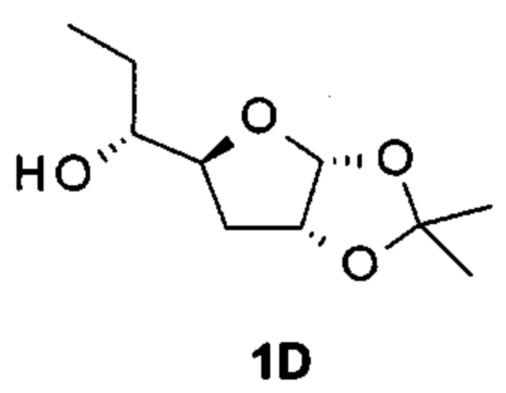
Настоящее изобретение относится к композициям и способам лечения вирусной инфекции гепатита В. А именно, настоящее изобретение относится к комбинированному лечению, включающему введение агониста TLR7 (от англ. "toll-like receptor" - толл-подобный рецептор) и ингибитора сборки капсида HBV (от англ. "hepatitis В virus" - вирус гепатита В), используемому в лечении пациента с хроническим гепатитом В.
Область техники
Хроническая инфекция вируса гепатита В (HBV) является серьезной проблемой здравоохранения во всем мире, поскольку более 240 миллионов людей в мире хронически инфицированы. HBV принадлежит семейству гепаднавирусов. После проникновения в гепатоцит его вирусный геном поступает в ядро, где ковалентно замкнутая кольцевая ДНК (кзкДНК) образуется посредством репарации ДНК частично двухцепочечного вирусного генома. кзкДНК служит в качестве матрицы для транскрипции вирусных РНК. Вирусная прегеномная РНК взаимодействует с другими двумя вирусными компонентами, капсидным белком и полимеразой, с образованием частиц капсида, где наблюдается репликация вирусной ДНК. HBV имеет икосаэдрическое ядро, состоящее из 240 копий капсидного (или ядерного) белка. Превалирующая биологическая функция капсидного белка состоит в выполнении функции структурного белка с заключением прегеномной РНК в капсид и образованием незрелых капсидных частиц в цитоплазме. Эта стадия является необходимым предварительным условием для репликации вирусной ДНК. Когда образуется почти полноразмерная релаксированная кольцевая ДНК в ходе обратной транскрипции вирусной прегеномной РНК, незрелый капсид становится зрелым капсидом. Большинство копий заключенного в капсид генома эффективно связаны с клеточными липидами и белками оболочки вируса (S, М и L) для сборки и секреции вириона. Однако также вырабатываются неинфекционные частицы, которые значительно превышают численно инфекционные вирионы. Эти пустые частицы в оболочке называются субвирусными частицами (SVPs, от англ. "subviral particles"). S, М и L белки оболочки экспрессированы из одной ORF (от англ. "open reading frame" - открытая рамка считывания), которая содержит три разных стартовых кодона. Все три белка делят 226аа последовательность, S-домен, на их С-конце. S-домен содержит эпитоп HBsAg (от англ. "hepatitis В surface antigen" - поверхностный антиген вируса гепатита В) (Lambert, С. & R. Prange. Virol J, 2007, 4, 45).
Многочисленные наблюдения показали, что некоторые вирусные белки HBV могут препятствовать первоначальной клеточной реакции организма, мешая сигнальной системе распознавания вируса и в дальнейшем противовирусной активности интерферонов (ИФН). Помимо этого чрезмерная секреция пустых субвирусных частиц HBV может участвовать в поддержании иммунологического устойчивого состояния, наблюдаемого у хронически инфицированных пациентов (ХИП). Непрерывное воздействие на HBsAg и другие вирусные антигены может приводить к HBV-специфической Т-клеточной делеции или к прогрессирующему функциональному нарушению (Kondo et al. Journal of Immunology 1993, 150, 4659-4671; Kondo et al. Journal of Medical Virology 2004, 74, 425-433; Fisicaro et al. Gastroenterology, 2010, 138, 682-93). Кроме того, сообщалось, что HBsAg подавляет функцию иммунных клеток, таких как моноциты, дендритные клетки (ДК) и естественные клетки-киллеры (NK, от англ. "natural killer") в ходе непосредственного взаимодействия (Ор den Brouw et al. Immunology, 2009b, 126, 280-9; Woltman et al. PLoS One, 2011, 6, e15324; Shi et al. J Viral Hepat. 2012, 19, e26-33; Kondo et al. ISRN Gasteroenterology, 2013, Article ID 935295).
Количественное выражение HBsAg представляет собой биомаркер для прогнозирования и отклика на лечение хронического гепатита В. Потеря и сероконверсия HBsAg являются целью клинического выздоровления, но редко наблюдаются у хронически инфицированных пациентов. Существующее на данный момент лечение, такое как нуклеоз(т)идные аналоги, которые ингибируют синтез ДНК HBV, не влияет напрямую на уровень HBsAg. Нуклеоз(т)идные аналоги даже при продолжительном лечении показывают очень низкие скорости выведения HBsAg, сопоставимые с теми, что наблюдаются в естественных условиях (Janssen et al. Lancet, 2005, 365, 123-9; Marcellin et al. N. Engl. J. Med., 2004, 351, 1206-17; Buster et al. Hepatology, 2007, 46, 388-94).
Толл-подобные рецепторы (TLRs) распознают большой диапазон консервативных патоген-ассоциированных молекулярных паттернов (ПАМП). Они играют важную роль в распознавании инвазивных патогенов и последующей инициации врожденных иммунных ответов. У человека существует 10 известных членов семейства TLR, которые представляют собой трансмембранные белки I типа, обладающие внеклеточным богатым лейцином доменом и цитоплазматическим концевым сегментом, который содержит консервативный рецепторный домен Toll/интерлейкин (ИЛ)-1 (TIR, от англ. "Toll/interleukin receptor"). В пределах этого семейства TLR3, TLR7, TLR8 и TLR9 расположены в эндосомах. TLR7 может быть активирован при связывании со специфическим низкомолекулярным лигандом (т.е. агонистом TLR7) или его природным лигандом (т.е. одноцепочечной РНК, оцРНК). Полагают, что после связывания оцРНК с TLR7 рецептор в его димеризованной форме подвергается структурному изменению, приводящему к последующему накоплению адаптерных белков в его цитоплазматическом домене, включая миелоидный дифференцировочный ген первичного ответа 88 (MyD88). После инициации сигнального каскада рецептора через путь MyD88 активируются цитоплазматические факторы транскрипции, такие как регуляторный фактор интерферона 7 (IRF-7, от англ. "interferon regulatory factor 7") и ядерный фактор каппа-В (NF-κВ, от англ. "nuclear factor kappa В"). Затем эти факторы транскрипции перемещаются к ядру и вызывают транскрипцию разных генов, например, ИФН-α и других противовирусных генов цитокинов. TLR7 преимущественно экспрессируется на плазмоцитоидных клетках и также на В-клетках. Измененная ответная реакция иммунных клеток могла бы вызывать пониженные врожденные иммунные ответы при хронических вирусных инфекциях. Следовательно, вызванная агонистом активация TLR7 могла бы представлять собой новый подход в лечении хронических вирусных инфекций. (D.J Connolly and L. AJ  , Current Opinion in Pharmacology 2012, 12: 510-518, P.A. Roethle et al, J. Med. Chem. 2013, 56, 7324-7333).
, Current Opinion in Pharmacology 2012, 12: 510-518, P.A. Roethle et al, J. Med. Chem. 2013, 56, 7324-7333).
Лечение пероральным агонистом TLR7 является многообещающим решением, которое обеспечит большую эффективность с лучшей переносимостью. В настоящее время используют пегилированный ИФН-α (ПЭГ-ИФН-α) для лечения хронического HBV, и он является альтернативой возможно пожизненного лечения противовирусными нуклеоз(т)идными аналогами. В подгруппе пациентов с хроническим HBV лечение с ПЭГ-ИФН-α может вызывать длительный иммунологический контроль над вирусом с последующей ограниченной продолжительностью лечения. Однако процент пациентов с HBV, которые достигли сероконверсии при лечении интерфероном, низкий (до 27% для HBeAg-положительных пациентов), и обычно лечение плохо переносится. Кроме того, функциональное излечение (определенное как потеря HBsAg и сероконверсия) также очень редко наблюдается как при лечении ПЭГ-ИФН-α, так и нуклеоз(т)идом. Учитывая эти ограничения, существует острая необходимость в улучшенных возможных способах лечения, чтобы лечить и вызывать функциональное излечение хронического HBV. Лечение пероральным низкомолекулярным агонистом TLR7 является многообещающим подходом, который обладает возможностью обеспечить большую эффективность и переносимость (T. Asselah et al, Clin Liver Dis 2007, 11, 839-849).
Капсидный белок HBV играет важную роль в репликации HBV. Гетероарилдигидропиримидины или ГАП, включая соединения, названные Bay 41-4109, Bay 38-7690 и Bay 39-5493, обнаружены при скрининге на основе тканевой культуры (Deres К. et al. Science 2003, 893). Эти аналоги ГАП действуют в качестве синтетических аллостерических активаторов и способны вызывать аберрантное образование капсида, которое приводит к разрушению ядерного белка. Также аналоги ГАП преобразовывали ядерный белок из предварительно собранных капсидов в некапсидные полимеры, предположительно в результате взаимодействия ГАП с димерами, освобожденными в ходе "дыхания" капсида, временного разрыва отдельных межсубъединичных связей. Bay 41-4109 вводили инфицированной HBV трансгенной мыши или гуманизированным мышиным моделям, и он демонстрировал эффективность in vivo со снижением ДНК HBV (Deres К. et al. Science 2003, 893; Brezillon N. et al. PLoS ONE 2011, e25096). Также было показано, что бис-АНС (4,4'-бис(1-анилинонафталин-8-сульфонат)) является малой молекулой, которая действует в качестве молекулярного "клина" и мешает нормальной геометрии капсид-белок и образованию капсида (Zlotnick A. et al. J. Virol. 2002, 4848-4854).
В настоящее время стандартом клинического излечения инфекции HBV является потеря и/или сероконверсия HBsAg. Несмотря на то, что ПЭГ-ИФН-α и нуклеоз(т)ид доступны пациентам с HBV, большинство (около или более 90%) получающих лечение пациентов неспособны достигнуть этой цели, главным образов вследствие того факта, что современные способы лечения не могут вызвать появление нейтрализующих антител против HBsAg (анти-HBs), признака разрушения инфекции HBV, у большинства хронически инфицированных пациентов. Следовательно, безусловно существует медицинская необходимость в способах лечения с улучшенным показателем эффективности вызывания потери и/или сероконверсии HBsAg и стимулирования выработки анти-HBs.
Краткое описание изобретения
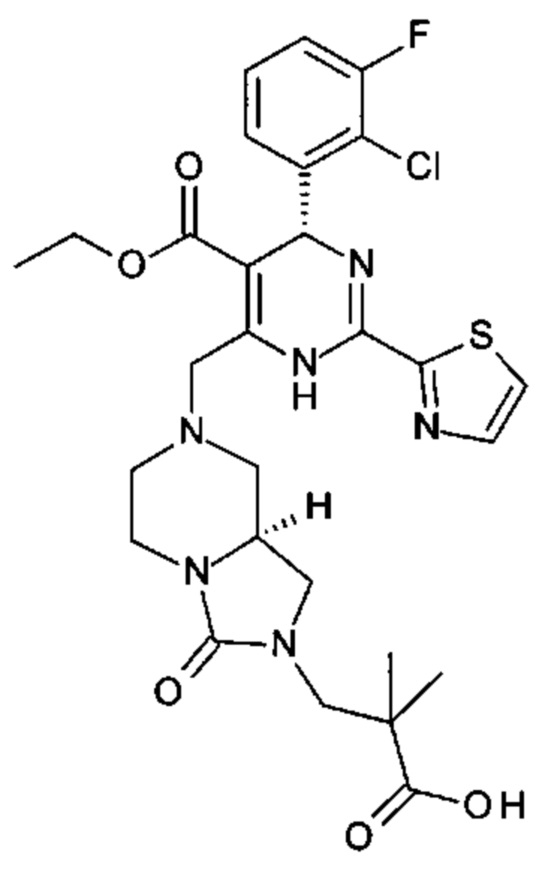
Настоящее изобретение относится к фармацевтической композиции, включающей агонист TLR7 и ингибитор сборки капсида HBV в фармацевтически приемлемом носителе. В данном документе "агонист TLR7" представляет собой соединение формулы (I), (II) или любое из соединений, раскрытых в патенте WO 2006/066080, в частности в данном документе "агонист TLR7" представляет собой [(1S)-1-[(2S,4R,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-4-гидрокси-тетрагидрофуран-2-ил]пропил]ацетат; [(S)-[(2S,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-1,3-оксатиолан-2-ил]-циклопропил-метил]ацетат; 5-амино-3-(3'-дезокси-β-D-рибофуранозил)-3H-тиазоло[4,5-d]пиримидин-2-он; 5-амино-3-(2'-O-ацетил-3'-дезокси-β-D-рибофуранозил)-3H-тиазоло[4,5-d]пиримидин-2-он или 5-амино-3-(3'-дезокси-β-D-рибофуранозил)-3H,6Н-тиазоло[4,5-d]пиримидин-2,7-дион, или их фармацевтически приемлемую соль, энантиомер или диастереомер. В данном документе ингибитор сборки капсида HBV представляет собой соединение формулы (III) или любое из соединений, раскрытых в патентах WO 2014/037480, WO 2014/184328 и WO 2015/132276, в частности в данном документе ингибитор сборки капсида HBV представляет собой 3-[(8aS)-7-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-этоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановую кислоту; 3-[(8aS)-7-[[(4S)-5-этоксикарбонил-4-(3-фтор-2-метил-фенил)-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановую кислоту; 2-[(1R,3S,5S)-8-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-6,6-дифтор-8-азабицикло[3.2.1]октан-3-ил]уксусную кислоту; 2-[(1S,3R,5R)-8-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-6,6-дифтор-8-азабицикло[3.2.1]октан-3-ил]уксусную кислоту или (S)-4-[(R)-6-(2-хлор-4-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-3,6-дигидро-пиримидин-4-илметил]-морфолин-3-карбоновую кислоту; или их фармацевтически приемлемую соль, энантиомер или диастереомер.
Краткое описание графических материалов
Фигура 1: Уровни ДНК HBV и HBsAg в сыворотке мышей в мышиной модели AAV (от англ. "adenoassociated virus" - аденоассоциированный вирус)-НВV. Результаты показаны на фигуре 1 для мышей с постоянным уровнем ДНК HBV и HBsAg, получающих наполнитель (показано ромбами), только соединение 1 при 100 мг/кг (показано кругами), только соединение 2 при 12 мг/кг (показано треугольниками) или комбинацию соединения 1 и соединения 2 (показано квадратами). Относительное уменьшение ДНК HBV и HBsAg после лечения вычисляли, нормируя их уровни к группе, получающей наполнитель, в качестве базовой линии. Синергетический противовирусный эффект в уменьшении HBsAg наблюдался у мышей, получающих комбинированное лечение, и что более важно, уменьшение ДНК HBV и HBsAg сохранялось в течение 2-недельного периода без лечения после комбинированного лечения. НПКО: нижний предел количественного определения.
Фигура 2: рентгеновская кристаллическая структура соединения 2А-2а.
Фигура 3: рентгеновская кристаллическая структура соединения 3J.
Фигура 4: ДНК HBV и HBsAg в AAV-HBV инфицированных мышах, получающих наполнитель, соединение 1 (100 мг/кг), соединение 4 (20 мг/кг) или комбинацию соединения 1 плюс соединение 4. Лечение начинали после того, как мышей заражали AAV-HBV в течение 4 недель. Они получали лечение в течение 6 недель, и следили за их состоянием в течение еще 6-недельного периода без лечения. ДНК HBV и HBsAg в мышиной сыворотке измеряли в указанные моменты времени с помощью количественной полимеразной цепной реакции с обратной транскрипцией (ОТ-кПЦР) и иммунохемилюминесцентного анализа (ИХЛА) HBsAg, соответственно. Результаты представлены в виде среднего значения ± СОС (стандартная ошибка среднего). НПКО: нижний предел количественного определения.
Фигура 5: ДНК HBV и HBsAg в AAV-HBV инфицированных мышах, получающих наполнитель, соединение 3 (30 мг/кг), соединение 4 (20 мг/кг) или комбинацию соединения 3 плюс соединение 4. Лечение начинали после того, как мышей заражали AAV-HBV в течение 4 недель. Они получали лечение в течение 6 недель, и следили за их состоянием в течение еще 6-недельного периода без лечения. ДНК HBV и HBsAg в мышиной сыворотке измеряли в указанные моменты времени с помощью ОТ-кПЦР и ИХЛА HBsAg, соответственно. Результаты представлены в виде среднего значения ± СОС. НПКО: нижний предел количественного определения.
Фигура 6: ДНК HBV и HBsAg в AAV-HBV инфицированных мышах, получающих наполнитель, соединение 1 (100 мг/кг), соединение 5 (12 мг/кг) или комбинацию соединения 1 плюс соединение 5. Лечение начинали после того, как мышей заражали AAV-HBV в течение 4 недель. Они получали лечение в течение 6 недель, и следили за их состоянием в течение еще 6-недельного периода без лечения. ДНК HBV и HBsAg в мышиной сыворотке измеряли в указанные моменты времени с помощью ОТ-кПЦР и ИХЛА HBsAg, соответственно. Результаты представлены в виде среднего значения ± СОС. НПКО: нижний предел количественного определения.
Фигура 7: Уровень анти-HBs антитела (антитело против HBsAg) в сыворотке каждой мыши, получающей лечение одним средством или комбинированное лечение, как описано на фигурах 4, 5 и 6. Образцы сыворотки собирали на 24 день после прекращения лечения, и анти-HBs измеряли в ходе ИХЛА анти-HBs. НПКО: нижний предел количественного определения.
Фигура 8: ДНК HBV и HBsAg в AAV-HBV инфицированных мышах, получающих наполнитель, соединение 8 (300 мг/кг), соединение 4 (20 мг/кг) или комбинацию соединения 8 плюс соединение 4. Лечение начинали после того, как мышей заражали AAV-HBV в течение по меньшей мере 38 дней. Они получали лечение в течение 6 недель, и следили за их состоянием в течение еще 6-недельного периода без лечения. ДНК HBV и HBsAg в мышиной сыворотке измеряли в указанные моменты времени с помощью ОТ-кПЦР и ИХЛА HBsAg, соответственно. Результаты представлены в виде среднего значения ± СОС. НПКО: нижний предел количественного определения.
Фигура 9: ДНК HBV и HBsAg в AAV-HBV инфицированных мышах, получающих наполнитель, соединение 8 (300 мг/кг), соединение 10 (20 мг/кг) или комбинацию соединения 8 плюс соединение 10. Лечение начинали после того, как мышей заражали AAV-HBV в течение по меньшей мере 38 дней. Они получали лечение в течение 6 недель, и следили за их состоянием в течение еще 6-недельного периода без лечения. ДНК HBV и HBsAg в мышиной сыворотке измеряли в указанные моменты времени с помощью ОТ-кПЦР и ИХЛА HBsAg, соответственно. Результаты представлены в виде среднего значения ± СОС. НПКО: нижний предел количественного определения.
Фигура 10: ДНК HBV и HBsAg в AAV-HBV инфицированных мышах, получающих наполнитель, соединение 1 (100 мг/кг), соединение 10 (20 мг/кг) или комбинацию соединения 1 плюс соединение 10. Лечение начинали после того, как мышей заражали AAV-HBV в течение по меньшей мере 38 дней. Они получали лечение в течение 6 недель, и следили за их состоянием в течение еще 6-недельного периода без лечения. ДНК HBV и HBsAg в мышиной сыворотке измеряли в указанные моменты времени с помощью ОТ-кПЦР и ИХЛА HBsAg, соответственно. Результаты представлены в виде среднего значения ± СОС. НПКО: нижний предел количественного определения.
Фигура 11: Уровень анти-HBs антитела (антитело против HBsAg) в сыворотке каждой мыши, получающей лечение одним средством или комбинированное лечение, как описано на фигурах 8, 9 и 10. Образцы сыворотки собирали на 31 день после прекращения лечения, и анти-HBs измеряли в ходе ИХЛА анти-HBs. НПКО: нижний предел количественного определения.
Подробное описание изобретения
Если не указано иное, то все технические и научные термины, используемые в данном документе, имеют те же самые значения, что обычно понимает под ними любой средний специалист в области техники, к которой данное изобретение относится.
Как используется в данном документе, термин "С1-6алкил" относится к одновалентной линейной или разветвленной насыщенной углеводородной группе от 1 до 6 атомов углерода. В конкретных воплощениях С1-6алкил имеет от 1 до 6 атомов углерода, и в особых воплощениях от 1 до 4 атомов углерода. Примеры С1-6алкила включают метил, этил, пропил, изопропил, н-бутил, изобутил, втор-бутил или трет-бутил.
Как используется в данном документе, термины "гало" или "галоген" используются в данном документе взаимозаменяемо и относятся к фтору, хлору, брому или йоду.
Как используется в данном документе, термин "С1-6алкокси" относится к группе С1-6алкил-О-, где "С1-6алкил" является таким, как определено выше; например метокси, этокси, пропокси, изопропокси, н-бутокси, изобутокси, 2-бутокси, трет-бутокси и подобным. Конкретные "С1-6алкокси" группы представляют собой метокси и этокси и особенно метокси.
Как используется в данном документе, термин "С3-7циклоалкил" относится к насыщенному углеродному кольцу, содержащему от 3 до 7 атомов углерода, особенно от 3 до 6 атомов углерода, например, циклопропилу, циклобутилу, циклопентилу, циклогексилу, циклогептилу и подобным. Конкретные группы "С3-7циклоалкил" представляют собой циклопропил, циклопентил и циклогексил.
Как используется в данном документе, термин "С2-6алкенил" относится к ненасыщенной, линейной или разветвленной алкенильной группе, содержащей от 2 до 6, особенно от 2 до 4 атомов углерода, например, винилу, пропенилу, аллилу, бутенилу и подобным. Конкретной группой "С2-6алкенил" является аллил.
Как используется в данном документе, термин "С2-6алкинил" относится к ненасыщенной, линейной или разветвленной алкинильной группе, содержащей от 2 до 6, особенно от 2 до 4 атомов углерода, например, этинилу, 1-пропинилу, пропаргилу, бутинилу и подобным. Конкретные группы "С2-6алкинил" представляют собой этинил, 1-пропинил и пропаргил.
Как используется в данном документе, термин "гетероциклическое" кольцо или "гетероциклил" относится к насыщенному или частично ненасыщенному моноциклическому или бициклическому кольцу, содержащему от 3 до 10 кольцевых атомов, которое может включать один, два или три атома, выбранных из азота, кислорода и/или серы. Примеры моноциклических гетероциклических колец, содержащих в частности от 3 до 7 кольцевых атомов, включают, но не ограничиваются этим, азиридинил, азетидинил, оксетанил, пиперидинил, пиперазинил, азепинил, диазепанил, пирролидинил, морфолинил, дигидрофурил, тетрагидрофурил, тетрагидропиранил, тетрагидротиопиранил и тиоморфолинил. Бициклический гетероциклил может представлять собой бициклическое конденсированное кольцо или бициклическое кольцо с внутренним мостиком. Примерами бициклического гетероциклила являются 8-аза-бицикло[3.2.1]октил, хинуклидинил, 8-окса-3-аза-бицикло[3.2.1]октил, 9-аза-бицикло[3.3.1]нонил, 3-окса-9-аза-бицикло[3.3.1]нонил, 3-тиа-9-аза-бицикло[3.3.1]нонил или дифторазабицикло[3.2.1]октил. Моноциклический и бициклический гетероциклил может быть дополнительно замещен галогеном, С1-6алкилом, циано, карбокси, карбоксиС1-6алкилом.
Термин "гетероциклический амино" относится к аминогруппе с атомом азота на гетероциклическом кольце, где "гетероциклическое" кольцо является такими, как определено выше.
Как используется в данном документе, термин "диастереомер" относится к стереоизомеру с двумя или более центрами хиральности, и молекулы которого не являются зеркальными изображениями друг друга. Диастереомеры обладают разными физическими свойствами, например температурами плавления, температурами кипения, спектральными свойствами, активностями и реакционными способностями.
Как используется в данном документе, термин "энантиомеры" относится к двум стереоизомерам соединения, которые представляют собой неналагающиеся зеркальные изображения друг друга.
Как используется в данном документе, термин "фармацевтически приемлемые соли" относится к солям, которые ни биологически или иным образом не являются нежелательными. Фармацевтически приемлемые соли включают соли присоединения как кислоты, так и основания.
Как используется в данном документе, термин "пролекарство" относится к форме или производному соединения, которое после введения преобразуется в процессе обмена веществ in vivo, например, под действием биологических жидкостей или ферментов у субъекта в фармакологически активную форму соединения, чтобы вызвать требуемый фармакологический эффект. Пролекарства описаны, например, в Organic Chemistry of Drug Design and Drug Action by Richard B. Silverman, Academic Press, San Diego, 2004, Chapter 8 Prodrugs and Drug Delivery Systems, pp. 497-558.
Термин "фармацевтически приемлемая соль присоединения кислоты" относится к тем фармацевтически приемлемым солям, что образованы с неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, серная кислота, азотная кислота, угольная кислота, фосфорная кислота, и органическими кислотами, выбранными из алифатического, циклоалифатического, ароматического, аралифатического, гетероциклического, карбонового и сульфонового классов органических кислот, такими как муравьиная кислота, уксусная кислота, пропионовая кислота, гликолевая кислота, глюконовая кислота, молочная кислота, пировиноградная кислота, щавелевая кислота, яблочная кислота, малеиновая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, винная кислота, лимонная кислота, аспарагиновая кислота, аскорбиновая кислота, глутаминовая кислота, антраниловая кислота, бензойная кислота, коричная кислота, миндальная кислота, эмбоновая кислота, фенилуксусная кислота, метансульфоновая кислота, этансульфоновая кислота, п-толуолсульфоновая кислота и салициловая кислота.
Термин "фармацевтически приемлемая соль присоединения основания" относится к тем фармацевтически приемлемым солям, что образованы органическим или неорганическим основанием. Примеры приемлемых неорганических оснований включают соли натрия, калия, аммония, кальция, магния, железа, цинка, меди, марганца и алюминия. Соли, полученные из фармацевтически приемлемых органических нетоксичных оснований, включают соли первичных, вторичных и третичных аминов, замещенные амины, включая встречающиеся в природе замещенные амины, циклические амины и основные ионообменные смолы, такие как смолы на основе изопропиламина, триметиламина, диэтиламина, триэтиламина, трипропиламина, этаноламина, 2-диэтиламиноэтанола, триметамина, дициклогексиламина, лизина, аргинина, гистидина, кофеина, прокаина, гидрабамина, холина, бетаина, этилендиамина, глюкозамина, метилглюкамина, теобромина, пуринов, пиперазина, пиперидина, N-этилпиперидина и полиамина.
Соединения общей формулы (I), которые содержат один или несколько хиральных центров, могут находиться в виде либо рацематов, либо диастереомерных смесей, либо оптически активных отдельных изомеров. Рацематы можно разделить в соответствии с известными способами на энантиомеры. 6 частности, диастереомерные соли, которые можно разделить в ходе кристаллизации, образуются из рацемических смесей в ходе реакции с оптически активной кислотой, такой как например D- или L-винная кислота, миндальная кислота, яблочная кислота, молочная кислота или камфорсульфоновая кислота.
Как используется в данном документе, "комбо" относится к комбинации.
Как используется в данном документе, "ОТ-ПЦР" относится к полимеразной цепной реакции с обратной транскрипцией.
Как используется в данном документе, "ИХЛА" относится к иммунохемилюминесцентному анализу.
Как используется в данном документе, "AAV" относится к аденоассоциированному вирусу.
Как используется в данном документе, "AAV-HBV" относится к рекомбинантному вирусу, который несет 1,3 копий генома HBV, размещенного в капсидах AAV. Мышиную модель хронической инфекции HBV можно получить в ходе инъекции мышам AAV-HBV через хвостовую вену. В этой мышиной модели активная репликация HBV приводит к устойчивым вирусным маркерам HBV (например ДНК HBV, HBsAg, HBeAg и т.п.).
Как используется в данном документе, "HBsAg" относится к поверхностному антигену гепатита В.
Как используется в данном документе, "HBeAg" относится к антигену "е" гепатита В.
Как используется в данном документе, "анти-HBs" относится к антителам против HBsAg.
Как используется в данном документе, "HBV специфичные праймеры" относятся к паре одноцепочечной нуклеиновой кислоты, которая служит в качестве начальной и конечной точек для специфической амплификации областей ДНК HBV.
Как используется в данном документе, "TLR7" относится к Toll-подобному рецептору 7 любых видов происхождения (например человека, мыши, сурка и т.п.).
Как используется в данном документе, "агонист TLR7" относится к соединению, которое действует в качестве агониста TLR7. Если не указано иное, то агонист TLR7 может включать соединение в любой фармацевтически приемлемой форме, включая любой изомер (например диастереомер или энантиомер), соль, сольват, полиморф и подобные. Агонизм TLR для конкретного соединения можно определить любым подходящим образом. Например, анализы для детектирования агонизма TLR исследуемых соединений описаны, например, в предварительной заявке на патент США с порядковым №60/432,650, поданной 11 декабря 2002, и рекомбинантные клеточные линии, подходящие для применения в таких анализах, описаны, например, в предварительной заявке на патент США с порядковым №60/432,651, поданной 11 декабря 2002.
Настоящее изобретение относится к фармацевтической композиции, включающей агонист TLR7 и ингибитор сборки капсида HBV в фармацевтически приемлемом носителе.
В одном воплощении настоящего изобретения "агонист TLR7" представляет собой соединение формулы (I):
 ,
,
где
R1 представляет собой гидрокси, С1-6алкил, галоС1-6алкил, С1-6алкилкарбонил-O-, С1-6алкил-S-, азидо, циано, С2-6алкенил, С1-6алкилсульфонил-NH-, (С1-6алкил)2N-, С1-6алкилкарбонил-NH- или гетероциклический амино;
R2 представляет собой водород, С1-6алкил, С1-6алкоксиС1-6алкил, С3-7циклоалкил, С2-6алкинил, С2-6алкенил, бензил и тиофенил;
R3 представляет собой водород или С1-6алкилкарбонил;
или его фармацевтически приемлемую соль, энантиомер или диастереомер.
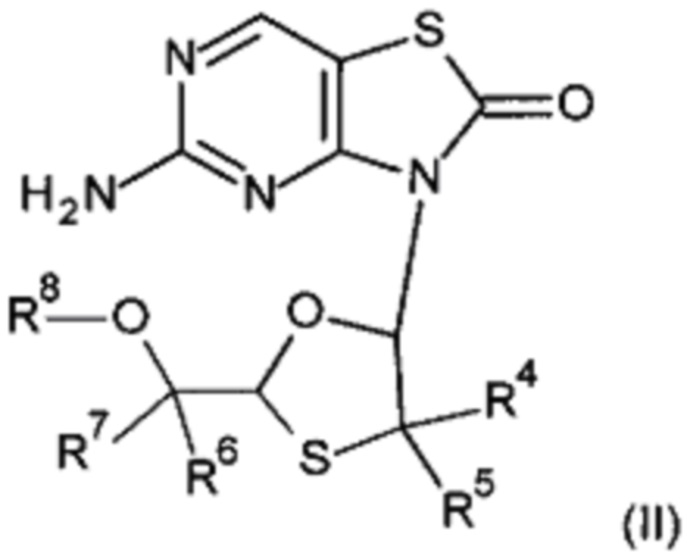
В другом воплощении настоящего изобретения "агонист TLR7" представляет собой соединение формулы (II):
 ,
,
где
R4 и R5 независимо выбраны из водорода, С2-6алкенила и С1-6алкила;
R6 и R7 независимо выбраны из водорода, С1-6алкила, С3-7циклоалкила, С3-7циклоалкилС2-6алкинила, С2-6алкенила, С2-6Элкинила и 2-тиофенила;
R8 представляет собой водород или С1-6алкилкарбонил;
или его фармацевтически приемлемую соль, энантиомер или диастереомер.
А именно, агонист TLR7 согласно настоящему изобретению относится к [(1S)-1-[(2S,4R,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-4-гидрокси-тетрагидрофуран-2-ил]пропил]ацетату; [(S)-[(2S,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-1,3-оксатиолан-2-ил]-циклопропил-метил]ацетату; 5-амино-3-(3'-дезокси-β-D-рибофуранозил)-3H-тиазоло[4,5-d]пиримидин-2-ону; 5-амино-3-(2'-O-ацетил-3'-дезокси-β-D-рибофуранозил)-3H-тиазоло[4,5-d]пиримидин-2-ону; 5-амино-3-(3'-дезокси-β-D-рибофуранозил)-3H,6Н-тиазоло[4,5-d]пиримидин-2,7-диону или [(2R,3R,5S)-5-[(1S)-1-ацетоксипропил]-2-(5-амино-2,7-диоксо-6H-тиазоло[4,5-d]пиримидин-3-ил)тетрагидрофуран-3-ил]ацетату; или их фармацевтически приемлемой соли, энантиомеру или диастереомеру. В другом воплощении "агонист TLR7" также относится к любому из соединений, раскрытых в патенте WO 2006/066080. После введения соединения формулы (I) или формулы (II) или соединения из патента WO 2006/066080 преобразуются в процессе обмена веществ в свои активные формы, которые являются полезными агонистами TLR7.
Как используется в данном документе, "вирус гепатита В" или "HBV" относится к члену семейства гепаднавирусов, имеющему небольшой геном двухцепочечной ДНК приблизительно из 3,200 пар оснований и тропизм к клеткам печени. "HBV" включает вирус гепатита В, который заражает любой организм из целого ряда млекопитающих (например человека, примата, отличного от человека, и т.п.) и птиц (утка и т.п.). "HBV" включает любой известный генотип HBV, например, серотип А, В, С, D, Е, F и G; любой серотип HBV или подтип HBV; любой изолят HBV; варианты HBV, например HBeAg-отрицательные варианты, лекарственно-устойчивые варианты HBV (например устойчивые к ламивудину варианты; устойчивые к адефовиру мутанты; устойчивые к тенофовиру мутанты; устойчивые к энтекавиру мутанты и т.п.) и подобные.
Как используется в данном документе, "ингибитор сборки капсида HBV" относится к соединению, которое ингибирует и/или разрушает, и/или ускоряет, и/или препятствует, и/или замедляет, и/или снижает, и/или модифицирует обычную сборку капсида HBV (например в процессе созревания) и/или обычную разборку капсида (например в ходе инфективности), и/или нарушает устойчивость капсида, тем самым вызывая аберрантную морфологию и функцию капсида.
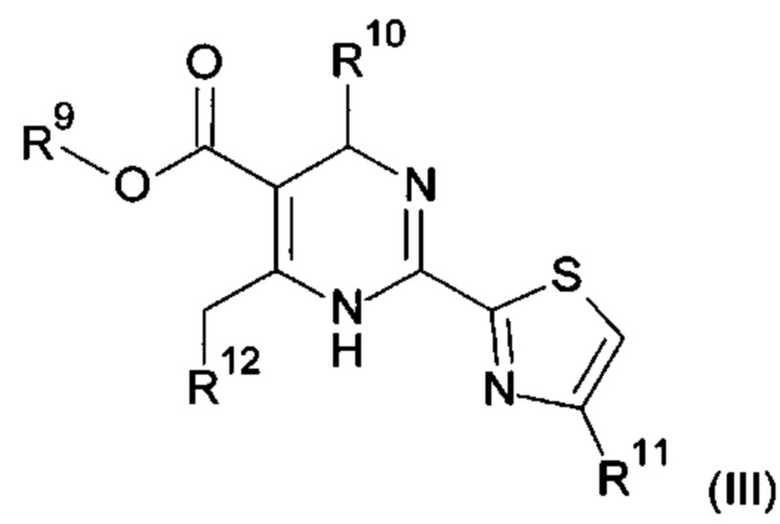
В одном воплощении настоящего изобретения ингибитор сборки капсида HBV представляет собой соединение формулы (III):
 ,
,
где
R9 представляет собой С1-6алкил;
R10 представляет собой фенил, который один раз или два раза, или три раза замещен галогеном или С1-6алкилом;
R11 представляет собой водород или С1-6алкил;
R12 представляет собой моноциклический, бициклический конденсированный или бициклический с внутренним мостиком гетероциклил;
или его фармацевтически приемлемую соль, энантиомер или диастереомер.
А именно, ингибитор сборки капсида HBV согласно настоящему изобретению относится к 3-[(8aS)-7-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-этоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановой кислоте; 3-[(8aS)-7-[[(4S)-5-этоксикарбонил-4-(3-фтор-2-метил-фенил)-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановой кислоте; 2-[(1R,3S,5S)-8-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-6,6-дифтор-8-азабицикло[3.2.1]октан-3-ил]уксусной кислоте; 2-[(1S,3R,5R)-8-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-6,6-дифтор-8-азабицикло[3.2.1]октан-3-ил]уксусной кислоте (раскрытой в патенте WO 2014/184328) или (S)-4-[(R)-6-(2-хлор-4-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-3,6-дигидро-пиримидин-4-илметил]-морфолин-3-карбоновой кислоте; или их фармацевтически приемлемой соли, энантиомеру или диастереомеру. В другом воплощении "ингибитор сборки капсида HBV" в частности является любым из соединений, раскрытых в патентах WO 2015/132276, WO 2014/184328 и WO 2014/037480.
В одном воплощении настоящего изобретения фармацевтическая композиция включает агонист TLR7 и ингибитор сборки капсида HBV, где агонист TLR7 и ингибитор сборки капсида HBV независимо выбраны из таблицы 1: (соединения 2 и 4 раскрыты в патенте WO 2015/132276; соединения 5 и 6 раскрыты в патенте WO 2014/184328; соединения 7, 8 и 9 раскрыты в патенте WO 2006/066080; соединение 10 раскрыто в патенте WO 2014/037480).
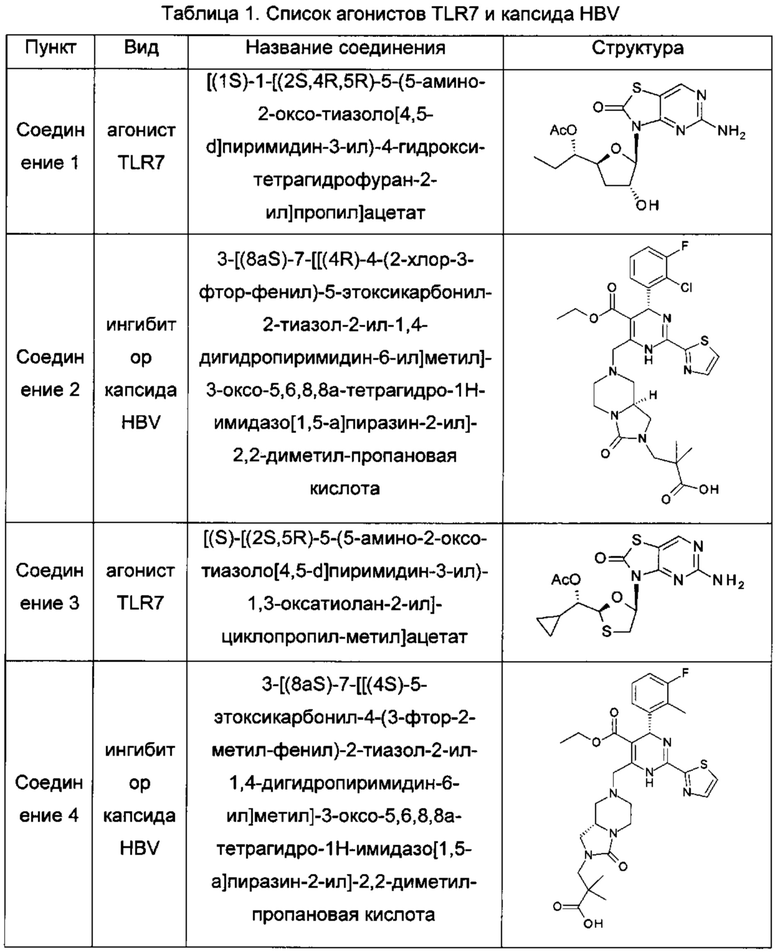
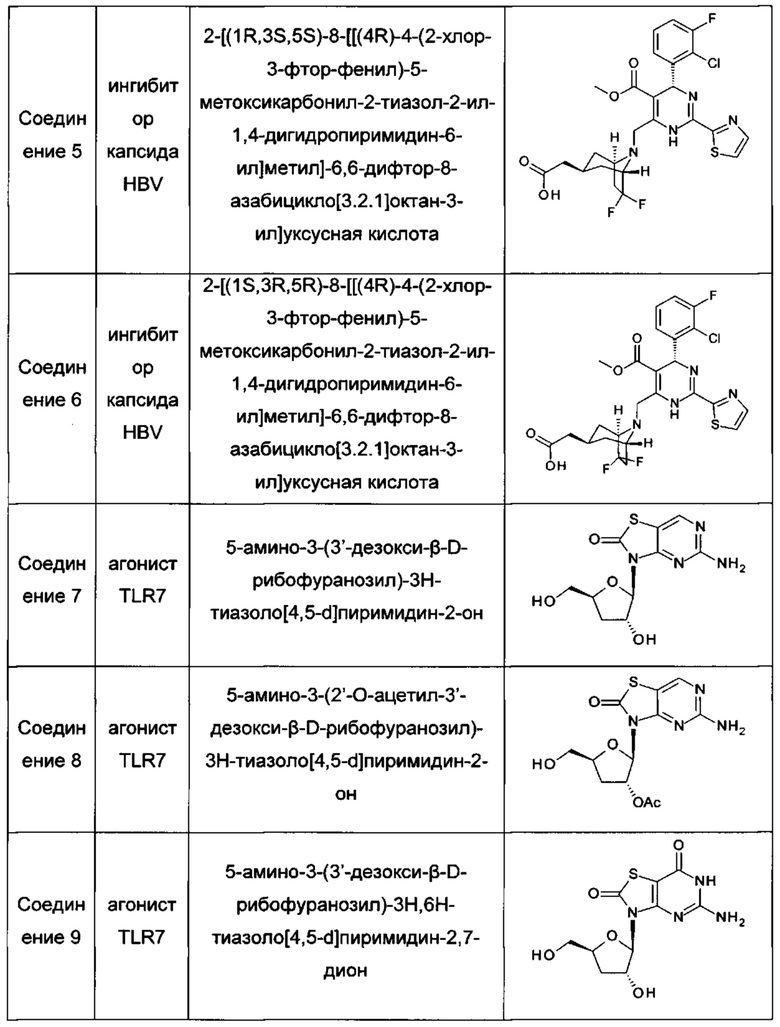
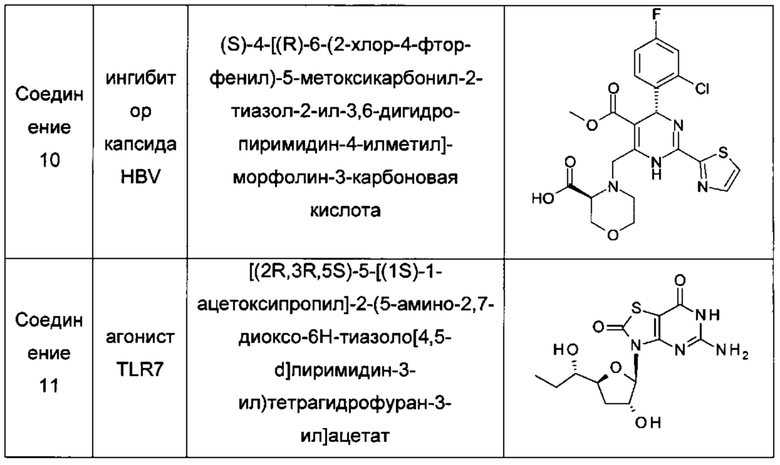
А именно, настоящее изобретение относится к фармацевтической композиции, включающей агонист TLR7 и ингибитор сборки капсида HBV, которая выбрана из любой следующей комбинации:
соединение 1 и соединение 2; соединение 1 и соединение 4;
соединение 1 и соединение 5; соединение 1 и соединение 6;
соединение 1 и соединение 10; соединение 3 и соединение 2;
соединение 3 и соединение 4; соединение 3 и соединение 5;
соединение 3 и соединение 6; соединение 3 и соединение 10;
соединение 7 и соединение 2; соединение 7 и соединение 4;
соединение 7 и соединение 5; соединение 7 и соединение 6;
соединение 7 и соединение 10; соединение 8 и соединение 2;
соединение 8 и соединение 4; соединение 8 и соединение 5;
соединение 8 и соединение 6; соединение 8 и соединение 10;
соединение 9 и соединение 2; соединение 9 и соединение 4;
соединение 9 и соединение 5; соединение 9 и соединение 6;
соединение 9 и соединение 10; соединение 11 и соединение 2;
соединение 11 и соединение 4; соединение 11 и соединение 5;
соединение 11 и соединение 6; и соединение 11 и соединение 10.
Соединение 1-11 вышеуказанной комбинации может быть заменено его соответствующей фармацевтически приемлемой солью, энантиомером или диастереомером, который является другим аспектом данного изобретения.
Соединение 1 вышеуказанной комбинации может быть заменено его соответствующими моно, двойными или тройными пролекарствами, такими как:
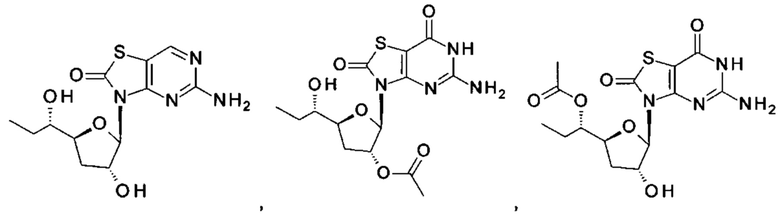 ,
,
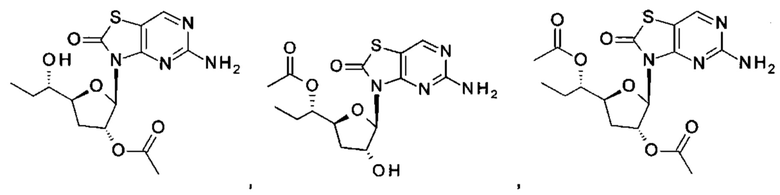 ,
,
и их фармацевтически приемлемой солью, энантиомером или диастереомером.
В одном воплощении настоящего изобретения фармацевтическая композиция состоит из агониста TLR7 и ингибитора сборки капсида HBV в фармацевтически приемлемом носителе. А именно, композиция состоит из:
[(1S)-1-[(2S,4R,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-4-гидрокси-тетрагидрофуран-2-ил]пропил]ацетата и 3-[(8aS)-7-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-этоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановой кислоты;
[(1S)-1-[(2S,4R,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-4-гидрокси-тетрагидрофуран-2-ил]пропил]ацетата и 3-[(8aS)-7-[[(4S)-5-этоксикарбонил-4-(3-фтор-2-метил-фенил)-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановой кислоты;
[(1S)-1-[(2S,4R,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-4-гидрокси-тетрагидрофуран-2-ил]пропил]ацетата и 2-[(1R,3S,5S)-8-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-6,6-дифтор-8-азабицикло[3.2.1]октан-3-ил]уксусной кислоты;
[(1S)-1-[(2S,4R,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-4-гидрокси-тетрагидрофуран-2-ил]пропил]ацетата и 2-[(1S,3R,5R)-8-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-6,6-дифтор-8-азабицикло[3.2.1]октан-3-ил]уксусной кислоты;
[(S)-[(2S,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-1,3-оксатиолан-2-ил]-циклопропил-метил]ацетата и 3-[(8aS)-7-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-этоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановой кислоты;
[(S)-[(2S,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-1,3-оксатиолан-2-ил]-циклопропил-метил]ацетата и 3-[(8aS)-7-[[(4S)-5-этоксикарбонил-4-(3-фтор-2-метил-фенил)-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановой кислоты;
[(S)-[(2S,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-1,3-оксатиолан-2-ил]-циклопропил-метил]ацетата и 2-[(1R,3S,5S)-8-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-6,6-дифтор-8-азабицикло[3.2.1]октан-3-ил]уксусной кислоты;
[(S)-[(2S,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-1,3-оксатиолан-2-ил]-циклопропил-метил]ацетата и 2-[(1S,3R,5R)-8-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-6,6-дифтор-8-азабицикло[3.2.1]октан-3-ил]уксусной кислоты;
[(1S)-1-[(2S,4R,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-4-гидрокси-тетрагидрофуран-2-ил]пропил]ацетата и (S)-4-[(R)-6-(2-хлор-4-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-3,6-дигидро-пиримидин-4-илметил]-морфолин-3-карбоновой кислоты;
[(S)-[(2S,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-1,3-оксатиолан-2-ил]-циклопропил-метил]ацетата и (S)-4-[(R)-6-(2-хлор-4-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-3,6-дигидро-пиримидин-4-илметил]-морфолин-3-карбоновой кислоты;
5-амино-3-(3'-дезокси-β-D-рибофуранозил)-3H-тиазоло[4,5-d]пиримидин-2-она и (S)-4-[(R)-6-(2-хлор-4-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-3,6-дигидро-пиримидин-4-илметил]-морфолин-3-карбоновой кислоты;
5-амино-3-(2'-O-ацетил-3'-дезокси-β-D-рибофуранозил)-3H-тиазоло[4,5-d]пиримидин-2-она и (S)-4-[(R)-6-(2-хлор-4-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-3,6-дигидро-пиримидин-4-илметил]-морфолин-3-карбоновой кислоты;
5-амино-3-(3'-дезокси-β-D-рибофуранозил)-3H,6Н-тиазоло[4,5-d]пиримидин-2,7-диона и (S)-4-[(R)-6-(2-хлор-4-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-3,6-дигидро-пиримидин-4-илметил]-морфолин-3-карбоновой кислоты;
5-амино-3-(3'-дезокси-β-D-рибофуранозил)-3H-тиазоло[4,5-d]пиримидин-2-она и 3-[(8aS)-7-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-этоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановой кислоты;
5-амино-3-(3'-дезокси-β-D-рибофуранозил)-3H-тиазоло[4,5-d]пиримидин-2-она и 3-[(8aS)-7-[[(4S)-5-этоксикарбонил-4-(3-фтор-2-метил-фенил)-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановой кислоты;
5-амино-3-(3'-дезокси-β-D-рибофуранозил)-3H-тиазоло[4,5-d]пиримидин-2-она и 2-[(1R,3S,5S)-8-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-6,6-дифтор-8-азабицикло[3.2.1]октан-3-ил]уксусной кислоты;
5-амино-3-(3'-дезокси-β-D-рибофуранозил)-3H-тиазоло[4,5-d]пиримидин-2-она и 2-[(18,3R,5R)-8-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-6,6-дифтор-8-азабицикло[3.2.1]октан-3-ил]уксусной кислоты;
5-амино-3-(2'-O-ацетил-3'-дезокси-β-D-рибофуранозил)-3H-тиазоло[4,5-d]пиримидин-2-она и 3-[(8aS)-7-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-этоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановой кислоты;
5-амино-3-(2'-O-ацетил-3'-дезокси-β-D-рибофуранозил)-3H-тиазоло[4,5-d]пиримидин-2-она и 3-[(8aS)-7-[[(4S)-5-этоксикарбонил-4-(3-фтор-2-метил-фенил)-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановой кислоты;
5-амино-3-(2'-O-ацетил-3'-дезокси-β-D-рибофуранозил)-3H-тиазоло[4,5-d]пиримидин-2-она и 2-[(1R,3S,5S)-8-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-6,6-дифтор-8-азабицикло[3.2.1]октан-3-ил]уксусной кислоты;
5-амино-3-(2'-O-ацетил-3'-дезокси-β-D-рибофуранозил)-3H-тиазоло[4,5-d]пиримидин-2-она и 2-[(1S,3R,5R)-8-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-6,6-дифтор-8-азабицикло[3.2.1]октан-3-ил]уксусной кислоты;
5-амино-3-(3'-дезокси-β-D-рибофуранозил)-3H,6Н-тиазоло[4,5-d]пиримидин-2,7-диона и 3-[(8aS)-7-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-этоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановой кислоты;
5-амино-3-(3'-дезокси-β-D-рибофуранозил)-3H,6Н-тиазоло[4,5-d]пиримидин-2,7-диона и 3-[(8aS)-7-[[(4S)-5-этоксикарбонил-4-(3-фтор-2-метил-фенил)-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановой кислоты;
5-амино-3-(3'-дезокси-β-D-рибофуранозил)-3H,6Н-тиазоло[4,5-d]пиримидин-2,7-диона и 2-[(1R,3S,5S)-8-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-6,6-дифтор-8-азабицикло[3.2.1]октан-3-ил]уксусной кислоты или
5-амино-3-(3'-дезокси-β-D-рибофуранозил)-3H,6Н-тиазоло[4,5-d]пиримидин-2,7-диона и 2-[(1S,3R,5R)-8-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-6,6-дифтор-8-азабицикло[3.2.1]октан-3-ил]уксусной кислоты; в фармацевтически приемлемом носителе.
В другом воплощении настоящего изобретения фармацевтическая композиция состоит из агониста TLR7 и ингибитора сборки капсида HBV в фармацевтически приемлемом носителе, а именно, композиция состоит из:
[(1S)-1-[(2S,4R,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-4-гидрокси-тетрагидрофуран-2-ил]пропил]ацетата и 3-[(8aS)-7-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-этоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановой кислоты;
[(1S)-1-[(2S,4R,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-4-гидрокси-тетрагидрофуран-2-ил]пропил]ацетата и 3-[(8aS)-7-[[(4S)-5-этоксикарбонил-4-(3-фтор-2-метил-фенил)-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановой кислоты;
[(S)-[(2S,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-1,3-оксатиолан-2-ил]-циклопропил-метил]ацетата и 3-[(8aS)-7-[[(4S)-5-этоксикарбонил-4-(3-фтор-2-метил-фенил)-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановой кислоты;
[(1S)-1-[(2S,4R,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-4-гидрокси-тетрагидрофуран-2-ил]пропил]ацетата и 2-[(1R,3S,5S)-8-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-6,6-дифтор-8-азабицикло[3.2.1]октан-3-ил]уксусной кислоты;
[(1S)-1-[(2S,4R,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-4-гидрокси-тетрагидрофуран-2-ил]пропил]ацетата и (S)-4-[(R)-6-(2-хлор-4-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-3,6-дигидро-пиримидин-4-илметил]-морфолин-3-карбоновой кислоты;
5-амино-3-(2'-O-ацетил-3'-дезокси-β-D-рибофуранозил)-3H-тиазоло[4,5-d]пиримидин-2-она и 3-[(8aS)-7-[[(4S)-5-этоксикарбонил-4-(3-фтор-2-метил-фенил)-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановой кислоты;
5-амино-3-(2'-O-ацетил-3'-дезокси-β-D-рибофуранозил)-3H-тиазоло[4,5-d]пиримидин-2-она и (S)-4-[(R)-6-(2-хлор-4-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-3,6-дигидро-пиримидин-4-илметил]-морфолин-3-карбоновой кислоты;
[(2R,3R,5S)-5-[(1S)-1-ацетоксипропил]-2-(5-амино-2,7-диоксо-6Н-тиазоло[4,5-d]пиримидин-3-ил)тетрагидрофуран-3-ил]ацетата и 3-[(8aS)-7-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-этоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановой кислоты;
[(2R,3R,5S)-5-[(1S)-1-ацетоксипропил]-2-(5-амино-2,7-диоксо-6Н-тиазоло[4,5-d]пиримидин-3-ил)тетрагидрофуран-3-ил]ацетата и 3-[(8aS)-7-[[(4S)-5-этоксикарбонил-4-(3-фтор-2-метил-фенил)-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановой кислоты;
[(2R,3R,5S)-5-[(18)-1-ацетоксипропил]-2-(5-амино-2,7-диоксо-6Н-тиазоло[4,5-d]пиримидин-3-ил)тетрагидрофуран-3-ил]ацетата и 2-[(1R,3S,5S)-8-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-6,6-дифтор-8-азабицикло[3.2.1]октан-3-ил]уксусной кислоты;
[(2R,3R,5S)-5-[(1S)-1-ацетоксипропил]-2-(5-амино-2,7-диоксо-6Н-тиазоло[4,5-d]пиримидин-3-ил)тетрагидрофуран-3-ил]ацетата и 2-[(1S,3R,5R)-8-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-6,6-дифтор-8-азабицикло[3.2.1]октан-3-ил]уксусной кислоты или
[(2R,3R,5S)-5-[(1S)-1-ацетоксипропил]-2-(5-амино-2,7-диоксо-6Н-тиазоло[4,5-d]пиримидин-3-ил)тетрагидрофуран-3-ил]ацетата и (S)-4-[(R)-6-(2-хлор-4-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-3,6-дигидро-пиримидин-4-илметил]-морфолин-3-карбоновой кислоты; в фармацевтически приемлемом носителе.
В другом воплощении настоящего изобретения другие агонисты TLR7 или ингибиторы сборки капсида HBV также можно использовать в фармацевтической композиции, включая малые молекулы или макромолекулы. Примеры других агонистов TLR7 включают, но не ограничиваются этим, имиквимод, резиквимод, PF-4878691, SM-276001, ANA975, ANA773 и GS9620. Примеры других ингибиторов сборки капсида HBV включают, но не ограничиваются этим, Bay 41-4109, Bay 38-7690, Bay 39-5493, GLS4, АТ-61 и АТ-130.
В другом воплощении настоящего изобретения фармацевтическая композиция может дополнительно включать один или более других противовирусных агентов, которые включают, но не ограничиваются этим, ламивудин, адефовир, тенофовир, телбивудин и энтекавир.
Обычные дозировки агониста TLR7 и/или ингибитора сборки капсида HBV могут находиться в диапазонах, рекомендованных изготовителем, и когда показано откликами in vitro на животных моделях, могут быть уменьшены вплоть до приблизительно одного порядка величины концентрации или количества. Таким образом, фактическая дозировка будет зависеть от решения врача, состояния пациента и эффективности терапевтического способа, исходя из ответной реакции in vitro соответствующих животных моделей.
Другое воплощение настоящего изобретения относится к способу изготовления лекарственного средства для лечения или профилактики вирусной инфекции гепатита В, отличающемуся тем, что агонист TLR7 и ингибитор сборки капсида HBV используют в лекарственном средстве.
Дополнительное воплощение настоящего изобретения относится к способу изготовления лекарственного средства для лечения или профилактики вирусной инфекции гепатита В, отличающемуся тем, что агонист TLR7 и ингибитор сборки капсида HBV совместно вводят в одном и том же препарате или разных препаратах.
Для целей настоящего изобретения "совместно вводить" относится к любому введению агониста TLR7 и ингибитора сборки капсида HBV в качестве двух активных агентов, либо по отдельности, либо совместно, когда два активных агента вводят как часть подходящего режима дозирования, предназначенного для получения положительного эффекта от комбинированного лечения. Таким образом, два активных агента могут быть введены либо как часть одной и той же фармацевтической композиции, либо в раздельных фармацевтических композициях. Также два активных агента могут быть введены либо в одно и то же время, либо последовательно.
Агонист TLR7 и ингибитор сборки капсида HBV могут быть введены с разными фармацевтически приемлемыми инертными носителями в форме таблеток, капсул, леденцов, пастилок, карамелек, порошков, спреев, кремов, бальзамов, суппозиториев, желе, гелей, паст, лосьонов, мазей, эликсиров, сиропов и подобных. Введение таких лекарственных форм можно осуществлять в виде однократной дозы или многократных доз. Носители включают твердые разбавители наполнителей, стерильную водную среду и разные нетоксичные органические растворители. Введение таких лекарственных форм можно выполнять посредством, но не ограничиваясь этим, перорального введения, парентерального введения, ветеринарного введения.
Дополнительное воплощение настоящего изобретения относится к способу изготовления лекарственного средства для лечения или профилактики вирусной инфекции гепатита В, отличающемуся тем, что агонист TLR7 и ингибитор сборки капсида HBV предназначены для введения субъекту одним и тем же путем или разными путями.
Дополнительное воплощение настоящего изобретения относится к способу изготовления лекарственного средства для лечения или профилактики вирусной инфекции гепатита В, отличающемуся тем, что агонист TLR7 и ингибитор сборки капсида HBV предназначены для введения субъекту путём парентерального или перорального введения.
Дополнительное воплощение настоящего изобретения относится к способу изготовления лекарственного средства для лечения или профилактики вирусной инфекции гепатита В, отличающемуся тем, что введение агониста TLR7 и ингибитора сборки капсида HBV субъекту происходит одновременно или последовательно. В любом из способов настоящего изобретения введение агентов одновременно можно выполнить, по отдельности или последовательно вводя агенты в одно и то же время, или вместе в виде комбинированного препарата. Также в любом из способов настоящего изобретения введение агентов по отдельности или последовательно может быть в любом порядке.
Другое воплощение настоящего изобретения относится к способу изготовления лекарственного средства для лечения или профилактики вирусной инфекции гепатита В, отличающемуся тем, что агонист TLR7 представляет собой соединение формулы (I) или формула (II), или его фармацевтически приемлемую соль, энантиомер или диастереомер. В частности агонист TLR7 представляет собой [(1S)-1-[(2S,4R,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-4-гидрокси-тетрагидрофуран-2-ил]пропил]ацетат; [(S)-[(2S,5R)-5-(5-амино-2-оксо-тиазоло[4,5-а]пиримидин-3-ил)-1,3-оксатиолан-2-ил]-циклопропил-метил]ацетат; 5-амино-3-(3'-дезокси-β-D-рибофуранозил)-3H-тиазоло[4,5-d]пиримидин-2-он; 5-амино-3-(2'-O-ацетил-3'-дезокси-β-D-рибофуранозил)-3H-тиазоло[4,5-d]пиримидин-2-он; 5-амино-3-(3'-дезокси-β-D-рибофуранозил)-3H,6Н-тиазоло[4,5-а]пиримидин-2,7-дион или [(2R,3S,5S)-5-[(1S)-1-ацетоксипропил]-2-(5-амино-2,7-диоксо-6Н-тиазоло[4,5-d]пиримидин-3-ил)тетрагидрофуран-3-ил]ацетат; или их фармацевтически приемлемую соль, энантиомер или диастереомер.
Другое воплощение настоящего изобретения относится к способу изготовления лекарственного средства для лечения или профилактики вирусной инфекции гепатита В, отличающемуся тем, что ингибитор сборки капсида HBV представляет собой соединение формулы (III) или его фармацевтически приемлемую соль, энантиомер или диастереомер. В частности ингибитор сборки капсида HBV представляет собой
3-[(8aS)-7-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-этоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановую кислоту;
3-[(8aS)-7-[[(4S)-5-этоксикарбонил-4-(3-фтор-2-метил-фенил)-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановую кислоту;
2-[(1R,3S,5S)-8-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-6,6-дифтор-8-азабицикло[3.2.1]октан-3-ил]уксусную кислоту;
2-[(1S,3R,5R)-8-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-6,6-дифтор-8-азабицикло[3.2.1]октан-3-ил]уксусную кислоту
или (S)-4-[(R)-6-(2-хлор-4-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-3,6-дигидро-пиримидин-4-илметил]-морфолин-3-карбоновую кислоту;
или их фармацевтически приемлемую соль, энантиомер или диастереомер.
Другое воплощение настоящего изобретения относится к способу изготовления лекарственного средства для лечения или профилактики вирусной инфекции гепатита В, отличающемуся тем, что лекарственное средство дополнительно включает один или более других противовирусных агентов, которые включают, но не ограничиваются этим, ламивудин, адефовир, тенофовир, телбивудин и энтекавир.
Другое воплощение настоящего изобретения относится к способу изготовления лекарственного средства для лечения или профилактики вирусной инфекции гепатита В, где агонист TLR7 и ингибитор сборки капсида HBV, используемые в лекарственном средстве, представляют собой:
[(1S)-1-[(2S,4R,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-4-гидрокси-тетрагидрофуран-2-ил]пропил]ацетат и 3-[(8aS)-7-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-этоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановую кислоту;
[(1S)-1-[(2S,4R,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-4-гидрокси-тетрагидрофуран-2-ил]пропил]ацетат и 3-[(8aS)-7-[[(4S)-5-этоксикарбонил-4-(3-фтор-2-метил-фенил)-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановую кислоту;
[(S)-[(2S,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-1,3-оксатиолан-2-ил]-циклопропил-метил]ацетат и 3-[(8aS)-7-[[(4S)-5-этоксикарбонил-4-(3-фтор-2-метил-фенил)-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановую кислоту;
[(1S)-1-[(2S,4R,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-4-гидрокси-тетрагидрофуран-2-ил]пропил]ацетат и 2-[(1R,3S,5S)-8-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-6,6-дифтор-8-азабицикло[3.2.1]октан-3-ил]уксусную кислоту;
[(1S)-1-[(2S,4R,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-4-гидрокси-тетрагидрофуран-2-ил]пропил]ацетат и (S)-4-[(R)-6-(2-хлор-4-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-3,6-дигидро-пиримидин-4-илметил]-морфолин-3-карбоновую кислоту;
5-амино-3-(2'-O-ацетил-3'-дезокси-β-D-рибофуранозил)-3H-тиазоло[4,5-d]пиримидин-2-он и 3-[(8aS)-7-[[(4S)-5-этоксикарбонил-4-(3-фтор-2-метил-фенил)-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановую кислоту или
5-амино-3-(2'-O-ацетил-3'-дезокси-β-D-рибофуранозил)-3H-тиазоло[4,5-d]пиримидин-2-он и (S)-4-[(R)-6-(2-хлор-4-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-3,6-дигидро-пиримидин-4-илметил]-морфолин-3-карбоновую кислоту; в фармацевтически приемлемом носителе.
Другое воплощение настоящего изобретения относится к набору, включающему контейнер, содержащий агонист TLR7 и ингибитор сборки капсида HBV, указанный набор может дополнительно включать стерильный разбавитель.
Дополнительное воплощение настоящего изобретения относится к указанному набору, где набор может дополнительно включать листок-вкладыш, содержащий напечатанные инструкции по применению комбинированного лечения с агонистом TLR7 и ингибитором сборки капсида HBV в качестве способа лечения или профилактики вирусной инфекции гепатита В.
Другое воплощение настоящего изобретения относится к указанному набору, где агонист TLR7 представляет собой [(1S)-1-[(2S,4R,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-4-гидрокси-тетрагидрофуран-2-ил]пропил]ацетат; [(S)-[(2S,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-1,3-оксатиолан-2-ил]-циклопропил-метил]ацетат; 5-амино-3-(3'-дезокси-β-D-рибофуранозил)-3H-тиазоло[4,5-d]пиримидин-2-он; 5-амино-3-(2'-O-ацетил-3'-дезокси-β-D-рибофуранозил)-3H-тиазоло[4,5-d]пиримидин-2-он; 5-амино-3-(3'-дезокси-β-D-рибофуранозил)-3H,6Н-тиазоло[4,5-d]пиримидин-2,7-дион или [(2R,3R,5S)-5-[(1S)-1-ацетоксипропил]-2-(5-амино-2,7-диоксо-6H-THa3ono[4,5-d]пиримидин-3-ил)тетрагидрофуран-3-ил]ацетат; или их фармацевтически приемлемую соль, энантиомер или диастереомер; и/или ингибитор сборки капсида HBV представляет собой 3-[(8aS)-7-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-этоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановую кислоту; 3-[(8aS)-7-[[(4S)-5-этоксикарбонил-4-(3-фтор-2-метил-фенил)-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановую кислоту; 2-[(1R,3S,5S)-8-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-6,6-дифтор-8-азабицикло[3.2.1]октан-3-ил]уксусную кислоту; 2-[(1S,3R,5R)-8-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-6,6-дифтор-8-азабицикло[3.2.1]октан-3-ил]уксусную кислоту или (S)-4-[(R)-6-(2-хлор-4-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-3,6-дигидро-пиримидин-4-илметил]-морфолин-3-карбоновую кислоту; или их фармацевтически приемлемую соль, энантиомер или диастереомер.
Другое воплощение настоящего изобретения относится к указанному набору, где агонист TLR7 и ингибитор сборки капсида HBV, используемые в контейнере, представляют собой:
[(1S)-1-[(2S,4R,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-4-гидрокси-тетрагидрофуран-2-ил]пропил]ацетат и 3-[(8aS)-7-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-этоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановую кислоту;
[(1S)-1-[(2S,4R,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-4-гидрокси-тетрагидрофуран-2-ил]пропил]ацетат и 3-[(8aS)-7-[[(4S)-5-этоксикарбонил-4-(3-фтор-2-метил-фенил)-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановую кислоту;
[(S)-[(2S,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-1,3-оксатиолан-2-ил]-циклопропил-метил]ацетат и 3-[(8aS)-7-[[(4S)-5-этоксикарбонил-4-(3-фтор-2-метил-фенил)-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановую кислоту;
[(1S)-1-[(2S,4R,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-4-гидрокси-тетрагидрофуран-2-ил]пропил]ацетат и 2-[(1R,3S,5S)-8-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-6,6-дифтор-8-азабицикло[3.2.1]октан-3-ил]уксусную кислоту;
[(1S)-1-[(2S,4R,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-4-гидрокси-тетрагидрофуран-2-ил]пропил]ацетат и (S)-4-[(R)-6-(2-хлор-4-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-3,6-дигидро-пиримидин-4-илметил]-морфолин-3-карбоновую кислоту;
5-амино-3-(2'-O-ацетил-3'-дезокси-β-D-рибофуранозил)-3H-тиазоло[4,5-d]пиримидин-2-он и 3-[(8aS)-7-[[(4S)-5-этоксикарбонил-4-(3-фтор-2-метил-фенил)-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановую кислоту или
5-амино-3-(2'-O-ацетил-3'-дезокси-β-D-рибофуранозил)-3H-тиазоло[4,5-d]пиримидин-2-он и (S)-4-[(R)-6-(2-хлор-4-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-3,6-дигидро-пиримидин-4-илметил]-морфолин-3-карбоновую кислоту; в фармацевтически приемлемом носителе.
Другое воплощение настоящего изобретения относится к способу лечения или профилактики вирусной инфекции гепатита В, согласно которому субъекту вводят эффективное первое количество агониста TLR7 или его фармацевтически приемлемой соли, энантиомера или диастереомера; и второе количество ингибитора сборки капсида HBV или его фармацевтически приемлемой соли, энантиомера или диастереомера; где агонист TLR7 представляет собой [(1S)-1-[(2S,4R,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-4-гидрокси-тетрагидрофуран-2-ил]пропил]ацетат; [(S)-[(2S,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-1,3-оксатиолан-2-ил]-циклопропил-метил]ацетат; 5-амино-3-(3'-дезокси-β-D-рибофуранозил)-3H-тиазоло[4,5-d]пиримидин-2-он; 5-амино-3-(2'-O-ацетил-3'-дезокси-β-D-рибофуранозил)-3H-тиазоло[4,5-d]пиримидин-2-он; 5-амино-3-(3'-дезокси-β-D-рибофуранозил)-3H,6Н-тиазоло[4,5-d]пиримидин-2,7-дион или [(2R,3R,5S)-5-[(1S)-1-ацетоксипропил]-2-(5-амино-2,7-диоксо-6H-тиазоло[4,5-d]пиримидин-3-ил)тетрагидрофуран-3-ил]ацетат; или их фармацевтически приемлемую соль, энантиомер или диастереомер; и/или ингибитор сборки капсида HBV представляет собой 3-[(8aS)-7-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-этоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановую кислоту; 3-[(8aS)-7-[[(4S)-5-этоксикарбонил-4-(3-фтор-2-метил-фенил)-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановую кислоту; 2-[(1R,3S,5S)-8-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-6,6-дифтор-8-азабицикло[3.2.1]октан-3-ил]уксусную кислоту; 2-[(1S,3R,5R)-8-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-6,6-дифтор-8-азабицикло[3.2.1]октан-3-ил]уксусную кислоту или (S)-4-[(R)-6-(2-хлор-4-фтор-фенил)-5-метоксикарбонил-2-тиазол-2-ил-3,6-дигидро-пиримидин-4-илметил]-морфолин-3-карбоновую кислоту; или их фармацевтически приемлемую соль, энантиомер или диастереомер.
Другое воплощение настоящего изобретения относится к применению фармацевтической композиции, указанной выше в данном документе, в качестве противовирусного лекарственного средства, в частности в качестве лекарственного средства для лечения или профилактики вирусной инфекции гепатита В.
Другое воплощение настоящего изобретения относится к применению агониста TLR7 и ингибитора сборки капсида HBV для изготовления фармацевтической композиции, указанной выше в данном документе, в качестве противовирусного лекарственного средства, в частности в качестве лекарственного средства для лечения или профилактики вирусной инфекции гепатита В.
Примеры
Изобретение станет более понятно с учетом следующих примеров. Однако их не следует истолковывать в качестве ограничения объема изобретения.
Пример 1
[(1S)-[(2S,4R,5R-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-4-гидрокси-тетрагидрофуран-2-ил]пропил]ацетат (соединение 1)
Соединение 1 получали по следующей схеме:
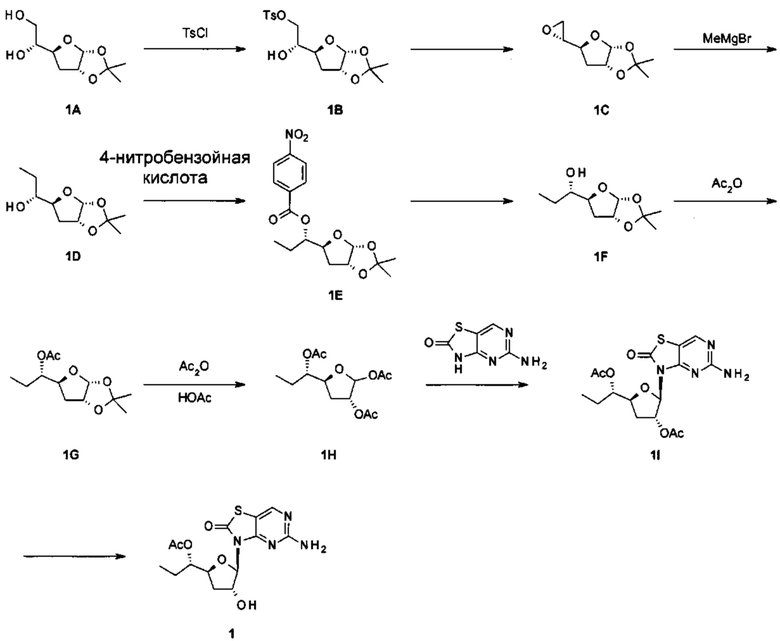
Получение [(2R)-2-[(3aR,5S,6aR)-2,2-диметил-3a,5,6,6а-тетрагидрофуро[2,3-d][1,3]диоксол-5-ил]-2-гидрокси-этил]-4-метилбензолсульфоната
К раствору (1R)-1-[(3aR,5S,6aR)-2,2-диметил-3а,5,6,6а-тетрагидрофуро[2,3-d][1,3]диоксол-5-ил]этан-1,2-диола (соединение 1А, 100 г, 490 ммоль) в безводном пиридине (1000 мл) добавляли п-толуолсульфонилхлорид (139 г, 735 ммоль) при 0°С. После перемешивания при комнатной температуре в течение 12 часов полученный в результате раствор гасили водой (100 мл) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (элюируя 1:10 до 1:3 EtOAc в петролейном эфире), получая 130 г [(2R)-2-[(3aR,5S,6aR)-2,2-диметил-3a,5,6,6а-тетрагидрофуро[2,3-d][1,3]диоксол-5-ил]-2-гидрокси-этил]-4-метилбензолсульфоната (соединение 1В) в виде светло-желтого масла.
Соединение 1В: 1Н ЯМР (ядерный магнитный резонанс) (400 МГц, CDCl3) δ ppm: 7,82 (d, J=8,00 Гц, 2Н), 7,38 (d, J=8,00 Гц, 2Н), 5,78 (d, J=3,76 Гц, 1Н), 4,75 (t, J=4,00 Гц, 1Н), 4,20-4,12 (m, 2Н), 4,03-3,97 (m, 2Н), 2,48 (s, 3Н), 2,39 (d, J=3,51 Гц, 1Н), 2,08-2,15 (m, 1Н), 1,75-1,80 (m, 1Н), 1,51 (s, 3Н), 1,33 (s, 3Н).
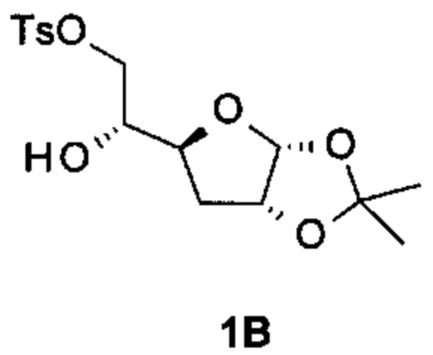
Получение (3aR,4S,6aR)-2,2-диметил-5-[(2R)-оксиран-2-ил]-3a,5,6,6а-тетрагидрофуро[2,3-d] [1,3]диоксола
К раствору [(2R)-2-[(3aR,5S,6aR)-2,2-диметил-3a,5,6,6а-тетрагидрофуро[2,3-d][1,3]диоксол-5-ил]-2-гидрокси-этил]-4-метилбензолсульфоната (соединение 1В, 100 г, 280 ммоль) в безводном тетрагидрофуране (ТГФ) (1500 мл), охлажденному при -70°С, добавляли бис(триметилсилил)амид калия (340 мл, 340 ммоль, 1 М в ТГФ) в атмосфере N2. После перемешивания при -70°С в течение 1 часа реакционную смесь выливали в насыщенный раствор NH4Cl. Органический слой отделяли и водную фазу экстрагировали EtOAc. Объединенные органические слои сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (элюируя 1:3 EtOAc в петролейном эфире), получая 40,5 г (3aR,5S,6aR)-2,2-диметил-5-[(2R)-оксиран-2-ил]-3a,5,6,6а-тетрагидрофуро[2,3-d][1,3]диоксола (соединение 1С) в виде светло-желтого масла.
Соединение 1С: 1Н ЯМР: (400 МГц, CDCl3) δ ppm: 5,87 (d, J=3,76 Гц, 1Н), 4,77 (t, J=4,00, 1Н), 4,20-4,28 (m, 1Н), 3,14-3,20 (m, 1Н), 2,83-2,88 (m, 1Н), 2,63 (dd, J=5,00, 2,80 Гц, 1Н), 2,09 (dd, J=12,00, 4,00 Гц, 1Н), 1,69-1,79 (m, 1Н), 1,52 (s, 3Н), 1,34 (s, 3Н).
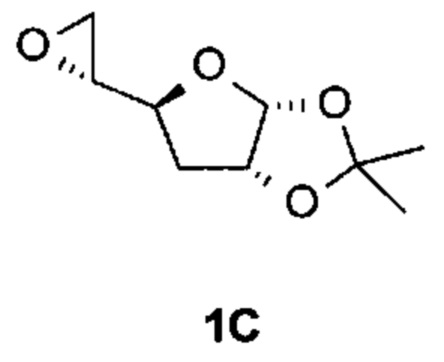
Получение (1R)-1-[(3aR,5S,6aR)-2,2-диметил-3a,5,6,6а-тетрагидрофуро[2,3-d][1,3]диоксол-5-ил]пропан-1-ола
К суспензии CuI (19,3 г, 107 ммоль) в безводном ТГФ (2000 мл) в атмосфере N2 добавляли метилмагнийбромид (3 M в диэтиловом эфире, 537 мл, 1,61 моль) при -70°С. После перемешивания при этой же температуре в течение 1 часа раствор (3aR,5S,6aR)-2,2-диметил-5-[(2R)-оксиран-2-ил]-3a,5,6,6a-тетрагидрофуро[2,3-d][1,3]диоксола (соединение 1С, 100 г, 537 ммоль, растворенное в 200 мл безводного ТГФ) добавляли к реакционной смеси по каплям. После перемешивания при -70°С в течение дополнительных 2 часов реакционную смесь выливали в насыщенный раствор NH4Cl. Органический слой отделяли и водную фазу экстрагировали дважды EtOAc. Объединенные органические слои сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (элюируя 1:3 EtOAc в петролейном эфире), получая 82 г (1R)-1-[(3aR,5S,6aR)-2,2-диметил-3a,5,6,6а-тетрагидрофуро[2,3-d][1,3]диоксол-5-ил]пропан-1-ола (соединение 1D) в виде светло-желтого твердого вещества.
Соединение 1D: 1Н ЯМР (400 МГц, CDCl3) δ ppm: 5,83 (d, J=3,76 Гц, 1Н), 4,81-4,73 (m, 1Н), 4,26-4,19 (m, 1Н), 3,91-3,82 (m, 1Н), 2,08-2,02 (m, 1Н), 1,93-1,89 (m, 1Н), 1,54 (s, 3Н), 1,49-1,39 (m, 2Н), 1,34 (s, 3Н), 1,02 (t, J=7,53 Гц, 3Н).
Получение [(1S)-1-[(3aR,5S,6aR)-2,2-диметил-3a,5,6,6а-тетрагидрофуро[2,3-d][1,3]диоксол-5-ил]пропил]-4-нитробензоата

К перемешиваемому раствору (1R)-1-[(3aR,5S,6aR)-2,2-диметил-3a,5,6,6а-тетрагидрофуро[2,3-d][1,3]диоксол-5-ил]пропан-1-ола (соединение 1D, 50 г, 245 ммоль), трифенилфосфина (195 г, 743 ммоль), 4-нитробензойной кислоты (124 г, 743 ммоль) в ТГФ (1200 мл) добавляли диэтилазодикарбоксилат (130 г, 743 ммоль) по каплям при 0°С в N2. После перемешивания при 18°С в течение 10 часов смесь гасили, добавляя насыщенный раствор NaHCO3, и экстрагировали EtOAc. Органические слои объединяли, сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (элюируя 1:3 EtOAc в петролейном эфире), получая 61 г [(1S)-1-[(3aR,5S,6aR)-2,2-диметил-3a,5,6,6а-тетрагидрофуро[2,3-d][1,3]диоксол-5-ил]пропил]-4-нитробензоата (соединение 1Е) в виде светло-желтого твердого вещества.
Соединение 1Е: 1Н ЯМР (400 МГц, CDCl3) δ ppm: 8,34-8,22 (m, 4Н), 5,85 (d, J=3,76 Гц, 1Н), 5,23-5,17 (m, 1Н), 4,76 (t, J=4,27 Гц, 1Н), 4,48-4,39 (m, 1Н), 2,12 (dd, J=13,30, 4,52 Гц, 1Н), 1,88-1,78 (m, 2Н), 1,71-1,62 (m, 1Н), 1,55 (s, 3Н), 1,34 (s, 3Н), 1,01 (t, J=7,40 Гц, 3Н).
Получение (1S)-1-[(3aR,5S,6aR)-2,2-диметил-3a,5,6,6а-тетрагидрофуро[2,3-d][1,3]диоксол-5-ил]пропан-1-ола

К раствору [(1S)-1-[(3aR,5S,6aR)-2,2-диметил-3a,5,6,6а-тетрагидрофуро[2,3-d][1,3]диоксол-5-ил]пропил]-4-нитробензоата (соединение 1Е, 100 г, 285 ммоль) в метаноле (1200 мл) добавляли K2CO3 (78,7 г, 570 ммоль). После перемешивания при комнатной температуре в течение 10 минут полученную в результате смесь фильтровали. Фильтрат концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (элюируя 1:8 EtOAc в петролейном эфире), получая 54,7 г (1S)-1-[(3aR,5S,6aR)-2,2-диметил-3a,5,6,6а-тетрагидрофуро[2,3-d][1,3]диоксол-5-ил]пропан-1-ола (соединение 1F) в виде светло-желтого твердого вещества.
Соединение 1F 1Н ЯМР (400 МГц, CDCl3) δ ppm: 5,81 (d, J=3,64 Гц, 1Н), 4,75 (t, J=4,20 Гц, 1Н), 4,18-4,11 (m, 1Н), 3,49-3,40 (m, 1Н), 2,07-2,00 (m, 2Н), 1,84-1,75 (m, 1Н), 1,59-1,47 (m, 5Н), 1,32 (s, 3Н), 1,01 (t, J=7,40 Гц, 3Н).
Получение [(1S)-1-[(3aR,5S,6aR)-2,2-диметил-3a,5,6,6а-тетрагидрофуро[2,3-d][1,3]диоксол-5-ил]пропил]ацетата

К перемешиваемому раствору (1S)-1-[(3aR,5S,6aR)-2,2-диметил-3a,5,6,6а-тетрагидрофуро[2,3-d][1,3]диоксол-5-ил]пропан-1-ола (соединение 1F, 13,5 г, 67 ммоль), триэтаноламина (ТЭА) (81 г, 804 ммоль), диметиламинопиридина (ДМАП) (1,6 г, 13 ммоль) в безводном дихлорметане (ДХМ) (150 мл) добавляли уксусный ангидрид (62 г, 603 ммоль). После перемешивания при 22°С в течение 10 часов реакцию гасили насыщенным раствором NaHCO3. Органический слой отделяли и водную фазу экстрагировали EtOAc. Объединенные органические слои сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (элюируя 1:8 EtOAc в петролейном эфире), получая 13 г [(1S)-1-[(3aR,5S,6R)-2,2-диметил-3a,5,6,6а-тетрагидрофуро[2,3-d][1,3]диоксол-5-ил]пропил]ацетата (соединение 1G) в виде бесцветного масла.
Соединение 1G: 1Н ЯМР (400 МГц, CDCl3) δ ppm: 5,83 (d, J=3,76 Гц, 1Н), 4,92 (dt, J=7,97, 5,18 Гц, 1Н), 4,74 (t, J=4,00 Гц, 1Н), 4,35-4,27 (m, 1Н), 2,12 (s, 3Н), 2,08-1,99 (m, 1Н), 1,74-1,56 (m, 3Н), 1,53 (s, 3Н), 1,34 (s, 3Н), 0,95 (t, J=7,40 Гц, 3Н).
Получение [(3R,5S)-2-ацетокси-5-[(1S)-1-ацетоксипропил]тетрагидрофуран-3-ил]ацетата
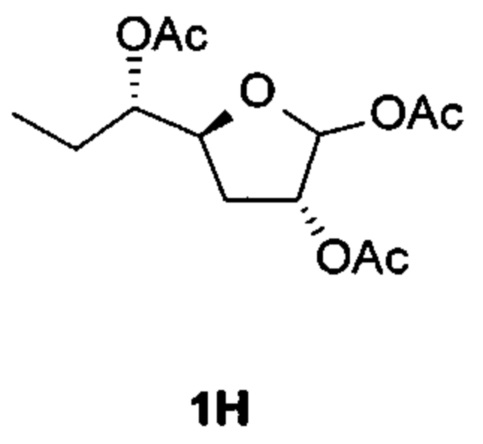
К раствору [(1S)-1-[(3aR,5S,6aR)-2,2-диметил-3a,5,6,6а-тетрагидрофуро[2,3-d][1,3]диоксол-5-ил]пропил]ацетата (соединение 1G, 4,8 г, 20 ммоль), уксусной кислоты (12,2 г, 200 ммоль) и уксусного ангидрида (10,2 г, 100 ммоль) в безводном ДХМ (100 мл) добавляли концентрированную H2SO4 (0,5 мл) при 0°С. После перемешивания при 22°С в течение 3 часов реакцию гасили, добавляя насыщенный раствор NaHCO3. Органический слой отделяли и водную фазу экстрагировали EtOAc. Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали на колонке с силикагелем (элюируя 1:8 EtOAc в петролейном эфире), получая 2,3 г [(3R,5S)-2-ацетокси-5-[(1S)-1-ацетоксипропил]тетрагидрофуран-3-ил]ацетата (соединение 1Н) в виде бесцветного масла.
Соединение 1Н: 1Н ЯМР (400 МГц, CDCl3) δ ppm: 6,12 (s, 1Н), 5,19 (d, J=4,52 Гц, 1Н), 4,83-4,91 (m, 1Н), 4,34-4,44 (m, 1Н), 2,09-2,19 (m, 9Н), 1,51-1,74 (m, 4Н), 0,94 (t, J=7,40 Гц, 3Н).
Получение [(2R,3R,5S)-5-[(1S)-1-ацетоксипропил]-2-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)тетрагидрофуран-3-ил]ацетата
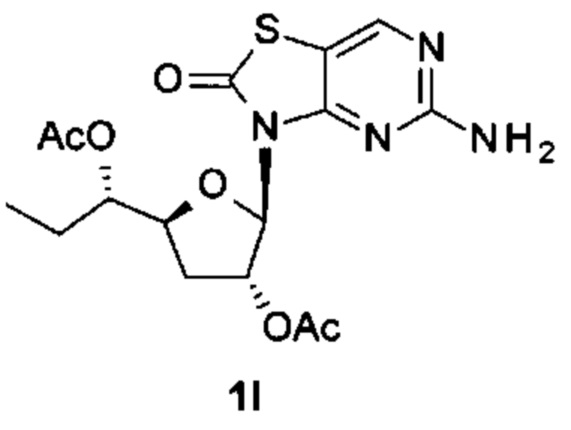
К суспензии 5-амино-3H-тиазоло[4,5-d]пиримидин-2-она (3,5 г, 20,8 ммоль) в ксилоле (160 мл) добавляли N,O-бис(триметилсилил)ацетамид (БСА) (21,2 г, 104 ммоль). Реакционную смесь перемешивали при 70°С в течение 1 часа в аргоне до образования прозрачного раствора. Затем выпаривали некоторое количество ксилола и лишний БСА, добавляли [(3R,5S)-2-ацетокси-5-[(1S)-1-ацетоксипропил]тетрагидрофуран-3-ил]ацетат (соединение 1Н, 3,0 г, 10,4 ммоль) и триметилсилил трифторметансульфонат (TMSOTf) (2,6 г, 11,6 ммоль) друг за другом при 0°С. После нагревания с перемешиванием при 65°С в течение 2 часов реакцию гасили водой (30 мл), экстрагировали этилацетатом (ЭА) (30 мл) три раза. Объединенные органические слои сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали на колонке с силикагелем (элюируя 1:10 до 1:1 EtOAc в петролейном эфире), получая 2,0 г [(2R,3R,5S)-5-[(1S)-1-ацетоксипропил]-2-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)тетрагидрофуран-3-ил]ацетата (соединение 11) в виде белого твердого вещества.
Соединение 1I: 1Н ЯМР (400 МГц, CDCl3) δ ppm: 8,15 (s, 1Н), 6,04 (d, J=1,51 Гц, 1Н), 5,80 (d, J=7,03 Гц, 1Н), 5,27 (br. s., 2Н), 4,98-5,04 (m, 1Н), 4,32-4,39 (m, 1Н), 2,63-2,77 (m, 1Н), 2,13 (s, 3Н), 2,09 (s, 3Н), 2,00-2,07 (m, 1Н), 1,61-1,75 (m, 2Н), 0,94 (t, J=7,40 Гц, 3Н).
Получение [(1S)-1-[(2S,4R,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-4-гидрокси-тетрагидрофуран-2-ил]пропил]ацетата
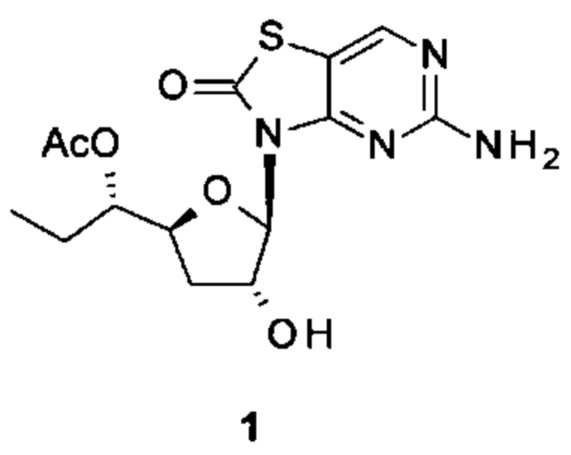
[(2R,3R,5S)-5-[(1S)-1-Ацетоксипропил]-2-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)тетрагидрофуран-3-ил]ацетат (соединение 1I, 3,2 г, 8,0 ммоль) и K2CO3 (2,2 г, 16,0 ммоль) суспендировали в безводном этаноле (85 мл) при комнатной температуре. Метанол (85 мл) добавляли по каплям в атмосфере N2. После добавления смесь перемешивали при комнатной температуре в течение 10 минут (контролировали с помощью тонкослойной хроматографии (ТСХ)). После реакции смесь выливали в насыщенный NH4Cl, экстрагировали ЭА (150 мл) четыре раза. Объединенные органические слои сушили над Na2SO4, концентрировали в вакууме. Остаток очищали на колонке с силикагелем (элюируя 1:100 до 1:70 МеОН в ДХМ), получая неочищенный продукт, который дополнительно очищали на флэш-колонке (элюируя ацетонитрилом и водой), получая 1,64 г [(1S)-1-[(2S,4R,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-4-гидрокси-тетрагидрофуран-2-ил]пропил]ацетата (соединение 1) в виде белого порошка.
Соединение 1: 1Н ЯМР (400 МГц, метанол-d4) δ ppm: 8,19 (s, 1Н), 6,02-6,05 (m, 1Н), 4,94-5,00 (m, 2Н), 4,33-4,40 (m, 1Н), 2,58-2,68 (m, 1Н), 2,03 (s, 3Н), 1,86-1,96 (m, 1Н), 1,56-1,77 (m, 2Н), 0,93 (t, J=7,40 Гц, 3Н). МС (масс-спектрометрия) наблюд. (ЭСИ- (электроспрей-ионизация)) [(М+Н)+]: 355,0.
Пример 2
3-[(8aS)-7-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-этоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановая кислота
Соединение 2 получали по следующей схеме:

К перемешиваемому раствору этил (4R)-6-(бромметил)-4-(2-хлор-3-фтор-фенил)-2-тиазол-2-ил-1,4-дигидропиримидин-5-карбоксилата (соединение 2А, 0,073 г, 0,16 ммоль) и 3-[(8aS)-3-оксо-1,5,6,7,8,8а-гексагидроимидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановой кислоты (соединение 2В, неочищенное, 0,25 ммоль) в 1,2-дихлорэтане (5 мл) добавляли по каплям диизопропилэтиламин (ДИПЭА) (0,078 мл, 0,45 ммоль). Реакционную смесь перемешивали при комнатной температуре до исчезновения соединения 2А. Затем смесь разбавляли EtOAc (50 мл) и промывали последовательно насыщенным водным раствором NH4Cl и солевым раствором. Органический слой отделяли и сушили над Na2SO4. Растворитель удаляли в вакууме и неочищенный продукт очищали с помощью препаративной высокоэффективной жидкостной хроматографии (преп-ВЭЖХ), получая 3-[(8aS)-7-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-этоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановую кислоту (соединение 2) в виде светло-желтого твердого вещества (12 мг). 1Н ЯМР (400 МГц, метанол-d4) ppm 7,92-8,02 (m, 1Н), 7,70-7,80 (m, 1Н), 7,21-7,36 (m, 2Н), 7,10-7,21 (m, 1Н), 6,19-6,28 (m, 1Н), 3,99-4,14 (m, 3Н), 3,81-3,96 (m, 3Н), 3,47-3,56 (m, 1Н), 3,38-3,44 (m, 1Н), 3,27-3,32 (m, 1Н), 3,15-3,25 (m, 1Н), 3,07-3,14 (m, 1Н), 2,79-2,96 (m, 2Н), 2,30-2,41 (m, 1Н), 2,13-2,23 (m, 1Н), 1,20 (t, J=2,76 Гц, 6Н), 1,13 (m, 3Н). МС: рассч. 619 (М+Н)+, измеренная 619 (М+Н)+.
Получение этил (4R)-6-(бромметил)-4-(2-хлор-3-фтор-фенил)-2-тиазол-2-ил-1,4-дигидропиримидин-5-карбоксилата (соединение 2А):
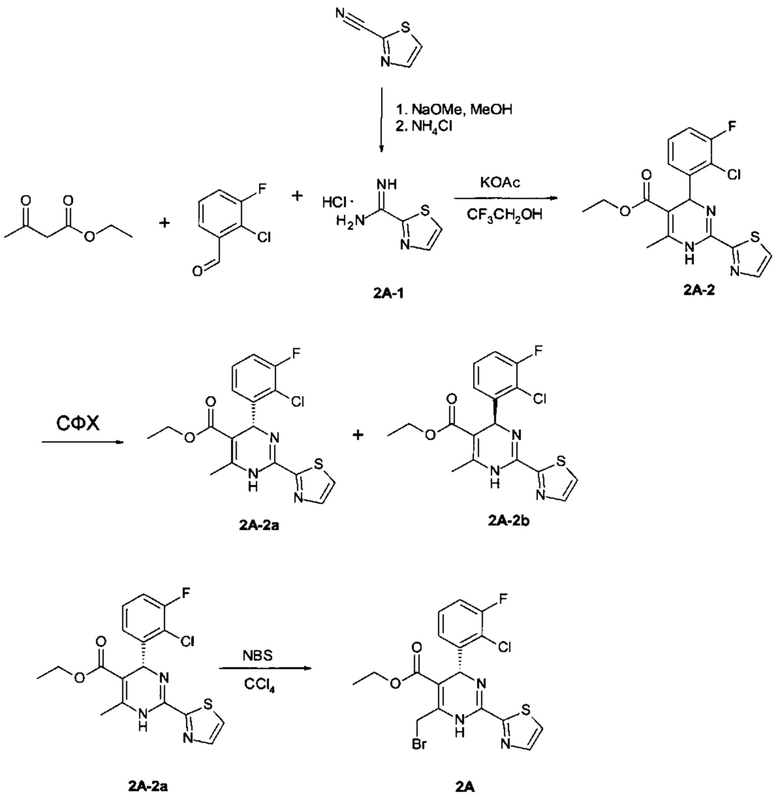
Получение тиазол-2-карбоксамидина гидрохлорида (соединение 2А-1): К перемешиваемому раствору тиазол-2-карбонитрила (1,5 г, 14 ммоль) в 5 мл безводного МеОН добавляли по каплям раствор метилата натрия (0,74 г, 14 ммоль) в 10 мл безводного метанола. Реакционную смесь перемешивали при комнатной температуре до исчезновения исходного вещества. Затем добавляли хлорид аммония (1,5 г, 28 ммоль) за один раз и реакционную смесь перемешивали в течение ночи. Нерастворившееся вещество удаляли фильтрованием, и фильтрат концентрировали, получая тиазол-2-карбоксамидина гидрохлорид (соединение 2А-1, 2,1 г) в виде серого твердого вещества, которое непосредственно использовали на следующей стадии без дополнительной очистки. МС: рассч. 128 (М+Н)+, измеренная 128 (М+Н)+.
Получение этил 4-(2-хлор-3-фтор-фенил)-6-метил-2-тиазол-2-ил-1,4-дигидропиримидин-5-карбоксилата (соединение 2А-2):
К перемешиваемому раствору тиазол-2-карбоксамидина гидрохлорида (1,3 г, 10 ммоль), этилацетоацетата (1,3 г, 10 ммоль) и 2-хлор-3-фторбензальдегида (1,6 г, 10 ммоль) в трифторэтаноле (30 мл) добавляли ацетат калия (2,0 г, 20 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 16 часов. Затем охлаждали до комнатной температуры, реакционную смесь концентрировали, и остаток растворяли в этилацетате и затем промывали солевым раствором. Органический слой сушили над Na2SO4. Растворитель удаляли в вакууме, и остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат/петролейный эфир: от 1/4 до 1/2), получая этил 4-(2-хлор-3-фтор-фенил)-6-метил-2-тиазол-2-ил-1,4-дигидропиримидин-5-карбоксилат (соединение 2А-2, 2,8 г) в виде желтого твердого вещества. МС: рассч. (М+Н)+ 380, измеренная (М+Н)+ 380.
Получение этил (4R)-4-(2-хлор-3-фтор-фенил)-6-метил-2-тиазол-2-ил-1,4-дигидропиримидин-5-карбоксилата (соединение 2А-2а):
Хиральное разделение рацемического соединения 2А-2, элюируемого смешанным растворителем 85% сверхкритического CO2/15% EtOH при скорости 100 мл/мин при использовании сверхкритической флюидной хроматографии (СФХ) (SFC-Multigram; IС: 5×250 мм, 5 мкм), давало два энантиомера этил (4R)-4-(2-хлор-3-фтор-фенил)-6-метил-2-тиазол-2-ил-1,4-дигидропиримидин-5-карбоксилат (соединение 2А-2а, быстрее элюируется) и этил (4S-4-(2-хлор-3-фтор-фенил)-6-метил-2-тиазол-2-ил-1,4-дигидропиримидин-5-карбоксилат (соединение 2А-2a, медленнее элюируется). Абсолютную конфигурацию требуемого (-)-энантиомера соединения 2А-2а ([α]D20 -46,6 (с 0,28, МеОН)) определяли в ходе рентгеноструктурного анализа (фигура 2).
Получение этил (4R)-6-(бромметил)-4-(2-хлор-3-фтор-фенил)-2-тиазол-2-ил-1,4-дигидропиримидин-5-карбоксилата (соединение 2А):
К перемешиваемому раствору этил (4R)-4-(2-хлор-3-фтор-фенил)-6-метил-2-тиазол-2-ил-1,4-дигидропиримидин-5-карбоксилата (соединение 2А-2а, 0,37 г, 1,0 ммоль) в CCl4 (5 мл) добавляли N-бромсукцинимид (NBS) (0,20 г, 1,1 ммоль) частями. Затем реакционную смесь перемешивали при комнатной температуре в течение 1 часа, растворитель удаляли в вакууме и остаток очищали с помощью колоночной хроматографии на силикагеле, получая этил (4R)-6-(бромметил)-4-(2-хлор-3-фтор-фенил)-2-тиазол-2-ил-1,4-дигидропиримидин-5-карбоксилат (соединение 2А, 0,35 г) в виде желтого твердого вещества. МС: рассч. 459 (М+Н)+, измеренная 459 (М+Н)+.
Получение 3-[(8aS)-3-оксо-1,5,6,7,8,8а-гексагидроимидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановой кислоты (соединение 2В):
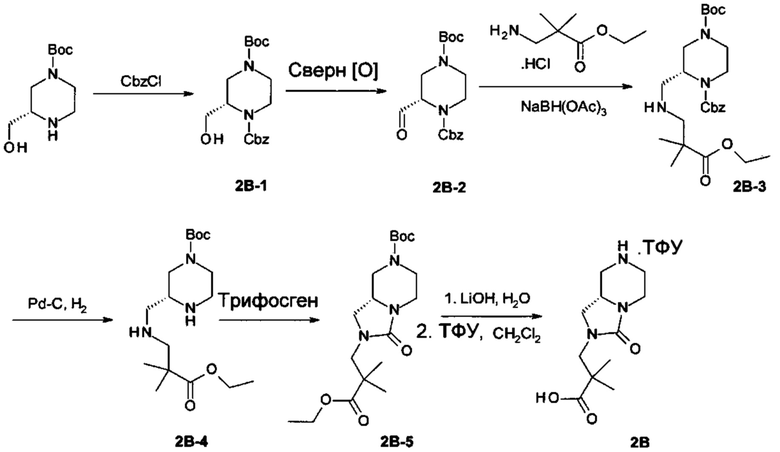
Получение O1-бензил O4-трет-бутил (2S)-2-(гидроксиметил)пиперазин-1,4-дикарбоксилата (соединение 2В-1):
К перемешиваемому раствору трет-бутил (3S)-3-(гидроксиметил)пиперазин-1-карбоксилата (CAS номер: 314741-40-7, Bepharm; с его синтезом просим ознакомиться в: Gao H., Renslo A.R. J. Org. Chem. 2007, 72, 8591-8592) (6 г, 27,8 ммоль) в насыщенном NaHCO3 (45 мл) и EtOAc (45 мл) добавляли бензилхлорформиат (7,1 г, 41,7 ммоль) по каплям при 0°С. Затем реакционную смесь перемешивали при комнатной температуре в течение 15 часов. Реакционную смесь разбавляли EtOAc (60 мл). Органический слой отделяли, и водный слой экстрагировали EtOAc (35 мл). Объединенные органические слои сушили над Na2SO4. Растворитель удаляли в вакууме, получая неочищенный продукт, который очищали с помощью колоночной хроматографии на силикагеле (петролейный эфир: EtOAc = 10:1 до 1:1), получая O1-бензил O4-трет-бутил (2S)-2-(гидроксиметил)пиперазин-1,4-дикарбоксилат (соединение 2В-1, 6,7 г). МС: рассч. 351 (М+Н)+, измеренная 351 (М+Н)+.
Получение O1-бензил O4-трет-бутил (2S)-2-формилпиперазин-1,4-дикарбоксилата (соединение 2В-2):
К перемешиваемому раствору оксалилхлорида (3,64 г, 28,6 ммоль) в безводном дихлорметане (80 мл) при -78°С добавляли по каплям диметилсульфоксид (4,47 г, 57,3 ммоль). Через 10 минут добавляли по каплям раствор O1-бензил O4-трет-бутил (2S)-2-(гидроксиметил)пиперазин-1,4-дикарбоксилата (соединение 2В-1, 6,7 г, 19,1 ммоль) в дихлорметане (20 мл). После того, как смесь перемешивали в течение 30 минут при -78°С, добавляли N,N-диизопропилэтиламин (14,78 г, 114,6 ммоль) и реакционную смесь перемешивали в течение 30 минут. После того, как реакционную смесь медленно нагревали до 0°С в течение 30 минут, ее разбавляли дихлорметаном (80 мл), промывали 5% водной лимонной кислотой (10 мл), солевым раствором и затем сушили над Na2SO4. После фильтрования смесь концентрировали в вакууме, получая неочищенный продукт O1-бензил O4-трет-бутил (2S)-2-формилпиперазин-1,4-дикарбоксилат (соединение 2В-2, 7,0 г). МС: рассч. 349 (М+Н)+, измеренная 349 (М+Н)+.
Получение O1-бензил O4-трет-бутил (2R)-2-[[(3-этокси-2,2-диметил-3-оксо-пропил)амино]метил]пиперазин-1,4-дикарбоксилата (соединение 2В-3):
К перемешиваемому раствору этил 3-амино-2,2-диметил-пропионата гидрохлорида (3,4 г, 18,6 ммоль) в безводном ДХМ (100 мл) добавляли ДИПЭА (2,6 г, 27,3 ммоль) при комнатной температуре. Затем добавляли O1-бензил О4-трет-бутил (2S)-2-формилпиперазин-1,4-дикарбоксилат (соединение 2В-2, неочищенное, 7,0 г, 20 ммоль), потом NaBH(ОАс)3 (6,3 г, 29,8 ммоль) и АсОН (1,5 мл) при 0°С. Реакционную смесь перемешивали в течение 16 часов при комнатной температуре. Добавляли воду (100 мл) и смесь экстрагировали ДХМ (100 мл). Органический слой сушили и концентрировали в вакууме, получая неочищенный продукт O1-бензил O4-трет-бутил (2R)-2-[[(3-этокси-2,2-диметил-3-оксо-пропил)амино]метил]пиперазин-1,4-дикарбоксилат (соединение 2В-3, 7,3 г). МС: рассч. 478 (М+Н)+, измеренная 478 (М+Н)+.
Получение трет-бутил (3R)-3-[[(3-этокси-2,2-диметил-3-оксо-пропил)амино]метил]пиперазин-1-карбоксилата (соединение 2В-4):
К раствору O1-бензил O4-трет-бутил (2R)-2-[[(3-этокси-2,2-диметил-3-оксо-пропил)амино]метил]пиперазин-1,4-дикарбоксилата (соединение 2В-3, неочищенное, 3,3 г, 6,9 ммоль) в EtOH (100 мл) добавляли 10% палладий на угле (1 г). Потом смесь перемешивали при 50°С в течение 3 часов в атмосфере водорода (50 пси). Реакционную смесь фильтровали и фильтрат концентрировали в вакууме, получая трет-бутил (3R)-3-[[(3-этокси-2,2-диметил-3-оксо-пропил)амино]метил]пиперазин-1-карбоксилат (соединение 2В-4, 1,8 г). МС: рассч. 344 (М+Н)+, измеренная 344 (М+Н)+.
Получение трет-бутил (8aR)-2-(3-этокси-2,2-диметил-3-оксо-пропил)-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-7-карбоксилата (соединение 2В-5):
К раствору трет-бутил (3R)-3-[[(3-этокси-2,2-диметил-3-оксо-пропил)амино]метил]пиперазин-1-карбоксилата (соединение 2В-4, 1,8 г, 5,2 ммоль) в безводном дихлорметане (60 мл) добавляли N,N-диизопропилэтиламин (3,4 г, 26,2 ммоль) при 0°С. Затем добавляли трифосген (783 мг, 2,6 ммоль) при 0°С и смесь перемешивали при 10-15°С в течение 16 часов. Добавляли воду (50 мл) и смесь экстрагировали дихлорметаном (60 мл). Органический слой сушили над Na2SO4, и растворитель удаляли в вакууме, получая неочищенный продукт. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (петролейный эфир: EtOAc = 5:1 до 1:1), получая трет-бутил (8aR)-2-(3-этокси-2,2-диметил-3-оксо-пропил)-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-7-карбоксилат (соединение 2В-5, 1,3 г). МС: рассч. 370 (М+Н)+, измеренная 370 (М+Н)+.
Получение 3-[(8aS)-3-оксо-1,5,6,7,8,8а-гексагидроимидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановой кислоты (соединение 2В):
К перемешиваемому раствору трет-бутил (8aR)-2-(3-этокси-2,2-диметил-3-оксо-пропил)-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-7-карбоксилата (соединение 2В-5, 94 мг, 0,25 ммоль) в ТГФ (3 мл) добавляли раствор LiOH⋅H2O (84 мг, 2,0 ммоль) в H2O (1 мл) при комнатной температуре. После перемешивания реакционной смеси при комнатной температуре в течение ночи ее подкисляли 1 н HCl до рН 3~4 при 0°С. Затем смесь концентрировали в вакууме и азеотропно сушили с толуолом, получая неочищенную кислоту, которую растворяли в дихлорметане (2 мл) и обрабатывали трифторуксусной кислотой (2 мл) при комнатной температуре. После перемешивания реакционной смеси при комнатной температуре в течение 1 часа растворитель удаляли в вакууме, получая 3-[(8aS)-3-оксо-1,5,6,7,8,8а-гексагидроимидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановую кислоту (соединение 2В), которую сразу же использовали. МС: рассч. 242 (М+Н)+, измеренная 242 (М+Н)+.
Пример 3
[(S)-[(2S,5R)-5-(5-амино-2-оксо-тиазоло[4,5-а]пиримидин-3-ил)-1,3-оксатиолан-2-ил]-циклопропил-метил]ацетат (соединение 3)
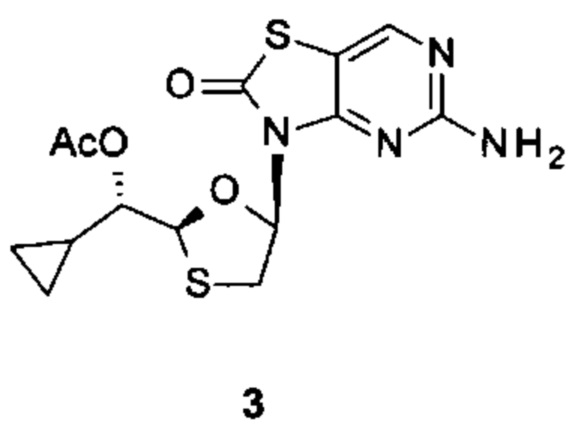
Соединение 3 получали по следующей схеме:
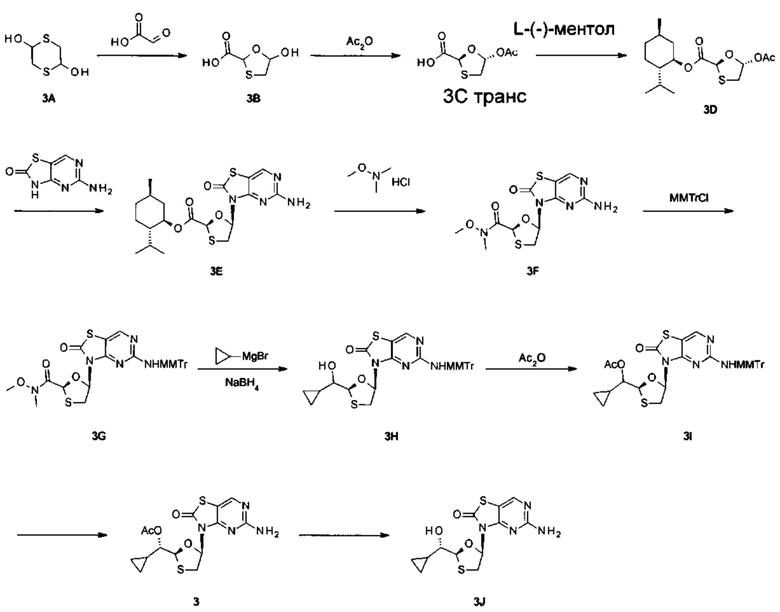
Получение 5-гидрокси-1,3-оксатиолан-2-карбоновой кислоты
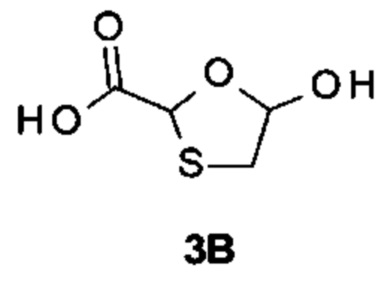
К перемешиваемому раствору 1,4-дитиан-2,5-диола (соединение 3А, 150 г, 0,98 моль) в метил-трет-бутиловом эфире (500 мл) и циклогексане (150 мл) добавляли глиоксиловую кислоту (180 г, 1,96 моль). Полученную в результате реакционную смесь перемешивали при 80°С в условиях Дина-Старка в течение 16 часов. Полученный в результате раствор концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (элюируя 1:7 этилацетата в петролейном эфире до 100% этилацетата), получая 220 г неочищенной 5-гидрокси-1,3-оксатиолан-2-карбоновой кислоты (соединение 3В), которую непосредственно использовали на следующей стадии без дополнительной очистки.
Получение транс-5-ацетокси-1,3-оксатиолан-2-карбоновой кислоты
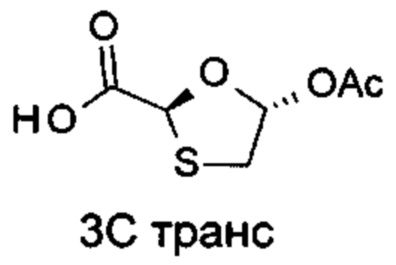
К раствору 5-гидрокси-1,3-оксатиолан-2-карбоновой кислоты (соединение 3В, 220 г, 1,5 моль) в НОАс (1,5 л) добавляли концентрированную серную кислоту (1 мл) и уксусный ангидрид (50 г, 0,5 моль). После перемешивания при комнатной температуре в течение 16 часов полученную в результате реакционную смесь разбавляли водой и экстрагировали EtOAc. Органическую фазу объединяли и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (элюируя 1:10 до 1:7 этилацетата в петролейном эфире), получая неочищенный продукт, который перекристаллизовывали из толуола, получая 10 г транс-5-ацетокси-1,3-оксатиолан-2-карбоновой кислоты (соединение 3С транс). (Также, пожалуйста, смотрите синтез в: J. Org. Chem. 1995, 60, 2621-2623.)
Соединение 3С транс: 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm: 13,26 (br, 1Н), 6,66 (d, J=4,0 Гц, 1Н), 5,66 (s, 1Н), 3,30-3,37 (т, 1Н), 3,19-3,25 (т, 1Н), 2,04 (s, 3Н).
Получение [(1R,2S,5R)-2-изопропил-5-метил-циклогексил]-(2S,5S)-5-ацетокси-1,3-оксатиолан-2-карбоксилата
Раствор дициклогексилкарбодиимида (12 г, 57 ммоль) в ДХМ (50 мл) добавляли в круглодонную колбу, содержащую раствор транс-5-ацетокси-1,3-оксатиолан-2-карбоновой кислоты (соединение 3С транс, 10 г, 52 ммоль), L-(-)-ментол (8,9 г, 57 ммоль) и ДМАП (0,6 г, 5,2 ммоль) в ДХМ (100 мл) при 0°С. После того, как реакционную смесь перемешивали при комнатной температуре в течение 16 часов, добавляли метанол (2 мл) и ледяную уксусную кислоту (2 мл). Реакционную смесь перемешивали в течение еще 10 минут и затем разбавляли гексаном (250 мл), фильтровали через целит, и фильтрат концентрировали, получая неочищенный продукт. (J. Org. Chem. 1995, 60, 2621-2623). Неочищенный продукт повторно растворяли в гексане (250 мл), фильтровали, и фильтрат концентрировали в вакууме. Остаток очищали с помощью сверхкритической флюидной хроматографии (СФХ), получая 3,2 г [(1R,2S,5R)-2-изопропил-5-метил-циклогексил]-(2S,5S)-5-ацетокси-1,3-оксатиолан-2-карбоксилата (соединение 3D) с диастереоизомерным избытком 85% в виде бесцветного масла. Значение диастереоизомерного избытка соединения 3D получали с помощью анализа ВЭЖХ (Agilent 1260 ВЭЖХ), используя хиральную колонку (CHIRALPAK IA-3 ODH (4,6 мм × 250 мм, 5 мкм)). Подвижная фаза для хирального анализа представляла собой 20:80 ацетонитрил в МеОН.
Соединение 3D: 1Н ЯМР (400 МГц, CDCl3) δ ppm: 6,81 (d, J=4,0 Гц, 1Н), 5,63 (s, 1Н), 4,76 (dt, J=10,9, 4,5 Гц, 1Н), 3,44 (dd, J=11,7, 4,1 Гц, 1Н), 3,17 (d, J=11,8 Гц, 1Н), 2,11 (s, 3Н), 2,00 (d, J=12,0 Гц, 1Н), 1,85 (dt, J=6,9, 2,5 Гц, 1Н), 1,69 (d, J=11,0 Гц, 2Н), 1,55-1,26 (m, 3Н), 1,11-1,00 (m, 2Н), 0,91 (dd, J=6,8, 9,8 Гц, 6Н), 0,76 (d, J=7,0 Гц, 3Н).
Получение [(1R,2S,5R)-2-изопропил-5-метил-циклогексил]-(2S,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-1,3-оксатиолан-2-карбоксилата

Суспензию 5-амино-3H-тиазоло[4,5-d]пиримидин-2-она (6,0 г, 36 ммоль) и БСА (24,0 г, 118 ммоль) в дихлорэтане (ДХЭ) (250 мл) нагревали при 85°С в течение 1 часа. Реакционную смесь охлаждали до 0°С, к вышеприведенной смеси добавляли раствор [(1R,2S,5R)-2-изопропил-5-метил-циклогексил]-(2S,5S)-5-ацетокси-1,3-оксатиолан-2-карбоксилата (соединение 3D, 9,0 г, 27 ммоль) в ДХЭ (10 мл), затем TMSI (14 г, 70 ммоль) по каплям. Реакционную смесь перемешивали при 60°С в течение 5 часов, гасили водным раствором NaHCO3 и затем экстрагировали EtOAc. Органический слой промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали, получая неочищенный продукт в виде масла, которое очищали с помощью колоночной хроматографии на силикагеле (элюируя 1:100 до 1:50 метанола в дихлорметане), получая 7,7 г смеси двух изомеров, которую дополнительно очищали и разделяли с помощью препаративной ВЭЖХ, получая требуемые 2,8 г бета-изомера [(1R,2S,5R)-2-изопропил-5-метил-циклогексил]-(2S,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-1,3-оксатиолан-2-карбоксилата (соединение 3Е) в виде белого твердого вещества. Конфигурацию соединения 3Е определяли с помощью спектроскопии ядерного эффекта Оверхаузера (NOESY, от англ. "Nuclear Overhauser Effect Spectroscopy").
Соединение 3Е: 1Н ЯМР (400 МГц, CDCl3) δ ppm: 8,17 (s, 1Н), 6,44 (m, 1Н), 5,51 (s, 1Н), 5,12 (bs, 2Н), 4,78 (m, 1Н), 4,47 (m, 1Н), 3,16 (m, 1Н), 2,00 (m, 1Н), 1,79 (m, 1Н), 1,62 (m, 2Н), 1,38 (m, 2Н), 0,98 (m, 2Н), 0,9-0,72 (m, 10Н). МС наблюд. (ЭСИ+) [(М+Н)+]: 439.
Получение (2S,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-N-метокси-N-метил-1,3-оксатиолан-2-карбоксамида
Раствор [(1R,2S,5R)-2-изопропил-5-метил-циклогексил]-(2S,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-1,3-оксатиолан-2-карбоксилата (соединение 3Е, 3,0 г, 7,5 ммоль) в 80% водной трифторуксусной кислоте (ТФУ) (20 мл) перемешивали при 50°С в течение 16 часов и затем концентрировали, получая неочищенную кислоту в виде белого твердого вещества, которое повторно растворяли в ТГФ (40 мл). К вышеприведенной смеси добавляли N-метоксиметиламина гидрохлорид (2,1 г, 22 ммоль), ДИПЭА (14,5 г, 112 ммоль) и 1-[бис(диметиламино)метилен]-1Н-1,2,3-триазоло[4,5-d]пиридиния 3-оксид гексафторфосфат (HATU) (8,36 г, 22 моль) при комнатной температуре. После перемешивания при комнатной температуре в течение 16 часов реакционную смесь разбавляли ДХМ, промывали водой и солевым раствором, сушили над безводным Na2SO4 и концентрировали, получая неочищенный продукт, который очищали с помощью флэш-хроматографии на силикагеле (элюируя 1:100 до 1:50 метанола в дихлорметане), получая 2,1 г (2S,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-N-метокси-N-метил-1,3-оксатиолан-2-карбоксамида (соединение 3F) в виде белого твердого вещества.
Соединение 3F: 1Н ЯМР (400 МГц, CDCl3) δ ppm: 8,16 (s, 1Н), 6,42 (m, 1Н), 5,83 (s, 1Н), 5,14 (bs, 2Н), 4,46 (t, J=9,6 Гц, 1Н), 3,72 (s, 3Н), 3,23 (s, 3Н), 3,15 (m, 1Н). МС наблюд. (ЭСИ+) [(М+Н)+]: 344.
Получение (2S,5R)-N-метокси-5-[5-[[(4-метоксифенил)-дифенил-метил]амино]-2-оксо-тиазоло[4,5-d]пиримидин-3-ил]-N-метил-1,3-оксатиолан-2-карбоксамида

К раствору (2S,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-N-метокси-N-метил-1,3-оксатиолан-2-карбоксамида (соединение 3F, 2,1 г, 6,1 ммоль) в ДХМ (30 мл) добавляли коллидин (1,45 г, 12 ммоль), AgNO3 (2,04 г, 12 ммоль) и 4-метокситрифенилметилхлорид (ММТrСl) (3,8 г, 12 ммоль) при комнатной температуре. После перемешивания при комнатной температуре в течение 16 часов реакционную смесь разбавляли ДХМ, фильтровали, чтобы удалить твердое вещество. Фильтрат промывали водой и солевым раствором, сушили над безводным Na2SO4 и концентрировали, получая неочищенный продукт, который очищали с помощью флэш-хроматографии на силикагеле (элюируя 1:100 до 2:1 этилацетата в петролейном эфире), получая 3,6 г (2S,5R)-N-метокси-5-[5-[[(4-метоксифенил)-дифенил-метил]амино]-2-оксо-тиазоло[4,5-d]пиримидин-3-ил]-N-метил-1,3-оксатиолан-2-карбоксамида (соединение 3G) в виде желтого твердого вещества. (ЭСИ+) [(М+Н)+]: 616.
Получение 3-[(2S,5R)-2-[циклопропил(гидрокси)метил]-1,3-оксатиолан-5-ил]-5-[[(4-метоксифенил)-дифенил-метил]амино]тиазоло[4,5-d]пиримидин-2-она
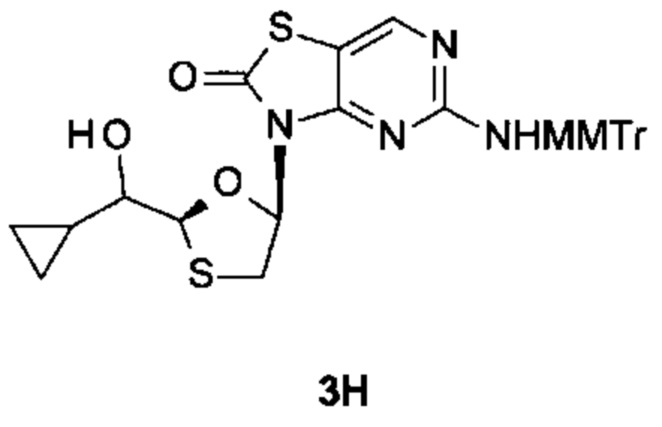
К раствору (2S,5R)-N-метокси-5-[5-[[(4-метоксифенил)-дифенил-метил]амино]-2-оксо-тиазоло[4,5-d]пиримидин-3-ил]-N-метил-1,3-оксатиолан-2-карбоксамида (соединение 3G, 3 г, 5 ммоль) в ТГФ (40 мл) добавляли реактив Гриньяра, циклопропилмагнийбромид (0,5 М, 25 мл), при 0°С. Реакционную смесь перемешивали при 0°С в течение 30 минут. Реакцию гасили насыщенным раствором NH4Cl и экстрагировали EtOAc. Органический слой сушили и концентрировали, получая неочищенный продукт, который повторно растворяли в МеОН (50 мл). К вышеприведенной смеси добавляли NaВН4 (2,0 г, 540 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Реакцию гасили насыщенным раствором NH4Cl и экстрагировали ДХМ. Органический слой сушили над безводным Na2SO4 и концентрировали, получая неочищенный продукт, который очищали с помощью флэш-хроматографии на силикагеле (элюируя 1:100 до 1:1 этилацетата в петролейном эфире), получая 1,8 г 3-[(2S,5R)-2-[циклопропил(гидрокси)метил]-1,3-оксатиолан-5-ил]-5-[[(4-метоксифенил)-дифенил-метил]амино]тиазоло[4,5-d]пиримидин-2-она (соединение 3Н) в виде желтого твердого вещества. (ЭСИ+) [(М+Н)+]: 599.
Получение [циклопропил-[(2S,5R)-5-[5-[[(4-метоксифенил)-дифенил-метил]амино]-2-оксо-тиазоло[4,5-d]пиримидин-3-ил]-1,3-оксатиолан-2-ил]метил]ацетата
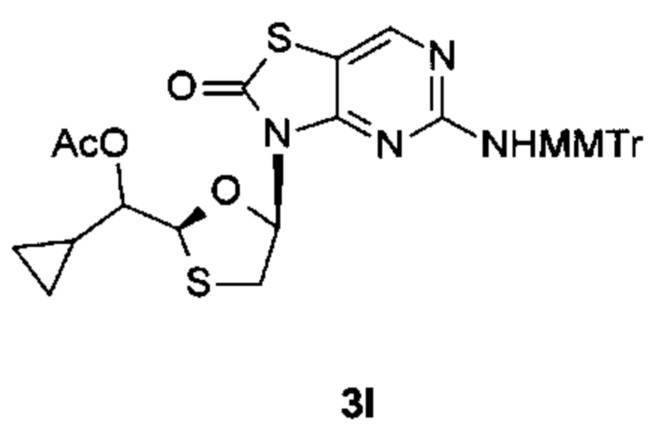
К раствору 3-[(2S,5R)-2-[циклопропил(гидрокси)метил]-1,3-оксатиолан-5-ил]-5-[[(4-метокси-фенил)-дифенил-метил]амино]тиазоло[4,5-d]пиримидин-2-она (соединение 3Н, 1,2 г, 2 ммоль) в ДХМ (10 мл) добавляли ТЭА (800 мг, 8 ммоль), ДМАП (30 мг, 0.2 ммоль) и Ас2O (400 мг, 4 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 48 часов. После завершения реакции реакционную смесь гасили водой, экстрагировали ДХМ. Органический слой сушили и концентрировали, получая 1,3 г неочищенного продукта [циклопропил-[(2S,5R)-5-[5-[[(4-метоксифенил)-дифенил-метил]амино]-2-оксо-тиазоло[4,5-а]пиримидин-3-ил]-1,3-оксатиолан-2-ил]метил]ацетат (соединение 3I) в виде белого твердого вещества, которое непосредственно использовали на следующей стадии без дополнительной очистки. (ЭСИ+) [(М+Н)+]: 641.
Получение [(S)-[(2S,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-1,3-оксатиолан-2-ил]-циклопропил-метил]ацетата
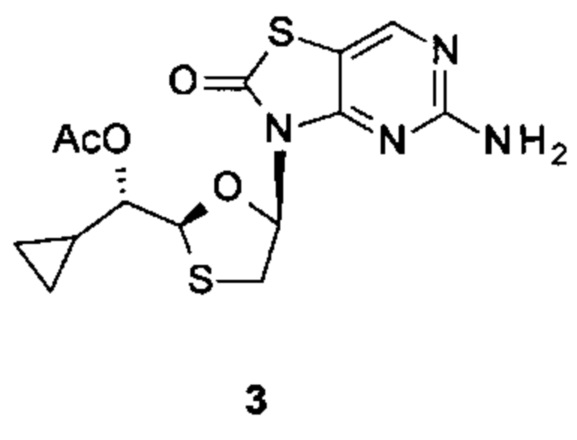
Раствор [циклопропил-[(2S,5R)-5-[5-[[(4-метоксифенил)-дифенил-метил]амино]-2-оксо-тиазоло[4,5-а]пиримидин-3-ил]-1,3-оксатиолан-2-ил]метил]ацетата (соединение 3I, 1,3 г, 2 ммоль) в 90% водном растворе НСООН (25 мл) перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь концентрировали, и остаток дополнительно очищали и разделяли с помощью препаративной ВЭЖХ, получая 114 мг [(S)-[(2S,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-1,3-оксатиолан-2-ил]-циклопропил-метил]ацетата (соединение 3) в виде белого твердого вещества.
Соединение 3: 1Н ЯМР (400 МГц, метанол-d4) δ ppm: 8,20 (s, 1Н), 6,34 (m, 1Н), 5,34 (d, J=6,4 Гц, 1Н), 4,54 (t, J=6,0 Гц, 1Н), 4,18 (t, J=8,4 Гц, 1Н), 3,31 (t, J=6,0 Гц, 1Н), 2,02 (s, 3Н), 1,13 (m, 1Н), 0,65-0,42 (m, 4Н). МС наблюд. (ЭСИ+) [(М+Na)+]: 391.
Получение 5-амино-3-[(2S,5R)-2-[(S)-циклопропил(гидрокси)метил]-1,3-оксатиолан-5-ил]тиазоло[4,5-а]пиримидин-2-она
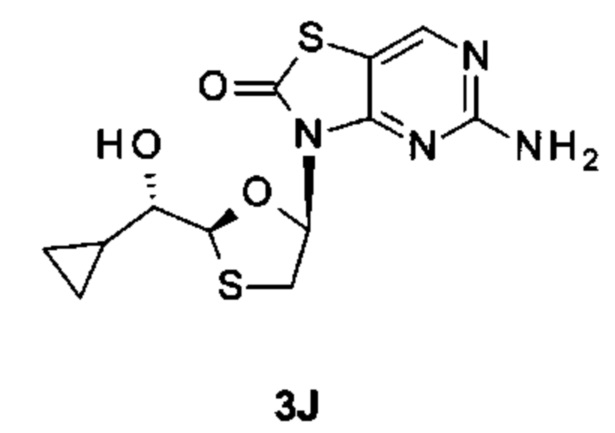
К раствору [(S)-[(2S,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-1,3-оксатиолан-2-ил]-циклопропил-метил]ацетата (соединение 3, 500 мг, 1,36 ммоль) в метаноле (5 мл) добавляли K2CO3 (94 мг, 0,68 ммоль). После перемешивания при комнатной температуре в течение 4 часов реакционную смесь гасили НОАс до рН 7 и затем концентрировали в вакууме. Остаток разбавляли EtOAc и фильтровали. Фильтрат концентрировали в вакууме. Остаток очищали и разделяли с помощью препаративной ВЭЖХ, получая 45 мг 5-амино-3-[(2S,5R)-2-[(S)-циклопропил(гидрокси)метил]-1,3-оксатиолан-5-ил]тиазоло[4,5-d]пиримидин-2-она (соединение 3J) в виде белого порошка.
Соединение 3J: Абсолютную структуру определяли с помощью 1Н ЯМР и рентгеноструктурного анализа монокристалла, как показано на фигуре 3. 1Н ЯМР (400 МГц, метанол-d4) δ ppm: 8,25 (s, 1Н), 6,39 (dd, J=9,16, 5,65 Гц, 1Н), 5,24 (d, J=5,27 Гц, 1Н), 4,06 (dd, J=10,29, 9,29 Гц, 1Н), 3,13-3,30 (m, 2Н), 0,37-1,04 (m, 5Н). МС наблюд. (ЭСИ+) [(М+Н)+]: 327,0.
Пример 4
3-[(8aS)-7-[[(4S)-5-этоксикарбонил-4-(3-фтор-2-метил-фенил)-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановая кислота (соединение 4)


Соединение 4 получали по следующей схеме:
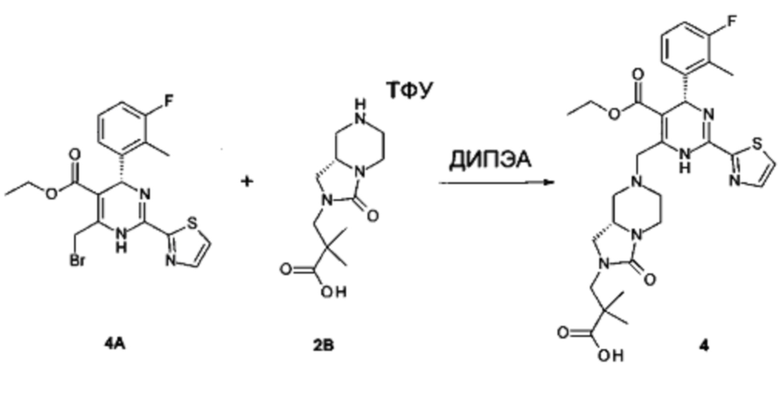
Указанное в заголовке соединение получали по аналогии с соединением 2, используя этил (4S)-6-(бромметил)-4-(3-фтор-2-метил-фенил)-2-тиазол-2-ил-1,4-дигидропиримидин-5-карбоксилат (соединение 4А) вместо этил (4R)-6-(бромметил)-4-(2-хлор-3-фтор-фенил)-2-тиазол-2-ил-1,4-дигидропиримидин-5-карбоксилата (соединение 2А). Соединение 4 получали в виде светло-желтого твердого вещества (132 мг). 1Н ЯМР (400 МГц, метанол-d4) δ ppm 7,95 (d, J=3,3 Гц, 1Н), 7,75 (d, J=3,3 Гц, 1Н), 7,08-7,23 (m, 2Н), 6,95 (t, J=8,8 Гц, 1Н), 5,99 (s, 1Н), 4,02-4,17 (m, 3Н), 3,79-4,00 (m, 3Н), 3,36-3,57 (m, 2Н), 3,26-3,33 (m, 1Н), 3,17-3,25 (m, 1Н), 3,11 (dd, J=9,3, 4,0 Гц, 1Н), 2,78-2,99 (m, 2Н), 2,53 (d, J=2,0 Гц, 3Н), 2,39 (dd, J=11,2, 8,2 Гц, 1Н), 2,14-2,26 (m, 1Н), 1,21 (d, J=2,8 Гц, 6Н), 1,15 ppm (t, J=7,2 Гц, 3Н). МС: рассч. 599 (М+Н)+, измеренная 599 (М+Н)+.
Получение этил (4S)-6-(бромметил)-4-(3-фтор-2-метил-фенил)-2-тиазол-2-ил-1,4-дигидропиримидин-5-карбоксилата (соединение 4А):
Соединение 4А получали по аналогии с соединением 2А, используя 2-метил-3-фторбензальдегид вместо 2-хлор-3-фторбензальдегида. Оптическое вращение соединения 4А: [α]D20-21,0 (с 0,10, МеОН).
Пример 5
5-амино-3-[(2R,3R,5S)-3-гидрокси-5-[(1S)-1-гидроксипропил]тетрагидрофуран-2-ил]-6Н-тиазоло[4,5-d]пиримидин-2,7-дион (соединение 11)
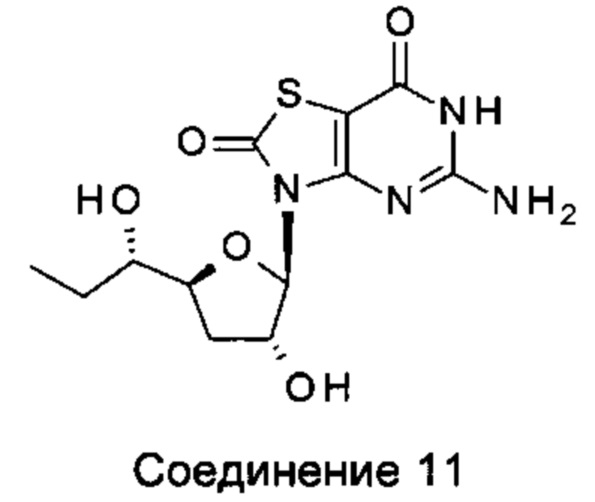
Соединение 11 получали согласно следующей схеме.
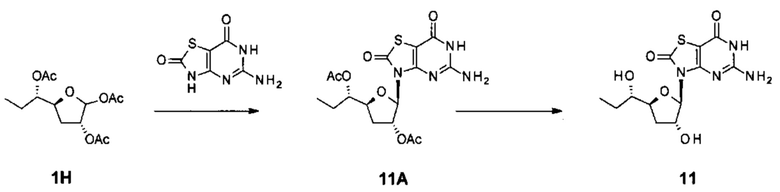
Получение [(2R,3R,5S)-5-[(1S)-1-ацетоксипропил]-2-(5-амино-2,7-диоксо-6Н-тиазоло[4,5-d]пиримидин-3-ил)тетрагидрофуран-3-ил]ацетата
К суспензии 5-амино-3,6-дигидротиазоло[4,5-d]пиримидин-2,7-диона (5,37 г, 29,1 ммоль) и [(3R,5S)-2-ацетокси-5-[(1S)-1-ацетоксипропил]тетрагидрофуран-3-ил]ацетата (соединение 1Н, 2,8 г, 9,7 ммоль) в ацетонитриле (140 мл) добавляли БСА (21,4 мл, 87,3 ммоль). Реакционную смесь перемешивали при 65°С в течение 1,5 часа в атмосфере аргона с образованием прозрачного раствора. Затем к раствору добавляли ТМSОТf (9,8 г, 43,7 ммоль) и смесь перемешивали при 65°С в течение еще 3 часов. Реакционную смесь концентрировали в вакууме. Остаток растворяли в EtOAc (200 мл) и экстрагировали насыщенным раствором NaHCO3 (50 мл). Осадок выпадал из органического слоя. Полученную в результате смесь фильтровали и фильтрат промывали солевым раствором (50 мл), сушили над Na2SO4 и концентрировали в вакууме, получая 2,3 г неочищенного продукта [(2R,3R,5S)-5-[(1S)-1-ацетоксипропил]-2-(5-амино-2,7-диоксо-6Н-тиазоло[4,5-d]пиримидин-3-ил)тетрагидрофуран-3-ил]ацетат (соединение 11А) в виде желтого твердого вещества. МС наблюд. (ЭСИ-) [(М+Н)+]: 413,1.
Получение 5-амино-3-[(2R,3R,5S)-3-гидрокси-5-[(1S)-1-гидроксипропил]тетрагидрофуран-2-ил]-6Н-тиазоло[4,5-d]пиримидин-2,7-диона
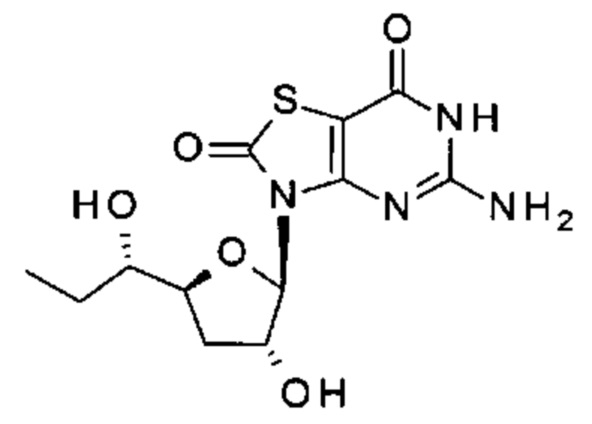
К раствору [(2Р,3R,53)-5-[(1S)-1-ацетоксипропил]-2-(5-амино-2,7-диоксо-6Н-тиазоло[4,5-d]пиримидин-3-ил)тетрагидрофуран-3-ил]ацетата (соединение 11А, 2,3 г, 5,58 ммоль) в метаноле (100 мл) добавляли метилат натрия (1,5 г, 27,9 ммоль). После добавления смесь перемешивали при комнатной температуре в течение 1,5 часа (контролировали с помощью ТСХ). После завершения реакции реакционную смесь гасили насыщенным водным NH4Cl (50 мл). Полученную в результате смесь концентрировали в вакууме, чтобы удалить большую часть МеОН. Остаток экстрагировали EtOAc (100 мл) десять раз. Объединенный органический слой промывали солевым раствором (100 мл), сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали на колонке с силикагелем, элюируя ДХМ/МеОН=20/1 до 10/1, получая 360 мг 5-амино-3-[(2R,3R,5S)-3-гидрокси-5-[(1S)-1-гидроксипропил]тетрагидрофуран-2-ил]-6Н-тиазоло[4,5-d]пиримидин-2,7-диона (соединение 11) в виде белого твердого вещества и 550 мг неочищенного продукта.
Соединение 11: 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 11,22 (br s, 1Н), 6,95 (br s, 2Н), 5,72 (d, J=2,26 Гц, 1Н), 5,42 (d, J=4,52 Гц, 1Н), 4,73 (m, 1Н), 4,53 (d, J=6,02 Гц, 1Н), 3,96 (m, 1Н), 3,25-3,32 (m, 1Н), 2,25-2,48 (m, 1Н), 1,66-1,74 (m, 1Н), 1,35-1,46 (m, 1Н), 1,19-1,31 (m, 1Н), 0,88 (t, J=7,28 Гц, 3Н).
Пример 6
Анализ клеток HEK293-Blue-hTLR-7:
Стабильную клеточную линию HEK293-Blue-hTLR-7 приобретали у InvivoGen (кат. №: hkb-htlr7, Сан-Диего, Калифорния, США). Эти клетки предназначались для изучения стимуляции TLR7 человека при контроле над активацией NF-κB. Размещали репортерный ген SEAP (от англ. "secreted embryonic alkaline phosphatase" - секретируемая эмбриональная щелочная фосфатаза) под контролем ИФН-β • минимальный стимулятор, соединенный с пятью NF-κB и АР-1-сайтами связывания. SEAP инициировали, активируя NF-κB и АР-1 в ходе стимуляции клеток HEK-Blue hTLR7 с лигандами TLR7. Таким образом, экспрессия репортера регулируется промотором NF-κВ при стимуляции TLR7 человека в течение 20 часов. Активность репортера SEAP в надосадочной клеточной культуре определяли, используя набор QUANTI-Blue™ (кат. №: rep-qb1, Invivogen, Сан-Диего, Калифорния, США) при длине волны 640 нм, среду для детектирования, которая становится пурпурной или синеет в присутствии щелочной фосфатазы.
Клетки HEK293-Blue-hTLR7 инкубировали при плотности 250,000~450,000 клеток/мл в объеме 180 мкл на 96-луночном планшете в модифицированной по способу Дульбекко среде Игла (DMEM, от англ. "Dulbecco's Modified Eagle's medium"), содержащей 4,5 г/л глюкозы, 50 ЕД/мл пенициллина, 50 мг/мл стрептомицина, 100 мг/мл Normocin, 2 мМ L-глютамин, 10% (об/об) термоинактивированной эмбриональной бычьей сыворотки, в течение 24 часов. Затем клетки HEK293-Blue-hTLR-7 инкубировали с добавлением 20 мкл исследуемого соединения при последовательном разведении в присутствии конечного ДМСО в количеств 1% и проводили инкубацию при 37°С в CO2 инкубаторе в течение 20 часов. Затем 20 мкл надосадочной жидкости из каждой лунки инкубировали со 180 мкл раствора субстрата Quanti-blue при 37°С в течение 2 часов, и поглощение считывали при 620~655 нм, используя спектрофотометр. Сигнальный путь, которым активация TLR7 приводит к активации NF-κB по ходу транскрипции, является общепризнанным, и, следовательно, подобный анализ репортера также широко используется для оценки агониста TLR7 (Tsuneyasu Kaisho and Takashi Tanaka, Trends in Immunology, Volume 29, Issue 7, July 2008, Pages 329.sci; Hiroaki Hemmi et al, Nature Immunology 3, 196-200 (2002)).
Активность агонизма TLR7 в анализе HEK293-hTLR-7 соединения 11 составляла 72 мкМ.
Пример 6
Комбинация агониста TLR7 (соединение 1) и ингибитора сборки капсида HBV (соединение 2) эффективно уменьшала ДНК HBV и HBsAg в мышиной модели AAV-HBV
Животная модель
Самцов мышей 4-недельного возраста C57BL/6 свободных от специфической патогенной микрофлоры приобретали в Shanghai Laboratory Animal Center (SLAC) Китайской академии наук и размещали в лаборатории по уходу за животными в отдельных вентилируемых клетках в условиях регулируемой температуры и света согласно ррекомендациям Институционального комитета по уходу за животными (Institutional Animal Care guidelines). Вирус AAV/HBV приобретали в Beijing FivePlus Molecular Medicine Institute (Пекин, Китай). Данный рекомбинантный вирус несет 1,3 копий генома HBV, который размещен в капсидах AAV серотипа 8 (AAV8). Мышам C57BL/6 вводили по 200 мкл рекомбинантного вируса, разбавленного солевым буфером, в ходе инъекции в хвостовую вену. У мышей брали кровь на 7 и 14 дни после инъекции, чтобы контролировать поверхностный антиген HBV (HBsAg), HBV e антиген (HBeAg), антитело HBs (HBsAb) и геномную ДНК HBV в сыворотке, и затем случайным образом группировали в соответствии с их биомаркерами HBV.
Измерение биомаркеров HBV
HBsAg и HBeAg в сыворотке измеряли, используя наборы для ИХЛА (Autobio Diagnostics Co., Ltd, Чжэнчжоу, Китай) в соответствии с инструкциями изготовителя. Нижний предел обнаружения HBsAg и HBeAg составлял 0,1 нг/мл и 0,25 NCU (от англ. "national clinical unit" - национальная клиническая единица)/мл, соответственно. Сыворотку разбавляли в 100 раз (для HBsAg) или 500 раз (для HBeAg), чтобы получить значения в пределах линейного диапазона градуировочной кривой. ДНК HBV в сыворотке экстрагировали, используя набор MagNA Pure 96 DNA and Viral NA Small Volume (Roche) в соответствии с инструкциями изготовителя. Образцы ДНК анализировали в ходе количественной ПЦР в реальном времени (кПЦР), используя HBV-специфичный праймер и набор зондов для специфической амплификации и детектирования 128 пн участка генома HBV от нуклеотида 2969 до 3096. Последовательности праймеров и зонда показаны ниже:
Прямой праймер: AAGAAAAACCCCGCCTGTAA;
Обратный праймер: CCTGTTCTGACTACTGCCTCTCC;
HBV-зонд: 5'TARMA-CCTGATGTGATGTTCTCCATGTTCAGC-BHQ2-3'.
Анти-HBs в сыворотке измеряли на 24 день после завершения обработки, используя анти-HBs наборы для ИХЛА (Autobio Diagnostics Co., Ltd, Чжэнчжоу, Китай) в соответствии с инструкциями изготовителя. Образцы сыворотки разбавляли в 3 раза и по 50 мкл разбавленных образцов использовали для анализа.
План эксперимента и результаты
10 мг/мл соединения 1 и 1,2 мг/мл соединения 2 получали в виде комплекса включения с 2% Klucel LF, 0,1% Полисорбата 80 и 0,1% парабенов в воде. Всем мышам дозы давали перорально всего в течение 6 недель с последующим 2-недельным периодом без лечения. В контрольном исследовании лечения одним лекарством пять мышей из группы, получающей соединение 1, получали соединение 1 в количестве 100 мг/кг один раз в два дня (QOD, от лат. "quaque other die"). Группа, получающая наполнитель, получала эквивалентный объем перорально один раз в день (QD, от лат. "quaque die") наполнителя в качестве плацебо (2% Klucel LF, 0,1% Полисорбата 80 и 0,1% парабенов в воде). В исследовании комбинированного лечения пяти мышам группы, получающей соединение 2, вводили по 12 мг/кг перорально один раз в день (QD). Группа Комбо получала 100 мг/кг соединения 1 QOD плюс 12 мг/кг соединения 2 QD. Группа, получающая наполнитель, получала эквивалентный объем перорально-QD наполнителя в качестве плацебо (2% Klucel LF, 0,1% Полисорбата 80 и 0,1% парабенов в воде).
Мышиную модель с экспрессией на высоком уровне как ДНК HBV, так и HBsAg получали при инъекции мышам C57BL/6 рекомбинантного аденоассоциированного вируса (AAV), несущего воспроизводимый геном HBV (AAV-HBV). Через 3 недели после инъекции вирусные маркеры персистирующего HBV, такие как геномная ДНК HBV, HBsAg и HBeAg, обнаруживали в сыворотке инфицированных мышей. При длительной виремии HBV и полностью компетентной иммунной системе модель AAV-HBV использовали для исследования отдельного и комбинированного эффекта соединения 1, пролекарства агониста TLR7, активная форма которого после превращения вызывает эффективные естественные иммунные ответы, и соединения 2, небольшой молекулы, которая ингибирует сборку капсида HBV. Как показано на фигуре 1, после 6-недельного лечения соединение 1 вызывало более чем 2-кратное логарифмическое уменьшение ДНК HBV и более чем 1-кратное логарифмическое уменьшение HBsAg. Одно соединение 2 снижало ДНК HBV более чем в 3 раза логарифмически и до уровня ниже НПКО (нижнего предела количественного определения), и незначительно уменьшало уровень HBsAg. Комбинация соединения 1 и соединения 2 давала в результате устойчивое снижение как ДНК HBV, так и HBsAg до уровня ниже НПКО даже в конце 2-недельного периода без лечения. Результаты доказывают синергический противовирусный эффект новой терапии с комбинированным лечением агонистом TLR7 и ингибитором сборки капсида HBV.
Пример 7
Комбинация агониста TLR7 (соединение 1 и 3) и ингибитора сборки капсида HBV (соединение 4 и 5) эффективно уменьшала ДНК HBV и HBsAg в мышиной модели AAV-HBV
В другом независимом исследовании проверяли лечение большим числом комбинаций агониста TLR7 плюс ингибитор капсида и соответствующим одним соединением (обобщено в таблице 2), используя ту же самую мышиную модель AAV-HBV и способ измерения биомаркеров HBV, что описаны в примере 5.
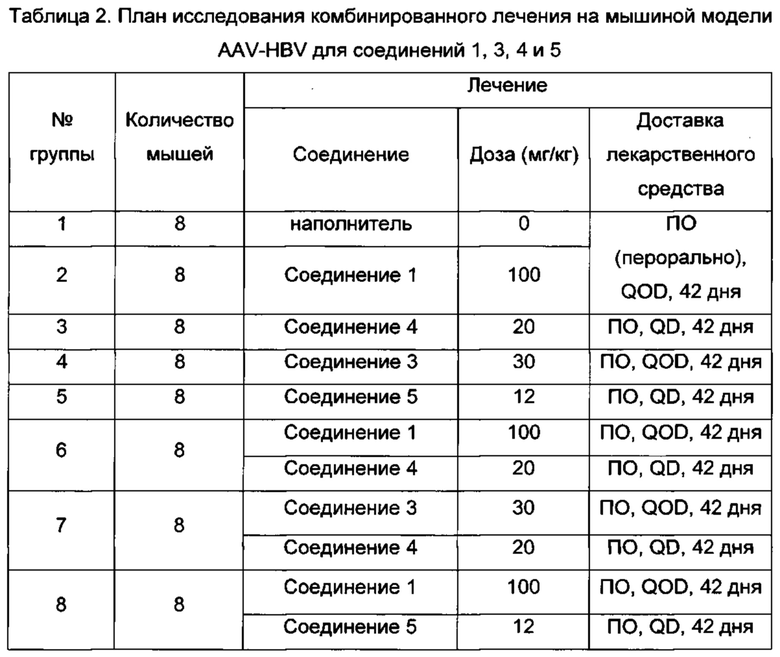
В этом исследовании по восемь мышей отбирали в каждую группу, и животные получали первую дозу на 28 день после заражения AAV-HBV. Исследуемые комбинации включали соединение 1 плюс соединение 4, соединение 3 плюс соединение 4 и соединение 1 плюс соединение 5. Все соединения получали в виде комплекса включения с 2% Klucel LF, 0,1% Полисорбата 80 и 0,1% парабенов в воде, и эквивалентный объем плацебо, содержащий 2% Klucel LF, 0,1% Полисорбата 80 и 0,1% парабенов, использовали в группе, получающей наполнитель. А именно, для комбинации соединения 1 плюс соединение 4 объединяли 10 мг/мл соединения 1 и 2 мг/мл соединения 4. Группе, получающей соединение 1, перорально вводили дозу 100 мг/кг QOD, тогда как группе, получающей соединение 4, перорально вводили дозу 20 мг/кг QD. Соответствующая Комбо группа получала 100 мг/кг соединения 1 QOD плюс 20 мг/кг соединения 4 QD. Для комбинации соединения 3 плюс соединение 4 объединяли 3 мг/мл соединения 3 и 2 мг/мл соединения 4. Группе, получающей соединение 3, перорально вводили дозу 30 мг/кг QOD, тогда как группе, получающей соединение 4, перорально вводили дозу 20 мг/кг QD. Соответствующая Комбо группа получала 30 мг/кг соединения 3 QOD плюс 20 мг/кг соединения 4 QD. Для комбинации соединения 1 плюс соединение 5 объединяли 10 мг/мл соединения 1 и 1,2 мг/мл соединения 5. Группе, получающей соединение 1, перорально вводили дозу 100 мг/кг QOD, тогда как группе, получающей соединение 5, перорально вводили дозу 12 мг/кг QD. Соответствующая Комбо группа получала 100 мг/кг соединения 1 QOD плюс 12 мг/кг соединения 5 QD. После первой дозы осуществляли подчелюстной забор крови у мышей (75 мкл крови/мышь) дважды в неделю для сбора сыворотки до окончания исследований. Собранную кровь оставляли при 37°С по меньшей мере на 30 минут до коагуляции и затем центрифугировали при 13,200 × g, 4°С в течение 3 минут, чтобы получить мышиную сыворотку. Эти образцы сыворотки подвергали анализу на биомаркеры HBV.
Как показано на фигуре 4, лечение только соединением 4 при 20 мг/кг ингибировало ДНК HBV и снижало HBsAg в 2 раза логарифмически в конце 6-недельного лечения. Комбинация соединения 1 (агонист TLR7) плюс соединение 4 (ингибитор капсида HBV) наглядно демонстрировала превосходный противовирусный эффект, особенно в регулировании HBsAg. У всех животных, получающих комбинированное лечение, их HBsAg снижался до уровня близкого или ниже НПКО в течение 4 недель лечения, и более чем 3,5-кратное логарифмическое снижение HBsAg в конце лечения могло продлиться в течение по меньшей мере 6 недель в период без лечения. Было установлено, что в период без лечения 6 из 8 мышей демонстрировали обнаруживаемые уровни анти-HBs, как показано на фигуре 7.
Как показано на фигуре 5, другой агонист TLR7 соединение 3 также снижал как ДНК HBV, так и HBsAg. Комбинация соединения 3 плюс ингибитор капсида соединение 4 давала дополнительное снижение ДНК HBV (>4 log) и HBsAg (2,7-log). Как показано на фигуре 7, 3 из 8 мышей, получающих соединение 3 плюс соединение 4, демонстрировали обнаруживаемые уровни анти-HBs в течение 6-недельного периода без лечения.
Как показано на фигуре 6, соединение 5 представляет собой другой ингибитор капсида, который снижал как ДНК HBV, так и HBsAg. Комбинация соединения 5 плюс агонист TLR7 соединение 1 дополнительно уменьшала HBsAg ниже НПКО в течение 4 недель после лечения, и снижение вирусной нагрузки сохранялось на протяжении исследования даже после прекращения лечения в течение 6 недель. Как показано на фигуре 7, 4 из 8 мышей, получающих соединение 1 плюс соединение 5, демонстрировали обнаруживаемые уровни анти-HBs в течение 6-недельного периода без лечения.
Пример 8
Комбинация агониста TLR7 (соединение 1 и 8) и ингибитора сборки капсида HBV (соединение 4 и 10) эффективно уменьшала ДНК HBV и HBsAg в мышиной модели AAV-HBV
В другом независимом исследовании проверяли лечение большим числом комбинаций агониста TLR7 плюс ингибитор капсида и соответствующим одним соединением (обобщено в таблице 3), используя ту же самую мышиную модель AAV-HBV и способы измерения биомаркеров HBV, что описаны в примере 5.
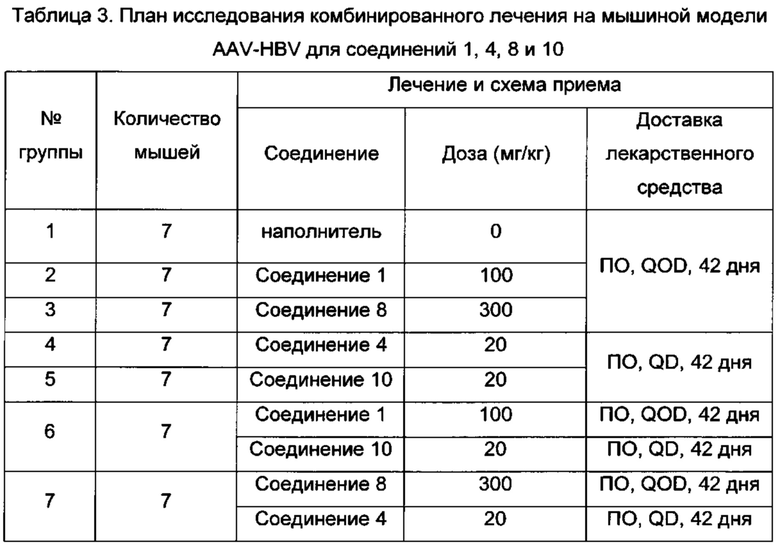

В этом конкретном исследовании по семь мышей отбирали в каждую группу, и животные получали первую дозу по меньшей мере на 38 день после заражения AAV-HBV. Исследуемые комбинации включали соединение 8 плюс соединение 4, соединение 8 плюс соединение 10 и соединение 1 плюс соединение 10. Все соединения получали в виде комплекса включения с 2% Klucel LF, 0,1% Полисорбата 80 и 0,1% парабенов в воде, и эквивалентный объем плацебо, содержащий 2% Klucel LF, 0,1% Полисорбата 80 и 0,1% парабенов, использовали в группе, получающей наполнитель. А именно, для комбинации соединения 8 плюс соединение 4 объединяли 30 мг/мл соединения 8 и 2 мг/мл соединения 4. Группе, получающей соединение 8, перорально вводили дозу 300 мг/кг QOD, тогда как группе, получающей соединение 4, перорально вводили дозу 20 мг/кг QD. И затем соответствующая Комбо группа получала 30 мг/кг соединения 8 QOD плюс 20 мг/кг соединения 4 QD. Для комбинации соединения 8 плюс соединение 10 объединяли 30 мг/мл соединения 8 и 2 мг/мл соединения 10. Группе, получающей соединение 8, перорально вводили дозу 300 мг/кг QOD, тогда как группе, получающей соединение 10, перорально вводили дозу 20 мг/кг QD. И затем соответствующая Комбо группа получала 300 мг/кг соединения 8 QOD плюс 20 мг/кг соединения 10 QD. Для комбинации соединения 1 плюс соединение 10 объединяли 10 мг/мл соединения 1 и 2 мг/мл соединения 10. Группе, получающей соединение 1, перорально вводили дозу 100 мг/кг QOD, тогда как группе, получающей соединение 10, перорально вводили дозу 20 мг/кг QD. И затем соответствующая Комбо группа получала 100 мг/кг соединения 1 QOD плюс 20 мг/кг соединения 10 QD. После первой дозы осуществляли подчелюстной забор крови у мышей (75 мкл крови/мышь) дважды в неделю для сбора сыворотки до окончания исследований. Собранную кровь оставляли при 37°С по меньшей мере на 30 минут до коагуляции и затем центрифугировали при 13,200 × g, 4°С в течение 3 минут, чтобы получить мышиную сыворотку. Эти образцы сыворотки подвергали анализу на биомаркеры HBV.
Результаты на фигуре 8 демонстрировали, что один агонист TLR7 соединение 8 снижал ДНК HBV и HBsAg приблизительно в 2 раза логарифмически и 1,5 раза логарифмически, соответственно, в конце лечения, тогда как комбинация соединения 8 плюс ингибитор капсида соединение 4 дополнительно снижала HBsAg до уровня ниже НПКО. В течение 6-недельного периода без лечения группа, получающая комбинацию соединений, демонстрировала устойчивое снижение HBsAg, минимальную отдачу ДНК HBV и высокие уровни анти-HBs, что не наблюдалось в группах, получающих наполнитель и одно соединение, как показано на фигуре 11. Также такие преимущества комбинированного лечения постоянно наблюдались в группах, получающих комбинацию соединения 8 плюс соединение 10 и соединения 1 плюс соединение 10, как показано на фигурах 9, 10 и 11.
Таким образом, вышеприведенные результаты впервые доказали, что комбинация агониста TLR7 плюс ингибитор капсида HBV является эффективным лечением, которое значительно снижает или даже удаляет ДНК HBV и HBsAg. Было показано, что после комбинированного лечения подавление вируса сохраняется в течение всех 6 недель без лечения. У большинства хронически HBV-инфицированных пациентов на данный момент доступные способы лечения редко могут приводить к сероконверсии HBsAg вследствие того, что большинство из этих способов лечения не могут добиться анти-HBs (антитело против HBsAg). В наших изучениях комбинаций неожиданно было установлено, что анти-HBs можно обнаружить в течение 6-недельного периода без лечения, и это было особо выражено у мышей, получающих комбинированное лечение, как показано на фигуре 7 и 11. Следовательно, комбинированное лечение агонистом TLR7 плюс ингибитор капсида HBV дает другое ключевое преимущество вызывать развитие анти-HBs. Поскольку длительное отсутствие HBsAg и/или сероконверсия анти-HBs является идеальной конечной точкой лечения хронического гепатита В, наше комбинированное лечение является новым способом достижения клинического излечения от хронической инфекции HBV.
--->
ПЕРЕЧЕНЬ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> F. Hoffmann-La Roche AG.
<120> Combined Treatment with a TLR7 agonist and an HBV capsid assembly
inhibitor
<130> P32687
<140> PCT/EP2016/055484
<141> 2015-03-15
<160> 3
<170> PatentIn version 3.5
<210> 1
<211> 20
<212> DNA
<213> Hepatitis B virus
<400> 1
aagaaaaacc ccgcctgtaa 20
<210> 2
<211> 23
<212> DNA
<213> Hepatitis B virus
<400> 2
cctgttctga ctactgcctc tcc 23
<210> 3
<211> 27
<212> DNA
<213> Hepatitis B virus
<400> 3
cctgatgtga tgttctccat gttcagc 27
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВОЕ ФОСФОРАМИДАТНОЕ ПРОИЗВОДНОЕ НУКЛЕОЗИДА И ЕГО ПРИМЕНЕНИЕ | 2014 |
|
RU2621709C2 |
| Макрогетероциклические нуклеозидные производные и их аналоги, получение и применение | 2017 |
|
RU2731385C1 |
| МОДУЛЯТОРЫ ПРОТЕОЛИЗА И СООТВЕТСТВУЮЩИЕ СПОСОБЫ ПРИМЕНЕНИЯ | 2019 |
|
RU2805511C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ АЦЕТАМИДА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ И ИНГИБИТОРЫ ПРОТЕАЗ НА ИХ ОСНОВЕ | 1997 |
|
RU2181360C2 |
| МОДУЛЯТОРЫ SHIP1 И ОТНОСЯЩИЕСЯ К НИМ СПОСОБЫ | 2014 |
|
RU2679805C2 |
| ЗАМЕЩЕННЫЕ (2R,3R,5R)-3-ГИДРОКСИ-(5-ПИРИМИДИН-1-ИЛ)ТЕТРАГИДРОФУРАН-2-ИЛМЕТИЛ АРИЛ ФОСФОРАМИДАТЫ | 2013 |
|
RU2553996C1 |
| НОВОЕ ПРОИЗВОДНОЕ ФЕНИЛПИРРОЛА | 2009 |
|
RU2470917C2 |
| ИНГИБИТОРЫ RMT5 | 2019 |
|
RU2814198C2 |
| ФЕНИКОЛОВЫЕ ПРОТИВОБАКТЕРИАЛЬНЫЕ СРЕДСТВА | 2013 |
|
RU2593204C2 |
| 7-ЗАМЕЩЕННЫЕ СУЛЬФОНИМИДОИЛПУРИНОНЫ ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ВИРУСНОЙ ИНФЕКЦИИ | 2017 |
|
RU2751349C2 |
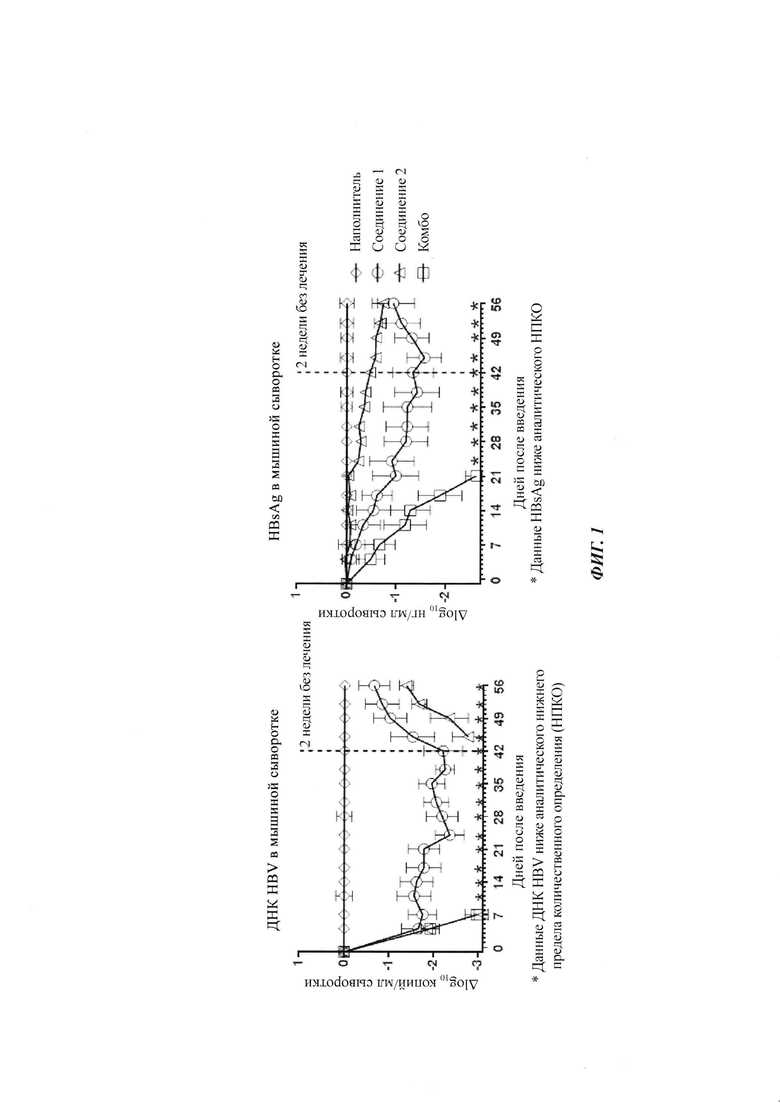
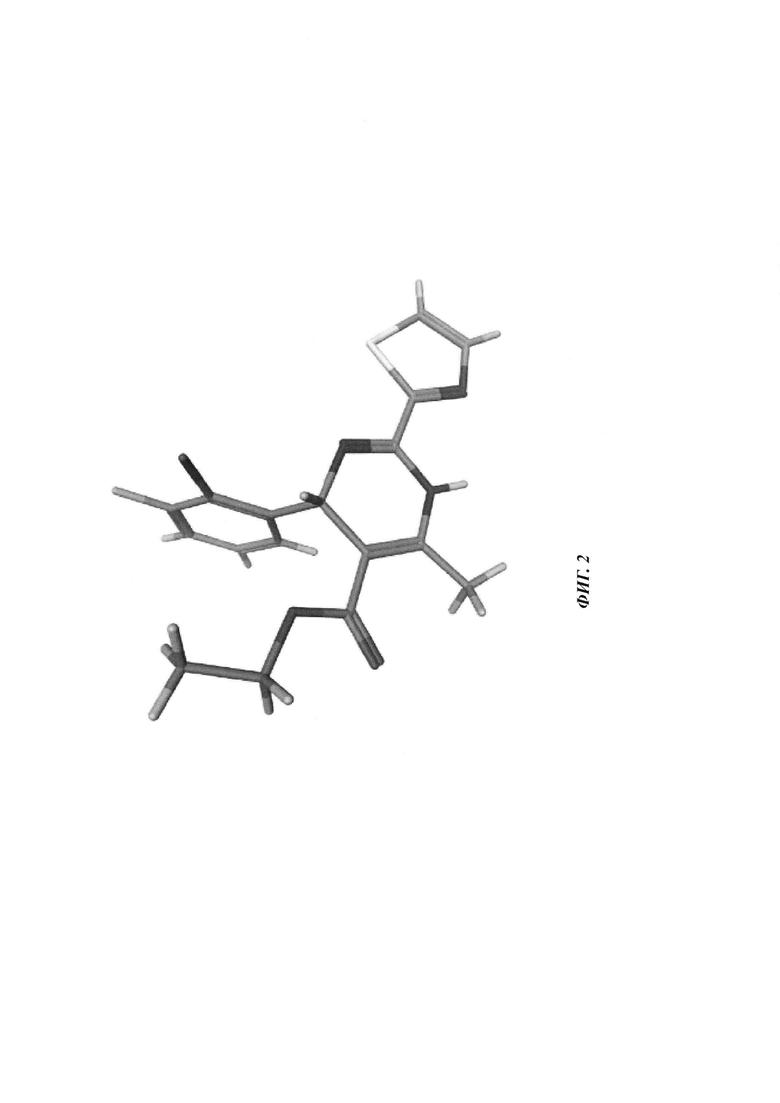
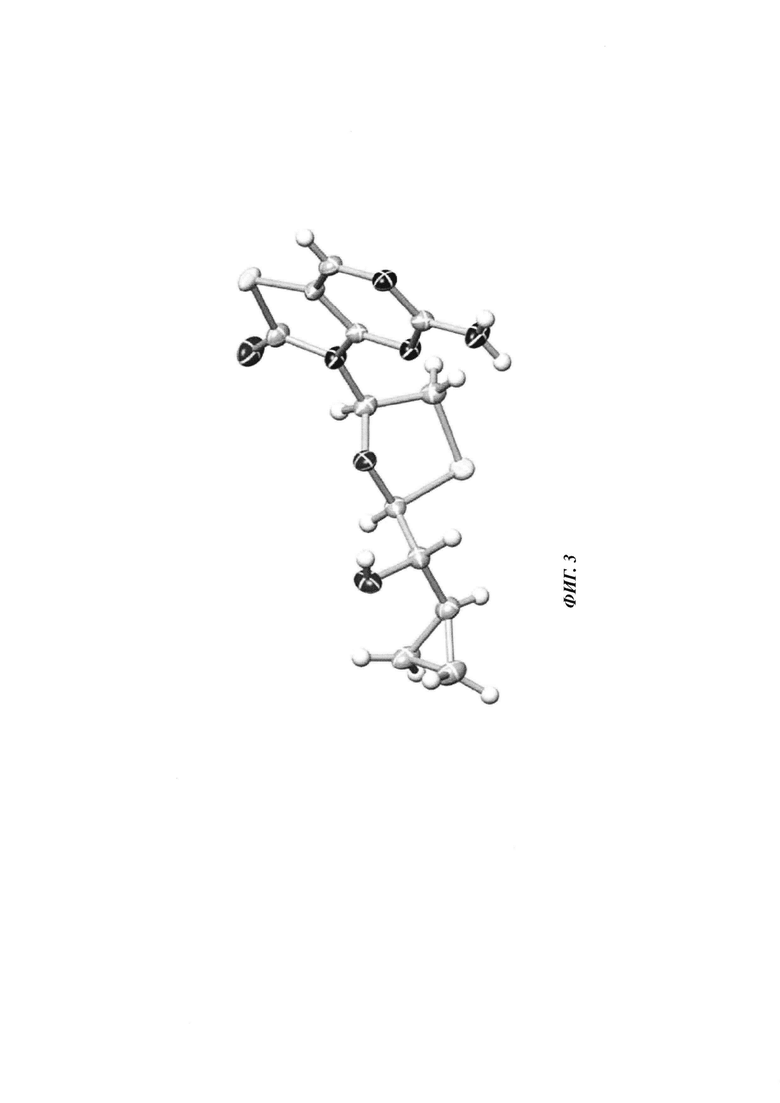
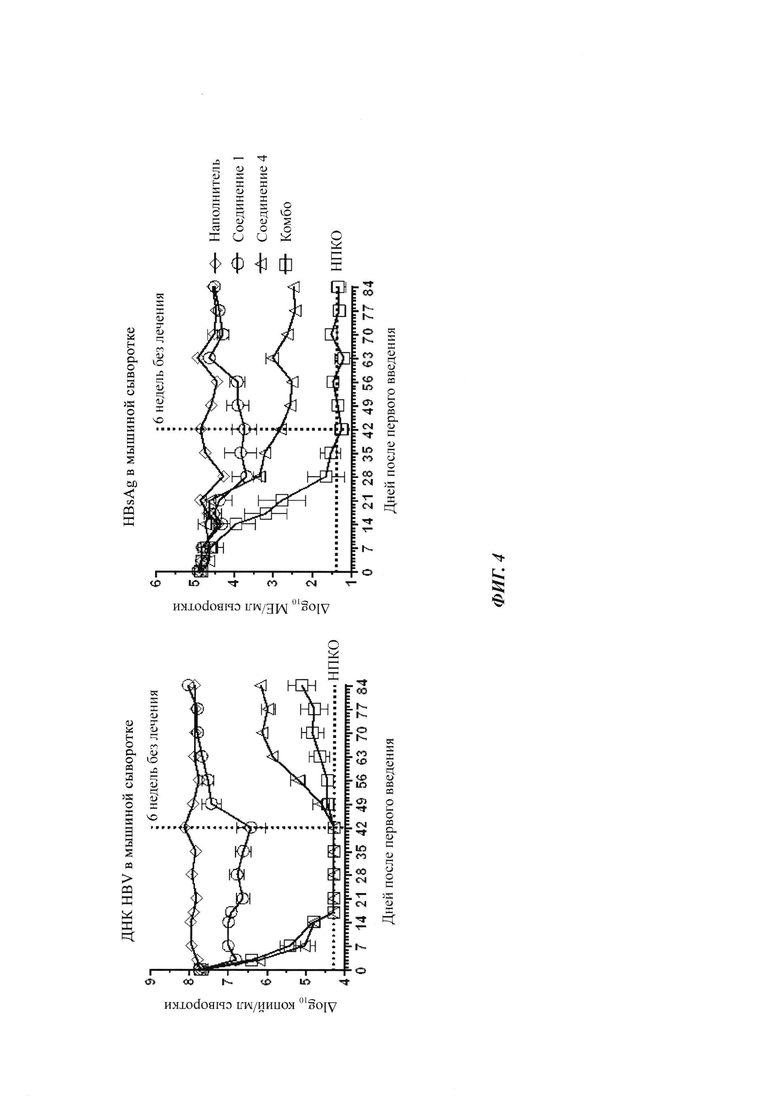
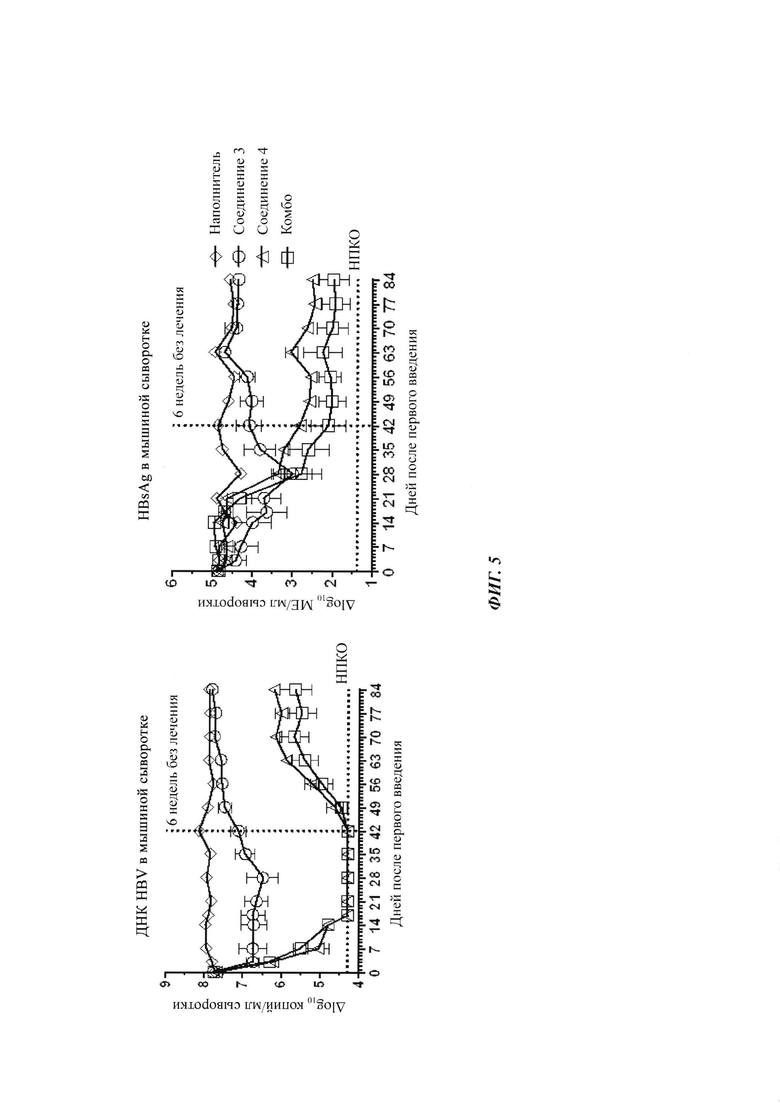
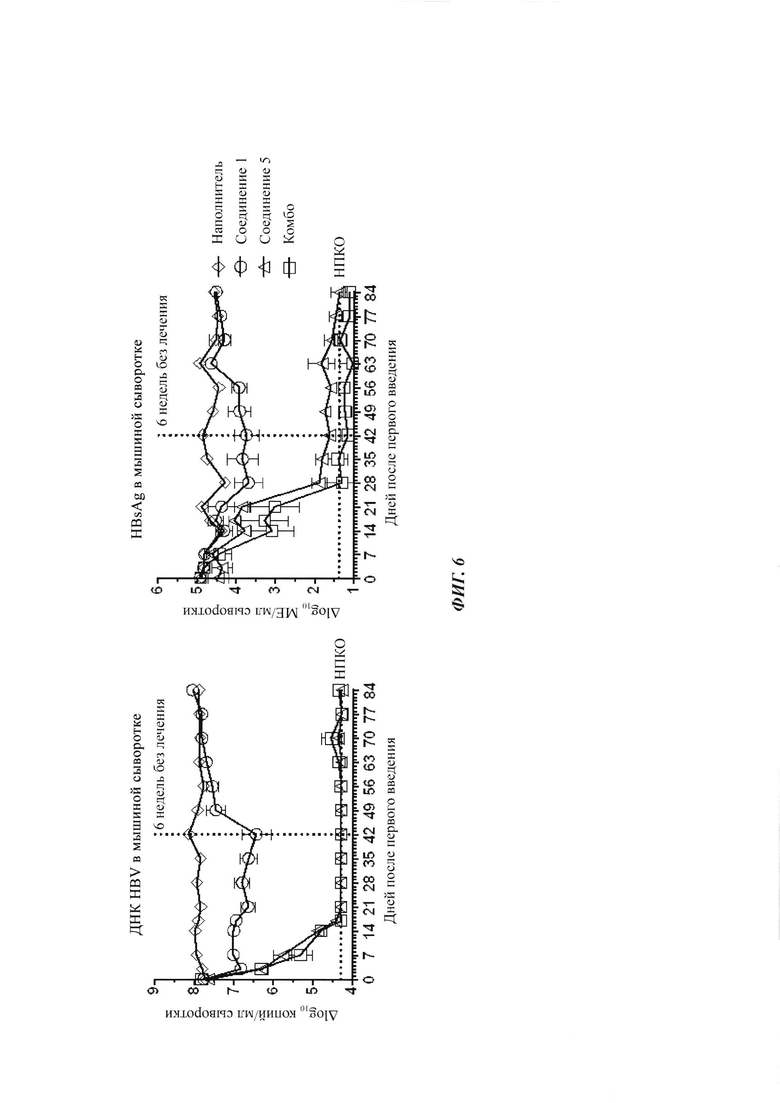

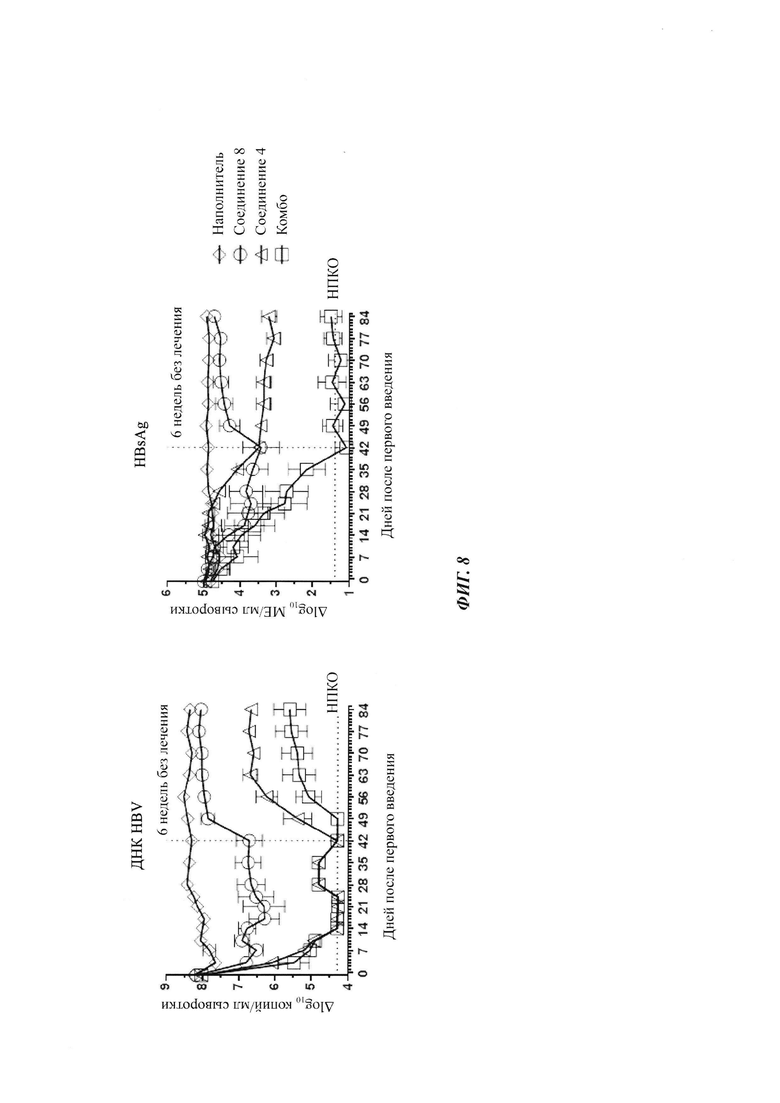


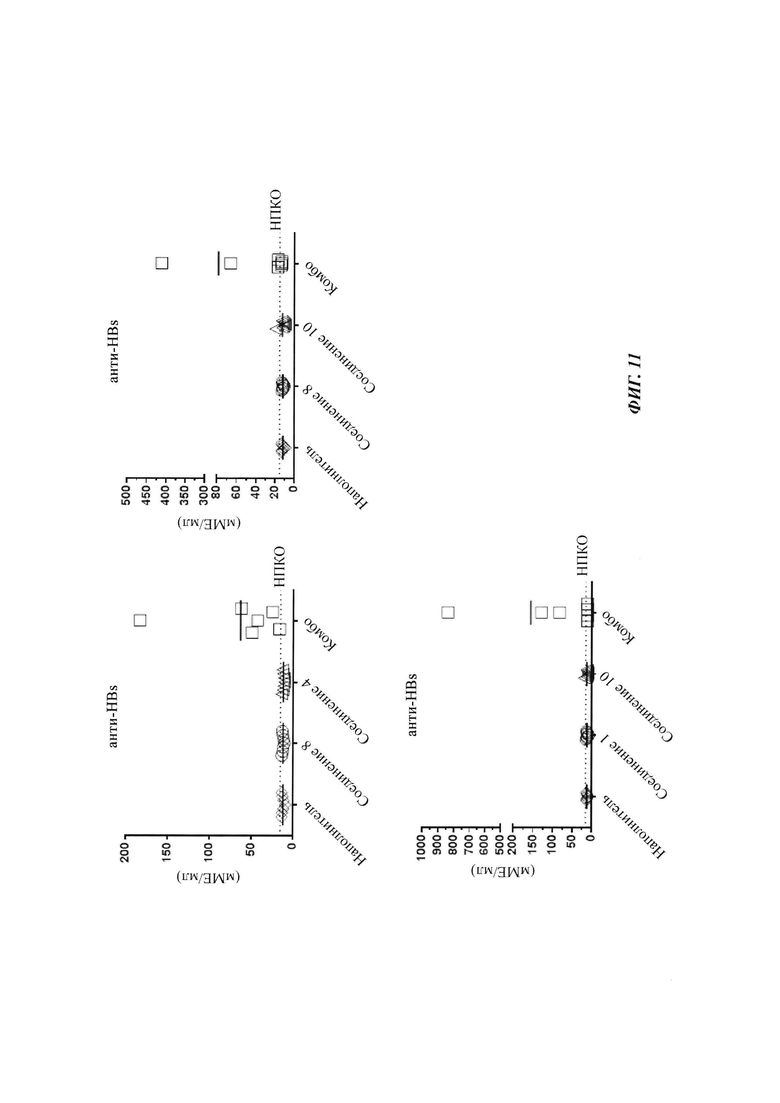
Группа изобретений относится к области медицины и фармацевтики, а именно к фармацевтической композиции, включающей агонист TLR7, выбранный из [(1S)-1-[(2S,4R,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-4-гидрокси-тетрагидрофуран-2-ил]пропил]ацетата и 5-амино-3-[(2R,3R,5S)-3-гидрокси-5-[(1S)-1-гидроксипропил]тетрагидрофуран-2-ил]-6Н-тиазоло[4,5-d]пиримидин-2,7-диона, и ингибитор сборки капсида HBV, выбранный из 3-[(8aS)-7-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-этоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1H-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановой кислоты и 3-[(8aS)-7-[[(4S)-5-этоксикарбонил-4-(3-фтор-2-метил-фенил)-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановой кислоты, в фармацевтически приемлемом носителе. Остальные объекты относятся к способу изготовления лекарственного средства, способу, набору и применению фармацевтической композиции для лечения или профилактики вирусной инфекции гепатита В. Технический результат заключается в синергетическом противовирусном эффекте комбинированной терапии с использованием конкретных комбинаций агонистов TLR7 и ингибиторов сборки капсида HBV. 5 н. и 8 з.п. ф-лы, 11 ил., 3 табл., 8 пр.
1. Фармацевтическая композиция, включающая агонист толл-подобного рецептора (TLR7) и ингибитор сборки капсида вируса гепатита В (HBV) в фармацевтически приемлемом носителе, где агонист TLR7 выбран из:
[(1S)-1-[(2S,4R,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-4-гидрокси-тетрагидрофуран-2-ил]пропил]ацетата, имеющего структуру
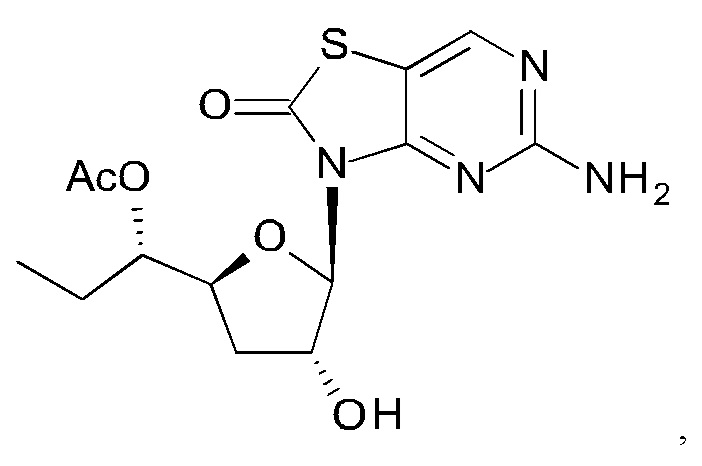
и
5-амино-3-[(2R,3R,5S)-3-гидрокси-5-[(1S)-1-гидроксипропил]тетрагидрофуран-2-ил]-6Н-тиазоло[4,5-d]пиримидин-2,7-диона, имеющего структуру
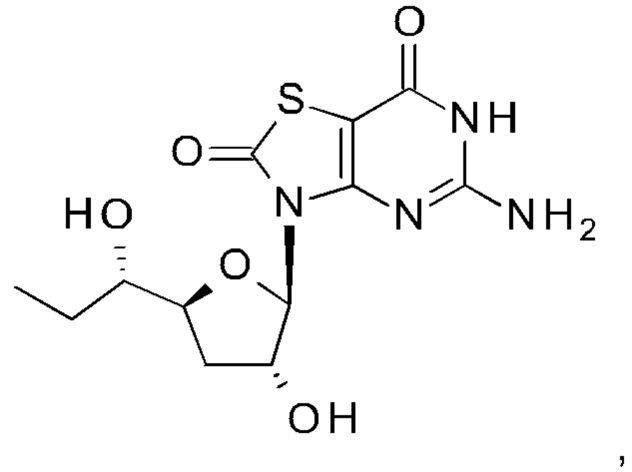
и где ингибитор сборки капсида HBV выбран из:
3-[(8aS)-7-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-этоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1H-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановой кислоты, имеющей структуру
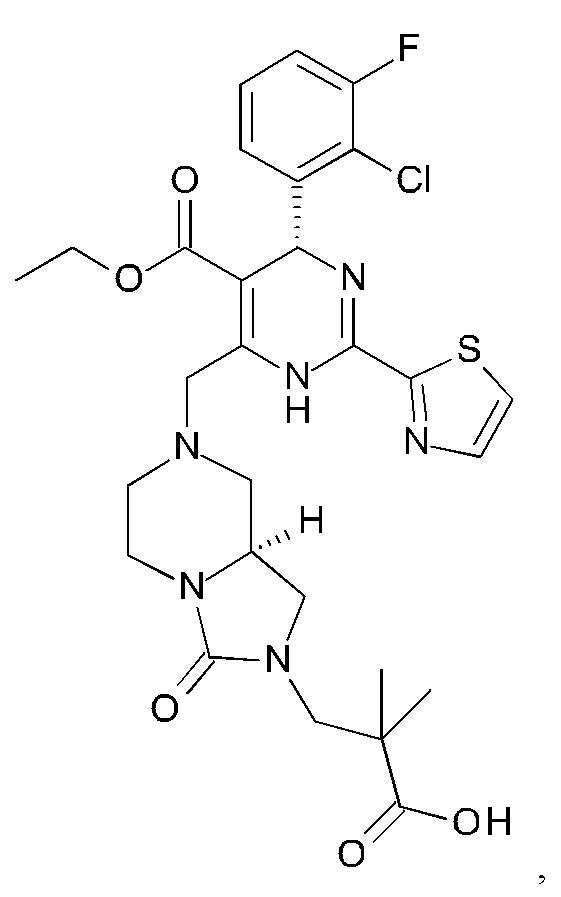
и
3-[(8aS)-7-[[(4S)-5-этоксикарбонил-4-(3-фтор-2-метил-фенил)-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановой кислоты, имеющей структуру
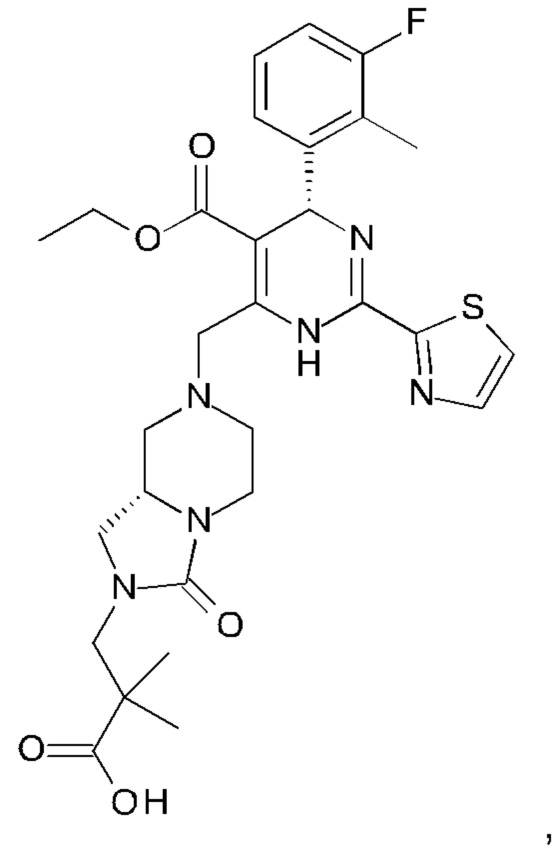
или их фармацевтически приемлемой соли, энантиомера или диастереомера.
2. Фармацевтическая композиция по п. 1, где композиция состоит из
[(1S)-1-[(2S,4R,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-4-гидрокси-тетрагидрофуран-2-ил]пропил]ацетата и
3-[(8aS)-7-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-этоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1H-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановой кислоты
в фармацевтически приемлемом носителе.
3. Фармацевтическая композиция по п. 1, где композиция состоит из
[(1S)-1-[(2S,4R,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-4-гидрокси-тетрагидрофуран-2-ил]пропил]ацетата и 3-[(8aS)-7-[[(4S)-5-этоксикарбонил-4-(3-фтор-2-метил-фенил)-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановой кислоты
в фармацевтически приемлемом носителе.
4. Фармацевтическая композиция по п. 1, где композиция состоит из
5-амино-3-[(2R,3R,5S)-3-гидрокси-5-[(1S)-1-гидроксипропил]тетрагидрофуран-2-ил]-6Н-тиазоло[4,5-d]пиримидин-2,7-диона и 3-[(8aS)-7-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-этоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1H-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановой кислоты
в фармацевтически приемлемом носителе.
5. Фармацевтическая композиция по п. 1, где композиция состоит из
5-амино-3-[(2R,3R,5S)-3-гидрокси-5-[(1S)-1-гидроксипропил]тетрагидрофуран-2-ил]-6Н-тиазоло[4,5-d]пиримидин-2,7-диона и 3-[(8aS)-7-[[(4S)-5-этоксикарбонил-4-(3-фтор-2-метил-фенил)-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановой кислоты
в фармацевтически приемлемом носителе.
6. Способ изготовления лекарственного средства для лечения или профилактики вирусной инфекции гепатита В, отличающийся тем, что в лекарственном средстве применяют агонист TLR7, выбранный из
[(1S)-1-[(2S,4R,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-4-гидрокси-тетрагидрофуран-2-ил]пропил]ацетата, имеющего структуру
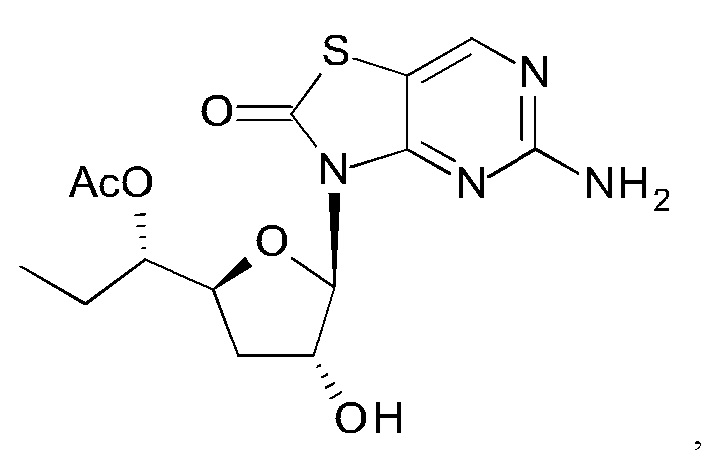
и
5-амино-3-[(2R,3R,5S)-3-гидрокси-5-[(1S)-1-гидроксипропил]тетрагидрофуран-2-ил]-6Н-тиазоло[4,5-d]пиримидин-2,7-диона, имеющего структуру
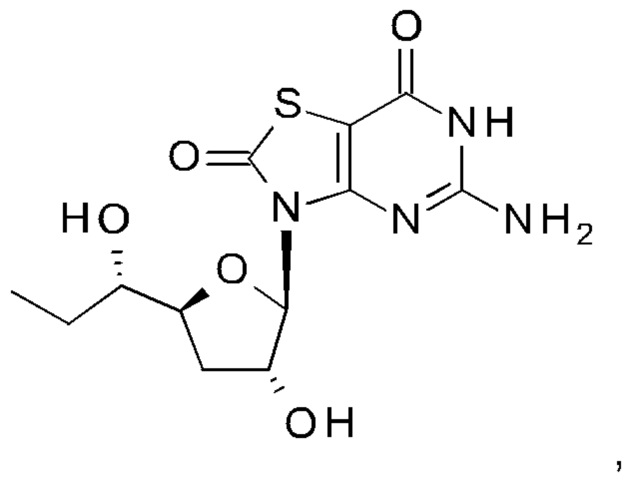
или их фармацевтически приемлемой соли, энантиомера или диастереомера,
и ингибитор сборки капсида HBV, выбранный из
3-[(8aS)-7-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-этоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановой кислоты, имеющей структуру
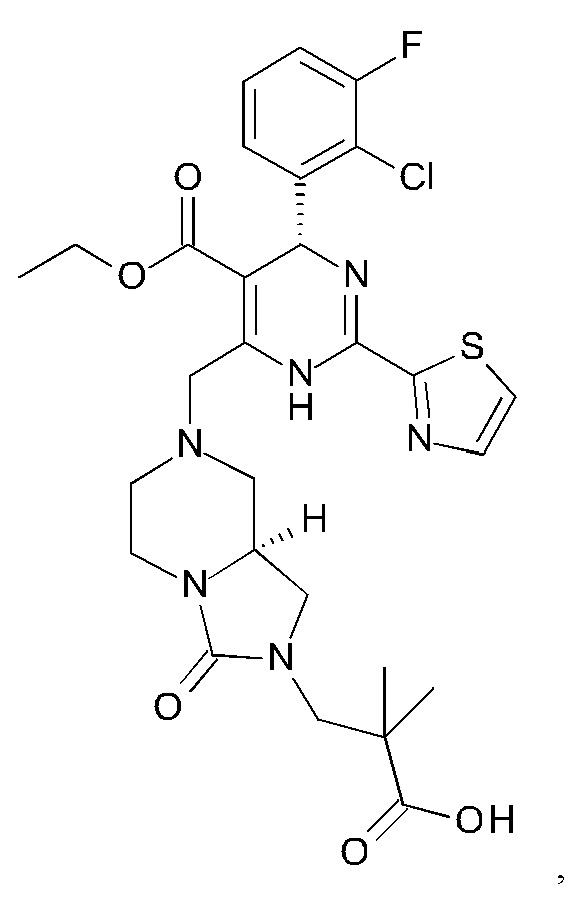
и
3-[(8aS)-7-[[(4S)-5-этоксикарбонил-4-(3-фтор-2-метил-фенил)-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановой кислоты, имеющей структуру
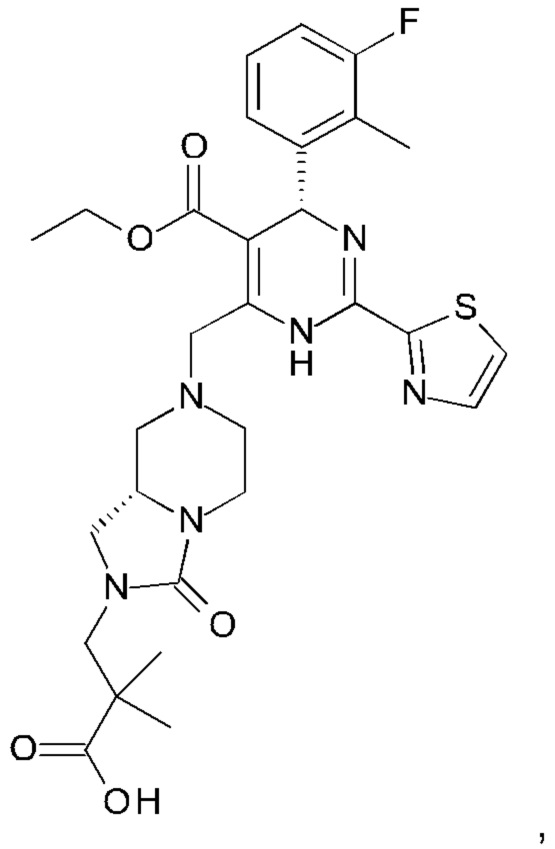
или их фармацевтически приемлемой соли, энантиомера или диастереомера.
7. Набор для лечения или профилактики вирусной инфекции гепатита В, включающий контейнер, содержащий агонист TLR7 и ингибитор сборки капсида HBV, где
агонист TLR7 выбран из
[(1S)-1-[(2S,4R,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-4-гидрокси-тетрагидрофуран-2-ил]пропил]ацетата, имеющего структуру
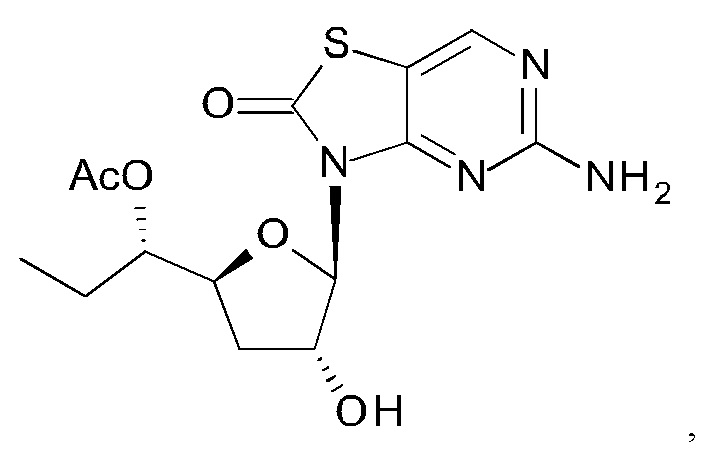
или
5-амино-3-[(2R,3R,5S)-3-гидрокси-5-[(1S)-1-гидроксипропил]тетрагидрофуран-2-ил]-6Н-тиазоло[4,5-d]пиримидин-2,7-диона, имеющего структуру
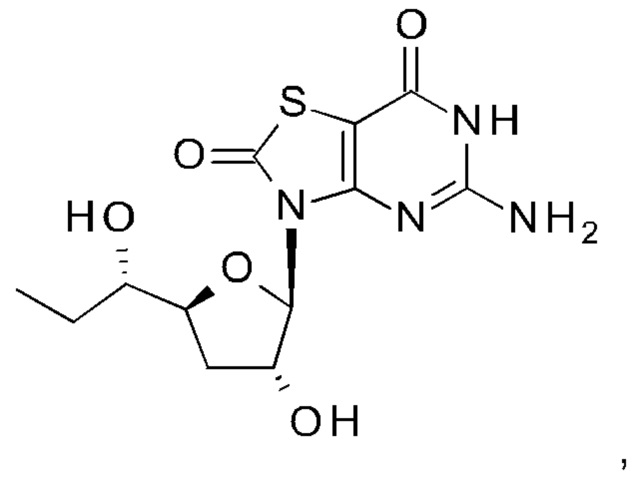
или их фармацевтически приемлемой соли, энантиомера или диастереомера; и
где ингибитор сборки капсида HBV выбран из
3-[(8aS)-7-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-этоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановой кислоты, имеющей структуру
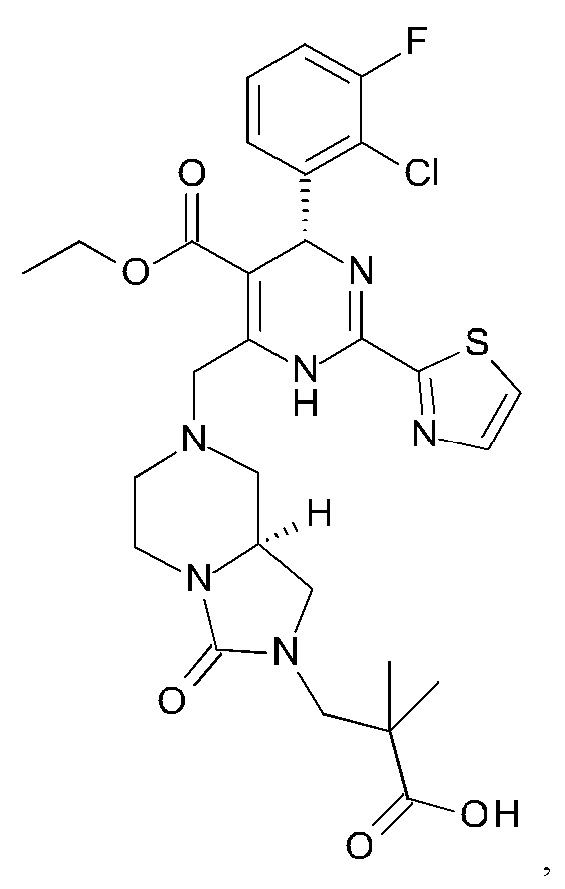
или
3-[(8aS)-7-[[(4S)-5-этоксикарбонил-4-(3-фтор-2-метил-фенил)-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановой кислоты, имеющей структуру
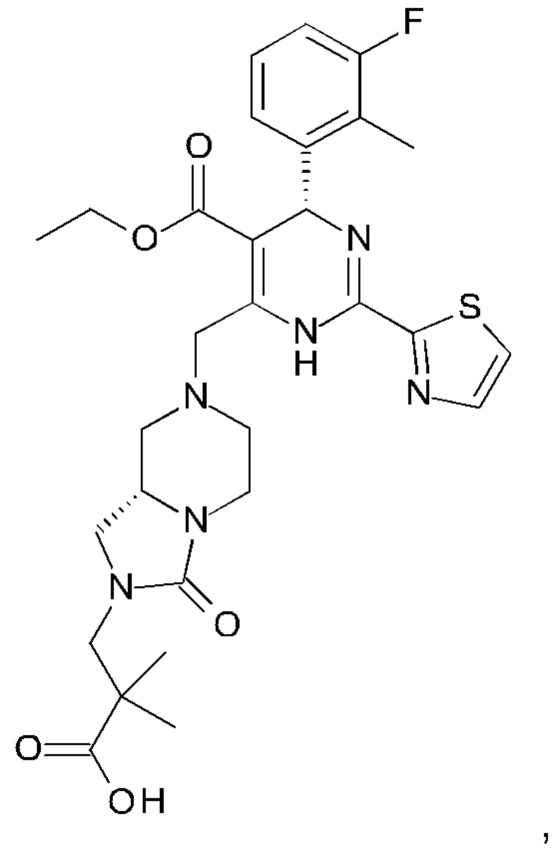
или их фармацевтически приемлемой соли, энантиомера или диастереомера.
8. Набор по п. 7, где агонист TLR7 и ингибитор сборки капсида HBV, используемые в контейнере, представляют собой:
[(1S)-1-[(2S,4R,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-4-гидрокси-тетрагидрофуран-2-ил]пропил]ацетат и 3-[(8aS)-7-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-этоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1H-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановую кислоту;
[(1S)-1-[(2S,4R,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-4-гидрокси-тетрагидрофуран-2-ил]пропил]ацетат и 3-[(8aS)-7-[[(4S)-5-этоксикарбонил-4-(3-фтор-2-метил-фенил)-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановую кислоту;
5-амино-3-[(2R,3R,5S)-3-гидрокси-5-[(1S)-1-гидроксипропил]тетрагидрофуран-2-ил]-6H-тиазоло[4,5-d]пиримидин-2,7-дион и 3-[(8aS)-7-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-этоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановую кислоту; или
5-амино-3-[(2R,3R,5S)-3-гидрокси-5-[(1S)-1-гидроксипропил]тетрагидрофуран-2-ил]-6Н-тиазоло[4,5-d]пиримидин-2,7-дион и 3-[(8aS)-7-[[(4S)-5-этоксикарбонил-4-(3-фтор-2-метил-фенил)-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановую кислоту;
в фармацевтически приемлемом носителе.
9. Набор по п. 7, дополнительно включающий стерильный разбавитель.
10. Набор по любому из пп. 7-9, дополнительно включающий листок-вкладыш, содержащий напечатанные инструкции по применению комбинированного лечения с агонистом TLR7 и ингибитором сборки капсида HBV в качестве способа лечения или профилактики вирусной инфекции гепатита В.
11. Способ лечения или профилактики вирусной инфекции гепатита В, согласно которому субъекту вводят эффективное первое количество агониста TLR7 или его фармацевтически приемлемой соли, энантиомера или диастереомера; и второе количество ингибитора сборки капсида HBV или его фармацевтически приемлемой соли, энантиомера или диастереомера; или наоборот,
где агонист TLR7 выбран из
[(1S)-1-[(2S,4R,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-4-гидрокси-тетрагидрофуран-2-ил]пропил]ацетата, имеющего структуру
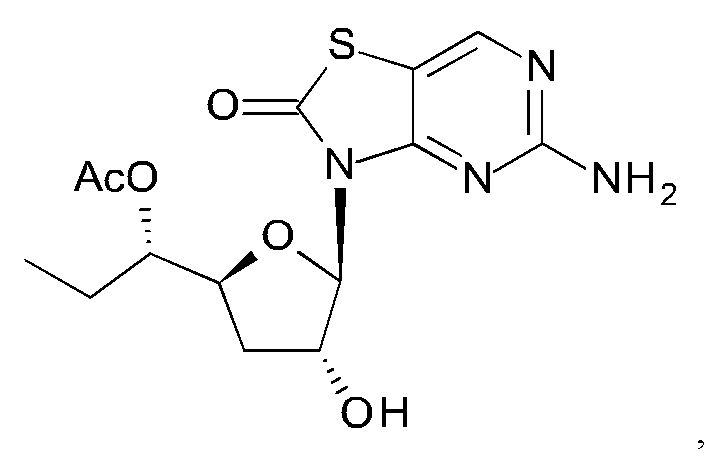
или
5-амино-3-[(2R,3R,5S)-3-гидрокси-5-[(1S)-1-гидроксипропил]тетрагидрофуран-2-ил]-6Н-тиазоло[4,5-d]пиримидин-2,7-диона, имеющего структуру

или их фармацевтически приемлемой соли, энантиомера или диастереомера;
и где ингибитор сборки капсида HBV выбран из
3-[(8aS)-7-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-этоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановой кислоты, имеющей структуру
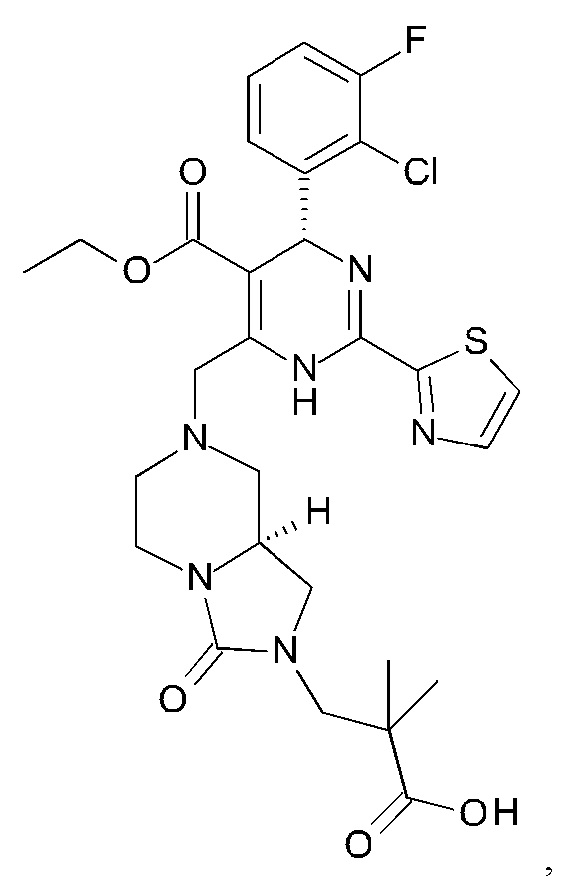
или
3-[(8aS)-7-[[(4S)-5-этоксикарбонил-4-(3-фтор-2-метил-фенил)-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановой кислоты, имеющей структуру
или их фармацевтически приемлемой соли, энантиомера или диастереомера.
12. Способ по п. 11, где агонист TLR7 и ингибитор сборки капсида HBV, используемые в способе, представляют собой:
[(1S)-1-[(2S,4R,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-4-гидрокси-тетрагидрофуран-2-ил]пропил]ацетат и 3-[(8aS)-7-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-этоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1H-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановую кислоту;
[(1S)-1-[(2S,4R,5R)-5-(5-амино-2-оксо-тиазоло[4,5-d]пиримидин-3-ил)-4-гидрокси-тетрагидрофуран-2-ил]пропил]ацетат и 3-[(8aS)-7-[[(4S)-5-этоксикарбонил-4-(3-фтор-2-метил-фенил)-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1H-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановую кислоту;
5-амино-3-[(2R,3R,5S)-3-гидрокси-5-[(1S)-1-гидроксипропил]тетрагидрофуран-2-ил]-6Н-тиазоло[4,5-d]пиримидин-2,7-дион и 3-[(8aS)-7-[[(4R)-4-(2-хлор-3-фтор-фенил)-5-этоксикарбонил-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановую кислоту; или
5-амино-3-[(2R,3R,5S)-3-гидрокси-5-[(1S)-1-гидроксипропил]тетрагидрофуран-2-ил]-6Н-тиазоло[4,5-d]пиримидин-2,7-дион и 3-[(8aS)-7-[[(4S)-5-этоксикарбонил-4-(3-фтор-2-метил-фенил)-2-тиазол-2-ил-1,4-дигидропиримидин-6-ил]метил]-3-оксо-5,6,8,8а-тетрагидро-1Н-имидазо[1,5-а]пиразин-2-ил]-2,2-диметил-пропановую кислоту;
в фармацевтически приемлемом носителе.
13. Применение фармацевтической композиции по любому из пп. 1-5 для лечения или профилактики вирусной инфекции гепатита В.
| WO 2013096744 A1, 27.06.2013 | |||
| US 2008032999 A1, 07.02.2008 | |||
| WO 2014029193 A1, 27.02.2014. |

