Родственные заявки
Настоящая заявка испрашивает приоритет и преимущества согласно дате подачи предварительной заявки на патент США 61/839729, поданной 26 июня 2013 года, и предварительной заявки на патент США 61/897577, поданной 30 октября 2013 года, содержание каждой включено в настоящее описание в качестве ссылки в полном объеме.
Предпосылки создания изобретения
Протеинкиназы представляют собой многочисленное семейство белков, которые играют центральную роль в регуляции широкого круга клеточных процессов и в поддержании клеточной функции. Неполный перечень таких киназ включает, без ограничения: нерецепторные тирозинкиназы, такие как семейство Tec (BTK, ITK, Tec, ETK/BMX и RLK/TXK), семейство киназы Janus (Jak1, Jak2, Jak3 и Tyk2); гибридные киназы, такие как BCR-Abl, киназа фокальной адгезии (FAK), Fes, Lck и Syk; рецепторные тирозинкиназы, такие как рецептор эпидермального фактора роста (EGFR), киназа рецептора фактора роста, полученного из тромбоцитов (PDGF-R), рецепторные киназы для фактора стволовых клеток, c-kit, рецептора фактора роста гепатоцитов, c-Met, и рецептора фактора роста фибробластов FGFR3; и серин/треонин киназы, такие как b-RAF, митоген-активированные протеинкиназы (например, MKK6) и SAPK2β. Абберантная киназная активность наблюдалась при многих болезненных состояниях, включая доброкачественные и злокачественные пролиферативные нарушения, также как заболевания, являющиеся результатом неадекватной активации иммунной и нервной систем. Новые соединения настоящего изобретения ингибируют активность одной или более из протеинкиназ, и поэтому ожидается, что они могут быть полезны при лечении опосредованных киназами заболеваний.
Тирозинкиназа Брутона (BTK) представляет собой нерецепторную тирозинкиназу с ключевой ролью в передачи сигнала иммунорецептора (BCR, FcεR, FcγR, DAP12, дектин-1, GPVI и т.д.) в множестве гематопоэтических клеток, включая B-клетки, тромбоциты, тучные клетки, базофилы, эозинофилы, макрофаги и нейтрофилы, а также остеокласты, участвующие в деструкции костной ткани (для обзора, см. Brunner et al., 2005 Histol. Histopathol., 20:945, Mohamed et al., 2009 Immunol. Rev., 228:58). Мутации в BTK, как известно, приводят к X-сцепленной агаммаглобулинемии (XLA) у людей и к X-сцепленному иммунодефициту (Xid) у мышей, которые характеризуются ограниченным продуцированием В-клеток и низкими титрами антитела (Lindvall et al., 2005 Immunol. Rev., 203:200). Комбинированное действие Btk в нескольких типах клеток делает ее перспективной мишенью при аутоиммунном заболевании. BTK связана с последовательностью гомологии другими киназами семейства Tec (ITK, Tec, ETK/BMX & RLK/TXK).
В B-лимфоцитах, BTK требуется для развития В-клеток и для активации Ca2+ вследствие вовлечения B-клеточного рецептора (BCR) (Khan et al., 1995 Immunity 3:283; Genevier et al., 1997 Clin. Exp. Immun., 110:286), где она находится ниже семейства киназ Src (например, Lyn), Syk и PI3K. BTK, как было показано, является важной для тимус-зависимого и тимус-независимого ответов типа 2 на антигены (Khan et al., Immunity 1995; 3; 283). В тучных клетках, исследования с использованием нокаутированной мыши по BTK (Hata et al., 1998 J. Exp. Med., 187:1235; Schmidt et al., 2009 Eur. J. Immun., 39:3228) указывают на роль BTK в индуцировании передачи сигнала FcεRI, высвобождении гистамина и продукции цитокинов, таких как TNF, IL-2 и IL-4. В тромбоцитах, BTK имеет важное значение для передачи сигналов через рецептор гликопротеина VI (GPVI), который отвечает на коллаген и, как было показано способствовует агрегации тромбоцитов и способствует продукции цитокинов из фибробластоподобных синовиоцитов (Hsu et al., 2013 Immun. Letters, 150:97). В моноцитах и макрофагах, действие BTK вовлечено в передачу сигнала, индуцированную FcγRI, а также может иметь роль в цитокиновых ответах, индуцированных Toll-подобным рецептором, включая TLR2, TLR4, TLR8 и TLR9 (Horwood et al., 2003 J. Exp. Med., 197:1603; Horwood et al., 2006 J. Immunol., 176:3635; Perez de Diego et al., 2006 Allerg. Clin. Imm., 117:1462; Doyle et al., 2007 J. Biol. Chem., 282:36959, Hasan et al., 2007 Immunology, 123:239; Sochorava et al., 2007 Blood, 109:2553; Lee et al., 2008, J. Biol. Chem., 283:11189).
Таким образом, ингибирование BTK, как ожидается, имеет место при нескольких важнейших стыках воспалительных реакций, приводящих к эффективному подавлению аутоиммунного ответа. Соотвественно заболевания, связанные с активацией В-клеточного рецептора, взаимодействием антитело-Fc рецептор и передачей сигнала рецептора GPVI, можно модулировать путем обработки ингибиторами BTK. Ингибирование BTK может действовать как на инициирование аутоиммунного заболевания, блокируя передачу сигналов BCR, и эффекторную фазу путем отмены передачи сигнала FcR в макрофаги, нейтрофилы, базофилы и тучные клетки. Кроме того, блокирование BTK будет обеспечивать дополнительное преимущество с помощью ингибирования созревания остеокластов и, следовательно, ослабит разрушение кости и общее разрушение суставов, связанные с ревматоидным артритом. Ингибирование BTK может быть полезно при лечении множества воспалительных и аллергических заболеваний - например (но ими не ограничиваясь), ревматоидный артрит (RA), системная красная волчанка (SLE), рассеянный склероз (MS) и реакции гиперчувствительности I типа, такие как аллергический ринит, аллергический конъюнктивит, атопический дерматит, аллергическая астма и общая анафилактическая реакция. Для обзора направленного действия BTK в качестве лечения воспалительных заболеваний и аутоиммунных заболеваний, а также лейкозов и лимфом, см. Uckun & Qazi, 2010 Expert Opin. Ther. Pat., 20:1457. Так как BTK сильно экспрессируется в раках гематопоэтической системы и BTK-зависимой передачи сигнала полагают приводит к дизрегуляции, ингибиторы BTK могут быть полезны для лечения B-клеточных лимфом/лейкозов и других онкологических заболеваний - например (но ими не ограничиваясь) острого лимфобластного лейкоза (ALL), хронического лимфоцитарного лейкоза (CLL), неходжкинской лимфомы (NHL), мелкоклеточной лимфоцитарной лимфомы (SLL) и острого миелоидного лейкоза (для обзора, см. Buggy & Elias 2012 Int Rev Immunol. 31:119). Вместе взятые, ингибиторы BTK обеспечивают серьезный способ лечения множества воспалительных заболеваний и иммунологических нарушений, а также гематологических злокачественных опухолей.
Сущность изобретения
В первом варианте осуществления настоящее изобретение относится к соединению формулы (I):
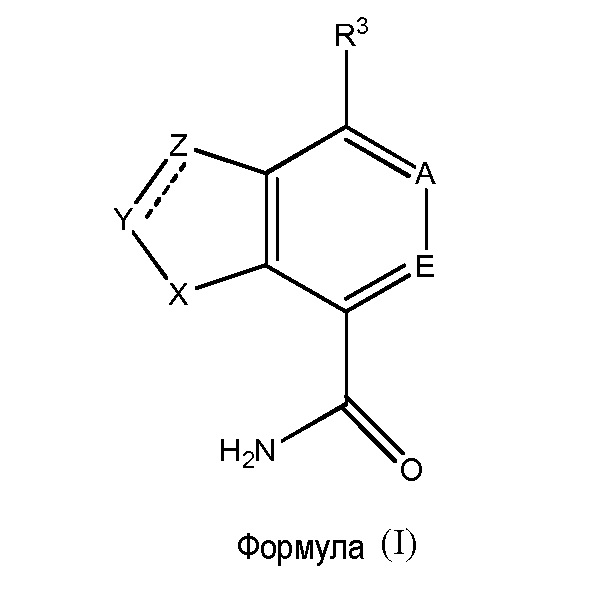
или к его фармацевтически приемлемой соли, пролекарству, биологически активному метаболиту, изомеру или стереоизомеру, где:
X представляет собой NR2 или S;
Y представляет собой N или CR1, и Z представляет собой N или CR1; или, Y представляет собой CR1R2 и Z представляет собой CR1R2;
A представляет собой N или CR4;
E представляет собой N или CR5;
R1 независимо представляет собой Н, дейтерий, CN, галоген, CF3, -NRcRc, -N(Ra)C(O)Rb, необязательно замещенный (C1-C6)алкил, необязательно замещенный (C2-C6)алкенил, необязательно замещенный арил, необязательно замещенный (C3-C6)циклоалкил, необязательно замещенный (C3-C6)циклоалкенил, необязательно замещенный гетероарил или необязательно замещенный насыщенный или частично насыщенный гетероциклил;
R2 независимо представляет собой Н, дейтерий, или необязательно замещенный (C1-C3)алкил;
R3 представляет собой галоген, -N(Ra)2, необязательно замещенный арил, необязательно замещенный (C3-C7)циклоалкил, необязательно замещенный насыщенный или частично насыщенный гетероциклил или необязательно замещенный гетероарил; или
R3 представляет собой -R301-L-R302, где
R301 представляет собой связь, -O-, -OCH2-, -NRd-, или необязательно замещенный (C1-C3)алкилен, и
L представляет собой необязательно замещенный фенил, необязательно замещенный (C3-C6)циклоалкил, необязательно замещенный гетероарил или насыщенный или частично насыщенный гетероциклил, содержащий один или несколько гетероатомов, по меньшей мере, один из которых представляет собой азот; или
L представляет собой -L1-L2, где L1 прикреплен к R301 и
L1 представляет собой необязательно замещенный фенил, необязательно замещенный гетероарил или необязательно замещенный насыщенный или частично насыщенный карбоцикл или насыщенный или частично насыщенный гетероциклил; и
L2 представляет собой связь, CH2, NRd, CH2N(H), S(O)2N(H), или -O-;
R302 представляет собой CN, -CH2CN, необязательно замещенный -C(=O)R302a, -(CH2)n-необязательно замещенный насыщенный или частично насыщенный гетероциклил или необязательно замещенный -S(O)2(C2)алкенил;
где R302a представляет собой необязательно замещенный (C1-C4)алкил, необязательно замещенный (C2-C4)алкенил, (C2-C4)алкинил, -C(O)-(C1-C4)алкил, необязательно замещенный насыщенный или частично ненасыщенный (C3-C6)циклоалкил, необязательно замещенный арил, необязательно замещенный гетероарил, -N(H)- необязательно замещенный гетероарил или -(CH2)n-необязательно замещенный ненасыщенный или частично насыщенный гетероциклил;
R4 представляет собой H, дейтерий, CN, необязательно замещенный (C1-C3)алкил, необязательно замещенный (C3-C6) циклоалкил или необязательно замещенный насыщенный или частично насыщенный гетероциклил или необязательно замещенный гетероарил;
где необязательно замещенный насыщенный или частично насыщенный гетероциклил; и необязательно замещенный гетероарил содержит, по меньшей мере, один атом азота; или
R3 и R4, вместе с атомами углерода, к которым они присоединены, образуют необязательно замещенное, насыщенное, ненасыщенное или частично ненасыщенное 5- или 6-членное карбоциклическое кольцо или необязательно замещенное, насыщенное или частично ненасыщенное 5- или 6-членное гетероциклическое кольцо, содержащее один или несколько гетероатомов, выбранных из N, S и O;
R5 представляет собой H, дейтерий, галоген или необязательно замещенный (C1-C3)алкил;
Ra независимо выбран из Н, -C(O)-необязательно замещенный (C2-C6)алкенил, необязательно замещенный (C1-C6)алкил, -(CH2)n-, необязательно замещенный (C3-C6)циклоалкил, -(CH2)n-необязательно замещенный гетероциклил или -(CH2)n-необязательно замещенный гетероарил;
Rb представляет собой H, необязательно замещенный (C1-C6)алкил, необязательно замещенный (C2-C6)алкенил, необязательно замещенный (C2-C6)алкинил, -CH2-O-необязательно замещенный арил, или -CH2-O-необязательно замещенный гетероарил;
Rc независимо представляет собой H, необязательно замещенный (C1-C6)алкил, необязательно замещенный (C3-C6)циклоалкил, необязательно замещенный насыщенный или частично насыщенный гетероциклил, необязательно замещенный арил или необязательно замещенный гетероарил;
Rd представляет собой H, необязательно замещенный гетероциклил, -(CH2)-необязательно замещенный (C3-C6)циклоалкил, -(CH2)-необязательно замещенный гетероарил или необязательно замещенный (C1-C3)алкил;
Rf представляет собой необязательно замещенный (C1-C3)алкил, необязательно замещенный (C2-C4)алкенил или необязательно замещенный (C2-C4)алкинил; и
n независимо равен 0 или 1.
Во втором варианте осуществления изобретение относится к соединению в соответствии с первым вариантом осуществления, в котором Y представляет CR1 и R1 Y представляет собой Н, необязательно замещенный этенил, необязательно замещенный этил, необязательно замещенный метил, необязательно замещенный 2,3-дигидробензофуранил, необязательно замещенный 1,4-диоксанил, необязательно замещенный 3,4-дигидро-2Н-бензо[b][1,4]оксазинил, необязательно замещенный 6,7-дигидро-4Н-пиразоло[5,1-c][1,4]оксазинил, необязательно замещенный хроманил, необязательно замещенный циклогексенил, необязательно замещенный циклопропил, необязательно замещенный тетрагидрофуранил, необязательно замещенный изохроманил, необязательно замещенный 1,2,3,4-тетрагидро-изохинолинил, необязательно замещенный изоксазолил, необязательно замещенный морфолинил, необязательно замещенный оксетанил, необязательно замещенный фенил, необязательно замещенный пиперидинил, необязательно замещенный пиперазинил, необязательно замещенный 3,6-дигидро-2H-пиранил, необязательно замещенный пирано[4,3-b]пиридинил, необязательно замещенный пиразолил, необязательно замещенный пиридинил, необязательно замещенный 3H-пиридин-1-он, необязательно замещенный 1,2,3,6-тетрагидропиридинил, необязательно замещенный пиримидинил, необязательно замещенный пирролидинил, необязательно замещенный 2,5-дигидропирролил, необязательно замещенный тетрагидропиранил или необязательно замещенный тетрагидро-2H-тиопиранил.
В третьем варианте осуществления настоящее изобретение относится к соединению по любому из предшествующих вариантов осуществления, где R1 представляет собой Н или R1 необязательно замещен одним или несколькими заместителями, независимо выбранными из группы, состоящей из CN, OH, =O, галогена, (C1-C4)алкила, (C1-C4)алкокси, -CH2CH2OH, -CH2C(CH3)2OH,-CH2CH(OH)CH2OH, -CH=CH2, -CH2NH2, -CH2N(H)C(O)Re, -C(O)(C1-C4)алкила, -C(O)(C1-C4)алкокси, -C(O)NH2, -C(O)N(CH3)2,-C(O)- необязательно замещенного гетероциклила, -N(H)C(O)CH3, N(CH3)2, -S(O)2(C1-C4)алкила, -S(O)2-пирролидинила, (C1-C4)алкокси, -CH2-морфолинила, -CH2CH2-морфолинила, морфолинила, тетрагидропиранила;
где Re представляет собой (C1-C3)алкил, -CH2Cl, -C≡CH, -C≡CCH3, -CH=CH2, -CH=CHCH3, -C(=CH2)CH3, -CH2CN, -CH2CH2N(CH3)2, -CH2CH2-пиперидинил, -CH2O-необязательно замещенный фенил.
В четвертом варианте осуществления настоящее изобретение относится к соединению по любому из предшествующих вариантов осуществления, где R3 представляет собой -N(H)C(O)CH=CH2, необязательно замещенный изоксазолил, необязательно замещенный фенил, необязательно замещенный пиразолил, необязательно замещенный пиридинил, необязательно замещенный пиримидинил, необязательно замещенный тиазолил или необязательно замещенный тиенил.
В пятом варианте осуществления настоящее изобретение относится к соединению по любому из предшествующих вариантов осуществления по п. 4, где R3 необязательно замещен одним или несколькими заместителями, независимо выбранными из -NH2, -NHCH3, (C1-C4)алкила и -C(O)(C2-C4)алкенила.
В шестом варианте осуществления изобретение относится к соединению по любому из предшествующих вариантов осуществления, где Х представляет собой NR2 и R2 представляет собой H.
В седьмом варианте осуществления изобретение относится к соединению по любому из предшествующих вариантов осуществления, где Y представляет собой CR1 и R1 из Y представляет собой Н, необязательно замещенный фенил, необязательно замещенный пиперазинил, необязательно замещенный пиразолил или необязательно замещенный 1,2,3,6 тетрагидропиридинил.
В восьмом варианте осуществления изобретение относится к соединению по любому из предшествующих вариантов осуществления, где Y представляет собой CR1 и R1 из Y необязательно замещен одним или несколькими заместителями, независимо выбранными из галогена, -(C1-C4)алкила, -C(O)(C1-C4)алкила, и -S(O)2(C1-C4)алкила.
В девятом варианте осуществления изобретение относится к соединению по любому из предшествующих вариантов осуществления, где
Z представляет собой N или Z представляет собой CR1 и R1 из Z представляет собой H; и
A представляет собой CR4 и R4 представляет собой H или азетидинил, замещенный -C(O)CH=CH2.
В десятом варианте осуществления изобретение относится к соединению по любому из предшествующих вариантов осуществления, где соединение представляет собой
4-(3-амино-2-метилфенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
2-(4-фторфенил)-4-(пиридин-3-ил)-1H-индол-7-карбоксамид;
4-(пиридин-3-ил)-2-п-толил-1H-индол-7-карбоксамид;
2-(4-фторфенил)-4-(пиридин-4-ил)-1H-индол-7-карбоксамид;
2-(4-фторфенил)-4-(1H-пиразол-5-ил)-1H-индол-7-карбоксамид;
4-(3,5-диметилизоксазол-4-ил)-2-п-толил-1H-индол-7-карбоксамид;
2-(1-ацетилпиперидин-4-ил)-4-(3-амино-2-метилфенил)-1H-индол-7-карбоксамид;
4-(пиридин-4-ил)-2-п-толил-1H-индол-7-карбоксамид;
4-(тиофен-2-ил)-2-п-толил-1H-индол-7-карбоксамид;
4-(2-аминофенил)-1H-индол-7-карбоксамид;
4-(3-амино-2-метилфенил)-1H-индол-7-карбоксамид;
4-(5-аминопиридин-3-ил)-1H-индол-7-карбоксамид;
4-(2-аминопиридин-4-ил)-1H-индол-7-карбоксамид;
4-(2-аминоэтиламино)-2-(4-фторфенил)-1H-индол-7-карбоксамид;
4-(2-аминоэтиламино)-2-п-толил-1H-индол-7-карбоксамид;
4-(пиримидин-5-ил)-2-п-толил-1H-индол-7-карбоксамид;
4-(1H-пиразол-4-ил)-2-п-толил-1H-индол-7-карбоксамид;
4-(1H-пиразол-5-ил)-2-п-толил-1H-индол-7-карбоксамид;
2-(4-фторфенил)-4-(пиримидин-5-ил)-1H-индол-7-карбоксамид;
4-(тиазол-2-ил)-2-п-толил-1H-индол-7-карбоксамид;
4-(пиридин-2-ил)-2-п-толил-1H-индол-7-карбоксамид;
4-(тиофен-3-ил)-2-п-толил-1H-индол-7-карбоксамид;
4-(1-метил-1H-пиразол-4-ил)-2-п-толил-1H-индол-7-карбоксамид;
4-(1H-пиразол-3-ил)-2-п-толил-1H-индол-7-карбоксамид;
4-(2-аминофенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-4-фенил-1H-индол-7-карбоксамид;
4-(3-амино-2-метилфенил)-2-(4,4-дифторциклогекс-1-енил)1H-индол-7-карбоксамид;
4-(3-амино-2-метилфенил)-1H-пирроло[2,3-c]пиридин-7-карбоксамид;
4-(1-акрилоилпиперидин-3-ил)-1H-индол-7-карбоксамид;
4-(1-акрилоилпиперидин-3-ил)-2-(1-метил-1H-пиразол-4-ил)-1H-индол-7-карбоксамид;
4-(2-аминоэтиламино)-2-п-толил-1H-индол-7-карбоксамид;
4-((1R,2R)-2-аминоциклогексиламино)-2-(4-фторфенил)-1H-индол-7-карбоксамид*;
4-(1-метил-1H-пиразол-5-иламино)-2-п-толил-1H-индол-7-карбоксамид;
4-йод-2-(пиридин-3-ил)-1H-индол-7-карбоксамид;
4-(3-амино-2-метилфенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
4-(3,5-диметилизоксазол-4-ил)-2-(4-фторфенил)-1H-индол-7-карбоксамид;
4-(2-аминофенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид; или
2-(1-ацетилпиперидин-4-ил)-4-(3-амино-2-метилфенил)-1H-индол-7-карбоксамид.
В одиннадцатом варианте осуществления изобретение относится к соединению по любому из с первого по третий вариантами осуществления, где R3 представляет собой -R301-L-R302, и R301 представляет собой связь, N(H), N(CH3), CH2, C(H)(необязательно замещенный (C1-C3)алкил), O, или OCH2.
В двенадцатом варианте осуществления изобретение относится к соединению в соответствии с любым из с первого по третий или одиннадцатым вариантами осуществления, где
L представляет собой необязательно замещенный азетидинил, необязательно замещенный циклопентил, необязательно замещенный 3,6-диазабицикло[3.2.0]гептанил, необязательно замещенный 1,4-диоксанил, необязательно замещенный морфолинил, необязательно замещенный [1,4]оксепанил, необязательно замещенный фенил, необязательно замещенный пиперидинил или необязательно замещенный пирролидинил; или
L представляет собой L1-L2, где
L1 представляет собой необязательно замещенный циклогексил, необязательно замещенный циклопентил, необязательно замещенный фенил, необязательно замещенный пиперидинил, необязательно замещенный пиридинил;
L2 представляет собой N(H), N(CH3), N(CH2CH2OH), N(CH2CH(CH3)2), N(оксетанил), N(CH2-циклопентил), N(CH2-тиазолил), O, S(O)2N(H) или CH2N(H).
В тринадцатом варианте осуществления изобретение относится к соединению по любому из с первого по третий, и одиннадцатого и двенадцатого варианты осуществления, где L или L1 необязательно замещен одним или несколькими заместителями, независимо выбранными из галогена, CN, ОН, (C1-C4)алкокси, (C1-C4)алкила, -CH2OH, -N(H)CH2-гетероарила, бензилокси и -OCH2-гетероарила.
В четырнадцатом варианте осуществления изобретение относится к соединению по любому из с первого по третий и с одиннадцатого по тринадцатый варианты осуществления, где R302 представляет собой -C(O)CH3, -C(O)C(O)CH3, -C(O)CF2(Cl), -CH(CH3)2, -CH2Cl, -CH2CN, -C(O)CH2CN, -C(O)CH2CH3, -C(O)CH2F, -C(O)CH(CH3)2, -C(O)-CH2CH(CH3)2, -C(O)CH(CH3)(Cl), -C(O)CH2CH(CH3)CH3, -C(O)CH(Cl)CH2CH3, -CH2CH2OH, -C(O)CH2CH2N(CH3)2, -C(O)CH=CH2, -C(O)C≡CH, -C(O)CH=CHCl, -C(O)CH=CHCH3, -C(O)C (=CH2)CH3, -C(O)C(CH2CH3)=CH2, -C(O)CH=CHCH(CH3)2, -C(O)CH=CHC(O)OH, -C(O)CH=CHC(O)N(H)CH2CH3, -C(O)CH=CHCH2N(CH3)2, -C(O)CH=CHC(O)OCH3, -C(O)CH=CHC(O)OCH2CH3, -C(O)CH=CHC(O)N(H)CH3, -C(O)CH=CHC(O)CH2CH2OCH3, -C(O)CH=CHC(O)N(CH3)2, -C(O)CH=CHC(O)N(H)CH2CH3, -C(O)CH=CHC(O)N(H)CH2CH2OCH3, -C(O)CH=CHCH2N(H)CH2CH2OCH3, -C(O)C(CN)=C(OH)(CH3), -C(O)CH=CH-необязательно замещенный пиразолил-C(O)CH=CHCH2N(H)-необязательно замещенный циклопропил, -C(O)CH=CHCH2N(H)CH2-необязательно замещенный тетрагидрофуранил, -C(O)CH=CHC(O)NH2,-C(O)CH=CHC(O)N(H)- необязательно замещенный циклопропил, -C(O)C(CH3)=CHCH3, -C(O)C(CH3)=CHCH2CH3, -C(O)C(=CH2)CH2N(CH3)2, -C(O)C(=CH2)CH2NH2, -C(O)C(=CH2)CH2N(H)(CH3), -C(O)C(=CH2)CH3, -C(O)C(=CH2)CH2- необязательно замещенный морфолинил, -C(O)C(=CH2)-необязательно замещенный фенил, -CH2- необязательно замещенный бензо[d]изотиазолил, -C(O)-CH2-O-необязательно замещенный фенил, -CH2-необязательно замещенный тиазолил, -CH2CH2-необязательно замещенный морфолинил, -C(O)CH2O-необязательно замещенный фенил, -C(O)CH2CH2-необязательно замещенный пиперазинил, -C(O)CH2CH2- необязательно замещенный пиперидинил, -C(O)CH2O-необязательно замещенный пиридинил, -C(O)CH2CH2 необязательно замещенный пирролидинил,-C(O)CH=CH необязательно замещенный циклопропил,-C(O)CH=CHCH2- необязательно замещенный морфолинил, -C(O)CH=CHCH2- необязательно замещенный пиперидинил, -C(O)CH=CH- необязательно замещенный пиразолил,-C(O)CH=CH-необязательно замещенный пиридинил, -C(O)CH=CH-необязательно замещенный тиазолил, -C(O)-необязательно замещенный циклогексенил, -C(=O)-необязательно замещенный циклогексил, -C(O)-необязательно замещенный циклопентенил, -C(O)-циклопентил, необязательно замещенный имидазо[1,2-a]пиразинил, необязательно замещенный тетрагидроимидазо[1,2-a]пиразинил, необязательно замещенный дигидроизоиндолил, необязательно замещенный 1,2,3,4-тетрагидро-изохинолинил, необязательно замещенный изохинолинил, -C(O)-необязательно замещенный изоксазолил, -C(O)-необязательно замещенный оксазолил, необязательно замещенный оксетанил,-C(=O)- необязательно замещенный фенил, необязательно замещенный пиперидинил, -C(O)-необязательно замещенный пиперидинил, необязательно замещенный пиразолил, -C(O)CH2O-необязательно замещенный пиридазинил, -C(O)-необязательно замещенный пиридинил, необязательно замещенный пиримидинил, необязательно замещенный хиназолинил, необязательно замещенный дигидрохинолинил, необязательно замещенный -C(O)-тетрагидробензо[b]тиофенил, -C(O)-необязательно замещенный тетрагидропиранил, -C(O)-необязательно замещенный тетрагидропиридинил, -C(O)-тиазолил, -C(O)N(H)-тиазолил, -C(O)NHCH2CN, или -S(O)2CH=CH2.
В пятнадцатом варианте осуществления изобретение относится к соединению по любому из с первого по третий или тринадцатого по четырнадцатый вариантов осуществления, где X представляет собой NR2 и R2 представляет собой H.
В шестнадцатом варианте осуществления изобретение относится к соединению по любому из с первого по третий или с тринадцатого по пятнадцатый варианты осуществления, где Y представляет собой CR1 и R1 из Y необязательно замещен одним или несколькими заместителями независимо выбранными из галогена, CN, =O, (C1-C4)алкила, (C2-C4)алкенила, -CH2NH2, -CH2CH2OH, -CH2CH(OH)CH2CH3, -CH2CH(OH)CH2OH, -CH2CH2OCH2CH3, -CH2C(OH)(CH3)2, -CH2NHC(O)(C1-C4)алкила, -CH2NHC(O)CH2Cl, -CH2NHC(O)CH2CN, -CH2NHC(O)CH2CH2N(CH3)2, -CH2NHC(O)C(=CH2)CH3, -CH2NHC(O)(C2-C4)алкинила, -CH2NHC(O)CH2CH2-пиперидинила, -(C1-C4)алкил-морфолинила, -CH2NHC(O)CH2O-фенила, где фенил необязательно замещен галогеном, (C1-C4)алкокси, -C(O)(C1-C4)алкилом, -C(O)(C1-C4)алкокси, -C(O)N(H)2, -C(O)N(CH3)2, -C(O)-морфолинилом, -C(O)-пирролидинилом, -N(CH3)2, -NHC(O)(C1-C4)алкилом, -NHC(O)(C2-C4)алкенилом, -NHC(O)CH2CN, -S(O)2(C1-C4)алкилом, -S(O)2-пирролидинилом, морфолинилом, тетрагидропиранилом или 4-метилпиперазинкарбонилом.
В семнадцатом варианте осуществления изобретение относится к соединению по любому из с первого по третий или с тринадцатого по шестнадцатый варианты осуществления, где Z представляет собой CR1 и R1 из Z представляет собой H, (C1-C4)алкил, -NHC(O)CH2Cl, -NHC(O)CH2CN, -NHC(O)(C2-C4)алкенил, -NHC(O)(C2-C4)алкинил, -NHC(O)C(=CH2)CH3, -NHC(O)CH2-фенил, где фенил необязательно замещен галогеном или пиразолил, замещенный CH3.
В восемнадцатом варианте осуществления изобретение относится к соединению по любому из с первого по третий или с тринадцатого по семнадцатый варианты осуществления, где R302 необязательно замещен одним или несколькими заместителями, независимо выбранными из галогена, CF3, OCF3, =O, CHF2, CN, C(O)OH, OH, (C1-C4)алкила, (C1-C4)алкокси, (C3-C6)циклоалкила, -(C1-C4)алкилCN, -(C1-C4)алкилC(O)NH2, -C(O)NH2, -C(O)N(H)(C1-C4)алкила, -C(O)N(C1-C4)алкил)2, -C(O)N(H)циклопропила, -C(O)(C1-C4)алкокси, NH2, N(H)CH3, N(CH3)2 или необязательно замещенного бензила.
В девятнадцатом варианте осуществления изобретение относится к соединению по любому из с первого по третий или с тринадцатого по восемнадцатый вариантах осуществления, где
X представляет собой NR2, где R2 представляет собой H;
Y представляет собой CR1, где R1 представляет собой H, CH3, замещенный пиразолил, 6,7-дигидро-4H-пиразоло[5,1-c][1,4]оксазинил или тетрагидрофуранил;
Z представляет собой CR1, где R1 представляет собой H;
E представляет собой CR5, где R5 представляет собой H;
R3 представляет собой -R301-L-R302, где
R301 представляет собой связь, -O-, -N(H)-, -N(CH3)- или -C(H)(CH3)-;
L представляет собой азетидинил, 3,6-диазабицикло[3.2.0]гептанил, морфолинил, [1,4]оксепанил, пиперидинил или пирролидинил;
где азетидинил необязательно замещен CH3; и
где пиперидинил необязательно замещен -CH2OH; и
R302 представляет собой -C(O)CH=CH2 или -C(O)C≡CH.
В двадцатом варианте осуществления изобретение относится к соединению по любому из с первого по третий или с тринадцатого по девятнадцатый варианты осуществления, где соединение представляет собой:
4-((1-акрилоилазетидин-3-ил)(метил)амино)-1H-индол-7-карбоксамид;
4-(5-ацетилтиофен-2-ил)-2-п-толил-1H-индол-7-карбоксамид;
4-(1-(4-метоксибензил)-1H-пиразол-5-иламино)-2-п-толил-1H-индол-7-карбоксамид;
4-(3-(6-фтор-4-оксохиназолин-3(4H)-ил)-2-метилфенил)-2-(пиридин-3-ил)-1H-индол-7-карбоксамид;
4-(3-(6-фтор-4-оксохиназолин-3(4H)-ил)-2-метилфенил)-2-(пиридин-3-ил)-1H-индол-7-карбоксамид;
4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-2-(пиридин-3-ил)-1H-индол-7-карбоксамид;
4-(2-метил-3-(4,5,6,7-тетрагидробензо[b]тиофен-2-карбоксамидо)фенил)-2-(пиридин-3-ил)-1H-индол-7-карбоксамид;
4-(2-метил-3-(1-оксоизоиндолин-2-ил)фенил)-2-(пиридин-3-ил)-1H-индол-7-карбоксамид;
4-(2-метил-3-(6-метил-1-оксоизоиндолин-2-ил)фенил)-2-(пиридин-3-ил)-1H-индол-7-карбоксамид;
4-(3-(6-фтор-1-оксоизоиндолин-2-ил)-2-метилфенил)-2-(пиридин-3-ил)-1H-индол-7-карбоксамид;
4-(3-(6-фтор-1-оксоизоиндолин-2-ил)-2-метилфенил)-2-(4-фторфенил)-1H-индол-7-карбоксамид;
2-(4-фторфенил)-4-(2-метил-3-(4,5,6,7-тетрагидробензо[b]тиофен-2-карбоксамидо)фенил)-1H-индол-7-карбоксамид;
N-(3-(7-карбамоил-2-(пиридин-3-ил)-1H-индол-4-ил)-4-метилфенил)тиазол-2-карбоксамид 2,2,2-трифторацетат;
N-(3-(7-карбамоил-2-(пиридин-3-ил)-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамид;
(R)-4-(3-(4-оксохиназолин-3(4H)-ил)пиперидин-1-ил)-1H-индол-7-карбоксамид*;
(R)-2-(4-фторфенил)-4-(3-(4-оксохиназолин-3(4H)-ил)пиперидин-1-ил)-1H-индол-7-карбоксамид*;
(R)-4-(3-(4-оксохиназолин-3(4H)-ил)пиперидин-1-ил)-2-(пиридин-3-ил)-1H-индол-7-карбоксамид*;
(R)-2-(1-метил-1H-пиразол-4-ил)-4-(3-(4-оксохиназолин-3(4H)-ил)пиперидин-1-ил)-1H-индол-7-карбоксамид*;
(R)-4-(3-(6-фтор-4-оксохиназолин-3(4H)-ил)пиперидин-1-ил)-2-(4-фторфенил)-1H-индол-7-карбоксамид*;
2-(1-метил-1H-пиразол-4-ил)-4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-1H-индол-7-карбоксамид;
4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-1H-индол-7-карбоксамид;
2-(1-ацетил-1,2,3,6-тетрагидропиридин-4-ил)-4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-1H-индол-7-карбоксамид;
(R)-4-(3-(4-трет-бутилбензамидо)пиперидин-1-ил)-2-(пиридин-3-ил)-1H-индол-7-карбоксамид*;
(R)-4-(3-(4-трет-бутилбензамидо)пиперидин-1-ил)-1H-индол-7-карбоксамид*;
(R)-N-(1-(7-карбамоил-1H-индол-4-ил)пиперидин-3-ил)-2-метилоксазол-4-карбоксамид*;
(R)-4-(3-(3-тиазол-2-илуреидо)пиперидин-1-ил)-1H-индол-7-карбоксамид*;
4-(3-(4-трет-бутилбензамидо)-2-метилфенил)-1H-индол-7-карбоксамид;
4-(3-(7-циклопропил-5-фтор-4-оксохиназолин-3(4H)-ил)пиперидин-1-ил)-1H-индол-7-карбоксамид;
(R)-4-(3-(4-трет-бутилбензамидо)пиперидин-1-ил)-2-(1-метил-1H-пиразол-4-ил)-1H-индол-7-карбоксамид*;
(R)-4-(3-(4-метоксибензамидо)пиперидин-1-ил)-2-(1-метил-1H-пиразол-4-ил)-1H-индол-7-карбоксамид*;
(R)-5-трет-бутил-N-(1-(7-карбамоил-1H-индол-4-ил)пиперидин-3-ил)изоксазол-3-карбоксамид*;
(R)-2-(1-метил-1H-пиразол-4-ил)-4-(3-(4-(трифторметил)бензамидо)пиперидин-1-ил)-1H-индол-7-карбоксамид*;
(R)-4-(3-(4-метоксибензамидо)пиперидин-1-ил)-1H-индол-7-карбоксамид*;
(R)-4-(3-(4-(трифторметил)бензамидо)пиперидин-1-ил)-1H-индол-7-карбоксамид*;
(R)-4-(3-(4-(дифторметил)бензамидо)пиперидин-1-ил)-2-(1-метил-1H-пиразол-4-ил)-1H-индол-7-карбоксамид*;
4-(3-(6-фтор-4-оксохиназолин-3(4H)-ил)-2-метилфенил)-2-(1-метил-1H-пиразол-4-ил)-1H-индол-7-карбоксамид;
2-(3,6-дигидро-2H-пиран-4-ил)-4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-1H-индол-7-карбоксамид;
2-(4-фторфенил)-4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-1H-индол-7-карбоксамид;
(R)-4-(3-(4-(1-амино-2-метил-1-оксопропан-2-ил)бензамидо)пиперидин-1-ил)-2-(1-метил-1H-пиразол-4-ил)-1H-индол-7-карбоксамид*;
(R)-2-(1-метил-1H-пиразол-4-ил)-4-(3-(4-(трифторметокси)бензамидо)пиперидин-1-ил)-1H-индол-7-карбоксамид*;
2-(1-(2-гидроксиэтил)-1H-пиразол-4-ил)-4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-1H-индол-7-карбоксамид;
(R)-4-(3-(6-фтор-1-оксоизоиндолин-2-ил)пиперидин-1-ил)-2-(1-метил-1H-пиразол-4-ил)-1H-индол-7-карбоксамид*;
2-(3,6-дигидро-2H-пиран-4-ил)-4-(3-(6-фтор-4-оксохиназолин-3(4H)-ил)-2-метилфенил)-1H-индол-7-карбоксамид;
2-(1-ацетил-1,2,3,6-тетрагидропиридин-4-ил)-4-(3-(6-фтор-4-оксохиназолин-3(4H)-ил)-2-метилфенил)-1H-индол-7-карбоксамид;
N-(3-(7-карбамоил-2-(1-метил-1H-пиразол-4-ил)-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамид;
4-(3-(6-фтор-4-оксохиназолин-3(4H)-ил)-2-(гидроксиметил)фенил)-2-(1-метил-1H-пиразол-4-ил)-1H-индол-7-карбоксамид;
2-(1-метил-1H-пиразол-4-ил)-4-(2-метил-3-(4,5,6,7-тетрагидробензо[b]тиофен-2-карбоксамидо)фенил)-1H-индол-7-карбоксамид;
(R)-4-(3-(4-циклопропилбензамидо)пиперидин-1-ил)-2-(1-метил-1H-пиразол-4-ил)-1H-индол-7-карбоксамид*;
2-(2,5-дигидро-1H-пиррол-3-ил)-4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-1H-индол-7-карбоксамид;
4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-2-(1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
2-(1-((R)-2,3-дигидроксипропил)-1H-пиразол-4-ил)-4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-1H-индол-7-карбоксамид*;
N-(3-(7-карбамоил-2-(1-метил-1H-пиразол-4-ил)-1H-индол-4-ил)-2-(гидроксиметил)фенил)тиазол-2-карбоксамид;
2-(1-ацетил-1,2,3,6-тетрагидропиридин-4-ил)-4-(3-(4-трет-бутилбензамидо)-2-метилфенил)-1H-индол-7-карбоксамид;
N-(3-(2-(1-ацетил-1,2,3,6-тетрагидропиридин-4-ил)-7-карбамоил-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамид;
2-(1-ацетил-1,2,3,6-тетрагидропиридин-4-ил)-4-(2-метил-3-(4,5,6,7-тетрагидробензо[b]тиофен-2-карбоксамидо)фенил)-1H-индол-7-карбоксамид;
2-(1-ацетил-1,2,3,6-тетрагидропиридин-4-ил)-4-(3-(4-циклопропилбензамидо)-2-метилфенил)-1H-индол-7-карбоксамид;
4-(2-метил-3-(1-оксо-3,4-дигидроизохинолин-2(1H)-ил)фенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
2-(1-метил-2,5-дигидро-1H-пиррол-3-ил)-4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-1H-индол-7-карбоксамид;
2-(1-ацетил-2,5-дигидро-1H-пиррол-3-ил)-4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-1H-индол-7-карбоксамид;
этил 3-(7-карбамоил-4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-1H-индол-2-ил)-2,5-дигидро-1H-пиррол-1-карбоксилат;
2-(1-метил-1,2,3,6-тетрагидропиридин-4-ил)-4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-1H-индол-7-карбоксамид;
4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
N-(3-(7-карбамоил-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамид;
N-(3-(7-карбамоил-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамид;
2-(1-((S)-2,3-дигидроксипропил)-1H-пиразол-4-ил)-4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-1H-индол-7-карбоксамид;
N-(3-(7-карбамоил-2-(1-метил-1H-пиразол-4-ил)-1H-индол-4-ил)-2-метилфенил)-N-метилтиазол-2-карбоксамид;
N-(3-(7-карбамоил-2-(1-метил-1H-пиразол-4-ил)-1H-индол-4-ил)-2-метилфенил)-N-(оксетан-3-ил)тиазол-2-карбоксамид;
2-(1-ацетил-1,2,3,6-тетрагидропиридин-4-ил)-4-(3-(4-(2-цианопропан-2-ил)бензамидо)-2-метилфенил)-1H-индол-7-карбоксамид;
4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-2-(пиримидин-5-ил)-1H-индол-7-карбоксамид;
4-(3-(6-фтор-4-оксохиназолин-3(4H)-ил)-2-метилфенил)-2-(пиримидин-5-ил)-1H-индол-7-карбоксамид;
4-(3-(4-(дифторметил)бензамидо)-2-метилфенил)-2-(пиримидин-5-ил)-1H-индол-7-карбоксамид;
4-(3-(4-циклопропилбензамидо)-2-метилфенил)-2-(пиримидин-5-ил)-1H-индол-7-карбоксамид;
4-(3-(6-фтор-4-оксохиназолин-3(4H)-ил)-2-метилфенил)-2-(1-(2-гидрокси-2-метилпропил)-1H-пиразол-4-ил)-1H-индол-7-карбоксамид;
(R)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-4-(3-(8-оксо-5,6-дигидроимидазо[1,2-a]пиразин-7(8H)-ил)пиперидин-1-ил)-1H-индол-7-карбоксамид*;
(R)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-4-(3-(8-oxoимидазо[1,2-a]пиразин-7(8H)-ил)пиперидин-1-ил)-1H-индол-7-карбоксамид*;
4-(2-метил-3-(оксетан-3-иламино)фенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
4-(2-метил-3-(1-оксо-3,4-дигидроизохинолин-2(1H)-ил)фенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
4-(3-(4-(дифторметил)бензамидо)-2-метилфенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
4-(3-(4-гидрокси-4-(трифторметил)циклогексанкарбоксамидо)-2-метилфенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
(R)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-4-(3-(1-оксо-3,4-дигидроизохинолин-2(1H)-ил)пиперидин-1-ил)-1H-индол-7-карбоксамид*;
2-(1-ацетилпиперидин-4-ил)-4-(3-(4-циклопропилбензамидо)-2-метилфенил)-1H-индол-7-карбоксамид;
(R)-N-(1-(7-карбамоил-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-4-ил)пиперидин-3-ил)-2-метилоксазол-4-карбоксамид*;
(R)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-4-(2-оксо-1,3'-бипиперидин-1'-ил)-1H-индол-7-карбоксамид*;
2-(1-метил-1H-пиразол-4-ил)-4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-1H-бензо[d]имидазол-7-карбоксамид;
4-(3-(4-(дифторметил)-N-(оксетан-3-ил)бензамидо)-2-метилфенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
4-(2-метил-3-(оксетан-3-иламино)фенил)-1H-индол-7-карбоксамид;
4-(3-(4-(дифторметил)бензамидо)-2-метилфенил)-1H-индол-7-карбоксамид;
4-(3-(2-гидроксиэтиламино)-2-метилфенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
(R)-N-(1-(7-карбамоил-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-4-ил)пиперидин-3-ил)тиазол-2-карбоксамид*;
4-(3-(циклогексанкарбоксамидо)-2-метилфенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
4-(3-(4-(дифторметил)-N-(2-гидроксиэтил)бензамидо)-2-метилфенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
N-(3-(7-карбамоил-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-4-ил)-2-метилфенил)изотиазол-4-карбоксамид;
4-(2-метил-3-(тетрагидро-2H-пиран-4-карбоксамидо)фенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
4-(2-метил-3-(1-метилпиперидин-3-карбоксамидо)фенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
4-(2-метил-3-(1-метилпиперидин-4-карбоксамидо)фенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
4-(3-(циклопентанкарбоксамидо)-2-метилфенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
N-(3-(7-карбамоил-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-4-ил)-2-метилфенил)-2-метилтиазол-4-карбоксамид;
4-(3-(3-метоксициклогексанкарбоксамидо)-2-метилфенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
4-(2-метил-3-(3-метилбутанамидо)фенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
4-(3-изобутирамидо-2-метилфенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
4-(2-метил-3-(никотинамидо)фенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
N-(3-(7-карбамоил-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-4-ил)-2-метилфенил)-5-метилтиазол-2-карбоксамид;
N-(3-(7-карбамоил-2-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамид;
N-((3R,4R)-1-(7-карбамоил-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-4-ил)-4-гидроксипиперидин-3-ил)тиазол-2-карбоксамид;
(R)-4-(3-акриламидопиперидин-1-ил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид*;
4-(2-метил-3-(тиазол-2-илметиламино)фенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
4-(2-метил-3-(N-(тиазол-2-илметил)акриламидо)фенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
(Z)-4-(2-метил-3-(2-метилбут-2-енамидо)фенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
(E)-4-(3-(4-(диметиламино)бут-2-енамидо)-2-метилфенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
4-(2-метил-3-(3-(пиперидин-1-ил)пропанамидо)фенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
4-(3-(2-цианоацетамидо)-2-метилфенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
4-(2-метил-3-пропионамидофенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
4-(3-метакриламидо-2-метилфенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
4-(3-(2-хлор-2,2-дифторацетамидо)-2-метилфенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
4-(3-(2-хлорпропанамидо)-2-метилфенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
(E)-4-(3-бут-2-енамидо-2-метилфенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
N1-(3-(7-карбамоил-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-4-ил)-2-метилфенил);
4-(3-(2-(4-фторфенокси)ацетамидо)-2-метилфенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
4-(2-метил-3-(3-(пирролидин-1-ил)пропанамидо)фенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
4-(3-(2-(4-цианофенокси)ацетамидо)-2-метилфенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
4-(2-метил-3-(2-(пиридин-3-илокси)ацетамидо)фенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
4-(3-(циклопент-1-енкарбоксамидо)-2-метилфенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
(E)-4-(2-метил-3-(2-метилпент-2-енамидо)фенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
(Z)-4-(3-(3-хлоракриламидо)-2-метилфенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
(E)-метил 4-(3-(7-карбамоил-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-4-ил)-2-метилфениламино)-4-оксобут-2-еноата;
4-(3-(циклогекс-1-енкарбоксамидо)-2-метилфенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
(E)-этил 4-(3-(7-карбамоил-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-4-ил)-2-метилфениламино)-4-оксобут-2-еноата;
4-(2-метил-3-(2-феноксиацетамидо)фенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
4-(3-(2-фторацетамидо)-2-метилфенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-(4,4-дифторциклогекс-1-енил)-1H-индол-7-карбоксамид;
4-(2-(акриламидометил)фенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
4-(3-(3-(диметиламино)пропанамидо)-2-метилфенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
4-(2-акриламидофенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
4-(3-(акриламидометил)фенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
4-(3-(акриламидометил)фенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
4-(3-(2-цианопиримидин-4-иламино)фенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
4-(3-(6-циклопропил-8-фтор-1-oxoизохинолин-2(1H)-ил)-2-(гидроксиметил)фенил)-2-(1-метил-1H-пиразол-4-ил)-1H-индол-7-карбоксамид;
4-(3-акриламидофенил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-метилфенил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-(2-метоксипиридин-3-ил)-1H-индол-7-карбоксамид;
4-(2-метил-3-(2-(пиридин-2-илокси)ацетамидо)фенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
N1-(3-(7-карбамоил-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-4-ил)-2-метилфенил)фумарамид;
4-(3-(2-хлорбутанамидо)-2-метилфенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
4-(2-метил-3-(3-(4-метилпиперазин-1-ил)пропанамидо)фенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
4-(2-метил-3-(2-(пиридазин-3-илокси)ацетамидо)фенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-4-(3-(тиазол-2-илметокси)фенил)-1H-индол-7-карбоксамид;
метил 3-(4-(3-акриламидо-2-метилфенил)-7-карбамоил-1H-индол-2-ил)бензоат;
4-(3-акриламидо-2-метилфенил)-2-(3-метоксифенил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-(4-метоксифенил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-(6-метилпиридин-3-ил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-(3-карбамоилфенил)-1H-индол-7-карбоксамид;
N-(3-(7-карбамоил-3-метил-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-(3,5-диметилизоксазол-4-ил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-(3,5-диметил-1H-пиразол-4-ил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-(1-изопропил-1H-пиразол-4-ил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-(1,3-диметил-1H-пиразол-4-ил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-(1-этил-1H-пиразол-4-ил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-(1-изобутил-1H-пиразол-4-ил)-1H-индол-7-карбоксамид;
(E)-N-(3-(3-бут-2-енамидо-7-карбамоил-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамид;
N-(3-(7-карбамоил-3-метакриламидо-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамид;
N-(3-(3-бут-2-инамидо-7-карбамоил-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамид;
N-(3-(7-карбамоил-3-(2-(4-фторфенокси)ацетамидо)-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-(2-фторпиридин-3-ил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-(1-этил-1H-пиразол-4-ил)-1H-индол-7-карбоксамид;
2-(3-ацетамидофенил)-4-(3-акриламидо-2-метилфенил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-(2-метоксипиридин-4-ил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-(3-цианофенил)-1H-индол-7-карбоксамид;
метил 4-(4-(3-акриламидо-2-метилфенил)-7-карбамоил-1H-индол-2-ил)бензоат;
4-(3-акриламидо-2-метилфенил)-2-(2,3-дигидробензофуран-5-ил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-(3-фторфенил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-(3-(диметиламино)фенил)-1H-индол-7-карбоксамид;
4-(2-(2-хлорацетамидо)фенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
4-(2-ацетамидофенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-(2-метил-5-(пирролидин-1-илсульфонил)фенил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-(2-фторфенил)-1H-индол-7-карбоксамид;
N-(3-(3-акриламидо-7-карбамоил-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамид;
N-(3-(7-карбамоил-3-(2-хлорацетамидо)-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-(1-метил-1H-пиразол-5-ил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-(пиридин-4-ил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-(1-(2-морфолиноэтил)-1H-пиразол-4-ил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-(6-морфолинопиридин-3-ил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-(3-(4-метилпиперазин-1-карбонил)фенил)-1H-индол-7-карбоксамид;
N-(3-(2-(2-(акриламидометил)фенил)-7-карбамоил-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамид;
N-(3-(2-(2-(ацетамидометил)фенил)-7-карбамоил-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамид;
N-(3-(7-карбамоил-2-(2-(пропионамидометил)фенил)-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамид;
N-(3-(2-(2-(бутирамидометил)фенил)-7-карбамоил-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамид;
(E)-N-(3-(2-(2-(бут-2-енамидометил)фенил)-7-карбамоил-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамид;
N-(3-(7-карбамоил-2-(2-(метакриламидометил)фенил)-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамид;
N-(3-(7-карбамоил-2-(2-(пропиоламидометил)фенил)-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамид;
N-(3-(2-(2-(бут-2-инамидометил)фенил)-7-карбамоил-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамид;
N-(3-(7-карбамоил-2-(2-((2-цианоацетамидо)метил)фенил)-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамид;
N-(3-(7-карбамоил-2-(2-((3-(диметиламино)пропанамидо)метил)фенил)-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамид;
N-(3-(7-карбамоил-2-(2-((3-(пиперидин-1-ил)пропанамидо)метил)фенил)-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамид;
N-(3-(7-карбамоил-2-(2-((2-феноксиацетамидо)метил)фенил)-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамид;
N-(3-(7-карбамоил-2-(2-((2-(4-фторфенокси)ацетамидо)метил)фенил)-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамид;
N-(3-(7-карбамоил-2-(2-((2-хлорацетамидо)метил)фенил)-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамид;
N-(3-(2-(2-(аминометил)фенил)-7-карбамоил-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-(4-фторфенил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-фенил-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-(2-(метилсульфонил)фенил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-(4-(диметилкарбамоил)фенил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-(пиримидин-5-ил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-(пиридин-3-ил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-(4-(морфолин-4-карбонил)фенил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-(4-(пирролидин-1-карбонил)фенил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-(4-(4-метилпиперазин-1-карбонил)фенил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-(4-(метилсульфонил)фенил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-(6-метоксипиридин-3-ил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-(4-цианофенил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-(2-метоксифенил)-1H-индол-7-карбоксамид;
N-(3-(7-карбамоил-3-(2-цианоацетамидо)-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамид;
4-(2-акриламидофенил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-(4-(морфолинометил)фенил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-(4-карбамоилфенил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-5-(тиазол-2-илметиламино)фенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
4-(2-метил-3-(N-метилакриламидо)фенил)-1H-индол-7-карбоксамид;
4-(3-(метиламино)фенил)-1H-индол-7-карбоксамид;
4-(3-(N-метилакриламидо)фенил)-1H-индол-7-карбоксамид;
4-(2-метил-3-(2-метиленбутанамидо)фенил)-1H-индол-7-карбоксамид;
4-(2-метил-3-(3-(пирролидин-1-ил)пропанамидо)фенил)-1H-индол-7-карбоксамид;
4-(3-метакриламидо-2-метилфенил)-1H-индол-7-карбоксамид;
(E)-4-(3-(3-циклопропилакриламидо)-2-метилфенил)-1H-индол-7-карбоксамид;
(E)-4-(2-метил-3-(3-(пиридин-2-ил)акриламидо)фенил)-1H-индол-7-карбоксамид;
(E)-4-(2-метил-3-(3-(1-метил-1H-пиразол-4-ил)акриламидо)фенил)-1H-индол-7-карбоксамид;
(E)-этил 4-(3-(7-карбамоил-1H-индол-4-ил)-2-метилфениламино)-4-оксобут-2-еноат;
(E)-4-(3-(4-(диметиламино)бут-2-енамидо)-2-метилфенил)-1H-индол-7-карбоксамид;
(E)-4-(2-метил-3-(3-(пиридин-3-ил)акриламидо)фенил)-1H-индол-7-карбоксамид;
(E)-4-(2-метил-3-(4-метилпент-2-енамидо)фенил)-1H-индол-7-карбоксамид;
N1-(3-(7-карбамоил-1H-индол-4-ил)-2-метилфенил)-N4-этилмалеамид;
4-(3-ацетамидо-2-метилфенил)-1H-индол-7-карбоксамид;
(E)-4-(3-бут-2-енамидо-2-метилфенил)-1H-индол-7-карбоксамид;
4-(2-метил-3-(3-морфолинопропанамидо)фенил)-1H-индол-7-карбоксамид;
(E)-4-(2-метил-3-(3-(тиазол-2-ил)акриламидо)фенил)-1H-индол-7-карбоксамид;
4-(2-метил-3-(2-фенилакриламидо)фенил)-1H-индол-7-карбоксамид;
(E)-4-(2-метил-3-(4-(пиперидин-1-ил)бут-2-енамидо)фенил)-1H-индол-7-карбоксамид;
(E)-4-(2-метил-3-(4-((тетрагидрофуран-2-ил)метиламино)бут-2-енамидо)фенил)-1H-индол-7-карбоксамид;
(E)-4-(3-(4-(2-метоксиэтиламино)бут-2-енамидо)-2-метилфенил)-1H-индол-7-карбоксамид;
(E)-4-(3-(4-(циклопропиламино)бут-2-енамидо)-2-метилфенил)-1H-индол-7-карбоксамид;
(E)-4-(2-метил-3-(4-морфолинобут-2-енамидо)фенил)-1H-индол-7-карбоксамид;
(E)-4-(2-метил-3-(4-(4-метилпиперазин-1-ил)бут-2-енамидо)фенил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-4-(бензилокси)фенил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-5-(бензилокси)фенил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-4-(тиазол-2-илметокси)фенил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-5-(тиазол-2-илметокси)фенил)-1H-индол-7-карбоксамид;
4-(2-акриламидо-4-(тиазол-2-илметокси)фенил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-метилфенил)-1H-пирроло[2,3-c]пиридин-7-карбоксамид;
4-(2-акриламидо-4-(бензилокси)фенил)-1H-индол-7-карбоксамид;
4-(5-акриламидопиридин-3-ил)-1H-индол-7-карбоксамид;
4-(2-акриламидопиридин-4-ил)-1H-индол-7-карбоксамид;
N1-(3-(7-карбамоил-1H-индол-4-ил)фенил)-N4-(2-метоксиэтил)малеамид;
N1-(3-(7-карбамоил-1H-индол-4-ил)фенил)-N4-этилмалеамид;
4-(3-(1-метил-1,2,5,6-тетрагидропиридин-3-карбоксамидо)фенил)-1H-индол-7-карбоксамид;
4-(3-(винилсульфонамидо)фенил)-1H-индол-7-карбоксамид;
4-(3-(2-оксопропанамидо)фенил)-1H-индол-7-карбоксамид;
(E)-метил 4-(3-(7-карбамоил-1H-индол-4-ил)фениламино)-4-оксобут-2-еноата;
4-(3-(цианометилкарбамоил)фенил)-1H-индол-7-карбоксамид;
N-(3-(7-карбамоил-1H-индол-4-ил)фенил)-5-метилизоксазол-4-карбоксамид;
N1-(3-(7-карбамоил-1H-индол-4-ил)фенил)-N4-метилфумарамид;
N1-(3-(7-карбамоил-1H-индол-4-ил)фенил)-N4,N4-диметилфумарамид;
N1-(3-(7-карбамоил-1H-индол-4-ил)фенил)-N4-этилфумарамид;
N1-(3-(7-карбамоил-1H-индол-4-ил)фенил)-N4-циклопропилфумарамид;
(E)-4-(3-(7-карбамоил-1H-индол-4-ил)фениламино)-4-оксобут-2-еновая кислота;
4-(3-(N-изобутилакриламидо)фенил)-1H-индол-7-карбоксамид;
амид 1-акрилоил-1,2,3,6-тетрагидро-пирроло[2,3-e]индол-5-карбоновой кислоты;
4-акриламидо-1H-индол-7-карбоксамид;
4-(3-(N-(цианометил)сульфамоил)фенил)-1H-индол-7-карбоксамид;
4-(3-акриламидофенил)-1H-пирроло[3,2-c]пиридин-7-карбоксамид;
4-(3-акриламидо-2-метилфенил)-1H-пирроло[3,2-c]пиридин-7-карбоксамид;
4-(3-((2-оксопропанамидо)метил)фенил)-1H-индол-7-карбоксамид;
4-(3-акриламидофенил)-1H-индазол-7-карбоксамид;
4-(3-акриламидо-2-метоксифенил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-фторфенил)-1H-индол-7-карбоксамид;
4-(5-акриламидо-2-фторфенил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-4-фторфенил)-1H-индол-7-карбоксамид;
4-(5-акриламидо-2-хлорфенил)-1H-индол-7-карбоксамид;
4-(5-акриламидо-2,4-дифторфенил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-4-цианофенил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2,6-дифторфенил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-5-метилфенил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-4-метилфенил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-4-метоксифенил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-5-метоксифенил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-4-хлорфенил)-1H-индол-7-карбоксамид;
4-(5-акриламидо-2,3-дифторфенил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-5-цианофенил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-цианофенил)-1H-индол-7-карбоксамид;
4-(3-акриламидофенил)-2-винил-1H-индол-7-карбоксамид;
4-(3-акриламидофенил)-2-этил-1H-индол-7-карбоксамид;
4-(3-(2-(морфолинометил)акриламидо)фенил)-1H-индол-7-карбоксамид;
4-(3-(2-((диметиламино)метил)акриламидо)фенил)-1H-индол-7-карбоксамид;
(E)-4-(3-(4-(диметиламино)бут-2-енамидо)-2-метилфенил)-1H-пирроло[2,3-c]пиридин-7-карбоксамид;
4-((1R,3S)-3-акриламидоциклогексил)-1H-индол-7-карбоксамид;
4-(цис-3-акриламидоциклогексил)-1H-индол-7-карбоксамид;
4-((1S,3S)-3-акриламидоциклогексил)-1H-индол-7-карбоксамид;
4-(транс-3-акриламидоциклогексил)-1H-индол-7-карбоксамид;
4-(цис-3-акриламидоциклогексил)-1H-индол-7-карбоксамид;
4-(3-(2-(аминометил)акриламидо)фенил)-1H-индол-7-карбоксамид;
4-((1R,3S)-3-акриламидоциклопентил)-1H-индол-7-карбоксамид;
4-(3-(2-((метиламино)метил)акриламидо)фенил)-1H-индол-7-карбоксамид;
4-(3-акриламидофенил)-2-метил-1H-индол-7-карбоксамид;
4-((1S,3S)-3-акриламидоциклопентил)-1H-индол-7-карбоксамид;
4-(3-акриламидофенил)-2-(2-этоксиэтил)-1H-индол-7-карбоксамид;
4-(3-акриламидофенил)-2-(2-гидроксиэтил)-1H-индол-7-карбоксамид;
4-(1-акрилоилпиперидин 3-ил)-2-(1-метил-1H-пиразол-4-ил)-1H-индол-7-карбоксамид;
4-(3-акриламидо-2-метилфенил)-2-(1-изопропил-1H-пиразол-4-ил)-1H-индол-7-карбоксамид;
4-(3-(4-циклопропилбензамидо)-2-метилфенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
4-(2-метил-3-(1-метилпиперидин-4-карбоксамидо)фенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид;
4-(3-(N-(циклопентилметил)акриламидо)фенил)-1H-индол-7-карбоксамид;
этил 4-(7-карбамоил-4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-1H-индол-2-ил)5,6-дигидропиридин-1(2H)-карбоксилат;
(R)-4-(3-(4-оксохиназолин-3(4H)-ил)пиперидин-1-ил)-1H-индол-7-карбонитрил;
4-(2,6-дихлорбензил)-2-(п-толил)-1H-индол-7-карбоксамид;
(E)-4-(3-(2-циано-3-гидроксибут-2-енамидо)фенил)-1H-индол-7-карбоксамид;
4-(цис-3-акриламидоциклопентил)-1H-индол-7-карбоксамид;
4-(транс-3-акриламидоциклопентил)-1H-индол-7-карбоксамид;
4-(транс-3-акриламидоциклопентил)-1H-индол-7-карбоксамид;
4-((1-акрилоилазетидин-3-ил)окси)-1H-индол-7-карбоксамид;
(S)-4-(1-(1-акрилоилазетидин-3-ил)этил)-1H-индол-7-карбоксамид;
(R)-4-(1-(1-акрилоилазетидин-3-ил)этил)-1H-индол-7-карбоксамид*;
4-((1-акрилоилазетидин-3-ил)(метил)амино)-1H-пирроло[2,3-c]пиридин-7-карбоксамид;
(R)-4-(1-акрилоилпиперидин-3-ил)-1H-индол-7-карбоксамид*;
(S)-4-(1-акрилоилпиперидин-3-ил)-1H-индол-7-карбоксамид*;
(S)-4-(1-акрилоилпиперидин-3-ил)-2-метил-1H-индол-7-карбоксамид;
(R)-4-(1-акрилоилпиперидин-3-ил)-2-метил-1H-индол-7-карбоксамид;
(R)-4-(4-акрилоилморфолин-2-ил)-2-(1-метил-1H-пиразол-4-ил)-1H-индол-7-карбоксамид;
(S)-4-(4-акрилоилморфолин-2-ил)-2-(1-метил-1H-пиразол-4-ил)-1H-индол-7-карбоксамид;
(R)-4-(1-акрилоилпирролидин-3-ил)-2-(6,7-дигидро-4H-пиразоло[5,1-c][1,4]оксазин-2-ил)-1H-индол-7-карбоксамид;
2-метил-4-(метил(1-пропиолоилазетидин-3-ил)амино)-1H-индол-7-карбоксамид;
(S)-4-(1-акрилоилпирролидин-3-ил)-2-(6,7-дигидро-4H-пиразоло[5,1-c][1,4]оксазин-2-ил)-1H-индол-7-карбоксамид;
(R)-4-(4-акрилоил-1,4-оксазепан-6-ил)-1H-пирроло[3,2-c]пиридин-7-карбоксамид;
(S)-4-(4-акрилоил-1,4-оксазепан-6-ил)-1H-пирроло[3,2-c]пиридин-7-карбоксамид;
(R)-4-(1-акрилоилпиперидин-3-ил)-1H-пирроло[3,2-c]пиридин-7-карбоксамид;
(S)-4-(1-акрилоилпиперидин-3-ил)-1H-пирроло[3,2-c]пиридин-7-карбоксамид;
(R)-7-(1-акрилоилпиперидин-3-ил)-2-(1-метил-1H-пиразол-4-ил)тиазоло[5,4-c]пиридин-4-карбоксамид;
(S)-7-(1-акрилоилпиперидин-3-ил)-2-(1-метил-1H-пиразол-4-ил)тиазоло[5,4-c]пиридин-4-карбоксамид;
(S)-4-(4-акрилоил-1,4-оксазепан-6-ил)-1H-индол-7-карбоксамид;
4-((3S,5R)-1-акрилоил-5-(гидроксиметил)пиперидин-3-ил)-1H-индол-7-карбоксамид;
4-((3S,5S)-1-акрилоил-5-(гидроксиметил)пиперидин-3-ил)-1H-индол-7-карбоксамид;
4-((3R,5S)-1-акрилоил-5-(гидроксиметил)пиперидин-3-ил)-1H-индол-7-карбоксамид;
4-((3R,5R)-1-акрилоил-5-(гидроксиметил)пиперидин-3-ил)-1H-индол-7-карбоксамид;
(R)-4-(1-акрилоилпирролидин-3-ил)-2-(1-метил-1H-пиразол-4-ил)-1H-индол-7-карбоксамид;
(S)-4-(1-акрилоилпирролидин-3-ил)-2-(1-метил-1H-пиразол-4-ил)-1H-индол-7-карбоксамид;
4-((1R,3R)-3-акриламидоциклопентил)-1H-индол-7-карбоксамид;
(S)-4-(1-акрилоилпиперидин-3-ил)-1H-пирроло[2,3-c]пиридин-7-карбоксамид;
(R)-4-(1-акрилоилпиперидин-3-ил)-1H-пирроло[2,3-c]пиридин-7-карбоксамид;
(R)-2-метил-4-(1-пропионилпирролидин-3-ил)-1H-индол-7-карбоксамид;
(S)-2-метил-4-(1-пропионилпирролидин-3-ил)-1H-индол-7-карбоксамид;
4-((1-акрилоилазетидин-3-ил)(метил)амино)-2-(изохроман-7-ил)-1H-индол-7-карбоксамид;
4-((1-акрилоилазетидин-3-ил)(метил)амино)-2-(6,7-дигидро-4H-пиразоло[5,1-c][1,4]оксазин-2-ил)-1H-индол-7-карбоксамид;
4-((1-акрилоилазетидин-3-ил)(метил)амино)-2-(4,4-дифторциклогекс-1-ен-1-ил)-1H-индол-7-карбоксамид;
4-((1-акрилоилазетидин-3-ил)(метил)амино)-2-(4-(метилсульфонил)циклогекс-1-ен-1-ил)-1H-индол-7-карбоксамид;
4-((1-акрилоилазетидин-3-ил)(метил)амино)-2-(6-морфолинопиридин-3-ил)-1H-индол-7-карбоксамид;
4-((1-акрилоилазетидин-3-ил)(метил)амино)-2-(7,8-дигидро-5H-пирано[4,3-b]пиридин-3-ил)-1H-индол-7-карбоксамид;
4-((1-акрилоилазетидин-3-ил)(метил)амино)-2-(хроман-7-ил)-1H-индол-7-карбоксамид;
4-((1-акрилоилазетидин-3-ил)(метил)амино)-2-(5-(морфолинометил)пиридин-2-ил)-1H-индол-7-карбоксамид;
4-((1-акрилоилазетидин-3-ил)(метил)амино)-2-(1-метил-1H-пиразол-4-ил)-1H-индол-7-карбоксамид;
4-((1-акрилоилазетидин-3-ил)(метил)амино)-2-(3,4-дигидро-2H-бензо[b][1,4]оксазин-6-ил)-1H-индол-7-карбоксамид;
4-((1-акрилоилазетидин-3-ил)(метил)амино)-2-(1-метил-1H-пиразол-5-ил)-1H-индол-7-карбоксамид;
4-((1-акрилоилазетидин-3-ил)(метил)амино)-2-(2-этил-1,2,3,4-тетрагидроизохинолин-6-ил)-1H-индол-7-карбоксамид;
4-((1-акрилоилазетидин-3-ил)(метил)амино)-2-(1,3-диметил-1H-пиразол-4-ил)-1H-индол-7-карбоксамид;
4-((1-акрилоилазетидин-3-ил)(метил)амино)-2-(1,1-диоксидтетрагидро-2H-тиопиран-4-ил)-1H-индол-7-карбоксамид;
4-((1-акрилоилазетидин-3-ил)(метил)амино)-2-(1-пропилпиперидин-4-ил)-1H-индол-7-карбоксамид;
4-((1-акрилоилазетидин-3-ил)(метил)амино)-2-(тетрагидрофуран-3-ил)-1H-индол-7-карбоксамид;
4-((1-акрилоилазетидин-3-ил)(метил)амино)-2-(3-гидроксиоксетан-3-ил)-1H-индол-7-карбоксамид;
4-((1-акрилоилазетидин-3-ил)(метил)амино)-2-метил-1H-индол-7-карбоксамид;
(R)-4-(4-акрилоил-1,4-оксазепан-6-ил)-1H-индол-7-карбоксамид;
(S)-4-(1-акрилоилпирролидин-3-ил)-2-метил-1H-индол-7-карбоксамид*;
(R)-4-(1-акрилоилпирролидин-3-ил)-2-метил-1H-индол-7-карбоксамид*;
4-((1R,5S)-6-акрилоил-3,6-диазабицикло[3.2.0]гептан-3-ил)-1H-индол-7-карбоксамид;
4-((1S,5R)-6-акрилоил-3,6-диазабицикло[3.2.0]гептан-3-ил)-1H-индол-7-карбоксамид;
(R)-4-(1-(1-акрилоилазетидин-3-ил)этил)-1H-пирроло[3,2-c]пиридин-7-карбоксамид;
(S)-4-(1-(1-акрилоилазетидин-3-ил)этил)-1H-пирроло[3,2-c]пиридин-7-карбоксамид;
4-((1-акрилоилазетидин-3-ил)амино)-1H-пирроло[2,3-c]пиридин-7-карбоксамид;
4-((1-акрилоил-3-метилазетидин-3-ил)(метил)амино)-1H-индол-7-карбоксамид;
4-((1-цианоазетидин-3-ил)(метил)амино)-2-метил-1H-индол-7-карбоксамид;
4-(2-хлор-6-фторбензил)-2-п-толил-1H-индол-7-карбоксамид;
(S)-4-((1-акрилоилазетидин-3-ил)(метил)амино)-2-(тетрагидрофуран-3-ил)-1H-индол-7-карбоксамид;
(R)-4-((1-акрилоилазетидин-3-ил)(метил)амино)-2-(тетрагидрофуран-3-ил)-1H-индол-7-карбоксамид;
(S)-4-(4-акрилоил-1,4-оксазепан-6-ил)-1H-пирроло[2,3-c]пиридин-7-карбоксамид;
(R)-4-(4-акрилоил-1,4-оксазепан-6-ил)-1H-пирроло[2,3-c]пиридин-7-карбоксамид;
(S)-4-(1-акрилоилпиперидин-3-ил)-2-(1-метил-1H-пиразол-4-ил)-1H-индол-7-карбоксамид; или
(R)-4-(1-акрилоилпиперидин-3-ил)-2-(1-метил-1H-пиразол-4-ил)-1H-индол-7-карбоксамид.
В двадцать первом варианте осуществления настоящее изобретение относится к способу лечения заболевания, включающему введение терапевтически эффективного количества соединения по п.1 пациенту, нуждающемуся в этом.
В двадцать втором варианте осуществления настоящее изобретение относится к соединению по любому из предшествующих вариантов осуществления, где заболевание представляет собой ревматоидный артрит, ювенильный ревматоидный артрит, остеоартрит, болезнь Крона, воспалительное заболевание кишечника, неспецифический язвенный колит, псориатический артрит, псориаз, анкилозирующий спондилоартрит, интерстициальный цистит, астму, системную красную волчанку, волчаночный нефрит, В-клеточный хронический лимфоцитарный лейкоз, рассеянный склероз, хронический лимфоцитарный лейкоз, мелкоклеточную лимфоцитарную лимфому, мантийноклеточную лимфому, В-клеточную неходжкинскую лимфому, активированных В-клеток, например, диффузную В-крупноклеточную лимфому, множественную миелому, диффузную В-крупноклеточную лимфому, фолликулярную лимфому, волосатоклеточный лейкоз или лимфобластную лимфому.
В двадцать третьем варианте осуществления изобретение относится к набору, содержащему упакованный продукт, содержащий компоненты, с которыми можно вводить соединение по любому из предшествующих вариантов осуществления для лечения аутоиммунных нарушений.
В двадцать четвертом варианте осуществления настоящее изобретение относится к набору в соответствии с двадцать третьим вариантом осуществления, в котором упакованный продукт содержит соединение по п.1 и инструкции по применению.
В двадцать пятом варианте осуществления изобретение относится к фармацевтической композиции, содержащей соединение по любому от первого по двадцатый варианты осуществления и одно или несколько фармацевтически приемлемых эксципиентов.
Подробное описание изобретения
Протеинкиназы представляют собой обширный и разнообразный класс, более чем 500 ферментов, который включает онкогены, рецепторы факторов роста, промежуточные соединения сигнальной трансдукции, связанные с апоптозом киназы и циклин-зависимые киназы. Они ответственны за перенос фосфатной группы на специфические тирозиновые, сериновые или треониновые аминокислотные остатки, и, в общем, классифицируются как тирозин и серин/треонин киназы, в результате их субстратной специфичности.
Протеинкиназы представляют собой многочисленное семейство белков, которые играют центральную роль в регуляции широкого круга клеточных процессов и в поддержании клеточной функции. Неполный перечень таких киназ включает, без ограничения: нерецепторные тирозинкиназы, такие как семейство Tec (BTK, ITK, Tec, ETK/BMX и RLK/TXK), семейство киназы Janus (Jak1, Jak2, Jak3 и Tyk2); гибридные киназы, такие как BCR-Abl, киназа фокальной адгезии (FAK), Fes, Lck и Syk; рецепторные тирозинкиназы, такие как рецептор эпидермального фактора роста (EGFR), киназа рецептора фактора роста, полученного из тромбоцитов (PDGF-R), рецепторные киназы для фактора стволовых клеток, c-kit, рецептора фактора роста гепатоцитов, c-Met, и рецептора фактора роста фибробластов FGFR3; и серин/треонин киназы, такие как b-RAF, митоген-активированные протеинкиназы (например, MKK6) и SAPK2β. Абберантная киназная активность наблюдалась при многих болезненных состояниях, включая доброкачественные и злокачественные пролиферативные нарушения, также как заболевания, являющиеся результатом неадекватной активации иммунной и нервной систем. Новые соединения настоящего изобретения ингибируют активность одной или более из протеинкиназ, и поэтому ожидается, что они могут быть полезны при лечении опосредованных киназами заболеваний.
Тирозинкиназа Брутона (BTK) представляет собой нерецепторную тирозинкиназу с ключевой ролью в передачи сигнала иммунорецептора (BCR, FcεR, FcγR, DAP12, дектин-1, GPVI и т.д.) в множестве гематопоэтических клеток, включая B-клетки, тромбоциты, тучные клетки, базофилы, эозинофилы, макрофаги и нейтрофилы, а также остеокласты, участвующие в деструкции костной ткани (для обзора, см. Brunner et al., 2005 Histol. Histopathol., 20:945, Mohamed et al., 2009 Immunol. Rev., 228:58). Мутации в BTK, как известно, приводят к X-сцепленной агаммаглобулинемии (XLA) у людей и к X-сцепленному иммунодефициту (Xid) у мышей, которые характеризуются ограниченным продуцированием В-клеток и низкими титрами антитела (Lindvall et al., 2005 Immunol. Rev., 203:200). Комбинированное действие Btk в нескольких типах клеток делает ее перспективной мишенью при аутоиммунном заболевании. BTK связана с последовательностью гомологии другими киназами семейства Tec (ITK, Tec, ETK/BMX и RLK/TXK).
В B-лимфоцитах, BTK требуется для развития В-клеток и для активации Ca2+ вследствие вовлечения B-клеточного рецептора (BCR) (Khan et al., 1995 Immunity 3:283; Genevier et al., 1997 Clin. Exp. Immun., 110:286), где она находится ниже семейства киназ Src (например, Lyn), Syk и PI3K. BTK, как было показано, является важной для тимус-зависимого и тимус-независимого ответов типа 2 на антигены (Khan et al., Immunity 1995; 3; 283). В тучных клетках, исследования с использованием нокаутированной мыши по BTK (Hata et al., 1998 J. Exp. Med., 187:1235; Schmidt et al., 2009 Eur. J. Immun., 39:3228) указывают на роль BTK в индуцировании передачи сигнала FcεRI, высвобождении гистамина и продукции цитокинов, таких как TNF, IL-2 и IL-4. В тромбоцитах, BTK имеет важное значение для передачи сигналов через рецептор гликопротеина VI (GPVI), который отвечает на коллаген и, как было показано способствовует агрегации тромбоцитов и способствует продукции цитокинов из фибробластоподобных синовиоцитов (Hsu et al., 2013 Immun. Letters, 150:97). В моноцитах и макрофагах, действие BTK вовлечено в передачу сигнала, индуцированную FcγRI, а также может иметь роль в цитокиновых ответах, индуцированных Toll-подобным рецептором, включая TLR2, TLR4, TLR8 и TLR9 (Horwood et al., 2003 J. Exp. Med., 197:1603; Horwood et al., 2006 J. Immunol., 176:3635; Perez de Diego et al., 2006 Allerg. Clin. Imm., 117:1462; Doyle et al., 2007 J. Biol. Chem., 282:36959, Hasan et al., 2007 Immunology, 123:239; Sochorava et al., 2007 Blood, 109:2553; Lee et al., 2008, J. Biol. Chem., 283:11189).
Таким образом, ингибирование BTK, как ожидается, имеет место при нескольких важнейших стыках воспалительных реакций, приводящих к эффективному подавлению аутоиммунного ответа. Соотвественно заболевания, связанные с активацией В-клеточного рецептора, взаимодействием антитело-Fc рецептор и передачей сигнала рецептора GPVI, можно модулировать путем обработки ингибиторами BTK. Ингибирование BTK может действовать как на инициирование аутоиммунного заболевания, блокируя передачу сигналов BCR, и эффекторную фазу путем отмены передачи сигнала FcR в макрофаги, нейтрофилы, базофилы и тучные клетки. Кроме того, блокирование BTK будет обеспечивать дополнительное преимущество с помощью ингибирования созревания остеокластов и, следовательно, ослабит разрушение кости и общее разрушение суставов, связанные с ревматоидным артритом. Ингибирование BTK может быть полезно при лечении множества воспалительных и аллергических заболеваний - например (но ими не ограничиваясь), ревматоидный артрит (RA), системная красная волчанка (SLE), рассеянный склероз (MS) и реакции гиперчувствительности I типа, такие как аллергический ринит, аллергический конъюнктивит, атопический дерматит, аллергическая астма и общая анафилактическая реакция. Для обзора направленного действия BTK в качестве лечения воспалительных заболеваний и аутоиммунных заболеваний, а также лейкозов и лимфом, см. Uckun & Qazi, 2010 Expert Opin. Ther. Pat., 20:1457. Так как BTK сильно экспрессируется в раках гематопоэтической системы и BTK-зависимой передачи сигнала полагают приводит к дизрегуляции, ингибиторы BTK могут быть полезны для лечения B-клеточных лимфом/лейкозов и других онкологических заболеваний - например (но ими не ограничиваясь) острого лимфобластного лейкоза (ALL), хронического лимфоцитарного лейкоза (CLL), неходжкинской лимфомы (NHL), мелкоклеточной лимфоцитарной лимфомы (SLL) и острого миелоидного лейкоза (для обзора, см. Buggy & Elias 2012 Int Rev Immunol. 31:119). Вместе взятые, ингибиторы BTK обеспечивают серьезный способ лечения множества воспалительных заболеваний и иммунологических нарушений, а также гематологических злокачественных опухолей.
Все киназы связывают общую молекулу, АТФ, и, следовательно, имеют структурно-подобные связывающие “карманы”. Таким образом, одна из задач любого ингибитора киназы является то, что они предрасположены к ингибированию более одной киназы благодаря гомологии связывающего “кармана”. Например, стауроспорин, хорошо изученный промискуитетный ингибитор киназы, было показано, ингибирует по меньшей мере 253 киназы с kd<3 мкМ из “кинома” человека (см. Nature Biotechnology, 208, 26, p. 127). Кроме того, несколько имеющихся в продаже ингибиторов киназы, как известно, ингибируют более чем одну определенную киназу, иматиниб (Gleevec®) воздействует на киназы ABL, ARG, PDGFR-α/β и c-KIT, сорафениб (Nexavar®) воздействует на B-RAF, VEGFR, PDGFR-α/β, FLT3 и c-KIT и сунитиниб (Sutent®) воздействует на VEGFR, PDGFR, CSF1R, FLT3 и c-KIT (Nature Reviews Drug Discovery 2011, 10, 111).
Ингибирование определенных киназ в “киноме” человека, как известно, имеет нежелательные эффекты при использовании в качестве фармацевтического лечения. Например, количество мишеней киназы были вовлечены играть роль в профиле кардиотоксичности для ингибиторов киназы, которые существуют в настоящее время на рынке. Эти киназы включают, но ими не ограничиваются, VEGFR2, PI3K, AKT, PDGFR-α/β, AMPK, GSK3, ERKs, CDK2, Aurora, PLK, JNK, CAMKII<PDK1, mTOR, LKB1, CAMKKβ, MEK1/2, PKA, PKCα, RAF1, B-RAF, EGFR, ERBB2, c-Kit, ABL, ARG, JAK2, FAK, DMPK, LTK, ROCK, LKB1, LDB3, PIM, GRK2, GRK5, ASK1 и PTEN (см. Nature Reviews Drug Discovery 2011, 10:111). Один пример из имеющегося в продаже ингибитора киназы, является то, что в клинических испытаниях с сунитинибом, пациенты оказались в группе повышенного риска гипертонии (см. The Lancet 2006, 368:1329; and J. Clin. Oncol. 2009, 27:3584). Последующие исследования механизма увеличения гипертензии указывают, что в то время PDGFR и VEGFR могут играть некоторую роль, нецелевое ингибирование киназы, такой как AMPK, также может быть одной из причин повышенного риска гипертензии от сунитиниба (Curr. Hypertens. Rep. 2011, 13:436). Кроме того, существует патентная заявка США 2011/0212461, которая была подана, которая представляет собой способ прогнозирования кардиотоксичности на основе активности по сравнению с перечнем киназ, включая CSF1R, KIT, FYN, PDGFR бета, FGR, LCK, рецептор Ephrin B2, FRK, ABL1, PDGFR1 альфа, HCK, ABL2, LYN, ZAK, YES1, MAP4K4, PKN1, BRAF, DDR2, MAP4K5 и STK24. Таким образом, идентификация ингибиторов киназы с селективным профилем Btk киназы желательна. Соединения по настоящему изобретению выбраны для ингибирования Btk по сравнению с другими киназами.
Было обнаружено, что многие киназы, будь то рецепторные или нерецепторные тирозинкиназы, или S/T киназы, участвуют в клеточных путях передачи сигналов, вовлеченных во множество патологических состояний, включая иммуномодуляцию, воспаления или пролиферативные нарушения, такие как рак.
Многие аутоиммунные заболевания и заболевания, связанные с хроническим воспалением, а также острыми реакциями, были связаны с избыточным или нерегулируемым продуцированием или активностью одного или нескольких цитокинов.
Соединения по настоящему изобретению также могут быть использованы для лечения ревматоидного артрита, астмы, аллергической астмы, остеоартрита, ювенильного артрита, волчанки, волчаночного нефрита, системной красной волчанки (SLE), анкилозирующего спондилита, заболевания глаз, интерстициального цистита, рака, солидной опухоли, саркомы, фибросаркомы, остеомы, меланомы, ретинобластомы, рабдомиосаркомы, глиобластомы, нейробластомы, тератокарциномы, реакций гиперчувствительности, гиперкинетических двигательных расстройств, гиперчувствительного пневмонита, гипертензии, двигательные расстройства гипокинетического характера, аортальной и периферической аневризмы, оценки гипоталамо-гипофизарно-надпочечниковой оси, расслоения аорты, артериальной гипертензии, артериосклероза, артериовенозной фистулы, атаксии, спинномозжечковой дегенерации, стрептококкового миозита, структурных поражений мозжечка, подострого склерозирующего панэцефалита, обморока, сифилитического поражения сердечно-сосудистой системы, системной анафилаксии, синдрома системного воспалительного ответа, системного проявления ювенильного ревматоидного артрита, T-клеточного или FAB ALL, телеангиоэктазии, облитерирующего тромбоангиита, трансплантатов, травмы/геморрагии, реакций гиперчувствительности III типа, гиперчувствительности IV типа, нестабильной стенокардии, уремии, уросепсиса, крапивницы, порока сердца, варикозного расширения вен, васкулита, заболевания вен, тромбоза вен, фибрилляции желудочков, вирусных и грибковых инфекций, витального энцефалита/асептического менингита, гемофагоцитарного синдрома, связанного с жизненно важными функциями, синдрома Вернике-Корсакова, болезни Вильсона, отторжения трансплантата любого органа или ткани, отторжения сердечного трансплантата, гемахроматоза, гемодиализа, гемолитического уремического синдрома/тромболитической тромбоцитопенической пурпуры, геморрагии, идиопатического легочного фиброза, опосредованной антителами цитотоксичности, астении, детской спинальной мышечной атрофии, воспаления аорты, гриппа A, воздействия ионизирующего излучения, иридоциклита/увеита/неврита зрительного нерва, ювенильной спинальной мышечной атрофии, лимфомы, миеломы, лейкоза, злокачественного асцита, злокачественных заболеваний гематопоэтической системы, диабетической патологии, такой как глаукома при инсулинзависимом сахарном диабете, диабетическая ретинопатия или микроангиопатия, серповидно-клеточной анемии, хронического воспаления, гломерулонефрита, отторжения трансплантата, болезни Лайма, болезни Гиппеля-Линдау, пемфигоида, болезни Педжета, фиброза, саркоидоза, цирроза, тироидита, синдрома повышенной вязкости крови, болезни Рандю-Вебера-Ослера, хронического окклюзионного заболевания легких, астмы или отека после ожогов, травмы, излучения, инсульта, гипоксии, ишемии, синдрома гиперстимуляции яичников, постперфузионного синдрома, постинфузионного синдрома, пост-MI кардиотомного синдрома, преэклампсии, менометроррагии, эндометриоза, легочной гипертензии, гемангиомы детей младшего возраста, или инфицирования Herpes simplex, Herpes Zoster, вирусом иммунодефицита человека, парапоксвирусами, простейшими или токсоплазмой, прогрессирующего супрануклеарного паралича, первичной легочной гипертензии, лучевой терапии, феномена и болезни Рейно, болезни Рефсума, привычной тахикардии с узкими комплексами QRS, реноваскулярная артериальная гипертензия, рестриктивной кардиомиопатии, саркомы, сенильной хореи, сенильной деменции типа телец Леви, шока, кожного аллотрансплантата, синдрома изменений кожи, отека зрительного нерва или макулярного отека, неоваскулярного заболевания глаз, склерита, радиальной кератотомии, увеита, воспаления стекловидного тела, миопии, врожденных ямок в диске зрительного нерва, хронического отслоения сетчатки, осложнений после лечения лазером, конъюнктивита, болезни Штаргардта, болезни Илза, ретинопатии, дегенерации желтого пятна, рестеноза, ишемии/реперфузионного повреждения, ишемического инсульта, окклюзии сосудов, обструкции сонных артерий, язвенного колита, воспалительного заболевания кишечника, диабета, сахарного диабета, инсулинзависимого сахарного диабета, аллергических заболеваний, дерматита склеродермии, заболевания «трансплантат против хозяина», отторжения органного трансплантата (в том числе, но не ограничиваясь перечисленным, отторжения трансплантата костного мозга и солидных органов), острого или хронического иммунологического заболевания, связанного с трансплантацией органов, саркоидоза, диссеминированного внутрисосудистого свертывания, болезни Кавасаки, нефротического синдрома, синдрома хронической усталости, грануломатоза Вегенера, пурпуры Геноха-Шенлейна, микроскопического васкулита почек, хронического активного гепатита, септического шока, синдрома токсического шока, септического синдрома, кахексии, инфекционных заболеваний, паразитарных заболеваний, синдрома приобретенного иммунодефицита, острого поперечного миелита, хореи Хантингтона, инсульта, первичного билиарного цирроза, гемолитической анемии, злокачественных заболеваний, болезни Аддисона, идиопатической болезни Аддисона, спорадической, полигландулярной недостаточности I типа и полигландулярной недостаточности II типа, синдрома Шмидта, (острого) респираторного дистресс синдрома взрослых, алопеции, очаговой алопеции, серонегативной артропатии, артропатии, болезни Рейтера, псоариатической артропатии, артропатии при язвенном колите, энтеропатического синовита, хламидийной, йерсиниозной и сальмонеллезной артропатии, атероматозного заболевания/артериосклероза, атопической аллергии, аутоиммунной буллезной болезни, обыкновенной пузырчатки, листовидной пузырчатки, пемфигоида, болезни линейных IgA, аутоиммунной гемолитической анемии, Кумбс позитивной гемолитической анемии, приобретенной пернициозной анемии, ювенильной перницитозной анемии, нарушений периферических сосудов, перитонита, пернициозной анемии, миалгического энцефалита/синдрома хронической усталости, хронического кандидоза кожи и слизистых, гигантоклеточного артериита, первичного склерозирующего гепатита, криптогенного аутоиммунного гепатита, синдрома приобретенного иммунодефицита, заболеваний, связанных с синдромом приобретенного иммунодефицита, гепатита A, гепатита B, гепатита C, аритмии, связанной с блокадой ножки пучка Гиса, ВИЧ инфекции/ВИЧ нейропатии, общего изменчивого иммунодефицита (общей вариабельной гипогаммаглобулинемии), дилятационной кардиомиопатии, женского бесплодия, угасания функции яичников, преждевременного угасания функции яичников, фиброзного заболевания легких, затяжного заживления ран, криптогенного фиброзирующего альвеолита, постинфекционной интерстициальной болезни легких, интерстициального пневмонита, пневмоцистной пневмонии, пневмонии, интерстициальной болезни легких, связанной с заболеванием соединительной ткани, заболевания легких, связанного со смешанным заболеванием соединительной ткани, интерстициальной болезни легких, связанной с системным склерозом, интерстициальной болезни легких, связанной с ревматоидным артритом, болезни легких, связанной с системной красной волчанкой, болезни легких, связанной с дерматомиозитом/полимиозитом, болезни легких, связанной с болезнью Шегрена, болезни легких, связанной с анкилозирующим спондилитом, васкулитного диффузного поражения легких, болезни легких, связанной с гемосидерозом, интерстициальной болезни легких, индуцированной лекарственными средствами, лучевого фиброза, облитерирующего бронхиолита, хронической эозинофильной пневмонии, лимфоцитарной инфильтрирующей болезни легких, постинфекционной интерстициальной болезни легких, подагрического артрита, аутоиммунного гепатита, аутоиммунного гепатита (классический аутоиммунный или волчаночный гепатит) типа-1, аутоиммунного гепатита (анти-LKM антительного гепатита) типа-2, аутоиммунной гипогликемии, инсулиновой резистентности В типа с акантокератодермией, гипопаратиреоза, острого иммунологического заболевания, связанного с трансплантацией органов, хронического иммунологического заболевания, связанного с трансплантацией органов, остеоартроза, первичного склерозирующего холангита, псориаза 1 типа, псориаза 2 типа, идиопатической лейкопении, аутоиммунной нейтропении, NOS почечной недостаточности, гломерулонефрита, микроскопического васкулита почек, болезни Лайма, дискоидной красной волчанки, мужского бесплодия идиопатического или NOS, аутоиммунных реакций на сперматозоиды, рассеянного склероза (всех подтипов), симпатической офтальмии, вторичной легочной гипертензии вследствие заболевания соединительной ткани, острой и хронической боли (различных форм боли), синдрома Гудпасчера, легочного проявления узелкового периартериита, острой ревматической лихорадки, ревматоидного спондилита, болезни Стилла, системного склероза, синдрома Шегрена, болезни Такаясу/артерита, аутоиммунной тромбоцитопении, токсичности, трансплантатов, и заболеваний, приводящих к неадекватной васкуляризации, например, диабетической ретинопатии, ретинопатии недоношенных, хороидальной неоваскуляризации вследствии возрастной дегенерации желтого пятна, и гемангиомы детей младшего возраста у человека. Кроме того, такие соединения могут быть использованы при лечении таких нарушений, как асцит, выпоты, и экссудаты, включая, например, макулярный отек, отек мозга, острое поражение легких, респираторный дистресс синдром взрослых (ARDS), пролиферативных нарушений, таких как рестеноз, фиброзных заболеваний, таких как цирроз печени и атеросклероз, мезангиальных клеточных пролиферативных нарушений, таких как диабетическая нефропатия, злокачественный нефросклероз, синдромов тромботической микроангиопатии, и гломерулопатий, миокардиального ангиогенеза, коронарных и церебральных коллатералей, ангиогенеза ишемизированных конечностей, ишемии/реперфузионного повреждения, пептидной язвы, заболеваний, связанных с Helicobacter, ангиогенных нарушений, индуцированных вирусами, преэклампсии, менометроррагии, болезни от кошачьих царапин, покраснения, неоваскулярной глаукомы и ретинопатий, например, связанных с диабетической ретинопатией, ретинопатии недоношенных, или возрастной дегенерации желтого пятна. Кроме того, эти соединения могут быть использованы в качестве активных средств против гиперпролиферативных нарушений, таких как гиперплазия щитовидной железы (особенно болезни Грейвса), и кист (например, гиперваскуляризации стромы яичников, характерной для синдрома поликистозных яичников (синдром Штейна-Левенталя) и поликистоза почек, поскольку такие заболевания нуждаются в пролиферации клеток кровеносных сосудов для роста и/или метастазирования.
В других вариантах осуществления соединения, описанные в настоящем документе, могут быть использованы для лечения рака, например, В-клеточны [пролиферативны [нарушений, которые включают, но ими не ограничиваются, диффузную крупноклеточную В-клеточную лимфому, фолликулярную лимфому, хроническую лимфоцитарную лимфому, хронический лимфоцитарный лейкоз, В-клеточный пролимфоцитарный лейкоз, лимфоплазмоцитарную лимфому/макроглобулинемию Вальденстрема, лимфому маргинальной зоны селезенки, плазмоклеточную миелому, плазмацитому, экстранодальную В-клеточную лимфому маргинальной зоны, нодальную В-клеточную лимфому маргинальной зоны, мантийноклеточную лимфому, медиастинальную (тимическую) В-крупноклеточную лимфому, внутрисосудистую В-крупноклеточную лимфому, первичную выпотную лимфому, лимфому/лейкоз Беркитта, лимфогранулематоз, рак поджелудочной железы, солидные или гематологические опухоли, доброкачественные или злокачественные опухоли, рак головного мозга, почек (например, почечно-клеточный рак (RCC)), плоскоклеточный рак, рак слюнной железы, печени, надпочечников, мочевого пузыря, молочной железы, желудка, опухоли желудка, яичников, толстой кишки, прямой кишки, простаты, поджелудочной железы, легких, влагалища, эндометрия, шейки матки, яичка, мочеполового тракта, пищевода, гортани, кожи, кости или щитовидной железы, саркому, глиобластомы, нейробластомы, множественную миелому или рак желудочно-кишечного тракта, в частности, рак толстой кишки или колоректальную аденому или опухоль шеи и головы, эпидермальную гиперпролиферацию, псориаз, гиперплазию предстательной железы, неоплазию, неоплазию эпителиального характера, аденому, аденокарциному, кератоакантому, эпидермоидный рак, крупноклеточный рак, немелкоклеточный рак легкого, лимфомы (включая, например, неходжкинскую лимфому (НХЛ) и лимфому Ходжкина (также называется Ходжкина или болезнь Ходжкина)), рак молочной железы, фолликулярный рак, недифференцированный рак, папиллярный рак, семиному, меланому или лейкоз.
В других вариантах осуществления соединения, описанные в настоящем документе, могут быть использованы для лечения болезни Бехчета, остеопороза, рака костей и метастазы в кости, системной склеродермии, контактный дерматит и другие экзематозные дерматиты, себорейный дерматит, красный плоский лишай, врожденный буллезный эпидермолиза, ангиодермита, васкулитов, кожной эозинофилии или весеннего конъюнктивита.
В других вариантах осуществления соединения, описанные в настоящем документе, могут быть использованы для лечения этих состояний, характеризующихся воспалением слизистой оболочки носа, включая острый ринит, аллергический, атрофический ринит и хронический ринит, включая казеозный ринит, гипертрофический ринит, гнойный ринит, сухой ринит и медикаментозный ринит; мембранозный ринит, включая крупозный, фибринозный и псевдомембранозный ринит и скрофулезный ринит, сезонный ринит, включая нервный ринит (поллиноз) и вазомоторный ринит, саркоидоз, “легкое фермера” и родственные заболевания, пневмофиброз и идиопатическую интерстициальную пневмонию.
Соединения формулы (I) по изобретению могут быть использованы отдельно или в комбинации с дополнительным средством, например, терапевтическим средством, указанное дополнительно средство выбирается специалистом для назначенной цели. Например, дополнительное средство может представлять собой терапевтическое средство, известное в данной области, как подходящее для лечения этого заболевания или состояния, которое лечат соединением по настоящему изобретению. Дополнительное средство может представлять собой средство, которое придает благоприятное свойство терапевтической композиции, например, средство, которое влияет на вязкость композиции.
Далее должно быть понятно, что комбинации, которые включены в настоящее изобретение, представляют собой комбинации, подходящие для назначенной цели. Средства, представленные ниже, иллюстрируют эти цели и не предназначены для ограничения. Комбинации, которые являются частью настоящего изобретения, могут представлять собой соединения по настоящему изобретению и по меньшей мере одно дополнительное средство, выбранное из представленного ниже списка. Комбинация может также включать более одного дополнительного средства, например, два или три дополнительных средства, если эта комбинация такая, что полученная композиция может выполнять заданную функцию.
Предпочтительными комбинациями являются нестероидное противовоспалительное лекарственное средство(а), также называемые NSAID, которые включают лекарственные средства наподобие ибупрофена. Другими предпочтительными комбинациями являются кортикостероиды, включающие преднизолон; хорошо известные побочные эффекты использования стероидов могут быть снижены или даже устранены путем снижения дозы стероида, необходимой при лечении пациентов, в комбинации соединениями по настоящему изобретению. Неограничивающие примеры терапевтических средств для лечения ревматоидного артрита, с которыми соединение формулы (I) по настоящему изобретению может быть объединено, включают следующие: цитокиновые супрессирующие противовоспалительные средства (CSAID); антитела к, или антагонисты других цитокинов человека или факторов роста, например TNF, LT, IL-1, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL-12, IL-15, IL-16, IL-21, IL-23, интерферонов, EMAP-II, GM-CSF, FGF, MMP-13 и PDGF. Соединения по настоящему изобретению могут быть объединены с антителами к молекулам клеточной поверхности, таким как CD2, CD3, CD4, CD8, CD25, CD28, CD30, CD40, CD45, CD69, CD80 (B7.1), CD86 (B7.2), CD90, CTLA или их лигандам, в том числе CD154 (gp39 или CD40L).
Предпочтительные комбинации терапевтических средств могут вмешиваться в различных точках в аутоиммунный и последующий воспалительный каскад; предпочтительные примеры включают антагонисты TNF наподобие химерных, гуманизированных антител или антител человека против TNF, D2E7 (HUMIRA®), (патент США № 6090382, HUMIRA™), CA2 (REMICADE™), SIMPONI™ (голимумаб), CIMZIA™, ACTEMRA™, CDP 571, и растворимых p55 или p75 TNF рецепторов, их производные, (p75TNFR1gG (ENBREL™) или p55TNFR1gG (Lenercept), а также ингибиторы TNFα превращающего фермента (TACE); аналогично ингибиторы IL-1 (ингибиторы интерлейкин-1-превращающего фермента, IL-1RA и т.д.) могут быть эффективными по той же причине. Другие предпочтительные комбинации включают интерлейкин 11. Еще другими предпочтительными комбинациями являются другие ключевые участники аутоиммунного ответа, которые могут действовать параллельно, зависимо или совместно с функцией IL-18; особенно предпочтительными являются антагонисты IL-12, включающие IL-12 антитела или растворимые IL-12 рецепторы, или IL-12 связывающие белки. Было показано, что IL-12 и IL-18 имеют перекрывающиеся, но различные функции и комбинация антагонистов к каждому из них может быть наиболее эффективной. Еще одни предпочтительные комбинации представляют собой неистощающие анти-CD4 ингибиторы. Еще одни предпочтительные комбинации включают антагонисты костимулирующего пути CD80 (B7.1) или CD86 (B7.2), в том числе антитела, растворимые рецепторы или антагонистические лиганды.
Соединение формулы (I) по настоящему изобретению также можно объединить с такими средствами, как метотрексат, 6-меркаптопурин, азатиоприн, сульфасалазин, месалазин, олсалазин хлорохин/гидроксихлорохин, пенцилламин, ауротиомалат (внутримышечно и перорально), азатиоприн, кохицин, кортикостероиды (перорально, ингаляционно или местной инъекцией), агонисты бета-2 адренорецептора (сальбутамол, тербуталин, салметерал), ксантины (теофиллин, аминофиллин), кромогликат, недокромил, кетотифен, ипратропий и окситропий, циклоспорин, FK506, рапамицин, микофенолят мофетил, лефлуномид, NSAID, например, ибупрофен, кортикостероиды, такие как преднизолон, ингибиторы фосфодиэстеразы, агонисты аденозина, антитромботические средства, ингибиторы комплемента, адренергические средства, средства, которые нарушают передачу сигнала провоспалительными цитокинами, такими как TNFα или IL-1 (например, NIK, IKK, JAK1, JAK2, JAK3, p38 или ингибиторы MAP киназы), ингибиторы IL-1β превращающего фермента, ингибиторы передачи сигнала Т-клеток, такие как ингибитры киназы, ингибиторы металлопротеиназы, сульфасалазин, 6-меркаптопурины, ингибиторы ангиотензин превращающего фермента, растворимые цитокиновые рецепторы и их производные (например, растворимые p55 или p75 TNF рецепторы и производные p75TNFRIgG (Enbrel™) и p55TNFRIgG (Lenercept), sIL-1RI, sIL-1RII, sIL-6R), противовоспалительные цитокины (например, IL-4, IL-10, IL-11, IL-13 и TGFβ), целекоксиб, фолиевая кислота, гидроксихлорохин сульфат, рофекоксиб, этанэрцепт, инфликсимаб, напроксен, валдекоксиб, сульфасалазин, метилпреднизолон, мелоксикам, метилпреднизолон ацетат, ауротиомалат натрия, аспирин, триамцинолон ацетонид, пропоксифен напсилат/апап, фолат, набуметон, диклофенак, пироксикам, этодолак, диклофенак натрия, оксапрозин, оксикодон HCl, гидрокодон битартрат/ацетаминофен, диклофенак натрия/мисопростол, фентанил, анакинра, трамадол HCl, салсалат, сулиндак, цианокобаламин/фа/пиридоксин, ацетаминофен, алендронат натрия, преднизолон, сульфат морфина, лидокаина гидрохлорид, индометацин, глюкозамин сульф/хондроитин, амитриптилин HCl, сульфадиазин, оксикодон HCl/ацетаминофен, олопатадин HCl мизопростол, напроксен натрия, омепразол, циклофосфамид, ритуксимаб, IL-1 TRAP, MRA, CTLA4-IG, IL-18 BP, анти-IL-12, анти-IL15, BIRB-796, SCIO-469, VX-702, AMG-548, VX-740, рофлумиласт, IC-485, CDC-801, агонисты S1P1 (как например, FTY720), ингибиторы семейства PKC (как например, Рубоксистаурин или AEB-071) и и мезопрам. Предпочтительные комбинации включают метотрексат или лефлуномид и в случаях умеренного или тяжелого ревматоидного артрита циклоспорин и анти-TNF антитела, как указано выше.
Неограничивающие примеры терапевтических средств для лечения воспалительного заболевания кишечника, с которыми соединение формулы (I) по настоящему изобретению могут быть объединены, включают следующие: буденозид; эпидермальный фактор роста; кортикостероиды; циклоспорин, сульфасалазин; аминосалицилаты; 6-меркаптопурин; азатиоприн; метронидазол; ингибиторы липоксигеназы; месаламин; олсалазин; балсалазид; антиоксиданты; ингибиторы тромбоксана; антагонисты рецептора IL-1; анти-IL-1β моноклональные антитела; анти-IL-6 моноклональные антитела; факторы роста; ингибиторы эластазы; соединения пиридинил-имидазола; антитела к или антагонисты других цитокинов человека или факторов роста, например TNF, LT, IL-1, IL-2, IL-6, IL-7, IL-8, IL-12, IL-15, IL-16, IL-23, EMAP-II, GM-CSF, FGF, и PDGF; молекулы клеточной поверхности, такие как CD2, CD3, CD4, CD8, CD25, CD28, CD30, CD40, CD45, CD69, CD90 или их лиганды; метотрексат; циклоспорин; FK506; рапамицин; микофенолят мофетил; лефлуномид; NSAID, например ибупрофен; кортикостероиды, такие как преднизолон; ингибиторы фосфодиэстеразы; агонисты аденозина; антитромботические средства; ингибиторы комплемента; адренергические средства; средства, которые препятствуют передаче сигнала провоспалительными цитокинами, такими как TNFα или IL-1 (например, ингибиторы NIK, IKK, p38 или MAP киназы); ингибиторы IL-1β превращающего фермента; ингибиторы TNFα превращающего фермента; ингибиторы T-клеточной передачи сигнала, такие как ингибиторы киназы; ингибиторы металлопротеиназы; сульфасалазин; азатиоприн; 6-меркаптопурины; ингибиторы ангиотензин-превращающего фермента; растворимые цитокиновые рецепторы и их производные (например, растворимые p55 или p75 TNF рецепторы, sIL-1RI, sIL-1RII, sIL-6R) и противовоспалительные цитокины (например, IL-4, IL-10, IL-11, IL-13 and TGFβ). Предпочтительные примеры терапевтических средств для лечения болезни Крона, с которыми соединение формулы (I) может быть объединено, включают следующие: антагонисты TNF, например, анти-TNF антитела, D2E7 (патент США № 6090382; HUMIRA®), CA2 (REMICADETM), CDP 571, конструкции TNFR-Ig, (p75TNFRIgG (ENBREL™) и p55TNFRIgG (LENERCEPT™) ингибиторы, и ингибиторы PDE4. Соединение формулы (I) может быть объединено с кортикостероидами, например, буденозидом и дексаметазоном; сульфасалазином, 5-аминосалициловой кислотой; олсалазином; и средствами, которые препятствуют синтезу или действию провоспалительных цитокинов, таких как IL-1, например ингибиторы IL-1β превращающего фермента и IL-1ra; ингибиторы T-клеточной передачи сигнала, например ингибиторы тирозинкиназы; 6-меркаптопурины; IL-11; месаламин; преднизон; азатиоприн; меркаптопурин; инфликсимаб; метилпреднизолон сукцинат натрия; дифеноксилат/атропсульфат; лоперамида гидрохлорид; метотрексат; омепразол; фолат; ципрофлоксацин/декстроза-вода; гидрокодон битартрат/ацетаминофен; тетрациклина гидрохлорид; флуоцинонид; метронидазол; тимеросал/борная кислота; холестирамин/сахароза; ципрофлоксацина гидрохлорид; гиосциамина сульфат; меперидина гидрохлорид; мидазолама гидрохлорид; оксикодона HCl/ацетаминофен; прометазина гидрохлорид; фосфат натрия; сульфаметоксазол/триметоприм; целекоксиб; поликарбофил; пропоксифен напсилат; гидрокортизон; мультивитамины; балсалазида динатрий; кодеинфосфат/ацетаминофен; колесевалам HCl; цианокобаламин; фолиевая кислота; левофлоксацин; метилпреднизолон; натализумаб и интерферон-гамма.
Неограничивающие примеры терапевтических средств для лечения рассеянного склероза, с которыми соединения формулы (I) могут быть объединены, включают следующие: кортикостероиды; преднизолон; метилпреднизолон; азатиоприн; циклофосфамид; циклоспорин; метотрексат; 4-аминопиридин; тизанидин; интерферон-β1a (Avonex®; Biogen); интерферон-β1b (Betaseron®; Chiron/Berlex); интерферон α-n3) (Interferon Sciences/Fujimoto), интерферон-α (Alfa Wassermann/J&J), интерферон β1A-IF (Serono/Inhale Therapeutics), ПЭГинтерферон α 2b (Enzon/Schering-Plough), Сополимер 1 (Cop-1; COPAXONE®; Teva Pharmaceutical Industries, Inc.); гипербарический кислород; внутривенный иммуноглобулин; кладрибин; антитела к или антагонисты других цитокинов человека или факторов роста или их рецепторов, например, TNF, LT, IL-1, IL-2, IL-6, IL-7, IL-8, IL-12, IL-23, IL-15, IL-16, EMAP-II, GM-CSF, FGF, и PDGF. Соединение формулы (I) может быть объединено с антителами к молекулам клеточной поверхности, таким как CD2, CD3, CD4, CD8, CD19, CD20, CD25, CD28, CD30, CD40, CD45, CD69, CD80, CD86, CD90 или их лигандам. Соединение формулы (I) также можно объединить с такими средствами, как метотрексат, циклоспорин, FK506, рапамицин, микофенолят мофетил, лефлуномид, агонистом S1P1, NSAID, например, ибупрофен, кортикостероиды, такие как преднизолон, ингибиторы фосфодиэстеразы, агонисты аденозина, антитроботические средства, ингибиторы комплемента, адренергические средства, средства, которые препятствуют передаче сигнала провоспалительными цитокинами, такими как TNFα или IL-1 (например, ингибиторы NIK, IKK, p38 или MAP киназы), ингибиторы IL-1β превращающего фермента, ингибиторы TACE, ингибиторы T-клеточной передачи сигнала, такие как ингибиторы киназы, ингибиторы металлопротеиназы, сульфасалазин, азатиоприн, 6-меркаптопурины, ингибиторы ангиотензин-превращающего фермента, растворимые цитокиновые рецепторы и их производные (например, растворимые p55 или p75 TNF рецепторы, sIL-1RI, sIL-1RII, sIL-6R) и противовоспалительные цитокины (например, IL-4, IL-10, IL-13 и TGFβ).
Предпочтительные примеры терапевтических средств для лечения рассеянного склероза, с которыми соединение формулы (I) может быть объединено, включают интерферон-β, например, IFNβ1a и IFNβ1b; копаксон, кортикостероиды, ингибиторы каспазы, например, ингибиторы каспазы-1, ингибиторы IL-1, ингибиторы TNF и антитела против CD40 лиганда и CD80.
Соединение формулы (I) также может быть объединено со средствами, такими как алемтузумаб, дронабинол, даклизумаб, митоксантрон, ксалипроден гидрохлорид, фампридин, глатирамер ацетат, натализумаб, синнабидол, α-иммунокин NNSO3, ABR-215062, AnergiX.MS, антагонисты хемокиновых рецепторов, BBR-2778, калагуалин, CPI-1189, LEM (инкапсулированный в липосому митоксантрон), THC.CBD (агонист каннабиноида), MBP-8298, мезопрам (ингибитор PDE4), MNA-715, антитело против IL-6 рецептора, нейровакс, пирфенидон аллотрап 1258 (RDP-1258), sTNF-R1, талампанел, терифлуномид, TGF-бета2, типлимотид, антагонисты VLA-4 (например, TR-14035, VLA4 Ultrahaler, Antegran-ELAN/Biogen), антагонисты интерферона гамма и агонисты IL-4.
Неограничивающие примеры терапевтических средств для лечения анкилозирующего спондилита, с которыми соединения формулы (I) могут быть объединены, включают следующие: ибупрофен, диклофенак, мизопростол, напроксен, мелоксикам, индометацин, диклофенак, целекоксиб, рофекоксиб, сульфасалазин, метотрексат, азатиоприн, миноциклин, преднизон и антитела против TNF, D2E7 (патент США No 6090382; HUMIRA™), CA2 (REMICADE™), CDP 571, конструкции TNFR-Ig, (p75TNFRIgG (ENBREL™) и p55TNFRIgG (LENERCEPT™).
Неограничивающие примеры терапевтических средств для лечения астмы, с которыми соединения формулы (I) могут быть объединены, включают следующие: альбутерол, салметерол/флутиказон, монтелукаст натрий, флутиказон пропионат, будесонид, преднизон, салметерол ксинафоат, левалбутерол HCl, альбутерол сульфат/ипратропий, преднизолон фосфат натрия, триацинолон ацетонид, беклометазон дипропионат, ипратропия бромид, азитромицин, пирбутерол ацетат, преднизолон, безводный теофиллин, метилпреднизолон сукцинат натрия, кларитромицин, зафирлукаст, формотерол фумарат, вакцину против вируса гриппа, амоксициллина тригидрат, флунизолид, аллергическую инъекцию, кромолин натрий, фексофенадин гидрохлорид, флунизолид/ментол, амоксициллин/клавуланат, левофлоксацин, ингаляционное вспомогательное устройство, гуаифенезин, дексаметазон натрия фосфат, моксифлоксацин HCl, доксициклин хиклат, гуаифенезин/d-меторфан, п-эфедрин/код/хлорфенир, гатифлоксацин, цетиризин гидрохлорид, мометазон фуроат, сальметерол ксинафоат, бензонатат, сефалексин, пе/гидрокодон/хлорфенир, цетиризин HCl/псевдоэфедрин, фенилэфрин/код/прометазин, кодеин/прометазин, цефпрозил, дексаметазон, гуаифенезин/псевдоэфедрин, хлорфенирамин/гидрокодон, недокромил натрия, тербуталин сульфат, эпинефрин, метилпреднизолон, антитела против IL-13 и метапротеренол сульфат.
Неограничивающие примеры терапевтических средств для лечения COPD, с которыми соединения формулы (I) могут быть объединены, включают следующие: альбутерол сульфат/ипратропий, ипратропия бромид, сальметерол/флутиказон, альбутерол, сальметерол ксинафоат, флутиказон пропионат, преднизон, безводный теофиллин, метилпреднизолон сукцинат натрия, монтелукаст натрий, будесонид, формотерол фумарат, триамцинолон ацетонид, левофлоксацин, гуаифенезин, азитромицин, беклометазон дипропионат, левалбутерол HCl, флунизолид, цефтриаксон натрий, амоксициллин тригидрат, гатифлоксацин, зафирлукаст, амоксициллин/клавуланат, флунизолид/ментол, хлорфенирамин/гидрокодон, метапротеренол сульфат, метилпреднизолон, мометазон фуроат, п-эфедрин/код/хлорфенир, пирбутерол ацетат, п-эфедрин/лоратадин, тербуталин сульфат, тиотропия бромид, (R,R)-формотерол, TgAAT, циломиласт и рофлумиласт.
Неограничивающие примеры терапевтических средств для лечения HCV, с которыми соединения формулы (I) могут быть объединены, включают следующие: интерферон-альфа-2α, интерферон-альфа-2β, интерферон-альфа con1, интерферон-альфа-n1, пегилированный интерферон-альфа-2α, пегилированый интерферон-альфа-2β, рибавирин, ПЭГинтерферон альфа-2b + рибавирин, урсодезоксихолиновую кислоту, глицирризиновую кислоту, тималфазин, максамин, VX-497 и любые соединения, которые используются для лечения HCV посредством воздействия на следующие мишени: HCV полимеразу, HCV протеазу, HCV геликазу и HCV IRES (внутренний сайт связывания рибосомы).
Неограничивающие примеры терапевтических средств для лечения идиопатического легочного фиброза, с которыми соединение формулы (I) могут быть объединены, включают следующие: преднизон, азатиоприн, албутерол, колхицин, албутерол сульфат, дигоксин, гамма интерферон, метилпреднизолон сукцинат натрия, лоразепам, фуросемид, лизиноприл, нитроглицерин, спиронолактон, циклофосфамид, ипратропия бромид, актиномицин d, альтеплаз, флутиказон проионат, левофлоксацин, метапротеренол сульфат, морфин сульфат, оксикодон HCl, хлорид калия, триамцинолон ацетонид, безводный такролимус, кальций, интерферон-альфа, метотрексат, микофенолят мофетил и интерферон-гамма-1β.
Неограничивающие примеры терапевтических средств для лечения инфаркта миокарда, с которыми соединение формулы (I) могут быть объединены, включают следующие: аспирин, нитроглицерин, метопролол тартрат, эноксапарин натрий, гепарин натрий, клопидогрел бисульфат, карведилол, атенолол, сульфат морфина, метопролол сукцинат, варфарин натрий, лизиноприл, изосорбид мононитрат, дигоксин, фуросемид, симвастатин, рамиприл, тенектеплаз, эналаприл малеат, торсемид, ретаваз, лозартан калий, хинаприл гидрохлорид/карбонат магния, буметанид, альтеплаз, эналаприлат, амиодарон гидрохлорид, тирофибан HCl м-гидрат, дилтиазем гидрохлорид, каптоприл, ирбесартан, валсартан, пропранолол гидрохлорид, фосиноприл натрий, лидокаина гидрохлорид, эптифибатид, цефазолин натрий, атропина сульфат, аминокапроновая кислота, спиронолактон, интерферон, соталол гидрохлорид, хлорид калия, докузат натрия, добутамин HCl, алпразолам, правастатин натрий, аторвастатин кальций, мидазолам гидрохлорид, меперидин гидрохлорид, изосорбид динитрат, эпинефрин, допамина гидрохлорид, бивалирудин, росувастатин, эзетимиб/симвастатин, авасимиб и карипорид.
Неограничивающие примеры терапевтических средств для лечения псориаза, с которыми соединение формулы (I) могут быть объединены, включают следующие: кальципротриен, клобетазол пропионат, триамцинолон ацетонид, галобетазол пропионат, тазаротен, метотрексат, флуоцинонид, усиленный бетаметазон дипроп, флуоцинолон ацетонид, ацитретин, дегтярный шампунь, бетаметазон валерат, мометазон фуроат, кетоконазол, прамоксин/флуоцинолон, гидрокортизон валерат, флурандренолид, мочевину, бетаметазон, клобетазол пропионат/эмолл, флутиказон пропионат, азитромицин, гидрокортизон, увлажняющий состав, фолиевую кислоту, дезонид, пимекролимус, деготь, дифлоразон диацетат, этанерцепт фолат, молочную кислоту, метоксален, hc/висмут субгал/znox/resor, метилпреднизолон ацетат, преднизон, солнцезащитное средство, галцинонид, салициловую кислоту, антралин, клокортолон пивалат, экстракт угля, деготь/салициловую кислоту, деготь/салициловую кислоту/серу, дезоксиметазон, диазепам, смягчающее средство для кожи, флуоцинонид/смягчающее средство для кожи, минеральное масло/касторовое масло/na lact, минеральное масло/арахисовое масло, нефть/изопропил миристат, псорален, салициловую кислоту, мыло/трибромсалан, тимеросал/борную кислоту, целекоксиб, инфликсимаб, циклоспорин, алефацепт, эфализумаб, такролимус, пимекролимус, PUVA, UVB, сульфасалазин, ABT-874 и устекинумаб.
Неограничивающие примеры терапевтических средств для лечения псориатического артрита, с которыми соединение формулы (I) могут быть объединены, включают следующие: метотрексат, этанерцепт, рофекоксиб, целекоксиб, фолиевую кислоту, сульфасалазин, напроксен, лефлуномид, метилпреднизолон ацетат, индометацин, гидроксихлорохин сульфат, преднизон, сулиндак, усиленный бетаметазон дипропионат, инфликсимаб, метотрексат, фолат, триамцинолон ацетонид, диклофенак, диметилсульфоксид, пироксикам, диклофенак натрий, кетопрофен, мелоксикам, метилпреднизолон, набуметон, толметин натрий, калципотриен, циклоспорин, диклофенак натрий/мизопростол, флуоцинонид, глюкозамин сульфат, ауротиомалят натрия, гидрокодон битартрат/ацетаминофен, ибупрофен, ризедронат натрия, сульфадиазин, тиогуанин, вальдекоксиб, алефацепт, D2E7 (патент США 6090382, HUMIRA™) и эфализумаб.
Неограничивающие примеры терапевтических средств для лечения рестеноза, с которыми соединение формулы (I) могут быть объединены, включают следующие: сиролимус, паклитаксел, эверолимус, такролимус, ABT-578 и ацетаминофен.
Неограничивающие примеры терапевтических средств для лечения воспаления седалищного нерва, с которыми соединение формулы (I) могут быть объединены, включают следующие: гидрокодон битартрат/ацетаминофен, рофекоксиб, циклобензаприн HCl, метилпреднизолон, напроксен, ибупрофен, оксикодон HCl/ацетаминофен, целекоксиб, вальдекоксиб, метилпреднизолон ацетат, преднизон, кодеин фосфат/ацетаминофен, трамадол HCl/ацетаминофен, метаксалон, мелоксикам, метокарбамол, лидокаин гидрохлорид, диклофенак натрий, габапентин, дексаметазон, каризопродол, кеторолак трометамин, индометацин, ацетаминофен, диазепам, набуметон, оксикодон HCl, тизанидин HCl, диклофенак натрий/мизопростол, пропоксифен н-pap, asa/оксикод/оксикодон ter, ибупрофен/гидрокодон бит, трамадол HCl, этодолак, пропоксифен HCl, амитриптилин HCl, каризопродол/кодеин phos/asa, морфина сульфат, мультивитамины, напроксен натрий, орфенадрин цитрат и темазепам.
Предпочтительные примеры терапевтических средств для лечения SLE (волчанки), с которыми соединения формулы (I) могут быть объединены, включают следующие: NSAID, например диклофенак, напроксен, ибупрофен, пироксикам, индометацин; ингибиторы COX2, например, целекоксиб, рофекоксиб, вальдекоксиб; противомалярийные средства, например, гидроксихлорохин; стероиды, например, преднизон, преднизолон, буденозид, дексаметазон; цитотоксические средства, например, азатиоприн, циклофосфамид, микофенолят мофетил, метотрексат; ингибиторы PDE4 или ингибиторы синтеза пуринов, например, Cellcept®. Соединение формулы (I) также можно объединить с такими средствами, как сульфасалазин, 5-аминосалициловая кислота, олсалазин, Imuran® и средствами, которые препятствуют синтезу, продукции или действию провоспалительных цитокинов, таких как IL-1, например, ингибиторы каспазы, наподобие ингибиторов IL-1β превращающего фермента и IL-1ra. Соединение формулы (I) также может быть использовано с ингибиторами Т-клеточной передачи сигнала, например, ингибиторами тирозинкиназы; или молекулами, которые направляют молекулы Т-клеточной активации, например, CTLA-4-IgG или анти-B7 семейство антител, анти-PD-1 семейство антител. Соединение формулы (I) может быть объединено с IL-11 или антицитокиновыми антителами, например, фонотолизумабом (анти-IFNg антитело), или антирецепторными антителами, например, анти-IL-6 рецепторным антителом и антителами к молекулам поверхности B-клеток. Соединение формулы (I) также может быть использовано с LJP 394 (абетимусом), средствами, которые истощают или инактивируют B-клетки, например ритуксимабом (анти-CD20 антителом), лимфостатом-B (анти-BlyS антителом), антагонистами TNF, например, анти-TNF антителами, D2E7 (патент США № 6090382; HUMIRA™), CA2 (REMICADE™), CDP 571, конструкциями TNFR-Ig, (p75TNFRIgG (ENBREL™) и p55TNFRIgG (LENERCEPT™).
В настоящем изобретении используются следующие определения.
«Терапевтически эффективное количество» представляет собой количество соединения формулы (I) или комбинации двух или нескольких таких соединений, которое ингибирует, полностью или частично, прогрессирование патологического состояния или облегчает, по меньшей мере частично, один или более симптомов патологического состояния. Терапевтически эффективным количеством может также быть количество, которое является эффективным для профилактики. Количество, которое является терапевтически эффективным, будет зависеть от размера тела пациента и пола, патологического состояния, подвергаемого лечению, тяжести состояния и желаемого результата. Для конкретного пациента терапевтически эффективное количество может быть определено способами, известными специалистам в данной области.
«Фармацевтически приемлемые соли» относятся к солям, которые сохраняют биологическую эффективность и свойства свободных оснований и которые получены путем взаимодействия с неорганическими кислотами, например, хлористоводородной кислотой, бромистоводородной кислотой, серной кислотой, азотной кислотой и фосфорной кислотой или органическими кислотами, такими как сульфоновая кислота, карбоновая кислота, органическая фосфорная кислота, метансульфоновая кислота, этансульфоновая кислота, п-толуолсульфоновая кислота, лимонная кислота, фумаровая кислота, малеиновая кислота, янтарная кислота, бензойная кислота, салициловая кислота, молочная кислота, винная кислота (например, (+) или (-)-винная кислота или их смеси), аминокислотами (например (+) или (-)аминокислотами или их смесями), и подобным. Эти соли могут быть получены способами, известными специалистам в данной области.
Некоторые соединения формулы (I), которые имеют кислые заместители, могут существовать в виде солей с фармацевтически приемлемыми основаниями. Настоящее изобретение включает такие соли. Примеры таких солей включают соли натрия, соли калия, соли лизина и соли аргинина. Эти соли могут быть получены способами, известными специалистам в данной области.
Некоторые соединения формулы (I) и их соли могут существовать в более чем одной кристаллической форме, и настоящее изобретение включает каждую кристаллическую форму или их смесь.
Некоторые соединения формулы (I) и их соли также могут существовать в форме сольватов, например, гидратов, и настоящее изобретение включает каждый сольват и их смеси.
Некоторые соединения формулы (I) могут содержать один или несколько хиральных центров и существовать в различных оптически активных формах. В тех случаях, когда соединения формулы (I) содержат один хиральный центр, соединения существуют в двух энантиомерных формах, и настоящее изобретение включает как энантиомеры, так и смеси энантиомеров, например, рацемические смеси. Энантиомеры можно разделить способами, известными специалистам в данной области, например, путем образования диастереоизомерных солей, которые могут быть разделены, например, кристаллизацией; путем образования диастереоизомерных производных или комплексов, которые могут быть разделены, например, путем кристаллизации, газожидкостной или жидкостной хроматографией; селективной реакцией одного энантиомера с энантиомер-специфичным реагентом, например, ферментативной этерификацией; или газожидкостной или жидкостной хроматографией в хиральном окружении, например, на хиральной подложке, например, диоксиде кремния со связанным хиральным лигандом или в присутствии хирального растворителя. Будет понятно, что в тех случаях, когда желаемый энантиомер превращается в другое химическое вещество под действием одной из процедур очистки, описанной выше, требуется дополнительная стадия для высвобождения желаемой энантиомерной формы. Альтернативно, специфические энантиомеры могут быть синтезированы путем асимметричного синтеза с использованием оптически активных реагентов, субстратов, катализаторов или растворителей или путем превращения одного энантиомера в другой путем асимметричной трансформации.
В тех случаях, когда соединение формулы (I) содержит более одного хирального центра, оно может существовать в диастереоизомерных формах. Диастереоизомерные соединения могут быть разделены способами, известными специалистам в данной области, например, хроматографически или кристаллизацией, и отдельные энантиомеры могут быть разделены, как описано выше. Настоящее изобретение включает каждый диастереоизомер соединений формулы (I) и их смеси.
Некоторые соединения формулы (I) могут существовать в различных таутомерных формах или в виде различных геометрических изомеров, и настоящее изобретение включает каждый таутомер и/или геометрический изомер соединений формулы (I) и их смеси.
Некоторые соединения формулы (I) могут существовать в различных стабильных конформационных формах, которые могут быть разделяемыми. Торсионная асимметрия вследствие ограниченного вращения около асимметричной одинарной связи, например, вследствие пространственного затруднения или напряжения кольца, может позволить разделить различные конформеры. Настоящее изобретение включает каждый конформационный изомер соединений формулы (I) и их смеси.
Некоторые соединения формулы (I) могут существовать в цвиттерионной форме, и настоящее изобретение включает каждую цвиттерионную форму соединений формулы (I) и их смеси.
Как используется в настоящем изобретении, термин «пролекарство» относится к средству, которое превращается в исходное лекарственное средство in vivo под действием некоторых физиологических химических процессов (например, пролекарство при приведении к физиологическому pH превращается в желаемую лекарственную форму). Пролекарства зачастую являются подходящими, поскольку в некоторых ситуациях их можно легче вводить, чем исходное лекарственное средство. Они могут, например, быть биодоступными посредством перорального введения, тогда как исходное лекарственное средство - нет. Пролекарство также может иметь улучшенную растворимость в фармакологических композициях по сравнению с исходным лекарственным средством. Например, без ограничения, пролекарством было бы соединение по настоящему изобретению, где его вводят в виде сложного эфира («пролекарство») для облегчения прохождения через клеточную мембрану, где водная растворимость не является благоприятной, но затем оно метаболически гидролизуется в карбоновую кислоту уже внутри клетки, где водная растворимость является благоприятной.
Пролекарства имеют многие полезные свойства. Например, пролекарство может быть более водорастворимым, чем конечное лекарственное средство, тем самым облегчая внутривенное введение лекарственного средства. Пролекарство также может иметь более высокий уровень пероральной биодоступности, чем конечное лекарственное средство. После введения пролекарство ферментативно или химически расщепляется для доставки конечного лекарственного средства в кровь или ткань.
Приводимые в качестве примера пролекарства при расщеплении высвобождают соответствующую свободную кислоту, и такие гидролизуемые эфиробразующие остатки соединений по изобретению включают, но ими не ограничиваются, заместители карбоновой кислоты, где свободный водород замещен (C1-C4)алкилом, (C2-C12)алканоилоксиметилом, (C4-C9)1-(алканоилокси)этилом, 1-метил-1-(алканоилокси)этилом, имеющим от 5 до 10 атомов углерода, алкоксикарбонилоксиметилом, имеющим от 3 до 6 атомов углерода, 1-(алкоксикарбонилокси)этилом, имеющим от 4 до 7 атомов углерода, 1-метил-1-(алкоксикарбонилокси)этилом, имеющим от 5 до 8 атомов углерода, N-(алкоксикарбонил)аминометилом, имеющим от 3 до 9 атомов углерода, 1-(N-(алкоксикарбонил)амино)этилом, имеющим от 4 до 10 атомов углерода, 3-фталидилом, 4-кротонолактонилом, гамма-бутиролактон-4-илом, ди-N,N-(C1-C2)алкиламино(C2-C3)алкилом (таким как β-диметиламиноэтил), карбамоил-(C1-C2)алкилом, N,N-ди(C1-C2)-алкилкарбамоил-(C1-C2)алкилом и пиперидино-, пирролидино- или морфолино(C2-C3)алкилом.
Другие приводимые в качестве примера пролекарства высвобождают спирт формулы (I), где свободный водород гидроксильного заместителя (например, R1 содержит гидроксил), замещен (C1-C6)алканоилоксиметилом, 1-((C1-C6)алканоилокси)этилом, 1-метил-1-((C1-C6)алканоилокси)этилом, (C1-C6)алкоксикарбонилоксиметилом, N-(C1-C6)алкоксикарбониламинометилом, сукциноилом, (C1-C6)алканоилом, α-амино(C1-C4)алканоилом, арилактилом и α-аминоацилом или α-аминоацил-α-аминоацилом, где указанные α-аминоацильные части независимо представляют собой любую из природных L-аминокислот, обнаруженных в белках, P(O)(OH)2, -P(O)(O(C1-C6)алкил)2 или гликозил (радикал, полученный в результате отсоединения гидроксила полуацеталя углевода).
Как используется в настоящем описании, термин “мостиковая (C5-C12)циклоалкильная группа” обозначает насыщенную или ненасыщенную, бициклическую или полициклическую мостиковую углеводородную группу, содержащую два или три C3-C10 циклоалкильных кольца. Немостиковые циклоалкилы исключены. Мостиковые циклические углеводороды могут включать такие, как бицикло[2.1.1]гексил, бицикло[2.2.1]гептил, бицикло[2.2.2]октил, бицикло[3.2.1]октил, бицикло[4.3.1]децил, бицикло[3.3.1]нонил, борнил, боренил, норборнил, норборненил, 6,6-диметилбицикло[3.1.1]гептил, трициклобутил и адамантил.
Как используется в настоящем описании, термин “мостиковый (C2-C10)гетероциклил” обозначает бициклические или полициклические аза-мостиковые углеводородные группы и может включать азанорборнил, хинуклидинил, изохинуклидинил, тропанил, азабицикло[3.2.1]октанил, азабицикло[2.2.1]гептанил, 2-азабицикло[3.2.1]октанил, азабицикло[3.2.1]октанил, азабицикло[3.2.2]нонанил, азабицикло[3.3.0]нонанил и азабицикло[3.3.1]нонанил.
Термин «гетероциклический», «гетероциклил» или «гетероциклилен», используемый в контексте настоящего изобретения, включает неароматические кольцевые системы, включающие, но ими не ограничивающиеся, моноциклические, бициклические, трициклические и спироциклические кольца, которые могут быть полностью насыщенными или которые могут содержать одну или более единиц ненасыщенности (для большей точности, степень ненасыщенности не приводит в результате к ароматической кольцевой системе), и имеет от 5 до 12 атомов, включающих по меньшей мере один гетероатом, такой как азот, кислород или сера. С иллюстрирующей целью, которую не следует рассматривать как ограничивающую объем настоящего изобретения, следующее представляет собой примеры гетероциклических колец: азепинил, азетидинил, индолинил, изоиндолинил, морфолинил, пиперазинил, пиперидинил, пирролидинил, хинуклидинил, тиоморфолинил, тетрагидропиранил, тетрагидрофуранил, тетрагидроиндолил, тиоморфолинил и тропанил.
Термин «гетероарил» или «гетероарилен», используемый в настоящем изобретении, включает ароматические кольцевые системы, в том числе, но не ограничиваясь перечисленным, моноциклические, бициклические и трициклические кольца, которые имеют от 5 до 12 атомов, содержащие по меньшей мере один гетероатом, такой как азот, кислород или сера. С целью иллюстрации, которую не следует считать ограничивающей объем настоящего изобретения: азаиндолил, бензо(b)тиенил, бензимидазолил, бензофуранил, бензоксазолил, бензотиазолил, бензотиадиазолил, бензоксадиазолил, фуранил, имидазолил, имидазопиридил, индолил, индазолил, изоксазол, изотиазолил, оксадиазолил, оксазолил, пуринил, пиранил, пиразинил, пиразолил, пиридинил, пиримидинил, пирролил, пирроло[2,3-d]пиримидинил, пиразоло[3,4-d]пиримидинил, хинолинил, хиназолинил, триазолил, тиазолил, тиофенил, тетразолил, тиадиазолил, тиенил, 6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразинил, 6H-имидазо[1,5-a]пирроло[2,3-e]пиразинил, 1,6-дигидропиразоло[3,4-d]пирроло[2,3-b]пиридин, 3H-3,4,6,8a-тетрааза-as-индаценил, 3H-имидазо[1,2-a]пирроло[2,3-e]пиразинил, пиразоло[3,4-d]пирроло[2,3-b]пиридинил, 1,6-дигидро-1,2,5,6-тетраза-as-индаценил, 3H-3,4,8a-триаза-as-индаценил, 6H-3-окса-2,5,6-триаза-as-индаценил, 3,6-дигидро-2,3,6-тетрааза-as-индаценил, 1,6-дигидро-дипирроло[2,3-b;2’3’-d]пиридинил, 6H-3-тиа-2,5,6-триаза-as-индаценил или 1,6-дигидроимидазо[4,5-d]пирроло[2,3-b]пиридин.
Как используется в контексте настоящего изобретения, “алкил”, “алкилен” или обозначения, такие как “(C1-C8)” включает углеводороды с прямой или разветвленной цепью, которые являются полностью насыщенными. Предпочтительными алкилами являются метил, этил, пропил, изопропил, бутил, пентил, гексил и их изомеры. Используемый в контексте настоящего изобретения “алкенил”, “алкенилен”, “алкинилен” и “алкинил” означает C2-C8 и включает углеводороды с прямой или разветвленной цепью, которые содержат одну или более единиц ненасыщенности, одну или более двойных связей для алкенила и одну или более тройных связей для алкинила.
Как используется в контексте настоящего изобретения, “ароматические” группы (или “арильные” или “ариленовые” группы) включают ароматические карбоциклические кольцевые системы (например, фенил) и конденсированные полициклические ароматические кольцевые системы (например, нафтил, бифенил и 1,2,3,4-тетрагидронафтил).
Как используется в настоящем изобретении, “циклоалкил” или “циклоалкилен” означают C3-C12 моноциклические или полициклические (например, бициклические, трициклические, спироциклические и т.д.) углеводороды, которые полностью насыщены. Примеры циклоалкильных групп представляют собой циклопропил, циклобутил, циклопентил и циклогексил.
Как используется в настоящем изобретении “циклоалкенил” означает C3-C12 моноциклические или полициклические (например, бициклические, трициклические, спироциклические и т.д.) углеводороды, которые имеют одну или более из ненасыщенных связей, но это количество не приводит к образованию ароматической группы. Примеры циклоалкенильных групп представляют собой циклопентенил и циклогексенил.
Как используется в настоящем изобретении, многие фрагменты или заместители именуют как или “замещенные” или “необязательно замещенные”. Если фрагмент обозначен одним из этих терминов, если нет других указаний, это означает, что любая часть фрагмента, который, как известно специалистам в данной области, доступен для замещения, может быть замещен, что включает один или более из заместителей, и где имеется более одного заместителя, тогда каждый заместитель выбирают независимо. Такие обозначения для замещения хорошо известны специалистам в данной области и/или приняты в настоящем описании. В качестве примеров, которые не следует рассматривать как ограничивающие объем настоящего изобретения, можно привести некоторые группы, которые являются заместителями: (C1-C8)алкильные группы, (C2-C8)алкенильные группы, (C2-C8)алкинильные группы, (C3-C10)циклоалкильные группы, галоген (F, Cl, Br или I), галогенированные (C1-C8)алкильные группы (например, но им не ограничиваясь, -CF3), -O-(C1-C8)алкильные группы, =O, =CH2, -OH, -CH2OH, -CH2NH2, (C1-C4)алкил-OH, -CH2CH2OCH2CH3, -S-(C1-C8)алкильные группы, -SH, -NH(C1-C8)алкильные группы, -N((C1-C8)алкил)2 группы, -NH2, -C(O)NH2, -CH2NHC(O)(C1-C4)алкил, -CH2NHC(O)CH2Cl, -CH2NHC(O)CH2CN, -CH2NHC(O)CH2CH2N(CH3)2, -CH2NHC(O)C(=CH2)CH3, -CH2NHC(O)(C2-C4)алкинил, -CH2NHC(O)CH2CH2-пиперидинил, -(C1-C4)алкил-морфолинил, -CH2NHC(O)CH2O-фенил, где фенил, необязательно замещен галогеном, (C1-C4)алкокси, -C(O)(C1-C4)алкилом, -C(O)(C1-C4)алкокси, -C(O)N(H)2, -C(O)N(CH3)2, -C(O)(C1-C6)гетероарилом, -N(CH3)2, -NHC(O)(C1-C4)алкилом, -NHC(O)(C2-C4)алкенилом, -NHC(O)CH2CN, -S(O)2(C1-C4)алкилом, -S(O)2(C1-C6)гетероарилом, -S(O)2(C1-C6) (C1-C6)гетероциклилом, 4-метилпиперазинкарбонилом, -(C1-C4)алкилC(O)NH2, -C(O)NH(C1-C8)алкильными группами, -C(O)N((C1-C8)алкил)2, -C(O)N(H)(C3-C8)циклоалкильными группами, -C(O)(C1-C4)алкокси, -NHC(O)H, -NHC(O)(C1-C8)алкильными группами, -NHC(O)(C3-C8)циклоалкильными группами, -N((C1-C8)алкил)C(O)H, -N((C1-C8)алкил)C(O)(C1-C8)алкильными группами, -NHC(O)NH2, -NHC(O)NH(C1-C8)алкильными группами, -N((C1-C8)алкил)C(O)NH2 группами, -NHC(O)N((C1-C8)алкил)2 группами, -N((C1-C8)алкил)C(O)N((C1-C8)алкил)2 группами, -N((C1-C8)алкил)C(O)NH((C1-C8)алкил), -NHCH2-гетероарилом, бензил, -OCH2-гетероарилом, -C(O)H, -C(O)(C1-C8) алкильными группами, -CN, -NO2, -S(O)(C1-C8) алкильными группами, -S(O)2(C1-C8)алкильными группами, -S(O)2N((C1-C8)алкил)2 группами, -S(O)2NH(C1-C8)алкильными группами, -S(O)2NH(C3-C8)циклоалкильными группами, -S(O)2NH2 группами, -NHS(O)2(C1-C8)алкильными группами, -N((C1-C8)алкил)S(O)2(C1-C8)алкильными группами, -(C1-C8)алкил-O-(C1-C8)алкильными группами, -O-(C1-C8)алкил-O-(C1-C8)алкильными группами, -C(O)OH, -C(O)O(C1-C8)алкильными группами, NHOH, NHO(C1-C8)алкильными группами, -O-галогенированные (C1-C8)алкильные группы (например, но им не ограничиваясь, -OCF3), -S(O)2-галогенированные (C1-C8)алкильные группы (например, но им не ограничиваясь, -S(O)2CF3), -S-галогенированные (C1-C8)алкильные группы (например, но им не ограничиваясь, -SCF3), -(C1-C6)гетероциклил (например, но им не ограничиваясь, пирролидин, тетрагидрофуран, пиран или морфолин), -(C1-C6)гетероарил (например, но им не ограничиваясь, тетразол, имидазол, фуран, пиразин или пиразол), -фенил, необязательно замещенный бензил, -NHC(O)O-(C1-C6)алкильные группы, -N((C1-C6)алкил)C(O)O-(C1-C6)алкильные группы, -C(=NH)-(C1-C6)алкильные группы, -C(=NOH)-(C1-C6)алкильные группы, или -C(=N-O-(C1-C6)алкил)-(C1-C6)алкильные группы.
Термин "набор", используемый в настоящем описании, относится к упакованному продукту, содержащему компоненты с которыми можно вводить соединение формулы (I) по настоящему изобретению для лечения аутоиммунных нарушений. Набор предпочтительно включает коробку или контейнер, который содержит компоненты набора. Коробка или контейнер снабжен меткой или одобренным управлением по контролю за продуктами и лекарствами протоколом. Коробка или контейнер содержит компоненты согласно изобретению, которые предпочтительно упакованы в пластиковые, полиэтиленовые, полипропиленовые, этиленовые или пропиленовые сосуды. Сосуды могут быть тюбиками с крышкой или бутылочками. Набор может также включать инструкции по введению соединения формулы (I).
Одно или более из соединений настоящего изобретения можно вводить пациентам-людям как они есть, или в фармацевтических композициях, где их смешивают с биологически подходящими носителями или эксципиентом (эксципиентами) в дозах, достаточных для лечения или облегчения заболевание или состояния, как раскрыто в настоящем описании. Смеси указанных соединений можно также вводить пациенту в виде простой смеси или в виде подходящим образом приготовленной фармацевтической композиции. Понятие терапевтически эффективная доза относится к такому количеству соединения или соединений, которого достаточно для обеспечения профилактики или для ослабления заболевания или состояния, как раскрыто в настоящем описании. Методики приготовления лекарственного средства и введения соединений рассматриваемой заявки можно найти в ссылках, которые хорошо известны специалистам в данной области, таких как “Remington's Pharmaceutical Sciences”, Mack Publishing Co., Easton, Pa., последнее издание.
Подходящие способы введения могут, например, включать пероральное, с помощью глазных капель, ректальное, трансмукозальное, местное или интестинальное введение; парентеральную доставку, включая внутримышечные, подкожные, интрамедуллярные инъекции, также как интратекальные, прямые интравентрикулярные, внутривенные, интраперитонеальные, интраназальные или интраокулярные инъекции.
Альтернативно, можно вводить указанное соединение локально, а не системно, например, за счет инъекции соединения непосредственно в отечную область, часто в виде депо-препаратов или композиций с замедленным выделением.
Кроме того, можно вводить лекарственное средство в виде направленной системы доставки лекарственного средства, например, в виде липосомы, покрытой эндотелиальным клетко-специфическим антителом.
Фармацевтические композиции по настоящему изобретению можно получить известными способами, например, с помощью процессов обычного перемешивания, растворения, гранулирования, изготовления драже, растирания в порошок, эмульгирования, инкапсулирования, включения или лиофилизации.
Таким образом, фармацевтические композиции для использования в соответствии с настоящим изобретением можно получать обычными способами, используя один или более из физиологически приемлемых носителей, включающих эксципиенты и вспомогательные вещества, которые облегчают переработку активных соединений в препараты, которые можно использовать фармацевтически. Подходящие формы зависят от выбранного способа введения.
Для инъекций средства по настоящему изобретению можно приготовить в виде водных растворов, предпочтительно в физиологически совместимых буферах, таких как раствор Хэнка, раствор Рингера или физиологический раствор. Для трансмукозального введения в препаратах используют пенетранты, подходящие для преодоления барьера. Такие пенетранты обычно известны специалистам в данной области.
Для перорального введения соединения могут быть получены просто путем объединения активных соединений с фармацевтически приемлемыми носителями, хорошо известными в данной области. Такие носители позволяют получать соединения по настоящему изобретению в виде таблеток, пилюль, драже, капсул, жидкостей, гелей, сиропов, густых суспензий, суспензий и тому подобного для перорального приема пациентом, проходящим лечение. Фармацевтические препараты для перорального применения могут быть получены путем объединения активного соединения с твердым эксципиентом, необязательно измельчения полученной в результате смеси, и обработки смеси гранул с последующим добавлением подходящих дополнительных веществ, по желанию, для получения ядер таблеток или драже. Подходящими эксципиентами являются, в частности, наполнители, такие как сахара, в том числе лактоза, сахароза, маннитол или сорбитол; препараты целлюлозы, например, такие как кукурузный крахмал, пшеничный крахмал, рисовый крахмал, картофельный крахмал, желатин, трагакантовая камедь, метилцеллюлоза, гидроксипропилметилцеллюлоза, карбоксиметилцеллюлоза натрия и/или поливинилпирролидон (PVP). При желании могут быть добавлены дезинтеграны, такие как поперечносшитый поливинилпирролидон, агар или альгиновая кислота, или их соли, такие как альгинат натрия.
Ядра драже обеспечивают подходящими покрытиями. Для этой цели могут быть использованы концентрированные растворы сахара, которые необязательно могут содержать аравийскую камедь, тальк, поливинилпирролидон, гель карбопола, полиэтиленкликоль и/или диоксид титана, растворы лака, и подходящие орагничекие растворители или смеси растворителей. Красители или пигменты могут быть добавлены в покрытия для таблеток или драже для идентификации или для обозначения различных комбинаций доз активного соединения.
Фармацевтические препараты, которые могут быть использованы перорально, включают твердые капсулы, изготовленные из желатина, а также мягкие герметично закрытые капсулы, изготовленные из желатина и пластификатора, такого как глицерин или сорбит. Твердые капсулы могут содержать активные ингредиенты в смеси с наполнителем, таким как лактоза, связующими, такими как крахмалы, и/или смазывающие вещества, такие как тальк или стеарат магния, и, необязательно, стабилизаторами. В мягких капсулах активные соединения могут быть растворены или суспендированы в подходящих жидкостях, таких как жирные масла, жидкий парафин или жидкие полиэтиленгликоли. Кроме того, могут быть добавлены стабилизаторы. Все композиции для перорального введения должны быть в дозах, подходящих для такого введения.
Для трансбуккального введения композиции могут принимать форму таблеток или пастилок для рассасывания, полученных обычным способом.
Для введения с помощью ингаляций соединения для использования в соответствии с настоящим изобретением, обычно поставляют в форме аэрозольных спреев, заключенных в контейнеры под давлением, или небулайзеров, в которых используют подходящий пропеллент, например, дихлордифторметан, трихлорфторметан, дихлортетрафторэтан, диоксид углерода или другой подходящий газ. В случае аэрозоля под давлением единичную дозу может обеспечивать клапан, отмеряющий определенное количество. Капсулы и картриджи, например, из желатина, для использования в ингаляторе или в инсуффляторе могут быть приготовлены так, чтобы содержать смесь порошков соединения и подходящего базового порошка, такого как лактоза или крахмал.
Соединения могут быть получены для парентерального введения путем инъекции, например, болюсной инъекции или непрерывной инфузии. Составы для инъекции могут быть представлены в единичной лекарственной форме, например, в ампулах или контейнерах с многократными дозами, с добавленным консервантом. Композиции могут принимать такие формы, как суспензии, растворы или эмульсии в масляных или водных носителях, и могут содержать вспомогательные средства, такие как суспендирующие, стабилизирующие и/или диспергирующие средства.
Фармацевтические составы для парентерального введения включают водные растворы активных соединений в водорастворимой форме. Дополнительно, суспензии активных соединений могут быть получены в виде соответствующих масляных инъекционных суспензий. Подходящие липофильные растворители или носители включают жирные масла, такие как кунжутное масло, или сложные эфиры синтетических жирных кислот, такие как этилолеат или триглицериды, или липосомы. Водные инъекционные суспензии могут содержать вещества, которые повышают вязкость суспензии, такие как карбоксиметилцеллюлоза натрия, сорбит или декстран. Необязательно суспензия также может содержать подходящие стабилизаторы или средства, которые повышают растворимость соединения для получения высококонцентрированных растворов.
Альтернативно, активный ингредиент может быть в форме порошка для разбавления подходящим наполнителем, например, стерильной апирогенной водой, до применения.
Соединения также могут быть получены в виде ректальных композиций, таких как суппозитории или удерживающие клизмы, например, содержащие обычные основания для суппозиториев, такие как масло какао или другие глицериды.
В дополнение к составам, описанным ранее, соединения также могут быть получены в виде депо препаратов. Такие составы пролонгированного действия можно вводить путем имплантации (например, подкожно или внутримышечно или путем внутримышечной инъекции). Таким образом, например, соединения могут быть получены с подходящими полимерными или гидрофобными веществами (например, в виде эмульсии в подходящем масле) или ионообменными смолами, или в виде умеренно растворимых производных, например, в виде умеренно растворимой соли.
Примером фармацевтического носителя для гидрофобных соединений по настоящему изобретению является система сорастворителей, содержащая бензиловый спирт, неполярное поверхностно-активное вещество, смешивающийся с водой органический полимер и водную фазу. Система сорастворителей может представлять собой систему сорастворителей VPD. VPD представляет собой раствор 3% масс./об. бензилового спирта, 8% масс./об. неполярного поверхностно-активного вещества полисорбат 80 и 65% масс./об. полиэтиленгликоля 300, доведенный до объема в абсолютном этаноле. Система сорастворителей VPD (VPD:5W) состоит из VPD, разведенного 1:1 5% декстрозой в водном растворе. Эта система сорастворителей хорошо растворяет гидрофобные соединения, и сама дает низкую токсичность при системном введении. Разумеется, пропорции системы сорастворителей могут варьироваться в значительной степени без нарушения ее растворимости и характеристик токсичности. Более того, идентичные компоненты сорастворителей могут быть изменены: например, могут быть использованы другие неполярные поверхностно-активные вещества с низкой токсичностью вместо полисорбата 80; размер фракции полиэтиленгликоля может быть изменен; другие биосовместимые полимеры могут заменять полиэтиленгликоль, например поливинилпирролидон; и другие сахара или полисахариды могут заменять декстрозу.
Альтернативно, могут быть использованы другие системы доставки для гидрофобных фармацевтических соединений. Липосомы и эмульсии являются хорошо известными примерами наполнителей или носителей системы доставки для гидрофобных лекарственных средств. Также могут быть использованы определенные органические растворители, такие как диметилсульфоксид, хотя обычно ценой большей токсичности. Дополнительно, соединения могут быть доставлены с использованием системы замедленного высвобождения, такой как полупроницаемые матрицы твердых гидрофобных полимеров, содержащих терапевтическое средство. Различные материалы для замедленного высвобождения были установлены и хорошо известны специалистам в данной области. Капсулы замедленного высвобождения могут в зависимости от их химической природы высвобождать соединения в течение от нескольких недель вплоть до 100 дней. В зависимости от химической природы и биологической стабильности терапевтического реагента могут быть использованы дополнительные стратегии стабилизации белков.
Фармацевтические композиции также могут содержать подходящие твердые или гелеобразные носители или эксципиенты. Примеры таких носителей или эксципиентов включают, но ими не ограничиваются, карбонат кальция, фосфат кальция, различные сахара, крахмалы, производные целлюлозы, желатин и полимеры, такие как полиэтиленгликоли.
Многие соединения по настоящему изобретению могут быть предоставлены в виде солей с фармацевтически приемлемыми противоионами. Фармацевтически совместимые соли могут быть образованы со многими кислотами, в том числе, но ими не ограничиваются, соляной, серной, уксусной, молочной, винной, яблочной, янтарной, и т.д. Соли имеют тенденцию быть более растворимыми в водных или других протонных растворителях, чем соответствующие формы свободных оснований.
Фармацевтические композиции, подходящие для использования в настоящем изобретении, включают композиции, где активные ингредиенты содержатся в эффективном количестве для достижения намеченной цели. Более конкретно, терапевтически эффективное количество означает количество, эффективное для профилактики развития или для облегчения существующих симптомов у пациента, проходящего лечение. Определение эффективных количеств находится в компетенции специалистов в данной области.
Для любого соединения, используемого в способе по настоящему изобретению, терапевтически эффективная доза может быть определена изначально на основании клеточных исследований. Например, доза может быть получена на клеточных или животных моделях для достижения диапазона циркулирующей концентрации, который включает IC50, определенную в клеточных исследованиях (например, концентрация испытуемого соединения, которое осуществляет половину максимального ингибирования заданной активности протеинкиназы). В некоторых случаях целесообразно определять IC50 в присутствии 3-5% сывороточного альбумина, поскольку такое определение аппроксимирует эффекты связывания белков плазмы на соединение. Такая информация может быть использована для более точного определения доз, применяемых у людей. Далее, наиболее предпочтительные соединения для системного введения эффективно ингибируют протеинкиназа передачу сигнала в интактных клетках на уровнях, которые безопасно достигаются в плазме.
Терапевтически эффективные дозы относятся к такому количеству соединения, которое приводит к облегчению симптомов у пациента. Токсичность и терапевтическая эффективность таких соединений может быть определена стандартными фармацевтическими процедурами в клеточных культурах или у экспериментальных животных, например, для определения максимально переносимой дозы (MTD) и ED50 (эффективной дозы для 50% максимального ответа). Отношение доз между токсическим и терапевтическим эффектами представляет собой терапевтический индекс и может быть выражено как соотношение между MTD и ED50. Соединения, которые демонстрируют высокие терапевтические индексы, являются предпочтительными. Данные, полученные в результате анализа клеточных культур и исследований на животных, могут быть использованы для составления интервала доз для применения у людей. Дозы таких соединений предпочтительно лежат в диапазоне циркулирующих концентраций, которые включают ED50 с небольшой токсичностью или без токсичности. Доза может варьировать в пределах этого интервала в зависимости от используемой лекарственной формы и пути введения. Точный состав, путь введения и доза могут быть выбраны персональным лечащим врачом с точки зрения состояния пациента. (см., например, Fingl et al., 1975, in The Pharmacological Basis of Therapeutics, Ch. 1, p. 1). При лечении кризов, экстренном введении болюса или инфузионном подходе MTD может потребоваться для получения быстрого ответа.
Количество доз и интервал могут быть подобраны индивидуально для обеспечения уровней в плазме активного вещества, которых достаточно для поддержания эффектов модулирования киназ, или минимальной эффективной концентрации (MEC). MEC будет варьироваться для каждого соединения, но может быть определена по данным in vitro; например, концентрация, необходимая для достижения 50-90% ингибирования протеинкиназ, с использованием анализов, описанных в настоящем изобретении. Дозы, необходимые для достижения MEC, будут зависеть от индивидуальных характеристик и пути введения. Однако ВЭЖХ анализ или биологические исследования могут быть использованы для определения концентраций в плазме.
Интервалы доз также можно определить, используя значение MEC. Соединения следует вводить, используя схему, которая поддерживает уровни в плазме выше MEC в течение 10-90% времени, предпочтительно между 30-90% и наиболее предпочтительно между 50-90% до достижения желаемого облегчения симптомов. В случаях местного введения или селективного захвата эффективная местная концентрация лекарственного средства может не быть сопоставимой с концентрацией в плазме.
Количество вводимой композиции будет, разумеется, зависеть от пациента, проходящего лечение, от массы тела пациента, тяжести заболевания, способа введения и мнения лечащего врача.
Композиции могут, при желании, быть представлены в упаковке или дозирующем устройстве, которые могут содержать одну или более единичных лекарственных форм, содержащих активный ингредиент. Упаковка может, например, содержать металлическую фольгу или полимерную пленку, такую как блистерная упаковка. Упаковка или дозатор могут быть снабжены инструкциями по применению. Композиции, содержащие соединение по настоящему изобретению, полученные в совместимом фармацевтическом носителе, также могут быть получены, помещены в соответствующий контейнер и маркированы для лечения указанного состояния.
В некоторых составах может быть полезным использовать соединения по настоящему изобретению в форме частиц очень маленького размера, например, полученные путем измельчения в кипящем слое.
Применение соединений по настоящему изобретению в производстве фармацевтических композиций проиллюстрировано следующим описанием. В настоящем описании термин «активное соединение» означает любое соединение по настоящему изобретению, но в особенности любое соединение, которое является конечным продуктом одного из следующих примеров.
a) Капсулы
При получении капсул 10 частей по массе активного соединения и 240 частей по массе лактозы могут быть дезагрегированы и перемешаны. Этой смесью можно заполнить твердые желатиновые капсулы, каждая капсула содержит единичную дозу или часть единичной дозы активного соединения.
b) Таблетки
Таблетки могут быть получены, например, из следующих ингредиентов.
Активное соединение, лактозу и некоторое количество крахмала, можно деагрегировать, перемешать, и полученную в результате смесь можно гранулировать с раствором поливинилпирролидона в этаноле. Сухой гранулят может быть перемешан со стеаратом магния и оставшимся крахмалом. Затем смесь прессуют в таблеточной машине с получением таблеток, каждая из которых содержит единичную дозу или часть единичной дозы активного соединения.
c) Таблетки, покрытые кишечнорастворимой оболочкой
Таблетки могут быть получены способом, описанным выше в пункте (b). На таблетки может быть нанесено кишечнорастворимое покрытие обычным способом, используя раствор 20% ацетатфталата целлюлозы и 3% диэтилфталата в этаноле:дихлорметане (1:1).
d) Суппозитории
При получении суппозиториев, например, 100 частей по массе активного соединения может быть включено в 1300 частей по массе триглицеридной основы для суппозиториев, и смесь формируют в суппозитории, каждый из которых содержит терапевтически эффективное количество активного ингредиента.
В композициях по настоящему изобретению активное соединение при желании может быть связано с другими совместимыми фармацевтически активными ингредиентами. Например, соединения по настоящему изобретению можно вводить в комбинации с другим терапевтическим средством, которое, как известно, лечит заболевание или патологическое состояние, описанное в настоящем изобретении. Например, с одним или более дополнительными фармацевтическими средствами, которые ингибируют или предотвращают продукцию VEGF или ангиопоэтинов, ослабляют внутриклеточные реакции на VEGF или ангиопоэтины, блокируют внутриклеточную передачу сигнала, ингибируют гиперпроницаемость сосудов, снижают воспаление или ингибируют или предотвращают образование отеков или неоваскуляризацию. Соединения по настоящему изобретению можно вводить до, после или одновременно с дополнительным фармацевтическим средством независимо от применяемого курса введения. Дополнительные терапевтические средства включают, но не ограничиваясь перечисленным, противоотечные стероиды, NSAID, ингибиторы ras, анти-TNF средства, анти-IL1 средства, антигистаминные средства, PAF-антагонисты, ингибиторы COX-1, ингибиторы COX-2, ингибиторы NO синтазы, ингибиторы Akt/PTB, ингибиторы IGF-1R, ингибиторы PKC, ингибиторы PI3 киназы, ингибиторы кальциневрина и иммуносупрессанты. Соединения по настоящему изобретению и дополнительные фармацевтические средства действуют либо суммарно, либо синергетически. Таким образом, введение такой комбинации веществ, которые ингибируют ангиогенез, сосудистую гиперпроницаемость и/или ингибируют образование отеков, может обеспечить большее облегчение повреждающих действий гиперпролиферативного нарушения, ангиогенеза, сосудистой гиперпроницаемости или отека, чем введение любого из веществ по отдельности. При лечении злокачественных заболеваний комбинации с антипролиферативными или цитотоксическими химиотерапевтическими средствами или излучением включены в объем настоящего изобретения.
Настоящее изобретение также включает применение соединения формулы (I) в качестве лекарственного средства.
Дополнительный аспект настоящего изобретения относится к применению соединения формулы (I) или его солей в производстве лекарственного средства для лечения сосудистой гиперпроницаемости, ангиогенез-зависимых нарушений, пролиферативных заболеваний и/или нарушений иммунной системы у млекопитающих, в частности, у людей.
Настоящее изобретение также относится к способу лечения сосудистой гиперпроницаемости, неадекватной неоваскуляризации, пролиферативных заболеваний и/или нарушений иммунной системы, который предусматривает введение терапевтически эффективного количества соединения формулы (I) млекопитающему, в частности, человеку, нуждающемуся в этом.
Сокращения
Ac: Ацетил
AcOH: Ледяная уксусная кислота
Bn: бензил
BnBr: бензилбромид
Boc: Трет-бутоксикарбонил
Boc2O: Ди-трет-бутилдикарбонат
BPO: перекись бензоила
ушир.: уширенный
t-BuOH: трет-бутанол
(CH2O)n: параформальдегид
д: Дублет
dba: дибензилиденацетон
DCAD: (Е)-бис(4-хлорбензил)диазен-1,2-дикарбоксилат
DCE: 1,2-дихлорэтан
DCM: Дихлорметан (метиленхлорид)
дд: Дублет дублетов
DIEA: N,N-диизопропилэтиламин
DMA: Диметилацетамид
DMAP: 4-диметиламинопиридин
DME: 1,2-диметоксиэтан
DMF: N,N-диметилформамид
DMSO: Диметилсульфоксид
dppf: 1,1'-бис(дифенилфосфино)ферроцен
EDC: 1-(3-диметиламинопропил)-3-этилкарбодиимид
EDC⋅HCl: N1-((этилимино)метилен)-N3,N3-диметилпропан-1,3-диамин гидрохлорид
эквив.: Эквивалент (эквиваленты)
EtOAc: Этилацетат
Et2O: Диэтиловый эфир
EtOH: Этанол
Fmoc: флуоренилметилоксикарбонил
г: грамм (граммы)
ч: час (часы)
HATU: 4-(3-Акриламидофенил)-2-этил-1H-индол-7-карбоксамид
HOBt: 1H-бензо[d][1,2,3]триазол-1-ол гидрата
HPLC: Высокоэффективная жидкостная хроматография
IPA: Изопропиловый спирт
KHMDS: Бис(триметилсилил)амид калия
KOAc: ацетат калия
KOt-Bu: Трет-бутоксид калия
ЖХ/МС: Жидкостной хроматографии/масс-спектрометрии
LDA: Литийдиизопропиламид
LiHMDS: Литий бис(триметилсилил)амид
м: Мультиплет
M: Молярный
MeCN: ацетонитрил
MeOH: Метиловый спирт
мин: минута (минуты)
ммоль: Миллимоль
МС: Масс-спектрометрия
MsCl: метансульфонилхлорид
MTBE: трет-бутилметиловый эфир
н-: Нормальный (неразветвленные)
н: Нормальный
NaBH(OAc)3: Натрий триацетоксигидроборат
NaHMDS: Бис(триметилсилил)амид натрия
n-BuLi: н-бутиллитий
NaOt-Bu: Трет-бутоксид натрия
NBS: N-бромсукцинимид
NCS: N-хлорсукцинимид
NH4OAc: ацетат аммония
NMP: N-метилпирролидинон
ЯМР: Ядерный магнитный резонанс
Pd2dba3: Трис(дибензилиденацетон)дипалладий (0)
Pd(OAc)2: Ацетат палладия(II)
Пет.эфир: петролейный эфир
pH: -log[H+]
Pd(PPh3)4: Тетракис(трифенилфосфин)палладий(0)
Pd(PPh3)2Cl2: бис(трифенилфосфин)палладий(II) хлорида
PMB: пара-метоксибензил
PPh3: трифенилфосфин
ppm: частей на миллион или м.д.
PrOH: пропанол
фунт на кв.дюйм: Фунты на квадратный дюйм
PyBOP: ((1H-бензо[d][1,2,3]триазол-1-ил)окси)три(пирролидин-1-ил)фосфония гексафторофосфат (V)
ВУ: время удерживания
К.т.: комнатная температура
с: синглет
SEM: 2-(триметилсилил)этоксиметил
SEMCl: 2-(триметилсилил)этоксиметил хлорид
SFC: сверхкритическая флюидная хроматография
SPE: твердофазная экстракция
т: триплет
трет-: третичный
TBAF: фторид тетрабутиламмония
TBME: трет-бутилметиловый эфир
TBDMS: трет-бутилдиметилсилан
TBSCl: трет-бутилдиметилсилил хлорид
TBTU: 2-(1H-бензо[d][1,2,3]триазол-1-ил)-1,1,3,3-тетраметил-изоуроний тетрафторборат
TEA: триэтиламин
трет-: третичный
трет-Бутил X-Phos: 2-Ди-трет-бутилфосфино-2',4',6'-триизопропилбифенил
TFA: Трифторуксусная кислота
THF: Тетрагидрофуран
TLC: Тонкослойная хроматография
TMS: триметилсилил
TMSCl: триметилсилилхлорид
TMSI: триметилсилил йодид
TsCl: п-толуолсульфонилхлорид
УФ: Ультрафиолет
масс.%: массовый процент
X-Phos: 2-дициклогексилфосфино-2',4',6'-триизопропилдифенил
ОБЩИЕ СХЕМЫ СИНТЕЗА
Соединения по настоящему изобретению можно получить, используя синтетические превращения, представленные на схемах I-XVIII. Исходные материалы коммерчески доступны, их можно получить раскрытыми в настоящем описании способами, раскрытыми в литературе способами или способами, которые хорошо известны специалистам в области органической химии.
Способы получения 1H-индол-7-карбоксамидных соединений 9 по настоящему изобретению проиллюстрированы на схеме I. На схеме I, стадия a, имеющаяся в продаже 4-бром-2-нитробензойная кислота 1 взаимодействует с винилмагнийбромидом путем синтеза индола Бартоли с использованием способов, известных специалисту в данной области (например, получение № 1, стадия а), с получением индола 2. Индол 2 можно алкилировать метилиодидом (схема I, стадия b), с получением метил 1H-индол-7-карбоксилата 3 с помощью способов, известных специалистам в данной области (например, получение № 1, стадия B). Полученный индол 3 можно защитить тозилом (Ts) (Схема I, стадия c), используя условия, такие как раскрыты в получении № 1, стадия C или способы, описанные в Greene, T.W. and Wuts, P.G.M. "Protective Groups in Organic Synthesis, 3rd Edition", 1999, Wiley-Interscience; Larock, R.C. "Comprehensive Organic Transformations: A Guide to Functional Group Preparations, 2nd edition", 1999, Wiley-VCH. На стадии d, направленное литирование метил-4-бром-1-тозил-1H-индол-7-карбоксилата 4 с последующим захватом аниона с йодом дает метил-4-бром-2-йод-1Н-индол-7-карбоксилаты 5, используя условия, такие как описаны в Получении №1, стадия D. Тозил-защищенные метил-4-бром-2-йод-1Н-индол-7-карбоксилаты 5 могут быть гидролизованными и незащищенными в водных основных условиях в одной стадии е с получением 4-бром-2-йод-1Н-индол-7-карбоновой кислоты 6, используя условия, такие как описаны в Получении №1, стадия Е или известны специалистам в данной области (например, книги от Larock, R.C. or Greene, T.W. and Wuts, P.G.M., упоминаемый выше). На стадии f, 4-бром-2-йод-1Н-индол-7-карбоновая кислота 6 можно быть превращена в первичный амид 7, как показано, с помощью условий, таких как описаны в общем способе D. 4-бром-2-йод-1Н-индол-7-карбоксамид 7 может вступать в различные реакции, известные специалисту в данной области (например, Larock, R.C., упоминаемый выше), включая, но ими не ограничиваясь, реакции сочетания Сузуки или Стилла, такие как описаны в общем способе А и Примере №22, стадия А. Альтернативно, на стадии i, тозил-защищенные индолы 5 могут вступать в различные реакции, известные специалисту в данной области (например, Larock, RC, упоминаемый выше), включая, но ими не ограничиваясь, реакции сочетания Сузуки или Стилла, описанные в общем способе А (например, получение №15 стадия А). Гидролиз сложных эфиров 10 дает кислоты 11 (Схема I, стадия j) с помощью хорошо известных условий, таких как те, которые описаны в Получении № 15, стадия B или общем способе С. На стадии k, карбоновая кислота 11 может быть превращена в первичные амиды 12, как показано с помощью условий, таких как описаны в общем способе D. Удаление сульфонамидной защитной группы у индолов 12 можно осуществить, используя условия, таких как описаны в общем способе N, или способами, известными специалистам в данной области (например, Larock, R.C. or Greene, T.W. and Wuts, P.G.M., упоминаемый выше), получая индолы 8 (схема I, стадия l). Индолы 8 подвергают взаимодействию с боронатным сложным эфиром или бороновой кислотой, либо коммерчески доступными, либо полученными способами, известными специалистам в данной области (см., например, Larock, R.C. "Comprehensive Organic Transformations: A Guide to Functional Group Preparations, 2nd edition", 1999, Wiley-VCH или общий способ P), используя условия реакции сочетания Сузуки, такие как описаны в общем способе А, получая 1H-индол-7-карбоксамидные соединения 9. Альтернативно, на стадии h, индолы 8 могут вступать в различные реакции, известные специалисту в данной области (например, Larock, R.C., упоминаемый выше), включая, но ими не ограничиваясь, условия сочетания Бухвальда или Негиши, как описано в Общих способах Т и U. Далее функционализацию группы R'' в индолы 9 можно осуществить, при желании, используя реакции, известные специалистам в данной области (например, Larock, R.C., упоминаемый выше). Например, индолы 9, содержащие двойную связь, могут быть восстановлены до насыщенных систем, используя условия гидрирования, такие как те, которые описаны в общем способе L. Эфиры могут быть получены из индолов 9, содержащих спирт, используя условие, такое как описано в общем способе Q. Кроме того амиды, мочевины, сульфонамиды, ариламины, гетероариламины или сульфонилмочевины можно получить из индолов 9, содержащих первичный или вторичный амин (например, Общие способы D, Е, I, H и J). Кроме того, удаление защитных групп у индолов 9, содержащих защитную группу в любом R' или R'', можно осуществить, используя условия, такие как описаны в Greene, T.W. and Wuts, P.G.M., упоминаемый выше, или в Общих способах G, M или N. Например, для R'', содержащей спирт, защищенный TBDMS, защитную группу можно удалить с получением незащищенного спирта (например, общий способ M) и соединения 9 с удаленной защитной группой можно затем подвергнуть дальнейшему взаимодействию, как описано выше. Альтернативно, соединение 4 можно сначала подвергнуть реакции сочетания на стадии m, включая, но ими не ограничиваясь, например, реакцию Сузуки, Бухвальда или Негиши с использованием условий, как описано в Общих способах А, Т и U с получением соединений 107 с последующей реакцией йодирования, как проиллюстрировано в общем способе Y с получением соединений 108 (стадия n). Индолы 108 можно подвергнуть различным реакциям, известным специалистам в данной области (например, Larock, R.C., упоминаемый выше), включая, но ими не ограничиваясь, реакции сочетания Сузуки или Стилла, такие как описаны в Общем способе A, с получением соединений 109. Можно в дальнейшем предусмотреть, что соединения 109 можно подвергнуть реакциям гидролиза, амидирования и де-тозилирования, аналогичным стадиям j, k и I с получением соединений 9.
Схема I
Альтернативный способ получения 1Н-индол-7-карбоксамидных соединений 9 по настоящему изобретению проиллюстрирован на схеме II. На стадии а, индол 3 из схемы I может быть защищен группой SEM с использованием условий, известных в литературе, таких как раскрыты Greene, TW и Wuts, P.G.M. указано выше, или, как описанные в получении № 10, стадии А. Полученный SEM-защищенный индол 13 можно подвергать направленному литиированию с последующим захватом аниона с электрофилом (например, йодметан) с образованием индола 14, как показано на стадии b c использованием условий, описанных в примере №19, стадия А или захватом аниона с йодом, как показано на стадии g с образованием метил 4-бром-2-йод-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоксилата 17 с использованием условий, таких как описаны в примере получения № 10, стадия В). На стадии h, индол 14 можно подвергать различным реакциям, известным специалистам в данной области, в том числе, но ими не ограничиваясь, реакциям сочетания Сузуки или Стилла, таким как описаны в Larock, R.C., указанные выше, общем способе А, и получении №10, стадия С. Гидролиз сложных эфиров 14 дает кислоты 15 (стадия с) с использованием хорошо известных условий, таких как описаны в получении № 10, стадия D, или общем способе C. Индол карбоновые кислоты 15 могут быть преобразованы в первичные амиды 16, как показано с использованием условий, таких как описаны в общем способе D. SEM-защитная группа 1H-индол-7-карбоксамидных соединений 16 может быть удалена с помощью способов, таких как описаны в получении № 10, стадия E или с использованием условий, таких как описаны в Greene, TW и Wuts, P.G.M. указанных выше, с получением 1H-индол-7-карбоксамидов 8. Индолы 8 можно затем подвергнуть дальнейшему взаимодействию, как описано выше (схема I) с получением целевых 1H-индол-7-карбоксамидных соединений 9.
Схема II
Дополнительный способ получения индол-7-карбоксамидных соединений 9 по настоящему изобретению проиллюстрирован на схеме III. Гидролиз сложного эфира 17 дает кислоту 18 (стадия а) с использованием хорошо известных условий, таких как описаны в получении № 10, стадия D, или Общем способе С. Кислота 18 может быть преобразована в первичный амид 19, как показано, с использованием условий, таких как описаны в Общем способе D. SEM-защитную группу индола 19 можно удалить способами, такими как описаны в примере № 19, стадия D или используя условия, такие как описаны в Greene, T.W. and Wuts, P.G.M., указанном выше, с получением 1H-индол-7-карбоксамидов 7. Индолы 7 можно затем подвергнуть дальнейшему взаимодействию, как описано выше, с получением целевых 1H-индол-7-карбоксамидных соединений 9.
Схема III
Альтернативный способ получения 1Н-индол-7-карбоксамидных соединений 9 по настоящему изобретению проиллюстрирован на схеме IV. Индол 19 можно подвергнуть различным взаимодействиям, известным специалисту в данной области (например, Larock, R.C., упоминаемый выше), включая, но ими не ограничиваясь, реакции сочетания Стилле, такие как те, что описаны в примере № 22, стадия А или реакция сочетания Сузуки, как описано в общем способе А. На стадии b, индол-7-карбоксамиды 16 взаимодействуют с боронатным сложным эфиром или бороновой кислотой, или коммерчески доступными, или могут быть получены способами, известными специалисту в данной области (см., например, Пример №22, стадия В; Larock, R.C. "Comprehensive Organic Transformations: A Guide to Functional Group Preparations, 2nd edition", 1999, Wiley-VCH; или общий способ А) с помощью условий сочетания Сузуки (например, Пример №19 или общий способ А). SEM-защитную группу индолов 20 можно удалить с помощью способов, таких как те, которые описаны в примере №22, стадия D, или с использованием условий, таких как описаны в Greene, T.W. and Wuts, P.G.M., указанные выше, с получением 1H-индол-7-карбоксамидов 9. Индолы 9 можно затем подвергнуть дальнейшему взаимодействию, как описано выше.
Схема IV
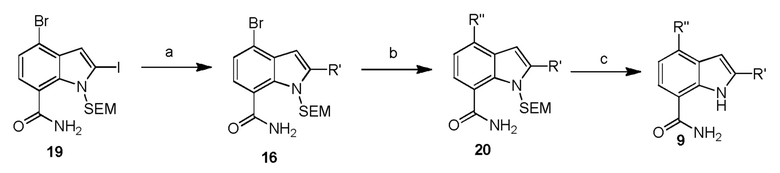
Индол-7-карбоксамидные соединения 9 по настоящему изобретению также могут быть получены с использованием пути, проиллюстрированного на схеме V. На стадии а, метиловый эфир 21 получали, используя стандартные условия, такие как описаны в общем способе F, или Larock, R.C., упоминаемом выше. Enolizable кетоны 23 взаимодействуют с м-нитроанилином 22 с образованием 4-нитроиндолов 24 (стадия b) с использованием стандартных условий, таких как те, которые описаны в общем способе F, или Tetrahedron, 2004, 60(2), 347. На стадии с кислоты 24 можно преобразовать в первичные амиды 25, как показано с использованием условий, таких как описаны в общем способе D или F. Аминоиндолы 26 получали восстановлением нитрогруппы первичных амидов 25 с помощью способов, известных специалистам в данной области (например, общий способ F, или Larock, R.C., упоминаемый выше). Диазотирование 26 с последующим йодированием дает 27 с использованием стандартных условий, таких как описаны в общем способе F, или Larock, R.C., упоминаемом выше. На стадии f, индолы 27 можно подвергнуть различным реакциям, известным специалисту в данной области (например, Larock, R.C., упоминаемый выше), включая, но ими не ограничиваясь, условия реакции сочетания Сузуки, Бухвальда или Негиши, как описано Общими способами А, Т и U. Индолы 9 можно затем подвергнуть дальнейшему взаимодействию, как описано выше.
Схема V
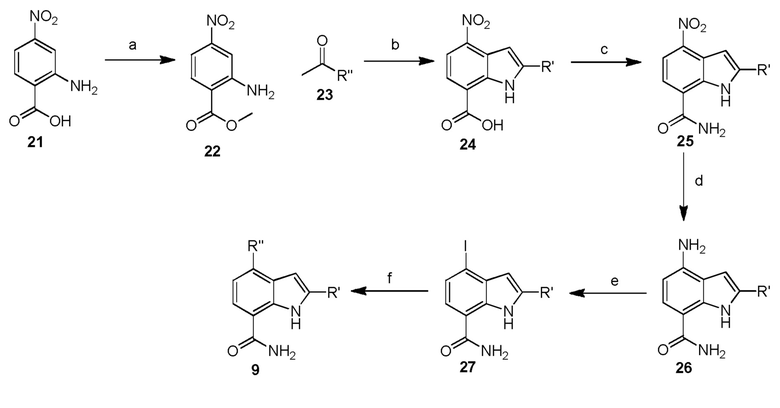
Способы получения 1H-индол-7-карбоксамидных соединений 30 по настоящему изобретению проиллюстрированы на схеме VI. На схеме VI, стадия а, коммерчески доступный 4-бром-1H-индол-7-карбонитрил [Sinova] 28 гидролизуют с получением первичного амида 29 с использованием условий, таких как те, которые описаны в Получении № 2 или известны специалистам в данной области (например, в книгах от Larock, R.C. or Greene, T.W. and Wuts, P.G.M., упоминаемых выше). На стадии b, индол 29 можно подвергнуть различным взаимодействиям, известным специалисту в данной области (например, Larock, R.C., упоминаемый выше), включая, но ими не ограничиваясь, условия реакции сочетания Сузуки, Бухвальда или Негиши, как описано Общими способами А, Т и U. В качестве альтернативы, индол 29 может быть превращен в боронатный сложный эфир 31 с использованием взаимодействий, таких, как описаны в Общем способе P. Индол 31 можно подвергнуть реакции сочетания Сузуки с использованием условий, таких как описаны в общем способе А или известных специалисту в данной области (например, Larock, R.C., упоминаемый выше). Дальнейшую функционализацию R’ группы в индолах 30 можно осуществить, при желании, используя реакции, известные специалистам в данной области (например, Larock, R.C., упоминаемый выше). Например, индолы 30, содержащие двойную связь, можно восстановить до насыщенных систем, используя условия гидрирования, такие как описаны в общем способе L. Простые эфиры можно получить из индолов 30, содержащих спирт, используя условие, такое как описано в общем способе Q. Кроме того, амиды, мочевины, сульфонамиды, ариламины, гетероариламины или сульфонилмочевины можно получить из индолов 30, в которых R' содержит первичный или вторичный амин (например, Общие способы D, Е, I, H и J). Кроме того, удаление защитных групп R' группы в 1Н-индол-7-карбоксамидных соединениях 30 до получения незащищенного соединения можно осуществить с использованием условий, таких как описаны в Greene, T.W. and Wuts, P.G.M., указано выше, или в общих способах G, M или N. Например, защитную группу, такую как Вос-группа, можно удалить из защищенного амина, получая незащищенный амин (например, общий способ G) и соединения 30 с удаленной защитной группой можно затем подвергнуть дальнейшему взаимодействию, как описано выше.
Схема VI

Способы получения 1H-индол-7-карбоксамидных соединений 35 по настоящему изобретению проиллюстрированы на схеме VII. Нитрование индола 29 (схема VII стадия а) можно осуществить с использованием условий, таких как описаны в Получении № 7, стадия С или известны специалистам в данной области (например, Larock, RC, упоминаемый выше). На стадии b, индол 32 можно подвергнуть различным взаимодействиям, известным специалисту в данной области (например, Larock, R.C., упоминаемый выше), включая, но ими не ограничиваясь, условия реакции сочетания Сузуки, Бухвальда или Негиши, как описано в Общих способах А, Т и U. Аминоиндолы 34 получали восстановлением нитроиндолов 33 с использованием способов, известных специалистам в данной области (например, Получение № 7, стадия E, или Larock, R.C., упоминаемый выше). Аминоиндолы 34 можно преобразовывать с получением амидов 35, как показано на стадии d, с использованием условий, таких как описаны в общем способе D или E.
Схема VII
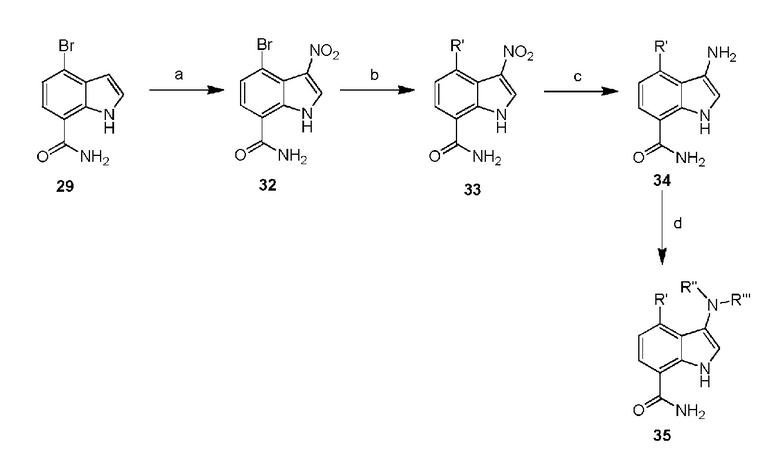
Способы получения 1H-пирроло[3,2-c]пиридин-7-карбоксамидов 39 по настоящему изобретению проиллюстрированы на схеме VIII. На схеме VIII, стадия a, 6-бром-4-нитроникотиновая кислота [European Journal of Medicinal Chemistry 1977, 12(6), 541] 36 взаимодействует с винилмагнийбромидом путем синтеза индола по Бартоли с использованием способов, известных специалистам в данной области (например, получение № 9, стадия А), с получением пирроло[3,2-с]пиридина 37. На стадии b, кислоту соединений 37 можно преобразовать в первичные амиды 38, как показано с использованием условий, таких как описаны в общем способе D. Пирроло[3,2-с]пиридин 38 можно подвергнуть различным реакциям, известным специалисту в данной области (например, Larock, R.C., упоминаемый выше), включая, но ими не ограничиваясь, условия реакции сочетания Сузуки, Бухвальда или Негиши, как описано в Общих способах A, T и U. Дальнейшую функционализацию R' группы в пирроло[3,2-c]пиридинах 39 можно осуществить, при желании, используя реакции, известные специалистам в данной области (например, Larock, R.C., упоминаемый выше). Например, индолы 39, содержащие двойную связь, можно восстановить до насыщенных систем, используя условия гидрирования, такие как описаны в общем способе L. Простые эфиры можно получить из индолов 39, содержащих спирт, используя условие, такое как описано в общем способе Q. Кроме того, амиды, мочевины, сульфонамиды, ариламины, гетероариламины или сульфонилмочевины можно получить из индолов 39, содержащих первичный или вторичный амин (например, Общие способы D, Е, I, H и J). Кроме того, удаление защитных групп у индолов 39, содержащих защитную группу в R' можно осуществить с использованием условий, таких как описаны в Greene, T.W. and Wuts, P.G.M., указано выше, или в Общих способах G, M или N. Например, для R'', содержащей спирт, защищенный TBDMS, защитную группу можно удалить с получением незащищенного спирта (например, общий способ M) и соединения 39 с удаленной защитной группой можно затем подвергнуть дальнейшему взаимодействию, как описано выше.
Схема VIII

Способы получения 1H-пирроло[2,3-c]пиридин-7-карбоксамидов 44 по настоящему изобретению проиллюстрированы на схеме IX. На схеме IX, стадия a, 5-бром-2-хлор-3-нитропиридин 40 взаимодействует с винилмагнийбромидом путем синтеза индола по Бартоли с использованием способов, известных специалистам в данной области (например, Пример № 2, стадия A) с получением пирроло[2,3-c]пиридина 41. На стадии b, пирроло[2,3-c]пиридин 41 можно подвергнуть различным взаимодействиям, известным специалисту в данной области (например, Larock, R.C., упоминаемый выше), включая, но ими не ограничиваясь, условия реакции сочетания Сузуки, Бухвальда или Негиши, как описано в Общих способах A, T и U с получением пирроло[2,3-c]пиридинов 42. На стадии c, карбонилирование пирроло[2,3-с]пиридинов 42 в присутствии Pd дает сложные эфиры 43 с использованием способов, известных специалистам в данной области, таких как описаны в Примере № 2, стадия C. Сложные эфиры 43 можно подвергнуть аммонолизу, как например, описано в Примере № 2, стадия D или известны специалистам в данной области (например, Larock, R.C., упоминаемый выше) с получением соединений 44. Дальнейшую функционализацию R' группы в пирроло[2,3-c]пиридинах 44 можно осуществить, при желании, используя реакции, известные специалистам в данной области (например, Larock, R.C., упоминаемый выше). Например, индолы 44, содержащие двойную связь, можно восстановить до насыщенных систем, используя условия гидрирования, такие как описаны в общем способе L. Простые эфиры можно получить из индолов 44, содержащих спирт, используя условие, такое как описано в общем способе Q. Кроме того, удаление защитных групп у индолов 44, содержащих защищенный спирт, можно осуществить с использованием условий, таких как описаны в Greene, T.W. and Wuts, P.G.M., указано выше, или в Общих способах M. Кроме того, амиды, мочевины, сульфонамиды, ариламины, гетероариламины или сульфонилмочевины можно получить из индолов 44, в которых R' содержит первичный или вторичный амин (например, Общие способы D, Е, I, H и J). Кроме того, удаление защитных групп R' группы в 1Н-индол-7-карбоксамидных соединениях 44 до получения незащищенного соединения можно осуществить с использованием условий, таких как описаны в Greene, T.W. and Wuts, P.G.M., указано выше, или в общих способах G, M или N. Например, защитную группу, такую как Вос-группа, можно удалить из защищенного амина, получая незащищенный амин (например, общий способ G) и соединения 44 с удаленной защитной группой можно затем подвергнуть дальнейшему взаимодействию, как описано выше.
Схема IX
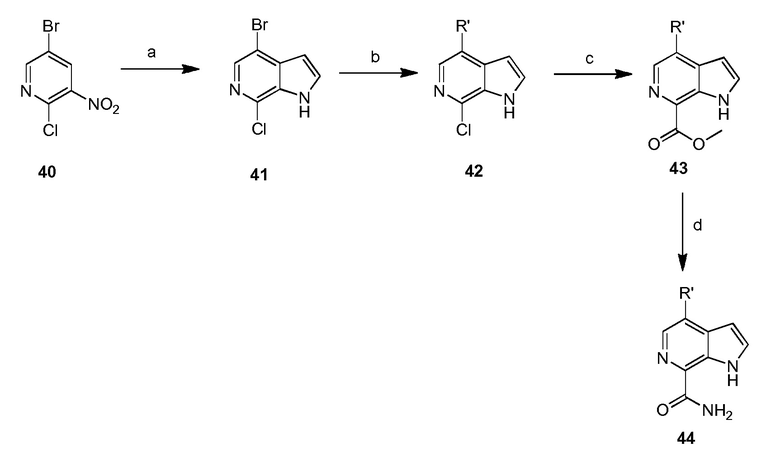
Способы получения 1H-индол-7-карбоксамидов 51 по настоящему изобретению проиллюстрированы на Схеме X. На Схеме X, стадия a, индол 45 при реакции Вильсмейера-Хаака с использованием способов, известных специалистам в данной области (например, Пример № 3, стадия А), дает альдегид 46. Восстановительное аминирование альдегида 46 с 4-метоксибензиламином (PMB) с использованием условий, такие как описаны в общем способе H приводит к получению амина 47 (схема X, стадия b). Гидролиз сложного эфира 47 дает кислоту 48 (стадия с) с использованием хорошо известных условий, таких как описаны в примере № 3, стадия С, или Общем способе С. Кислота 48 может быть превращена в первичный амид 49, как показано с использованием условий, таких как описаны в Общем способе D. Индол 49 можно подвергнуть различным взаимодействиям, известным специалистам в данной области (например, Larock, R.C., упоминаемый выше), включая, но ими не ограничиваясь, условия реакции сочетания Сузуки, Бухвальда или Негиши, как описано в Общих способах A, T и U. Индолы 50 можно преобразовать с получением метил индолов 51 с использованием условий, таких как описаны в Примере № 3, стадия F.
Схема X
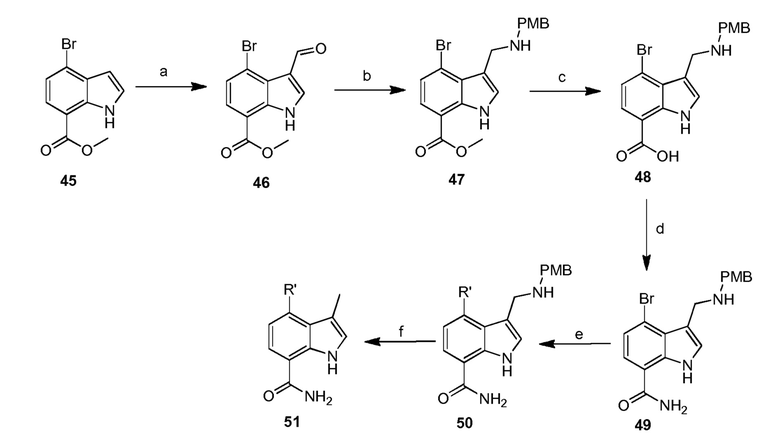
Способы получения 1,2,3,6-тетрагидропирроло[2,3-e]индол-5-карбоксамидов 58 по настоящему изобретению проиллюстрированы на схеме XI. Нитрование 5-броминдолина 52 (схема XI, стадия а) можно осуществить с использованием условий, таких как описаны в примере № 4, стадия А или известны специалистам в данной области (например, Larock, R.C., упоминаемый выше). Полученный индолин 53 может быть защищен (Схема XI, стадия b) с использованием условий, описанных в Greene, T.W. and Wuts, P.G.M., упоминаемый выше (например, Boc-защитной группой, с использованием условий, таких как описаны в Примере № 4, стадия В или которые описаны в Greene, T.W. and Wuts, P.G.M., упоминаемый выше). На схеме XI, стадия c, индолин 54 взаимодействует с винилмагнийбромидом путем синтеза индола по Бартоли с использованием способов, известных специалистам в данной области, с получением индола 55 с использованием условий, описанных в Примере № 4, стадия С. На стадии d, цианирование бромида 55 в присутствии Pd дает соответствующий нитрил 56 (например, Пример № 4, стадия D или Tetrahedron Letters 1999, 40(47), 8193-8195). Последующий гидролиз нитрила 56 дает первичный амид 57 (схема XI, стадия е), используя способы, известные специалисту в данной области (например, Общий способ O). Первичный амид 57 можно преобразовать с получением амидов 58, как показано на стадии f, используя условия, такие как описаны в общем способе D или Е.
Схема XI
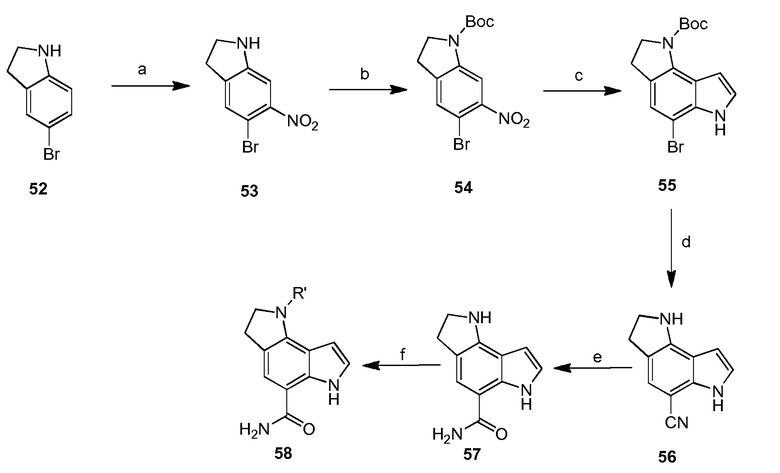
Способы получения бензимидазолов 64 по настоящему изобретению проиллюстрированы на схеме XII. На стадии a, 4,7-дибромбензо[c][1,2,5]тиадиазол 59 может вступать в различные реакции, известные специалисту в данной области (например, Larock, R.C., упоминаемый выше) включая, но ими не ограничиваясь, условия реакции сочетания Сузуки, Бухвальда или Негиши, как описано в Общих способах A, T и U. На стадии b, цианирование бромида 60 в присутствии Pd дает соответствующие нитрилы 61 (например, Tetrahedron Letters 1999, 40(47), 8193-8195). Нитрилы 61 можно подвергнуть реакции раскрытия кольца, с получением диамина 62, используя условия, такие как описаны в Примере № 14, стадия С. Как показано на схеме XII, стадия d, циклизацию диамина 62 можно осуществить взаимодействием с альдегидами (например, Пример № 14, стадия D). Гидролиз нитрила 63 дает бензимидазолы 64 (Схема XII, стадия e) с использованием способов, известных специалистам в данной области, таких как описаны в общем способе O.
Схема XII
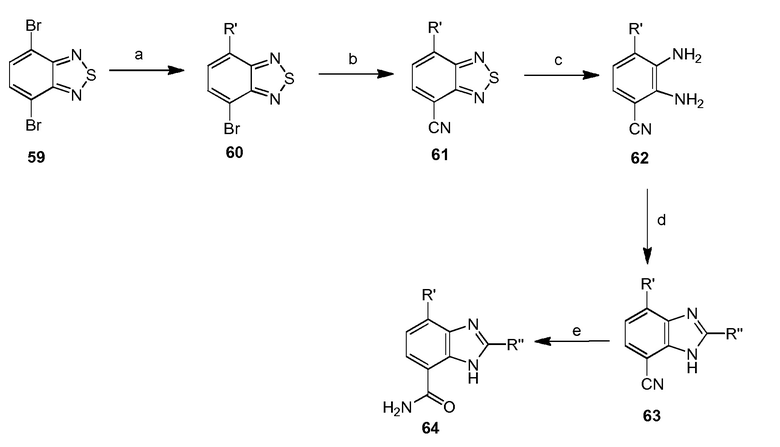
Способы получения индазолов 70 по настоящему изобретению проиллюстрированы на схеме XIII. На схеме XIII, стадия a, 2-амино-4-хлор-3-метилбензойную кислоту [Енамин] 65 этерифицируют с использованием стандартных условий, таких как описаны в Общем способе F или Larock, R.C., упоминаемый выше. На стадии b, циклизация сложного эфира 66 дает индазол 67 с помощью способов, известных специалистам в данной области (например, Пример № 18, стадия B или WO2007/113596). Гидролиз сложного эфира 67 дает кислоту 68 (Схема XIII, стадия c) с использованием хорошо известных условий, таких как те, которые описаны в общем способе C. Кислота 68 могут быть превращена в амид 69, как показано на стадии d, используя условия, такие как описаны в общем способе D. Индол 69 можно подвергать различным реакциям, известным специалисту в данной области (например, Larock, R.C., упоминаемый выше) включая, но ими не ограничиваясь, условия реакции сочетания Сузуки, Бухвальда или Негиши, как описано в Общих способах A, T и U. Дальнейшую функционализацию R’ группы в индолах 70 можно осуществить, при желании, используя реакции, известные специалистам в данной области (например, Larock, R.C., упоминаемый выше). Например, индолы 70, содержащие двойную связь, можно восстановить до насыщенных систем, используя условия гидрирования, такие как описаны в общем способе L. Простые эфиры можно получить из индолов 70, содержащих спирт, используя условие, такое как описано в общем способе Q. Кроме того, амиды, мочевины, сульфонамиды, ариламины, гетероариламины или сульфонилмочевины можно получить из индолов 70, в которых R' содержит первичный или вторичный амин (например, Общие способы D, Е, I, H и J). Кроме того, удаление защитных групп R' группы в 1Н-индол-7-карбоксамидных соединен иях 70 до получения незащищенного соединения можно осуществить с использованием условий, таких как описаны в Greene, T.W. and Wuts, P.G.M., упоминаемый выше, или в общих способах G, M или N. Например, защитную группу, такую как Вос-группа, можно удалить из защищенного амина, получая незащищенный амин (например, общий способ G) и соединения 70 с удаленной защитной группой можно затем подвергнуть дальнейшему взаимодействию, как описано выше.
Схема XIII

Способы получения 1H-индол-7-карбоксамидных соединений 77 по настоящему изобретению проиллюстрированы на Схеме XIV. На схеме XIV, стадия a, индол 71 можно защитить тозилом (Ts) (схема I, стадия c), используя условия, такие как описаны в Получении № 1 стадия C или те, которые описаны в Greene, T.W. and Wuts, P.G.M. or Larock, R.C., упоминаемый выше). На стадии b, направленное литирование 4-фтор-1-тозил-1H-индол-7-карбонитрила 72 с последующим захватом аниона с йодом дает индол 73, используя условия, такие как описаны в Получении № 1, стадия D. 4-Фтор-2-йод-1-тозил-1H-индол-7-карбонитрил 73 можно подвергуть различным реакциям, известным специалистам в данной области (например, Larock, R.C., упоминаемый выше), включая, но ими не ограничиваясь, реакции сочетания Сузуки, такие как описаны в общем способе A. Дальнейшую функционализацию R' группы в карбонитрилах 74, защищенных тозилом, можно осуществить, при желании, используя реакции, известные специалистам в данной области (например, Larock, R.C., упоминаемый выше). Например, амиды, эфиры, мочевины, сульфонамиды, ариламины, гетероариламины или сульфонилмочевины можно получить из соединений 74, в которых R' содержит первичный или вторичный амин (например, Общие способы D, Е, I, H и J). Кроме того, удаление защитных групп у R' группы в соединениях 74 до получения незащищенного соединения можно осуществить с использованием условий, таких как описаны в Greene, T.W. and Wuts, P.G.M., упоминаемый выше, или в общих способах G, M или N. Например, защитную группу, такую как Вос-группа, можно удалить из защищенного амина, получая незащищенный амин (например, Получение № 27, стадия D или общий способ G) и соединения 74 с удаленной защитной группой можно затем подвергнуть дальнейшему взаимодействию, как описано выше, амин. Индол карбонитрилы 74, показанные на стадии d, можно подвергнуть взаимодействию с аминами с помощью химии смещения с использованием условий, известных специалистам в данной области, таких как описаны в Общем способе В с получением соединений 75. Тозил-защищенные 1H-индол-7-карбонитрилы 75 могут быть незащищенными в водных основных условиях, в одной стадии с получением соединения 76, используя условия, такие как описаны в Примере № 12, стадия B или известны специалистам в данной области (например, книги от Larock, R.C. or Greene, T.W. and Wuts, P.G.M., упоминаемые выше). На стадии f, 1H-индол-7-карбонитрилы 76 гидролизовали с получением первичного амида 77 с использованием условий, таких как описаны в Получении № 2 или известны специалистам в данной области (например, книги от Larock, R.C. or Greene, T.W. and Wuts, P.G.M., упоминаемые выше). Кроме того, амиды, карбаматы, мочевины или замещенные амины можно получить из 1H-индол-7-карбоксамидных соединений 77, содержащих первичный или вторичный амин (например, Общие способы). Кроме того, удаление защитной группы 1H-индол-7-карбоксамидных соединений 77, содержащих защищенный первичный или вторичный амин, может быть выполнена с использованием условий, таких как описаны в Greene, T.W. and Wuts, P.G.M., упоминаемый выше или в Общих способах. Например, для R'' или R''', содержащих защитную группу (например, Boc-группу), защитная группа может быть удалена с получением амина незащищенного амина (например, общий способ G) и соединения 3 с удаленной защитной группой можно затем подвергнуть дальнейшему взаимодействию, как описано выше.
Схема XIV
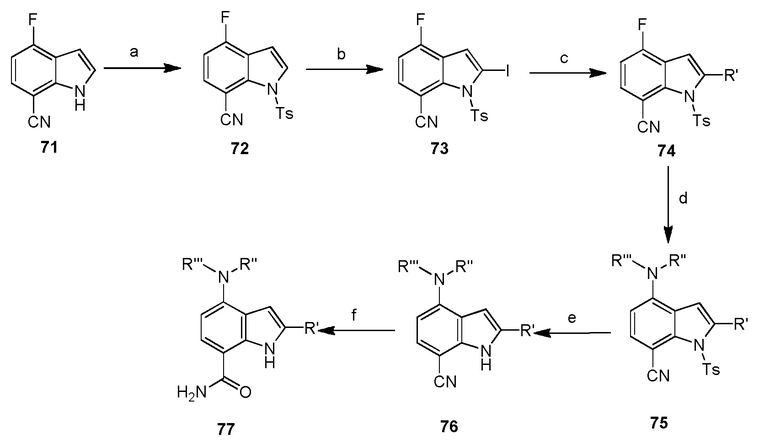
Способы получения 7-хлортиазоло[5,4-c]пиридин-4-карбоксамидов 87 по настоящему изобретению проиллюстрированы на Схеме XV. Реакцию Виттига альдегида 78 (стадия a) осуществляют с илидом трифенилфосфония с использованием стандартных условий, известных на специалисту в данной области, таких как описаны в Получении № 46, стадия A или Larock, R.C., упоминаемый выше, с получением α, β-ненасыщенного метилового сложного эфира 79. Это промежуточное соединение подвергают взаимодействию с боронатом или бороновой кислотой посредством реакции Сузуки на стадии b, с использованием условий, таких, как проиллюстрированы в Получении № 46, стадия B. Промежуточное соединение 80 гидролизуют с получением кислоты, как показано в Получении № 46, стадия B (стадия c). На стадии d, кислота преобразуется в ацилазида с помощью in situ формирования ацилхлорида с использованием стандартных условий, таких как описаны в Получении № 46, стадия D или WO 2012/035039. Промежуточное соединения ацилазид затем можно подвергнуть перегруппировки Курциуса и циклизации с получением пиридона 83 на стадии е, при высоких температурах (например, Получение № 46, стадия E или WO 2012/035039). При обработке POCl3, на стадии f, пиридин-2-хлорид образуется (например, Получение № 46, стадия F или WO 2012/035039), который впоследствии можно быть обработан NCS на стадии g, с получением промежуточного соединения 4-бром-7-хлортиазоло[5,4-c]пиридин 85, как показано в Получении № 46, стадия G. Преобразование бромгруппы в 85 до функциональности циано выполняют с помощью реакции Pd-катализируемого цианирования и последующим гидролизом цианогруппы, получая 7-хлортиазоло[5,4-c]пиридин-4-карбоксамид, как показано в Получении № 46, стадия H. На стадии j, тиазоло[5,4-c]пиридин-4-карбоксамид 87 может подвергаться различным реакциям, известным специалисту в данной области (например, Larock, R.C., упоминаемый выше), включая, но ими не ограничиваясь, условия реакции сочетания Сузуки, Бухвальда или Негиши, как описано в Общих способах A, T и U с получением тиазоло[5,4-c]пиридин-4-карбоксамидов 88.
Схема XV
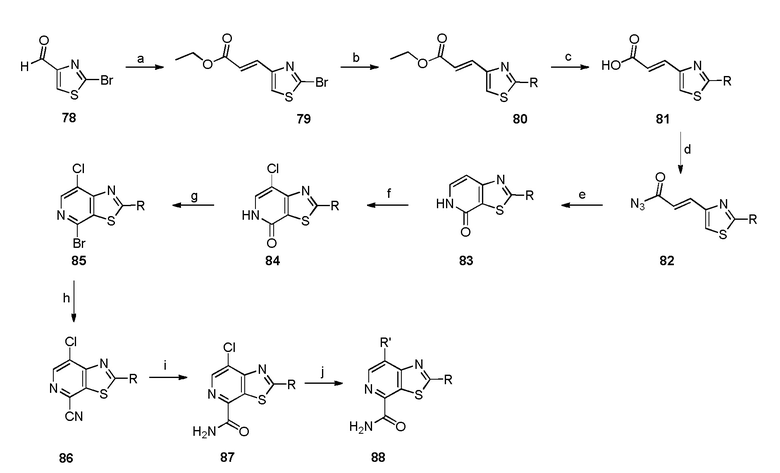
Второй вариант получения 1H-пирроло[3,2-c]пиридин-7-карбоксамидов 39 по отношению к пути, показанному на схеме VIII показан на схеме XVI, где 1H-пирроло[3,2-c]пиридин-7-карбоксамиды 39 также могут быть получены из коммерчески доступного метил 1H-пирроло[3,2-c]пиридин-7-карбоксилата 89, который сначала тозилировали на стадии а, с использованием стандартных условий, известных специалистам в данной области, как показано в общем способе AH. Тозилированное промежуточное соединение 90 затем окисляли (стадия b) с использованием условий, таких как те, которые описаны в общем способе AC с получением N-оксид промежуточного соединения 91. На стадии с материал галогенировали как показано в Получении № 45, стадия С, с последующим гидролизом с использованием основания, с удалением тозил-группы и гидролизом сложного эфира до кислоты с использованием условий, таких как описаны в общем способе Х. Кислоту затем можно подвергнуть стандартной реакции связывания амина, как проиллюстрировано в общим способе D, с получением амида на стадии е. Пирроло[3,2-c]пиридин 94 можно подвергнуть различным реакциям, известным специалисту в данной области (например, Larock, R.C., упоминаемый выше), включая, но ими не ограничиваясь, условия реакции сочетания Сузуки, Бухвальда или Негиши, как описано в Общих способах A, T и U с получением соединений 39. Дальнейшую функционализацию R' группы в пирроло[3,2-c]пиридинах 39 можно осуществить, при желании, используя реакции, известные специалистам в данной области (например, Larock, R.C., упоминаемый выше). Например, пирроло[3,2-c]пиридины 39, содержащие двойную связь, можно восстановить до насыщенных систем, используя условия гидрирования, такие как описаны в общем способе L. Простые эфиры можно получить из индолов 39, содержащих спирт, используя условие, такое как описано в общем способе Q. Кроме того, амиды, мочевины, сульфонамиды, ариламины, гетероариламины или сульфонилмочевины можно получить из индолов 39, содержащих первичный или вторичный амин (например, Общие способы D, Е, I, H и J). Кроме того, удаление защитных групп у индолов 39, содержащих защитную группу в R' можно осуществить с использованием условий, таких как описаны в Greene, T.W. and Wuts, P.G.M., указано выше, или в Общих способах G, M или N. Например, для R'', содержащей спирт, защищенный TBDMS, защитную группу можно удалить с получением незащищенного спирта (например, общий способ M) и соединения 39 с удаленной защитной группой можно затем подвергнуть дальнейшему взаимодействию, как описано выше.
Схема XVI
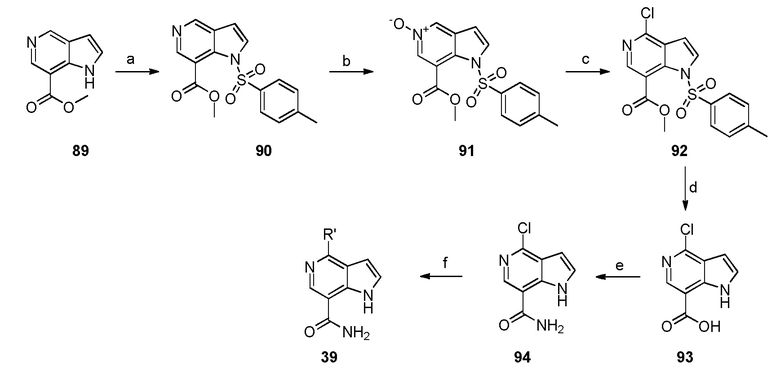
Третья альтернатива путям, показанным на схемах VIII и XVI, получения 1H-пирроло[3,2-c]пиридин-7-карбоксамидов 39 показана на схеме XVII. На стадии a, (4-метоксифенил)метанамин обрабатывали диметил 3-оксопентандиоат с получением промежуточного соединения 96, который не выделяли. На стадии b, соединение циклизовали in situ путем обработки с помощью хлорацетальдегида с использованием условий, таких как показаны в Получении № 37, стадия A или WO 2005121140. Депротонирование кислотного водорода 97 и реакцию с метилформиата, на стадии с, осуществляют с использованием способов, известных специалистам в данной области (например, Получение № 37, стадия B, или WO 2005121140) с получением промежуточного соединения 98. На стадии d, циклизацию промежуточного соединения 98 выполняют с использованием условий, таких как показаны в Получении № 37, стадия C или WO 2005121140 с получением промежуточного пиридинона 99. Последующую ароматизацию и галогенирование промежуточного пиридинона 99 на стадии е осуществляют с помощью хорошо известных условий (например, получение № 37, стадия D или WO 2005121140) с получением пирроло[3,2-c]пиридина 100. Гидролиз сложноэфирной функциональности в 100 дает кислоты 93 (стадия f) с использованием стандартных условий, таких как описаны в общем способе С. Кислоту затем можно подвергнуть реакции сочетания амина, как показано в Общем способе D, с получением амида на стадии е. Пирроло[3,2-c]пиридин 94 можно подвергнуть различным реакцим, известным специалисту в данной области (например, Larock, R.C., упоминаемый выше), включая, но ими не ограничиваясь, условия реакции сочетания Сузуки, Бухвальда или Негиши, как описано в Общих способах A, T и U с получением соединений 39. Дальнейшую функционализацию R' группы в пирроло[3,2-c]пиридинах 39 можно осуществить, при желании, используя реакции, известные специалистам в данной области (например, Larock, R.C., упоминаемый выше). Например, пирроло[3,2-c]пиридины 39, содержащие двойную связь, можно восстановить до насыщенных систем, используя условия гидрирования, такие как описаны в общем способе L. Простые эфиры можно получить из индолов 39, содержащих спирт, используя условие, такое как описано в общем способе Q. Кроме того, амиды, мочевины, сульфонамиды, ариламины, гетероариламины или сульфонилмочевины можно получить из индолов 39, содержащих первичный или вторичный амин (например, Общие способы D, Е, I, H и J). Кроме того, удаление защитных групп у индолов 39, содержащих защитную группу в R', можно осуществить с использованием условий, таких как описаны в Greene, T.W. and Wuts, P.G.M., указано выше, или в Общих способах G, M или N. Например, для R'', содержащей спирт, защищенный TBDMS, защитную группу можно удалить с получением незащищенного спирта (например, общий способ M) и соединения 39 с удаленной защитной группой можно затем подвергнуть дальнейшему взаимодействию, как описано выше.
Схема XVII
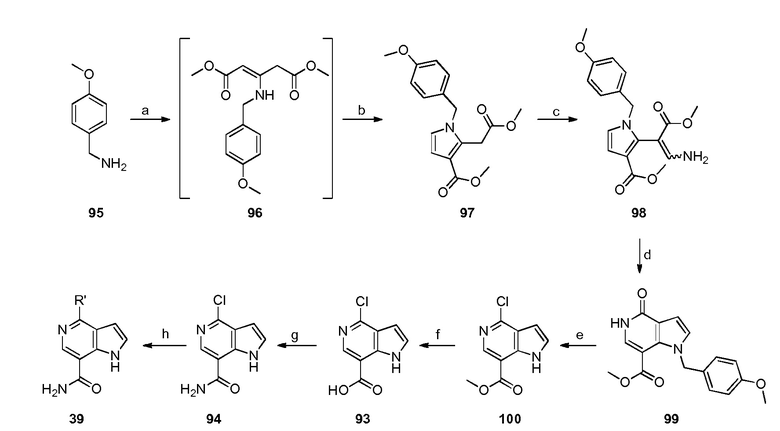
Альтернативные способы получения 1H-пирроло[2,3-c]пиридин-7-карбоксамидов 44 по настоящему изобретению проиллюстрированы на схеме XVIII. 4-бром-1H-пирроло[2,3-c]пиридин 101 окисляют до промежуточного N-оксида, используя способы, известные специалисту в данной области (например, общий способ AC). Цианирование N-оксида 102 на стадии b осуществляли с использованием условий, таких, как показаны в Общем способе AD с получением карбонитрила 103. Карбонитрил 103 можно подвергнуть различным реакциям, известным специалисту в данной области (например, Larock, R.C., упоминаемый выше), включая, но ими не ограничиваясь, условия реакции сочетания Сузуки, Бухвальда или Негиши, как описано в Общих способах A, T и U с получением пирроло[2,3-c]пиридинов 106. Последующий гидролиз пирроло[2,3-c]пиридинов 106 на стадии f, с использованием стандартных условий (например, общий способ О) дает соединения 44. Альтернативно, карбонитрил 103 можно сначала гидролизовать, как показано на стадии с, с получением амида 104 при воздействии на известных условиях (например, общий способ O). Амид 104 можно затем подвергнуть различным реакциям, известным специалисту в данной области (например, Larock, R.C., упоминаемый выше), включая, но ими не ограничиваясь, условия реакции сочетания Сузуки, Бухвальда или Негиши, как описано в Общих способах A, T и U, с получением соединений 44. Дальнейшую функционализацию R' группы в пирроло[2,3-c]пиридинах 44 можно осуществить, при желании, используя реакции, известные специалистам в данной области (например, Larock, R.C., упоминаемый выше). Например, пирроло[2,3-c]пиридины 44, содержащие двойную связь, можно восстановить до насыщенных систем, используя условия гидрирования, такие как описаны в общем способе L. Простые эфиры можно получить из пирроло[2,3-c]пиридинов 44, содержащих спирт, используя условие, такое как описано в общем способе Q. Кроме того, амиды, мочевины, сульфонамиды, ариламины, гетероариламины или сульфонилмочевины можно получить из пирроло[2,3-c]пиридинов 44, содержащих первичный или вторичный амин (например, Общие способы D, Е, I, H и J). Также, удаление защитной группы пирроло[2,3-c]пиридинов 44, содержащих защитную группу в R' можно осуществить с использованием условий, таких как описаны в Greene, TW и Wuts, P.G.M., указаны выше, или в Общих способах G, M, или N. Например, для R'', содержащей спирт, защищенный TBDMS, защитную группу можно удалить с получением незащищенного спирта (например, общий способ M) и соединения 44 с удаленной защитной группой можно затем подвергнуть дальнейшему взаимодействию, как описано выше.
Схема XVIII
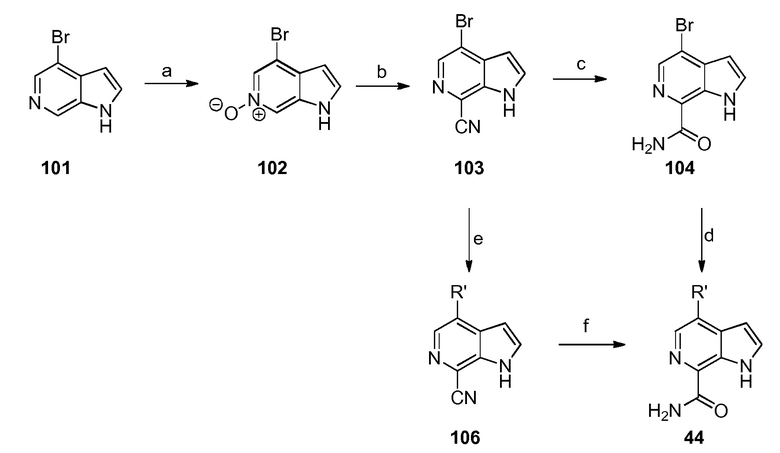
При желании, хиральное разделение любого из хиральных соединений в Схемах I-XVIII может быть сделано с помощью способов, известных специалистам в данной области, такими как хиральная препаративная ВЭЖХ, хиральная SFC или кристаллизация диастереомерных солей.
ОБЩИЕ СПОСОБЫ И ПРИМЕРЫ
Далее на схемах 1-34 представлены общие схемы синтеза, которые использованы для получения большинства соединений, раскрытых в рассматриваемой заявке. Указанные схемы представлены только с иллюстративными целями и их не следует рассматривать как ограничивающие настоящее изобретение.
Схема 1. Реакция Сузуки арил- или гетероарил- галогенида с арил- или гетероарил-бороновой кислотой или боронатом (Общий способ A)
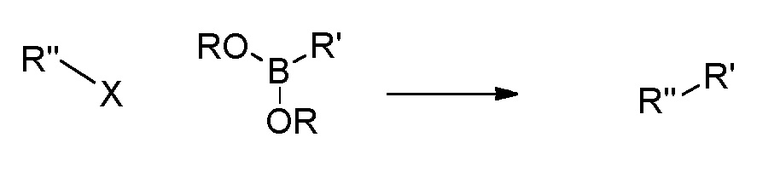
Схема 2. Нуклеофильное замещение арилгалогенида с амином (Общий способ B)
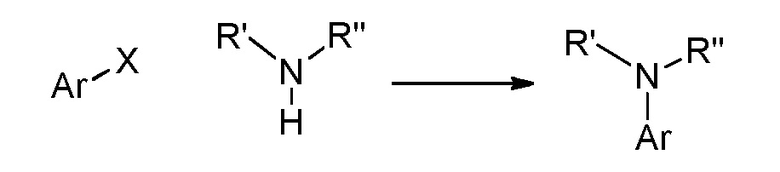
Схема 3. Гидролиз сложного эфира до карбоновой кислоты (Общий способ C)
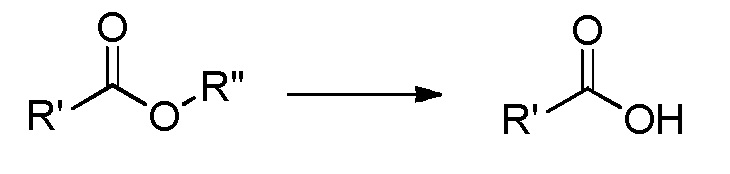
Схема 4. Получение амида из амина и карбоновой кислоты (Общий способ D)
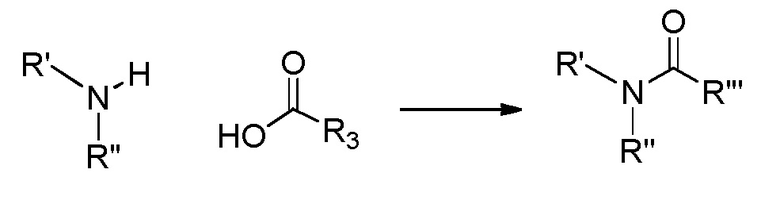
Схема 5. Получение амида из амина и галоидангидрида или ангидрида (Общий способ E)

Схема 6. Получение 4-йодиндол-7-карбоксамида (Общий способ F)
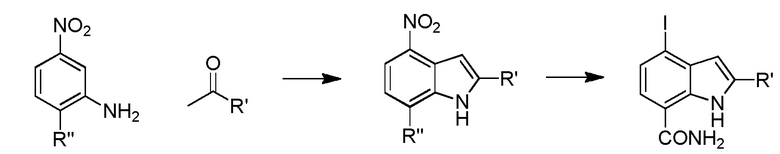
Схема 7. Кислотное расщепление Вос-защищенного амина (Общий способ G)

Схема 8. Восстановительное аминирование альдегида или кетона с первичным или вторичным амином (Общий способ H)
Схема 9. Получение сульфонамида из амина и сульфонилхлорида (Общий способ I)

Схема 10. Замещение алкилгалогенида амином нуклеофилом (Общий способ J)
Схема 11. Гидролиз ацетонида (Общий способ K)
Схема 12. Гидрирование алкена (Общий способ L)
Схема 13. Удаление силильной группы у О-силилового эфира (Общий способ M)
Схема 14. Гидролиз сульфонамида (Общий способ N)
Схема 15. Гидролиз нитрила до первичного амида (Общий способ O)
Схема 16. Получение бороната из арилгалогенида или гетероарилгалогенида (Общий способ P)
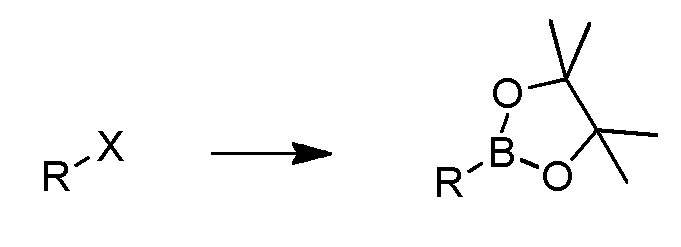
Схема 17. Реакция Мицунобу спирта (Общий способ Q)
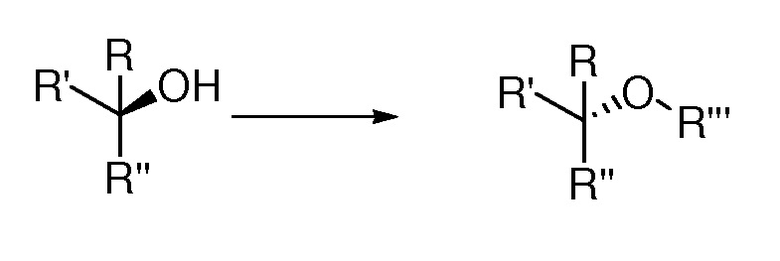
Схема 18. Восстановление нитрогруппы до амина с использованием Fe (Общий способ R)
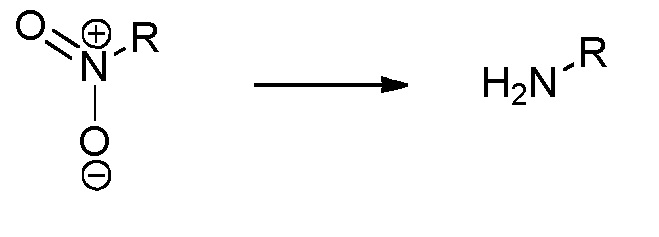
Схема 19. Деметилирование арил метилового эфира (Общий способ S)
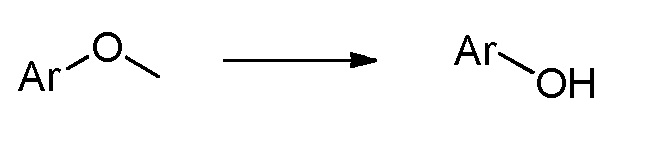
Схема 20. Реакция Бухвальда арилгалогенида или гетероарилгалогенида с амином (Общий способ T)
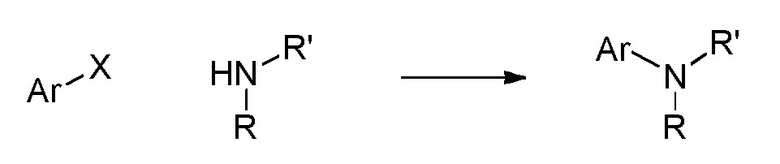
Схема 21. Реакция Негиши арилгалогенида или гетероарилгалогенида с цинкорганическим соединением (Общий способ U)
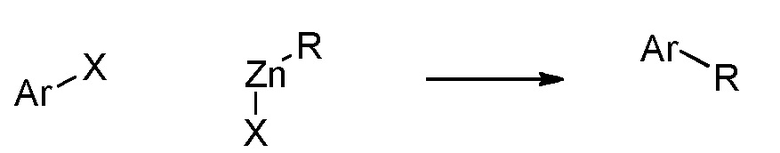
Схема 22. Получение амида из Вос-защищенного амина и карбоновой кислоты (Общий способ V)
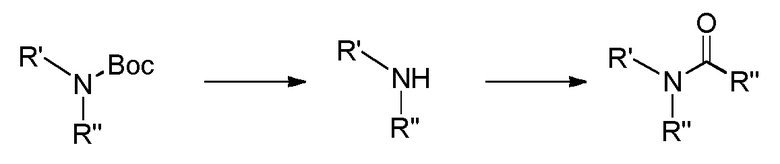
Схема 23. Превращение винилтрифлата в винил-боронат или бороновую кислоту (Общий способ W)
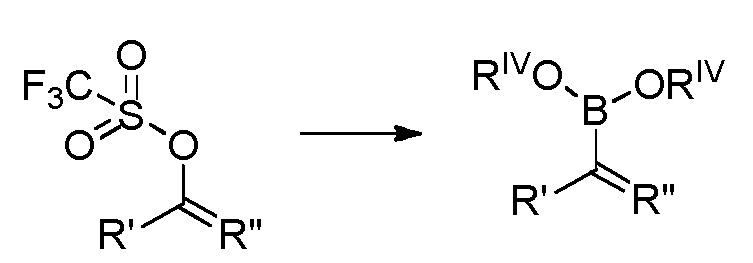
Схема 24. Гидролиз сложного эфира до карбоновой кислоты в основных условиях и удаление тозил-группы из N-тозил-защищенного гетероарильного кольца (Общий способ X)
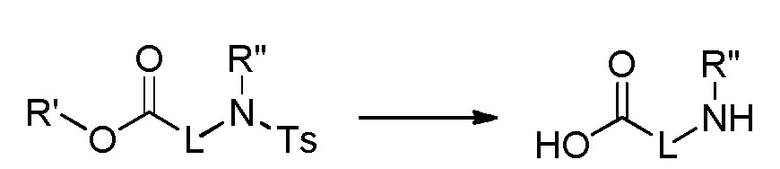
Схема 25. Йодирование 1H-индола или 1Н-азаиндольного кольца с получением 2-йод-1H-индола или 2-йод-1H-азаиндольного кольца (Общий способ Y)

Схема 26. Получение N-Вос защищенного амина (Общий способ Z)
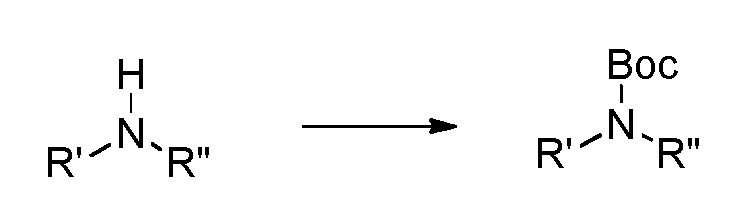
Схема 27. Превращение кетона в винилтрифлат (Общий способ AA)
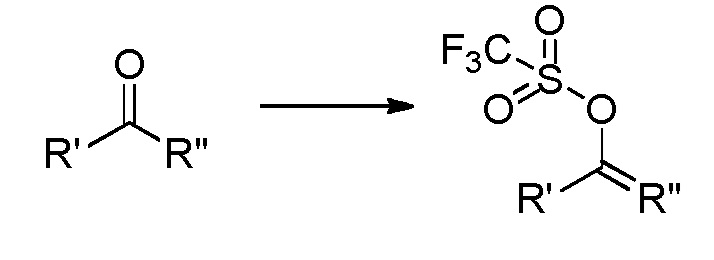
Схема 28. Восстановление двойной связи и удаление CBZ-группы у CBZ-защищенного амина (Общий способ AB)
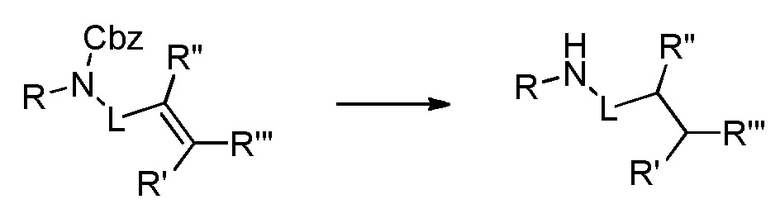
Схема 29. N-Окисление N-содержащего гетероароматического кольца (Общий способ AC)

Схема 30. Цианирование N-оксида, содержащего гетероарильное кольцо (Общий способ AD)
Схема 31. Восстановление сложного эфира до спирта (Общий способ AE)
Схема 32. Восстановление пиридинового кольца до пиперидинового кольца (Общий способ AF)
Схема 33. Борилирование винилтрифлата и реакция Сузуки in situ образованного бороната с арилгалогенидом (Общий способ AG)
Схема 34. Получение N-тозил защищенного гетероароматического кольца (Общий способ AH)
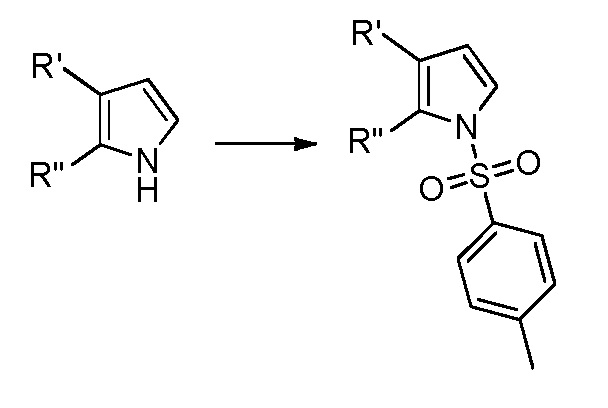
Список общих способов
Общий способ A: Реакция Сузуки арил- или гетероарил- галогенида с арил- или гетероарил-бороновой кислотой или боронатом
Общий способ B: Нуклеофильное замещение арилгалогенида с амином
Общий способ C: Гидролиз сложного эфира до карбоновой кислоты
Общий способ D: Получение амида из амина и карбоновой кислоты
Общий способ E: Получение амида из амина и галоидангидрида или ангидрида
Общий способ F: Получение 4-йодиндол-7-карбоксамида
Общий способ G: Кислотное расщепление Вос-защищенного амина
Общий способ H: Восстановительное аминирование альдегида или кетона с первичным или вторичным амином
Общий способ I: Получение сульфонамида из амина и сульфонилхлорида
Общий способ J: Замещение алкилгалогенида амином нуклеофилом
Общий способ K: Гидролиз ацетонида
Общий способ L: Гидрирование алкена
Общий способ M: Удаление силильной группы у О-силилового эфира
Общий способ N: Гидролиз сульфонамида
Общий способ O: Гидролиз нитрила до первичного амида
Общий способ P: Получение бороната из арилгалогенида или гетероарилгалогенида
Общий способ Q: Реакция Мицунобу спирта
Общий способ R: Восстановление нитрогруппы до амина с использованием Fe
Общий способ S: Деметилирование арил метилового эфира
Общий способ T: Реакция Бухвальда арилгалогенида или гетероарилгалогенида с амином
Общий способ U: Реакция Негиши арилгалогенида или гетероарилгалогенида с цинкорганическим соединением
Общий способ V: Образования амида из Вос-защищенного амина и карбоновой кислоты
Общий способ W: Конверсия винилтрифлата до винил-бороната или бориновой кислоты
Общий способ X: Гидролиз сложного эфира до карбоновой кислоты в основных условиях и удаление тозил-группы из N-тозил-защищенного гетероарильного кольца
Общий способ Y: Йодирование 1H-индола или 1Н-азаиндольного кольца с получением 2-йод-1H-индола или 2-йод-1H-азаиндольного кольца
Общий способ Z: Получение N-Вос защищенного амина
Общий способ AA: Превращение кетона в винилтрифлат
Общий способ AB: Восстановление двойной связи и удаление CBZ-группы у CBZ-защищенного амина
Общий способ AC: N-Окисление N-содержащего гетероароматического кольца
Общий способ AD: Цианирование N-оксида, содержащего гетероарильное кольцо
Общий способ AE: Восстановление сложного эфира до спирта
Общий способ AF: Восстановление пиридинового кольца до пиперидинового кольца
Общий способ AG: Борилирование винилтрифлата и реакция Сузуки вновь образованного бороната с арилгалогенидом
Общий способ AH: Получение N-тозил защищенного гетероароматического кольца
Следующие примеры расположены в соответствии с конечным общим способом, используемым при их получении. Способы синтеза всех новых промежуточных соединений детализированы с помощью последующего перечисления общих способов (буквенные обозначения) в скобках после их названия с дополнительными соответствующими реактантами и реагентами. Рабочий пример указанного протокола приведен далее с использованием примера № A.3.7 в качестве неограничивающей иллюстрации. Пример № A.3.7 представляет собой 2-(1-ацетил-1,2,3,6-тетрагидропиридин-4-ил)-4-(3-(4-(дифторметил)бензамидо)-2-метилфенил)-1H-индол-7-карбоксамид, который получают из 2-(1-ацетил-1,2,3,6-тетрагидропиридин-4-ил)-4-бром-1H-индол-7-карбоксамида, используя общий способ A с 4-(дифторметил)-N-(2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)бензамидом как представлено на схеме A.
Схема A
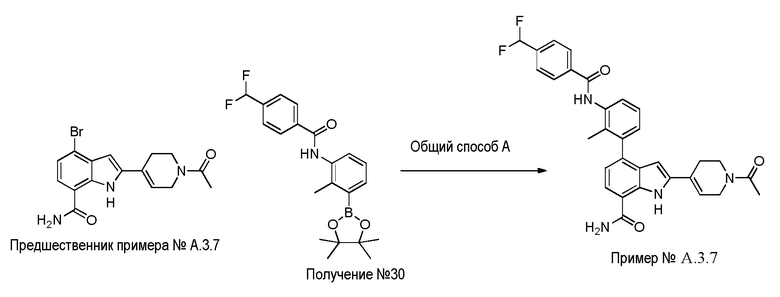
Предшественник примера № A.3.7, 2-(1-ацетил-1,2,3,6-тетрагидропиридин-4-ил)-4-бром-1H-индол-7-карбоксамид, получали (как показано на схеме B) взаимодействием 4-бром-2-иод-1H-индол-7-карбоксамида (Получение № 1) с 1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2H)-ил)этаноном, производимым компанией Combi-Blocks, в соответствии с условиями, представленными в общем способе A. Таким образом, Пример № A.3.7 может быть записан как: Пример № A.3.7 получали из 4-(дифторметил)-N-(2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)бензамида (Получение № 29) и 2-(1-ацетил-1,2,3,6-тетрагидропиридин-4-ил)-4-бром-1H-индол-7-карбоксамида (получали с использованием A с 4-бром-2-йодо-1H-индол-7-карбоксамидом [Получение №1] и 1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2H)-ил)этаноном [Combi-Blocks]), с помощью общего способа A. В таблицах после общего способа, это представлено одним реагентом в заголовке таблицы и одним в отдельной колонке в той же строке в качестве продукта.
Схема B
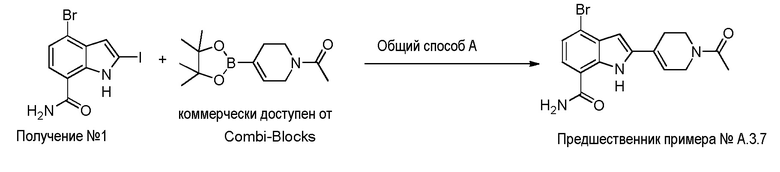
In vitro активность Btk киназы определяли с помощью метода резонансного переноса энергии флуоресценции с временным разрешением (trFRET)
BTK соответствует рекомбинантному каталитическому домену человека (а.к. 393-659), который экспрессируется в клетках SF9 с N-концевой гистидиновой меткой и очищали с помощью афинной хроматографии с использованием иммобилизованных металлов. BTK смешивали с пептидным субстратом (биотин-TYR1, последовательность: биотин-(Ahx)-GAEEEIYAAFFA-COOH, 0,2 мкМ конечная) при различных концентрациях ингибитора в реакционном буфере: 50 мM MOPSO pH 6,5, 10 мМ MgCl2, 2 мM MnCl2, 2,5 мM DTT, 0,01% BSA, 0,1 мM Na3VO4 и 0,001 мM ATP. После приблизительно 60 минут инкубации при комнатной температуре, реакцию гасили добавлением EDTA (конечная концентрация: 100 мМ) и обнаруживали добавлением детектирующих реагентов (окончательные приблизительные концентрации: 30 мM HEPES pH 7,0, 0,06% BSA, 0,006% Tween-20, 0,24 M KF, 80 нг/мл PT66K (меченное европием антитело против фосфотирозина кат. № 61T66KLB Cisbio, Bedford, MA) и 0,6 мкг/мл SAXL (Phycolink стрептавидин-аллофикоцианин акцептор, кат. №PJ25S, Prozyme, San Leandro, CA). Подготовленную реакционную смесь инкубировали в темноте в течение приблизительно 60 минут при комнатной температуре, а затем считывали показания, испрользуя флуоресцентный детектор с временным разрешением (Rubystar, BMG) с помощью лазера с длиной волны возбуждения 337 нм и мониторинга длинами волн испускания при 665 нм. В линейном интервале анализа отношение наблюдаемого сигнала при 665 нм непосредственно связано с фосфорилированным продуктом, и эти значения используют для расчета величин IC50.
В рамках Таблиц и Примеров, ниже, Btk IC50 каждого соединения выражается следующим образом: A = соединение с IC50 менее чем 0,1 мкМ, B = соединение с IC50 в диапазоне от 0,1 мкМ до 1 мкМ, и C = соединение с IC50 Btk в диапазоне от 1 мкМ до 10 мкМ.
Аналитические способы
Аналитические результаты включены в раскрытые далее способы, в иллюстрации общих способов, или в таблицы примеров. Если нет других указаний, все результаты 1H-ЯМР получены на приборах Varian Mercury Plus 400 МГц, Inova, или 400-MR и химические сдвиги выражены как части на миллион (м.д.). Результаты ЖХ/МС и ВЭЖХ относятся к условиям ЖХ/МС и ВЭЖХ, приведенным в таблице, и обозначены мелкими буквами, указанными далее в таблице 1.
Способы ЖХ/МС и ВЭЖХ
Подвижная фаза: A: 15 мл TFA в 20 л H2O; B: MeCN
Колонка: Luna 100×30,0 мм, 5 мк; Скорость потока: 25 мл/мин;
Мониторинг длины волны: 220 и 254 нм
Градиент: начальная выдержка при 21% B в течение 1 минуты, градиент 21% до 51% B в 12 мин
Колонка: Synergi Max-RP C18 250×80 мм внутренний диаметр 10 мк
Подвижная фаза: A для H2O (0,09% TFA) и B для CH3CN
Градиент: B от 15% до 43% в 25 мин
Скорость потока: 40 мл/мин
Количество инъекции: 50 мг на инъекцию
Подвижная фаза: A: TFA/H2O=0,075% об./об.; B: MECN
Колонка: Luna C18 100×30,0 мм, 5 мк
Скорость потока: 25 мл/мин
Мониторинг длины волны: 220 и 254 нм
Градиент:
Подвижная фаза: A: TFA/H2O=0,075% об./об.; B: MeCN
Колонка: Luna C18 200×21,2 мм, 5 мкм
Скорость потока: 25 мл/мин
Мониторинг длины волны: 220 и 254 нм
Градиент:
Подвижная фаза: A: 15 мл TFA в 20 л H2O; B: MeCN
Колонка: Luna 100×30,0 мм, 5 мкм
Скорость потока: 25 мл/мин
Мониторинг длины волны: 220 и 254 нм
Градиент: начальная выдержка при 8% B в течение 1 мин, градиент 8% до 38% B в 12 мин
Подвижная фаза: A: TFA/H2O=0,075% об./об.; B: MeCN
Колонка: Luna C18 100×30,0 мм, 5 мкм
Скорость потока: 25 мл/мин
Мониторинг длины волны: 220 и 254 нм
Градиент:
Подвижная фаза: A: 8 мл NH3.H2O в 20 л H2O; B: MeCN
Колонка: waters Xbridge130×21,2 мм, 5 мкм
Скорость потока: 25 мл/мин
Мониторинг длины волны: 220 и 254 нм
Градиент: начальная выдержка при 27% B в течение 1 мин, градиент 27% до 57% B в 12 мин
Колонка: Luna(2) C18 250×50 мм внутренний диаметр 10 мк
Подвижная фаза: A для H2O (0,09% TFA) и B для CH3CN
Градиент: B от 82% до 82%
Скорость потока: 100 мл/мин
Количество инъекции: 0,7 г на инъекцию
Колонка: waters X-bridge ODS C 18 19×250 мм,10 мкм
Подвижная фаза: A: вода (10 ппM NH4HCO3); B: ACN
Скорость потока: 30 мл/мин
Мониторинг длины волны: 220 и 254 нм
Градиент: 10-60% B в 8 мин, остановка 15 мин
Способы хиральной ВЭЖХ
Общие способы очистки
Для общих способов, окончательные соединения могут быть очищены любым способом или комбинацией способов, известных специалистам в данной области. Некоторые примеры, которые являются неограничивающими, включают колоночную хроматографию с твердой фазой (т.е. силикагель, оксид алюминия, и тому подобное) и растворителем (или комбинацией растворителей), который элюирует желаемые соединения (т.е. гексаны, гептан, EtOAc, DCM, MeOH, EtOH, MeCN, вода и тому подобное); препаративную ТСХ с твердой фазой (т.е. силикагель, оксид алюминия и тому подобное) и растворителем (или комбинацией растворителей), который элюирует желаемые соединения (т.е. гексан, гептан, EtOAc, DCM, MeOH, EtOH, MeCN, вода и тому подобное); ВЭЖХ с обращенной фазой (см. Таблица 1 для некоторых, не ограничивающих условий); перекристаллизацию из подходящего растворителя (например, MeOH, EtOH, IPA, EtOAc, толуол и тому подобное) или комбинации растворителей (т.е. EtOAc/гептан, EtOAc/МеОН, и тому подобное); хиральную LC с твердой фазой и соответствующим растворителем (т.е. EtOH/гептан, МеОН/гептан, IPA/ гептан и тому подобное, с или без модификатора, такого как диэтиламин, TFA, и тому подобное) для элюирования желаемого соединения; хиральная SFC с твердой фазой и СО2 с соответствующим модификатором (т.е. МеОН, EtOH, IPA с или без дополнительного модификатора, такого как диэтиламин, TFA, и тому подобное); осаждение из комбинации растворителей (т.е. DMF/вода, DMSO/DCM, EtOAc/гептан и тому подобное); растирание с соответствующим растворителем (т.е. EtOAc, DCM, MeCN, MeOH, EtOH, IPA, н-PrOH и тому подобное); экстракцию растворением соединения в жидкости и промывку соответствующейся несмешивающейся жидкостью (т.е. DCM/вода, EtOAc/вода, DCM/насыщенный NaHCO3, EtOAc/насыщенный раствор NaHCO3, DCM/10%-ный водный раствор HCl, EtOAc/10%-ный водный раствор HCl, и тому подобное); дистилляцию (т.е. простую, фракционную, Кугельрофа и тому подобное); газовую хроматографию с использованием соответствующей температуры, газа-носителя и скорости потока; сублимацию при соответствующей температуре и давлении; фильтрование через среду (т.е. Florosil®, глинозем, целит, силикагель и тому подобное) с растворителем (т.е. гептан, гексан, EtOAc, DCM, MeOH и тому подобное) или комбинацией растворителей; образование соли с твердой подложкой (смолой на основе, то есть ионный обмен) или без. Некоторые описания этих способов можно найти в следующих ссылках, Gordon, A. J. and Ford, R. A. "The Chemist’s Companion”, 1972; Palleros, D. R. “Experimental Organic Chemistry”, 2000; Still, W. C., Kahn, M. and Mitra, A. J. Org. Chem. 1978, 43, 2923; Yan, B. “Analysis and Purification Methods in Combinatorial Chemistry” 2003; Harwood, L. M., Moody, C. J. and Percy, J. M. “Experimental Organic Chemistry: Standard and Microscale, 2nd Edition”, 1999; Stichlmair, J. G. and Fair, J. R. “Distillation; Principles and Practices” 1998; Beesley T. E. and Scott, R. P. W. “Chiral Chromatography”, 1999; Landgrebe, J. A. “Theory and Practice in the Organic Laboratory, 4th Edition”, 1993; Skoog, D. A. and Leary, J. J. “Principles of Instrumental Analysis, 4th Edition” 1992; Subramanian, G. "Chiral Separation Techniques 3rd Edition" 2007; Kazakevich, Y. и Lobrutto, R. "HPLC for Pharmaceutical Scientists" 2007. Конечные или промежуточные соединения, полученные с помощью любого из следующих общих способов, могут быть необязательно очищены с помощью одного или нескольких способов очистки, описанных выше.
Получения и Примеры
Далее приводятся общие способы синтеза, которые используют в каждой общей процедуре и которые включают иллюстрацию соединения, которое синтезируют, используя обозначенный общий способ. Ни одно из перечисленных здесь конкретных условий и ни один из конкретных реагентов не следует рассматривать как ограничивающие объем настоящего изобретения, и они представлены здесь только для иллюстративных целей. Все исходные материалы коммерчески доступны от Sigma-Aldrich (включая Fluka и Discovery CPR) если только нет других указаний после химического наименования. Названия реагентов даны в соответствии с названиями на коммерческих флаконах или в соответствии с номенклатурой IUPAC, CambridgeSoft® ChemDraw Ultra 9.0.7, CambridgeSoft® Chemistry E-Notebook v9.0.127 или v11.0.3.68, или AutoNom 2000. Соединения, обозначенные как соли (например, гидрохлорид, ацетат) могут содержать более одного молярного эквивалента соли. Соединения по настоящему изобретению, где абсолютная стереохимия была определена с использованием коммерчески доступного энантиомерно чистого исходного вещества или стереохимически определенного промежуточного соединения, или путем дифракции рентгеновских лучей, обозначены звездочкой после номера примера.
Получение № 1. 4-бром-2-йод-1H-индол-7-карбоксамид
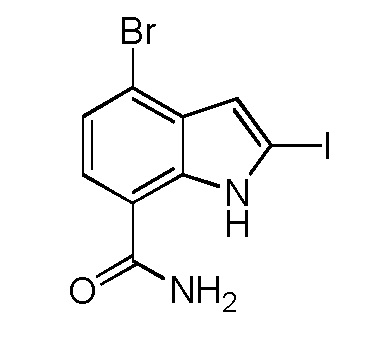
Стадия A: 4-бром-1H-индол-7-карбоновая кислота

К раствору 4-бром-2-нитробензойной кислоты (30 г, 122 ммоль) в безводном THF (500 мг), добавляли по каплям раствор винилмагнийбромида (51,2 мл, 512 ммоль, 1 н) в THF при температуре приблизительно -30 до -50°C. Реакционную смесь перемешивали при приблизительно от -30 до -40°C в течение приблизительно 2 часов. Затем реакционную смесь выливали в насыщенный водный раствор NH4Cl и смесь экстрагировали EtOAc (200 мл×2). Объединенные органические слои промывали насыщенным солевым раствором, сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением 4-бром-1H-индол-7-карбоновой кислоты (33 г сырой), которую использовали непосредственно на следующей стадии без дополнительной очистки. 1H-ЯМР (400 МГц, DMSO-d6) δ 11,42 (м, 1H), 8,11 (ушир.с, 1H), 7,63 (дд, J=17,4, 8,0 Гц, 1H), 7,45 (дт, J=14,2, 2,8 Гц, 1H), 7,32 (дд, J=21,9, 8,0 Гц, 1H), 6,47 (ддд, J=25,5, 3,1, 2,1 Гц, 1H).
Стадия B: Метил 4-бром-1H-индол-7-карбоксилат
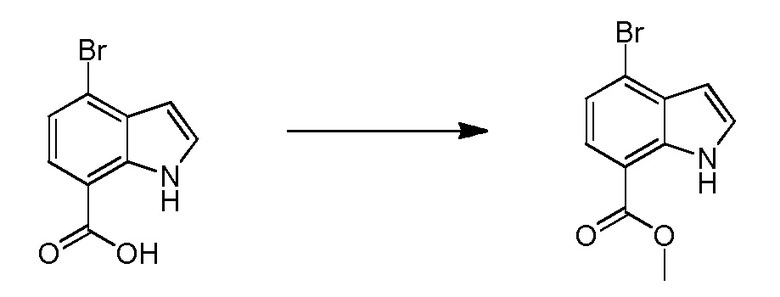
К раствору 4-бром-1H-индол-7-карбоновой кислоты (33 г, 137 ммоль) в DMF (300 мг), добавляли Cs2CO3 (90 г, 276 ммоль) и перемешивали при комнатной температуре в течение 1 часа. Затем добавляли по каплям йодметан (29,3 г, 206 ммоль) при температуре приблизительно 0°C. Реакционную смесь нагревали до комнатной температуры в течение приблизительно 3 часов. Смесь выливали в воду и экстрагировали EtOAc (200 мл×2). Объединенные органические слои промывали насыщенным солевым раствором, сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, с получением метил 4-бром-1H-индол-7-карбоксилата (13,8 г, 20%): 1H-ЯМР (CDCl3) δ 9,98 (с, 1H), 7,76-7,74 (д, J=8, 1H), 7,39-7,34 (м, 2H), 6,68-6,66 (м, 1H), 4,00 (с, 3H).
Стадия C: Метил 4-бром-1-тозил-1H-индол-7-карбоксилат
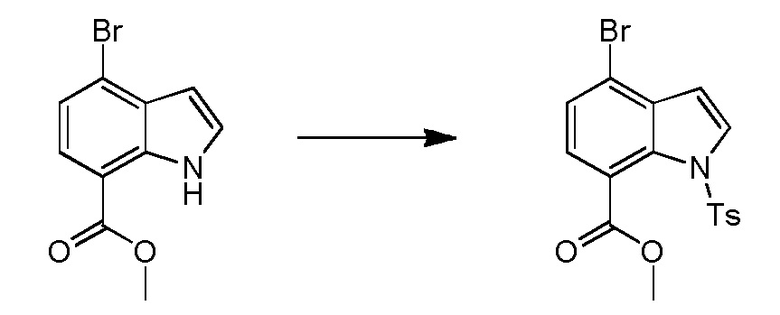
К раствору метил 4-бром-1H-индол-7-карбоксилата (130 г, 512 ммоль) в безводном THF (1500 мг) добавляли NaH (18,4 г, 767 ммоль) порциями при приблизительно 0°C и перемешивали в течение приблизительно 0°C и перемешивали в течение приблизительно 1 часа при 0°C. Затем TsCl (117 г, 614 ммоль) добавляли порциями при приблизительно 0°C. Реакционную смесь нагревали до комнатной температуры в течение приблизительно 2 часов. Реакционную смесь выливали в ледяную воду и экстрагировали EtOAc (1000 мл×2). Объединенные органические слои промывали насыщенным солевым раствором, сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, получая метил 4-бром-1-тозил-1H-индол-7-карбоксилат (150 г, 72%): 1H-ЯМР (CDCl3) δ 7,60-7,58 (д, J=8,4, 2H), 7,54-7,53 (д, J=3,6, 1H), 7,46-7,44 (д, J=8, 1H), 7,37-7,35 (д, J=8,4, 1H), 7,21-7,18 (д, J=8,4, 2H), 6,77-6,76 (м, 1H), 3,93 (с, 3H), 2,35 (с, 3H).
Стадия D: Метил 4-бром-2-йод-1-тозил-1H-индол-7-карбоксилат

К раствору диизопропиламина (6,2 г, 61,2 ммоль) в безводном THF (100 мг), перемешанный в трет-BuLi (3,92 г, 61,2 ммоль) в пентане добавляли при приблизительно 0°C в атмосфере N2 и смесь перемешивали в течение приблизительно 10 минут. Раствор метил 4-бром-1-тозил-1H-индол-7-карбоксилата (10 г, 24,49 ммоль) в безводном THF (100 мг) добавляли при приблизительно -70°C в атмосфере N2. После приблизительно 30 минут добавляли раствор I2 (9,33 г, 36,7 ммоль) в безводном THF (50 мг). После приблизительно 30 минут, охлаждающую баню удаляли и смесь перемешивали в течение приблизительно одного часа. Смесь гасили насыщенным водным раствором Na2S2O3. К смеси добавляли воду и EtOAc. Слои разделяли и водный слой экстрагировали EtOAc (300 мл×2). Объединенные органические слои промывали насыщенным солевым раствором, сушили над безводным Na2SO4, фильтровали, концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле с получением метил 4-бром-2-йод-1-тозил-1H-индол-7-карбоксилата (7,5 г, 38%): 1H-ЯМР (CDCl3): δ 7,64-7,59 (м, 2H), 7,55-7,53 (м, 2H), 7,30-7,27 (м, 2H), 7,17-7,17 (м, 1H), 4,06-4,05 (д, J=1,2, 3H), 2,49 (с, 3H).
Стадия E: 4-бром-2-йод-1H-индол-7-карбоновая кислота
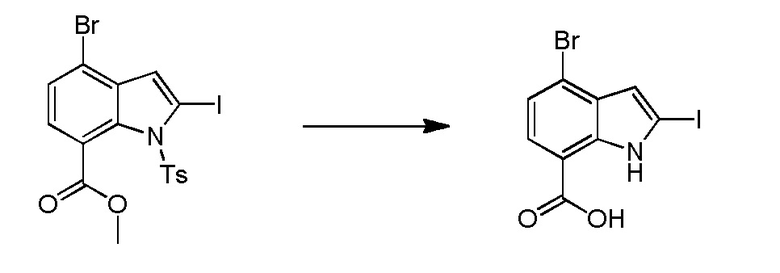
К раствору метил 4-бром-2-йод-1-тозил-1H-индол-7-карбоксилата (75 г, 23,4 ммоль) в MeOH (750 мг), THF (1500 мг) и воде (750 мг), LiOH (67 г, 280 ммоль) добавляли, реакционную смесь нагревали при температуре приблизительно 45°C в течение приблизительно 3 часов. Полученный раствор концентрировали при пониженном давлении, для удаления MeOH и THF, затем раствор доводили до рН=6-7 с помощью HCl (1 н), осадок отфильтровывали и сушили в глубоком вакууме, получая 4-бром-2-йод-1H-индол-7-карбоновую кислоту (45 г, 88%): 1H-ЯМР (DMSO-d6) δ 11,60 (с, 1H), 7,56 (д, J=8,0, 1H), 7,31 (м, J=8,0, 1H), 6,72 (с, 1H).
Стадия F: 4-бром-2-йод-1H-индол-7-карбоксамид
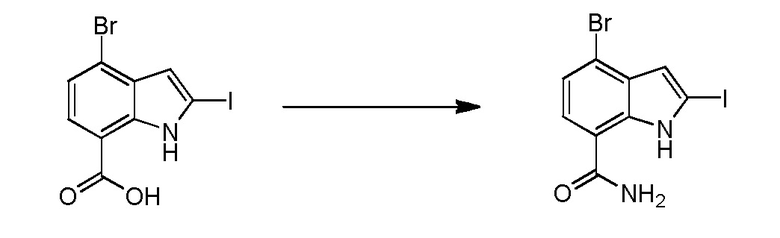
К раствору 4-бром-2-йод-1H-индол-7-карбоновой кислоты (45 г, 123 ммоль) в DMF (450 мг) добавляли HOBt (28,2 г, 184 ммоль), PyBOP (96 г, 184 ммоль), NH4Cl (10 г, 184,5 ммоль) и DIEA (63,6 г, 492 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение приблизительно 2 часов. Добавляли воду, реакционную смесь экстрагировали EtOAc (1000 мл×2), органическую фазу сушили над безводным Na2SO4 и концентрировали при пониженном давлении и остаток очищали колоночной хроматографией с петролейным эфиром:EtOAc (20:1 до 1:1) с получением 4-бром-2-йод-1H-индол-7-карбоксамида (25 г, 56%): 1H-ЯМР (DMSO-d6) δ 11,62 (с, 1H), 8,24 (с, 1H), 7,62-7,60 (д, J=8, 2H), 7,38-7,36 (д, J=8, 1H), 6,77 (с, 1H): ЖХ/МС (Таблица 1, способ d) Время удерживания = 3,07 минуты; MS m/z: 366 (M-H)-.
Получение № 2. 4-бром-1H-индол-7-карбоксамид

К раствору 4-бром-1H-индол-7-карбонитрила (3 г, 13,57 ммоль, Sinova) в EtOH (36,2 мг)/DMSO (9,05 мг) медленно добавляли пероксид водорода (28,0 мл, 274 ммоль) и NaOH (28,0 мл, 28,0 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение приблизительно 1 часа. Добавляли воду и осадок собирали фильтрованием, промывали водой и сушили в вакууме с получением 4-бром-1H-индол-7-карбоксамида (2,85 г, 88%). ЖХ/МС (Таблица 1, способ f) Время удерживания = 1,42 минуты; MS m/z: 280 (M+MeCN)+.
Получение № 3. 2-(2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-3,4-дигидроизохинолин-1(2H)-он
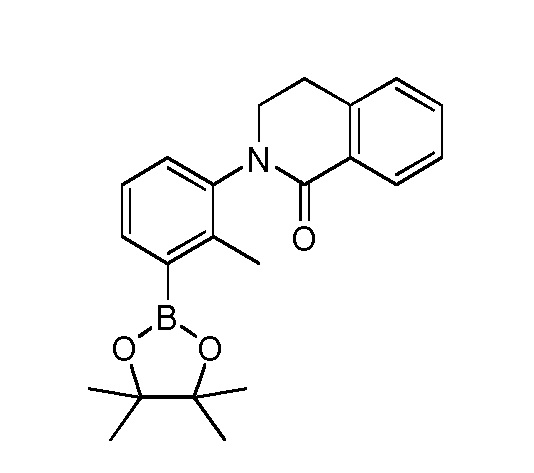
Стадия A: 3,4-Дигидроизохинолин-1(2H)-он
К раствору 2,3-дигидро-1H-инден-1-она (30 г, 227 ммоль) в DCM (300 мг) добавляли метансульфоновую кислоту (300 мг) и раствор охлаждали приблизительно до 0°C. Азид натрия (30 г, 461 ммоль) добавляли к раствору порциями при приблизительно 0°C и реакционную смесь перемешивали в течение ночи при комнатной температуре. Реакционную смесь нейтрализовали 20%-ным водным NaOH и экстрагировали DCM (2×1 л). Органическую фазу сушили безводным Na2SO4 и концентрировали с получением остатка, который очищали с помощью колоночной хроматографии на силикагеле с получением 3,4-дигидроизохинолин-1(2H)-она (5 г, 15%): 1H-ЯМР (MeOD) δ 7,93-7,91 (м, 1H), 7,49-7,45 (м, 1H), 7,36-7,45 (м, 1H), 7,28-7,26 (д, 1H), 3,50-3,46 (т, 2H), 2,97-2,94 (т, 2H).
Стадия B: 2-(3-бром-2-метилфенил)-3,4-дигидроизохинолин-1(2H)-он
Смесь 3,4-дигидроизохинолин-1(2H)-она (3,5 г, 13,6 ммоль), 1,3-дибром-2-метилбензола (17,5 г, 70,5 ммоль) и K2CO3 (9,85 г, 71,3 ммоль) в DMSO (40 мг) продували N2, обрабатывали CuI (1,75 г, 9 ммоль) и нагревали до приблизительно 160°C в течение приблизительно 4 часов. Реакционную смесь разбавляли DCM и фильтровали через целит. Фильтрат промывали 5% гидроксидом аммония, сушили и концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле с получением 2-(3-бром-2-метилфенил)-3,4-дигидроизохинолин-1(2H)-она (6 г, 80%): 1H-ЯМР (CDCl3) δ 8,16-8,14 (д, 1H), 7,56-7,54 (д, 2H), 7,49-7,41 (т, 1H), 7,26 (д, 1H), 7,25-7,18 (д, 1H), 7,15-7,13 (д, 1H), 3,98-3,92 (м, 1H), 3,76-3,70 (м, 1H), 3,30-3,22 (м, 1H), 3,13-3,07(м, 1H) 2,36 (с, 3H).
Стадия C: 2-(2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-3,4-дигидроизохинолин-1(2H)-он
Смесь 2-(3-бром-2-метилфенил)-3,4-дигидроизохинолин-1(2H)-она (4,6 г, 14,6 ммоль), бис(пинаколато)диборона (8,8 г, 34,6 ммоль) и CH3COOK (9 г, 91,8 ммоль) в 1,4-диоксане (100 мг) и DMSO (20 мг), PdCl2 (dppf) (1 г, 1,4 ммоль) добавляли. Реакционную смесь нагревали при приблизительно 120°C в течение ночи под защитой N2. После охлаждения до комнатной температуры реакционную смесь фильтровали через целит, твердое вещество промывали EtOAc и фильтрат промывали водой и насыщенным солевым раствором, сушили над Na2SO4, концентрировали и остаток очищали с помощью колоночной хроматографии на силикагеле, с получением 2-(2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-3,4-дигидроизохинолин-1(2H)-она (1,5 г, 28%): 1H-ЯМР (CDCl3) δ 8,19-8,17 (дд, 1H), 7,80-7,78 (дд, 1H), 7,51-7,47 (т, 1H), 7,42-7,38 (т, 1H), 7,32-7,25 (м, 3H), 3,96-3,89 (м, 1H), 3,77-3,71 (м, 1H), 3,27-3,23 (м, 1H), 3,14-3,08 (м, 1H), 2,50 (с, 3H), 1,36 (с, 12H); ЖХ/МС (Таблица 1, способ o) Время удерживания = 3,34 мин; MS m/z: 364 (M+H)+.
Получение № 4. N-(2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)тиазол-2-карбоксамид
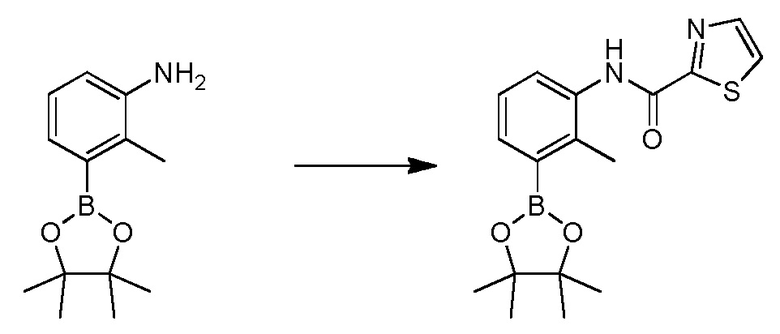
К раствору 2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина (1,9 г, 8,15 ммоль, CombiBlocks) в DCM (50 мг), DIEA (2,1 г, 16,3 ммоль) и HATU (4,03 г, 10,6 ммоль) добавляли при комнатной температуре. После приблизительно 5 минут добавляли тиазол-2-карбоновую кислоту (1,9 г, 8,15 ммоль) и раствор перемешивали в течение приблизительно 3 часов при комнатной температуре. Реакционную смесь выливали в воду, экстрагировали DCM (100 мл×2) и органическую фазу промывали насыщенным солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (элюируя петролейным эфиром:EtOAc = от 10:1 до 3:1), получая N-(2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)тиазол-2-карбоксамид (1 г, 36%): 1H-ЯМР (CDCl3) δ 9,07 (с, 1H), 8,16-8,14 (д, J=8 Гц, 1H), 7,87-7,86 (т, J=3,2 Гц, 1H), 7,57-7,55 (м, 2H), 7,20-7,18 (м, 1H), 2,53 (с, 3H), 1,29 (с, 12H).
Получение № 5. 1-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2(1H)-он
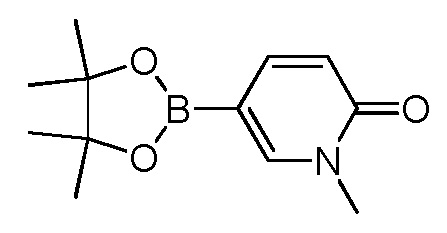
Стадия A: 5-бром-1-метилпиридин-2(1H)-он
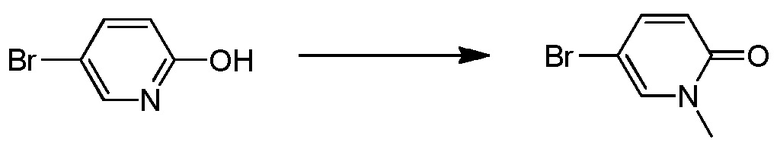
К раствору 5-бромпиридин-2-ола (4 г, 23 ммоль) в THF (200 мг) при приблизительно 0°C добавляли NaH (0,83 г, 34,7 ммоль) порциями. Реакционную смесь перемешивали при комнатной температуре в течение приблизительно 15 минут с последующим добавлением иодметана (9,8 г, 69 ммоль). Смесь перемешивали в течение ночи при комнатной температуре. После завершения реакции (ТСХ мониторинг), реакционную смесь охлаждали до приблизительно 0°C, добавляли воду, экстрагировали EtOAc (100 мл×2). Органический слой промывали насыщенным раствором соли, сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением 5-бром-1-метилпиридин-2-(1H)-она (3 г, 69%): 1H-ЯМР (MeOD) δ 7,87 (с, 1 H), 7,58-7,55 (м, 1 H), 6,47 (д, J=9,6 Гц, 1 H), 3,53 (с, 3 H).
Стадия B: 1-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2(1H)-он
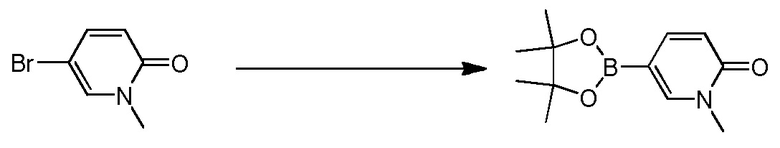
К смеси 5-бром-1-метилпиридин-2(1H)-она (1,0 г, 5,32 ммоль), KOH (0,78 г, 7,98 ммоль) и бис(пинаколато)диборона (0,162 г, 6,38 ммоль) в 1,4-диоксане (20 мг), трициклогексилфосфин (149 мг, 0,532 ммоль), Pd2dba3 (487 мг, 0,532 ммоль) добавляли в атмосфере N2. Смесь перемешивали при температуре приблизительно 80°C в течение приблизительно 5 часов. Затем добавляли воду, водный слой экстрагировали EtOAc (50 мл×2), и органический слой сушили над безводным Na2SO4, концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле с получением 1-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2(1H)-она (0,80 г, 64%): 1H-ЯМР (CDCl3) δ 7,70 (с, 1 H), 7,54 (д, J=8,8 Гц, 1 H), 6,47 (д, J=8,8 Гц, 1 H), 3,49 (с, 3 H), 1,24 (с, 12 H).
Получение № 6. 4-(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фениламино)пиримидин-2-карбонитрил
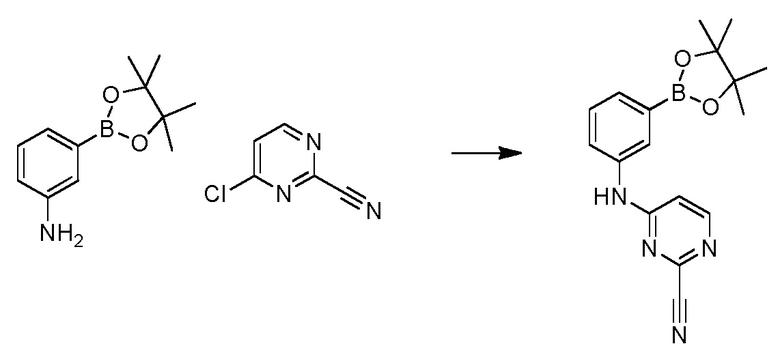
В сосуд для микроволновой печи добавляли 4-хлорпиримидин-2-карбонитрил (100 мг, 0,717 ммоль, CombiPhos), 3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилин (314 мг, 1,433 ммоль), и N-этил-N-изопропилпропан-2-амин (0,250 мл, 1,433 ммоль) в MeCN (7 мл). Сосуд герметизировали и нагревали в микроволновой печи при приблизительно 150°C в течение приблизительно 20 минут при перемешивании. Реакционную смесь охлаждали до комнатной температуры и растворитель удаляли в токе теплого азота. Остаток растворяли в DCM (10 мг) и промывали водой (10 мг). Смесь разделяли с помощью разделителя фаз Biotage и органические слои концентрировали в вакууме с получением сырого продукта. Сырой продукт помещали в колонку с силикагелем и элюировали смесью 10-60% EtAcO/гептан с получением 4-(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фениламино)пиримидин-2-карбонитрила (0,11 г, 48%): ЖХ/МС (Таблица 1, способ f) Время удерживания = 1,89 минут; MS m/z: 323 (M+H)+.
Получение № 7. N-(3-(3-амино-7-карбамоил-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамид

Стадия A: 4-бром-1H-индол-7-карбоновая кислота
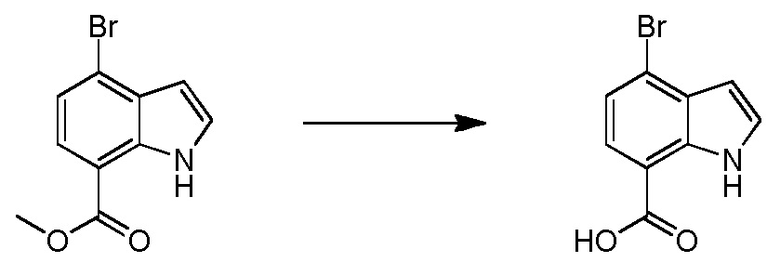
К раствору метил 4-бром-1H-индол-7-карбоксилата (6 г, 23 ммоль, Получение № 1 стадия B) в THF (300 мг), воды (60 мг) и MeOH (60 мг) добавлеяли гидроксид лития (2,83 г, 118 ммоль). Затем смесь нагревали с обратным холодильником в течение ночи. После охлаждения до комнатной температуры, растворитель удаляли при пониженном давлении, водный слой подкисляли добавлением 4 н HCl до приблизительно рН 6. Осадок фильтровали и твердое вещество сушили с получением 4-бром-1H-индол-7-карбоновой кислоты (5,5 г, 97%): 1H-ЯМР (DMSO-d6) δ 11,39 (ушир., 1H), 7,65-7,63 (д, J=8,0 Гц, 1H), 7,46-7,44 (м, 1H), 7,33-7,31 (д, J=8,0 Гц, 1H), 6,49-6,48 (м, 1H).
Стадия B: 4-бром-1H-индол-7-карбоксамид

Раствор 4-бром-1H-индол-7-карбоновой кислоты (5,5 г, 22,91 ммоль) EDC (6,59 г, 34,4 ммоль) и HOBt (5,26 г, 34,4 ммоль) в THF (150 мг) и DCM (180 мг) перемешивали при комнатной температуре в течение 1 часа. Затем смесь барботировали газом NH3 в течение 15 минут и полученную смесь перемешивали при комнатной температуре в течение ночи. Смесь разбавляли добавлением воды и экстрагировали DCM. Органическую фазу промывали насыщенным солевым раствором, сушили и концентрировали с получением остатка, который суспендировали в простом эфире и фильтровали с получением 4-бром-1H-индол-7-карбоксамида (5,3 г, 97%): 1H-ЯМР (DMSO-d6) δ 11,40 (ушир., 1H), 8,08 (ушир., 1H), 7,29-7,57 (д, J=7,6 Гц, 1H), 7,43-7,42 (м, 2H), 7,28-7,26 (д, J=7,6 Гц, 1H), 6,43-6,42 (м, 1H).
Стадия C: 4-бром-3-нитро-1H-индол-7-карбоксамид
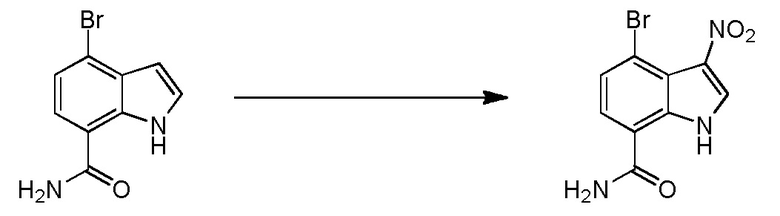
К раствору 4-бром-1H-индол-7-карбоксамида (5,3 г, 22,17 ммоль) и AgNO3 (11,30 г, 66,5 ммоль) в CH3CN (100 мг) добавляли бензоилхлорид (9,35 г, 66,5 ммоль) в CH3CN (20 мг) при температуре приблизительно 0°C и смесь перемешиваюли при температуре приблизительно 0°C в течение 1 часа в темноте. Добавляли воду и EtOAc. Органическую фазу концентрировали с получением остатка, который промывали DCM с получением 4-бром-3-нитро-1H-индол-7-карбоксамида (2,6 г, 41%): 1H-ЯМР (DMSO-d6) δ 12,46 (ушир., 1H), 8,39-8,38 (д, J=3,6 Гц, 1H), 8,33 (ушир., 1H), 7,77-7,73 (м, 2H), 7,67-7,62 (м, 1H). ЖХ/МС (Таблица 1, способ l) Время удерживания = 2,41 мин; MS m/z: 285 (M+H)+.
Стадия D: N-(3-(7-карбамоил-3-нитро-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамид
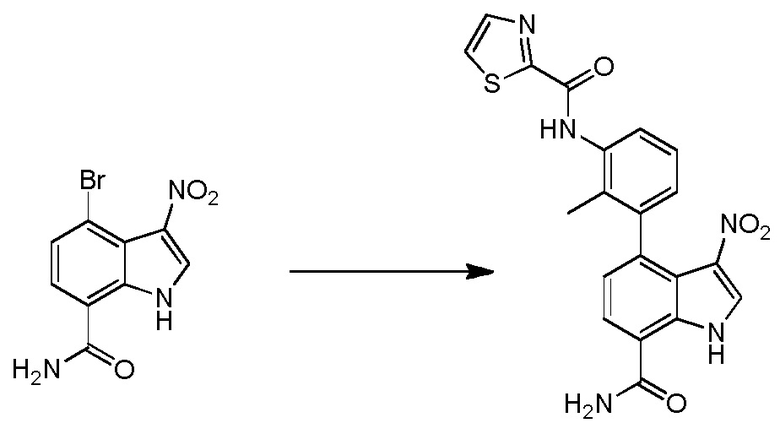
К раствору 4-бром-3-нитро-1H-индол-7-карбоксамида (4 г, 14 ммоль), N-(2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)тиазол-2-карбоксамида (5,8 г, 16,9 ммоль, Получение № 4) в 1,4-диоксане (100 мг) и воде (25 мг) добавляли Pd(PPh3)4 (0,81 г, 0,7 ммоль) и CsF (6,4 г, 42 ммоль) и смесь перемешивали при температуре приблизительно 120°C в течение ночи в атмосфере N2. После охлаждения до комнатной температуры смесь разбавляли добавлением воды и экстрагировали EtOAc. Органическую фазу сушили и концентрировали при пониженном давлении с получением остатка, который очищали с помощью препаративной ВЭЖХ (таблица 1, способ ah) с получением сырого N-(3-(7-карбамоил-3-нитро-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамида (2 г, 33%): ЖХ/МС (Таблица 1, способ l) Время удерживания = 1,44 мин; MS m/z: 422 (M+H)+.
Стадия E: N-(3-(3-амино-7-карбамоил-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамид
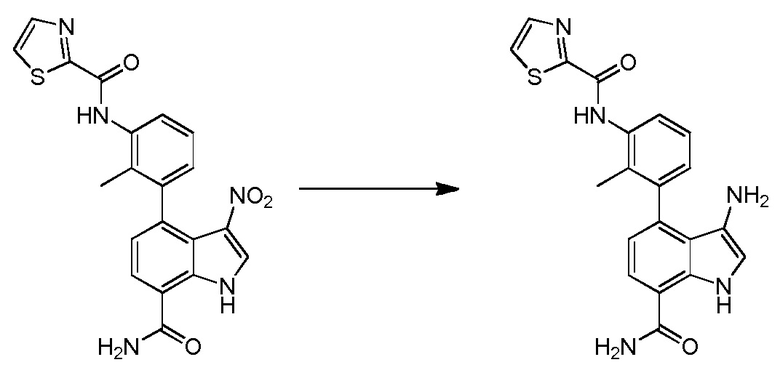
К раствору N-(3-(7-карбамоил-3-нитро-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамида (0,20 г, 0,48 ммоль) в EtOH (20 мг) добавляли никель Ренея (0,10 г) и смесь перемешивали при комнатной температуре в атмосфере H2 50 фунт на кв.дюйм в течение приблизительно 6 часов. Смесь фильтровали и фильтрат концентрировали при пониженном давлении с получением сырого N-(3-(3-амино-7-карбамоил-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамида (0,11 г, 59%), который использовали без дальнейшей очистки: ЖХ/МС (Таблица 1, способ l) Время удерживания = 1,54 мин; MS m/z: 392 (M+H)+.
Получение № 8. 4-Гидрокси-N-(2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-4-(трифторметил)циклогексанкарбоксамид
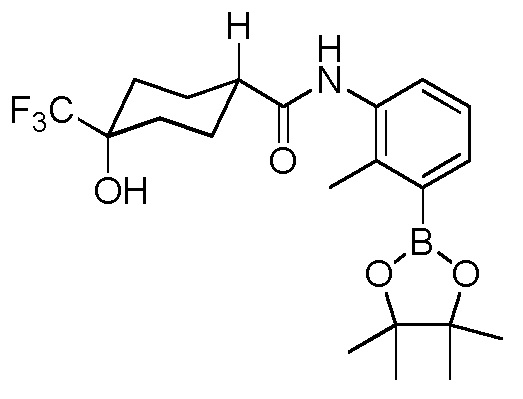
Стадия A: Этил 4-гидрокси-4-(трифторметил)циклогексанкарбоксилат
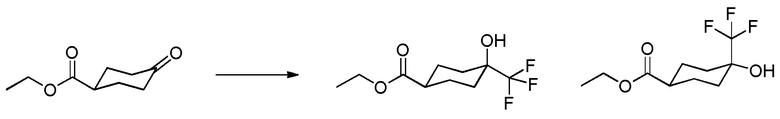
Круглодонную колбу загружали этил 4-оксоциклогексанкарбоксилатом (10,0 г, 58,8 ммоль) и CsF (8,92 г, 58,8 ммоль) в DME (100 мг) при температуре приблизительно 23°C. Реакционную смесь охлаждали на бане со льдом до приблизительно 5°C, затем добавляли триметил(трифторметил)силан (8,35 г, 58,8 ммоль) по каплям с такой скоростью, чтобы поддерживать температуру реакционной смеси ниже 8°C. Реакционную смесь перемешивали приблизительно 18 часов при температуре приблизительно 23°C. TBAF (19,4 мл, 1M раствор в THF, 19,39 ммоль) добавляли по каплям и смесь перемешивали приблизительно 20 минут. Смесь разбавляли EtOAc (200 мг) и промывали водой (3×200 мг). Органический слой сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали на силикагеле, используя градиент от 10 до 50% EtOAc в гептане, с получением этил 4-гидрокси-4-(трифторметил)циклогексанкарбоксилата (9,27 г, 67%). Продукт брали в качестве смеси изомеров на следующей стадии без дополнительной очистки: 1H-ЯМР (DMSO-d6) δ 5,73 (с, 0,5H), 5,72 (с, 0,5H), 4,13-4,01 (м, 2H), 2,70-2,64 (м, 0,55H), 2,37-2,27 (м, 0,45H), 1,90-1,45(м, 8H), 1,21-1,14 (м, 3H).
Стадия B: (1s,4s)-4-Гидрокси-4-(трифторметил)-циклогексанкарбоновая кислота

Сухой EtOH (90 мг) обрабатывали натрием (1,03 г, 45,0 ммоль) при комнатной температуре и смесь перемешивали до полного растворения натрия. Раствор этил 4-гидрокси-4-(трифторметил)циклогексанкарбоксилата (9,00 г, 37,5 ммоль) в EtOH (90 мг) добавляли и смесь нагревали при приблизительно 70°C в атмосфере азота в течение приблизительно 18 часов. К смеси добавляли 2 н водный NaOH (18,7 мл, 37,5 ммоль) и смесь перемешивали при нагревании при приблизительно 70°C течение приблизительно 4 часов. Реакционную смесь охлаждали до комнатной температуры и концентрировали для удаления большей части этанола. Полученную суспензию разбавляли водой (50 мг) с получением прозрачного раствора. Раствор подкисляли концентрированной HCl до рН=2. Раствор концентрировали до объема приблизительно 50 мл и выпавший в осадок продукт собирали фильтрованием. Осадок промывали водой (2×8 мг) и сушили в течение приблизительно 18 часов при пониженном давлении с получением (1s,4s)-4-гидрокси-4-(трифторметил)циклогексанкарбоновой кислоты в виде белого твердого вещества (5,99 г, 75%): ЖХ/МС (Таблица 1, способ a) Время удерживания = 1,35 мин; MS m/z 211 (M-H)-, 1H-ЯМР (DMSO-d6) δ 12,10 (с, 1H), 5,69 (с, 1H), 2,26-2,16 (м, 1H), 1,79-1,69 (м, 4H), 1,69-1,56 (м, 2H), 1,55-1,44 (м, 2H).
Стадия C: (1s,4s)-4-Гидрокси-N-(2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-4-(трифторметил)циклогексанкарбоксамид
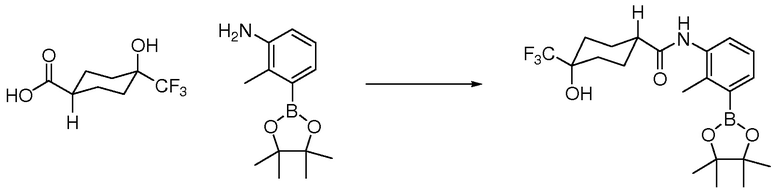
Раствор, содержащий (1s,4s)-4-гидрокси-4-(трифторметил)циклогексанкарбоновую кислоту (100 мг, 0,471 ммоль) и 2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилин (110 мг, 0,471 ммоль, CombiBlocks) в DMF (2,0 мг) обрабатывали DIEA (0,082 мл, 0,471 ммоль) и 2-(3H-[1,2,3]триазоло[4,5-b]пиридин-3-ил)-1,1,3,3-тетраметилизоурония гексафторофосфатом(V) (179 мг, 0,471 ммоль) и смесь перемешивали при комнатной температуре в течение приблизительно 1 часа. Смесь разбавляли водой (5 мг), растирали в порошок и супернатант декантировали. Остаток растворяли в EtOAc (10 мг), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали на силикагеле, используя градиент 25-75% EtOAc в гептане. Фракции продукта объединяли, концентрировали и сушили до твердого вещества при пониженном давлении с получением (1s,4s)-4-гидрокси-N-(2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-4-(трифторметил)циклогексанкарбоксамида в виде твердого вещества (135 мг, 67%): ЖХ/МС (Таблица 1, способ b) Время удерживания = 1,56 мин; MS m/z 428 (M+H)+, 1H-ЯМР (DMSO-d6) δ 9,23 (с, 1H), 7,46 (дд, J=7,4, 1,4 Гц, 1H), 7,35 (дд, J=7,9, 1,4 Гц, 1H), 7,14 (т, J=7,6 Гц, 1H), 5,74 (с, 1H), 2,44-2,34 (м, 1H), 2,32 (с, 3H), 1,90-1,67 (м, 6H), 1,60-1,42 (м, 2H), 1,30 (с, 12H).
Получение № 9: 4-бром-1H-пирроло[3,2-c]пиридин-7-карбоксамид
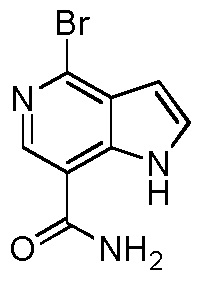
Стадия A: 4-бром-1H-пирроло[3,2-c]пиридин-7-карбоновая кислота
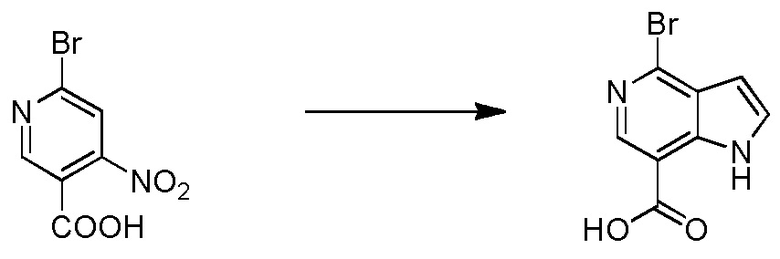
Раствор 6-бром-4-нитроникотиновой кислоты (3,8 г, 15,4 ммоль, Eur. J. Med. Chem. 1977, 12(6), 541) в безводной THF (100 мг) перемешивали между приблизительно -40 и -50°C в течение приблизительно 5 минут. Затем винилмагнийбромид (1 н в THF, 69,2 мл, 69,2 ммоль) добавляли по каплям. Смесь перемешиваюли между приблизительно -40 и -50°C в течение приблизительно 4 часов. Смесь гасили насыщенным водным NH4Cl (2 мг). Растворитель удаляли при пониженном давлении, чтобы получить остаток, который очищали с помощью препаративной ВЭЖХ (таблица 1, способ w) с получением 4-бром-1H-пирроло[3,2-c]пиридин-7-карбоновой кислоты (1 г, 27%): 1H-ЯМР (DMSO-d6) δ 11,90 (ушир.с, 1 H), 8,46 (с, 1 H), 7,54 (т, J=2,65 Гц, 1 H), 6,56 (ушир., 1 H).
Стадия B: 4-бром-1H-пирроло[3,2-c]пиридин-7-карбоксамид

К раствору 4-бром-1H-пирроло[3,2-c]пиридин-7-карбоновой кислоты (100 мг, 0,42 ммоль) в DMF (2 мг) добавляли HOBt (95 мг, 0,62 ммоль) и EDCI (119 мг, 0,62 ммоль). После перемешивания реакционной смеси при комнатной температуре в течение приблизительно 1 часа, добавляли NH3/THF (10 мг) и полученную смесь перемешивали при комнатной температуре в течение ночи. Затем суспензию фильтровали и фильтрат концентрировали при пониженном давлении. Добавляли воду и экстрагировали EtOAc. Объединенную органическую фазу промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением 4-бром-1H-пирроло[3,2-c]пиридин-7-карбоксамида (60 мг, 42%). Продукт использовали без дальнейшей очистки: 1H-ЯМР (DMSO-d6) δ 11,89 (ушир., 1H), 8,51 (с, 1H), 8,27 (ушир., 1H), 7,68 (ушир., 1H), 7,52-7,51 (д, J=2,8 Гц, 1H), 6,52-6,51 (д, J=3,2 Гц, 1H).
Получение № 10. 4-бром-2-(1-метил-1H-пиразол-4-ил)-1H-индол-7-карбоксамид
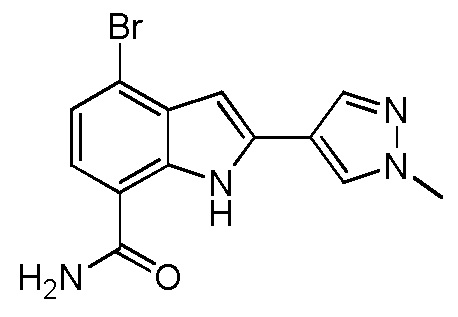
Стадия A: Метил 4-бром-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоксилат
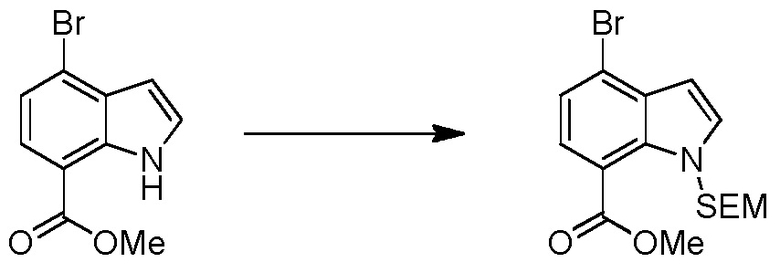
К раствору метил 4-бром-1H-индол-7-карбоксилата (35 г, 138 ммоль, получение № 1 стадия B) в безводном THF (1500 мг) порциями добавляли NaH (10 г, 250 ммоль) при температуре приблизительно 0°C и перемешивали в течение 1 часа при температуре приблизительно 0°C. Затем SEMCl (31,9 мл, 180 ммоль) добавляли порциями при температуре приблизительно 0°C. Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение приблизительно 12 часов. Затем к реакционной смеси добавляли насыщенный водный NH4Cl и экстрагируовали EtOAc. Объединенные органические слои промывали насыщенным солевым раствором, сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка, который очищаюли колоночной хроматографией на силикагеле с получением метил 4-бром-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоксилата (32 г, 60%): 1H-ЯМР (CDCl3) δ 7,62-7,60 (д, J=8,4 Гц, 1H), 7,46-7,44 (д, J=8,0 Гц, 1H), 7,36-7,35 (д, J=3,2 Гц, 1H), 6,77-6,76 (д, J=3,6 Гц, 1H), 5,80 (с, 2H), 4,06 (с, 3H), 3,32-3,28 (т, J=8,0 Гц, 2H), 0,89-0,85 (т, J=8,0 Гц, 2H), 0,00 (с, 9H).
Стадия B: Метил 4-бром-2-йод-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоксилат
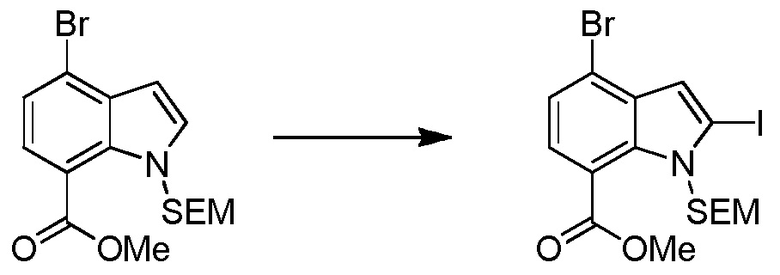
К раствору метил 4-бром-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоксилата (10 г, 26 ммоль, получение № 1 стадия B) в безводном THF (200 мг) добавлеяли диизопропиламид лития (18 мл, 36 ммоль) при приблизительно -70°C и перемешивали в течение приблизительно 2 часов. Затем раствор I2 (10 г, 39 ммоль) в безводном THF (50 мг) добавляли к полученному выше раствору по каплям при температуре приблизительно -70°C и затем перемешивали в течение приблизительно 2 часов. Смесь выливали в водный раствор Na2S2O3 и экстрагировали EtOAc. Объединенные органические фазы промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали под давлением с получением остатка, который очищали с помощью колоночной хроматографии (элюируя петролейным эфиром:EtOAc = 200:1) с получением метил 4-бром-2-йод-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоксилата (6,2 г, 47%): 1H-ЯМР (CDCl3) δ 7,50-7,48 (д, J=8,0 Гц, 1H), 7,42-7,40 (д, J=8,0 Гц, 1H), 7,10 (с, 1H), 5,90 (с, 2H), 4,06 (с, 3H), 3,29-3,25 (т, J=8,0 Гц, 2H), 0,87-0,83 (т, J=8,0 Гц, 2H), 0,00 (с, 9H).
Стадия C: Метил 4-бром-2-йод-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоксилат
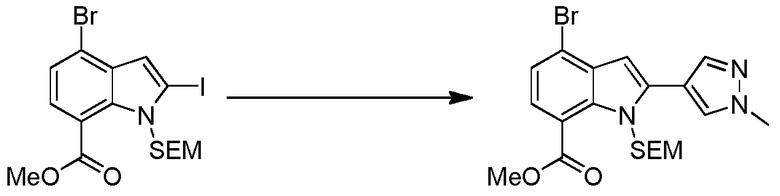
К раствору метил 4-бром-2-йод-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоксилата (1,1 г, 2,2 ммоль) в DME (20 мг) и воды (5 мг) добавляли 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол (0,49 г, 2,37 ммоль), PdCl2(dppf) (0,176 г, 0,216 ммоль) и Na2CO3 (0,894 г, 6,47 ммоль). Смесь нагревали с обратным холодильником в течение приблизительно 3 часов. После охлаждения до комнатной температуры, воду (20 мг) добавляли к раствору и экстрагировали EtOAc (50 мг). Органическую фазу сушили над Na2SO4 и концентрировали в вакууме, получая неочищенный продукт, который очищали с помощью колоночной хроматографии на силикагеле (элюируя петролейным эфиром:EtOAc = 10:1), с получением метил 4-бром-2-йод-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоксилата (0,65 г, 65%): 1H-ЯМР (CDCl3) δ 7,84 (с, 1H), 7,77 (с, 1H), 7,61-7,59 (д, J=7,2 Гц, 1H), 7,49-7,40 (д, J=8,0 Гц, 1H), 6,79 (с, 1H), 5,84 (с, 2H), 4,14 (с, 3H), 4,11 (с, 3H), 3,20-3,16 (т, J=8,4 Гц, 2H), 0,82-0,78 (т, J=8,4 Гц, 2H), 0,00 (с, 9H).
Стадия D: 4-бром-2-(1-метил-1H-пиразол-4-ил)-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоновая кислота
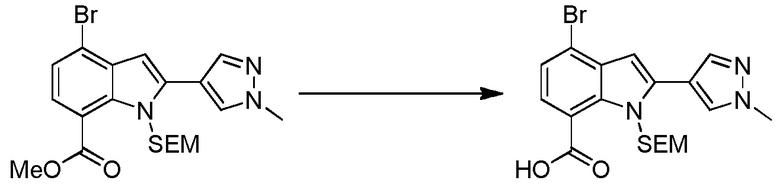
К раствору метил 4-бром-2-йод-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоксилата (0,65 мг, 1,41 ммоль) в THF (10 мг), MeOH (2 мг) и воды (2 мг) добавляли LiOH (0,17 мг, 7,04 ммоль). Смесь нагревали с обратным холодильником в течение приблизительно 4 часов. После охлаждения до комнатной температуры, растворитель удаляли при пониженном давлении и водный слой подкисляли водной HCl (1 н) до рН=4, экстрагировали EtOAc (10 мг), сушили над Na2SO4 и концентрировали при пониженном давлении с получением 4-бром-2-(1-метил-1H-пиразол-4-ил)-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоновой кислоты (0,63 г, 99%): 1H-ЯМР (CDCl3) δ 7,90 (с, 1H), 7,81 (с, 1H), 7,80-7,79 (д, J=2,4 Гц, 1H), 7,54-7,52 (д, J=8,0 Гц, 1H), 6,84 (с, 1H), 5,95 (с, 2H), 4,18 (с, 3H), 3,25-3,20 (т, J=7,2 Гц, 2H), 0,82-0,78 (т, J=7,2 Гц, 2H), 0,00 (с, 9H).
Стадия E: 4-бром-2-(1-метил-1H-пиразол-4-ил)-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоксамид
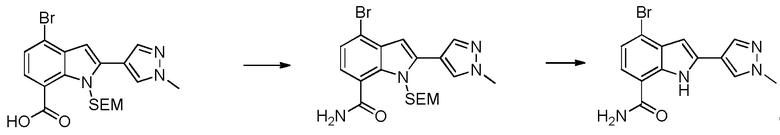
К раствору 4-бром-2-(1-метил-1H-пиразол-4-ил)-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоновой кислоты (0,63 г, 1,4 ммоль) в DMF (10 мг) добавляли PyBOP (1,46 г, 2,80 ммоль), HOBt (0,43 г, 2,80 ммоль), NH4Cl (0,11 г, 2,10 ммоль) и DIEA (0,72 г, 5,60 ммоль). Смесь перемешивали при комнатной температуре в течение приблизительно 2 часов. К смеси добавляли воду (20 мг) и экстрагировали EtOAc (30 мг). Органическую фазу сушили над Na2SO4 и концентрировали при пониженном давлении с получением сырого продукта, который очищали с помощью колоночной хроматографии на силикагеле (элюируя петролейным эфиром:EtOAc = 3:1) с получением сырого 4-бром-2-(1-метил-1H-пиразол-4-ил)-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоксамида. Полученное вещество растворяли в безводном THF (10 мг), добавляли (2,02 г, 12,2 ммоль) и этан-1,2-диамин (2,20 г, 36,7 ммоль) и нагревали до приблизительно 100°C в течение приблизительно 2 часов. После охлаждения до комнатной температуры добавляли воду для разбавления смеси, экстрагировали EtOAc, органическую фазу сушили над Na2SO4, и концентрировали при пониженном давлении с получением остатка, который очищали с помощью колоночной хроматографии на силикагеле (элюируя петролейным эфиром:EtOAc = 3:1) с получением 4-бром-2-(1-метил-1H-пиразол-4-ил)-1H-индол-7-карбоксамида (0,20 г, 51%): 1H-ЯМР (CDCl3) δ 10,40 (ушир., 1 H), 7,87 (с, 1 H), 7,75 (с, 1 H), 7,30-7,28 (д, J=8, 1 H), 7,20-7,18 (д, J=8, 1 H), 6,64 (с, 1 H), 6,05 (ушир., 2 H), 3,99 (с, 3 H).
Получение № 11. 3-(2-(((трет-бутилдиметилсилил)окси)метил)-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-6-фторхиназолин-4(3H)-он
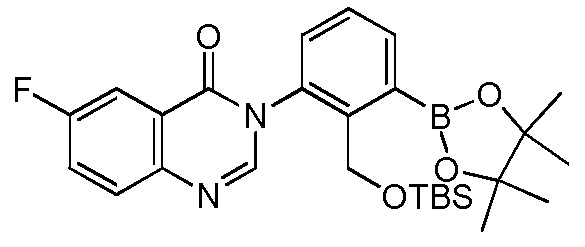
Стадия A: (2-амино-6-бромфенил)метанол
Раствор 2-амино-6-бромбензойной кислоты (19,8 г, 91,7 ммоль) в THF (190 мг) добавляли к суспензии LiAlH4 (7,00 г, 183 ммоль) в THF (190 мг) по каплям при температуре приблизительно 0°C. После того, как добавление было завершено, смесь перемешивали при комнатной температуре в течение приблизительно 4 часов. Затем смесь гасили EtOAc (180 мг). Смесь выливали в H2O (1,1 л) и фильтровали. Фильтрат экстрагировали EtOAc (3×900 мг). Объединенный органический слой сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле (элюируя петролейным эфиром:EtOAc = 50:1-5:1) с получением (2-амино-6-бромфенил)метанола (10 г, 54%): 1H-ЯМР (CDCl3) δ 1,77 (с, 1H), 4,34 (с, 2H), 4,92 (с, 2H), 6,64 (м, 1H), 6,95 (м, 2H).
Стадия B: 3-бром-2-(((трет-бутилдиметилсилил)окси)метил)анилин
К раствору (2-амино-6-бромфенил)метанола (3,02 г, 15 ммоль) и имидазола (1,83 г, 27 ммоль) в DMF (40 мг) добавляли TBSCl (3,39 г, 22,5 ммоль) частями при температуре приблизительно 0°C. Затем полученную смесь перемешивали при комнатной температуре в течение ночи. Смесь выливали в H2O (80 мг), экстрагировали МТВЕ (3×80 мг). Объединенную органическую фазу промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (элюируя петролейным эфиром:EtOAc=15:1) с получением 3-бром-2-(((трет-бутилдиметилсилил)окси)метил)анилина (4,2 г, 89%): 1H-ЯМР (CDCl3) δ 0,00 (с, 6H), 0,80 (с, 9H), 4,38 (с, 2H), 4,85 (с, 2H), 6,48 (м, 1H), 6,79 (м, 2H).
Стадия C: 3-(3-бром-2-(((трет-бутилдиметилсилил)окси)метил)фенил)-6-фторхиназолин-4(3H)-он
Смесь 3-бром-2-(((трет-бутилдиметилсилил)окси)метил)анилина (3,5 г, 11 ммоль), 2-амино-5-фтор-бензойной кислоты (1,7 г, 11 ммоль) и CH(OMe)3 (1,8 г, 16,5 ммоль) в THF (30 мг) нагревали при температуре приблизительно 120°C в запаянной трубке в течение ночи. Смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток промывали EtOAc с получением 3-(3-бром-2-(((трет-бутилдиметилсилил)окси)метил)фенил)-6-фторхиназолин-4(3H)-она (1,3 г, 25%): 1H-ЯМР (CDCl3) δ 0,00 (д, J=8 Гц, 6H), 0,85 (с, 9H), 4,57 (д, J=11,6 Гц, 1H), 4,98 (д, J=11,6 Гц, 1H), 7,35 (м, 1H), 7,43 (т, J=8 Гц, 1H), 7,62 (м, 1H), 7,83 (м, 2H), 8,06 (м, 2H).
Стадия D: 3-(2-(((трет-бутилдиметилсилил)окси)метил)-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-6-фторхиназолин-4(3H)-он

Смесь 3-(3-бром-2-(((трет-бутилдиметилсилил)окси)метил)фенил)-6-фторхиназолин-4(3H)-она (4 г, 8,6 ммоль), 4,4,5,5,4',4',5',5'-октаметил-[2,2']ди[1,3,2]диоксабороланила (2,6 г, 10,4 ммоль), KOAc (1,7 г, 17,2 ммоль) и Pd(dppf)Cl2 (0,8 г) в DMSO/1,4-диоксане (8 мл: 40 мг) нагревали до приблизительно 110°C в атмосфере N2 в течение приблизительно 2 часов. Смесь охлаждали до комнатной температуры, разбавляли EtOAc (100 мг), фильтровади и фильтрат промывали H2O (30 мг) и насыщенным солевым раствором (30 мг) последовательно. Органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением сырого продукта, который очищали с помощью колоночной хроматографии на силикагеле (петролейный эфир/EtOAc, от 30:1 до 5:1) с получением 3-(2-(((трет-бутилдиметилсилил)окси)метил)-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-6-фторхиназолин-4(3H)-она (1,7 г, 38%): 1H-ЯМР (CDCl3) δ 0,00 (д, J=2 Гц, 6H), 0,92 (с, 9H), 1,52 (с, 12H), 4,70 (д, J=1,6 Гц, 1H), 5,43 (д, J=1,6 Гц, 1H), 7,63 (м, 1H), 7,70 (м, 2H), 7,93 (м, 1H), 8,16 (м, 3H).
Получение № 12: гидрохлорид (R)-7-(пиперидин-3-ил)имидазо[1,2-a]пиразин-8(7H)-она
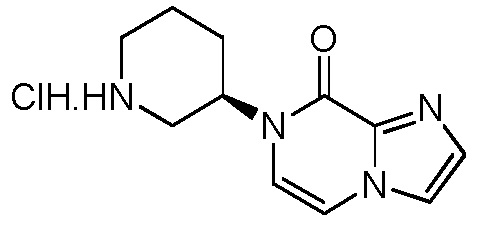
Стадия A: (R)-трет-Бутил (1-бензилпиперидин-3-ил)карбамат
К раствору (R)-трет-бутил пиперидин-3-илкарбамата (40,0 г, 0,2 моль, 1,0 эквив.) и TEA (22,22 г, 0,22 моль, 1,1 эквив.) в DCM (500 мг) по каплям добавляли бромметил-бензол (37,62 г, 0,22 моль, 1,1 эквив.) при 0°C. После перемешивания в течение ночи при температуре приблизительно 25°C, раствор разбавляли DCM и промывали водой. Органический слой сушили и выпаривали с получением (R)-трет-бутил (1-бензилпиперидин-3-ил)карбамата (58,0 г, 100%), который использовали на следующей стадии без дополнительной очистки: 1H-ЯМР (CDCl3) 7,15-7,26 (м, 5H), 4,92 (с, 1H), 3,67 (с, 1H), 3,39 (с, 2H), 2,16-2,45 (м, 4H), 1,41-1,61(м, 4H), 1,37 (с, 9H)
Стадия B: гидрохлорид (R)-1-бензилпиперидин-3-амина
К раствору (R)-трет-бутил (1-бензилпиперидин-3-ил)карбамата (58,0 г, 0,2 моль, 1,0 эквив.) в MeOH (200 мг) добавляли HCl/MeOH (4,0 M, 200 мг) и смесь перемешивали в течение приблизительно 2 часов. Растворитель удаляли в вакууме с получением гидрохлорида (R)-1-бензилпиперидин-3-амина (50 г): 1H-ЯМР (MeOD) δ 7,64 (д, J=2,4 Гц, 2H), 7,50 (с, 3H), 4,42-4,52 (кв, 2H), 3,64-3,66 (д, J=10,8 Гц, 2H), 3,51-3,54 (д, J=12 Гц, 1H), 3,01-3,16 (м, 2H), 2,20-2,22 (д, J=11,2 Гц, 1H), 2,00-2,11 (м, 2H), 1,66-1,74 (м, 1H)
Стадия C: (R)-N-(1-Бензилпиперидин-3-ил)-1H-имидазол-2-карбоксамид
К раствору 1H-имидазол-2-карбоновой кислоты (16,8 г, 0,15 моль) в DMF (500 мг) добавляли HATU (57 г, 0,15 моль) и смесь перемешивали в течение приблизительно 2 часов при комнатной температуре. Затем (R)-трет-бутил (1-бензилпиперидин-3-ил)карбамат (39,45 г, 0,15 моль) добавляли к раствору и смесь перемешивали в течение ночи. Добавляли дополнительное количество 1Н-имидазол-2-карбоновой кислоты (5,2 г, 46 ммоль) и HATU (17,6 г, 46 ммоль, 0,3 эквив.) и смесь перемешивали при комнатной температуре в течение 3 суток. Растворитель удаляли и остаток растворяли в EtOAc, промывали водой, сушили и концентрировали. Остаток очищали колоночной хроматографией на силикагеле с получением сырого (R)-N-(1-бензилпиперидин-3-ил)-1H-имидазол-2-карбоксамида (50 г): ЖХ/МС (Таблица 1, способ k) Время удерживания = 1,15 мин; MS m/z: 285 (M+H)+.
Стадия D: (R)-N-(1-Бензилпиперидин-3-ил)-1-(2,2-диэтоксиэтил)-1H-имидазол-2-карбоксамид
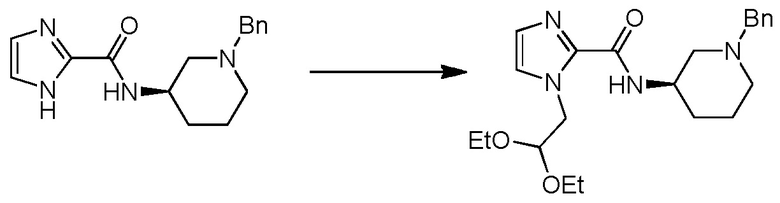
Смесь (R)-7-(1-бензилпиперидин-3-ил)имидазо[1,2-a]пиразин-8(7H)-она (73,0 г, 150 ммоль, сырой), 2-бром-1,1-диэтокси-этана (30 г, 150 ммоль), K2CO3 (41,4 г, 300 ммоль) и KI (1 г) в DMF (500 мг) нагревали до температуры приблизительно 120°C в течение 3 дней. Растворитель удаляли. Остаток растворяли в DCM, промывали водой, сушили и упаривали с получением (R)-N-(1-бензилпиперидин-3-ил)-1-(2,2-диэтоксиэтил)-1H-имидазол-2-карбоксамида (30 г, 75 ммоль) в виде масла: ЖХ/МС (Таблица 1, Способ k) Время удерживания = 1,81 мин; MS m/z: 401 (M+H)+.
Стадия E: (R)-7-(1-Бензилпиперидин-3-ил)имидазо[1,2-a]пиразин-8(7H)-он
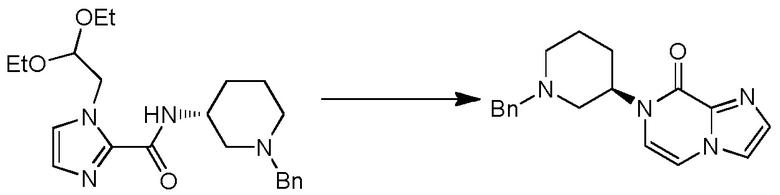
Смесь (R)-N-(1-бензилпиперидин-3-ил)-1-(2,2-диэтоксиэтил)-1H-имидазол-2-карбоксамида (30,0 г, 75 ммоль, сырой) в 2 н HCl (200 мг) нагревали с обратным холодильником в течение ночи. Растворитель удаляли и остаток разбавляли водой (50 мг), который подщелачивали насыщенным Na2CO3 до рН 10. Водную фазу экстрагировали DCM, высушивали и выпаривали. Остаток очищали колоночной хроматографией на силикагеле с получением (R)-7-(1-бензилпиперидин-3-ил)имидазо[1,2-a]пиразин-8(7H)-она (3,0 г, 9,7 ммоль): 1H-ЯМР (CDCl3) δ 7,44 (с, 1H), 7,17-7,24 (м, 7H), 7,01-7,02 (д, J=6 Гц, 1H), 5,00-5,05 (м, 1H), 3,45-3,47 (д, J=5,6 Гц, 2H), 2,78-2,80 (м, 1H), 2,55-2,58 (м, 1H), 2,31-2,36 (м, 1H), 2,25 (с, 1H), 1,81 (с, 1H), 1,16-1,69 (м, 3H)
Стадия F: (R)-трет-Бутил 3-(8-оксоимидазо[1,2-a]пиразин-7(8H)-ил)пиперидин-1-карбоксилат

К раствору (R)-7-(1-бензилпиперидин-3-ил)имидазо[1,2-a]пиразин-8(7H)-ону (2,13 г, 6,9 ммоль) в MeOH (40 мг) добавляли (Boc)2O (3,09 г, 13,8 ммоль) и Pd/C (1,5 г). Смесь гидрировали в атмосфере H2 из баллона в течение ночи и затем фильтровали. Фильтрат концентрировали и очищали с помощью колоночной хроматографии на силикагеле с получением (R)-трет-бутил 3-(8-оксоимидазо[1,2-a]пиразин-7(8H)-ил)пиперидин-1-карбоксилата (1,4 г, 64%): 1H-ЯМР (MeOD) δ 7,69-7,70 (д, J=1,2 Гц, 1H), 7,52-7,54 (д, J=6,4 Гц, 1H), 7,50 (с, 1H), 7,12-7,14 (д, J=6 Гц, 1H), 4,74-4,82 (м, 1H), 4,12-4,15 (д, J=11,6 Гц, 1H), 4,04-4,05 (м, 1H), 3,05-3,11 (м, 1H), 2,83 (с, 1H), 1,91-2,02 (м, 2H), 1,86-1,90 (м, 1H), 1,60-1,71 (м, 1H), 1,46 (с, 9H)
Стадия G: гидрохлорид (R)-7-(пиперидин-3-ил)имидазо[1,2-a]пиразин-8(7H)-она
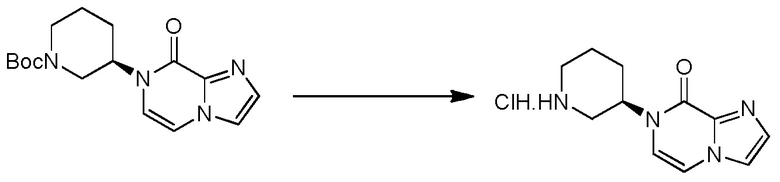
К раствору (R)-трет-бутил 3-(8-оксоимидазо[1,2-a]пиразин-7(8H)-ил)пиперидин-1-карбоксилата (1,4 г, 4,4 ммоль) в MeOH (10 мг) добавляли HCl/MeOH (4 M, 10 мг) и смесь перемешивали в течение приблизительно 1 часа при комнатной температуре. Растворитель удаляли с получением гидрохлорида (R)-7-(пиперидин-3-ил)имидазо[1,2-a]пиразин-8(7H)-она (1,35 г, 100%): 1H-ЯМР (DMSO-d6) δ 10,06 (с, 1H), 9,67 (с, 1H), 8,18-8,21 (м, 1H), 8,00-8,03 (м, 1H), 7,89-7,93 (м, 1H), 7,69-7,74 (м, 1H), 5,12-5,18 (м, 1H), 3,20-3,34 (м, 3H), 2,82-2,90 (м, 1H), 2,02-2,08 (м, 1H), 1,84-1,93 (м, 3H).
Получение № 13: гидрохлорид (R)-7-(пиперидин-3-ил)-6,7-дигидроимидазо[1,2-a]пиразин-8(5H)-она
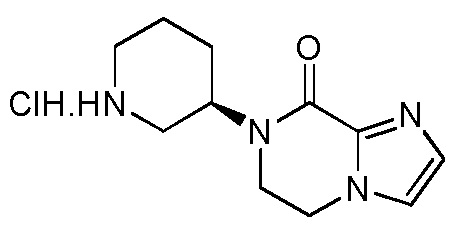
Стадия A: (R)-трет-Бутил 3-(8-оксо-5,6-дигидроимидазо[1,2-a]пиразин-7(8H)-ил)пиперидин-1-карбоксилат

К раствору (R)-7-(1-бензилпиперидин-3-ил)имидазо[1,2-a]пиразин-8(7H)-она (0,77 г, 2,5 ммоль) в MeOH (20 мг) добавляли (Boc)2O (1,09 г, 5,0 ммоль) и Pd(OH)2 (0,5 г). Смесь гидрировали в атмосфере H2 из баллона в течение ночи и затем фильтровали. Фильтрат упаривали и очищали колоночной хроматографией на силикагеле с получением (R)-трет-бутил 3-(8-оксо-5,6-дигидроимидазо[1,2-a]пиразин-7(8H)-ил)пиперидин-1-карбоксилата (0,5 г, 60%): 1H-ЯМР (MeOD) δ 7,16 (с, 1H), 7,06 (с, 1H), 4,22-4,33 (м, 1H), 4,19-4,20 (м, 2H), 3,93-3,96 (м, 2H), 3,64-3,78 (м, 2H), 2,86-2,89 (м, 1H), 2,61 (с, 1H), 168-1,79 (м, 3H), 1,47-1,53 (м, 1H), 1,46 (с, 9H).
Стадия B: гидрохлорид (R)-7-(пиперидин-3-ил)-6,7-дигидроимидазо[1,2-a]пиразин-8(5H)-она
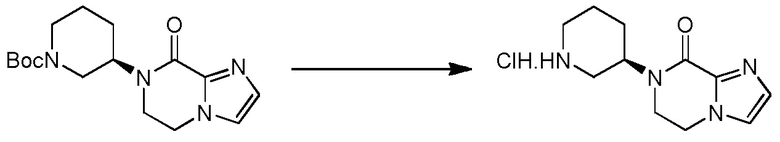
К раствору (R)-трет-бутил 3-(8-оксо-5,6-дигидроимидазо[1,2-a]пиразин-7(8H)-ил)пиперидин-1-карбоксилата (0,5 г, 1,5 ммоль, 1 эквив.) в MeOH (5 мг) добавляли HCl/MeOH (4,0 M, 5 мг) и смесь перемешивали в течение 1 часа при комнатной температуре. Растворитель удаляли с получением гидрохлорида (R)-7-(пиперидин-3-ил)-6,7-дигидроимидазо[1,2-a]пиразин-8(5H)-она (0,45 г, 100%): 1H-ЯМР (MeOD) δ 7,75-7,78 (кв, J=9,6 Гц, 2H), 4,66-4,74 (м, 1H), 4,56-4,59 (кв, J=7,2 Гц, 2H), 3,99-4,03 (т, J=6 Гц, 2H), 3,32-3,45 (м, 3H), 2,96-3,03 (м, 1H), 1,85-2,14 (м, 4H).
Получение № 14: (Z)-4-((3-(7-карбамоил-1H-индол-4-ил)фенил)амино)-4-оксобут-2-еновая кислота
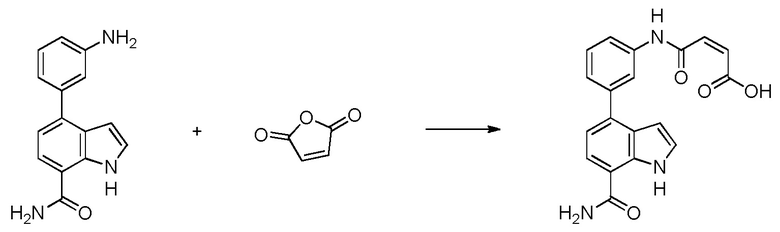
К раствору 4-(3-аминофенил)-1H-индол-7-карбоксамида (0,25 г, 0,995 ммоль, получение № A.1), фуран-2,5-дион (0,117 г, 1,19 ммоль), и N-этил-N-изопропилпропан-2-амин (0,521 мл, 2,98 ммоль) в DMF (10,0 мг) добавляли. Смесь перемешивали в течение ночи при комнатной температуре. Растворитель удаляли в высоком вакууме и остаток очищали препаративной ВЭЖХ (таблица 2, способ y) с получением (Z)-4-((3-(7-карбамоил-1H-индол-4-ил)фенил)амино)-4-оксобут-2-еновой кислоты (0,32 г, 92%) в виде твердого вещества. ЖХ/МС (Таблица 1, способ g) Время удерживания = 1,37 мин; MS m/z 350 (M+H)+.
Получение № 15. трет-Бутил 3-(7-карбамоил-4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-1H-индол-2-ил)-2,5-дигидро-1H-пиррол-1-карбоксилат
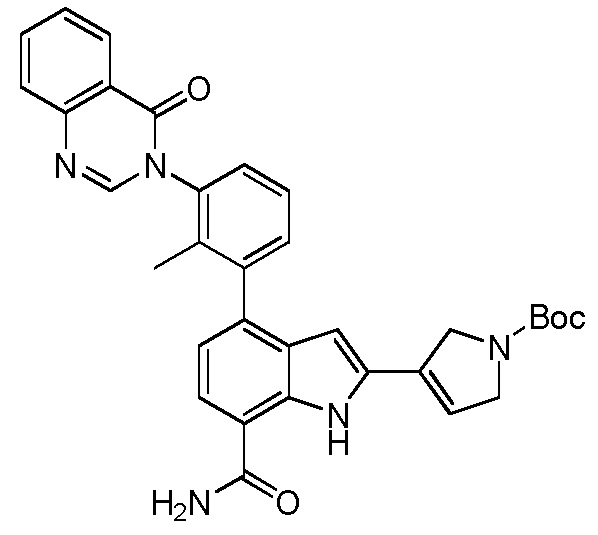
Стадия A. Метил 4-бром-2-(1-(трет-бутоксикарбонил)-2,5-дигидро-1H-пиррол-3-ил)-1-тозил-1H-индол-7-карбоксилат
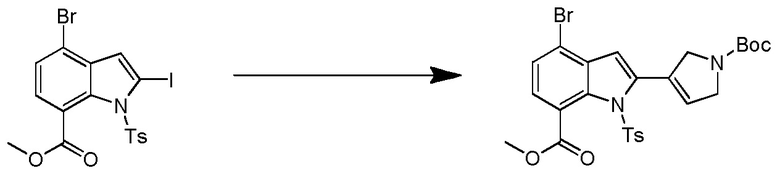
К смеси метил 4-бром-2-йод-1-тозил-1H-индол-7-карбоксилата (1 г, 1,9 ммоль, получение № 1, Стадия D) в DME (20 мг)/вода (5 мг) добавляли трет-бутил 3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2,5-дигидро-1H-пиррол-1-карбоксилат (0,72 г, 2,4 ммоль), Na2CO3 (0,6 г, 5,6 ммоль) и Pd(dppf)Cl2 (0,2 г, 0,28 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 10 часов в атмосфере N2. После фильтрации фильтрат концентрировали при пониженном давлении с получением остатка, который очищали с помощью колоночной хроматографии на силикагеле (элюируя гексаном:EtOAc = 5:1) с получением метил 4-бром-2-(1-(трет-бутоксикарбонил)-2,5-дигидро-1H-пиррол-3-ил)-1-тозил-1H-индол-7-карбоксилата (0,6 г, 56%) в виде желтого твердого вещества: 1H-ЯМР (CDCl3) δ 7,68-7,56 (д, J=8,22 Гц, 1H), 7,55-7,54 (м, 1H), 7,14-7,05 (м, 4H), 6,45-6,37 (м, 2H), 4,37-4,31 (м, 2H), 4,05 (с, 3H), 3,89-3,84 (м, 2H), 2,38-2,34 (м, 3H), 1,53 (м, 9H).
Стадия B: 4-бром-2-(1-(трет-бутоксикарбонил)-2,5-дигидро-1H-пиррол-3-ил)-1H-индол-7-карбоновая кислота

К раствору метил 4-бром-2-(1-(трет-бутоксикарбонил)-2,5-дигидро-1H-пиррол-3-ил)-1-тозил-1H-индол-7-карбоксилата (2,5 г, 4,34 ммоль) в THF (20 мг)/MeOH (5 мг)/вода (5 мг) добавляли LiOH•H2O (2,5 г, 59,5 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение приблизительно 3 часов. Реакционную смесь концентрировали и остаток подкисляли добавлением 2 н HCl до рН 5 и экстрагировали EtOAc (3×50 мг). Объединенный органический слой сушили и концентрировали с получением твердого вещества, которое промывали EtOAc и МТВЕ, получая 4-бром-2-(1-(трет-бутоксикарбонил)-2,5-дигидро-1H-пиррол-3-ил)-1H-индол-7-карбоновую кислоту (1 г, 56,5%) в виде белого твердого вещества: 1H-ЯМР (CDCl3) δ 9,84 (м, 1 H), 7,77-7,75 (т, J=5,6 Гц, 1H), 7,34-7,32 (д, J=8 Гц, 1H), 6,54-6,49 (д, J=16,8 Гц, 1H), 6,18-6,14 (д, J=18 Гц, 1H), 4,58-4,51 (д, J=30,4 Гц, 2H), 4,38-4,32 (д, J=22 Гц, 2H), 1,54 (с, 9H).
Стадия C: трет-Бутил 3-(4-бром-7-карбамоил-1H-индол-2-ил)-2,5-дигидро-1H-пиррол-1-карбоксилат

К раствору 4-бром-2-(1-(трет-бутоксикарбонил)-2,5-дигидро-1H-пиррол-3-ил)-1H-индол-7-карбоновой кислоты (1 г, 2, 5 ммоль) в DMF (6 мг) добавляли PyBOP (2,6 г, 4,9 ммоль), HOBt (0,75 г, 4,91 ммоль), DIEA (1,7 мл, 9,82 ммоль) и NH4Cl (0,2 г, 3,7 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. После гашения водой, водный слой экстрагировали EtOAc (3×25 мг). Объединенные органические слои сушили и концентрировали при пониженном давлении с получением остатка, который очищали с помощью препаративной ВЭЖХ (таблица 1, способ ad) с получением трет-бутил 3-(4-бром-7-карбамоил-1H-индол-2-ил)-2,5-дигидро-1H-пиррол-1-карбоксилата (0,6 г, 54%) в виде белого твердого вещества: 1H-ЯМР (CDCl3) δ 10,42 (с, 1 H), 7,26-7,25 (м, 2H), 6,48 (с, 1H), 6,19-6,13 (д, J=22,4 Гц, 1H), 4,55-4,51 (д, J=16 Гц, 2H), 4,37-4,32 (д, J=18 Гц, 2H), 1,54 (с, 9H).
Стадия D: трет-Бутил 3-(7-карбамоил-4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-1H-индол-2-ил)-2,5-дигидро-1H-пиррол-1-карбоксилат
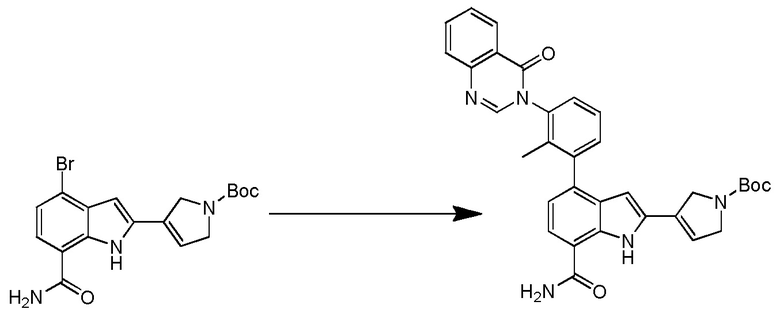
Раствор трет-бутил 3-(4-бром-7-карбамоил-1H-индол-2-ил)-2,5-дигидро-1H-пиррол-1-карбоксилата (0,6 г, 1,48 ммоль), 3-(2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)хиназолин-4(3H)-она (1 г, 2,95 ммоль, WO 2011159857), K2CO3 (0,816 г, 5,91 ммоль) и Pd(dppf)Cl2 (0,22 г, 0,3 ммоль) в THF (20 мг/MeOH (5 мг)/вода (5 мг) перемешивали при температуре около 60°C в течение приблизительно 2 часов в атмосфере N2. Растворитель удаляли с получением остатка, который очищали с помощью колоночной хроматографии на силикагеле (элюируя гексанами:EtOAc = 2:1) с получением трет-бутил 3-(7-карбамоил-4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-1H-индол-2-ил)-2,5-дигидро-1H-пиррол-1-карбоксилата (0,6 г, 72%) в виде твердого вещества: 1H-ЯМР (MeOD) δ 10,44 (с, 1H), 8,40-8,38 (д, J=8 Гц, 1H), 8,15-8,10 (с, J=21,6 Гц, 1H), 7,83-7,81 (м, 2H), 7,59-7,35 (м, 5H), 7,09-6,98 (м, 1H), 6,31-6,11 (м, 4H), 4,49-4,36 (м, 4H), 2,04 (с, 3H), 1,51 (с, 9H).
Получение № 16. трет-Бутил 4-(7-карбамоил-4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-1H-индол-2-ил)-5,6-дигидропиридин-1(2H)-карбоксилат
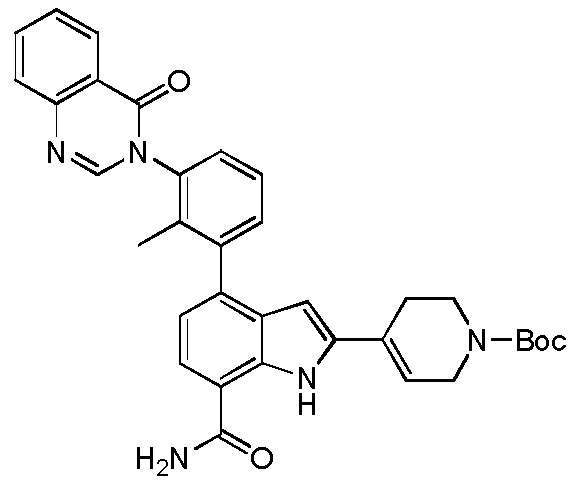
Стадия A: трет-Бутил 2-(4-бром-7-карбамоил-1H-индол-2-ил)бензилкарбамат
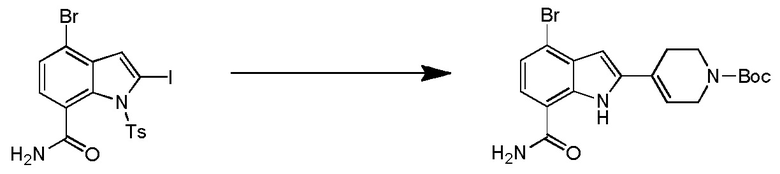
К раствору соединения метил 4-бром-2-йод-1-тозил-1H-индол-7-карбоксилата (2,4 г, 6,58 ммоль, Получение № 1) и трет-бутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2H)-карбоксилата (2,0 г, 6,58 ммоль) в THF (50 мг), MeOH (10 мг) и воде (10 мг) добавляли Na2CO3 (2,1 г, 19,73 ммоль) и Pd(dppf)Cl2 (0,481 г, 0,658 ммоль), смесь нагревали до температуры приблизительно 80°C приблизительно 3 часа. Полученный раствор разбавляли EtOAc (100 мг), и промывали водой (30 мг). Органическую фазу сушили над Na2SO4, и концентрировали при пониженном давлении с получением сырого продукта, который очищали с помощью колоночной хроматографии на силикагеле (элюируя петролейным эфиром:EtOAc = 1:1) с получением трет-бутил 4-(4-бром-7-карбамоил-1H-индол-2-ил)-5,6-дигидропиридин-1(2H)-карбоксилата (2 г, 72%) в виде твердого вещества: 1H-ЯМР (DMSO-d6) δ 10,87 (с, 1H), 8,15 (с, 1H), 7,59-7,57 (д, J=8,0 Гц, 1H), 7,52 (с, 1H), 7,27-7,25 (д, J=8,0 Гц, 1H), 6,47 (с, 1H), 6,42 (с, 1H), 4,03 (с, 2H), 3,55 (с, 2H), 2,52 (с, 2H), 1,41 (с, 9H).
Стадия B: трет-Бутил 4-(7-карбамоил-4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-1H-индол-2-ил)-5,6-дигидропиридин-1(2H)-карбоксилат
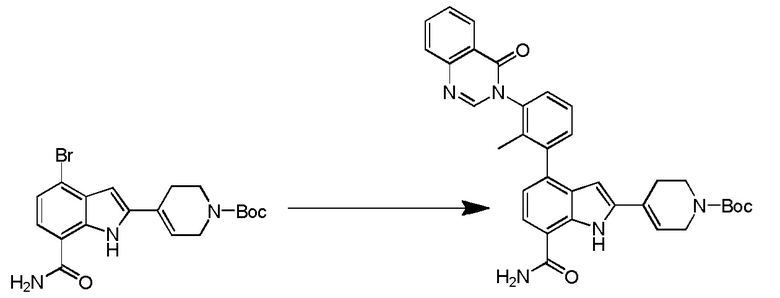
К раствору трет-бутил 4-(4-бром-7-карбамоил-1H-индол-2-ил)-5,6-дигидропиридин-1(2H)-карбоксилата (2 г, 4,76 ммоль) и 3-(2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)хиназолин-4(3H)-ону (2,59 г, 7,14 ммоль, WO 2011159857) в THF (40 мг), MeOH (10 мг) и воды (10 мг) добавляли Na2CO3 (1,513 г, 14,28 ммоль) и Pd(dppf)Cl2 (0,348 г, 0,476 ммоль). Смесь нагревали приблизительно до 80°C в течение приблизительно 4 часов. Полученный раствор разбавляли EtOAc (100 мг), и промывали водой и насыщенным солевым раствором (30 мл каждого). Органическую фазу сушили над Na2SO4 и концентрировали с получением сырого продукта, который очищали с помощью колоночной хроматографии на силикагеле (элюируя петролейным эфиром:EtOAc = 1:1) с получением трет-бутил 4-(7-карбамоил-4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-1H-индол-2-ил)-5,6-дигидропиридин-1(2H)-карбоксилата (1,4 г, 51%) в виде твердого вещества: 1H-ЯМР (CDCl3) 10,43 (с, 1H), 8,42-8,40 (д, J=7,6 Гц, 1H), 8,15 (с, 1H), 7,85-7,83 (м, 2H), 7,61-7,59 (м, 1H), 7,49-7,45 (м, 3H), 7,37-7,34 (м, 1H), 7,04-7,01 (м, 1H), 6,20 (с, 2H), 3,65 (с, 2H), 2,55 (с, 2H), 2,00 (с, 3H), 1,76 (с, 2H), 1,50 (с, 9H).
Получение № 17: 1-(Метилсульфонил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,6-тетрагидропиридин
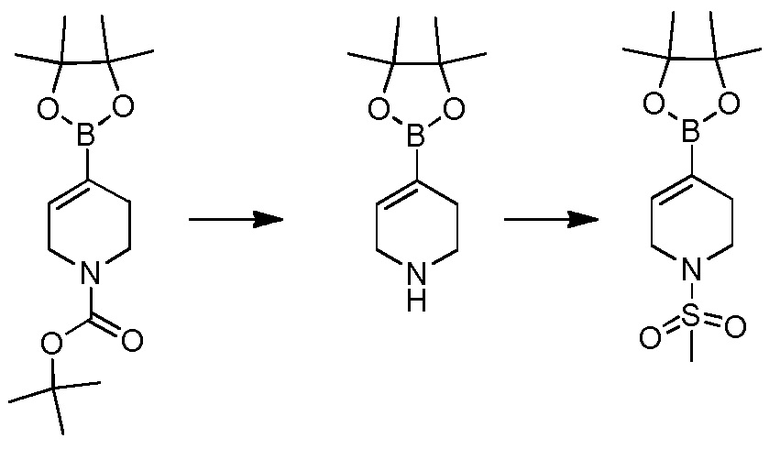
Раствор трет-бутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2H)-карбоксилата (4,03 г, 13,03 ммоль, Carbocore) в HCl (4 M в диоксане, 19,55 мл, 78 ммоль) перемешивали при комнатной температуре в течение приблизительно 2 часов. Раствор концентрировали при пониженном давлении, затем растворяли в DCM (20,05 мг) и добавляли TEA (12,72 мл, 91 ммоль). Смесь охлаждали до приблизительно 0°C и добавляли по каплям метансульфонилхлорид (1,83 мл, 23,5 ммоль). Смесь перемешивали при комнатной температуре в течение приблизительно 2 часов. К смеси добавляли 1 н HCl (60 мг) и органический слой экстрагировали. Органический слой с насыщенным водным раствором бикарбоната натрия (60 мг), сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Остаток растирали со смесью EtOAc и гептана, фильтровали и сушили (1,477 г). Фильтрат концентрировали и остаток растирали со смесью EtOAc и гептана, отфильтровывали и сушили с получением второй партии (0,940 г). Партии объединяли с получением 1-(метилсульфонил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,6-тетрагидропиридина (2,41 г, 64%). ЖХ/МС (Таблица 1, способ a) Время удерживания = 2,18 мин: MS m/z: 288 (M+H)+.
Получение № 18: 4-бром-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид
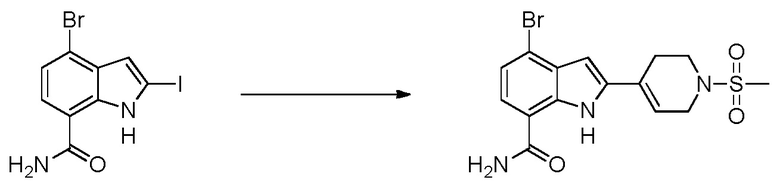
Колбу, содержащую 1-(метилсульфонил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,6-тетрагидропиридин (0,446 г, 1,55 ммоль, получение № 17), 4-бром-2-йод-1H-индол-7-карбоксамид (0,54 г, 1,48 ммоль, получение № 1), карбонат натрия (0,470 г, 4,44 ммоль) и 1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II) (0,108 г, 0,148 ммоль) продували азотом. Смесь THF (15,0 мг), MeOH (2,10 мг), и воды (2,10 мг) добавляли. Смесь перемешивали в течение приблизительно 2 часов при температуре приблизительно 70°C. Смесь фильтровали через целит, промывая EtOAc и концентрировали при пониженном давлении. Остаток растирали с DCM, фильтровали, промывали DCM и EtOAc с получением твердого вещества (0,315 г). Фильтрат концентрировали и очищали с помощью колоночной хроматографии на силикагеле (40-100% EtOAc/гептан). Полученный остаток растирают с DCM, фильтровали и сушили с получением твердого вещества (0,125 г). Твердые вещества объединяли с получением 4-бром-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамида (0,44 г, 75%). ЖХ/МС (Таблица 1, способ a) Время удерживания = 1,92 мин: MS m/z: 400 (M+H)+.
Получение № 19: N-метил-N-(2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)тиазол-2-карбоксамид
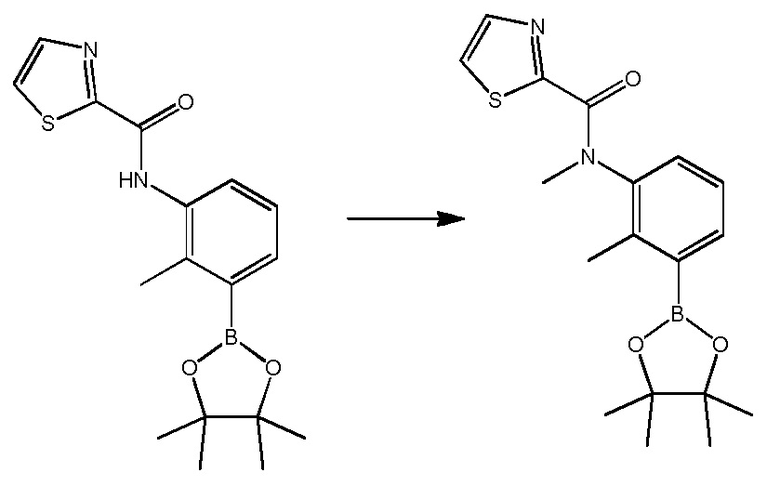
К N-(2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)тиазол-2-карбоксамиду (502 мг, 1,46 ммоль, получение № 4) в THF (10 мг) добавляли гидрид натрия (70,0 мг, 1,75 ммоль) при приблизительно 0°C и перемешивали в течение приблизительно 25 минут. К смеси добавляли йодметан (0,363 мл, 5,83 ммоль) при температуре приблизительно 0°C. Реакционную смесь доводили до комнатной температуры и затем перемешивали при комнатной температуре в течение приблизительно 18 часов. К смеси добавляли воду, экстрагировали дважды DCM и слои разделяли. Объединенные органические слои выпаривали, а остаток очищали с помощью хроматографии с нормальной фазой с получением N-метил-N-(2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)тиазол-2-карбоксамида (0,406 г, 59%). ЖХ/МС (Таблица 1, способ f) Время удерживания = 1,97 мин: MS m/z: 359 (M+H)+.
Получение № 20. (R)-1-((2,2-Диметил-1,3-диоксолан-4-ил)метил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол
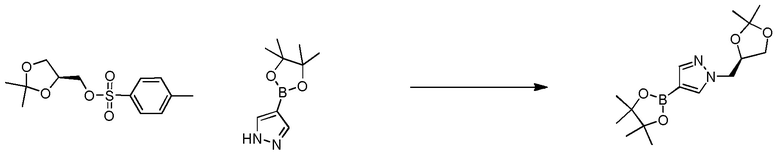
К смеси 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (1 г, 5,15 ммоль) в DMF (25,8 мг) добавляли гидрид натрия (0,206 г, 5,15 ммоль). Смесь перемешивали при комнатной температуре в течение примерно 10 минут в атмосфере азота. Добавляли (S)-(+)-2,2-диметил-1,3-диоксолан-4-илметил п-толуолсульфонат (1,62 г, 5,67 ммоль) и смесь перемешивали при приблизительно 90°C в течение ночи в атмосфере азота. Реакционную смесь охлаждали до комнатной температуры и разделяли между EtOAc и водой. Водный слой повторно экстрагировали EtOAc (2×) и органические слои объединяли, промывали водой, насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле с EtOAc/гексаном (30-75%) с получением (R)-1-((2,2-диметил-1,3-диоксолан-4-ил)метил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (0,66 г, 42%): ЖХ/МС (Таблица 1, способ f) Время удерживания = 1,41 мин; MS m/z: 309 (M+H)+.
Получение № 21. (S)-1-((2,2-Диметил-1,3-диоксолан-4-ил)метил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол
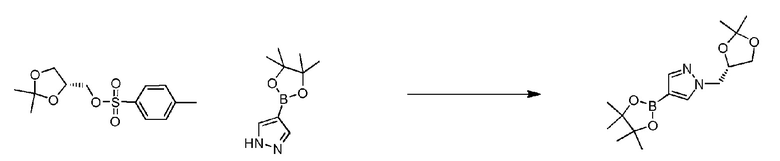
К смеси 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (1,0 г, 5,2 ммоль) в DMF (25,8 мг) добавляли гидрид натрия (0,206 г, 5,15 ммоль). Смесь перемешивали при комнатной температуре в течение приблизительно 10 минут в атмосфере азота. Добавляли (R)-(2,2-диметил-1,3-диоксолан-4-ил)метил 4-метилбензолсульфонат (1,62 г, 5,67 ммоль) и смесь перемешивали при температуре приблизительно 90°C в течение ночи в атмосфере азота. Реакционную смесь охлаждали до комнатной температуры, разделяли между EtOAc и водой. Водный слой повторно экстрагировали EtOAc (2×) и органические слои объединяли, промывали водой, насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле с EtOAc/гексаном (30-75%) с получением (S)-1-((2,2-диметил-1,3-диоксолан-4-ил)метил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (0,83 г, 52%): ЖХ/МС (Таблица 1, способ f) Время удерживания = 1,35 мин; MS m/z: 251 (M-(CH3)2CHO +H)+.
Получение № 22: N-(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)акриламид
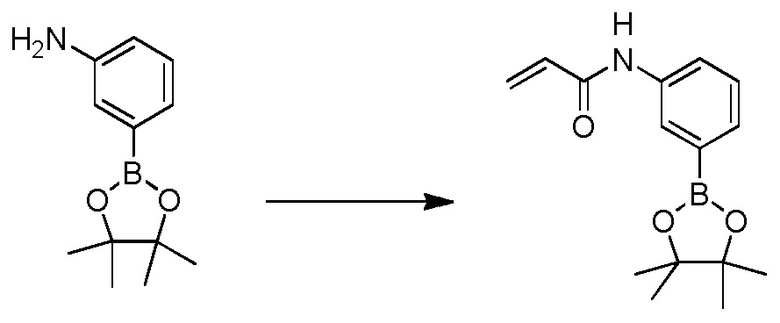
В сосуд добавляли 3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилин (0,30 г, 1,37 ммоль) в DCM (10 мг), и DIEA (0,72 мл, 4,11 ммоль). Смесь охлаждали до приблизительно 0°C и добавляли акрилоил хлорида (0122 мл, 1,51 ммоль) при перемешивании. Смесь перемешивали в течение приблизительно 20 минут, нагревая до комнатной температуры. Смесь разбавляли с и допонительным DCM (10 мг) промывали водой (2×10 мг), фильтровали через разделитель фаз Biotage и концентрировали в токе теплого азота с получением N-(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)акриламида (0,375 г, 100%): ЖХ/МС (Таблица 1, способ f) Время удерживания = 1,70 мин; MS m/z: 274 (M+H)+.
Получение № 23: N-(транс-4-Гидроксипиперидин-3-ил)тиазол-2-карбоксамид
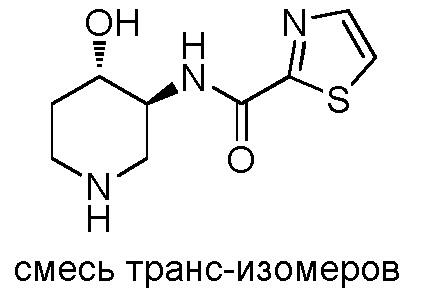
Стадия A. Бензил 4-(гидроксиимино)пиперидин-1-карбоксилат
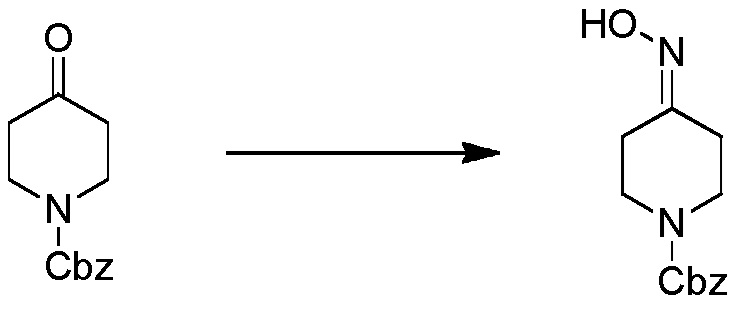
Смесь бензил 4-оксопиперидин-1-карбоксилата (10 г, 42,9 ммоль), NH2OH HCl (5,9 г, 86 ммоль) и K2CO3 (11,8 г, 86 ммоль) в EtOH (45 мл) нагревали при температуре приблизительно 50°C в течение приблизительно 0,5 часов. Затем растворитель удаляли при пониженном давлении. Воду и EtOAc добавляли к остатку. Водную фазу экстрагировали EtOAc (3×75 мг). Органический слой промывали насыщенным солевым раствором и сушили над Na2SO4, фильтровали и концентрировали с получением бензил 4-(гидроксиимино)пиперидин-1-карбоксилата (10 г, 94%). 1H-ЯМР (CDCl3) δ 2,36 (ушир., 2H), 2,63 (ушир., 2H), 3,63-3,58 (м, 4H), 5,15 (с, 2H), 7,36-7,35 (м, 5H), 9,05 (ушир., 1H).
Стадия B. Бензил 4-((тозилокси)имино)пиперидин-1-карбоксилат
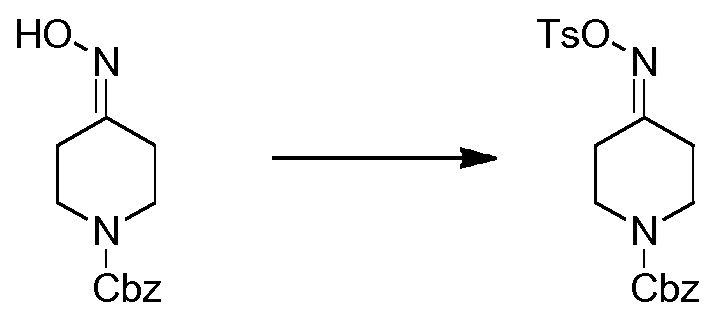
К раствору бензил 4-(гидроксиимино)пиперидин-1-карбоксилата (12,2 г, 49,1 ммоль) в пиридине (75 мг) медленно добавляли TsCl (12,2 г, 64 ммоль) при приблизительно 0°C. Реакционную смесь перемешивали при этой температуре в течение приблизительно 0,5 часа и перемешивали при комнатной температуре в течение еще 2 часов. Затем растворитель удаляли при пониженном давлении. Воду и EtOAc добавляли к остатку. Водную фазу экстрагировали EtOAc (3×125 мг). Органический слой промывали насыщенным солевым раствором и сушили над Na2SO4. Растворитель концентрировали с получением сырого продукта, который очищали с помощью колоночной хроматографии на силикагеле (петролейный эфир:EtOAc = 15:1) с получением бензил 4-((тозилокси)имино)пиперидин-1-карбоксилата (5 г, 25,3%): 1H-ЯМР (CDCl3) δ 2,37 (ушир., 2H), 2,44 (с, 3H), 2,63 (ушир., 2H), 3,62-3,55 (м, 4H), 5,13 (с, 2H), 7,35-7,32 (м, 7H), 7,85 (д, J=8,0 Гц, 2H).
Стадия C. Бензил 3-амино-4-оксопиперидин-1-карбоксилат гидрохлорид
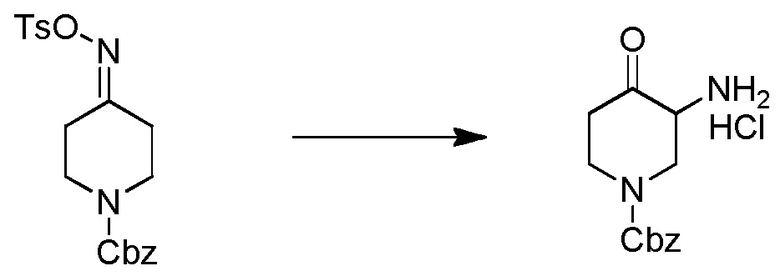
Na (28,6 мг, 1,243 ммоль) добавляли к EtOH (6,5 мг) и смесь перемешивали до тех пор, пока Na полностью не растворился. MgSO4 (0,98 г) добавляли к раствору, затем бензил 4-((тозилокси)имино)пиперидин-1-карбоксилат (0,5 г, 1,242 ммоль) добавляли к раствору при приблизительно 0°C. После того как реакционную смесь нагревали при температуре приблизительно 30°C в течение приблизительно 2 часов, смесь фильтровали и 1 н HCl (6,5 мг) добавляли к фильтрации. Фильтрат перемешивали при комнатной температуре в течение приблизительно 0,5 часов и концентрировали. Остаток смешивали с EtOH (3 мг) и фильтровали. Фильтрат концентрировали с получением сырого бензил 3-амино-4-оксопиперидин-1-карбоксилат гидрохлорида (200 мг, 0,702 ммоль): 1H-ЯМР (MeOD) δ=7,33 (м, 5 H), 5,12 (ущир. с, 2H), 3,75-3,95 (м, 1H), 3,6-3,7 (м, 1H), 3,5 (м, 2H), 3,1-3,2 (м, 1H), 1,95-2,10 (м, 1H), 1,7-1,8 (м, 1H).
Стадия D. Бензил 4-оксо-3-(тиазол-2-карбоксамидо)пиперидин-1-карбоксилат

Раствор тиазол-2-карбоновой кислоты (189 мг, 14,6 ммоль) и HATU (723 мг, 1,9 ммоль) в DMF (20 мг) перемешивали при комнатной температуре в течение 0,5 часов, затем DIEA (945 мг, 7,31 ммоль) и бензил 3-амино-4-оксопиперидин-1-карбоксилат гидрохлорид (500 мг, 1,76 ммоль) добавляли к смеси. Реакционный раствор перемешивали при комнатной температуре в течение приблизительно 4 часов. Воду добавляли к смеси, экстрагировали EtOAc (3×45 мг). Объединенный органический слой промывали насыщенным солевым раствором несколько раз, сушили над Na2SO4, фильтровали и концентрировали с получением сырого продукта, который очищали с помощью препаративной ВЭЖХ (Таблица 1, способ ai) с получением бензил 4-оксо-3-(тиазол-2-карбоксамидо)пиперидин-1-карбоксилата (82 мг, 12%). 1H-ЯМР (CDCl3) δ 2,68-2,62 (ушир., 2H), 2,93-2,86 (м, 1H), 3,16 (ушир., 1H), 4,7-5,9 (ушир., 2H), 5,08-5,05 (м, 1H), 5,31-5,22 (м, 2H), 7,43-7,38 (м, 5H), 7,60 (кв,J=1,2 Гц, 1H), 7,92-7,90 (м, 1H), 8,08 (с, 1H).
Стадия E. транс-Бензил 4-гидрокси-3-(тиазол-2-карбоксамидо)пиперидин-1-карбоксилат
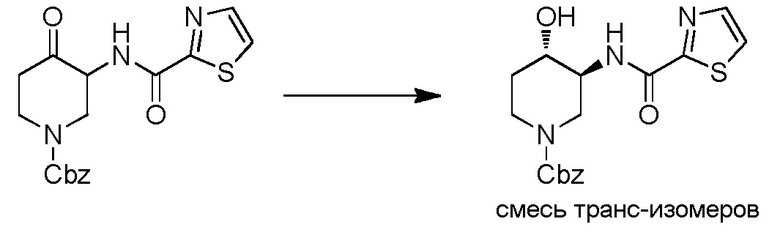
К раствору бензил 4-оксо-3-(тиазол-2-карбоксамидо)пиперидин-1-карбоксилата (6,9 г, 19,2 ммоль) в MeOH (50 мг) добавляли NaBH4 (0,726 г, 0,019 ммоль) периодически, и смесь перемешивали при комнатной температуре в течение приблизительно 0,5 часов. Затем добавляли воду (50 мг) к реакционной смеси и экстрагировали DCM (3×60 мг). Органический слой промывали насыщенным солевым раствором и сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением сырого продукта, который очищали с помощью колоночной хроматографией на силикагеле с получением транс-бензил 4-гидрокси-3-(тиазол-2-карбоксамидо)пиперидин-1-карбоксилата (3 г, 43%). 1H-ЯМР (MeOD) δ 1,56-1,51 (м, 1H), 2,00 (т, J=5,2 Гц, 1H), 3,10-2,97 (м, 2H), 3,85-3,75 (м, 2H), 4,16-3,99 (м, 1H), 4,21-4,20 (м, 1H), 5,12 (с, 2H), 7,34-7,31 (м, 5H), 7,85 (кв, J=3,2 Гц, 1H), 7,94 (т, J=3,2 Гц, 1H).
Стадия F. N-(транс-4-Гидроксипиперидин-3-ил)тиазол-2-карбоксамид

К перемешиваемому раствору транс-бензил 4-гидрокси-3-(тиазол-2-карбоксамидо)пиперидин-1-карбоксилата (0,7 г, 1,937 ммоль) в MeCN (15 мг) добавляли TMSI (1,55 г, 775 ммоль) медленно при температуре приблизительно 0°С, затем смесь перемешивали при комнатной температуре в течение приблизительно 1 часа. Воду выливали в смесь и MeCN удаляли при пониженном давлении. 1 н HCl добавляли к остатку и смесь экстрагировали МТВЕ (3×30 мг). Затем водную фазу подщелачивали NaOH (3 н) до приблизительно рН=12 и экстрагировали DCM (6×45 мг). Органическую фазу промывали насыщенным солевым раствором и сушили над Na2SO4, фильтровали и концентрировали с получением сырого продукта, который очищали с помощью препаративной ТСХ (1:1 МеОН/DCM) с получением N-(транс-4-гидроксипиперидин-3-ил)тиазол-2-карбоксамида (50 мг, 11%): 1H-ЯМР (MeOD) δ 1,86-1,77 (м, 1H), 2,28-2,22 (м, 1H), 3,29-309 (м, 2H), 3,56-3,44 (м, 2H), 4,84-3,90 (м, 2H), 7,88 (кв, J=3,2 Гц, 1H), 7,97 (кв, J=3,2 Гц, 1H).
Получение № 24: 4-бром-2-йод-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоксамид
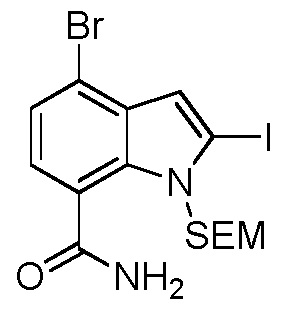
Стадия A. 4-бром-2-йод-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоновая кислота
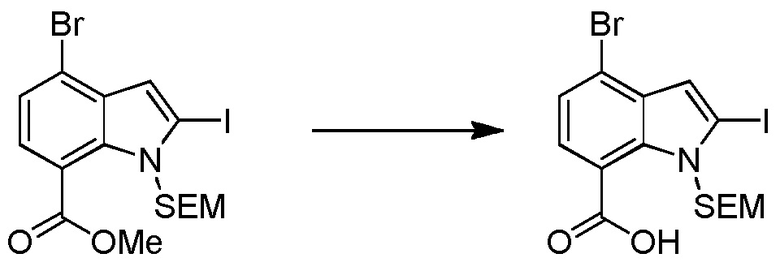
К раствору метил 4-бром-2-йод-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоксилата (10 г, 19,6 ммоль, получение № 10, стадия B) в MeOH (150 мг), THF (300 мг) и воды (150 мг) добавляли гидрат гидроксида лития (12 г, 286 ммоль). Полученную смесь нагревали при приблизительно 45°C в течение приблизительно 3 часов. Затем смесь концентрировали при пониженном давлении для удаления большей части растворителя, остаток растворяли в воде. Водную смесь подкисляли добавлением водного HCl (1 н) до приблизительно рН 6. Осадок фильтровали и твердое вещество сушили с получением 4-бром-2-йод-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоновой кислоты (9,1 г, 94%) в виде твердого вещества: 1H-ЯМР (CDCl3) δ 13,44 (ушир., 1H), 7,57-7,51 (м, 2H), 7,09 (с, 1H), 5,95 (с, 2H), 3,35-3,11 (т, J=8,0 Гц, 2H), 0,87-0,83 (т, J=8,0 Гц, 2H), 0,00 (с, 9H).
Стадия B. 4-бром-2-йод-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоксамид
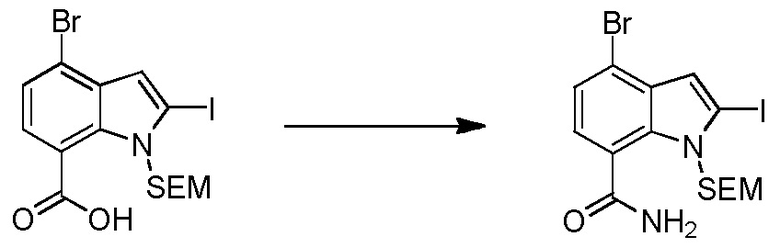
Раствор 4-бром-2-йод-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоновой кислоты (8 г, 16 ммоль), EDCI (4,6 г, 24 ммоль) и HOBt (3,7 г, 24 ммоль) в THF (240 мг) и DCM (280 мг) перемешивали при комнатной температуре в течение приблизительно 1 часа. Реакционную смесь затем барботировали газом NH3 в течение 15 минут и перемешивали при комнатной температуре в течение ночи. Затем смесь концентрировали и разделяли между водным раствором NaHCO3 и EtOAc. Органическую фазу промывали насыщенным солевым раствором, сушили и концентрировали с получением остатка, который суспендировали в петролейном эфире и твердое вещество собирали фильтрованием с получением 4-бром-2-йод-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоксамида (7,2 г, 90%) в виде белого твердого вещества: 1H-ЯМР (CDCl3) δ 7,36-7,33 (м, 1H), 7,26-7,24 (д, J=8,0 Гц, 1H), 7,05 (с, 1H), 6,08 (ушир., 1H), 5,82 (ушир., 1H)5,82 (с, 2H), 3,48-3,41 (м, 2H), 0,90-0,86 (м, 2H), 0,00 (с, 9H).
Получение № 25: 4-(Дифторметил)-N-(2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-N-(оксетан-3-ил)бензамид
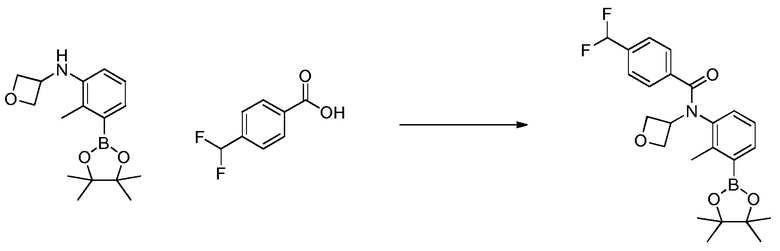
Раствор 4-(дифторметил)бензойной кислоты (0,089 г, 0,519 ммоль, Oakwood) в DCM (3,46 мг) в атмосфере азота обрабатывали дихлоридом серы (0,075 мл, 1,037 ммоль) и 1 каплей DMF. Смесь перемешивали при температуре приблизительно 35°C в течение приблизительно 16 часов. Реакционную смесь концентрировали при пониженном давлении, растирали остаток гептаном и концентрировали. Остаток растворяли в DCM (3,46 мг) и добавляли N-(2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)оксетан-3-амин (0,100 г, в 0,346 ммоль, полученный с использованием H из 2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина [Combi-Blocks] и 3-оксетанона [(Molbridge]) и TEA (0,193 мл, 1,383 ммоль). Смесь перемешивали при комнатной температуре в течение приблизительно 4 часов, затем разбавляли DCM (10 мг) и гасили насыщенным водным раствором бикарбоната натрия (10 мг). Органические фазы объединяли и промывали 30 мл насыщенного водного раствора бикарбоната натрия. Органический слой сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением сырого продукта, который очищали колоночной хроматографией на силикагеле (0-40% EtOAc/гептан) с получением желтого масла, которое затвердевало при стоянии с получением 4-(дифторметил)-N-(2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-N-(оксетан-3-ил)бензамида (0,092 г, 60%). LCMS (Таблица 1, способ a) Время удерживания = 2,51 мин: MS m/z: 444 (M+H)+.
Получение № 26: 2-метил-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)пропан-2-ол

К раствору 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (2,0 г, 10,31 ммоль) в 2,2-диметилоксиране (11,96 мл, 134 ммоль) в 30 мл сосуде для микроволновой печи добавляли карбонат цезия (0,521 г, 1,60 ммоль). Смесь нагревали в микроволновой печи при температуре приблизительно 120°C в течение приблизительно 30 минут. Реакционную смесь охлаждали и фильтровали. Полученный раствор выпаривали досуха с получением 2-метил-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)пропан-2-ола в виде белого твердого вещества. (2,7 г, 99%); (Таблица 1, способ g) Время удерживания = 1,34 мин.; MS m/z: 267 (M+H)+
Получение № 27: 4-фтор-2-йод-1-тозил-1H-индол-7-карбонитрил
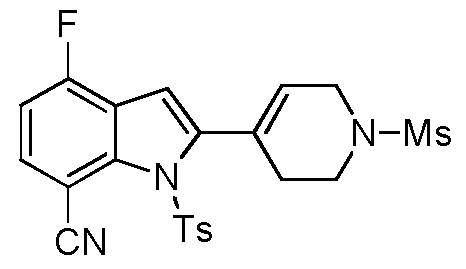
Стадия A. 4-фтор-1-тозил-1H-индол-7-карбонитрил
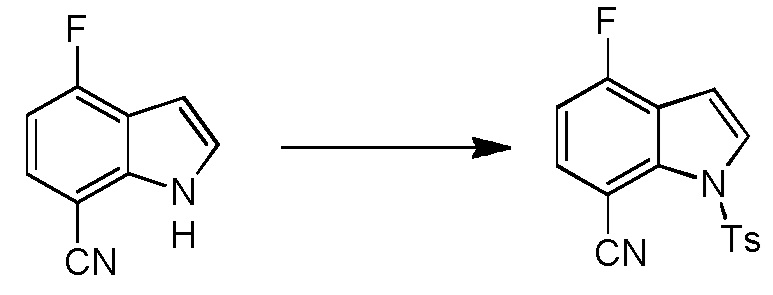
К раствору 4-фтор-1H-индол-7-карбонитрила (5,3 г, 33,1 ммоль, Sinova) в DMF (92 мг) добавляли NaH (2,0 г, 49,6 ммоль) при 0°C в атмосфере N2 и перемешивали в течение приблизительно 30 минут. Затем TsCl (9,46 г, 49,6 ммоль) добавляли к указанной выше смеси и перемешивали при комнатной температуре в течение приблизительно 5 часов. Смесь выливали в насыщенный водный раствор NH4Cl (200 мг), экстрагировали этилацетатом EtOAc (100 мл×3). Объединенную органическую фазу промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали с получением сырого продукта, который промывали MTBE с получением 4-фтор-1-тозил-1H-индол-7-карбонитрила (7 г, 67,3%) в виде твердого вещества: 1H-ЯМР (CDCl3) δ 2,39 (с, 3H), 6,86 (д, J=4 Гц, 1H), 6,99 (т, J=8,4 Гц, 1H), 7,33 (д, J=8,4 Гц, 2H), 7,62 (м, 1H), 7,84 (д, J=3,6 Гц, 1H), 7,92 (д, J=8,4 Гц, 2H).
Стадия B. 4-фтор-2-йод-1-тозил-1H-индол-7-карбонитрил
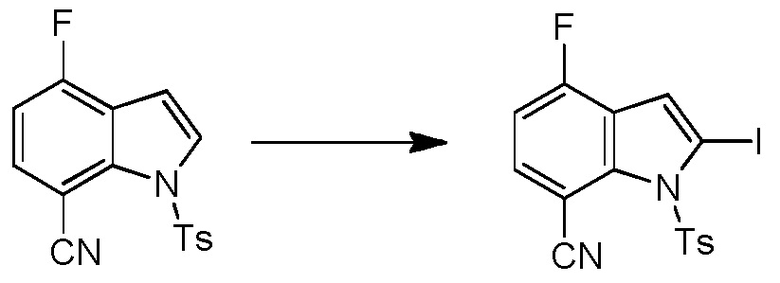
Свежеприготовленный LDA (67 мл, 38,2 ммоль) добавляли по каплям к раствору 4-фтор-1-тозил-1Н-индол-7-карбонитрила (10 г, 31,8 ммоль) в THF (50 мг) при температуре приблизительно -78°C. После того, как добавление было завершено, смесь перемешивали в течение еще 45 минут. Затем раствор I2 (9,69 г, 38,2 ммоль) в THF (50 мг) добавляли по каплям к смеси при приблизительно -78°C. После добавления смесь перемешивали в течение еще приблизительно 1 часа. Раствор выливали в насыщенный водный раствор Na2S2O3 (400 мг), экстрагировали EtOAc (100 мл×3). Объединенную органическую фазу промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали с получением сырого продукта, который промывали EtOAc с получением 4-фтор-2-йод-1-тозил-1H-индол-7-карбонитрила (8,5 г, 61%) в виде твердого вещества: 1H-ЯМР (CDCl3) δ 2,45 (с, 3H), 7,01 (т, J=8,4 Гц, 1H), 7,20 (с, 1H), 7,33 (д, J=8,4 Гц, 2H), 7,64 (м, 1H), 8,05 (д, J=8,4 Гц, 2H).
Стадия C. трет-Бутил 4-(7-циано-4-фтор-1-тозил-1H-индол-2-ил)-5,6-дигидропиридин-1(2H)-карбоксилат
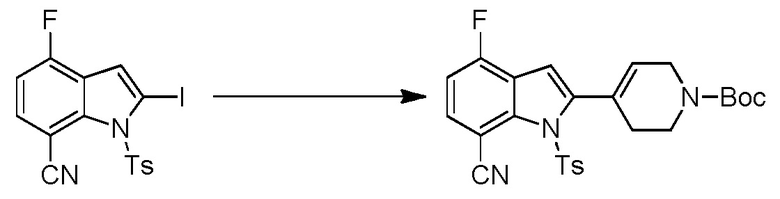
К раствору 4-фтор-2-йод-1-тозил-1H-индол-7-карбонитрила (2,92 г, 6,63 ммоль) и трет-бутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2H)-карбоксилата (2,05 г, 6,63 ммоль) в смеси THF (20 мг), MeOH (4 мг) и воды (4 мг) добавляли Na2CO3 (2,108 г, 19,89 ммоль) и PdCl2(dppf) DCM (0,541 г, 0,663 ммоль). Смесь нагревали при температуре приблизительно 80°C в течение приблизительно 3 часов. Затем реакционную смесь охлаждали и разбавляли EtOAc (30 мг) и промывали водой (3×10 мг). Органическую фазу сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением сырого продукта, который очищали с помощью колоночной хроматографии на силикагеле (элюируя петролейным эфиром:EtOAc = 10:1) с получением трет-бутил 4-(7-циано-4-фтор-1-тозил-1H-индол-2-ил)-5,6-дигидропиридин-1(2H)-карбоксилата (2,5 г, 76%): 1H-ЯМР (CDCl3) δ 1,25 (с, 2H), 1,52 (с, 9H), 2,38 (с, 3H), 3,63 (т, J=5,6 Гц, 2H), 4,09 (д, J=2,8 Гц, 2H), 5,83 (д, J=2,8 Гц, 1H), 6,56 (с, 1H), 7,04 (т, J=8,4 Гц, 1H), 7,20 (д, J=8,0 Гц, 2H), 7,48 (с, 2H), 7,68 (кв, J=5,2 Гц, 1H).
Стадия D. гидрохлорид 4-фтор-2-(1,2,3,6-тетрагидропиридин-4-ил)-1-тозил-1H-индол-7-карбонитрила
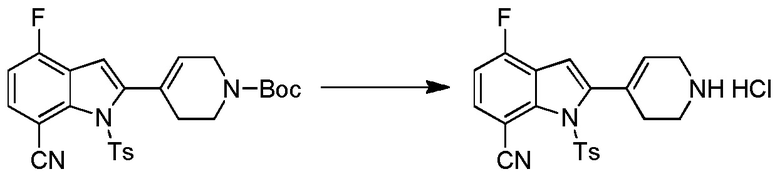
К раствору трет-бутил 4-(7-циано-4-фтор-1-тозил-1H-индол-2-ил)-5,6-дигидропиридин-1(2H)-карбоксилата (2,7 г, 5,45 ммоль) в EtOAc (30 мг) по каплям добавляли HCl/EtOAc (30 мг) при приблизительно 0°C, затем реакционную смесь перемешивали при комнатной температуре в течение приблизительно 3 часов. Смесь фильтровали и осадок на фильтре промывали EtOAc с получением гидрохлорида 4-фтор-2-(1,2,3,6-тетрагидропиридин-4-ил)-1-тозил-1H-индол-7-карбонитрила (1,96 г, 83%): 1H-ЯМР (MeOD) δ 2,35 (с, 3H), 2,78 (с, 2H), 3,48 (т, J=5,6 Гц, 2H), 3,94 (с, 2H), 6,04 (с, 1H), 6,86(с, 1H), 7,23-7,29 (м, 3H), 7,43 (д, J=8,0 Гц, 2H), 7,84 (т, J=5,2 Гц, 1H).
Стадия E. 4-фтор-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1-тозил-1H-индол-7-карбонитрил
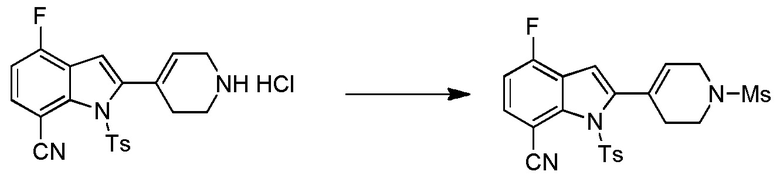
К раствору трет-бутил 4-(7-циано-4-фтор-1-тозил-1H-индол-2-ил)-5,6-дигидропиридин-1(2H)-карбоксилата (1,96 г, 4,54 ммоль) и TEA (1,84 г, 18,2 ммоль) в DCM (30 мг) добавляли MsCl (0,623 г, 5,44 ммоль), затем смесь перемешивали при комнатной температуре в течение приблизительно 24 часов. Затем добавляют воду к смеси и реакционную смесь экстрагировали DCM (3×30 мг). Объединенный органический слой промывали насыщенным солевым раствором и сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением 4-фтор-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1-тозил-1H-индол-7-карбонитрила (1,35 г, 63%), который использовали на следующей стадии без дополнительной очистки. ЖХ/МС (Таблица 1, способ f) Время удерживания = 2,15 мин; MS m/z: 474 (M+H)+.
Получение № 28: 3-бром-N-(цианометил)бензолсульфонамид
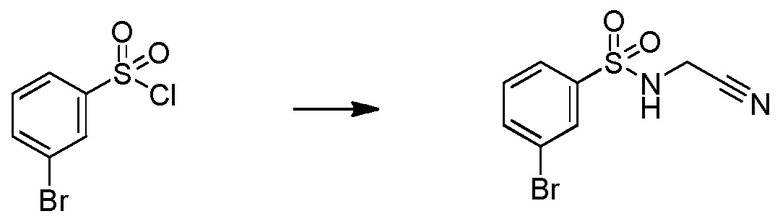
К охлажденному (0°C) раствору гидрохлорида 2-аминоацетонитрила (0,50 г, 5,40 ммоль) в пиридине (27,0 мг) медленно добавляли хлорид 3-бромбензол-1-сульфонила (0,779 мл, 5,40 ммоль). Смесь медленно нагревали до комнатной температуры и перемешивали в течение приблизительно 16 часов. Смесь концентрировали при пониженном давлении и остаток растворяли в DCM и промывали 1 н HCl, насыщенным бикарбонатом натрия и насыщенным солевым раствором и фильтровали через разделитель фаз Biotage после каждой стадии промывки. Органические вещества концентрировали при пониженном давлении, получая сырой продукт. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, элюируя EtOAc/гептан (0-40%) с получением 3-бром-N-(цианометил)бензолсульфонамида (0,61 г, 41%): 1H-ЯМР (DMSO-d6): δ 8,73 (ушир., 1H), 7,98 (т, J=1,79, 1H), 7,91 (д, J=8,02, 1H), 7,84 (д, J=8,02, 1H), 7,60 (т, J=7,92, 1H), 4,18 (с, 2H).
Получение № 29: 4-Циклопропил-N-(2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)бензамид

К раствору 2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина (0,350 г, 1,501 ммоль) и HATU (0,856 г, 2,252 ммоль) в DCM (2 мг) добавляли TEA (0,628 мл, 4,50 ммоль) и 4-(дифторметил)бензойную кислоту (0,336 г, 1,952 ммоль). Смесь перемешивали при комнатной температуре в течение приблизительно 18 часов. Смесь выпаривали и полученный остаток очищали с помощью хроматографии на силикагеле, элюируя градиентом 30-50% EtOAc в гексане, получая 4-циклопропил-N-(2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)бензамид (0,52, 89%); ЖХ/МС (Таблица 1, способ c) Время удерживания = 2,10 мин.; MS m/z: 388 (M+H)+
Получение № 30: гидрохлорид (R)-6-фтор-2-(пиперидин-3-ил)изоиндолин-1-она

Стадия A: Метил 5-фтор-2-метилбензоат
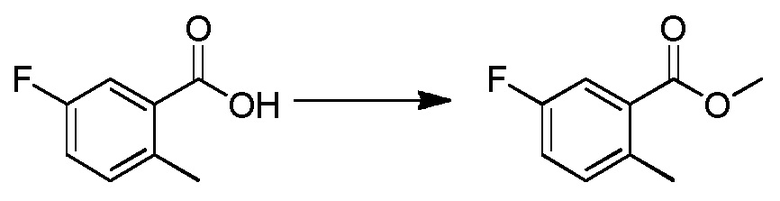
К раствору 5-фтор-2-метилбензойной кислоты (20 г, 0,13 моль) в безводном MeOH (200 мг) добавляли SOCl2 (38,9 г, 0,33 моль) по каплям. Полученную смесь перемешивали при комнатной температуре в течение ночи. Растворитель выпаривали досуха с получением метил 5-фтор-2-метилбензоата (24 г, 99%) в виде масла. 1H-ЯМР (CDCl3): δ 7,62-7,59 (д, J=9,6 Гц, 1H), 7,21-7,18 (д, J=8,4 Гц, 1H), 7,12-7,09 (д, J=8,0 Гц, 1H), 3,89 (с, 3 H), 2,55 (с, 3H).
Стадия B: Метил 2-(бромметил)-5-фторбензоат
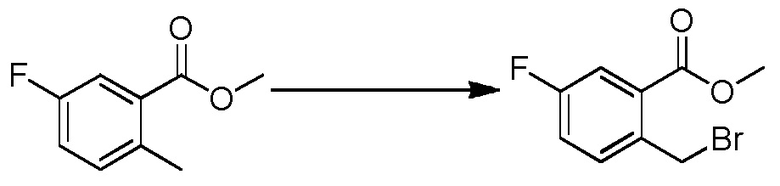
К раствору метил 5-фтор-2-метилбензоата (24 г, 0,14 моль) в CCl4 (250 мг) добавляли NBS (28 г, 0,16 моль) и BPO (1,7 г, 7,2 ммоль). Реакционную смесь нагревали с обратным холодильником в течение приблизительно 18 часов. Горячую реакционную смесь фильтровали и фильтрат концентрировали в вакууме с получением метил 2-(бромметил)-5-фторбензоата (35 г, сырой), который использовали на следующей стадии реакции непосредственно без дальнейшей очистки. 1H-ЯМР (DMSO-d6): δ 7,67-7,60 (м, 2H), 7,48-7,45 (д, J=8,4 Гц, 1H), 4,98 (с, 2H), 3,86 (с, 3H).
Стадия C: (R)-трет-Бутил 3-(6-фтор-1-оксоизоиндолин-2-ил)пиперидин-1-карбоксилат
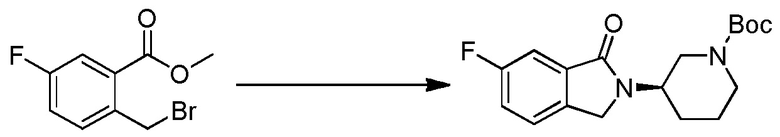
К раствору метил 2-(бромметил)-5-фторбензоата (35 г) в MeCN (400 мг) добавляли K2CO3 (39 г, 0,29 моль) и 3-(R)-амино-пиперидин-1-карбоновой кислоты трет-бутиловый эфир (20 г, 0,10 моль). Реакционную смесь нагревали с обратным холодильником в течение приблизительно 3 часов, а затем перемешивали при комнатной температуре в течение ночи. Полученную суспензию фильтровали и фильтрат концентрировали в вакууме с получением остатка, который растворяли в EtOAc (300 мг) и промывали насыщенным солевым раствором (2×100 мг). Органическую фазу сушили над Na2SO4 и концентрировали. Полученный остаток очищали колоночной хроматографией на силикагеле (элюируя 15:1 петролейный эфир:EtOAc) с получением (R)-трет-бутил 3-(6-фтор-1-оксоизоиндолин-2-ил)пиперидин-1-карбоксилата (12 г, 25%) в виде твердого вещества: 1H-ЯМР (CDCl3): δ 7,46-7,43 (д, J=7,6 Гц, 1H), 7,35-7,32 (д, J=8,0 Гц, 1H), 7,20-7,14 (м, 1H), 4,36-4,26 (м, 2H), 4,18 (м, 1H), 4,06-3,89 (м, 2H), 2,99-2,93 (м, 1H), 2,75 (с, 1H), 1,95-1,92 (м, 1H), 1,74-1,65 (м, 2H), 1,56-1,54 (м, 1H), 1,39 (с, 9H).
Стадия D: гидрохлорид (R)-6-фтор-2-(пиперидин-3-ил)изоиндолин-1-она
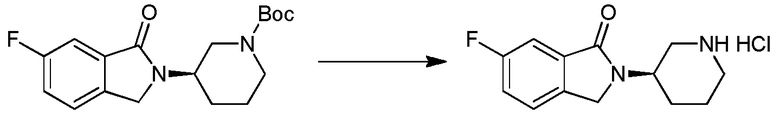
К раствору (R)-трет-бутил 3-(6-фтор-1-оксоизоиндолин-2-ил)пиперидин-1-карбоксилата (12 г, 0,036 моль) в DCM (100 мг) добавляли 1M HCl в MeOH (150 мг). Полученную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали в вакууме с получением гидрохлорида (R)-6-фтор-2-(пиперидин-3-ил)изоиндолин-1-она B (9,0 г, 100%) в виде твердого вещества. LCMS (ESI+): m/z 235 (M+H)+, Время удерживания: 1,90 мин; 1H-ЯМР (D2O): δ 7,43-7,40 (м, 1H), 7,28-7,21 (м, 2H), 4,39-4,37 (д, J=5,6 Гц, 2H), 4,33-4,31 (м, 1H), 3,38-3,34 (м, 2H), 3,12-3,06 (т, J=12,0 Гц, 1H), 2,88-2,85 (м, 1H), 2,00-1,95 (м, 2H), 1,87-1,77 (м, 2H).
Получение № 31: (R)-3-(Пиперидин-3-ил)хиназолин-4(3H)-он
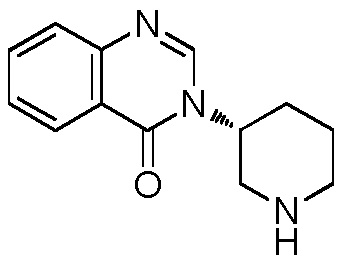
Стадия A: (R)-трет-Бутил 3-(4-оксохиназолин-3(4H)-ил)пиперидин-1-карбоксилат
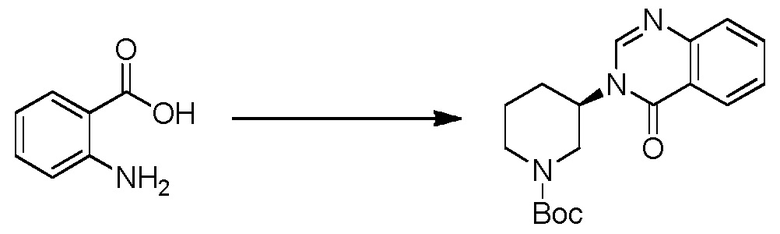
К раствору 2-аминобензойной кислоты (7,5 г, 54,7 ммоль) и 3-(R)-амино-пиперидин-1-карбоновой кислоты трет-бутилового эфира (10,9 г, 54,7 ммоль) в THF (20 мг) добавляли триэтилортоформиат (8,1 г, 54,7 ммоль). Реакционную смесь нагревали до температуры приблизительно 110°C в запаянной трубке в течение ночи. После охлаждения до комнатной температуры смесь разбавляли водой и экстрагировали EtOAc. Объединенную органическую фазу промывали насыщенным солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением остатка, который очищали с помощью колоночной хроматографии на силикагеле (элюирование 10:1 петролейный эфир:EtOAc) с получением (R)-трет-бутил 3-(4-оксохиназолин-3(4H)-ил)пиперидин-1-карбоксилата (7,5 г, 42%) в виде желтого твердого вещества. 1H-ЯМР (CDCl3): δ 8,34-8,32 (м, 1H), 8,11 (с, 1H), 7,80-7,71 (м, 2H), 7,55-7,51 (м, 1H), 4,75 (ушир., 1H), 4,23-4,11 (ушир., 2H), 3,24-3,18 (т, 1H), 2,87 (ушир., 1H), 2,18-1,98 (м, 2H), 1,91-1,87 (ушир., 1H), 1,77-1,71 (м, 1H), 1,48 (с, 9H).
Стадия B: (R)-3-(Пиперидин-3-ил)хиназолин-4(3H)-он

Реакционный раствор (R)-трет-бутил 3-(4-оксохиназолин-3(4H)-ил)пиперидин-1-карбоксилата (12,5 г, 36 ммоль) в 1M HCl/MeOH (150 мг) перемешивали при приблизительно комнатной температуре в течение приблизительно 2,5 часов. Смесь фильтровали. Твердое вещество промывали EtOAc и сушили с получением (R)-3-(пиперидин-3-ил)хиназолин-4(3H)-она (10 г, 98%) в виде белого твердого вещества. LCMS (ESI+): m/z 248 (M+H)+, ВРЕМЯ УДЕРЖИВАНИЯ: 1,90 мин. 1H-ЯМР (D2O): δ 8,55-8,54 (д, J=2,8 Гц, 1H), 7,80-7,77 (дд, J=3,2 Гц, J=2,8 Гц, 1H), 7,68-7,60 (м, 2H), 4,95-4,89 (м, 1H), 3,61-3,57 (м, 1H), 3,46-3,43 (д, J=12,4 Гц, 1H), 3,37-3,31 (т, 1H), 3,04-2,97 (м, 1H), 2,24-2,14 (м, 3H), 1,94-1,87 (м, 1H).
Получение № 32: гидрохлорид (R)-6-фтор-3-(пиперидин-3-ил)хиназолин-4(3H)-она
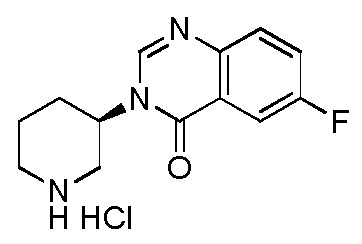
Стадия A: (R)-трет-Бутил 3-(6-фтор-4-оксохиназолин-3(4H)-ил)пиперидин-1-карбоксилат
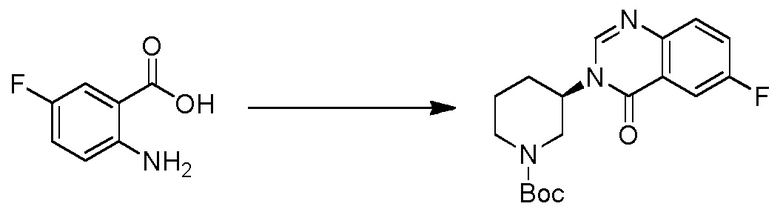
Реакционный раствор 2-амино-5-фторбензойной кислоты (7,5 г, 48,4 ммоль), трет-бутилового эфира 3-(R)-амино-пиперидин-1-карбоновой кислоты (9,68 г, 48,4 ммоль) и триэтилортоформиата (7,2 г, 48,4 ммоль) в THF (20 мг) нагревали до приблизительно 110°C в запаянной трубке в течение ночи. После охлаждения до комнатной температуры смесь разбавляли водой. Водный слой экстрагировали EtOAc. Объединенную органическую фазу промывали насыщенным солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением остатка, который очищали с помощью колоночной хроматографии на силикагеле (элюирование 10:1 петролейный эфир:EtOAc) с получением (R)-трет-бутил 3-(6-фтор-4-оксохиназолин-3(4H)-ил)пиперидин-1-карбоксилата (6,25 г, 37%) в виде твердого вещества. 1H-ЯМР (CDCl3): δ 8,08 (с, 1H), 7,97-7,95 (м, 1H), 7,76-7,72 (м, 1H), 7,53-7,48 (м, 1H), 4,74 (ушир., 1H), 4,24-4,12 (ушир., 2H), 3,24-3,19 (т, 1H), 2,89 (ушир., 1H), 2,14-2,10 (м, 2H), 2,04-2,01 (м, 1H), 1,91-1,71 (м, 1H), 1,49 (с, 9H).
Стадия B: гидрохлорид (R)-6-фтор-3-(пиперидин-3-ил)хиназолин-4(3H)-она
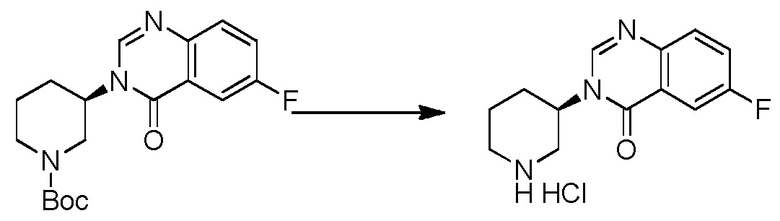
Раствор (R)-трет-бутил 3-(6-фтор-4-оксохиназолин-3(4H)-ил)пиперидин-1-карбоксилата (12,5 г, 36 ммоль) в 1M HCl/MeOH (150 мг) перемешивали при приблизительно комнатной температуре приблизительно в течение приблизительно 2,5 часов. Смесь фильтровали, твердое вещество промывали EtOAc и сушили с получением гидрохлорида (R)-6-фтор-3-(пиперидин-3-ил)хиназолин-4(3H)-она (10 г, 98%) в виде твердого вещества. ЖХ/МС (ESI+): m/z 248 (M+H)+, ВРЕМЯ УДЕРЖИВАНИЯ: 1,90 мин. 1H-ЯМР (D2O): δ 8,55-8,54 (д, J=2,8 Гц, 1H), 7,80-7,77 (дд, J=3,2 Гц, J=2,8 Гц, 1H), 7,68-7,60 (м, 2H), 4,95-4,89 (м, 1H), 3,61-3,57 (м, 1H), 3,46-3,43 (д, J=12,4 Гц, 1H), 3,37-3,31 (т, 1H), 3,04-2,97 (м, 1H), 2,24-2,14 (м, 3H), 1,94-1,87 (м, 1H).
Получение № 33: гидрохлорид 7-циклопропил-5-фтор-3-(пиперидин-3-ил)хиназолин-4(3H)-она
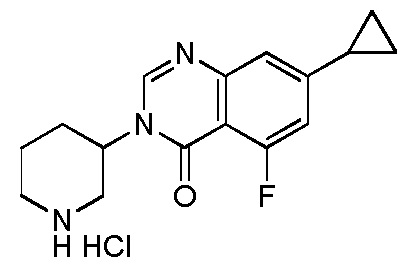
Стадия A: трет-Бутил 3-(7-бром-5-фтор-4-оксохиназолин-3(4H)-ил)пиперидин-1-карбоксилат
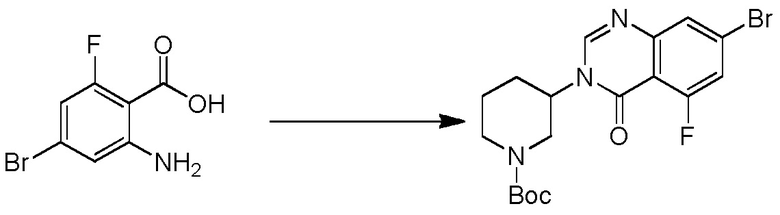
К раствору 2-амино-4-бром-6-фторбензойной кислоты (7 г, 0,03 моль, полученный в соответствии с WO 2011075699) и трет-бутилового эфира 3-амино-пиперидин-1-карбоновой кислоты (6,6 г, 0,033 моль) в THF (50 мг) добавляли триэтилортоформиат (6,6 г, 0,044 моль). Реакционную смесь нагревали при приблизительно 110°C в запаянной трубке в течение ночи. После охлаждения до приблизительно комнатной температуры, смесь разбавляли водой. Водный слой экстрагировали EtOAc. Объединенную органическую фазу промывали насыщенным солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением остатка, который очищали с помощью колоночной хроматографии на силикагеле (элюируя 50:1 петролейный эфир:EtOAc) с получением трет-бутил 3-(7-бром-5-фтор-4-оксохиназолин-3(4H)-ил)пиперидин-1-карбоксилата (6,4 г, 50%) в виде твердого вещества. 1H-ЯМР (CDCl3): δ 8,1 (с, 1H), 7,54-7,52 (дд, J=2,4 Гц, 1H), 7,35-7,32 (дд, J=2,8 Гц, 1H), 4,7 (ушир., 1H), 4,2-4,16 (ушир., 1H), 4,07-4,03 (ушир., 1H), 3,24-3,18 (т, 1H), 2,92-2,89 (ушир., 1H), 2,11-2,09 (ушир., 1H), 1,98-1,96 (ушир., 1H), 1,89-1,85 (ушир., 1H), 1,74-1,64 (ушир., 1H), 1,45 (с, 9H).
Стадия B: трет-Бутил 3-(7-циклопропил-5-фтор-4-оксохиназолин-3(4H)-ил)пиперидин-1-карбоксилат
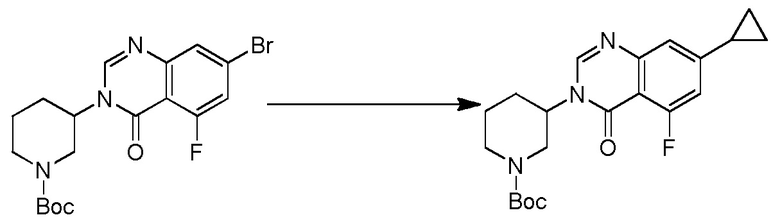
К смеси трет-бутил 3-(7-бром-5-фтор-4-оксохиназолин-3(4H)-ил)пиперидин-1-карбоксилата (20 г, 0,047 моль), Pd(OAc)2 (0,526 г, 0,002 моль), трициклогексилфосфина (1,31 г, 0,005 моль), безводного K3PO4 (50 г, 0,236 моль) и воды (40 мг) в толуоле (200 мг) добавляли циклопропилбороновую кислоту (6,06 г, 0,07 моль). Реакционную смесь нагревали с обратным холодильником в течение ночи в атмосфере N2. После охлаждения до комнатной температуры, смесь разбавляли водой. Водный слой экстрагировали EtOAc. Объединенную органическую фазу промывали насыщенным солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением остатка, который очищали с помощью колоночной хроматографии на силикагеле (элюируя 50:1 петролейный эфир:EtOAc) с получением трет-бутил 3-(7-циклопропил-5-фтор-4-оксохиназолин-3(4H)-ил)пиперидин-1-карбоксилата (15 г, 83%) в виде твердого вещества. 1H-ЯМР (CDCl3): δ 7,96 (с, 1H), 7,07-7,04 (дд, J=2,4 Гц, 1H), 6,71-6,67 (дд, J=2,4 Гц, 1H), 4,68-4,65 (ушир., 1H), 4,16 (ушир., 1H), 4,06-4,02 (ушир., 1H), 3,37-3,33 (м, 1H), 3,08-3,02 (м, 1H), 2,82-2,76 (ушир., 1H), 2,06-2,01 (м, 1H), 1,90-1,69 (м, 2H), 1,64-1,60 (м, 1H), 1,40 (с, 9H), 1,20-1,06 (м, 2H), 0,712-0,608 (м, 2H).
Стадия C: гидрохлорид 7-циклопропил-5-фтор-3-(пиперидин-3-ил)хиназолин-4-(3H)-она

Раствор трет-бутил 3-(7-циклопропил-5-фтор-4-оксохиназолин-3(4H)-ил)пиперидин-1-карбоксилата (15 г, 0,039 ммоль) в 1M HCl/MeOH (150 мг) перемешивали при приблизительно комнатной температуре в течение приблизительно 2,5 часов. Смесь фильтровали и твердое вещество промывали EtOAc и сушили с получением гидрохлорида 7-циклопропил-5-фтор-3-(пиперидин-3-ил)хиназолин-4(3H)-она (10 г, 91%) в виде твердого вещества. LCMS (ESI+): m/z 288 (M+H)+, Время удерживания: 2,916 мин. 1H-ЯМР (D2O): δ 8,56 (с, 1H), 6,99-6,96 (м, 1H), 6,85-6,82 (дд, J=1,6 Гц, 1H), 4,87-4,83 (м, 1H), 3,54-3,51 (м, 1H), 3,41-3,38 (д, 1H), 3,24-3,18 (т, 1H), 2,96-2,89 (т, 1H), 2,84-2,81 (м, 1H), 2,13-2,09 (м, 3H), 1,89-1,82 (м, 1H), 0,96-094 (ушир., 2H), 0,61 (ушир., 2H).
Получение № 34: 2-(Бензилокси)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилин
Стадия A: 1-(Бензилокси)-4-бром-2-нитробензол
К раствору 4-бром-2-нитрофенола (5 г, 22,9 ммоль) в ацетоне (100 мг) добавляли (бромметил)бензол (4,7 г, 27,5 ммоль) и K2CO3 (6,3 г, 45,9 ммоль). Смесь кипятили с обратным холодильником в течение ночи. После охлаждения до комнатной температуры, смесь фильтровали. Фильтрат концентрировали при пониженном давлении с получением остатка, который промывали TBME с получением 1-(бензилокси)-4-бром-2-нитробензола (6,3 г, 89%): 1H-ЯМР (CDCl3) δ 8,00 (д, J=2,2 Гц, 1H), 7,60 (дд, J=2,6, 8,8 Гц, 1H), 7,49-7,31 (м, 5H), 7,03 (д, J=8,8 Гц, 1H), 5,24 (с, 2H).
Стадия B: 2-(Бензилокси)-5-броманилин
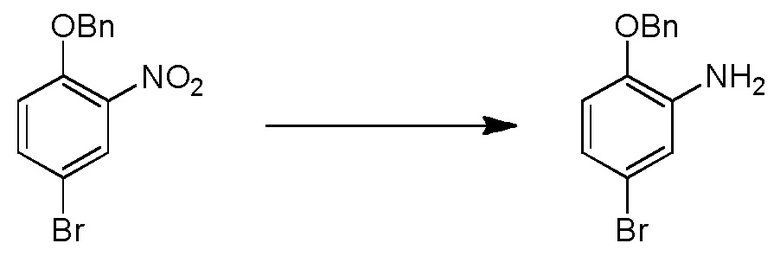
К раствору 1-(бензилокси)-4-бром-2-нитробензола (2 г, 6,5 ммоль) в EtOH (80 мг) и воды (20 мг) добавляли железо (1,8 г, 32,5 ммоль) и NH4Cl (1,7 г, 32,5 ммоль). Полученную смесь нагревали с обратным холодильником в течение 3 часов. Смесь фильтровали. Фильтрат разбавляли водой и экстрагировали этилацетатом. Органический слой концентрировали с получением 2-(бензилокси)-5-броманилина (1,6 г, 89%): 1H-ЯМР (CDCl3) δ 7,51-7,30 (м, 5H), 6,86 (д, J=2,2 Гц, 1H), 6,83-6,76 (м, 1H), 6,74-6,66 (м, 1H), 5,07 (с, 2H), 3,91 (ушир., 2H).
Стадия C: 2-(Бензилокси)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилин

К раствору 2-(бензилокси)-5-броманилина (2,0 г, 7,19 ммоль) в DMSO (30 мг) добавляли 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (2,2 г, 8,6 ммоль), Pd(dppf)Cl2 (0,53 г, 0,72 ммоль) и ацетат калия (2,1 г, 21,6 ммоль). Смесь перемешивали при 80°C в течение ночи в атмосфере N2. После охлаждения до комнатной температуры, смесь разбавляли водой и экстрагировали EtOAc. Органический слой концентрировали и очищали на колонке с получением 2-(бензилокси)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина (1,5 г, 64%): 1H-ЯМР (CDCl3) δ 7,55-7,29 (м, 5H), 7,23-7,12 (м, 2H), 6,86 (д, J=7,9 Гц, 1H), 5,11 (с, 2H), 3,80 (ушир., 2H), 1,32 (с, 12H).
Получение № 35: 3-(Бензилокси)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилин

Стадия A: 3-бром-5-нитрофенол
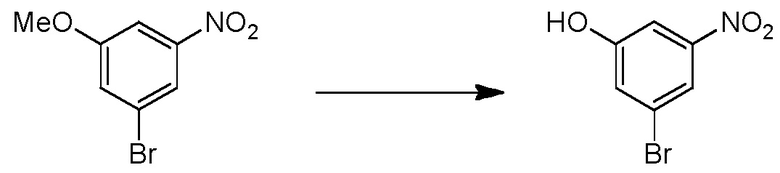
К раствору 1-бром-3-метокси-5-нитробензола (19 г, 82 ммоль) в DCM (800 мг) по каплям добавляли BBr3 (27,9 мл, 295 ммоль) в DCM (120 мг). Полученную смесь нагревали с обратным холодильником в течение ночи. После охлаждения в воде со льдом, смесь разбавляли добавлением воды. Затем смесь промывали насыщенным солевым раствором. Органическую фазу сушат над Na2SO4, концентрировали при пониженном давлении с получением остатка, который очищали колоночной хроматографией на силикагеле с получением 3-бром-5-нитрофенола (8 г, 44%) в виде твердого вещества: 1H-ЯМР (CDCl3) δ 7,89 (с, 1H), 7,57 (с, 1H), 7,27 (с, 1H), 5,27 (с, 1H).
Стадия B: 1-(Бензилокси)-3-бром-5-нитробензол
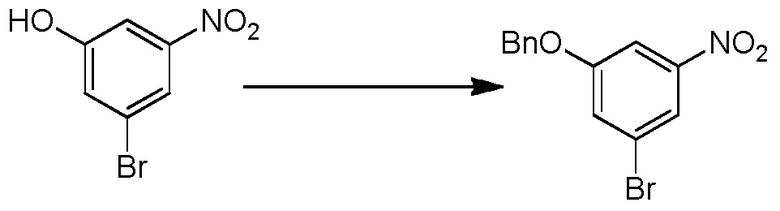
К раствору 3-бром-5-нитрофенола в ацетоне (50 мг) добавляли (бромметил)бензол (2,4 г, 13,8 ммоль) и K2CO3 (3,2 г, 22,9 ммоль). Полученную смесь нагревали с обратным холодильником в течение ночи. Смесь фильтровали. Фильтрат концентрировали при пониженном давлении с получением остатка, который промывали TBME с получением 1-(бензилокси)-3-бром-5-нитробензола (1,3 г, 37%) в виде твердого вещества: 1H-ЯМР (CDCl3) δ 8,00 (с, 1H), 7,78-7,77 (м, 1H), 7,64-7,40 (м, 6H), 5,15 (с, 2H).
Стадия C: 3-(Бензилокси)-5-броманилин

К раствору 1-(бензилокси)-3-бром-5-нитробензола (1,3 г, 4,2 ммоль) в EtOH (30 мг) и воде (7,5 мг) добавляли железо (1,2 г, 21,1 ммоль) и NH4Cl (1,1 г, 21,1 ммоль). Смесь нагревали с обратным холодильником в течение ночи. Смесь фильтровали. Фильтрат концентрировали при пониженном давлении с получением остатка, который разбавляли добавлением воды и экстрагировали EtOAc. Органический слой концентрировали при пониженном давлении с получением 3-(бензилокси)-5-броманилина (1 г, 85%): 1H-ЯМР (CDCl3) δ 7,33-7,31 (м, 5H), 6,48 (с, 1H), 6,39 (с, 1H), 6,14 (с, 1H), 4,92 (с, 2H), 3,63 (ушир., 2H).
Стадия D: 3-(Бензилокси)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилин
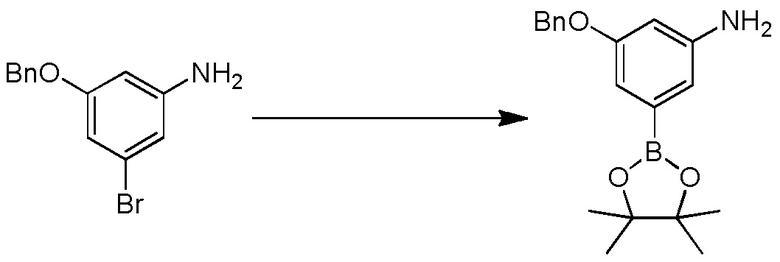
К раствору 3-(бензилокси)-5-броманилина (1 г, 3,6 ммоль) и 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (1,1 г, 4,3 ммоль) в DMSO (1 мг) добавляли Pd(dppf)Cl2 (0,26 г, 0,36 ммоль) и ацетат калия (1,1 г, 10,8 ммоль). Смесь нагревали до приблизительно 80°C в течение ночи в атмосфере N2. После охлаждения до комнатной температуры, смесь разбавляли добавлением воды и экстрагировали EtOAc. Органический слой концентрировали при пониженном давлении с получением остатка, который очищали колоночной хроматографией на силикагеле с получением 3-(бензилокси)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина (1 г, 86%) в виде твердого вещества: 1H-ЯМР (CDCl3) δ 7,43-7,31 (м, 5H), 6,87 (с, 1H), 6,77 (с, 1H), 6,43-6,42 (м, 1H), 5,05 (с, 2H), 3,64 (ушир., 2H), 1,34 (с, 12H).
Получение № 36: 4-(Бензилокси)-1-бром-2-нитробензол
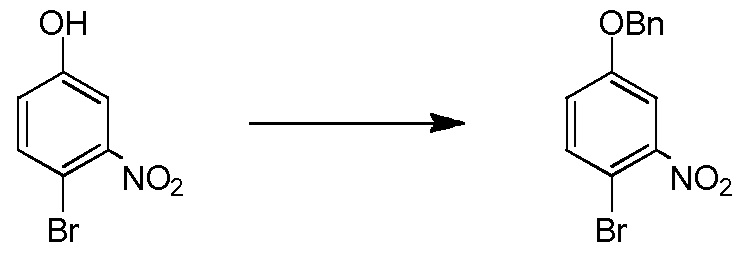
К раствору 4-бром-3-нитрофенола (2 г, 9,17 ммоль, Получение № S.1) в ацетоне (50 мг) добавляли BnBr (1,9 г, 11,0 ммоль) и K2CO3 (2,5 г, 18,4 ммоль). Смесь фильтровали. Фильтрат концентрировали при пониженном давлении с получением остатка, который промывали TBME с получением 4-(бензилокси)-1-бром-2-нитробензола (2,6 г, 92%): 1H-ЯМР (CDCl3) δ 7,62 (д, J=8,8 Гц, 1H), 7,48 (д, J=2,6 Гц, 1H), 7,45-7,35 (м, 5H), 7,07 (дд, J=2,9, 9,0 Гц, 1H), 5,12 (с, 2H).
Получение № 37: 4-(Бензилокси)-1-бром-2-нитробензол
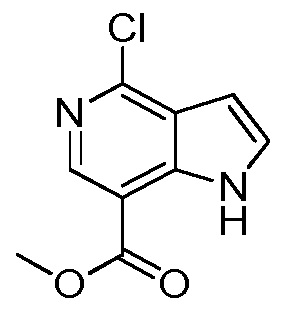
Стадия A: Метил 2-(2-метокси-2-оксоэтил)-1-(4-метоксибензил)-1H-пиррол-3-карбоксилат
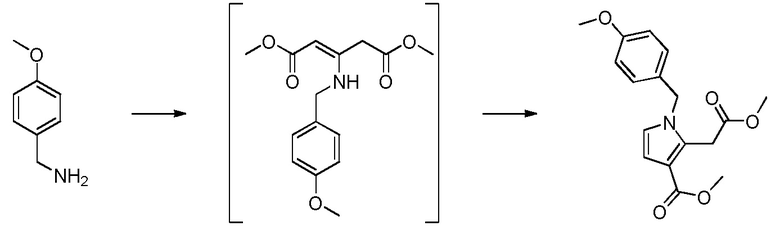
В сосуд загружали диметил 3-оксопентандиоат (77,0 г, 442 ммоль), (4-метоксифенил)метанамин (60,1 мл, 460 ммоль) и безводный NaOAc (72,5 г, 884 ммоль) в диоксане (100 мг). Реакционную смесь перемешивали при приблизительно комнатной температуре в течение приблизительно 30 минут, затем нагревали до приблизительно 50°C и перемешивали в течение приблизительно 16 часов. Реакционную смесь охлаждали до комнатной температуры и добавляли диоксан (250 мг). 2-хлорацетальдегид (51,9 мл, 442 ммоль) добавляли через капельную воронку. После приблизительно 7 часов добавляли дополнительный 2-хлорацетальдегид (17,4 г, 221 ммоль) и перемешивали в течение приблизительно 16 часов. Добавляли дополнительное количество 2-хлорацетальдегида (17,4 г, 221 ммоль) и перемешивали в течение приблизительно 5 часов, добавляли еще 2-хлорацетальдегид (25,9 мл, 221 ммоль), конечную часть 2-хлорацетальдегида (25,9 мл, 221 ммоль) добавляли примерно через 2 часа и оставляли перемешиваться в течение приблизительно 72 часов. Добавляли NaOAc (36,3 г, 442 ммоль) и раствор и перемешивали в течение приблизительно 16 часов. Реакционную смесь охлаждали на бане со льдом и добавляли к этому воду со льдом (около 500 мг). Смесь экстрагировали DCM (850 мг). Органический слой промывали водой (4×700 мг). Органический слой сушили над MgSO4, фильтровали и концентрировали с получением вязкого масла. Сырой продукт очищали с помощью флэш-хроматографии (используя гептан для 3 объемов колонки, 0-25% EtOAc/гептан в 4 объемах колонки, 20-35% в 4 объемах колонки). Чистые фракции объединяли и концентрировали, и добавляли минимальное Et2O для осаждения первой партии продукта, который собирали фильтрованием. Фильтрат объединяли с неочищенными фракциями, концентрировали в вакууме и перекристаллизовывали из изопропанола с получением твердого вещества, которое собирали фильтрованием и объединяли с первой партией продукта. Материал сушили в вакуумной печи при температуре приблизительно 70°C в течение приблизительно 16 часов с получением метил 2-(2-метокси-2-оксоэтил)-1-(4-метоксибензил)-1H-пиррол-3-карбоксилата (28,5 г, 20%): ЖХ/МС (Таблица 1, способ as) Время удерживания = 2,20 мин; MS m/z: 318 (M+H)+.
Стадия B: Метил 2-(1-амино-3-метокси-3-оксопроп-1-ен-2-ил)-1-(4-метоксибензил)-1H-пиррол-3-карбоксилат

В сосуд загружали NaH (23,3 г, 582 ммоль) и THF (500 мг). Смесь охлаждали приблизительно до 0°C и метил 2-(2-метокси-2-оксоэтил)-1-(4-метоксибензил)-1H-пиррол-3-карбоксилат (28 г, 88 ммоль) добавляли порциями. Внутреннюю температуру поддерживали ниже 10°C во время добавления. Суспензию перемешивали при приблизительно 0°C в течение приблизительно 1 часа. Добавили метилформиат (7,62 мл, 124 ммоль). Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение приблизительно 16 часов. Добавляли дополнительное количество метилформиата (1,09 мл, 17,6 ммоль) и смесь перемешивали при комнатной температуре в течение приблизительно от 4 до 5 часов, в этот момент все исходное вещество было израсходовано. Реакционную смесь охлаждали на льду и гасили добавлением МеОН (5 мг), и аккуратно добавляли воду до тех пор, пока кипение не прекратилось. Затем смесь подкисляли до рН приблизительно 1 водным раствором 6 н HCl, сохраняя при этом сосуд на бане со льдом. Реакционную смесь разбавляли EtOAc (100 мг) и водой (100 мг). Водный слой отделяли и экстрагировали этилацетатом (3×50 мг). Затем объединенные органические слои сушили над MgSO4 и фильтровали. Растворитель упаривали с получением масла, состоящего из двух слоев. Более тонкий верхний слой очищали и отделяли с помощью пипетки и отбрасывали. Оставшийся нижний слой представлял собой сырой промежуточный продукт, метил 2-(1-гидрокси-3-метокси-3-оксопроп-1-ен-2-ил)-1-(4-метоксибензил)-1H-пиррол-3-карбоксилат. В сосуд загружали этот сырой метил 2-(1-гидрокси-3-метокси-3-оксопроп-1-ен-2-ил)-1-(4-метоксибензил)-1H-пиррол-3-карбоксилат (30 г, 87 ммоль) и МеОН (300 мг). Добавляли ацетат аммония (33,5 г, 434 ммоль) и реакционную смесь кипятили с обратным холодильником в течение приблизительно 4 часов и перемешивали при приблизительно 60°C в течение приблизительно 72 часов. Реакционную смесь концентрировали в вакууме и разбавляли водой (200 мг) и EtOAc (200 мг). Часть продукта выпадало в осадок и его собирали фильтрацией. Органический слой отделяли. Водный слой снова экстрагировали этилацетатом (2×80 мг). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали. Остаток суспендировали в Et2O (200 мг) и перемешивали в течение приблизительно 10 минут и фильтровали для сбора продукта. Данную порцию объединяли с предыдущим осадком и сушили в вакуумной печи при температуре приблизительно 70°C в течение приблизительно 4 часов с получением метил 2-(1-амино-3-метокси-3-оксопроп-1-ен-2-ил)-1-(4-метоксибензил)-1H-пиррол-3-карбоксилата (25,7 г, 82%): ЖХ/МС (Таблица 1, способ as) Время удерживания = 1,88 мин; MS m/z: 345 (M+H)+.
Стадия C: Метил 1-(4-метоксибензил)-4-оксо-4,5-дигидро-1H-пирроло[3,2-c]пиридин-7-карбоксилат
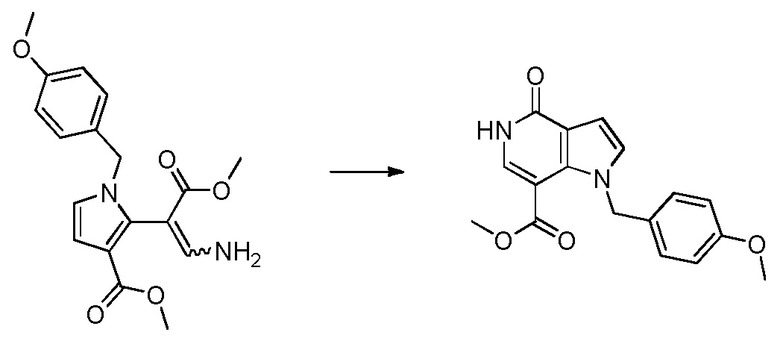
В сосуд загружали метил 2-(1-амино-3-метокси-3-оксопроп-1-ен-2-ил)-1-(4-метоксибензил)-1H-пиррол-3-карбоксилат (24,6 г, 71,4 ммоль) и t-BuONa (6,87 г, 71,4 ммоль) в DMA (100 мг). Раствор нагревали при приблизительно 150°C в течение приблизительно 10 минут и охлаждали до комнатной температуры. Затем раствор выливали на воду со льдом (250 мг) и разбавляли EtOAc (200 мг). Смесь перемешивали при комнатной температуре в течение приблизительно 45 минут. Осадок, который образовывался, отфильтровывали и промывали водой, затем сушили в вакуумной печи при температуре приблизительно 70°C в течение приблизительно 16 часов с получением метил 1-(4-метоксибензил)-4-оксо-4,5-дигидро-1H-пирроло[3,2-c]пиридин-7-карбоксилата (18,9 г, 85%): ЖХ/МС (Таблица 1, способ as) Время удерживания = 1,76 мин; MS m/z: 313 (M+H)+.
Стадия D: Метил 4-хлор-1H-пирроло[3,2-c]пиридин-7-карбоксилат

Смесь метил 1-(4-метоксибензил)-4-оксо-4,5-дигидро-1H-пирроло[3,2-c]пиридин-7-карбоксилата (24 г, 76 ммоль) в фенил фосфородихлоридате (30,8 мл, 206 ммоль) нагревали при температуре приблизительно 150°C в течение приблизительно 30 минут. LCMS показала полную конверсию в смеси эфира и кислоты. Реакционную смесь охлаждали до приблизительно 0°C и 50%-ный водный раствор NaOH медленно добавляли до рН приблизительно 7. Реакционную смесь экстрагировали DCM (3×100 мг). Органические слои объединяли и концентрировали при пониженном давлении. Остаток суспендировали в Et2O (100 мг), перемешивали при приблизительно 30°C в течение приблизительно 1 часа, охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали с получением сырого метил 4-хлор-1-(4-метоксибензил)-1H-пирроло[3,2-c]пиридин-7-карбоксилата (22,5 г, 75%) в виде черного масла. Смесь этого сырого метил 4-хлор-1-(4-метоксибензил)-1H-пирроло[3,2-c]пиридин-7-карбоксилата (21,76 г, 65,8 ммоль) и трифторметансульфонового ангидрида (7,50 мл, 44,4 ммоль) в TFA (50 мг) перемешивали при приблизительно 50°C в течение приблизительно 16 часов. Реакционную смесь охлаждали до комнатной температуры и добавляли к охлажденному льдом раствору NaHCO3. Водный NaOH медленно добавляли для доведения рН до приблизительно 9. Твердое вещество отфильтровывали и обрабатывали ультразвуком в Et2O. Осадок фильтровали и фильтрат концентрировали с получением метил 4-хлор-1H-пирроло[3,2-c]пиридин-7-карбоксилата (9,4 г, 68%): LCMS (Таблица 1, способ a) Время удерживания = 1,83 мин; MS m/z: 211 (M+H)+.
Получение № 38: Метил 4-(1-(трет-бутоксикарбонил)пирролидин-3-ил)-2-метил-1H-индол-7-карбоксилат
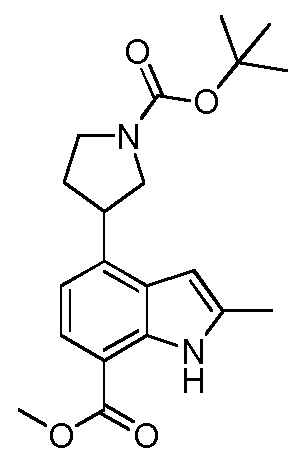
Стадия A: 1-трет-Бутил 7-метил 4-(1-(трет-бутоксикарбонил)пирролидин-3-ил)-2-метил-1H-индол-1,7-дикарбоксилат
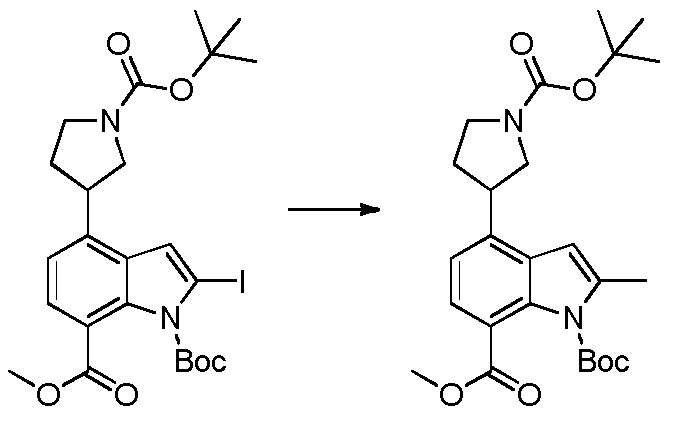
К раствору 1-трет-бутил 7-метил 4-(1-(трет-бутоксикарбонил)пирролидин-3-ил)-2-йод-1H-индол-1,7-дикарбоксилата (2,0 г, 3,5 ммоль, получение № Y.1) в THF (35 мг) добавляли Zn(Me)2 (1 M в гексане, 21,04 мл, 21,04 ммоль). Смесь дегазировали с использованием азота и Pd(dppf)Cl2 (0,257 г, 0,351 ммоль) добавляли в виде одной порции и перемешивали при комнатной температуре в течение приблизительно 19 ч. Реакционную смесь нагревали до приблизительно 45°C и перемешивали в течение приблизительно 22 часов. Реакционную смесь осторожно гасили добавлением насыщенного водного NaHCO3 (50 мг) и разбавляли EtOAc (50 мг) и насыщенным солевым раствором (20 мг). Слои разделяли и водную фазу экстрагировали EtOAc (2×50 мг). Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали, концентрировали при пониженном давлении и очищали с помощью колоночной хроматографии на силикагеле (0-50% EtOAc/гептан) с получением 1-трет-бутил 7-метил 4-(1-(трет-бутоксикарбонил)пирролидин-3-ил)-2-метил-1H-индол-1,7-дикарбоксилата (1,45 г, 79%): LCMS (Таблица 1, способ ba) Время удерживания = 3,02 мин; MS m/z: 476 (M+H)+.
Стадия B: Метил 4-(1-(трет-бутоксикарбонил)пирролидин-3-ил)-2-метил-1H-индол-7-карбоксилат
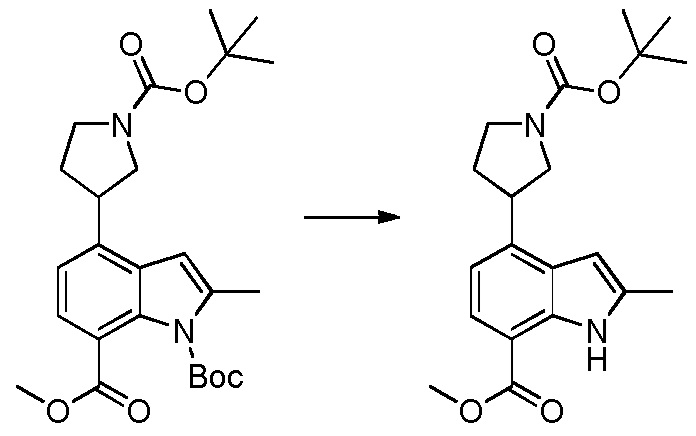
Раствор 1-трет-бутил 7-метил 4-(1-(трет-бутоксикарбонил)пирролидин-3-ил)-2-метил-1H-индол-1,7-дикарбоксилата (1,40 г, 3,05 ммоль) в MeOH (7 мг) добавляли в реакционный сосуд для микроволновой печи, и раствор нагревали до приблизительно 120°C в течение приблизительно 30 минут. Реакционную смесь адсорбировали на силикагеле и очищали с помощью хроматографии на силикагеле (0-50% EtOAc/гептан) с получением метил 4-(1-(трет-бутоксикарбонил)пирролидин-3-ил)-2-метил-1H-индол-7-карбоксилата (1 г, 86%): LCMS (Таблица 1, способ as) Время удерживания = 2,58 мин; MS m/z: 359 (M+NH4)+.
Получение № 39: Метил 4-(1-(трет-бутоксикарбонил)-1,2,5,6-тетрагидропиридин-3-ил)-1-тозил-1H-индол-7-карбоксилат
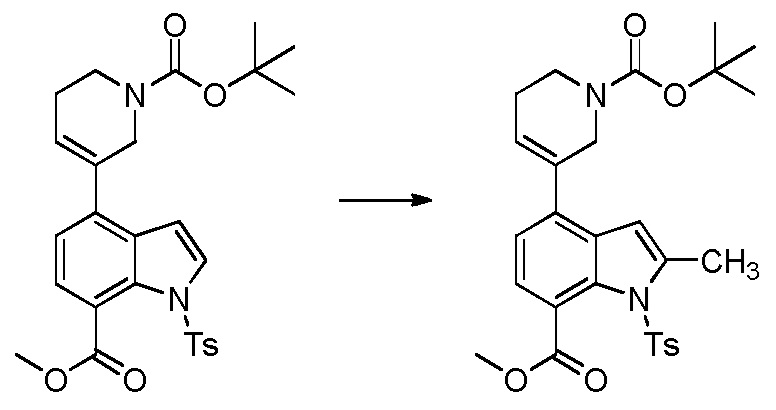
В сосуд загружали метил 4-(1-(трет-бутоксикарбонил)-1,2,5,6-тетрагидропиридин-3-ил)-1-тозил-1H-индол-7-карбоксилат (2,00 г, 3,92 ммоль, полученный с использованием A из Получения № 1, стадия B с трет-бутил 3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2H)-карбоксилатом) в THF (39,2 мг). Раствор охлаждали до приблизительно -71°C. LDA (1M раствор в гексане/THF, 5,88 мл, 5,88 ммоль) добавляли по каплям в течение приблизительно 5 минут при поддержании температуры ниже -65°C. Раствор перемешивали при температуре приблизительно -72°C в течение приблизительно 45 минут. Добавляли CH3I (0,367 мл, 5,88 ммоль). Смесь перемешивали при температуре приблизительно -70°C еще в течение 2,5 часов, а затем гасили насыщенным водным раствором Na2CO3 (150 мг). Смесь экстрагировали (2×200 мг) и DCM (1×100 мг). Объединенные органические слои сушили над Na2SO4, фильтровали, концентрировали при пониженном давлении и очищали с помощью колоночной хроматографии на силикагеле (25-75% EtOAc/гептан), с получением метил 4-(1-(трет-бутоксикарбонил)-1,2,5,6-тетрагидропиридин-3-ил)-1-тозил-1H-индол-7-карбоксилата (1,67 г, 57%, 70% чистота): LCMS (Таблица 1, способ as) Время удерживания = 2,88 мин; MS m/z: 542 (M+NH4)+.
Получение № 40: трет-бутил 3-((7-карбамоил-2-йод-1H-индол-4-ил)(метил)амино)азетидин-1-карбоксилат
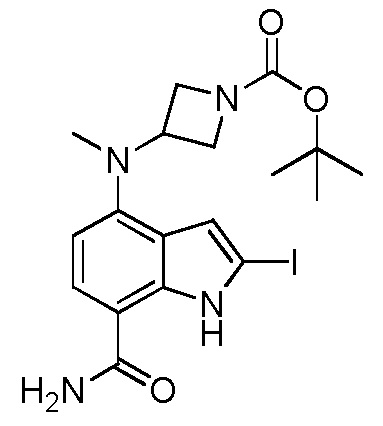
Стадия A: Метил 4-((1-(трет-бутоксикарбонил)азетидин-3-ил)(метил)амино)-2-йод-1-тозил-1H-индол-7-карбоксилат
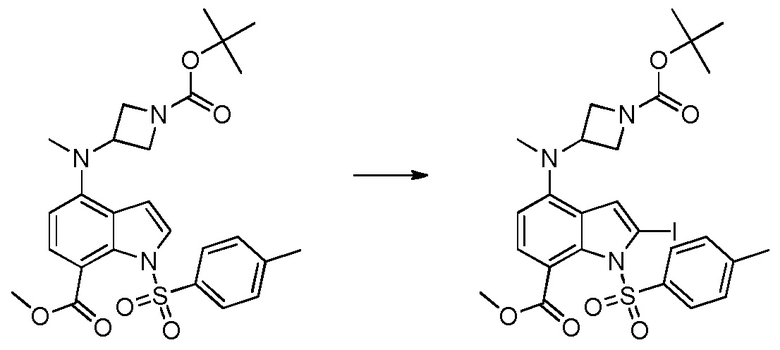
К раствору метил 4-((1-(трет-бутоксикарбонил)азетидин-3-ил)(метил)амино)-1-тозил-1H-индол-7-карбоксилата (4,00 г, 7,79 ммоль, полученный с использованием T из Получения № 1, стадия C с трет-бутил-3-аминоазетидин-1-карбоксилатом и J с CH3I) в THF (60 мг) при температуре приблизительно -78°C медленно добавляли LDA (2 M раствор в THF, 5,84 мл, 11,7 ммоль). Реакционную смесь перемешивали при температуре приблизительно -78°C в течение приблизительно 1 часа и медленно добавляли раствор I2 (2,97 г, 11,7 ммоль) в THF (10 мг) и реакционную смесь перемешивали при температуре приблизительно -78°C в течение приблизительно 4 часов. Охлаждающую баню удаляли для нагревания реакционной смеси до комнатной температуры и реакцию гасили добавлением насыщенного водного Na2S2O3 (120 мг), экстрагировали дополнительным EtOAc (2×150 мг) и промывали насыщенным солевым раствором (2×150 мг). Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением сырого продукта, метил 4-((1-(трет-бутоксикарбонил)азетидин-3-ил)(метил)амино)-2-йод-1-тозил-1H-индол-7-карбоксилата (4,1 г, 80%): ЖХ/МС (Таблица 1, способ aa) Время удерживания = 1,87 мин; MS m/z: 640 (M+H)+.
Стадия B: 4-((1-(трет-Бутоксикарбонил)азетидин-3-ил)(метил)амино)-2-йод-1H-индол-7-карбоновая кислота
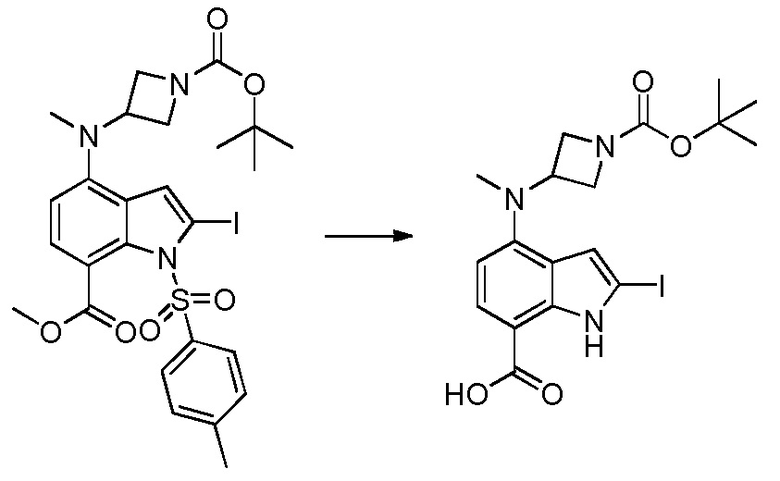
К раствору метил 4-((1-(трет-бутоксикарбонил)азетидин-3-ил)(метил)амино)-2-йод-1-тозил-1H-индол-7-карбоксилата (15,5 г, 24,2 ммоль) в MeOH (75 мг):THF (75 мг):вода (30 мг) добавляли KOH (9,52 г, 170 ммоль). Смесь перемешивали при приблизительно 60°C в течение приблизительно 16 часов, охлаждали и подкисляли водной 2 н HCl. Экстрагировали этилацетатом (2×350 мг) и промывали насыщенным солевым раствором (2×300 мг). Объединенные органические вещества сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением сырого продукта 4-((1-(трет-бутоксикарбонил)азетидин-3-ил)(метил)амино)-2-йод-1H-индол-7-карбоновой кислоты (11,4 г, 99%): ЖХ/МС (Таблица 1, способ aa) Время удерживания = 1,86 мин; MS m/z: 416 (M +H-tBu)+.
Стадия C: трет-Бутил 3-((7-карбамоил-2-йод-1H-индол-4-ил)(метил)амино)азетидин-1-карбоксилат

4-((1-(трет-Бутоксикарбонил)азетидин-3-ил)(метил)амино)-2-йод-1H-индол-7-карбоновую кислоту (13,7 г, 29,1 ммоль), HOBt (8,90 г, 58,1 ммоль) и EDC (11,2 г, 58,1 ммоль) растворяли в DMF (260 мг) и добавляли DIEA (25,4 мл, 145 ммоль). Смесь перемешивали при комнатной температуре в течение приблизительно 10 минут и добавляли NH4Cl (12,4 г, 233 ммоль). Смесь перемешивали при комнатной температуре в течение приблизительно 16 часов и добавляли насыщенный водный NH4Cl (1 л). Твердое вещество собирали фильтрацией, промывали водой и сушили с получением сырого продукта трет-бутил 3-((7-карбамоил-2-йод-1H-индол-4-ил)(метил)амино)азетидин-1-карбоксилата (13,4 г, 97%): ЖХ/МС (Таблица 1, способ aa) Время удерживания = 1,81 мин; MS m/z: 471 (M+H)+.
Получение № 41: 4-(Азетидин-3-ил(метил)амино)-2-(тетрагидрофуран-3-ил)-1H-индол-7-карбоксамид
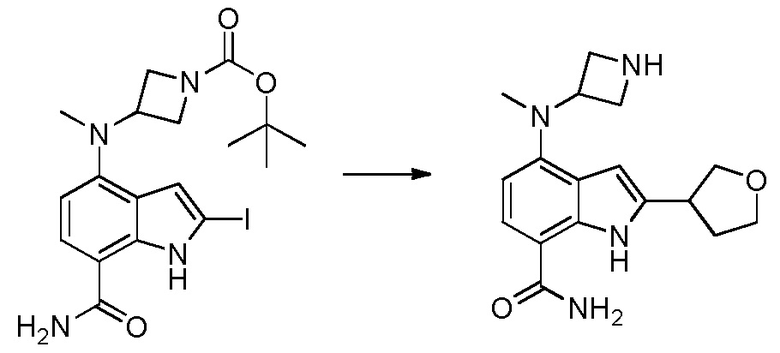
Реакционный сосуд загружали трет-бутил 3-((7-карбамоил-2-йод-1H-индол-4-ил)(метил)амино)азетидин-1-карбоксилатом (0,050 г, 0,11 ммоль, Получение № 40), (Z)-бут-2-ен-1,4-диолом (0,014 г, 0,16 ммоль), NaHCO3 (10,7 мг, 0,128 ммоль) и PdCl2 (1,885 мг, 10,63 мкмоль) в NMP (1,2 мг). Смесь продували азотом и нагревали при приблизительно 130°C в течение приблизительно 1 часа. Экстрагировали EtOAc (2×20 мг) и промывали насыщенным солевым раствором (2×20 мг). Объединенные органические вещества сушили над безводным Na2SO4, фильтровали, концентрировали при пониженном давлении и очищали с помощью препаративной ТСХ (EtOAc) с получением сырого трет-бутил 3-((7-карбамоил-2-(2,3-дигидрофуран-3-ил)-1H-индол-4-ил)(метил)амино)азетидин-1-карбоксилата (0,028 г, 39%). Смесь трет-бутил 3-((7-карбамоил-2-(2,3-дигидрофуран-3-ил)-1H-индол-4-ил)(метил)амино)азетидин-1-карбоксилата 0,055 г, 0,081 ммоль) в DCM (1,5 мг) перемешивали при приблизительно 0°C на бане со льдом. Добавляли триэтилсилан (0,014 г, 0,12 ммоль), а затем добавляли по каплям BF3.OEt2 (0,015 мл, 0,122 ммоль). Смесь перемешивали при приблизительно 0°C в течение приблизительно 1 часа и гасили насыщенным водным раствором Na2CO3 до рН приблизительно 8, затем фильтровали. Фильтрат очищали препаративной ВЭЖХ (таблица 1, способ bc) с получением 4-(азетидин-3-ил(метил)амино)-2-(тетрагидрофуран-3-ил)-1H-индол-7-карбоксамида (0,008 мг, 28%): ЖХ/МС (Таблица 1, способ av) Время удерживания = 1,03 мин; MS m/z: 315 (M+H)+.
Получение № 42: Метил 4-((1-(трет-бутоксикарбонил)азетидин-3-ил)(метил)амино)-2-(3-гидроксиоксетан-3-ил)-1-тозил-1H-индол-7-карбоксилат
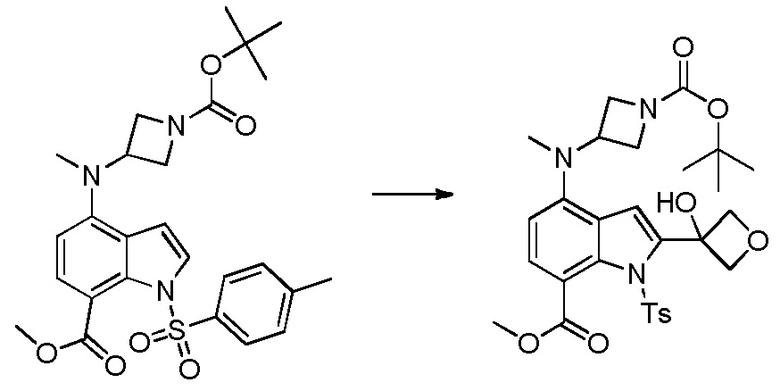
К холодному раствору метил 4-((1-(трет-бутоксикарбонил)азетидин-3-ил)(метил)амино)-1-тозил-1H-индол-7-карбоксилата (0,80 г, 1,56 ммоль, полученному с использованием T из Получения № 1, стадия C с трет-бутил-3-аминоазетидин-1-карбоксилатом и J с CH3I) в THF (12 мг) при приблизительно -78°C медленно добавляли LDA (2M раствор в THF, 1,168 мл, 2,336 ммоль). Реакционную смесь перемешивали при приблизительно -78°C в течение приблизительно 1 часа, затем раствор оксетан-3-она (0,168 г, 2,34 ммоль) в THF (1 мг) медленно добавляли и реакционную смесь перемешивали при приблизительно -78°C в течение приблизительно 4 часов. Охлаждающую баню удаляли и реакцию гасили насыщенным водным раствором NH4CI. Смесь экстрагировали EtOAc (2×50 мг) и промывали насыщенным солевым раствором (2×50 мг). Объединенные органические вещества сушили над безводным Na2SO4, фильтровали, концентрировали при пониженном давлении и очищали с помощью препаративной ТСХ (1:1 EtOAc/пет. Et2O) с получением метил 4-((1-(трет-бутоксикарбонил)азетидин-3-ил)(метил)амино)-2-(3-гидроксиоксетан-3-ил)-1-тозил-1H-индол-7-карбоксилата (0,55 г, 59%): ЖХ/МС (Таблица 1, способ av) Время удерживания = 1,67 мин; MS m/z: 586 (M+H)+.
Получение № 43: трет-Бутил 2-(7-циано-1-тозил-1H-индол-4-ил)морфолин-4-карбоксилат
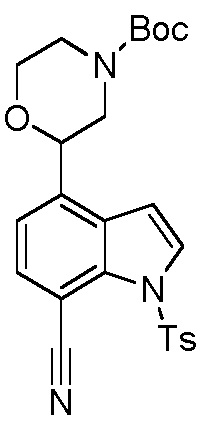
Стадия A: 4-бром-1-тозил-1H-индол-7-карбонитрил
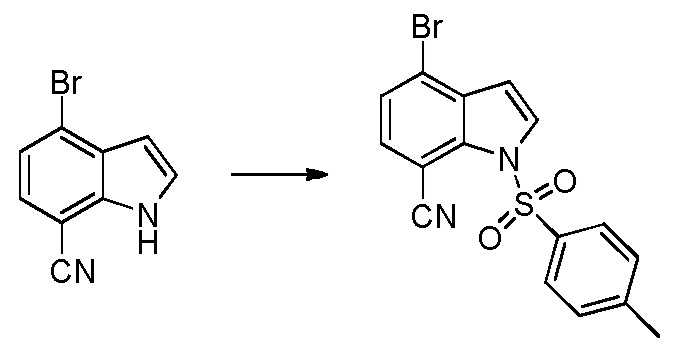
Круглодонную колбу загружали 4-бром-1H-индол-7-карбонитрилом (4,50 г, 20,4 ммоль) и THF (75 мг). Раствор охлаждали до приблизительно 0°C с последующим добавлением NaH (60% дисперсия в минеральном масле, 1,22 г, 30,5 ммоль). Раствор перемешивали при приблизительно 0°C в течение приблизительно 40 минут с последующим добавлением 4-метилбензол-1-сульфонилхлорид (4,66 г, 24,4 ммоль). Ледяную баню удаляли и смесь перемешивали при комнатной температуре в течение приблизительно 15 часов. Смесь выливали в воду со льдом (~150 мг) и продукт экстрагировали EtOAc (4×75 мг). Объединенные экстракты промывали водой (75 мг), сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением 4-бром-1-тозил-1H-индол-7-карбонитрила (5,74 г, 75%): 1H-ЯМР (400 МГц, DMSO-d6) δ 8,21 (д, J=3,9 Гц, 1H), 7,97-7,89 (м, 2H), 7,80-7,64 (м, 2H), 7,56-7,42 (м, 2H), 7,00 (д, J=3,8 Гц, 1H), 2,38 (с, 3H).
Стадия B: 1-тозил-4-винил-1H-индол-7-карбонитрил
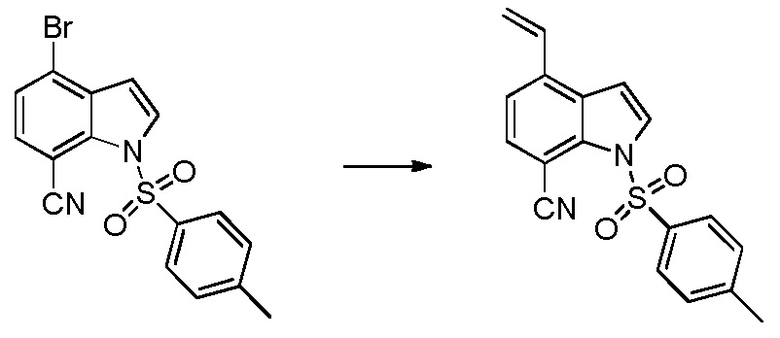
Круглодонную колбу загружали 4-бром-1-тозил-1H-индол-7-карбонитрилом (8,54 г, 22,8 ммоль), Na2CO3 (7,24 г, 68,3 ммоль) и PdCl2(dppf) (1,665 г, 2,276 ммоль) с последующим добавлением THF (70,2 мг): MeOH (10,03 мг):вода (10,03 мг). Реакционную смесь продували N2 в течение приблизительно 15 минут, добавляли 4,4,5,5-тетраметил-2-винил-1,3,2-диоксаборолан (4,63 мл, 27,3 ммоль) и смесь нагревали до приблизительно 70°C в течение приблизительно 5 часов. Смесь охлаждали до комнатной температуры и DCM (75 мг) и воду (50 мг) добавляли. Слои разделяли и водный слой экстрагировали DCM (50 мг). Объединенные экстракты сушили над MgSO4, фильтровали, концентрировали при пониженном давлении и пропускали через слой силикагеля, элюируя дихлорметаном, и концентрировали в вакууме. Остаток суспендировали в смеси Et2O/EtOAc, фильтровали и затем промывали осадок небольшим количеством EtOAc/Et2O. Полученный таким образом материал сушили в вакуумной печи с получением 1-тозил-4-винил-1H-индол-7-карбонитрила (5,62 г, 77%): ЖХ/МС (Таблица 1, способ as) Время удерживания = 2,57 мин; MS m/z: 323 (M+H)+.
Стадия C: 4-(Оксиран-2-ил)-1-тозил-1H-индол-7-карбонитрил
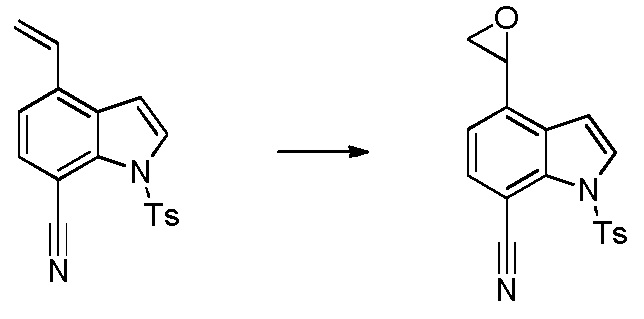
К суспензии 1-тозил-4-винил-1H-индол-7-карбонитрила (0,40 г, 1,241 ммоль) в диоксане (16 мг) и воде (8 мг) добавляли AcOH (0,0710 мл, 1,24 ммоль). Смесь охлаждали до приблизительно 0°C. NBS (0,243 г, 1,36 ммоль) добавляли в виде одной порции. Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение приблизительно 2 часов. NaOH (2М водный раствор, 8,0 мл, 16 ммоль) добавляли в виде одной порции. Образовавшееся твердое вещество собирали фильтрованием, промывали водой и сушили в вакуумной печи при приблизительно 60°C в течение приблизительно 16 часов с получением 4-(оксиран-2-ил)-1-тозил-1H-индол-7-карбонитрила (0,29 г, 68%): ЖХ/МС (Таблица 1, способ as) Время удерживания = 2,36 мин; MS m/z: 339 (M+H)+.
Стадия C: 4-(1-Гидрокси-2-((2-гидроксиэтил)амино)этил)-1-тозил-1H-индол-7-карбонитрил
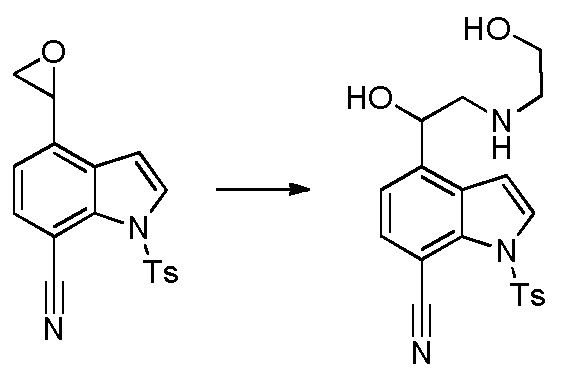
К суспензии 4-(оксиран-2-ил)-1-тозил-1H-индол-7-карбонитрила (0,285 г, 0,841 ммоль) в IPA (8 мг) добавляли TEA (0,586 мл, 4,21 ммоль) с последующим 2-аминоэтанолом (0,253 мл, 4,21 ммоль). Смесь нагревали при приблизительно 75°C в течение приблизительно 3 часов и концентрировали при пониженном давлении. Остаток разделяли между этилацетатом и водой. Смесь экстрагировали EtOAc (2×10 мг). Объединенные органические слои сушили над Na2SO4, фильтровали, концентрировали и сушили, используя вакуумный насос, с получением 4-(1-гидрокси-2-((2-гидроксиэтил)амино)этил)-1-тозил-1H-индол-7-карбонитрила (0,39 г, 94%): ЖХ/МС (Таблица 1, способ as) Время удерживания = 1,53 мин; MS m/z: 400 (M+H)+.
Стадия D: трет-Бутил (2-(7-циано-1-тозил-1H-индол-4-ил)-2-гидроксиэтил)(2-гидроксиэтил)карбамат
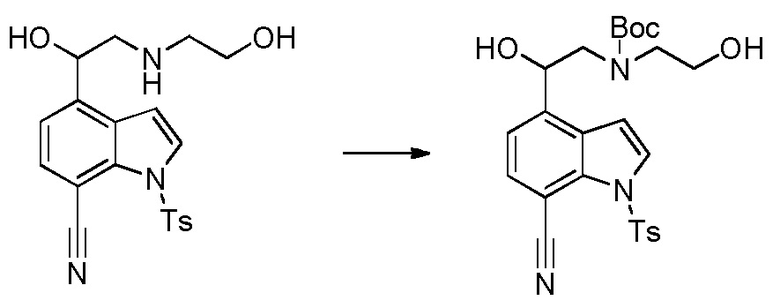
К раствору 4-(1-гидрокси-2-((2-гидроксиэтил)амино)этил)-1-тозил-1H-индол-7-карбонитрила (0,336 г, 0,673 ммоль) в EtOAc (3 мг) добавляли DIEA (0,176 мл, 1,01 ммоль) с последующим добавлением по каплям раствора ди-трет-бутил-дикарбоната (0,220 г, 1,01 ммоль) в EtOAc (1 мг) при комнатной температуре. THF (1 мг) добавляли к улучшающей растворение смеси и перемешивали при комнатной температуре в течение приблизительно 2 часов. Добавляли дополнительное количество DIEA (0,060 мл, 0,34 ммоль) и ди-трет-бутил-дикарбонат (0,073 г, 0,34 ммоль). Смесь перемешивали при комнатной температуре в течение приблизительно еще 2 часов. Растворитель удаляли при пониженном давлении и очищали с помощью флэш-хроматографии (25-50% EtOAc/гептан), затем с помощью ВЭЖХ (таблица 1, способ bd), с получением трет-бутил (2-(7-циано-1-тозил-1H-индол-4-ил)-2-гидроксиэтил)(2-гидроксиэтил)карбамата (0,25 г, 74%): ЖХ/МС (Таблица 1, способ as) Время удерживания = 2,22 мин; MS m/z: 500 (M+H)+.
Стадия E: трет-Бутил 2-(7-циано-1-тозил-1H-индол-4-ил)морфолин-4-карбоксилат
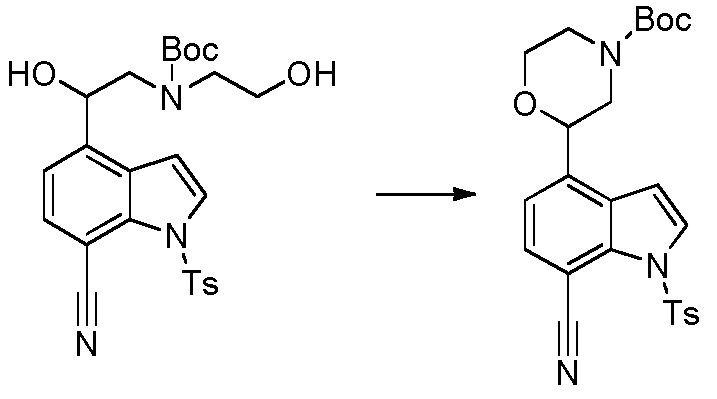
В сосуд загружали трет-бутил-(2-(7-циано-1-тозил-1H-индол-4-ил)-2-гидроксиэтил)(2-гидроксиэтил)карбамат (0,50 г, 1,0 ммоль) и PPh3 (0,315 г, 1,20 ммоль) в толуоле (10 мг) при приблизительно 0°C добавляли TEA (0,367 мл, 2,63 ммоль) с последующим добавлением DCAD (0,441 г, 1,20 ммоль). Раствор перемешивали при приблизительно 0°C в течение приблизительно 5 минут, а затем перемешивали при комнатной температуре в течение приблизительно 16 часов. Дополнительное количество PPh3 (0,131 г, 0,500 ммоль) и DCAD (0,184 г, 0,500 ммоль) добавляли при комнатной температуре и смесь перемешивали при приблизительно комнатной температуре в течение приблизительно 6 часов. Реакционную смесь фильтровали и фильтрат концентрировали и очищали флэш-хроматографией (0-30% EtOAc/гептан) с получением трет-бутил 2-(7-циано-1-тозил-1H-индол-4-ил)морфолин-4-карбоксилата (0,41 г, 84%): ЖХ/МС (Таблица 1, способ as) Время удерживания = 2,72 мин; MS m/z: 499 (M+H2O)+.
Получение № 44: 2-Йод-6,7-дигидро-4H-пиразоло[5,1-c][1,4]оксазин
Стадия A: 2-нитро-6,7-дигидро-4H-пиразоло[5,1-c][1,4]оксазин
Смесь (1-(2-бромэтил)-3-нитро-1H-пиразол-5-ил)метанол (4,0 г, 12 ммоль) [Princeton] в NMP (7,7 мг) нагревали при приблизительно 130°C в течение приблизительно 16 часов. Смесь разбавляли DCM и промывали водой и насыщенным солевым раствором. Органический слой сушили, концентрировали и очищали с помощью хроматографии на силикагеле (0-5% МеОН/DCM) с получением 2-нитро-6,7-дигидро-4H-пиразоло[5,1-c][1,4]оксазина (1 г, 49%): 1H-ЯМР (400 МГц, DMSO-d6) δ 6,88 (с, 1H), 4,83 (с, 2H), 4,24 (т, J=5,2 Гц, 2H), 4,13 (дд, J=5,9, 4,6 Гц, 2H).
Стадия B: 6,7-дигидро-4H-пиразоло[5,1-c][1,4]оксазин-2-амин
Сосуд загружали Pd/C (10% масс., 0,755 г, 0,709 ммоль) в атмосфере азота перед добавлением раствора 2-нитро-6,7-дигидро-4Н-пиразоло[5,1-с][1,4]оксазина (4,0 г, 24 ммоль) в EtOAc (59,1 мг) и MeOH (59,1 мг). Реакционную смесь перемешивали при комнатной температуре в течение приблизительно 16 часов. Реакционную смесь фильтровали через слой целита и фильтрат концентрировали при пониженном давлении с получением 6,7-дигидро-4H-пиразоло[5,1-c][1,4]оксазин-2-амина (3,2 г, 97%): ЖХ/МС (Таблица 1, способ as) Время удерживания = 0,61 мин; MS m/z: 140 (M+H)+.
Стадия C: 2-Йод-6,7-дигидро-4H-пиразоло[5,1-c][1,4]оксазин
В 50-мл круглодонную колбу загружали 6,7-дигидро-4H-пиразоло[5,1-c][1,4]оксазин-2-амин (1,5 г, 11 ммоль) и концентрированную HCl (2,43 мл, 29,6 ммоль). Смесь охлаждали до приблизительно 0°C. Добавляли раствор NaNO2 (0,707 г, 10,2 ммоль) в воде (10 мг) и реакционную смесь перемешивали в течение приблизительно 15 минут. Раствор KI (2,86 г, 17,3 ммоль) в воде (10 мг) осторожно добавляли и реакционную смесь перемешивали при приблизительно 0°C в течение приблизительно 1 часа и перемешивали при комнатной температуре в течение приблизительно 30 минут. Реакционную смесь разбавляли EtOAc (20 мг) и водой (20 мг), а затем отделяют от водного слоя. Раствор очищали с помощью хроматографии на силикагеле (0-50% EtOAc/гептан) с получением 2-йод-6,7-дигидро-4H-пиразоло[5,1-c][1,4]оксазина (0,996 г, 37%): ЖХ/МС (Таблица 1, способ as) Время удерживания = 1,58 мин; MS m/z: 251 (M+H)+.
Получение № 45: Метил 4-хлор-1-тозил-1H-пирроло[3,2-c]пиридин-7-карбоксилат
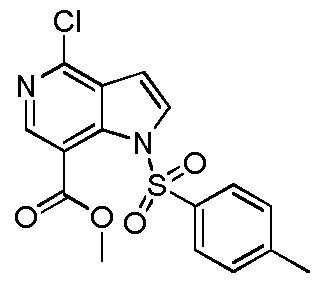
Стадия A: Метил 1-тозил-1H-пирроло[3,2-c]пиридин-7-карбоксилат

Круглодонную колбу загружали метил 1H-пирроло[3,2-c]пиридин-7-карбоксилатом (14 г, 79 ммоль) и THF (225 мг) [Pharmablock] и раствор охлаждали до приблизительно 5°C с последующим добавлением KHMDS (1 М в THF, 79 мл, 79 ммоль). Затем раствор перемешивали в течение приблизительно 1 часа с последующим добавлением раствора 4-метилбензол-1-сульфонилхлорида (15,2 г, 79,0 ммоль) в THF (25 мг). Смесь перемешивали в течение приблизительно 2 часов при приблизительно от 0 до 5°C с последующим добавлением насыщенного водного NH4Cl и DCM. Слои разделяли и органический раствор сушили над MgSO4, фильтровали, концентрировали при пониженном давлении и очищали с помощью хроматографии на силикагеле (0-50% EtOAc/DCM), получая метил 1-тозил-1H-пирроло[3,2-c]пиридин-7-карбоксилат (18,8 г, 72%): ЖХ/МС (Таблица 1, Способ as) Время удерживания = 2,10 мин; MS m/z: 331 (M+H)+.
Стадия B: 7-(Метоксикарбонил)-1-тозил-1H-пирроло[3,2-c]пиридин 5-оксид
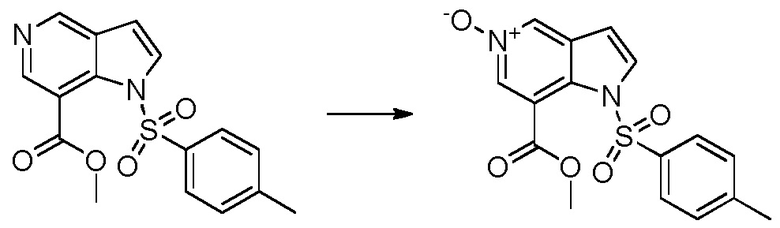
Круглодонную колбу загружали метил 1-тозил-1H-пирроло[3,2-c]пиридин-7-карбоксилатом (16,0 г, 48,4 ммоль) и EtOAc (150 мг). К реакционному раствору добавляли раствор 3-хлорбензопероксидной кислоты (14,2 г, 82 ммоль) в EtOAc (80 мг) и перемешивали при комнатной температуре в течение приблизительно 16 часов. К реакционной смеси добавляли насыщенный водный Na2CO3 (50 мг) и слои разделяли. Водный слой экстрагировали EtOAc (2×30 мг) и DCM (2×30 мг). Объединенные экстракты сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением густого масла, которое сушили, с помощью вакуумного насоса с получением 7-(метоксикарбонил)-1-тозил-1H-пирроло[3,2-c]пиридин-5-оксида (11,6 г, 69%): ЖХ/МС (Таблица 1, Способ as) Время удерживания = 1,73 мин; MS m/z: 347 (M+H)+.
Стадия C: Метил 4-хлор-1-тозил-1H-пирроло[3,2-c]пиридин-7-карбоксилат
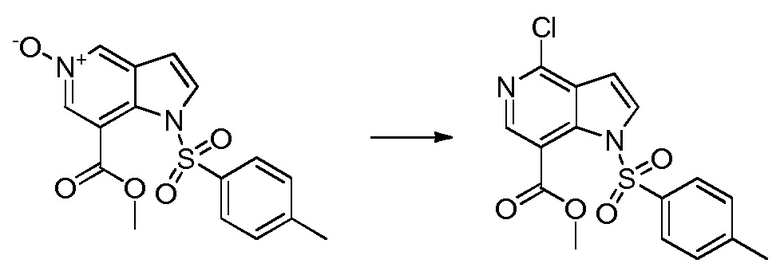
Круглодонную колбу загружали 7-(метоксикарбонил)-1-тозил-1H-пирроло[3,2-c]пиридин 5-оксидом (11,6 г, 33,5 ммоль) и PCl3 (26,5 мл, 285 ммоль) и нагревали до приблизительно 60°C в течение приблизительно 2 часов. Раствор охлаждали до комнатной температуры и медленно выливали в ледяную воду при перемешивании, и полученную смесь нейтрализовали добавлением насыщенного водного Na2CO3. Водную смесь экстрагировали EtOAc (3×40 мг) и объединенные экстракты сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением метил 4-хлор-1-тозил-1H-пирроло[3,2-c]пиридин-7-карбоксилата (8,47 г, 69%): ЖХ/МС (Таблица 1, способ as) Время удерживания = 2,46 мин; MS m/z: 365 (M+H)+.
Получение № 46: 7-Хлор-2-(1-метил-1H-пиразол-4-ил)тиазоло[5,4-c]пиридин-4-карбоксамид
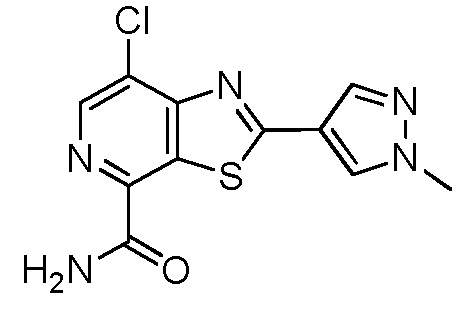
Стадия A: (E)-Этил 3-(2-бромтиазол-4-ил)акрилат
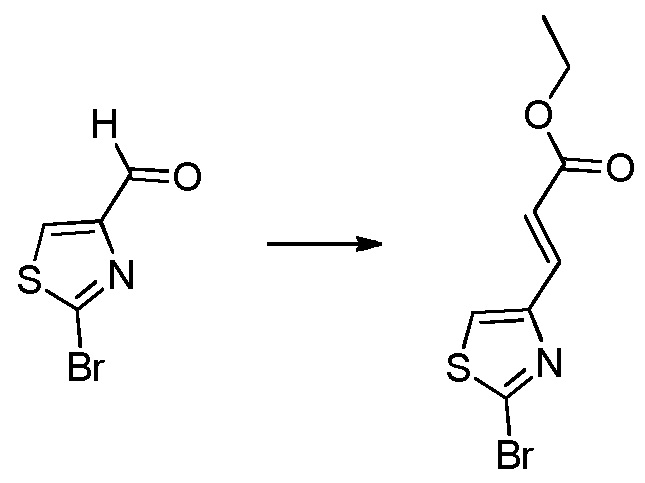
Круглодонную колбу, 1 л, загружали этил 2-(трифенилфосфоранилиден)ацетатом (37,2 г, 107 ммоль) в DCM (130 мг) с получением бесцветного раствора. Раствор охлаждали до приблизительно 0°C и добавляли раствор 2-бромтиазол-4-карбальдегида (20,5 г, 107 ммоль) [ArkPharm] в DCM (500 мг) по каплям через капельную воронку. Реакционную смесь медленно нагревали до комнатной температуры и перемешивали в течение приблизительно 2 часов, затем концентрировали при пониженном давлении. Смесь переносили в Et2O (300 мг) и перемешивали при приблизительно 40°C в течение приблизительно 30 минут. Затем охлаждали, фильтровали и промывали Et2O (50 мг). Осадок отбрасывали и фильтрат концентрировали до половины объема. Образовавшийся осадок собирали с помощью фильтрации с получением первой партии продукта. Фильтрат концентрировали и добавляли Et2O (60 мг), смесь перемешивали при комнатной температуре в течение приблизительно 20 минут и вновь образованный осадок снова фильтровали для сбора второй партии продукта. Фильтрат из данной партии концентрировали при пониженном давлении и очищали хроматографией на силикагеле (0-10% EtOAc/гептан). Полученный таким образом продукт перекристаллизовывали из Et2O, получая третью и последнюю партию продукта. Все партии объединяли, получая белое кристаллическое вещество, (E)-этил 3-(2-бромтиазол-4-ил)акрилат (20,1 г, 72%): ЖХ/МС (Таблица 1, способ as) Время удерживания = 2,26 мин; MS m/z: 262, 264 (M+H)+.
Стадия B: (E)-Этил 3-(2-(1-метил-1H-пиразол-3-ил)тиазол-4-ил)акрилат
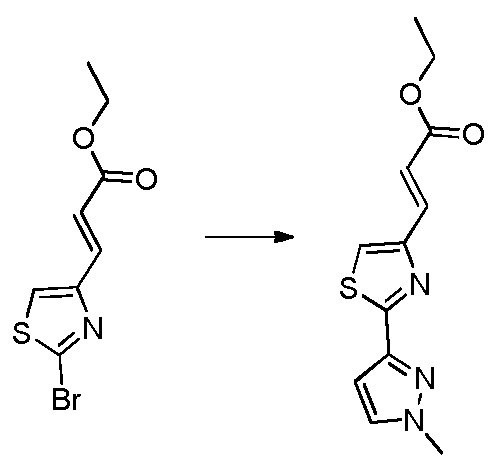
Круглодонную колбу, 500 мл, загружали 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразолом (20,7 г, 100 ммоль), (E)-этил 3-(2-бромтиазол-4-ил)акрилатом (20,1 г, 77,0 ммоль), Na2CO3 (24,4 г, 230 ммоль), PdCl2(dppf) (5,61 г, 7,67 ммоль) и (E)-этил 3-(2-бромтиазол-4-ил)акрилатом (20,1 г, 77,0 ммоль). К твердой смеси добавляли THF (150 мг): MeOH (21,00 мг): воду (21 мг) и суспензию дегазировали и продували N2 в течение приблизительно 20 минут. Реакционную смесь нагревали при приблизительно 75°C в течение приблизительно 15 часов. Реакционную смесь фильтровали и промывали EtOAc (100 мг) и фильтрат промывали водой (70 мг). Водный слой экстрагировали EtOAc (2×70 мг) и объединенные органические слои сушили над MgSO4, фильтровали и концентрировали. К остатку добавляли DCM (50 мг) и гептан (150 мг). Всю суспензию фильтровали, промывали ацетоном и изопропанолом и сушили в вакуумной печи с получением первой партии продукта. Фильтрат концентрировали, растворяли в DCM (40 мг) и пропускали через слой силикагеля (элюент: 50% EtOAc/гептан). Фильтрат концентрировали и кипятили с обратным холодильником в ацетоне (35 мг) и охлаждали. Осадок отфильтровывали, промывали изопропанолом, объединяли с первой партией и сушили в вакуумной печи при приблизительно 70°C в течение приблизительно 16 часов, с получением (E)-этил 3-(2-(1-метил-1H-пиразол-3-ил)тиазол-4-ил)акрилата (15,2 г, 75%): ЖХ/МС (Таблица 1, способ as) Время удерживания = 1,94 мин; MS m/z: 264(M+H)+.
Стадия C: (E)-3-(2-(1-метил-1H-пиразол-4-ил)тиазол-4-ил)акриловая кислота
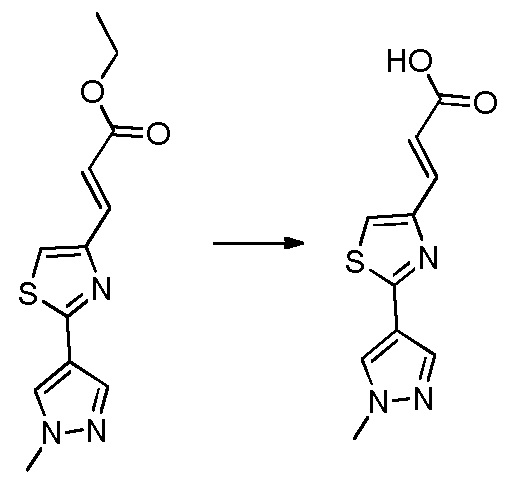
В 20-мл реакционный сосуд добавляли (E)-этил 3-(2-(1-метил-1H-пиразол-4-ил)тиазол-4-ил)акрилат (15,2 г, 57,7 ммоль) и LiOH (4,15 г, 173 ммоль) в MeOH (60 мг):вода (12 мг). Реакционную смесь перемешивали при приблизительно 40°C в течение 2 часов. Реакционную смесь концентрировали, разбавляли водой (50 мг) и промывали DCM (50×3 мг). Водный слой подкисляли 1 н HCl до прекращения образования осадка. Осадок собирали путем фильтрации и сушили в вакуумной печи при приблизительно 60°C в течение приблизительно 16 часов с получением (E)-3-(2-(1-метил-1H-пиразол-4-ил)тиазол-4-ил)акриловой кислоты (12,3 г, 91%): 1H-ЯМР (400 МГц, DMSO-d6) δ 12,42 (с, 1H), 8,38 (с, 1H), 7,94 (с, 2H), 7,56 (с, 1H), 6,56 (с, 1H), 3,90 (с, 3H).
Стадия D: (E)-3-(2-(1-метил-1H-пиразол-4-ил)тиазол-4-ил)акрилоил азид
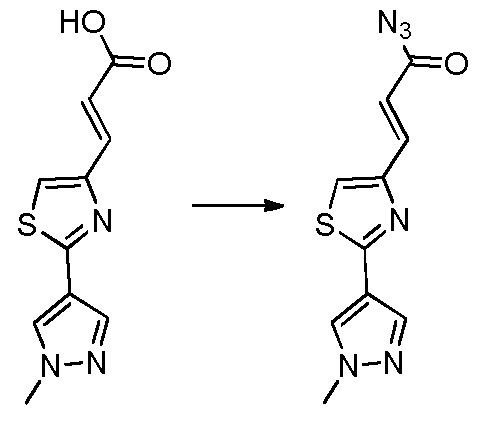
К суспензии (E)-3-(2-(1-метил-1H-пиразол-4-ил)тиазол-4-ил)акриловой кислоты (11,2 г, 47,4 ммоль) в ацетоне (170 мг) добавляли TEA (6,61 мл, 47,4 ммоль) и смесь охлаждали на бане со льдом. Изобутилхлорформиат (6,22 мл, 47,4 ммоль) добавляли по каплям. После приблизительно 3,5 часов раствор NaN3 (3,85 г, 59,2 ммоль) в воде (15 мг) осторожно добавляли и реакционную смесь перемешивали в течение приблизительно 3 часов при температуре приблизительно 0°C. Реакционную смесь выливали на лед и перемешивали в течение приблизительно 5 минут, фильтровали и промывали водой (50 мг). Осадок сушили в вакуумной печи при приблизительно 60°C в течение приблизительно 16 часов с получением (E)-3-(2-(1-метил-1H-пиразол-4-ил)тиазол-4-ил)акрилоил азида (9,6 г, 78%): ЖХ/МС (Таблица 1, способ as) Время удерживания = 1,91 мин; MS m/z: 261(M+H)+.
Стадия E: 2-(1-метил-1H-пиразол-4-ил)тиазоло[5,4-c]пиридин-4(5H)-он
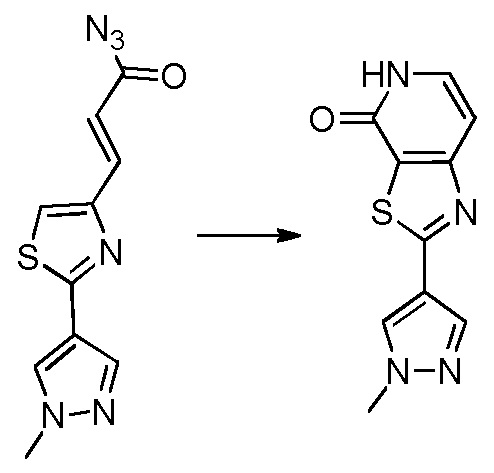
В 3-х горлую круглодонную колбу на 250 мл загружали трибутиламин (6,10 мл, 25,6 ммоль) в дифениловом эфире (30 мг). Реакционную смесь нагревали до приблизительно 190°C и добавляли осторожно раствор (E)-3-(2-(1-метил-1H-пиразол-4-ил)тиазол-4-ил)акрилоил азида (5,60 г, 21,5 ммоль) в дифениловом эфире (80 мг) и и реакционную смесь перемешивали в течение приблизительно 5 часов при приблизительно 190°C. Реакционную смесь охлаждали и выливали в петролейный эфир (300 мг) и перемешивали в течение приблизительно 5 минут и фильтровали. Осадок сушили в вакуумной печи при приблизительно 70°C в течение приблизительно 30 минут. Материал суспендировали в Et2O (100 мг) и нагревали при приблизительно 50°C в течение приблизительно 20 минут. Затем материал фильтровали и промывали холодным Et2O. Осадок сушили в вакуумной печи при приблизительно 70°C в течение приблизительно 10 часов с получением 2-(1-метил-1H-пиразол-4-ил)тиазоло[5,4-c]пиридин-4(5H)-она (3,8 г, 76 %): ЖХ/МС (Таблица 1, способ as) Время удерживания = 1,13 мин MS m/z: 233 (M+H)+.
Стадия F: 7-Хлор-2-(1-метил-1H-пиразол-4-ил)тиазоло[5,4-c]пиридин-4(5H)-он
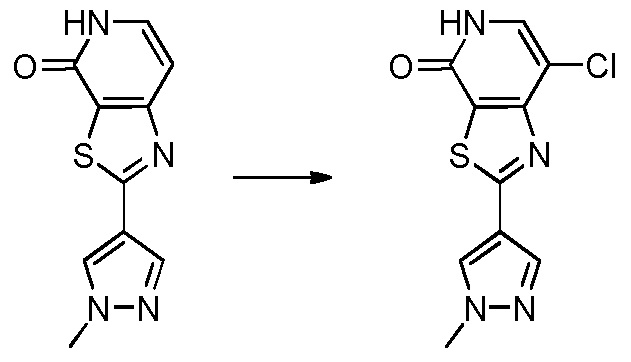
В круглодонную колбу на 250 мл добавляли 2-(1-метил-1H-пиразол-4-ил)тиазоло[5,4-c]пиридин-4(5H)-он (3,7 г, 16 ммоль) в MeCN (80 мг) с получением суспензии. Реакционную смесь нагревали при перемешивании до приблизительно 80°C. Раствор NCS (3,19 г, 23,9 ммоль)) в MeCN (25 мг) добавляли по каплям через капельную воронку, и реакционную смесь перемешивали в течение приблизительно 5 часов при приблизительно 80°C. Смесь разбавляли водой (100 мг), фильтровали и промывали водой (40 мг). Осадок сушили в вакуумной печи при приблизительно 70°C в течение приблизительно 16 часов с получением 7-хлор-2-(1-метил-1H-пиразол-4-ил)тиазоло[5,4-c]пиридин-4(5H)-она (3,55 г, 84%): ЖХ/МС (Таблица 1, способ as) Время удерживания = 1,27 мин; MS m/z: 267 (M+H)+.
Стадия G: 4-бром-7-хлор-2-(1-метил-1H-пиразол-4-ил)тиазоло[5,4-c]пиридин
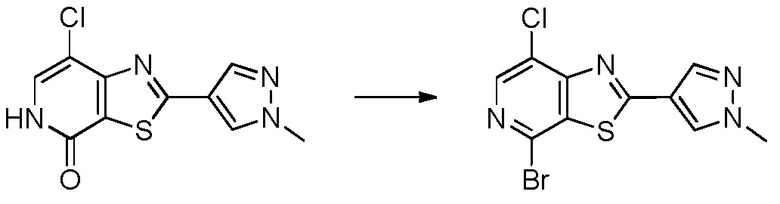
В 3-х горлой круглодонной колбе на 100 мл нагревали смесь 7-хлор-2-(1-метил-1H-пиразол-4-ил)тиазоло[5,4-c]пиридин-4(5H)-она (1,30 г, 4,87 ммоль) и POBr3 (3,91 г, 13,6 ммоль) до приблизительно 70°C в течение приблизительно 10 минут, затем нагревали до приблизительно 120°C в течение приблизительно 45 минут. Добавляли дополнительное количество POBr3 (1,40 г, 4,87 ммоль) и нагревали в течение приблизительно 50 минут. Смесь охлаждали на бане со льдом и к ней осторожно добавляли смесь измельченного льда и воды (40 мг). Смесь перемешивали при комнатной температуре в течение приблизительно 16 часов. К суспензии добавляли DCM (60 мг) и перемешивали в течение приблизительно 30 минут, затем фильтровали для удаления некоторых черных твердых частиц. Слой DCM отделяли и водный слой экстрагировали DCM (2×20 мг). Объединенные органические слои сушили над MgSO4, фильтровали и адсорбировали на силикагеле (4-6 г). Материал очищали с помощью хроматографии на силикагеле (1-3% EtOAc/гептан) с получением 4-бром-7-хлор-2-(1-метил-1H-пиразол-4-ил)тиазоло[5,4-c]пиридина (0,85 г, 53%): ЖХ/МС (Таблица 1, способ as) Время удерживания = 2,20 мин; MS m/z: 331 (M+H)+.
Стадия H: 7-Хлор-2-(1-метил-1H-пиразол-4-ил)тиазоло[5,4-c]пиридин-4-карбонитрил

В 50-мл круглодонную колбу добавляли 4-бром-7-хлор-2-(1-метил-1H-пиразол-4-ил)тиазоло[5,4-c]пиридин (0,770 г, 2,13 ммоль), Zn(CN)2 (0,168 г, 1,44 ммоль) и Pd(PPh3)4 (0,174 г, 0,151 ммоль) в DMF (10 мг). Колбу дегазировали и продували азотом и затем нагревали термически в атмосфере азота при приблизительно от 110°C до 120°C в течение приблизительно 50 минут. Реакционную смесь разбавляли водой (25 мг) и перемешивали в течение приблизительно 5 минут, фильтровали и промывали водой (6 мг). Осадок сушили в вакуумной печи при приблизительно 70°C в течение приблизительно 16 часов с получением сырого 7-хлор-2-(1-метил-1H-пиразол-4-ил)тиазоло[5,4-c]пиридин-4-карбонитрила (0,67 г, 98%). В колбу, загруженную NaOH (1 М водного раствора, 7,29 мл, 7,29 ммоль) в МеОН (12 мг) добавляли H2O2 (30% водный раствор, 1,24 мл, 12,2 ммоль). Этот раствор добавляли в колбу, содержащую 7-хлор-2-(1-метил-1H-пиразол-4-ил)тиазоло[5,4-c]пиридин-4-карбонитрил (0,670 г, 2,43 ммоль) и перемешивали при приблизительно 30°C в течение приблизительно 5 минут. Реакционную смесь разбавляли водой (51 мг) и перемешивали при комнатной температуре в течение приблизительно 5 минут и фильтровали. Осадок растирали в порошок с Et2O, фильтровали и сушили в вакуумной печи в течение приблизительно 16 часов с получением 7-хлор-2-(1-метил-1H-пиразол-4-ил)тиазоло[5,4-c]пиридин-4-карбоксамида (0,597 г, 84%): ЖХ/МС (Таблица 1, способ as) Время удерживания = 1,58 мин; MS m/z: 294(M+H)+.
Получение № 47: Метил 4-((1R,3R)-3-((трет-бутоксикарбонил)амино)циклопентил)-1-тозил-1H-индол-7-карбоксилат

Стадия A: Метил 4-(3-оксоциклопент-1-ен-1-ил)-1-тозил-1H-индол-7-карбоксилат
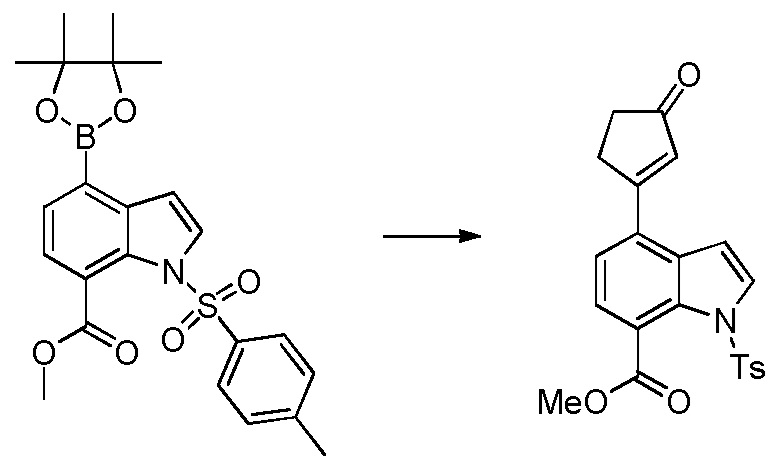
В колбу загружали метил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1-тозил-1H-индол-7-карбоксилат (1,74 г, 3,82 ммоль, полученный с использованием А из Получения № 1, стадия С, с бис(пинаколато)дибороном) в 2-метил-THF (18,64 мг) и воду (12,43 мг). Смесь охлаждали до приблизительно 10°C на бане с холодной водой. Добавляли NaIO4 (1,23 г, 5,73 ммоль), реакционную смесь перемешивали в течение приблизительно 30 минут и добавляли по каплям водный 1 М HCl (8,41 мл, 8,41 ммоль). Смесь перемешивали при комнатной температуре в течение приблизительно 16 часов. Добавляли дополнительное количество 2-метил-THF (50 мг), водный слой разделяли и органический слой промывали 10%-ным водным Na2S2O3 (2×30 мг), насыщенным водным NaHCO3 (30 мг) и насыщенным солевым раствором (20 мг). Органический слой сушили над Na2SO4, фильтровали и концентрировали с получением сырой (7-(метоксикарбонил)-1-тозил-1H-индол-4-ил)бороновой кислоты. В круглодонную колбу на 100 мл добавляли сырую (7-(метоксикарбонил)-1-тозил-1H-индол-4-ил)бороновую кислоту (1,59 г, 4,26 ммоль) в диоксане (17 мг). Добавляли раствор Cs2CO3 (3,47 г, 10,7 ммоль) в воде (4,26 мг), смесь дегазировали азотом с последующим добавлением PdCl2(PPh3)2 (0,209 г, 0,298 ммоль) и 3-бромциклопент-2-енона (1,4 мл, 12,8 ммоль) в инертной атмосфере. Смесь нагревали при приблизительно 80°C в течение приблизительно 3 часов, затем охлаждали до комнатной температуры и добавляли DCM (100 мг) и воду (50 мг). Слои разделяли и водный слой экстрагировали DCM (2×50 мг). Объединенные органические слои сушили над MgSO4. Растворитель удаляли в вакууме и остаток очищали с помощью хроматографии на силикагеле (0-60% EtOAc/гептан) с получением метил 4-(3-оксоциклопент-1-ен-1-ил)-1-тозил-1H-индол-7-карбоксилата (1,2 г, 69%): 1H-ЯМР (400 МГц, DMSO-d6) δ 7,92 (д, J=3,9 Гц, 1H), 7,71 (д, J=7,9 Гц, 1H), 7,67-7,62 (м, 2H), 7,58 (д, J=7,9 Гц, 1H), 7,39-7,31 (м, 2H), 7,23 (д, J=3,9 Гц, 1H), 6,67 (т, J=1,8 Гц, 1H), 3,83 (с, 3H), 3,12 (дт, J=6,9, 1,9 Гц, 2H), 2,47 (дд, J=4,9, 2,5 Гц, 2H), 2,33 (с, 3H).
Стадия B: (R)-метил 4-(3-оксоциклопентил)-1-тозил-1H-индол-7-карбоксилат
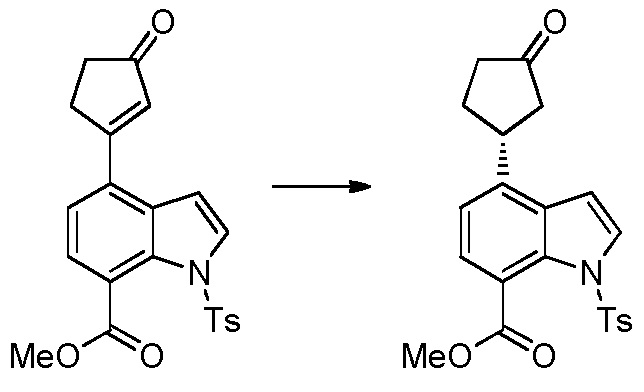
В реакционный сосуд на 40 мл, (2S,5S)-5-бензил-3-метил-2-(5-метилфуран-2-ил)имидазолидин-4-он (0,190 г, 0,703 ммоль) и метил 4-(3-оксоциклопент-1-ен-1-ил)-1-тозил-1H-индол-7-карбоксилат (3,05 г, 7,45 ммоль) в THF (5,67 мг) добавляли. Смесь охлаждали до приблизительно 0°C и дегазировали азотом. Ди-трет-бутил 2,6-диметил-1,4-дигидропиридин-3,5-дикарбоксилат (1,05 г, 3,40 ммоль) и трихлоруксусную кислоту (0,071 мл, 0,70 ммоль) добавляли в атмосфере инертного газа. Реакционную смесь перемешивали при приблизительно 4°C в течение 16 часов. Добавляли дополнительное количество ди-трет-бутил 2,6-диметил-1,4-дигидропиридин-3,5-дикарбоксилата (0,420 г, 1,36 ммоль), и реакционную смесь перемешивали при охлаждении в течение приблизительно 72 часов. Сырой продукт адсорбировали на силикагеле и очищали с помощью хроматографии на силикагеле (0-45% EtAOc/гептан) с получением (R)-метил 4-(3-оксоциклопентил)-1-тозил-1H-индол-7-карбоксилата (1 г, 79%). 1H-ЯМР (400 МГц, CDCl3-d) δ 7,67-7,58 (м, 2H), 7,58-7,45 (м, 2H), 7,23-7,10 (м, 3H), 6,75 (д, J=4,2 Гц, 1H), 3,91 (с, 3H), 3,73 (тдд, J=10,1, 7,6, 6,0 Гц, 1H), 2,73-2,61 (м, 1H), 2,51-2,24 (м, 7H), 2,16-1,98 (м, 1H).
Стадия C: Метил 4-((1R,3S)-3-гидроксициклопентил)-1-тозил-1H-индол-7-карбоксилат
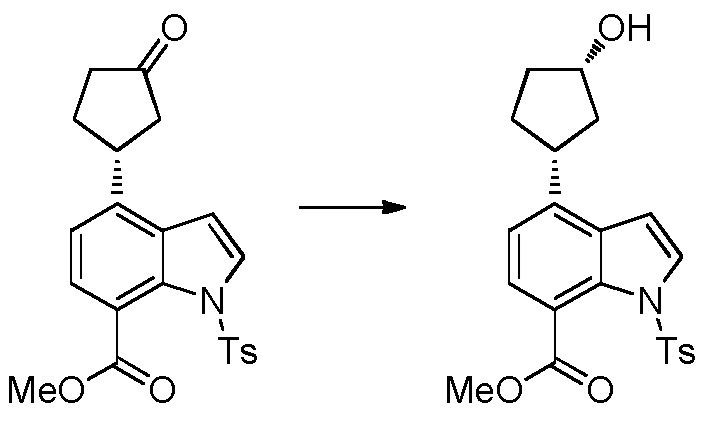
В круглодонную колбу 200 мл, добавляли (R)-метил 4-(3-оксоциклопентил)-1-тозил-1H-индол-7-карбоксилат (1,60 г, 3,89 ммоль) в THF (32,4 мг). Раствор охлаждали до приблизительно -78°C. L-Селектрид (7,78 мл, 7,78 ммоль) добавляли по каплям в течение приблизительно 20 минут и смесь перемешивали в течение приблизительно 16 часов. Реакционную смесь охлаждали на бане со льдом, добавляли насыщенный водный NH4Cl (60 мг) по каплям, затем добавляли EtOAc (100 мг) и воду (20 мг). Органический слой отделяли, промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали, концентрировали и очищали с помощью хроматографии на силикагеле (0-65% EtOAc/гептан). Полученный остаток очищали с помощью хиральной хроматографии (таблица 2, способ 19), с получением метил 4-((1R,3S)-3-гидроксициклопентил)-1-тозил-1H-индол-7-карбоксилата ( 0,36 г, 22%): ЖХ/МС (Таблица 1, способ a) Время удерживания = 2,21 мин; MS m/z: 431(M+H2O)+.
Стадия D: Метил 4-((1R,3R)-3-((трет-бутоксикарбонил)амино)циклопентил)-1-тозил-1H-индол-7-карбоксилат
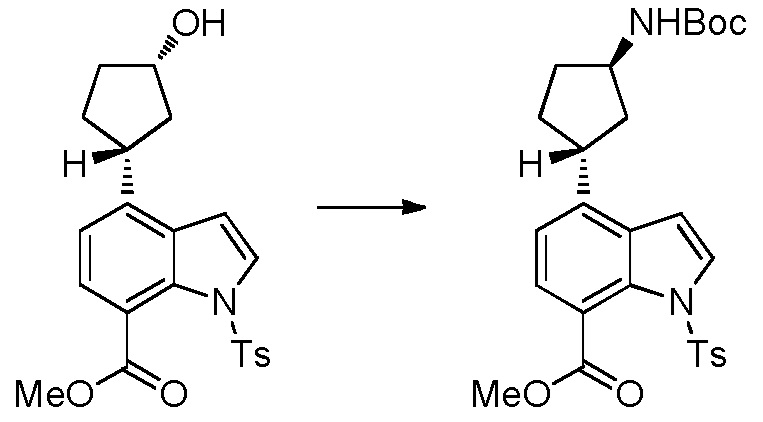
В 40 мл реакционный сосуд, добавляли метил 4-((1R,3S)-3-гидроксициклопентил)-1-тозил-1H-индол-7-карбоксилат (0,35 г, 0,85 ммоль) и PPh3 (0,266 г, 1,02 ммоль) в THF (3,4 мг). Раствор охлаждали до приблизительно 10°C, добавляли DIEA (0,148 мл, 0,846 ммоль) с последующим добавлением по каплям DIAD (0197 мл, 1,02 ммоль) и реакционную смесь перемешивали в течение приблизительно 30 минут. Дифенилфосфоразидат (0,219 мл, 1,02 ммоль) добавляли по каплям и перемешивали при комнатной температуре в течение приблизительно 3 часов. Раствор PPh3 (0,289 г, 1,10 ммоль) в THF (0,6 мг) добавляли по каплям и смесь перемешивали в течение приблизительно 18 часов. Добавляли воду (0,183 мл, 10,2 ммоль) и смесь нагревали при приблизительно 45°C в течение приблизительно 72 часов. К реакционной смеси добавляли DCM (10,7 мл, 166 ммоль) и раствор гидрофосфата калия (0,737 г, 4,23 ммоль) в воде (2,14 мл, 119 ммоль). Раствор ди-трет-бутил дикарбоната (0,393 мл, 1,69 ммоль) в DCM (2,14 мл, 33,2 ммоль) добавляли по каплям и перемешивали при комнатной температуре в течение приблизительно 1 часа. Добавляли насыщенный солевой раствор (2 мг), органический слой отделяли и промывали насыщенным солевым раствором (3 мг), сушили над Na2SO4, фильтровали, концентрировали и очищали с помощью хроматографии на силикагеле (0-40% EtOAc/гептан) с получением метил 4-((1R,3R)-3-((трет-бутоксикарбонил)амино)циклопентил)-1-тозил-1H-индол-7-карбоксилата (0,396 г, 59%): ЖХ/МС (Таблица 1, способ a) Время удерживания = 2,72 мин; MS m/z: 530 (M+H2O)+.
Получение № 48: трет-Бутил 3-((7-карбамоил-2-(5-(морфолинометил)пиридин-2-ил)-1H-индол-4-ил)(метил)амино)азетидин-1-карбоксилат
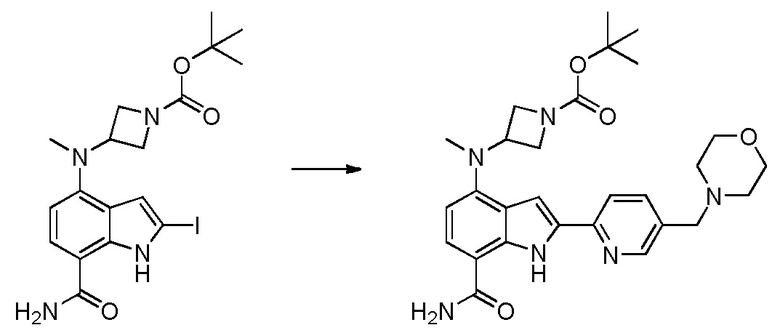
К смеси 4-((6-бромпиридин-3-ил)метил)морфолина (0,300 г, 1,17 ммоль) в THF (5 мл) добавляли н-BuLi (1,17 мл, 2,92 ммоль). Смесь перемешивали при приблизительно -78°C в течение приблизительно 1 часа, а затем медленно добавляли трибутилхлорстаннан (0,949 г, 2,92 ммоль). Смеси давали нагреться до комнатной температуры в течение приблизительно 1 часа, и добавляли насыщенный раствор NH4Cl. Смесь экстрагировали EtOAc и объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением сырого 4-((6-(трибутилстаннил)пиридин-3-ил)метил)морфолина. Раствор, содержащий трет-бутил 3-((7-карбамоил-2-йод-1H-индол-4-ил)(метил)амино)азетидин-1-карбоксилат (0,300 г, 0,638 ммоль, получение № 40) в DMF (2 мл) обрабатывали LiCl (0,270 г, 6,38 ммоль), аддуктом PdCl2(dppf)-CH2Cl2 (0,156 г, 0,191 ммоль) и 4-((6-(трибутилстаннил)пиридин-3-ил)метил)морфолином (0,894 г, 1,91 ммоль). Смесь нагревали при приблизительно 100°C в течение приблизительно 16 часов, охлаждали, фильтровали через целит и разделяли между EtOAc и водой. Органическую фазу промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали с помощью хроматографии на силикагеле (0-5% MeOH/DCM) с получением трет-бутил 3-((7-карбамоил-2-(5-(морфолинометил)пиридин-2-ил)-1H-индол-4-ил)(метил)амино)азетидин-1-карбоксилата (0,172 г, 11%): LCMS (Таблица 1, способ av) Время удерживания = 1,24 мин; MS m/z: 521 (M+H)+.
Получение № 49: трет-Бутил 6-(7-карбамоил-1H-пирроло[2,3-c]пиридин-4-ил)-2,3-дигидро-1,4-оксазепин-4(7H)-карбоксилат
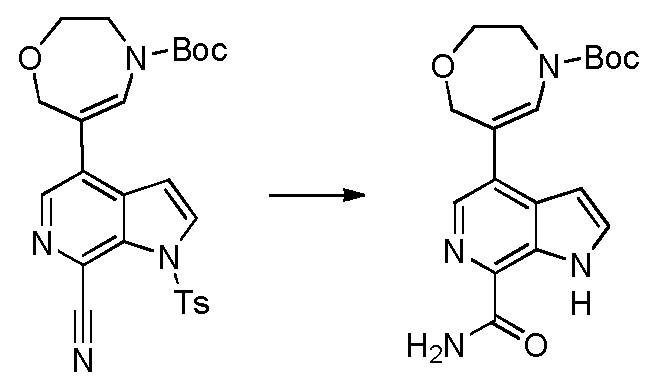
К раствору трет-бутил 6-(7-циано-1-тозил-1H-пирроло[2,3-c]пиридин-4-ил)-2,3-дигидро-1,4-оксазепин-4(7H)-карбоксилата (0,973 г, 1,97 ммоль, полученный с использованием AG из трет-бутил 6-(((трифторметил)сульфонил)окси)-2,3-дигидро-1,4-оксазепин-4(7H)-карбоксилата (Получение № W.1) с 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) и Получение № AH.1) в EtOH (3,93 мг) при приблизительно 0°C добавляли NaOH (1 н водный раствор, 7,87 мл, 7,87 ммоль) с последующим добавлением H2O2 (30% водный раствор, 1,12 мл, 9,84 ммоль). После приблизительно 10 минут баню со льдом удаляли. После приблизительно 1 часа добавляли дополнительное количество NaOH (1 н водный раствор, 7 мл, 7 ммоль) и H2O2 (30% водный раствор, 1,00 мл, 8,82 ммоль) и DCM (3 мг). Реакционную смесь оставляли перемешиваться в течение приблизительно 1 часа и концентрировали до приблизительно 15 мл и разбавляли водой (10 мг) и DCM (20 мг). Суспензию фильтровали для удаления любых твердых веществ. Слой DCM отделяли, сушили над MgSO4, фильтровали, концентрировали и очищали с помощью хроматографии на силикагеле с получением трет-бутил 6-(7-карбамоил-1H-пирроло[2,3-c]пиридин-4-ил)-2,3-дигидро-1,4-оксазепин-4(7H)-карбоксилата (0,138 г, 20%): ЖХ/МС (Таблица 1, способ as) Время удерживания = 1,90 мин; MS m/z: 359 (M+H)+.
Общий способ A: Реакция Сузуки арил- или гетероарилгалогенида с арил- или гетероарилбороновой кислотой или боронатом
К смеси арилгалогенида (предпочтительно, 1 эквивалент), бороновой кислоты или боронатного эфира (1-2 эквивалента, предпочтительно 1,1 эквивалент) и неорганического основания (например, KF, Na2CO3, K2CO3 или Cs2CO3, предпочтительно, Na2CO3 или Cs2CO3) (1,1-16 эквивалентов, предпочтительно, 2 эквивалента) в растворителе (например, THF, DME, DMF, 1,4-диоксан, 1,4-диоксан/вода, DME/вода, 1,4-диоксан/вода, толуол/EtOH/вода, 1,4-диоксан/EtOH/вода или THF/MeOH/вода, предпочтительно, THF/MeOH/вода, 1,4-диоксан/вода, DME/вода или 1,4-диоксан/EtOH/вода) добавляли палладиевый катализатор (например, Pd(OAc)2, Pd2dba3, Pd(PPh3)4, бис(ацетато)трифенилфосфинпалладия(II), полимер-связанный FibreCat™ 1032, SiliaCat DPP-Pd, PdCl2(dppf), (1,1'-бис(дифенилфосфино)ферроцен)дихлорпалладий(II) или Pd(PPh3)2Cl2; предпочтительно, PdCl2(dppf), (1,1'-бис(дифенилфосфино)ферроцен)дихлорпалладий(II) или SiliaCat DPP-Pd 0,01-0,20 эквивалент, предпочтительно, 0,1 эквивалент) и лиганд (например, трициклогексилфосфин, три-трет-бутил-фосфин; предпочтительно, нет или трициклогексилфосфин; 0,01-1,0 эквивалент, предпочтительно, 0,16 эквивалентов) добавляли необязательно. Смесь нагревали при температуре приблизительно от 40 до 120°C (предпочтительно, приблизительно 70-85°C) в течение приблизительно от 1 до 48 часов (предпочтительно, приблизительно 24 часа) термически, или при температуре приблизительно 100-200°C (предпочтительно, приблизительно 120-150°C) в течение приблизительно 5-60 минут (предпочтительно, приблизительно 20-45 минут) в микроволновой печи (предпочтительно, время вывода установки в рабочий режим 5 минут, максимальная мощность 300 Вт, максимальное давление 250 фунт на кв.дюйм). Смесь оставляли охлаждаться до комнатной температуры и обрабатывали с помощью одного из следующих способов. Способ 1. Для реакций, содержащих воду, смесь может быть разбавлена органическим растворителем (например, DCM или EtOAc). Слои разделяли, органический раствор необязательно промывали водой и/или насыщенным солевым раствором, сушили над безводным MgSO4 или Na2SO4, фильтровали и растворитель удаляли при пониженном давлении с получением желаемого соединения. Способ 2. Смесь концентрировали при пониженном давлении. Способ 3. Катализатор удаляли фильтрованием и фильтрат концентрировали при пониженном давлении.
Иллюстрация общего способа A
Получение № A.1: 4-(3-Аминофенил)-1H-индол-7-карбоксамид
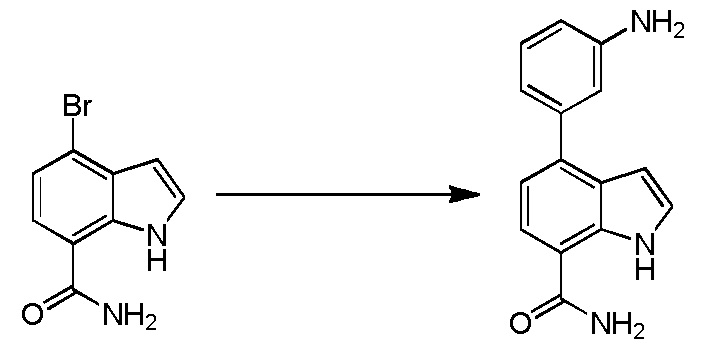
В сосуд загружали 4-бром-1H-индол-7-карбоксамид (2,08 г, 8,70 ммоль, Получение № 2), 3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилин (2,10 г, 9,57 ммоль), карбонат натрия (2,77 г, 26,1 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) (0,637 г, 0,870 ммоль) и продували азотом. Добавляли смесь THF (71,4 мг), MeOH (10 мг) и воды (10 мг) и реакционную смесь перемешивали при приблизительно 70°C в течение приблизительно 24 часов. Смесь фильтровали через целит, растворитель удаляли при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле, элюируя MeOH/DCM (0-10%) с получением твердого вещества. Твердое вещество растирали с эфиром с получением 4-(3-аминофенил)-1H-индол-7-карбоксамида (1,37 г, 63%): ЖХ/МС (Таблица 1, способ f) Время удерживания = 0,76 мин; MS m/z: 293(M+MeCN+H)+.
Примеры, полученные из N-(2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)акриламида (полученные с использованием E из 2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина и акрилоилхлорида) с использованием общего способа A
(Таблица 1, Способ)
(полученный с использованием A из Получения № 1 и пинаколового эфира 3,5-диметилизоксазол-4-бороновой кислоты)
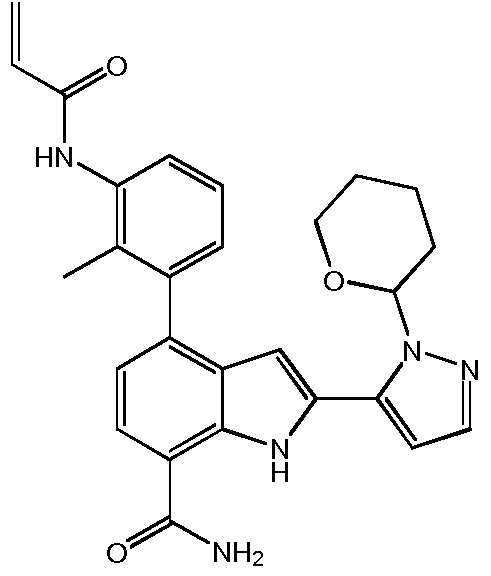
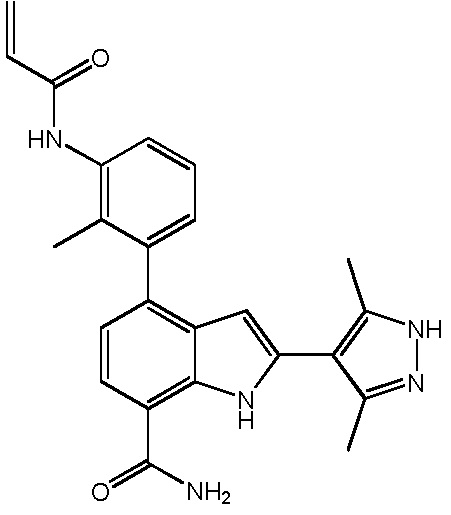

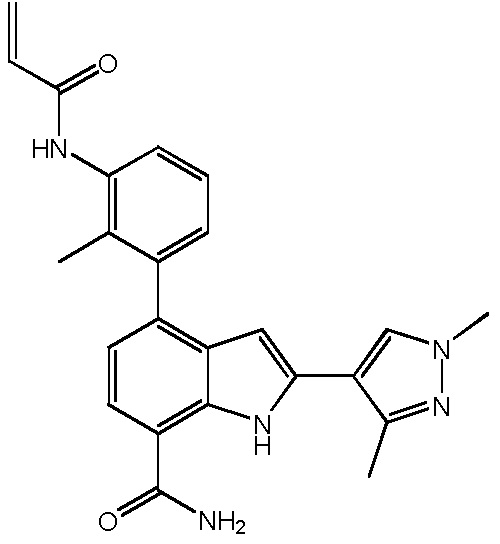
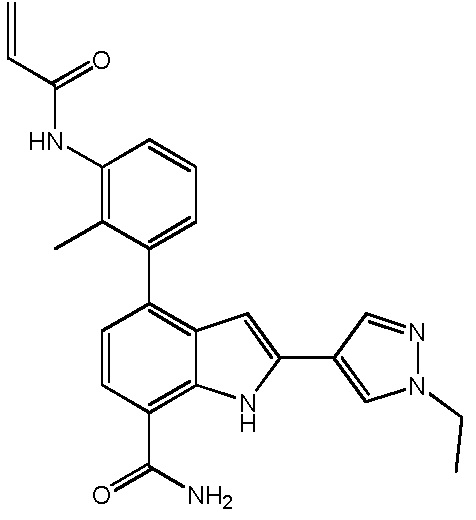
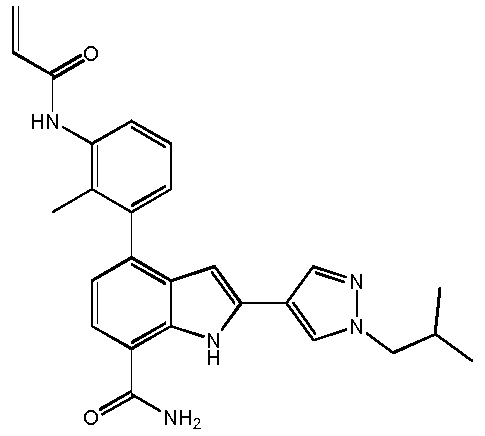
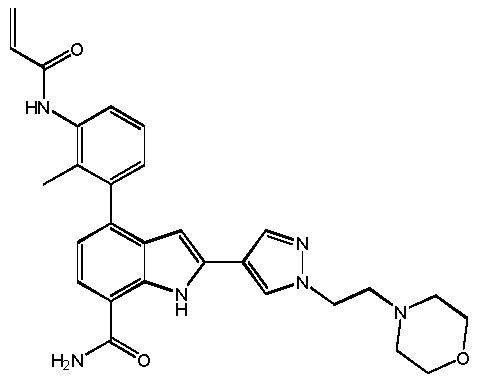
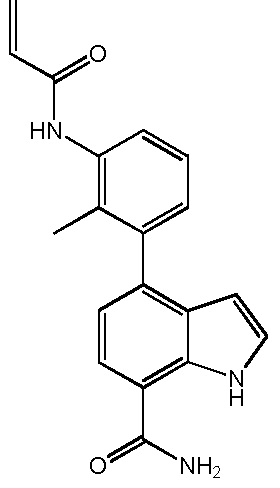

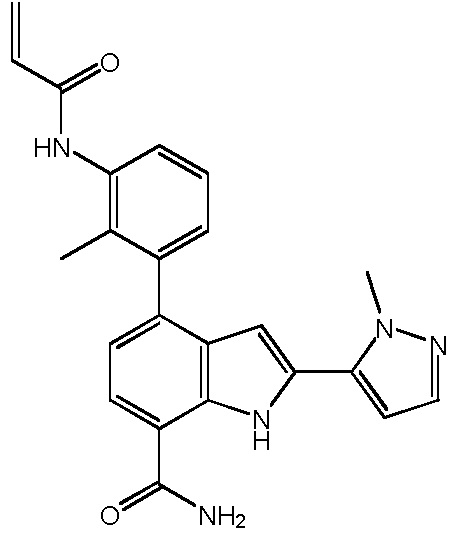

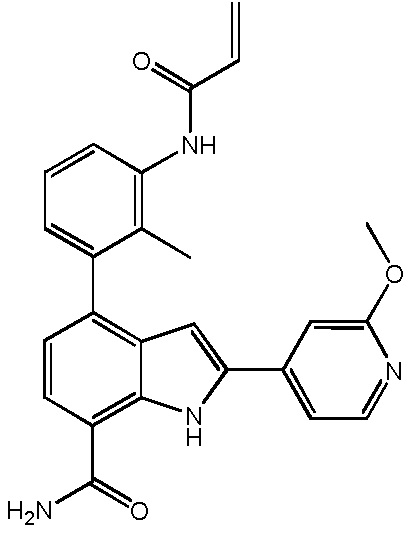

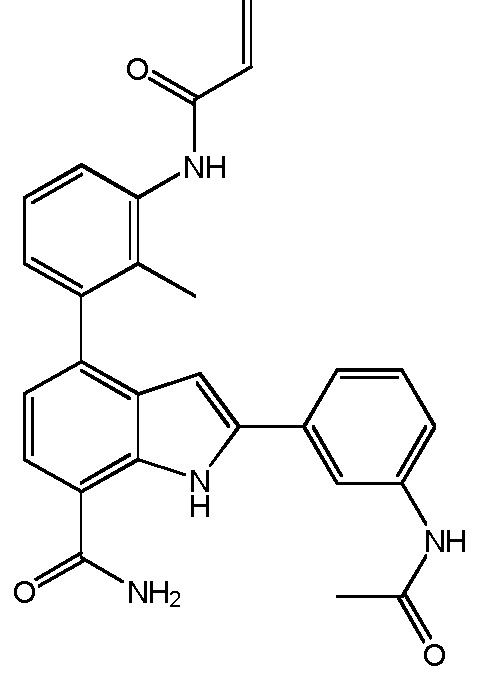
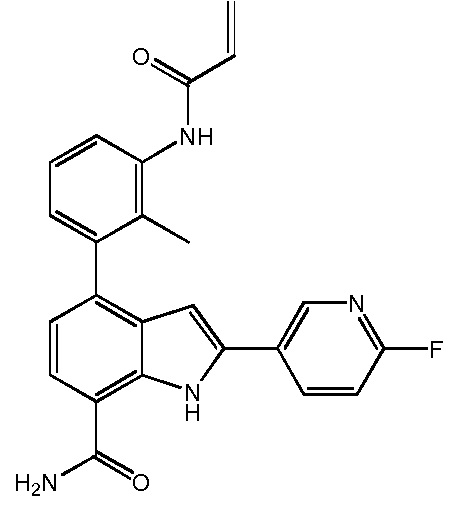
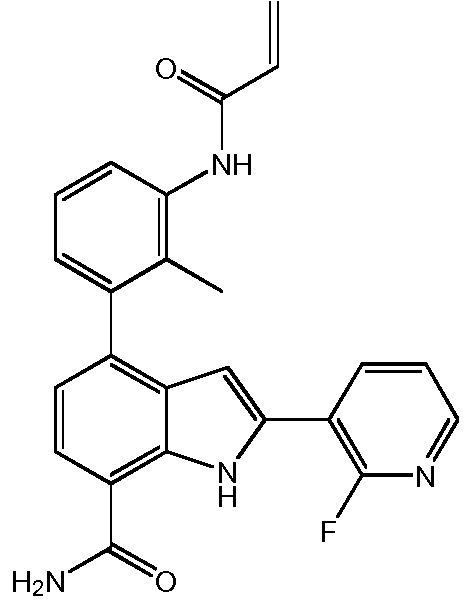
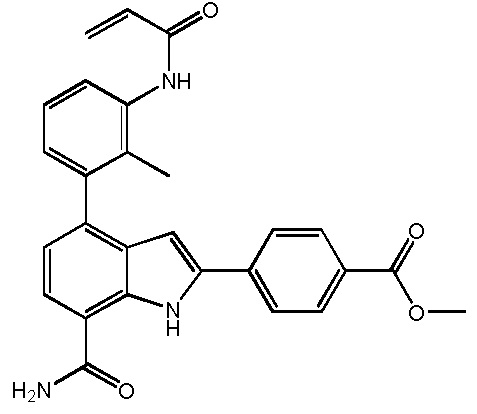
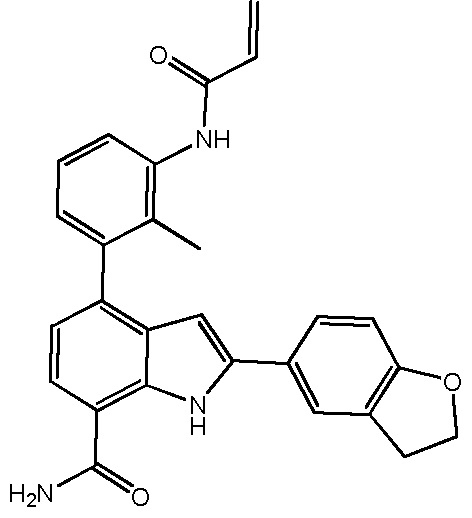
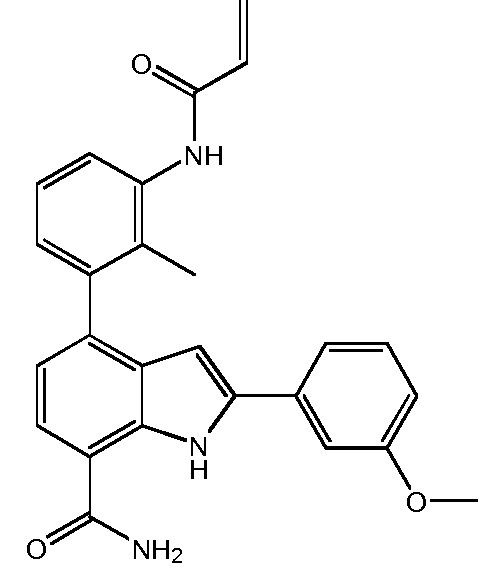
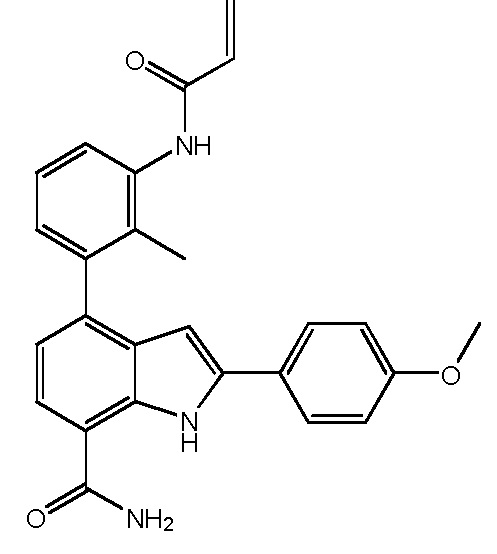
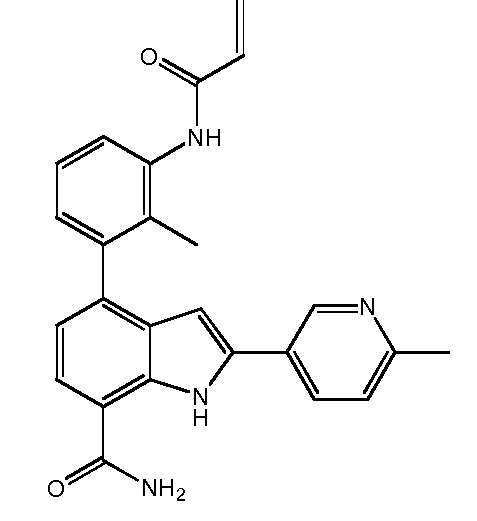
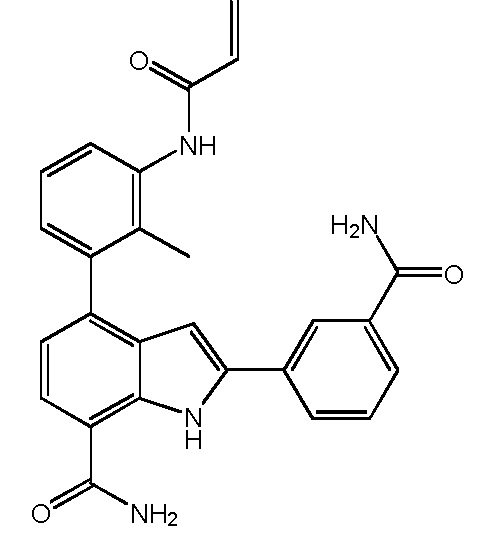
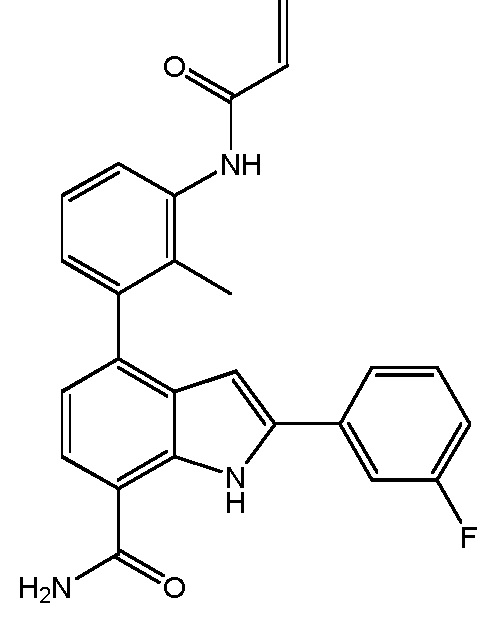
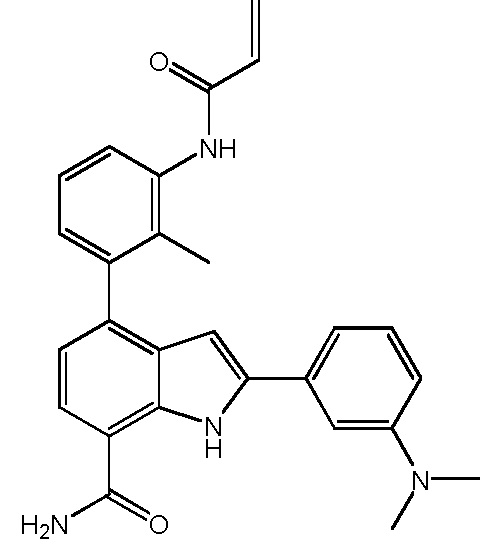
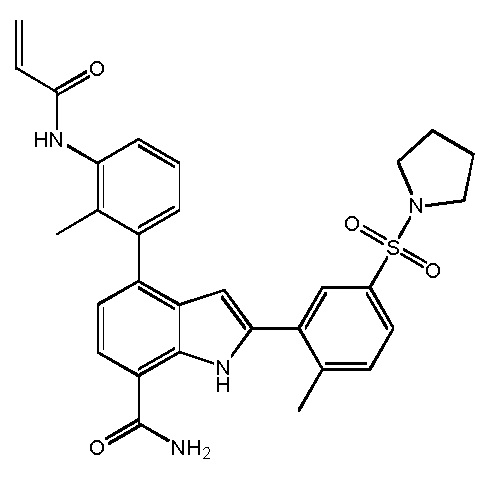

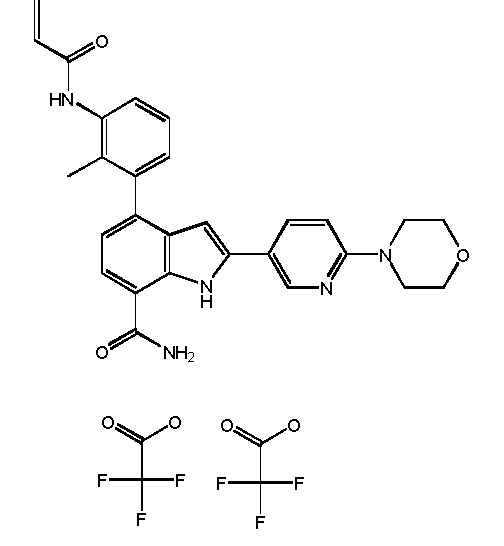
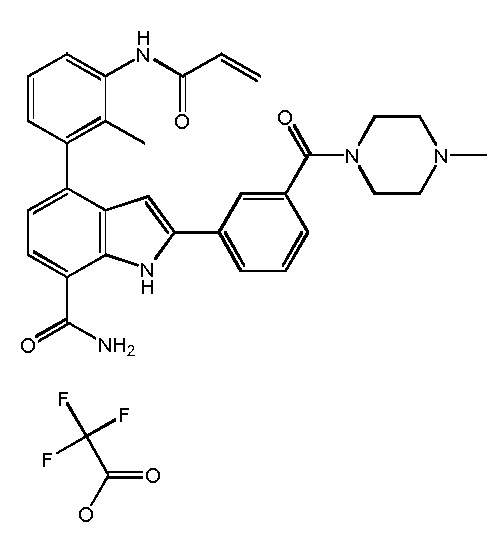

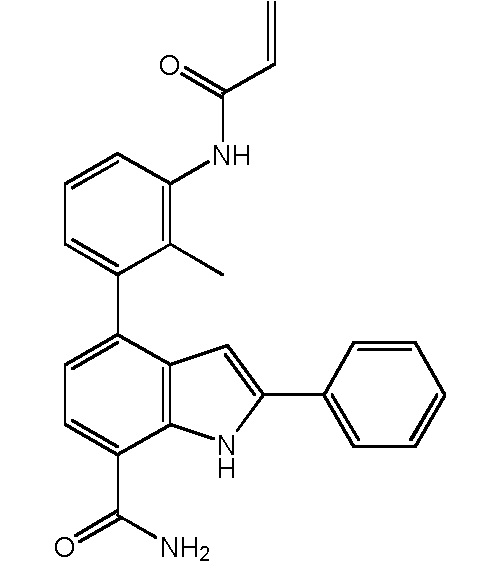
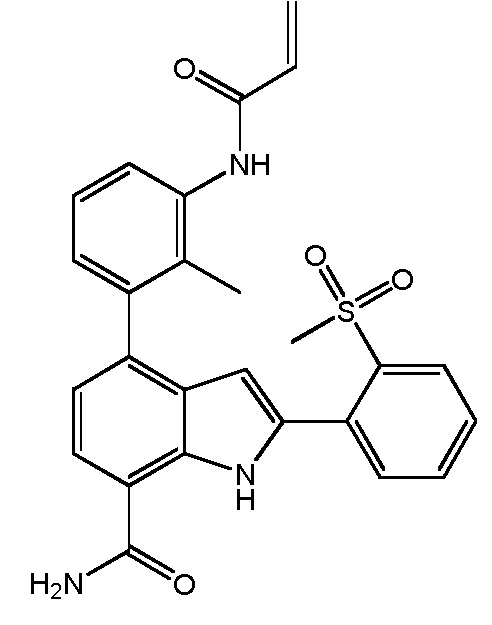
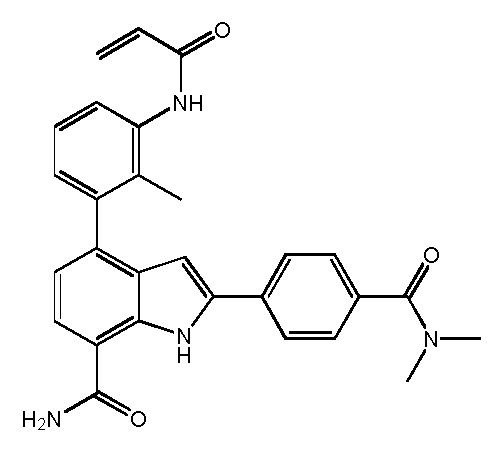

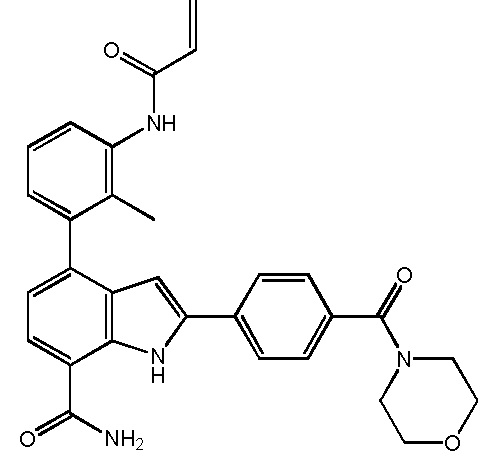

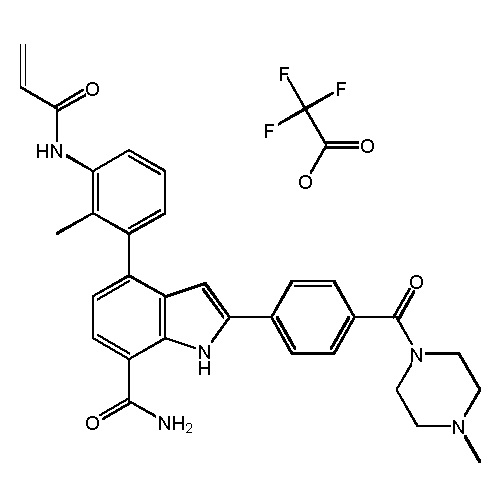
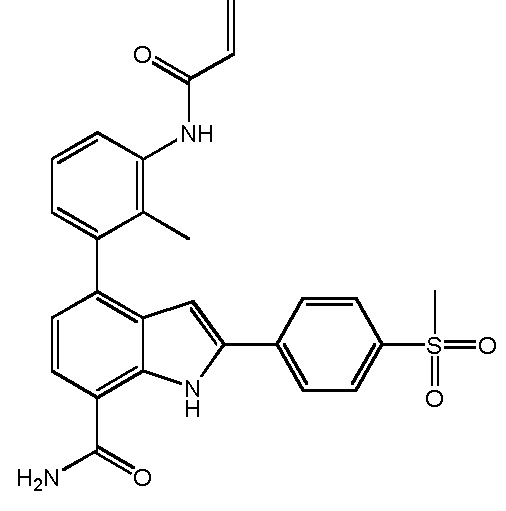
2-метокси-5-пиридинбороновой кислоты)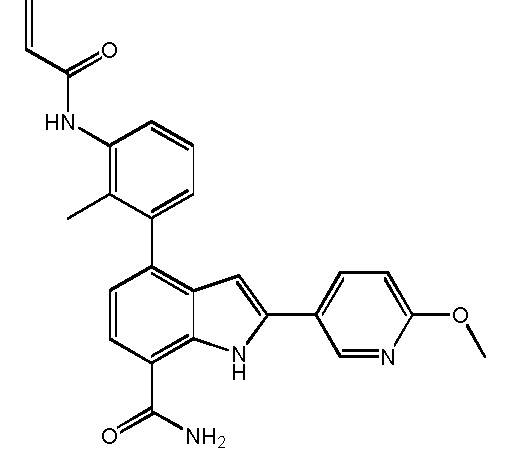

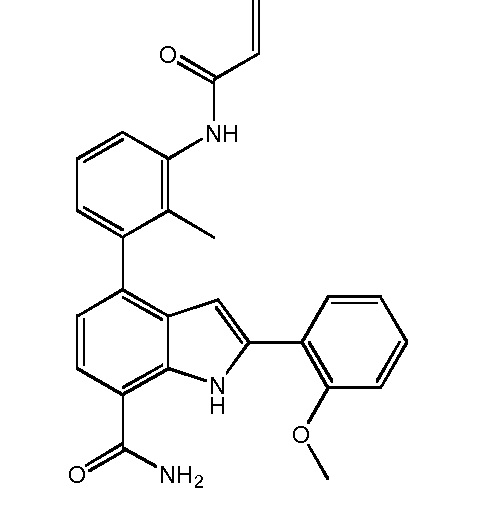
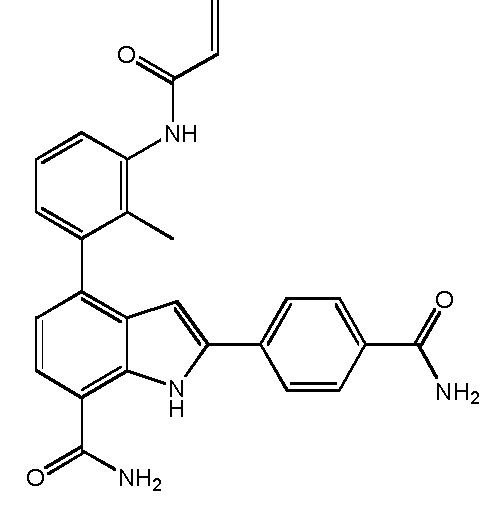
Примеры, полученные из N-(2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)тиазол-2-карбоксамида (Получение № 4) с использованием общего способа А
(Таблица 1, Способ)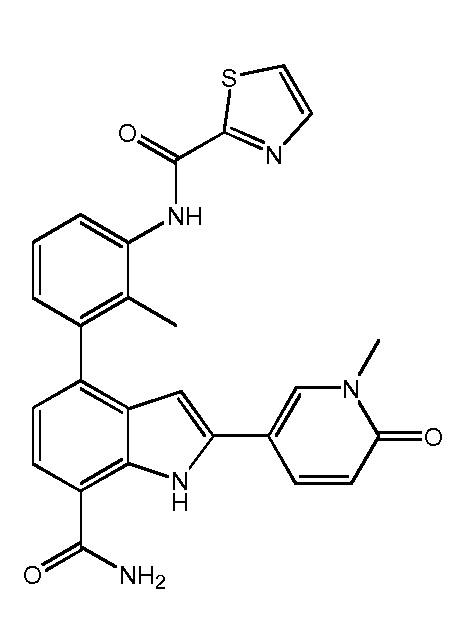
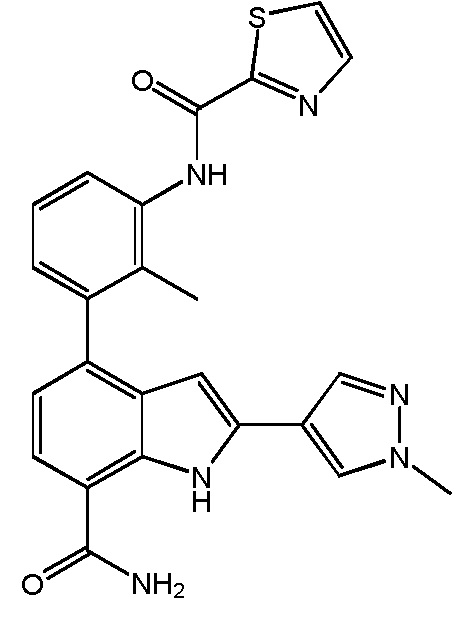
Примеры получены из 2-(1-ацетил-1,2,3,6-тетрагидропиридин-4-ил)-4-бром-1H-индол-7-карбоксамида (полученные с использованием A с 4-бром-2-йод-1H-индол-7-карбоксамидом (Получение № 1) и 1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2H)-ил)этанона [Combi-Blocks]) с использованием общего способа A
(Таблица 1, Способ)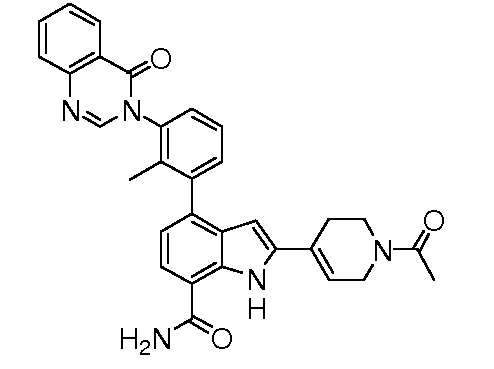
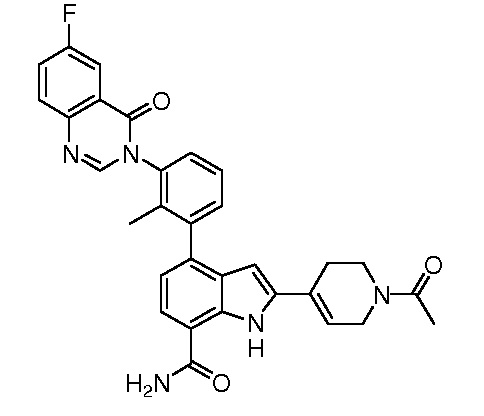
[WO 2006/099075]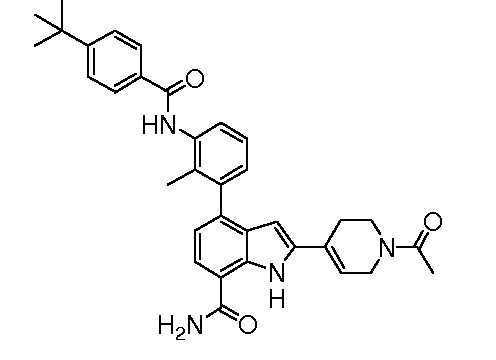
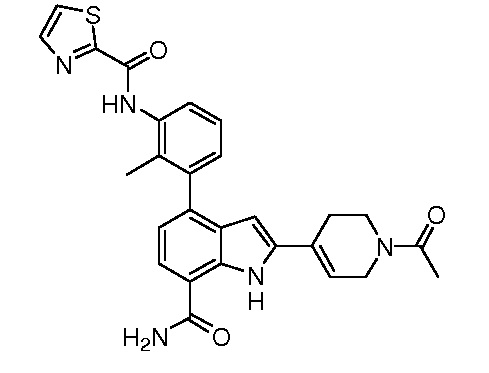
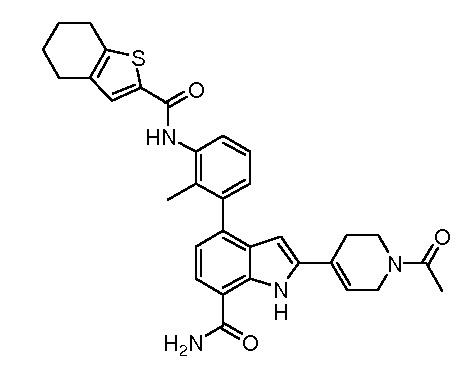

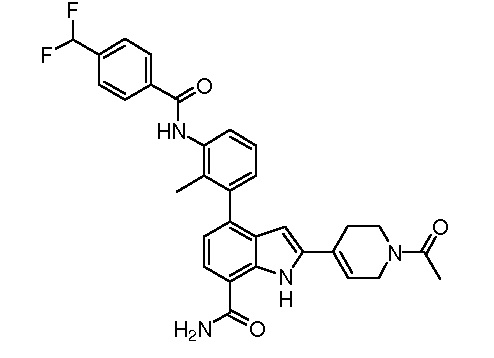
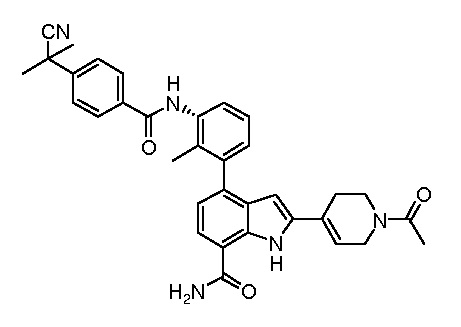
Примеры, полученные из 4-бром-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид (Получение № 18) с использованием общего способа A
(Таблица 1, Способ)
(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)метанамина [ChemMaker] и акрилоилхлорида)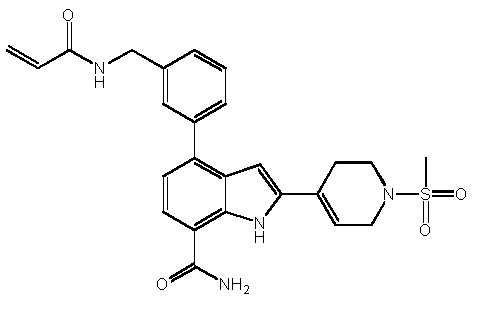
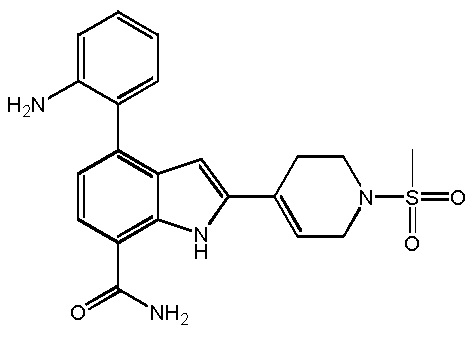
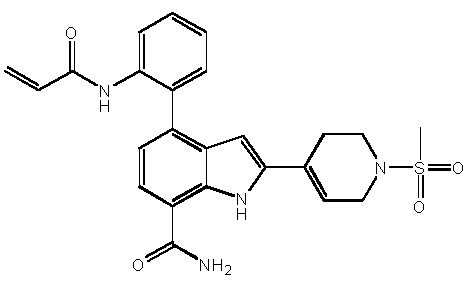
3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенола и
тиазол-2-илметанола)
[CombiBlocks]

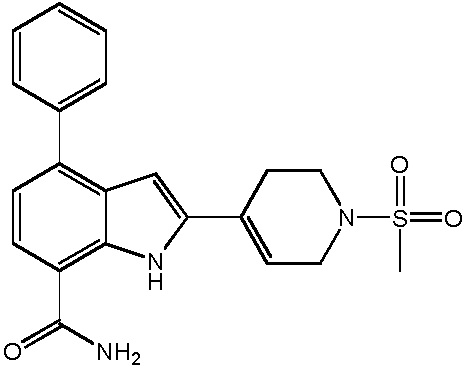
(Получение № 6)
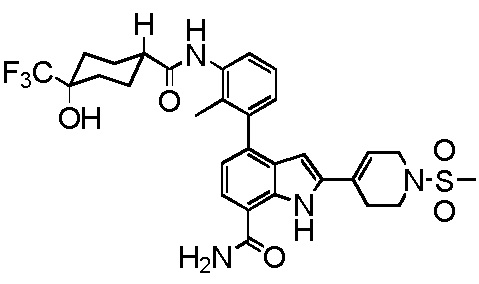
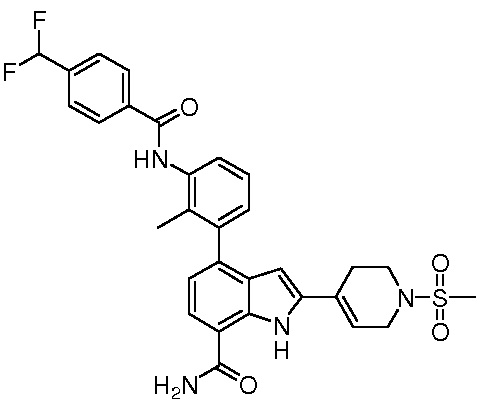
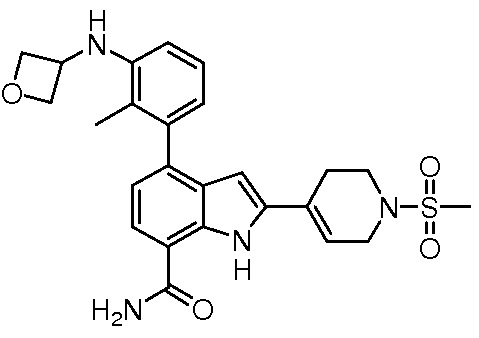
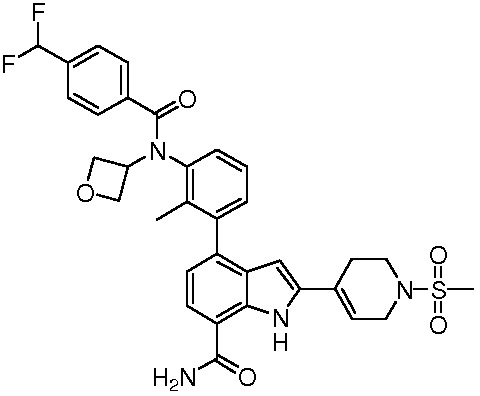
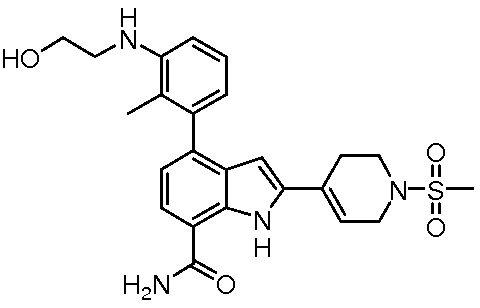
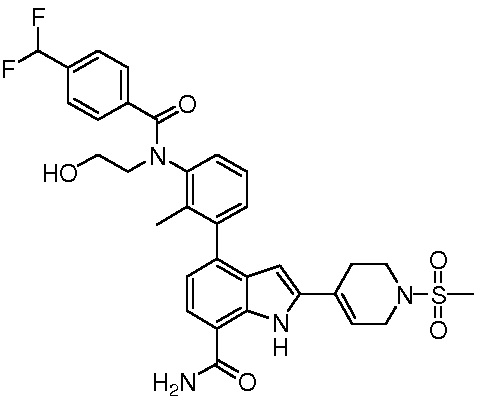

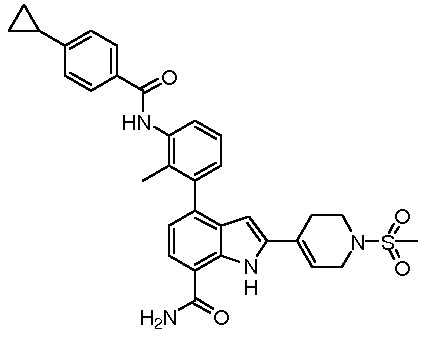
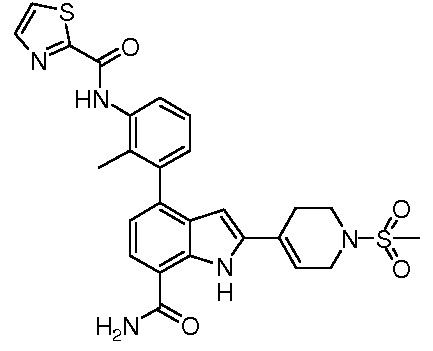
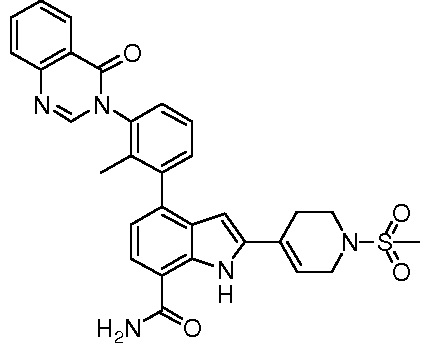
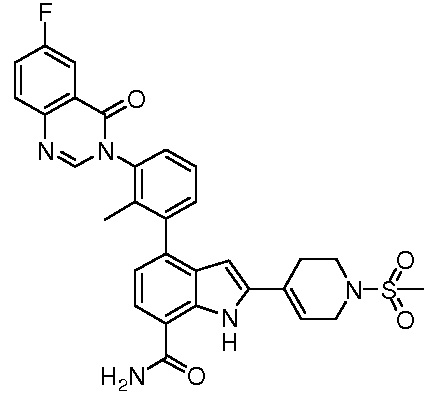
Примеры, полученные из 4-бром-1H-индол-7-карбоксамида (Получение № 2) с использованием общего способа A
(Таблица 1, Способ)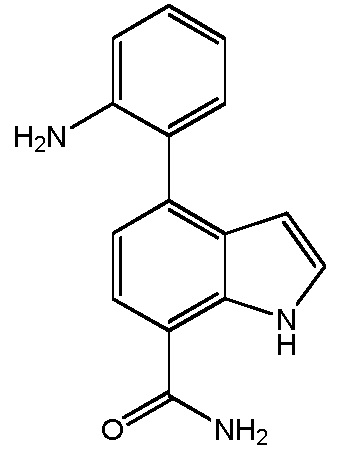
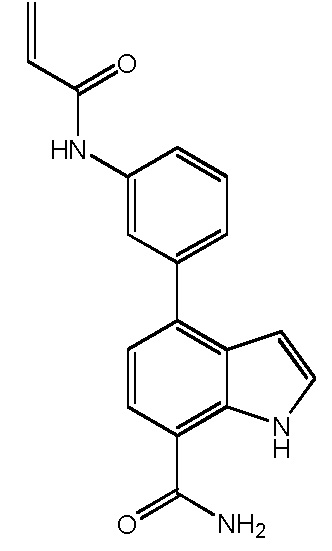
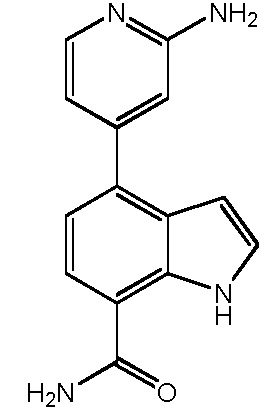
[Maybridge]

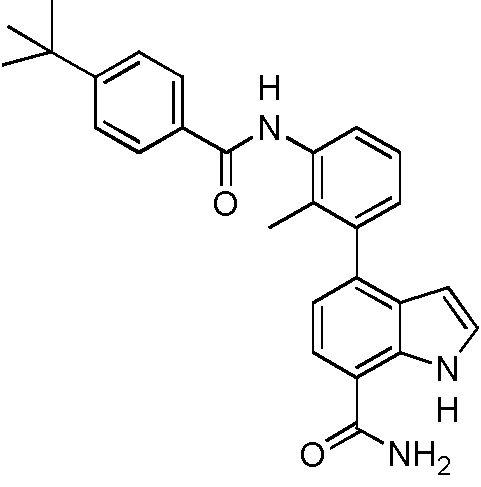
Примеры, полученные из 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-7-карбоксамида (Получение № P.1) с использованием общего способа A
(Таблица 1, Способ)
(Получение № 29)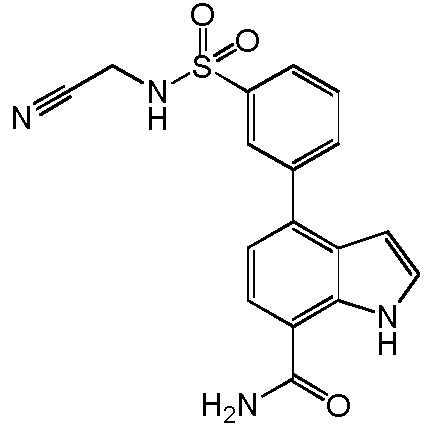
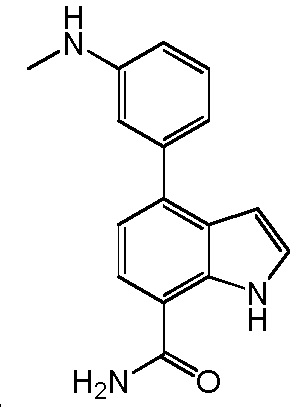
Примеры, полученные из 4-йод-2-(пиридин-3-ил)-1H-индол-7-карбоксамида (Пример № F.1) с использованием общего способа A
(Таблица 1, Способ)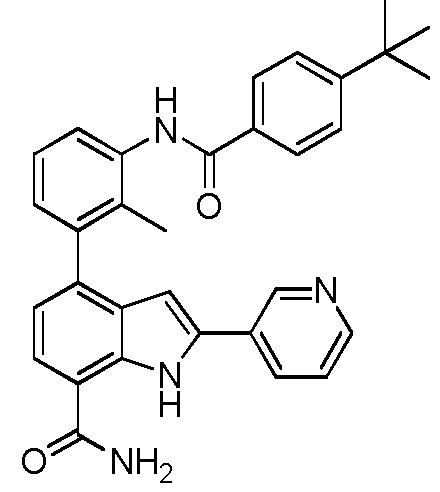
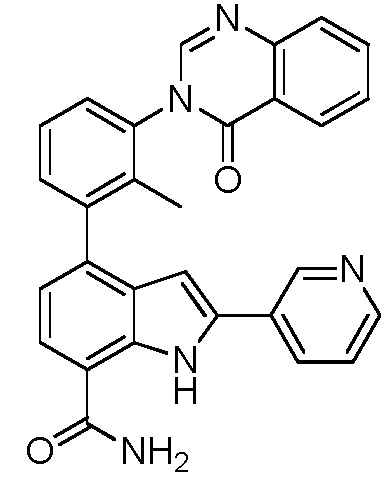

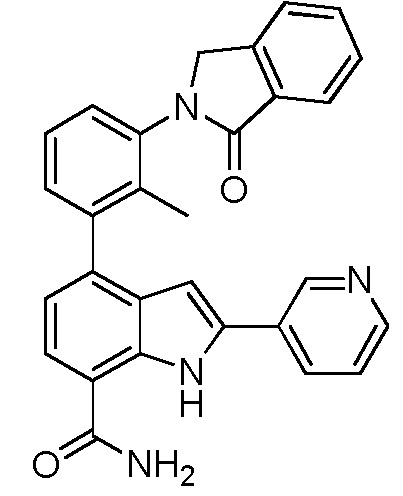
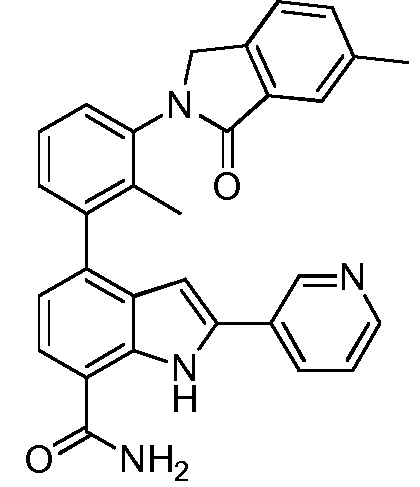
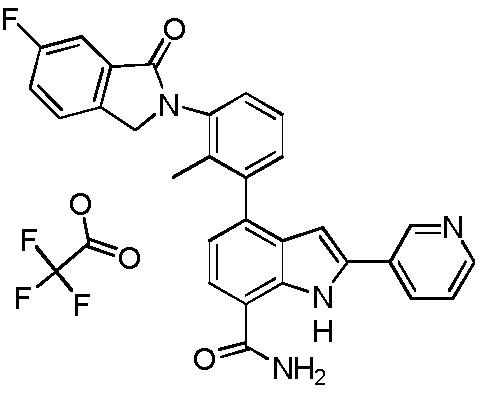
5-амино-2-метилфенилбороновой кислоты, пинаколового эфира и
1,3-тиазол-2-карбонил хлорида [Maybridge-International])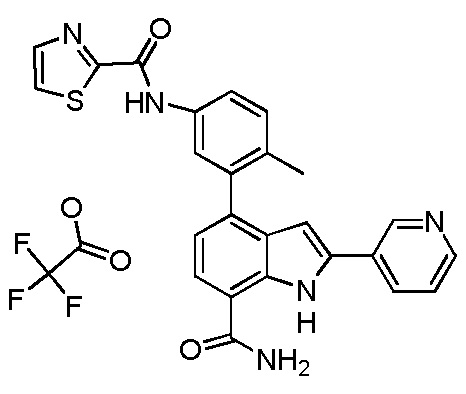
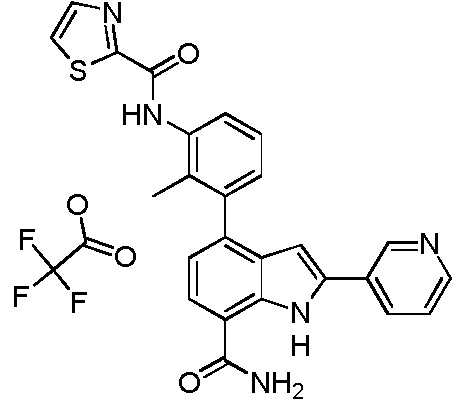
Примеры, полученные из 4-йод-2-(п-толил)-1H-индол-7-карбоксамид (полученный с использованием F из 1-(п-толил)этанона) с использованием общего способа A
(Таблица 1, Способ)
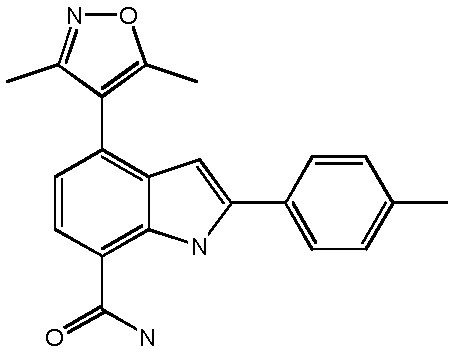
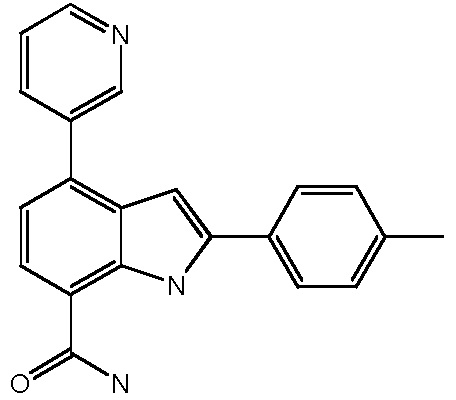
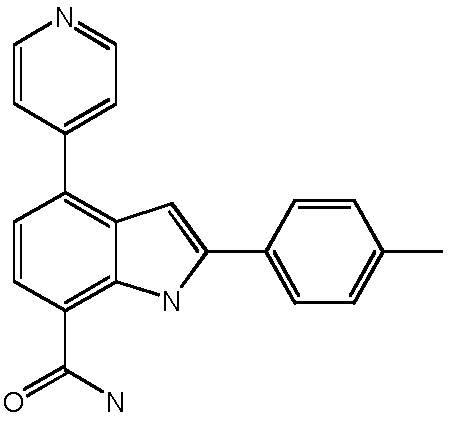
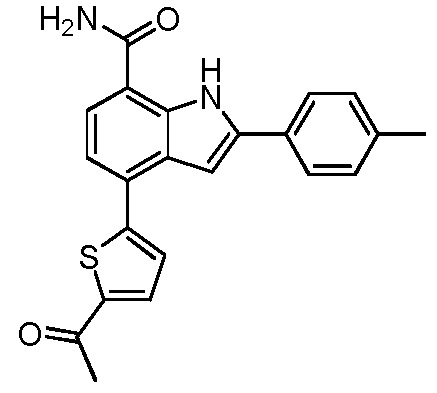
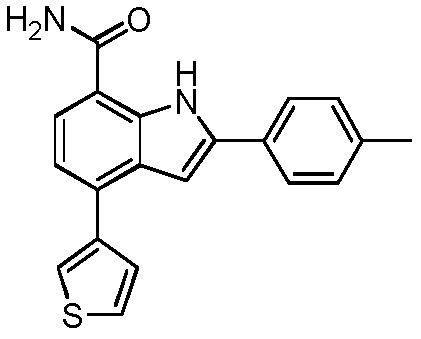
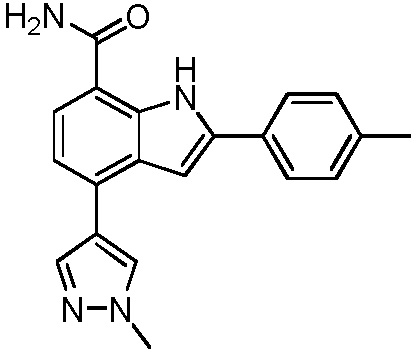
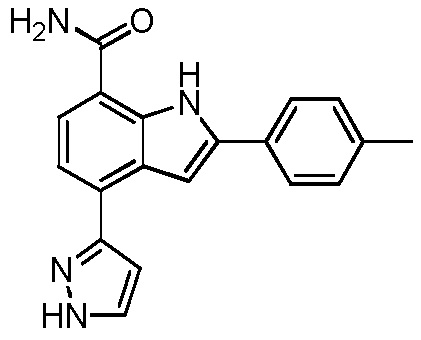
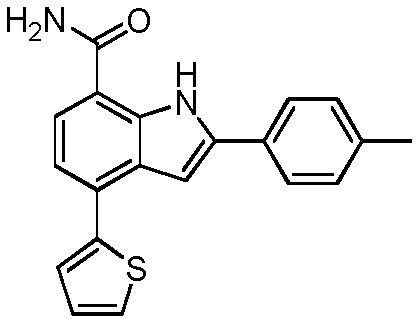
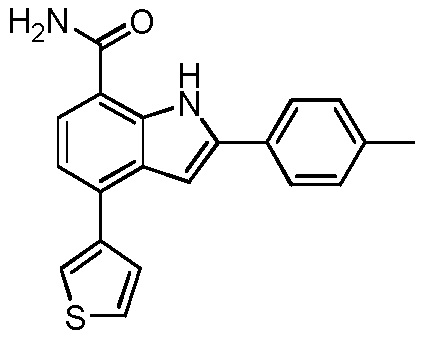
Примеры, полученные из 4-йод-2-(п-толил)-1H-индол-7-карбоксамида (полученный с F с использованием 1-(4-фторфенил)этанона) с использованием общего способа A
(Таблица 1, Способ)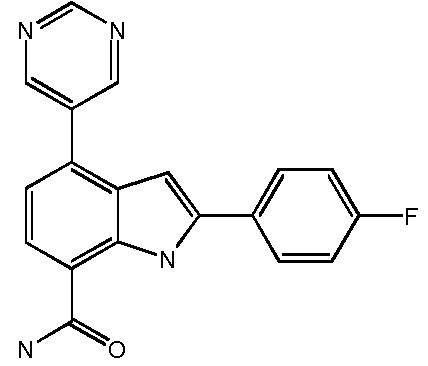

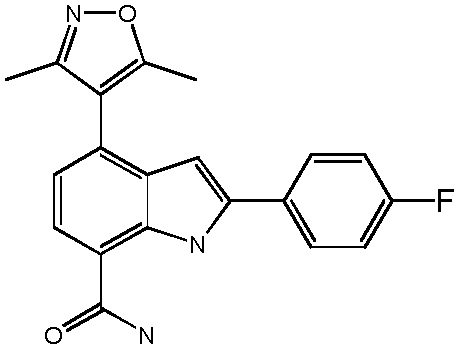
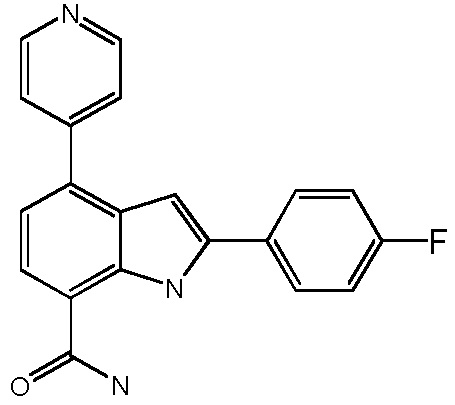
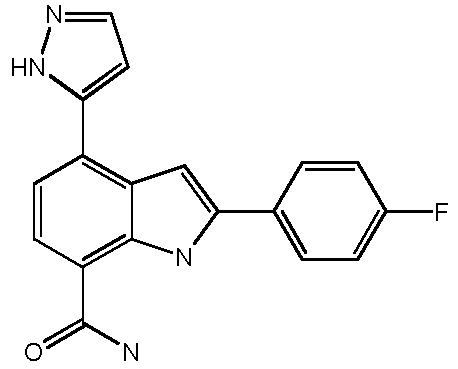
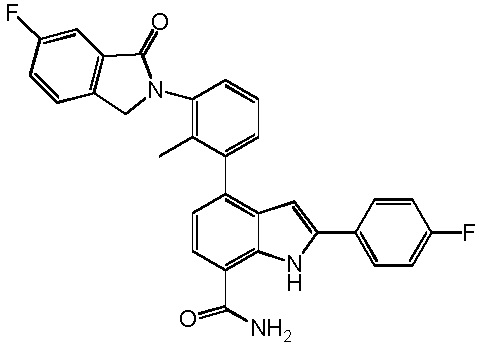
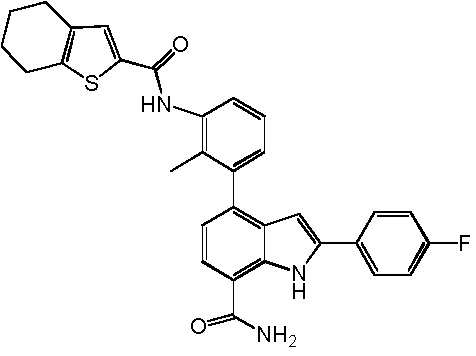
Примеры, полученные из 4-бром-2-(1-метил-1H-пиразол-4-ил)-1H-индол-7-карбоксамида (полученный с использованием A из 4-бром-2-йод-1H-индол-7-карбоксамида (Получение № 1) с 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразолом) с использованием общего способа A
(Таблица 1, Способ)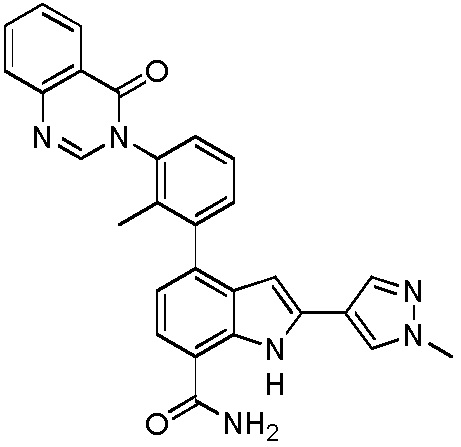
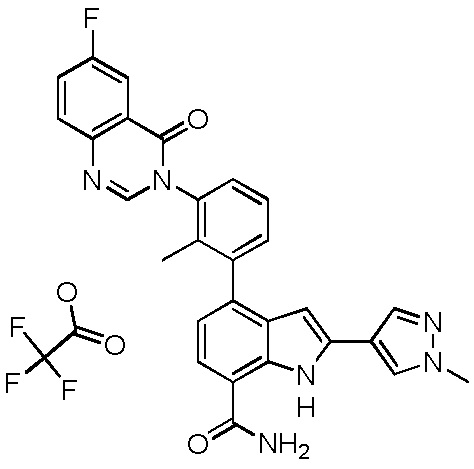
(полученный с использованием H из 2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина [Combi-Blocks] и 3-оксетанона [Molbridge]), E с тиазол-2-карбонил хлоридом [Maybridge])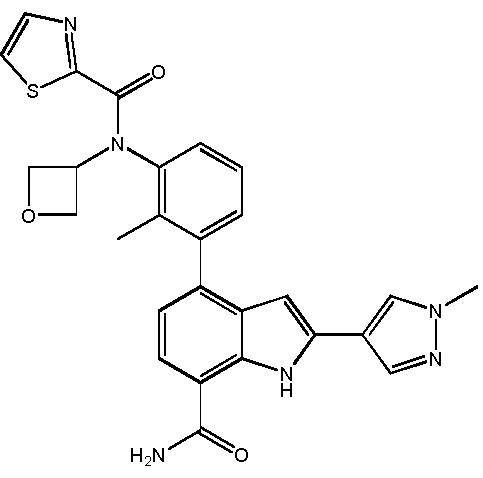
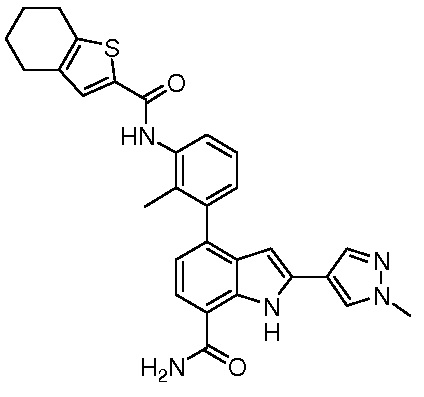
Примеры, полученные из 4-бром-2-(3,6-дигидро-2H-пиран-4-ил)-1H-индол-7-карбоксамид (полученный с использованием A из 4-бром-2-йод-1H-индол-7-карбоксамида (Получение № 1) и 2-(3,6-дигидро-2H-пиран-4-ил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана) с использованием общего способа A
(Таблица 1, Способ)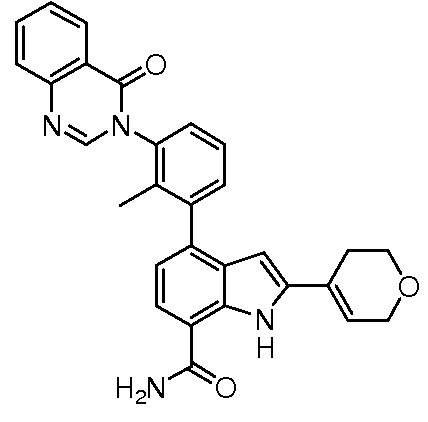
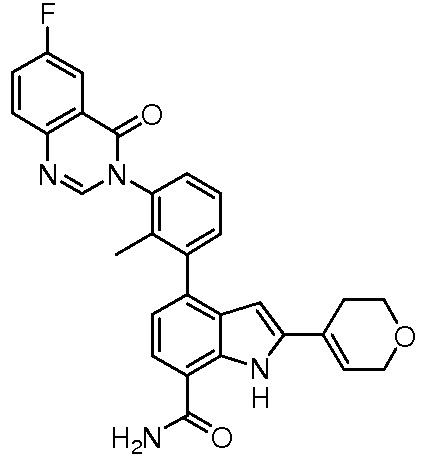
Примеры, полученные из 4-бром-2-(4-фторфенил)-1H-индол-7-карбоксамид (полученный с использованием A из 4-бром-2-йод-1H-индол-7-карбоксамида (Получение № 1) и 2-(4-фторфенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана) с использованием общего способа A
(Таблица 1, Способ)
Примеры, полученные из 4-бром-2-(пиримидин-5-ил)-1H-индол-7-карбоксамида (полученный с использованием A из 4-бром-2-йод-1H-индол-7-карбоксамида (Получение № 1) и 5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиримидина) с использованием общего способа A
(Таблица 1, Способ)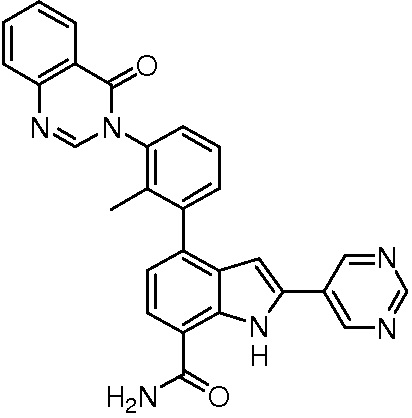
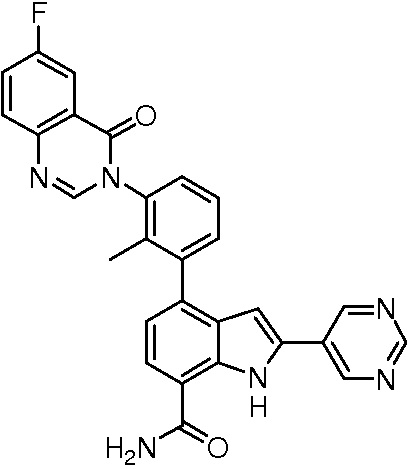

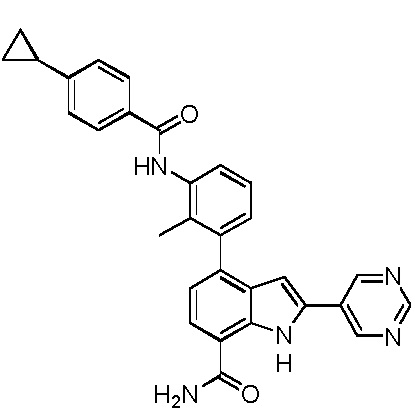
Примеры, полученные из 4-бром-2-(1-(2-гидрокси-2-метилпропил)-1H-пиразол-4-ил)-1H-индол-7-карбоксамида (полученный с использованием A из 4-бром-2-йод-1H-индол-7-карбоксамида (Получение № 1) и 2-метил-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)пропан-2-ола (Получение № 26) с использованием общего способа A
(Таблица 1, Способ)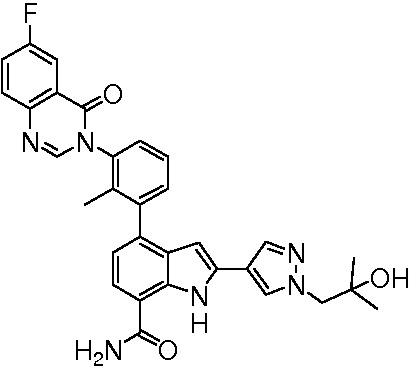
Примеры, полученные из 2-(3-хлор-2-(гидроксиметил)фенил)-6-циклопропил-8-фторизохинолин-1(2H)-она [U.S. 20100222325] с использованием общего способа A
(Таблица 1, Способ)
-2-ил)-1H-пиразолом, и P с 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксабороланом))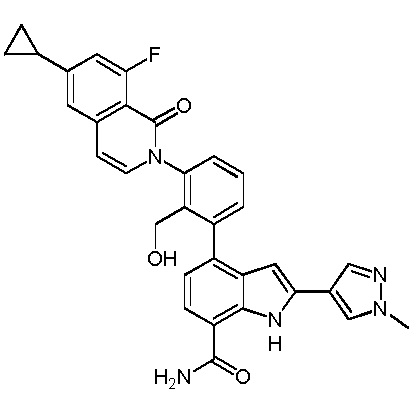
Общий способ В: Нуклеофильное замещение арилгалогенида амином
К раствору арилгалогенида или гетероарилгалогенида и соответствующего органического растворителя (например, DMSO, 1,4-диоксан, н-бутанол, THF, пиридин, предпочтительно, DMSO или пиридин) добавляли амин (1-10 эквивалента, предпочтительно 1 эквивалент) и основание (например, TEA, пиридин, DIEA, K2CO3, предпочтительно TEA; 1-5 эквивалента, предпочтительно 1 эквивалент). Полученный раствор перемешивали при температуре приблизительно от 20 до 150°C (предпочтительно, приблизительно 130-150°C) термически в течение периода приблизительно от 1 часа до 72 часов (предпочтительно, приблизительно 24 часа) или в микроволновой печи в течение приблизительно от 5 минут до 2 часов (предпочтительно, приблизительно 30 минут). Смесь необязательно концентрировали в вакууме или в потоке теплого азота с получением промежуточных соединений или целевого соединения или необязательно фильтровали через среду (например, SiCO3 или целит), которую промывали соответствующим растворителем (например, EtOAc, 1,4-диоксан, THF, MeCN, DCM, Et2O, MeOH, EtOH, DMSO, 1:1 MeOH/DMSO, 2:1 MeOH/DMSO) и затем, необязательно, концентрировали в вакууме или в потоке теплого азота, с получением целевого соединения.
Иллюстрация общего способа B
Получение № B.1: (R)-трет-Бутил 1-(7-циано-1H-индол-4-ил)пиперидин-3-илкарбамат
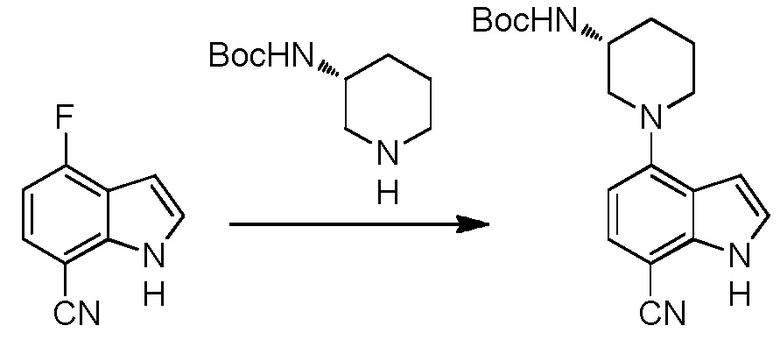
Смесь (R)-трет-бутил пиперидин-3-илкарбамата (1,501 г, 7,49 ммоль) и 4-фтор-1H-индол-7-карбонитрила (0,6 г, 3,75 ммоль) в пиридине (3,02 мл, 37,5 ммоль) нагревали при температуре приблизительно 150°C в течение приблизительно 30 минут в микроволновой печи. Смесь выпаривали досуха и полученный остаток очищали с помощью хроматографии на силикагеле при элюировании с градиентом от 30 до 100% EtOAc в гексанах с получением (R)-трет-бутил 1-(7-циано-1H-индол-4-ил)пиперидин-3-илкарбамата (0,4 г, 31%); ЖХ/МС (Таблица 1, способ g) Время удерживания = 1,69 мин; MS m/z: 341 (M+H)+
Общий способ C: Гидролиз сложного эфира до карбоновой кислоты
В сосуд, содержащий сложный эфир (предпочтительно, 1 эквивалент) в чистом виде или в органическом растворителе (например, 1,4-диоксане, MeOH или THF/MeOH, предпочтительно, 1,4-диоксане), добавляли водный раствор основания (такой как, водный раствор NaOH или LiOH; 1-10 эквивалентов, предпочтительно 2-6 эквивалентов). Смесь перемешивали при приблизительно 0-100°C (предпочтительно, приблизительно 25-60°C) в течение приблизительно от 1 до 48 часов (предпочтительно, приблизительно от 4 до 24 часов). Органический растворитель, необязательно концентрировали в вакууме. Затем смесь подкисляли добавлением подходящего водного раствора кислоты (такого как водный раствор HCl). Если образуется осадок, его можно собрать фильтрованием с получением продукта. Смесь или фильтрат, если твердое вещество не представляет собой продукт, можно необязательно концентрировать в вакууме с получением целевого соединения в виде карбоксилатной соли. Альтернативно, смесь необязательно фильтровали через среду (например, силикагель или целит), которую промывали соответствующим растворителем (например, EtOAc, 1,4-диоксан, THF, MeCN, DCM, Et2O, MeOH, EtOH) и затем, необязательно, концентрировали в вакууме с получением остатка в качестве целевого соединения. Или остаток или раствор необязательно разделяли между водой и органическим растворителем (таким как EtOAc, Et2O или DCM). Органический слой выделяли и необязательно промывали в произвольном порядке водой и/или водными растворами, содержащими кислоту (такую как HCl, AcOH или NH4Cl) и/или водными растворами, содержащими основание (такое как NaHCO3, Na2CO3, NaOH, KOH или NH4OH) и/или водными растворами, содержащими неорганическую соль (такую как NaCl, Na2SO3 или Na2S2O3). Органический раствор затем необязательно сушили с высушивающим веществом (например, безводным MgSO4 или Na2SO4), фильтровали и концентрировали в вакууме с получением целевого соединения.
Иллюстрация общего способа C
Пример № C.1: (E)-4-((3-(7-карбамоил-1H-индол-4-ил)фенил)амино)-4-оксобут-2-еновая кислота
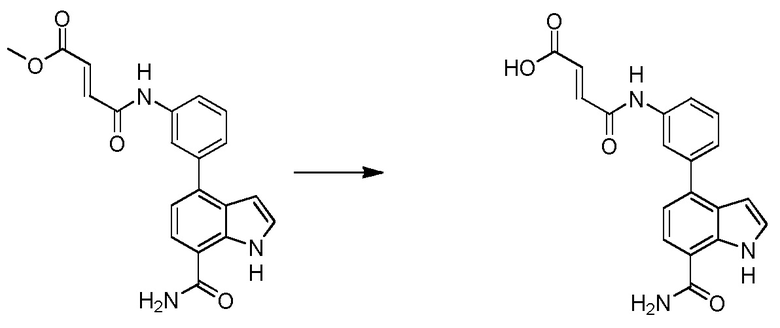
(E)-Метил 4-((3-(7-карбамоил-1H-индол-4-ил)фенил)амино)-4-оксобут-2-еноат (0,610 г, 1,68 ммоль, Пример № D.1) суспендировали в 1,4-диоксане (8,39 мг). Добавляли гидроксида лития (1M в воде, 8,39 мл, 8,39 ммоль) и смесь перемешивали при приблизительно 60°C в течение приблизительно 1 часа. Реакционную смесь концентрировали до приблизительно 8 мл и разбавляли водой (10 мг). Величину рН доводили до приблизительно 4, используя 1 н HCl. Твердые вещества собирали, промывали водой и сушили в вакууме с получением (E)-4-((3-(7-карбамоил-1H-индол-4-ил)фенил)амино)-4-оксобут-2-еновой кислоты (0,45 г, 77%) в виде твердого вещества. 50 мг сырого продукта очищали далее путем препаративной ВЭЖХ (Таблица 1, способ af) для получения 30,9 мг для получения аналитически чистой (E)-4-((3-(7-карбамоил-1H-индол-4-ил)фенил)амино)-4-оксобут-2-еновой кислоты: ЖХ/МС (Таблица 1, способ f) Время удерживания = 1,64 мин; MS m/z: 350 (M+H)+ (Btk IC50=C)
Общий способ D: Образование амида из амина и карбоновой кислоты
В колбу добавляли в произвольном порядке карбоновую кислоту или карбоксилатную соль (1-5 эквивалента, предпочтительно 1,1-1,5 эквивалента), амин (1-5 эквивалента, предпочтительно, 1-1,5 эквивалента), органический растворитель (такой как DCM, DCE, THF или 1,4-диоксан, DMF, DMF/пиридин, предпочтительно, DCM или DMF/пиридин), связующий пептидный реагент (такой как BOP-Cl, HATU, EDC, DCI, PyBOP, or EDC•HCl, предпочтительно, HATU или EDC; 1-10 эквивалента, предпочтительно, 1-2,5 эквивалента), основание (такое как TEA, DIEA, пиридин или DIEA, предпочтительно, DIEA; 1-20 эквивалента, предпочтительно, 1-5 эквивалента) и, необязательно, HOBt (0-5 эквивалента, предпочтительно, 0-1 эквивалент). Затем смесь перемешивали при температуре 10-60°C (предпочтительно, приблизительно 25-50°C) в течение приблизительно от 5 минут до 48 часов (предпочтительно приблизительно от 5 минут до 24 часов). Необязательно, дополнительные количества вышеуказанных реагентов добавляли для доведения реакции до конца. Смесь необязательно концентрировали в вакууме с получением целевого соединения. Смесь необязательно фильтровали через среду (такую как силикагель или целит), которую промывали соответствующим растворителем (таким как EtOAc, 1,4-диоксан, THF, MeCN, DCM, Et2O, MeOH, EtOH), а затем при необходимости концентрировали в вакууме с получением остатка. Или остаток или раствор необязательно разделяли между водой и органическим растворителем (таким как EtOAc, Et2O или DCM). Если продукт не разделяли, смесь перемешивали в течение от 5 минут до 1 часа (предпочтительно, 30 минут) и твердое вещество собирали с помощью вакуумной фильтрации. Альтернативно, органический слой выделяли и необязательно промывали в произвольном порядке водой и/или водными растворами, содержащими кислоту (такую как HCl, AcOH или NH4Cl) и/или водными растворами, содержащими основание (такое как NaHCO3, Na2CO3, NaOH, KOH или NH4OH) и/или водными растворами, содержащими неорганическую соль (такую как NaCl, Na2SO3 или Na2S2O3). Органический раствор затем необязательно сушили с высушивающим веществом (например, безводным MgSO4 или Na2SO4), фильтровали и концентрировали в вакууме с получением целевого соединения.
Иллюстрация общего способа D:
Пример № D.1: (E)-Метил 4-((3-(7-карбамоил-1H-индол-4-ил)фенил)амино)-4-оксобут-2-еноат

К раствору (E)-4-метокси-4-оксобут-2-еновой кислоты (0,43 г, 3,28 ммоль) в DCM (40 мг) и DIEA (0,59 мл, 3,58 ммоль) добавляли HATU (1,362 г, 3,58 ммоль). Смесь перемешивали при комнатной температуре в течение 5 минут, затем добавляли 4-(3-аминофенил)-1H-индол-7-карбоксамид (0,75 г, 2,98 ммоль, Получение № A.1). Смесь перемешивали при комнатной температуре в течение приблизительно 3 часов. Смесь концентрировали и остаток суспендировали между водой и EtOAc. Смесь перемешивали при комнатной температуре в течение приблизительно 30 минут, фильтровали для сбора твердого вещества, которое промывали водой и EtOAc и сушили в вакууме с получением (E)-метил 4-((3-(7-карбамоил-1H-индол-4-ил)фенил)амино)-4-оксобут-2-еноата (0,64 г, 59%): ЖХ/МС (Таблица 1, способ f) Время удерживания = 1,45 мин; MS m/z: 364 (M+H)+ (Btk IC50=A)
Примеры, полученные из N-(3-(2-(2-(аминометил)фенил)-7-карбамоил-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамида (Пример № 1) с использованием общего способа D
(Таблица 1, Способ)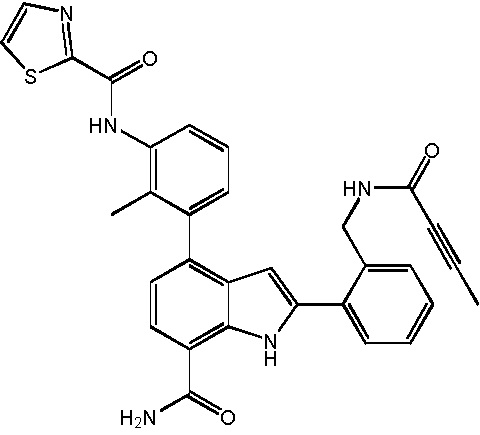
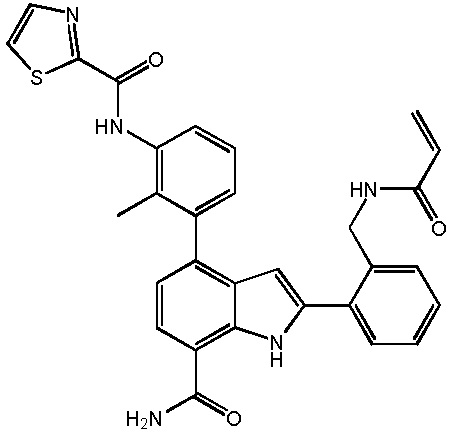

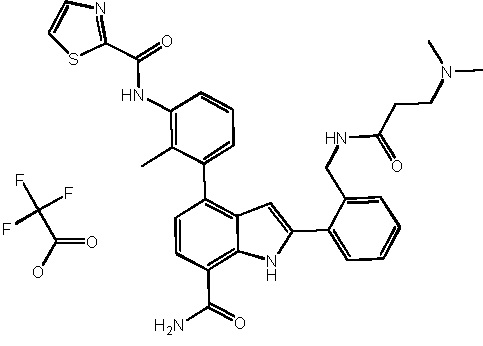
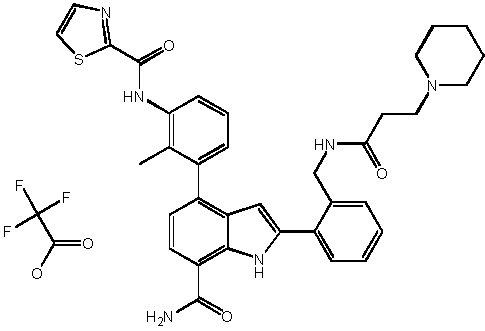
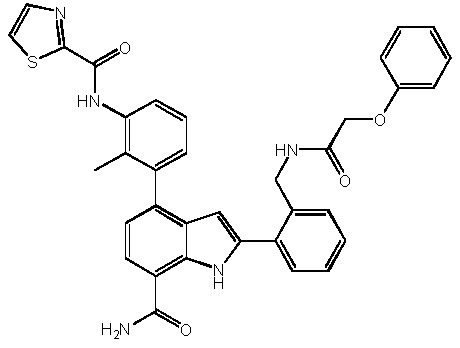
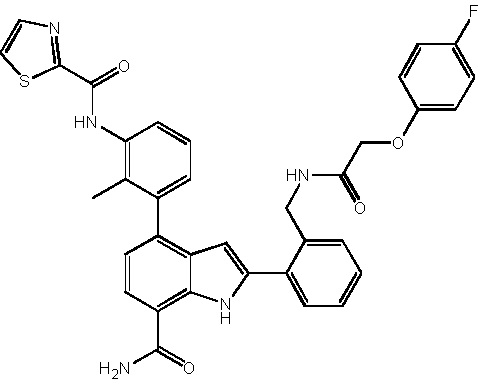
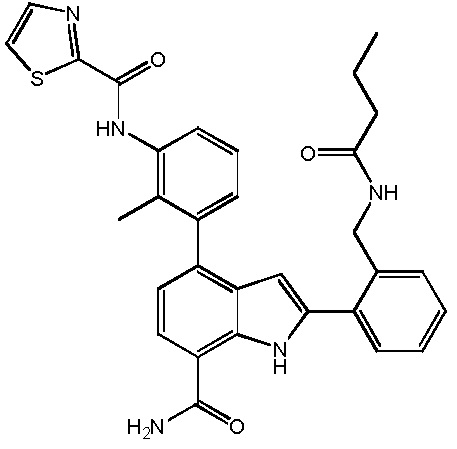
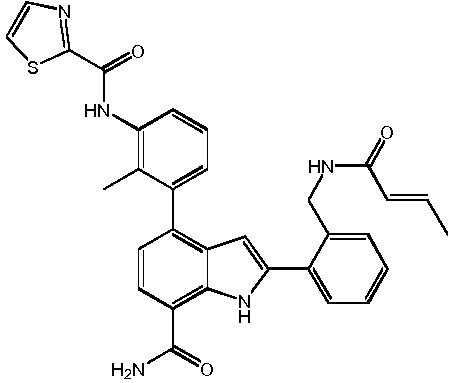


Примеры, полученные из амина и 2-(3-оксобензо[d]изотиазол-2(3H)-ил)уксусной кислоты [Matrix] с использованием общего способа D
(Таблица 1, Способ)
4-бром-1H-индол-7-карбоксамида
(Получение № 2) и
2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина)
Примеры, полученные из N-(3-(3-амино-7-карбамоил-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамида (Получение № 7) с использованием общего способа D
(Таблица 1, Способ)
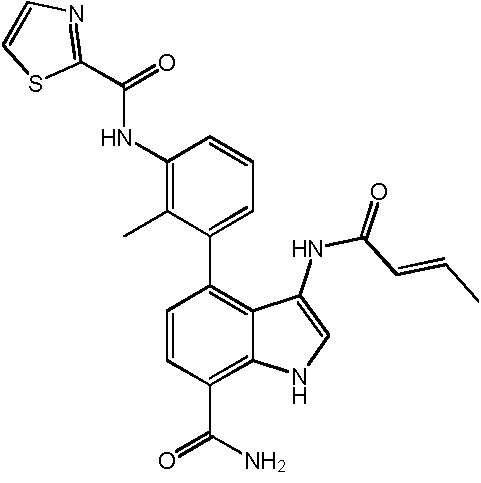
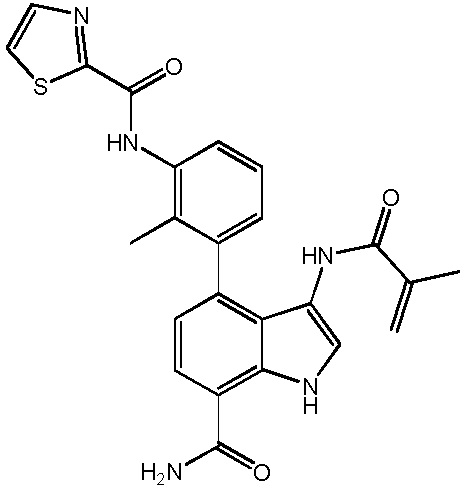
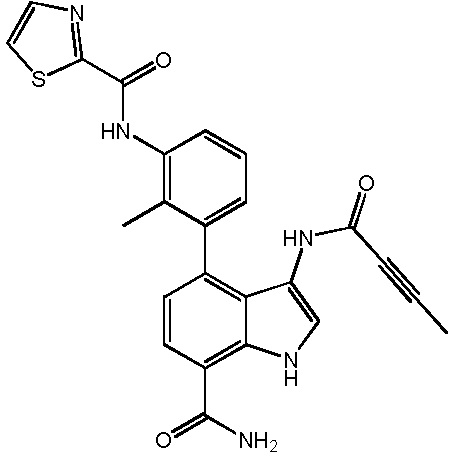
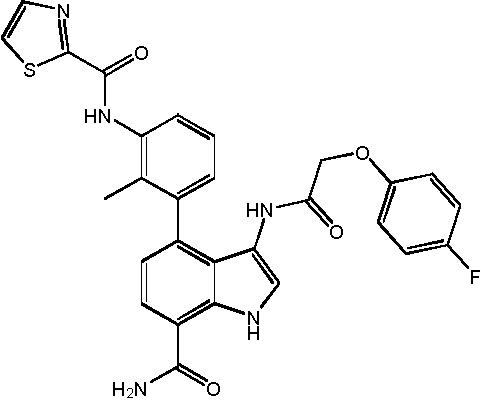
Примеры, полученные из (E)-4-((3-(7-карбамоил-1H-индол-4-ил)фенил)амино)-4-оксобут-2-еновой кислоты (Пример № C.1) с использованием общего способа D
(Таблица 1, Способ)
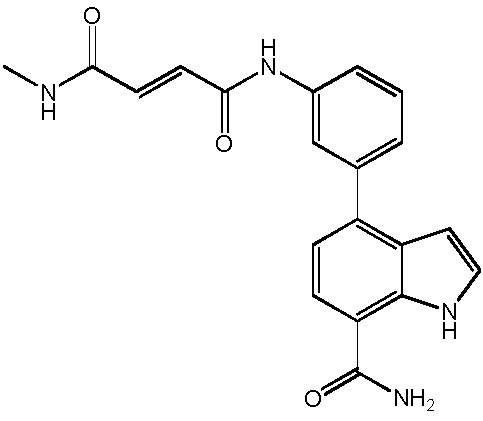
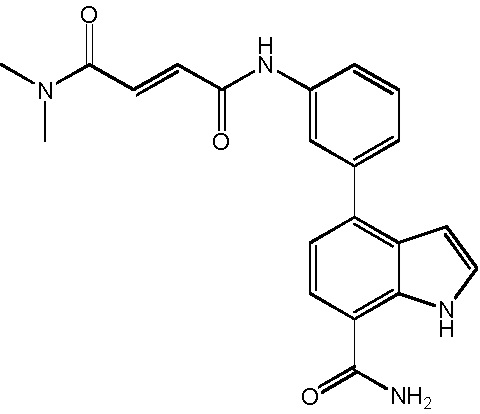
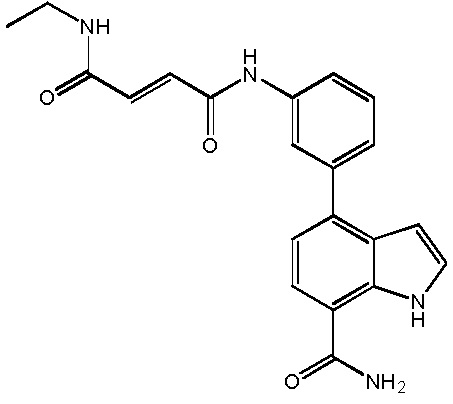
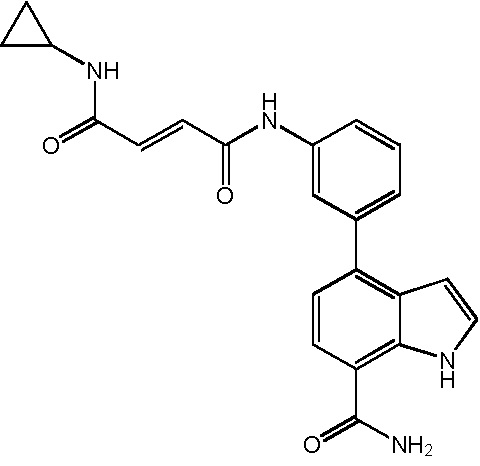
Примеры, полученные из кислоты и 2-(1-ацетилпиперидин-4-ил)-4-(3-амино-2-метилфенил)-1H-индол-7-карбоксамида (Пример № L.1) с использованием общего способа D
(Таблица 1, Способ)
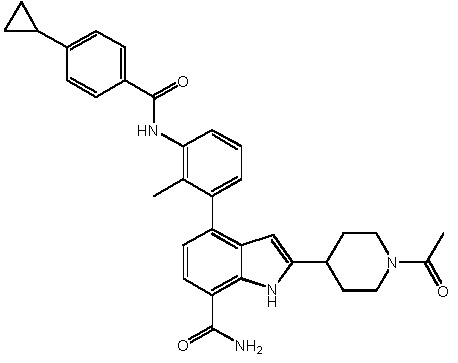
Примеры, полученные из 4-(3-аминофенил)-1H-индол-7-карбоксамид (Получение № A.1) с использованием общего способа B
(Таблица 1, Способ)
Примеры, полученные из 4-(3-амино-2-метилфенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид (Пример № A.4.5) с использованием общего способа D.

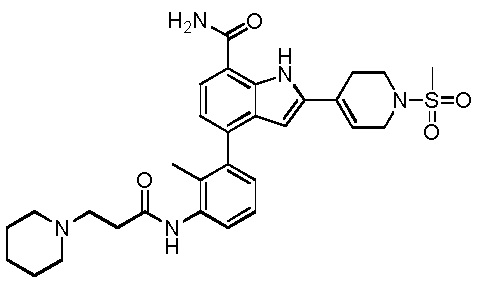
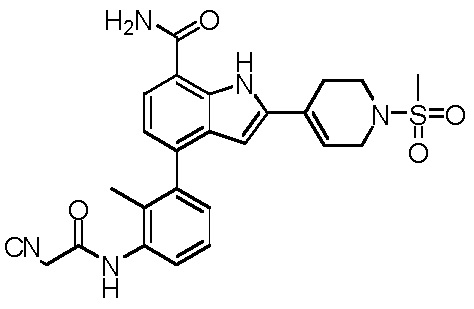
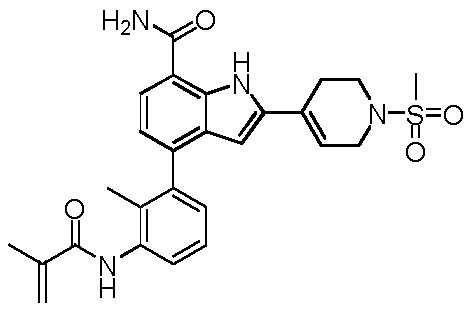
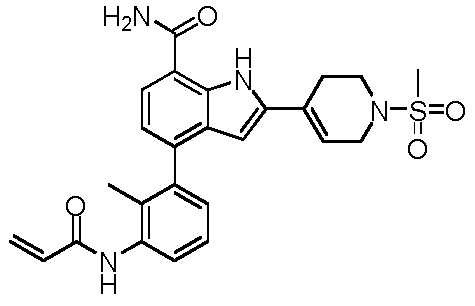
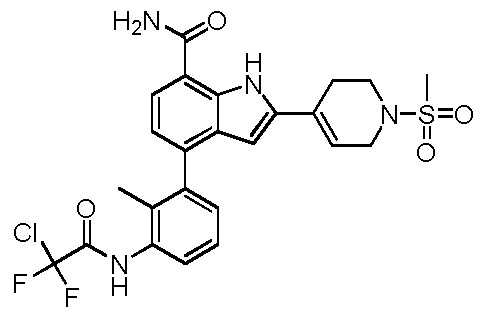
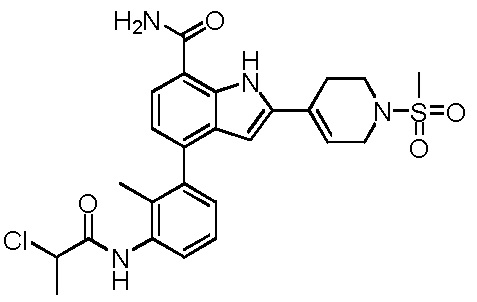
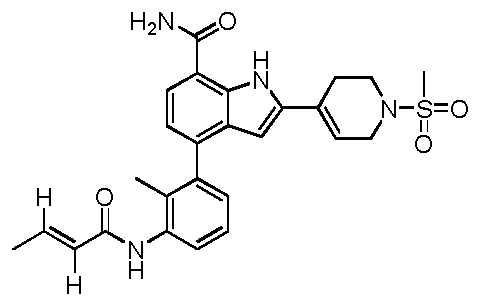
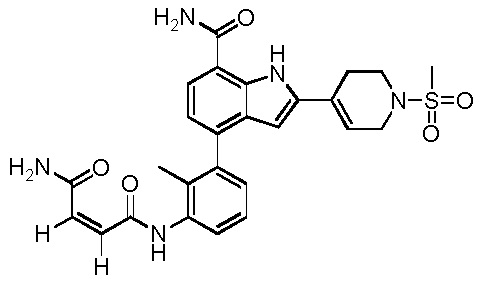
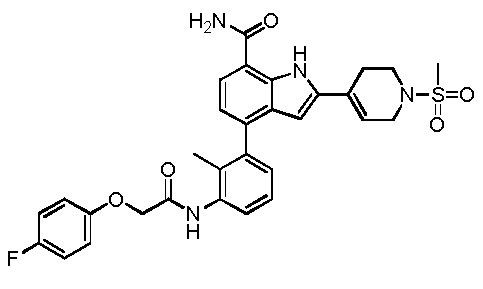
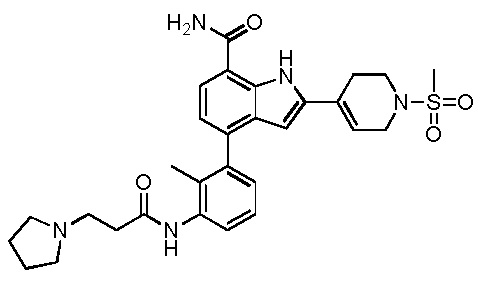

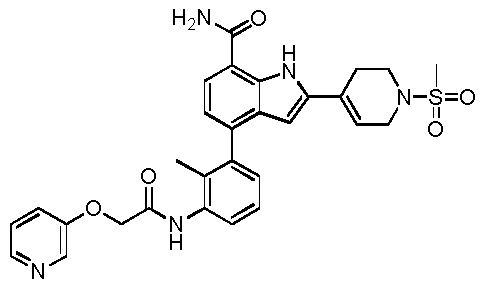
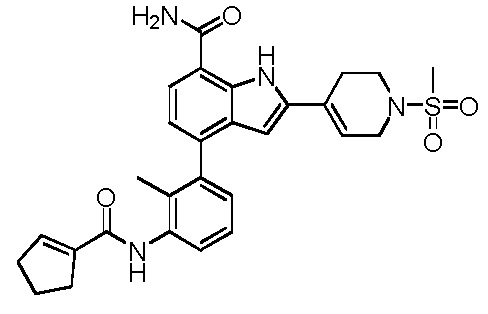
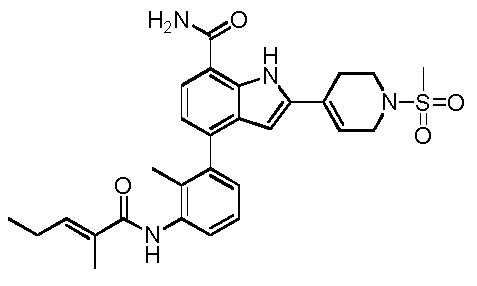
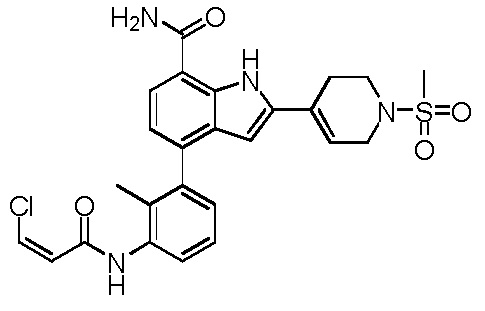
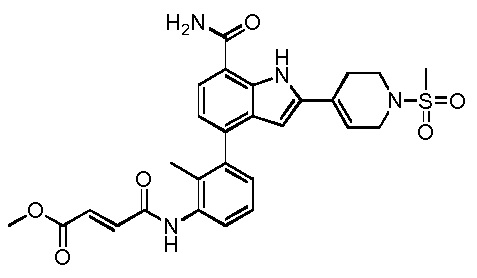

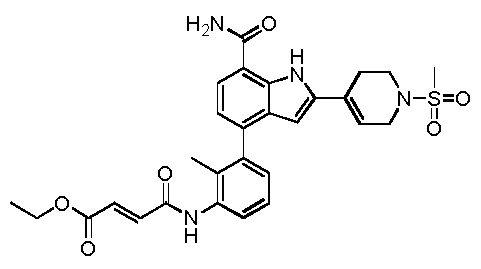
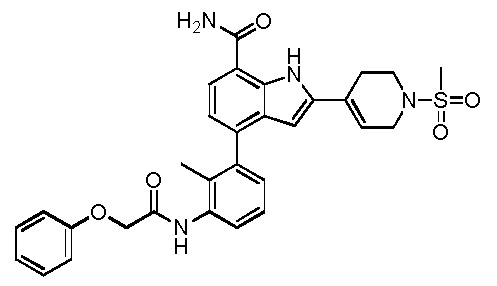
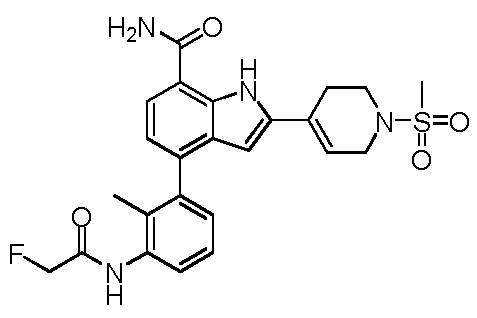
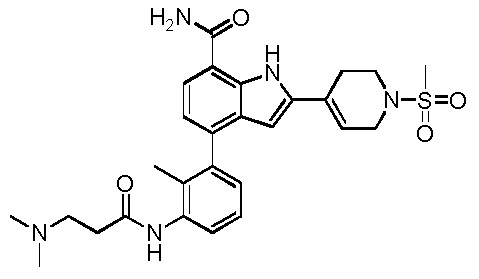
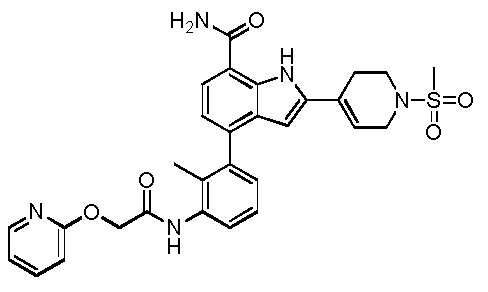
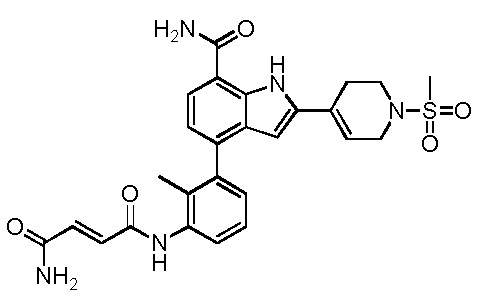
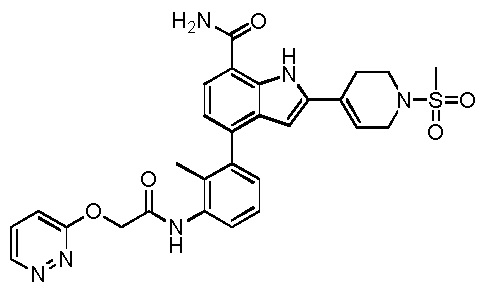

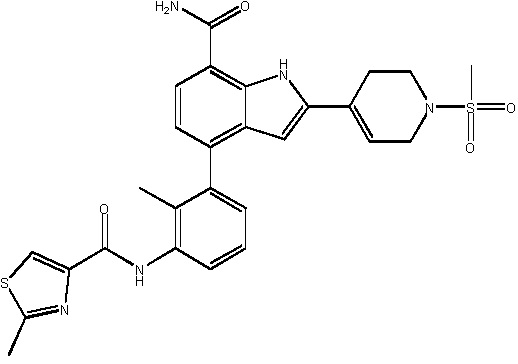
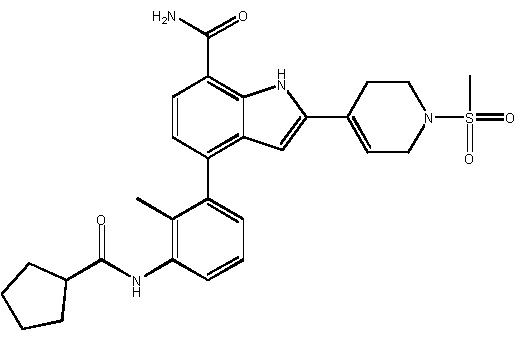
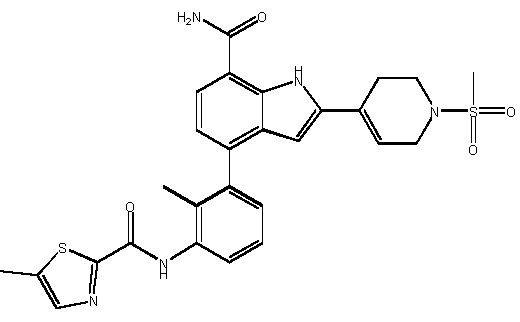
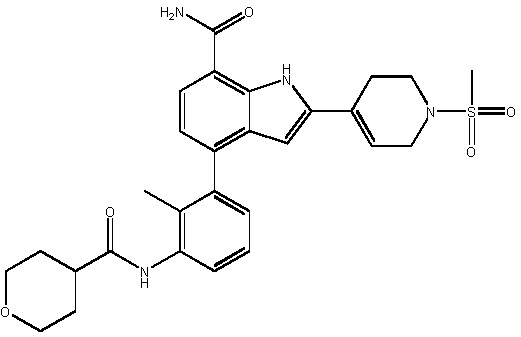

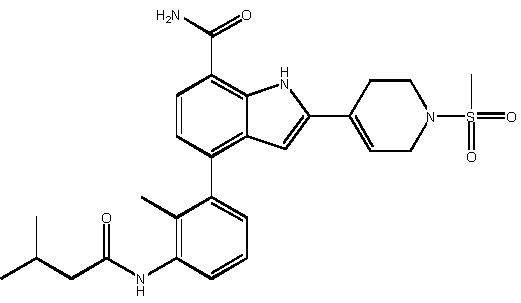
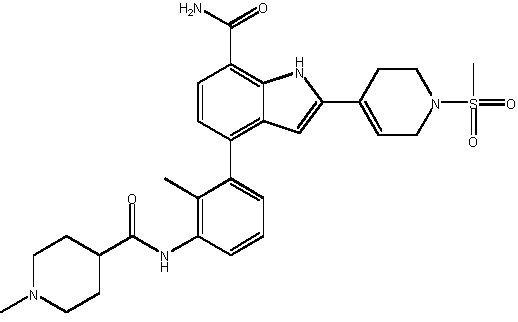
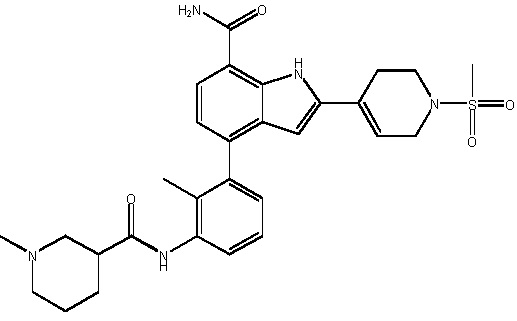
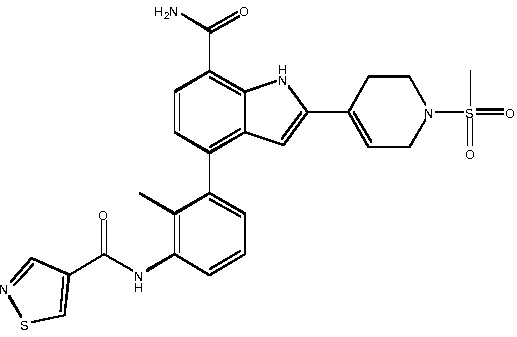

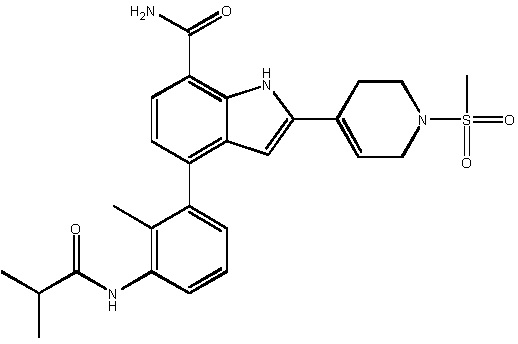
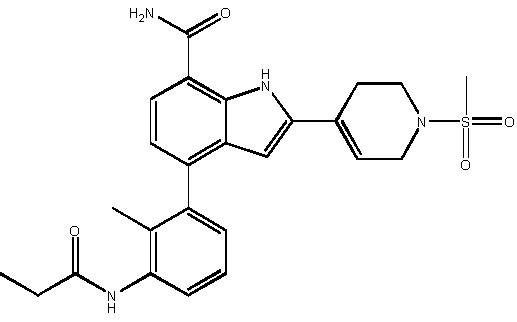
Соединение, полученные из 4-(3-амино-2-метилфенил)-1H-индол-7-карбоксамида (Пример № 16) с использованием общего способа D.
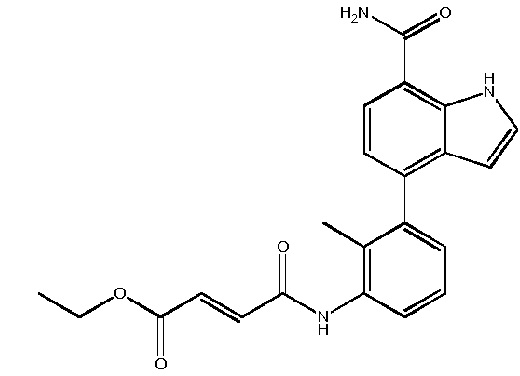
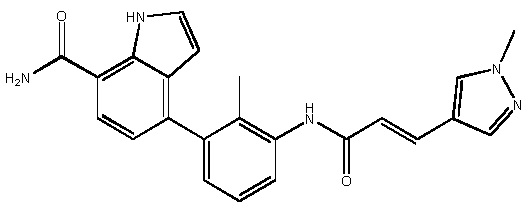

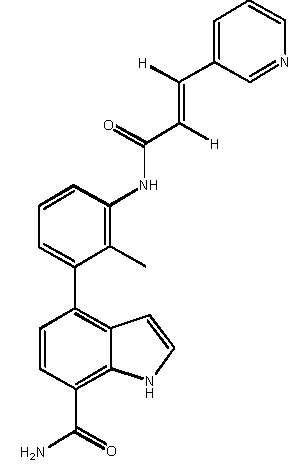
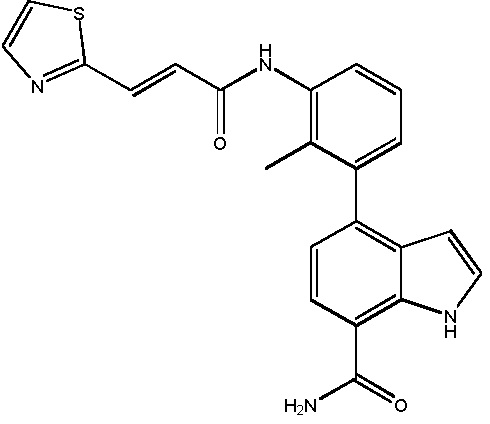
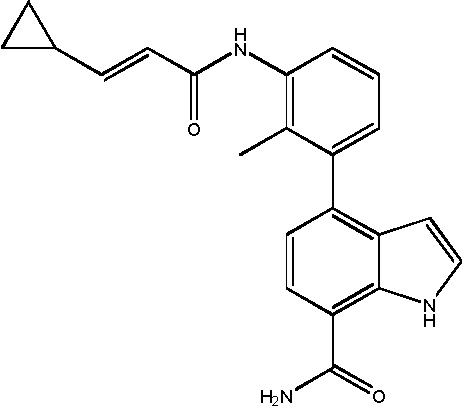
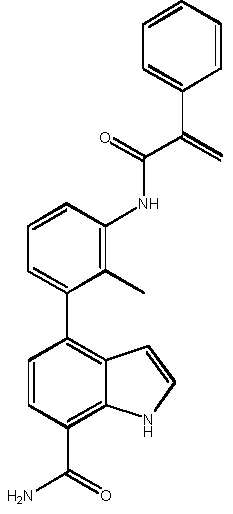
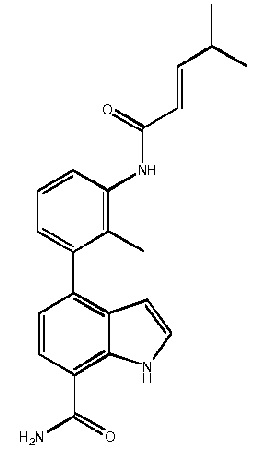
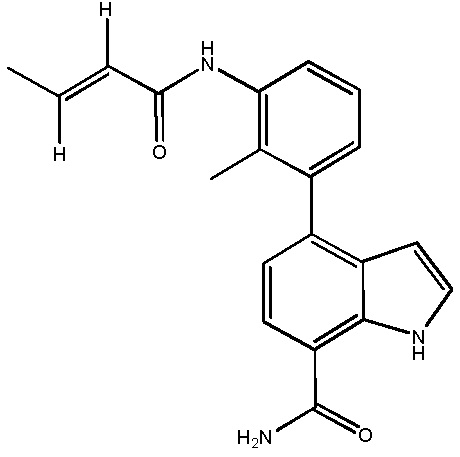
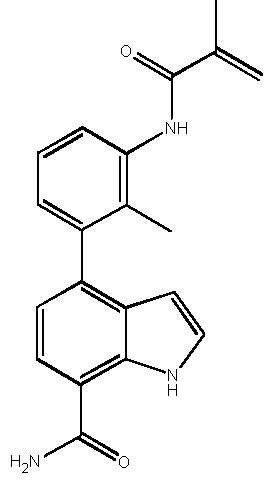
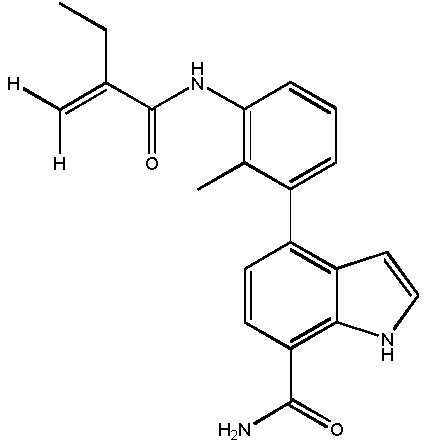
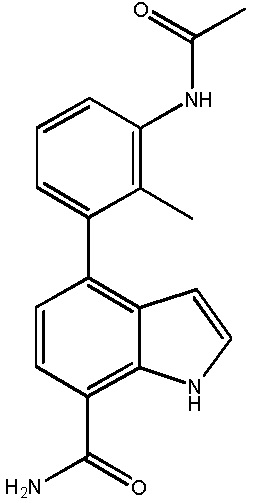
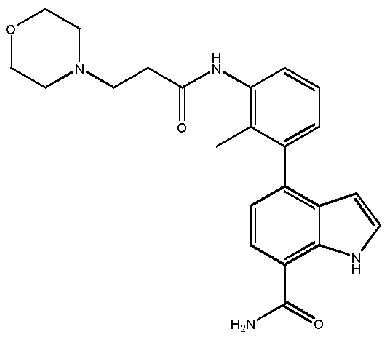
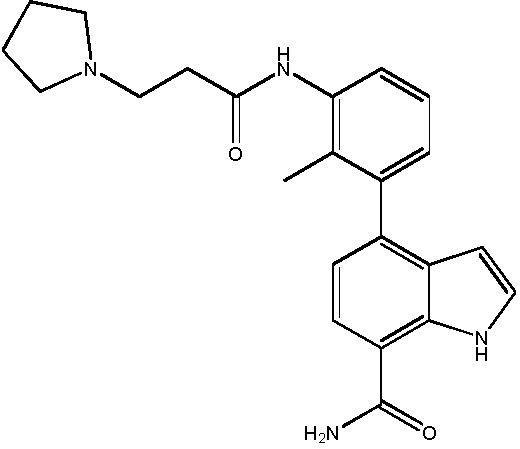
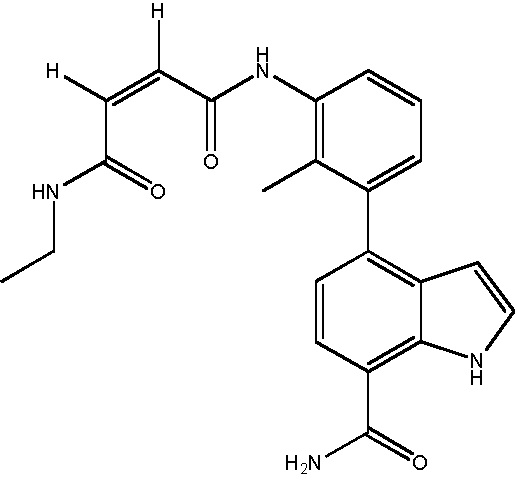
Примеры, полученные из (Z)-4-((3-(7-карбамоил-1H-индол-4-ил)фенил)амино)-4-оксобут-2-еновой кислоты (Получение № 14) с использованием общего способа D
(Таблица 1, Способ)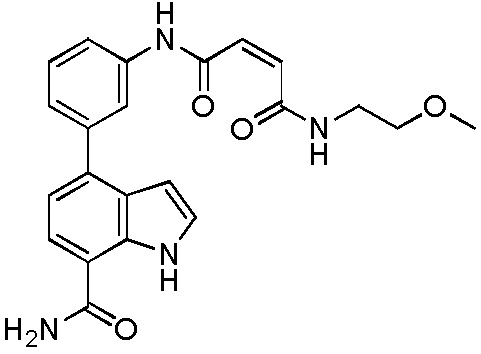
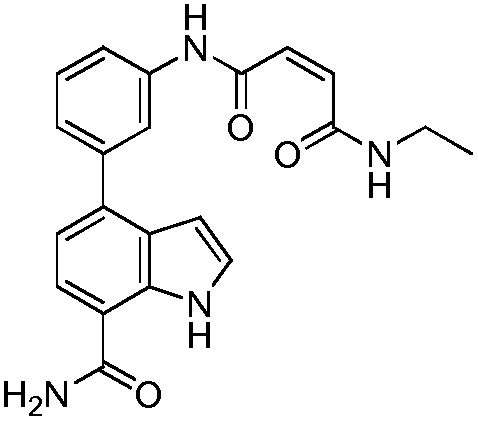
Примеры, полученные из пропиоловой кислоты с амином с использованием общего способа D
(Таблица 1, Способ)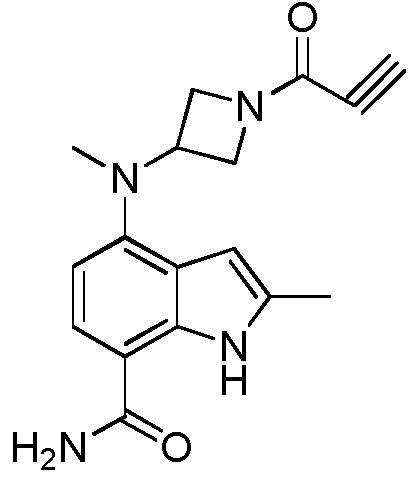
Общий способ E: Получение амида из амина и галоидангидрида или ангидрида
К раствору амина (1-3 эквив., предпочтительно, 1-3 эквив.), необязательно в виде хлористоводородной соли, в органическом растворителе (таком как DCM, DCE, DMF, DMA, NMP, THF, Et2O или 1,4-диоксан, предпочтительно DMF, DMA, или DCM), добавляли основание (такое как TEA, DIEA или пиридин; 1-4 эквив., предпочтительно, TEA или DIEA 1-3 эквив.) и галоидангидрид или ангидрид (1-4 эквив., предпочтительно, 1-4 эквив.). Смесь необязательно охлаждали до приблизительно 0°C перед добавлением галоидангидрида или ангидрида. Смесь оставляли перемешиваться при температуре приблизительно 0-60°C (предпочтительно, приблизительно 0-50°C) в течение приблизительно от 5 минут до 20 часов (предпочтительно, приблизительно от 20 минут до 2 часов). Смесь необязательно нейтрализовали AcOH. Смесь необязательно концентрировали в вакууме с получением конечного соединения. Смесь необязательно фильтровали через среду (такую как силикагель или целит), которую промывали соответствующим растворителем (таким как, EtOAc, 1,4-диоксан, THF, MeCN, DCM, Et2O, MeOH, EtOH), а затем необязательно концентрировали в вакууме с получением остатка. Или остаток или раствор необязательно разделяли между водой и органическим растворителем (таким как EtOAc, Et2O или DCM). Органический слой выделяли и необязательно промывали в произвольном порядке водой и/или водными растворами, содержащими кислоту (такую как HCl, AcOH или NH4Cl) и/или водными растворами, содержащими основание (такое как NaHCO3, Na2CO3, NaOH, KOH или NH4OH) и/или водными растворами, содержащими неорганическую соль (такую как NaCl, Na2SO3 или Na2S2O3). Органический раствор затем необязательно сушили с высушивающим веществом (например, безводным MgSO4 или Na2SO4), фильтровали и концентрировали в вакууме с получением целевого соединения. Альтернативно, остаток от концентрации реакционной смеси суспендировали в воде, обрабатывали ультразвуком и собирали вакуумным фильтрованием.
Иллюстрация общего способа E:
Пример № E.1. 4-(3-акриламидо-2-метилфенил)-2-(4,4-дифторциклогекс-1-ен-1-ил)-1H-индол-7-карбоксамид
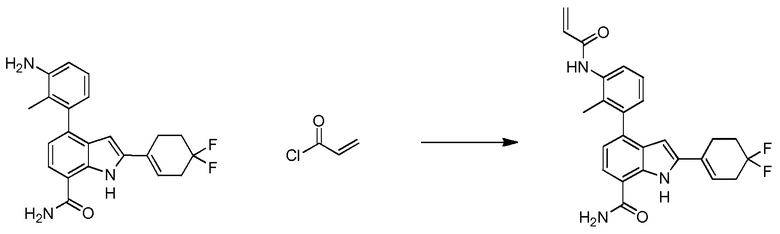
В сосуд добавляли 4-(3-амино-2-метилфенил)-2-(4,4-дифторциклогекс-1-ен-1-ил)-1H-индол-7-карбоксамид (0,189 г, 0,496 ммоль, Пример № 21) в DCM (5 мг), и DIEA (0,129 мл, 0,743 ммоль). Смесь охлаждали до приблизительно 0°C и акрилоилхлорид (0,044 мл, 0,545 ммоль) добавляли при перемешивании. Смесь нагревали до комнатной температуры в течение примерно 20 минут, затем концентрировали и остаток суспендировали в воде (30 мг). Суспензию обрабатывали ультразвуком в течение 5 минут, фильтровали, промывали водой, простым эфиром и сушили в вакууме. Сырой продукт добавляли в колонку с силикагелем и элюировали гептаном/EtOAc (0-100%) с получением 4-(3-акриламидо-2-метилфенил)-2-(4,4-дифторциклогекс-1-ен-1-ил)-1H-индол-7-карбоксамида (0,16 г, 74%): ЖХ/МС (Таблица 1, способ g) Время удерживания = 3,02 мин; MS m/z: 436 (M+H)+. (BTK IC50=A)
Примеры, полученные из акрилоилхлорида с использованием общего способа E
(Таблица 1, способ)
Получения № 18 и
трет-бутил 2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензилкарбамата
[JW] и G с HCl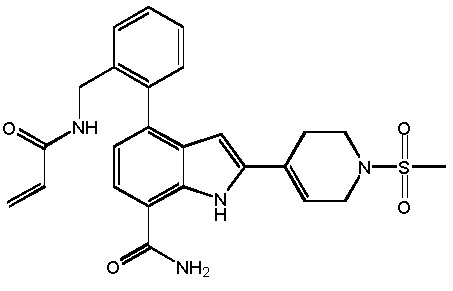

(Пример № A.5.3)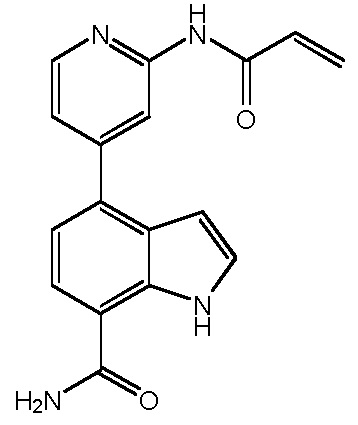
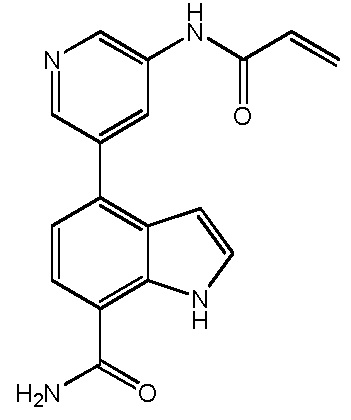
(полученный с использованием A из
Получения № P.1 и
3-бром-N-метиланилина)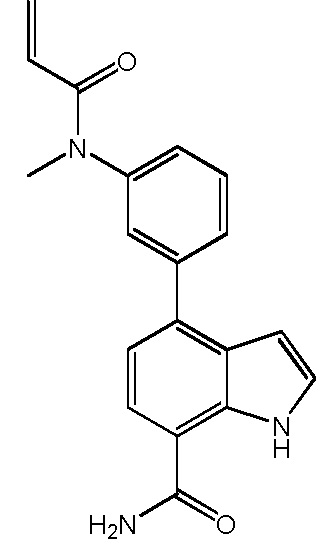
Получения № P.1 и
3-бром-N,2-диметиланилина [Beta Pharm])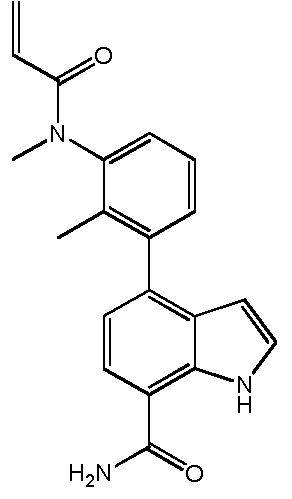
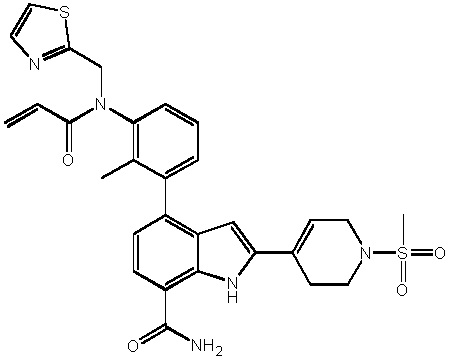
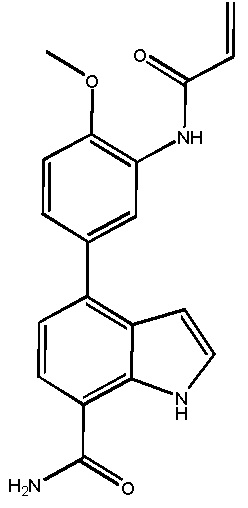
2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина [CombiBlocks])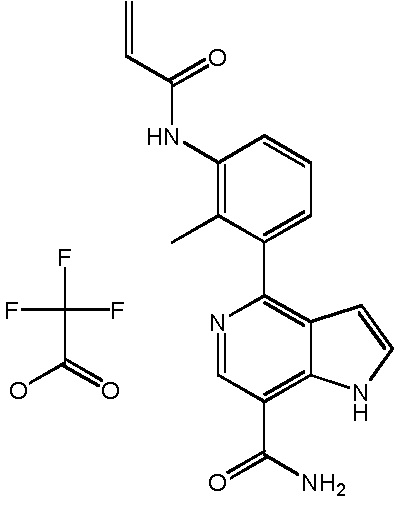
3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина)
(полученный с использованием B из Получения № 27 и
(R)-трет-бутил пиперидин-3-илкарбамата, N с Cs2CO3, G с HCl, и O)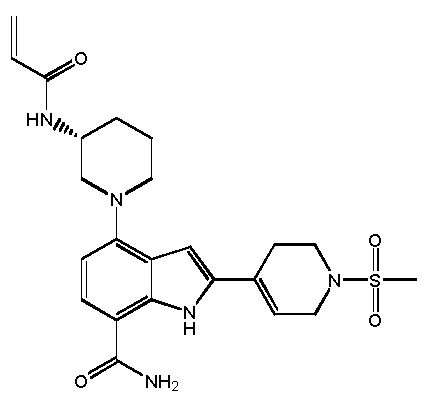
Получение № 2 и Получение № 34)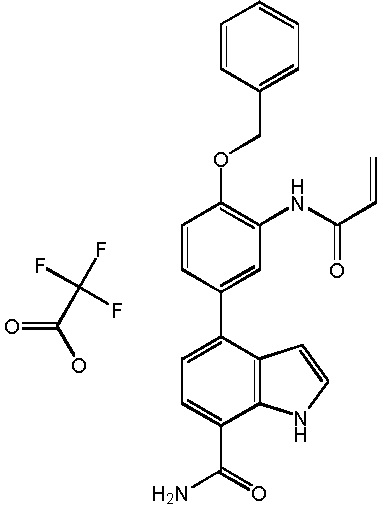
(полученный с использованием R из
Получения № Q.1, A из Получения № P.1)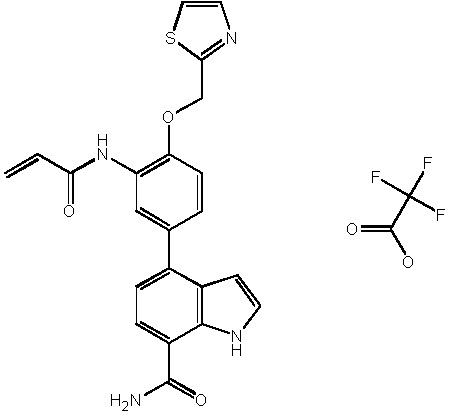
Получение № 2 и Получение № 35)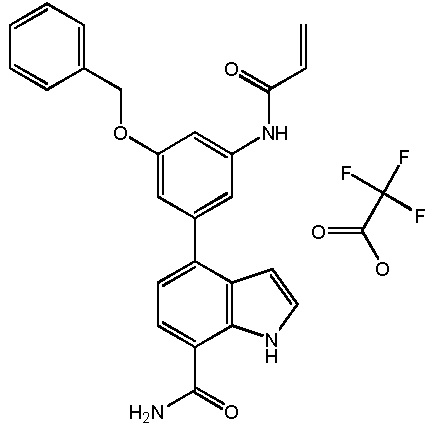
1-бром-3-метокси-5-нитробензола с BBr3, Q из тиазол-2-илметанола, R с Fe, P с
4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксабороланом), и A из Получения № 2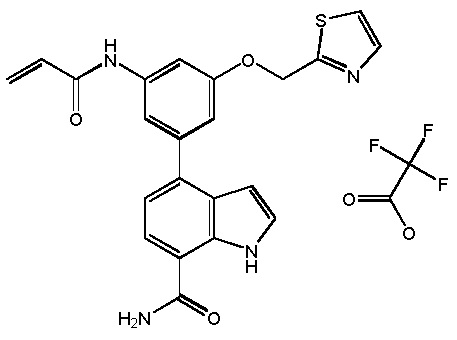
Получения № R.1)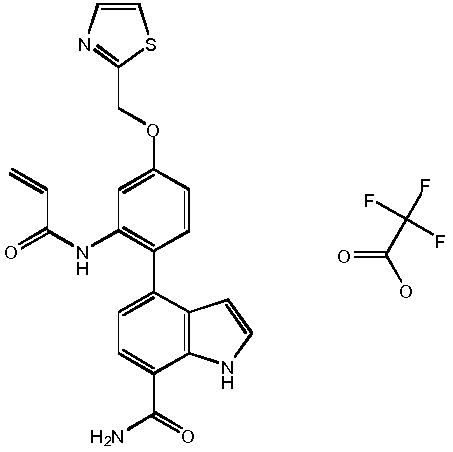
Получения № 36
с Fe, и A из Получения № P.1)
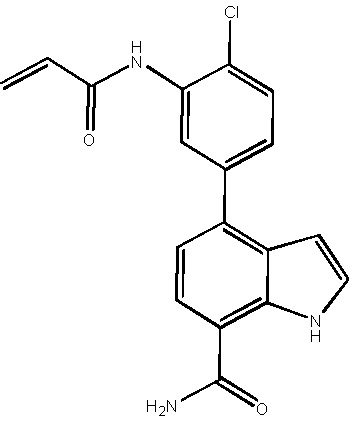
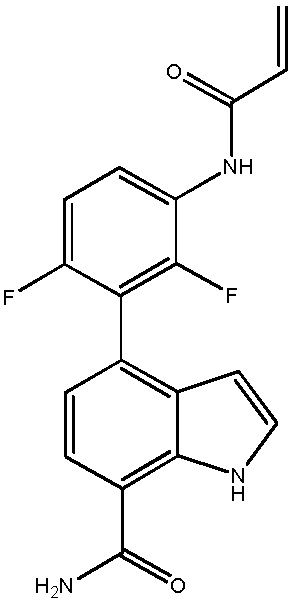
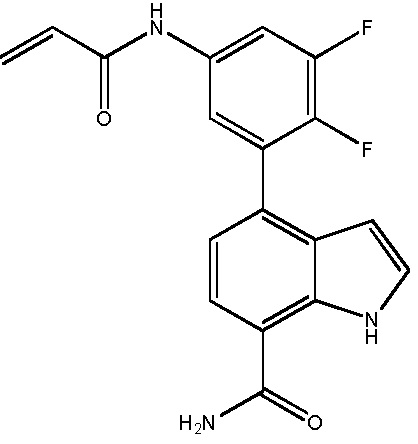
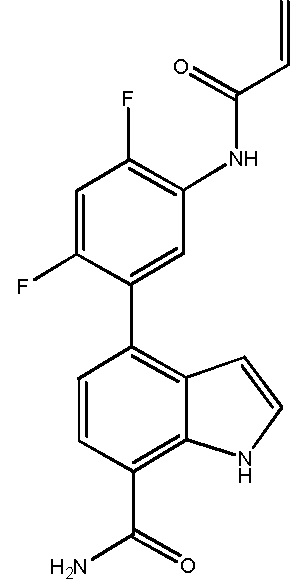
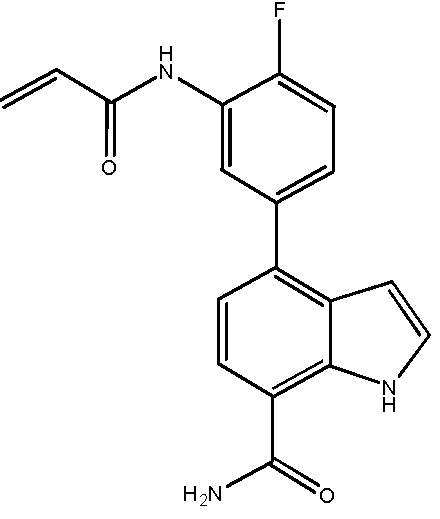
3-бром-4-хлоранилина)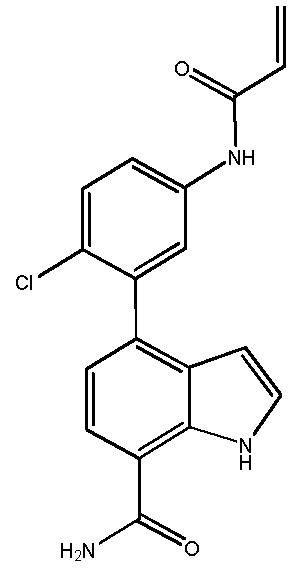
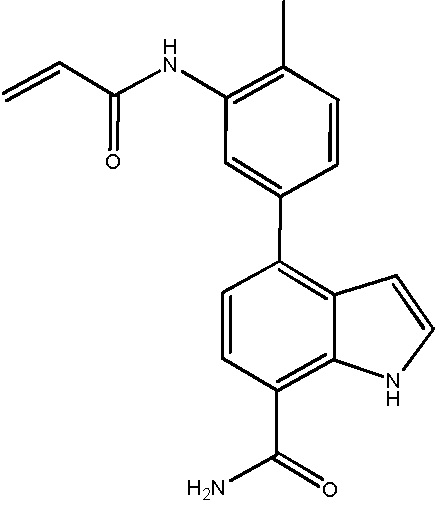
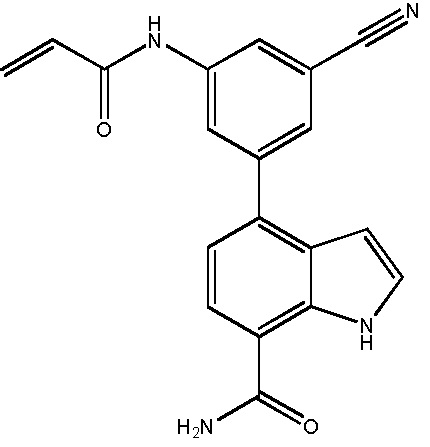

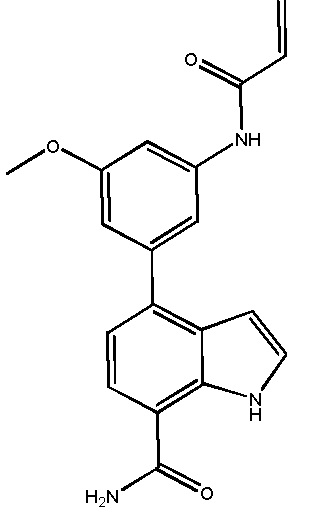
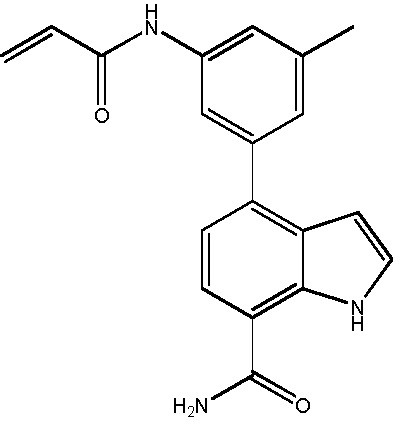

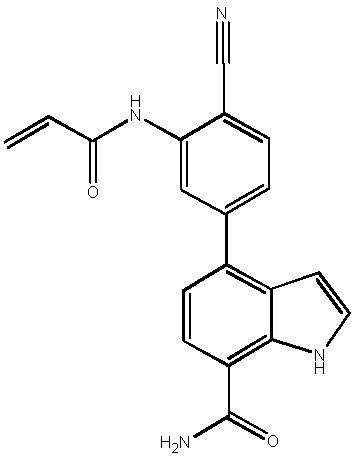
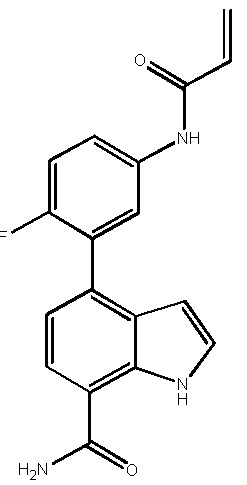
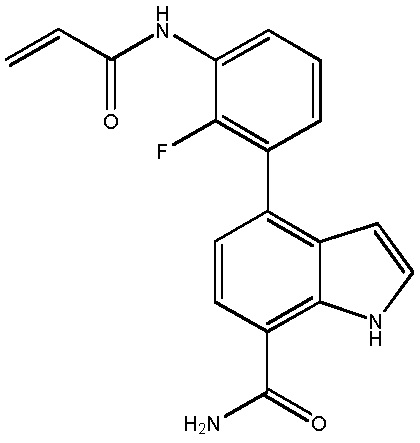
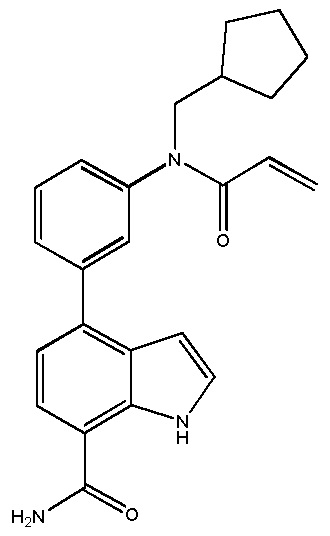
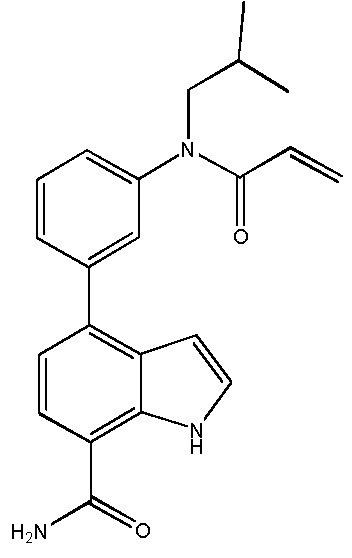
Примеры, полученные из 4-(3-аминофенил)-1H-индол-7-карбоксамид
(Получение № A.1) с использованием общего способа E
(Таблица 1, Способ)
[J. Med. Chem., 1980, 23 (8) 865]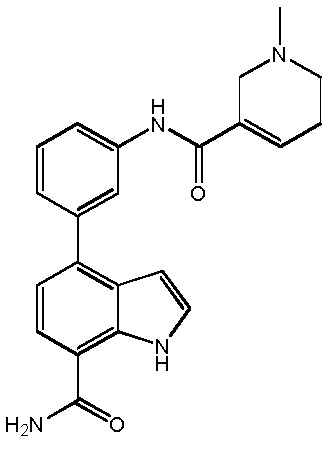
Примеры, полученные из 4-(2-аминофенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамида (Пример № A.4.2) с использованием общего способа E
(Таблица 1, Способ)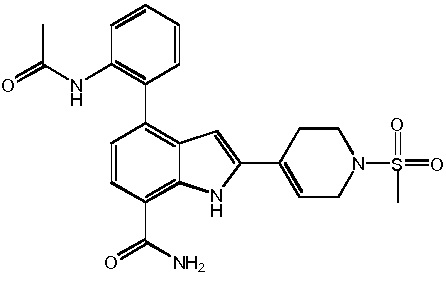
Примеры, полученные из N-(3-(2-(2-(аминометил)фенил)-7-карбамоил-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамид (Пример № 1) с использованием общего способа E
(Таблица 1, Способ)
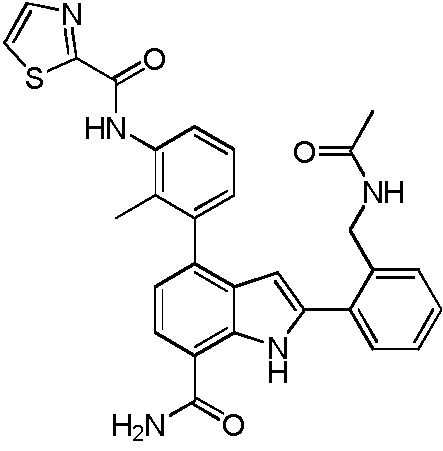
Примеры, полученные из N-(3-(3-амино-7-карбамоил-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамид (Получение № 7) с использованием общего способа E
(Таблица 1, Способ)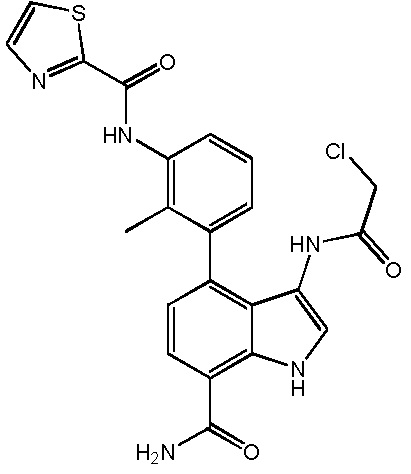
Примеры, полученные из этил карбоно-хлоридата с использованием общего способа E
(Таблица 1, Способ)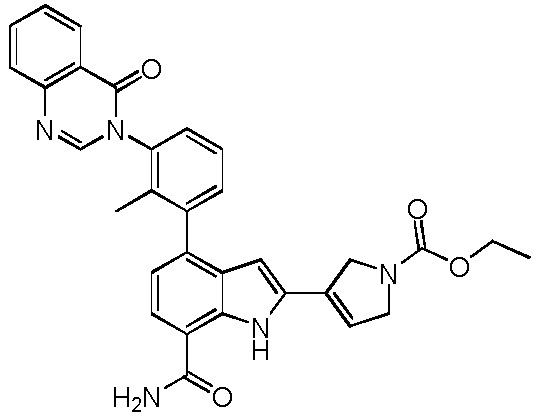
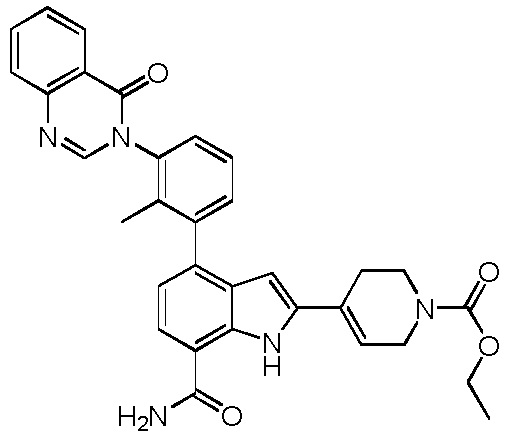
Примеры, полученные из 2-оксопропаноил хлорида (полученный из пировиноградной кислоты и 1,1-дихлордиметилового эфира [Synthesis, 1975, 3 163-164]) с использованием общего способа E
(Таблица 1, Способ)
(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)метанамина с 4-бром-1H-индол-7-карбоксамидом [Получение № 2])
Примеры, полученные из ацетилхлорида с использованием общего способа E
(Таблица 1, Способ)
Примеры, полученные из акрилоилхлорида с амином с использованием общего способа E
(Таблица 1, Способ)
Получения № 18 с
трет-бутил 2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензилкарбаматом
[JW] и G с HCl)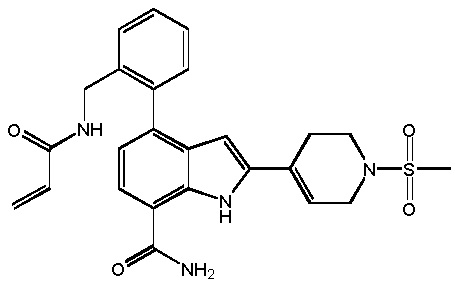
Получения № 40 с
циклопропилбороновой кислотой [SCRC] и G с HCl)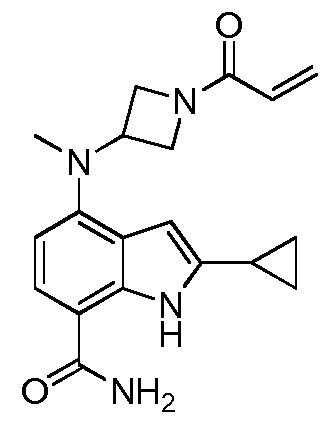
Получения № 40 с
2-(изохроман-7-ил)-4,4,5,5-тетраметил-1,3,2-диоксабороланом
[полученный с использованием P и
7-бромизохроман] и G с HCl)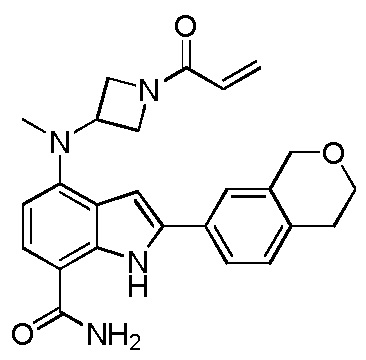
4,4,5,5-тетраметил-1,3,2-диоксабороланом, A с Получением № 44, C с LiOH, D с NH4Cl и G с HCl)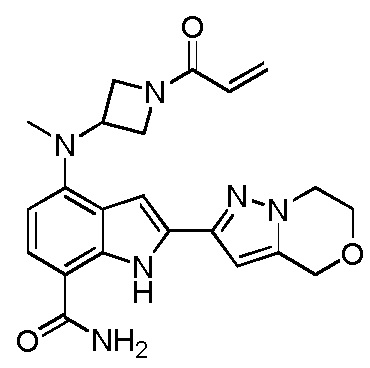
(полученный с использованием A из
Получения № 40 с
2-(4,4-дифторциклогекс-1-ен-1-ил)-4,4,5,5-тетраметил-1,3,2-диоксабороланом
[Syngene] и G с HCl)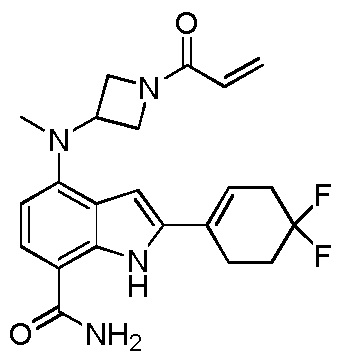
Получения № 40 с
4,4,5,5-тетраметил-2-(4-(метилсульфонил)-циклогекс-1-ен-1-ил)-1,3,2-диоксабороланом (WO2005/73206 A1)
и G с HCl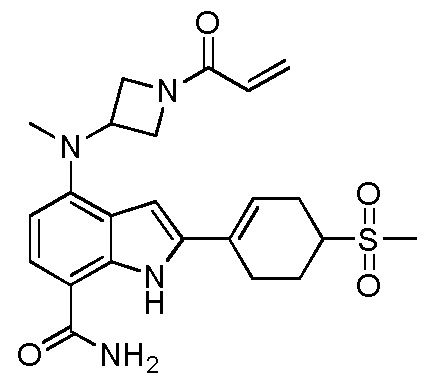
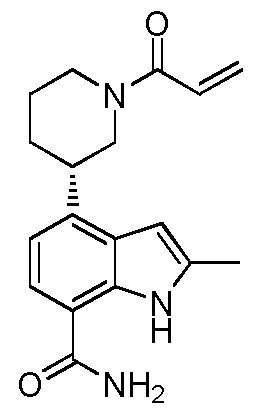
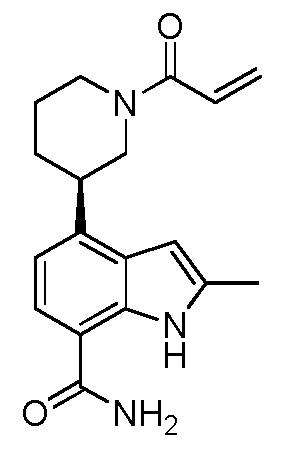
4-(5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)морфолином и G с HCl)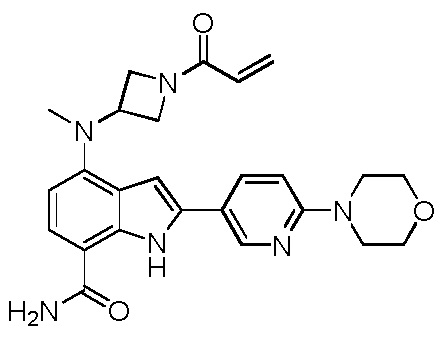
(Полученный с использованием A из Получения № 40 с
(7,8-дигидро-5H-пирано[4,3-b]пиридин-3-ил)бороновой кислотой [Anichem]) и G с HCl)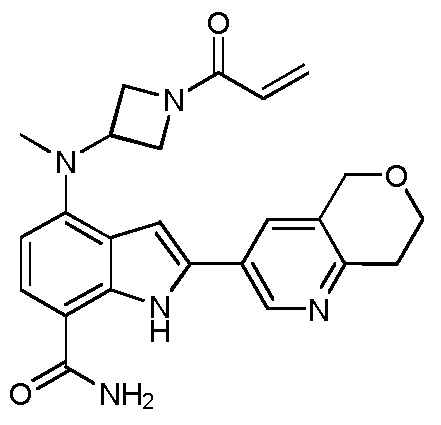
(Полученный с использованием P из 7-бромхромана [Arkpharm] с бис(пинаколато)-дибороном, A с Получением № 40 и G с HCl)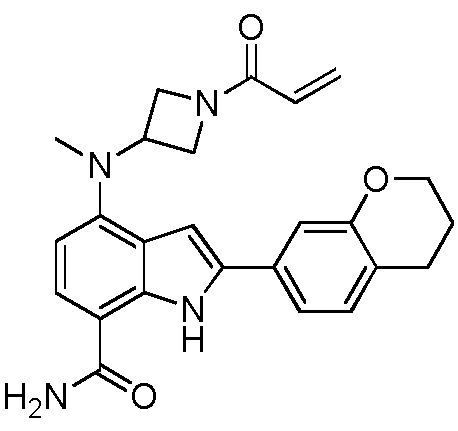
G из Получения № 48 с HCl)
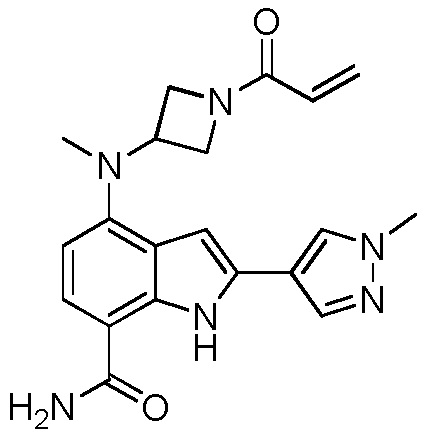
(Полученный с использованием A из Получения № 40 с трет-бутил 3-((7-карбамоил-2-йод-1H-индол-4-ил)(метил)амино)-азетидин-1-карбоксилатом [Arkpharminc] и G с HCl)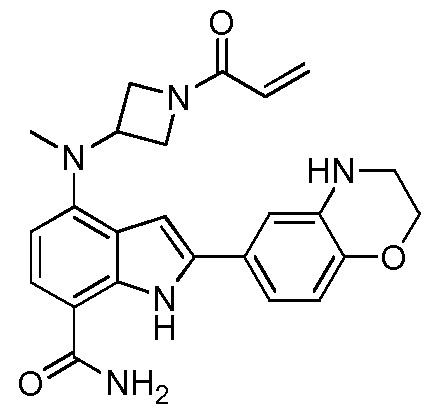
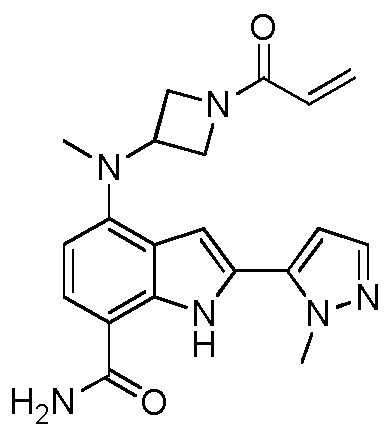
(Полученный с использованием A из Получения № 40 с 1-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразолом и G с HCl)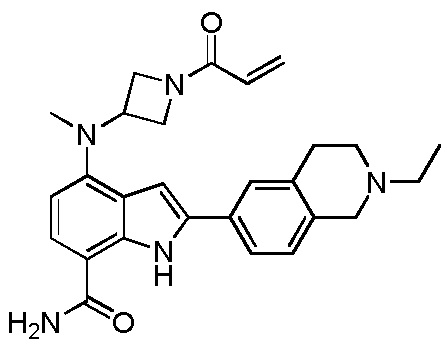
(Полученный с использованием A из Получения № 40 с 1,3-диметил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразолом и G с HCl)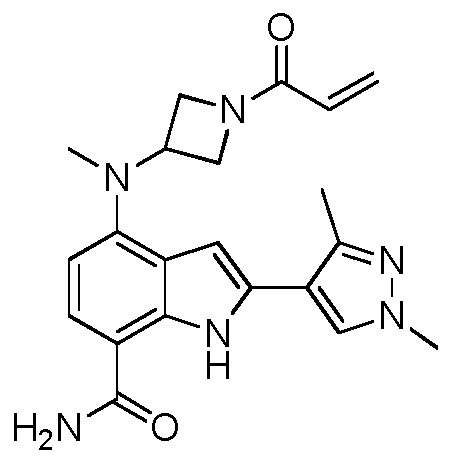
(Полученный с использованием A из Получения № 40 с 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,6-дигидро-2H-тиопираном 1,1-диоксидом
[JWpharmlab], L с Pd/C и G с HCl)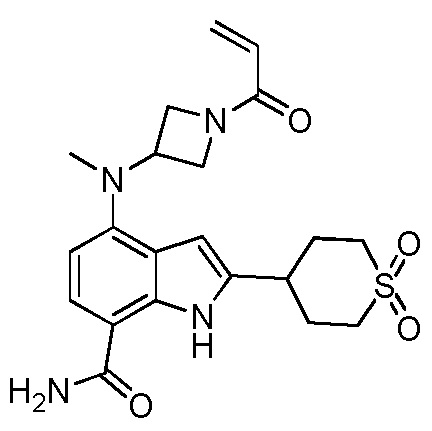
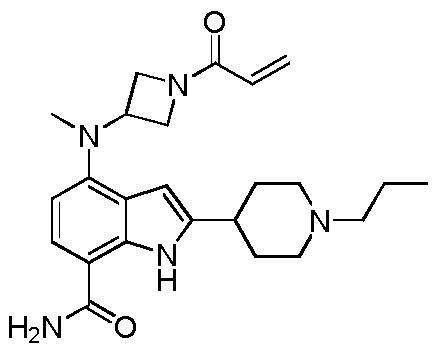

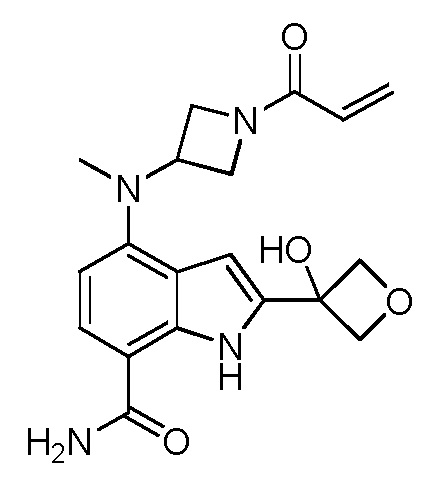
(Полученный с использованием Y из Получения № 43, A с 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразолом [Arkpharm], O, хиральное разделение (Таблица 2, способ 4) и G с HCl)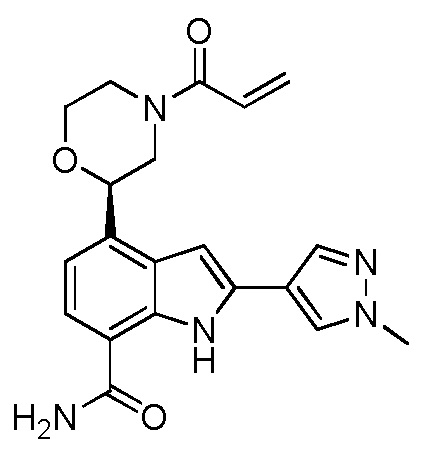
(Полученный с использованием Y из Получения № 43, A с 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразолом [Arkpharm], O, хиральное разделение (Таблица 2, способ 4) с G с HCl))
P из Получения № Y.1 с
4,4,5,5-тетраметил-1,3,2-диоксабороланом, A с Получением № 44, хиральное разделение (Таблица 2, Способ 6), C с LiOH, D с NH3 и G с HCl)
P из Получения № Y.1 с
4,4,5,5-тетраметил-1,3,2-диоксабороланом, A с Получением № 44, хиральное разделение (Таблица 2, Способ 6), C с LiOH, D с NH3 и G с HCl)
трет-бутил 3-ацетилазетидин-1-карбоксилата [JWpharm] с N-(5-хлорпиридин-2-ил)-1,1,1-трифтор-N-((трифторметил)-сульфонил)метан-сульфонамидом, W с 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан)], A с Получением № 37, L с Pd/C, C с LiOH, D с NH4Cl, хиральное разделение (Таблица 2, Способ 7) и G с HCl)

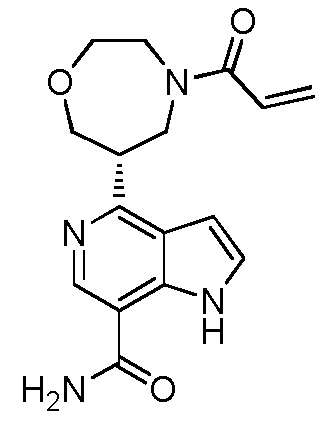
C из Получение № AH.1 с LiOH, D с NH4Cl, L с Pd(OH)2, хиральное разделение (Таблица 2, Способ 8) и G с HCl)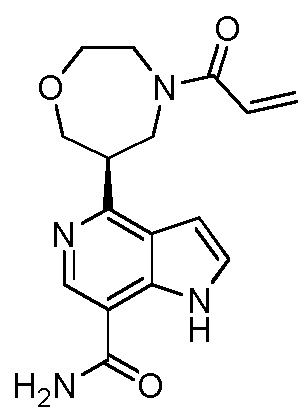
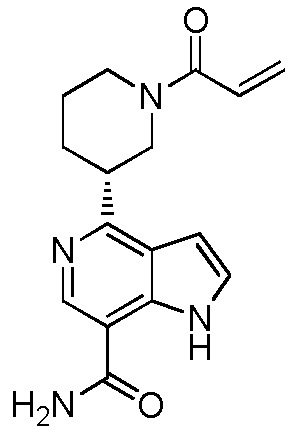
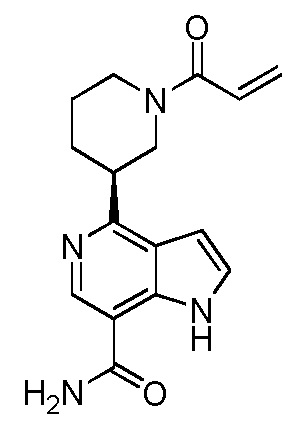
трет-бутил 3-аминоазетидин-1-карбоксилат[arkpharm] и G с HCl)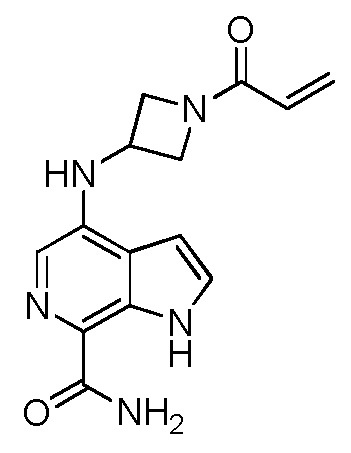
(Полученный с использованием T из Получения № 1, Стадия C и трет-бутил 3-амино-3-метилазетидин-1-карбоксилата [AKSCI], J с CH3I, X с LiOH, D с NH4Cl и G с HCl)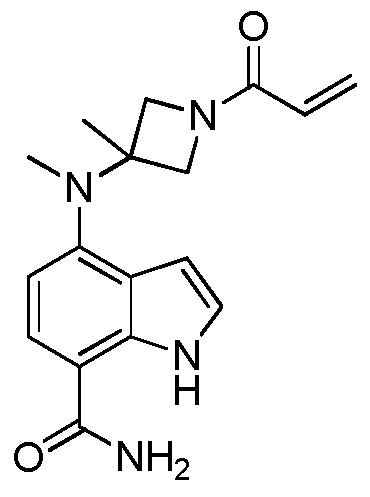
трет-бутил 3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2H)-карбоксилатом, L с Pd/C, хиральное разделение (Таблица 2, Способ 10) и G с HCl)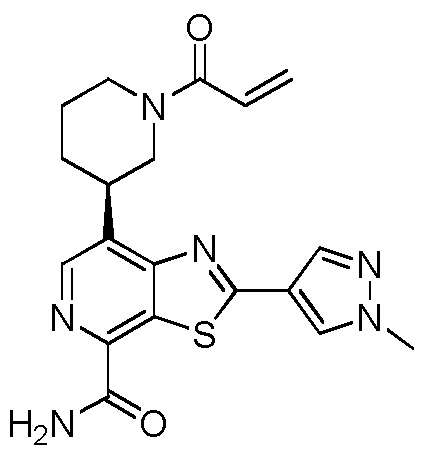
трет-бутил 3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2H)-карбоксилатом, L с Pd/C, хиральное разделение (Таблица 2, Способ 10) и G с HCl)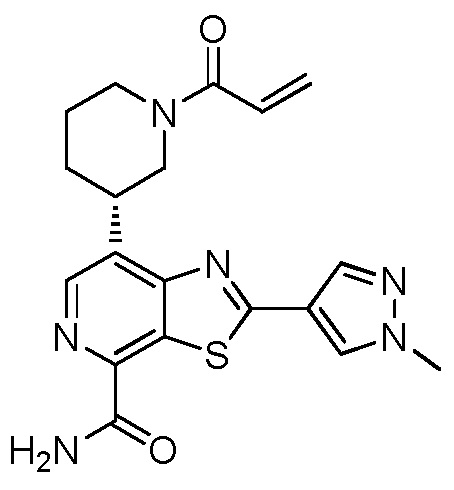
трет-бутил 6-оксо-1,4-оксазепан-4-карбоксилатом [Arkpharm] и
1,1,1-трифтор-N-фенил-N-((трифторметил)-сульфонил)метан-сульфонамидом, A с Получением № P.1, L с Pd/C, хиральное разделение (Таблица 2, Способ 11) и G с HCl)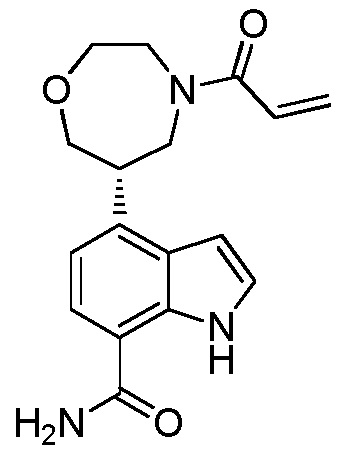
трет-бутил 6-оксо-1,4-оксазепан-4-карбоксилатом [Arkpharm] и
1,1,1-трифтор-N-фенил-N-((трифторметил)-сульфонил)метан-сульфонамид, A с Получением № P.1, L с Pd/C, хиральное разделение (Таблица 2, Способ 11) и G с HCl)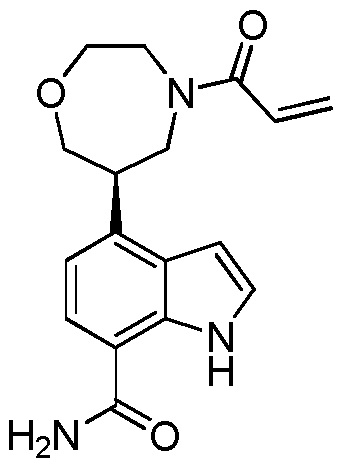
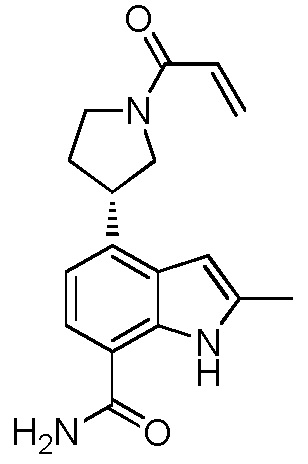
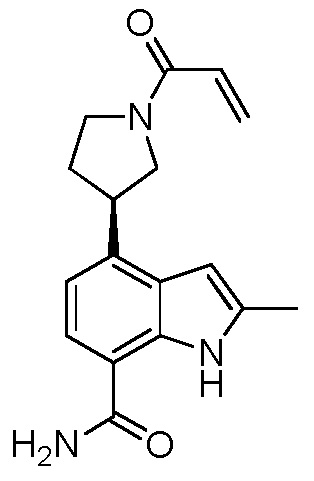

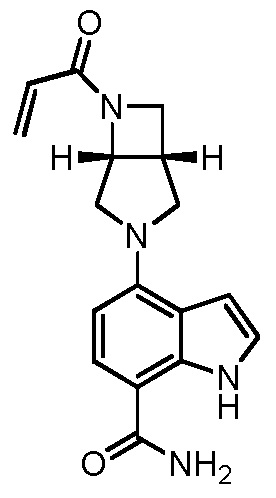
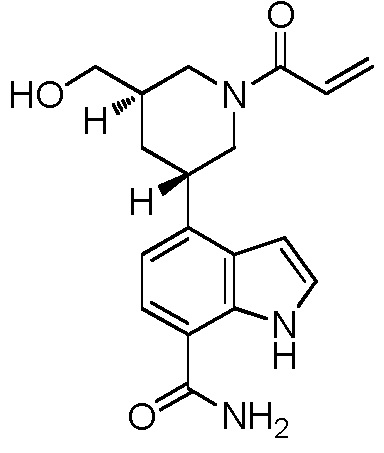
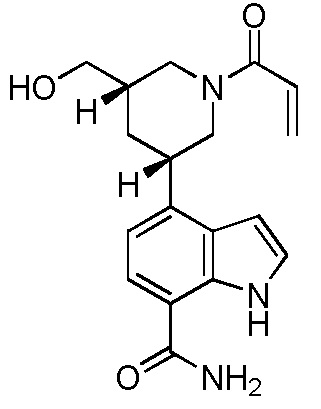
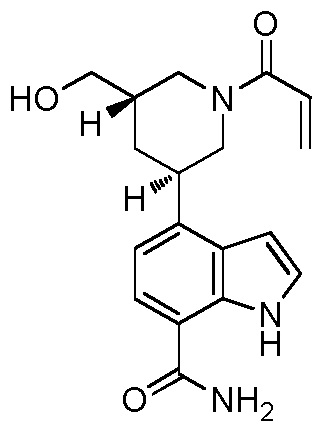
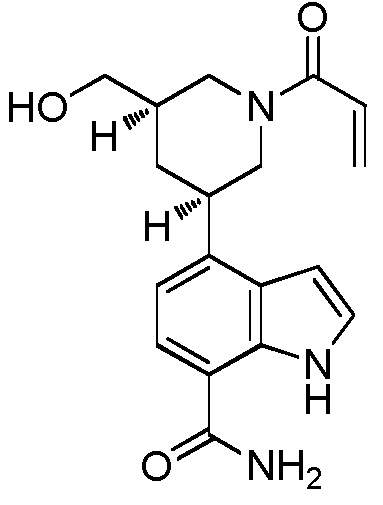
1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразолом [arkpharm], хиральное разделение (Таблица 2, Способ 17), C с LiOH, D с NH3 и G с HCl)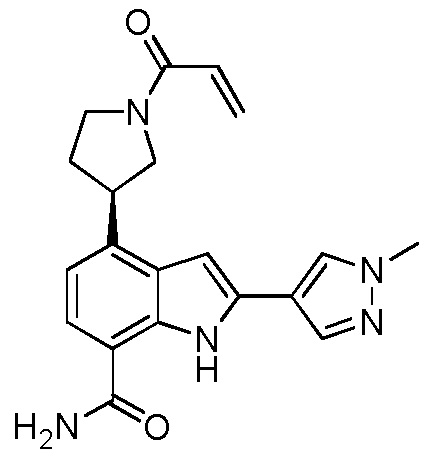
1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразолом [arkpharm], хиральное разделение (Таблица 2, Способ 17), C с LiOH, D с NH3 и G с HCl)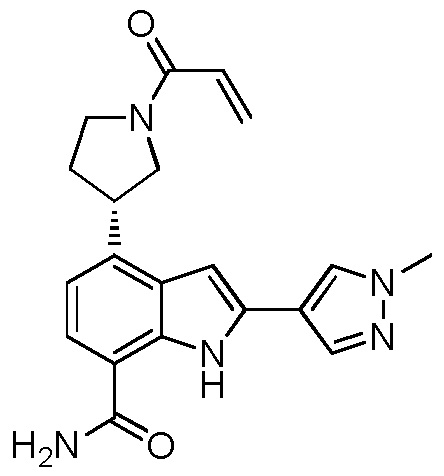
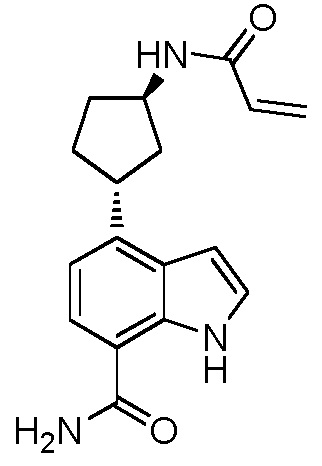
трет-бутил 3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2H)-карбоксилатом, O, L с Pd/C, хиральное разделение (Таблица 2, Способ 18) и G с ацетилхлоридом)
трет-бутил 3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2H)-карбоксилатом, O, L с Pd/C, хиральное разделение (Таблица 2, способ 18) и G с ацетилхлоридом)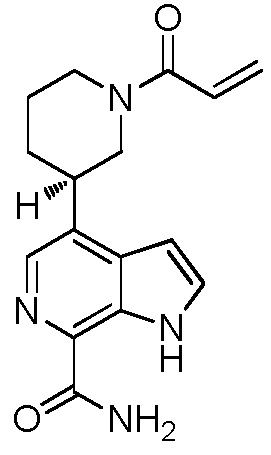
Примеры, полученные из акрилоилхлорида с амином с использованием общего способа E
(Таблица 1, Способ)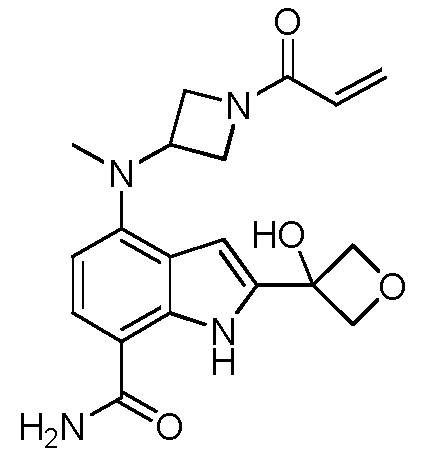
Примеры, полученные из пропионилхлорида с амином с использованием общего способа E
(Таблица 1, Способ)
(полученный с использованием хирального разделения (Таблица 2, способ 12) из Получения № 38, C с LiOH, D с NH3 и G с HCl)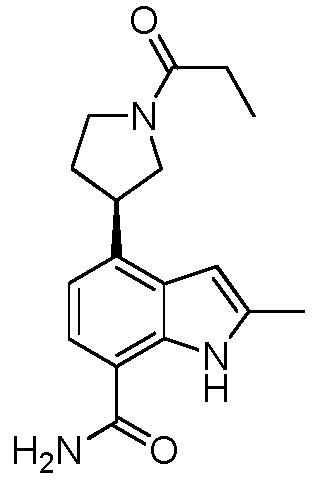
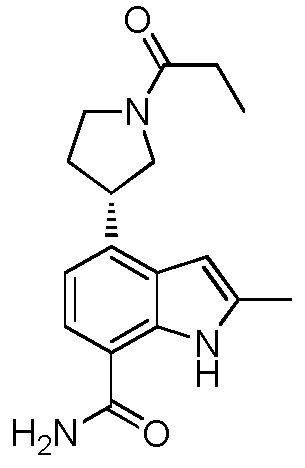
Общий способ F: Образование 4-йодиндол-7-карбоксамида
К раствору 2-амино-4-нитробензойной кислоты (предпочтительно, 1 эквив.) в MeOH медленно добавляли концентрированную серную кислоту (предпочтительно, 1 эквив.). Полученный раствор нагревали при приблизительно 75°C в течение приблизительно 3 дней. После охлаждения реакционную смесь нейтрализовали добавлением водного раствора NaOH до рН~10. Реакционную смесь экстрагировали этилацетатом, сушили над безводным сульфатом натрия, фильтровали и концентрировали. К этому промежуточному соединению (предпочтительно 1 эквив.) добавляли метилкетон (1-2 эквивалента, предпочтительно 2 эквив.) и органический растворитель (предпочтительно, диметилсульфоксид). Реакционную смесь охлаждали до приблизительно -15°C. Добавляли основание (предпочтительно, трет-бутоксид калия, 2 эквив.). После перемешивания в течение приблизительно 2,5 часов при комнатной температуре, реакционную смесь гасили насыщенным водным раствором хлорида аммония и затем перемешивали в течение приблизительно 1 часа при комнатной температуре. Полученную суспензию фильтровали, промывали водой и твердое вещество сушили в высоком вакууме. К промежуточному соединению (предпочтительно 1 эквивалент) добавляли ((1H-бензо[d][1,2,3]триазол-1-ил)окси)три(пирролидин-1-ил)фосфония гексафторофосфат (V) (предпочтительно, 2 эквив.), гидроксибензотриазол гидрат (предпочтительно, 2 эквив.) и хлорид аммония (предпочтительно, 1,5 эквив.) и органический растворитель (предпочтительно, DMF). Добавляли органическое основание (предпочтительно, диизопропилэтиламин, 4 эквив.). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь выливали в воду и полученный осадок отфильтровывали, промывали водой и EtOAc и собирали. К этому промежуточному соединению (предпочтительно, 1 эквив.) добавляли органический растворитель (предпочтительно, MeOH), и раствор продували азотом. К этому раствору добавляли 10% палладий на угле (предпочтительно 0,1 эквив.). Полученную суспензию обрабатывали водородом (30 фунт на кв.дюйм). После перемешивания в течение ночи при комнатной температуре, реакционную смесь фильтровали, и твердое вещество промывали МеОН. Фильтрат концентрировали. Раствор нитрита натрия (предпочтительно 2,2 эквив.) в воде добавляли к охлажденной льдом суспензии этого промежуточного соединения (предпочтительно, 1 эквив.) в органическом растворителе (предпочтительно, MeCN) и 2 н HCl (предпочтительно 5,4 эквив.) при перемешивании, поддерживая температуру ниже приблизительно -5°C. После перемешивания в течение приблизительно 30 минут, холодный раствор водного иодида калия (предпочтительно 2,5 эквив.) добавляли к реакционной смеси и полученную смесь перемешивали при комнатной температуре в течение приблизительно 30 минут. Реакционную смесь нагревали при приблизительно 85°C в течение приблизительно 5 минут. Реакционную смесь охлаждали до комнатной температуры и нейтрализовали насыщенным водным раствором бикарбоната натрия до рН 8. Смесь экстрагировали DCM. Органический слой промывали насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали флэш-хроматографией (предпочтительно, силикагель, петролейный эфир) с получением целевого соединения.
Иллюстрация общего способа F
Пример № F.1: 4-Йод-2-(пиридин-3-ил)-1H-индол-7-карбоксамид
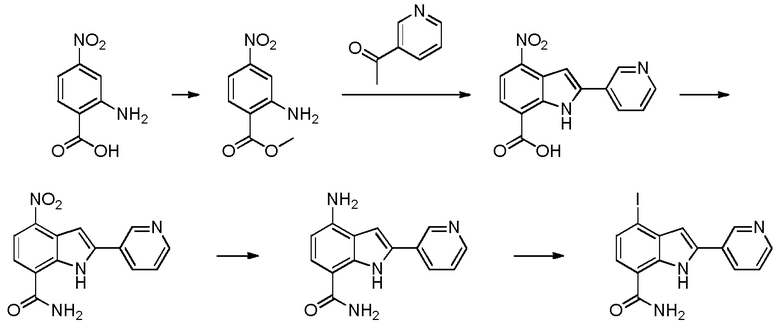
К раствору 2-амино-4-нитробензойной кислоты (102 г, 560 ммоль) в MeOH (1,5 л) медленно добавляли концентрированную серную кислоту (0,030 л, 560 ммоль). Полученный раствор нагревали при температуре приблизительно 75°C в течение приблизительно 3 дней. После охлаждения продукт нейтрализовали добавлением водного раствора NaOH до рН~10. Сырой продукт экстрагировали EtOAc, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением метил 2-амино-4-нитробензоата (100 г, 91%). ЖХ/МС (Таблица 1, Способ ar) Время удерживания = 1,85 минут; MS m/z 197,1 (M+H)+. К части этого вещества (25 г, 127 ммоль) и 1-(пиридин-3-ил)этанона (30,9 г, 255 ммоль) в диметилсульфоксиде (150 мг) при температуре приблизительно -15°C добавляли трет-бутоксид калия (28,6 г, 255 ммоль). После перемешивания в течение приблизительно 2,5 часов при комнатной температуре, реакцию гасили насыщенным водным раствором хлорида аммония (100 мг) и затем перемешивали в течение приблизительно 1 часа при комнатной температуре. Полученную суспензию фильтровали, промывали водой и сушили в высоком вакууме с получением 4-нитро-2- (пиридин-3-ил)-1H-индол-7-карбоновой кислоты (22,4 г, 34%). ЖХ/МС (Таблица 1, Способ ab) Время удерживания = 1,50 минуты; MS m/z 284,1 (M+H)+. К смеси этого материала (26,9 г, 95 ммоль), ((1Н-бензо[d][1,2,3]триазол-1-ил)окси)три(пирролидин-1-ил)фосфония гексафторфосфат(V), (99 г, 190 ммоль), гидроксибензотриазолгидрату (29,1 г, 190 ммоль) и хлориду аммония (7,62 г, 142 ммоль) в DMF (150 мл) добавляли диизопропилэтиламин (66,3 мл, 380 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь выливали в 1000 мл воды и осадок отфильтровывали, промывали водой и EtOAc, и собирали с получением 4-нитро-2-(пиридин-3-ил)-1H-индол-7-карбоксамид (17,48 г, 56%). ЖХ/МС (Таблица 1, Способ ar) Время удерживания = 1,44 минуты; MS m/z 283,1 (M+H)+. К продутому азотом перемешиваемому раствору этого вещества (17,5 г, 52,6 ммоль) в МеОН (1,5 л) добавляли 10% палладий на угле (5,60 г, 5,26 ммоль). Полученную суспензию обрабатывали водородом (30 фунт на кв.дюйм). После перемешивания в течение ночи при комнатной температуре, реакционную смесь фильтровали и твердые вещества промывали МеОН. Фильтрат концентрировали, чтобы получить 4-амино-2-(пиридин-3-ил)-1H-индол-7-карбоксамид (10 г, 75%). ЖХ/МС (Таблица 1, Способ ar) Время удерживания = 1,10 минут; MS m/z 253,1 (M+H)+. Раствор нитрита натрия (7,82 г, 113 ммоль) в воде (20 мг) добавляли к охлажденной льдом суспензии этого вещества (13 г, 51,5 ммоль) в MeCN (150 мг) и хлорида водорода (2 н 188 мл, 376 ммоль) при перемешивании, поддерживая температуру ниже приблизительно -5°C. После перемешивания в течение примерно 30 минут, холодный раствор водного иодида калия (21,4 г, 129 ммоль) добавляли к реакционной смеси и полученную смесь перемешивали при комнатной температуре в течение 30 минут. Реакционную смесь нагревали на водяной бане (85°C) в течение 5 минут. Реакционную смесь охлаждали до комнатной температуры и нейтрализовали насыщенным водным раствором бикарбоната натрия до рН 8. Смесь экстрагировали DCM. Органический слой промывали насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали путем флэш-хроматографии (силикагель, петролейный эфир), получая 4-йод-2-(пиридин-3-ил)-1H-индол-7-карбоксамид (2,0 г, 9%). ЖХ/МС (Таблица 1, Способ ab) Время удерживания = 1,88 мин; MS m/z 364,0 (M+H)+. (IC50 Btk=B)
Общий способ G: Кислотное расщепление Boc-защищенного амина
К раствору N-Boc амина (1 эквив.) в органическом растворителе (таком, как DCM, EtOAc или 1,4-диоксан) добавляли кислоту (такую, как TFA или HCl, предпочтительно, TFA; 2-35 эквив., предпочтительно, 15-25 эквив.). Смесь перемешивали при температуре приблизительно 0-100°C (предпочтительно, приблизительно 20-60°C) в течение приблизительно от 1 до 24 часов (предпочтительно, приблизительно 1-6 часов). Необязательно дополнительное количество кислоты (2-35 эквив., предпочтительно, 20-25 эквив.) добавляли и смесь перемешивали при приблизительно 0-100°C (предпочтительно, приблизительно 15-60°C) в течение приблизительно от 1 до 24 часов (предпочтительно, приблизительно от 1 до 6 часов). Если в смеси присутствует твердый продукт, полученную реакционную смесь можно необязательно фильтровать, и твердый продукт промывали органическим растворителем, таким как 1,4-диоксан или Et2O. Полученную твердую часть затем необязательно сушили при пониженном давлении с получением целевого соединения. Альтернативно, смесь необязательно концентрировали в вакууме с получением конечного соединения. Альтернативно, смесь необязательно фильтровали через среду (например, силикагель или целит), которую промывали соответствующим растворителем (таким как, EtOAc, 1,4-диоксан, THF, MeCN, DCM, Et2O, MeOH, EtOH) и затем, необязательно, концентрировали в вакууме с получением остатка. Или остаток или раствор необязательно разделили между водой и органическим растворителем (таким как EtOAc, Et2O или DCM). Органический слой выделяли и необязательно промывали в произвольном порядке водой и/или водными растворами, содержащими кислоту (такую как HCl, AcOH или NH4Cl) и/или водными растворами, содержащими основание (такое как NaHCO3, Na2CO3, NaOH, KOH или NH4OH) и/или водными растворами, содержащими неорганическую соль (такую как NaCl Na2SO3 или Na2S2O3). Органический раствор затем необязательно сушили высушивающим веществом (например, безводным MgSO4 или Na2SO4), фильтровали и концентрировали в вакууме с получением целевого соединения.
Иллюстрация общего способа G
Пример № G.1. 2-(2,5-Дигидро-1H-пиррол-3-ил)-4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-1H-индол-7-карбоксамид
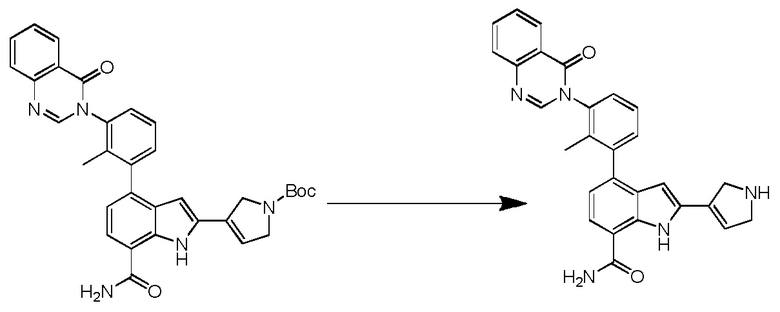
К раствору трет-бутил 3-(7-карбамоил-4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-1H-индол-2-ил)-2,5-дигидро-1H-пиррол-1-карбоксилата (0,6 г, 1 ммоль, Получение № 15) в EtOAc (20 мг) добавляли HCl/EtOAc при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Твердое вещество собирали в виде соли путем фильтрации и высушивали с получением гидрохлорида 2-(2,5-дигидро-1H-пиррол-3-ил)-4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-1H-индол-7-карбоксамида (0,5 г, 94%): ЖХ/МС (Таблица 1, способ d) Время удерживания = 2,39 мин; MS m/z: 462 (M+H)+ (Btk IC50=A).
Примеры, полученные с использованием процедуры общего способа G
(Таблица 1, способ)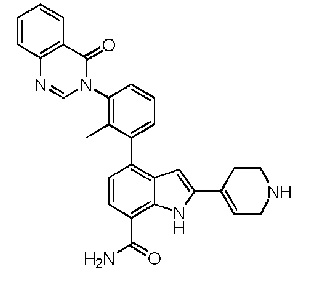
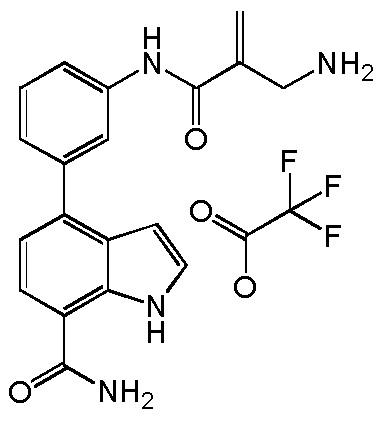
-(метил)карбамат (полученный с использованием J из 2-(бромметил)акриловой кислоты и трет-бутил метилкарбамата, D из Получения № A.1)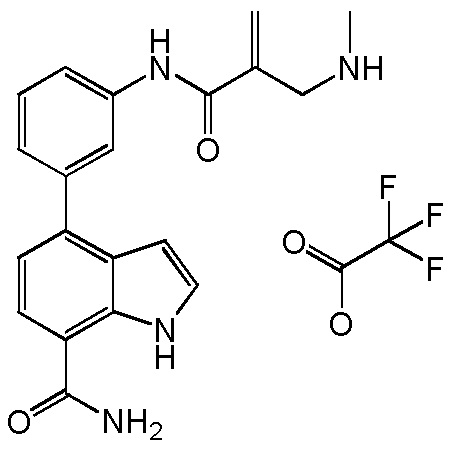
Общий способ H: Восстановительное аминирование альдегида или кетона с первичным или вторичным амином
Альдегид или кетон (предпочтительно, 1,0 эквив. до 1,3 эквив.) и амин или соль амина (предпочтительно, 1,0 до 2,2 эквив.) добавляют в органическом растворителе или смеси органических растворителей (таких, как DCM, DCE или MeOH, или смесь DCE и MeOH, предпочтительно, DCE, MeOH, или 1:1 MeOH/DCM) при приблизительно комнатной температуре до приблизительно 80°C (предпочтительно, приблизительно комнатная температура). Если использовали соль амина, затем необязательно добавляли аминное основание (такое как TEA или DIEA, 1,0-2,2 эквив.). AcOH (от 0,1 эквив. до 5,0 эквив.) необязательно добавляли. Смесь перемешивали при комнатной температуре в течение приблизительно 1-90 минут (предпочтительно, 5-30 минут). Восстанавливающее средство (такое как NaBH(OAc)3, Na(CN)BH3, NaBH4, MP-цианоборгидрид от Biotage™, 0,5-5,0 эквив., предпочтительно, 2,5-3,0 эквив. NaBH(OAc)3), добавляли в виде твердого вещества или в виде раствора в органическом растворителе (например, DCM, DCE или MeOH, или смесь DCE и MeOH). Смесь перемешивали при комнатной температуре в течение приблизительно от 30 минут до 72 часов (предпочтительно, 1-24 часа). Сырую смесь можно концентрировать при пониженном давлении или необязательно распределяли между водой и органическим растворителем (таким как EtOAc, Et2O или DCM). Органический слой выделяли и необязательно промывали водой и/или водными растворами, содержащими кислоту (такую как HCl, AcOH или NH4Cl) и/или водными растворами, содержащими основание (такое как NaHCO3, Na2CO3, NaOH, KOH или NH4OH) и/или водными растворами, содержащими неорганическую соль (такую как NaCl или Na2SO3). Органический раствор затем необязательно сушили с высушивающим веществом (таким как MgSO4 или Na2SO4), фильтровали и концентрировали в вакууме с получением целевого соединения.
Иллюстрация общего способа H
Пример № H.1. 2-(1-метил-2,5-дигидро-1H-пиррол-3-ил)-4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-1H-индол-7-карбоксамид
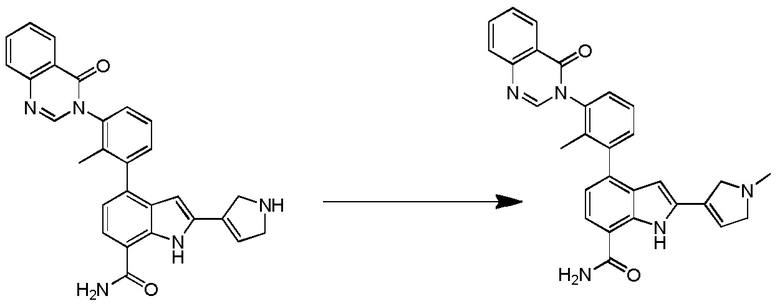
К раствору 2-(2,5-дигидро-1H-пиррол-3-ил)-4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-1H-индол-7-карбоксамида (50 мг, 0,1 ммоль, Пример № G.1) в MeOH (1 мг) добавляли (CH2O)n (1,6 мг, 0,054 ммоль) при комнатной температуре. После перемешивания при комнатной температуре в течение 1 часа в атмосфере N2, добавляли NaBH(OAc)3 (60 мг, 0,27 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 2 часов. Растворитель удаляли при пониженном давлении с получением остатка, который очищали с помощью препаративной ВЭЖХ с получением 2-(1-метил-2,5-дигидро-1H-пиррол-3-ил)-4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-1H-индол-7-карбоксамида (15 мг, 32%): ЖХ/МС (Таблица 1, способ o) Время удерживания = 2,05 мин; MS m/z: 476 (M+H)+ (Btk IC50 A).
Примеры получены из 4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-2-(1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамида (Пример № G.1.1) с использованием Общего способа H
Примеры, полученные из 4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-2-(1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид (Пример № G.1.1) с использованием общего способа H
(Таблица 1, Способ)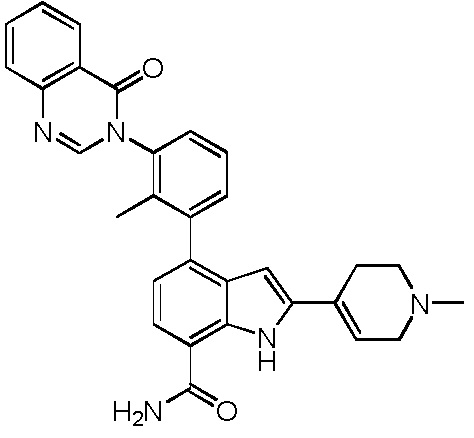
Примеры, полученные из 4-(3-амино-2-метилфенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид (Пример № A.4.5) с использованим Общего способа H
(Таблица 1, Способ)
Общий способ I: Получение сульфонамида из амина и сульфонилхлорида
В сосуд добавляли амин (1,0 эквив.), необязательно в виде хлористоводородной соли, растворителя или смеси растворителей (таких, как DCM, DCE, EtOAc, THF, 1,4-диоксан, пиридин, DME, или пиридин/DCM, предпочтительно, THF, необязательно с основанием (таким, как TEA, DIEA, предпочтительно, DIEA; 1-5 эквив., предпочтительно, 1-2 эквив.) и сульфонилхлорид (0,9-2,0 эквив., предпочтительно 1,0-1,25 эквив.). Смесь перемешивали при приблизительно 0-80°C (предпочтительно, приблизительно 0-35°C) в течение приблизительно от 1 часа до 24 часов (предпочтительно, 5-16 часов). Смесь необязательно концентрировали в вакууме с получением остатка в виде целевого соединения. Или остаток или раствор необязательно разделяли между водой и органическим растворителем (таким как EtOAc, Et2O или DCM). Органический слой выделяли и необязательно промывали в произвольном порядке водой и/или водными растворами, содержащими кислоту (такую как HCl, AcOH или NH4Cl) и/или водными растворами, содержащими основание (такое как NaHCO3, Na2CO3, NaOH, KOH или NH4OH) и/или водными растворами, содержащими неорганическую соль (такую как NaCl, Na2SO3 или Na2S2O3). Органический раствор затем необязательно сушили с высушивающим веществом (например, безводным MgSO4 или Na2SO4), фильтровали и концентрировали в вакууме с получением целевого соединения.
Иллюстрация общего способа I
Пример № I.1: 4-(3-(винилсульфонамидо)фенил)-1H-индол-7-карбоксамид
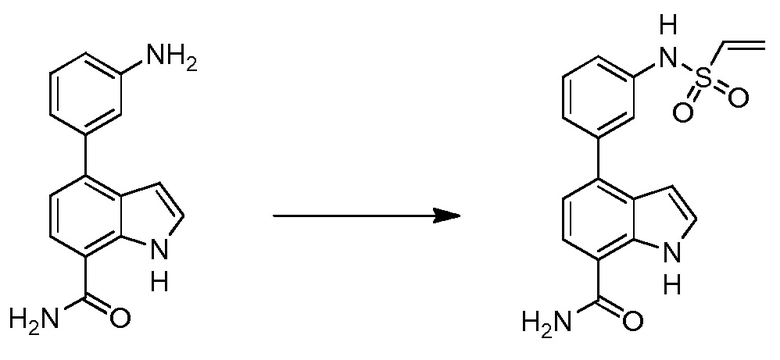
К смеси 4-(3-аминофенил)-1H-индол-7-карбоксамида (0,11 г, 0,438 ммоль, Получение № A.1), THF (4 мг) и DIEA (0,152 мл, 0,876 ммоль) при приблизительно 0°C (баня со льдом) добавляли этенсульфонилхлорид (0,058 г, 0,460 ммоль, FCH Группа). Баню со льдом удаляли и смесь перемешивали в течение приблизительно 6 часов при комнатной температуре. Реакционную смесь концентрировали при пониженном давлении и остаток растворяли в DCM и промывали водой (2×), насыщенным солевым раствором, и пропускали через разделитель фаз Biotage. Смесь концентрировали при пониженном давлении и остаток очищали на силикагеле, используя градиент 0-10% МеОН в DCM с получением твердого вещества. Твердое вещество растирали с эфиром (3×, обрабатывали ультразвуком после каждого добавления эфира). Твердое вещество сушили в течение ночи при пониженном давлении при 75°C с получением 4-(3-(винилсульфонамидо)фенил)-1H-индол-7-карбоксамида (29 мг, 19%): ЖХ/МС (Таблица 1, Способ c) Время удерживания = 2,34 мин; MS m/z 342 (M+H)+. (Btk IC50=A)
Общий способ J: Замена алкилгалогенида амином нуклеофила
В сосуд загружали алкилгалогенид (предпочтительно 1 эквив.) и органический растворитель (такой как THF, MeCN, DMF, DMA, NMP или DMSO; предпочтительно, THF или MeCN). В сосуд добавляли в произвольном порядке амин нуклеофила (1-25 эквив., предпочтительно 1,2-20 эквив.) и необязательно основание (такое как LiHMDS, NaH, K2CO3, NaHMDS, NaOt-Bu, KHMDS или KOt-Bu, предпочтительно, нет, NaH или K2CO3; 1-5 эквив., предпочтительно, 1-3 эквив.). Смесь перемешивали при приблизительно 0-100°C (предпочтительно, приблизительно 0-40°C) в течение приблизительно 1-24 часов (предпочтительно, приблизительно 3-20 часов). Смесь необязательно концентрировали в вакууме с получением остатка в виде целевого соединения. Или остаток или раствор необязательно разделяли между водой и органическим растворителем (таким как EtOAc, Et2O или DCM). Органический слой выделяли и необязательно промывали в произвольном порядке водой и/или водными растворами, содержащими кислоту (такую как HCl, AcOH или NH4Cl) и/или водными растворами, содержащими основание (такое как NaHCO3, Na2CO3, NaOH, KOH или NH4OH) и/или водными растворами, содержащими неорганическую соль (такую как NaCl, Na2SO3 или Na2S2O3). Органический раствор затем необязательно сушили с высушивающим веществом (например, безводным MgSO4 или Na2SO4), фильтровали и концентрировали в вакууме с получением целевого соединения. Альтернативно, остаток от концентрации реакционной смеси суспендировали в воде, обрабатывали ультразвуком и собирали вакуумным фильтрованием.
Иллюстрация общего способа J
Пример № J.1: (E)-4-(3-(4-(Диметиламино)бут-2-енамидо)-2-метилфенил)-1H-индол-7-карбоксамид
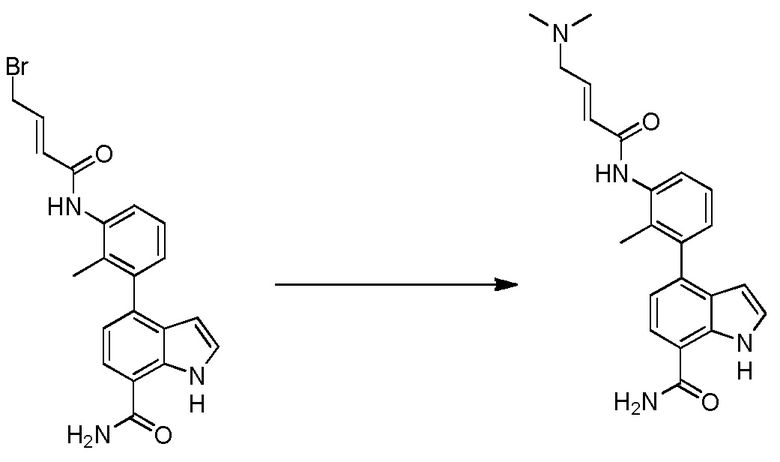
К раствору (E)-4-(3-(4-бромбут-2-енамидо)-2-метилфенил)-1H-индол-7-карбоксамида (1,4 г, 3,40 ммоль, полученный с использованием E из 4-(3-амино-2-метилфенил)-1H-индол-7-карбоксамида (Пример № 16) и (E)-4-бромбут-2-еноил хлорида [J.Org.Chem. 2011, 76, 4467]) в THF (24 мл) при 0°C добавляли 2 M диметиламина в THF (34,0 мл, 67,9 ммоль). Смесь перемешивали в течение 3 часов при нагревании до комнатной температуры. Смесь концентрировали при пониженном давлении и к остатку добавляли воду (15 мг). Смесь обрабатывают ультразвуком в течение приблизительно 20 минут при комнатной температуре, фильтровали, промывали водой и сушили при пониженном давлении. Остаток помещали в колонку с силикагелем и элюировали смесью MeOH/DCM (0-15%) с получением сырого продукта (0,650 г). Сырой продукт растворяли в DMA (5 мг) и добавляли воду (100 мг) при перемешивании в течение 20 минут при комнатной температуре. Смесь фильтровали, промывали водой (50 мл×3), и сушили при пониженном давлении с получением (E)-4-(3-(4-(диметиламино)бут-2-енамидо)-2-метилфенил)-1H-индол-7-карбоксамида (0,40 г, 31%): ЖХ/МС (Таблица 1, Способ f) Время удерживания = 1,05 мин; MS m/z 377 (M+H)+. (Btk IC50 B)
Примеры, полученные из (E)-4-(3-(4-бромбут-2-енамидо)-2-метилфенил)-1H-индол-7-карбоксамида (полученный с использованием E из 4-(3-амино-2-метилфенил)-1H-индол-7-карбоксамида (Пример № 16) и (E)-4-бромбут-2-еноил хлорида [J.Org.Chem. 2011, 76, 4467]) с использованием общего способа J
(Таблица 1, Способ)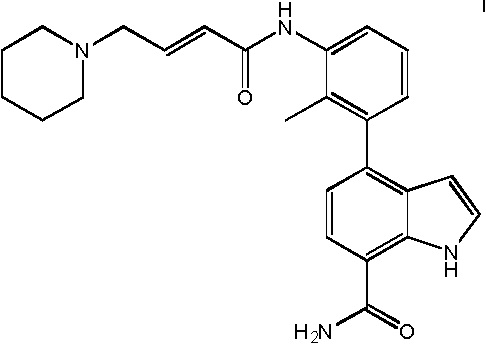
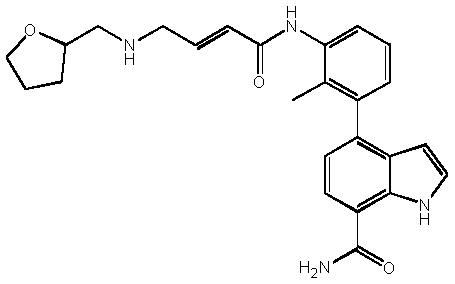
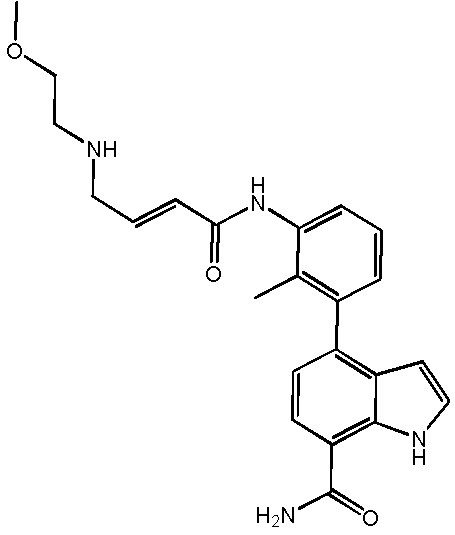


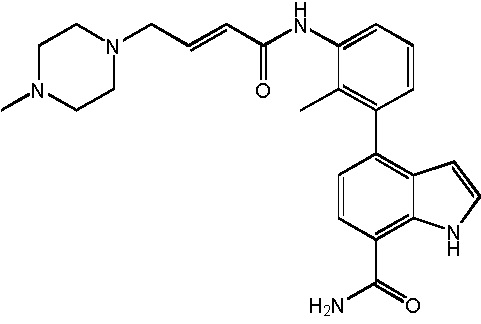
Пример, полученный из (E)-4-(3-(4-бромбут-2-енамидо)-2-метилфенил)-1H-пирроло[2,3-c]пиридин-7-карбоксамида (полученный с использованием E из 4-(3-амино-2-метилфенил)-1H-пирроло[2,3-c]пиридин-7-карбоксамида (Пример № 2) и (E)-4-бромбут-2- еноил хлорида [J.Org.Chem. 2011, 76, 4467]) с использованием общего способа J
(Таблица 1, Способ)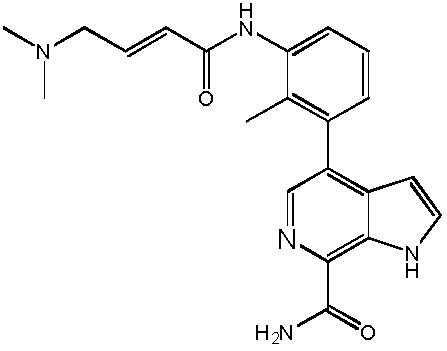
Пример, полученный из цианового бромида с амином с использованием общего способа J
(Таблица 1, Способ)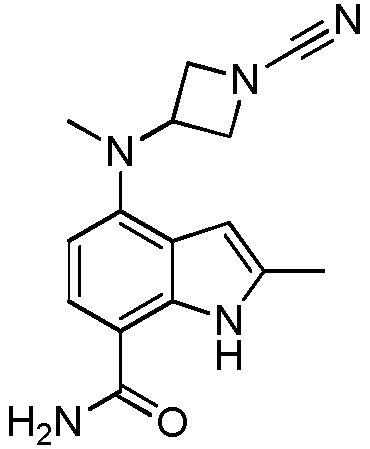
Общий способ K: Гидролиз ацетонида
К раствору ацетонида (предпочтительно 1 эквив.) в органическом растворителе (таком как 1,4-диоксан и THF, предпочтительно, THF) добавляли кислоту, такую как 4 M HCl в 1,4-диоксане (3-100 эквив., предпочтительно, 30-40 эквив.). Реакционную смесь нагревали при приблизительно 20-120°C (предпочтительно, приблизительно при комнатной температуре с использованием обычного нагрева; приблизительно 120°C с использованием микроволнового излучения) в течение приблизительно 0,25-24 часов (предпочтительно, приблизительно 4 часов с использованием обычного нагрева; приблизительно 20 минут с использованием микроволнового излучения). Реакционную смесь оставляли охлаждаться до температуры окружающей среды, прежде чем смесь необязательно распределяли между органическим растворителем (таким как EtOAc или DCM) и водным основанием (таким как NaHCO3, Na2CO3 или NaOH, предпочтительно, NaHCO3) и водный слой дополнительно экстрагировали с дополнительным органическим растворителем (таким как EtOAc или DCM). Органический слой сушили над безводным MgSO4 или Na2SO4, фильтровали и концентрировали при пониженном давлении. Альтернативно растворитель удаляли при пониженном давлении, получая требуемое соединение.
Иллюстрация общего способа K:
Пример № K.1*: 2-(1-((R)-2,3-дигидроксипропил)-1H-пиразол-4-ил)-4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-1H-индол-7-карбоксамид

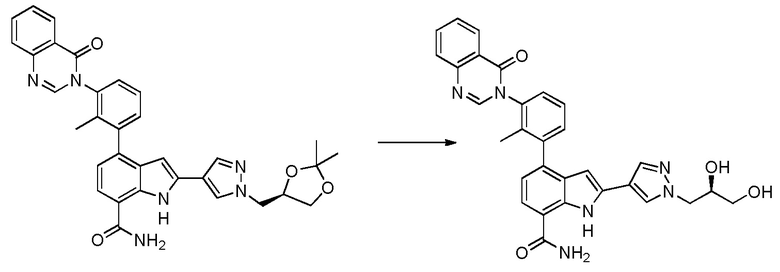
К раствору 2-(1-(((R)-2,2-диметил-1,3-диоксолан-4-ил)метил)-1H-пиразол-4-ил)-4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-1H-индол-7-карбоксамида (0,047 г, 0,082 ммоль, полученный с использованием A из 4-бром-2-йод-1H-индол-7-карбоксамида и (R)-1-((2,2-диметил-1,3-диоксолан-4-ил)метил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (Получение № 20), A из 3-(2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)хиназолин-4(3H)-она [PCT Int. Appl., WO 2011159857]) в THF (5 мг) добавляли 4 M HCl в 1,4-диоксане (0,5 мг). Смесь перемешивали при комнатной температуре в течение приблизительно 4 часов. Реакционную смесь концентрировали при пониженном давлении и остаток очищали с помощью препаративной ВЭЖХ (Таблица 1, способ af) с получением 2-(1-((R)-2,3- дигидроксипропил)-1H-пиразол-4-ил)-4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-1H-индол-7-карбоксамида (0,035 г, 80%): ЖХ/МС (Таблица 1, способ a) Время удерживания = 1,65 мин; MS m/z 535. (Btk IC50=A)
Примеры, полученные из ацетонида с использованием общего способа K
(Таблица 1, Способ)
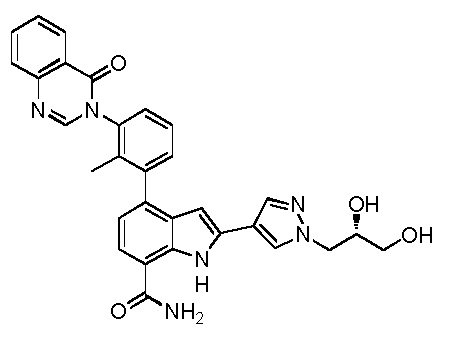
Общий способ L: Гидрирование алкена
В круглодонную колбу загружали палладиевый катализатор, такой как Pd/C или Pd(OH)2 (10-20% масс., приблизительно 0,005-1,0 эквивалента, предпочтительно 0,5-1,0 эквивалента). Колбу откачивали, затем продували азотом от 2 до 5 раз (предпочтительно, 3 раза) перед добавлением органического растворителя или смеси растворителей (такого как EtOAc, MeOH, EtOH или MeOH/AcOH,, предпочтительно МеОН/АсОН) в атмосфере азота. К смеси добавляли алкен (предпочтительно, 1 эквивалент), в чистом виде или при необходимости в виде раствора в органическом растворителе или смеси растворителей (таких, как EtOAc, MeOH, EtOH или MeOH/AcOH, предпочтительно, MeOH). Смесь перемешивали в атмосфере водорода (приблизительно, 30-50 фунт на кв.дюйм) в течение приблизительно 1-60 часов (предпочтительно, приблизительно 4-5 часов). Необязательно, реакцию можно проводить с использованием устройства H-cube или с Pd/C или с Pd(OH)2 картриджами (10 или 20% масс.) и исходное вещество проходит через систему в виде раствора в предпочтительном растворителе(ях). В тех случаях, когда реакция не протекает до конца, как контролировали с помощью ТСХ, ЖХ/МС или ВЭЖХ, смесь необязательно нагревали до приблизительно 30-80°C (предпочтительно, приблизительно 50°C) в течение приблизительно 1-24 часов (предпочтительно, приблизительно 16 часов) и в случаях, когда H-cube использовали для проведения реакции, давление может быть увеличено (25-50 бар, предпочтительно, 40-50 бар). Затем смесь фильтровали и осадок на фильтре промывали органическим растворителем (таким как EtOAc, MeOH или EtOH, предпочтительно, реакционный растворитель), и фильтрат концентрировали при пониженном давлении с получением сырого продукта.
Иллюстрация общего способа L
Пример № L.1: 2-(1-ацетилпиперидин-4-ил)-4-(3-амино-2-метилфенил)-1H-индол-7-карбоксамид
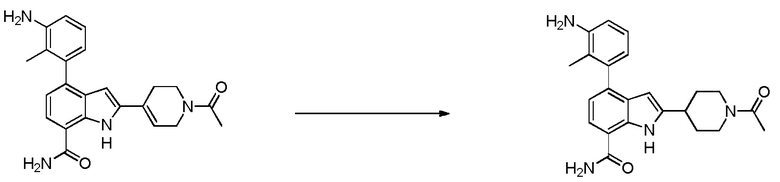
2-(1-Ацетил-1,2,3,6-тетрагидропиридин-4-ил)-4-(3-амино-2-метилфенил)-1H-индол-7-карбоксамид (300 мг, 0,772 ммоль, полученный с использованием A с 4-бром-2-йод-1H-индол-7-карбоксамидом (Получение № 1) и 1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2H)-ил)этанона [Combi-Blocks], A с 3-амино-2-метилфенилбороновой кислоты, пинаколовый эфир [Combi-Blocks]) и растворитель MeOH (72 мг) добавляли к 20 масс.% Pd/C (60,0 мг, 0,564 ммоль) в 250 мл емкость из нержавеющей стали и перемешивали в течение приблизительно 4,5 часов при 30 фунт на кв.дюйм, затем при температуре около 50°C в течение приблизительно 16 часов. Реакционную смесь фильтровали, концентрировали в вакууме и остаток очищали на силикагеле, используя градиент 0-10% MeOH в DCM с получением 2-(1-ацетилпиперидин-4-ил)-4-(3-амино-2-метилфенил)-1H-индол-7-карбоксамида (77,1 мг, 0,197 ммоль): ЖХ/МС (Таблица 1, способ f) Время удерживания = 1,06 мин; MS m/z 391. (Btk IC50=B)
Общий способ M: Удаление силильной группы у О-силилового эфира
Способ 1:
К раствору O-силилового эфира (1 эквив.) в органическом растворителе (например, DMF, 1,4-диоксан, или DCM, предпочтительно, DCM) добавляли кислоту (такую как TFA или HCl, 5-50 эквив., предпочтительно, 30 эквив.) и смесь перемешивали при температуре приблизительно от 0 до 50°C (предпочтительно, приблизительно 15-25°C) в течение приблизительно 1-48 часов (предпочтительно, приблизительно 4-16 часов). Альтернативно, может быть добавлено дополнительное количество кислоты (5-20 эквив., предпочтительно, 10 эквив.) и смесь нагревали при приблизительно от 30 до 100°C (предпочтительно, приблизительно 50-80°C) в течение приблизительно 0,5-10 часов (предпочтительно, приблизительно 1-5 часов).
Способ 2:
К раствору O-силилового эфира (1 эквив.) в органическом растворителе (например, DMF, 1,4-диоксан, или DCM, предпочтительно, DMF) добавляли источник фторида, такой как HF, TBAF (1-10 эквив., предпочтительно, 4 эквив.), и смесь перемешивали при приблизительно 20-110°C (предпочтительно, приблизительно 25-60°C) в течение приблизительно 1-20 часов (предпочтительно, приблизительно 2-8 часов).
Для любого способа, целевое соединение при необходимости может быть выделено путем охлаждения смеси и фильтрации осадка. Альтернативно, смесь необязательно концентрировали в вакууме с получением целевого соединения. Альтернативно, смесь необязательно фильтровали через среду (например, силикагель или целит), которую промывали соответствующим растворителем (таким как EtOAc, 1,4-диоксан, THF, MeCN, DCM, Et2O, MeOH или EtOH) и затем, необязательно, концентрировали в вакууме с получением остатка. Или остаток или раствор необязательно разделяли между водой и органическим растворителем (таким как EtOAc, Et2O или DCM). Органический слой выделяли и необязательно промывали в произвольном порядке водой и/или водными растворами, содержащими кислоту (такую как HCl, AcOH или NH4Cl) и/или водными растворами, содержащими основание (такое как NaHCO3, Na2CO3, NaOH, KOH или NH4OH) и/или водными растворами, содержащими неорганическую соль (такую как NaCl, Na2SO3 или Na2S2O3). Органический раствор затем необязательно сушили с высушивающим веществом (таким как безводный MgSO4 или Na2SO4), фильтровали и концентрировали в вакууме с получением целевого соединения.
Иллюстрация общего способа M:
Пример № M.1: N-(3-(7-карбамоил-2-(1-метил-1H-пиразол-4-ил)-1H-индол-4-ил)-2-(гидроксиметил)фенил)тиазол-2-карбоксамид
К раствору N-(2-(((трет-бутилдиметилсилил)окси)метил)-3-(7-карбамоил-2-(1-метил-1H-пиразол-4-ил)-1H-индол-4-ил)фенил)тиазол-2-карбоксамида (100 мг, 0,170 ммоль, полученный с использованием D из тиазол-2-карбоновой кислоты и 2-((трет-бутилдиметилсилилокси)метил)-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина [Matrix], A и Получения № 10) в 1,4-диоксане (2 мг) добавляли 3 н водной HCl (2 мл, 6,00 ммоль) и смесь перемешивали при приблизительно 25°C течение приблизительно 3 часов. Полученный раствор разбавляли EtOAc (5 мг) и промывали водой (3 мг). Органическую фазу сушили над Na2SO4 и концентрировали с получением сырого продукта, который очищали с помощью препаративной ТСХ (DCM:MeOH=20:1) с получением N-(3-(7-карбамоил-2-(1-метил-1H-пиразол-4-ил)-1H-индол-4-ил)-2-(гидроксиметил)фенил)тиазол-2-карбоксамида (36 мг, 45%): 1H-ЯМР (DMSO-d6) δ 11,16 (с, 1H), 10,92 (с, 1H), 8,32 (с, 1H), 8,27-8,25 (д, J=8,4 Гц, 1H), 8,14-8,07 (м, 3H), 7,94 (с, 1H), 7,67-7,65 (д, J=6,4 Гц, 1H), 7,46-7,43 (м, 2H), 7,14-7,12 (д, J=7,6 Гц, 1H), 6,96-6,94 (д, J=7,6 Гц, 1H), 6,31 (с, 1H), 5,78 (с, 1H), 4,54-4,47 (м, 2H), 3,82 (с, 3H). ЖХ/МС (Таблица 1, способ o) Время удерживания = 2,73 мин; MS m/z: 473 (M-H)+. (Btk IC50=A)
Примеры, полученные из O-силилового эфира с использованием общего способа M
(Таблица 1, Способ)
(полученный с использованием J из
4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола с
A c
(2-бромэтокси)-трет-бутилдиметилсиланом,
4-бром-2-йод-1H-индол-7-карбоксамидом,
A с
3-(2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)хиназолин-4(3H)-оном [WO 2011159857])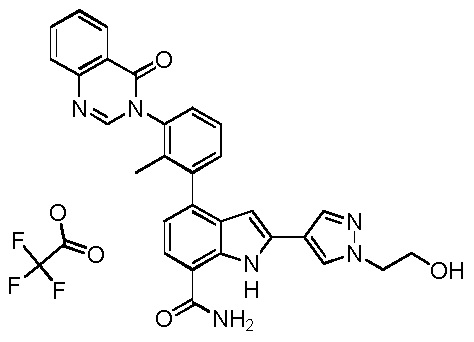
Общий способ N: Гидролиз сульфонамида
В сосуд, содержащий сульфонамид, например, сульфонил-защищенный индол, (предпочтительно 1 эквив.) в органическом растворителе (таком как 1,4-диоксан, MeOH, или THF/MeOH, предпочтительно 1,4-диоксан) добавляли основание (такое как K2CO3, Cs2CO3, водный Na2CO3 или водный NaOH, 1-30 эквив.; предпочтительно, 1-5 эквив. для Cs2CO3,). Смесь перемешивали при приблизительно 25-100°C (предпочтительно, приблизительно 60°C) в течение приблизительно 1-72 часов (предпочтительно, приблизительно 1-18 часов). В тех случаях, когда реакция не протекала до конца, как контролировали с помощью ТСХ, ЖХ/МС или ВЭЖХ, добавляли дополнительное основание (такое, как K2CO3, Cs2CO3, водный раствор Na2CO3 или водный раствор NaOH, предпочтительно, 1-5 эквив. для Cs2CO3,) и/или сорастворитель (такой, как EtOH). Реакцию продолжали при приблизительно 25-100°C (предпочтительно, приблизительно 60°C) в течение приблизительно 0,25-3 часов (предпочтительно, приблизительно 1-2 часов). В любом случае, где присутствует дополнительное основание неустойчивая группа (например, сложный эфир или цианогруппа), эта группа также может быть гидролизована. Реакционную смесь обрабатывали с помощью одного из следующих способов. Способ 1. Органический растворитель необязательно удаляли при пониженном давлении и водный раствор нейтрализовали добавлением подходящего водного раствора кислоты (такой, как водный раствор HCl). Добавляли подходящий органический растворитель (например, EtOAc или DCM) и воду, слои разделяли и органический раствор сушили над безводным Na2SO4 или MgSO4, фильтровали и концентрировали досуха при пониженном давлении с получением целевого соединения. Способ 2. Органический растворитель, необязательно удаляли при пониженном давлении, подходящий органический растворитель (такой, как EtOAc или DCM) и воду добавляли, слои разделяли, и органический раствор сушили над безводным Na2SO4 или MgSO4, фильтровали и концентрировали досуха при пониженном давлении с получением целевого соединения. Способ 3. Реакционную смесь концентрировали при пониженном давлении и непосредственно очищали одним из последующих способов.
Иллюстрация общего способа N:
Получение № N.1: : (R)-4-(3-(4-Оксохиназолин-3(4H)-ил)пиперидин-1-ил)-1H-индол-7-карбонитрил.

К смеси (R)-4-(3-(4-оксохиназолин-3(4H)-ил)пиперидин-1-ил)-1-тозил-1H-индол-7-карбонитрила (0,12 г, 0,229 ммоль, полученный с использованием B из 4-фтор-1-тозил-1H-индол-7-карбонитрила (Получение № 27, стадия A) и (R)-3-(пиперидин-3-ил)хиназолин-4(3H)-она (Получение № 31) в THF (2 мг) и MeOH (1 мг) добавляли карбонат цезия (0,128 мл, 1,60 ммоль) и перемешивали при комнатной температуре в течение приблизительно 18 часов. Реакционную смесь разбавляли водой (60 мг) и перемешивали еще в течение 20 минут. Смесь экстрагировали DCM, сушили путем пропускания через разделитель фаз Biotage для удаления остаточной воды и выпаривали досуха с получением (R)-4-(3-(4-оксохиназолин-3(4H)-ил)пиперидин-1-ил)-1H-индол-7-карбонитрила (0,044 г, 52%); ЖХ/МС (Таблица 1, способ g) Время удерживания = 1,50 мин; MS m/z: 370 (M+H)+
Общий способ O: Гидролиз нитрила до первичного амида
В сосуд, содержащий нитрил (предпочтительно, 1 эквив.) в органическом растворителе (таком как MeOH, EtOH, DMSO, DMSO/MeOH, или DMSO/EtOH, предпочтительно, DMSO/EtOH) добавляли основание (такое как KOH, водный раствор КОН или водный раствор NaOH, 1-30 эквив., предпочтительно, 3-5 эквив. для КОН, предпочтительно, 10-15 эквив. для водного раствора NaOH). Смесь перемешивали при приблизительно комнатной температуре в течение приблизительно 1-30 минут (предпочтительно, приблизительно 1-10 минут), затем 30% H2O2 (5-30 эквив., предпочтительно, 9-27 эквив.) добавляли медленно к смеси и реакционную смесь перемешивали при комнатной температуре в течение приблизительно 10-30 минут. В тех случаях, когда реакция не протекала до конца, как контролировали с помощью ТСХ, ЖХ/МС или ВЭЖХ, реакцию продолжали при комнатной температуре в течение приблизительно 0,25-1 часов (предпочтительно, приблизительно 0,25-0,5 часов). Реакционную смесь обрабатывали с помощью одного из следующих способов. Способ 1. Смесь разбавляли насыщенным NH4Cl и водой, перемешивали при комнатной температуре в течение приблизительно 1-30 минут. Полученную суспензию собирали фильтрованием, промывали подходящим растворителем (таким как MeOH, EtOH или вода), и осадок на фильтре сушили в вакууме с получением целевого соединения. Способ 2. Органический растворитель, необязательно удаляли при пониженном давлении, подходящий органический растворитель (такой как EtOAc или DCM) и воду добавляли, слои разделяли, и органический раствор сушили над безводным Na2SO4 или MgSO4, фильтровали и концентрировали досуха при пониженном давлении с получением целевого соединения. Способ 3. Реакционную смесь концентрировали при пониженном давлении и непосредственно очищали одним из последующих способов.
Иллюстрация общего способа O:
Пример № O.1: N-(транс-1-(7-карбамоил-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-4-ил)-4-гидроксипиперидин-3-ил)тиазол-2-карбоксамид

К перемешиваемому раствору N-(транс-1-(7-циано-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-4-ил)-4-гидроксипиперидин-3-ил)тиазол-2-карбоксамида (36 мг, 0,068 ммоль, полученный с использованием B Получение № 27 и Получение № 23, N с Cs2CO3) в DMSO (0,8 мг) добавляли EtOH (4,8 мг) и KOH (12,81 мг, 0,228 ммоль). Смесь перемешивали при комнатной температуре в течение приблизительно 10 минут, затем 30% H2O2 (0,070 мг, 0,615 мкмоль) добавляли к смеси медленно и реакционную смесь перемешивали при комнатной температуре в течение приблизительно 15 минут. Затем добавляли воду (6 мг) к смеси и раствор экстрагировали EtOAc (3×20 мг). Органический слой промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали с получением сырого продукта, который очищали флэш-хроматографией с получением N-(транс-1-(7-карбамоил-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-4-ил)-4-гидроксипиперидин-3-ил)тиазол-2-карбоксамида (15 мг, 40%): ЖХ/МС (Таблица 1, способ d) Время удерживания = 2,52 мин.; MS m/z: 545 (M+H)+. (Btk IC50=A)
Примеры, полученные с использованием общего способа O
(Таблица 1, Способ)
(полученный с использованием A из
4-бром-1H-индол-7-карбонитрила
и
2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина [Combi-Blocks]), N с Cs2CO3
(полученный с использованием A из
4-бром-1H-индол-7-карбонитрила
и
2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина [CombiBlocks]), H из оксетан-3-она, N с Cs2CO3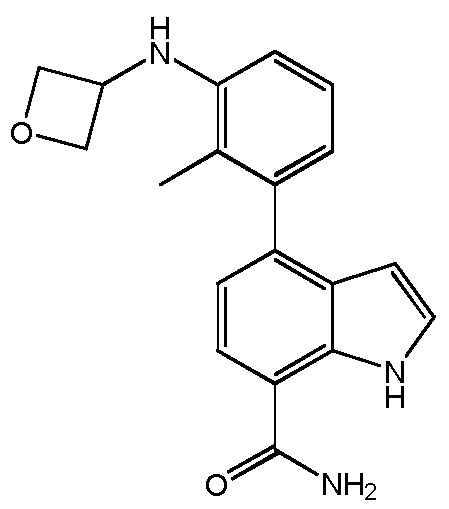
Получения № 27 и
Получения № 13, N с Cs2CO3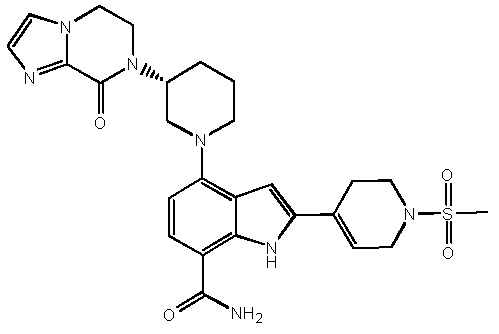
Получения № 27 и Получения № 12, N с Cs2CO3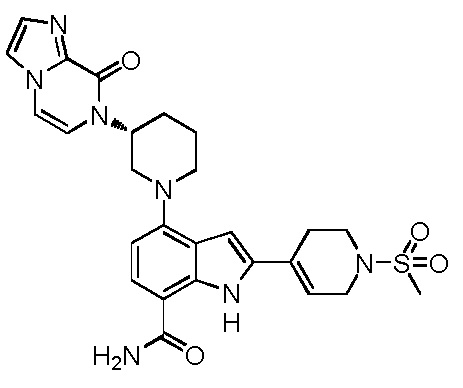
(R)-трет-бутил пиперидин-3-илкарбамата, G с HCl, и D с
2-метилоксазол-4-карбоновой кислотой, N с Cs2CO3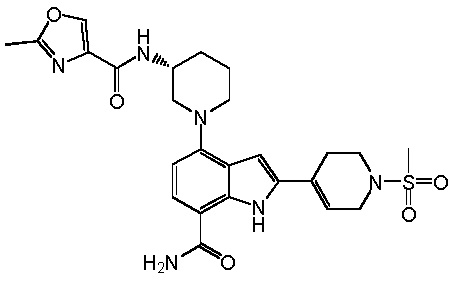
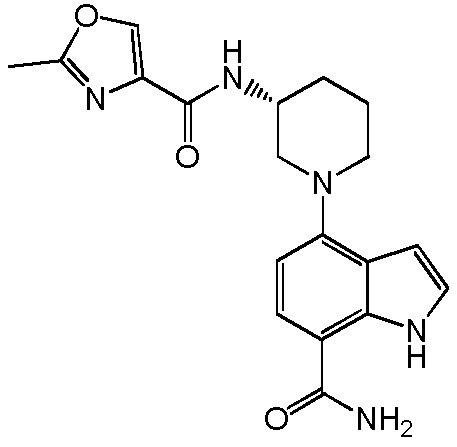
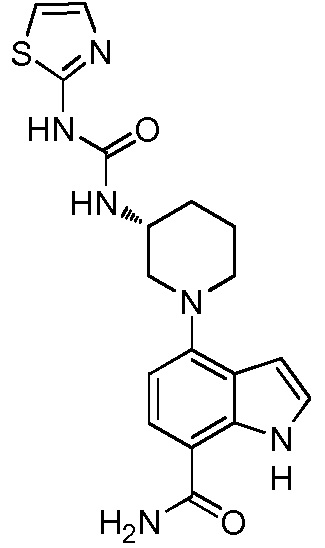
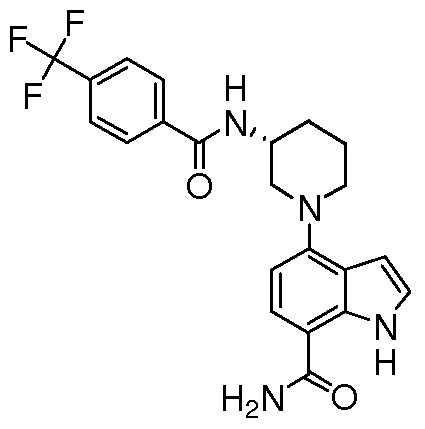
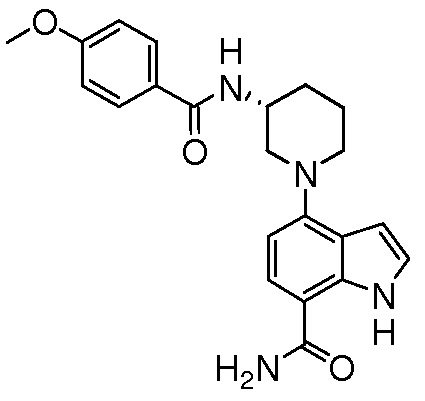
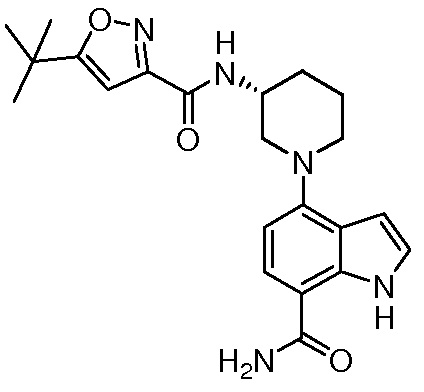
4-трет-бутилбензойной кислотой и Получения № B.1, N с Cs2CO3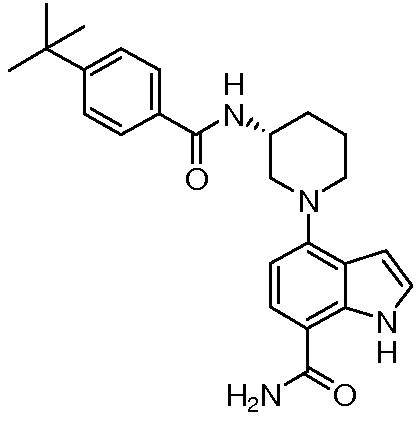
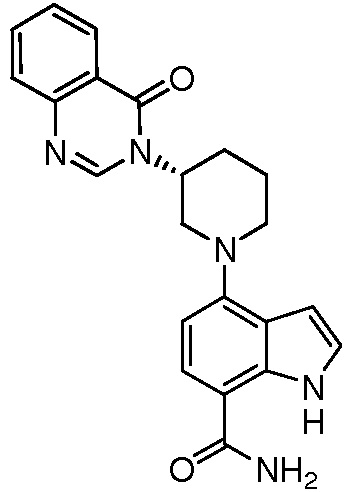
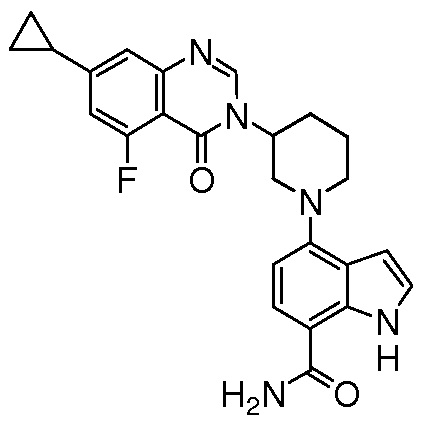
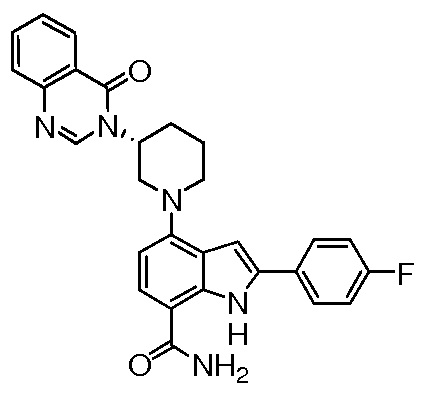
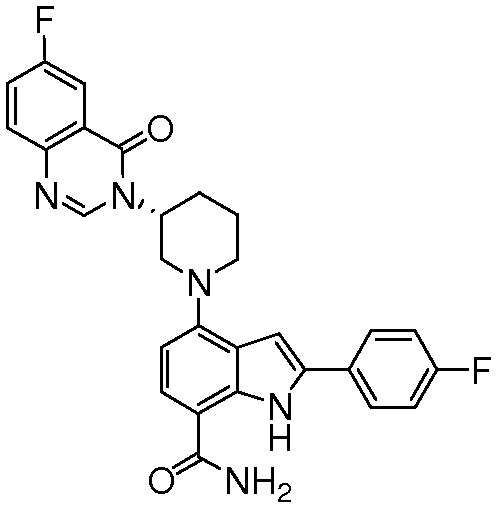
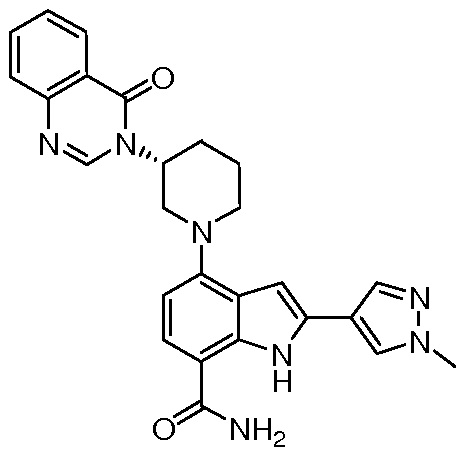
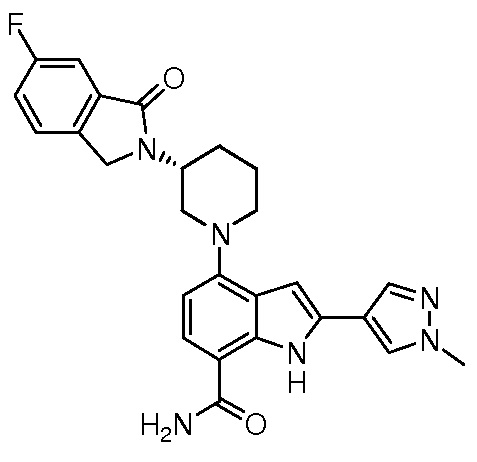
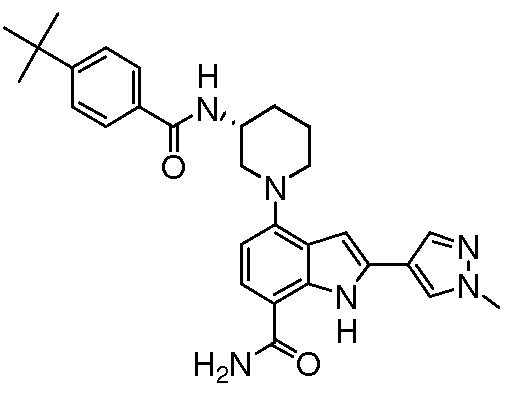
(полученный с использованием A из Получения № 27, стадия B и 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола, B с (R)-трет-бутил пиперидин-3-илкарбаматом, V с 4- метоксибензойной кислотой, N с Cs2CO3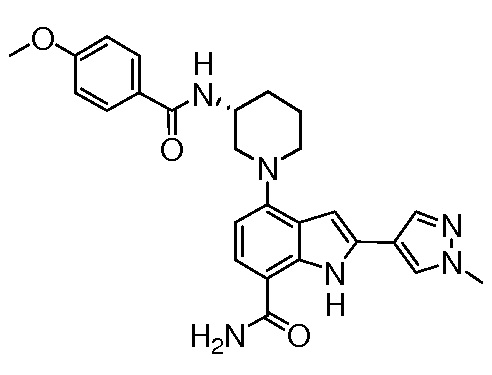
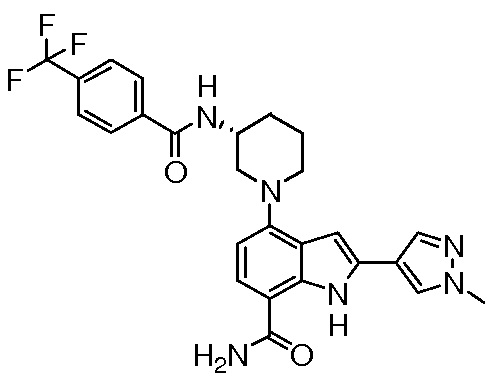
(полученный с использованием A из Получения № 27, стадия B и 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола, B с (R)-трет-бутил пиперидин-3-илкарбаматом, V с 4-(дифторметил)-бензойной кислотой, N с Cs2CO3
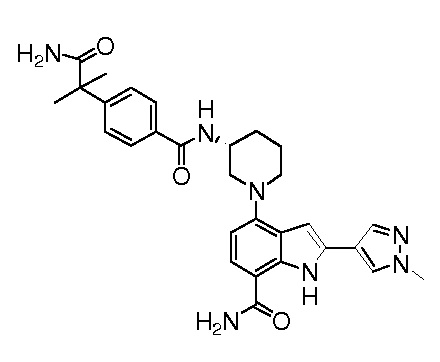
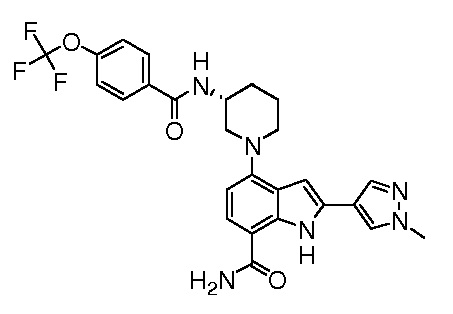
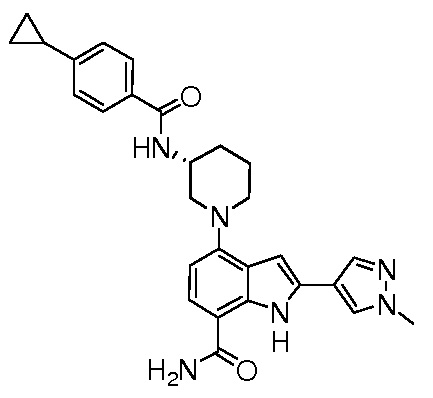
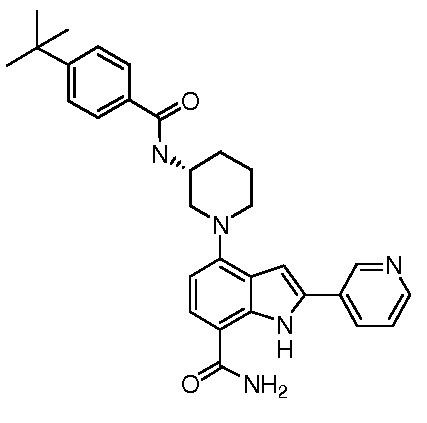
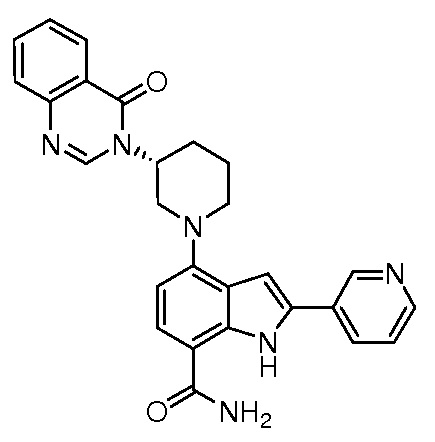
Общий способ P: Образование бороната из арилгалогенида или гетероарилгалогенида
К смеси галогенида, например, броминдолу (предпочтительно 1 эквив.), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (1-3 эквив., предпочтительно 1,2 эквив.), ацетату калия (2-5 эквив., предпочтительно 3 эквив.), и в растворителе (таком как THF или 1,4-диоксан; предпочтительно, 1,4-диоксан) добавляли палладиевый катализатор (например, Pd2dba3 или (1,1'-бис(дифенилфосфино)ферроцен)дихлорпалладий(II), комплекс с DCM; предпочтительно, 1,1'-бис(дифенилфосфино)ферроцен)дихлорпалладий(II) комплекс с DCM, 0,01-0,20 эквив., предпочтительно, 0,1 эквив.). Смесь нагревали при приблизительно 40-120°C (предпочтительно, приблизительно 80°C) в течение приблизительно 1-24 часов (предпочтительно, приблизительно 16 часов). Смесь оставляли охлаждаться до комнатной температуры и обрабатывали с помощью одного из следующих способов. Способ 1. Смесь возможно разбавляли органическим растворителем (таким как DCM или EtOAc) и органический раствор необязательно промывали водой и/или насыщенным солевым раствором, сушили над безводным MgSO4 или Na2SO4, фильтровали и растворитель удаляли при пониженном давлении с получением целевого соединения. Способ 2. Смесь концентрировали при пониженном давлении и необязательно очищали с использованием одного или нескольких способов очистки, описанных выше, с получением желаемого соединения. Способ 3. Катализатор удаляли фильтрованием и фильтрат концентрировали при пониженном давлении.
Иллюстрация общего способа P
Получение № P.1: 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-7-карбоксамид
Смесь 4-бром-1H-индол-7-карбоксамида (5 г, 20,9 ммоль, Получение № 2), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (6,37 г, 25,1 ммоль), ацетата калия (6,16 г, 62,7 ммоль) и Pd(dppf)Cl2-DCM (0,85 г, 1,05 ммоль) в 1,4-диоксане (2 мг) нагревали при температуре приблизительно 80°C в атмосфере N2 в течение ночи. Растворитель удаляли при пониженном давлении с получением остатка, который очищали колоночной хроматографией на силикагеле с получением 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-7-карбоксамида (3 г, 50%): 1H-ЯМР (CDCl3) δ 10,30 (ушир., 1H), 7,64-7,62 (д, J=8 Гц, 1H), 7,40-7,38 (м, 2H), 7,08-7,07 (м, 1H), 1,42 (с, 12H).
Общий способ Q: Реакция Мицунобу спирта
К спирту (предпочтительно, 1 эквив.) в органическом растворителе (таком как THF, бензол, толуол или 1,4-диоксан, предпочтительно, толуол или 1,4-диоксан) добавляли подходящий кислотный реагент (такой как карбоновая кислота, фенол или гетероарил спирта, 1-3 эквив., предпочтительно 1 эквив.), затем три-н-бутилфосфин, трифенилфосфин или полимер-связанный трифенилфосфин (предпочтительно, трифенилфосфин, 1-3 эквив., предпочтительно, 1,2 эквив.) и TMAD, 1,1'-(азодикарбонил)дипиперидин, DIAD или DEAD (предпочтительно, DEAD, 1-3 эквив., предпочтительно 1,2 эквив.) добавляли по каплям при температуре приблизительно 0-120°C (предпочтительно, 0-25°C). Реакционную смесь перемешивали при приблизительно 5-120°C в течение приблизительно 5-48 часов (предпочтительно, приблизительно 16 часов). Кроме того, после приблизительно 0,1-24 часов, дополнительное количество фосфинового реагента (0,2-2 эквив.) и TMAD, 1,1'-(азодикарбонил)дипиперидина, DIAD или DEAD (0,2-1 эквив.) добавляли для доведения реакции до конца. Способ 1. При использовании полимер-связанного реагента, реакционную смесь фильтровали и промывали смесью растворителей, таких как DCM, EtOAc и МеОН (предпочтительно, DCM, затем МеОН). Фильтрат концентрировали при пониженном давлении. Способ 2. Когда не использовали полимер-связанный реагент, реакционную смесь необязательно разбавляли органическим растворителем, таким как DCM или EtOAc, а затем промывали водой, насыщенным водным раствором NaHCO3, насыщенным солевым раствором и сушили над безводным Na2SO4 или MgSO4, фильтровали и концентрировали при пониженном давлении. Кроме того, реакционную смесь непосредственно концентрировали при пониженном давлении.
Иллюстрация общего способа Q
Получение № Q.1: 2-((4-бром-3-нитрофенокси)метил)тиазол
К раствору 4-бром-3-нитрофенола (2 г, 9,17 ммоль, Получение № S.1), тиазол-2-илметанола (1,01 г, 9,17 ммоль) и трифенилфосфина (2,9 г, 11,01 ммоль) в безводном толуоле (50 мг) добавляли DEAD (1,7 мл, 11,01 ммоль) при приблизительно 0°C под N2. Затем смесь нагревали с обратным холодильником в течение ночи. После охлаждения до комнатной температуры смесь концентрировали при пониженном давлении с получением остатка, который очищали колоночной хроматографией на силикагеле с получением 2-((4-бром-3-нитрофенокси)метил)тиазола (2 г, 69%): 1H-ЯМР (CDCl3) δ 7,83 (д, J=3,1 Гц, 1H), 7,63 (д, J=8,8 Гц, 1H), 7,53 (д, J=3,1 Гц, 1H), 7,42 (д, J=3,1 Гц, 1H), 7,12 (дд, J=3,1, 8,8 Гц, 1H), 5,43 (с, 2H).
Общий способ R: Восстановление нитрогруппы до амина с использованием Fe
К смеси нитросодержащего соединения в растворителе (таком как MeOH, EtOH, MeOH/вода или EtOH/вода, предпочтительно, EtOH/вода) добавляли Fe (3-5 эквив., предпочтительно 5 эквив.) и NH4Cl (3-5 эквивалентов, предпочтительно 5 эквив.). Смесь нагревали при приблизительно 40-100°C (предпочтительно, приблизительно 80°C) в течение приблизительно 2-24 часов (предпочтительно, приблизительно 16 часов). Смесь оставляли охлаждаться до комнатной температуры и обрабатывали с помощью одного из следующих способов. Способ 1. Смесь возможно разбавляли органическим растворителем (таким как DCM или EtOAc) и органический раствор необязательно промывали водой и/или насыщенным солевым раствором, сушили над безводным MgSO4 или Na2SO4, фильтровали и растворитель удаляли при пониженном давлении с получением целевого соединения. Способ 2. Смесь концентрировали при пониженном давлении и необязательно очищали с использованием одного или нескольких из способов очистки, описанных выше, с получением желаемого соединения. Способ 3. Катализатор удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Промежуточные и конечные соединения, полученные с помощью этого общего способа можно необязательно очистить с использованием одного или нескольких способов очистки, описанных выше.
Иллюстрация общего способа R
Получение № R.1: 2-бром-5-(тиазол-2-илметокси)анилин
К раствору 2-((4-бром-3-нитрофенокси)метил)тиазола (1 г, 3,2 ммоль) в EtOH (40 мг) и воды (20 мг) добавляли железо (0,88 г, 15,8 ммоль) и NH4Cl (0,85 г, 15,8 ммоль). Смесь нагревали с обратным холодильником в течение ночи. Смесь фильтровали и фильтрат концентрировали при пониженном давлении с получением остатка, который разбавляли добавлением воды и экстрагировали EtOAc. Органический слой концентрировали при пониженном давлении с получением 2-бром-5-(тиазол-2-илметокси)анилина (0,7 г, 77%): ЖХ/МС (Таблица 1, способ l) Время удерживания = 1,46 мин; MS m/z 285 (M+H)+.
Общий способ S: Деметилирование арил метилового эфира
К смеси метоксильного соединения в растворителе (таком как DCM, DCE, THF, бензол, толуол или 1,4-диоксан, предпочтительно DCM) медленно добавляли BBr3 (2-24 эквив., предпочтительно 2,5 эквив.). Смесь нагревали при приблизительно 30-110°C (предпочтительно, приблизительно 45°C) в течение приблизительно 2-24 часов (предпочтительно, приблизительно 4-24 часов). Смесь оставляли охлаждаться до 0-10°C (предпочтительно, приблизительно 0°C) и разбавляли водой. Смесь может быть разбавлена органическим растворителем (таким как DCM или EtOAc) и органический раствор, необязательно промывали водой и/или насыщенным NaHCO3 и/или насыщенным солевым раствором, сушили над безводным MgSO4 или Na2SO4, фильтровали и растворитель удаляли при пониженном давлении с получением желаемого соединения.
Иллюстрация общего способа S
Получение № S.1: 4-бром-3-нитрофенол
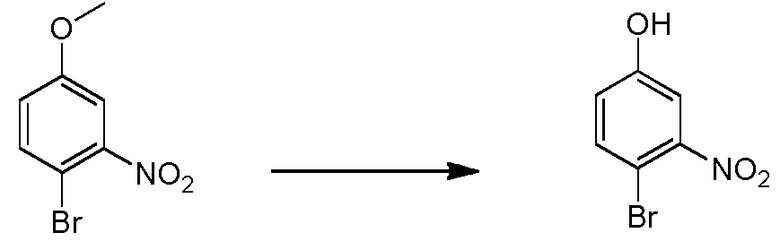
К раствору 1-бром-4-метокси-2-нитробензола (20 г, 82 ммоль) в DCM (800 мг) добавляли по каплям BBr3 (19 мл, 207 ммоль) в DCM (120 мг). Полученную смесь нагревали с обратным холодильником в течение ночи. Смесь охлаждали в ледяной воде и разбавляли добавлением воды. Затем смесь промывали насыщенным раствором NaHCO3 и насыщенным солевым раствором. Органическую фазу сушили над Na2SO4, концентрировали при пониженном давлении с получением остатка, который очищали с помощью колоночной хроматографии на силикагеле, с получением 4-бром-3-нитрофенола (6 г, 31%) в виде твердого вещества: 1H-ЯМР (CDCl3): δ 7,57 (д, J=8,8 Гц, 1H), 7,35 (д, J=2,6 Гц, 1H), 6,94 (дд, J=2,9, 8,6 Гц, 1H), 5,90 (ушир., 1H).
Общий способ T: Реакция Бухвальда арилгалогенида или гетероарилгалогенида с амином
Смесь арилгалогенида или гетероарилгалогенида (1,0 эквивалент), амина (1-2,2 эквивалента, предпочтительно 1-1,2 эквивалента), палладиевого катализатора (такого как Pd2dba3 или Pd(OAc)2, предпочтительно, Pd2dba3; 0,01-1,0 эквивалент, предпочтительно, 0,04-0,1 эквивалент), лиганда (такого как X-phos, Xanthphos или трет-бутил-X-phos, предпочтительно, трет-бутил-X-phos или X-Phos, 0,01-2,0 эквивалента, предпочтительно 0,04-0,1 эквивалента) и основания (такого как K2CO3, Na2CO3, Cs2CO3, K3PO4, NaOt-Bu, KOt-Bu, KOAc, KOH, предпочтительно K2CO3; 1-5 эквивалента, предпочтительно 1-3 эквивалента) добавляли к растворителю (такому как 1,4-диоксан, t-BuOH, предпочтительно, t-BuOH). Смесь дегазировали в атмосфере инертного газа (например, азота или аргона, предпочтительно, азота) и нагревали общепринятым нагреванием при приблизительно 80-100°C (предпочтительно, приблизительно 85-95°C) в течение приблизительно 2-24 часов (предпочтительно, приблизительно 18 часов) или нагревали за счет микроволнового излучения при приблизительно 100-150°C в течение приблизительно от 30 минут до 2 часов. Смесь охлаждали до комнатной температуры. Смесь необязательно фильтровали через среду (например, силикагель или целит), которую промывали соответствующим растворителем (таким как EtOAc, 1,4-диоксан, THF, MeCN, DCM, Et2O, MeOH, EtOH, DMSO, 1:1 MeOH/DMSO или 2:1 MeOH/DMSO, предпочтительно, MeOH/DMSO) и затем фильтрат необязательно концентрировали в вакууме или в потоке теплого азота с получением остатка.
Иллюстрация общего способа T
Получение № T.1: 4-(1-метил-1H-пиразол-5-иламино)-2-п-толил-1H-индол-7-карбоксамид
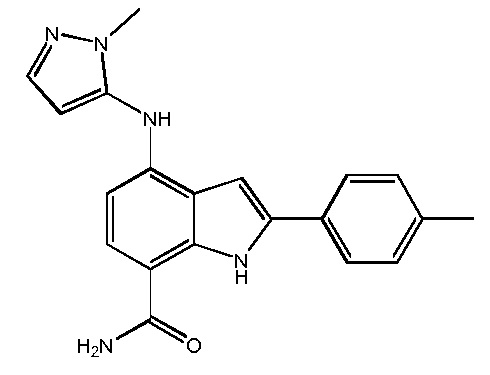
4-Йод-2-(п-толил)-1H-индол-7-карбоксамид (99 мг, 0,26 ммоль, полученный с использованием F с 1-(п-толил)этанона), 1-метил-1H-пиразол-5-иламин (27 мг, 0,26 ммоль, Maybridge-Int), X-Phos (7,53 мг, 0,016 ммоль), K2CO3 (44 мг, 0,316 ммоль) и Pd2dba3 (14 мг, 0,016 ммоль) смешивали в t-BuOH (1,32 мг) в запаянном сосуде для микроволновой печи. Сосуд дегазировали и продували N2 и нагревали при приблизительно 85°C в течение 18 часов. Реакционную смесь охлаждали до комнатной температуры и фильтровали через целит. Фильтрат дважды экстрагировали DCM. Объединенные органические слои концентрировали. Остаток продукта очищали на колонке с нормальной фазой (18 мг, 20%): ЖХ/МС (Таблица 1, Способ f) Время удерживания = 1,48 мин; MS m/z 346 (M+ H)+. (Btk IC50=B)
Примеры, полученные из 4-йод-2-(п-толил)-1H-индол-7-карбоксамид (полученный с использованием F с 1-(п-толил)этаноном) с использованием общего способа T
(Таблица 1, способ)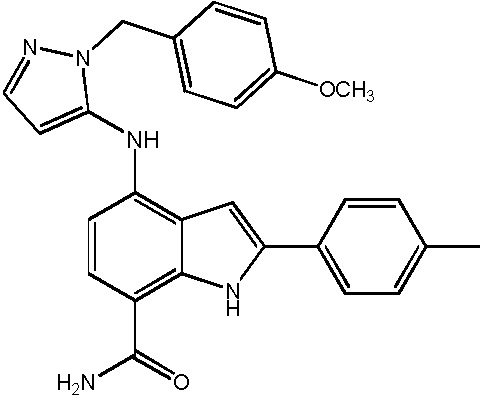
Общий способ U: Реакция кросс-сочетания Негиши арилгалогенида или гетероарилгалогенида с цинкорганическим соединением
Смесь арилгалогенида или гетероарилгалогенида (предпочтительно, 1,0 эквив.) органического растворителя или смеси растворителей (таких как THF, Et2O или 1,4-диоксан, предпочтительно, THF), цинкорганического соединения (0,67-1,5 эквив., предпочтительно 0,9-1,2 эквив.), палладиевого катализатора (такого как Pd(PPh3)4, 0,01-1,0 эквив., предпочтительно 0,025-0,10 эквив.) перемешивали при от приблизительно комнатной температуры до 90°C (предпочтительно, приблизительно 85°C) в течение приблизительно 1-24 часов (предпочтительно, приблизительно 18 часов). Смесь охлаждали до комнатной температуры. Смесь необязательно фильтровали через среду (такую как силикагель или целит), которую промывали соответствующим растворителем (таким как, EtOAc, 1,4-диоксан, THF, MeCN, DCM, Et2O, MeOH, EtOH), а затем необязательно концентрировали в вакууме с получением остатка. Или остаток или раствор необязательно разделяли между водой и органическим растворителем (таким как EtOAc, Et2O или DCM). Органический слой выделяли и необязательно промывали в произвольном порядке водой и/или водными растворами, содержащими кислоту (такую как HCl, AcOH или NH4Cl) и/или водными растворами, содержащими основание (такое как NaHCO3, Na2CO3, NaOH, KOH или NH4OH) и/или водными растворами, содержащими неорганическую соль (такую как NaCl, Na2SO3 или Na2S2O3). Органический раствор затем необязательно сушили с высушивающим веществом (например, безводным MgSO4 или Na2SO4), фильтровали и концентрировали в вакууме с получением целевого соединения.
Иллюстрация общего способа U
Получение № U.1: 4-(2-Хлор-6-фторбензил)-2-п-толил-1H-индол-7-карбоксамид
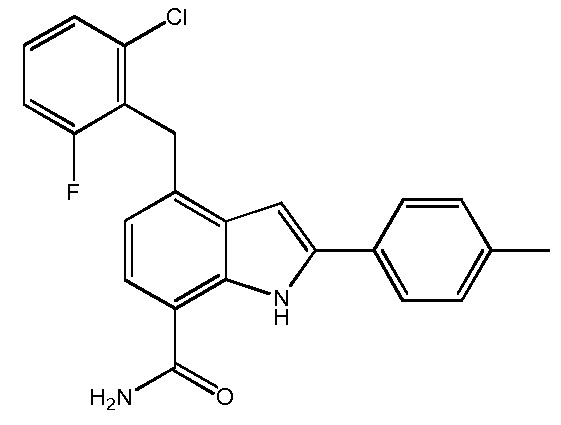
4-Йод-2-(п-толил)-1H-индол-7-карбоксамид (97 мг, 0,258 ммоль, полученный с использованием F из 1-(п-толил)этанона), (2-хлор-6-фторбензил)цинк(II) бромид (0,77 мл, 0,387 ммоль) и тетракис (трифенилфосфин) палладий(0) (15 мг, 0,013 ммоль) растворяли в THF (0,82 мг) в запаянном сосуде для микроволновой печи и нагревали термически при 85°C в течение приблизительно 18 часов. Реакционную смесь охлаждали до комнатной температуры и фильтровали через целит. Фильтрат концентрировали с получением остатка. Остаток очищали на колонке с нормальной фазой, элюируя EtOAc в гексане с получением 4-(2-хлор-6-фторбензил)-2-п-толил-1H-индол-7-карбоксамида (30 мг, 30%): ЖХ/МС (Таблица 1, Способ f) Время удерживания = 2,09 мин; MS m/z 393 (M+H)+.
Примеры, полученные из 4-йод-2-(п-толил)-1H-индол-7-карбоксамида (полученный с использованием F с 1-(п-толил)этаноном) с использованием общего способа U
(Таблица 1, Способ)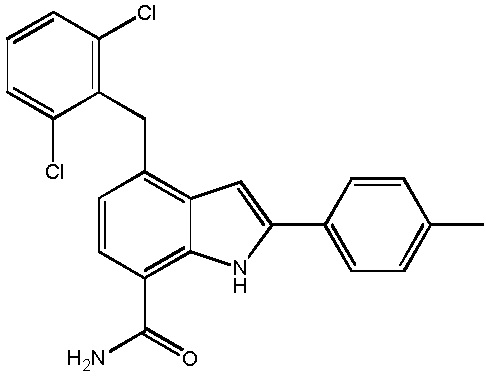
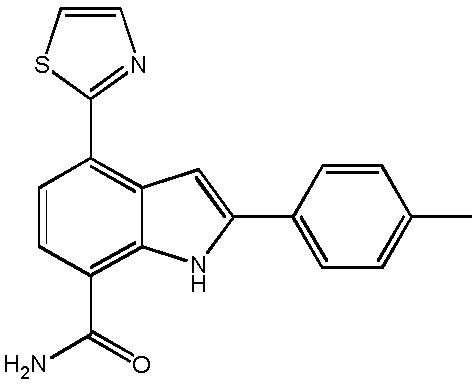

Общий способ V: Получение амида из Boc-защищенного амина и карбоновой кислоты
К раствору N-Boc амина (1 эквив.) в органическом растворителе (таком как DCM, DCE, 1,4-диоксан или MeOH, предпочтительно, DCM или 1,4-диоксан) добавляли кислоту (такую как TFA или HCl, предпочтительно, TFA; 2-100 эквив., предпочтительно, 25-50 эквив.). Смесь перемешивали при приблизительно 0-100°C (предпочтительно, приблизительно 20-60°C) в течение приблизительно 0,5-24 часов (предпочтительно, приблизительно 0,5-6 часов). Необязательно, добавляли дополнительное количество кислоты (2-35 эквив., предпочтительно, 20-25 эквив.) и смесь перемешивали при приблизительно 0-100°C (предпочтительно, приблизительно 20-60°C) в течение приблизительно 1-24 часов (предпочтительно, приблизительно 1-6 часов). Если твердое вещество присутствовало в смеси, смесь необязательно фильтровали и твердое вещество промывали органическим растворителем, таким как 1,4-диоксан или Et2O. Полученное твердое вещество затем, необязательно, сушили при пониженном давлении. Альтернативно, реакционную смесь концентрировали при пониженном давлении. К остатку в сосуде добавляли в произвольном порядке карбоновую кислоту или карбоксилатную соль (1-5 эквив., предпочтительно, 1,1-1,5 эквив.), органический растворитель (такой как DCM, DCE, DMF, THF или 1,4-диоксан, предпочтительно, DCM или DMF), связующий пептидный реагент (такой как BOP-Cl, IBCF, HATU, DCI, PyBOP, или EDC•HCl, предпочтительно, HATU; 1-10 эквив., предпочтительно 1-2 эквив.), основание (такое как TEA, DIEA, пиридин или DIEA, предпочтительно, DIEA; 1-20 эквив., предпочтительно 1-5 эквив.) и необязательно HOBt (0-5 эквив., предпочтительно 0-1 эквив.). Затем смесь перемешивали при температуре 10-60°C (предпочтительно, приблизительно 25-50°C) в течение приблизительно от 15 минут до 48 часов (предпочтительно, приблизительно от 15 минут до 24 часов). Необязательно, дополнительные количества вышеуказанных реагентов добавляли для доведения реакции до конца. Смесь необязательно концентрировали в вакууме с получением целевого соединения. Смесь необязательно фильтровали через среду (такую как силикагель или целит), которую промывали соответствующим растворителем (таким как EtOAc, 1,4-диоксан, THF, MeCN, DCM, Et2O, MeOH, EtOH), а затем при необходимости концентрировали в вакууме с получением остатка. Или остаток или раствор необязательно разделяли между водой и органическим растворителем (таким как EtOAc, Et2O или DCM). Органический слой выделяли и необязательно промывали в произвольном порядке водой и/или водными растворами, содержащими кислоту (такую как HCl, AcOH или NH4Cl) и/или водными растворами, содержащими основание (такое как NaHCO3, Na2CO3, NaOH, KOH или NH4OH) и/или водными растворами, содержащими неорганическую соль (такую как NaCl, Na2SO3 или Na2S2O3). Органический раствор затем необязательно сушили с высушивающим веществом (например, безводным MgSO4 или Na2SO4), фильтровали и концентрировали в вакууме с получением целевого соединения.
Иллюстрация общего способа V
Получение № V.1: (R)-N-(1-(7-циано-1H-индол-4-ил)пиперидин-3-ил)-2-метилоксазол-4-карбоксамид

К раствору (R)-трет-бутил 1-(7-циано-1H-индол-4-ил)пиперидин-3-илкарбамата (0,11 г, 0,333 ммоль, Получение № B.1) в DCM (1 мг) добавляли TFA (1 мг) и раствор перемешивали при приблизительно 25°C в течение приблизительно 30 минут. Смесь упаривали досуха с последующим добавлением DMF (2 мг), TEA (0,139 мл, 0,999 ммоль), HATU (190 мг, 0,499 ммоль) и 2-метилоксазол-4-карбоновой кислоты (0,055 г, 0,433 ммоль). Смесь перешивали при приблизительно комнатной температуре в течение приблизительно 18 часов. Реакционную смесь выпаривали и полученный остаток очищали с помощью хроматографии на силикагеле при элюировании с градиентом 30-100% EtOAc в гексане с получением (R)-N-(1-(7-циано-1H-индол-4-ил)пиперидин-3-ил)-2-метилоксазол-4-карбоксамида (0,092 г, 79%); ЖХ/МС (Таблица 1, способ g) Время удерживания = 1,35 мин.; MS m/z: 350 (M+H)+
Общий способ W: Превращение винилтрифлата в винил-боронат или бороновую кислоту
К смеси бороновой кислотой или бороната (1-2 эквив., предпочтительно, 1,1 эквив.) палладиевого катализатора (например, Pd(OAc)2, Pd2dba3, Pd(PPh3)4, бис(ацетато)трифенилфосфинпалладий(II), PdCl2(dppf), (1,1'-бис(дифенилфосфино)ферроцен)дихлорпалладий(II), или Pd(PPh3)2Cl2; предпочтительно PdCl2(dppf) или Pd(PPh3)2Cl2; 0,01-0,20 эквив., предпочтительно, 0,05-0,1 эквив.), основания (такого как KF, KOAc, Na2CO3, K2CO3 или Cs2CO3, предпочтительно K2CO3 или KOAc) (1,1-16 эквив., предпочтительно, 1,5-2 эквив.) и необязательно фосфиновой добавки (предпочтительно, PPh3; 0,01-0,1 эквив., предпочтительно, 0,06 эквив.) в органическом растворителе (таком как диоксан, DME или DCE, предпочтительно, диоксан) добавляли винилтрифлат (1 эквив.). Смесь нагревали в атмосфере инертного газа при приблизительно 60-90°C (предпочтительно, 70-80°C) в течение приблизительно 1-20 часов (предпочтительно, 8-16 часов). Смесь необязательно концентрировали в вакууме с получением целевого соединения. Альтернативно, смесь необязательно фильтровали через среду (такую как, силикагель или целит), которую промывали соответствующим растворителем (таким как, EtOAc, 1,4-диоксан, THF, ACN, DCM, Et2O, MeOH или EtOH) и затем, необязательно, концентрировали в вакууме с получением остатка. Или остаток или раствор необязательно разделяли между водой и органическим растворителем (таким как EtOAc, Et2O или DCM). Органический слой выделяли и необязательно промывали в произвольном порядке водой и/или водными растворами, содержащими кислоту (такую как HCl, AcOH или NH4Cl) и/или водными растворами, содержащими основание (такое как NaHCO3, Na2CO3, NaOH, KOH или NH4OH) и/или водными растворами, содержащими неорганическую соль (такую как NaCl, Na2SO3 или Na2S2O3). Органический раствор затем необязательно сушили с высушивающим веществом (например, безводным MgSO4 или Na2SO4), фильтровали и концентрировали в вакууме с получением целевого соединения.
Иллюстрация общего способа W
Получение № W.1: трет-Бутил 6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2,3-дигидро-1,4-оксазепин-4(7H)-карбоксилат

В 3-горлую круглодонную колбу, емкостью 100 мл, помещали 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (1,10 г, 4,34 ммоль, Получение № AA.1), PPh3 (0,062 г, 0,24 ммоль), Pd(PPh3)2Cl2 (0,138 г, 0,197 ммоль) и K2CO3 (0,818 г, 5,92 ммоль). К этой смеси добавляли раствор трет-бутил 6-(((трифторметил)сульфонил)окси)-2,3-дигидро-1,4-оксазепин-4(7H)-карбоксилата (1,37 г, 3,94 ммоль) в диоксане (30 мг). Всю смесь дегазировали в течение приблизительно 5 минут и продували азотом. Смесь нагревали при приблизительно 75°C в течение приблизительно 15 часов. Смесь разбавляли EtOAc (30 мг) и водой (30 мг). Органический слой разделяли, сушили над MgSO4, фильтровали и концентрировали. Полученную смесь очищали хроматографией на силикагеле (10-40% EtOAc/гептан) с получением трет-бутил 6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2,3-дигидро-1,4-оксазепин-4(7H)-карбоксилата (0,57 г, 44%): ЖХ/МС (Таблица 1, Способ as) Время удерживания = 2,65 мин; MS m/z: 226 (M+H-Boc)+
Общий способ X: Гидролиз сложного эфира до карбоновой кислоты в основных условиях и удаление тозил-группы из N-тозил-защищенного гетероарильного кольца
В сосуд, содержащий соединение с и функциональностью сложного эфира и тозил-защищенным гетероароматическим кольцом (1 эквив.) или в чистом виде или в органическом растворителе (таком как 1,4-диоксан, MeOH или THF/MeOH, THF/вода/MeOH, предпочтительно, THF/вода/MeOH) добавляли основание или комбинацию оснований (таких как водный или твердый Na2CO3, KOH, Cs2CO3, K2CO3, NaOH или LiOH, предпочтительно, LiOH или KOH; 1-10 эквив., предпочтительно, 5-10 эквив.). Смесь перемешивали при приблизительно 0-100°C (предпочтительно, приблизительно 40-85°C) в течение приблизительно 1-48 часов (предпочтительно, приблизительно 1-24 часов). Необязательно, добавляли дополнительное основание (такое как водный или твердый Na2CO3, KOH, Cs2CO3, K2CO3, NaOH или LiOH, предпочтительно, LiOH или NaOH, 1-10 эквив., предпочтительно, 2-6 эквив.) и смесь перемешивали при приблизительно 0-100°C (предпочтительно, приблизительно 10-100°C) в течение приблизительно 1-48 часов (предпочтительно, приблизительно 4-24 часов). Затем смесь подкисляли добавлением подходящей водной кислоты (такой как водная HCl, AcOH или лимонная кислота, предпочтительно, лимонная кислота). Смесь необязательно концентрировали в вакууме с получением целевого соединения. Альтернативно, смесь необязательно фильтровали через среду (например, силикагель или целит), которую промывали соответствующим растворителем (таким как EtOAc, 1,4-диоксан, THF, ACN, DCM, Et2O, MeOH, или EtOH) и затем, необязательно, концентрировали в вакууме с получением остатка. Или остаток или раствор необязательно разделяли между водой и органическим растворителем (таким как EtOAc, Et2O или DCM). Органический слой выделяли и необязательно промывали в произвольном порядке водой и/или водными растворами, содержащими кислоту (такую как HCl, AcOH или NH4Cl) и/или водными растворами, содержащими основание (такое как NaHCO3, Na2CO3, NaOH, KOH или NH4OH) и/или водными растворами, содержащими неорганическую соль (такую как NaCl, Na2SO3 или Na2S2O3). Органический раствор затем необязательно сушили с высушивающим веществом (например, безводным MgSO4 или Na2SO4), фильтровали и концентрировали в вакууме с получением целевого соединения.
Иллюстрация общего способа X
Получение № X.1: 4-(1-(трет-Бутоксикарбонил)-1,2,5,6-тетрагидропиридин-3-ил)-2-метил-1H-индол-7-карбоновая кислота
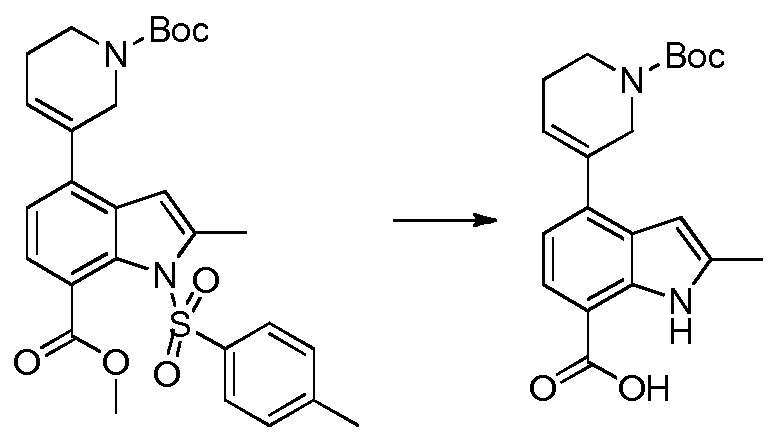
В круглодонную колбу загружали метил 4-(1-(трет-бутоксикарбонил)-1,2,5,6-тетрагидропиридин-3-ил)-2-метил-1-тозил-1H-индол-7-карбоксилат (1,67 г, 2,30 ммоль, Получение № 39) в THF (12 мг), воду (4 мг) и MeOH (4 мг). Добавляли LiOH (моногидрат, 0,468 г, 11,1 ммоль). Смесь перемешивали при приблизительно 60°C. После приблизительно 7 часов добавляли дополнительное количество LiOH (моногидрат, 0, 234 г, 5,57 ммоль) и смесь оставляли перемешиваться в течение приблизительно 24 часов при приблизительно 60°C. Смесь разбавляли 5% лимонной кислотой (200 мг) и экстрагировали DCM (2×100 мг) и 3:1, CHCl3: изопропанол (100 мг). Объединенные органические слои промывали водой (50 мг) и сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением 4-(1-(трет-бутоксикарбонил)-1,2,5,6-тетрагидропиридин-3-ил)-2-метил-1H-индол-7-карбоновой кислоты (1,16 г, 93%): ЖХ/МС (Таблица 1, Способ as) Время удерживания = 2,33 мин; MS m/z: 355 (M-H)-.
Общий способ Y: Йодирование 1H-индола или 1Н-азаиндольного кольца с получением 2-йод-1H-индола или 2-йод-1H-азаиндольного кольца
К раствору индола или азаиндола (1 эквив.) в органическом растворителе (таком как THF или Et2O, предпочтительно, THF) при температуре приблизительно от -60 до -78°C (предпочтительно, приблизительно от -70 до -78°C) добавляли основание (такое как BuLi или LDA, предпочтительно, LDA; 1-2 эквив., предпочтительно 1,1-1,5 эквив.). Реакционную смесь затем перемешивали в течение приблизительно 30-45 минут и затем добавляли йод (1-2 эквив., предпочтительно, 1,4-1,6 эквив.). Реакционную смесь перемешивали в течение приблизительно 10-60 минут (предпочтительно, приблизительно 10-30 минут). Смесь необязательно гасили Na2S2O3. Смесь необязательно концентрировали в вакууме с получением целевого соединения. Или остаток или раствор необязательно разделяли между водой и органическим растворителем (таким как EtOAc, Et2O или DCM). Органический слой выделяли и необязательно промывали в произвольном порядке водой и/или водными растворами, содержащими кислоту (такую как HCl, AcOH или NH4Cl) и/или водными растворами, содержащими основание (такое как NaHCO3, Na2CO3, NaOH, KOH или NH4OH) и/или водными растворами, содержащими неорганическую соль (такую как NaCl, Na2SO3 или Na2S2O3). Органический раствор затем необязательно сушили с высушивающим веществом (например, безводным MgSO4 или Na2SO4), фильтровали и концентрировали в вакууме с получением целевого соединения.
Иллюстрация общего способа Y
Получение № Y.1: 1-трет-Бутил 7-метил 4-(1-(трет-бутоксикарбонил)пирролидин-3-ил)-2-йод-1H-индол-1,7-дикарбоксилат

Раствор безводного 1-трет-бутил 7-метил 4-(1-(трет-бутоксикарбонил)пирролидин-3-ил)-1H-индол-1,7-дикарбоксилата (10,0 г, 22,5 ммоль, (Получение № Z.1) в THF (136 мг) охлаждали до приблизительно -78°C и LDA (1M в THF, 33,7 мл, 33,7 ммоль) добавляли по каплям. После приблизительно 45 минут, раствор йода (7,99 г, 31,5 ммоль) в ТГФ (15 мг) добавляли по каплям при поддержании температуры при приблизительно -71°C. Реакционную смесь затем гасили вливанием в водный раствор Na2S2O3 и NaHCO3 (10:1, 150 мг). Смесь разбавляли EtOAc и слои разделяли. Водную фазу экстрагировали EtOAc (3×50 мг). Объединенные органические слои промывали насыщенным солевым раствором, сушили над MgSO4 и фильтровали. Растворитель удаляли при пониженном давлении с получением метил 4-(1-(трет-бутоксикарбонил)-2,5-дигидро-1H-пиррол-3-ил)-1H-индол-7-карбоксилата (10,4 г, 97%): ЖХ/МС (Таблица 1, Способ as) Время удерживания = 2,90 мин; MS m/z: 588 (M+NH4)+.
Общий способ Z: Получение N-Вос защищенного амина
К раствору амина или соли амина (предпочтительно, 1 эквив.) в органическом растворителе (таком как ACN, 1,4-диоксан, DCM, DMF или THF, предпочтительно, DCM) добавляли водное основание, такое как Na2CO3, NaOH, K2CO3 или NaHCO3, предпочтительно, Na2CO3 (2-20 эквив., предпочтительно, 2-0 эквив.) или органическое основание, такое, как TEA или DIEA, предпочтительно, TEA (1-5 эквив., предпочтительно, 1-2 эквив.) с последующим добавлением реагента переноса Вос, такого как BoC2O, Boc ON, Boc-азид или Boc-OSu, предпочтительно, Boc2O (1-4 эквив., предпочтительно, 1-2 эквив.). Необязательно, добавляли добавку, такую как, DMAP (0,01-0,1 эквив., предпочтительно, 0,05 эквив.). Добавление основания является обязательным, если соль амина не используют. Смесь перемешивали при приблизительно 0-40°C (предпочтительно, приблизительно 0-25°C) в течение приблизительно 2-24 часов (предпочтительно, приблизительно 2-16 часов). Смесь необязательно концентрировали в вакууме с получением целевого соединения. Альтернативно, смесь необязательно фильтровали через среду (например, силикагель или целит), которую промывали соответствующим растворителем (таким как EtOAc, 1,4-диоксан, THF, ACN, DCM, Et2O, MeOH, EtOH) и затем, необязательно, концентрировали в вакууме с получением остатка в качестве целевого соединения. Или остаток или раствор необязательно разделяли между водой и органическим растворителем (таким как EtOAc, Et2O или DCM). Органический слой выделяли и необязательно промывали в произвольном порядке водой и/или водными растворами, содержащими кислоту (такую как HCl, AcOH или NH4Cl) и/или водными растворами, содержащими основание (такое как NaHCO3, Na2CO3, NaOH, KOH или NH4OH) и/или водными растворами, содержащими неорганическую соль (такую как NaCl, Na2SO3 или Na2S2O3). Органический раствор затем необязательно сушили с высушивающим веществом (например, безводным MgSO4 или Na2SO4), фильтровали и концентрировали в вакууме с получением целевого соединения.
Иллюстрация общего способа Z
Получение № Z.1: 1-трет-Бутил 7-метил 4-(1-(трет-бутоксикарбонил)пирролидин-3-ил)-1H-индол-1,7-дикарбоксилат
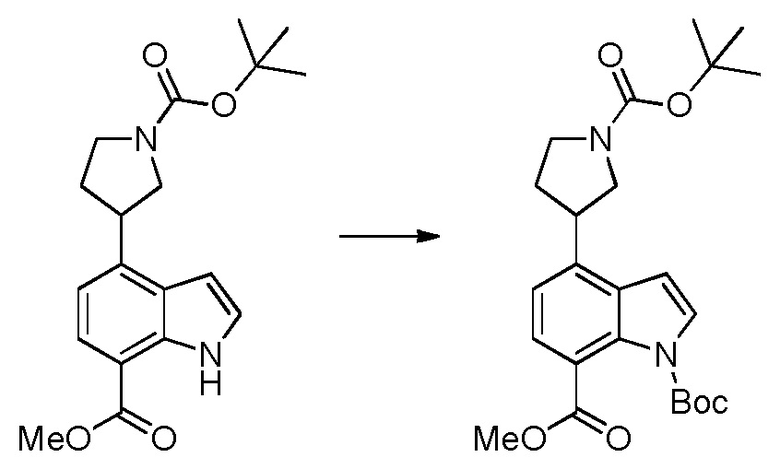
В круглодонную колбу 200 мл добавляли метил 4-(1-(трет-бутоксикарбонил)пирролидин-3-ил)-1H-индол-7-карбоксилат (12,4 г, 36,0 ммоль, полученный с использованием A из метил 4-бром-1H-индол-7-карбоксилата [Anthem] с трет-бутил 3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2,5-дигидро-1H-пиррол-1-карбоксилатом [AKSCI] и L с Pd/C) и ди-трет-бутил дикарбонат (9,43 г, 43,2 ммоль)) в ACN (100 мг). Добавляли DMAP (0,22 г, 1,8 ммоль), реакционную смесь перемешивали при комнатной температуре в течение приблизительно 18 часов, TEA (10 мл, 72 ммоль) и ди-трет-бутил дикарбонат (1,60 мл, 6,87 ммоль) добавляли. Реакционную смесь перемешивали при комнатной температуре в течение приблизительно 16 часов. Смесь экстрагировали разбавленной уксусной кислотой и EtOAc. Объединенные органические слои сушили над MgSO4, концентрировали при пониженном давлении и очищали с помощью хроматографии на силикагеле (0-25% EtOAC/гептан) с получением 1-трет-бутил 7-метил 4-(1-(трет-бутоксикарбонил)пирролидин-3-ил)-1H-индол-1,7-дикарбоксилата (12,5 г, 70%, чистота 89%): ЖХ/МС (Таблица 1, Способ as) Время удерживания = 2,79 мин; MS m/z: 462 (M+NH4)+.
Общий способ AA: Превращение циклического кетона в циклический винилтрифлат
Раствор кетона (1 эквив.) в органическом растворителе (таком как THF, диоксан или простой эфир, предпочтительно, THF) охлаждали до приблизительно от -60 до -78°C (предпочтительно, приблизительно от -65 до -75°C). Затем медленно добавляли основание (такое как LiHMDS, KHMDS или NaHMDS, предпочтительно, KHMDS). После приблизительно 20-60 минут (предпочтительно, 60 минут) добавляли раствор трифлатирующего реагента, такого как, N-(5-хлор-2-пиридил)бис(трифторметансульфонимид)) или 1,1,1-трифтор-N-фенил-N-((трифторметил)сульфонил)метансульфонамид в THF. Реакционную смесь затем оставляли нагреваться до комнатной температуры в течение приблизительно 1-1,5 часов. Затем реакционную смесь необязательно гасили насыщенным раствором NH4Cl или водой и разбавляли органическим растворителем (таким как DCM или EtOAc). Слои разделяли, органический раствор необязательно промывали водой и/или насыщенным солевым раствором, сушили над безводным MgSO4 или Na2SO4, фильтровали и растворитель удаляли при пониженном давлении с получением желаемого соединения.
Иллюстрация общего способа AA
Получение № AA.1: трет-Бутил 6-(((трифторметил)сульфонил)окси)-2,3-дигидро-1,4-оксазепин-4(7H)-карбоксилат
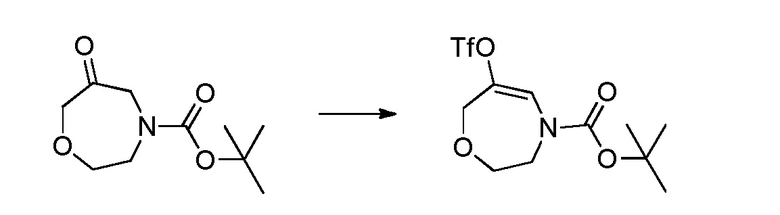
К раствору трет-бутил 6-оксо-1,4-оксазепан-4-карбоксилата (5,00 г, 23,2 ммоль) [Arkpharm] в THF (51,6 мг) при приблизительно -78°C добавляли KHMDS (1 M в THF, 30,2 мл, 30,2 ммоль) по каплям, поддерживая внутреннюю температуру приблизительно от -72 до -74°C. Затем смесь перемешивали при приблизительно -77°C в течение приблизительно 1 часа. Раствор 1,1,1-трифтор-N-фенил-N-((трифторметил)сульфонил)метансульфонамида (7,88 г, 22,1 ммоль) в THF (25,8 мг) добавляли по каплям. Смесь постепенно нагревали до приблизительно 0°C в течение приблизительно 1-2 часов. Реакционную смесь гасили насыщенным водным раствором NH4Cl и экстрагировали EtOAc (2×75 мг). Объединенные органические слои промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали, концентрировали при пониженном давлении и пропускали через слой нейтрального оксида алюминия (EtOAc/гептан в качестве элюента) с получением (((трифторметил)сульфонил)окси)-2,3-дигидро-1,4-оксазепин-4(7H)-карбоксилата (5,1 г, 63,2%); 1H-ЯМР (400 МГц, DMSO-d6) δ 7,17 (с, 1H), 4,41 (с, 2H), 3,77 (кв, J=2,3 Гц, 4H), 1,45 (с, 9H).
Общий способ AB: Восстановление двойной связи и удаление CBZ-группы у CBZ-защищенного амина
В круглодонную колбу загружали палладиевый катализатор, такой как Pd/C или Pd(OH)2 (10 или 20% масс., приблизительно 0,005-1,0 эквив., предпочтительно, 0,5-1,0 эквив.). Колбу откачивали, затем продували азотом 2-5 раз (предпочтительно, 3 раза) перед добавлением органического растворителя или смеси растворителей (такого как EtOAc, MeOH, EtOH или MeOH/AcOH, предпочтительно MeOH/AcOH) в атмосфере азота. К смеси добавляли соединение с функциональностью алкена и N-CBZ-защищенный амин (предпочтительно, 1 эквивалент), в чистом виде или при необходимости в виде раствора в органическом растворителе или смеси растворителей (таком как EtOAc, MeOH, EtOH или MeOH/AcOH, предпочтительно, MeOH). Смесь перемешивали в атмосфере водорода (приблизительно 30-50 фунт на кв.дюйм) в течение приблизительно 1-60 часов (предпочтительно, приблизительно 4-5 часов). Необязательно, реакцию можно проводить с использованием устройства H-cube или с Pd/C или с Pd(OH)2 картриджами (10 или 20% масс.) и исходное вещество проходит через систему в виде раствора в предпочтительном растворителе(ях). В тех случаях, когда реакция не протекает до конца, как контролировали с помощью ТСХ, ЖХ/МС или ВЭЖХ, смесь необязательно нагревали до приблизительно 30-80°C (предпочтительно, приблизительно 50°C) в течение приблизительно 1-24 часов (предпочтительно, приблизительно 16 часов) и в случаях, когда H-cube использовали для проведения реакции, давление может быть увеличено (25-50 бар, предпочтительно, 40-50 бар). Затем смесь фильтровали и осадок на фильтре промывали органическим растворителем (таким как EtOAc, MeOH или EtOH, предпочтительно, реакционный растворитель), и фильтрат концентрировали при пониженном давлении с получением сырого продукта.
Иллюстрация общего способа AB
Получение № AB.1: 4-(Пиперидин-3-ил)-1H-пирроло[3,2-c]пиридин-7-карбоксамид
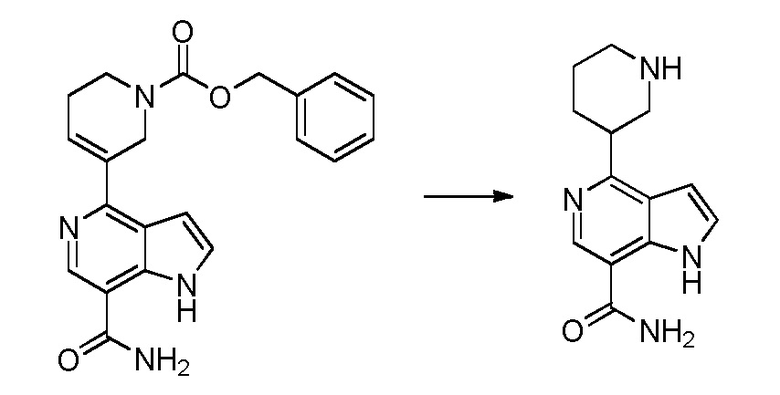
Круглодонную колбу загружали Pd(OH)2 (20% масс., 0,336 г, 0,478 ммоль) с последующим медленным добавлением раствора бензил 3-(7-карбамоил-1H-пирроло[3,2-c]пиридин-4-ил)-5,6-дигидропиридин-1(2H)-карбоксилата (1,8 г, 4,8 ммоль, полученный с использованием A из Получения № 45 и бензил 3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2H)-карбоксилата [Arkpharm], Y с LiOH и D с NH4Cl) в MeOH (30 мг) и AcOH (10 мг). Колбу продували N2, затем заполняли Н2 с помощью баллона. Реакционную смесь затем нагревали при приблизительно 45°C в течение приблизительно 3 часов. Реакционную смесь охлаждали до комнатной температуры и фильтровали через слой целита, промывали MeOH. Фильтрат концентрировали при пониженном давлении, растворяли в МеОН и затем обрабатывали МР-карбонатными шариками перемешиванием при комнатной температуре в течение приблизительно 2 часов. Шарики отфильтровывали и фильтрат концентрировали при пониженном давлении с получением 4-(пиперидин-3-ил)-1H-пирроло[3,2-c]пиридин-7-карбоксамида (0,84 г, 72%): ЖХ/МС (Таблица 1, Способ as) Время удерживания = 0,58 мин.; MS m/z: 245 (M+H)+.
Общий способ AC: N-Окисление N-содержащего гетероароматического кольца
Раствор N-содержащего гетероароматического соединения (1 эквив.) в органическом растворителе (таком как DCE, DME, DCM или EtOAc, предпочтительно, DCM) охлаждали до приблизительно 0°C и окисляющий реагент, такой как 3-хлорбензопероксидная кислота или магния монопероксифталат гексагидрат (1-3 эквив., предпочтительно, 2 эквив.). Раствор перемешивали при комнатной температуре в течение приблизительно 2-24 часов (предпочтительно, приблизительно 10-16 часов). Смесь необязательно фильтровали с получением желаемого продукта или необязательно концентрировали в вакууме с получением остатка, или остаток или раствор необязательно разделяли между водой и органическим растворителем (таким как EtOAc, Et2O или DCM). Органический слой выделяли и необязательно промывали в произвольном порядке водой и/или водными растворами, содержащими кислоту (такую как HCl, AcOH или NH4Cl) и/или водными растворами, содержащими основание (такое как NaHCO3, Na2CO3, NaOH, KOH или NH4OH) и/или водными растворами, содержащими неорганическую соль (такую как NaCl, Na2SO3 или Na2S2O3). Органический раствор затем необязательно сушили с высушивающим веществом (например, безводным MgSO4 или Na2SO4), фильтровали и концентрировали в вакууме с получением целевого соединения.
Иллюстрация общего способа AC
Получение № AC.1: 4-бром-1H-пирроло[2,3-c]пиридин 6-оксид
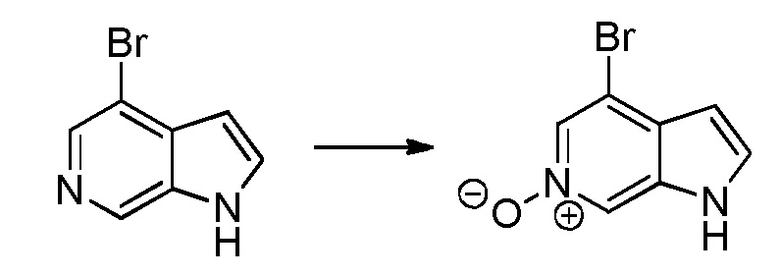
В сосуд загружали 4-бром-1H-пирроло[2,3-c]пиридин (10,0 г, 50,8 ммоль) [Combiblocks] и растворяли в EtOAc (254 мг). Сосуд охлаждали до приблизительно 0°C и раствор 3-хлорбензопероксидной кислоты (10,5 г, 60,9 ммоль) в EtOAc (254 мг) медленно добавляли. Реакционную смесь перемешивали, нагревая до комнатной температуры, в течение приблизительно 16 часов. Осадок, который образовался, собирали фильтрованием и сушили в вакуумной печи с получением 4-бром-1H-пирроло[2,3-c]пиридин 6-оксида (0,85 г, 79%): ЖХ/МС (Таблица 1, способ as) Время удерживания = 1,18 мин; MS m/z: 213, 215(M+H)+.
Общий способ AD: Цианирование N-оксида, содержащего гетероарильное кольцо
В сосуд загружали гетероароматическое соединение N-оксида (1 эквив.) в подходящем органическом растворителе, таком как ACN. Добавляли TEA (1-2 эквив., предпочтительно 1,5 эквив.). TMSCN (2-5 эквив., предпочтительно, 3-4 эквив.) затем добавляли с помощью шприца. Реакционную смесь нагревали с обратным холодильником до полного расхода исходного материала наблюдается с помощью ТСХ или ЖХ/МС. Реакционную смесь охлаждали до комнатной температуры и гасили соответствующим образом, предпочтительно водным раствором NaOH и экстрагировали органическим растворителем, таким как DCM или EtOAC. Органический слой выделяли и необязательно промывали в произвольном порядке водой и/или водными растворами, содержащими кислоту (такую как HCl, AcOH или NH4Cl) и/или водными растворами, содержащими основание (такое как NaHCO3, Na2CO3, NaOH, KOH или NH4OH) и/или водными растворами, содержащими неорганическую соль (такую как NaCl, Na2SO3 или Na2S2O3). Органический раствор затем необязательно сушили с высушивающим веществом (например, безводным MgSO4 или Na2SO4), фильтровали и концентрировали в вакууме с получением целевого соединения.
Иллюстрация общего способа AD
Получение № AD.1: 4-бром-1H-пирроло[2,3-c]пиридин-7-карбонитрил

В сосуд загружали 4-бром-1H-пирроло[2,3-c]пиридин-6-оксид 3-хлорбензоат (6,25 г, 16,91 ммоль, Получение № AC.1) в ACN (97 мг) и TEA (3,56 мл, 25,4 ммоль). TMSCN (9,02 мл, 67,6 ммоль) добавляли в виде одной порции с помощью шприца, смесь кипятили с обратным холодильником в течение приблизительно 45 минут. Реакцию гасили осторожным добавлением 50 мл водного раствора 1 М NaOH, переносили в делительную воронку и разбавляли водным раствором 1 М NaOH (200 мг) и EtOAc (200 мг). Слои разделяли и органическую фазу промывали снова 50 мл водного раствора 1 М NaOH. Объединенные водные экстракты промывали EtOAc (4×75 мг) и затем 1 M NaOH (2×20 мг) и насыщенным солевым раствором (1×50 мг), сушили над Na2SO4, фильтровали и растворитель удаляли с получением 4-бром-1H-пирроло[2,3-c]пиридин-7-карбонитрила (3,84 г, 93%): 1H-ЯМР (400 МГц, DMSO-d6) δ 8,27 (с, 1H), 7,90 (д, J=2,8 Гц, 1H), 6,60 (д, J=2,8 Гц, 1H).
Общий способ AE: Восстановление сложного эфира до образования спирта
К раствору сложного эфира в подходящем органическом растворителе (таком как THF, диоксан, DCM или EtOAc, предпочтительно, THF) необязательно добавляли воду (1-4 эквив., предпочтительно, 2 эквив.). Затем смесь охлаждали до приблизительно 0°C и добавляли восстанавливающий агент (такой как LiBH4 или LAH, предпочтительно, LiBH4; 2-12 эквив., предпочтительно, 6 эквив.). Реакционную смесь перемешивали в течение приблизительно 5-24 часов до полного расхода сложного эфира. Необязательно добавляли дополнительное количество восстанавливающего агента по мере необходимости. Реакционную смесь затем гасили водным раствором NH4Cl. Органический слой выделяли и необязательно промывали в произвольном порядке водой и/или водными растворами, содержащими кислоту (такую как HCl, AcOH или NH4Cl) и/или водными растворами, содержащими основание (такое как NaHCO3, Na2CO3, NaOH, KOH или NH4OH) и/или водными растворами, содержащими неорганическую соль (такую как NaCl, Na2SO3 или Na2S2O3). Органический раствор затем необязательно сушили с высушивающим веществом (например, безводным MgSO4 или Na2SO4), фильтровали и концентрировали в вакууме с получением целевого соединения.
Иллюстрация общего способа AE
Получение № AE.1: трет-Бутил 3-(7-карбамоил-1H-индол-4-ил)-5-(гидроксиметил)пиперидин-1-карбоксилат
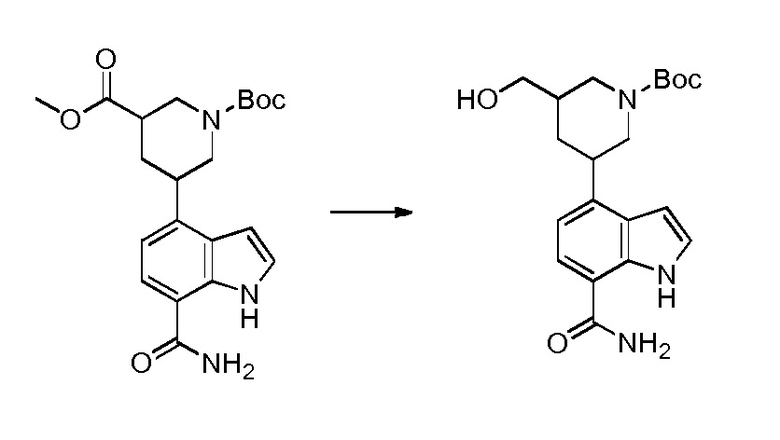
В круглодонную колбу 500 мл добавляли 1-трет-бутил 3-метил 5-(7-карбамоил-1H-индол-4-ил)пиперидин-1,3-дикарбоксилат (6,75 г, 16,8 ммоль, полученный с использованием Z из Получения № AF.1) в THF (150 мг). Реакционную смесь охлаждали до приблизительно 0°C и добавляли воду (0,606 мл, 33,6 ммоль). Добавляли LiBH4 (2,93 г, 135 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение приблизительно 12 часов. Добавляли дополнительное количество LiBH4 (2,93 г, 135 ммоль) и реакционную смесь перемешивали в течение приблизительно 3 часов. Реакционную смесь осторожно добавляли к насыщенному водному раствору NH4Cl (800 мг) при приблизительно -10°C. Смесь экстрагировали DCM (500 мг). Слой DCM сушили над MgSO4, фильтровали и концентрировали с получением сырого трет-бутил 3-(7-карбамоил-1H-индол-4-ил)-5-(гидроксиметил)пиперидин-1-карбоксилата (6,35 г, 101%): ЖХ/МС (Таблица 1, Способ as) Время удерживания = 1,74 мин; MS m/z: 374 (M+H)+.
Общий способ AF: Восстановление пиридинового кольца до пиперидинового кольца
К раствору пиридина (1 эквив.) в уксусной кислоте добавляли восстанавливающий реагент (такой как PtO2, Pd(OH)2 или Pd/C, предпочтительно, PtO2; 0,05-0,5 эквив., предпочтительно, 0,1-0,2 эквив.). Реакционную смесь нагревали при приблизительно 50°C при приблизительно 20-50 фунт на кв.дюйм (предпочтительно, приблизительно 30 фунт на кв.дюйм) в течение приблизительно 6-12 часов (предпочтительно, приблизительно 10 часов). Реакционную смесь концентрировали при пониженном давлении с получением желаемого соединения.
Иллюстрация общего способа AF
Получение № AF.1: Метил 5-(7-карбамоил-1H-индол-4-ил)пиперидин-3-карбоксилат
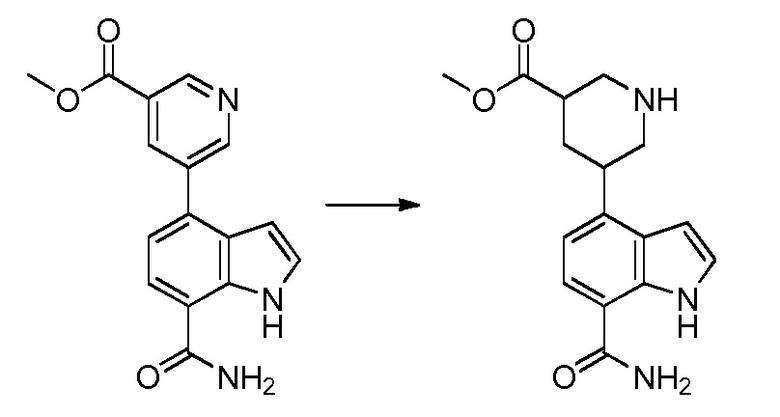
Метил 5-(7-карбамоил-1H-индол-4-ил)никотинат (6,25 г, 23,7 ммоль, полученный с использованием A из Получения № P.1 с метил 5-бромникотинатом) и AcOH (70 мг) добавляли к PtO2 (1,26 г, 5,55 ммоль) в 50 мл сосуде для реакций под давлением и встряхивали в течение приблизительно 10 часов при приблизительно 50°C при приблизительно 30 фунт на кв.дюйм. Полученный черный раствор концентрировали при пониженном давлении и фильтровали через слой целита и промывали DCM. Затем фильтрат концентрировали до густого вязкого маслянистого остатка черного цвета. Это вещество растворяли в 15% MeOH/EtOAc и пропускали через большой слой силикагеля. Слой продували 10% MeOH/EtOAc (250 мг), затем 35-40% MeOH/EtOAc (1,5 л) с получением метил 5-(7-карбамоил-1H-индол-4-ил)пиперидин-3-карбоксилата (6,3 г, 79%): ЖХ/МС (Таблица 1, Способ a) Время удерживания = 0,96 мин; MS m/z: 302 (M+H)+.
Общий способ AG: Одностадийное борилирование трифлата и реакция Сузуки in situ образованного бороната с арилгалогенидом
К смеси винилтрифлата (предпочтительно, 1 эквив.), бороновой кислоты или боронатного эфира (1-2 эквив., предпочтительно, 1,1 эквив.), и неорганического основания (например, KF, Na2CO3, K2CO3 или Cs2CO3, предпочтительно, Na2CO3 или Cs2CO3; 1,1-16 эквив., предпочтительно, 2 эквив.) в растворителе (таком как THF, DME, DMF, 1,4-диоксан, 1,4-диоксан, предпочтительно, диоксан) добавляли палладиевый катализатор (такой как Pd(OAc)2, Pd2dba3, Pd(PPh3)4, бис(ацетато)трифенилфосфинпалладия(II), полимер-связанный FibreCat™ 1032, SiliaCat DPP-Pd, PdCl2(dppf) или Pd(PPh3)2Cl2; предпочтительно, PdCl2(dppf) или Pd(PPh3)2Cl2; 0,01-0,20 эквив., предпочтительно, 0,05-0,1 эквив.) и лиганд (например, трициклогексилфосфин, три-трет-бутил-фосфин; предпочтительно, нет или PPh3; 0,01-1,0 эквив., предпочтительно, 0,01-0,03 эквив.) добавляли необязательно. Смесь нагревали при приблизительно 40-120°C (предпочтительно, приблизительно 70-85°C) в течение приблизительно 1-48 часов (предпочтительно, приблизительно 2-4 часов) термически, или при приблизительно 100-200°C (предпочтительно, приблизительно 120-150°C) в течение приблизительно 5-60 минут (предпочтительно, приблизительно 20-45 минут) в микроволновой печи (предпочтительно, время вывода установки в рабочий режим 5 минут, максимальная мощность 300 Вт, максимальное давление 250 фунт на кв.дюйм). Смеси необязательно давали остыть до комнатной температуры и фильтровали. К реакционной смеси добавляли арилгалогенид (1-2 эквив.), воду (приблизительно 1/3-1/4 объема используемого исходного органического растворителя) и необязательно дополнительный катализатор, основание и лиганд добавляли (предпочтительно, такие же, как используемые в первой реакции) и нагревали при той же температуре в течение приблизительно 3-24 часов (предпочтительно, приблизительно 8-10 часов) и обрабатывали с помощью одного из следующих способов. Способ 1. Для реакций, содержащих воду, смесь может быть разбавлена органическим растворителем (например, DCM или EtOAc). Слои разделяли, органический раствор необязательно промывали водой и/или насыщенным солевым раствором, сушили над безводным MgSO4 или Na2SO4, фильтровали и растворитель удаляли при пониженном давлении с получением желаемого соединения. Способ 2. Смесь концентрировали при пониженном давлении. Способ 3. Катализатор удаляли фильтрованием и фильтрат концентрировали при пониженном давлении.
Иллюстрация общего способа AG
Получение № AG.1: трет-Бутил 6-(7-(метоксикарбонил)-1H-пирроло[3,2-c]пиридин-4-ил)-2,3-дигидро-1,4-оксазепин-4(7H)-карбоксилат
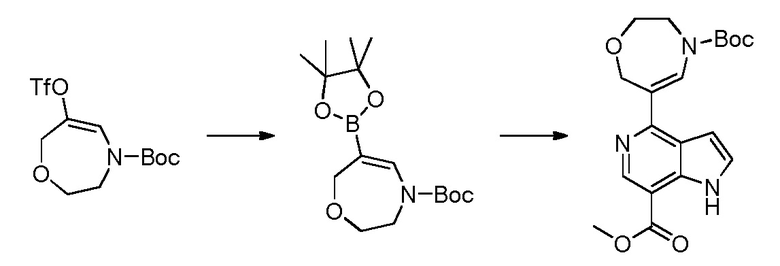
Реакционную пробирку для микроволной печи, емкостью 40 мл, заполняли 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (0,995 г, 3,92 ммоль), PPh3(0,056 г, 0,214 ммоль), Pd(PPh3)2Cl2 (0,125 г, 0,178 ммоль) и K2CO3 (0,738 г, 5,34 ммоль). К этой смеси добавляли раствор трет-бутил 6-(((трифторметил)сульфонил)окси)-2,3-дигидро-1,4-оксазепин-4(7H)-карбоксилата (1,24 г, 3,56 ммоль, Получение № AA.1) в диоксане (13 мг). Всю смесь дегазировали в течение приблизительно 5 минут и продували азотом. Смесь нагревали при приблизительно 75°C в течение приблизительно 2 часов. К реакционной смеси добавляли метил 4-хлор-1H-пирроло[3,2-c]пиридин-7-карбоксилат (0,600 г, 2,85 ммоль), Pd(PPh3)2Cl2 (125 мг, 0,178 ммоль), K2CO3 (0,492 г, 3,56 ммоль) и воду (3,25 мг). Всю суспензию дегазировали азотом в течение приблизительно 10 минут и нагревали при приблизительно 75°C в течение приблизительно 8 часов. Реакционную смесь охлаждали, фильтровали через плотный слой целита и MgSO4, концентрировали и очищали хроматографией на силикагеле (0-40% EtOAc/гептан) с получением трет-бутил 6-(7-(метоксикарбонил)-1H-пирроло[3,2-c]пиридин-4-ил)-2,3-дигидро-1,4-оксазепин-4(7H)-карбоксилата (0,3 г, 23%): ЖХ/МС (Таблица 1, Способ as) Время удерживания = 2,04 мин; MS m/z: 374(M+H)+.
Общий способ AH: Образование N-тозил защищенного гетероароматического кольца
Раствор соединения с N-гетероароматическим кольцом, таким как индол или азаиндол (1 эквив.) в соответствующем органическом растворителе (таком как THF, DMF, DCE, толуол или диоксан, предпочтительно, THF) необязательно охлаждали до приблизительно 0°C и добавляли основание (такое как NaH, NaOH или КОН, предпочтительно NaH; 1-2 эквив., предпочтительно 1,1-1,3 эквив.). Реакционную смесь перемешивали в течение приблизительно 10-30 минут и добавляли 4-метил- бензолсульфонилхлорид (1-3 эквив., предпочтительно, 1-1,5 эквив.). Реакционной смеси необязательно давали нагреться до комнатной температуры при охлаждении или необязательно нагревали при приблизительно 30-90°C до полного расходования исходного N-гетероароматического соединения. Дополнительное основание и тозилирующий агент необязательно добавляли по мере необходимости. Реакционную смесь гасили добавлением воды и экстрагировали органическим растворителем (таким как EtOAc или DCM). Органический слой выделяли и необязательно промывали в произвольном порядке водой и/или водными растворами, содержащими кислоту (такую как HCl, AcOH или NH4Cl) и/или водными растворами, содержащими основание (такое как NaHCO3, Na2CO3, NaOH, KOH или NH4OH) и/или водными растворами, содержащими неорганическую соль (такую как NaCl, Na2SO3 или Na2S2O3). Органический раствор затем необязательно сушили с высушивающим веществом (например, безводным MgSO4 или Na2SO4), фильтровали и концентрировали в вакууме с получением целевого соединения.
Иллюстрация общего способа AH
Получение № AH.1: 4-бром-1-тозил-1H-пирроло[2,3-c]пиридин-7-карбонитрил
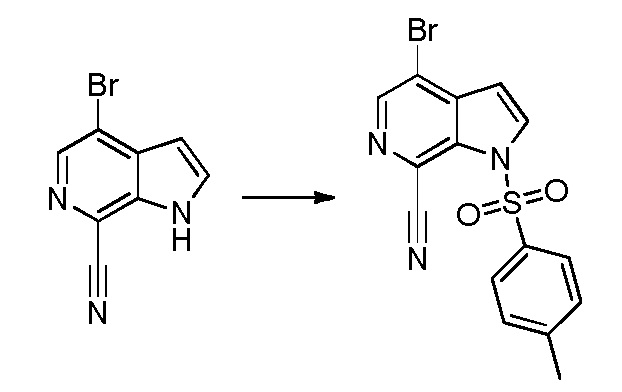
Сосуд заполняли 4-бром-1H-пирроло[2,3-c]пиридин-7-карбонитрилом (0,985 г, 4,44 ммоль, Получение № AD.1) в THF (30 мг). NaH (60% дисперсия в минеральном масле, 0,213 г, 5,32 ммоль) добавляли порциями при температуре приблизительно 0°C. Смесь перемешивали в течение приблизительно 15 минут, затем 4-метил-бензолсульфонилхлорид (0,930 г, 4,88 ммоль) добавляли в виде одной порции и реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали или приблизительно 16 часов. Дополнительное количество NaH (60% дисперсия в минеральном масле, 0,355 г, 0,89 ммоль) и 4-метилбензол-1-сульфонилхлорида (0,254 г, 1,33 ммоль) добавляли в последовательности и перемешивали при комнатной температуре в течение приблизительно 1 часа. Реакционную смесь разбавляли водой (30 мг) и экстрагировали EtOAc (60 мг). Органический слой сушили над MgSO4, фильтровали, концентрировали и очищали с помощью хроматографии на силикагеле (0-35% EtOAc/гептан) с получением 4-бром-1-тозил-1H-пирроло[2,3-c]пиридин-7-карбонитрила (1,35 г, 81%): ЖХ/МС (Таблица 1, Способ as) Время удерживания = 2,51 мин; MS m/z: 376, 378(M+H)+.
Пример № 1: трет-Бутил 2-(4-бром-7-карбамоил-1H-индол-2-ил)бензилкарбаматметилфенил)тиазол-2-карбоксамид
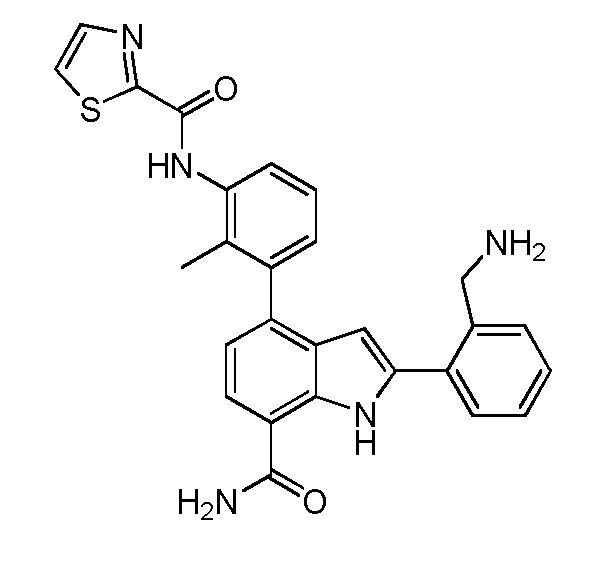
Стадия A: трет-Бутил 2-(4-бром-7-карбамоил-1H-индол-2-ил)бензилкарбамат
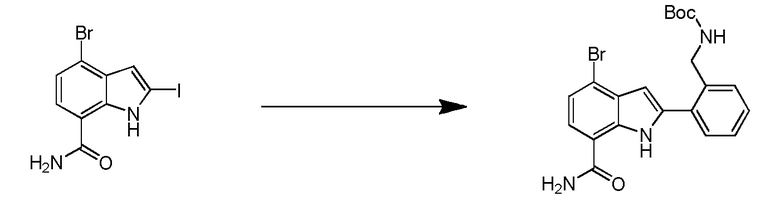
К раствору 4-бром-2-йод-1H-индол-7-карбоксамида (2,5 г, 6,8 ммоль, Получение № 1) в THF (185 мг), MeOH (25 мг) и воды (25 мг) добавляли трет-бутил 2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензилкарбамат (2,7 г, 8,2 ммоль, JW), Pd(dppf)Cl2 (0,5 г, 0,7 ммоль) и Na2CO3 (2,2 г, 20,6 ммоль). Смесь перемешивали при приблизительно 80°C в течение ночи в атмосфере азота. Растворитель удаляли при пониженном давлении с получением остатка, который очищали с помощью колоночной хроматографии на силикагеле с получением сырого трет-бутил 2-(4-бром-7-карбамоил-1H-индол-2-ил)бензилкарбамата (2,5 г, 5,6 ммоль).
Стадия B: трет-Бутил 2-(7-карбамоил-4-(2-метил-3-(тиазол-2-карбоксамидо)фенил)-1H-индол-2-ил)бензилкарбамат
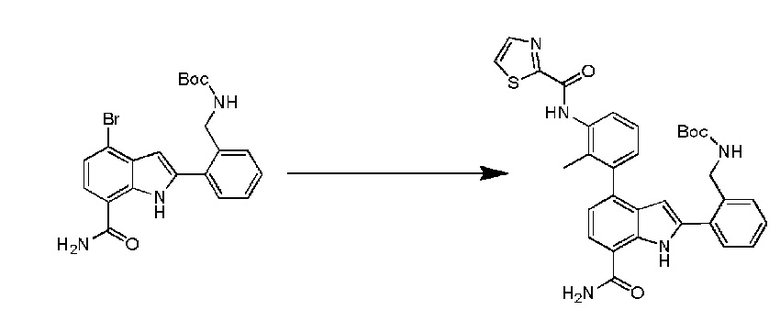
К раствору 2-(4-бром-7-карбамоил-1H-индол-2-ил)бензилкарбамата (2,5 г, 5,6 ммоль) в THF (185 мг), MeOH (25 мг) и воды (25 мг) добавляли N-(2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)тиазол-2-карбоксамид (2,3 г, 6,8 ммоль, Получение № 4), Pd(dppf)Cl2 (0,4 г, 0,6 ммоль) и Na2CO3 (1,8 г, 16,9 ммоль). Смесь перемешивали при приблизительно 80°C в течение ночи в атмосфере азота. Растворитель удаляли при пониженном давлении с получением остатка, который очищали с помощью колоночной хроматографии на силикагеле, с получением трет-бутил 2-(7-карбамоил-4-(2-метил-3-(2-оксо-2-(тиазол-2-ил)этил)фенил)-1H-индол-2-ил)бензилкарбамата (3 г, 92%): 1H-ЯМР (CDCl3) δ 10,57 (с, 1H), 9,25 (с, 1H), 8,22-8,20 (д, J=7,6 Гц, 1H), 7,92-7,91 (д, J=3,2 Гц, 1H), 7,64-7,63 (д, J=3,2 Гц, 1H), 7,50-7,45 (м, 3H), 7,37-7,35 (м, 3H), 7,26-7,24 (м, 2H), 7,04-7,02 (д, J=3,6 Гц, 1H), 6,32 (с, 1H), 4,43 (с, 2H), 2,25 (с, 3H), 1,38 (с, 9H).
Стадия C: N-(3-(2-(2-(аминометил)фенил)-7-карбамоил-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамид
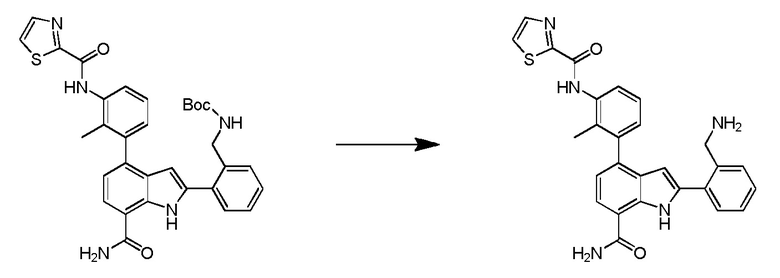
Раствор трет-бутил 2-(7-карбамоил-4-(2-метил-3-(2-оксо-2-(тиазол-2-ил)этил)фенил)-1H-индол-2-ил)бензилкарбамата (3 г, 5,2 ммоль) в DCM (50 мл) и TFA (10 мл) перемешивали при приблизительно 25°C в течение приблизительно 6 часов. Растворитель удаляли при пониженном давлении. Добавляли воду и раствор подщелачивали добавлением насыщенного водного NaHCO3 до pH 9. Смесь экстрагировали EtOAc. Органическую фазу концентрировали с получением N-(3-(2-(2-(аминометил)фенил)-7-карбамоил-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамида (2,2 г, 89%): ЖХ/МС (Таблица 1, Способ b) Время удерживания = 2,53 мин; MS m/z: 482(M+H)+. (Btk IC50=B)
Пример № 2: 4-(3-амино-2-метилфенил)-1H-пирроло[2,3-c]пиридин-7-карбоксамид
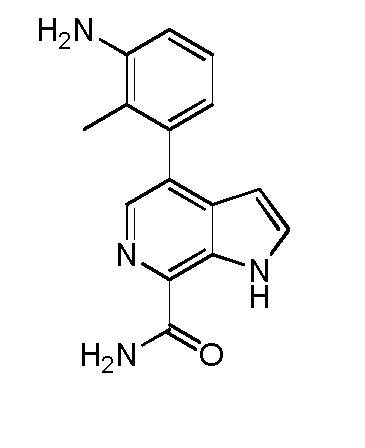
Стадия A: 4-бром-7-хлор-1H-пирроло[2,3-c]пиридин
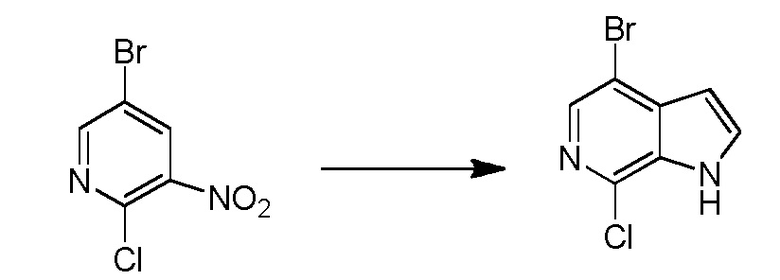
К раствору 5-бром-2-хлор-3-нитропиридина (10 г, 0,042 моль) в безводном THF (150 мг) по каплям добавляли раствор винилмагнийбромида (17 г, 0,127 моль) в THF при температуре от -30 до -50°C. Реакционную смесь перемешивали при температуре от -30 до -40°C в течение 2 часов. Затем реакционную смесь выливали в насыщенный водный раствор NH4Cl и смесь экстрагировали EtOAc (50 мл×3). Объединенные органические слои промывали насыщенным солевым раствором, сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении и остаток очищали колоночной хроматографией, с получением 4-бром-7-хлор-1H-пирроло[2,3-c]пиридина (3 г, 31%): 1H-ЯМР: (DMSO-d6) δ 12,45 (с, 1H), 8,04 (с, 1H), 7,79-7,78 (м, 1H), 6,59-6,58 (д, J=2,0, 1H).
Стадия B: 3-(7-Хлор-1H-пирроло[2,3-c]пиридин-4-ил)-2-метиланилин

К смеси 4-бром-7-хлор-1H-пирроло[2,3-c]пиридина [Matrix] (5 г, 21,6 ммоль), 2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина (7,55 г, 32,4 ммоль, CombiBlocks) и карбоната натрия (1,6 г, 64,8 ммоль) в THF (80 мг), MeOH (80 мг) и воды (20 мг), Pd(dppf)Cl2 (1,6 г, 2,16 ммоль) добавляли и смесь дегазировали несколько раз и нагревали до приблизительно 70°C в течение ночи под N2. Реакционную смесь фильтровали через целит и концентрировали при пониженном давлении и остаток очищали колоночной хроматографией с получением 3-(7-хлор-1H-пирроло[2,3-c]пиридин-4-ил)-2-метиланилина (2,2 г, 40%): 1H-ЯМР (DMSO-d6) δ 12,05 (с, 1H), 7,71 (с, 1H), 7,64 (д, J=2,4, 1H), 6,99-6,96 (м, 1H), 6,72-6,70 (д, J=8,0, 1H), 6,48 (д, J=6,8, 1H), 6,2 (д, J=2,8, 1H), 4,95 (с, 2H), 1,82 (с, 3H).
Стадия C: Метил 4-(3-амино-2-метилфенил)-1H-пирроло[2,3-c]пиридин-7-карбоксилат
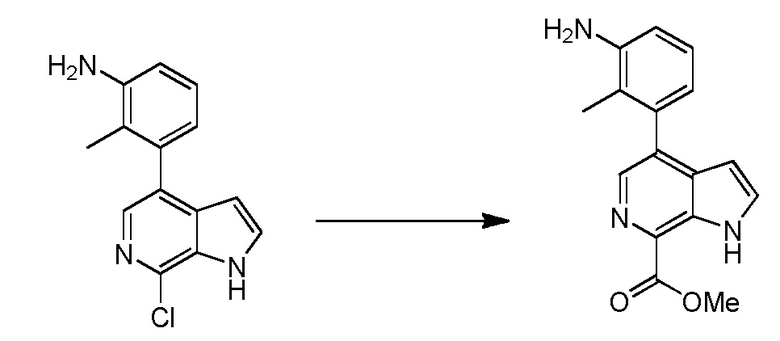
К раствору 3-(7-хлор-1H-пирроло[2,3-c]пиридин-4-ил)-2-метиланилина (800 мг, 3,1 ммоль) в безводном MeOH (80 мг), Et3N (3,1 г, 31 ммоль) и Pd(dppf)Cl2 (0,45 г, 0,62 ммоль) добавляли и реакционную смесь нагревали до температуры приблизительно 130°C в течение приблизительно 24 часов под CO. Реакционную смесь концентрировали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем с получением метил 4-(3-амино-2-метилфенил)-1H-пирроло[2,3-c]пиридин-7-карбоксилата (0,60 г, 69%): 1H-ЯМР (DMSO-d6): δ 11,65 (ушир. с., 1H), 8,09 (с, 1H) 7,65 (с, 1H) 7,02 (т, J=7,72 Гц, 1H), 6,74 (д, J=7,94 Гц, 1H), 6,52 (д, J=7,50 Гц, 1H) 6,26 (д, J=2,65 Гц, 1H), 5,02 (с, 2H), 4,0 (с, 3H), 1,83 (с, 3H)
Стадия D: 4-(3-амино-2-метилфенил)-1H-пирроло[2,3-c]пиридин-7-карбоксамид
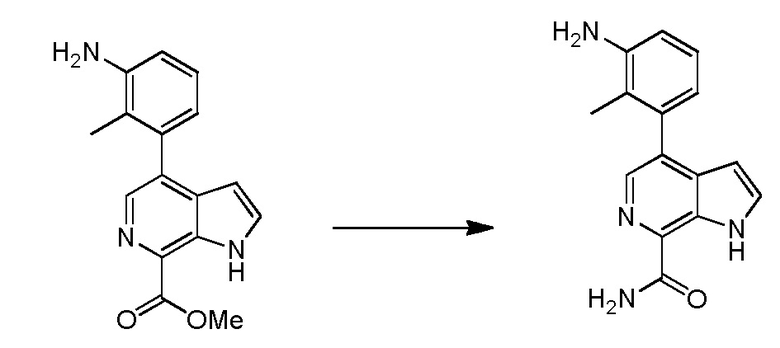
К раствору метил 4-(3-амино-2-метилфенил)-1H-пирроло[2,3-c]пиридин-7-карбоксилата (600 мг, 2,13 ммоль) в MeOH (10 мг), добавляли аммиак (2 мг) и реакционную смесь перемешивали в течение ночи при комнатной температуре. Смесь концентрировали и остаток очищали с помощью препаративной ТСХ (30:1 DCM/MeOH) с получением 4-(3-амино-2-метилфенил)-1H-пирроло[2,3-c]пиридин-7-карбоксамида (320 мг, 56%): 1H-ЯМР (DMSO-d6): δ 11,56 (с, 1H), 8,2 (с, 1H), 7,97 (с, 1H), 7,64 (с, 1H), 7,55 (с, 1H), 7,0-6,97 (м, 1H), 6,71 (д, J=7,6, 1H), 6,50 (д, J=4,4, 1H), 6,17 (с, 1H), 4,97 (с, 2H), 1,82 (с, 3H); (Таблица 1, Способ d) Время удерживания = 1,95 мин; MS m/z: 267 (M+H)+. (BtkIC50 = C)
Пример № 3: N-(3-(7-карбамоил-3-метил-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамид
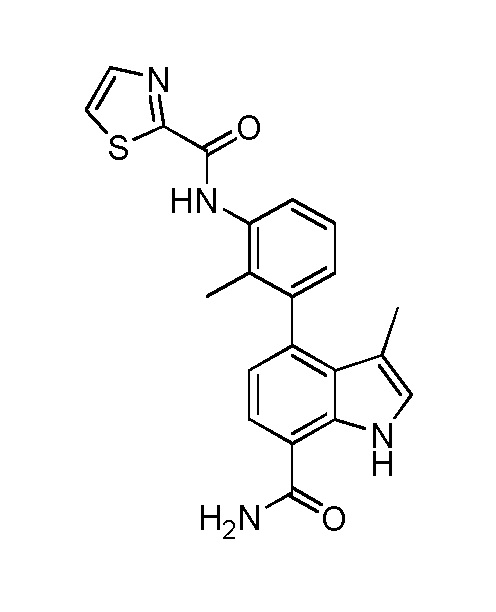
Стадия A: Метил 4-бром-3-формил-1H-индол-7-карбоксилат

POCl3 (2,4 мл, 26 ммоль) добавляли в DMF (60 мг) раствор по каплям при 0°C и перемешивали в течение приблизительно 30 минут. Затем раствор метил 4-бром-1H-индол-7-карбоксилата (5 г, 13 ммоль, Получение № 1, стадия B) в DMF (60 мг) добавляли по каплям в указанную выше реакционную смесь при приблизительно 0°C и перемешивали в течение приблизительно 20 минут. Полученную реакционную смесь нагревали до приблизительно 90°C в течение приблизительно 3 часов. После охлаждения до комнатной температуры смесь выливали в воду со льдом и подщелачивали добавлением водного раствора NaOH до рН=8-9. Водную смесь экстрагировали EtOAc. Объединенную органическую фазу промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка, который очищали с помощью колоночной хроматографии на силикагеле с получением метил 4-бром-3-формил-1H-индол-7-карбоксилата (3,5 г, 95%): 1H-ЯМР (DMSO-d6): δ 12,33 (ушир., 1H), 10,69 (с, 1H), 8,20 (д, J=2,0 Гц, 1H), 7,76-7,74 (д, J=8,0 Гц, 1H), 7,61-7,59 (д, J=8,4 Гц, 1H), 3,94 (с, 3H).
Стадия B: Метил 4-бром-3-(((4-метоксибензил)амино)метил)-1H-индол-7-карбоксилат
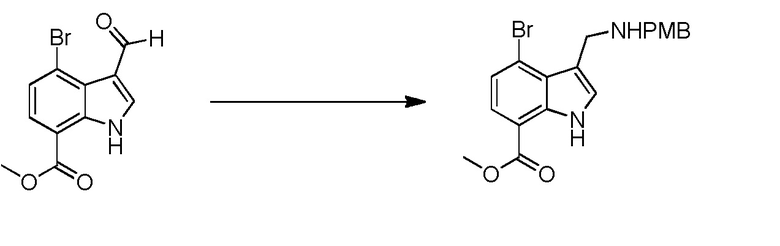
К раствору метил 4-бром-3-формил-1H-индол-7-карбоксилата (3,5 г, 12,4 ммоль) в безводном DCE (50 мг) добавляли (4-метоксифенил)метанамин (2,6 г, 18,6 ммоль) и количество катализатора AcOH. Реакционную смесь перемешивали при комнатной температуре в течение приблизительно 1 часа. Затем NaBH(OAc)3 (13,2 г, 62 ммоль) добавляли порциями и перемешивали в течение ночи при комнатной температуре. Когда реакция была завершена, добавляли воду, чтобы погасить реакцию. Водную фазу экстрагировали DCM. Объединенную органическую фазу концентрировали при пониженном давлении с получением остатка, который очищали с помощью колоночной хроматографии на силикагеле с получением метил 4-бром-3-(((4-метоксибензил)амино)метил)-1H-индол-7-карбоксилата (4 г, 80%): 1H-ЯМР (DMSO-d6): δ 11,25 (ушир., 1H), 7,61-7,59 (д, J=8,4 Гц, 1H), 7,41 (с, 1H), 7,30-7,23 (м, 3H), 6,85-6,83 (д, J=8,4 Гц, 2H), 4,02 (с, 2H), 3,90 (с, 3H), 3,70-3,69 (м, 5H), 1,88 (с, 1H).
Стадия C: 4-бром-3-(((4-метоксибензил)амино)метил)-1H-индол-7-карбоновая кислота
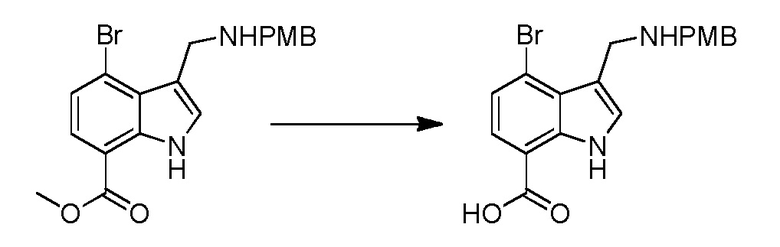
К раствору метил 4-бром-3-(((4-метоксибензил)амино)метил)-1H-индол-7-карбоксилата (5,4 г, 13,4 ммоль) в THF (250 мг), MeOH (50 мг) и воды (50 мг) добавляли LiOH (1,6 г, 67,0 ммоль) и нагревали с обратным холодильником в течение приблизительно 6 часов. После охлаждения до комнатной температуры, органический растворитель удаляли при пониженном давлении. Водную фазу подкисляли 1 н HCl до рН=5-6. Затем суспензию фильтровали и осадок на фильтре промывали водой и сушили с получением 4-бром-3-(((4-метоксибензил)амино)метил)-1H-индол-7-карбоновой кислоты (4 г, 77%): 1H-ЯМР (DMSO-d6) δ 11,40 (ушир., 1H), 7,58-7,56 (д, J=8,0 Гц, 1H), 7,53 (с, 1H), 7,40-7,38 (д, J=8,4 Гц, 2H), 7,27-7,25 (д, J=8,0 Гц, 1H), 6,94-6,92 (д, J=8,4 Гц, 2H), 4,31 (с, 2H), 3,98 (с, 2H), 3,74 (с, 3H).
Стадия D: 4-бром-3-(((4-метоксибензил)амино)метил)-1H-индол-7-карбоксамид
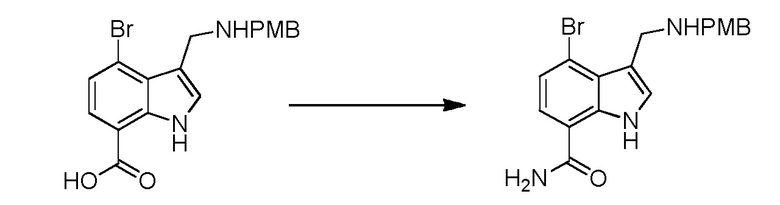
Смесь 4-бром-3-(((4-метоксибензил)амино)метил)-1H-индол-7-карбоновой кислоты (9,3 г, 23,9 ммоль), EDCI (5,5 г, 28,7 ммоль) и HOBt (4,4 г, 28,7 ммоль) в THF (350 мг) и DCM (420 мг) перемешивали при комнатной температуре в течение приблизительно 1 часа. Затем реакционную смесь барботировали газообразным аммиаком в течение 15 минут при приблизительно -60°C, затем нагревали до комнатной температуры и перемешивали в течение ночи. Растворитель удаляли при пониженном давлении и добавляли MeOH. Суспензию фильтровали и отфильтрованное концентрировали при пониженном давлении с получением остатка, который очищали с помощью препаративной ВЭЖХ (таблица 1, способ S) с получением 4-бром-3-(((4-метоксибензил)амино)метил)-1H-индол-7-карбоксамида (2,1 г, 23%): ЖХ/МС (Таблица 1, способ d) Время удерживания = 2,31 мин; MS m/z: 388 (M+H)+
Стадия E: N-(3-(7-карбамоил-3-(((4-метоксибензил)амино)метил)-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамид
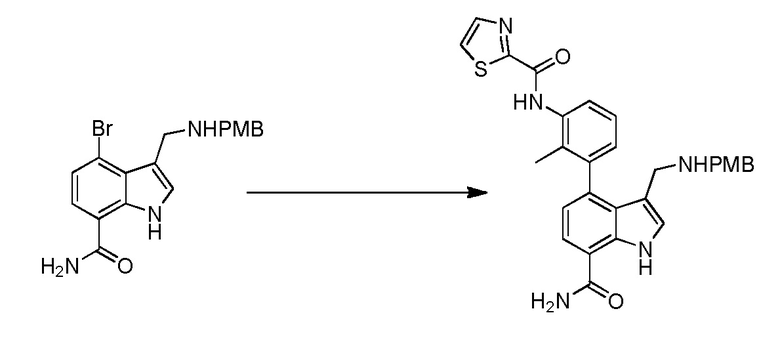
К раствору 4-бром-3-(((4-метоксибензил)амино)метил)-1H-индол-7-карбоксамида (100 мг, 0,26 ммоль), N-(2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)тиазол-2-карбоксамида (116 мг, 0,39 ммоль, Получение № 4) и CsF (39 мг, 0,26 ммоль) в 1,4-диоксане (2 мг) и воды (0,4 мг) добавляли Pd(PPh3)4 (29,8 мг, 0,03 ммоль). Затем реакционную смесь нагревали до приблизительно 100°C в атмосфере азота в течение приблизительно 12 часов. После охлаждения до комнатной температуры, добавляли воду и экстрагировали EtOAc. Объединенную органическую фазу промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением сырого продукта, который очищали с помощью препаративной ВЭЖХ (Таблица 1, Способ r) с получением N-(3-(7-карбамоил-3-(((4-метоксибензил)амино)метил)-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамида (10 мг, 8%): 1H-ЯМР (DMSO-d6): δ 11,05 (ушир., 1H), 10,23 (ушир., 1H), 8,14-8,10 (м, 3H), 7,72-7,65 (м, 2H), 7,27 (ушир., 1H), 7,26-7,24 (м, 2H), 7,11-7,09 (м, 1H), 7,02-7,00 (д, J=8,8 Гц, 2H), 6,77-6,71 (м, 3H), 3,63 (с, 3H), 3,24-3,21 (м, 4H), 1,88 (с, 3H), 1,83 (с, 1H)
Стадия F: N-(3-(7-карбамоил-3-метил-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамид
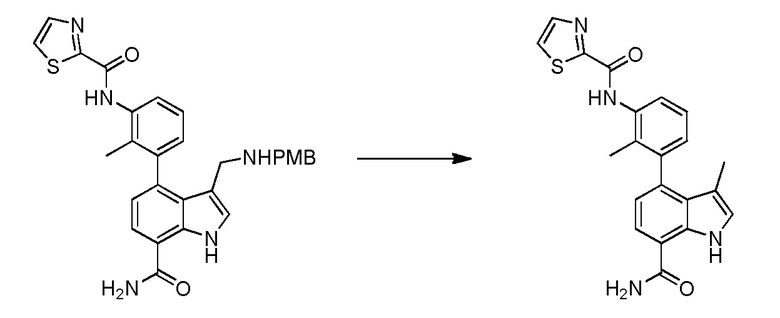
К раствору N-(3-(7-карбамоил-3-(((4-метоксибензил)амино)метил)-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамида (10 мг, 0,02 ммоль) в безводном MeOH (5 мг) добавляли сухой Pd/C (5 мг) и перемешивали при комнатной температуре в атмосфере водорода (50 фунт на кв.дюйм) в течение ночи. Затем реакционную смесь фильтровали и фильтрат концентрировали при пониженном давлении с получением остатка, который очищали с помощью препаративной ВЭЖХ (Таблица 1, способ q) с получением N-(3-(7-карбамоил-3-метил-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамида (1,1 мг, 15%): ЖХ/МС (Таблица 1, способ j) Время удерживания = 3,05 мин; MS m/z: 391 (M+H)+. (Btk IC50=B)
Пример № 4: N-(3-(7-карбамоил-3-метил-1H-индол-4-ил)-2-метилфенил)тиазол-2-карбоксамид
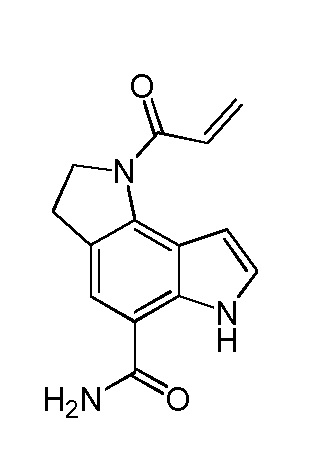
Стадия A: 5-бром-6-нитроиндолин
К раствору 5-броминдолина (12,33 г, 83 ммоль) в H2SO4 (60 мг) добавляли KNO3 (7,55 мл, 74,7 ммоль) при приблизительно 0°C. Раствор перемешивали при 0-10°C в течение приблизительно 1 часа, и затем смесь перемешивали в течение ночи при комнатной температуре. Смесь выливали в ледяную воду, подщелачивали NaCO3 до приблизительно рН 8. Смесь экстрагировали EtOAc (300 мл×3), органическую фазу сушили над NaSO4, концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (петролейный эфир:EtOAc=20:1) с получением 5-бром-6-нитроиндолина (12,3 г, 81%): 1H-ЯМР (CDCl3) δ 7,25 (с, 1H), 6,91 (с, 1H), 3,98 (с, 1H), 3,66-3,56 (м, 2H), 3,08-2,96 (м, 2H).
Стадия B: трет-Бутил 5-бром-6-нитроиндолин-1-карбоксилат
К раствору 5-бром-6-нитроиндолина (7,5 г, 30,9 ммоль) в DCM (750 мг) добавляли (Boc)2O (13,47 г, 61,7 ммоль) при 0°C. Затем к смеси добавляли Et3N (9,37 г, 93 ммоль) и DMAP (0,337 г, 3,09 ммоль). Смесь перемешивали в течение ночи при комнатной температуре. Реакционную смесь выливали в воду, экстрагировали DCM (300 мл×3) и органическую фазу сушили над NaSO4, концентрировали при пониженном давлении и остаток очищали на колонке с силикагелем (петролейный эфир:EtOAc = 30:1) с получением трет-бутил 5-бром-6-нитроиндолин-1-карбоксилата (6,7 г, 63%): 1H-ЯМР (CDCl3) δ 8,29 (с, 1H), 7,42 (с, 1H), 4,06 (с, 2H), 3,18-3,13 (м, 2H) 1,57 (с, 9H).
Стадия C: трет-Бутил 5-бром-2,3-дигидропирроло[2,3-e]индол-1(6H)-карбоксилат

К смеси трет-бутил 5-бром-6-нитроиндолин-1-карбоксилата (4 г, 11,66 ммоль) в THF (60 мл) добавляли винилмагнийбромид (6,43 г, 49,0 ммоль) при температуре от -40 до 50°C, затем полученную смесь перемешивали при температуре от -20 до -30°C в течение приблизительно 2 часов, и затем при комнатной температуре в течение ночи. Смесь выливали в насыщенный раствор NH4Cl и экстрагировали EtOAc (100 мл×3). Органическую фазу сушили над NaSO4, концентрировали при пониженном давлении и остаток очищали с помощью хроматографии на силикагеле (петролейный эфир:EtOAc=50:1) с получением трет-бутил 5-бром-2,3-дигидропирроло[2,3-e]индол-1(6H)-карбоксилата (0,7 г, 18%): 1H-ЯМР (CDCl3) δ 8,17 (с, 1H), 7,13-7,10 (м, 2H),7,07 (м, 1H), 4,05-4,00 (т, J=8,4 Гц, 2H), 3,07-3,03 (т, J=8,4 Гц, 2H), 1,5 (с, 9H).
Стадия D: 1,2,3,6-Тетрагидропирроло[2,3-e]индол-5-карбонитрил

К раствору трет-бутил 5-бром-2,3-дигидропирроло[2,3-e]индол-1(6H)-карбоксилата (60 мг, 0,178 ммоль) в DMF (2 мг) добавляли Zn(CN)2 (12,53 мг, 0,107 ммоль) и Pd(PPh3)4 (20,56 мг, 0,018 ммоль). Раствор нагревали при приблизительно 145°C в течение приблизительно 50 минут в микроволновой печи под N2. Смесь концентрировали при пониженном давлении и остаток очищали с помощью препаративной ВЭЖХ (Таблица 1, Способ aj) с получением 1,2,3,6-тетрагидропирроло[2,3-e]индол-5-карбонитрила (20 мг, 61%): 1H-ЯМР (MeOD): δ 7,34 (с, 1H), 7,30 (д, J=3,2, 1H), 6,51 (д, J=3,2, 1H), 3,82-3,78 (т, J=8 Гц, 2H), 3,23-3,18 (т, J=8,4 Гц, 2H).
Стадия E: 1,2,3,6-Тетрагидропирроло[2,3-e]индол-5-карбоксамид
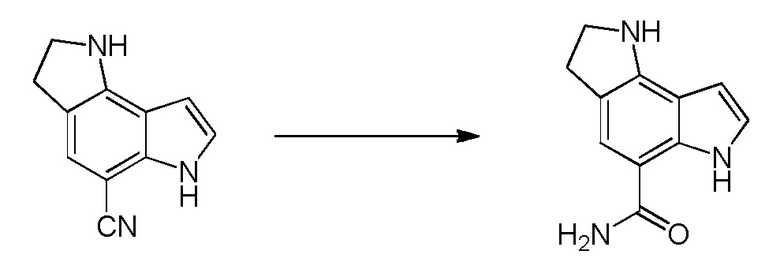
К раствору 1,2,3,6-тетрагидропирроло[2,3-e]индол-5-карбонитрила (160 мг, 0,873 ммоль) в DMSO (4 мг) добавляли K2CO3 (300 мг, 2,171 ммоль), затем H2O2 (4 мл, 39,2 ммоль) добавляли по каплям при комнатной температуре. И реакционную смесь перемешивали в течение ночи при комнатной температуре. Смесь выливали в воду, экстрагировали EtOAc (20 мл×3) и органическую фазу промывали насыщенным водным раствором Na2S2O3, сушили и концентрировали и остаток очищали с помощью препаративной ВЭЖХ (Таблица 1, Способ ak) с получением 1,2,3,6-тетрагидропирроло[2,3-e]индол-5-карбоксамида (70 мг, 40%): ЖХ/МС (Таблица 1, Способ d) Время удерживания = 1,43 мин; MS m/z: 202 (M+H)+.
Стадия F: 1-акрилоил-1,2,3,6-тетрагидропирроло[2,3-e]индол-5-карбоксамид
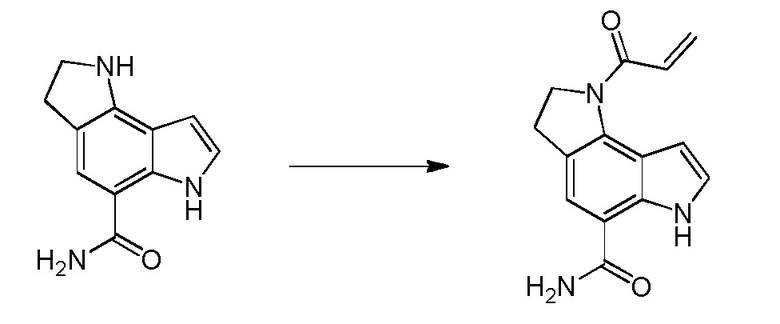
К раствору 1,2,3,6-тетрагидропирроло[2,3-e]индол-5-карбоксамида (15 мг, 0,075 ммоль) в DCM (10 мг) добавляли Et3N (1 мл, 7,17 ммоль), и затем раствор акрилоилхлорида (10 мг, 0,11 ммоль) в DCM (0,5 мг) добавляли по каплям при 0°C. Реакционную смесь перемешивали в течение ночи при комнатной температуре. Реакционный раствор концентрировали при пониженном давлении и остаток очищали с помощью препаративной ВЭЖХ (Таблица 1, Способ t) с получением 1-акрилоил-1,2,3,6-тетрагидропирроло[2,3-e]индол-5-карбоксамида (12 мг, 63%): 1H-ЯМР: (DMSO-d6) δ 11,13 (с, 1H), 7,93 (с, 1H), 7,61 (с, 1H), 7,21 (с, 2H), 6,8-6,73 (м, 2H), 6,34-6,30 (м, 1H), 5,84-5,82 (д, J=10,4, 1H), 4,25-4,21 (т, J=8,0, 2H), 3,21-3,13 (м, 2H); ЖХ/МС (Таблица 1, Способ d) Время удерживания = 2,39 мин; MS m/z: 256 (M+H)+. (Btk IC50=B)
Пример № 5: 4-акриламидо-1H-индол-7-карбоксамид
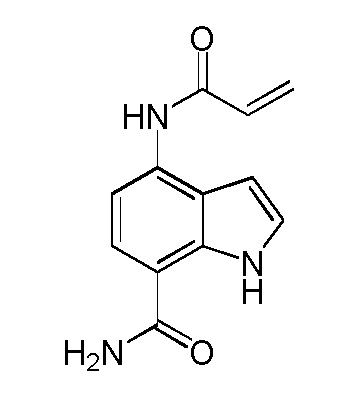
Стадия A: 4-амино-1-тозил-1H-индол-7-карбонитрил
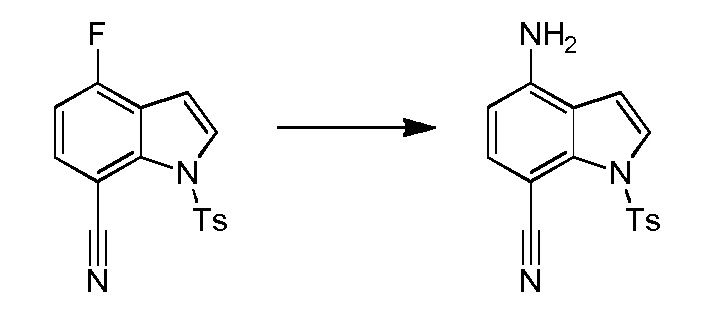
К раствору 4-фтор-1-тозил-1H-индол-7-карбонитрила (500 мг, 1,59 ммоль, Получение № 27, стадия A) в 1,4-диоксане (5 мг) добавляли аммиак (2,5 мл, 116 ммоль). Смесь перемешивали при приблизительно 120°C в течение ночи. Реакционную смесь концентрировали при пониженном давлении и остаток очищали на колонке с силикагелем с получением 4-амино-1-тозил-1H-индол-7-карбонитрила (100 мг, 20%): 1H-ЯМР (DMSO-d6): δ 7,86-7,84 (м, 2H), 7,77-7,76 (д, J=4, 1H), 7,46-7,44 (д, J=8, 2H), 7,37-7,35 (д, J=8, 1H), 7,12 (с, 1H), 6,70 (с, 2H), 6,46-6,44 (д, J=8, 1H), 2,37 (с, 3H).
Стадия B: 4-амино-1H-индол-7-карбонитрил
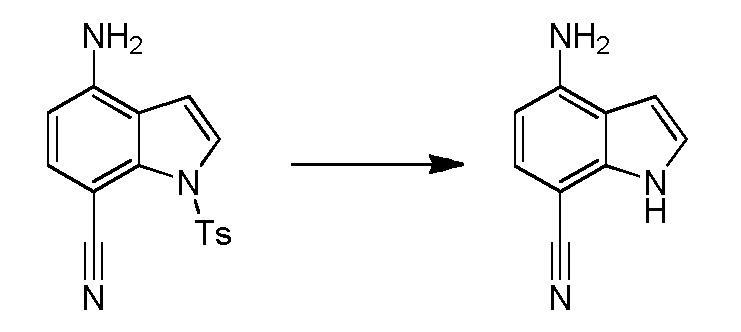
К раствору 4-амино-1-тозил-1H-индол-7-карбонитрила (90 мг, 0,289 ммоль) в THF (2 мг), MeOH (1 мг) и воды (1 мг) добавляли LiOH (69 мг, 2,89 ммоль). Смесь перемешивали при приблизительно 40°C в течение ночи. Реакционную смесь концентрировали при пониженном давлении, добавляли воду и экстрагировали EtOAc (20 мл×3). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением 4-амино-1H-индол-7-карбонитрила (40 мг, 88%): 1H-ЯМР (DMSO-d6): δ 11,43 (с, 1H), 7,21-7,19 (д, J=8, 1H), 7,13-7,12 (м, 1H), 6,67-6,62 (м, 1H), 6,20-6,18 (д, J=8, 1H).
Стадия C: 4-амино-1H-индол-7-карбоксамид
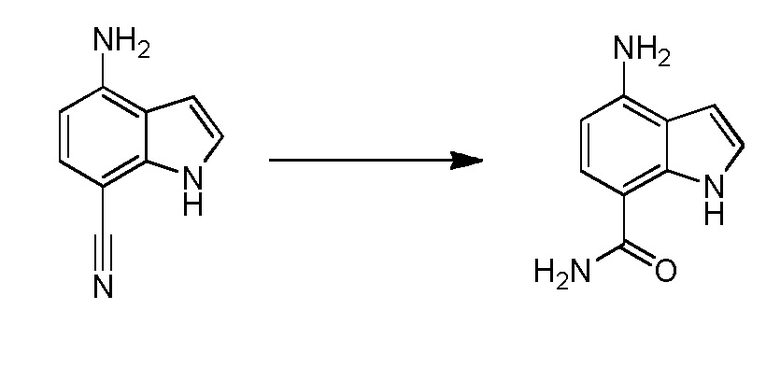
К раствору 4-амино-1H-индол-7-карбонитрила (40 мг, 0,254 ммоль) в DMSO (2 мг), K2CO3 (52,8 мг, 0,382 ммоль) и 30% H2O2 (2 мг) добавляли при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 5 часов. Воду добавляли к реакционной смеси и смесь экстрагировали EtOAc (20 мл×3) и органическую фазу сушили над Na2SO4, концентрировали при пониженном давлении и остаток очищали с помощью препаративной ТСХ (DCM:MeOH=15:1) с получением 4-амино-1H-индол-7-карбоксамида (30 мг, 67%): 1H-ЯМР (DMSO-d6) δ 10,79 (с, 1H), 7,43-7,41 (д, J=8, 1H), 7,04 (с, 1H), 6,52 (с, 1H), 6,10-6,08 (д, J=8, 1H), 5,83 (с, 2H).
Стадия D: 4-акриламидо-1H-индол-7-карбоксамид
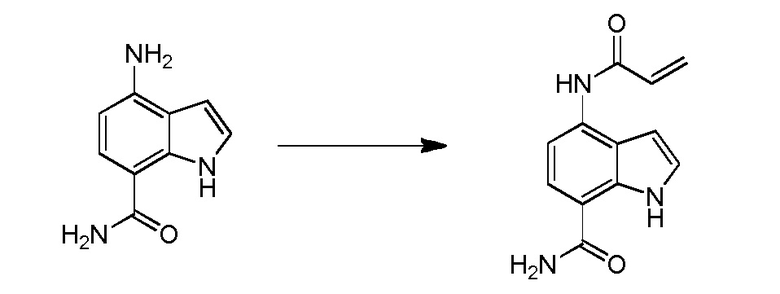
К раствору 4-амино-1H-индол-7-карбоксамида (30 мг, 0,171 ммоль) в DCM (3 мг), DIEA (0,060 мл, 0,342 ммоль) и акрилоилхлорид (18,60 мг, 0,205 ммоль) добавляли и реакционную смесь перемешивали в течение ночи при комнатной температуре. Затем реакционную смесь концентрировали при пониженном давлении и остаток очищали с помощью препаративной ВЭЖХ (Таблица 1, Способ u) с получением 4-акриламидо-1H-индол-7-карбоксамида (17 мг, 43%): ЖХ/МС (Таблица 1, Способ d) Время удерживания = 2,10 мин; MS m/z: 230 (M+H)+. (Btk IC50 = C)
Пример № 6: 4-акриламидо-1H-индол-7-карбоксамид
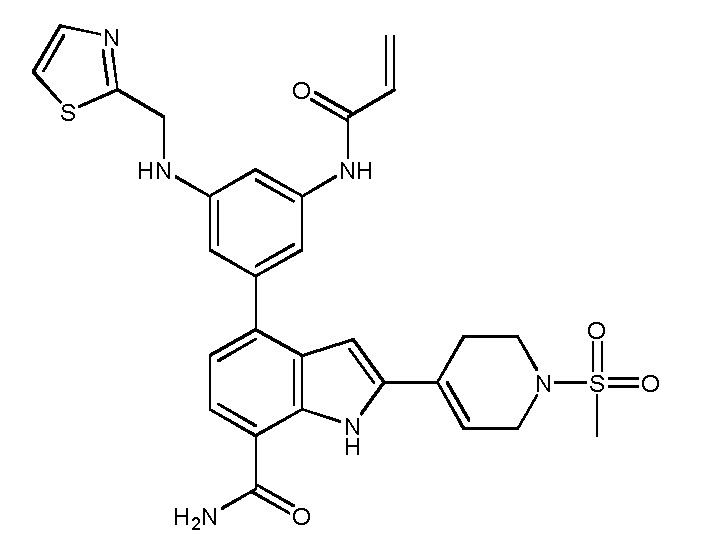
Стадия A: 4-(3-акриламидо-5-аминофенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид

В круглодонную колбу добавляли 4-(3-акриламидо-5-нитрофенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид (0,175 г, 0,343 ммоль, полученный с использованием A из 4-бром-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамида (Получение № 18) и гидрохлорида 3-амино-5-нитрофенилбороновой кислоты [CombiBlocks], E и акрилоилхлорида) в NMP (2 мг) и HCl, 37% (0,222 мг) с получением красной суспензии. Реакционную смесь нагревали до приблизительно 85°C и добавляли хлорид олова (II) (0,600 г, 0,316 ммоль). Реакционную смесь перемешивали при приблизительно 85°C в течение приблизительно 1,5 часов. Добавляи дополнительное количество хлорида олова (II) (2,39 г, 1,26 ммоль) и реакционную смесь дополнительно перемешивали при приблизительно 85°C в течение приблизительно 2 часов. Реакционную смесь охлаждали до комнатной температуры и DCM (30 мг), MeOH (10 мг), и 1 н NaOH (15 мг) добавляли. Смесь энергично перемешивали в течение приблизительно 2 часов, фильтровали, и фильтрат экстрагировали DCM (3×). Органические слои объединяли и растворитель удаляли в вакууме. Воду и EtOAc добавляли к остатку и экстрагировали EtOAc (4×). Органические слои объединяли и промывали водой и насыщенным солевым раствором. Органические слои объединяли и растворитель удаляют в вакууме. Сырой продукт помещали в колонку с силикагелем и элюировали смесью 0-10% MeOH в DCM. Вещество подвергали дальнейшей очистке с помощью препаративной ВЭЖХ (Таблица 1, Способ ag) с получением 4-(3-акриламидо-5-аминофенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамида (20 мг, 12%): ЖХ/МС (Таблица 1, Способ g) Время удерживания = 1,12 мин.; MS m/z: 480 (M+H)+.
Стадия B: 4-(3-акриламидо-5-(тиазол-2-илметиламино)фенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамид

К перемешиваемому раствору 4-(3-акриламидо-5-аминофенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамида (20 мг, 0,042 ммоль) и тиазол-2-карбальдегида (4,03 мкл, 0,046 ммоль) в MeOH (1 мг) добавляли MP-цианоборгидрид (88 мг, 0,167 ммоль) и уксусную кислоту (9,55 мкл, 0,167 ммоль). Суспензию перемешивали при приблизительно 40°C в течение приблизительно 40 часов. Суспензию фильтровали и смолу промывали DCM и MeOH. Фильтрат пропускали через слой Si-карбоната. Фильтрат концентрировали при пониженном давлении и остаток очищали с помощью препаративной ТСХ (10% MeOH/DCM) с последующей второй очисткой с помощью препаративной ТСХ (5% MeOH/DCM) с получением 4-(3-акриламидо-5-(тиазол-2-илметиламино)фенил)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-7-карбоксамида (7,2 мг, 25%): ЖХ/МС (Таблица 1, Способ g) Время удерживания = 1,56 мин.; MS m/z: 577 (M+H)+. (Btk IC50=A)
Пример № 7. (E)-4-(3-(2-Циано-3-гидроксибут-2-енамидо)фенил)-1H-индол-7-карбоксамид
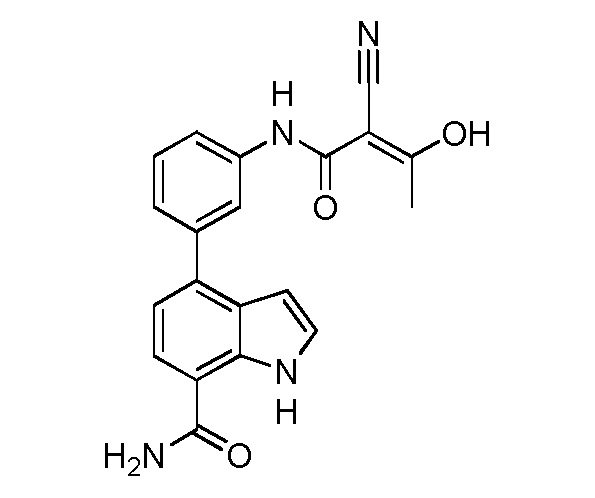
Смесь N-(3-(7-карбамоил-1H-индол-4-ил)фенил)-5-метилизоксазол-4-карбоксамида (0,060 г, 0,166 ммоль, Пример № E.2.1) и NaOH (0,008 г, 0,200 ммоль) в MeOH (1,9 мг) нагревали в сосуде при температуре приблизительно 60°C. После приблизительно 2 часов, реакционную смесь охлаждали до комнатной температуры и добавляли 1 н водный раствор HCl для подкисления. Полученный осадок собирали вакуумным фильтрованием с получением (E)-4-(3-(2-циано-3-гидроксибут-2-енамидо)фенил)-1H-индол-7-карбоксамида (0,047 г, 78%) в виде твердого вещества после сушки в вакууме при температуре приблизительно 55°C: ЖХ/МС (Таблица 1, Способ c) Время удерживания = 2,79 мин.; MS m/z: 361 (M+H)+. (Btk IC50 = C)
Пример № 8. 4-(цис-3-Акриламидоциклогексил)-1H-индол-7-карбоксамид и Пример № 9. 4-(транс-3-Акриламидоциклогексил)-1H-индол-7-карбоксамид
Стадия A: трет-Бутил (3-(7-карбамоил-1H-индол-4-ил)циклогекс-2-ен-1-ил)карбамат и трет-Бутил (3-(7-карбамоил-1H-индол-4-ил)циклогекс-3-ен-1-ил)карбамат
К раствору 4-бром-1H-индол-7-карбоксамида (296 мг, 1,237 ммоль, Получение № 2), смеси трет-бутилового сложного эфира [3-(4,4,5,5-тетраметил[l,3,2]диоксаборолан-2-ил)-циклогекс-3-енил]-карбаминовой кислоты и трет-бутилового сложного эфира [3-(4,4,5,5-тетраметил-[l,3,2]диоксаборолан-2-ил)-циклогекс-2-енил]-карбаминовой кислоты (400 мг, 1,237 ммоль, U.S. 2009/0197864), Na2CO3 (328 мг, 3,09 ммоль), аддукт PdCl2(dppf)-DCM (101 мг, 0,124 ммоль) в THF:MeOH:H2O (отношение: 4:2:2, 20 мг) в атмосфере N2, смесь нагревали при приблизительно 100°C в течение ночи. Реакционную смесь фильтровали через слой целита. Полученную смесь разбавляли EtOAc (30 мг), промывали H2O (20 мл×2), сушили Na2SO4, концентрировали при пониженном давлении и остаток очищали с помощью препаративной ВЭЖХ (Таблица 1, Способ x) с получением смеси трет-бутил (3-(7-карбамоил-1H-индол-4-ил)циклогекс-2-ен-1-ил)карбамата и трет-бутил (3-(7-карбамоил-1H-индол-4-ил)циклогекс-3-ен-1-ил)карбамата (300 мг, 68%): ЖХ/МС (Таблица 1, Способ l) Время удерживания = 1,67 мин; MS m/z: 356 (M+H)+.
Стадия B: трет-Бутил (3-(7-карбамоил-1H-индол-4-ил)циклогексил)карбамат
К раствору трет-бутил (3-(7-карбамоил-1H-индол-4-ил)циклогекс-2-ен-1-ил)карбамата и трет-бутил (3-(7-карбамоил-1H-индол-4-ил)циклогекс-3-ен-1-ил)карбамата (300 мг, 0,844 ммоль) в THF (20 мг), добавляли Pd/C (44,9 мг, 0,422 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение приблизительно 3 часов в атмосфере H2. Смесь фильтровали и концентрировали при пониженном давлении с получением сырого продукта трет-бутил (3-(7-карбамоил-1H-индол-4-ил)циклогексил)карбамата (290 мг, 96%), который использовали на следующей стадии непосредственно. ЖХ/МС (Таблица 1, Способ l) Время удерживания = 1,53 мин; MS m/z: 358 (M+H)+.
Стадия C: 4-(3-Аминоциклогексил)-1H-индол-7-карбоксамид
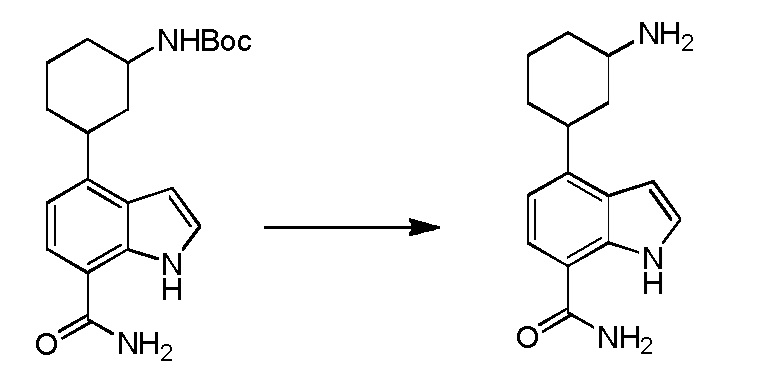
К раствору трет-бутил (3-(7-карбамоил-1H-индол-4-ил)циклогексил)карбамата (220 мг, 0,615 ммоль) в MeOH (10 мг) добавляли MeOH/HCl (10 мг) при приблизительно 0°C, затем реакционную смесь перемешивали при комнатной температуре в течение приблизительно 3 часов. Реакционную смесь концентрировали при пониженном давлении с получением сырого продукта 4-(3-аминоциклогексил)-1H-индол-7-карбоксамида (100 мг, 63%), который использовали на следующей стадии непосредственно. ЖХ/МС (Таблица 1, Способ l) Время удерживания = 0,54 мин; MS m/z: 258 (M+H)+.
Стадия D: 4-(цис-3-Акриламидоциклогексил)-1H-индол-7-карбоксамид и 4-(транс-3-Акриламидоциклогексил)-1H-индол-7-карбоксамид

К раствору 4-(3-аминоциклогексил)-1H-индол-7-карбоксамида (120 мг, 0,466 ммоль) в DCM (3 мг) добавляли DIEA (120 мг, 0,933 ммоль), добавляли акрилоилхлорид (42,2 мг, 0,466 ммоль) по каплям при приблизительно 0°C и смесь перемешивали при приблизительно 0°C в течение приблизительно 10 минут, затем концентрировали при пониженном давлении и остаток очищали с помощью препаративной ВЭЖХ (Таблица 1, Способ y) с получением 4-(цис-3-акриламидоциклогексил)-1H-индол-7-карбоксамида (27 мг, 19%) 1H-ЯМР: (MeOD) δ 7,59 (д, J=8, 1H), 7,33 (д, J=3,2, 1H), 6,95 (д, J=8, 1H), 6,64 (д, J=4, 1H), 6,26-6,17 (м, 2H), 5,67-5,58 (м, 1H), 4,01-3,96 (м, 1H), 3,22-3,13 (м, 1H), 2,19-1,97 (м, 4H), 1,65-1,59 (м, 3H),1,37-1,34 (м, 1H); ЖХ/МС (Таблица 1, Способ d) Время удерживания = 2,56 мин; MS m/z: 312 (M+H)+. (Btk IC50=A) и 4-(транс-3-акриламидоциклогексил)-1H-индол-7-карбоксамида (33 мг, 23%): 1H-ЯМР: (MeOD) δ 7,58 (д, J=8, 1H), 7,31 (д, J=3,2, 1H), 6,98 (д, J=8, 1H), 6,59 (д, J=2,8, 1H), 6,52-6,46 (м, 1H), 6,28-6,24 (м, 1H), 5,69-5,64 (м, 1H), 4,35 (с, 1H), 3,42-3,36 (м, 1H), 2,13-1,72 (м, 8H); ЖХ/МС (Таблица 1, Способ d) Время удерживания = 2,56 мин; MS m/z: 312 (M+H)+. (Btk IC50=B)
Пример № 10 и № 11: 4-(цис-3-Акриламидоциклопентил)-1H-индол-7-карбоксамид и 4-(транс-3-Акриламидоциклопентил)-1H-индол-7-карбоксамид
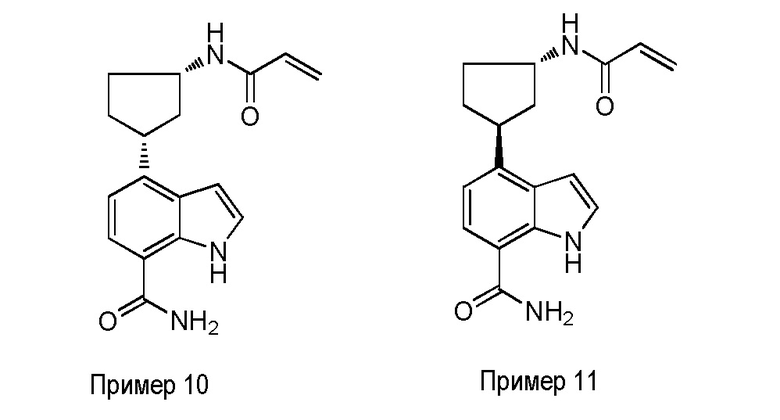
Стадия A: 3-((трет-Бутоксикарбонил)амино)циклопент-1-ен-1-ил трифторметансульфонат и 4-((трет-бутоксикарбонил)амино)циклопент-1-ен-1-ил трифторметансульфонат
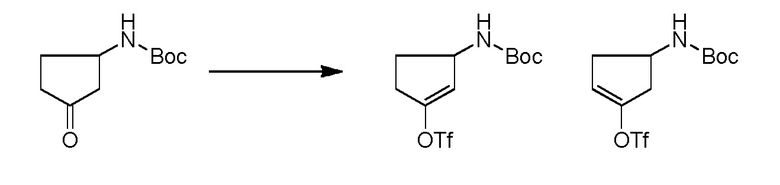
К свежеприготовленному раствору LDA (2 M в THF, 9,38 мг) добавляли по каплям трет-бутил (3-оксоциклопентил)карбамат (2,00 г, 10,0 ммоль) в THF (4 мг) при приблизительно -78°C. Смесь нагревали до комнатной температуры в течение приблизительно 30 минут и затем снова охлаждали до приблизительно -78°C. Раствор 1,1,1-трифтор-N-фенил-N-((трифторметил)сульфонил)метансульфонамида (5,38 г, 15,1 ммоль) в THF (10 мг) добавляли по каплям к реакционной смеси при приблизительно -78°C. Полученную смесь нагревали до комнатной температуры и перемешивали еще в течение 3 часов. Обрабатывали EtOAc (30 мг), смесь промывали H2O (20 мл×3) и насыщенным солевым раствором (10 мг), сушили Na2SO4, концентрировали при пониженном давлении и остаток очищали хроматографией на силикагеле, получая смесь 3-((трет-бутоксикарбонил)амино)циклопент-1-ен-1-ил трифторметансульфоната и 4-((трет-бутоксикарбонил)амино)циклопент-1-ен-1-ил трифторметансульфоната (0,82 г, 25%), который использовали на следующей стадии без дополнительной очистки.
Стадия B: трет-Бутил (3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)циклопент-2-ен-1-ил)карбамат и трет-бутил (3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)циклопент-3-ен-1-ил)карбамат
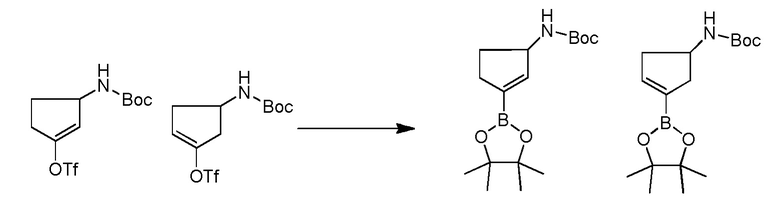
Смесь 3-((трет-бутоксикарбонил)амино)циклопент-1-ен-1-ил трифторметансульфоната и 4-((трет-бутоксикарбонил)амино)циклопент-1-ен-1-ил трифторметансульфоната (720 мг, 2,173 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (662 мг, 2,61 ммоль), PdCl2(dppf)-DCM аддукт (177 мг, 0,217 ммоль) и KOAc (427 мг, 4,35 ммоль) в 1,4-диоксане (20 мг) в атмосфере N2 нагревали при приблизительно 100°C в течение ночи. Полученную смесь разбавляли DCM (30 мг), промывали H2O (20 мл×2), концентрировали при пониженном давлении и остаток очищали на силикагеле с получением сырой смеси трет-бутил (3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)циклопент-2-ен-1-ил)карбамата и трет-бутил (3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)циклопент-3-ен-1-ил)карбамата (0,42 г, 63%), который использовали на следующей стадии без дополнительной очистки.
Стадия C: трет-Бутил (3-(7-карбамоил-1H-индол-4-ил)циклопент-2-ен-1-ил)карбамат и трет-бутил (3-(7-карбамоил-1H-индол-4-ил)циклопент-3-ен-1-ил)карбамат
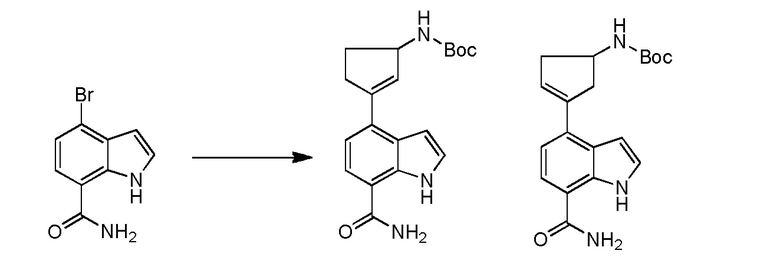
К раствору 4-бром-1H-индол-7-карбоксамида (325 мг, 1,36 ммоль, Получение № 2), трет-бутил (3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)циклопент-2-ен-1-ил)карбамата и трет-бутил (3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)циклопент-3-ен-1-ил)карбамата (420 мг, 1,36 ммоль), Na2CO3 (360 мг, 3,4 ммоль), PdCl2(dppf)-DCM аддукта (111 мг, 0,136 ммоль) в THF:MeOH:H2O (отношение: 4:2:2, 15 мг) в атмосфере N2, смесь перемешивали при приблизительно 100°C в течение ночи. Реакционную смесь фильтровали для удаления Pd комплекса. Полученную смесь разбавляли EtOAc (30 мг), промывали H2O (20 мл×2), сушили Na2SO4, концентрировали и очищали с помощью препаративной ВЭЖХ (Таблица 1, Способ y) с получением смеси трет-бутил (3-(7-карбамоил-1H-индол-4-ил)циклопент-2-ен-1-ил)карбамата и трет-бутил (3-(7-карбамоил-1H-индол-4-ил)циклопент-3-ен-1-ил)карбамата (0,32 г, 69%): ЖХ/МС (Таблица 1, Способ l) Время удерживания = 1,65 мин; MS m/z: 342 (M+H)+.
Стадия D: трет-Бутил (3-(7-карбамоил-1H-индол-4-ил)циклопентил)карбамат
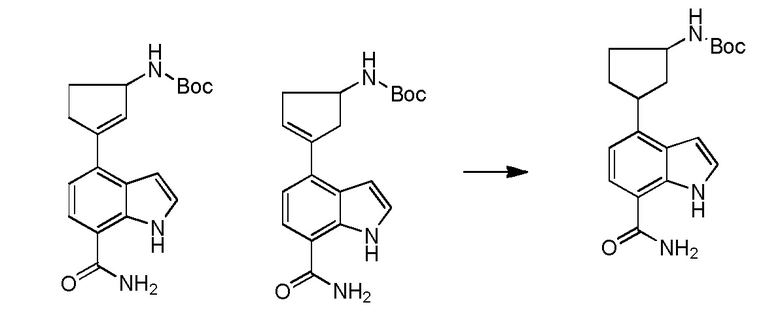
К раствору трет-бутил (3-(7-карбамоил-1H-индол-4-ил)циклопент-2-ен-1-ил)карбамата и трет-бутил (3-(7-карбамоил-1H-индол-4-ил)циклопент-3-ен-1-ил)карбамата (300 мг, 0,844 ммоль) в THF (20 мг), добавляли Pd/C (44,9 мг, 0,422 ммоль) и смесь перемешивали в течение приблизительно 3 часов при комнатной температуре под H2. Смесь фильтровали и концентрировали при пониженном давлении с получением сырого трет-бутил (3-(7-карбамоил-1H-индол-4-ил)циклопентил)карбамата (0,29 г, 96%), который использовали на следующей стадии непосредственно без дополнительной очистки. ЖХ/МС (Таблица 1, Способ l) Время удерживания = 1,50 мин; MS m/z: 344 (M+H)+.
Стадия E: 4-(цис-3-Аминоциклопентил)-1H-индол-7-карбоксамид и 4-(транс-3-аминоциклопентил)-1H-индол-7-карбоксамид
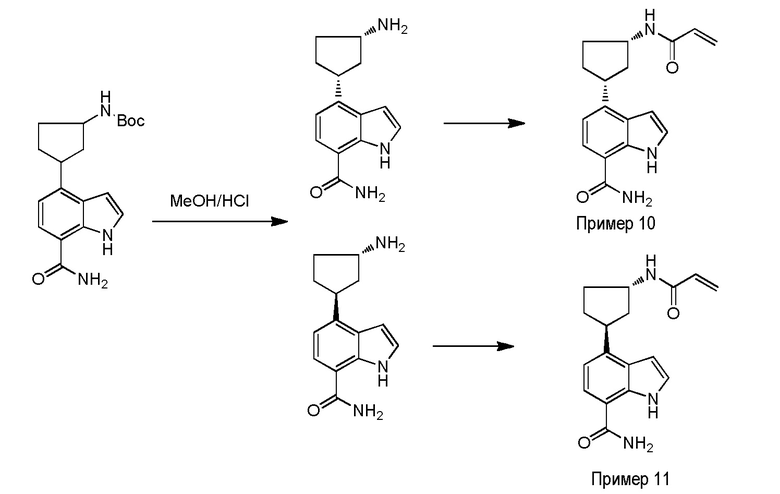
К раствору трет-бутил (3-(7-карбамоил-1H-индол-4-ил)циклопентил)карбамата (250 мг, 0,728 ммоль) в MeOH (10 мг) добавляли MeOH/HCl (10 мг) при приблизительно 0°C и смесь перемешивали в течение приблизительно 3 часов при комнатной температуре. Смесь концентрировали при пониженном давлении и остаток очищали с помощью препаративной ВЭЖХ (Таблица 1, Способ t) с получением 4-(транс-3-аминоциклопентил)-1H-индол-7-карбоксамида (10 мг, 6%) и 4-(цис-3-аминоциклопентил)-1H-индол-7-карбоксамида (50 мг, 28%). К раствору 4-(цис-3-аминоциклопентил)-1H-индол-7-карбоксамида (50 мг, 0,206 ммоль) в DCM (3 мг) добавляли DIEA (53 мг, 0,411 ммоль), затем добавляли по каплям акрилоилхлорид (18,60 мг, 0,206 ммоль) при приблизительно 0°C, смесь перемешивали при приблизительно 0°C в течение приблизительно 10 минут, затем концентрировали при пониженном давлении и остаток очищали с помощью препаративной ВЭЖХ (Таблица 1, Способ z) с получением 4-(цис-3-акриламидоциклопентил)-1H-индол-7-карбоксамида (20 мг, 33%): 1H-ЯМР (MeOD) δ 7,59 (д, J=7,2, 1H), 7,33 (с, 1H), 7,02 (д, J=8, 1H), 6,64 (с, 1H), 6,30-6,20 (м, 2H), 5,64 (д, J=8,8, 1H), 4,51-4,40 (м, 1H), 3,60-3,58 (м, 1H), 2,56-2,51 (м, 1H), 2,26-2,21 (м, 2H), 2,07-2,02 (м, 1H), 1,86-1,78 (м, 2H): ЖХ/МС (Таблица 1, Способ d) Время удерживания = 2,48 мин; MS m/z: 298 (M+H)+. (Btk IC50=A) К раствору 4-(транс-3-аминоциклопентил)-1H-индол-7-карбоксамида (10 мг, 0,041 ммоль) в DCM (1 мг) добавляли DIEA (11 мг, 0,082 ммоль), затем добавляли по каплям акрилоилхлорид (3,72 мг, 0,041 ммоль), смесь перемешивали при приблизительно 0°C в течение приблизительно 10 минут, концентрировали и очищали с помощью препаративной ВЭЖХ (Таблица 1, Способ z) с получением 4-(транс-3-акриламидоциклопентил)-1H-индол-7-карбоксамида (1,1 мг, 9%): 1H-ЯМР (MeOD) δ 7,60 (д, J=7,6, 1H), 7,33 (д, J=2,8, 1H), 7,00 (д, J=7,6, 1H), 6,62 (д, J=3,2, 1H), 6,33-6,20 (м, 2H), 5,67-5,64 (м, 1H), 4,50-4,49 (м, 1H), 3,81-3,72 (м, 1H), 2,34-2,28 (м, 3H), 2,26-2,23 (м, 1H), 2,07-1,89 (м, 1H), 1,88-1,74 (м, 1H); ЖХ/МС (Таблица 1, Способ d) Время удерживания = 2,47 мин; MS m/z: 298 (M+H)+. (BtkIC50=A)
Пример № 12*: (R)-2-(1-(Метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-4-(2-оксо-1,3'-бипиперидин-1'-ил)-1H-индол-7-карбоксамид
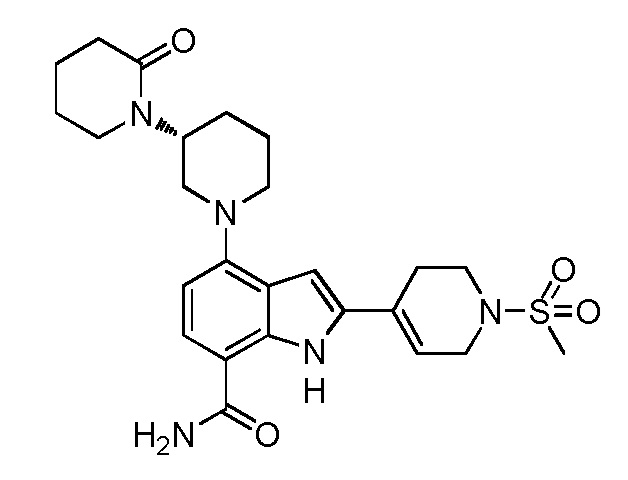
Стадия A: (R)-2-(1-(Метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-4-(2-оксо-1,3'-бипиперидин-1'-ил)-1-тозил-1H-индол-7-карбонитрил
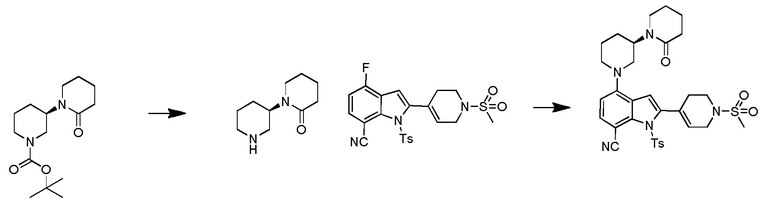
К раствору (R)-трет-бутил 2-оксо-1,3'-бипиперидин-1'-карбоксилата (100 мг, 0,354 ммоль, WO 2011/029046) в DCM (4 мг) добавляли TFA (1,000 мг). Реакционную смесь перемешивали в течение приблизительно 4 часов при комнатной температуре. Растворитель отгоняли и смесь 4-фтор-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1-тозил-1H-индол-7-карбонитрила (168 мг, 0,354 ммоль, Получение № 27) и TEA (0,197 мл, 1,417 ммоль) в DMSO (2 мг) добавляли. Пробирку герметично укупоривали и реакционную смесь нагревали в микроволновой печи при приблизительно 120°C в течение приблизительно 30 минут. Добавляли воду (20 мг) и экстрагировали DCM, затем промывали насыщенным солевым раствором и пропускали через разделитель фаз для удаления остаточной воды. Выпаривали и хроматографировали на силикагеле, элюируя градиентом 0-100% EtOAc/гексан, с получением сырого (R)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-4-(2-оксо-1,3'-бипиперидин-1'-ил)-1-тозил-1H-индол-7-карбонитрила (0,041 г, 18,21%).
Стадия B: (R)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-4-(2-оксо-1,3'-бипиперидин-1'-ил)-1H-индол-7-карбоксамид

Смесь Cs2CO3 (20,50 мг, 0,063 ммоль) и (R)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-4-(2-оксо-1,3'-бипиперидин-1'-ил)-1-тозил-1H-индол-7-карбонитрила (40 мг, 0,063 ммоль) в THF (2 мг) и MeOH (1,000 мг) перемешивали при комнатной температуре в течение ночи. Раствор разбавляли водой (15 мг) и перемешивали в течение приблизительно 20 минут. Добавляли DCM для расстворения суспензии и смесь фильтровали через разделитель фаз Biotage. Органические слои собирали и концентрировали. Промежуточное соединение растворяли в трет-бутаноле (1 мг) и DMSO (0,500 мг) и NaOH (0,377 мл, 0,755 ммоль) и перекись водорода (0,175 мл, 1,699 ммоль) добавляли. Смесь перемешивали в течение приблизительно 20 минут при комнатной температуре и добавляли насыщенный NH4Cl (1 мг). Смесь разбавляли водой (15 мг) и перемешивали в течение приблизительно 15 минут. Твердые вещества собирали фильтрованием, промывкой несколько раз водой и сушили в вакууме и очищали с помощью препаративной ВЭЖХ (Таблица 1, Способ aq). Образцы возвращали и растворяли в DCM. Органические слои объединяли и промывали насыщенным раствором бикарбоната натрия, фильтровали через разделитель фаз Biotage и концентрировали с получением (R)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-4-(2-оксо-1,3'-бипиперидин-1'-ил)-1H-индол-7-карбоксамида (3 мг, 9,54%): ЖХ/МС (Таблица 1, Способ f) Время удерживания = 1,37 мин; MS m/z: 500 (M+H)+. (Btk IC50=B)
Пример № 13*: (R)-2-(1-(Метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-4-(3-(1-оксо-3,4-дигидроизохинолин-2(1H)-ил)пиперидин-1-ил)-1H-индол-7-карбоксамид
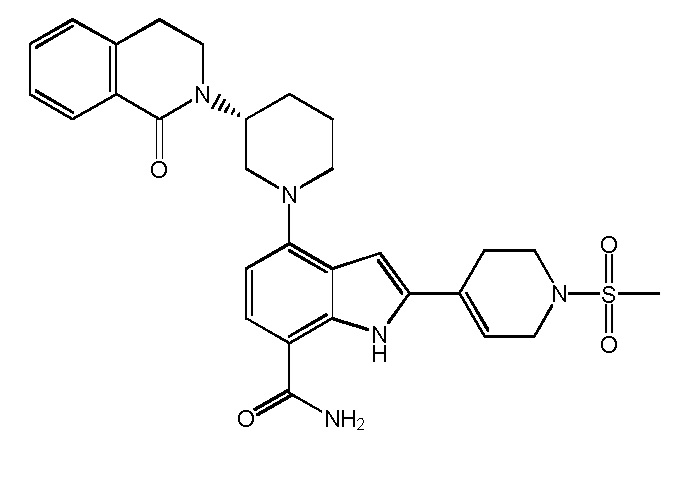
Стадия A: (R)-2-метил-N-(пиперидин-3-ил)бензамид
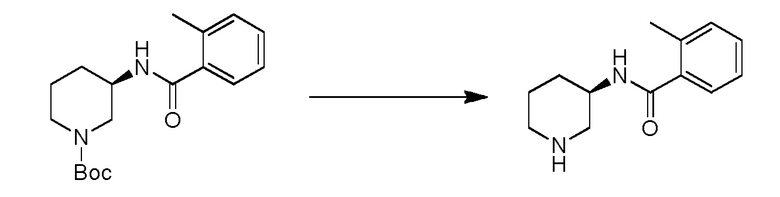
Смесь (R)-трет-бутил 3-(2-метилбензамидо)пиперидин-1-карбоксилата (19,0 г, 59,7 ммоль, полученный с использованием D из (R)-трет-бутил 3-аминопиперидин-1-карбоксилата и 2-метилбензойная кислота) в HCl (2 н в MeOH, 300 мл, 600 ммоль) перемешивали при комнатной температуре в течение приблизительно 4 часов, затем концентрировали при пониженном давлении с получением сырого (R)-2-метил-N-(пиперидин-3-ил)бензамида (20,0 г), который использовали непосредственно на следующей стадии без дополнительной очистки.
Стадия B: (R)-N-(1-Бензилпиперидин-3-ил)-2-метилбензамид
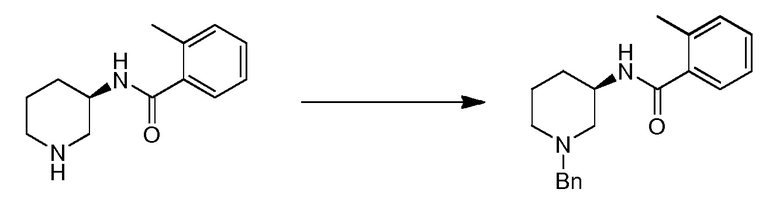
К раствору (R)-2-метил-N-(пиперидин-3-ил)бензамида (20,0 г, сырой) и TEA (30,1 г, 298,5 ммоль) в DCM (260 мг) по каплям добавляли BnBr (11,2 г, 65,7 ммоль) при комнатной температуре в течение приблизительно 30 минут. Затем смесь перемешивали в течение ночи при комнатной температуре. После завершения добавляли DCM (1 л), и смесь промывали H2O (3×100 мг). Органическую фазу сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением (R)-N-(1-бензилпиперидин-3-ил)-2-метилбензамида (12,0 г, 65% после двух стадий): ЖХ/МС (Таблица 1, Способ l) Время удерживания = 0,91 мин; MS m/z: 309 (M+H)+.
Стадия C: (R)-2-(1-Бензилпиперидин-3-ил)изохинолин-1(2H)-он
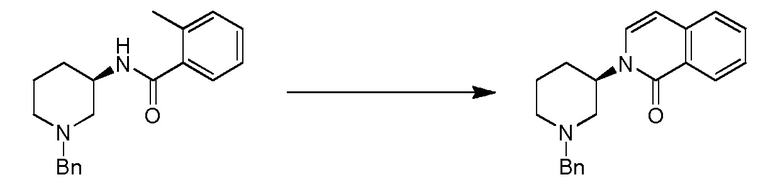
К раствору (R)-N-(1-бензилпиперидин-3-ил)-2-метилбензамида (12,0 г, 38,9 ммоль) в THF по каплям добавляли n-BuLi (2,5 M, 32,7 мг) между -22 и -14°C, в течение приблизительно 30 минут. Полученный темно-красный раствор перемешивали при приблизительно -22°C в течение приблизительно 30 минут и добавляли DMF ниже приблизительно -14°C (внутренняя). После того, как добавление закончивали, раствор перемешивали при приблизительно -22°C в течение приблизительно 30 минут. Затем HCl (6 н водный, 25 мл, 150 ммоль) медленно добавляли, поддерживая температуру ниже 5°C. Смесь подщелачивали добавлением насыщенного NaOH при приблизительно 0°C до pH 14 и экстрагировали DCM (3×500 мг). Органическую фазу сушили над Na2SO4 и концентрировали при пониженном давлении с получением (R)-2-(1-бензилпиперидин-3-ил)изохинолин-1(2H)-она (12,0 г, 97%) в виде твердого вещества: ЖХ/МС (Таблица 1, Способ l) Время удерживания = 1,35 мин; MS m/z: 319 (M+H)+.
Стадия D: (R)-2-(Пиперидин-3-ил)-3,4-дигидроизохинолин-1(2H)-он
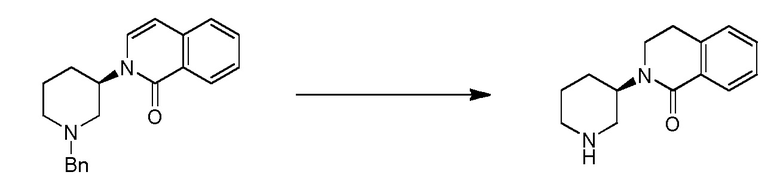
Смесь (R)-2-(1-бензилпиперидин-3-ил)изохинолин-1(2H)-она (12 г, 37,7 ммоль) и Pd(OH)2 (0,5 г) в MeOH перемешивали при температуре приблизительно 50°C в атмосфере H2 (50 фунт на кв.дюйм) в течение ночи. Затем смесь фильтровали через целит, и фильтрат концентрировали. Сырой продукт очищали флэш-хроматографией с получением 6,3 г сырого продукта, который перекристаллизовывали в смеси MTBE (15 мг) и HCl/MeOH (5 мг) с получением (R)-2-(пиперидин-3-ил)-3,4-дигидроизохинолин-1(2H)-она (HCl соль) в виде твердого вещества (2,1 г, 21%): 1H-ЯМР (MeOD) 7,95 (д, J=8, 1H), 7,51-7,47 (м, 1H), 7,38-7,34 (м, 1H), 7,29 (д, J=7,6, 1H), 4,86-4,80 (м, 1H), 3,61-3,58 (м, 2H), 3,39-3,35 (м, 2H), 3,28-3,22 (м, 1H), 3,03-2,95 (м, 3H), 2,12-1,87 (м, 4H); ЖХ/МС (Таблица 1, Способ d) Время удерживания = 2,05 мин; MS m/z: 231 (M+H)+.
Стадия E: (R)-2-(1-(Метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-4-(3-(1-оксо-3,4-дигидроизохинолин-2(1H)-ил)пиперидин-1-ил)-1-тозил-1H-индол-7-карбонитрил
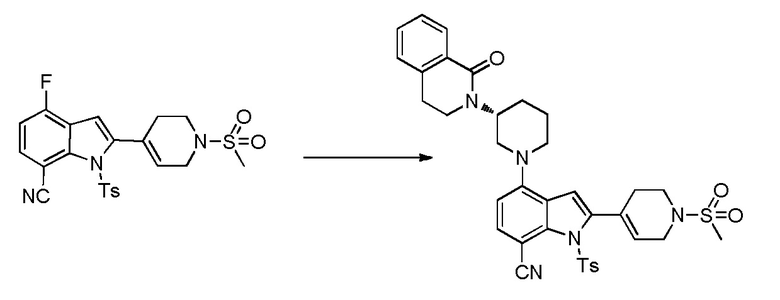
Смесь 4-фтор-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1-тозил-1H-индол-7-карбонитрила (318 мг, 0,672 ммоль, Получение № 27), гидрохлорида (R)-2-(пиперидин-3-ил)-3,4-дигидроизохинолин-1(2H)-она (179 мг, 0,672 ммоль) и TEA (0,374 мл, 2,69 ммоль) в DMSO (4 мг) нагревали в микроволновой печи при приблизительно 120°C в течение приблизительно 20 минут. Реакционную смесь нагревали в микроволновой печи при приблизительно 120°C в течение дополнительных 30 минут. Добавляли воду (50 мг) и экстрагировали DCM. Раствор промывали насыщенным солевым раствором и пропускали через разделитель фаз для удаления остаточной воды. Органические слои концентрировали и хроматографировали на силикагеле, элюируя градиентом 0-100% EtOAc/гексан с получением сырого (R)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-4-(3-(1-оксо-3,4-дигидроизохинолин-2(1H)-ил)пиперидин-1-ил)-1-тозил-1-индол-7-карбонитрила (110 мг, 24%). Материал использовали без дальнейшей очистки.
Стадия F: (R)-2-(1-(Метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-4-(3-(1-оксо-3,4-дигидроизохинолин-2(1H)-ил)пиперидин-1-ил)-1H-индол-7-карбоксамид

Смесь Cs2CO3 (51,9 мг, 0,159 ммоль) и (R)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-4-(3-(1-оксо-3,4-дигидроизохинолин-2(1H)-ил)пиперидин-1-ил)-1-тозил-1H-индол-7-карбонитрила (109 мг, 0,159 ммоль) в THF (2 мг) и MeOH (1,000 мг) перемешивали в течение ночи при комнатной температуре. Смесь разбавляли водой (15 мг) и перемешивали в течение приблизительно 20 минут. Осадок собирали фильтрованием и осадок на фильтре промывали водой. Осадок на фильтре растворяли в трет-бутаноле (1 мг) и DMSO (0,500 мг) добавляли NaOH (0,956 мл, 1,91 ммоль) и перекись водорода (0,444 мл, 4,30 ммоль). Смесь перемешивали в течение приблизительно 20 минут при комнатной температуре и добавляли насыщенный NH4Cl (1 мг). Смесь разбавляли водой (15 мг) и перемешивали в течение приблизительно 15 минут. Твердые вещества собирали фильтрованием, промывали несколько раз водой и сушили в вакууме. Полученные твердые вещества очищали с помощью препаративной ВЭЖХ (Таблица 1, Способ ap). Образцы возвращали и растворяли в DCM. Органические слои объединяли и промывали насыщенным раствором бикарбоната натрия, фильтровали через разделитель фаз Biotage, и концентрировали. Остаток дополнительно сушили в вакуумной печи при приблизительно 50°C в течение приблизительно 48 часов с получением (R)-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-4-(3-(1-оксо-3,4-дигидроизохинолин-2(1H)-ил)пиперидин-1-ил)-1H-индол-7-карбоксамида (30 мг, 34%): ЖХ/МС (Таблица 1, Способ f) Время удерживания = 1,63 мин; MS m/z: 548 (M+H)+. (Btk IC50=A)
Пример № 13A*: (R)-N-(1-(7-карбамоил-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-4-ил)пиперидин-3-ил)тиазол-2-карбоксамид
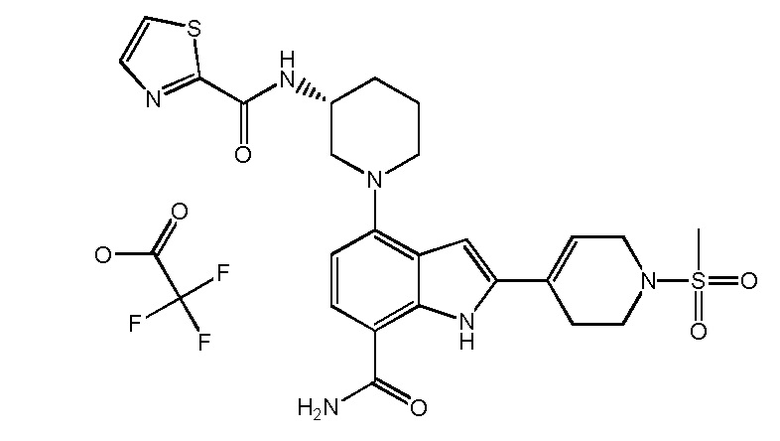
Стадия A: (R)-трет-Бутил 3-(тиазол-2-карбоксамидо)пиперидин-1-карбоксилат
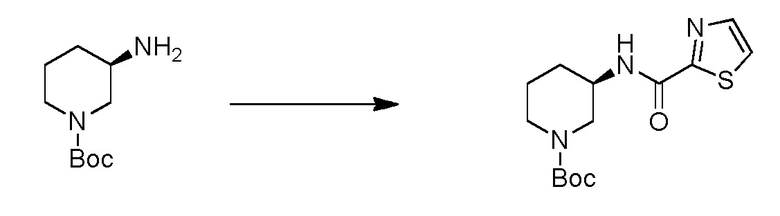
К раствору (R)-трет-бутил 3-аминопиперидин-1-карбоксилата (2 г, 9,99 ммоль) и тиазол-2-карбоновой кислоты (1,29 г, 9,99 ммоль) в DCM (40 мг) добавляли HATU (4,85, 12,5 ммоль) и DIEA (3,87 г, 29,9 ммоль) и смесь перемешивают в течение ночи при комнатной температуре. Затем смесь выливали в воду и экстрагировали DCM (3×80 мг). Объединенные органические слои промывали насыщенным водным раствором NaHCO3 (80 мг) и насыщенным солевым раствором (80 мг), и сушили над Na2SO4. Растворитель концентрировали при пониженном давлении с получением сырого продукта, который очищали с помощью колоночной хроматографии на силикагеле, с получением (R)-трет-бутил 3-(тиазол-2-карбоксамидо)пиперидин-1-карбоксилата (2,2 г, 71%): 1H-ЯМР (CDCl3) δ 1,45 (с, 9H), 1,78-1,73 (м, 2H), 1,94-1,91 (м, 1H), 2,80 (с, 2H), 3,42 (ушир., 2H), 3,66 (д, J=13,2 Гц, 1H), 4,11 (с, 1H), 7,36 (ушир., 1H), 7,57 (т, J=3,2 Гц, 1H), 7,84 (т, J=3,2 Гц, 1H).
Стадия B: (R)-N-(Пиперидин-3-ил)тиазол-2-карбоксамид

К раствору (R)-трет-бутил 3-(тиазол-2-карбоксамидо)пиперидин-1-карбоксилата (1,9 г, 6,1 ммоль) в EtOAc (20 мг) добавляли по каплям HCl/EtOAc (20 мг) при приблизительно 0°C, затем реакционную смесь перемешивали при комнатной температуре в течение приблизительно 3 часов. Смесь фильтровали и осадок на фильтре являлся гигроскопичным. Осадок на фильтре растворяли в воде и насыщенным водным раствором NaHCO3. Смесь экстрагировали DCM (3×50 мг) и объединенные органические слои промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали с получением (R)-N-(пиперидин-3-ил)тиазол-2-карбоксамида (1,2 г, 5,68 ммоль, 93%): 1H-ЯМР (CDCl3) δ 1,79-1,66 (м, 3H), 1,92-1,86 (м, 1H), 2,04 (с, 1H), 2,87-2,70 (м, 3H), 3,15-2,88 (м, 1H), 4,12-4,06 (м, 1H), 7,54-7,53 (м, 2H), 7,84 (т, J=2,8 Гц, 1H).
Стадия C: (R)-N-(1-(7-Циано-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1-тозил-1H-индол-4-ил)пиперидин-3-ил)тиазол-2-карбоксамид
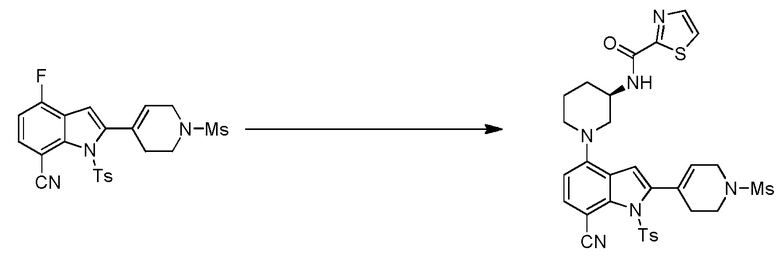
Смесь 4-фтор-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1-тозил-1H-индол-7-карбонитрила (200 мг, 0,422 ммоль, Получение № 27), (R)-N-(пиперидин-3-ил) тиазол-2-карбоксамида (178 мг, 0,842 ммоль) и TEA (170 мг, 1,680 ммоль) в DMSO (2 мг) нагревали в микроволновой печи при приблизительно 120°C в течение приблизительно 1 часа. Добавляли воду (10 мг) к смеси и экстрагировали DCM (3×20 мг). Органический слой промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением сырого продукта, который очищали с помощью препаративной ТСХ (DCM:MeOH = 75:1) с получением (R)-N-(1-(7-циано-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1-тозил-1H-индол-4-ил)пиперидин-3-ил)тиазол-2-карбоксамида (20 мг, 7%): ЖХ/МС (Таблица 1, Способ m) Время удерживания = 2,24 мин; MS m/z: 665 (M+H)+.
Стадия D: (R)-N-(1-(7-карбамоил-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-4-ил)пиперидин-3-ил)тиазол-2-карбоксамид
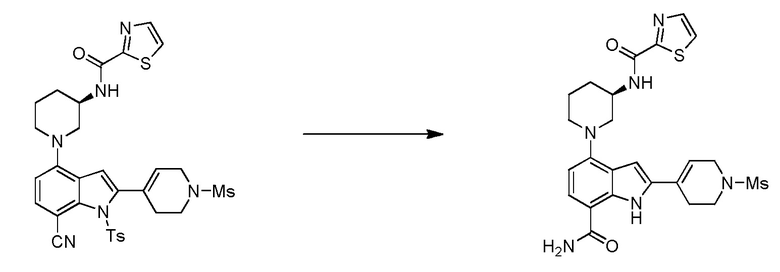
Смесь (R)-N-(1-(7-циано-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1-тозил-1H-индол-4-ил)пиперидин-3-ил)тиазол-2-карбоксамида (76 мг, 0,114 ммоль), NaOH (54,9 мг, 1,37 ммоль) и 30% H2O2 (350 мг, 3,09 ммоль) в смеси DMSO (1 мг) и н-бутанола (2 мг) перемешивали при комнатной температуре в течение приблизительно 24 часов. Затем добавляли насыщенный водный раствор NH4Cl (2 мг) и разбавляли водой (30 мг) и перемешивали в течение 30 минут. Твердое вещество собирали фильтрацией и промывали несколько раз водой и сырой продукт очищали с помощью препаративной ТСХ (50:1 DCM/MeOH) с получением (R)-N-(1-(7-карбамоил-2-(1-(метилсульфонил)-1,2,3,6-тетрагидропиридин-4-ил)-1H-индол-4-ил)пиперидин-3-ил)тиазол-2-карбоксамида (32 мг, 53%): ЖХ/МС (Таблица 1, Способ d) Время удерживания = 2,90 мин; MS m/z: 529 (M+H)+. (Btk IC50=A)
Пример № 14: 2-(1-метил-1H-пиразол-4-ил)-4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-1H-бензо[d]имидазол-7-карбоксамид
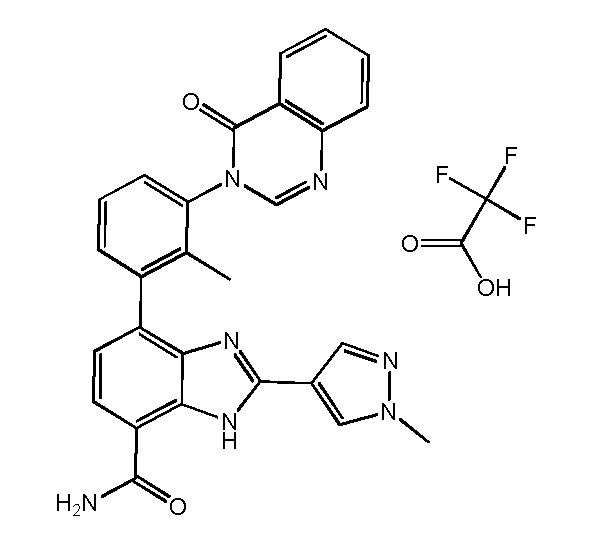
Стадия A: 3-(3-(7-бромбензо[c][1,2,5]тиадиазол-4-ил)-2-метилфенил)хиназолин-4(3H)-он
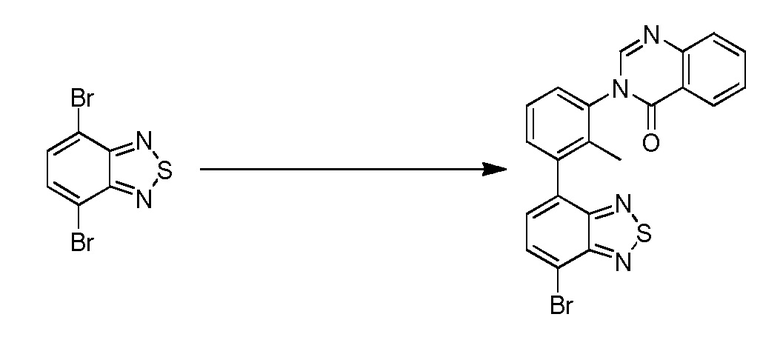
К раствору 4,7-дибромбензо[c][1,2,5]тиадиазола (1,029 г, 3,5 ммоль) и 3-(2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)хиназолин-4(3H)-она (1,141 г, 3,15 ммоль, WO 2011159857) в смеси толуола (40 мг), MeOH (10 мг) и воды (10 мг) добавляли Na2CO3 (0,742 г, 7,00 ммоль) и Pd(PPh3)4 (0,081 г, 0,070 ммоль). Смесь нагревали примерно до 100°C в течение 24 часов. Полученный раствор охлаждали до комнатной температуры и разбавляли EtOAc, промывали водой и насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали с получением сырого продукта, который очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир:EtOAc = 5:1-1:1) с получением 3-(3-(7-бромбензо[c][1,2,5]тиадиазол-4-ил)-2-метилфенил)хиназолин-4(3H)-она (1,0 г, 64%): 1H-ЯМР (CDCl3) δ 8,40-8,38 (д, J=8,0 Гц, 1H), 8,13 (с, 1H), 7,95-7,93 (д, J=7,6 Гц, 1H), 7,82-7,80 (м, 2H), 7,58-7,56 (м, 1H), 7,51-7,46 (м, 3H), 7,41-7,39 (т, J=4,8 Гц, 1H), 1,95 (с, 3H).
Стадия B: 7-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)бензо[c][1,2,5]тиадиазол-4-карбонитрил
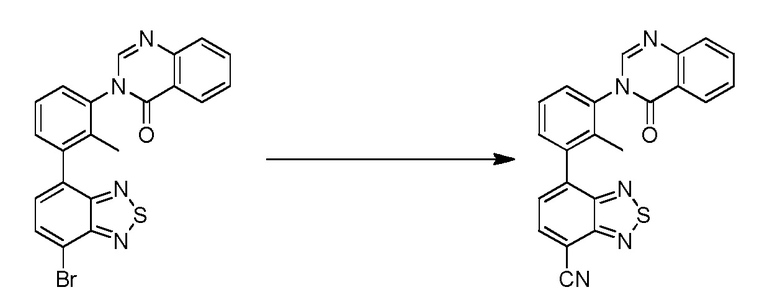
К раствору 3-(3-(7-бромбензо[c][1,2,5]тиадиазол-4-ил)-2-метилфенил)хиназолин-4(3H)-она (0,449 г, 1 ммоль) в DMF (12 мг) добавляли Zn(CN)2 (0,076 г, 0,650 ммоль) и Pd(PPh3)4 (0,046 г, 0,040 ммоль). Смесь нагревали примерно до приблизительно 160°C в течение приблизительно 15 минут в атмосфере N2 в микроволновом реакторе. Полученный раствор разбавляли EtOAc и промывали насыщенным солевым раствором (4×). Органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением сырого продукта, который очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир:EtOAc = 5:1-1:1), с получением 7-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)бензо[c][1,2,5]тиадиазол-4-карбонитрила (0,3 г, 76%): 1H-ЯМР (CDCl3) δ 8,33-8,03 (д, J=8,0 Гц, 1H), 8,10-8,06 (т, J=7,2 Гц, 2H), 7,77-7,74 (м, 2H), 7,63-7,61 (т, J=7,2 Гц, 1H), 7,53-7,45 (м, 3H), 7,39-7,37 (д, J=7,2 Гц, 1H), 1,90 (с, 3H).
Стадия C: 2,3-Диамино-2'-метил-3'-(4-оксохиназолин-3(4H)-ил)-[1,1'-дифенил]-4-карбонитрил
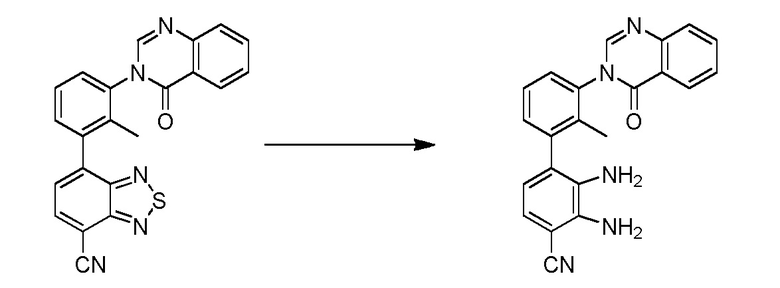
К раствору 2,3-диамино-2'-метил-3'-(4-оксохиназолин-3(4H)-ил)-[1,1'-дифенил]-4-карбонитрила (0,53 мг, 1,34 ммоль) в AcOH (50 мг) добавляли цинк (1,75 г, 26,8 ммоль), смесь нагревали до приблизительно 120°C в течение приблизительно 2 часов. Растворитель концентрировали, и остаток переносили в EtOAc, промывали насыщенным водным раствором NaHCO3 и насыщенным солевым раствором. Органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением сырого продукта, который очищали с помощью колоночной хроматографии на силикагеле (элюировали петролейным эфиром:EtOAc=1:1-0:1) с получением 2,3-диамино-2'-метил-3'-(4-оксохиназолин-3(4H)-ил)-[1,1'-дифенил]-4-карбонитрила (0,4 г, 81%): ЖХ/МС (Таблица 1, Способ l) Время удерживания = 1,33 мин; MS m/z: 368 (M+H)+.
Стадия D: 2-(1-метил-1H-пиразол-4-ил)-4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-1H-бензо[d]имидазол-7-карбонитрил
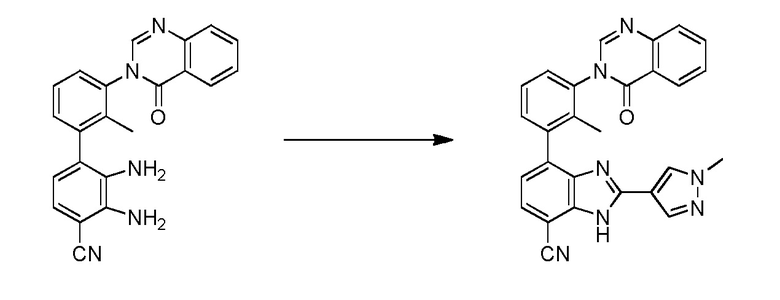
К раствору 2,3-диамино-2'-метил-3'-(4-оксохиназолин-3(4H)-ил)-[1,1'-дифенил]-4-карбонитрила (400 мг, 1,09 ммоль) в DMF (15 мг) добавляли 1-метил-1Н-пиразол-4-карбальдегид (240 мг, 2,18 ммоль) и TMSCl (0,417 мл, 3,27 ммоль). Смесь нагревали до приблизительно 100°C в течение приблизительно 30 минут в микроволновом реакторе. Полученный раствор разбавляли EtOAc и промывали насыщенным солевым раствором (4×). Органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением сырого продукта, который очищали с помощью колоночной хроматографии на силикагеле (элюировали петролейный эфир:EtOAc = 1:1, затем EtOAc:MeOH=50:1), с получением 2-(1-метил-1H-пиразол-4-ил)-4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-1H-бензо[d]имидазол-7-карбонитрила (200 мг, 40%): ЖХ/МС (Таблица 1, Способ m) Время удерживания = 1,78 минут; MS m/z: 458 (M+H)+.
Стадия E: 2-(1-метил-1H-пиразол-4-ил)-4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-1H-бензо[d]имидазол-7-карбоксамид

К раствору 2-(1-метил-1H-пиразол-4-ил)-4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-1H-бензо[d]имидазол-7-карбонитрила (278 мг, 0,608 ммоль) в смеси бутанола (6 мг) и DMSO (3 мг) добавляли NaOH (292 мг, 7,29 ммоль) и H2O2 (1,68 мл, 16,4 ммоль). Смесь перемешивали в течение приблизительно 24 часов при температуре приблизительно 25°C. Полученный раствор гасили насыщенным водным раствором NH4Cl, экстрагировали EtOAc. Органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением сырого продукта, который очищали с помощью препаративной ВЭЖХ (таблица 1, способ n), с получением 2-(1-метил-1H-пиразол-4-ил)-4-(2-метил-3-(4-оксохиназолин-3(4H)-ил)фенил)-1H-бензо[d]имидазол-7-карбоксамида (140 мг, 48%): LCMS (Таблица 1, Способ d) Время удерживания = 2,53 мин; MS m/z: 476 (M+H)+. (IC50 Btk=B)
Пример № 15: 4-(3-Акриламидофенил)-1H-индазол-7-карбоксамид
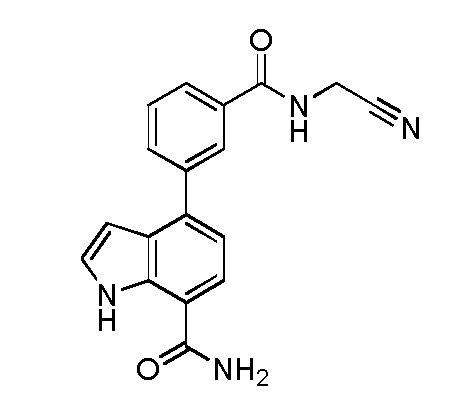
Стадия A: 3-(7-карбамоил-1H-индол-4-ил)бензойная кислота
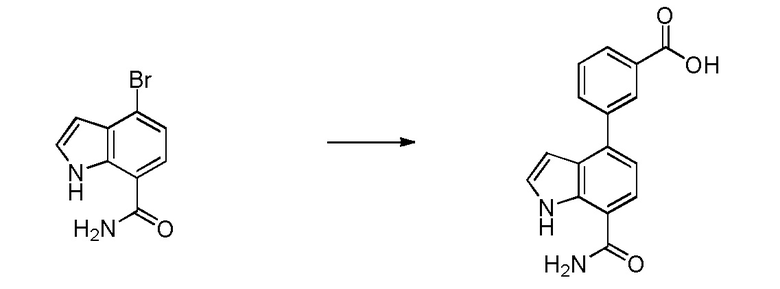
Смесь 4-бром-1H-индол-7-карбоксамида (0,5 г, 2,091 ммоль, Получение № 2), (3-(метоксикарбонил)фенил)бороновой кислоты (0,565 г, 3,14 ммоль), карбоната натрия (2,61 мл, 5,23 ммоль) в DME (10,00 мг) дегазировали и продували азотом в течение приблизительно 5 минут, затем добавляли тетракис(трифенилфосфин)палладий(0) (0,121 г, 0,105 ммоль). Реакционный сосуд герметично закрывали и нагревали в микроволновой печи (Biotage Initiator) при приблизительно 110°C в течение приблизительно 45 минут. Смесь охлаждали до комнатной температуры с последующим добавлением приблизительно 50 мл воды. Осадок отфильтровывали, сушили на воздухе и использовали без дальнейшей очистки. Этот сырой продукт растворяли в THF (25 мг) и обрабатывали раствором гидроксидом лития (0,250 г, 10,46 ммоль) в воде (25 мг). Реакционную смесь перемешивали при комнатной температуре в течение ночи. THF удаляли и водный слой экстрагировали DCM для удаления трифенилфосфиноксида. Водную фазу подкисляли раствором 1 н HCl до рН 2. Осадок отфильтровывали и сушили с получением 0,58 г сырой 3-(7-карбамоил-1H-индол-4-ил)бензойной кислоты в виде твердого вещества. ЖХ/МС (Таблица 1, Способ g) Время удерживания = 1.37 мин; MS m/z 281 (M+H)+.
Стадия B: 4-(3-((Цианометил)карбамоил)фенил)-1H-индол-7-карбоксамид

Смесь 3-(7-карбамоил-1H-индол-4-ил)бензойной кислоты (0,1 г, 0,357 ммоль), TBTU (0,172 г, 0,535 ммоль) и DIEA (0,249 мл, 1,43 ммоль) в DMF (5,0 мг) перемешивали при комнатной температуре в течение приблизительно 5 минут, с последующим добавлением 2-аминоацетонитрил, соляной кислоты (0,040 г, 0,43 ммоль). Реакционную смесь перемешивали при той же температуре в течение ночи. Добавляли воду и водную фазу экстрагировали EtOAc. Органический слой промывали насыщенным солевым раствором, сушили над сульфатом магния и фильтровали. Фильтрат сушили и сырой продукт очищали с помощью препаративной ВЭЖХ (таблица 1, способ i) с получением цианометил)карбамоил)фенил)-1H-индол-7-карбоксамида (0,065 г, 57%) в виде твердого вещества. ЖХ/МС (Таблица 1, способ g) Время удерживания = 1,30 мин; MS m/z 319 (M+H)+ (Btk IC50=C)
Пример № 16: 4-(3-амино-2-метилфенил)-1H-индол-7-карбоксамид
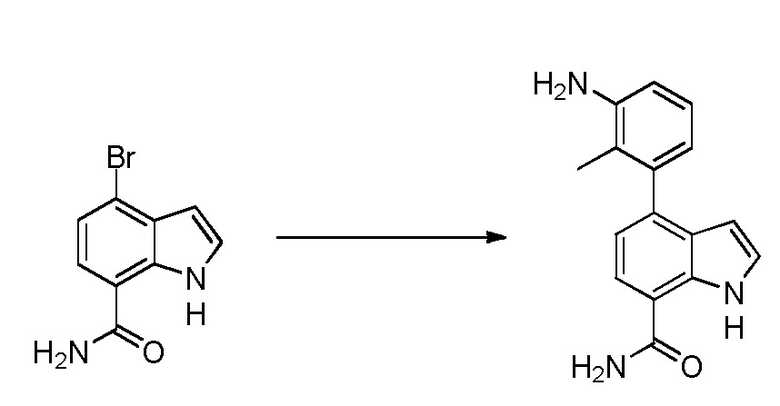
Смесь 4-бром-1H-индол-7-карбоксамида (1,28 г, 5,35 ммоль, Получение № 2), 2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина (1,37 г, 5,89 ммоль, Combi-Blocks), Na2CO3 (1,70 г, 16,06 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) (0,392 г, 0,535 ммоль) в THF (41,8 мг), MeOH (5,86 л Ll) и воды (5,86 мг) перемешивали при температуре приблизительно 70°C в течение приблизительно 16 часов в атмосфере азота. Смесь фильтровали через целит и концентрировали при пониженном давлении. Сырой продукт очищали на колонке с силикагелем с 0-10% МеОН в DCM с получением неочищенного продукта. Сырой продукт очищали на колонке с силикагелем с 0-10% МеОН в DCM с получением сырого продукта. Остаток растирали с DCM (2×с ультразвуком в течение 5 минут), фильтровали, промывали DCM и сушили при пониженном давлении с получением 4-(3-амино-2-метилфенил)-1H-индол-7-карбоксамида (0,86 г, 61%): ЖХ/МС (Таблица 1, Способ g) Время удерживания = 1,03 мин; MS m/z: 266 (M+H)+. (Btk IC50=C)
Пример № 17: 4-(3-акриламидо-2-метилфенил)-1H-пирроло[2,3-c]пиридин-7-карбоксамид
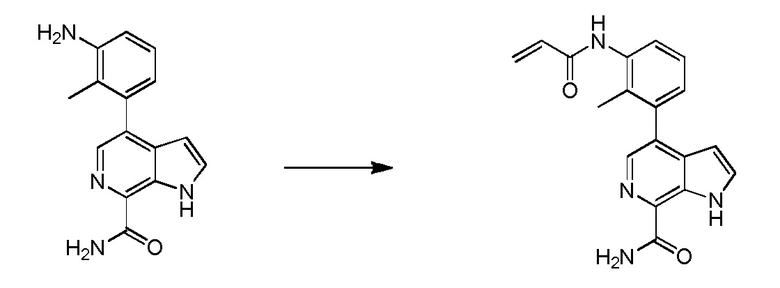
К раствору 4-(3-амино-2-метилфенил)-1H-пирроло[2,3-c]пиридин-7-карбоксамида (3,0 г, 11,3 ммоль, Пример № 2) и TEA (3,14 мл, 22,5 ммоль) в THF (113 мг) медленно добавляли акрилоилхлорид (1,01 мл, 12,4 ммоль) при 0°C. Реакционную смесь перемешивали при приблизительно 0°C в течение приблизительно 20 минут. Смесь концентрировали при пониженном давлении и добавляли воду (100 мг) и суспензию обрабатывали ультразвуком в течение 30 минут, фильтровали, промывали водой (100 мг), простым эфиром (100 мг) и сушили с получением 4-(3-акриламидо-2-метилфенил)-1H-пирроло[2,3-c]пиридин-7-карбоксамида (3,05 г, 85%): ЖХ/МС (Таблица 1, Способ f) Время удерживания = 1,27 мин; MS m/z: 321 (M+H)+. (Btk IC50=A)
Пример № 18: 4-(3-Акриламидофенил)-1H-индазол-7-карбоксамид
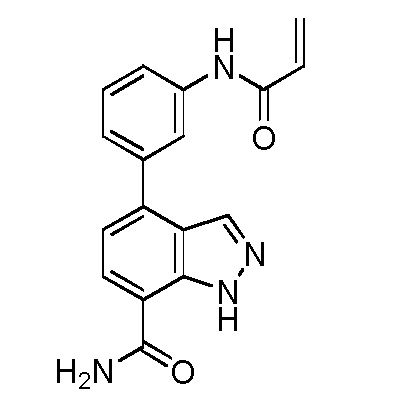
Стадия A: Метил 2-амино-4-хлор-3-метилбензоат

К смеси 2-амино-4-хлор-3-метилбензойной кислоты (5,0 г, 26,9 ммоль, Енамин) и карбоната цезия (13,2 г, 40,4 ммоль) в DMF (100 мг) добавляли йодметан (1,77 мл, 28,3 ммоль). Затем смесь перемешивали при комнатной температуре в течение приблизительно 16 часов. Добавляли воду и экстрагировали EtOAc. Органический слой промывали насыщенным солевым раствором, сушили над сульфатом магния и фильтровали. Фильтрат концентрировали и очищали с помощью хроматографии на силикагеле (5-60% EtOAc в гептане) с получением метил 2-амино-4-хлор-3-метилбензоата (4,48 г) в виде твердого вещества. ЖХ/МС (Таблица 1, Способ g) Время удерживания = 1,74 мин; MS m/z 200 (M+H)+.
Стадия B: Метил 4-хлор-1H-индазол-7-карбоксилат
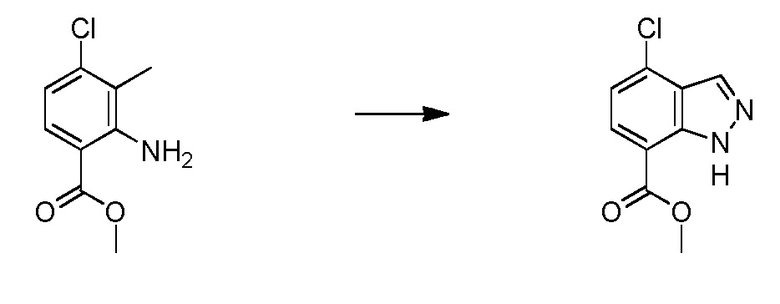
К раствору метил 2-амино-4-хлор-3-метилбензоата (4,5 г, 22,5 ммоль) в CHCl3 (100 мг) добавляли ангидрид уксусной кислоты (4,89 мл, 51,8 ммоль). Затем смесь перемешивали при комнатной температуре в течение приблизительно 2 часов, с последующим добавлением изопентилнитрита (6,68 мл, 49,6 ммоль) и ацетата калия (0,664 г, 6,76 ммоль). Реакционную смесь нагревали с обратным холодильником в течение приблизительно 18 часов. Реакционную смесь разбавляли DCM и промывали насыщенным раствором бикарбоната натрия и сушили над сульфатом магния. Фильтрат концентрировали с получением сырого метил 4-хлор-1H-индазол-7-карбоксилата (4,46 г): ЖХ/МС (Таблица 1, способ g) Время удерживания = 1,47 мин; MS m/z 211 (M+H)+.
Стадия C: 4-Хлор-1H-индазол-7-карбоксамид
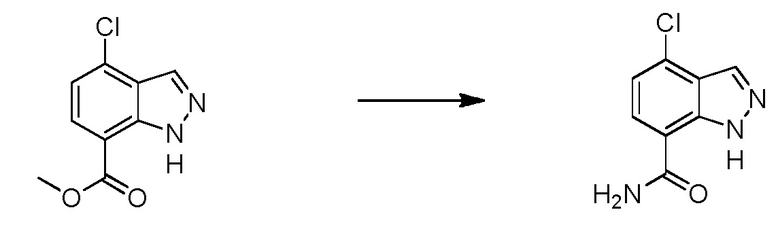
К суспензии метил 4-хлор-1H-индазол-7-карбоксилата (4,3 г, 20,4 ммоль) в 1,4-диоксане (75 мг) добавляли раствор KOH (1,69 г, 26,5 ммоль) в воде (75 мг). Реакционную смесь затем перемешивали при комнатной температуре в течение приблизительно 16 часов с получением прозрачного раствора. Растворитель удаляли и остаток обрабатывали 1 н HCl, для осаждения неочищенной кислоты, которую использовали без дальнейшей очистки. Смесь этой неочищенной кислоты (0,5 г, 2,54 ммоль), гидрохлорида N1-((этилимино)метилен)-N3,N3-диметилпропан-1,3-диамина (0,731 г, 3,82 ммоль) и HOBt (0,584 г, 3,82 ммоль) в DMF (15 мг) перемешивали при комнатной температуре в течение приблизительно 60 минут, затем добавляли аммиак (0,5 н раствора в 1,4-диоксане, 50,9 мл, 25,4 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение приблизительно 6 часов. Суспензию фильтровали и промывали EtOAc. Фильтрат концентрировали и обрабатывали водой. Осадок отфильтровывали, промывали водой и сушили на воздухе с получением 4-хлор-1H-индазол-7-карбоксамида (0,43 г) в виде твердого вещества; ЖХ/МС (Таблица 1, Способ g) Время удерживания = 1,00 мин; MS m/z 196 (M+H)+.
Стадия D: 4-(3-Аминофенил)-1H-индазол-7-карбоксамид
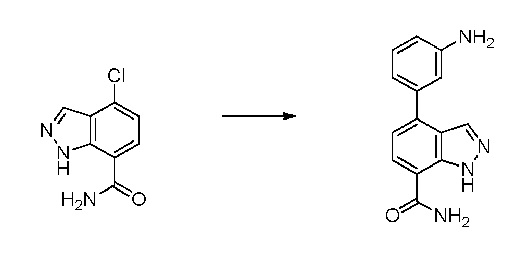
Суспензию 4-хлор-1H-индазол-7-карбоксамида (0,15 г, 0,767 ммоль), трет-бутил (3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)карбамата (0,367 г, 1,15 ммоль), карбоната цезия (0,75 г, 2,3 ммоль) в DME (4,0 мг) и воды (2,0 мг) дегазировали и продували азотом в течение 5 минут. Затем трис(дибензилиденацетон)дипалладия(0) (0,07 г, 0,077 ммоль) и 2-(дициклогексилфосфино)-2',4',6'-триизопропилдифенил (0,037 г, 0,077 ммоль) добавляли. Реакционный сосуд герметично закрывали и нагревали при помощи Biotage Initiator при приблизительно 140°C в течение приблизительно 30 минут. Смесь охлаждали до комнатной температуры и фильтровали через слой целита. Фильтрат разделяли между водой и EtOAc. Органический слой промывали насыщенным солевым раствором, сушили над сульфатом магния и фильтровали. Фильтрат концентрировали и очищали с помощью хроматографии на силикагеле (30-100% EtOAc/гептан). Этот продукт растворяли в DCM (2 мг) и обрабатывали TFA (5 мл, 64,9 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Избыток TFA и растворителя удаляли с получением сырого 4-(3-аминофенил)-1H-индазол-7-карбоксамид, трифторуксусная кислота (0,195 г) в виде твердого вещества. ЖХ/МС (Таблица 1, способ g) Время удерживания = 0,25 мин; MS m/z 253(M+H)+.
Стадия E: 4-(3-Акриламидофенил)-1H-индазол-7-карбоксамид
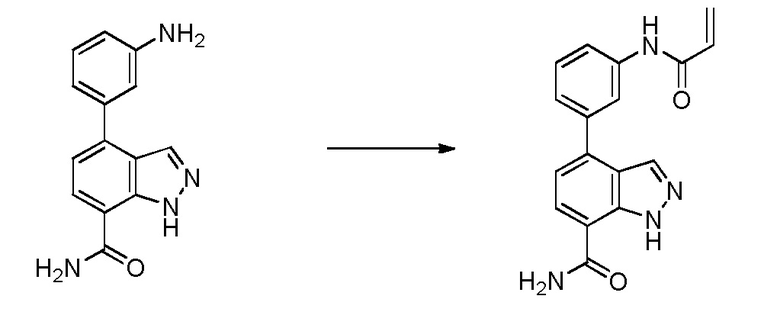
Суспензию 4-(3-аминофенил)-1H-индазол-7-карбоксамида, TFA (0,1 г, 0,27 ммоль), DIEA (0,143 мл, 0,819 ммоль) в THF (2,5 мг) охлаждали на бане со льдом и медленно добавляли акрилоилхлорид (0,026 мл, 0,31 ммоль). Через 30 минут реакционную смесь обрабатывали МеОН и перемешивали в течение приблизительно 5 минут. Растворитель затем удаляли в вакууме и остаток растирали с DCM с получением 4-(3-акриламидофенил)-1H-индазол-7-карбоксамида (56 мг) в виде твердого вещества: 1H-ЯМР (d-DMSO-d6) δ 13,17 (с, 1 H) 10,34 (с, 1 H) 8,28 (с, 1 H) 8,21 (с, 1 H) 8,17 (с, 1 H) 8,00 (д, J=7,48 Гц, 1 H) 7,73 (д, J=7,70 Гц, 1 H) 7,40-7,59 (м, 3 H) 7,30 (д, J=7,59 Гц, 1 H) 6,39-6,58 (м, 1 H) 6,17-6,36 (м, 1 H) 5,60-5,97 (м, 1 H). (Btk IC50=A)
Пример № 19: 4-(3-акриламидофенил)-1H-индазол-7-карбоксамид
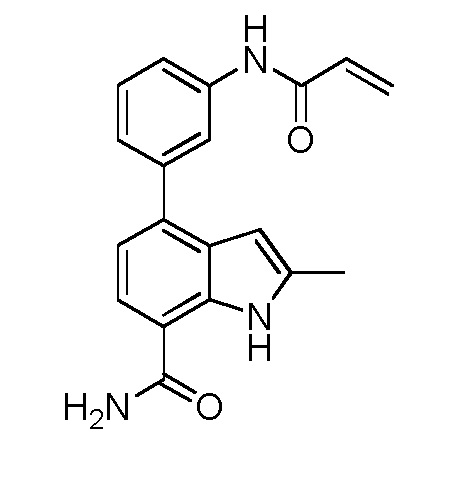
Стадия A: Метил 4-бром-2-метил-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоксилат

К раствору диизопропиламина (1,45 мл, 10,1 ммоль) и безводного THF (30 мг) добавляли раствор t-BuLi (11 мл, 11,7 ммоль) в пентане при приблизительно -78°C в атмосфере азота реакционную смесь перемешивали в течение приблизительно 30 минут. Затем раствор метил 4-бром-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоксилата (3 г, 7,81 ммоль, Получение № 10, стадия A) в безводном THF (10 мг) добавляли при приблизительно -78°C. Приблизительно через 2 часа, добавляли раствор йодометана (2,216 г, 15,61 ммоль) в безводном THF (10 мг) при приблизительно -78°C. Смесь продолжали перемешивать в течение приблизительно 2 часов при приблизительно -78°C. Реакционную смесь гасили водным NH4Cl, экстрагировали EtOAc (500 мл×3). Органическую фазу сушили над Na2SO4, концентрировали при пониженном давлении, и остаток очищали с помощью препаративной ВЭЖХ (Таблица 1, способ ao) с получением метил 4-бром-2-метил-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоксилата (1 г, 32%) в виде твердого вещества: 1H-ЯМР (CDCl3) δ 7,51-7,49 (д, J=8,0, 1H), 7,39-7,37 (д, J=8, 1H), 6,55 (с, 1H), 5,77 (с, 2H), 4,06 (с, 3H), 3,31-3,27 (м, 2H), 2,60 (с, 3H), 0,87-0,83 (м, 2H), 0,00 (с, 9H).
Стадия B: 4-бром-2-метил-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоновая кислота
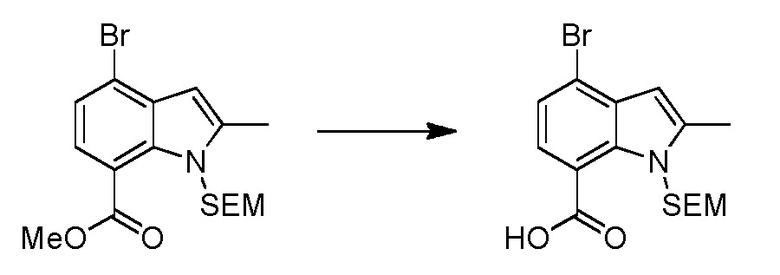
К раствору метил 4-бром-2-метил-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоксилата (0,6 г, 1,5 ммоль) в MeOH (3 мг), THF (6 мг) и воды (3 мг), LiOH (0,361 г, 15,1 ммоль) добавляли и реакционную смесь нагревали до приблизительно 45°C в течение приблизительно 3 часов. Реакционную смесь доводили до рН<3 добавлением 1 н HCl, затем экстрагировали EtOAc (300 мл×3), и органическую фазу концентрировали при пониженном давлении с получением 4-бром-2-метил-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоновой кислоты (0,5 г, 86%) в виде твердого вещества: 1H-ЯМР (DMSO-d6) δ 13,32 (с, 1H), 7,53-7,42 (м, 2H), 6,56 (с, 1H), 5,86 (с, 2H), 3,36-3,32 (м, 2H), 2,63 (с, 3H), 0,90-0,82 (м, 2H), 0,00 (с, 9H).
Стадия C: 4-бром-2-метил-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоксамид
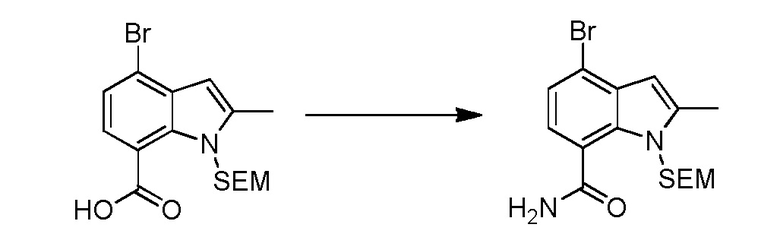
К раствору 4-бром-2-метил-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоновой кислоты (0,5 г, 1,30 ммоль) в THF (10 мг) и DCM (12 мг), HOBt (0,299 г, 1,95 ммоль) и EDCI (0,374 г, 1,95 ммоль) добавляли при приблизительно 0°C. Затем реакционную смесь перемешивали в течение приблизительно 1 часа при комнатной температуре, затем барботировали газом NH3 в течение приблизительно 20 минут, и перемешивание продолжали в течение ночи при комнатной температуре. Добавляли водный раствор NaHCO3 и смесь экстрагировали EtOAc (200 мл×3), и органическую фазу сушили над Na2SO4, концентрировали при пониженном давлении с получением 4-бром-2-метил-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоксамида (0,45 г, 90%) в виде твердого вещества: 1H-ЯМР (DMSO-d6) δ 8,10 (с, 1H), 7,67 (с, 1H), 7,36-7,34 (д, J=8, 1H), 7,20-7,18 (д, J=8, 1H), 6,46 (с, 1H), 5,74 (с, 2H), 3,46-3,38 (м, 2H), 2,56 (с, 3H), 0,90-0,83 (м, 2H), 0,00 (с, 9H).
Стадия D: 4-бром-2-метил-1H-индол-7-карбоксамид
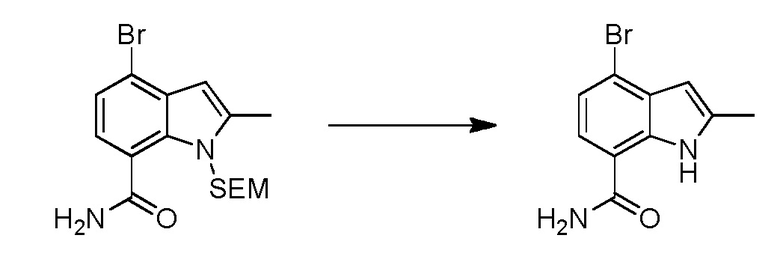
К раствору 4-бром-2-метил-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоксамида (350 мг, 0,913 ммоль) в THF (15 мг) добавляли TBAF (2,4 г, 9,13 ммоль) и этан-1,2-диамин (1,1 г, 18,3 ммоль). Смесь кипятили с обратным холодильником в течение ночи. Реакционную смесь концентрировали при пониженном давлении и остаток очищали на колонке с силикагелем с получением 4-бром-2-метил-1H-индол-7-карбоксамида (180 мг, 78%) в виде твердого вещества: 1H-ЯМР (DMSO-d6) δ 11,18 (с, 1H), 8,05 (с, 1H), 7,48-7,42 (м, 2H), 7,20-7,18 (д, J=8, 1H), 6,14 (с, 1H), 2,41 (с, 3H).
Стадия E: 4-(3-Аминофенил)-2-метил-1H-индол-7-карбоксамид
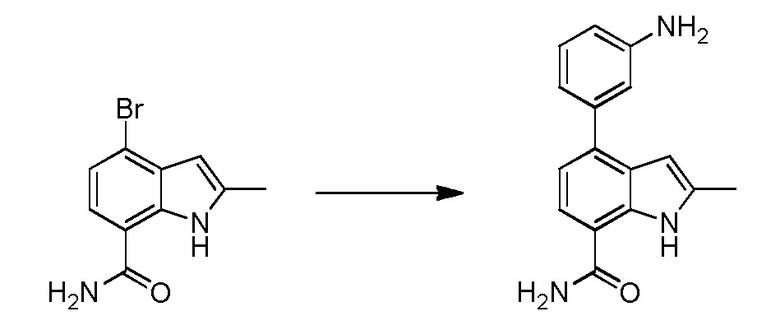
К раствору 4-бром-2-метил-1H-индол-7-карбоксамида (180 мг, 0,711 ммоль) в THF (8 мг) и воде (4 мг) и MeOH (4 мг) добавляли 3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилин (187 мг, 0,853 ммоль), Pd(dppf)Cl2 (104 мг, 0,142 ммоль) и Na2CO3 (226 мг, 2,13 ммоль), и раствор нагревали при приблизительно 90°C в течение приблизительно 2 часов. Реакционную смесь концентрировали при пониженном давлении и очищали на колонке с силикагелем с получением 4-(3-аминофенил)-2-метил-1H-индол-7-карбоксамида (80 мг, 42%) в виде твердого вещества: 1H-ЯМР (MeOD) δ 10,92 (с, 1H), 7,99 (с, 1H), 7,66-7,63 (д, J=12, 2H), 7,61 (с, 1H), 7,13-7,09 (м, 1H), 6,99-6,97 (д, J=8, 1H), 6,88 (с, 1H), 6,78-6,73 (м, 2H), 6,58-6,56 (д, J=8, 1H), 6,29 (с, 1H), 2,42 (с, 3H).
Стадия F: 4-(3-Акриламидофенил)-2-метил-1H-индол-7-карбоксамид
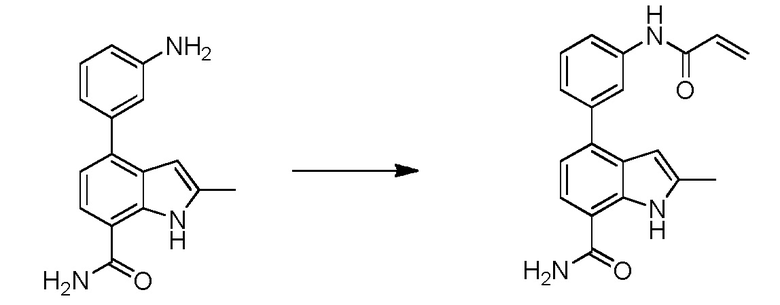
К раствору 4-(3-аминофенил)-2-метил-1H-индол-7-карбоксамида (80 мг, 0,302 ммоль) в DCM (6 мг), акрилоилхлорид (40,9 мг, 0,452 ммоль) и DIEA (0,105 мл, 0,603 ммоль) добавляли при приблизительно 0°C. Смесь перемешивали в течение приблизительно 1 часа при комнатной температуре. Реакционную смесь концентрировали при пониженном давлении и остаток очищали с помощью препаративной ВЭЖХ (Таблица 1, Способ an) с получением 4-(3-акриламидофенил)-2-метил-1H-индол-7-карбоксамида (10 мг, 11%) в виде твердого вещества: ЖХ/МС (Таблица 1, Способ j) Время удерживания = 2,07 мин; MS m/z: 320 (M+H)+. (Btk IC50=A)
Пример № 20: 4-(3-Акриламидофенил)-2-этил-1H-индол-7-карбоксамид
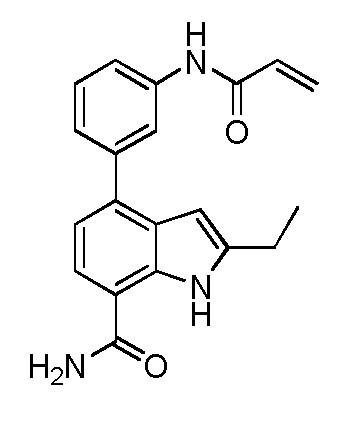
Стадия A: 4-бром-2-йод-1H-индол-7-карбоксамид

К раствору 4-бром-2-йод-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоксамида ((1,5 г, 3,03 ммоль, Получение № 24) в THF (20 мг), TBAF (15,84 г, 60,6 ммоль) и этан-1,2-диамин (1,82 г, 30,3 ммоль) добавляли, и раствор нагревали с обратным холодильником в течение ночи. Раствор концентрировали при пониженном давлении и добавляли воду (30 мг) и EtOAc (30 мг), и органическую фазу сушили и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии (петролейный эфир:EtOAc = 10:1-1:1) с получением 4-бром-2-йод-1H-индол-7-карбоксамида (700 мг, 63%): ЖХ/МС (Таблица 1, Способ k) Время удерживания = 1,91 мин; MS m/z: 367 (M+H)+.
Стадия B: 4-бром-2-винил-1H-индол-7-карбоксамид
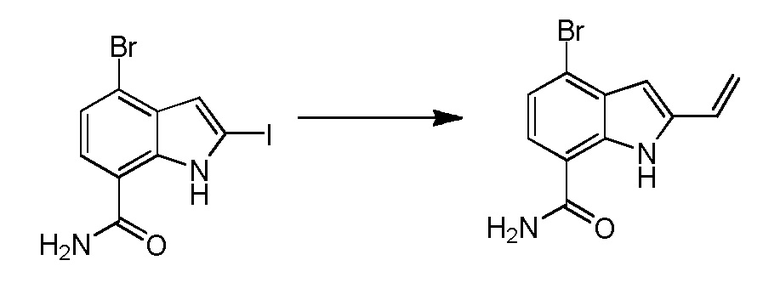
К раствору 4-бром-2-йод-1H-индол-7-карбоксамида (0,630 г, 1,726 ммоль) в 1,4-диоксане (4,5 мг) и воды (0,5 мг), CsF (0,787 г, 5,18 ммоль), Pd(PPh3)2Cl2 (0,242 г, 0,345 ммоль) и калия трифтор(винил)борат (254 мг, 1,899 ммоль) добавляли. Реакционную смесь нагревали до приблизительно 90°C в течение приблизительно 2 часов в атмосфере N2. Смесь концентрировали при пониженном давлении, и остаток очищали с помощью колоночной хроматографии с получением 4-бром-2-винил-1H-индол-7-карбоксамида (0,140 г, 31%): 1H-ЯМР (CDCl3) δ 10,36 (с, 1H), 7,2-7,12 (м, 2H), 6,72-6,65 (м, 1H), 6,50 (с, 1H), 6,25-5,78 (м, 2H), 5,69 (д, J=17,6, 1H), 5,33 (д, J=10,8, 1H).
Стадия C: 4-(3-Аминофенил)-2-винил-1H-индол-7-карбоксамид
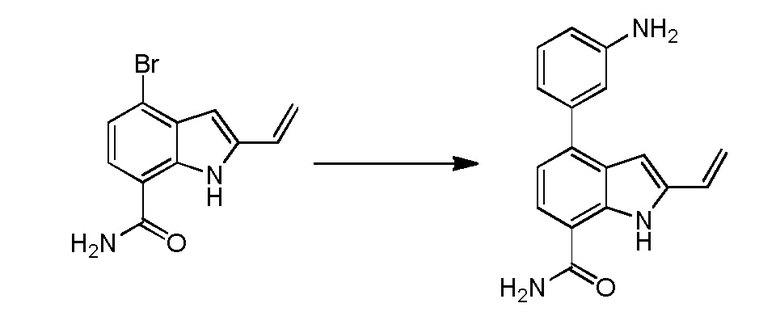
К раствору 4-бром-2-винил-1H-индол-7-карбоксамида (0,12 г, 0,45 ммоль) в THF (10 мг), воды (5 мг) и MeOH (5 мг), 3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина (119 мг, 0,543 ммоль), PdCl2(dppf) (66,2 мг, 0,091 ммоль) и Na2CO3 (144 мг, 1,358 ммоль) добавляли. Реакционную смесь нагревали при приблизительно 90°C в течение приблизительно 2 часов. Смесь концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле с получением 4-(3-аминофенил)-2-винил-1H-индол-7-карбоксамида (80 мг, 75%): ЖХ/МС (Таблица 1, Способ l) Время удерживания = 1,06 мин; MS m/z: 278 (M+H)+.
Стадия C: 4-(3-Аминофенил)-2-этил-1H-индол-7-карбоксамид
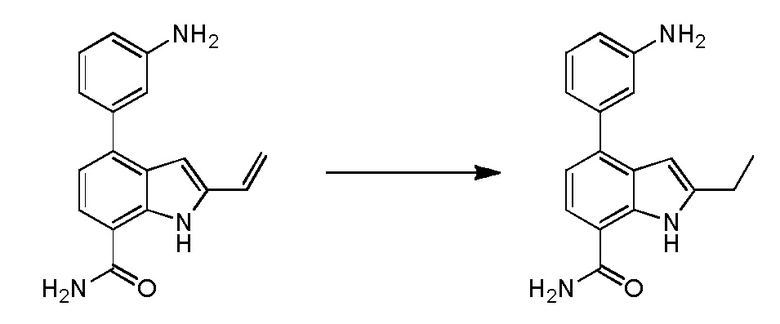
К раствору 4-(3-аминофенил)-2-винил-1H-индол-7-карбоксамида (46 мг, 0,116 ммоль) в THF (10 мг) добавляли Pd/C (10 мг, 0,094 ммоль). Смесь перемешивали в течение приблизительно 1,5 часов при комнатной температуре. Смесь фильтровали через слой целита и фильтрат концентрировали при пониженном давлении с получением 4-(3-аминофенил)-2-этил-1H-индол-7-карбоксамида (40 мг, 70%), который использовали на следующей стадии непосредственно: ЖХ/МС (Таблица 1, Способ l) Время удерживания = 1,21 мин; MS m/z: 280 (M+H)+.
Стадия D: 4-(3-Акриламидофенил)-2-этил-1H-индол-7-карбоксамид
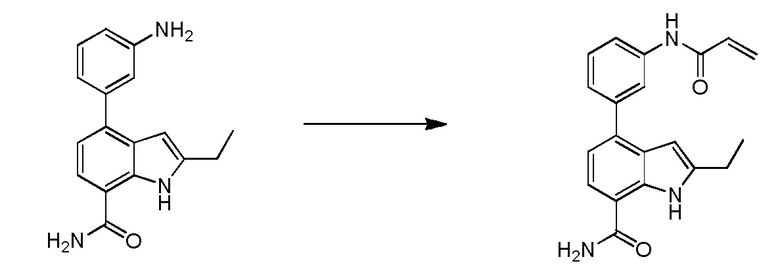
К раствору 4-(3-аминофенил)-2-этил-1H-индол-7-карбоксамида (20 мг, 0,072 ммоль) в DCM (15 мг), TEA (29 мг, 0,288 ммоль) и акрилоилхлорид (13,05 мг, 0,144 ммоль) добавляли при приблизительно 0°C. Раствор перемешивали в течение ночи при комнатной температуре. Раствор концентрировали при пониженном давлении, и остаток очищали с помощью препаративной ВЭЖХ (Таблица 1, Способ am) с получением 4-(3-акриламидофенил)-2-этил-1H-индол-7-карбоксамида (9 мг, 38%): ЖХ/МС (Таблица 1, Способ d) Время удерживания = 2,91 мин; MS m/z: 334 (M+H)+. (Btk IC50=A)
Пример № 21: 4-(3-амино-2-метилфенил)-2-(4,4-дифторциклогекс-1-енил)-1H-индол-7-карбоксамид

Стадия A: 4-бром-2-(4,4-дифторциклогекс-1-ен-1-ил)-1H-индол-7-карбоксамид
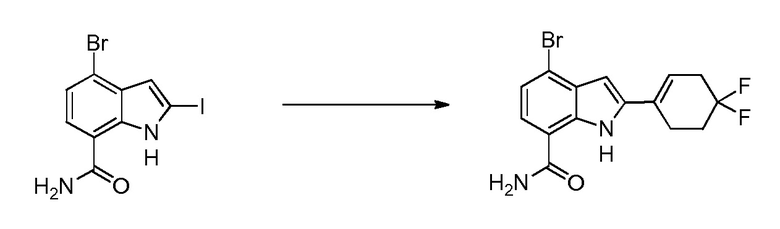
К смеси 2-(4,4-дифторциклогекс-1-ен-1-ил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (0,267 г, 1,09 ммоль, Syngene), 4-бром-2-йод-1H-индол-7-карбоксамида (0,363 г, 0,995 ммоль, Получение № 1), Na2CO3 (0,316 г, 2,98 ммоль) в THF (7 мг), MeOH (0,98 мг), и воды (0,98 мг) добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) (0,073 г, 0,099 ммоль). Смесь барботировали азотом и сосуд герметично закрывали и нагревали при температуре приблизительно 80°C в течение приблизительно 4 часов. Реакционную смесь охлаждали до комнатной температуры, фильтровали через целит и концентрировали при пониженном давлении. Остаток очищали на колонке с силикагелем с EtOAc/гексаны (30-100%) с получением сырого продукта, который затем очищали на колонке с силикагелем, элюируя градиентом 30-70% EtOAc/гексаны с получением 4-бром-2-(4,4-дифторциклогекс-1-ен-1-ил)-1H-индол-7-карбоксамида (0,25 г, 71%): ЖХ/МС (Таблица 1, Способ f) Время удерживания = 1,82 мин; MS m/z: 357 (M+H)+.
Стадия B: 4-(3-амино-2-метилфенил)-2-(4,4-дифторциклогекс-1-енил)-1H-индол-7-карбоксамид
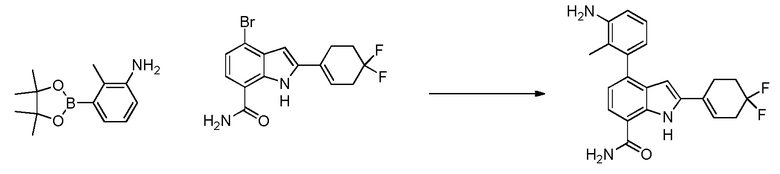
Смесь 4-бром-2-(4,4-дифторциклогекс-1-енил)-1H-индол-7-карбоксамида (0,48 г, 0,622 ммоль), 2-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина (0,203 г, 0,870 ммоль, Combi-Blocks), Na2CO3 (0,198 г, 1,865 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) (0,045 г, 0,062 ммоль) в THF (5 мг), MeOH (0,700 мг), и воды (0,700 мг) перемешивали при температуре приблизительно 70°C в течение приблизительно 16 часов в атмосфере азота. Смесь фильтровали через целит и концентрировали при пониженном давлении. Остаток пропускали через колонку с силикагелем с EtOAc/гептан (50-75%) с получением сырого продукта. Сырой продукт растирали с DCM (2× с ультразвуком в течение приблизительно 5 минут), фильтровали, промывали DCM и сушили при пониженном давлении с получением 4-(3-амино-2-метилфенил)-2-(4,4-дифторциклогекс-1-енил)-1H-индол-7-карбоксамида (134 мг, 57%): ЖХ/МС (Таблица 1, Способ f) Время удерживания = 1,36 мин; MS m/z: 382 (M+H)+. (Btk IC50=A)
Пример № 22: 4-(3-Акриламидофенил)-2-(2-этоксиэтил)-1H-индол-7-карбоксамид

Стадия A: (E)-4-бром-2-(2-этоксивинил)-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоксамид
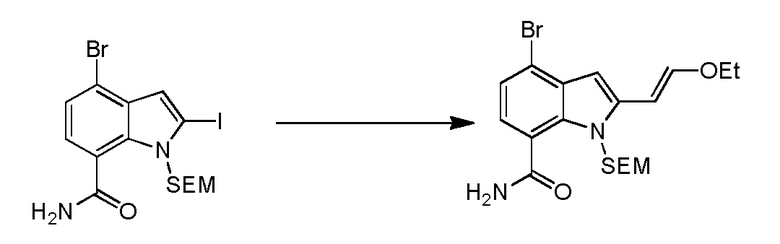
Пять реакционных сосудов загружали раствором 4-бром-2-йод-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоксамида (1 г, 2,02 ммоль, Получение № 24) в толуоле (100 мг) добавляли (E)-трибутил(2-этоксивинил)станнан (1,09 г, 3,03 ммоль), Pd(PPh3)2Cl2 (0,142 г, 0,202 ммоль) и LiCl (0,428 г, 10,1 ммоль). Смеси нагревали при температуре приблизительно 90°C в течение ночи в атмосфере N2. Все пять реакционные смеси объединяли, концентрировали при пониженном давлении, и остаток очищали на колонке с силикагелем с получением (E)-4-бром-2-(2-этоксивинил)-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоксамида (2 г, 45%) в виде желтого твердого вещества: 1H-ЯМР (DMSO-d6) δ 8,11 (с, 1H), 7,69 (с, 1H), 7,37-7,35 (д, J=8, 1H), 7,17-7,15 (д, J=8, 1H), 6,96 (с, 1H), 6,78-6,76 (д, J=8, 1H), 5,80-5,78 (д, J=8, 2H), 5,69-5,68 (д, J=4, 1H), 4,24-4,08 (м, 2H), 3,42-3,36 (м, 2H), 1,43-1,34 (м, 3H), 0,86-0,82 (м, 2H), 0,00 (с, 9H).
Стадия B: (E)-4-(3-Аминофенил)-2-(2-этоксивинил)-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоксамид
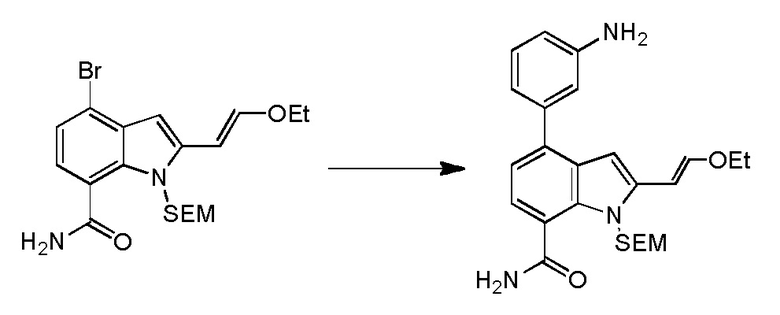
К раствору (E)-4-бром-2-(2-этоксивинил)-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоксамида (1,5 г, 3,41 ммоль) в THF (20 мг), воды (10 мг) и MeOH (10 мг) добавляли 3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилин (0,897 г, 4,10 ммоль), Pd(dppf)Cl2 (0,5 г, 0,683 ммоль) и Na2CO3 (1,085 г, 10,24 ммоль). Раствор нагревали при приблизительно 90°C в течение приблизительно 2 часов. Реакционную смесь концентрировали при пониженном давлении и очищали на колонке с силикагелем с получением (E)-4-(3-аминофенил)-2-(2-этоксивинил)-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоксамида (0,80 г, 52%): 1H-ЯМР (DMSO-d6) δ 8,06 (с, 1H), 7,62 (с, 1H), 7,30-7,22 (м, 2H), 7,15 (с, 1H), 7,10-7,08 (д, J=8, 1H), 6,93 (с, 1H), 6,83-6,81 (д, J=8, 1H), 6,68-6,65 (м, 2H), 5,82-5,80 (д, J=8, 2H), 5,67-5,66 (д, J=4, 1H), 5,28 (с, 2H), 4,18-4,06 (м, 2H), 3,43-3,37 (м, 2H), 1,39-1,33 (м, 3H), 0,86-0,82 (м, 2H), 0,00 (с, 9H).
Стадия C: 4-(3-Аминофенил)-2-(2-этоксиэтил)-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоксамид
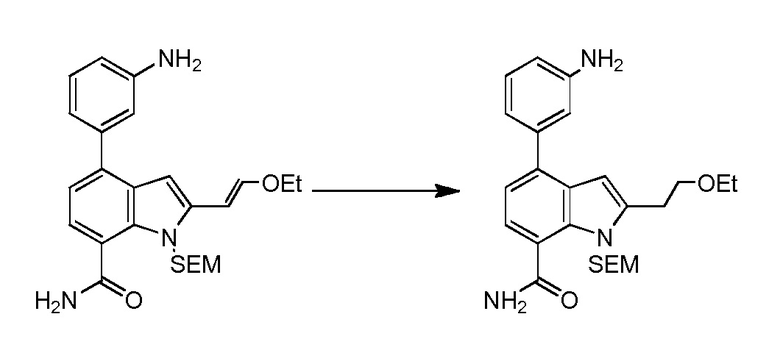
Два реакционных сосуда загружали раствором (E)-4-(3-аминофенил)-2-(2-этоксивинил)-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоксамида (400 мг, 0,886 ммоль) в MeOH (60 мг), и Pd/C (400 мг, 10%). Смеси перемешивали в течение приблизительно 1 часа при комнатной температуре в атмосфере Н2 (14 фунт на кв.дюйм). Две реакционные смеси объединяли, фильтровали и концентрировали при пониженном давлении с получением 4-(3-аминофенил)-2-(2-этоксиэтил)-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоксамида (600 мг, 75%) в виде твердого вещества, которое использовали непосредственно на следующей стадии: 1H-ЯМР (DMSO-d6) δ 8,05 (с, 1H), 7,61 (с, 1H), 7,32-7,31 (д, J=4, 1H), 7,23-7,09 (м, 2H), 6,90 (с, 1H), 6,81-6,79 (д, J=8, 1H), 6,68-6,66 (д, J=8, 1H), 6,58 (с, 1H), 5,78 (с, 2H), 5,26 (с, 2H), 3,79-3,76 (м, 2H), 3,55-3,52 (м, 2H), 3,45-3,41 (м, 2H), 3,15-3,12 (м, 2H), 1,26-1,15 (м, 3H), 0,87-0,83 (м, 2H), 0,01 (с, 9H).
Стадия D: 4-(3-Аминофенил)-2-(2-этоксиэтил)-1H-индол-7-карбоксамид
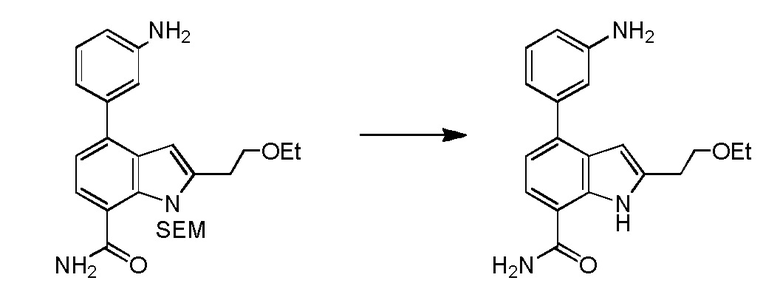
К раствору 4-(3-аминофенил)-2-(2-этоксиэтил)-1-((2-(триметилсилил)этокси)метил)-1H-индол-7-карбоксамида (500 мг, 1,10 ммоль) в THF (20 мг) добавляли TBAF (2,88 г, 11,0 ммоль) и этан-1,2-диамин (1,33 г, 22,0 ммоль). Смесь перемешивали в течение приблизительно 5 часов при приблизительно 80°C. Реакционную смесь концентрировали при пониженном давлении, и остаток очищали на колонке с силикагелем с получением 4-(3-аминофенил)-2-(2-этоксиэтил)-1H-индол-7-карбоксамида (267 мг, 75%) в виде твердого вещества: 1H-ЯМР (DMSO-d6) δ 11,09 (с, 1H), 8,12 (с, 1H), 7,76-7,74 (д, J=8, 1H), 7,46-7,44 (д, J=8, 1H), 7,24-7,19 (м, 1H), 7,09-7,07 (д, J=8, 1H), 6,96 (с, 1H), 6,87-6,85 (д, J=8, 1H), 6,67-6,66 (д, J=4, 1H), 6,45 (с, 1H), 5,25 (с, 2H), 3,76-3,73 (м, 2H), 3,59-3,54 (м, 2H), 3,13-3,09 (м, 2H), 1,27-1,23 (м, 3H).
Стадия E: 4-(3-Акриламидофенил)-2-(2-этоксиэтил)-1H-индол-7-карбоксамид
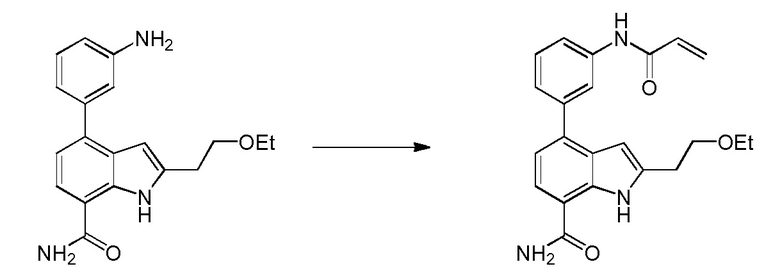
Два реакционных сосуда загружали раствором 4-(3-аминофенил)-2-(2-этоксиэтил)-1H-индол-7-карбоксамида (60 мг, 0,186 ммоль) в DCM (2 мг). Добавляли DIEA (0,065 мл, 0,371 ммоль) и акрилоилхлорид (25,2 мг, 0,278 ммоль) и смеси перемешивали в течение приблизительно 1 часа при комнатной температуре. Две реакционные смеси объединяли, концентрировали при пониженном давлении, и остаток очищали с помощью препаративной ВЭЖХ (Таблица 1, Способ w) с получением 4-(3-акриламидофенил)-2-(2-этоксиэтил)-1H-индол-7-карбоксамида (21,6 мг, 26,4%) в виде твердого вещества: ЖХ/МС (Таблица 1, Способ d) Время удерживания = 2,95 мин; MS m/z: 378 (M-H)-. (Btk IC50=A)
Пример № 23: 4-(3-Акриламидофенил)-2-(2-гидроксиэтил)-1H-индол-7-карбоксамид
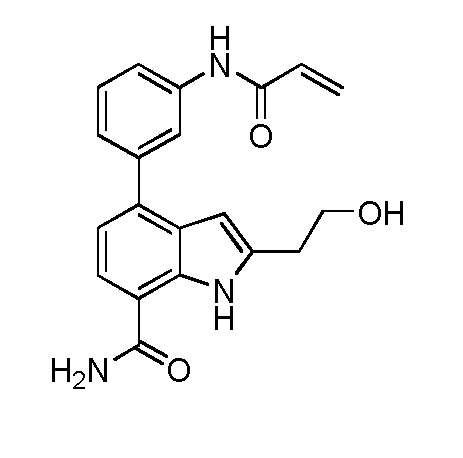
Стадия A: 4-(3-Аминофенил)-2-(2-гидроксиэтил)-1H-индол-7-карбоксамид
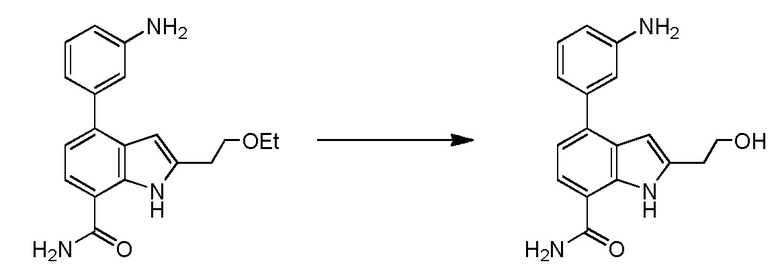
Два реакционных сосуда загружали раствором 4-(3-аминофенил)-2-(2-этоксиэтил)-1H-индол-7-карбоксамида (100 мг, 0,309 ммоль, Пример № 22, Стадия D) в DCM (10 мг) по каплям добавляли трибромборан (387 мг, 1,55 ммоль) при приблизительно -78°C. Смеси перемешивают в течение приблизительно 2 часов при приблизительно 0°C. Две реакционные смеси объединяли и добавляли водный раствор NaHCO3 и смесь экстрагировали DCM (100 мл×3). Органическую фазу сушили над Na2SO4, концентрировали при пониженном давлении с получением 4-(3-аминофенил)-2-(2-гидроксиэтил)-1H-индол-7-карбоксамида (160 мг, 88%) в виде желтого твердого вещества: 1H-ЯМР (DMSO-d6) δ 10,96 (с, 1H), 8,04 (с, 1H), 7,67-7,65 (д, J=8, 1H), 7,38-7,34 (д, J=16, 1H), 7,16-7,12 (м, 1H), 7,01-6,99 (д, J=8, 1H), 6,91 (с, 1H), 6,81-6,80 (д, J=4, 1H), 6,62-6,59 (д, J=12, 1H), 6,36 (с, 1H), 5,33 (с, 2H), 4,87 (с, 1H), 3,73-3,70 (м, 2H), 2,96-2,93 (м, 2H).
Стадия B: 4-(3-Аминофенил)-2-(2-гидроксиэтил)-1H-индол-7-карбоксамид
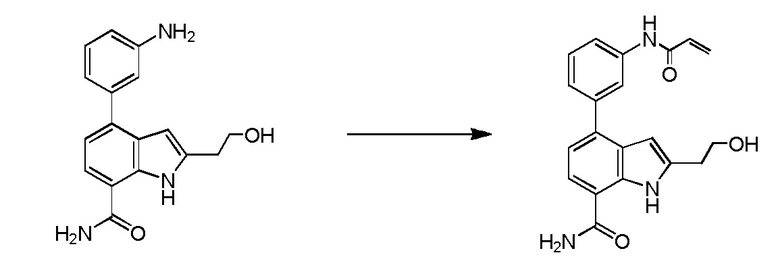
К раствору 4-(3-аминофенил)-2-(2-гидроксиэтил)-1H-индол-7-карбоксамида (40 мг, 0,135 ммоль) в пиридине (4 мг) добавляли EDCI (31 мг, 0,163 ммоль) и акриловую кислоту (9,8 мг, 0,135 ммоль). Смесь перемешивали в течение приблизительно 3 часов при приблизительно 110°C. Реакционную смесь концентрировали при пониженном давлении и остаток очищали с помощью препаративной ВЭЖХ (Таблица 1, Способ al) с получением 4-(3-акриламидофенил)-2-(2-гидроксиэтил)-1H-индол-7-карбоксамида (4,5 мг, 10%) в виде твердого вещества: ЖХ/МС (Таблица 1, Способ j) Время удерживания = 2,46 мин; MS m/z: 350 (M+H)+. (Btk IC50=A)
Пример № 24: 4-((1-акрилоилазетидин-3-ил)(метил)амино)-1H-индол-7-карбоксамид
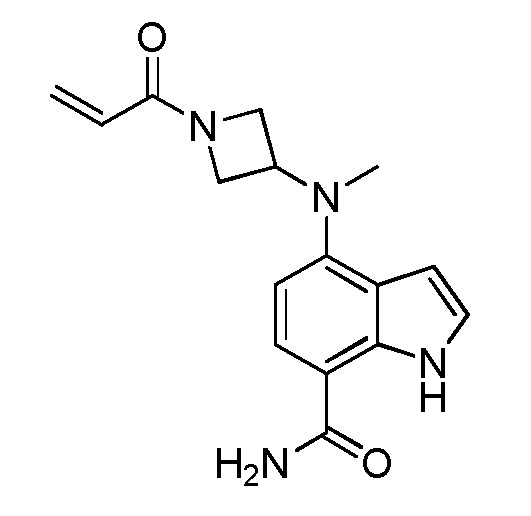
Стадия A: трет-Бутил-3-((7-циано-1H-индол-4-ил)амино)азетидин-1-карбоксилат
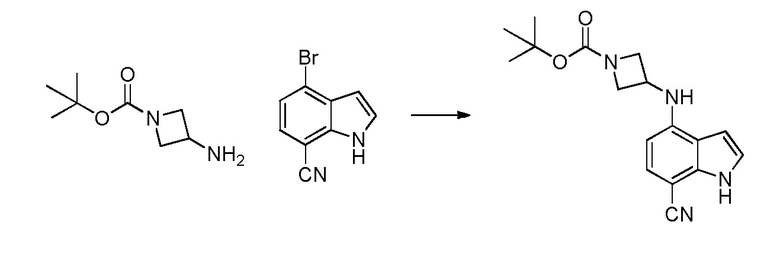
В 4 мл реакционный сосуд, добавляли 4-бром-1H-индол-7-карбонитрил (200 мг, 0,905 ммоль, Sinova), хлор[2-(дициклогексилфосфино)-3,6-диметокси-2',4',6'-триизопропил-1,1'-дифенил][2-(2-аминоэтил)фенил]палладий (II) (9,03 мг, 0,011 ммоль), и дициклогексил(2',4',6'-триизопропил-3,6-диметокси-[1,1'-дифенил]-2-ил)фосфин (6,07 мг, 0,011 ммоль). Твердую смесь удаляли и заполняли азотом. Добавляли бис(триметилсилил)амид лития (2,17 мл, 2,17 ммоль) с последующим добавлением трет-бутил-3-аминоазетидин-1-карбоксилата (170 мкл, 1,09 ммоль). Реакционную смесь нагревали при приблизительно 65°C в течение приблизительно 2,5 часов. Реакционную смесь гасили добавлением нескольких капель 1 н HCl и разбавляли EtOAc (10 мг). Слой EtOAc промывали насыщенным водным раствором NaHCO3 и сушили над MgSO4, фильтровали и концентрировали под вакуумом. Неочищенный материал очищали флэш-хроматографией, используя градиент 5-40% EtOAc в гептане с получением трет-бутил-3-((7-циано-1H-индол-4-ил)амино)азетидин-1-карбоксилата (160 мг, 57%); ЖХ/МС (Таблица 1, Способ as) Время удерживания = 2,13 мин.; MS m/z: 311 (M-H)-.
Стадия B: трет-Бутил 4-((1-(трет-бутоксикарбонил)азетидин-3-ил)амино)-7-циано-1H-индол-1-карбоксилат
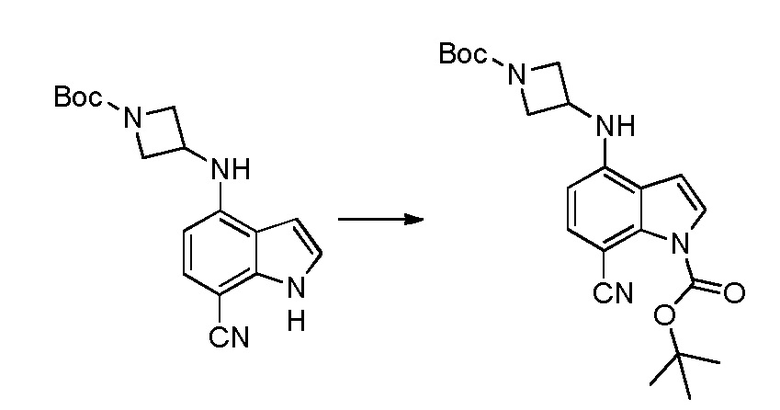
В 100 мл круглодонную колбу добавляли трет-бутил 3-((7-циано-1H-индол-4-ил)амино)азетидин-1-карбоксилат (200 мг, 0,640 ммоль) в MeCN (5 мг) с получением коричневого раствора. Добавляли DMAP (15,6 мг, 0,128 ммоль) и BOC2O (419 мг, 1,92 ммоль). Реакционную смесь перемешивали в течение приблизительно 18 часов при комнатной температуре. Реакционную смесь разбавляли водой (2 мг) и EtOAC (3 мг). Всю суспензию фильтровали и промывали EtOAc (5 мг). Белый осадок собирали высушиванием в вакуумной печи при температуре приблизительно 70°C в течение приблизительно 2 часов с получением трет-бутил 4-((1-(трет-бутоксикарбонил)азетидин-3-ил)амино)-7-циано-1H-индол-1-карбоксилата (154 мг, 58,3%). ЖХ/МС (Таблица 1, Способ as) Время удерживания = 2,54 мин.; MS m/z: 411 (M-H)-.
Стадия C: трет-Бутил-4-((1-(трет-бутоксикарбонил)азетидин-3-ил)(метил)амино)-7-циано-1H-индол-1-карбоксилат
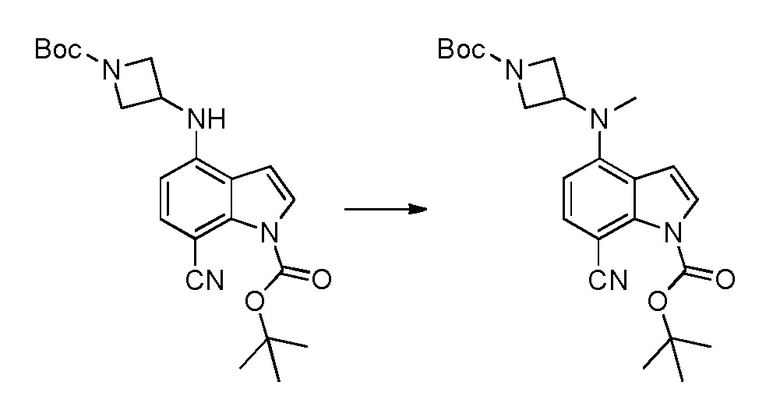
В 4 мл реакционный сосуд, гидрид натрия (23,9 мг, 0,598 ммоль, 60% дисперсия в минеральном масле) в DMF (1 мг) добавляли с получением белой суспензии. Реакционную смесь охлаждали до приблизительно 0°C и трет-бутил 4-((1-(трет-бутоксикарбонил)азетидин-3-ил)амино)-7-циано-1H-индол-1-карбоксилат (145 мг, 0,352 ммоль) добавляли в виде раствора в DMF (4 мг). Через приблизительно 30 минут добавляли йодметан (33 мкл, 0,528 ммоль). Перемешивание продолжали при 0°C в течение приблизительно 1 часа. Реакцию гасили водой (15 мг) и экстрагировали EtOAc (20 мг). Органический слой сушили над MgSO4, фильтровали и концентрировали. Вещество очищали флэш-хроматографией с использованием градиента 0-25% EtOAc/гептан в течение 5 мин, затем выдерживали при 25% EtOAc/гептан в течение 5 минут, с получением сырого трет-бутил-4-((1-(трет-бутоксикарбонил)азетидин-3-ил)(метил)амино)-7-циано-1H-индол-1-карбоксилата (148 мг, 71,1%); ЖХ/МС (Таблица 1, Способ as) Время удерживания = 2,71 мин.; MS m/z: 427 (M+H)+.
Стадия D: трет-Бутил 3-((7-карбамоил-1H-индол-4-ил)(метил)амино)азетидин-1-карбоксилат
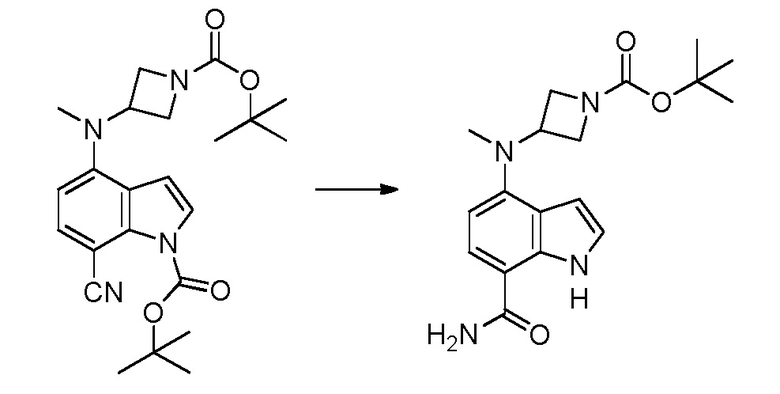
К раствору трет-бутил 4-((1-(трет-бутоксикарбонил)азетидин-3-ил)(метил)амино)-7-циано-1H-индол-1-карбоксилата (148 мг, 0,250 ммоль) в этаноле (2 мг)/DMSO (0,500 мг) добавляли пероксид водорода (0,515 мл, 5,04 ммоль) и NaOH (1 M, 0,515 мл, 0,515 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение приблизительно 2 часов. К реакционной смеси добавляли воду (5 мг) и осадок собирали фильтрованием, промывали водой (5 мг) и сушили в вакуумной печи при приблизительно 70°C в течение приблизительно 2 часов с получением трет-бутил 3-((7-карбамоил-1H-индол-4-ил)(метил)амино)азетидин-1-карбоксилата (60 мг, 52%); ЖХ/МС (Таблица 1, способ as) Время удерживания = 1,97 мин.; MS m/z: 345 (M+H)+.
Стадия E: 4-(Азетидин-3-ил(метил)амино)-1H-индол-7-карбоксамид
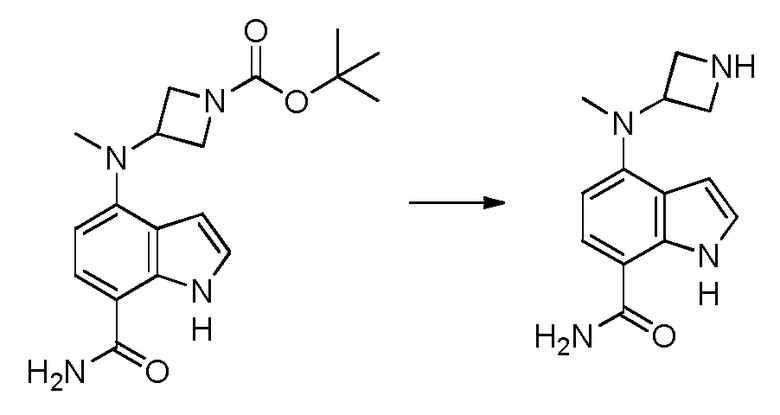
В 4 мл реакционный сосуд добавляли трет-бутил 3-((7-карбамоил-1H-индол-4-ил)(метил)амино)азетидин-1-карбоксилат (60 мг, 0,129 ммоль) в 1,4-диоксане (2 мг) с получением не совсем белого раствора. Добавляли 4 M HCl в диоксане (0,129 мл, 0,516 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение приблизительно 2 часов. Смесь нагревали при приблизительно 50°C в течение приблизительно 2 часов. Добавляли дополнительное количество 4 M HCl в диоксане (0,129 мл, 0,516 ммоль) и перемешивание продолжали при приблизительно 50°C в течение приблизительно 45 минут. Реакционную смесь фильтровали и промывали DCM с получением осадка. Осадок растворяли в воде (2 мг) и подщелачивали с добавлением нескольких капель 5 н водного раствора NaOH. Водный слой затем экстрагировали DCM (2×7 мг) и EtOAC (2×8 мг). Органические слои объединяли и сушили над MgSO4, фильтровали и концентрировали с получением 4-(азетидин-3-ил(метил)амино)-1H-индол-7-карбоксамида (29 мг, 66%); ЖХ/МС (Таблица 1, Способ as) Время удерживания = 0,73 мин; MS m/z: 245 (M+H)+.
Стадия F: 4-((1-акрилоилазетидин-3-ил)(метил)амино)-1H-индол-7-карбоксамид
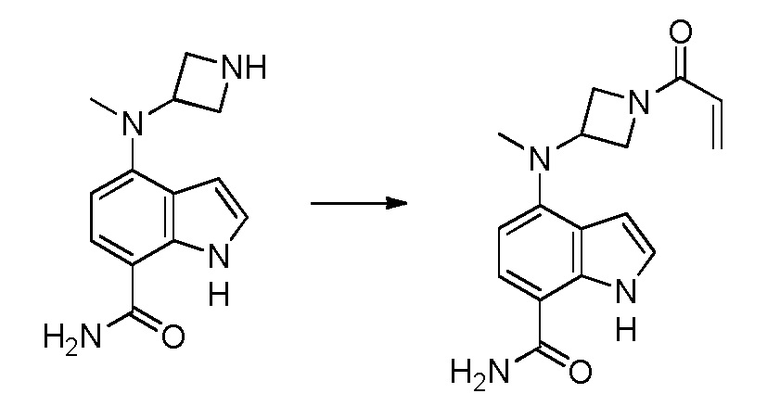
Сосуд заполняли 4-(азетидин-3-ил(метил)амино)-1H-индол-7-карбоксамидом (28 мг, 0,083 ммоль) и N-этил-N-изопропилпропан-2-амином (65 мкл, 0,373 ммоль) в DCM (5 мг). Смесь охлаждали до 0°C на бане со льдом. Добавляли акрилоилхлорид (7,38 мкл, 0,091 ммоль) и смесь перемешивали в течение 20 минут. Реакционную смесь концентрировали. Вещество очищали флэш-хроматографией с использованием градиента 1,0-3,3% MeOH/DCM в течение 7 минут, затем выдерживали при 3,3% в течение 5 минут с получением 4-((1-акрилоилазетидин-3-ил)(метил)амино)-1H-индол-7-карбоксамида (10,5 мг, 43%); ЖХ/МС (Таблица 1, Способ a) Время удерживания = 1,31 мин.; MS m/z: 299 (M+H)+. (Btk IC50=A)
Пример № 25: 4-(1-Акрилоилпиперидин-3-ил)-1H-индол-7-карбоксамид
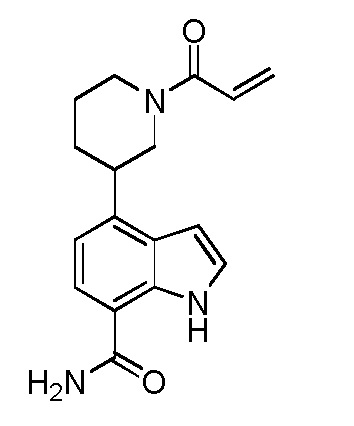
Стадия A: трет-Бутил 3-(7-карбамоил-1H-индол-4-ил)-5,6-дигидропиридин-1(2H)-карбоксилат
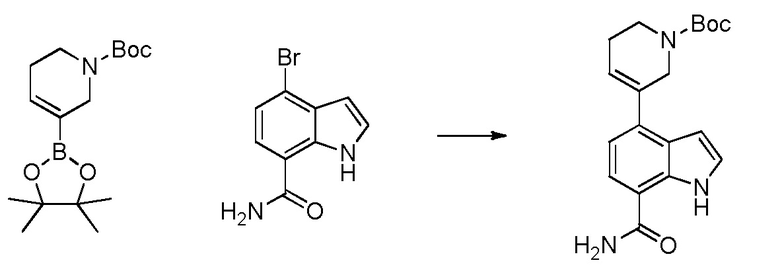
В сосуд 20 мл загружали 4-бром-1H-индол-7-карбоксамид (300 мг, 1,255 ммоль), трет-бутил 3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2H)-карбоксилат (466 мг, 1,506 ммоль), (1,1-бис(дифенилфосфино)ферроцен)дихлорпалладий (92 мг, 0,125 ммоль) и карбонат натрия (399 мг, 3,76 ммоль). К твердой смеси добавляли THF (6 мг):MeOH (0,840 мг):воду (0,840 мг). Суспензию продували азотом в течение приблизительно 5 минут. Реакционную смесь нагревали при приблизительно 70°C в течение ночи. Реакционную смесь фильтровали через слой целита, концентрировали и очищали на колонке с силикагелем (30-60% EtOAc/гептан) с получением трет-бутил 3-(7-карбамоил-1H-индол-4-ил)-5,6-дигидропиридин-1(2H)-карбоксилата (355 мг, 83%); ЖХ/МС (Таблица 1, Способ as) Время удерживания = 2,14 мин.; MS m/z: 340 (M-H)-.
Стадия B: трет-Бутил 3-(7-карбамоил-1H-индол-4-ил)пиперидин-1-карбоксилат
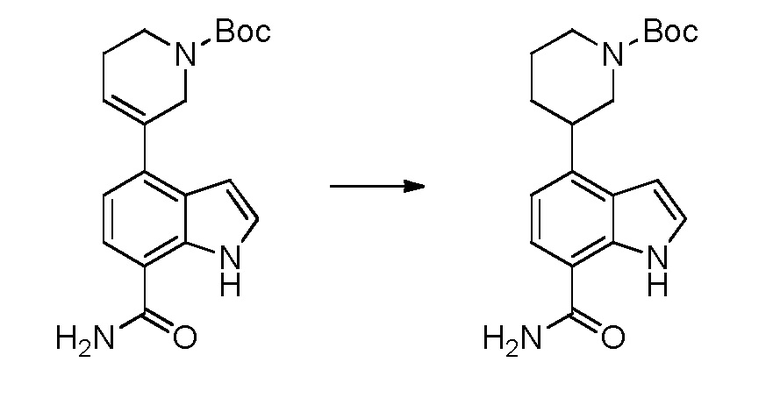
В сосуд загружали трет-бутил 3-(7-карбамоил-1H-индол-4-ил)-5,6-дигидропиридин-1(2H)-карбоксилат (355 мг, 1,04 ммоль) и палладий (55,3 мг, 0,520 ммоль). Добавляли этилацетат (10 мг) под вакуумом и смесь перемешивали в атмосфере баллонного H2 при комнатной температуре в течение приблизительно 5 часов. Реакционную смесь фильтровали через слой целита и промывали MeOH (20 мг) и EtOAc (30 мг). Фильтрат концентрировали при пониженном давлении с получением трет-бутил 3-(7-карбамоил-1H-индол-4-ил)пиперидин-1-карбоксилата (357 мг, 100%); ЖХ/МС (Таблица 1, Способ as) Время удерживания = 2,14 мин.; MS m/z: 342 (M-H)-.
Стадия C: 4-(Пиперидин-3-ил)-1H-индол-7-карбоксамид
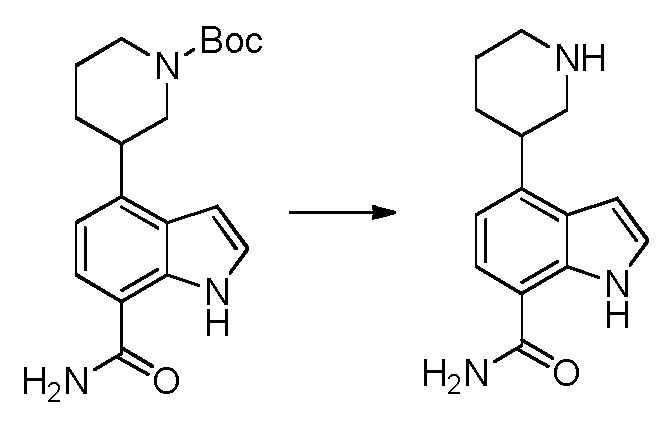
В сосуд загружали метанол (5 мг) и охлаждали до 0°C. Ацетилхлорид (0,828 мл, 11,6 ммоль) добавляли по каплям, и ледяную баню удаляли. Смесь перемешивали при комнатной температуре в течение приблизительно 25 минут. Затем раствор добавляли к трет-бутил 3-(7-карбамоил-1H-индол-4-ил)пиперидин-1-карбоксилату (100 мг, 0,291 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение приблизительно 4 часов. Смесь концентрировали в вакууме. Остаток растворяли в воде (10 мг) и промывали EtOAc (7 мг). Водный слой подщелачивали несколькими каплями 50% масс./масс. раствора NaOH и экстрагировали EtOAC (12 мг). Слой EtOAc сушили над MgSO4, фильтровали и концентрировали с получением 4-(пиперидин-3-ил)-1H-индол-7-карбоксамида (40 мг, 56%); материал использовали сырым на следующей стадии без дальнейшего исследования.
Стадия D: 4-(1-Акрилоилпиперидин-3-ил)-1H-индол-7-карбоксамид
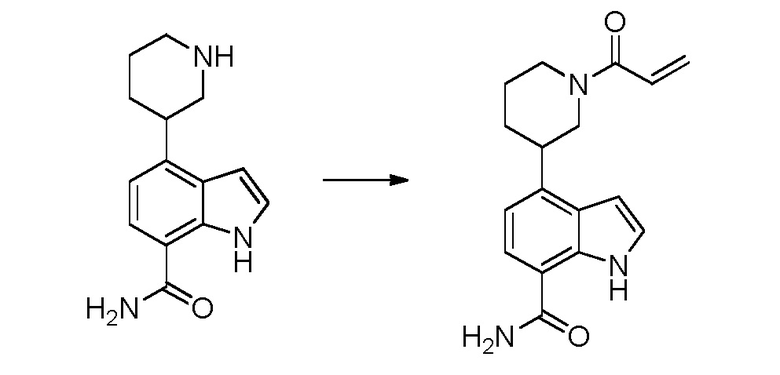
В сосуд загружали 4-(пиперидин-3-ил)-1H-индол-7-карбоксамид (40 мг, 0,164 ммоль) и N-этил-N-изопропилпропан-2-амин (43 мкл, 0,247 ммоль) в DCM (5 мг). Смесь охлаждали до 0°C. Добавляли акрилоилхлорид (14,69 мкл, 0,181 ммоль) и смесь перемешивали в течение приблизительно 20 минут. Реакционную смесь концентрировали. Вещество очищали на колонке с силикагелем с использованием градиента 1,0-5,5% MeOH/CH2Cl2 в течение 10 минут; с получением 4-(1-акрилоилпиперидин-3-ил)-1H-индол-7-карбоксамида (41 мг, 84%); ЖХ/МС (Таблица 1, способ a) Время удерживания = 1,53 мин.; MS m/z: 298 (M+H)+. (Btk IC50=B)
Пример № 26: 4-(1-Акрилоилпиперидин 3-ил)-2-(1-метил-1H-пиразол-4-ил)-1H-индол-7-карбоксамид
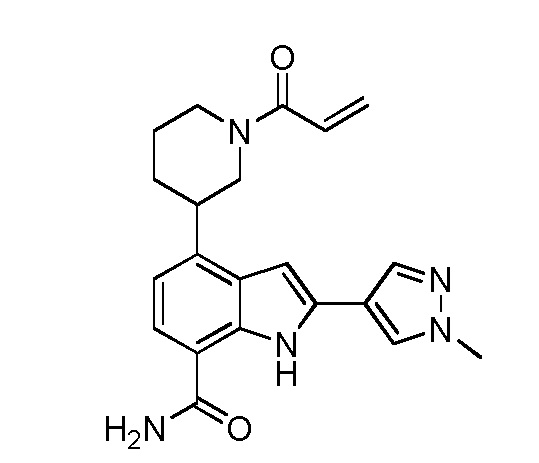
Стадия A: трет-Бутил 3-(7-карбамоил-2-(1-метил-1H-пиразол-4-ил)-1H-индол-4-ил)-5,6-дигидропиридин-1(2H)-карбоксилат
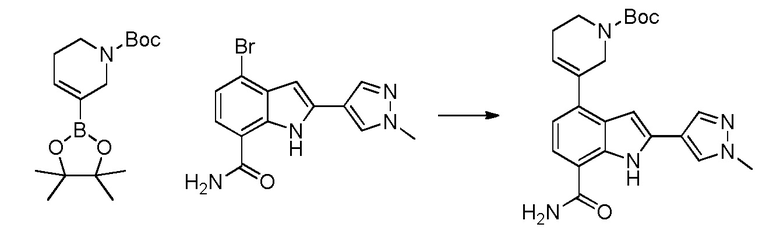
В сосуд 20 мл загружали 4-бром-2-(1-метил-1H-пиразол-4-ил)-1H-индол-7-карбоксамид (216 мг, 0,677 ммоль, Получение № 10), трет-бутил 3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2H)-карбоксилат (251 мг, 0,812 ммоль), (1,1-бис(дифенилфосфино)ферроцен)дихлорпалладий (1:1) комплекс с DCM (55,3 мг, 0,068 ммоль) и карбонат натрия (215 мг, 2,03 ммоль). К твердой смеси добавляли THF (3 мг):MeOH (0,420 мг):воду (0,420 мг). Суспензию продували N2 в течение приблизительно 5 минут. Реакционную смесь нагревали при приблизительно 70°C в течение ночи. Реакционную смесь фильтровали через слой целита, концентрировали и очищали с помощью колонки с силикагелем (0-2% MeOH/DCM) с получением трет-бутил 3-(7-карбамоил-2-(1-метил-1H-пиразол-4-ил)-1H-индол-4-ил)-5,6-дигидропиридин-1(2H)-карбоксилата (227 мг, 80%); ЖХ/МС (Таблица 1, Способ as) Время удерживания = 2,09 мин.; MS m/z: 422 (M+H)+.
Стадия B: трет-бутил 3-(7-карбамоил-2-(1-метил-1H-пиразол-4-ил)-1H-индол-4-ил)пиперидин-1-карбоксилат
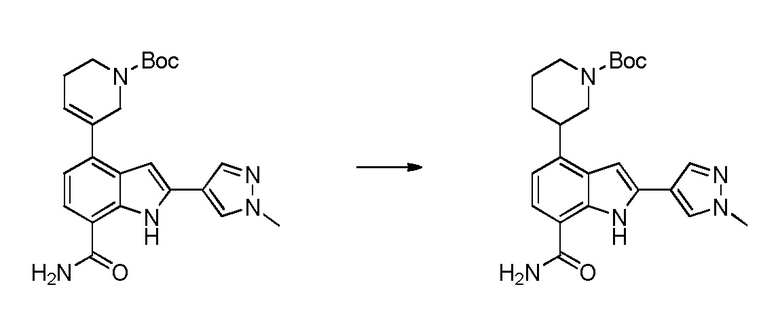
В сосуд загружали трет-бутил 3-(7-карбамоил-2-(1-метил-1H-пиразол-4-ил)-1H-индол-4-ил)-5,6-дигидропиридин-1(2H)-карбоксилат (227 мг, 0,539 ммоль) и 10% палладий на угле (28,7 мг, 0,027 ммоль). Этилацетат (5 мг) добавляли в вакууме и смесь перемешивали в атмосфере баллонного H2 при комнатной температуре в течение приблизительно 5 часов. Реакционную смесь фильтровали через слой целита и промывали МеОН (20 мг) и EtOAc (30 мг). Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (177 мг, 78%); ЖХ/МС (Способ as) Время удерживания = 2,08 мин.; MS m/z: 424 (M+H)+.
Стадия C: 2-(1-метил-1H-пиразол-4-ил)-4-(пиперидин-3-ил)-1H-индол-7-карбоксамид

В сосуд загружали MeOH (2 мг) и охдаждали до 0°C. Ацетилхлорид (0,151 мл, 2,12 ммоль) добавляли по каплям, и ледяную баню удаляли. Смесь перемешивали при комнатной температуре в течение приблизительно 25 минут. Затем раствор добавляли к трет-бутил 3-(7-карбамоил-2-(1-метил-1H-пиразол-4-ил)-1H-индол-4-ил)пиперидин-1-карбоксилату (30 мг, 0,071 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь концентрировали под вакуумом. Остаток растворяли в воде (3 мг) и промывали DCM (3 мг). Водный слой подщелачивали добавлением нескольких капель 5 н NaOH с получением суспензии, к которой добавляли DCM. Слой DCM отделяли. Водный слой образовывал осадок, который собирали фильтрованием и промывали смесью DCM/EtOAC/MeOH (1:1:1) (6 мг). Это фильтрат объединяли со слоем DCM и концентрировали в вакууме с получением 2-(1-метил-1H-пиразол-4-ил)-4-(пиперидин-3-ил)-1H-индол-7-карбоксамида (18 мг, 79%); ЖХ/МС (Таблица 1, Способ as) Время удерживания = 1,03 мин.; MS m/z: 324 (M+H)+.
Стадия D: 4-(1-Акрилоилпиперидин 3-ил)-2-(1-метил-1H-пиразол-4-ил)-1H-индол-7-карбоксамид
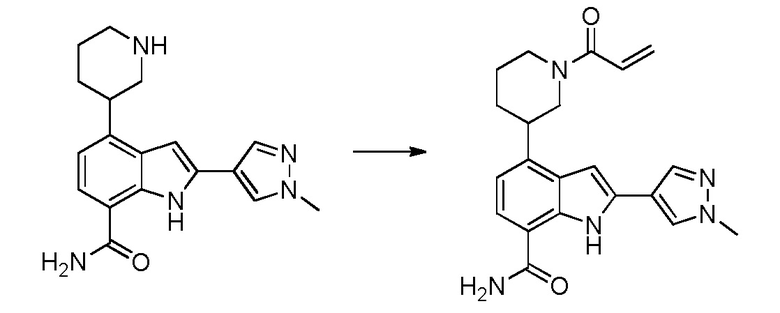
В сосуд загружали 2-(1-метил-1H-пиразол-4-ил)-4-(пиперидин-3-ил)-1H-индол-7-карбоксамид (18 мг, 0,056 ммоль) и N-этил-N-изопропилпропан-2-амин (0,044 мл, 0,250 ммоль) в DCM (5 мг). Смесь охлаждали до 0°C на бане со льдом. Добавляли акрилоилхлорид (4,97 мкл, 0,061 ммоль) и смесь перемешивали в течение приблизительно 20 минут. Реакционную смесь концентрировали. Вещество очищали на колонке с силикагелем (2,0-6,5% MeOH/DCM) с получением 4-(1-акрилоилпиперидин 3-ил)-2-(1-метил-1H-пиразол-4-ил)-1H-индол-7-карбоксамида (9 мг, 43%); ЖХ/МС (Таблица 1, способ a) Время удерживания = 1,56 мин.; MS m/z: 378 (M+H)+. (Btk IC50=A)
Пример № 27: 4-((1-акрилоилазетидин-3-ил)окси)-1H-индол-7-карбоксамид
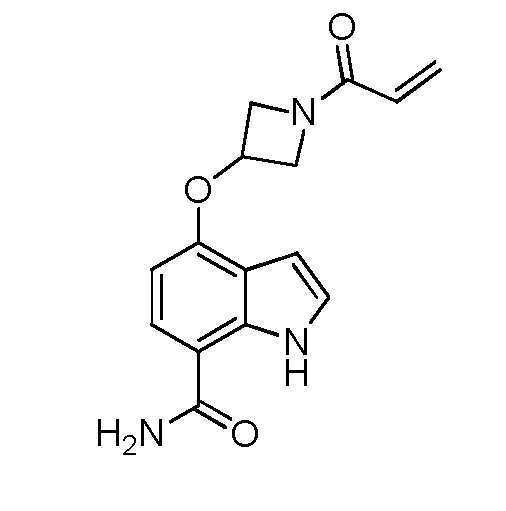
Стадия A: трет-бутил 3-(4-бром-3-нитрофенокси)азетидин-1-карбоксилат
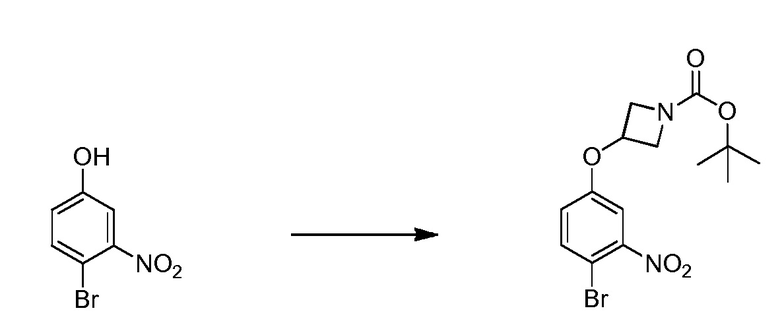
Карбонат цезия (2,038 г, 6,26 ммоль) добавляли в DMF (12 мг), получая белую суспензию. Молекулярные сита (4 Å, 8-12 меш, гранулы, 100 мг) 4-бром-3-нитрофенол (1 г, 4,59 ммоль) и трет-бутил 3-((метилсульфонил)окси)азетидин-1-карбоксилат (1,048 г, 4,17 ммоль) добавляли, и смесь нагревали при приблизительно 85°C в течение приблизительно 18 часов. Сырую смесь распределяли между EtOAc (50 мг) и насыщенным водным раствором хлорида аммония (30 мг). Органический слой промывали насыщенным солевым раствором (30 мг), сушили над сульфатом натрия, фильтровали и концентрировали с получением трет-бутил 3-(4-бром-3-нитрофенокси)азетидин-1-карбоксилата (0,799 г, 2,14 ммоль, выход 46,7%): ЖХ/МС (Таблица 1, Способ a) Время удерживания = 2,62 мин; MS m/z 373, 375 (M+H)+.
Стадия B: трет-бутил 3-((7-бром-1H-индол-4-ил)окси)азетидин-1-карбоксилат
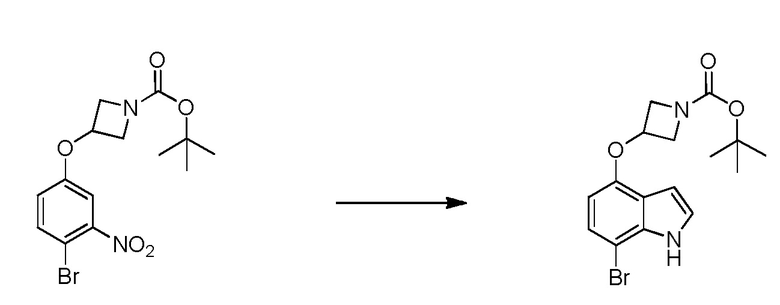
Круглодонную колбу емкостью 100 мл дегазировали азотом и охлаждали до приблизительно -70°С на бане сухой лед/ацетон. Раствор винилмагний бромида в THF (1,0 M, 21,59 мл, 21,59 ммоль) добавляли в колбу. Затем раствор трет-бутил 3-(4-бром-3-нитрофенокси)азетидин-1-карбоксилата (2,65 г, 5,40 ммоль) в 2-метил-THF (18 мг) добавляли по каплям в течение 8 минут, смесь перемешивали при приблизительно -70°C в течение приблизительно 1 часа, и реакционную смесь гасили насыщенным водным раствором хлорида аммония (22 мг) при приблизительно -60°C. Полученную смесь нагревали до комнатной температуры и добавляли EtOAc (50 мг) и воду (40 мг). Слои разделяли, водный слой экстрагировали EtOAc (50 мг), объединенные органические слои промывали насыщенным солевым раствором (50 мг), сушили над сульфатом натрия, фильтровали и концентрировали с получением оранжевого масла, которое очищали с помощью хроматографии на силикагеле, элюируя градиентом 0-40% EtOAc/гептан с получением трет-бутил 3-((7-бром-1H-индол-4-ил)окси)азетидин-1-карбоксилата (0,87 г, 2,37 ммоль, выход 43,9%): ЖХ/МС (Таблица 1, Способ a) Время удерживания = 2,52 мин; MS m/z 367, 369 (M+H)+.
Стадия C: трет-бутил 3-((7-циано-1H-индол-4-ил)окси)азетидин-1-карбоксилат
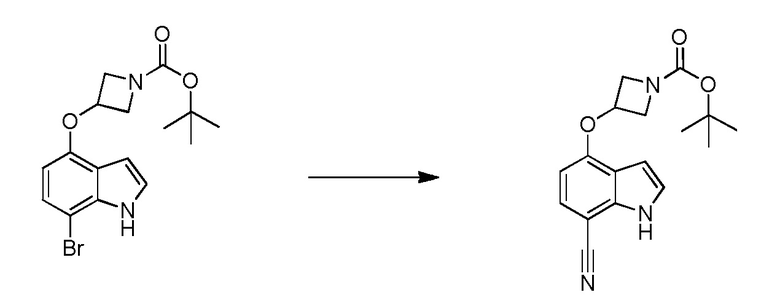
В 20 мл реакционный сосуд для микроволновой печи добавляли трет-бутил 3-((7-бром-1H-индол-4-ил)окси)азетидин-1-карбоксилат (0,8 г, 2,178 ммоль), цианид цинка (0,512 г, 4,36 ммоль) и DMF (12 мг) с получением желтой суспензии. Сосуд дегазировали азотом, добавляли тетракис(трифенилфосфин)палладий(0) (0,755 г, 0,654 ммоль). Смесь дегазировали азотом, а затем реакционную смесь нагревали в микроволновом реакторе Biotage® при температуре приблизительно 160°C в течение приблизительно 30 минут (2 фунт на кв.дюйм максимальное давление, 235 максимум Ватт). Полученную оранжевую суспензию фильтровали через целит, промывали DMF (10 мг) и 2-метил-THF (3×10 мг), фильтрат концентрировали в вакууме для удаления большей части DMF, затем распределяли между 2-метил-THF (50 мг) и насыщенным водным раствором хлорида аммония (50 мг). Органический слой промывали водой (30 мг) и насыщенным солевым расствором (30 мг), сушили над сульфатом натрия, фильтровали и концентрировали с получением оранжевого масла, которое очищали хроматографией на силикагеле, элюируя градиентом 0-50% EtOAc/гептан, с получением трет-бутил 3-((7-циано-1H-индол-4-ил)окси)азетидин-1-карбоксилата (0,28 г, 0,894 ммоль, выход 41,0%): ЖХ/МС (Таблица 1, Способ a) Время удерживания = 2,29 мин; MS m/z 314 (M+H)+.
Стадия D: 4-((1-акрилоилазетидин-3-ил)окси)-1H-индол-7-карбоксамид
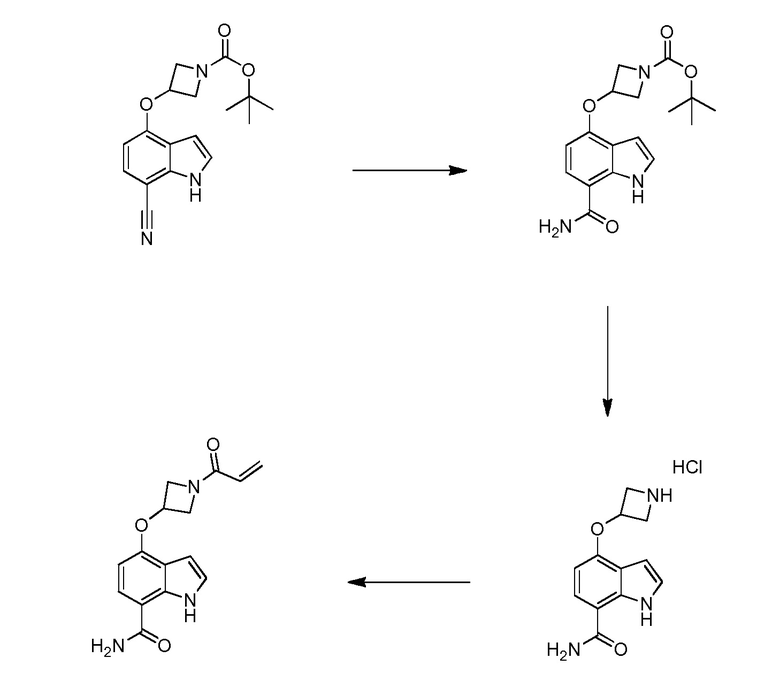
Смесь трет-бутил 3-((7-циано-1H-индол-4-ил)окси)азетидин-1-карбоксилата (0,28 г, 0,894 ммоль) и карбоната калия (0,309 г, 2,234 ммоль) в DMSO (2,98 мг) охлаждали до приблизительно 10°C с помощью ледяной водяной бани, затем добавляли перекись водорода (0,091 мл, 0,894 ммоль) по каплям. Реакционную смесь перемешивали при комнатной температуре в течение приблизительно 18 часов, добавляли пероксид водорода (0,023 мл, 0,225 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение приблизительно дополнительных 9 часов. Воду (30 мг) добавляли к реакционной смеси и смесь экстрагировали EtOAc (2×30 мг) и объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали с получением сырого трет-бутил 3-((7-карбамоил-1H-индол-4-ил)окси)азетидин-1-карбоксилата, который использовали непосредственно на следующей стадии.
К суспензии трет-бутил 3-((7-карбамоил-1H-индол-4-ил)окси)азетидин-1-карбоксилата (0,27 г, 0,815 ммоль) в MeOH (4,45 мг) добавляли водород хлорид (4,0 M в диоксане, 4,07 мл, 16,30 ммоль) по каплям, смесь перемешивали при комнатной температуре в течение приблизительно 30 минут, затем смесь концентрировали в вакууме с получением сырого гидрохлорида 4-(азетидин-3-илокси)-1H-индол-7-карбоксамида, который использовали непосредственно на следующей стадии.
Суспензию гидрохлорида 4-(азетидин-3-илокси)-1H-индол-7-карбоксамида (0,218 г, 0,815 ммоль) в DCM (13,0 мг) охлаждали до приблизительно -10°C на бане лед/хлорид натрия, TEA (0,568 мл, 4,08 ммоль) добавляли по каплям; затем раствор акрилоилхлорида (0,075 мл, 0,897 ммоль) в DCM (3,26 мг) добавляли по каплям с помощью шприца и реакционную смесь перемешивали в течение приблизительно 30 минут. Реакционную смесь концентрировали в вакууме, сырой продукт очищали с помощью хроматографии на силикагеле, элюируя градиентом 0-10% MeOH/DCM с получением 4-((1-акрилоилазетидин-3-ил)окси)-1H-индол-7-карбоксамида (0,16 г, 0,555 ммоль, выход 68,1%): ЖХ/МС (Таблица 1, способ a) Время удерживания = 1,37 мин; MS m/z 286 (M+H)+. (Btk IC50=A)
Пример № 28*: (S)-4-(1-(1-акрилоилазетидин-3-ил)этил)-1H-индол-7-карбоксамид и (R)-4-(1-(1-акрилоилазетидин-3-ил)этил)-1H-индол-7-карбоксамид
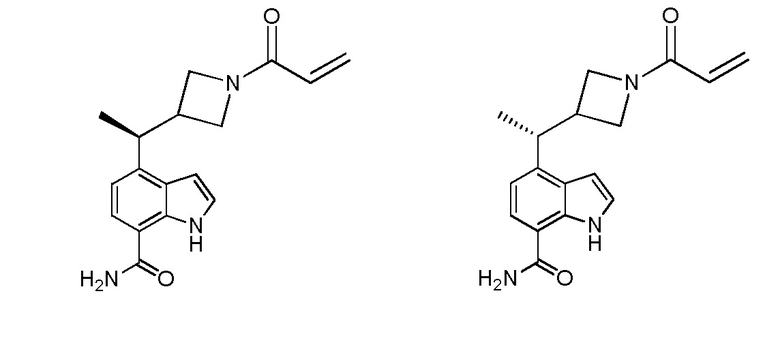
Стадия A: трет-бутил 3-(1-(((трифторметил)сульфонил)окси)винил)азетидин-1-карбоксилат и трет-бутил 3-(1-(((трифторметил)сульфонил)окси)этилиден)азетидин-1-карбоксилат
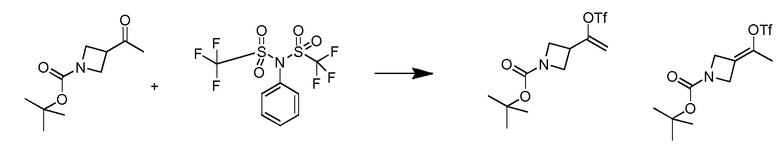
К раствору диизопропиламина (0,646 мл, 4,57 ммоль) в THF (3,8 мг) при приблизительно 0°C добавляли раствор н-бутиллития (2 M в гексанах) (2,28 мл, 4,57 ммоль) по каплям (поддерживая внутреннюю температуру ниже приблизительно 3°C). Реакционную смесь перемешивали при приблизительно 0°C в течение приблизительно 30 минут, и охлаждали до приблизительно -78°C. Раствор трет-бутил 3-ацетилазетидин-1-карбоксилата (0,758 г, 3,81 ммоль) в THF (7,6 мг) добавляли по каплям (поддерживая внутреннюю температуру ниже приблизительно -70°C), и затем реакционную смесь перемешивали при -78°C в течение приблизительно 30 минут. Раствор 1,1,1-трифтор-N-фенил-N-((трифторметил)сульфонил)метансульфонамида (1,42 г, 4,00 ммоль) в THF (7,6 мг) добавляли по каплям (поддерживая внутреннюю температуру ниже приблизительно -70°C). После добавления смесь оставляли нагреваться до приблизительно 0°C в течение приблизительно 4 часов, и реакционную смесь гасили насыщенным NH4Cl и экстрагировали EtOAc (3×50 мг), концентрировали и очищали хроматографией на силикагеле, элюируя градиентом 0-15% EtOAc/гептан с получением смеси трет-бутил 3-(1-(((трифторметил)сульфонил)окси)винил)азетидин-1-карбоксилата и трет-бутил 3-(1-(((трифторметил)сульфонил)окси)этилиден)азетидин-1-карбоксилата в виде желтого масла (0,398 г, 31%):1H-ЯМР (400 МГц, CDCl3) трет-бутил 3-(1-(((трифторметил)сульфонил)окси)винил)азетидин-1-карбоксилат: δ 5,32 (д, J=4,2 Гц, 1H), 5,16 (дд, J=4,2, 1,0 Гц, 1H), 4,15 (т, J=8,8 Гц, 2H), 3,93 (дд, J=8,8, 6,1 Гц, 2H), 3,49-3,37 (м, 1H), 1,44 (с, 9H); трет-бутил 3-(1-(((трифторметил)сульфонил)окси)этилиден)азетидин-1-карбоксилат: δ 4,58-4,53 (м, 2H), 4,52-4,49 (м, 2H), 1,98-1,94 (м, 3H), 1,45 (с, 9H)
Стадия B: трет-бутил 3-(1-(7-карбамоил-1H-индол-4-ил)винил)азетидин-1-карбоксилат и трет-бутил 3-(1-(7-карбамоил-1H-индол-4-ил)этилиден)азетидин-1-карбоксилат
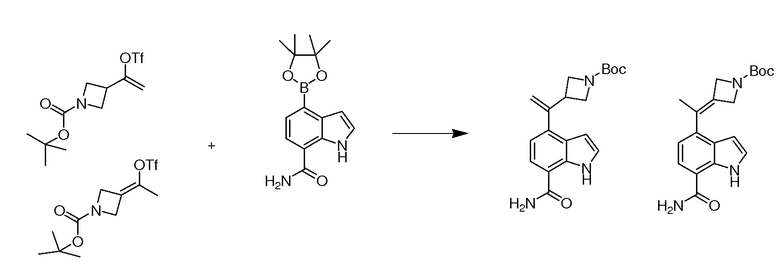
В сосуд, загруженный смесью трет-бутил 3-(1-(((трифторметил)сульфонил)окси)винил)азетидин-1-карбоксилата и трет-бутил 3-(1-(((трифторметил)сульфонил)окси)этилиден)азетидин-1-карбоксилата (0,388 г, 1,17 ммоль), 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-7-карбоксамида (0,279 г, 0,975 ммоль), Pd(dppf)Cl2 (0,043 г, 0,059 ммоль) и карбоната натрия (0,31 г, 2,93 ммоль) добавляли 1,4-диоксан (3 мг) и воду (1 мг). Реакционную смесь откачивали и заполняли азотом (повторяли 3 раза). Затем смесь нагревали при приблизительно 80°C в течение приблизительно 1 часа. Реакционную смесь концентрировали и разбавляли MeOH/DCM. Смесь фильтровали и промывали MeOH/DCM и фильтрат концентрировали досуха. Сырой продукт очищали с помощью хроматографии на силикагеле, элюируя градиентом 0-3% MeOH/DCM с получением смеси трет-бутил-3-(1-(7-карбамоил-1H-индол-4-ил)винил)азетидин-1-карбоксилата и трет-бутил-3-(1-(7-карбамоил-1H-индол-4-ил)этилиден)азетидин-1-карбоксилата (0,277 г, 83%) в виде желтого масла: ЖХ/МС (Таблица 1, Способ a) Время удерживания = 2,08, 2,13 мин.; MS m/z: 340 (M-H)-.
Стадия C: трет-бутил 3-(1-(7-карбамоил-1H-индол-4-ил)этил)азетидин-1-карбоксилат
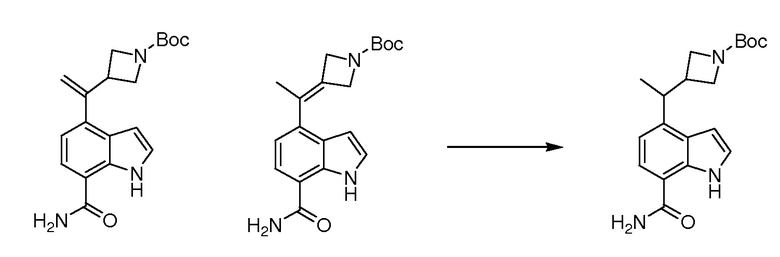
В сосуд, заполненный 10% масс. Pd/C (0,026 г, 0,024 ммоль), добавляли раствор трет-бутил 3-(1-(7-карбамоил-1H-индол-4-ил)винил)азетидин-1-карбоксилата и трет-бутил 3-(1-(7-карбамоил-1H-индол-4-ил)этилиден)азетидин-1-карбоксилата (0,26 г, 0,76 ммоль) в EtOAc (10 мг) и приблизительно 2 капли MeOH. Смесь гидрогенизировали водородом из баллона при приблизительно комнатной температуре в течение приблизительно 2 часов. Реакционную смесь фильтровали через слой целита и промывали EtOAc. Фильтрат концентрировали досуха с получением трет-бутил 3-(1-(7-карбамоил-1H-индол-4-ил)этил)азетидин-1-карбоксилата (0,212 г, 81%) в виде светло-желтой пены: ЖХ/МС (Таблица 1, Способ a) Время удерживания = 2,08 мин.; MS m/z: 342 (M-H)-.
Стадия D: (S)-4-(1-(1-акрилоилазетидин-3-ил)этил)-1H-индол-7-карбоксамид и (R)-4-(1-(1-акрилоилазетидин-3-ил)этил)-1H-индол-7-карбоксамид
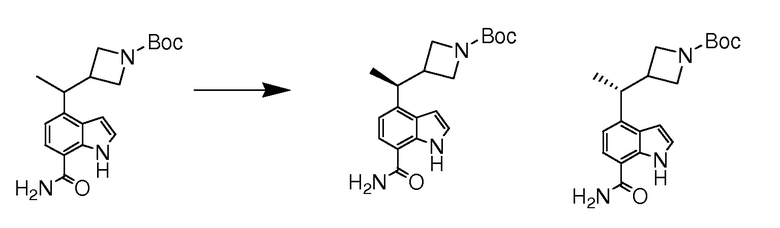
трет-Бутил 3-(1-(7-карбамоил-1H-индол-4-ил)этил)азетидин-1-карбоксилат (0,17 г, 0,495 ммоль) очищали с помощью препаративной хиральной ВЭЖХ (Таблица 2, Способ 1) с получением (S)-4-(1-(1-акрилоилазетидин-3-ил)этил)-1H-индол-7-карбоксамида (0,063 г, 37%) (Время удерживания = 12,339 мин, или = положительный) и (R)-4-(1-(1-акрилоилазетидин-3-ил)этил)-1H-индол-7-карбоксамида (0,066 г, 39%) (Время удерживания = 18,959 мин, или = отрицательный).
Стадия E.1: (S)-4-(1-(1-акрилоилазетидин-3-ил)этил)-1H-индол-7-карбоксамид
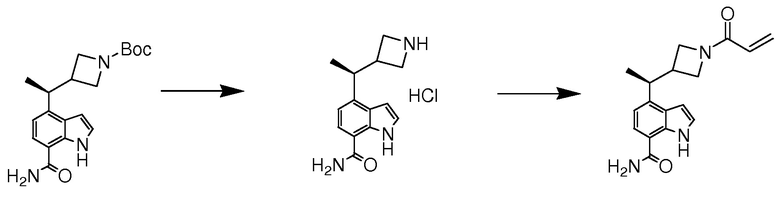
В сосуд, заполненный (S)-трет-бутил 3-(1-(7-карбамоил-1H-индол-4-ил)этил)азетидин-1-карбоксилатом (0,063 г, 0,183 ммоль) и MeOH (1 мг) добавляли водород хлорид (4 M в диоксане, 0,92 мл, 3,67 ммоль) при приблизительно комнатной температуре. Смесь перемешивали в течение приблизительно 30 минут, затем смесь концентрировали в вакууме с получением сырого гидрохлорида (S)-трет-бутил 3-(1-(7-карбамоил-1H-индол-4-ил)этил)азетидин-1-карбоксилата, который использовали без дополнительной очистки.
К суспензии гидрохлорида (S)-4-(1-(азетидин-3-ил)этил)-1H-индол-7-карбоксамида (0,051, 0,183 ммоль) в THF (2 мг) и DCM (1 мг) при приблизительно 0°C добавляли N-этил-N-изопропилпропан-2-амин (0,096 мл, 0,550 ммоль) с последующим добавлением акрилоилхлорида (0,017 мл, 0,202 ммоль). Смесь перемешивали при приблизительно 0°C в течение приблизительно 30 минут. Смесь гасили МеОН и летучие вещества удаляли при пониженном давлении. Остаток разделяли между DCM и насыщенным водным раствором NaHCO3. Органический слой концентрировали, и сырой продукт очищали с помощью хроматографии на силикагеле, элюируя градиентом 0-5% MeOH/DCM с получением (S)-4-(1-(1-акрилоилазетидин-3-ил)этил)-1H-индол-7-карбоксамида (0,039 г, 69,9 %) в виде белого твердого вещества: ЖХ/МС (Таблица 1, Способ a) Время удерживания = 1,50 мин.; MS m/z: 298 (M+H)+. (Btk IC50=B)
Стадия E.2: (R)-4-(1-(1-акрилоилазетидин-3-ил)этил)-1H-индол-7-карбоксамид
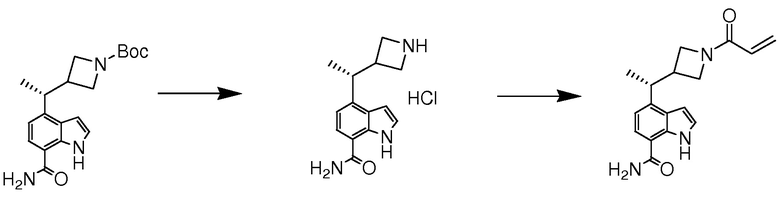
В сосуд, заполненный (R)-трет-бутил 3-(1-(7-карбамоил-1H-индол-4-ил)этил)азетидин-1-карбоксилатом (0,066 г, 0,192 ммоль) и MeOH (1 мг) добавляли водород хлорид (4 M в диоксане, 0,96 мл, 3,84 ммоль) при приблизительно комнатной температуре. Смесь перемешивали при комнатной температуре в течение приблизительно 1 часа, затем смесь концентрировали в вакууме с получением сырого гидрохлорида (R)-трет-бутил 3-(1-(7-карбамоил-1H-индол-4-ил)этил)азетидин-1-карбоксилата, который использовали без дополнительной очистки.
К суспензии гидрохлорида (R)-4-(1-(азетидин-3-ил)этил)-1H-индол-7-карбоксамида (0,054 г, 0,192 ммоль) в THF (2 мг) и DCM (1 мг) при приблизительно 0°C добавляли N-этил-N-изопропилпропан-2-амин (0,1 мл, 0,577 ммоль) с последующим добавлением по каплям акрилоилхлорида (0,018 мл, 0,212 ммоль). Смесь перемешивали при приблизительно 0°C в течение приблизительно 30 минут. Смесь гасили МеОН и летучие вещества удаляли при пониженном давлении. Остаток разделяли между DCM и насыщенным водным раствором NaHCO3. Органический слой концентрировали, и сырой продукт очищали с помощью хроматографии на силикагеле, элюируя градиентом 0-5% MeOH/DCM с получением (R)-4-(1-(1-акрилоилазетидин-3-ил)этил)-1H-индол-7-карбоксамида (0,042 г, 73,2%) в виде белого твердого вещества. ЖХ/МС (Таблица 1, Способ a) Время удерживания = 1,50 мин.; MS m/z: 298 (M+H)+. (Btk IC50=A)
Пример № 29: 4-((1-акрилоилазетидин-3-ил)(метил)амино)-1H-пирроло[2,3-c]пиридин-7-карбоксамид
Стадия A: 4-бром-1H-пирроло[2,3-c]пиридин-7-карбонитрил
К раствору 4-бром-1H-пирроло[2,3-c]пиридина [ChemTec] (10,4 г, 52,8 ммоль) в DCM (66,0 мг) и DME (66,0 мг) добавляли 3-хлорбензопероксидную кислоту (21,29 г, 95 ммоль, 77% по массе) в виде одной порции и смесь оставляли перемешиваться в течение приблизительно 16 часов. Органические растворители удаляли при пониженном давлении, твердый остаток растирали с DCM и твердое вещество отфильтровывали с получением смеси продукта и бензойной кислоты. Фильтрат еще содержит дополнительный продукт и его концентрировали дополнительно при пониженном давлении, для обеспечения второй фильтрации. Объединенные отфильтрованные осадки сушили и переносили в 1 л круглодонную колбу, содержащую магнитную мешалку. MeCN (264 мг) и TEA (14,8 мл, 106 ммоль) добавляли с получением не совсем белой взвеси. Триметилсилилцианид (24,64 мл, 185 ммоль) добавляли в виде одной порции с помощью шприца и смесь нагревали с обратным холодильником. После приблизительно 2 часов нагревания смеси давали остыть до комнатной температуры. Реакцию гасили путем добавления 100 мл 1 M NaOH, разбавляли 100 мл EtOAc, переносили в делительную воронку и дополнительно разбавляли 100 мл 1 M NaOH и 100 мл EtOAc. Слои разделяли и водную фазу экстрагировали EtOAc (3×150 мг). Объединенные органические экстракты промывали смесью 1:1 насыщенного солевого раствора и 1 M NaOH (2×50 мг), сушили над Na2SO4, фильтровали и растворитель удаляли с получением 4-бром-1H-пирроло[2,3-c]пиридин-7-карбонитрила в виде коричнево-желтого твердого вещества (10,28 г, 80%). 1H-ЯМР (400 МГц, DMSO) δ 8,44 (с, 1H), 7,96 (д, J=3,1 Гц, 1H), 6,71 (д, J=3,1 Гц, 1H).
Стадия B: 4-бром-1H-пирроло[2,3-c]пиридин-7-карбоксамид
К раствору 4-бром-1H-пирроло[2,3-c]пиридин-7-карбонитрила (10,2 г, 45,9 ммоль) в EtOH (104 мг) добавляли 1 M водный раствор NaOH (115 мл, 115 ммоль) и 30% пероксид водорода (80 мл, 781 ммоль) и реакционную смесь нагревали до приблизительно 45°C и перемешивали в течение приблизительно 30 минут. Органический растворитель удаляли при пониженном давлении. Смесь разбавляли 30 мл воды и фильтровали с получением 4-бром-1H-пирроло[2,3-c]пиридин-7-карбоксамида в качестве светло-желтого твердого вещества (9,87 г, 83%). ЖХ/МС (Таблица 1, Способ as): Время удерживания = 1,81 мин; MS m/z: 240, 242 (M+H)+.
Стадия C: трет-бутил 3-((7-карбамоил-1H-пирроло[2,3-c]пиридин-4-ил)(метил)амино)азетидин-1-карбоксилат
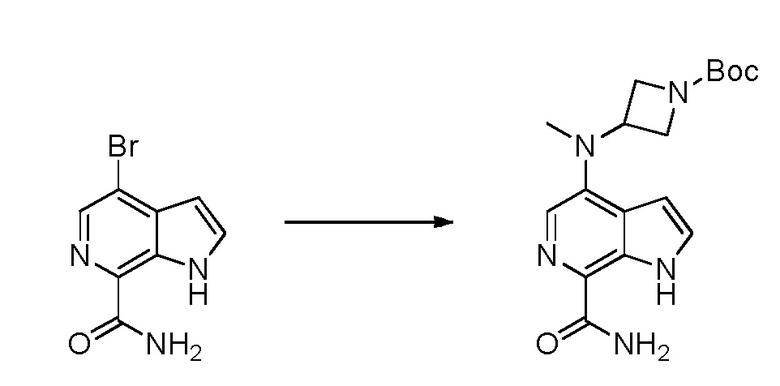
4-бром-1H-пирроло[2,3-c]пиридин-7-карбоксамид (580 мг, 2,416 ммоль) растворяли в 12 мл безводного диоксана и сушили в течение приблизительно 1 часа над Na2SO4. Раствор затем фильтровали в высушенном в печи 75 мл сосуде под давлением. и высушивающее вещество промывали с помощью 3 мл диоксана. Раствор дегазировали с помощью потока аргона и добавляли гидрохлорид трет-бутил 3-(метиламино)азетидин-1-карбоксилата (0,969 г, 4,35 ммоль, Synthonix) с последующим добавлением хлор(2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-дифенил)[2-(2-аминоэтил)фенил)]палладия (II) (0,089 г, 0,12 ммоль) и X-Phos (0,057 г, 0,12 ммоль). Смесь дегазировали в течение приблизительно 10 минут и LiHMDS (1 M в THF, 10,87 мл, 10,87 ммоль) добавляли по каплям с помощью шприца, флакон герметично закрывали и нагревали до приблизительно 90°C в течение приблизительно 19 часов. Реакционную смесь охлаждали до комнатной температуры и гасили добавлением водного раствора NaHCO3 (20 мг) и разбавляли EtOAc (50 мг). Дальнейшее разбавление с использованием воды (10 мг) и насыщенного солевого раствора (10 мг) привело к полному растворению и слои разделяли. Водную фазу экстрагировали EtOAc (3×20 мг). Объединенные органические экстракты промывали 1:1 насыщенный солевой раствор и водный раствор NaHCO3 (20 мг), сушили над Na2SO4, фильтровали и растворитель удаляли при пониженном давлении. Сырой продукт наносили на силикагеле и очищали с использованием колонки с силикагелем (40 г), элюируя 0-5% MeOH/DCM. Фракции, содержащие продукт, концентрировали при пониженном давлении с получением трет-бутил 3-((7-карбамоил-1H-пирроло[2,3-c]пиридин-4-ил)(метил)амино)азетидин-1-карбоксилата в виде светло-желтого твердого вещества (0,61 г, 69%). 1H-ЯМР (400 МГц, DMSO) δ 11,41 (ушир.с, 1H), 7,90 (ушир.с, 1H), 7,48-7,43 (м, 1H), 7,43-7,39 (м, 2H), 6,60 (дд, J=3,1, 2,0 Гц, 1H), 4,61-4,51 (м, 1H), 4,23-4,14 (м, 2H), 3,86 (дд, J=8,9, 5,2 Гц, 2H), 3,06 (с, 3H), 1,38 (с, 9H).
Стадия D: гидрохлорид 4-(азетидин-3-ил(метил)амино)-1H-пирроло[2,3-c]пиридин-7-карбоксамида
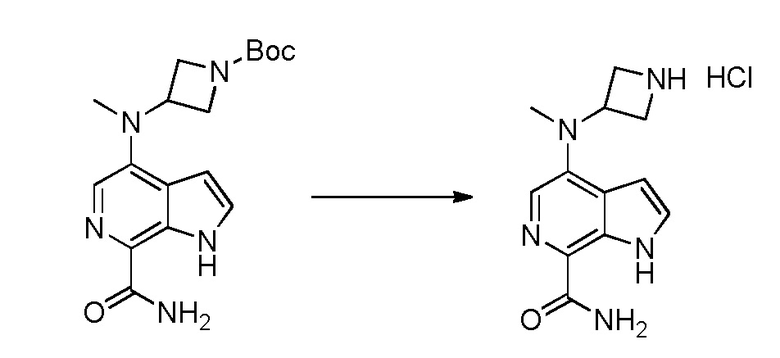
В 50 мл круглодонную колбу, содержащую магнитную мешалку и MeOH (1,97 мг) добавляли ацетилхлорид (1307 мкл, 18,38 ммоль) при приблизительно 0°C с помощью шприца. После приблизительно 10 минут, смесь нагревали до комнатной температуры и перемешивали в течение приблизительно 1 часа. Затем, раствор трет-бутил 3-((7-карбамоил-1H-пирроло[2,3-c]пиридин-4-ил)(метил)амино)азетидин-1-карбоксилата (127 мг, 0,368 ммоль) в MeOH (1970 мкл) и DCM (657 мкл) добавляли по каплям с помощью шприца и реакционную смесь перемешивали в течение приблизительно 5 часов при комнатной температуре. Растворители удаляли при пониженном давлении с получением гидрохлорида 4-(азетидин-3-ил(метил)амино)-1H-пирроло[2,3-c]пиридин-7-карбоксамида (128 мг, 99%). ЖХ/МС (Таблица 1, Способ at): Время удерживания = 0,93 мин.; MS m/z: 246 (M+H)+.
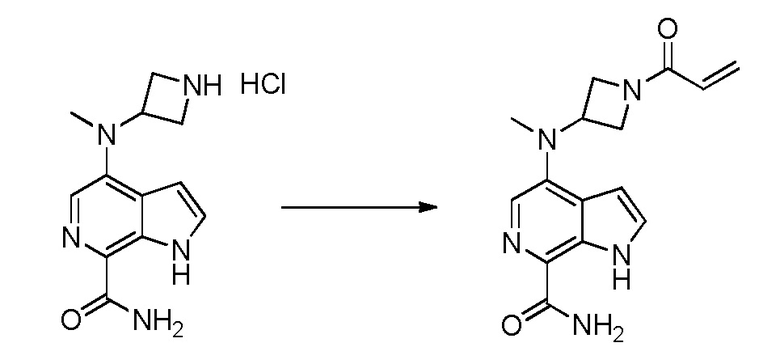
Стадия E: 4-((1-акрилоилазетидин-3-ил)(метил)амино)-1H-пирроло[2,3-c]пиридин-7-карбоксамид
К охлажденному раствору гидрохлорида 4-(азетидин-3-ил(метил)амино)-1H-пирроло[2,3-c]пиридин-7-карбоксамида (101 мг, 0,36 мг) в DCM (5760 мкл) и этилдиизопропиламина (258 мкл, 1,440 ммоль) добавляли раствор акрилоилхлорида (50 мг, 0,552 ммоль) в DCM (1440 мкл) по каплям с помощью шприца, поддерживая внутреннюю температуру на уровне или ниже -4°C. Смесь перемешивали в течение 15 минут. Реакцию гасили добавлением 0,3 мл воды, объем растворителя снижали до 1,5 мл, и смесь загружаали на 4 г силикагеля. Материал очищали, используя колонку с силикагелем 24 г, 0-10% MeOH/DCM. Фракции, содержащие продукт, концентрировали при пониженном давлении с получением 4-((1-акрилоилазетидин-3-ил)(метил)амино)-1H-пирроло[2,3-c]пиридин-7-карбоксамида в виде белого твердого вещества (89 мг, 78%). 1H-ЯМР (400 МГц, DMSO) δ 11,43 (ушир.с, 1H), 7,98-7,88 (м, 1H), 7,49-7,44 (м, 2H), 7,42 (с, 1H), 6,64-6,58 (м, 1H), 6,40-6,29 (м, 1H), 6,11 (дд, J=17,0, 2,2 Гц, 1H), 5,68 (дд, J=10,2, 2,2 Гц, 1H), 4,72-4,62 (м, 1H), 4,60-4,52 (м, 1H), 4,31-4,18 (м, 2H), 3,97 (дд, J=10,5, 5,2 Гц, 1H), 3,08 (с, 3H); MS m/z: 300 (M+H)+. (Btk IC50=A)
Пример № 30*: (R)-4-(1-Акрилоилпиперидин-3-ил)-1H-индол-7-карбоксамид и (S)-4-(1-акрилоилпиперидин-3-ил)-1H-индол-7-карбоксамид

Образец 4-(1-акрилоилпиперидин-3-ил)-1H-индол-7-карбоксамида (0,03 г, 0,10 ммоль) очищали с помощью препаративной хиральной ВЭЖХ (Таблица 2, Способ 2) с получением (R)-4-(1-акрилоилпиперидин-3-ил)-1H-индол-7-карбоксамида (0,012 г, 40%) (Время удерживания=17,14 мин, или = положительный) (Btk IC50=B) и (S)-4-(1-акрилоилпиперидин-3-ил)-1H-индол-7-карбоксамида (0,013 г, 43%) (Время удерживания=20,46 мин, или = отрицательный) (Btk IC50=A): ЖХ/МС (Таблица 1, Способ a) Время удерживания = 1,47 мин.; MS m/z: 298 (M+H)+.
Примеры, полученные из акрилоил амида с использованием способа хиральной ВЭЖХ: Таблица 2, Способ 4
(Таблица 1, Способ)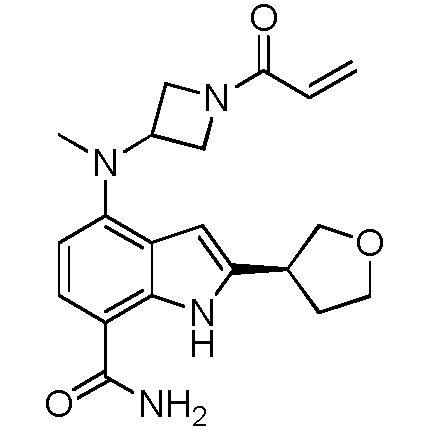
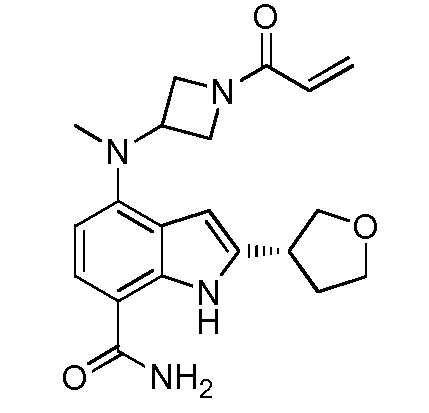
Примеры, полученные из акрилоил амида с использованием способа хиральной ВЭЖХ: Таблица 2, Способ 15
(Таблица 1, Способ)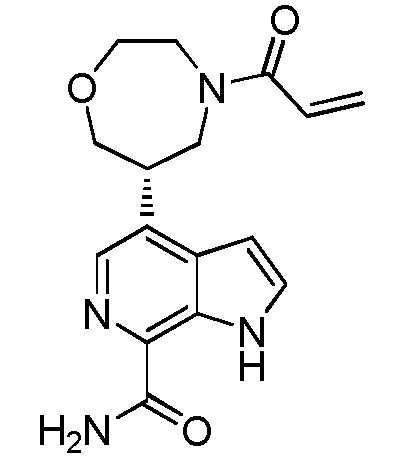
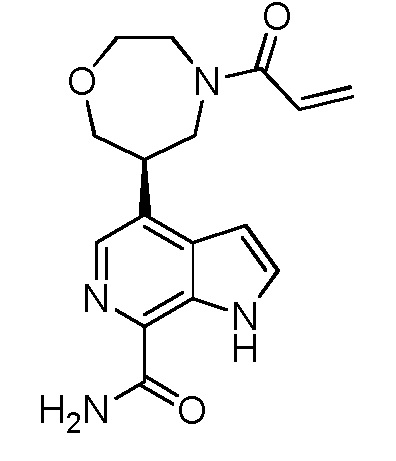
Примеры, полученные из акрилоил амида с использованием способа хиральной ВЭЖХ: Таблица 2, Способ 16
(Таблица 1, Способ)
трет-бутил 5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,4-дигидропиридин-1(2H)-карбоксилатом [Anisyn], L с Pd/C, G с ацетилхлоридом, E с акрилоилхлоридом)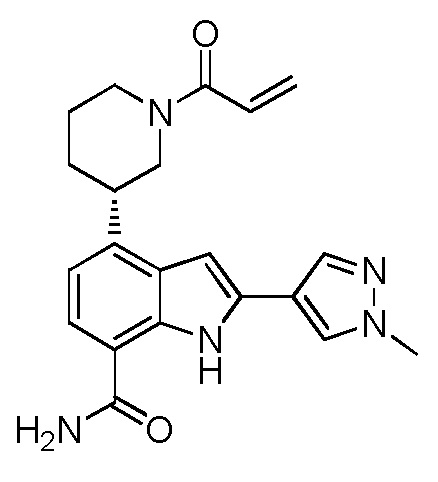
трет-бутил 5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,4-дигидропиридин-1(2H)-карбоксилатом [Anisyn], L с Pd/C, G с ацетилхлоридом, E с акрилоилхлоридом)

| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ЗАМЕЩЕННЫЕ БИАРИЛОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ИНДОЛАМИН-2,3-ДИОКСИГЕНАЗЫ (IDO) | 2018 |
|
RU2786586C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ PDK1 | 2010 |
|
RU2615130C2 |
| БИАРИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ БЕЛОК-БЕЛКОВОГО ВЗАИМОДЕЙСТВИЯ YAP/TAZ-TEAD | 2021 |
|
RU2830596C1 |
| АНАЛОГИ СОЕДИНЕНИЙ 4Н-ПИРАЗОЛО[1,5-А]БЕНЗИМИДАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРОВ PARP | 2015 |
|
RU2672722C2 |
| ИНГИБИТОРЫ ВИРУСА ГЕПАТИТА С, ИМЕЮЩИЕ ЖЕСТКУЮ ВЫТЯНУТУЮ ЦЕПЬ И СОДЕРЖАЩИЕ ФРАГМЕНТ { 2-[4-(БИФЕНИЛ-4-ИЛ)-1Н-ИМИДАЗО-2-ИЛ]ПИРРОЛИДИН-1-КАРБОНИЛМЕТИЛ} АМИНА | 2012 |
|
RU2625787C2 |
| ПОЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ЦЕЛЕНАПРАВЛЕННОЙ ДЕГРАДАЦИИ ПОЛИПЕПТИДОВ БЫСТРО УСКОРЕННОЙ ФИБРОСАРКОМЫ | 2019 |
|
RU2830173C2 |
| БИАРИЛЬНОЕ ПРОИЗВОДНОЕ В КАЧЕСТВЕ АГОНИСТА GPR120 | 2015 |
|
RU2683648C2 |
| Новые 3-индол замещенные производные, фармацевтические композиции и способы применения | 2016 |
|
RU2672252C1 |
| СПОСОБ ЛЕЧЕНИЯ | 2012 |
|
RU2621148C2 |
| ЗАМЕЩЕННЫЕ ПИРИДОПИРАЗИНЫ КАК НОВЫЕ ИНГИБИТОРЫ Syk | 2012 |
|
RU2569635C9 |
Изобретение относится к конкретным соединениям, представляющим собой (S)-4-(1-акрилоилпиперидин-3-ил)-1Н-индол-7-карбоксамид, (R)-4-(1-акрилоилпиперидин-3-ил)-1Н-индол-7-карбоксамид, 4-(1-акрилоилпиперидин-3-ил)-2-(1-метил-1Н-пиразол-4-ил)-1Н-индол-7-карбоксамид, 4-((1-акрилоилазетидин-3-ил)(метил)амино)-1Н-индол-7-карбоксамид, 4-(3-акриламидофенил)-2-(2-гидроксиэтил)-1Н-индол-7-карбоксамид, 4-(3-акриламидофенил)-2-этил-1Н-индол-7-карбоксамид, или их фармацевтически приемлемой соли. Соединения по настоящему изобретению являются полезными в качестве ингибиторов BTK. 6 н.п. ф-лы, 52 табл., 30 пр.
1. Соединение, где соединение представляет собой (S)-4-(1-акрилоилпиперидин-3-ил)-1Н-индол-7-карбоксамид или его фармацевтически приемлемую соль.
2. Соединение, где соединение представляет собой (R)-4-(1-акрилоилпиперидин-3-ил)-1Н-индол-7-карбоксамид или его фармацевтически приемлемую соль.
3. Соединение, где соединение представляет собой 4-(1-акрилоилпиперидин-3-ил)-2-(1-метил-1Н-пиразол-4-ил)-1Н-индол-7-карбоксамид или его фармацевтически приемлемую соль.
4. Соединение, где соединение представляет собой 4-((1-акрилоилазетидин-3-ил)(метил)амино)-1Н-индол-7-карбоксамид или его фармацевтически приемлемую соль.
5. Соединение, где соединение представляет собой 4-(3-акриламидофенил)-2-(2-гидроксиэтил)-1Н-индол-7-карбоксамид или его фармацевтически приемлемую соль.
6. Соединение, где соединение представляет собой 4-(3-акриламидофенил)-2-этил-1Н-индол-7-карбоксамид или его фармацевтически приемлемую соль.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Способ гальванического снятия позолоты с серебряных изделий без заметного изменения их формы | 1923 |
|
SU12A1 |
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |

