ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Изобретение относится к соединениям, которые могут использоваться в качестве ингибиторов Аврора-протеинкиназ. Изобретение также относится к фармацевтически приемлемым композициям, включающим соединения по изобретению, способам применения соединений, к композициям для лечения различных расстройств и к способам получения соединений.
УРОВЕНЬ ТЕХНИКИ
Аврора-протеины представляют собой семейство трех родственных серин/треонинкиназ (обозначаемых как Аврора-A, -B и -C), которые играют существенную роль в прохождении через фазу митоза клеточного цикла. Более точно, Аврора-A играет ключевую роль в созревании и расщеплении центросомы, образовании митотического веретена и точного расщепления хромосом. Аврора-B представляет собой хромосомный белок-пассажир, который играет центральную роль в регуляции выравнивания хромосом в метафазе, моменте сборки веретена и правильном завершении клеточного деления.
Сверхэкспрессия Авроры-A, -B или -C наблюдается при некоторых видах рака у человека, включая колоректальный рак, рак яичников, рак желудка и инвазивные аденокарциномы протоков.
В ряде исследований было продемонстрировано, что сокращение количества или ингибирование Авроры-A или -B в линиях человеческих раковых клеток под действием siРНК, доминирующих отрицательных антител или нейтрализующих антител нарушает прохождение через митоз с накоплением клеток, имеющих 4N ДНК, что в некоторых случаях приводит к эндоредупликации и смерти клетки.
Аврора-киназы являются привлекательными мишенями благодаря их участию в развитии многочисленных видов рака у человека и той роли, которую они играют в пролиферации раковых клеток. Было бы желательно иметь ингибитор Аврора-киназы с привлекательными свойствами потенциального лекарственного средства, такими как стабильность в микросомах печени человека. В связи с этим существует потребность в соединениях, которые ингибируют Аврора-киназы и также проявляют привлекательные свойства потенциального лекарственного средства.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение предоставляет соединения и их фармацевтически приемлемые композиции, которые могут использоваться в качестве ингибиторов Аврора-протеинкиназ. Эти соединения представлены формулой I:
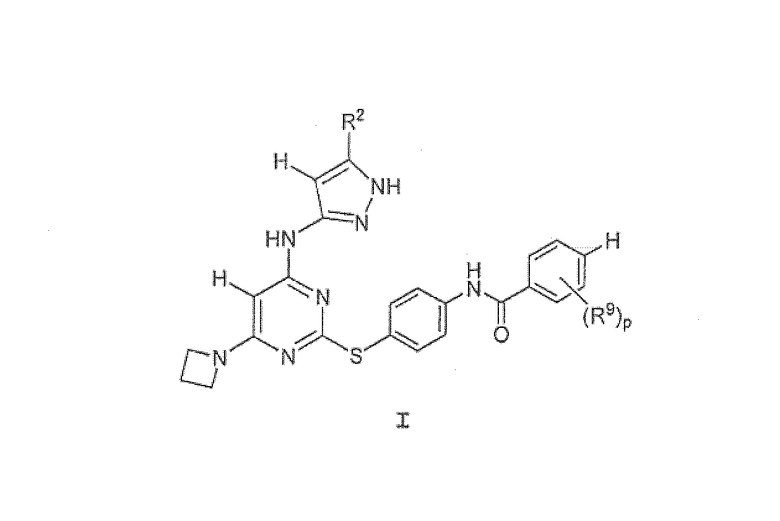
или их фармацевтически приемлемые соли, где R2, R9 и p определены здесь.
Эти соединения и их фармацевтически приемлемые композиции являются применимыми для ингибирования киназ in vitro, in vivo и ex vivo. Такие применения включают лечение или предотвращение миелопролиферативных расстройств и пролиферативных расстройств, таких как меланома, миелома, лейкемия, лимфома, нейробластома и рак. Другие применения включают исследования роли киназ в биологических и патологических процессах; исследование внутриклеточных путей сигнальной трансдукции, опосредованных такими киназами; и сравнительные исследования новых ингибиторов киназ.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение предоставляет соединение формулы I:
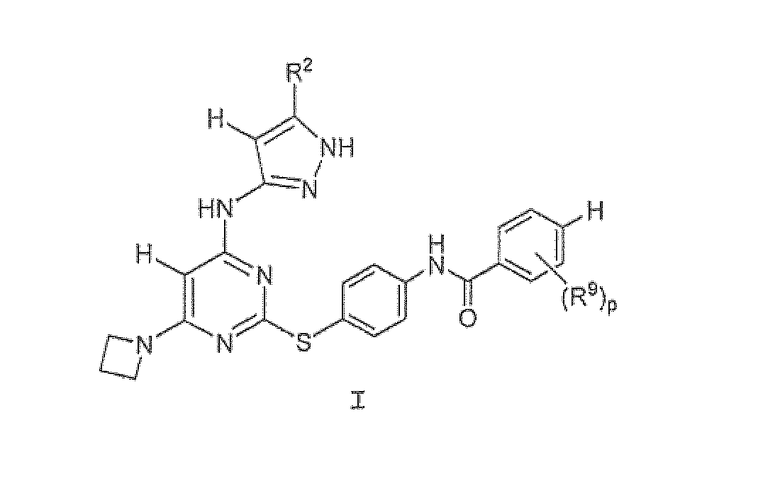
или его фармацевтически приемлемую соль, где
R2 представляет собой C1-3-алкил или циклопропил;
R9 представляет собой галоген, C1-3-алкил, -O-(C1-3-алкил), -S-(C1-3-алкил), -OCF3, или CF3 и p равно 1-2.
В некоторых вариантах осуществления R2 представляет собой метил.
В других вариантах осуществления p равно 1.
В некоторых вариантах осуществления R9 является заместителем в орто-положении.
В некоторых аспектах изобретения R9 представляет собой CF3, галоген, C1-3-алкил или -S-(C1-3-алкил). В некоторых вариантах осуществления, R9 представляет собой F, Cl или CF3.
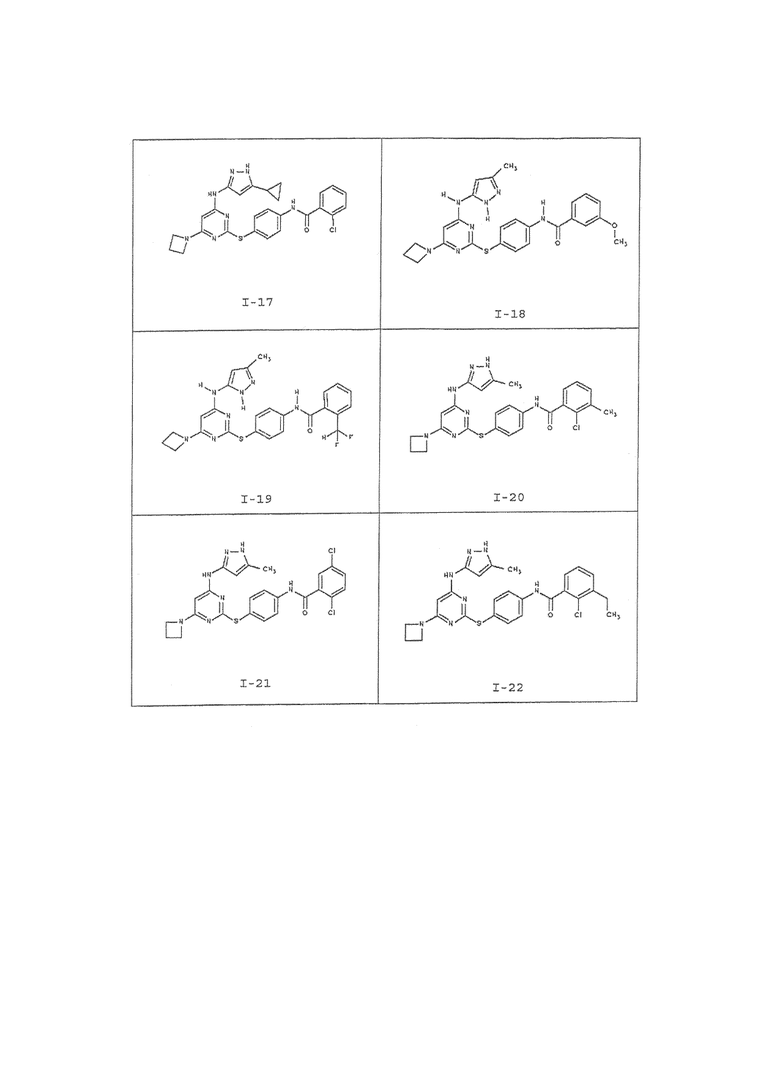
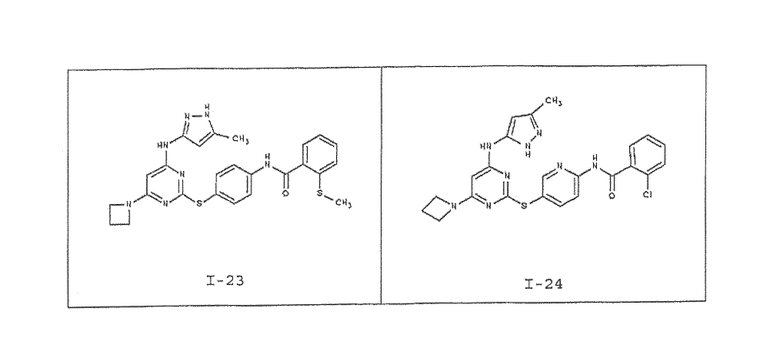
В одном аспекте предоставлено соединение, выбранное из таблицы 1 (или его фармацевтически приемлемая соль):
Таблица 1:
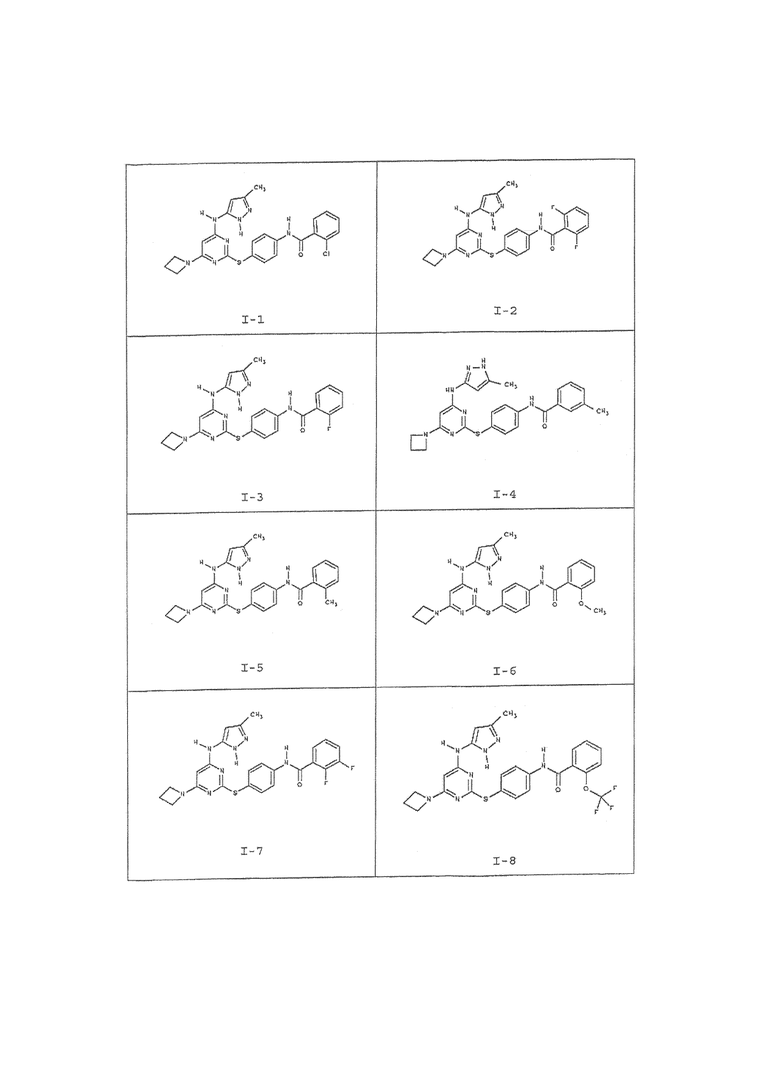
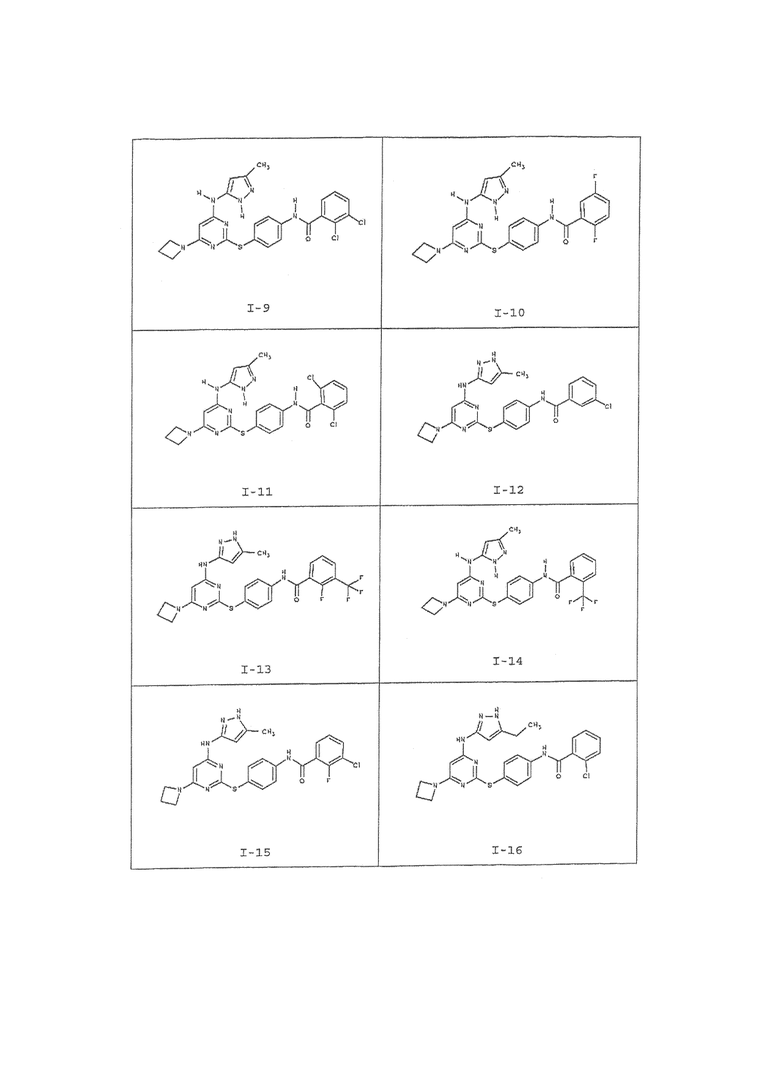
Для целей настоящего изобретения химические элементы идентифицировали как в Периодической таблице элементов, версия CAS, Handbook of Chemistry and Physics, 75 th Ed. Кроме того, общие принципы органической химии описаны в документах, известных для специалистов в данной области, включая, например, "Organic Chemistry", Thomas Sorrell, University Science Books, Sausalito: 1999, и "March's Advanced Organic Chemistry", 5 th Ed., Ed.: Smith, M.B. and March, J., John Wiley & Sons, New York: 2001, полное содержание которых включено сюда в виде ссылки.
Как описано здесь, определенный диапазон числа атомов включает любое целое число в пределах этого диапазона. Например, группа, имеющая от 1 до 4 атомов, может иметь 1, 2, 3 или 4 атома.
Как описано здесь, соединения по изобретению могут быть необязательно замещены одним или несколькими заместителями, такими как представленные в общем выше, или как проиллюстрировано на примерах отдельных классов, подклассов и примеров по изобретению. Подразумевается, что фраза "необязательно замещенный" применяется как взаимозаменяемая с фразой "замещенный или незамещенный". В целом, термин "замещенный", любо с предваряющим термином "необязательно", либо нет, относится к замене водородных радикалов в данной структуре на радикал определенного заместителя. Если не указано иное, необязательно замещенная группа может иметь заместитель в каждом замещаемом положении группы, и если более чем одно положение в любой данной структуре может быть замещено более чем одним заместителем, выбранным из определенной группы, заместитель может быть или таким же или различным при каждом положении. Комбинации заместителей, включенные в данное изобретение, предпочтительно являются такими, которые приводят к образованию стабильных или химически допустимых соединений.
Термин "стабильный", применяемый здесь, относится к соединениям, которые не изменяются существенным образом, когда подвергаются действию условий, допускающих их получение, идентификацию и, предпочтительно, их выделение, очистку и применение для одной или нескольких впервые описанных здесь целей. В некоторых вариантах осуществления стабильное соединение или химически допустимое соединение представляет собой соединение, которое не изменяется существенным образом, когда хранится при температуре 40°C или менее, в отсутствие влаги или других химически реакционно-способных условий, в течение по меньшей мере недели.
Термин "алкил", применяемый здесь, обозначает неразветвленный или разветвленный линейный углеводород, который является полностью насыщенным и имеет одну точку присоединения к остатку молекулы. Специфические примеры алкильных групп включают, без ограничения, метил, этил, изопропил, н-пропил и втор-бутил.
Термин "циклоалкил" относится к моноциклическому углеводороду, который является полностью насыщенным и имеет одну точку присоединения к остатку молекулы. Подходящие циклоалкильные группы включают, без ограничения, циклопропил, циклобутил и циклопентил.
Термин "ненасыщенный", применяемый здесь, обозначает, что фрагмент имеет одно или несколько ненасыщенных звеньев.
Термин "галогеналкил" обозначает алкил, замещенный одним или несколькими атомами галогена. Термин включает перфторированные алкильные группы, такие как CF3.
Термин "галоген" обозначает F, Cl, Br или I.
Термин "защитная группа", применяемый здесь, относится к реагенту, применяемому для временной блокировки одного или нескольких желательных реакционно-способных фрагментов в полифункциональном соединении. В определенных вариантах осуществления защитная группа имеет одну или несколько, или предпочтительно все, из следующих характеристик: a) реагирует селективно с хорошим выходом, что дает защищенный субстрат, который является стабильным в реакциях, имеющих место на одном или нескольких других реакционно-способных фрагментах; и b) селективно удаляется с хорошим выходом под действием реагентов, которые не атакуют высвобождаемую функциональную группу. Типичные защитные группы детально описаны в Greene, T.W., Wuts, P.G. in "Protective Groups in Organic Synthesis", Third Edition, John Wiley & Sons, New York: 1999, и других изданиях этой книги, полное содержание которой включено сюда в виде ссылки. Термин "защитная группа азота", применяемый здесь, относится к реагентам, применяемым для временной защиты одного или нескольких желательных реакционно-способных фрагментов с азотом в полифункциональном соединении. Предпочтительные защитные группы азота также обладают характеристиками, иллюстрированными на примерах выше, и определенные типичные защитные группы азота также детально описаны в главе 7 в Greene, T.W., Wuts, P.G. in "Protective Groups in Organic Synthesis", Third Edition, John Wiley & Sons, New York: 1999, полное содержание которой включено сюда в виде ссылки.
Если не указано иное, подразумевается, что структуры, описанные здесь, также включают все изомерные (например, энантиомерные, диастереомерные и геометрические (или конформационные)) формы структуры; например, R- и S-конфигурации для каждого асимметрического центра, (Z)- и (E)-изомеры по двойной связи и (Z)- и (E)-конформационные изомеры. Следовательно, отдельные стереохимические изомеры, также как энантиомерные, диастереомерные и геометрические (или конформационные) смеси настоящих соединений находятся в пределах объема изобретения.
Если не указано иное, все таутомерные формы соединений по изобретению находятся в пределах объема изобретения. Как должно быть понятно специалисту в данной области, пиразольная группа может быть представлена множеством способов. Например, структура, представленная как 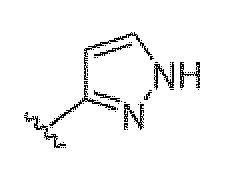 , также представляет другие возможные таутомеры, такие как
, также представляет другие возможные таутомеры, такие как 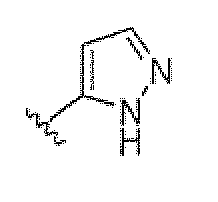 .
.
Аналогичным образом, структура, представленная как 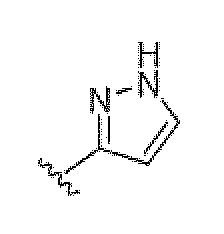 , также представляет другие возможные таутомеры, такие как
, также представляет другие возможные таутомеры, такие как 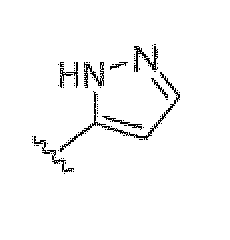 .
.
Если не указано иное, заместитель может свободно вращаться вокруг любых подходящих к вращению связей. Например, заместитель, изображенный как  , также изображает
, также изображает  .
.
Аналогичным образом, заместитель, изображенный как 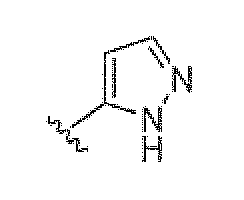 , также изображает
, также изображает 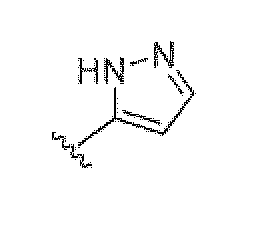 .
.
Кроме того, если не указано иное, подразумевается, что структуры, описанные здесь, также включают соединения, которые отличаются только присутствием одного или нескольких изотопно обогащенных атомов. Например, соединения, которые имеют структуры по настоящему изобретению, за исключением того, что атом водорода заменен на дейтерий или тритий, или углерод заменен на 13C- или 14C- обогащенный углерод, находятся в пределах объема настоящего изобретения. Такие соединения могут быть применены, например, для аналитических целей или в качестве зондов в биологических исследованиях.
В рамках приведенного описания соединения по этому изобретению могут быть получены с применением стадий, в целом известных для специалистов в данной области. Такие соединения могут быть проанализированы известными способами, включая без ограничения, LCMS (жидкостная хроматомасс-спектрометрия) и ЯМР (ядерный магнитный резонанс). Должно быть понятно, что специфические условия, представленные ниже, представляют собой только примеры и не предназначены для ограничения диапазона условий, которые могут быть применены для получения соединений по этому изобретению. Напротив, изобретение также включает условия, которые должны быть очевидны для специалистов в данной области для получения соединений по этому изобретению в рамках описания. Если не указано иное, все переменные на следующих схемах определены выше.
Применяются следующие аббревиатуры:
Общая схема:
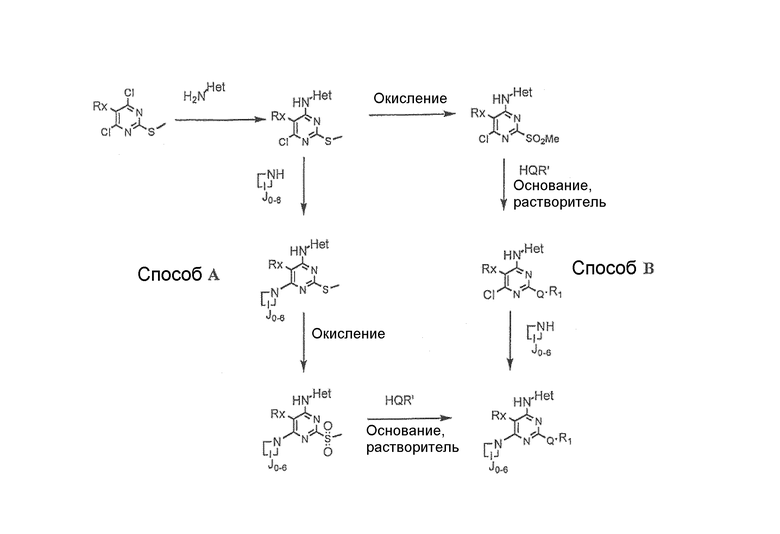
Общая схема выше показывает некоторые способы получения соединений по этому изобретению.
Схема I
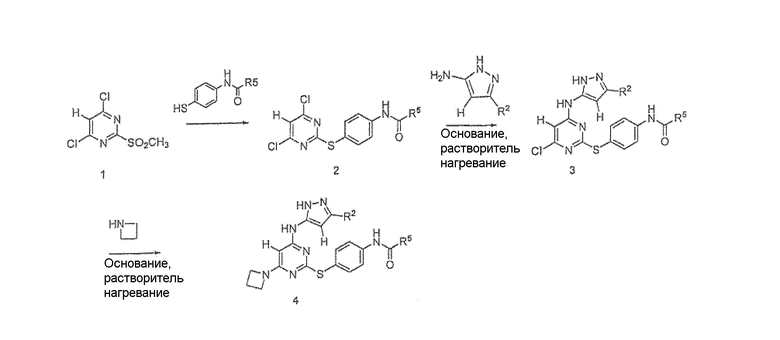
Схема I выше показывает общий путь получения соединений формулы 4 (на схеме I), где переменные определены выше. Дихлорированный пиримидин формулы 1 объединяли с HQ-R1 для получения соединения формулы 2. В некоторых вариантах осуществления два соединения нагревали в присутствии приемлемого растворителя (например, трет-BuOH) в течение 16 часов. В других вариантах осуществления два соединения перемешивали при 0°C в присутствии ацетонитрила и триэтиламина в течение 1 часа. Соединение формулы 2 затем нагревали в присутствии приемлемого растворителя (например, ДМФА) и подходящего основания (например, DIPEA/NaI) с необязательно замещенным аминопиразолом для образования соединения формулы 3, которое нагревали в присутствии азетидина в присутствии приемлемого растворителя (например, н-BuOH) для образования соединения формулы 4.
Схема II
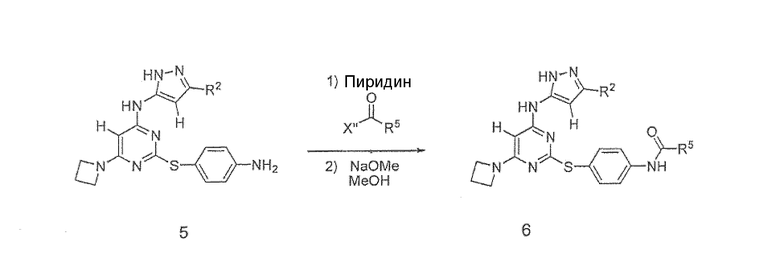
Схема II выше показывает общий путь получения соединений формулы 6 (на схеме II), где R2 и R5 определены выше. Соединение формулы 5 объединяли с приемлемым хлорангидридом (где X" представляет собой Cl) в присутствии пиридина для получения промежуточного соединения, которое при смешивании в присутствии метоксида натрия и метанола образует соединение формулы 6. В некоторых вариантах осуществления X" может представлять собой OH, в таком случае подходящий конденсирующий реагент для кислот применяется для конденсации кислоты с амином. Примеры подходящих конденсирующих реагентов для кислот включают, без ограничения, EDC, DCI и HOBT. Подходящие растворители для этих реакций конденсации включают, без ограничения, ТГФ, CH2Cl2 и диоксан.
Таким образом, настоящее изобретение относится к способам получения соединений по этому изобретению.
Способы исследования активности соединений по этому изобретению (например, киназные исследования) известны из уровня техники и также описаны в примерах, приведенных ниже.
Активность соединений в качестве ингибиторов протеинкиназ может быть исследована in vitro, in vivo или на клеточной линии. Исследования in vitro включают исследования, которые определяют ингибирование киназной активности или АТФазной активности активированной киназы. Альтернативные исследования in vitro количественно определяют способность ингибитора связываться с протеинкиназой и могут быть проведены путем введения радиоактивной метки в ингибитор перед связыванием, выделения комплекса ингибитор/киназа и определения количества связанной радиоактивной метки; или путем проведения конкурентного эксперимента, где новые ингибиторы инкубируют с киназой, связанной с известными радиолигандами.
Другой аспект изобретения относится к ингибированию киназной активности в биологическом образце, который включает контактирование указанного биологического образца с соединением формулы I или с композицией, включающей указанное соединение. Термин "биологический образец", применяемый здесь, обозначает образец in vitro или ex vivo, включая, без ограничения, клеточные культуры или их экстракты; биопсический материал, полученный из млекопитающего или его экстрактов; крови, слюны, мочи, кала, спермы, слез или других телесных жидкостей или их экстрактов.
Ингибирование киназной активности в биологическом образце может быть использовано для различных целей, которые известны специалисту в данной области. Примеры таких целей включают, без ограничения, переливание крови, трансплантацию органа, хранение биологического образца и биологические исследования.
Ингибирование киназной активности в биологическом образце также может быть использовано для исследования роли киназ в биологических и патологических процессах; исследования путей внутриклеточной сигнальной трансдукции, опосредованной такими киназами, и сравнительного исследования новых ингибиторов киназ.
Ингибиторы Аврора-протеинкиназы или их фармацевтические соли могут быть включены в фармацевтические композиции для введения животным или людям. Эти фармацевтические композиции, которые включают количество ингибитора Аврора-протеинкиназы, эффективное для лечения или предотвращения опосредованного Аврора-киназой состояния и фармацевтически приемлемый носитель представляют собой другой вариант осуществления настоящего изобретения.
Термин "опосредованное Аврора-киназой состояние" или "опосредованное Аврора-киназой заболевание" применяется здесь для обозначения любого заболевания или другого опасного состояния, в котором Аврора (Аврора-A, Аврора-B и Аврора-C), как известно, принимает участие. Такие состояния включают, без ограничения, рак, пролиферативные расстройства и миелопролиферативные расстройства.
Примеры миелопролиферативных расстройств включают, без ограничения, истинную полицитемию, тромбоцитемию, миелоидную метаплазию с миелофиброзом, хроническую миелогенную лейкемию (CML), хроническую миеломоноцитную лейкемию, гиперэозинофильный синдром, ювенильную миеломоноцитную лейкемию и системный мастоцитоз.
Термин "рак" также включает, без ограничения, следующие виды рака: эпидермальные ротовые: щечного кармана, губы, языка, рта, зева; сердца: саркому (ангиопластическую саркому, фибросаркому, рабдомиосаркому, липосаркому), миксому, рабдомиому, фиброму, липому и тератому; легкого: бронхогенную карциному (сквамозно-клеточную или эпидермоидную, недифференцированную мелкоклеточную, недифференцированную крупноклеточную, аденокарциному), альвеолярную карциному (аденоматоз легких), бронхиальную аденому, саркому, лимфому, хондроматозную гамартому, мезотелиому; желудочно-кишечные: пищевода (сквамозно-клеточную карциному гортани, аденокарциному, лейомиосаркому, лимфому), желудка (карциному, лимфому, лейомиосаркому), поджелудочной железы (аденокарциному протоков, инсулиному, глюкагонома, ульцерогенную аденому поджелудочной железы, карциноидные опухоли, випому), тонкой кишки или тонких кишок (аденокарциному, лимфому, карциноидные опухоли, саркому Капоши, лейомиому, гемангиому, липому, нейрофиброму, фиброму), толстой кишки или толстых кишок (аденокарциному, трубчатую аденому, ворсинчатую аденому, гамартому, лейомиому), ободочной кишки, ободочной кишки-прямой кишки, колоректальный; прямой кишки, мочеполового тракта: почки (аденокарциному, опухоль Уилма (Wilm) [нефробластому], лимфому, лейкемию), мочевого пузыря и уретры (сквамозно-клеточную карциному, переходно-клеточную карциному, аденокарциному), простаты (аденокарциному, саркому), яичка (сперматоцитому, тератому, эмбриональную карциному, тератокарциному, хориокарциному, саркому, интерстициально-клеточную карциному, фиброму, фиброаденому, аденоматоидные опухоли, липому); печени: гепатому (печеночно-клеточную карциному), холангиокарциному, гепатобластому, ангиосаркому, печеночно-клеточную аденому, гемангиому, желчных протоков; кости: остеогенную саркому (остеосаркому), фибросаркому, злокачественную фиброзную гистиоцитому, хондросаркому, саркому Юинга (Ewing), злокачественную лимфому (ретикуло-клеточную саркому), множественную миелому, злокачественную гигантско-клеточную хордому, остеохондроматоз (костно-хрящевой экзостоз), доброкачественную хондрому, хондробластому, хондромиксоидную фиброму, остеоид-остеому и гигантско-клеточные опухоли; нервной системы: черепные (остеому, гемангиому, гранулему, ксантому, деформирующий остоз), мягких мозговых оболочек (менингиому, менингиосаркому, глиоматоз), мозга (астроцитому, медуллобластому, глиому, эпендимальную глиому, герминому [аденому шишковидного тела], многообразные глиобластомы, олигодендроглиому, шванному, ретинобластому, внутренние опухоли), нейрофиброму спинного мозга, менингиому, глиому, саркому); гинекологические: матки (внутриматочную карциному), шейки матки (карциному шейки матки, предопухолевую дисплазию шейки матки), яичников (карциному яичников [серозную цистаденокарциному, слизеобразующую цистаденокарциному, неклассифицированную карциному], гранулезоклеточные опухоли, опухоли клеток Сертоли-Лейдинга (Sertoli-Leydig), семиному яичника, злокачественную тератому), вульвы (сквамозно-клеточную карциному, внутриэпителиальную карциному, аденокарциному, фибросаркому, меланому), влагалища (прозрачно-клеточную карциному, сквамозно-клеточную карциному, ботриоидную саркому (эмбриональную рабдомиосаркому), фаллопиевых труб (карцинома), груди; гематологические: крови (миелоидную лейкемию [острую и хроническую], острую лимфобластную лейкемию, хроническую лимфоцитарную лейкемию, миелопролиферативные заболевания, множественную миелому, миелодисплазивный синдром), болезнь Ходжкина, не-ходжкинскую лимфому [злокачественную лимфому] волосковых клеток; лимфатические расстройства; кожи: злокачественную меланому, базально-клеточную карциному, сквамозно-клеточную карциному, саркому Капоши, кератоакантому, синдром бородавочного диспластического невуса, липому, ангиому, дерматофиброму, келоиды, псориаз, щитовидной железы: папиллярную карциному щитовидной железы, фолликулярную карциному щитовидной железы; медуллярную карциному щитовидной железы, недифференцированный рак щитовидной железы, множественную эндокринную неоплазию типа 2A, множественную эндокринную неоплазию типа 2B, наследственный медуллярный рак щитовидной железы, хромаффиноцитому, параганглиому; и надпочечников: нейробластому. Таким образом, термин "раковая клетка", как здесь представлено, включает клетку, пораженную любым из идентифицированных выше состояний. В некоторых вариантах осуществления рак выбран из колоректального рака щитовидной железы или рака груди.
В некоторых вариантах осуществления соединения по этому изобретению являются применимыми для лечения рака, такого как колоректальный, щитовидной железы, груди, и рак легкого; и миелопролиферативных расстройств, таких как истинная полицитемия, тромбоцитемия, миелоидная метаплазия с миелофиброзом, хроническая миелогенная лейкемия, хроническая миеломоноцитная лейкемия, гиперэозинофильный синдром, ювенильная миеломоноцитная лейкемия и системный мастоцитоз.
В некоторых вариантах осуществления соединения по этому изобретению являются применимыми для лечения гемопоэтических расстройств, в частности, острой миелогенной лейкемии (AML), хронической миелогенной лейкемии (CML), острой промиелоцитарной лейкемии (APL) и острой лимфоцитарной лейкемии (ALL).
В добавление к соединениям по этому изобретению в композициях для лечения или предотвращения идентифицированных выше расстройств также могут быть применены фармацевтически приемлемые производные или пролекарства соединений по этому изобретению.
"Фармацевтически приемлемое производное или пролекарство" обозначает любой фармацевтически приемлемый сложный эфир, соль сложного эфира или другое производное соединения по этому изобретению, которое при введении пациенту способно предоставлять, или прямо или косвенно, соединение по этому изобретению или активный в качестве ингибитора метаболит, или его остаток. Такие производные или пролекарства включают те, что увеличивают биодоступность соединений по этому изобретению, когда такие соединения вводят пациенту (например, при пероральном введении соединения для более легкого абсорбирования в кровь) или которые усиливают доставку исходного соединения в биологический отдел (например, в мозг или лимфатическую систему) по сравнению с исходными образцами.
Примеры фармацевтически приемлемых пролекарств соединений по этому изобретению включают, без ограничения, сложные эфиры, сложные эфиры аминокислот, фосфатные эфиры, соли с металлами и сульфонатные эфиры.
Соединения по этому изобретению для лечения могут существовать в свободной форме или, если необходимо, в виде фармацевтически приемлемой соли.
Применяемый здесь термин "фармацевтически приемлемая соль" относится к солям соединения, которые, с точки зрения медицинской практики, являются подходящими для применения в контакте с тканями людей и низших животных без непомерной токсичности, раздражения, аллергической реакции и подобного, и соответствуют благоприятному отношению польза/риск.
Фармацевтически приемлемые соли соединений по этому изобретению включают полученные из приемлемых неорганических и органических кислот и оснований. Эти соли могут быть получены in situ при конечном выделении и очистке соединений. Соли присоединения кислоты могут быть получены путем 1) реакции очищенного соединения в форме его свободного основания с подходящей органической или неорганической кислотой и 2) выделения образуемой таким образом соли.
Примеры приемлемых солей с кислотами включают ацетат, адипат, альгинат, аспартат, бензоат, бензолсульфонат, бисульфат, бутират, цитрат, камфорат, камфорсульфонат, циклопентанпропионат, диглюконат, додецилсульфат, этансульфонат, формиат, фумарат, глюкогептаноат, глицерофосфат, гликолят, полусульфат, гептаноат, гексаноат, гидрохлорид, гидробромид, гидроиодид, 2- гидроксиэтансульфонат, лактат, малеат, малонат, метансульфонат, 2-нафталинсульфонат, никотинат, нитрат, оксалат, пальмоат, пектинат, персульфат, 3-фенилпропионат, фосфат, пикрат, пивалат, пропионат, салицилат, сукцинат, сульфат, тартрат, тиоцианат, тозилат и ундеканоат. Другие кислоты, такие как щавелевая, в то время как они не являются сами по себе фармацевтически приемлемыми, могут быть применены при получении солей, которые могут быть применены в качестве промежуточных соединений при получении соединений по изобретению и их фармацевтически приемлемых солей присоединения кислоты.
Соли присоединения основания могут быть получены путем 1) реакции очищенного соединения в форме кислоты с приемлемым органическим или неорганическим основанием и 2) выделения образуемой таким образом соли.
Соли, полученные из соответствующих оснований, включают соли со щелочным металлом (например, натрий и калий), щелочноземельным металлом (например, магний), аммонием и N+(C1-4-алкил)4. Это изобретение также включает кватернизацию любых основных азотсодержащих групп соединений, впервые описанных здесь. Путем такой кватернизации могут быть получены водо- или маслорастворимые или диспергируемые продукты.
Соли присоединения основания также включают соли с щелочным или щелочноземельным металлом. Репрезентативные соли с щелочным или щелочноземельным металлом включают натриевые, литиевые, калиевые, кальциевые, магниевые и подобные. Далее, фармацевтически приемлемые соли включают, если необходимо, нетоксичные аммониевые, четвертичные аммониевые и аминные катионы, образуемые с использованием противоионов, таких как галогенид, гидроксид, карбоксилат, сульфат, фосфат, нитрат, низший алкилсульфонат и арилсульфонат. Другие кислоты и основания, в то время как они сами не являются фармацевтически приемлемыми, могут быть применены для получения солей, которые могут быть применены в качестве промежуточных соединений при получении соединений по изобретению и их фармацевтически приемлемых солей присоединения кислоты или основания.
Фармацевтически приемлемые носители, которые могут быть применены в этих фармацевтических композициях, включают, без ограничения, ионообменники, оксид алюминия, стеарат алюминия, лецитин, протеины сыворотки, такие как человеческий сывороточный альбумин, буферные вещества, такие как фосфаты, глицин, сорбиновую кислоту, сорбат калия, смеси частично этерефицированных глицеридов насыщенных растительных жирных кислот, воду, соли или электролиты, такие как сульфат протамина, гидрогенфосфат динатрия, гидрогенфосфат калия, хлорид натрия, цинковые соли, коллоидный диоксид кремния, трисиликат магния, поливинилпирролидон, основанные на целлюлозе вещества, полиэтиленгликоль, натрий-карбоксиметилцеллюлозу, полиакрилаты, воски, блок-сополимеры полиэтилена и полиоксипропилена, полиэтиленгликоль и ланолин.
Композиции по настоящему изобретению могут быть введены перорально, парентерально, в виде спрея для ингаляции, местно, ректально, назально, буккально, вагинально или путем имплантируемой емкости. Термин "парентерально", применяемый здесь, включает подкожные, внутривенные, внутримышечные, внутрисуставные, внутрисиновиальные, надчревные, интратекальные, внутрибрюшинные, внутрипеченочные, внутрь пораженных тканей и внутричерепные методы инъекции или инфузии.
Стерильные инъецируемые формы композиции по этому изобретению могут представлять собой водную или масляную суспензию. Эти суспензии могут быть приготовлены согласно методикам, известным в технике, с применением приемлемых диспергирующих или смачивающих агентов и суспендирующих агентов. Стерильная инъецируемая препаративная форма также может представлять собой стерильный инъецируемый раствор или суспензию в нетоксичном парентерально-приемлемом разбавителе или растворителе, например, в виде раствора в 1,3-бутандиоле. Приемлемые носители и растворители, которые могут быть применены, представляют собой воду, раствор Рингера и изотонический раствор хлорида натрия. В добавление, стерильные нелетучие масла обычно применяются в качестве растворителя или суспендирующей среды. Для этой цели может быть применено безвкусное нелетучее масло, включая синтетические моно- или диглицериды. Жирные кислоты, такие как олеиновая кислота и ее глицеридные производные, могут быть применены при получении инъецируемых растворов, поскольку они представляют собой природные фармацевтически приемлемые масла, такие как оливковое масло или касторовое масло, особенно в их полиоксиэтилированных вариантах. Эти масляные растворы или суспензии также могут содержать длинноцепочечный спиртовой разбавитель или дисперсант, такой как карбоксиметилцеллюлоза или подобные диспергирующие агенты, которые обычно применяются при приготовлении композиции фармацевтически приемлемых лекарственных форм, включая эмульсии и суспензии. Для целей приготовления композиции также могут быть применены другие обычно применяемые поверхностно-активные вещества, такие как Tweens, Spans, другие эмульгирующие агенты или усиливающие биодоступность агенты, которые обычно применяются при производстве фармацевтически приемлемых твердых, жидких или других форм дозировки.
Фармацевтические композиции по этому изобретению могут быть введены перорально в любой орально приемлемой лекарственной форме, включая, без ограничения, капсулы, таблетки, водные суспензии или растворы. В случае таблеток для перорального применения обычно применяемые носители могут включать лактозу и кукурузный крахмал. Также могут быть добавлены смазывающие агенты, такие как стеарат магния. Для перорального введения в капсульной форме применяемые разбавители могут включать лактозу и высушенный кукурузный крахмал. Когда водные суспензии требуются для перорального применения, активный компонент может быть объединен с эмульгирующими и суспендирующими агентами. Если желательно, могут также быть добавлены определенные посластители, отдушки или окрашивающие агенты.
Альтернативно, фармацевтические композиции по этому изобретению могут быть введены в форме суппозиториев для ректального введения. Они могут быть получены путем смешивания агента с подходящим не вызывающим раздражения наполнителем, который представляет собой твердое вещество при комнатной температуре, но превращается в жидкость при ректальной температуре и, следовательно, будет расплавляться в прямой кишке для высвобождения лекарственного средства. Такие вещества могут включать кокосовое масло, воск и полиэтиленгликоли.
Фармацевтические композиции по этому изобретению также могут быть введены местно, особенно когда мишень для лечения включает области или органы, легко доступные путем местного применения, включая болезни глаз, кожи или нижний отдел кишечного тракта. Подходящие местные препаративные формы могут быть приготовлены для каждого из этих областей или органов.
Местное применение для нижнего отдела кишечного тракта может быть осуществлено в форме ректального суппозитория (см. выше) или в форме, подходящей для клизмы. Также могут быть применены местные чрескожные пластыри.
Для местных применений фармацевтические композиции могут быть составлены в виде приемлемой мази, содержащей активный компонент, суспендированный или растворенный в одном или нескольких носителях. Носители для местного нанесения соединений по этому изобретению могут включать, без ограничения, минеральное масло, жидкой вазелин, белый вазелин, пропиленгликоль, полиоксиэтилен, полиоксипропиленовое соединение, эмульгированный воск и воду. Альтернативно, фармацевтические композиции могут быть приготовлены в виде подходящего лосьона или крема, содержащего активные компоненты, суспендированные или растворенные в одном или нескольких фармацевтически приемлемых носителях. Подходящие носители могут включать, без ограничения, минеральное масло, моностеарат сорбита, полисорбат 60, цетиловый эфирный воск, цетеариловый спирт, 2-октилдодеканол, бензиловый спирт и воду.
Для офтальмологического применения фармацевтические композиции могут быть приготовлены в виде тонкоизмельченных суспензий в изотоническом, с установленным pH, стерильном физиологическом растворе или в виде растворов в изотоническом, с установленным pH, стерильном физиологическом растворе, или с консервантом, или без, таким как хлорид бензилалкония. Альтернативно, для офтальмологических применений фармацевтические композиции могут быть приготовлены в виде мази, такой как вазелин.
Фармацевтические композиции по этому изобретению также могут быть введены путем назального аэрозоля или ингаляции. Такие композиции могут быть получены в виде растворов в физиологическом растворе, использующем бензиловый спирт или другие подходящие консерванты, промоторы абсорбции для усиления биодоступности, фторуглероды и/или другие традиционные способствующие растворению или диспергированию агенты.
Количество ингибитора киназы, который может быть объединен с материалами носителей для приготовления единичной лекарственной формы, будет изменяться в зависимости от состояния излечиваемого пациента, особенности режима введения и симптомов. В варианте осуществления композиции должны быть составлены так, чтобы дозировка ингибитора в диапазоне 0,01-100 мг/кг массы тела/день могла быть введена пациенту, получающему эти композиции. В другом варианте осуществления композиции должны быть составлены так, чтобы дозировка ингибитора в диапазоне 0,1-100 мг/кг массы тела/день могла быть введена пациенту, получающему эти композиции.
Также должно быть понятно, что конкретные дозировки и режим лечения для любого отдельного пациента будут зависеть от разнообразных факторов, включая активность конкретного применяемого соединения, возраста, массы тела, общего состояния здоровья, пола, диеты, времени введения, скорости выделения, лекарственной комбинации, мнения лечащего врача и тяжести конкретной излечиваемой болезни. Количество ингибитора будет также зависеть от конкретного соединения в композиции.
Согласно другому варианту осуществления изобретение предоставляет способы лечения или предотвращения рака, пролиферативного расстройства или миелопролиферативного расстройства, включающие стадию введения пациенту одного из описанных здесь соединений или фармацевтических композиций.
Применяемый здесь термин "пациент" обозначает животное, включая человека.
В некоторых вариантах осуществления указанный способ применяется для лечения или предотвращения гемопоэтического расстройства, такого как острая миелогенная лейкемия (AML), острая промиелоцитарная лейкемия (APL), хроническая миелогенная лейкемия (CML) или острая лимфоцитарная лейкемия (ALL).
В других вариантах осуществления указанный способ применяется для лечения или предотвращения миелопролиферативных расстройств, таких как истинная полицитемия, тромбоцитемия, миелоидная метаплазия с миелофиброзом, хроническая миелогенная лейкемия (CML), хроническая миеломоноцитная лейкемия, гиперэозинофильный синдром, ювенильная миеломоноцитная лейкемия и системный мастоцитоз.
В следующих вариантах осуществления указанный способ применяется для лечения или предотвращения рака, такого как рак груди, толстой кишки, простаты, кожи, поджелудочной железы, мозга, мочеполового тракта, лимфатической системы, желудка, гортани и легкого, включая аденокарциному легкого, мелкоклеточный рак легкого и немелкоклеточный рак легкого.
Другой вариант осуществления предоставляет способ лечения или предотвращения рака, включающий стадию введения пациенту соединения формулы I или композиции, включающей указанное соединение.
Другой аспект изобретения относится к ингибированию киназной активности у пациента, где способ включает введение пациенту соединения формулы I или композиции, включающей указанное соединение. В некоторых вариантах осуществления упомянутая киназа представляет собой Аврора-киназу (Аврора-A, Аврора-B, Аврора-C), Abl, Arg, FGFR1, MELK, MLK1, MuSK, Ret или TrkA.
В зависимости от конкретных излечиваемых или предотвращаемых состояний дополнительные лекарственные средства могут быть назначены совместно с соединениями по этому изобретению. В некоторых случаях эти дополнительные лекарственные средства обычно назначаются для лечения или предотвращения того же состояния. Например, химиотерапевтические агенты или другие антипролиферативные агенты могут быть объединены с соединениями по этому изобретению для лечения пролиферативных заболеваний.
Другой аспект этого изобретения направлен на способ лечения рака у пациента, нуждающегося в таком лечении, включающий последовательное или совместное введение соединения по этому изобретению или его фармацевтически приемлемой соли, и другого терапевтического агента. В некоторых вариантах осуществления указанный дополнительный терапевтический агент выбран из противоракового агента, антипролиферативного агента или химиотерапевтического агента.
В некоторых вариантах осуществления указанный дополнительный терапевтический агент выбран из камптотецина, ингибитора MEK: U0126, ингибитора KSP (белок движения веретена), адриамицина, интерферонов и производных платины, таких как цисплатин.
В других вариантах осуществления указанный дополнительный терапевтический агент выбран из таксанов; ингибиторов bcr-abl (таких как гливек, дасатиниб и нилотиниб); ингибиторов EGFR (таких как тарсева и иресса); повреждающих ДНК агентов (таких как цисплатин, оксалиплатин, карбоплатин, ингибиторов топоизомеразы, и антарациклинов); и антиметаболитов (таких как AraC и 5-FU).
В следующих вариантах осуществления указанный дополнительный терапевтический агент выбран из камптотецина, доксорубицина, идарубицина, цисплатина, таксола, таксотера, винкристина, тарсева, ингибитора MEK, U0126, ингибитора KSP, вориностата, гливека, дасатиниба и нилотиниба.
В другом варианте осуществления указанный дополнительный терапевтический агент выбран из ингибиторов Her-2 (такого как Herceptin); ингибиторов HDAC (таких как вориностат), ингибиторов VEGFR (таких как авастин), ингибиторов c-KIT и FLT-3 (таких как сунитиниб), ингибиторов BRAF (таких как BAY 43-9006 от Bayer) ингибиторов MEK (таких как PD0325901 от Pfizer); веществ, повреждающих веретено деления (таких как эпотилоны) и мелкодисперсного связанного с белком паклитаксела (такого как Abraxane®).
Другие терапевтические средства или противораковые агенты, которые могут быть применены в комбинации с противораковыми агентами по настоящему изобретению, включают хирургическое вмешательство, лучевую терапию (в качестве отдельных примеров можно привести гамма-облучение, лучевую терапию нейтронным пучком, лучевую терапию электронным пучком, протонную терапию, близкофокусную лучевую терапию и системное действие радиоактивных изотопов), эндокринную терапию, модификаторы биологического отклика (в частности, интерфероны, интерлейкины и фактор некроза опухолей (TNF)), гипертермию и криотерапию, агенты для ослабления любых побочных эффектов (например, противорвотные средства) и другие разрешенные химиотерапевтические лекарственные средства, включая, без ограничения, алкилаторные лекарственные средства (мехлоретамин, хлорамбуцил, циклофосфамид, мелфалан, ифосфамид), антиметаболиты (метотрексат), антагонисты пуринов и антагонисты пиримидинов (6-меркаптопурин, 5-фторурацил, цитарабил, гемцитабин), веретенные яды (винбластин, винкристин, винорелбин, паклитаксел), подофилотоксины (этопозид, иринотекан, топотекан), антибиотики (доксорубицин, блеомицин, митомицин), нитрозомочевины (кармустин, ломустин), неорганические ионы (цисплатин, карбоплатин), ферменты (аспарагиназа), и гормоны (тамоксифен, лейпролид, флутамид и мегестрол), GleevecТМ, дексаметазон и циклофосфамид.
Соединение по настоящему изобретению также может быть применено для лечения рака в комбинации со следующими терапевтическими агентами: абареликс (Phenaxis depot®); алдеслейкин (Prokine®); алдеслейкин (Proleukin®); алемтузумаб (Campath®); алитретиноин (Panretin®); аллопуринол (Zyloprim®); алтретамин (Hexalen®); амифостин (Ethyol®); анастрозол (Arimidex®); триоксид мышьяка (Trisenox®); аспарагиназа (Elspar®); азацитидин (Vidaza®); бевацузимаб (Avastin®); бексаротен в капсулах (Targretin®); бексаротен-гель (Targretin®); блеомицин (Blenoxane®); бортезомиб (Velcade®); бусулфан внутривенный (Busulfex®); бусулфан оральный (Myleran®); калустерон (Methosarb®); капецитабин (Xeloda®); карбоплатин (Paraplatin®); кармустин (BCNU®, BiCNU®); кармустин (Gliadel®); кармустин с имплантантом полифепросан 20 (Gliadel Wafer®); целекоксиб (Celebrex®); цетуксимаб (Erbitux®); хлорамбуцил (Leukeran®); цисплатин (Platinol®); кладрибин (Leustatin®, 2-CdA®); клофарабин (Clolar®); циклофосфамид (Cytoxan®, Neosar®); циклофосфамид (Cytoxan Injection®); циклофосфамид (Cytoxan Tablet®); цитарабин (Cytosar-U®); цитарабин липосомальный (DepoCyt®); дакарбазин (DTIC-Dome®); дактиномицин, актиномицин D (Cosmegen®); дарбепоэтин альфа (Aranesp®); даунорубицин липосомальный (Danuoxome®); даунорубицин, дауномицин (Daunorubicine®); даунорубицин, дауномицин (Cerubidine®); денилейкин дифтитокc (Ontak®); дексразоксан (Zinecard®); доцетаксел (Taxotere®); доксорубицин (Adriamycin PFS®); доксорубицин (Adriamycin®, Rubex®); доксорубицин (Adriamycin PFS Injection®); доксорубицин липосомальный (Doxil®); дромостанолон пропионат (Dromostanolone®); дромостанолон пропионат (Masterone Injection®); раствор B по Эллиоту (Elliott) (Elliott's B Solution®); эпирубицин (Ellence®); эпоэтин альфа (Epogen®); эрлотиниб (Tarceva®); эстрамустин (Emcyt®); этопозид фосфат (Etopophos®); этопозид, VP-16 (Vepesid®); эксеместан (Aromasin®); филграстим (Neupogen®); флоксуридин (внутриартериальный) (FUDR®); флударабин (Fludara®); фторурацил, 5-FU (Adrucil®); фулвестрант (Faslodex®); гефитиниб (Iressa®); гемцитабин (Gemzar®); гемтузумаб озогамицин (Mylotarg®); госерелин ацетат (Zoladex Implant®); госерелин ацетат (Zoladex®); гистрелин ацетат (Histrelin Implant®); гидроксимочевина (Hydrea®); ибритумомаб тиуксетан (Zevalin®); идарубицин (Idamycin®); ифосфамид (IFEX®); мезилат иматиниба (Gleevec®); интерферон альфа-2a (Roferon A®); Интерферон альфа-2b (Intron A®); иринотекан (Camptosar®); леналидомид (Revlimid®); летрозол (Femara®); лейковорин (Wellcovorin®, Leucovorin®); Лейпролид ацетат (Eligard®); левамизол (Ergamisol®); ломустин, CCNU (CeeBU®); меклоретамин, азотное горчичное масло (Mustargen®); мегестрол ацетат (Megace®); мелфалан, L-PAM (Alkeran®); меркаптопурин, 6-MP (Purinethol®); месна (Mesnex®); месна (Mesnex tabs®); метотрексат (Methotrexate®); метоксален (Uvadex®); митомицин C (Mutamycin®); митотан (Lysodren®); митоксантрон (Novantrone®); фенпропионат нандролона (Durabolin-50®); неларабин (Arranon®); нофетумомаб (Verluma®); опрелвекин (Neumega®); оксалиплатин (Eloxatin®); паклитаксель (Paxene®); паклитаксель (Taxol®); мелкодисперсный паклитаксель, связанный с белком (Abraxane®); палифермин (Kepivance®); памидронат (Aredia®); пегадемаза (Adagen (Pegademase Bovine)®); пегаспаргаза (Oncaspar®); пегфилграстим (Neulasta®); пеметрекс-динатрий (Alimta®); пентостатин (Nipent®); пипоброман (Vercyte®); пликамицин, митрамицин (Mithracin®); порфимер натрий (Photofrin®); прокарбазин (Matulane®); хинакрин (Atabrine®); расбуриказа (Elitek®); Ритуксимаб (Rituxan®); сарграмостим (Leukine®); сарграмостим (Prokine®); зорафениб (Nexavar®); стрептозоцин (Zanosar®); малеат сунитиниба (Sutent®); тальк (Sclerosol®); тамоксифен (Nolvadex®); темозоломид (Temodar®); тенипозид, VM-26 (Vumon®); тестолактон (Teslac®); тиогуанин, 6-TG (Тhiоguanine®); тиотепа (Thioplex®); топотекан (Hycamtin®); торемифен (Fareston®); тоситумомаб (Bexxar®); Тоситумомаб/I-131, тоситумомаб (Bexxar®); трастузумаб (Herceptin®); третиноин, ATRA (Vesanoid®); урацильное горчичное масло (Uracil Mustard Capsules®); валрубицин (Valstar®); винбластин (Velban®); винкристин (Oncovin®); винорелбин (Navelbine®); золедронат (Zometa®) и вориностат (Zolinza®).
Для более подробного обсуждения современных противораковых терапий см. http://www.nci.nih.gov/, перечень противораковых лекарственных средств, утвержденных Управлением по контролю за продуктами и лекарственными средствами (FDA) на сайте http://www.fda.gov/cder/cancer/druglistframe.htm, и справочник Merck, Seventeenth Ed. 1999, полное содержание которых включено сюда в виде ссылки.
Другой вариант осуществления предоставляет одновременное, раздельное или последовательное применение комбинированного препарата.
Эти дополнительные агенты могут быть назначены отдельно, в виде части режима с множественным дозированием, от соединения или композиции, содержащей ингибитор киназы. Альтернативно, такие агенты могут представлять собой часть единичной лекарственной формы, смешанной совместно с ингибитором киназы в единой композиции.
Для того чтобы это изобретение было более полно понятно, далее представлены следующие препаративные примеры и примеры тестирования. Эти примеры даны только для целей иллюстрации и не должны обсуждаться, любым способом, как ограничивающие объем изобретения. Все документы, цитированные здесь, таким образом являются включенными сюда в виде ссылки.
ПРИМЕРЫ
Применяемый здесь термин "Rt(мин)" относится ко времени удерживания в методе ВЭЖХ, приведенному для данного соединения в минутах. Если не указано иное, метод ВЭЖХ использовали для получения указанного времени удерживания, как указано ниже:
колонка: колонка ACE C8, 4,6×150 мм
градиент: 0-100% ацетонитрил + метанол 60:40 (20 мМ Трисфосфата)
скорость потока: 1,5 мл/мин
анализ: 225 нм.
Образцы для масс-спектроскопии анализировали на масс-спектрометре MicroMass Quattro Micro, функционирующем в режиме одиночного масс-спектра с электроспрей-ионизацией. Образцы вводили в масс-спектрометр с применением хроматографии. Подвижная фаза для всех масс-спектрометрических анализов состояла из 10 мМ раствора, при pH 7, ацетата аммония и смеси 1:1 ацетонитрил-метанол, условия градиента на колонке: 5%-100% ацетонитрил-метанол в течение 3,5 минут времени градиента и 5 минут времени элюирования на колонке ACE C8 3,0×75 мм. Скорость потока составляла 1,2 мл/мин.
1H-ЯМР спектры измеряли при 400 МГц с применением прибора Bruker DPX 400. Следующие соединения формулы I получали и анализировали, как описано ниже.
Пример 1
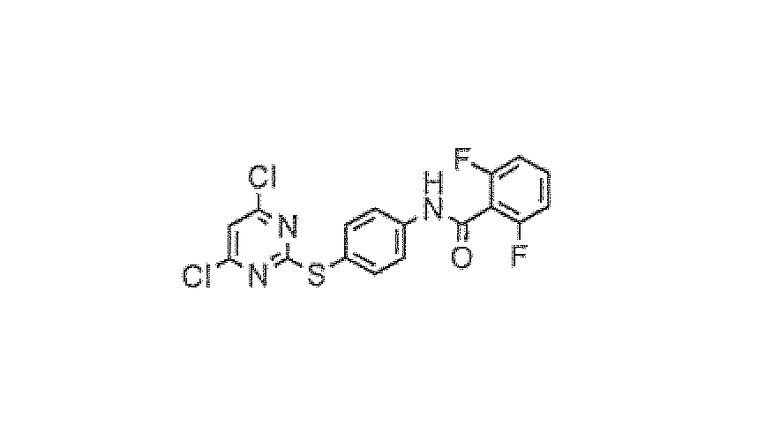
2,6-Дифтор-N-[4-(4,6-дихлорпиримидин-2-илсульфанил)фенил]бензамид
В 250-мл круглодонную колбу, снабженную обратным холодильником, помещали 4,6-дихлор-2-метансульфонилпиримидин (4,2 г, 18,6 ммоль), 2,6-дифтор-N-(4-меркаптофенил)бензамид (4,98 г, 18,8 ммоль) и трет-бутанол (75 мл) в атмосфере азота. Реакционную смесь тщательно дегазировали и затем нагревали при 90°C в течение 2 часов. Реакционной смеси позволяли охладиться до комнатной температуры и концентрировали в вакууме. Твердый остаток переводили в этилацетат (50 мл), промывали насыщенным раствором бикарбоната натрия и насыщенным солевым раствором. Органическую фазу сушили над сульфатом магния, фильтровали и концентрировали до начала выпадения продукта в осадок. Смесь затем охлаждали и выдерживали в течение 12 часов. Продукт собирали путем фильтрования, промывали холодным этилацетатом и высушивали. Это дало соединение, обозначенное в заголовке примера, в виде не совсем белого твердого вещества (2,7 г, 35%).
1H-ЯМР (ДМСО) 7,32 (2H, м), 7,61 (3H, м), 7,79 (1H, с), 7,82 (2H, д), 10,9 (1H, с). MS (ES+): 412,19.
Пример 2
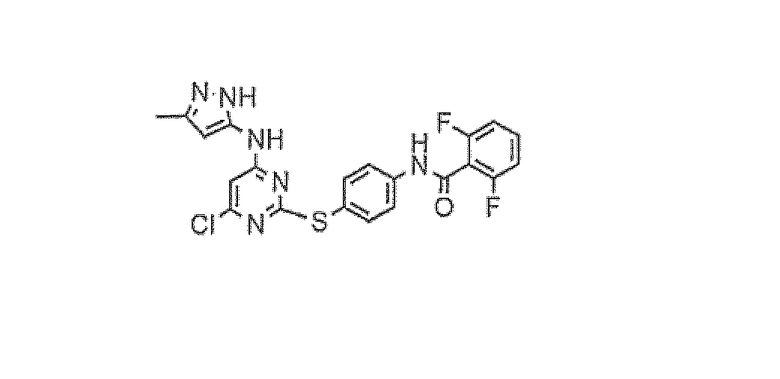
2,6-Дифтор-N-{4-[4-хлор-6-(5-метил-2Н-пиразол-3-иламино)пиримидин-2-илсульфанил]фенил}бензамид
В 50-мл круглодонную колбу помещали 2,6-дифтор-N-[4-(4,6-дихлорпиримидин-2-илсульфанил)фенил]бензамид (1,0 г, 2,3 ммоль), 5-метил-2H-пиразол-3-иламин (250 мг, 2,58 ммоль), иодид натрия (351 мг, 2,34 ммоль), диизопропилэтиламин (333 мг, 2,58 ммоль) и диметилформамид (5 мл) в атмосфере азота. Реакционную смесь перемешивали при 90°C в течение 18 часов, затем позволяли охладиться до комнатной температуры. Реакционную смесь разбавляли этилацетатом (25 мл), промывали насыщенным раствором бикарбоната натрия и насыщенным солевым раствором. Органическую фазу сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Соединение очищали методом флэш-хроматографии (75 до 80% этилацетат/петролейный эфир) с получением соединения, обозначенного в заголовке примера (1,08 г, 98%).
1H-ЯМР (ДМСО) 2,00 (3H, с), 5,25 (1H, ушир.с), 6,48 (1H, ушир.с), 7,30-7,97 (7H, м), 10,28 (1H, с), 10,89 (1H, с), 11,90 (1H, с); MS(ES+): 473,4.
Пример 3

N-{4-[4-азетидин-1-ил-6-(5-метил-2Н-пиразол-3-иламино)пиримидин-2-илсульфанил]фенил}-2,6-дифторбензамид
(Соединение I-2)
В 10-мл круглодонную колбу помещали 2,6-дифтор-N-{4-[4-хлор-6-(5-метил-2H-пиразол-3-иламино)пиримидин-2-илсульфанил]фенил}бензамид (150 мг, 0,31 ммоль), азетидин (35 мг, 0,62 ммоль), диизопропилэтиламин (80 мг, 0,62 ммоль) и н-бутанол (1,5 мл). Реакционную смесь перемешивали при 80°C в течение 4 часов, затем охлаждали и концентрировали в вакууме. Соединение очищали методом препаративной ВЭЖХ (MeCN/вода + 0,05% TFA 10/90 до 100/0 в течение 10 мин), что дало соединение, обозначенное в заголовке примера в виде соли с трифторуксусной кислотой (54 мг, 29%).
1H-ЯМР (ДМСО) 2,05 (3H, с), 2,25-2,36 (2H, м), 3,70-3,98 (4H, м, наложение), 5,31 (1H, с), 5,52 (1H, ушир.с), 7,39 (2H, м), 7,46-7,67 (3H, м), 7,79 (2H, д), 9,35 (1H, ушир.с), 11,04 (1H, с), 11,80 (1H, ушир.с); MS (ES+):494,5.
В таблице 2 ниже представлены данные для соединений, полученных согласно способу, описанному на схеме I и в примерах 1-3. Номера соединений соответствуют тем соединениям, что приведены в таблице 1.
(наблюдаемый)
Пример 4
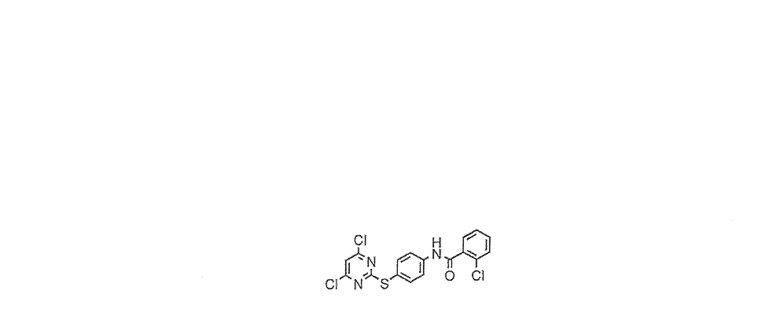
2-Хлор-N-[4-(4,6-дихлорпиримидин-2-илсульфанил)фенил]бензамид
В 250-мл круглодонную колбу помещали 4,6-дихлор-2-метансульфонилпиримидин (7,00 г, 26,6 ммоль), 2-хлор-N-(4-меркаптофенил)бензамид (6,33 г, 27,9 ммоль) и ацетонитрил (100 мл) в атмосфере азота. После растворения твердого вещества, реакционную смесь охлаждали до 0°C и по каплям добавляли триэтиламин (3,7 мл, 26,6 ммоль). Раствор перемешивали при 0°C в течение 10 мин, затем позволяли нагреться до комнатной температуры и перемешивали в течение 1 часа. После этого времени добавляли воду (50 мл), белое твердое вещество выпадало в осадок и реакционную смесь перемешивали в течение еще 4 часов. После этого времени реакционную смесь фильтровали, твердое вещество промывали ацетонитрилом (2×10 мл) с получением указанного в заголовке соединения в виде белого твердого вещества (8,03 г, 74%).
1H-ЯМР (ДМСО) 7,4-7,6 (5H, м), 7,7 (1H, с), 7,80-7,85 (2H, д), 10,9 (1H, с). MS (ES+): 412.
Пример 5
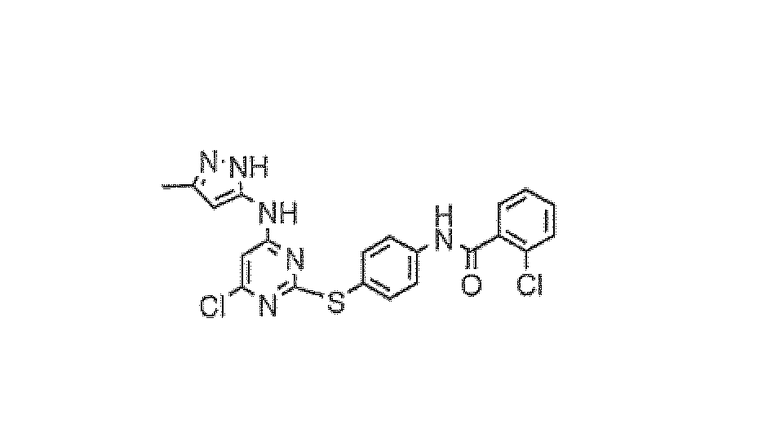
2-Хлор-N-{4-[4-хлор-6-(5-метил-2H-пиразол-3-иламино)пиримидин-2-илсульфанил]фенил}бензамид
В 250-мл круглодонную колбу помещали 2-хлор-N-[4-(4,6-дихлорпиримидин-2-илсульфанил)фенил]бензамид (12,5 г, 30,4 ммоль), 5-метил-2H-пиразол-3-иламин (3,55 г, 36,5 ммоль), иодид натрия (4,56 г, 30,4 ммоль), N,N-диизопропилэтиламин (6,9 мл, 40,0 ммоль) и N,N-диметилформамид (125 мл) в атмосфере азота. Реакционную смесь перемешивали при 90°C в течение 5 часов, затем позволяли охладиться до комнатной температуры. Добавляли воду (600 мл) и полученную суспензию перемешивали при комнатной температуре в течение 2 часов и твердое вещество собирали путем фильтрования и высушивания. Полученное белое твердое вещество растирали с горячим этилацетатом (50 мл), фильтровали и промывали этилацетатом (1×20 мл) с получением указанного в заголовке соединения в виде белого твердого вещества (11,76 г, 82%).
1H-ЯМР (ДМСО): 2,16 (3H, с), 5,30 (1H, с), 6,48 (1H, с), 7,49-7,62 (6H, м), 7,89 (2H, м), 10,28 (1H, с), 10,84 (1H, с), 11,93 (1H, с); MS (ES+): 471.
Пример 6
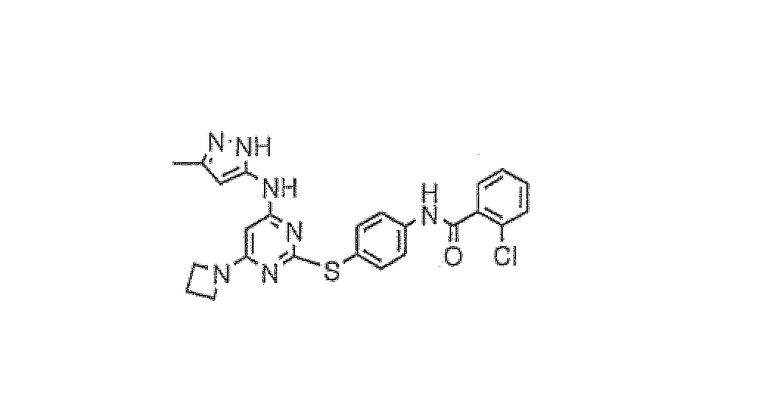
N-{4-[4-азетидин-1-ил-6-(5-метил-2H-пиразол-3-иламино)пиримидин-2-илсульфанил]фенил}-2-хлорбензамид
(Соединение I-1)
В 500-мл круглодонную колбу помещали 2-хлор-N-{4-[4-хлор-6-(5-метил-2Н-пиразол-3-иламино)пиримидин-2-илсульфанил]фенил}бензамид (16,0 г, 34,0 ммоль), азетидин (3,87 г, 68,0 ммоль), N,N-диизопропилэтиламин (13,0 мл, 74,7 ммоль) и н-бутанол (250 мл). Реакционную смесь перемешивали при 90°C в течение 5 часов. Реакционную смесь охлаждали и концентрировали в вакууме. Добавляли диэтиловый эфир (200 мл) и в осадок выпадало светло-коричневое твердое вещество. Раствор фильтровали и твердое вещество перекристаллизовывали из этанола с получением чистого продукта в виде белого твердого вещества (9,42 г, 52%)
1H-ЯМР (ДМСО): 2,04 (3H, с), 2,32 (2H, м), 3,87 (4H, м), 5,39 (1H, с), 5,66 (1H, ушир.с), 7,48-7,59 (6H, м), 7,82 (2H, м), 9,87 (1H, с), 10,74 (1H, с), 11,68 (1H, с); MS (ES+): 492.
Другой способ, примененный для получения примера 6, описан ниже:
К суспензии 2-хлор-N-{4-[4-хлор-6-(5-метил-2Н-пиразол-3-иламино)пиримидин-2-илсульфанил]фенил}бензамида (169 г, 0,36 моль) в 2-пропаноле (1,3 л) порциями добавляли азетидин (100 г, 1,76 моль). Реакционную смесь нагревали до 80-82°C. Через 24 часа добавляли диизопропилэтиламин (73,4 г, 0,57 моль). Протекание реакции контролировали методом ВЭЖХ. Реакционную смесь концентрировали при пониженном давлении досуха, азеотропно отгоняли с метанолом три раза (3×650 мл), перемешивали в течение 2 часов в метаноле (1 л) при 40°C и охлаждали до 10°C. Фильтровали полученное не совсем белое твердое вещество. Выделенное вещество суспендировали в ацетонитриле при кипячении с обратным холодильником в течение 3 часов, охлаждали до 20-25°C, фильтровали и высушивали в вакуумной печи в течение ночи. Вещество суспендировали в ацетонитриле снова при кипячении с обратным холодильником в течение 3 часов, охлаждали до 20-25°C и фильтровали. Веществу позволяли высохнуть до постоянной массы. Желаемый продукт выделяли в виде не совсем белого твердого вещества (154 г, 86%).
В таблице 3 ниже представлены данные для определенных типичных соединений, полученных согласно способу, описанному на схеме I и в примерах 4-6. Номера соединений соответствуют тем соединениям, что приведены в таблице 1.
соединения
(наблюдаемый)
Пример 7
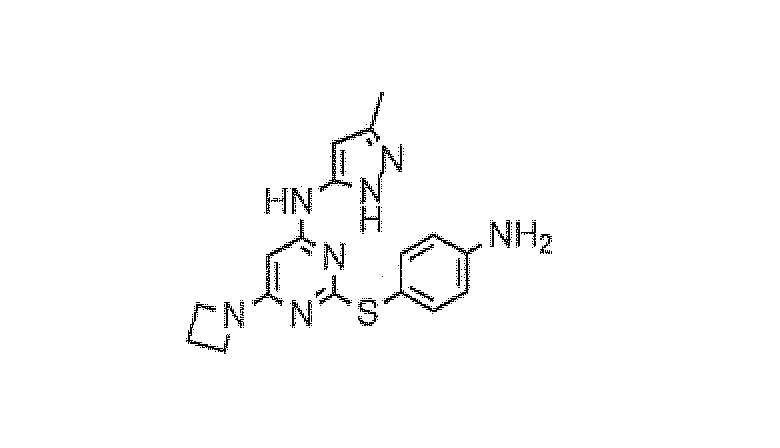
2-(4-аминофенилтио)-6-(азетидин-1-ил)-N-(3-метил-1H-пиразол-5-ил)пиримидин-4-амин
Трет-бутил 4-(4-(5-метил-1H-пиразол-3-иламино)-6-(азетидин-1-ил)пиримидин-2-илтио)фенилкарбамат (полученный с применением способа, аналогичного тому, что описан для примера 6) (2,53 г, 5,6 ммоль) растворяли в 1:1 TFA-DCM (20 мл) и полученному раствору позволяли стоять в течение ночи при комнатной температуре. Раствор концентрировали в вакууме. Остаток суспендировали в EtOAc и промывали насыщенным водным раствором бикарбоната натрия (×2), затем насыщенным солевым раствором и высушивали над сульфатом натрия. Полученное желто-коричневое твердое вещество (1,8 г, 91%) [MS (ES+) 354] применяли без дополнительной очистки или охарактеризовывания на следующей стадии.
Пример 8
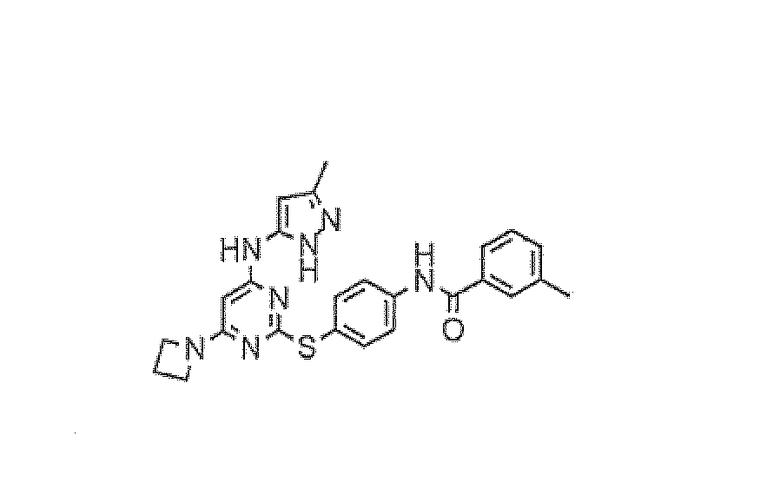
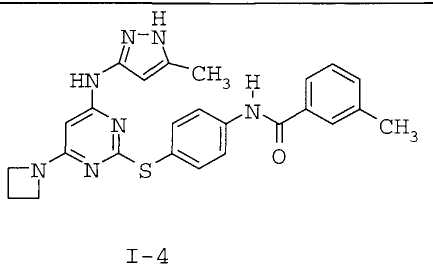
N-(4-(4-(3-метил-1H-пиразол-5-иламино)-6-(азетидин-1-ил)пиримидин-2-илтио)фенил)-3-метилбензамид (I-4)
2-(4-Аминофенилтио)-6-(азетидин-1-ил)-N-(3-метил-1H-пиразол-5-ил)пиримидин-4-амин (200 мг, 0,57 ммоль) суспендировали в пиридине (2 мл) и по каплям при комнатной температуре добавляли м-толуоилхлорид (0,187 мл, 1,42 ммоль). Через 15 минут реакционную смесь концентрировали в вакууме и остаток суспендировали в метаноле (3 мл). Добавляли метоксид натрия (25% мас./мас. раствор в MeOH, 1 мл) и полученный мутный раствор перемешивали при комнатной температуре в течение 15 минут. Реакционную смесь прямо очищали методом хроматографии (оксид кремния, 5-100% EtOAc-петролейный эфир, градиентное элюирование) с получением указанного в заголовке соединения (89 мг, 33%) в виде белого твердого вещества.
1H-ЯМР (400 МГц, ДМСО) 1,99 (3H, ушир.с), 2,31 (2H, кв.), 2,42 (3H, с), 3,88 (4H, т), 5,39 (1H, ушир.с), 5,67 (1H, оч.шир.с), 7,43-7,46 (2H, м), 7,54 (2H, д), 7,73-7,76 (2H, м), 7,91 (2H, д), 9,23 (1H, ушир.с), 10,42 (1H, с), 11,67 (1H, ушир.с). ES+ 472.
В таблице 4 ниже представлены данные для определенных типичных соединений, полученных согласно способу, описанному на схеме II и в примерах 7-8. Номера соединений соответствуют тем соединениям, что приведены в таблице 1.
соединения
(наблюдаемый)
Экспериментальные данные, представленные ниже, описывают получение некоторых соединений, примененных в описанных здесь примерах.
Соединение a:
2-хлор-N-(4-меркаптофенил)бензамид
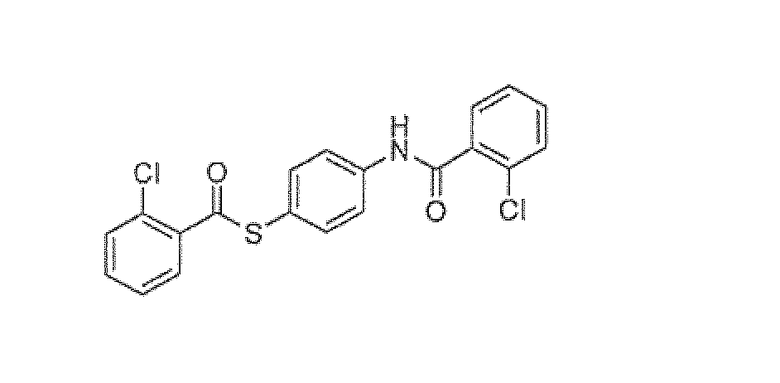
S-4-(2-хлорбензамидо)фенил 2-хлорбензотиоат
Дегазированный EtOAc (3,2 л) помещали в колбу. Растворитель охлаждали до 0°C в атмосфере азота. 4-аминобензолтиол (435 г, 3,48 моль) расплавляли и добавляли непосредственно в колбу. Добавляли триэтиламин (773 г, 7,65 моль) в течение 30 минут образования осадка. Затем добавляли в чистом виде 2-хлорбензоилхлорид (1340 г, 7,65 моль) при поддерживании температуры ниже 5°C. После завершения добавления смесь нагревали до 20°C в течение одного часа. Суспензию фильтровали и остаток промывали EtOAc (780 мл). Вещество сушили при 50°C в вакууме в токе азота до получения постоянной массы. Соединение вводили в следующую реакцию без дополнительной очистки.
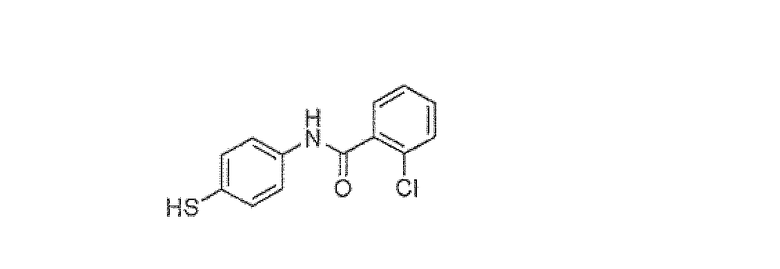
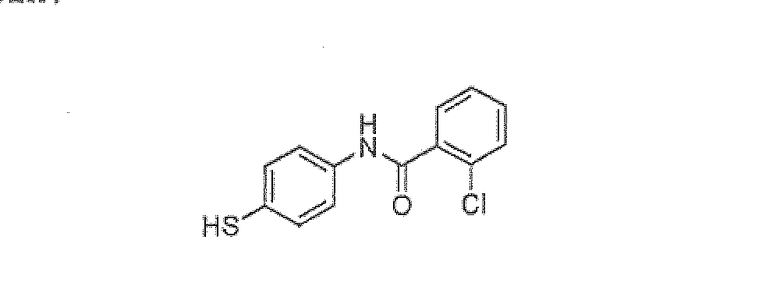
2-Хлор-N-(4-меркаптофенил)бензамид
S-4-(2-хлорбензамидо)фенил 2-хлорбензотиоат (305 г, 0,76 моль), EtOAc (325 мл) и воду (65 мл) помещали в колбу, снабженную обратным холодильником. Добавляли раствор NaOH (3 экв., 50% водный) и смесь нагревали до 70°C в течение 30-40 минут. EtOAc удаляли методом дистилляции при 100 мм Hg и смесь охлаждали до 5°C. Смесь подкисляли 6N HCl до pH 2. Затем вакуумным фильтрованием собирали твердое вещество и промывали водой (390 мл). Твердое вещество суспендировали в CH2Cl2 (520 мл) и промывали насыщенным водным NaHCO3. Органический слой сушили над Na2SO4, фильтровали и концентрировали с получением желаемого вещества (174 г, 87%).
Пример 10: Исследование ингибирования Авроры-2 (Авроры A)
Соединения подвергали скринингу на их способность ингибировать Аврору-2 с применением стандартного сопряженного ферментного анализа (Fox et al., Protein Sci. (1998) 7, 2249). Исследования проводили в смеси 100 мМ Hepes (pH 7,5), 10 мМ MgCl2, 1 мМ DTT, 25 мМ NaCl, 2,5 мМ фосфоенолпирувата, 300 мкМ NADH, 30 мкг/мл пируват-киназы и 10 мкг/мл лактат-дегидрогеназы. Конечные концентрации субстрата в исследовании составляли 400 мкМ АТФ (Sigma Chemicals) и 570 мкМ пептида (Kemptide, American Peptide, Sunnyvale, CA). Исследования проводили при 30°C и в присутствии 40 нМ Авроры-2.
Исходный буферный раствор для исследования содержал все реагенты, перечисленные выше, за исключением Авроры-2 и тестируемого интересующего соединения. 55 мкл исходного раствора помещали в 96-луночный планшет с последующим добавлением 2 мкл исходного раствора в ДМСО, содержащего серийные разбавления тестируемого соединения (типично исходя из конечной концентрации 7,5 мкМ). Планшет предварительно инкубировали в течение 10 минут при 30°C и реакцию инициировали путем добавления 10 мкл Авроры-2. Начальные скорости реакции определяли на ридере планшетов Molecular Devices SpectraMax Plus в течение 10-минутного отрезка времени. Данные для IC50 и Ki вычисляли методом нелинейного регрессионного анализа с применением пакета программного обеспечения Prism (GraphPad Prism версия 3,0cx для Macintosh, GraphPad Software, San Diego California, USA).
Пример 11: Исследование ингибирования Авроры-1 (Авроры-B) (радиометрическое)
Приготавливали буферный раствор для исследования, который состоял из 25 мМ HEPES (pH 7,5), 10 мМ MgCl2, 0,1% BSA и 10% глицерина. В буфере для исследования приготавливали 22 нМ раствор Авроры-B, также содержащий 1,7 мМ DTT и 1,5 мМ Kemptide (LRRASLG). К 22 мкл раствора Авроры-B в 96-луночном планшете добавляли 2 мкл исходного раствора соединения в ДМСО и смеси позволяли приходить к состоянию равновесия в течение 10 минут при 25°C. Ферментативную реакцию инициировали путем добавления 16 мкл исходного раствора [γ-33P]-АТФ (-20 nCi/мкл), полученного в буфере для исследования, до конечной концентрации для исследования 800 мкМ. Реакцию останавливали через 3 часа путем добавления 16 мкл 500 мМ фосфорной кислоты и уровни включения 33P в пептидный субстрат определяли следующим способом.
Фосфоцеллюлозный 96-луночный планшет (Millipore, Cat no. MAPHNOB50) предварительно обрабатывали 100 мкл 100 мМ фосфорной кислоты перед добавлением ферментативной реакционной смеси (40 мкл). Раствор оставляли впитываться на фосфоцеллюлозной мембране в течение 30 минут и планшет затем промывали четыре раза 200 мкл 100 мМ фосфорной кислоты. К каждой лунке сухого планшета добавляли 30 мкл жидкого коктейля Optiphase "SuperMix" для сцинтилляционного измерения (Perkin Elmer) перед сцинтилляционным считыванием (счетчик сцинтилляции жидкости 1450 Microbeta, Wallac). Уровни некатализируемой ферментом фоновой радиоактивности определяли путем добавления 16 мкл 500 мМ фосфорной кислоты к контрольным лункам, содержащим все компоненты для исследования (которые действуют в направлении денатурации фермента), перед добавлением раствора [γ-33P]-АТФ. Уровни катализируемого ферментом включения 33P вычисляли путем вычитания измеренных фоновых значений из тех значений, что измерены для каждой концентрации ингибитора. Для каждого измерения Ki получали по 8 точек, типично покрывающих диапазон концентраций соединения 0-10 мкМ, в виде двух дублирующих экспериментов (исходные растворы в ДМСО получали из начального 10 мМ раствора соединения с последующими 1:2,5 серийными разбавлениями). Величины Ki вычисляли из данных начальной скорости путем нелинейной регрессии с применением пакета программного обеспечения Prism (Prism 3.0, GraphPad Software, San Diego, CA).
Пример 12: Исследование ингибирования Itk: радиоизотопное исследование
Соединения по настоящему изобретению исследовали в качестве ингибиторов Itk-киназы человека с использованием радиоизотопного исследования.
Исследования проводили в смеси 20 мМ MOPS (pH 7,0), 10 мМ MgCl2, 0,1% BSA и 1 мМ DTT. Конечные концентрации субстрата при исследовании составляли 7,5 мкМ [γ-33P]АТФ (400 мкCi 33P АТФ/мкмоль АТФ, Amersham Pharmacia Biotech/Sigma Chemicals) и 3 мкМ пептида (SAM68, белок Δ332-443). Исследования проводили при 25°C, в присутствии 50 нМ Itk. Исходный буферный раствор для исследования содержал все реагенты, перечисленные выше, за исключением АТФ и тестируемого интересующего соединения. 50 мкл исходного раствора помещали в 96-луночный планшет с последующим добавлением 2 мкл исходного раствора в ДМСО, содержащего серийные разбавления тестируемого соединения (типично исходя из конечной концентрации 50 мкМ с 2-кратными серийными разбавлениями) в виде двух дублирующих экспериментов (конечная концентрация в ДМСО 2%). Планшет предварительно инкубировали в течение 10 минут при 25°C и реакцию инициировали путем добавления 50 мкл [γ-33P]АТФ (конечная концентрация 7,5 мкМ).
Реакцию останавливали через 10 минут путем добавления l00 мкл 0,2 М фосфорной кислоты + 0,01% TWEEN 20. Многооконный фосфоцеллюлозный фильтрующий 96-луночный планшет (Millipore, Cat no. MAPHN0B50) предварительно обрабатывали l00 мкл 0,2 М фосфорной кислоты + 0,01% TWEEN 20 перед добавлением 170 мкл остановленной смеси для исследования. Планшет промывали 4×200 мкл 0,2 М фосфорной кислоты + 0,01% TWEEN 20. После высушивания добавляли 30 мкл жидкого коктейля Optiphase "SuperMix" для сцинтилляционного измерения (Perkin Elmer) к лунке перед сцинтилляционным считыванием (счетчик сцинтилляции жидкости 1450 Microbeta, Wallac).
Величины Ki (аппроксимированные) вычисляли путем нелинейного регрессионного анализа данных начальной скорости с применением пакета программного обеспечения Prism (GraphPad Prism версия 3,0cx для Macintosh, GraphPad Software, San Diego California, USA).
Пример 13: Исследование ингибирования JAK3
Соединения подвергали скринингу на их способность ингибировать JAK с использованием исследования, описанного ниже. Реакции проводили в киназном буфере, содержащем 100 мМ HEPES (pH 7,4), 1 мМ DTT, 10 мМ MgCl2, 25 мМ NaCl и 0,01% BSA. Концентрации субстрата в исследовании составляли 5 мкМ АТФ (200 мкCi/мкмоль АТФ) и 1 мкМ поли(Glu)4Tyr. Реакции проводили при 25°C и 1 нМ JAK3.
К каждой лунке 96-луночного поликарбонатного планшета добавляли 1,5 мкл тестируемого ингибитора JAK3 в соответствии с 50 мкл киназного буфера, содержащего 2 мкМ поли(Glu)4Tyr и 10 мкМ АТФ. Смесь затем перемешивали и для начала реакции добавляли 50 мкл киназного буфера, содержащего 2 нМ фермента JAK3. Через 20 минут при комнатной температуре (25°C) реакцию останавливали действием 50 мкл 20% трихлоруксусной кислоты (TCA), которая также содержала 0,4 мМ АТФ. Все содержимое каждой лунки затем переносили в 96-луночный стекловолоконный планшет для фильтрования с использованием клеточного харвестера TomTek. После промывания добавляли 60 мкл сцинтилляционной жидкости и измеряли включение 33P на счетчике Perkin Elmer TopCount.
Пример 14: Исследование ингибирования JAK2
Исследования проводили, как описано выше в примере 13, за исключением того, что применяли фермент JAK-2, конечная концентрация поли(Glu)4Tyr составляла 15 мкМ, конечная концентрация АТФ составляла 12 мкМ.
Пример 15: Исследование ингибирования FLT-3
Соединения подвергали скринингу на их способность ингибировать активность FLT-3 с применением радиометрического анализа по степени связывания вещества с мембранным фильтром. Это исследование измеряет включение 33P в субстрат поли(Glu, Tyr) 4:1 (pE4Y). Реакции проводили в растворе, содержащем 100 мМ HEPES (pH 7,5), 10 мМ MgCl2, 25 мМ NaCl, 1 мМ DTT, 0,01% BSA и 2,5% ДМСО. Конечные концентрации субстрата в исследовании составляли 90 мкМ АТФ и 0,5 мг/мл pE4Y (оба от Sigma Chemicals, St Louis, MO). Конечная концентрация соединения по настоящему изобретению находилась обычно между 0,01 и 5 мкМ. Типично титрование по 12-ти точкам проводили путем проведения серийных разбавлений от 10 мМ исходного раствора тестового соединения в ДМСО. Реакции проводили при комнатной температуре.
Получали два раствора для исследования. Раствор 1 содержит 100 мМ HEPES (pH 7,5), 10 мМ MgCl2, 25 мМ NaCl, 1 мг/мл pE4Y и 180 мМ АТФ (содержит 0,3 mCi [γ-33P] АТФ для каждой реакции). Раствор 2 содержит 100 мМ HEPES (pH 7,5), 10 мМ MgCl2, 25 мМ NaCl, 2 мМ DTT, 0,02% BSA и 3 нМ FLT-3. Исследование проводили в 96-луночном планшете путем смешивания 50 мкл каждого раствора 1 и 2,5 мл раствора соединения по настоящему изобретению. Реакцию инициировали действием раствора 2. После инкубирования в течение 20 минут при комнатной температуре реакцию останавливали действием 50 мкл 20% TCA, содержащего 0,4 мМ АТФ. Весь реакционный объем затем переносили на фильтрующий планшет и промывали 5% TCA в аппарате Harvester 9600 от TOMTEC (Hamden, CT). Количество включенного 33P в pE4y анализировали на микропланшетном сцинтилляционном счетчике Packard Top Count (Meriden, CT). Для получения величин IC50 или Ki данные аппроксимировали с использованием программного обеспечения Prism.
Пример 16: Исследование микросомальной стабильности
Микросомальную стабильность измеряли путем измерения профиля исчерпания в микросомах из различных источников (самец мыши CD-1, самец крысы Sprague-Dawley, самец собаки Бигль, самец макаки-крабоеда и человека, объединенный от разных полов). Пиковые растворы соединения приготавливали путем разбавления исходного раствора соединения в ДМСО (типично 10 мМ), что давало раствор в ацетонитриле (0,5 мМ). Соединение (что приводило к конечной концентрации 5 мкМ) инкубировали с конечной реакционной смесью (1000 мкл), состоящей из белка микросом печени (1 мг/мл) и β-никотинамидаденин динуклеотидфосфата, системы, регенерирующей восстановленную форму (NADPH) (RGS) [состоящей из 2 мМ β-никотинамидаденин динуклеотидфосфата (NADP), 20,5 мМ изолимонной кислоты, 0,5 U изоцитратдегидрогеназы/мл, 30 мМ хлорида магния и 0,1 М фосфатного буфера (PB) pH 7,4] в присутствии 0,1 М PB (pH 7,4).
Реакцию инициировали путем добавления (250 мкл) предварительно инкубированной RGS к предварительно инкубированной смеси микросом/VRT/PB (предварительная инкубация в обоих случаях составляла 10 минут при 37°C). Образцы инкубировали в сосудах Эппендорфа (Eppendorf) (1,5 мл) на нагревающем вибростенде (DPC Micromix 5 (установки: форма 20, амплитуда 4), модифицированном для нагревания до 37°C, двумя планшетными нагревателями, закрепленными на деке, и с контролем от Packard Manual Heater), присоединенными к автоматическому жидкостному манипулятору Multiprobe II HT Ex. Жидкостной манипулятор был запрограммирован (программное обеспечение WinPREP) на отбор образца из смеси микросомальной инкубации через 0, 2, 10, 30 и 60 мин инкубации и перенесение аликвоты (100 мкл) в останавливающий модуль (96-луночный модуль), содержащий 100 мкл охлажденного метанола. Процент органики в останавливаемой смеси оптимизировали для анализа путем добавления соответствующих объемов водно-органической смеси (типично 100 мкл 50:50 метанол:вода).
Перед анализом остановленный модуль помещали на вибростенд (DPC Micromix 5; 10 мин, форма 20, амплитуда 5) для осаждения белков. Модуль затем центрифугировали (Jouan GR412; 2000 об/мин, 15 мин, 4°C). Затем аликвоту образца (200 мкл) переносили в модуль для анализа и модуль центрифугировали повторно (Jouan GR412; 2000 об/мин, 5 мин, 4°C) перед отправкой на анализ. Профиль уменьшения определяли путем контроля исчезновения VRT методом жидкостной хроматографии, совмещенной с масс-спектрометрией (LC-MS/MS). Образцы инжектировали в аналитическую колонку (20 мкл; жидкостная хроматографическая система Agilent 1100, снабженная автоматическим пробоотборником). Подвижная фаза состояла из воды + 0,05% (об./об.) муравьиной кислоты (A) и метанола + 0,05% (об./об.) муравьиной кислоты (B).
Пропускание в градиентном способе, оптимизированном для интересующего соединения, проводили путем элюирования соединения из аналитической колонки. Общее время пропускания составляло 6 минут при скорости потока 0,35 мл/мин. Полный поток, исходящий с колонки, между 0,5 и 5,9 мин пробега, вводился в источник электроспрей-ионизации (режим положительного иона) на Micromass Quattro LC, соединенного с масс-спектрометром. Масс-спектрометрию оптимизировали для интересующего соединения. Все инкубации проводили как два дублирующих эксперимента и результаты выражали в виде % остатка от исходного при или 30 мин, или 60 мин относительно образца при 0 мин.
Пример 17: Анализ клеточной пролиферации и жизнеспособности
Соединения подвергали скринингу на их способность ингибировать клеточную пролиферацию и их влияние на жизнеспособность клеток с использованием клеток Colo205, полученных из ECACC с применением исследования, описанного ниже.
Клетки Colo205 высевали в 96-луночные планшеты и серийно разбавленное соединение добавляли в лунки в виде двух дублирующих экспериментов. Контрольные группы включали необработанные клетки, разбавитель соединения (0,1% чистого ДМСО) и культуральную среду без клеток. Затем клетки инкубировали в течение 72 или 96 часов при 37°C в атмосфере 5% CO2/95% влажности.
Для измерения пролиферации к каждой лунке добавляли за 3 часа до конца эксперимента 0,5 мкCi 3H-тимидина. Затем клетки собирали и включенная радиоактивность измерялась на бета-счетчике микропланшетов от Wallac. Жизнеспособность клеток оценивали с применением Promega CellTiter 96AQ для измерения конверсии MTS. Кривые доза-отклик вычисляли с применением или программного обеспечения Prism 3.0 (GraphPad), или SoftMax Pro 4.3.1 LS (Molecular Devices).
Пример 18: Исследование ингибирования активности Abl-киназы и определение константы ингибирования Ki
Соединения подвергали скринингу на их способность ингибировать активность усеченной на N-конце (Δ27) Abl-киназы с применением стандартной сопряженной ферментативной системы (Fox et al., Protein Sci., 7, pp. 2249 (1998)). Реакции проводили в растворе, содержащем 100 мМ HEPES (pH 7,5), 10 мМ MgCl2, 25 мМ NaCl, 300 мкМ NADH, 1 мМ DTT и 3% ДМСО. Конечные концентрации субстрата в исследовании составляли 110 мкМ АТФ (Sigma Chemicals, St. Louis, MO) и 70 мкМ пептида (EAIYAAPFAKKK, American Peptide, Sunnyvale, CA). Реакции проводили при 30°C и 21 нМ Abl-киназы. Конечные концентрации компонентов сопряженной ферментативной системы составляли: 2,5 мМ фосфоенолпирувата, 200 мкМ NADH, 60 мкг/мл пируваткиназы и 20 мкг/мл лактатдегидрогеназы.
Исходный буферный раствор для исследования содержал все реагенты, перечисленные выше, за исключением АТФ и тестового интересующего соединения. Исходный буферный раствор для исследования (60 мкл) инкубировали в 96-луночном планшете с 2 мкл тестового интересующего соединения при конечных концентрациях типично в небольшом диапазоне от 0,002 мкМ до 30 мкМ при 30°C в течение 10 мин. Типично, титрование по 12-ти точкам получали путем серийных разбавлений (из 1 мМ исходных растворов соединения) с тестовыми соединениями в ДМСО в дочерних планшетах. Реакцию инициировали путем добавления 5 мкл АТФ (конечная концентрация 110 мкМ). Скорости реакции измеряли с применением ридера планшетов Molecular Devices Spectamax (Sunnyvale, CA) в течение 10 мин при 30°C. Величины Ki определяли из данных по остаточной скорости как функцию концентрации ингибитора с применением нелинейной регрессии (Prism 3,0. GraphPad Software, San Diego, CA).
Соединение 14, как было найдено, ингибирует Abl-киназу.
Пример 19: Исследование ингибирования активности мутантной Abl киназы (T315I) и определение константы ингибирования IC50
Соединения подвергали скринингу на их способность ингибировать T315I мутантную форму Abl человека на приборе Upstate Cell Signaling Solutions (Dundee, UK). В конечном реакционном объеме 25 мкл, мутантный Abl человека T315I (5-10 mU) инкубировали с 8 мМ MOPS pH 7,0, 0,2 мМ EDTA, 50 мкМ EAIYAAPFAKKK, 10 мМ ацетата Mg, [γ-33P-АТФ] (специфическая активность приблизительно 500 cpm/пмоль, при конечной концентрации для исследования 10 мМ) и тестовое интересующее соединение при конечных концентрациях в диапазоне 0-4 µнМ. Реакцию инициировали путем добавления смеси Mg-АТФ. После инкубирования в течение 40 минут при комнатной температуре реакцию останавливали путем добавления 5 мкл 3% раствора фосфорной кислоты. 10 мкл реакционной смеси затем наносили компактным пятном на плоский фильтр P30 и промывали три раза в течение 5 минут в 75 мМ фосфорной кислоты и один раз в метаноле перед высушиванием и измерением сцинтилляции. Величины ингибирования IC50 определяли методом нелинейного регрессионного анализа остаточных активностей фермента как функцию концентрации ингибитора. (Prism 3,0, GraphPad Software, San Diego, CA).
Пример 20: Исследование ингибирования Arg (Abl-2), FGFR1, MELK, MLK1, MuSK, Ret и TrkA
Соединение I-1 подвергали скринингу на его способность ингибировать Arg (Abl-2), FGFR1, MELK, MLK1, MuSK, Ret и TrkA с применением способов скрининга, известных специалисту в данной области. Все из приведенных выше ферментов подвергали скринингу при концентрациях АТФ, равных Km для АТФ, или близких к ней.
В таблице 5 ниже представлены Ki величины, полученные из примера 20:
(мкМ)
(мкМ)
(мкМ)
(мкМ)
(мкМ)
(мкМ)
(мкМ)
В таблице 6 ниже представлены данные из описанных выше примеров 10-11 и 16.
для
Аврора A (мкМ)
для
Аврора B (мкМ)
В таблице 7 ниже представлены данные из примеров 13-15 и 18-19.
Для FLT (мкМ)
для
JAK-2 (мкМ)
для
JAK-3
(мкМ)
для Abl
(T315I)
(мкМ)
для Abl (немутантный) (мкМ)
В то время как авторы описали ряд вариантов осуществления по настоящему изобретению, очевидно, что основные примеры, приведенные в настоящем описании, могут быть модифицированы для предоставления других вариантов осуществления, которые применяют или включают соединения, способы и процессы по настоящему изобретению. Следовательно, подразумевается, что объем этого изобретения определен в прилагаемой формуле изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| АМИНОПИРИМИДИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2006 |
|
RU2423361C2 |
| 4, 5-ДИГИДРО-[1, 2, 4]ТРИАЗОЛО[4, 3-F]ПТЕРИДИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ Plk1 ДЛЯ ЛЕЧЕНИЯ ПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2007 |
|
RU2441006C2 |
| ПИРАЗОЛО[1,5-a]ПИРИМИДИНЫ, ПРИМЕНЯЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗ | 2005 |
|
RU2417996C2 |
| ТРИАЗОЛЫ, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗ | 2005 |
|
RU2393155C2 |
| ДИГИДРОДИАЗЕПИНЫ, КОТОРЫЕ МОЖНО ИСПОЛЬЗОВАТЬ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗ | 2007 |
|
RU2475488C2 |
| НОВЫЕ ПИРИМИДИНАМИДНЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2003 |
|
RU2378267C2 |
| ПИРИД-2-ОНЫ, ПРИМЕНИМЫЕ КАК ИНГИБИТОРЫ ПРОТЕИНКИНАЗ СЕМЕЙСТВА ТЕС ДЛЯ ЛЕЧЕНИЯ ВОСПАЛИТЕЛЬНЫХ, ПРОЛИФЕРАТИВНЫХ И ИММУНОЛОГИЧЕСКИ-ОПОСРЕДОВАННЫХ ЗАБОЛЕВАНИЙ | 2005 |
|
RU2423351C2 |
| АЗАИНДОЛЫ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ЯНУС-КИНАЗ | 2007 |
|
RU2453548C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗО [1,2-b]ПИРИДАЗИНА И ПИРАЗОЛО[1,5-a]ПИРИМИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА ПРОТЕИНКИНАЗ | 2007 |
|
RU2487875C2 |
| ЗАМЕЩЕННЫЕ 4-(СЕЛЕНОФЕН-2(ИЛИ 3)-ИЛАМИНО)ПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2011 |
|
RU2597609C2 |
Изобретение относится к соединению формулы I или его таутомеру или их фармацевтически приемлемым солям, где R2 представляет собой С1-3-алкил или циклопропил; R9 представляет собой галоген, С1-3-алкил, -O-(C1-3-алкил), -S-(С1-3-алкил) или CF3 и р равно 1-2. Предлагаемые соединения являются ингибиторами Аврора-протеинкиназ. Они могут использоваться в способе ингибирования активности Аврора-протеинкиназы. Изобретение также предоставляет фармацевтически приемлемые композиции, включающие такие соединения; и способ лечения пролиферативного расстройства у пациента с применением указанного соединения. 4 н. и 7 з.п. ф-лы, 7 табл.
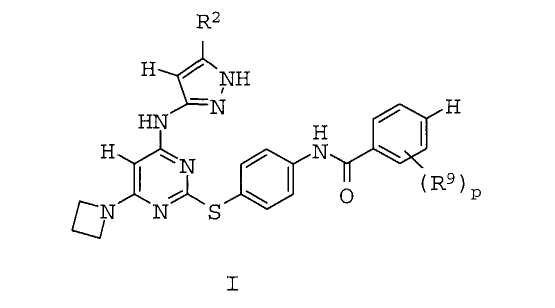
1. Соединение формулы I:
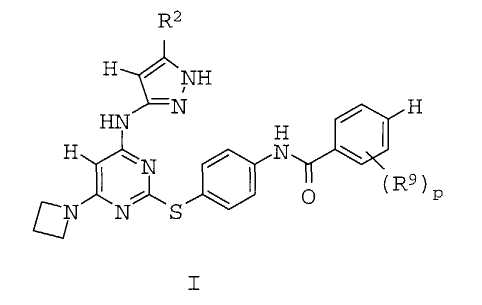
или его таутомер, или их фармацевтически приемлемая соль, где
R2 представляет собой C1-3-алкил или циклопропил;
R9 представляет собой галоген, С1-3-алкил, -О-(С1-3-алкил), -S-(С1-3-алкил) или CF3 и
р равно 1-2.
2. Соединение по п.1, в котором R2 представляет собой метил.
3. Соединение по п.2, в котором р равно 1.
4. Соединение по п.2, в котором R9 является заместителем в орто-положении.
5. Соединение по п.3, в котором R9 является заместителем в орто-положении.
6. Соединение по п.5, где R9 представляет собой F, Cl или CF3.
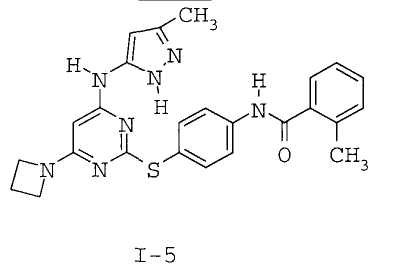
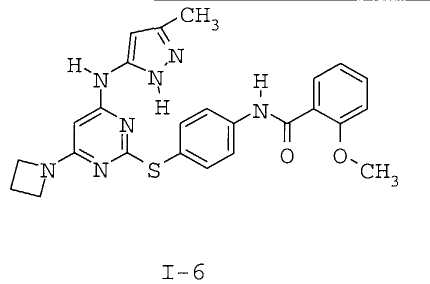
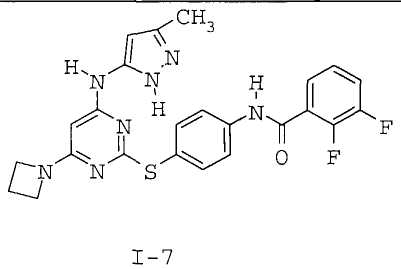
7. Соединение по п.1, выбранное из следующих:
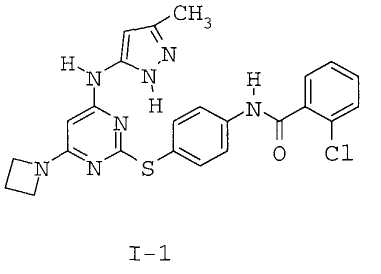
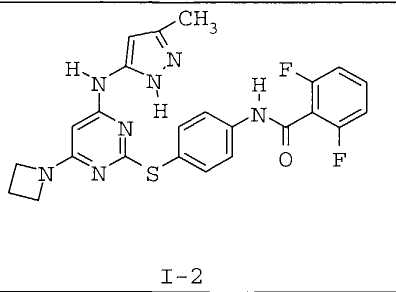
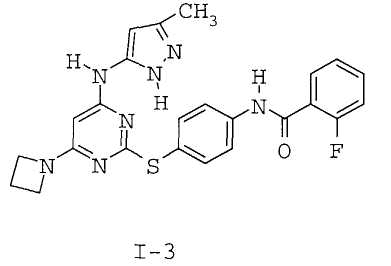
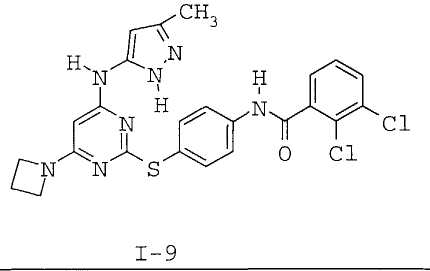
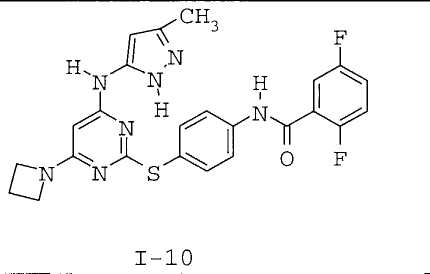
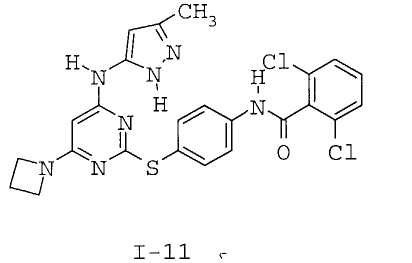
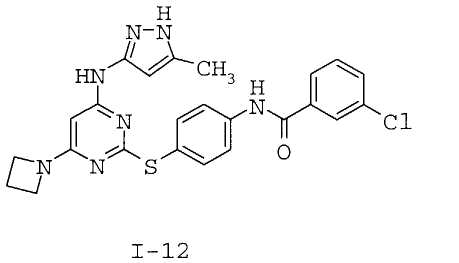
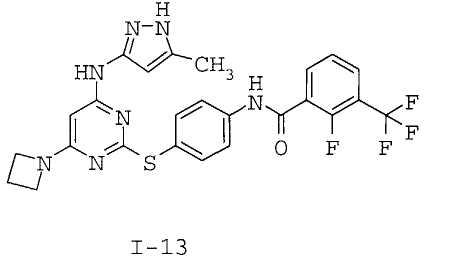
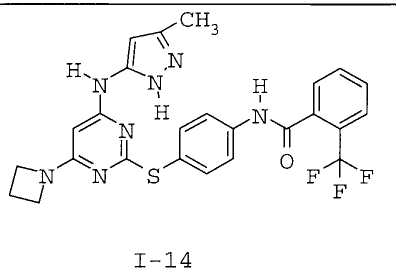
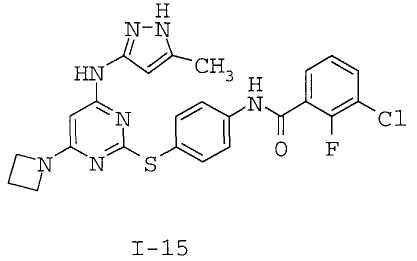
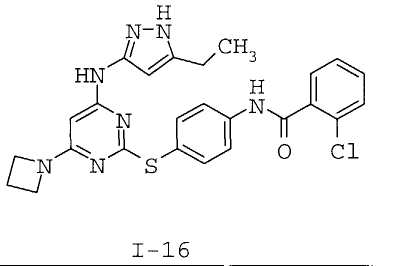
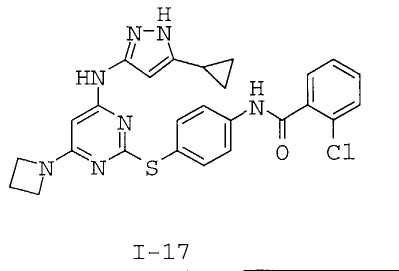
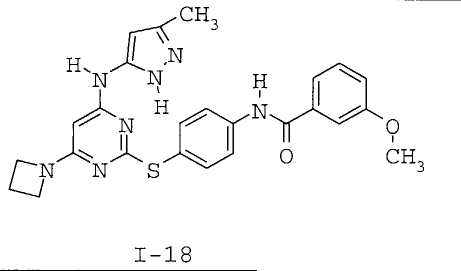
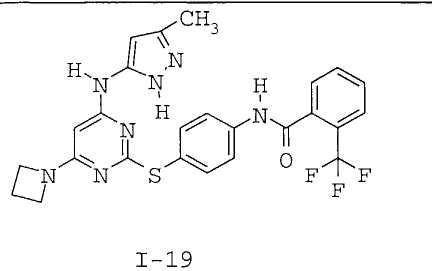
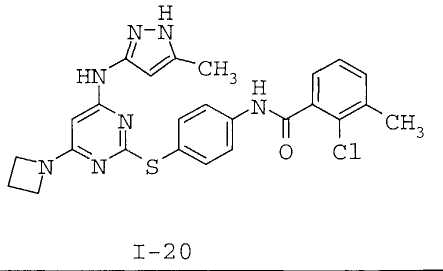
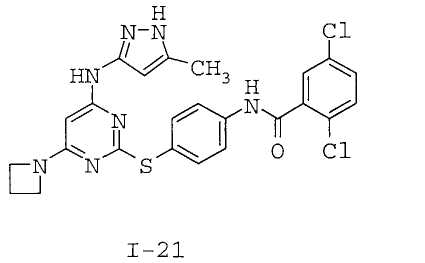
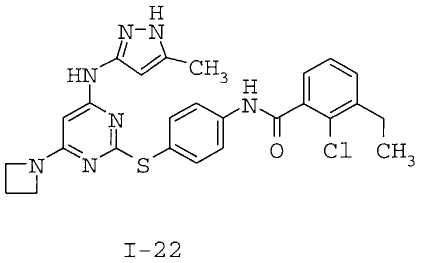
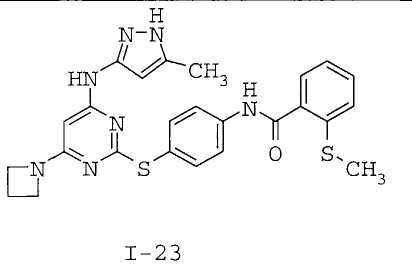

8. Фармацевтическая композиция, обладающая антипролиферативным действием, включающая соединение по любому из пп.1-7 и фармацевтически приемлемый носитель, вспомогательное вещество или разбавитель.
9. Способ ингибирования активности Аврора-протеинкиназы в биологическом образце, включающий контактирование указанного биологического образца с соединением по любому из пп.1-7.
10. Способ лечения пролиферативного расстройства у пациента, включающий стадию введения указанному пациенту соединения по любому из пп.1-7.
11. Способ по п.10, где указанное пролиферативное расстройство выбрано из меланомы, миеломы, лейкемии, лимфомы, нейробластомы, или рака, выбранного из рака толстой кишки, груди, желудка, яичников, шейки матки, легкого, центральной нервной системы (ЦНС), почки, простаты, мочевого пузыря, поджелудочной железы, мозга (глиомы), головы и шеи, почек, печени, меланомы, саркомы или рака щитовидной железы у пациента, нуждающегося в лечении, где указанный способ включает введение указанному пациенту соединения по любому из пп.1-7.
| RU 2003122209 А, 10.02.2005 | |||
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| US 6696452 В2, 24.02.2004. | |||

