным соединениям формулы (IA), где R2, R3 и R5 каждый независимо выбран из водорода или метила, или их фармацевтически приемлемым солям. Соединения формулы (IA) являются ингибитором деацетилазы гистонов. Изобретение также относится к способу получения соединений формулы (IA), фармацевтической композиции, которая их содержит, и их применению для приготовления лекарственного средства для применения в качестве антипролиферативного средства для предотвращения или лечения опухолей или других пролиферативных состояний, которые чувствительны к ингибированию деацетилазы гистонов (HDAC). 7 н. и 10 з.п. ф-лы, 3 табл., 11 пр.
1. Соединение формулы (1А)
где R2, R3 и R5 каждый независимо выбран из водорода или метила;
или его фармацевтически приемлемая соль.
2. Соединение по п.1, где по меньшей мере одна группа, выбранная из R2, R3 и R5, отличается от водорода.
3. Соединение по п.1, которое представляет собой N-(2-аминофенил)-4-{1-[(1-метил-1Н-пиразол-4-ил)метил]пиперидин-4-ил}бензамид или его фармацевтически приемлемая соль.
4. Соединение по п.1, которое представляет собой N-(2-аминофенил)-4-{1-[(1,3-диметил-1Н-пиразол-4-ил)метил]пиперидин-4-ил}бензамид или его фармацевтически приемлемая соль.
5. Соединение по п.1, которое представляет собой N-(2-аминофенил)-4-{1-[(1,3,5-триметил-1Н-пиразол-4-ил)метил]пиперидин-4-ил}бензамид или его фармацевтически приемлемая соль.
6. Соединение по п.1, которое представляет собой N-(2-аминофенил)-4-{1-[(1,5-диметил- 1Н-пиразол-4-ил)метил]пиперидин-4-ил}бензамид или его фармацевтически приемлемая соль.
7. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении гистондеацетилазы (HDAC), которая содержит соединение по любому из пп.1-6 или его фармацевтически приемлемую соль, в сочетании с фармацевтически приемлемым разбавителем или носителем.
8. Соединение по любому из пп.1-6 или его фармацевтически приемлемая соль, для применения в качестве лекарственного средства, обладающего ингибирующей активностью в отношении гистондеацетилазы (HDAC).
9. Соединение по любому из пп.1-6 или его фармацевтически приемлемая соль для применения для лечения злокачественного новообразования.
10. Соединение по любому из пп.1-6 или его фармацевтически приемлемая соль для применения для лечения рака легких, рака прямой кишки, рака молочной железы, рака предстательной железы, лимфомы и/или лейкоза.
11. Соединение по любому из пп.1-6 или его фармацевтически приемлемая соль для применения для получения ингибирующего действия по отношению к HDAC у теплокровного животного, такого как человек.
12. Способ получения ингибирующего действия по отношению к HDAC у теплокровного животного, такого как человек, нуждающегося в таком лечении, который включает введение указанному животному соединения по любому из пп.1-6 в эффективном количестве.
13. Применение соединения по любому из пп.1-6 или его фармацевтически приемлемой соли для приготовления лекарственного средства для применения для получения действия, ингибирующего клеточный цикл (действия, подавляющего пролиферацию клеток) у теплокровного животного, такого как человек.
14. Способ ингибирования клеточного цикла (подавления пролиферации клеток) у теплокровного животного, такого как человек, нуждающегося в таком лечении, который включает введение указанному животному соединения по любому из пп.1-6 или его фармацевтически приемлемой соли в эффективном количестве.
15. Способ лечения злокачественного новообразования у теплокровного животного, такого как человек, нуждающегося в таком лечении, который включает введение указанному животному соединения по любому из пп.1-6 или его фармацевтически приемлемой соли в эффективном количестве.
16. Применение соединения по любому из пп.1-6 или его фармацевтически приемлемой соли, как определено выше, для приготовления лекарственного средства для применения для лечения злокачественного новообразования.
17. Способ получения соединения по п.1 или его фармацевтически приемлемой соли, которой включает:
взаимодействие соединения формулы (II)
в котором анилиновая часть может быть подходяще защищена;
с соединением формулы (III)
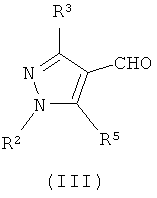
в присутствии восстановителя, где R2, R3 и R5 имеют значения, указанные в п.1;
и затем, при необходимости, удаление любых оставшихся защитных групп, которые могут присутствовать.
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| RU 2004135066 A, 17.04.2003. | |||

