Настоящее изобретение относится к парентеральной полутвердой композиции с задержкой высвобождения и с контролируемым высвобождением, содержащей олигомер с конечными нереакционноспособными группами и активное вещество без какого-либо вспомогательного агента для понижения вязкости или наполнителя.
Кроме того, настоящее изобретение предусматривает непрерывное высвобождение активного вещества в течение периода, по меньшей мере, в одну неделю, когда полутвердую композицию помещают в водную физиологическую окружающую среду.
Более конкретно, настоящее изобретение относится к фармацевтической композиции в форме полутвердой композиции, содержащей, по меньшей мере, один биологически деградируемый полимер с конечными нереакционноспособными группами с очень низкой молекулярной массой и, по меньшей мере, одно активное вещество. Такая фармацевтическая композиция используется парентеральным путем, таким как подкожная или внутримышечная инъекция, и образует композицию импланта/депо, когда вводится в водную физиологическую окружающую среду.
В течение долгого времени была известна ценность введения активного вещества в форме композиций с задержкой высвобождения.
Обсуждаются различные подходы для контроля скорости высвобождения активного вещества.
Среди таких стратегий обнаружены два различных подхода для разработки препаратов имплантов или микрочастиц, содержащих биологически деградируемые полимеры, такие как поли(лактид-со-гликолид), в которые инкорпорируется активное вещество.
Другой подход заключается в разработке композиций депо для инъекций, содержащих биологически деградируемые полимеры и растворители/пластификаторы, которые являются очень или относительно растворимыми в водной телесной жидкости. В некоторых случаях, диффузия растворителей/пластификаторов способствует быстрому затвердеванию полимера в области имплантирования и, таким образом, вызывает медленное высвобождение лекарственного средства.
В Европейском патенте EP 1126822 показано, что посредством объединения термопластичного полимера или сополимера, органического растворителя, активного вещества и полимерной добавки с контролируемым высвобождением, такой как блок-сополимер поли(лактид-со-гликолид)/полиэтиленгликоль (PLGA/PEG), может контролироваться скорость высвобождения активного вещества. Когда он приводится в контакт с водной окружающей средой, такой как телесные или тканевые жидкости, органический растворитель, как предполагается, рассеивается или диспергируется в телесных жидкостях и одновременно с этим по существу нерастворимый термопластичный основной полимер преципитирует с формированием твердой матрицы или импланта.
Кроме того, композиция депо, как описано в заявке на Международный патент WO 2004/012703, может вводиться в виде инъекции в желаемое положение в организме пациента с формированием импланта. Такие композиции депо для инъекций требуют использования органических растворителей, таких как бензиловый спирт или бензилбензоат, и тиксотропного агента и спирта, подобного этанолу, в дополнение к лекарственному средству и полимеру. Тем не менее, эти добавки инкорпорируются в композицию депо, и это может приводить к низкой биологической совместимости продукта или к меньшей стабильности инкорпорированного активного агента в нем.
Кроме того, является нежелательным нахождение остатков органического растворителя, как правило, используемого во время получения полимера или вовлеченного в режим работы самого импланта.
Тем не менее, без добавления агента для понижения вязкости, такого как органический растворитель, композиция либо является недостаточно текучей, чтобы делать инъекцию, либо инъекция становится сложной и требует шприцевых устройств с очень широкой иглой и поэтому очень болезненных.
Другой подход может осуществляться с помощью фармацевтических композиций, которые вводятся посредством инъекций и самопроизвольно образуют препараты гелей с задержкой высвобождения. О таких препаратах сообщается в заявке на Международный патент WO 96/07398. Как описывается в тексте, эти соединения приготавливают как парентеральные препараты гелей с задержкой высвобождения без какого-либо добавления биологически деградируемых полимеров или другой матрицы носителя для контроля профиля высвобождения пептидов.
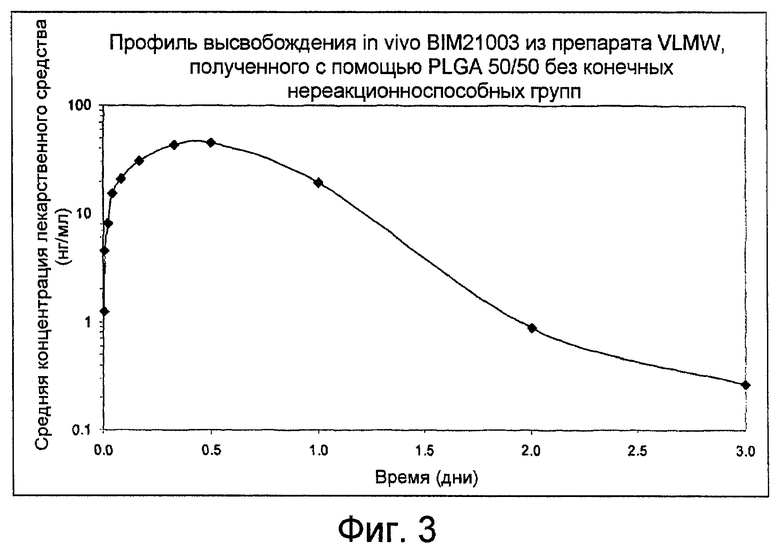
В рамках использования полимеров VLMW (с очень низкой молекулярной массой), предыдущий анализ с использованием PLGС при отношении 50/50 (то есть, сополимера молочной кислоты и гликолевой кислоты, содержащего 50% единиц молочной кислоты и 50% гликолевой кислоты) и имеющего молекулярную массу примерно 2300 Дальтон, заканчивающегося группой карбоновой кислоты, не дает ожидаемых результатов. Наблюдается быстрая деградация полимера, когда он вводится в водную физиологическую окружающую среду, как иллюстрируется на фиг. 3 и в Примере 12. В дополнение к этому, одна из главных сложностей, встречающихся для таких препаратов, заключается в проблеме вязкости, и таким образом требуется нагрев компонентов примерно при 50-60°C в течение процесса получения и также перед инъекцией.
Неожиданно обнаружено, что полутвердые импланты, полученные в соответствии с различными способами и изготовленные из модифицированного полимера с очень низкой молекулярной массой, например полилактида или сополимера, такого как PLA, PLGA, или их смеси, и с нереакционноспособным конечным остатком на карбоксильном окончании, могут образовывать импланты подкожных/внутримышечных депо без какой-либо вспомогательной добавки до и после инъекции, когда помещаются в водную физиологическую окружающую среду. Наблюдают, что активное вещество высвобождается в течение периода, по меньшей мере, одной недели, и даже в течение одного месяца, когда вводится в водную физиологическую окружающую среду. Кроме того, неожиданная сыпучесть таких полимеров/препаратов в их сухой форме делает возможным использование обычных устройств, например, типа шприцов, для парентерального введения. Другое ценное преимущество настоящего изобретения заключается в получении композиций, которые могут использоваться как предварительно заполненные (например, предварительно заполненный шприц) или готовые к употреблению препараты.
Присоединение конечных нереакционноспособных групп на концах полимерных цепей увеличивает их текучесть, таким образом, облегчая получение полутвердых композиций и делая возможным их инъекцию с помощью обычного устройства, например, типа шприца. Устройства для введения могут представлять собой 0,3-1 мл пластиковые шприцы с иглами, имеющими размер 16G, и с более тонкими иглами, с более толстыми иглами 19G или еще толще.
Целью настоящего изобретения является создание полутвердых фармацевтических композиций, используемых как препараты с задержкой высвобождения, которые высвобождают активное вещество в течение, по меньшей мере, одной недели и даже более одного месяца, когда полутвердую композицию вводят в водную физиологическую окружающую среду.
Настоящее изобретение может также рассматриваться как конкретная стабильная суспензия твердого активного вещества (порошка) в сплошной матрице с очень низкой молекулярной массой.
Цель настоящего изобретения достигается посредством исследований in-vivo и in-vitro и с помощью способа по настоящему изобретению.
Если не указано иного, следующие далее определения приводятся для иллюстрирования и определения значения и рамок различных терминов, используемых здесь для описания изобретения.
Термины "очень низкая молекулярная масса" и "модифицированная очень низкая молекулярная масса" относятся к полимеру или сополимеру с конечными нереакционноспособными группами, со средневзвешенной молекулярной массой в пределах между 500 и 5000 Дальтон, предпочтительно, между 700 и 3000 Дальтон, а более предпочтительно, между 800 и 2000 Дальтон.
Термин конечная нереакционноспособная группа или конечной нереакционноспособный остаток относится к прививке не нереакционноспособной химической группы на конец полимерной цепи, придавая, таким образом, полимеру лучшую стабильность по отношению к деградирующим агентам.
Термин "алкил" сам по себе или в сочетании с другими группами, как здесь используется, относится к насыщенной углеводородной группе, включая линейный, разветвленный, циклический, незамещенный, замещенный алкил.
Термин "гетероалкил" относится к алкилу, у которого, по меньшей мере, один атом углерода заменен гетероатомом.
Термин "алкокси" относится к алкильной группе, связанной с атомом кислорода.
Термин "арил" относится к ароматическому заместителю, содержащему одно ароматическое кольцо или множество ароматических колец, которые конденсированы друг с другом, связаны ковалентно или связаны с общей группой, такой как этиленовый или метиленовый остаток.
Термин "замещенный арил" относится к арильному остатку, замещенному одной или несколькими группами заместителями.
Термин гетероарил относится к арильной группе, в которой, по меньшей мере, один атом углерода заменен гетероатомом.
Термин аралкил относится к алкильной группе, замещенной арильной группой.
Термин гетероаралкил относится к алкильной группе, замещенной гетероарильной группой.
Предпочтительные конечные группы представляют собой алкильные группы, включая линейные, разветвленные, циклические, незамещенные и замещенные производные.
Под бинарной полутвердой фармацевтической композицией подразумевается биологически деградируемый фармацевтически приемлемый полимер или их смесь в дополнение, по меньшей мере, к одному активному веществу или к их смеси, предпочтительно, в их высушенной форме.
Пригодные для использования биологически деградируемые и фармацевтически приемлемые полимеры представляют собой, как правило, производные олигомерных гидрокарбоновых кислот, в частности молочной кислоты и/или гликолевой кислоты, производные лактида-гликолида или их смеси.
Термин фармацевтически приемлемый означает в этом контексте физиологически хорошо переносимый млекопитающим или человеком.
Термин "распределение молекулярных масс (MW)", включая "средневзвешенную молекулярную массу (MW)" и "полидисперсность (lp)", полимера относится к измерениям, осуществляемым с помощью любого стандартного способа, известного специалисту в данной области, например, с помощью эксклюзионной хроматографии (SEC), также называемой гель-проникающей хроматографией (GPC).
Описание фигур:
Фиг. 1: показывает профиль высвобождения аналога GLP-1 (BIM51077) in-vitro из препарата VLMW, полученного с помощью PLA с конечными нереакционноспособными группами, как получено в Примере 9.
Фиг. 2: показывает профиль высвобождения трипторелина (BIM21003) in-vitro из препарата VLMW, полученного с помощью PLGA с конечными нереакционноспособными группами, 80/20, как получено в Примере 10.
Фиг. 3: показывает профиль высвобождения трипторелина (BIM21003) in-vivo из препарата VLMW, полученного с помощью PLGA без конечных нереакционноспособных групп, 50/50, как получено в Примере 12.
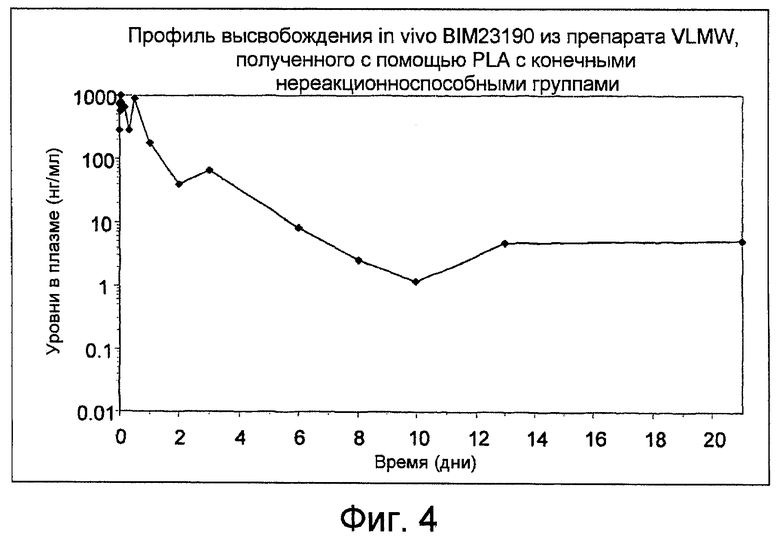
Фиг. 4: показывает профиль высвобождения аналога соматостатина (BIM23190) in-vivo из препарата VLMW, полученного с помощью PLA с конечными нереакционноспособными группами, как получено в Примере 13.
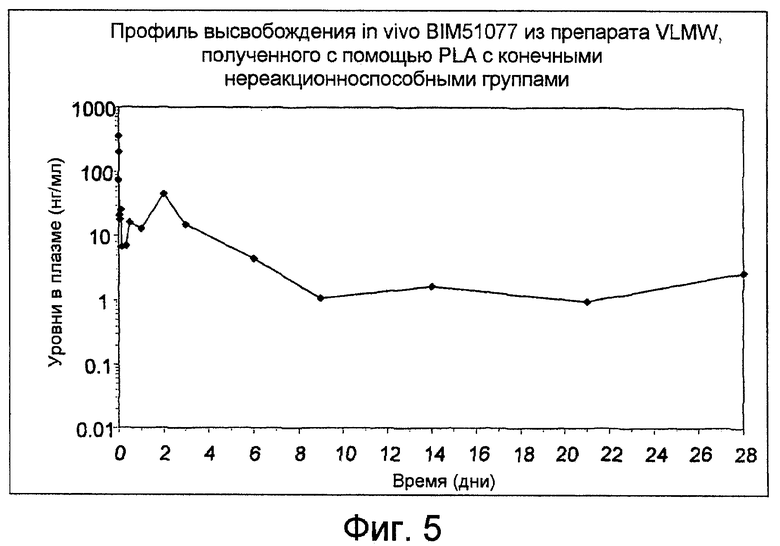
Фиг. 5: показывает профиль высвобождения аналога GLP-1 (BIM 51077) in-vivo из препарата VLMW, полученного с помощью PLA с конечными нереакционноспособными группами, как получено в Примере 13.
Фиг. 6: показывает профили высвобождения трипторелина (BIM 21003) in-vivo из препаратов VLMW, полученного с помощью PLA с конечными нереакционноспособными группами, как получено в Примере 13.
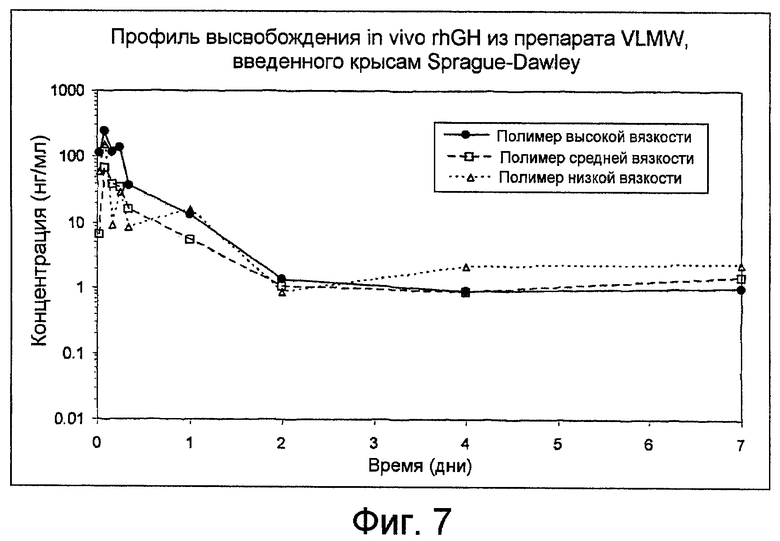
Фиг. 7: показывает профиль высвобождения рекомбинантного гормона роста человека (rhGH) in-vivo из препаратов VLMW, вводимых крысам Sprague-Dawley, как получено в Примере 14.
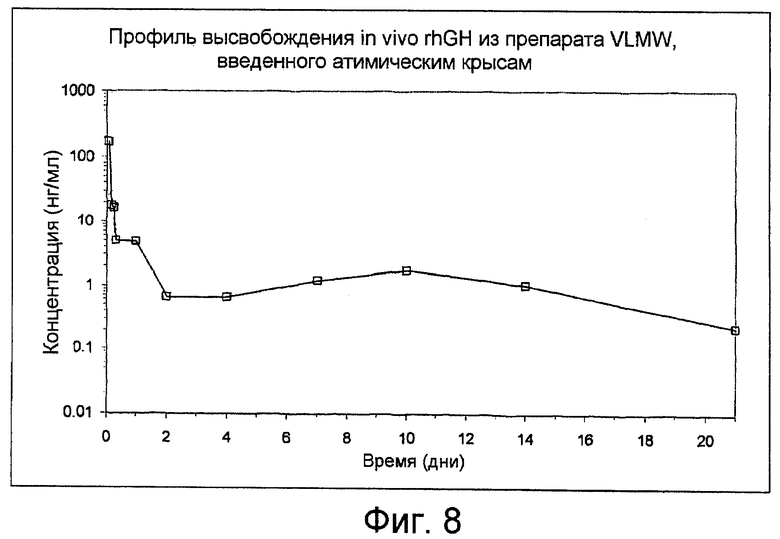
Фиг. 8: показывает профиль высвобождения рекомбинантного гормона роста человека (rhGH) in-vivo из препарата VLMW, вводимого атимическим крысам, как получено в Примере 15.
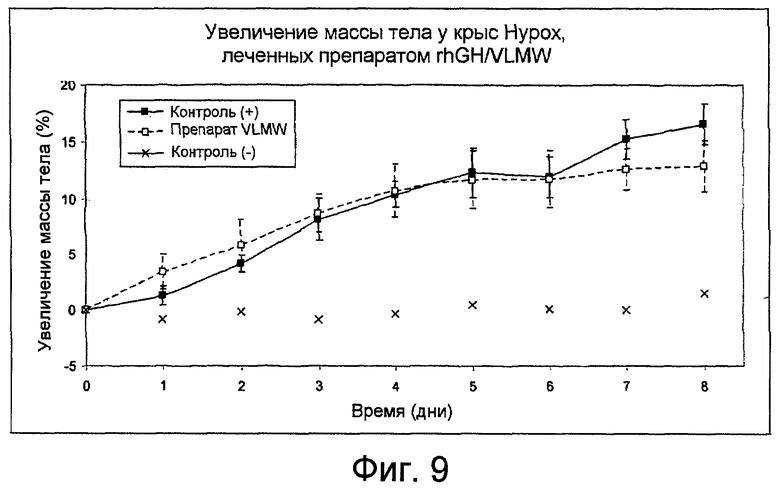
Фиг. 9: показывает увеличение массы тела крыс Hypox, леченных препаратом rhGH/VLMW, как получено в Примере 16.
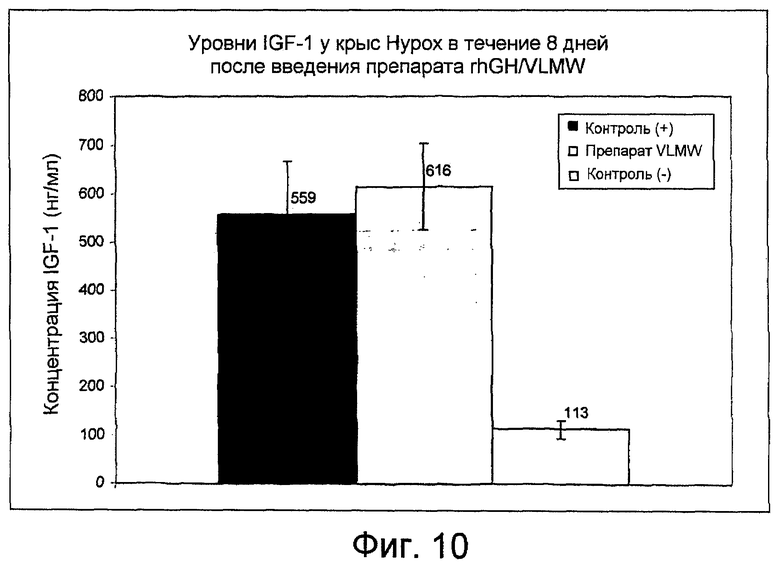
Фиг. 10: показывает уровни IGF-1 у крыс Hypox через 8 дней после введения препарата rhGH/VLMW, как получено в Примере 16.
Соответственно, настоящее изобретение относится к полутвердой композиции/импланту депо с задержкой высвобождения и с контролируемым высвобождением, содержащему:
Полутвердая композиция с задержкой высвобождения и с контролируемым высвобождением, содержащая:
a) Модифицированный биологически деградируемый полимер или сополимер с конечными нереакционноспособными группами с очень низкой молекулярной массой или их смесь, имеющая средневзвешенную молекулярную массу от 500 до 5000 Дальтон,
b) По меньшей мере, одно активное веществе или их смесь,
где указанный препарат получают без вспомогательных или дополнительных разбавителей, пластификаторов, растворителей или наполнителей.
Композиция, кроме того, может содержать соответствующие биологически деградируемые и фармацевтически приемлемые низкомолекулярные полимеры, которые могут использоваться в препаратах, описанных в настоящем изобретении, и выбираются из списка, но, не ограничиваясь этим: полилактиды, полигликолиды, поли(лактид-со-гликолиды), поли(молочные кислоты), поли(гликолевые кислоты), поликапролактоны, полиангидриды, полиамины, полиамиды, амиды сложных полиэфиров, простые полиэфиры, сложные полиэфирэфиры, сложные полиортоэфиры, полидиоксаноны, полиацетали, поликетали, поликарбонаты, полииминокарбонаты, сложные полифосфоэфиры, полиортокарбонаты, полифосфазаны, поли(алкиленалкилаты), полиуретаны, сукцинаты, поли(малеиновые кислоты), поли(аминокислоты), поливинилпирролидон, полиэтиленгликоль, полигидроксицеллюлоза, полисахариды, поли(пропиленфумараты) и их сополимеры, терполимеры и смеси.
Предпочтительная форма композиции содержит соответствующий биологически деградируемый и фармацевтически приемлемый низкомолекулярный полимер, такой как полилактиды, полигликолиды, поли(лактид-со-гликолиды), поли(молочные кислоты), поли(гликолевые кислоты) или их смесь.
Соответствующие активные вещества, которые могут добавляться к препаратам, описанным в настоящем изобретении, выбираются из следующего далее списка, но, не ограничиваясь этим: белки, ферменты, гормоны, полинуклеотиды, нуклеопротеины, полисахариды, гликопротеины, липопротеины, пептиды, полипептиды, стероиды, анальгетики, локальные анестетики, антибиотики, химиотерапевтические агенты, иммуносупрессивные агенты, противовоспалительные агенты, антипролиферативные агенты, антимитотические агенты, ангиогенные агенты, антипсихотические агенты, агенты для центральной нервной системы (CNS), антикоагулянты, фибринолитические агенты, факторы роста, антитела, антигены, глазные лекарственные средства, гормон роста человека, метионин - гормон роста человека, дес-фенилаланин - гормон роста человека, глюкагон, кальцитонин, инсулин, гепарин, интерлейкин-1, интерлейкин-2, Фактор Viol, Фактор IX, лютеинизирующий гормон, релаксин, грелин, фолликулостимулирующий гормон, атриальный натрийуретический фактор, филграстим - эпидермальные факторы роста (EGF), полученные из тромбоцитов факторы роста (PDGFs), инсулиноподобные факторы роста (IGF), факторы роста фибробластов (FGF), трансформирующие факторы роста (TGF), интерлейкины (IL), колониестимулирующие факторы (CSF, MCF, GCSF, GMCSF), интерфероны (IFN), эндотелиальные факторы роста (VEGF, EGF), эритропоэтины (EPO), ангиопоэтины (ANG), факторы роста, полученные из плаценты (PIGF), токсины, такие как токсин ботулизма, и индуцируемые гипоксией регуляторы транскрипции (HIF), и фармацевтически приемлемые соли этих соединений или их аналоги, фрагменты или производные.
Предпочтительно, активное вещество композиции может представлять собой пептид, полипептид, белок, такой как лютеинизирующий гормон (LHRH) или аналоги LHRH, тиреоид-стимулирующий гормон (TSH), фолликулостимулирующий гормон (FSH), паратиреоидный гормон (PTH), инсулин, аналоги и производные соматостатина, гормон роста, гормон высвобождения гормона роста (GHRH), пептид высвобождения гормона роста, кальцитонин и фармацевтически приемлемые соли этих соединений, их аналоги, фрагменты или производные.
В соответствии с настоящим изобретением более предпочтительные активные вещества композиции представляет собой пептид, производное пептида, полипептид или белок, такой как рекомбинантный гормон роста человека (rhGH), гормон высвобождения гормона роста (GHRH), инсулиноподобные факторы роста (IGFs), такие как IGF-1, аналог GLP-1, лютеинизирующий гормон (LHRH) или аналоги LHRH и аналоги соматостатина.
Фармацевтические соли активных веществ, пригодные для композиций в соответствии с настоящим изобретением, получают с помощью кислотно-аддитивных солей посредством органических и неорганических кислот. Примеры кислотно-аддитивных солей соединений представляют собой соли с минеральными кислотами, например, галогенводородных кислот, таких как хлористоводородная кислота, бромистоводородная и йодистоводородная кислота, соли серной кислоты, азотной кислоты, фосфорной кислоты и тому подобное, соли с органическими сульфоновыми кюлотами, например, с алкил- и арилсульфоновыми кислотами, такими как метансульфоновая кислота, п-толуолсульфоновая кислота, бензолсульфоновая кислота и тому подобное, а также соли с органическими карбоновыми кислотами, например, с уксусной кислотой, винной кислотой, малеиновой кислотой, лимонной кислотой, бензойной кислотой, фумаровой кислотой, щавелевой кислотой, стеариновой кислотой, салициловой кислотой, аскорбиновой кислотой или нерастворимые соли, такие как соли памовой кислоты, и тому подобное.
Когда активное вещество содержит карбоксильную группу, она также образует фармацевтически приемлемые соли с основаниями. Примеры таких солей представляют собой соли щелочных металлов, например, соли натрия и калия, соли аммония, соли с органическими основаниями, например, с амином, таким как диизопропиламин, бензиламин, дибензиламин, триэтаноламин, триэтиламин, N,N-дибензилэтилендиамин, N-метилморфолин, пиридин, пиперазин, N-этилпиперидин, N-метил-D-глюкамин и прокаин, или с аминокислотами, такими как аргинин и лизин.
Предпочтительная соль пептида представляет собой соль, образованную с органической кислотой.
В предпочтительной форме, настоящее изобретение относится к модифицированным полимерам или сополимерам с очень низкой молекулярной массой, которые содержат единицы молочной кислоты и/или гликолевой кислоты, имеющим молекулярную массу от 500 до 5000 Дальтон.
Предпочтительная молекулярная масса полимеров с очень низкой молекулярной массой составляет от 700 до 3000 Дальтон, особенно предпочтительные молекулярные массы находятся в пределах между 800 и 2000.
В соответствии с настоящим изобретением, полимер может иметь отношение мономеров молочной кислоты/гликолевой кислоты или лактида/гликолида от 100/0 до 50/50, предпочтительно, от 100/0 до 80/20.
Предпочтительно полимеры с очень низкой молекулярной массой описаны в настоящем изобретении и представляют собой полимеры с конечными нереакционноспособными группами для улучшения их стабильности по отношению к деградирующим агентам, и также для увеличения их физической текучести, для облегчения, таким образом, процессов получения и инъекции. Примеры пригодных для использования конечных групп представляют собой алкил, замещенный алкил, гетероалкил, алкокси, арил, замещенный арил, гетероарил, аралкил, гетероаралкил или другой заместитель. Предпочтительные конечные группы представляют собой алкильные группы, включая линейные, разветвленные, циклические, незамещенные и замещенные производные.
Более предпочтительные конечные группы представляют собой алкильные линейные группы.
Настоящее изобретение, кроме того, охватывает полутвердую фармацевтическую композицию/имплант депо, содержащую, по меньшей мере, биологически деградируемый полимер с очень низкой молекулярной массой, с конечными нереакционноспособными группами с C5-C18 алкильным остатком вместо его окончания карбоновой кислоты и активное вещество, указанная фармацевтическая композиция является пригодной для формирования депо/импланта при температуре в пределах между 15 и 50°C, который непрерывно высвобождает активное вещество в течение, по меньшей мере, одной недели, когда фармацевтическая композиция вводится в водную физиологическую окружающую среду.
В соответствии с настоящим изобретением предпочтительные конечные группы полимера представляют собой алкильные группы, среди них могут находиться, в частности, алкильные группы, содержащие от 5 до 18 атомов углерода, и более предпочтительно, алкильные группы, содержащие от 5 до 12 атомов углерода.
В дополнение к этому предпочтительная температура для парентерального применения полутвердой фармацевтической композиции/импланта депо, как здесь описано, составляет от 25 до 40°C. В соответствии с настоящим изобретением, как здесь описано, количество активного вещества, инкорпорируемого в полимерный низкомолекулярный препарат, зависит от желаемого профиля высвобождения по отношению к концентрации активного вещества, необходимого для оказания биологического воздействия, и продолжительности времени, в течение которого должно высвобождаться вещество.
Кроме того, настоящее изобретение относится к способу получения бинарной полутвердой фармацевтической композиции/импланта депо для парентерального применения, содержащей биологически деградируемый полимер с очень низкой молекулярной массой, смешанный, по меньшей мере, с одним активным веществом.
Первый способ в соответствии с настоящим изобретением для получения смеси полутвердой композиции/импланта депо может осуществляться следующим образом:
a) Введение активного вещества и полимера в два различных шприца и вставка поршней.
b) Соединение двух заполненных шприцов с 3-конусным коннектором из нержавеющей стали.
c) Удаление воздуха из шприца, заполненного активным веществом, в вакууме.
d) Смешивание двух компонентов с помощью способа замешивания из двух шприцов, необязательно, при контролируемой температуре в пределах между 5°C и 60°C, предпочтительно, при комнатной температуре.
В соответствии с настоящим изобретением фармацевтические препараты приготавливают соответствующим образом:
- посредством смешивания соответствующего количества модифицированного полимера с очень низкой молекулярной массой и одного активного вещества или смеси активных веществ, включая их физиологически безопасную соль, отличающегося тем, что смесь может быть получена посредством шприцов.
- активное вещество необязательно выбирают как высушенный порошок, но оно может также представлять собой жидкость. Высушенный порошок или порошкообразная форма является предпочтительной.
- после этого заполненные шприцы закрывают с помощью поршней.
- после этого заполненные шприцы соединяют посредством коннектора.
- удаление воздуха из устройства с помощью соответствующего способа, известного специалисту в данной области, например, с помощью вакуума, сонификации или микроволн. Способ предпочтительно осуществляют в вакууме, когда это требуется.
- после этого смешивание двух компонентов посредством смешивания их через коннектор.
- необязательное ожижение смеси посредством нагрева шприцов известным способом.
Настоящее изобретение, кроме того, относится ко второму способу получения полутвердой композиции/импланта депо, как здесь описано.
Второй способ по настоящему изобретению, пригодный для получения полутвердой композиции/импланта депо, включает в себя следующие стадии:
a) Смешивание полимера в блендере.
b) Добавление активного вещества.
c) Cмешивание компонентов при комнатной температуре, необязательно, при контролируемой температуре в пределах между 5°C и 60°C.
d) Последняя стадия способа может осуществляться в вакууме для удаления пузырьков воздуха.
В соответствии с настоящим изобретением фармацевтические препараты приготавливают соответствующим образом:
- инициация смешивания компонентов посредством конкретного устройства, отличающегося тем, что репрезентативное устройство, используемое для смешивания в иллюстративном способе, включает в себя, например, устройство в форме цилиндров, с контролируемой скоростью перемещения.
- активное вещество необязательно выбирают как высушенный порошок, но оно также может представлять собой жидкость. Высушенный порошок или порошкообразная форма является предпочтительной.
- смешивание компонентов фармацевтического препарата может осуществляться при контролируемой температура, смешивание предпочтительно осуществляют при комнатной температуре.
Настоящее изобретение, кроме того, относится к третьему способу для соответствующего получения полутвердой композиции/импланта депо, как здесь описывается.
Третий способ, пригодный для получения полутвердой композиции/импланта депо, может осуществляться следующим образом:
a) Растворение полимера с очень низкой молекулярной массой в малом объеме метиленхлорида.
b) Добавление активного вещества к полимерному раствору.
c) Добавление соответствующего объема гептана во время перемешивания раствора/суспензии полимера и активного вещества.
d) Последняя стадия способа может осуществляться при соответствующей температуре и в вакууме для удаления растворителей из объема.
В соответствии с настоящим изобретением, фармацевтические препараты приготавливают следующим образом:
- растворяют модифицированный полимер с очень низкой молекулярной массой в соответствующем растворителе или поддерживают полимер растворенным в растворителе, используемом для синтеза и/или очистки.
- добавляют к полимерному раствору соответствующее количество одного активного вещества или смеси активных веществ, включая их физиологически безопасную соль.
- активное вещество необязательно выбирают как высушенный порошок, но оно также представляет собой жидкость. Высушенный порошок или порошкообразная форма является предпочтительной.
- затем добавляют к смеси плохой растворитель для полимера с очень низкой молекулярной массой, вызывают его преципитацию и частичное инкорпорирование активного вещества.
- смешивание компонентов фармацевтического препарата может осуществляться при контролируемой температуре, смешивание предпочтительно осуществляют при комнатной температуре.
- удаляют растворители из объема непосредственно во время или после преципитации с помощью соответствующего способа, известного специалистам в данной области, например нагрева в вакууме.
В дополнение к этому, способы приготовления могут полностью контролироваться относительно температуры, давления, периодов нагрева и охлаждения с помощью оборудования, известного специалисту в данной области.
Активное вещество может включаться в количестве от 0,001 до 70% (масс/масс), предпочтительно, в количестве от 0,1 до 30% (масс/масс), более предпочтительно, в количестве от 2 до 30% (масс/масс).
Активное вещество может суспендироваться, диспергироваться или растворяться внутри полутвердой композиции.
Еще в одном варианте осуществления настоящего изобретения, активное вещество может предварительно обрабатываться с помощью лиофилизации, сушки, измельчения, гранулирования, экструзии, микроинкапсулирования, комплексообразования или любого другого соответствующего способа, известного специалисту в данной области, перед приготовлением композиции.
В дополнение к этому, любой наполнитель, который не используется для уменьшения вязкости или улучшения композиции для инъекций и который не действует на полимер, может необязательно использоваться. Например, могут добавляться любые классические наполнители, используемые в композициях для инъекций, подобные стабилизаторам и поверхностно-активным веществам.
Любая технология, известная специалисту в данной области, такая как радиоактивная стерилизация, автоклавная стерилизация или стерильное фильтрование, может использоваться для полутвердой композиции/имплантов депо, как описано в настоящем изобретении для получения стерильного препарата.
Также охватываемое настоящим изобретением получение полутвердой композиции может реализовываться при асептических условиях.
Получение полутвердой композиции может реализовываться экстемпорально, то есть перед инъекцией посредством способа замешивания с помощью двух предварительно заполненных шприцов.
В качестве дополнительного варианта осуществления полутвердая композиция по настоящему изобретению может кондиционироваться в предварительно заполненных шприцах как готовая для использования.
В качестве конкретного варианта осуществления настоящего изобретения полутвердая композиция может предназначаться для лечения и/или предотвращения хронических расстройств или заболеваний.
В конкретном варианте осуществления настоящего изобретения, препарат с задержкой высвобождения и с контролируемым высвобождением предназначается для инъекций.
Следующие далее примеры служат в качестве иллюстрации настоящего изобретения без его ограничения.
Пример 1
Определение средневзвешенной молекулярной массы полимеров с помощью гель-проникающей хроматографии
Измерения осуществляют посредством гель-проникающей хроматографии с помощью колонки Styragel HR1 (Waters) при 40°C, с тетрагидрофураном (качество для ВЭЖХ) в качестве элюента с потоком 0,2 мл/мин различные полистирольные стандарты, имеющие диапазон молекулярных масс в пределах между 382 и 4,920 г/моль (Polymer Laboratories), используют для калибровки. Растворы полимеров приготавливают при 0,4 мг/мл в тетрагидрофуране. Анализы осуществляют с помощью хроматографической системы Waters Alliance 2695, снабженной испарительным детектором рассеяния света (PL-ELS 1000, Polymer Laboratories).
Результаты приводятся в Таблице 1.
Пример 2
Определение температуры стеклования полимеров с помощью дифференциальной сканирующей калориметрии
Температуры стеклования измеряют с использованием дифференциального сканирующего калориметра (DSC7, Perkin Elmer Instruments), снабженного контроллером термического анализа (TAC7/DX, Perkin Elmer Instruments) и охлаждающим оборудованием (Intracooler 2, Perkin Elmer Instruments). Температуру и энтальпию инструмента калибруют с помощью индия и н-октана, используемых в качестве стандартов. 5-10 мг полимера вводят в алюминиевый поддон и после этого закрывают крышкой с отверстиями. Пустой поддон используют в качестве этанола во всех случаях и газообразный азот используют в качестве продувочного газа.
Образцы подвергают воздействию программы охлаждения-нагрева от температуры окружающей среды до -70°C, выдерживают в течение 10 мин при -70°C и затем нагревают до 20°C со скоростью 5°C/мин. Программу охлаждения-нагрева повторяют. Температуру стеклования берут как начало перехода, измеряемое при первой и второй стадиях нагрева.
Результаты приводятся в Таблице 1.
Пример 3
Получение препарата BIM23190C/VLMW (загрузка N174055)
0,6 г аналога соматостатина BIM23190C (ацетатная соль) вводят в шприц и 6 г полутвердого PLA (загрузка FB341, смотри Таблицу 1) вводят во второй шприц. Два шприца соединяют с 3-конусным коннектором из нержавеющей стали. Воздух удаляют из активного вещества, прикладывая вакуум к первому шприцу. Смешивание BIM23190C с полимером наполнителем выполняют посредством процесса замешивания возвратно-поступательного типа между двумя шприцами. Систему подогревают до 50-60°C для облегчения процесса смешивания.
Смесь (объемный продукт) собирают в одном шприце, затем разделяют в 0,3 мл одноразовых шприцах, соединенных с иголками 19x0,8 мм, и упаковывают в индивидуальные алюминиевые герметичные пакеты. Стерилизацию упакованного продукта осуществляют с помощью гамма-облучения при 25 kGy в сухом льду. Готовый продукт хранят при -22°C.
Содержание и чистоту активного вещества контролируют с помощью жидкостной хроматографии с обращенной фазой. Препараты растворяют в системе растворителей, в ацетонитриле и растворе уксусной кислоты 0,1% (20/80 объем/объем). Измерения осуществляют с помощью колонки Symmetry C18 (Waters) при 40°C, с использованием градиента элюирования с помощью ацетонитрила/раствора трифторуксусной кислоты (0,1%). Анализы осуществляют с помощью насосов ВЭЖХ (Waters 515), снабженных модулем контроля насоса Waters, автосэмплером Waters 717Plus, печкой Spark Mistral и перестраиваемым детектором поглощения Waters 486, на 280 нм.
Содержание BIM23190 в готовом продукте согласно определению составляет примерно 7,7% (масс/масс) и площадь пика BIM23190 составляет 97,1% от общей площади.
Результаты приводятся в Таблице 2.
(%)
(%)
Пример 4
Получение препарата BIM51077C/VLMW (загрузка N182054)
7,0 г полутвердого PLA (загрузка MG02.013, смотри Таблицу 1) помещают между двумя цилиндрами блендера (AR400, Erweka). В то время как полимер медленно перемешивают с помощью системы, 0,70 мг аналога GLP-1 BIM51077C (ацетатная соль) медленно вводят через воронку. Гомогенную композицию получают при комнатной температуре после 2 часов медленного перемешивания с помощью этой системы. После сбора смеси (объемного продукта) в 10 мл пластиковый шприц объем помещают в вакуум при комнатной температуре в течение 30 минут для удаления пузырьков воздуха, введенных во время процесса смешивания.
Затем объемный продукт разделяют в 0,3-мл одноразовые шприцы, соединенные с иглами 19 x 1,2 мм, и упаковывают в индивидуальные алюминиевые герметичные пакеты. Стерилизацию упакованного продукта осуществляют с помощью гамма-облучения при 25 kGy в сухом льду. Готовый продукт хранят при -22°C.
Содержание и чистоту активного вещества контролируют с помощью жидкостной хроматографии с обращенной фазой. Препараты растворяют в системе растворителей, в ацетонитриле и растворе уксусной кислоты 0,1 M (20/80 объем/объем). Измерения осуществляют с помощью колонки Symmetry C18 (Waters) при 50°C, с использованием градиента элюирования с помощью триэтиламинфосфатного буфера (pH 2,3)/ацетонитрила. Анализы осуществляют с помощью хроматографической системы Waters Alliance 2695, снабженной детектором поглощения с двумя длинами волн Waters 2487, на 220 нм.
Содержание BIM51077 в готовом продукте согласно определению составляет примерно 8,9% (масс/масс) и площадь пика BIM51077 составляет 95,1% от общей площади.
Другую 10% полутвердую композицию BIM51077 (загрузка N174088) также получают, как описано выше, вместе с VLMW PLA (загрузка FB341).
Результаты приводятся в Таблице 2.
Пример 5
Получение препарата BIM21003C/VLMW (загрузка N182045)
3,8 г полутвердого PLGA 80/20 (загрузка FB342, смотри Таблицу 1) помещают между двумя цилиндрами блендера (AR400, Erweka). В то время как полимер медленно перемешивают с помощью системы, 0,42 мг трипторелина BIM21003C (ацетатная соль) медленно вводят через воронку. Гомогенную композицию получают при комнатной температуре после 2 часов медленного перемешивания с помощью этой системы. Смесь (объемный продукт) собирают в 10-мл пластиковый шприц, затем разделяют в 0,3-мл одноразовые шприцы, соединенные с иглами 19 x 1,2 мм, и упаковывают в индивидуальные алюминиевые герметичные пакеты. Стерилизацию упакованного продукта осуществляют с помощью гамма-облучения при 25 kGy в сухом льду. Готовый продукт хранят при -22°C.
Содержание и чистоту активного вещества контролируют с помощью жидкостной хроматографии с обращенной фазой. Препараты растворяют в системе растворителей, в ацетонитриле и растворе уксусной кислоты 0,1% (20/80 объем/объем). Измерения осуществляют с помощью колонки Symmetry C18 (Waters) при 40°C, с использованием градиента элюирования с помощью ацетонитрила/раствора трифторуксусной кислоты (0,1%). Анализы осуществляют с помощью насосов для ВЭЖХ (Waters 515), снабженных модулем контроля насоса Waters, автосэмплерером Waters 717Plus, печкой Spark Mistral и перестраиваемым детектором поглощения Waters 486, на 280 нм.
Содержание BIM21003 в готовом продукте согласно определению составляет примерно 9,1% (масс/масс) и площадь пика BIM21003 составляет 98,8% от общей площади.
Результаты приводятся в Таблице 2.
Пример 6
Получение препарата BIM21003C/ LMW (загрузка N193075)
0,55 г трипторелина BIM21003C (ацетатная соль) вводят в шприц и 5,0 г полутвердого PLA (загрузка MG03.035, смотри Таблица 1) вводят во второй шприц. После соединения шприцов перемешивание BIM21003C с полимерным наполнителем осуществляют посредством возвратно-поступательного процесса замешивания с помощью двух шприцов примерно при 55°C.
Смесь (объемный продукт) собирают в один шприц, затем разделяют в 0,3-мл одноразовые шприцы, соединенные с иглами 19x1,2 мм, и упаковывают в индивидуальные алюминиевые герметичные пакеты. Стерилизацию упакованного продукта осуществляют с помощью гамма-облучения при 25 kGy в сухом льду. Готовый продукт хранят при -22°C.
Содержание и чистоту активного вещества контролируют с помощью жидкостной хроматографии с обращенной фазой, как описано в Примере 5.
Содержание BIM21003 в готовом продукте согласно определению составляет примерно 10,7% (масс/масс) и площадь пика BIM21003 составляет 98,7% от общей площади.
Результаты приводятся в Таблице 2.
Пример 7
Получение препарата BIM21003C/VLMW
0,19 г трипторелина BIM21003C (ацетатная соль) вводят в шприц и 7,8 г полутвердого PLA (загрузка MG04.110, смотри Таблицу 1) вводят во второй шприц. После соединения шприцов смешивание B1M21003C с полимерным наполнителем осуществляют посредством возвратно-поступательного процесса замешивания с помощью двух шприцов при комнатной температуре.
Смесь (объемный продукт) собирают в один шприц, затем разделяют в 0,3-мл одноразовые шприцы, соединенные с иглами 19x1,2 мм, и упаковывают в индивидуальные алюминиевые герметичные пакеты. Стерилизацию упакованного продукта осуществляют с помощью гамма-облучения при 25 kGy в сухом льду. Готовый продукт хранят при -22°C.
Содержание и чистоту активного вещества контролируют с помощью жидкостной хроматографии с обращенной фазой, как описано в Примере 5.
Содержание BIM21003 в готовом продукте согласно определению составляет примерно 2,0% (масс/масс) и площадь пика BIM21003 составляет 98,7% от общей площади.
Результаты приводятся в Таблице 2.
Пример 8
Оценка свойств инъецируемости с помощью опосредованной оценки вязкости
Относительную вязкость при инъекции препаратов определяют посредством динамометра (L1000R, Lloyd Instruments), снабженного силовой ячейкой (NLC 100 N, Lloyd Instruments). Готовые композиции, упакованные в 0,3-мл одноразовые шприцы, соединенные с иглами 19 x 1,2 мм, выдерживают перед измерением 2 часа при комнатной температуре.
Предварительно заполненные шприцы подвергают нажатию на поршень при 100 мм/мин, в то время как регистрируется сила инъекции. Максимальное значение силы регистрируется как максимальная сила инъекции шприца (SIFmax) в ньютонах.
Результаты приводятся в Таблице 2.
Пример 9
Анализ in-vitro высвобождения для препарата BIM51077C/VLMW (загрузка N174088)
Анализ in-vitro осуществляют с полутвердым препаратом 10% BIM51077 с использованием PLA с C5 алкильными группами на концах. Получение этой композиции, ее кондиционирование, стерилизация и аналитический контроль описываются в Примере 4.
Статический анализ высвобождения in-vitro осуществляют с помощью устройства для диализа в фосфатном буферном растворе pH 7,4 (Европейская Фармакопея) при 37°C. Примерно 140 мг готового продукта вводят в цилиндрическую мембрану для диализа (диаметр 3,5 мм, M.W.C.O 25 кДа, Spectra Por), после этого устройство для диализа помещают в 20 мл фосфатного буфера при 37°C. Инкубационные среды собирают для анализа в течение анализа и заменяют 20 мл буфера при 37°C. Затем инкубационные среды анализируют с помощью ОФ-ВЖЭХ (как описано в Примере 4) для определения высвобождаемого количества активного вещества. Каждый эксперимент осуществляют независимо по три раза.
Профиль высвобождения BIM51077 in-vitro показан на фиг. 1.
Пример 10
Анализ высвобождения in-vitro для препарата BIM21003C/VLMW (загрузка N182045)
Анализ in-vitro осуществляют для полутвердой композиции 9% BIM21003 с использованием PLGA 80/20 с C5 алкильными группами на концах. Получение этой композиции, ее кондиционирование, стерилизация и аналитический контроль описаны в Примере 5.
Статический анализ высвобождения in-vitro осуществляют с помощью устройства для диализа в фосфатном буферном растворе, pH 7,4 (Европейская Фармакопея), при 37°C. Примерно 130 мг готового продукта вводят в цилиндрическую мембрану для диализа (диаметр 3,5 мм, M.W.C.O 25 kD, Spectra Por) и устройство для диализа помещают в 20 мл буфера при 37°C. Инкубационные среды собирают для анализа в течение анализа и заменяют 20 мл буфера при 37°C. Затем инкубационные среды анализируют с помощью спектрофотометрии УФ-видимого света на 280 нм для определения высвобождаемого количества активного вещества. Каждый эксперимент осуществляют независимо по три раза.
Профиль высвобождения in-vitro BIM21003 показан на фиг. 2.
Пример 11
Препарат BIM21003C/VLMW, полученный с помощью преципитации
Полимер VLMW, используемый в этой композиции, представляет собой модифицированный PLC C5 алкильными конечными группами и со средневзвешенной молекулярной массой Mw примерно 1,140 Дальтон. Примерно 180 мг трипторелина BIM21003C (ацетатная соль) добавляют к 1,0 г полимера VLMW, растворенного в 0,5 мл метиленхлорида. Пока суспензию BIM21003C перемешивают, 7,5 мл гептана добавляют для преципитации полимера. После этого суспензию фильтруют и преципитированную смесь сушат в вакууме при 50°C.
Содержание и чистоту активного вещества контролируют с помощью жидкостной хроматографии с обращенной фазой, как описано в Примере 5. Содержание BIM21003 в объемном продукте согласно определению составляет примерно 7,2% (масс/масс) и площадь пика BIM21003 составляет 98,7% от общей площади.
Статический анализ высвобождения in-vitro осуществляют по два раза, как описано в Примере 10.
Пример 12
Анализ высвобождения in-vivo на собаках Бигль для полимера VLMW без конечных нереакционноспособных групп
Анализ in-vivo осуществляют для полутвердой композиции 10% BIM21003 с использованием PLGA 50/50 с группами карбоновой кислоты на концах (загрузка J001/3000013, смотри Таблицу 1). Получение препарата осуществляют с помощью возвратно-поступательного процесса замешивания с помощью 2 шприцов примерно при 50°C, как описано в Примере 6. Смесь (объемный продукт) кондиционируют в 0,3-мл одноразовых шприцах, соединенных с иглами 20×1,4 мм, затем упаковывают в индивидуальные алюминиевые герметичные пакеты и стерилизуют с помощью гамма-облучения при 25 kGy в сухом льду.
Содержание и чистоту активного вещества контролируют с помощью жидкостной хроматографии с обращенной фазой, как описано в Примере 5. Содержание BIM21003 в готовом продукте согласно определению составляет примерно 11,2% (масс/масс), и площадь пика BIM21003 составляет 95,9% от общей площади.
Шести самцам собаки породы Бигль осуществляют внутримышечную инъекцию с помощью одноразовых шприцов, предварительно нагретых при 50°C в течение 3 мин, для улучшения текучести и облегчения инъекции. Шприцы взвешивают до и после введения, и доза BIM21003, вводимая посредством инъекции согласно вычислениям составляет примерно 3,6 мг на животное.
Четырехмиллилитровые образцы крови получают из цефалических вен через заданные интервалы времени после введения. Образцы крови помещают в пробирки, содержащие антикоагулянт (15% водный раствор EDTAK3) и консервант (Trasylol®). После центрифугирования плазму удаляют и образцы хранят при температуре ниже -20°C до анализа RIA. Полученные уровни BIM21003 в плазме показаны на фиг. 3.
Через 3 дня после введения уровни BIM-21003 в плазме ниже предела количественного определения. Таким образом, наблюдают относительно быстрое поглощение лекарственного средства. Эти результаты могут быть связаны с быстрой деградацией полимера, как наблюдается в экспериментах in-vitro, осуществляемых при 37°C в водной физиологической среде.
Пример 13
Анализ высвобождения in-vivo для крыс Sprague-Dawley, для нескольких препаратов пептидов
Несколько анализов in-vivo осуществляют для полутвердых композиций, содержащих следующие пептиды: BIM23190C, BIM51077C или BIM21003C (ацетатные соли). Получение этих композиций, их кондиционировнаие, стерилизация, аналитический контроль и анализы in-vitro описаны в предыдущих примерах. Их характеристики приводятся в Таблице 2.
Двенадцати самцам крыс Sprague-Dawley делают подкожно инъекцию с помощью одноразовых шприцов. Шприцы взвешивают до и после введения для проверки общей введенной дозы полутвердой композиции. Образцы крови 1,5 мл получают через ретроорбитальный синус посредством микропипетки Пастера, у анестезированных изофлураном животных (по 4 отбора пробы для каждой крысы) через заданные интервалы времени после введения. Образцы крови помещают в пробирках, содержащих антикоагулянт (15% водный раствор EDTAK3) и консервант (Trasylol®). После центрифугирования плазму удаляют и образцы хранят ниже -20°C до анализа RIA или SPE-ВЭЖХ-MS/MS (способ анализа зависит от терапевтического агента).
Уровни в плазме, полученные с помощью BIM23190, высвобождаемого из препарата N174055, показаны на фиг. 4. Задержку высвобождения, по меньшей мере, в течение 21 дня наблюдают после введения дозы 3,3 мг.
Уровни в плазме, полученные для BIM51077, высвобождаемого из препарата N182054, показаны на фиг. 5. Задержку высвобождения, по меньшей мере, до 28 дней наблюдают в случае введения 4,6 мг BIM51077.
Уровни в плазме, полученные для BIM21003, высвобождаемого из препаратов N193075 и D009099, показаны на фиг. 6. В случае препарата N193075 содержание BIM21003 составляет примерно 10,7% (масс/масс) в препарате, и высвобождение дозы 3,1 мг наблюдают в течение 14 дней после введения. В случае препарата D009099 содержание BIM21003 составляет примерно 2,0% (масс/масс) в препарате, и высвобождение дозы 2,8 мг наблюдают в течение 21 дней после введения.
Пример 14
Анализ высвобождения in-vivo для крыс Sprague-Dawley для препаратов гормона роста
Несколько полутвердых композиций приготавливают с rhGH (рекомбинантный гормон роста человека). Используемый гормон роста содержит бикарбонат натрия, сахарозу и полисорбат 20 для его стабилизации. Используют три загрузки PLA с очень низкой молекулярной массой, для различных вязкостей: высокой, средней и низкой. Характеристики полимеров приводятся в Таблице 1.
Полутвердые полимеры предварительно стерилизуют с помощью гамма-облучения при 25 kGy в сухом льду. При асептических условиях соответствующее количество rhGH (полученного сушкой вымораживанием) и полутвердого PLA вводят в два различных шприца. После соединения шприцов гомогенную смесь получают с помощью возвратно-поступательного процесса замешивания с помощью двух шприцов. Смесь (объемный продукт) собирают в одном шприце, а затем разделяют в 0,3-мл одноразовых шприцах, соединенных с иглами. Готовый продукт хранят при 5°C.
Композиция этих препаратов, их кондиционирование и вводимая доза приводятся в Таблице 3.
(высокая вязкость)
(средняя вязкость)
(низкая вязкость)
(средняя вязкость)
25 (Hypox)
Шести самцам крыс Sprague-Dawley делают инъекцию подкожно с помощью одноразовых шприцов. Шприцы взвешивают до и после введения для проверки общего количества введенной полутвердой композиции. Образцы крови по 0,4 мл получают через ретроорбитальный синус посредством микропипеток Пастера, для анестезированных изофлураном животных через заданные интервалы времени в течение 7 дней после введения. Образцы крови помещают в пробирки и центрифугируют. Затем образцы сыворотки декантируют и быстро помещают в морозилку при -80°C до их анализа посредством метода ELISA.
Концентрации в сыворотке, полученные для гормона роста, показаны на фиг. 7.
Задержку высвобождения наблюдают в течение, по меньшей мере, 7 дней, при этом концентрация rhGH в сыворотке остаются почти постоянными от 2 до 7 дня после введения.
Пример 15
Анализ высвобождения in-vivo для атимических крыс, для препарата гормона роста
Для оценки профиля высвобождения гормона роста в течение 7 дней после введения препарата VLMW осуществляют анализ in-vivo для атимических крыс, у которых конгенитально отсутствует вилочковая железа.
Другую композицию с гормоном роста приготавливают, как описано в Примере 14 (загрузка N203069), с PLA средней вязкости. Его композиция, кондиционирование и вводимая доза приводятся в Таблице 3.
Десяти самкам атимических крыс делают инъекцию подкожно с помощью одноразовых шприцов. Шприцы взвешивают до и после введения для проверки общего количества вводимой полутвердой композиции (смотри Таблицу 3).
Полученные концентрации в сыворотке, для гормона роста, показаны на фиг. 8.
Задержанное высвобождение наблюдают в течение 21 дня после введения.
Пример 16
Анализ высвобождения in-vivo для крыс Hypox для препарата гормона роста
Для оценки эффективности гормона роста, высвобождаемого из препаратов VLMW, композицию N203069 (смотри Таблицу 3 и Пример 15) также вводят крысам Sprague-Dawley после гипофизоэктомии (Hypox).
Пяти самкам крыс Hypox делают инъекцию подкожно с помощью одноразовых шприцов. Шприцы взвешивают до и после введения для проверки общего количества введенной полутвердой композиции (смотри Таблицу 3).
Увеличение массы тела показано на фиг. 9 и концентрации IGF-1 показаны на фиг. 10. Результаты сравнивают со значениями, полученными после введения плацебо "контроль (-)", и ежедневного введения водного раствора rhGH (2 мг/кг/день) "контроль (+)".
В течение 8 дней после введения профиль массы тела и уровень IGF-1, полученный для гормона роста, высвобождаемого из препарата VLMW, близки к значениям контроля (+).
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, ИМЕЮЩИЕ ВЫБРАННУЮ ПРОДОЛЖИТЕЛЬНОСТЬ ВЫСВОБОЖДЕНИЯ | 2017 |
|
RU2756514C1 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ С ПОВЫШЕННОЙ СТАБИЛЬНОСТЬЮ | 2007 |
|
RU2427383C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ С УЛУЧШЕННОЙ СТАБИЛЬНОСТЬЮ | 2015 |
|
RU2728786C2 |
| ЖИДКИЕ ДЕПО-ПРЕПАРАТЫ | 2005 |
|
RU2390331C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2009 |
|
RU2504360C2 |
| Фармацевтическая композиция с пролонгированным действием гонадотропинов для проведения индукции суперовуляции у самок млекопитающих | 2016 |
|
RU2633079C2 |
| ДЕПО-ФОРМА АНАЛОГА СОМАТОСТАТИНА И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2005 |
|
RU2411031C2 |
| КОМПОЗИЦИИ ПРОЛОНГИРОВАННОГО ДЕЙСТВИЯ С КОНТРОЛИРУЕМЫМ ВЫСВОБОЖДЕНИЕМ | 2003 |
|
RU2355385C2 |
| КОМПОЗИЦИЯ ИНСУЛИНА ДЛИТЕЛЬНОГО ДЕЙСТВИЯ | 2010 |
|
RU2556340C2 |
| ДЕПО-ПРЕПАРАТ, СОДЕРЖАЩИЙ СЛОЖНЫЙ ЭФИР ЛИМОННОЙ КИСЛОТЫ | 2016 |
|
RU2749952C2 |



Группа изобретений относится к медицине, в частности к полутвердой композиции с высвобождением с задержкой и с контролируемым высвобождением и способам ее получения. Композиция содержит а) биологически деградируемый полимер или сополимер с конечными нереакционноспособными группами молекулярной массой от 500-5000 Дальтон и имеющий алкильный остаток вместо его окончания карбоновой кислоты, в) по меньшей мере одно активное вещество или их смесь и где композиция высвобождает активное вещество в течение периода, по меньшей мере, в одну неделю, без какого-либо вспомогательного агента для понижения вязкости или наполнителя. Группа изобретений обеспечивает образование импланта/депо, когда вводится парентеральным путем и таким образом вызывает медленное высвобождение лекарственного средства. 4 н. и 20 з.п. ф-лы, 16 пр., 3 табл., 10 ил.
1. Полутвердая фармацевтическая композиция с задержкой высвобождения и с контролируемым высвобождением, содержащая:
а) биологически деградируемый полимер или сополимер с конечными нереакционноспособными группами с очень низкой молекулярной массой или их смесь, имеющая средневзвешенную молекулярную массу от 500 до 5000 Да, и имеющий алкильный остаток вместо его окончания карбоновой кислоты,
б) по меньшей мере, одно активное вещество или их смесь,
где указанную композицию получают без вспомогательных или дополнительных разбавителей, пластификаторов, растворителей или наполнителей и,
где указанная композиция с задержкой высвобождения высвобождает активное вещество в течение периода, по меньшей мере, в одну неделю, когда полутвердую композицию помещают в водную физиологическую окружающую среду.
2. Композиция по п.1, где указанная композиция с задержкой высвобождения высвобождает активное вещество в течение периода, по меньшей мере, в один месяц, когда полутвердую композицию помещают в водную физиологическую окружающую среду.
3. Композиция по п.2, в которой алкильный остаток представляет собой алкильную группу, содержащую от 5 до 18 атомов углерода.
4. Композиция по п.2, где алкильный остаток представляет собой алкильную группу, содержащую от 5 до 12 атомов углерода.
5. Композиция по п.2, где алкильный остаток представляет собой алкильную группу, содержащую 5 атомов углерода.
6. Композиция по п.1, для которой соответствующая температура для нанесения с целью получения полутвердого препарата находится в пределах между 15°С и 50°С.
7. Композиция с задержкой высвобождения по п.6, для которой соответствующая температура для нанесения с целью получения полутвердого препарата находится в пределах между 25°С и 40°С.
8. Композиция с задержкой высвобождения по п.1, в которой молекулярная масса находится в пределах между 700 и 3000 Да, предпочтительно между 800 и 2000 Да.
9. Композиция по п.1, в которой биологически деградируемый полимер с очень низкой молекулярной массой выбирают из списка, включающего: полилактиды, полигликолиды, поли(лактид-со-гликолиды), поли(молочные кислоты), поли(гликолевые кислоты), поликапролактоны, полиангидриды, полиамины, полиамиды, амиды сложных полиэфиров, простые полиэфиры, сложные полиэфирэфиры, сложные полиортоэфиры, полидиоксаноны, полиацетали, поликетали, поликарбонаты, полииминокарбонаты, сложные полифосфоэфиры, полиортокарбонаты, полифосфазаны, поли(алкиленалкилаты), полиуретаны, сукцинаты, поли(малеиновые кислоты), поли(аминокислоты), поливинилпирролидон, полиэтиленгликоль, полигидроксицеллюлоза, полисахариды, поли(пропиленфумараты) и их сополимеры, терполимеры и смеси.
10. Композиция по п.9, в которой биологически деградируемый полимер с очень низкой молекулярной массой представляет собой: полилактиды, полигликолиды, поли(лактид-со-гликолиды), поли(молочные кислоты), поли(гликолевые кислоты) и их смеси.
11. Композиция по п.1, в которой активное вещество композиции выбирается из списка, включающего: белки, ферменты, гормоны, полинуклеотиды, нуклеопротеины, полисахариды, гликопротеины, липопротеины, пептиды, полипептиды, стероиды, анальгетики, локальные анестетики, антибиотики, химиотерапевтические агенты, иммуносупрессивные агенты, противовоспалительные агенты, антипролиферативные агенты, антимитотические агенты, ангиогенные агенты, антипсихотические агенты, агенты для центральной нервной системы (CNS), антикоагулянты, фибринолитические агенты, факторы роста, антитела, антигены, глазные лекарственные средства, гормон роста человека, метионин - гормон роста человека, дес-фенилаланин - гормон роста человека, глюкагон, кальцитонин, инсулин, гепарин, интерлейкин-1, интерлейкин-2, Фактор Viol, Фактор IX, лютеинизирующий гормон, релаксин, грелин, фолликулостимулирующий гормон, атриальный натрийуретический фактор, филграстим - эпидермальные факторы роста (EGF), полученные из тромбоцитов факторы роста (PDGFs), инсулиноподобные факторы роста (IGF), факторы роста фибробластов (FGF), трансформирующие факторы роста (TGF), интерлейкины (IL), колониестимулирующие факторы (CSF, MCF, GCSF, GMCSF), интерфероны (IFN), эндотелиальные факторы роста (VEGF, EGF), эритропоэтины (ЕРО), ангиопоэтины (ANG), факторы роста, полученные из плаценты (PIGF), токсины, такие как токсин ботулизма, и индуцируемые гипоксией регуляторы транскрипции (HIF), и фармацевтически приемлемые соли этих соединений или их аналоги, фрагменты или производные.
12. Композиция по п.11, в которой активное вещество композиции выбирается из списка, включающего: пептид, полипептид, белок, такой как лютеинизирующий гормон (LHRH) или аналоги LHRH, тиреоид-стимулирующий гормон (TSH), фолликулостимулирующий гормон (FSH), паратиреоидный гормон (РТН), инсулин, аналоги и производные соматостатина, гормон роста, гормон, высвобождающий гормон роста (GHRH), пептид, высвобождающий гормон роста, кальцитонин и фармацевтически приемлемые соли этих соединений или их аналоги, фрагменты или производные.
13. Композиция по п.12, в которой активное вещество композиции представляет собой рекомбинантный гормон роста человека (rhGH) или гормон высвобождения гормона роста (GHRH), инсулиноподобные факторы роста (IGF), такие как IGF-1, аналог GLP-1, лютеинизирующий гормон (LHRH) или аналоги LHRH и аналоги соматостатина.
14. Композиция по п.13, в которой активное вещество композиции представляет собой рекомбинантный гормон роста человека (rhGH) или гормон высвобождения гормона роста (GHRH).
15. Композиция по п.13, в которой активное вещество композиции представляет собой IGF-1 и его фармацевтически приемлемые соли.
16. Композиция по п.13, в которой активное вещество композиции представляет собой аналог GLP-1 и его фармацевтически приемлемые соли.
17. Композиция по п.13, в которой активное вещество композиции представляет собой лютеинизирующий гормон (LHRH) или аналоги LHRH и их фармацевтически приемлемые соли.
18. Композиция по п.13, в которой активное вещество композиции представляет собой аналог соматостатина и его фармацевтически приемлемые соли.
19. Композиция по п.1, в которой композиция содержит некоторое количество активного вещества, в пределах от 0,001 до 70% мас./мас., предпочтительно от 0,1 до 30% (мас./мас.), более предпочтительно от 2 до 30% мас./мас.
20. Композиция по любому из пп.4 или 5, где композиция представлена в виде импланта депо.
21. Композиция по любому из пп.1-5, для которой соответствующая температура для инъекции импланта депо находится в пределах между 15°С и 50°С.
22. Композиция по п.21, для которой соответствующая температура для инъекции импланта депо находится в пределах между 25°С и 40°С.
23. Способ получения полутвердой фармацевтической композиции по п.1, включающий в себя следующие стадии:
a) введение активного вещества и полимера в два различных шприца и вставку поршней;
b) соединение двух заполненных шприцов с 3-конусным коннектором из нержавеющей стали;
c) удаление воздуха из шприца, заполненного активным веществом, в вакууме;
d) смешивание двух компонентов посредством способа замешивания с помощью двух шприцов.
24. Способ по п.23, отличающийся тем, что его необязательно осуществляют при контролируемой температуре в пределах между 5°С и 60°С, предпочтительно при комнатной температуре.
25. Способ получения полутвердой фармацевтической композиции по п.1, включающий в себя следующие стадии:
a) смешивание полимера в блендере;
b) добавление активного вещества;
c) смешивание компонентов при комнатной температуре, необязательно, при контролируемой температуре в пределах между 5°С и 60°С.
26. Способ по п.25, отличающийся тем, что активное вещество находится в форме порошка.
27. Фармацевтическая композиция, которую получают с помощью способа по любому из пп.25 и 26.
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| МИКРОЧАСТИЦЫ, ВКЛЮЧАЮЩИЕ СОЛИ ПЕПТИДОВ С ПОЛИЭФИРАМИ, ИМЕЮЩИМИ КОНЦЕВЫЕ КАРБОКСИГРУППЫ, И СОДЕРЖАЩИЕ ИХ КОМПОЗИЦИИ | 1993 |
|
RU2152225C1 |
| ЕР 1489126 А, 22.12.2004. | |||

