Изобретение относится к области химии биологически активных веществ, в частности к способу получения N-пиразиноил-L-фенилаланил-L-лейцинбороновой кислоты (1) или ее ангидрида, которые могут быть использованы для лечения некоторых видов рака (N.A.Petasis. Aust. J. Chem., 2007, 60, 795-798).

В международной заявке WO 2005/097809 описан конвергентный способ синтеза N-пиразиноил-L-фенилаланил-L-лейцинбороновой кислоты (1) или ее ангидрида, основанный на стереоселективной перегруппировке «ониевой» соли, получаемой из дихлорметиллития и (1S,2S,3R,5S)-пинандиолового эфира изобутилбороновой кислоты, под действием кислоты Льюиса, такой как ZnCl2 (перегруппировка Маттесона; D.S.Matteson, Chem. Rev., 1989, 89, 1535-1551; E.J.Corey, D.Barnes-Seeman, T.W.Lee. Tetrahedron Asymm., 1997, 8 (22), 3711-3713), с получением соответствующего хирального α-галогенборонового эфира, при этом реакционная смесь содержит эфирный растворитель, слабо смешивающийся с водой, или его смесь с координирующим со-растворителем, обработке α-галогенборонового эфира дисилиламидом щелочного металла с образованием соединения, в котором аминогруппа защищена двумя силильными группами, удалению силильных групп с применением трифторуксусной кислоты с получением соответствующей соли амина (2) (Фиг.2), которую далее вводили в реакцию с N-пиразиноил-L-фенилаланином (3) и N,N'-дициклогексилкарбодиимидом, получая (1S,2S,3R,5S)-пинандиол-N-пиразиноил-L-фенилаланил-L-лейцинборонат с выходом 95,5% (содержит примесь 2% N,N-дициклогексилмочевины), который взаимодействием с изобутилбороновой кислотой в присутствии соляной кислоты превращали в ангидрид N-пиразиноил-L-фенилаланил-L-лейцинбороновой кислоты с выходом 81,8% на сырец или 57,2% после высаживания метил-трет-бутиловым эфиром из этилацетата (при этом чистота полученного таким образом продукта не указана), а суммарный выход на последние две стадии, включая очистку, составил 54,6% (см. международную заявку WO 2005/097809, п.84 формулы изобретения).
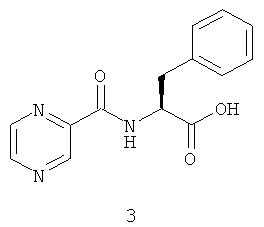
Дальнейшее усовершенствование конвергентного подхода к синтезу соединения (1) основано на конденсации N-пиразиноил-L-фенилаланина (3) с трифторацетатом известного амина (2) под действием (3-оксидо-1H-1,2,3-бензотриазол-1-ил)-N,N,N',N'-тетраметилформамидиния тетрафторбората (TBTU), обеспечивающего низкую степень рацемизации, и диизопропилэтиламина в хлористом метилене с последующим удалением (1S,2S,3R,5S)-пинандиолового индуктора в кислой среде (Tetrahedron, 2009, 65, 7105-7108, см. Фиг.2). В результате суммарный выход на двух последних стадиях синтеза после очистки составил 71%.
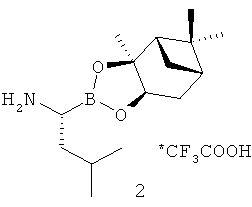
Недостатки способа, изложенного в международной заявке WO 2005/097809, состоят в том, что целевой продукт получают с примесью (2%) N,N'-дициклогексилмочевины. Загрязнение продуктов пептидного синтеза этой примесью наряду с N-ацил-N,N'-дициклогексилмочевинами неизбежно при использовании N,N'-дициклогексилкарбодиимида в качестве конденсирующего агента. При этом часто требуется дополнительная очистка продукта (J.S.Albert, A.D.Hamilton, 1,3-dicyclohexylcarbodiimide, in "Encyclopedia of Reagents for Organic Synthesis", L.A.Paquette Ed. - in - Chief; John Wiley & Sons, Inc., Chichester, 2005 (electronic version)), что приводит к усложнению технологии. Другим существенным недостатком описанного подхода является кристаллизация вышеупомянутого трифторацетата аммония с использованием значительного избытка трифторуксусной кислоты. Трифторуксусная кислота является чрезвычайно едким коррозионным веществом, требующим специальных мер безопасности при работе (S.Nova, Trifluoroacetic acid, in "Encyclopedia of Reagents for Organic Synthesis", L.A.Paquette Ed. - in - Chief; John Wiley & Sons, Inc., Chichester, 2005 (electronic version)).
К недостаткам усовершенствованного конвергентного способа получения целевого соединения (Tetrahedron, 2009, 65, 7105-7108) следует отнести то, что TBTU, используемый в качестве конденсирующего агента при сочетании амина (2) и карбоновой кислоты (3), нерастворим в хлористом метилене, в котором проводится реакция, что приводит к более медленному протеканию реакции, затрудняет ее масштабирование при промышленном использовании.
Таким образом, задача изобретения состоит в разработке нового способа, позволяющего получить целевой продукт с более высокой чистотой, с большим выходом и по более эффективной и безопасной технологии.
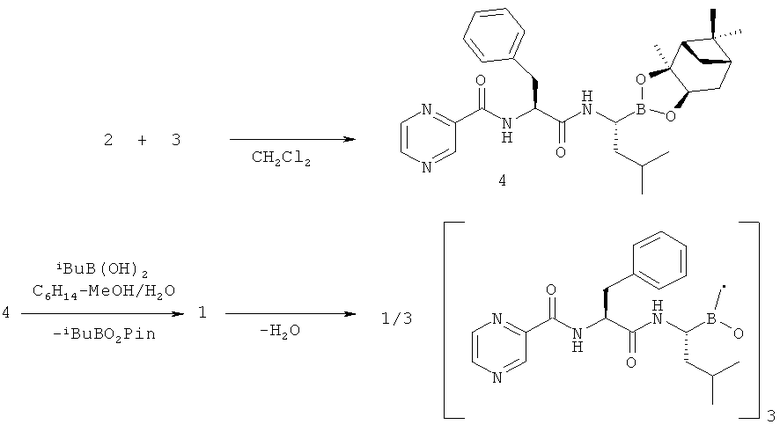
Вышеуказанная задача решается предложенным способом получения N-пиразиноил-L-фенилаланил-L-лейцинбороновой кислоты (1) или ее ангидрида с использованием реакции трифторацетата (1S,2S,3R,5S)-пинандиол-(R)-1-аммоний-3-метилбутан-1-бороната (2) с N-пиразиноил-L-фенилаланином (3) при использовании активации ультразвуком под действием подходящего конденсирующего агента из класса урониевых соединений, например (3-оксидо-1H-1,2,3-бензотриазол-1-ил)-N,N,N',N'-тетраметилформамидиния тетрафторбората (TBTU), гексафторфосфата O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HBTU), ангидридов фосфоновых кислот, например циклического ангидрида 2-пропанфосфоновой кислоты (T3R), карбодиимидов, фосфониевых соединений, смешаных ангидридов и других классов конденсирующих реагентов, применяемых для создания амидной связи, в присутствии подходящего основания, предпочтительно третичного амина, такого как диизопропилэтиламин, N-метилморфолин или триэтиламин, с образованием (1S,2S,3R,5S)-пинандиол-N-пиразиноил-L-фенилаланил-L-лейцинбороната (4), который далее подвергают кислотному гидролизу по известному методу с образованием N-пиразиноил-1-фенилаланил-1-лейцинбороновой кислоты или ее ангидрида.
Синтез целевого соединения (1) был осуществлен в две стадии из соединений (2) и (3), полученных известными методами. На первой стадии соль амина (2) реагировала с карбоновой кислотой (3) при облучении ультразвуком под действием конденсирующего агента в присутствии диизопропилэтиламина. На второй стадии хиральный индуктор пинандиола был удален при помощи обменной реакции промежуточного соединения (4) с изобутилбороновой кислотой (3) в кислой среде по известному методу. Суммарный выход на двух последних стадиях синтеза (после очистки) составил 74-77%, что выше, чем в источниках, описанных в уровне технике. Кроме того, реакция конденсации протекала быстрее благодаря использованию ультразвуковой активации.
Реакцию проводят по следующей схеме:
Изобретение иллюстрируется следующими примерами, которые не ограничивают его объем.
Пример 1 (1S,2S,3R,5S)-Пинандиол N-пиразиноил-L-фенилаланил-L-лейцинборонат (4) (сравнения)
Смесь 7,1 г (0,0187 моль) соли амина (2), 5,08 г (0,019 моль) N-пиразиноил-L-фенилаланина (3) и 6,62 г (0,021 моль) (3-оксидо-1H-1,2,3-бензотриазол-1-ил)-N,N,N',N'-тетраметилформамидиния тетрафторбората (TBTU) суспендировали в 75 мл хлористого метилена. В реакционную массу прибавляли при интенсивном перемешивании 9,5 мл (6,80 г, 0,058 моль) диизопропилэтиламина в 35 мл хлористого метилена при температуре -5°С в течение 2 часов. Перемешивание продолжали при -5°С в течение 1,5 часов, а затем нагрели до комнатной температуры. Растворитель удалили в вакууме, а остаток растворили в 110 мл этилацетата. Полученный раствор промыли последовательно 75 мл воды, 4×50 мл 3% водного раствора поташа, 75 мл воды, 3×50 мл 3% водного раствора лимонной кислоты и 75 мл воды. Сушили над сульфатом натрия, а затем растворитель удалили в вакууме. Остаток растворили в 50 мл диэтилового эфира, хроматографировали через слой силикагеля 2,5×1,5 см (элюировали 100 мл диэтилового эфира). Растворитель удалили в вакууме. Получили 8,14 г соединения (4) (выход 84%). ESI-MS: m/z 519.8 [М+Н]+, 1038.1 [2М+Н]+. ЯМР 1H (ацетон-d6): δ 3.05-0.7 (м, 25Н, αСН-В, 5СН3, 3CH2, 3CH); 3.2 (т, 2Н, CH 2Ph); 4.30 (дд, 1Н, J=8.64, 1.8 Hz, CH pinane-О); 4.93 (м, 1Н, αCHPhe); 7.10-7.33 (м, 5Н, CHPh); 7.61 (уш с, 1Н, NH), 8.40 (уш д, 1Н, NH); 8.63 (м, 1Н, CHpyrazine); 8.82 (д, 1Н, J=2.16 Hz, CHpyrazine); 9.20 (д, 1Н, J=1.44 Hz, CHpyrazine).
Пример 2 (1S,2S,3R,5S)-Пинандиол N-пиразиноил-L-фенилаланил-L-лейцинборонат (4)
Смесь 7,1 г (0,0187 моль) соли амина (2), 5,08 г (0,019 моль) N-пиразиноил-L-фенилаланина (3) и 6,62 г (0,021 моль) (3-оксидо-1H-1,2,3-бензотриазол-1-ил)-N,N,N',N'-тетраметилформамидиния тетрафторбората (TBTU) суспендировали в 75 мл хлористого метилена. В реакционную массу прибавляли при интенсивном перемешивании и облучении ультразвуком 9,5 мл (6.80 г, 0,058 моль) диизопропилэтиламина в 35 мл хлористого метилена при температуре -5°С в течение 1 часа. Перемешивание и облучение ультразвуком продолжали при -5°С в течение 1 часа, после чего реакционную массу нагрели до комнатной температуры. Растворитель удалили в вакууме, а остаток растворили в 110 мл этилацетата. Полученный раствор промыли последовательно 75 мл воды, 4×50 мл 3% водного раствора поташа, 75 мл воды, 3×50 мл 3% водного раствора лимонной кислоты и 75 мл воды. Сушили над сульфатом натрия, а затем растворитель удалили в вакууме. Остаток растворили в 50 мл диэтилового эфира, хроматографировали через слой силикагеля 2,5×1,5 см (элюировали 100 мл диэтилового эфира). Растворитель удалили в вакууме. Получили 8,92 г соединения (4) (выход 92%).
Пример 3 (1S,2S,3R,5S)-Пинандиол N-пиразиноил-1-фенилаланил-L-лейцинборонат (4)
К 12 мл хлористого метилена прибавили 1.0 г (2,64 ммоль) соли амина (2), 715 мг (2,64 ммоль) N-пиразиноил-L-фенилаланина (3) и 3.164 ммоль ангидрида 2-пропанфосфоновой кислоты в виде 50% раствора в N,N-диметилформамиде. Реакционную массу охладили до 0 - -5°С и прибавляли при интенсивном перемешивании 1,02 г (7,91 ммоль) диизопропилэтиламина в 6 мл хлористого метилена при температуре -5°С в течение 2 часов. Перемешивание продолжали при -5°С в течение 30 минут, затем нагрели до комнатной температуры. Растворитель удалили в вакууме, а остаток растворили в 12 мл этилацетата. Полученный раствор промыли последовательно 8 мл воды, 4×6 мл 3% водного раствора поташа, 8 мл воды, 3×6 мл 3% водного раствора лимонной кислоты и 8 мл воды. Сушили над сульфатом натрия, а затем растворитель удалили в вакууме. Остаток растворили в 10 мл диэтилового эфира, хроматографировали через слой силикагеля (элюент - диэтиловый эфир). Растворитель удалили в вакууме. Получили 1.09 г соединения (4) (выход 79%).
Пример 4 (1S,2S,3R,5S)-Пинандиол N-пиразиноил-L-фенилаланил-L-лейцинборонат (4)
К 12 мл хлористого метилена прибавили 1.0 г (2,64 ммоль) соли амина (2), 715 мг (2,64 ммоль) N-пиразиноил-L-фенилаланина (3) и 3.164 ммоль ангидрида 2-пропанфосфоновой кислоты в виде 50% раствора в N,N-диметилформамиде. Реакционную массу охладили до 0 - -5°С и прибавляли при облучении ультразвуком и интенсивном перемешивании 1.02 г (7,91 ммоль) диизопропилэтиламина в 6 мл хлористого метилена при температуре -5°С в течение 45 минут. Перемешивание и облучение ультразвуком реакционной смеси продолжали при -5°С в течение 30 минут, после чего ее нагрели до комнатной температуры. Растворитель удалили в вакууме, а остаток растворили в 12 мл этилацетата. Полученный раствор промыли последовательно 8 мл воды, 4×6 мл 3% водного раствора поташа, 8 мл воды, 3×6 мл 3% водного раствора лимонной кислоты и 8 мл воды. Сушили над сульфатом натрия, а затем растворитель удалили в вакууме. Остаток растворили в 10 мл диэтилового эфира, хроматографировали через слой силикагеля (элюент - диэтиловый эфир). Растворитель удалили в вакууме. Получили 1.22 г (2.43 ммоль) соединения (4) (выход 88%).
Пример 5. Ангидрид N-пиразиноил-L-фенилаланил-L-лейцинбороновой кислоты (1)
6,8 г (13,12 ммоль) эфира (4) растворили в 50 мл метанола и 50 мл гексана и добавили 29 мл 1N соляной кислоты. Раствор охладили до 10°С, при перемешивании добавили 2,4 г (23,54 моль) изобутилбороновой кислоты (3) и продолжили перемешивание в течение 17 часов при комнатной температуре. Верхний слой отделили, а нижний промыли 3×10 мл гексана. Из объединенных гексановых фракций регенерировали пинандиоловый эфир изобутилбороновой кислоты, содержащий ценный хиральный индуктор. К кислому водно-метанольному раствору добавили 3 г гидрокарбоната натрия и перемешивали 30 минут. Полученную смесь экстрагировали 850 мл этилацетата, органическую фазу сушили над сульфатом натрия и хроматографировали на колонке, содержащей 20 г силикагеля. После упаривания получили 4.5 г (выход 94%) продукта-сырца (1). После кристаллизации из этилацетата получили 4.0 г (выход 84%) продукта с чистотой 98.8% по данным ВЭЖХ. ЯМР 1H (CDCl3): 0.76 (д, 6Н, J=6.60, 2СН3); 1.30, 1.17 (2м, 2Н, СН 2-iPr); 1.52 (м, 1Н, СН(СН3)2); 2.64 (ш м, 1Н, αСН-В); 3.15 (м, 2Н, CH 2Ph): 4.86 (м, 1Н, αCHPhe); 7.10-7.30 (м, 5Н, CHPh); 8.70 (м, 1Н); 8.80 (с, 1Н); 8.82 (д, 2Н, J=2.4); 9.08 (д, 1Н, J=1.2). ЯМР 13С (CDCl3): δ 22.28, 22.77, 24.87, 37.18 (CH2), 39.80 (CH2), 42.70, 51.53, 126.23, 127.79, 127.86, 129.08, 136.64, 143.09, 143.17, 143.29, 143.89, 147.49, 162.30, 172.49.
Предложенный способ позволяет усовершенствовать технологию, повысить выход и чистоту, время проведения синтеза N-пиразиноил-L-фенилаланил-L-лейцинбороновой кислоты или ее ангидрида за счет применения ультразвуковой активации на стадии сочетания N-пиразиноил-1-фенилаланина и трифторацетата (13,2S,3R,5S)-пинандиол-1-аммоний-3-метилбутан-1-бороната в присутствии подходящего основания. Представленные в примерах конденсирующие агенты обеспечивают легкое выделение продукта в чистом виде с помощью экстракции, достигаемое благодаря ионной природе побочных продуктов.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ N-ПИРАЗИНОИЛ-(L)-ФЕНИЛАЛАНИЛ-(L)-ЛЕЙЦИНБОРОНОВОЙ КИСЛОТЫ ИЛИ ЕЕ АНГИДРИДА | 2008 |
|
RU2402564C2 |
| СПОСОБ ТОРМОЖЕНИЯ РЕТРОВИРУСНЫХ ПРОТЕАЗ | 1991 |
|
RU2028155C1 |
| СОЕДИНЕНИЯ ЗАМЕЩЕННОЙ ТРИАЗОЛБОРОНОВОЙ КИСЛОТЫ | 2013 |
|
RU2625801C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ПЕПТИДОВ ИЛИ ИХ ФИЗИОЛОГИЧЕСКИ ПРИЕМЛЕМЫХ СОЛЕЙ | 1990 |
|
RU2047621C1 |
| НОВЫЕ КОНЪЮГАТЫ СВЯЗЫВАЮЩЕЕ СОЕДИНЕНИЕ - АКТИВНОЕ СОЕДИНЕНИЕ (ADC) И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2610336C2 |
| ПРОИЗВОДНЫЕ АДАМАНТИЛА, ПОЛЕЗНЫЕ ДЛЯ ЛЕЧЕНИЯ JNK-ОПОСРЕДОВАННОГО РАССТРОЙСТВА | 2012 |
|
RU2626890C2 |
| КОНЪЮГАТ ЛИГАНДА С ЦИТОТОКСИЧЕСКИМ ЛЕКАРСТВЕННЫМ СРЕДСТВОМ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2016 |
|
RU2708461C2 |
| ЦИТОТОКСИЧЕСКИЕ ПЕПТИДЫ И ИХ КОНЪЮГАТЫ АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2012 |
|
RU2586885C2 |
| КОМБИНИРОВАННОЕ ЛЕЧЕНИЕ АГОНИСТОМ ТОЛЛ-ПОДОБНОГО РЕЦЕПТОРА (TLR7) И ИНГИБИТОРОМ СБОРКИ КАПСИДА ВИРУСА ГЕПАТИТА В | 2016 |
|
RU2718917C2 |
| ПРОИЗВОДНЫЕ ДОЛАСТАТИНА 10 И АУРИСТАТИНОВ | 2014 |
|
RU2662951C2 |
Изобретение относится способу получения N-пиразиноил-L-фенилаланил-L-лейцинбороновой кислоты или ее ангидрида. Способ включает проведение реакции трифторацетата (1S,2S,3R,5S)-пинандиол-(R)-1-аммоний-3-метилбутан-1-бороната с N-пиразиноил-L-фенилаланином при активации ультразвуком с использованием подходящего конденсирующего агента в присутствии подходящего основания. При этом образовавшийся (1S,2S,3R,5S)-пинандиол N-пиразиноил-L-фенилаланил-L-лейцинборонат подвергают реакции обмена с изобутилбороновой кислотой в кислой среде с образованием N-пиразиноил-L-фенилаланил-L-лейцинбороновой кислоты или ее ангидрида. Способ позволяет получить целевой продукт с более высокой чистотой, с большим выходом и по более эффективной и безопасной технологии. 6 з.п. ф-лы, 5 пр.
1. Способ получения N-пиразиноил-L-фенилаланил-L-лейцинбороновой кислоты или ее ангидрида, отличающийся тем, что реакция трифторацетата (1S,2S,3R,5S)-пинандиол-(R)-1-аммоний-3-метилбутан-1-бороната с N-пиразиноил-L-фенилаланином проводится при активации ультразвуком с использованием подходящего конденсирующего агента, в присутствии подходящего основания, при этом образовавшийся (1S,2S,3R,5S)-пинандиол N-пиразиноил-L-фенилаланил-L-лейцин-боронат подвергают реакции обмена с изобутилбороновой кислотой в кислой среде с образованием N-пиразиноил-L-фенилаланил-L-лейцинбороновой кислоты или ее ангидрида.
2. Способ по п.1, отличающийся тем, что в качестве конденсирующего агента используют соединения урониевого типа.
3. Способ по п.2, отличающийся тем, что в качестве конденсирующего агента урониевого типа используют TBTU или HBTU.
4. Способ по п.1, отличающийся тем, что в качестве конденсирующего агента используют ангидриды фосфоновых кислот.
5. Способ по п.4, отличающийся тем, что в качестве конденсирующего агента из группы ангидридов фосфоновых кислот используют циклический ангидрид 2-пропанфосфоновой кислоты.
6. Способ по п.1, отличающийся тем, что в качестве основания используют третичные амины.
7. Способ по п.6, отличающийся тем, что в качестве третичного амина используют диизопропилэтиламин, триэтиламин или N-метилморфолин.
| IVANOV A.S | |||
| ET AL, A CONVERGENT APPROACH TO SYNTHESIS OF BORTEZOMIB: THE USE OF TBTU SUPPRESSES RACEMIZATION IN THE FRAGMENT CONDENSATION, TETRAHEDRON, 2009, v.65, p.7105-7108 | |||
| RU 2008139637 A, 20.04.2010 | |||
| Устройство для указания уровня воды в резервуаре | 1928 |
|
SU12927A1 |

