Область техники
Настоящее изобретение относится к фармацевтической композиции, содержащей соединение инсулина, применению композиции, способу лечения, набору, а также к комбинированному лечению с соединением GLP-1, таким как агонист GLP-1. Фармацевтическую композицию можно вводить менее часто относительно существующих препаратов инсулина длительного действия, и она отличается высвобождением структурно интактного инсулина в течение всего периода времени между введения по существу без выброса соединения инсулина. Этот метод лечения может помочь пациентам снизить частоту инъекций, сохраняя при этом возможность оптимального контроля уровня инсулина в плазме и, соответственно, уровня глюкозы в крови.
Предпосылки создания изобретения
Лечение инсулином характеризуется тем, что высвобождение препарата инсулина необходимо поддерживать в очень ограниченном диапазоне концентраций из-за узкого «терапевтического окна», а побочные эффекты гиперинсулинемии потенциально опасны для жизни. В продаже имеется множество препаратов инсулина с различными профилями действия, удовлетворяющих определенным потребностям популяции людей с диабетом. Быстродействующие аналоги инсулина вводят непосредственно перед едой для контроля пиковых концентраций глюкозы в плазме после принятия пищи, в то время как аналоги инсулина длительного действия обычно принимаются один или два раза в день для обеспечения постоянного базального уровня инсулина.
Современные разработки также включают пероральный инсулин и ингаляционный инсулин. Однако вследствие того что инсулин представляет собой белок, при пероральном приеме он легко расщепляется в желудке и желудочно-кишечном тракте. В качестве альтернативы в течение короткого периода времени в продаже был доступен ингаляционный инсулин, вводимый в легкие с помощью ингалятора (Эксубера®, компания Pfizer прекратила его выпуск в 2007 году). Этот состав обеспечивал инсулин в течение длительного периода времени (часы), но тем не менее требовал введения пациентами базального инсулина длительного действия с помощью инъекций. Другие недостатки ингаляционного инсулина включали сложность изготовления, приводящую к непомерно дорогой системе доставки. В результате все доступные в продаже составы инсулина необходимо вводить с помощью инъекций, подкожных либо внутривенных.
Различные доступные в продаже инсулины отличаются различными профилями их концентрации в плазме. Указанные профили концентрации в плазме можно описать как профили с максимальной и минимальной концентрацией в плазме, которые зависят от состава и типа используемого инсулина. Для того чтобы было возможно прогнозировать эффект снижения уровня глюкозы в плазме в результате действия введенного инсулина, очень важно получить профиль концентрации в плазме, воспроизводимый как от пациента к пациенту, так и у одного пациента. Кроме того, при многократном введении базального инсулина, желательно чтобы отличие между максимальной и минимальной концентрацией в плазме было минимально возможным. Это приводит к более постоянной концентрации инсулина в плазме, и, поэтому, к более унифицированному эффекту снижения уровня глюкозы в течение всего интервала дозирования.
Существующая на сегодняшний день базальная инсулиновая терапия состоит из ежедневного введения или введения дважды в день базальных инсулинов длительного действия, таких как NPH-инсулин, инсулин гларгин или инсулин детемир. Хотя при разработке новых инсулиновых аналогов целью являлось снижение вариабельности инсулинотропных эффектов, эффект снижения уровня глюкозы у этих составов длительного действия все еще отличается сильной вариабельностью как у отдельных субъектов, так и между ними, что описано в работе Heise et al. (Diabetes, 2004 (53), 1614-1620). В этом исследовании инсулин детемир проявлял наименьшую фармацевтическую вариабельность с коэффициентом вариабельности (CV) 15 по сравнению с CV для NPH-инсулина и инсулина гладрина, составляющими 26 и 34, соответственно. Эта достаточно высокая вариабельность является основным препятствием к достижению оптимального контроля уровня глюкозы, так как трудно предсказать уровень доступности инсулина.
В этом же исследовании изучали фармакодинамическую вариабельность, оцениваемую по скорости инфузии глюкозы (GIR). В этом исследовании также было продемонстрировано, что инсулин детемир имел меньшую вариабельность у индивидуальных субъектов по сравнению с NPH-инсулином и инсулином гладрин в отношении фармакодинамического маркера - GIR. Кроме того, в этом исследовании было продемонстрировано, что эффект инсулина на скорость инфузии глюкозы не длился в течение всего периода дозирования 24 часов, что указывает на явную необходимость в инсулине длительного действия, который будет полноценно снижать уровень глюкозы на протяжении всего интервала времени между дозами. Чтобы преодолеть проблему того, что существующие базальные инсулины для ежедневного приема не действуют на протяжении 24 часов, некоторые пациенты разбивают свою дозировку базального инсулина на две ежедневные инъекции для лучшего контроля уровня глюкозы в течение дня.
Поэтому, очевидно необходимы новые препараты инсулина длительного действия, которые постоянно высвобождают инсулин на протяжении всего периода времени между введениями препарата.
Кроме того, даже если пациенты могут регулировать свой уровень глюкозы в крови с помощью ежедневных инъекций базального инсулина, то из-за ежедневных инъекций они сопротивляются началу инсулиновой терапии. Это является нежелательным, так как Американская Ассоциация Диабета (ADA) и Европейская Ассоциация по Исследованиям Диабета (EASD) называют инсулин препаратом первой линии после перорального метформина, как обеспечивающий наилучший результат лечения (DM Nathan et al., Diabetologica (2008) 51: 8-11).
При снижении частоты введения инсулина вероятно снижение физиологического барьера перед началом инсулиновой терапии, что дает возможность пациентам начать терапию инсулином на более ранней стадии и значительно улучшает их состояние здоровья.
Проблема при разработке составов базального инсулина длительного действия заключается в узком терапевтическом диапазоне концентраций инсулина и высоком соотношении между максимальной и минимальной концентрациями в фармакокинетическом профиле инсулина, кроме того, во всех случаях необходимо избегать выброса инсулина.
Для снижения частоты введения с сохранением высвобождения инсулина в узком диапазоне концентраций были предложены несколько подходов, но они не увеличивали продолжительность эффекта снижения уровня глюкозы более чем на пару дней, хотя отличались низким соотношением между максимальной и минимальной концентрацией инсулина в плазме.
В публикации WO 06003014 описан способный высвобождать инсулин гидрогель с возможностью снижения частоты дозирования по сравнению со стандартными ежедневными инъекциями базального инсулина. Однако высвобождение инсулина происходит со слишком большой скоростью для гарантии жесткого инсулинотропного контроля в течение периодов времени больше 2 дней. В действительности, время высвобождения инсулина составляет приблизительно 30 часов, поэтому, пролекарственное соединение необходимо вводить по меньшей мере каждые 30 часов для того чтобы соотношение между максимальной и минимальной концентрациями было ниже 2 в равновесном состоянии.
Концепция получения обратимого полимерного конъюгата инсулина исследовалась группой Shechter и др. и описана в научных статьях и патентных заявках (например, в European Journal of Pharmaceutics and Biopharmaceutics 2008(70), 19-28 и WO 2004/089280). Инсулин конъюгирован с ПЭГ-полимером 40 кДа через спейсерную молекулу 9-гидроксиметил-7-(амино-3-малеимидопропионат)-флуорен-N-гидроксисукцинимид. Гидролиз указанной спейсерной молекулы высвобождает инсулин с временем полужизни приблизительно 30 часов, то есть пролекарственное соединение необходимо вводить по меньшей мере каждые 30 часов, для того чтобы соотношение между максимальной и минимальной концентрациями было ниже 2 в равновесном состоянии.
Были проведены другие попытки снижения частоты дозирования инсулина. Группой Hinds и др. (Journal of Controlled Release, 2005 (104), 447-460) был описан способ получения инсулина для введения один раз в неделю, в результате первоначального необратимого ПЭГилирования молекулы инсулина и затем последующего заключения ПЭГилированного инсулина в микрокапсулы (в PLGA-микрочастицы). Было показано, что заключение белков в PLGA вызывает побочные реакции сложных эфиров полимера с аминогруппами пептида или белка. После воздействия на составы буферных растворов с нейтральным рН наблюдались продукты ацилирования молочной кислоты. (Nat. Biotechnol. 18 (2000) 52-57; Pharm. Res. 11 (1994) 865-868; Pharm. Res. 19 (2002) 175-181).
Конкретно в случае инсулина было продемонстрировано негативное воздействие полимерных составов (Pharm. Dev. Technol. 4 (1999) 633-642; Pharm. Dev. Technol. 5 (2000) 1-9).
В указанном выше случае инсулин претерпевал значительные структурные изменения в результате перманентной модификации высокомолекулярным полимером, так как ПЭГилирование инсулина, по-видимому, служит для защиты пептида от разрушения в составе PLGA-полимера.
К сожалению, такие высокомолекулярные инсулины могут проявлять меньшую эффективность в результате ослабления связывания с рецепторами, а также могут давать реакцию в месте инъекций, такую как липодистрофия, вследствие более длительного присутствия высоких концентраций высокомолекулярного инсулина в подкожной ткани. Кроме того, такие ПЭГилированные инсулины будут иметь меньший объем распределения, что является значительным недостатком в лечении диабета.
Кроме того, фармакокинетический профиль высвобождаемого конъюгата инсулина отличается первоначальным выбросом непосредственно после введения, за которым следует падение концентрации конъюгата инсулина в плазме с последующим увеличением концентрации конъюгата инсулина в плазме в течение следующих дней. Этот фармакокинетический профиль типичен для микрокапсулированных составов лекарственных средств и может привести к непрогнозируемой реакции на глюкозу у субъектов, получающих лечение таким составом.
Поэтому остается проблемой разработка инсулина длительного действия без нарушения фармакодинамики инсулина в результате перманентного присоединения высокомолекулярного соединения.
Ситуация дополнительно осложняется тем, что инсулин легко вступает в побочные реакции, что связано с присутствием в его молекуле трех дисульфидных связей. Например, инсулин может разбиваться на А и В-цепи в результате расщепления дисульфидных связей, или могут формироваться димеры или олигомеры вследствие обменных реакций между дисульфидными группами. Такая «перетасовка» дисульфидных связей особенно вероятна, если молекулы инсулина принудительно находятся в близком контакте случайным образом ("Stability of insulin: studies on the physical and chemical stability of insulin in pharmaceutical formulation", Jens Brange, Springer, 1994). Такая природная лабильность молекулы инсулина существенно затрудняет прогресс в разработке депо-составов длительного действия и не позволяет использовать другие полимерные составы, в которых инсулин заключен в форме, подобной аморфному осадку, который, как известно, дает различные продукты деградации, возникающие в результате интенсивных реакций обмена между дисульфидными группами.
На скорость побочных реакций дополнительно влияет концентрация инсулина - скорость повышается при высокой концентрации инсулина. Поэтому, проблемой является получение составов с высокой концентрацией инсулина длительного действия, в которых инсулин не претерпевает нежелательных побочных реакций.
Поэтому, существует явная необходимость в новых препаратах инсулина длительного действия, которые постоянно высвобождают структурно интактный инсулин в течение всего периода времени между введениями препарата и, кроме того, сохраняют низкое соотношение между максимальной и минимальной концентрацией инсулина в плазме для исключения слишком высоких или слишком низких концентраций инсулина, потенциально вредных для пациента.
Количество инсулина, требуемое пациентам с диабетом, является весьма индивидуальным, причем доза зависит от нескольких физиологических факторов, включая функцию панкреатических бета-клеток, чувствительность к инсулину, массу тела и пищевой рацион. Довольно часто пациентам требуется 40 МЕ инсулина (или больше) в день. Это равняется 280 МЕ в неделю, что соответствует 12,6 мг инсулина человека в неделю. Для минимизации дискомфорта у пациента это количество должно быть введено в маленьком объеме, например, одном миллилитре. Поэтому, задача настоящего изобретения относится к составу инсулина, в котором концентрация инсулина составляет по меньшей мере 10 мг/мл, одновременно высвобождающему структурно интактный инсулин и имеющему фармакокинетический профиль по существу без выброса инсулина. Кроме того, в результате вышеуказанного, задача настоящего изобретения заключается в возможности введения одной дозы инсулина длительного действия в виде одной инъекции состава, содержащего по меньшей мере 10 мг соединения инсулина.
В US2007/0207210 A1 описан способ получения аморфных микрочастиц высокомолекулярных белков и, в частности, антител. Согласно описанию в примере упомянут инсулин, введенный в состав в концентрации до 400 мг/мл. Однако задача изобретения относится к составу с фармакокинетическим профилем, аналогичным нативному белку, то есть не относится к пролонгированному высвобождению. Поэтому, US2007/0207210 A1 не предлагает решения снижения частоты введения инсулина.
Определения
Ниже в настоящем документе изложены некоторые определения, важные для понимания настоящего изобретения.
Предполагается, что используемый в настоящем документе термин «по существу нет выброса» или «по существу без выброса» (оба термина используются взаимозаменяемо в настоящем описании) означает, что после введения соединения инсулина, которое может представлять собой пролекарственное соединение или действующее соединение инсулина, соотношение пиковой концентрации детектируемого соединения инсулина в плазме крови в течение первых 48 часов после введения, например, подкожного или внутримышечного, к минимальной концентрации детектируемого соединения инсулина в плазме крови после пиковой концентрации в течение первых 48 часов после введения составляет меньше 2 (по существу нет детектируемого выброса), предпочтительно - меньше 1,5 (нет детектируемого выброса).
Предполагается, что используемый в настоящем документе термин «выброс» означает, что после введения соединения инсулина, которое может представлять собой пролекарственное соединение или действующее соединение инсулина, соотношение пиковой концентрации детектируемого соединения инсулина в плазме крови в течение 48 часов после введения, например, подкожного или внутримышечного, к минимальной концентрации детектируемого соединения инсулина в плазме крови после пиковой концентрации в течение 48 часов после введения составляет 2 или выше.
В отношении детекции соединения инсулина в плазме крови, такое соединение инсулина может представлять собой структурно интактную форму введенного соединения инсулина, или в случае когда введенное соединение инсулина представляет собой пролекарственное соединение, детектируемым соединением инсулина будет интактное соединение инсулина, высвобождаемое из пролекарственного соединения, например, инсулин человека, аналоги инсулина, производные инсулина и их фрагменты.
Предполагается, что используемый в настоящем документе термин «соединение GLP-1» означает любое соединение GLP-1, например GLP-1(7-37), GLP-1(7-36)NH2 и аналог GLP-1, включая агониста GLP-1. Примерами агонистов GLP-1, используемых в настоящем изобретении, являются агонисты GLP-1, эксендина-3 или эксендина-4, в том числе без ограничений:
(i) аналоги эксендина-4 и амидированные аналоги эксендина-4, в последовательностях которых один или несколько аминокислотных остатков были заменены другими аминокислотными остатками, включая N-концевые модификации,
(ii) укороченный эксендин-4 и амидированные укороченные формы,
(iii) укороченный эксендин-4 и амидированные укороченные формы, в последовательностях которых один или несколько аминокислотных остатков были заменены другими аминокислотными остатками,
(iv) GLP-1 и амидированный GLP-1,
(v) аналоги GLP-1 и амидированные аналоги GLP-1, в последовательностях которых один или несколько аминокислотных остатков были заменены другими аминокислотными остатками, включая N-концевые модификации,
(vi) укороченный GLP-1 и амидированные укороченные формы,
(vii) укороченный GLP-1 и амидированные укороченные формы, в последовательностях которых один или несколько аминокислотных остатков были заменены другими аминокислотными остатками,
(viii) известные вещества AVE-0010(ZP-10) (Sanofi-Aventis Zealand Pharma), BAY-73-7977 (Bayer), TH-0318, BIM-51077 (Ipsen, Tejin, Roche), NN-2211 (Novo Nordisk), LY315902.
Агонисты GLP-1 имитируют функции нативного GLP-1, в результате связывания рецепторов, на которые оказывает действие GLP-1, являющееся полезным в качестве инсулинотропного, а также при лечении сахарного диабета, или в результате имитации эффектов эксендина на выделение мочи, замедление эвакуации содержимого желудка, повышение чувства сытости, повышение выведения натрия с мочой и/или снижение концентрации калия в моче в результате связывания рецептора(ов), через которые эксендин осуществляет это действие.
Предполагается, что используемый в настоящем документе термин «отношение пиковой концентрации к остаточной» означает соотношение между максимальной концентрацией в плазме и минимальной концентрацией в плазме соединения инсулина, например, инсулина человека, в заданный период времени между введениями.
Предполагается, что используемый в настоящем документе термин «структурно интактное соединение инсулина» означает интактный инсулин, состоящий из двух пептидов, называемых А и В-цепями, соединенными двумя дисульфидными мостиками. Кроме того, А-цепь содержит внутримолекулярный дисульфидный мостик. Потеря внутри- или межмолекулярных дисульфидных мостиков или перестановка двух цепей, например, А-А или В-В гомодимеры, могут вызывать инактивацию инсулина. Структурную целостность измеряют, расщепляя инсулин соответствующей эндопротеазой, например, эндопротеиназой Glu-C, и анализируя полученные фрагменты с помощью масс-спектрометрии. Отсутствие сигналов, возникающих от одиночных инсулиновых цепей, указывает на интактный инсулин. Предполагается, что используемый в настоящем документе термин «пролекарственное соединение» означает соединение инсулина, которое претерпевает биотрансформацию перед проявлением своих фармакологических эффектов. Поэтому, пролекарственные соединения можно рассматривать в качестве биологически активных остатков, содержащих специализированные нетоксичные защитные группы, используемые временно для изменения или устранения нежелательных свойств исходной молекулы. Например, пролекарственным соединением может быть биогидролизуемый амид и биогидролизуемый сложный эфир, а также это понятие охватывает: а) соединения, в которых биогидролизуемая функциональная группа в таком пролекарственном соединении включена в соединение, и b) соединения, которые могут биологически окисляться или восстанавливаться по данной функциональной группе. Типичные пролекарственные соединения могут представлять собой связанное с носителем пролекарственное соединение, содержащее временную связь данного действующего вещества с временной группой носителя, которая обеспечивает улучшенные физико-химические или фармакокинетические свойства, и которая легко удаляется in vivo, обычно в результате гидролитического расщепления; или это может быть каскадное пролекарственное соединение, для которого отщепление носителя становится эффективным только после демаскировки активирующей группы.
Для усиления физико-химических или фармакокинетических свойств лекарственного соединения, например, инсулина, такое лекарственное соединение можно конъюгировать с носителем. Если лекарственное соединение транзиентно соединено с носителем и/или линкером, то такие системы обычно называют связанные с носителем пролекарственные соединения. По определению, данному IUPAC (цитата из http://www.chem.qmul.ac.uk/iupac.medchem, по состоянию на 22 июля, 2009 года), связанное с носителем пролекарственное соединение представляет собой пролекарственное соединение, которое содержит временную связь данного действующего вещества с транзиентной группой-носителем, которая обеспечивает улучшенные физико-химические или фармакокинетические свойства, и которая легко удаляется in vivo, обычно в результате гидролитического расщепления.
Используемые в таких связанных с носителями пролекарственных соединениях линкеры являются транзиентными, то есть они гидролитически деградируют (расщепляются) без привлечения ферментов при физиологических условиях (водный буфер при рН 7,4, 37°C) с временем полужизни в диапазоне, например, от одного часа до трех месяцев.
Подходящими носителями являются полимеры, и они могут быть либо конъюгированы с линкером напрямую, либо через нерасщепляемый спейсер. «Пролекарственное соединение инсулин-гидрогеля» относится к пролекарственному соединению, в котором инсулин транзиентно соединен с гидрогелевым носителем. Термины «гидрогелевое пролекарственное соединение» и «связанное с гидрогелем пролекарственное соединение» относятся к пролекарственным соединениям биологически активных агентов, транзиентно связанных с гидрогелем, и используются в качестве синонимов.
Термины «лекарственное соединение», «биологически активная молекула», «биологически активная часть молекулы», «биологически активный агент», «действующий агент» и т.п. означают любое вещество, которое может влиять на физически или биохимические свойства биологического организма, включая без ограничений, вирусы, бактерии, грибы, растения, животных и человека. В частности, используемые в настоящем изобретении биологически активные молекулы, включают любое вещество, предназначенное для диагностики, излечения, смягчения, лечения или предупреждения заболевания у человека или животных, или в противном случае - для усиления физического или душевного здоровья человека или животного.
Используемое в настоящем документе выражение «терапевтически эффективное количество» инсулина означает количество, достаточное для излечения, ослабления или частичной остановки клинических проявлений данного заболевания или его осложнений. Количество, достаточное для этого, определяют как «терапевтически эффективное количество». Эффективное количество для каждой цели будет зависеть от тяжести заболевания или поражения, а также от массы и общего состояния субъекта. Понятно, что соответствующую дозировку можно определить с использованием стандартных экспериментов путем создания матрицы значений и тестирования различных элементов в матрице, что входит в компетенцию опытного терапевта.
«Стабильный» и «стабильность» означает, что в пределах указанного времени хранения конъюгаты остаются конъюгированными и не гидролизуются в значительной степени, а также имеют приемлемый состав примесей относительно инсулина. Стабильной считается композиция, содержащая менее 5% лекарственного соединения в свободной форме.
Используемый в настоящем документе термин «биогидролизуемый сложный эфир» представляет собой сложный эфир соединения, который либо (а) не нарушает биологическую активность исходного вещества, но придает данному веществу предпочтительные свойства in vivo, такие как длительность действия, начало действия и т.п., либо b) является биологически неактивным, но легко превращается in vivo субъектом в биологически активное начало.
Используемый в настоящем документе термин «биогидролизуемый амид» представляет собой амид соединения, который либо (а) не нарушает биологическую активность исходного вещества, но придает данному веществу предпочтительные свойства in vivo, такие как длительность действия, начало действия и т.п., либо b) является биологически неактивным, но легко превращается in vivo субъектом в биологически активное начало.
Предполагается, что используемый в настоящем документе термин «гидрогель» означает трехмерную, гидрофильную или амфифильную полимерную сель, способную поглощать большое количество воды. Сети состоят из гомополимеров или сополимеров, являются нерастворимыми вследствие присутствия ковалентных химических или физических (ионных, гидрофобных взаимодействий, переплетений) сшивок. Сшивки обеспечивают структуру сети и физическую целостность. Гидрогели проявляют термодинамическую совместимость с водой, которая позволяет им набухать в водной среде. Цепи соединены в сети, образуя поры, и размер значительной части данных пор находится между 1 нм и 1000 нм.
Термин «гель» относится к несшитому, гелеподобному раствору полимера.
Предполагается, что используемый в настоящем документе термин «депо-препарат» означает систему доставки для лекарственного соединения инсулина, обычно вводимую с помощью подкожной или внутримышечной инъекции и способную к постоянному высвобождению действующего соединения в течение продолжительного периода времени.
Предполагается, что используемый в настоящем документе термин «пиковая концентрация» означает максимальную концентрацию, полученную после введения соединения инсулина.
Предполагается, что используемый термин «соединение инсулина» означает любой инсулин млекопитающих, например, инсулин человека, свиной инсулин или бычий инсулин с дисульфидными мостиками между CysA7 и CysB7 и между CysA20 и CysB19, а также с внутримолекулярным дисульфидным мостиком между CysA6 и CysA11, рекомбинантный инсулин млекопитающих, например, рекомбинантный инсулин человека, аналоги инсулина, производные инсулина и их фрагменты, типичными примерами являются rh инсулин, инсулин гларгин, инсулин детемир, инсулин глулизин, инсулин аспартат, инсулин лизпро, инсулин, конъюгированный с низкомолекулярным ПЭГ, где низкомолекулярный ПЭГ имеет молекулярную массу меньше 10 кДа. Соединение инсулина может находиться в форме пролекарственного соединения, в случае которого соединение, высвобождаемое в плазму, является активным инсулином, которое образуется после введения пролекарственного соединения.
Под «аналогом инсулина», используемым в настоящем изобретении, понимается полипептид, имеющий молекулярную структуру, которую формально можно получить из структуры природного инсулина, например, инсулина человека, в результате удаления и/или замены по меньшей мере одного аминокислотного остатка, присутствующего в природном инсулине, и/или в результате добавления по меньшей мере одного аминокислотного остатка. Добавляемые и/или используемые для замены аминокислотные остатки могут представлять собой либо кодируемые аминокислотные остатки, либо другие природные остатки, либо чисто синтетические аминокислотные остатки.
Аналогами инсулина могут быть белки, в которых природный остаток Pro в позиции 28 в В-цепи может быть изменен на один из Asp, Lys или Ile. В другом аспекте Lys в позиции B29 изменен на Pro. Также Asn в позиции A21 может быть изменен на Ala, Gln, Glu, Gly, His, Ile, Leu, Met, Ser, Thr, Trp, Tyr или Val, в частности, на Gly, Ala, Ser или Thr и, предпочтительно, на Gly. Кроме того, Asn в позиции B3 может быть изменен на Lys или Asp. Дополнительными примерами аналогов инсулина являются инсулин des(B30) человека; аналоги инсулина des(B30) человека; аналоги инсулина, в которых удален PheB1; аналоги инсулина с удлиненной с N-конца А-цепью и/или В-цепью и аналоги инсулина с удлиненной с С-конца А-цепью и/или В-цепью. Таким образом, один или два остатка Arg могут быть добавлены к позиции B1.
Под «инсулин desB30», «инсулин desB30 человека» понимается природный инсулин или его аналог, у которого отсутствует аминокислотный остаток В30. Аналогичным образом «инсулин desB29desB30» или «инсулин desB29desB30 человека» означает природный инсулин или его аналог, у которого отсутствуют аминокислотные остатки В29 и В30.
Под «В1», «А1» и т.д. понимается аминокислотный остаток в позиции 1 в В-цепи инсулина (считая с N-конца) и аминокислотный остаток в позиции 1 в А-цепи инсулина (считая с N-конца), соответственно. Также может быть указан аминокислотный остаток в указанной позиции, как например, PheB1, означающий, что аминокислотным остатком в позиции В1 является остаток фенилаланина.
Используемый в настоящем документе термин «неактивный линкер» означает линкер, который не оказывает фармакологических эффектов лекарственного соединения, полученного из биологически активного агента.
Используемый в настоящем документе термин «алкил» означает прямую или разветвленную углеродную цепь. Каждый водород углерода в алкиле может быть замещен заместителем.
Используемый в настоящем документе термин «С1-4алкил» означает алкильную цепь, имеющую 1-4 атомов углерода, например, на конце молекулы: метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил или, например, -CH2-, -CH2-CH2-, -CH(CH3)-, -CH2-CH2-CH2-, -CH(C2H5)-, -C(CH3)2-, когда две части молекулы соединены алкильной группой. Каждый водород углерода в С1-4алкиле может быть замещен заместителем.
Используемый в настоящем документе термин «С1-6алкил» означает алкильную цепь, имеющую 1-6 атомов углерода, например, на конце молекулы: С1-4алкил, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, н-гексил или, например, -CH2-, -CH2-CH2-, -CH(CH3)-, -CH2-CH2-CH2-, -CH(C2H5)-, -C(CH3)2-, когда две части молекулы соединены алкильной группой. Каждый водород углерода в С1-6алкиле может быть замещен заместителем.
Соответственно, «С1-18алкил» означает алкильную цепь, имеющую от 1 до 18 атомов углерода, а «С8-18алкил» означает алкильную цепь, имеющую от 8 до 18 атомов углерода. Соответственно, «С1-50алкил» означает алкильную цепь, имеющую от 1 до 50 атомов углерода.
Используемый в настоящем документе термин «С2-50алкенил» означает прямую или разветвленную алкенильную цепь, имеющую от 2 до 50 атомов углерода, например, на конце молекулы: -CH=CH2, -CH=CH-CH3, -CH2-CH=CH2, -CH=CH-CH2-CH3, -CH=CH-CH=CH2, или, например, -CH=CH-, когда две части молекулы соединены алкенильной группой. Каждый водород углерода в С2-50алкениле может быть замещен дополнительно указанным заместителем. Соответственно, термин «алкенил» относится к углеродной цепи по меньшей мере с одной двойной углеродной связью. Необязательно, могут присутствовать одна или несколько тройных связей.
Используемый в настоящем документе термин «С2-50алкинил» означает прямую или разветвленную алкинильную цепь, имеющую от 2 до 50 атомов углерода, например, на конце молекулы: -C≡CH, -CH2-C=CH, CH2-CH2-C=CH, CH2-C≡C-CH3, или, например, -C≡C-, когда две части молекулы соединены алкинильной группой. Каждый водород углерода в С2-50алкиниле может быть замещен дополнительно указанным заместителем. Соответственно, термин «алкинил» относится к углеродной цепи по меньшей мере с одной тройной углеродной связью. Необязательно, могут присутствовать одна или несколько двойных связей.
Используемый в настоящем документе термин «С3-7циклоалкил» или «С3-7циклоалкильное кольцо» означает циклическую алкильную цепь, имеющую от 3 до 7 атомов углерода, которая может иметь углеродные двойные связи, при этом по меньшей мере частично насыщенную, например, циклопропил, циклобутил, циклопентил, циклогексил, циклогексенил, циклогептил. Каждый водород углерода в циклоалкиле может быть замещен заместителем. Термин «С3-7циклоалкил» или «С3-7циклоалкильное кольцо» также включает соединенные мостиками бициклы, такие как норбонан или норбонен. Соответственно, «С3-5циклоалкил» означает циклический алкил, имеющий от 3 до 5 атомов углерода.
Соответственно, «С3-10циклоалкил» означает циклический алкил, имеющий от 3 до 10 атомов углерода, например, «С3-7циклоалкил», циклопропил, циклобутил, циклопентил, циклогексил, циклогексенил, циклогептил, циклооктил, циклононил, циклодецил. Термин «С3-10циклоалкил» также включает по меньшей мере частично насыщенные карбомоно- и -бициклы.
Используемый в настоящем документе термин «галоген» означает фтор, хлор, бром или йод. Обычно предпочтительно, чтобы галогеном являлся фтор или хлор.
Используемый в настоящем документе термин «4-7-членный гетероциклил» или «4-7-членный гетероцикл» означает кольцо с 4, 5, 6 или 7 атомами в кольце, которое может содержать до максимального количества двойных связей (ароматическое или неароматическое кольцо, полностью насыщенное, частично насыщенное или ненасыщенное), в котором по меньшей мере один атом в кольце (и до 4 атомов в кольце) заменен гетероатомом, выбранным из группы, состоящей из серы (включая -S(O)-, -S(O)2-), кислорода и азота (включая =N(O)-), и когда кольцо присоединено к остальной молекуле через атом углерода или азота. Примерами 4-7-членных гетероциклов являются азетидин, оксетан, тиетан, фуран, тиофен, пиррол, пирролидин, имидазол, имидазолин, пиразол, пиразолин, оксазол, оксазолин, изоксазол, изоксазолин, тиазол, тиазолин, изотиазол, изотиазолин, тиадиазол, тиадиазолин, тетрагидрофуран, тетрагидротиофен, пирролидин, имидазолидин, пиразолидин, оксазолидин, изоксазолидин, тиазолидин, изотиазолидин, тиадиазолидин, сульфолан, пиран, дигидропиран, тетрагидропиран, имидазолидин, пиридин, пиридазин, пиразин, пиримидин, пиперазин, пиперидин, морфолин, тетразол, триазол, триазолидин, тетразолидин, диазепан, азепин или гомопиперазин.
Используемый в настоящем документе термин «9-11-членный гетеробициклил» или «9-11-членный гетеробицикл» означает гетероциклическую систему из двух колец с 9-11 атомами в кольце, причем по меньшей мере один кольцевой атом входит в оба кольца, и которая может содержать до максимального количества двойных связей (ароматическое или неароматическое кольцо, полностью насыщенное, частично насыщенное или ненасыщенное), в которой по меньшей мере один атом в кольце (и до 6 атомов в кольце) замещен гетероатомом, выбранным из группы, состоящей из серы (включая -S(O)-, -S(O)2-), кислорода и азота (включая =N(O)-), и когда кольцо присоединено к остальной молекуле через атом углерода или азота. Примерами 9-11-членных гетеробициклов являются индол, индолин, бензофуран, бензотиофен, бензоксазол, бензизоксазол, бензотиазол, бензизотиазол, бензимидазол, бензимидазолин, хинолин, хиназолин, дигидрохиназолин, хинолин, дигидрохинолин, тетрагидрохинолин, декагидрохинолин, изохинолин, декагидроизохинолин, тетрагидроизохинолин, дигидроизохинолин, бензазепин, пурин или птеридин. Термин 9-11-членный гетеробицикл также включает спироструктуры из двух колец, такие как 1,4-диокса-8-азаспиро[4.5]декан или соединенные мостиком гетероциклы, такие как 8-аза-бицикло[3.2.1]октан.
Используемый в настоящем документе термин «фармацевтически приемлемый» означает одобренный надзорными органами, такими как EMEA (Европа) и/или FDA (США), и/или любыми другими национальными надзорными органами для применения у животных, предпочтительно у человека.
Используемый в настоящем документе термин «фармацевтическая композиция» или «композиция» означает одни или несколько действующих ингредиентов, и один или несколько инертных ингредиентов, а также любой продукт, который получается прямо или непрямо в результате объединения, образования комплексов или агрегации любых двух или нескольких ингредиентов, или в результате диссоциации одного или нескольких ингредиентов, или в результате других типов реакций или взаимодействий одного или нескольких ингредиентов. Соответственно, фармацевтические композиции по настоящему изобретению охватывают любую композицию, изготовленную путем смешивания соединения по настоящему изобретению и фармацевтически приемлемого вспомогательного вещества (фармацевтически приемлемого носителя).
«Свободная форма» лекарственного соединения относится к лекарственному соединению в его немодифицированной фармакологически активной форме, например, после высвобождения из полимерного конъюгата.
«Сухая композиция» означает, что композиция пролекарственного соединения инсулина с гидрогелем представлена в сухой форме в контейнере. Подходящими способами высушивания являются сушка распылением и лиофилизация (сушка при замораживании). Такая сухая композиция пролекарственного соединения инсулина с гидрогелем имеет остаточное содержание воды максимум 10%, предпочтительно, меньше 5% и, еще более предпочтительно, меньше 2% (определяемое по Karl Fischer). Предпочтительным способом сушки является лиофилизация. «Лиофилизированная композиция» означает, что композицию пролекарственного соединения инсулина в гидрогеле исходно замораживают и далее удаляют воду, понижая давление. Эта терминология не исключает дополнительные стадии сушки, которые присутствуют в процессе изготовления до заполнения композицией конечного контейнера.
«Лиофилизация» (сушка при замораживании) представляет собой процесс удаления воды, отличающийся замораживанием композиции и затем снижением давления и, необязательно, нагреванием для прямой возгонки замороженной воды в композиции из твердой фазы в газ. Обычно, пары воды собирают десублимацией.
«Восстановление» означает добавление жидкости для восстановления исходной формы композиции.
«Раствор для восстановления» относится к жидкости, используемой для восстановления сухой композиции пролекарственного соединения инсулина с гидрогелем перед введением нуждающемуся в нем пациенту.
«Контейнер» означает любой контейнер, который содержит композицию пролекарственного соединения инсулина в гидрогеле, и в котором она может храниться до восстановления.
Используемое в настоящем документе выражение «терапевтически эффективное количество» соединения инсулина означает количество, достаточное для излечения, ослабления или частичной остановки клинических проявлений данного заболевания и его осложнений. Количество, достаточное для этого, определяют как «терапевтически эффективное количество». Эффективное количество для каждой цели будет зависеть от тяжести заболевания или поражения, а также от массы и общего состояния субъекта. Понятно, что соответствующую дозировку можно определить с использованием стандартных экспериментов путем создания матрицы значений и тестирования различных элементов в матрице, что входит в компетенцию опытного терапевта или ветеринара.
Термин «буфер» или «буферный агент» относится к химическим соединениям, которые поддерживают рН в заданном диапазоне. Физиологически совместимыми буферами являются, например, натрия фосфат, сукцинат, гистидин, бикарбонат, цитрат и ацетат, сульфат, нитрат, хлорид, пируват. Также можно использовать антациды, такие как Mg(OH)2 или ZnCO3. Буферную емкость можно подводить, чтобы соответствовать условиям, наиболее чувствительным к стабильности рН.
Термин «вспомогательные вещества» относится к соединениям, вводимым совместно с терапевтическим агентом, например, буферными агентами, модификаторами изотоничности, консервантами, стабилизиторами, анти-адсорбционными агентами, агентами, защищающими от окисления, и с другими вспомогательными агентами. Однако в некоторых случаях одно вспомогательное вещество может иметь двойные или тройные функции.
«Лиопротектант» представляет собой молекулу, которая при объединении с целевым белком, в значительной степени предупреждает или снижает химическую и/или физическую нестабильность белка при высушивании в общем, и, особенно, в ходе лиофилизации и последующего хранения. Примеры лиопротектантов включают сахара, такие как сахароза или трегалоза; аминокислоты, такие как мононатрия глутамат или гистидин; метиламины, такие как бетаин; лиотропные соли, такие как сульфат магния; многоатомные спирты, такие как трехатомные или полиатомные сахарные спирты, например, глицерин, эритрит, глицерин, арабит, ксилит, сорбит и маннит; этиленгликоль; пропиленгликоль; полиэтиленгликоль; плюроники; гидроксиалкильные крахмалы, например, гидроксиэтиловый крахмал (HES) и их комбинации.
«Поверхностно-активное вещество» относится к смачивающим агентам, снижающим поверхностное натяжение жидкости.
«Модификаторы изотоничности» относится к соединениям, минимизирующим боль, которая может возникнуть в результате повреждения клеток вследствие разницы осмотического давления в месте депо-инъекции.
Термин «стабилизаторы» относится к соединениям, используемым для стабилизации полимерного пролекарственного соединения. Стабилизация достигается в результате повышения сил, стабилизирующих белок, дестабилизации денатурированного состояния или в результате прямого связывания вспомогательных веществ с белком.
«Анти-адсорбционные агенты» относится в основном к ионным или неионным поверхностно-активным веществам или другим белкам или растворимым полимерам, используемым для конкурентного покрытия внутренней поверхности или адсорбции на внутреннюю поверхность контейнера, содержащего композицию. Выбор концентрации и типа вспомогательного вещества зависят от эффекта, которого следует избегать, но обычно монослой поверхностно-активного вещества образуется на поверхности раздела фаз немного выше значения критической концентрации мицеллообразования.
«Агенты, защищающие от окисления» относится к антиоксидантам, таким как аскорбиновая кислота, эктоин, метионин, глутатион, монотиолглицерин, морин, полиэтиленимин (PEI), пропилгаллат, витамин Е, хелатирующие агенты, такие как лимонная кислота, EDTA, гексафосфат, тиогликолевая кислота.
«Противомикробные агенты» относится к химическому веществу, которое убивает или ингибирует рост микроорганизмов, такие как бактерии, грибы, дрожжи, простейшие, и/или разрушает вирусы.
«Герметизация контейнера» означает, что контейнер закрыт таким образом, что он является воздухонепроницаемым, не позволяя газообмен между внешней средой и внутренней средой контейнера, и сохраняет содержимое в стерильном состоянии.
Подразумевается, что выражение «фармацевтически приемлемый» охватывает любое вспомогательное вещество и/или добавку, которые не нарушают эффективность биологической активности действующего ингредиента и не токсичны для хозяина, которому их вводят.
Термин «реагент» относится к промежуточному соединению или исходному материалу, используемому в способе изготовления, приводящему к пролекарственному соединению по настоящему изобретению.
Термин «химическая функциональная группа» относится к карбоновой кислоте и активированным производным, аминогруппе, малеимидной группе, тиольной группе и производным, сульфоновой кислоте и производным, карбонату и производным, карбамату и производным, гидроксилу, альдегиду, кетону, гидразину, изоцианату, изотиоцианату, фосфорной кислоте и производным, фосфоновой кислоте и производным, галоацетилу, алкилгалидам, акрилоильной группе и другим альфа-бета-ненасыщенным акцепторам Михаэля, арилирующим агентам, таким как арилфториды, гидроксиламину, дисульфидам, таким как пиридилдисульфид, винилсульфон, винилкетон, диазоалканам, диазоацетильным соединениям, оксирану и азиридину.
Если химическая функциональная группа соединена с другой химической функциональной группой, полученная в результате химическая структура именуется «связью». Например, реакция аминогруппы с карбоксильной группой дает амидную связь.
«Реакционно-способные функциональные группы» являются химическими функциональными группам каркасной части молекулы, которые соединены с гипер-разветвленной частью молекулы.
«Функциональная группа» является собирательным термином, используемым для «реакционно-способной функциональной группы», «деградируемой сопряженной (образующей межмолекулярную связь) функциональной группы» или «конъюгирующей функциональной группы».
«Деградируемая сопряженная функциональная группа» представляет собой соединение, содержащее биодеградируемую связь, которая с одной стороны соединена со спейсером, связанным с каркасной частью молекулы, а с другой стороны соединена с сшивающей частью молекулы. Термины «деградируемая сопряженная функциональная группа», «биодеградируемая сопряженная функциональная группа», «сопряженная биодеградируемая функциональная группа» и «сопряженная функциональная группа» используются в качестве синонимов.
Термины «блокирующая группа» или «кэпирующая группа» используются в качестве синонимов и относятся к частям молекул, которые необратимо присоединены к реакционно-способным функциональным группам для блокирования их способности вступать в реакции, например, с химическими функциональными группами.
Термины «защищающая группа» или «защитная группа» относятся к частям молекул, обратимо присоединенным к реакционно-способным функциональным группам для блокирования их способности вступать в реакции, например, с химическими функциональными группами.
Термин «способная образовывать межмолекулярную связь функциональная группа» относится к химическим функциональным группам, которые участвуют в реакции радикальной полимеризации и являются частью сшивающего реагента или каркасного реагента.
Термин «полимеризуемая функциональная группа» относится к химическим функциональным группам, которые участвуют в реакции полимеризации сшивающего типа и являются частью сшивающего реагента или каркасного реагента.
Для сопряженных функциональных групп термин «гидролитически деградируемые» относится в контексте настоящего изобретения к соединениям, которые гидролитически деградируют без участия ферментов при физиологических условиях (водный буфер с рН 7,4, 37°C) с временем полужизни в диапазоне от одного часа до трех месяцев, и которые включают в себя, но не ограничены этими соединениями, аконитилы, ацетали, ангидриды карбоновых кислот, сложные эфиры, имины, гидразоны, амиды малеаминовой кислоты, ортоэфиры, фосфамиды, фосфоэфиры, силильные эфиры фосфора, силильные эфиры, сульфоновые эфиры, ароматические карбаматы, их комбинации и т.п. Предпочтительными биодеградируемыми соединениями являются карбоновые эфиры, карбонаты, фосфоэфиры и эфиры сульфоновой кислоты, а наиболее предпочтительными являются карбоновые эфиры или карбонаты. Очевидно, что в исследованиях in vitro в практических целях можно использовать ускоренные условия, например, рН 9, 37°C, водный буфер.
Каркасная часть молекулы может содержать спейсерную часть молекулы, которая одним концом присоединена к каркасной части молекулы, а другим концом к сшивающей части молекулы.
Термин «производные» относится к химическим функциональным группам, соответственно замещенным защищающими и/или активирующими группами или к активированным формам соответствующей химической функциональной группы, известным опытному специалисту в данной области. Например, активированные формы карбоксильной группы включают, но не ограничены эти, реакционно-способные сложные эфиры, такие как сукцинимидный эфир, бензотриазольный эфир, нитрофенильный эфир, пентафторфенильный эфир, азабензотриазольный эфир, ацилгалогениды, смешанные и симметричные ангидриды, ацилимидазол.
Термин «линкер, расщепляемый без участия ферментов» относится к линкерам, гидролитически деградируемым при физиологических условиях без ферментативной активности.
«Биологически неактивный линкер» означает линкер, который не показывает фармакологических эффектов лекарственного соединения (D-H), полученного из биологически активной молекулы.
Термины «спейсер», «спейсерная группа», «спейсерная молекула» и «спейсерная часть молекулы» используются взаимозаменяемо, и при использовании для описания молекулы, присутствующей в гидрогелевом носителе по изобретению, относятся к любой молекуле, подходящей для соединения двух частей молекулы, например, C1-50алкилу, C2-50алкенилу или C2-50алкинилу, которая необязательно прерывается одной или несколькими группами, выбранными из -NH-, -N(C1-4алкил)-, -O-, -S-, -C(O)-, -C(O)NH-, -C(O)N(C1-4алкил)-, -O-C(O)-, -S(O)-, -S(O)2-, 4-7-членного гетероцикла, фенила или нафтила.
Термины «концевой», «конец» или «дальний конец» относится к позиции функциональной группы или связи в молекуле или части молекулы, в соответствии которыми такая функциональная группа может быть химической функциональной группой, а связь может быть деградируемой или постоянной связью, которые характеризуются тем, что они расположены близко к соединению или в соединении между двумя частями молекулы либо на конце олигомерной или полимерной цепи.
Фразы «в связанной форме» или «часть молекулы» относятся к субструктурам, которые являются частью более крупной молекулы. Фразу «в связанной форме» используют для упрощения ссылки на части молекул путем упоминания или перечисления реагентов, исходных материалов или возможных исходных материалов, хорошо известных в данной области, и в соответствии с которой «в связанной форме» означает, что, например, один или несколько радикалов водорода (-Н) или одна или несколько активирующих или защитных групп, присутствующих в реагенте или исходных веществах, не присутствуют в части молекулы.
Очевидно, что все реагенты и молекулы, содержащие полимерные молекулы, относятся к макромолекулярным структурам, которые, как известно, обладают вариабельностью по своей молекулярной массе, длине цепей или степени полимеризации, или числу функциональных групп. Поэтому, структуры, показанные для каркасных реагентов, каркасных частей молекул, реагентов для сшивания и сшивок, являются только представительными примерами.
Реагент или часть молекулы могут быть линейными или разветвленными. Если реагент или часть молекулы имеют две концевых группы, они именуются линейным реагентом или частью молекулы. Если реагент или часть молекулы имеют более двух концевых групп, они считаются разветвленными или многофункциональными реагентом или частью молекулы.
Термин «полимерная цепь на основе полиэтиленгликоля» «цепь на основе ПЭГ» относится к олигомерной или полимерной молекулярной цепи.
Предпочтительно, чтобы такая полимерная цепь на основе полиэтиленгликоля была связана с ядром (точкой разветвления), и представляла собой линейную полиэтиленгликолевую цепь, у которой один конец присоединен к ядру, а другой к гиперразветвленной дендритной части. Очевидно, что цепь на основе ПЭГ может оканчиваться (или прерываться) алкильной или арильной группами, необязательно замещенными гетероатомами и химическими функциональными группами.
Если термин «полимерная цепь на основе полиэтиленгликоля» используется в применении к сшивающему реагенту, он относится к сшивке или цепи, содержащим по меньшей мере 20% (по массе) молекул этиленгликоля.
Используемое в настоящем документе единственное число и аналогичные ссылки следует истолковывать как ссылки на единственное и множественное число, если в настоящем документе прямо не указано иное, или если этому прямо не противоречит контекст.
Описание
Настоящее изобретение относится к препарату инсулина длительного действия для обеспечения базального инсулина. Базальные инсулины представляют собой составы инсулина или аналогов инсулина длительного действия, разработанные для имитации секреции базального инсулина бета-клетками поджелудочной железы. Оптимально, чтобы таким образом осуществлялся непрерывный контроль содержания глюкозы в крови в течение всего интервала дозирования.
Авторами изобретения было открыто, что соединение инсулина может непрерывно высвобождаться из инъекционного депо-препарата, (например, для подкожного введения) в структурно интактной форме в интервале между введениями без какого-либо эффекта выброса. Структурную целостность высвобождаемого соединения инсулина обеспечивает высоко гидратированная полимерная матрица, минимизирующая межмолекулярные контакты молекул инсулина. Кроме того, в результате отсутствия выброса инсулина снижается риск вредных побочных эффектов у пациента.
Настоящее изобретение снижает риск гипогликемии после введения инсулина благодаря отсутствию «взрывоподобного» высвобождения инсулина, снижает риск гипергликемии в конце периода дозирования, снижает частоту инъекций и обеспечивает предсказуемый уровень инсулина в плазме крови пациента.
Дополнительная задача настоящего изобретения относится к новому базальному инсулину, требующему менее частого введения относительно существующих схем ежедневного введения базального инсулина и обеспечивающему высокий уровень безопасности в результате высвобождения структурно интактного инсулина в течение всего интервала времени межу инъекциями и, обычно, имеющему соотношение пиковой концентрации к остаточной концентрации меньше 2.
Дополнительные преимущества будут очевидны из настоящего описания.
В первом аспекте настоящее изобретение относится к фармацевтической композиции, содержащей соединение инсулина в концентрации, достаточной для поддержания терапевтически эффективного уровня соединения инсулина в плазме крови в течение по меньшей мере 3 дней, обычно, по меньшей мере 80 часов, например, в течение недели, характеризуются тем, что она имеет фармакокинетический профиль in vivo по существу без выброса соединения инсулина.
Такая концентрация будет варьировать от субъекта к субъекту и зависит от терапевтического окна у отдельного субъекта, но для продолжительности терапевтического эффекта в течение по меньшей мере 3 дней, например, недели (то есть, примерно 7 дней), концентрация обычно составляет по меньшей мере около 10 мг/мл, например, более 10 мг/мл.
Соответственно, в дополнительном аспекте настоящее изобретение относится к фармацевтической композиции, содержащей соединение инсулина в концентрации по меньшей мере 10 мг/мл, характеризуются тем, что она имеет фармакокинетический профиль in vivo по существу без выброса соединения инсулина.
В дополнительном аспекте настоящее изобретение относится к фармацевтической композиции, содержащей соединение инсулина с концентрацией по меньшей мере 11 мг/мл, для введения в разовой дозе по меньшей мере 10 мг соединения инсулина.
Используемая в настоящем описании разовая доза соединения инсулина дана в мг, а концентрация соединения инсулина в фармацевтической композиции дана в мг/мл. Если соединение инсулина является пролекарственным соединением, концентрация основана на количественном высвобождении свободного инсулина из пролекарственного соединения. С помощью известных в данной области способов аликвоту композиции помещают в условия для высвобождения инсулина (водный буфер, рН 7,4, 37°C, или ускоренные условия при повышенном рН) до прекращения заметного увеличения концентрации инсулина и определяют общее количество высвобожденного инсулина. Очевидно, что в случае растворимых носителей, количественное высвобождение является синонимом количественного гидролиза.
В одном варианте осуществления настоящего изобретения концентрация соединения инсулина составляет по меньшей мере 11 мг/мл, например от 11 мг/мл до 35 мг/мл, более предпочтительно, от 15 мг/мл до 25 мг/мл, еще более предпочтительно, около 20 мг/мл, и, еще более предпочтительно, концентрация составляет около 24 мг/мл.
Объем, который используют для введения эффективной дозы, например, с помощью шприца, субъекту, например, человеку, предпочтительно составляет меньше 1,5 мл, обычно - 1,0 мл или меньше.
В дополнительном варианте осуществления изобретения разовая доза соединения инсулина составляет по меньшей мере 5 мг, например, от 5 мг до 100 мг, более предпочтительно, от 5 мг до 50 мг и, еще более предпочтительно, от 5 мг до 25 мг.
В дополнительном варианте осуществления изобретения соотношение пиковой концентрации соединения инсулина в плазме крови в течение первых 48 часов после введения, например, с помощью подкожной или внутримышечной инъекции, к минимальной концентрации соединения инсулина в плазме крови после пиковой концентрации в течение первых 48 часов после введения, составляет меньше 2, обычно меньше 1,5.
Вышеуказанные варианты осуществления изобретения, а также варианты осуществления, описанные ниже, следует рассматривать относительно любого из аспектов, описанных в настоящем документе, а также относительно любого из вариантов осуществления, описанных в настоящем документе, если не указано, что вариант осуществления относится к определенному аспекту или аспектам настоящего изобретения.
Обычно фармацевтическая композиция представляет собой регулируемую систему доставки, которая включает в себя соединение инсулина и характеризуется тем, что при доставке соединения инсулина в плазму крови млекопитающих по существу не происходит выброса инсулина.
В еще одном дополнительном варианте осуществления изобретения фармакокинетический профиль измеряют в плазме крови млекопитающих, например в плазме крови человека. Концентрацию инсулина в плазме можно измерить с помощью доступных в продаже наборов для ELISA, сравнивая результаты измерений с калибровочной кривой, полученной с использованием инсулиновых стандартов. Для статистической достоверности эксперимент проводят с достаточным числом биологических и технических повторов и вычисляют средние и медианные значения для учета биологической и технической вариабельности.
В дополнительном варианте осуществления изобретения композиция характеризуется тем, что имеет соотношение пиковой концентрации к остаточной концентрации меньше 2, обычно - меньше 1,75, предпочтительно, меньше 1,5 или, еще более предпочтительно - меньше 1,25.
В еще одном дополнительном варианте осуществления изобретения композиция отличается постоянным высвобождением соединения структурно интактного инсулина в течение всего интервала времени между введениями.
«Постоянное высвобождение» относится к непрерывному высвобождению инсулина.
В дополнительном варианте осуществления изобретения полный интервал времени между введениями составляет по меньшей мере примерно 80 часов, например, примерно 110 часов, обычно, по меньшей мере - неделю, например, 1-2 недели или даже больше.
В еще одном дополнительном варианте осуществления изобретения соединение инсулина является пролекарственным соединением.
В дополнительном варианте осуществления изобретения соединение инсулина является связанным с носителем пролекарственным соединением.
В еще одном дополнительном варианте осуществления изобретения соединение инсулина является каскадным пролекарственным соединением.
Пролекарственное соединение можно вводить в виде жидкости, например, раствора или геля, или оно может содержаться в депо-препарате, или даже может быть встроено в депо-препарат, так что депо-препарат действует в качестве пролекарственного соединения.
В дополнительном варианте осуществления изобретения соединение инсулина полностью содержится в депо-препарате.
Термин «полностью содержится» относится к депо-препарату, в котором менее 10% лекарственного соединения, то есть инсулина, присутствует в водной фракции после добавления 1 мл воды к 1 мл депо-препарата, смешивания и отделения депо-препарата от воды.
В еще одном дополнительном варианте осуществления изобретения депо-препаратом является полимерный гель, например, гидрогель.
В дополнительном варианте осуществления изобретения депо-препаратом является высоко гидратированный полимерный матрикс.
Соединение инсулина может содержаться в депо-препарате в различных вариантах, например, соединение инсулина может быть нековалентно связано с депо-препаратом или ковалентно связано с депо-препаратом, причем депо-препарат (без ограничений) можно выбрать из полимерного геля, гидрогеля или высоко гидратированного полимерного матрикса. Неограничивающими примерами подходящих полимеров являются полимеры, способные образовывать квази-бесконечные трехмерные высоко гидратированные молекулярные сети. Такими гидрогелями являются химически или физически перекрестно-сшитые функционализированные или нефункционализированные полиалкоксильные полимеры, например, полипропиленгликоль или полиэтиленгликоль, декстран, хитозан, гиалуроновая кислота и производные, альгинат, ксилан, маннан, каррагинан, агароза, целлюлоза, крахмал, гидроксиэтилкрахмал (HES) и другие углеводные полимеры, поливиниловые спирты, полиоксазолины, полиангидриды, полиортоэфиры, поликарбонаты, полиуретаны, полиакриловые кислоты, полиакриламиды, например, полигидроксипропилметакриламид (HMPA), полиакрилаты, полиметакрилаты, например, полигидроксиэтилметакрилат, полиорганофосфазены, полисилоксаны, поливинилпирролидон, полицианоакрилаты, полиэфиры, например, полимолочная кислота или полигликолевые кислоты, полииминокарбонаты, полиаминокислоты, например, полиглутаминовая кислота или полилизин, коллаген, желатин, сополимеры, привитые сополимеры, перекрестно сшитые полимеры, гидрогели и блок-сополимеры из вышеперечисленных полимеров. Эти полимеры могут служить в качестве каркасных частей молекул или сшивающих частей молекул, а также возможна комбинация различных полимеров в виде сополимеров при условии высокой степени гидратации молекулярной сети. Кроме олигомерных или полимерных сшивающих частей молекул перечисленных выше типов полимеров, можно использовать низкомолекулярные сшивки, особенно, при использовании высокомолекулярных каркасных частей молекул для образования гидрогелевых носителей пролекарственных соединений.
Одним путем минимизации контактов между молекулами инсулина является гомогенное распределение молекул инсулина в высоко гидратированном полимерном матриксе. Гомогенное распределение можно получить с помощью ковалентного связывания инсулина с полимером, а в результате использования линкеров, расщепляющихся в водной среде при нейтральном рН, гарантируется медленное высвобождение структурно интактного инсулина.
Предпочтительным является связанное с полимером пролекарственное соединение инсулина, которое по существу не имеет биологической активности, причем указанное свойство обуславливает то, что вся инсулинотропная активность относится к высвобождаемому инсулину. Поэтому, путем моделирования свойств высвобождения пролекарственного соединения можно достичь высокой степени контроля уровня инсулина в плазме.
Инсулин может быть присоединен через все значимые функциональные группы в молекуле, и такие предпочтительные функциональные группы в природных аминокислотах обычно выбраны из гуанидиновой, имидазольной, индольной, карбоксильной, карбоксамидной, первичной и вторичной гидроксильной, фенольной, и первичной аминогрупп. Например, инсулин человека имеет следующие важные функциональные группы: карбоксильную, карбоксиамидную, первичную и вторичную гидроксильную, фенольную, имидазольную и первичную аминогруппу.
В дополнительном варианте осуществления изобретения соединение инсулина связано с носителем пролекарственного соединения либо ковалентно с любым из остатков лизина, либо с N-концом любой из А- или В-цепи соединения инсулина.
В конкретном варианте осуществления изобретения пролекарственное соединение или его фармацевтически приемлемая соль содержат конъюгат инсулина с линкером D-L, в котором
D представляет собой молекулу инсулина; а
-L является биологически неактивной линкерной молекулой -L1, представленной формулой (I),
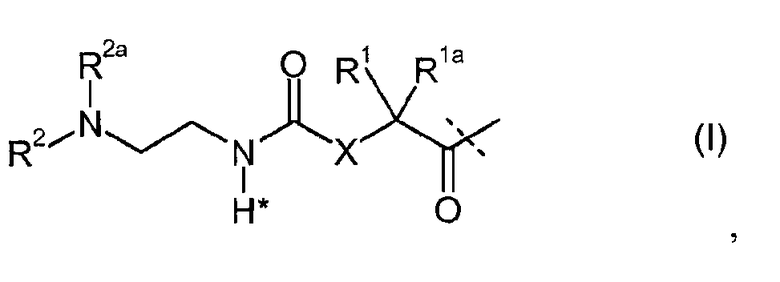
в которой пунктирная линия указывает присоединение одной из аминогрупп инсулина с образованием амидной связи;
X является C(R3R3a); или N(R3);
R1a, R3a независимо выбраны из группы, состоящей из H, NH(R2b), N(R2b)C(O)R4 и C1-4алкила;
R1, R2, R2a, R2b, R3, R4 независимо выбраны из группы, состоящей из H и C1-4алкила,
в которой L1 замещена одним заместителем L2-Z и необязательно дополнительно замещена, при условии, что атом водорода, обозначенный звездочкой в формуле (I) не замещен заместителем, и в которой
L2 является одиночной химической связью или спейсером; а
Z является гидрогелем.
В случае, когда соединения по формуле (I) содержат одну или несколько кислотных или основных групп, изобретение также содержит их соответствующие фармацевтически или токсикологически приемлемые соли, в частности, их фармацевтически используемые соли. Поэтому, соединения по формуле (I), которые содержат кислотные группы, можно использовать по изобретению, например, в качестве солей щелочных металлов, солей щелочноземельных металлов или солей аммония. Более конкретные примеры таких солей включают соли натрия, соли калия, соли кальция, соли магния или соли аммония или органических аминов, таких как, например, этиламин, этаноламин, триэтаноламин или аминокислоты. Соединения по формуле (I), которые содержат одну или несколько основных групп, то есть групп, которые могут протонироваться, могут присутствовать, и их можно использовать по изобретению в форме их солей присоединения с неорганическими или органическими кислотами. Примеры подходящих кислот включают хлорид водорода, бромид водорода, фосфорную кислоту, серную кислоту, азотную кислоту, метансульфоновую кислоту, п-толуолсульфоновую кислоту, нафталиндисульфоновую кислоту, щавелевую кислоту, уксусную кислоту, винную кислоту, молочную кислоту, салициловую кислоту, бензойную кислоту, муравьиную кислоту, пропионовую кислоту, пивалиновую кислоту, диэтилуксусную кислоту, малоновую кислоту, янтарную кислоту, пимелиновую кислоту, фумаровую кислоту, малеиновую кислоту, яблочную кислоту, сульфаминовую кислоту, фенилпропионовую кислоту, глюконовую кислоту, аскорбиновую кислоту, изоникотиновую кислоту, лимонную кислоту, адипиновую кислоту и другие кислоты, известные опытным специалистам в данной области. Если соединения по формуле (I) одновременно содержат кислые и основные группы в молекуле, то изобретение также включает (в дополнение к указанным формам солей) внутренние соли или бетаины (цвиттер-ионы). Соответствующие соли по формуле (I) можно получить обычными способами, известными опытному специалисту в данной области, например, путем контакта с органическими или неорганическими кислотой или основанием в растворителе или диспергирующем веществе или в результате анионообмена или катионообмена с другими солями. Настоящее изобретение также включает все соли соединений по формуле (I), которые вследствие низкой физиологической совместимости, напрямую не подходят для использования в фармацевтических препаратах, но которые можно использовать, например, в качестве промежуточных соединений для химических реакций или для получения фармацевтически приемлемых солей.
Предпочтительно, в формуле (I) R2 замещен L2-Z.
Предпочтительно, в формуле (I) R1 замещен L2-Z.
Предпочтительно, в формуле (I) X является N(R3).
Предпочтительно, в формуле (I) X является C(R3R3a), а R3a является N(R2b)C(O)R4.
Предпочтительно, Х является C(R3R3a), R3a является N(R2b)-L2-Z.
Предпочтительные пролекарственные соединения по настоящему изобретению содержат конъюгат инсулина с линкером D-L, в котором L1 из формулы (I) представлена формулами (Ia), (Ib), (Ic) или (Id):
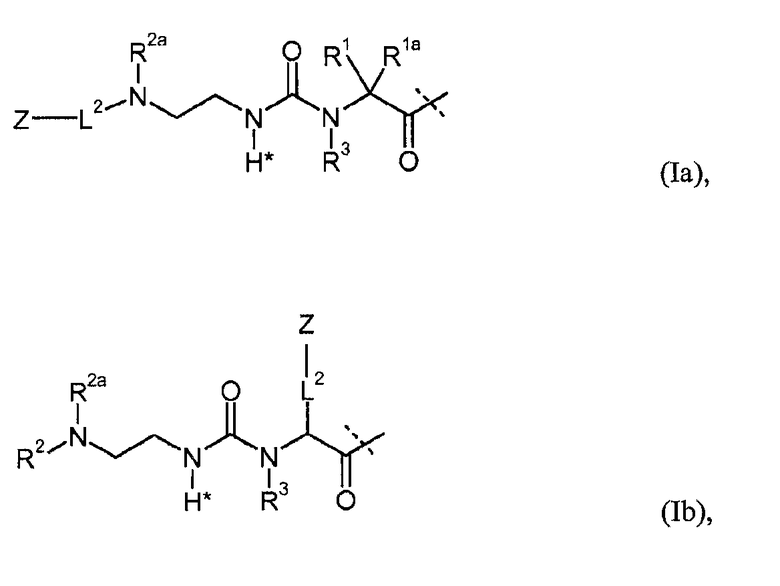
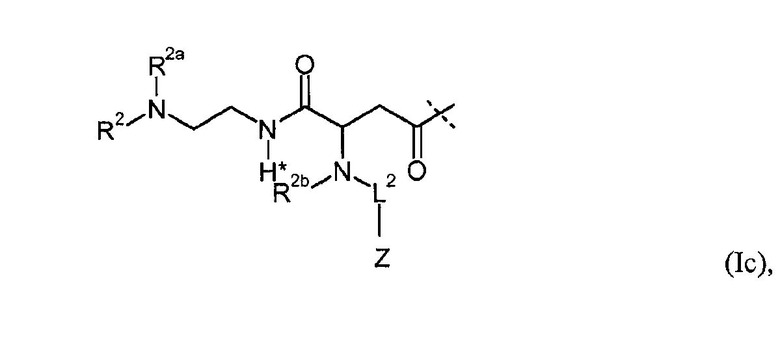
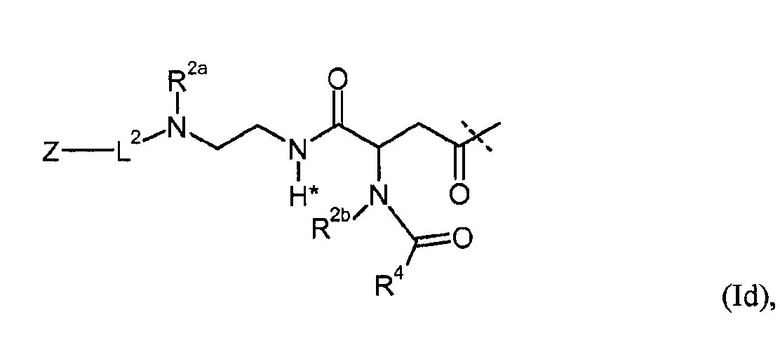
в которых R1, R1a, R2, R2a, R2b, R3, R4, L2, Z имеют значения, указанные в настоящем документе, и в которых L1 необязательно дополнительно замещена при условии, что атом водорода, обозначенный звездочкой в формулах (Ia)-(Id), не замещен заместителем.
Предпочтительно, L1 дополнительно не замещена (кроме обязательного заместителя L2-Z).
Как показано, например, в формулах (Ia)-(Id) один водород замещен группой L2-Z.
В общем, L2 может быть присоединена к L1 по любому положению, кроме замещения водорода, обозначенного звездочкой в формуле (I). Предпочтительно, чтобы один из атомов водорода из R1, R1a, R2, R2a, R2b, R3, R4 прямо или в качестве водорода из C1-4алкила или дополнительных групп был замещен L2-Z.
Кроме того, L1 необязательно может быть замещена. В общем, можно использовать любой заместитель, при условии что не затрагивается принцип расщепления. Однако предпочтительно, чтобы L1 не была дополнительно замещена.
Предпочтительно, чтобы один или несколько дополнительных заместителей были независимо выбраны из группы, состоящей из галогена; CN; COOR9; OR9; C(O)R9; C(O)N(R9R9a); S(O)2N(R9R9a); S(O)N(R9R9a); S(O)2R9; S(O)R9; N(R9)S(O)2N(R9aR9b); SR9; N(R9R9a); NO2; OC(O)R9; N(R9)C(O)R9a; N(R9)S(O)2R9a; N(R9)S(O)R9a; N(R9)C(O)OR9a; N(R9)C(O)N(R9aR9b); OC(O)N(R9R9a); T; C1-50алкила; C2-50алкенила или C2-50алкинила, где T; C1-50алкил, C2-50алкенил и C2-50алкинил необязательно замещены одним или несколькими R10, которые могут быть одинаковыми или различными, и где C1-50алкил, C2-50алкенил и C2-50алкинил необязательно прерваны одной или несколькими группами, выбранными из группы, состоящей из T, -C(O)O-; -O-; -C(O)-; -C(O)N(R11)-; -S(O)2N(R11)-; -S(O)N(R11)-; -S(O)2-; -S(O)-; -N(R11S(O)2N(R11a)-; -S-; -N(R11)-; -OC(O)R11; -N(R11)C(О)-; -N(R11)S(О)2-; -N(R11)S(О)-; -N(R11)C(O)O-; -N(R11)C(O)N(R11a)- и -OC(O)N(R11R11a);
R9, R9a, R9b независимо выбраны из группы, состоящей из H; T; C1-50алкила; C2-50алкенила или C2-50алкинила, где T; C1-50алкил, C2-50алкенил и C2-50алкинил необязательно замещены одним или несколькими R10, которые могут быть одинаковыми или различными, и где C1-50алкил, C2-50алкенил и C2-50алкинил необязательно прерваны одной или несколькими группами, выбранными из группы, состоящей из T, -C(O)O-; -O-; -C(O)-; -C(O)N(R11)-; -S(O)2N(R11)-; -S(O)N(R11)-; -S(O)2-; -S(O)-; -N(R11S(O)2N(R11a)-; -S-; -N(R11)-; -OC(O)R11; -N(R11)C(О)-; -N(R11)S(О)2-; -N(R11)S(О)-; -N(R11)C(O)O-; -N(R11)C(O)N(R11a)- и -OC(O)N(R11R11a);
Т выбрана из группы, состоящей из фенила; нафтила; инденила; инданила; тетралинила; С3-10циклоалкила; 4-7-членного гетероциклила; или 9-10-членного гетеробициклила, причем Т необязательно замещена одним или несколькими из R10, которые могут быть одинаковыми или различными;
R10 является галогеном; CN; оксогруппой (=О); COOR12; OR12; C(O)R12; C(O)N(R12R12a); S(O)2N(R12R12a); S(O)N(R12R12a); S(O)2R12; S(O)R12; N(R12)S(O)2N(R12aR12b); SR12; N(R12R12a); NO2; OC(O)R12; N(R12)C(O)R12a; N(R12)S(O)2R12a; N(R12)S(O)R12a; N(R12)C(O)OR12a; N(R12)C(O)N(R12aR12b); OC(O)N(R12R12a); или С1-6алкилом, причем С1-6алкил необязательно замещен одним или несколькими галогенами (одинаковыми или различными);
R11, Rl1a, R12, R12a, R12b независимо выбраны из группы, состоящей из H или С1-6алкила, причем С1-6алкил необязательно замещен одним или несколькими галогенами (одинаковыми или различными).
Термин «прерван» означает, что между двумя атомами углерода или на конце углеродной цепи между атомами углерода и водорода вставлена группа.
L2 представляет собой одиночную химическую связь или спейсер. В том случае, когда L2 является спейсером, то он предпочтительно, описан как один или несколько необязательных заместителей, описанных выше, при условии что L2 замещен Z.
Соответственно, когда L2 не является одиночной химической связью, то L2-Z является COOR9; OR9; C(O)R9; C(O)N(R9R9a); S(O)2N(R9R9a); S(O)N(R9R9a); S(O)2R9; S(O)R9; N(R9)S(O)2N(R9aR9b); SR9; N(R9R9a); OC(O)R9; N(R9)C(O)R9a; N(R9)S(O)2R9a; N(R9)S(O)R9a; N(R9)C(O)OR9a; N(R9)C(O)N(R9aR9b); OC(O)N(R9R9a); T; C1-50алкилом; C2-50алкенилом или C2-50алкинилом, причем Т, C1-50алкил, C2-50алкенил и C2-50алкинил необязательно замещены одним или несколькими R10, которые могут быть одинаковыми или различными, и где C1-50алкил, C2-50алкенил и C2-50алкинил необязательно прерваны одной или несколькими группами, выбранными из группы, состоящей из -T-, -C(O)O-; -O-; -C(O)-; -C(O)N(R11)-; -S(O)2N(R11)-; -S(O)N(R11)-; -S(O)2-; -S(O)-; -N(R11S(O)2N(R11a)-; -S-; -N(R11)-; -OC(O)R11; -N(R11)C(О)-; -N(R11)S(О)2-; -N(R11)S(О)-; -N(R11)C(O)O-; -N(R11)C(O)N(R11a)- и -OC(O)N(R11R11a);
R9, R9a, R9b независимо выбраны из группы, состоящей из H; Z; T; и C1-50алкила; C2-50алкенила или C2-50алкинила, причем Т, C1-50алкил, C2-50алкенил и C2-50алкинил необязательно замещены одним или несколькими R10, которые могут быть одинаковыми или различными, и где C1-50алкил, C2-50алкенил и C2-50алкинил необязательно прерваны одной или несколькими группами, выбранными из группы, состоящей из T, -C(O)O-; -O-; -C(O)-; -C(O)N(R11)-; -S(O)2N(R11)-; -S(O)N(R11)-; -S(O)2-; -S(O)-; -N(R11S(O)2N(R11a)-; -S-; -N(R11)-; -OC(O)R11; -N(R11)C(О)-; -N(R11)S(О)2-; -N(R11)S(О)-; -N(R11)C(O)O-; -N(R11)C(O)N(R11a)- и -OC(O)N(R11R11a);
Т выбрана из группы, состоящей из фенила; нафтила; инденила; инданила; тетралинила; С3-10циклоалкила; 4-7-членного гетероциклила; или 9-10-членного гетеробициклила, причем Т необязательно замещена одним или несколькими из R10, которые могут быть одинаковыми или различными;
R10 является Z; галогеном; CN; оксогруппой (=О); COOR12; OR12; C(O)R12; C(O)N(R12R12a); S(O)2N(R12R12a); S(O)N(R12R12a); S(O)2R12; S(O)R12; N(R12)S(O)2N(R12aR12b); SR12; N(R12R12a); NO2; OC(O)R12; N(R12)C(O)R12a; N(R12)S(O)2R12a; N(R12)S(O)R12a; N(R12)C(O)OR12a; N(R12)C(O)N(R12aR12b); OC(O)N(R12R12a); или С1-6алкилом, причем С1-6алкил необязательно замещен одним или несколькими галогенами (одинаковыми или различными);
R11, Rl1a, R12, R12a, R12b независимо выбраны из группы, состоящей из H; Z; или С1-6алкила, причем С1-6алкил необязательно замещен одним или несколькими галогенами (одинаковыми или различными);
при условии что один из R9, R9a, R9b, R10, R11, R11a, R12, R12a, R12b является Z.
Более предпочтительно, чтобы L2 представляла собой C1-20алкильную цепь, которая необязательно прерывалась одной или несколькими группами, независимо выбранными из -O- и C(O)N(R3aa); необязательно была замещена одной или несколькими группами, независимо выбранными из OH; и C(O)N(R3aaR3aaa), в которых R3aa, R3aaa независимо выбраны из группы, состоящей из H и C1-4алкила.
Предпочтительно, чтобы молекулярная масса L2 находился в диапазоне от 14 г/моль до 750 г/моль.
Предпочтительно, чтобы L2 была присоединена к Z через концевую группы, выбранную из
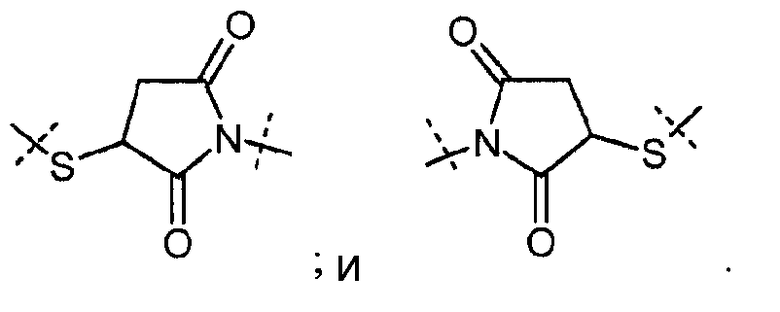
В случае, когда L2 имеет такую концевую группу, более предпочтительно, чтобы молекулярная масса L2 находился в диапазоне от 14 г/моль до 500 г/моль, рассчитанный без данной концевой группы.
Предпочтительно, чтобы ковалентная связь, образуемая между линкером и гидрогелем Z, была постоянной.
Предпочтительно, чтобы гидрогель Z представлял собой биодеградируемый водонерастворимый гидрогель на основе полиэтиленгликоля (ПЭГ). В настоящем документе термин «на основе ПЭГ» означает, что массовое соотношение цепей ПЭГ в гидрогеле составляет по меньшей мере 10% по массе, предпочтительно, 25%, от общей массы гидрогеля. Остальную часть геля могут составлять другие спейсеры и/или олигомеры или полимеры, такие как олиго- или полилизинны.
Кроме того, термин «водонерастворимый» относится к набухающей трехмерно-сшитой молекулярной сети, образующей гидрогель. Гидрогель при ресуспендировании в большом избытке воды или водного буфера с физиологической осмоляльностью может включить в себя значительное количество воды, например, 10-кратная масса, и, поэтому, является набухающим, но после удаления избытка воды сохраняет физическую стабильность и форму геля. Гель может принимать любую геометрическую форму, и очевидно, что такой отдельный гидрогель следует рассматривать, как единую молекулу, состоящую из компонентов, в которой каждый компонент соединен с другим посредством химических связей.
По изобретению гидрогель может состоять из каркасных частей, соединенных друг с другом гидролитически расщепляемыми связями.
Предпочтительно, чтобы каркасная часть молекулы имела молекулярную массу в диапазоне от 1 кДа до 20 кДа, более предпочтительно, от 1 кДа до 15 кДа, и еще более предпочтительно, от 1 кДа до 10 кДа. Предпочтительно, чтобы основой каркасной части молекулы был ПЭГ, и каркасная часть содержала одну или несколько цепей ПЭГ.
В гидрогеле, несущем конъюгаты лекарственного соединения и линкера по изобретению, каркасная часть молекулы характеризуется тем, что она несет ряд функциональных групп, включающих в себя биодеградируемые сопряженные функциональные группы, и соединенные с гидрогелем конъюгаты лекарственного соединения и линкера, и, необязательно - кэпирующие группы. Это означает, что каркасная часть молекулы характеризуется рядом соединенных с гидрогелем конъюгатов лекарственного соединения и линкера; функциональными группами, содержащими биодеградируемые сопряженные функциональные группы; и, необязательно, кэпирующими группами. Предпочтительно, чтобы сумма сопряженных биодеградируемых функциональных групп, конъюгатов лекарственного соединения с линкером и кэпирующих групп составляла 16-128, предпочтительно, 20-100, более предпочтительно, 24-80 и, наиболее предпочтительно, 30-60.
Предпочтительно, чтобы сумма сопряженных функциональных групп, конъюгатов лекарственного соединения с линкером и кэпирующих групп в каркасной молекуле была поровну разделена между числом полимерных цепей на основе ПЭГ, выступающих из точки разветвления. Например, если число сопряженных функциональных групп и конъюгатов лекарственного соединения с линкером и кэпирующих групп составляет 32, то на каждую из четырех полимерных цепей на основе ПЭГ, выступающих из точки разветвления, приходится по 8 групп, предпочтительно посредством дендритных частей молекулы, присоединенных на конце каждой полимерной цепи на основе ПЭГ. В качестве альтернативы, на каждую из восьми полимерных цепей на основе ПЭГ, выступающих из ядра, приходится по 4 группы, или по 2 группы приходится на каждую из шестнадцати полимерных цепей на основе ПЭГ. Если число полимерных цепей на основе ПЭГ, выступающих из точки разветвления, не позволяет равного распределения, то предпочтительно, чтобы отклонение от среднего от суммы сопряженных функциональных групп, конъюгатов лекарственного соединения с линкером и кэпирующих групп на полимерную цепь на основе ПЭГ сохранялось минимальным.
В таких соединенных с носителем пролекарственных соединениях по изобретению желательно, чтобы высвобождение почти всего лекарственного соединения (>90%) происходило до высвобождения каркасных молекул в значительном количестве (<10%). Этого можно достичь, модифицируя время полужизни соединенного с носителем пролекарственного соединения относительно кинетики деградации гидрогеля по изобретению.
Предпочтительно, каркасная часть молекулы характеризуется тем, что она имеет точку разветвления из которой выступают по меньшей мере три полимерные цепи на основе ПЭГ. Соответственно, в предпочтительном варианте каркасный реагент включает в себя точку разветвления, из которой выступают по меньшей мере три полимерные цепи на основе ПЭГ. Такие точки разветвления могут состоять из поли- или олигоспиртов в связанной форме, предпочтительно, пентаэритрита, трипентаэритрита, гексаглицерина, сахарозы, сорбита, фруктозы, маннита, глюкозы, целлюлозы, амилоз, крахмалов, гидроксиалкилкрахмалов, поливиниловых спиртов, декстранов, гиалуронатов; или точки разветвления могут состоять из поли- или олигоаминов, таких как орнитин, диаминомасляная кислота, трилизин, тетрализин, пентализин, гексализин, гептализин, октализин, нонализин, декализин, ундекализин, додекализин, тридекализин, тетрадекализин, пентадекализин или олиголизины, полиэтиленимины, поливиниламины в связанной форме.
Предпочтительно, из точки разветвления выступают от трех до шестнадцати полимерных цепей на основе ПЭГ, более предпочтительно, от четырех до восьми. Предпочтительные точки разветвления могут состоять из, пентаэритрита, орнитина, диаминомасляной кислоты, трилизина, тетрализина, пентализина, гексализина, гептализина или олиголизина, низкомолекулярного PEI, гексаглицерина, трипентаэритрита в связанной форме. Предпочтительно, чтобы из точки разветвления выступали от трех до шестнадцати полимерных цепей на основе ПЭГ, более предпочтительно, от четырех до восьми. Предпочтительно, чтобы полимерная цепь на основе ПЭГ была линейной полиэтиленгликолевой цепью, у которой один конец соединен с точкой разветвления, а другой конец присоединен к гиперразветвленной дендритной части. Очевидно, что полимерная цепь на основе ПЭГ может оканчиваться или прерываться алкильными или арильными группами, необязательно, замещенными гетероатомами и химическими функциональными группами.
Предпочтительно, чтобы полимерная цепь на основе ПЭГ представляла собой соответственно замещенное производное полиэтиленгликоля (на основе ПЭГ).
Предпочтительными структурами для соответствующих полимерных цепей на основе ПЭГ, выступающих из точки разветвления, содержащейся в каркасной части молекулы, являются многолучевые производные ПЭГ, такие как, например описанные в списке продукции JenKem Technology, USA (доступном на сайте www.jenkemusa.com 28 июля, 2009 года), 4-лучевые ПЭГ-производные (пентаэритритное ядро), 8-лучевые ПЭГ-производные (гексаглицериновое ядро) и 8-лучевые ПЭГ-производные (трипентаэритритное ядро). Наиболее предпочтительными являются 4-лучевой ПЭГ-амин (пентаэритритное ядро) и 4-лучевой ПЭГ-карбоксил (пентаэритритное ядро), 8-лучевой ПЭГ-амин (гексаглицериновое ядро), 8-лучевой ПЭГ-карбоксил (гексаглицериновое ядро), 8-лучевой ПЭГ-амин (трипентаэритритное ядро). Предпочтительная молекулярная масса для таких многолучевых ПЭГ-производных в каркасной части молекулы составляет от 1 кДа до 20 кДа, более предпочтительно, от 1 кДа до 15 кДа и, еще более предпочтительно, от 1 кДа до 10 кДа.
Очевидно, что концевые аминогруппы вышеуказанных многолучевых молекул присутствуют в связанной форме в каркасной молекуле, обеспечивая дополнительные сопряженные функциональные группы и реакционно-способные функциональные группы каркасной части молекулы.
Предпочтительно, чтобы сумма сопряженных функциональных групп и реакционно-способных функциональных групп в каркасной молекуле была поровну разделена между числом полимерных цепей на основе ПЭГ, выступающих из точки разветвления. Если число полимерных цепей на основе ПЭГ, выступающих из точки разветвления, не позволяет равного распределения, то предпочтительно, чтобы отклонение от среднего от суммы сопряженных и реакционно-способных функциональных групп на полимерную цепь на основе ПЭГ сохранялось минимальным.
Более предпочтительно, чтобы сумма сопряженных и реакционно-способных функциональных групп в каркасной молекуле была поровну разделена между числом полимерных цепей на основе ПЭГ, выступающих из точки разветвления. Например, если число сопряженных функциональных групп и реакционно-способных функциональных групп составляет 32, то на каждую из четырех полимерных цепей на основе ПЭГ, выступающих из точки разветвления, приходится по 8 групп, предпочтительно посредством дендритных молекул, присоединенных на конце каждой полимерной цепи на основе ПЭГ. В качестве альтернативы, на каждую из восьми полимерных цепей на основе ПЭГ, выступающих из ядра, приходится по 4 группы, или по 2 группы приходится на каждую из шестнадцати полимерных цепей на основе ПЭГ.
Такие дополнительные функциональные группы могут обеспечиваться дендритными молекулами. Предпочтительно, чтобы каждая дендритная молекула имела молекулярную массу в диапазоне от 0,4 кДа до 4 кДа, более предпочтительно, от 0,4 кДа до 2 кДа. Предпочтительно, чтобы каждая дендритная молекула имела по меньшей мере 3 разветвления и по меньшей мере 4 реакционно-способных функциональных группы, и самое большее 63 разветвления и 64 реакционно-способные функциональные группы, предпочтительно, по меньшей мере 7 разветвлений и по меньшей мере - 8 реакционно-способных функциональных групп и самое большее - 31 разветвление и 32 реакционно-способные функциональные группы.
Примеры таких дендритных молекул включают в себя трилизин, тетрализин, пентализин, гексализин, гептализин, октализин, нонализин, декализин, ундекализин, додекализин, тридекализин, тетрадекализин, пентадекализин, гексадекализин, гептадекализин, октадекализин, нонадекализин в связанной форме. Примеры таких предпочтительных дендритных молекул включают в себя трилизин, тетрализин, пентализин, гексализин, гептализин в связанной форме, наиболее предпочтительно - трилизин, пентализин или гептализин, орнитин, диаминомасляную кислоту в связанной форме.
Наиболее предпочтительно, чтобы гидрогелевый носитель по настоящему изобретению отличался тем, что его каркасная часть имела четвертичный углерод из формулы C(A-Hyp)4, в котором каждая А независимо представляла собой полимерную цепь на основе полиэтиленгликоля, одним концом присоединенную четвертичному углероду постоянной ковалентной связью, а дальний конец полимерной цепи на основе ПЭГ был ковалентно присоединен к дендритной молекуле Hyp, причем каждая дендритная молекула Hyp имеет по меньшей мере четыре функциональные группы, представляющие сопряженные функциональные группы и реакционно-способные функциональные группы.
Предпочтительно, чтобы каждая А была независимо выбрана из формулы -(CH2)n1(OCH2CH2)nX-, в которой n1 равняется 1 или 2; n представляет собой целое число в диапазоне от 5 до 50; а Х является химической функциональной группой, ковалентно соединяющей А и Hyp.
Предпочтительно, чтобы А и Hyp были ковалентно соединены амидной связью.
Предпочтительно, чтобы дендритная молекула Hyp представляла собой гиперразветвленный полипептид. Предпочтительно, чтобы гиперразветвленный полипептид содержал лизин в связанной форме. Предпочтительно, чтобы каждая дендритная молекула Hyp имела молекулярную массу в диапазоне от 0,4 кДа до 4 кДа. Очевидно, что каркасная часть C(A-Hyp)4 может состоять из одинаковых или различных дендритных молекул Hyp, и что каждая молекула Hyp может быть выбрана независимо. Каждая молекула Hyp состоит из 5-32 лизинов, предпочтительно, по меньшей мере 7 лизинов, то есть каждая молекула Hyp состоит из 5-32 лизинов в связанной форме, предпочтительно, по меньшей мере 7 лизинов в связанной форме. Наиболее предпочтительно, чтобы Hyp состояла из гептализинила.
В результате реакции полимеризуемых функциональных групп из каркасного реагента, более конкретно - Hyp, с полимеризуемыми функциональными группами из сшивающих реагентов на основе полиэтиленгликоля образуется постоянная амидная связь.
Предпочтительно, молекулярная масса C(A-Hyp)4 находился в диапазоне от 1 кДа до 20 кДа, более предпочтительно, от 1 кДа до 15 кДа, и еще более предпочтительно, от 1 кДа до 10 кДа.
Одна предпочтительная каркасная часть молекулы приведена ниже, пунктирные линии указывают сопряженные биодеградируемые связи с сшивающими молекулами, а n является целым числом от 5 до 50:
Способность к биодеградации гидрогелей по настоящему изобретению достигается введением гидролитически расщепляемых связей.
Термины «гидролитически деградируемый», «биодеградируемый» или «гидролитически расщепляемый», «авторасщепляемый» или «саморасщепление», «саморасщепляемый», «транзиентный» или «временный» относятся в контексте настоящего изобретения к связям и соединениям, которые неферментативно гидролитически деградируют или расщепляются при физиологических условиях (водный буфер с рН 7,4, 37°C) с временем до трех месяцев, включающим, но не ограниченным этими соединениями, аконитилы, ацетали, амиды, ангидриды карбоновых кислот, сложные эфиры, имины, гидразоны, амиды малеаминовой кислоты, ортоэфиры, фосфамиды, фосфоэфиры, силильные эфиры фосфора, силильные эфиры, сульфоновые эфиры, ароматические карбаматы, их комбинации и т.п.
В качестве деградируемой межмолекулярной функциональной группы, присутствующей в гидрогеле по изобретению, предпочтительными биодеградируемыми связями являются сложные эфиры, карбонаты, фосфоэфиры и эфиры сульфоновой кислоты, а наиболее предпочтительными являются сложные эфиры или карбонаты.
Постоянные связи представляют собой связи, неферментативно гидролитически деградируемые при физиологических условиях (водный буфер при рН 7,4, 37°C), с временем полужизни шесть месяцев или больше, например, амиды.
Для введения гидролитически расщепляемых связей в гидрогелевый носитель по изобретению, каркасные части молекулы можно напрямую соединить друг с другом посредством биодеградируемых связей.
В одном варианте осуществления изобретения каркасные молекулы биодеградируемого гидрогелевого носителя могут быть соединены вместе напрямую, то есть без участия сшивок. Гиперразветвленные дендритные части двух каркасных частей молекулы такого биодеградируемго гидрогеля могут быть либо напрямую соединены через сопряженную функциональную группу, которая соединяет две гиперразветвленные дендритные части. В качестве альтернативы, две гиперразветвленные дендритные части двух различных каркасных частей молекулы могут быть соединены друг с другом через два спейсера, соединенных с каркасной частью, а другой стороной - соединенных со сшивкой, разделенными сопряженными функциональными группами.
В качестве альтернативы, каркасные части молекулы могут быть соединены вместе через сшивающие части молекулы, причем каждая сшивка оканчивается, по меньшей мере, двумя гидролитически деградируемыми связями. Кроме деградирумых связей на концах, сшивки могут содержать дополнительные биодеградируемые связи. Таким образом, каждый конец сшивающей части молекулы, соединенный с каркасной частью молекулы, содержит гидролитически расщепляемую связь, и, необязательно, в сшивающей части молекулы могут присутствовать дополнительные биодеградируемые связи.
Предпочтительно, чтобы биодеградируемый гидрогелевый носитель состоял из каркасных частей молекулы, соединенных друг с другом гидролитически деградируемыми связями, и каркасные части молекулы были соединены вместе через сшивки.
Биодеградируемый гидрогелевый носитель может содержать один или несколько типов сшивок, предпочтительно - один тип. Сшивающая часть молекулы может быть линейной или разветвленной молекулой, и предпочтительно - линейной молекулой. В предпочтительном варианте осуществления изобретения сшивающая часть молекулы соединена с каркасными частями молекулы по меньшей мере двумя биодеградируемыми связями.
Предпочтительно, сшивающие части молекулы имели молекулярную массу в диапазоне от 60 Да до 5 кДа, более предпочтительно, от 0,5 кДа до 4 кДа, еще более предпочтительно, от 1 кДа до 4 кДа, еще более предпочтительно, от 1 кДа до 3 кДа. В одном варианте осуществления изобретения сшивающая часть состоит из полимера.
В дополнение к олигомерным или полимерным сшивающим частям молекулы, можно использовать низкомолекулярные сшивки, особенно когда для образования биодеградируемого гидрогеля по изобретению используются гидрофильные высокомолекулярные каркасные части молекулы.
Предпочтительно, чтобы сшивками на основе полиэтиленгликоля были углеводородные цепи, содержащие этиленгликолевые единицы, необязательно, содержащие дополнительные химические функциональные группы, причем сшивающие части на основе полиэтиленгликоля содержат по меньшей мере m этиленгликолевых единиц, где m является целым числом в диапазоне от 3 до 100, предпочтительно, от 10 до 70. Предпочтительно, чтобы сшивки на основе полиэтиленгликоля имели молекулярную массу в диапазоне от 0,5 кДа до 5 кДа.
При использовании в связи с сшивающей частью или полимерной цепью на основе ПЭГ, соединенных с точкой разветвления, термин «на основе ПЭГ» относится к сшивающей части молекулы или полимерной цепи на основе ПЭГ, содержащим по меньшей мере 20% по массе молекул этиленгликоля.
В одном варианте осуществления изобретения мономеры, составляющие полимерные сшивающие части, соединены биодеградируемыми связями. Такие полимерные сшивающие части могут содержать до 100 биодеградируемых связей или больше, в зависимости от молекулярной массы сшивающей части и молекулярной массы мономерных единиц. Примерами таких сшивающих частей молекулы являются полимеры на основе полимолочной кислоты или полигликолевой кислоты. Очевидно, что такая цепь полимолочной кислоты или полигликолевой кислоты может оканчиваться или прерываться алкильными или арильными группами, и они могут (необязательно) быть замещены гетероатомами и химическими функциональными группами.
Предпочтительно, чтобы сшивающие части молекулы были на основе ПЭГ, предпочтительно, представленные только одной молекулярной цепью на основе ПЭГ. Предпочтительно, чтобы сшивающие части молекулы на основе полиэтиленгликоля были углеводородными цепями, содержащими этиленгликолевые звенья, необязательно, содержащими дополнительные химические функциональные группы, причем сшивающие части на основе полиэтиленгликоля содержат по меньшей мере m этиленгликолевых единиц, где m является целым числом в диапазоне от 3 до 100, предпочтительно, от 10 до 70. Предпочтительно, чтобы сшивающие части молекулы на основе полиэтиленгликоля имели молекулярную массу в диапазоне от 0,5 кДа до 5 кДа.
В предпочтительном варианте осуществления настоящего изобретения сшивающая часть молекулы состоит из ПЭГ, который симметрично соединен через эфирные связи с двумя альфа,омега-алифатическими дикарбоновыми спейсерами, при условии что каркасные части молекулы соединены с гиперразветвленной дендритной частью через постоянные амидные связи.
Спейсерные дикарбоновые кислоты, соединенные с каркасной частью молекулы, а на другой стороне - с сшивающей частью, состоят из 3-12 атомов углерода, наиболее предпочтительно, 5-8 атомов углерода, и могут быть замещены по одному или нескольким атомам углерода. Предпочтительными заместителями являются алкильные группы, гидроксильные группы или амидогруппы, или замещенные аминогруппы. Одна или несколько алифатических метиленовых групп в дикарбоновых кислотах может быть необязательно замещена O или NH или алкил-замещенным N. Предпочтительным алкилом является линейный или разветвленный алкил с 1-6 атомами углерода.
Предпочтительно наличие постоянной амидной связи между гиперразветвленной дендритной частью и спейсером, соединенным с каркасной частью и с другой стороны соединенным с сшивающей молекулой.
Одна, предпочтительна сшивающая часть молекулы показана ниже; пунктирные линии указывают на сопряженные биодеградируемые связи с каркасными частями:
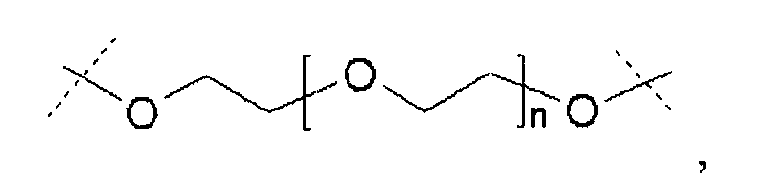
в которой n является целым числом от 5 до 50.
Предпочтительно, чтобы гидрогелевый носитель состоял из каркасных частей, соединенных гидролитически деградируемыми связями.
Более предпочтительно, каркасные части содержат точку разветвления со следующей формулой:
в которой пунктирная линия указывает на присоединение к остальной части каркаса.
Более предпочтительно, каркасные части молекулы содержат структуру со следующей формулой:
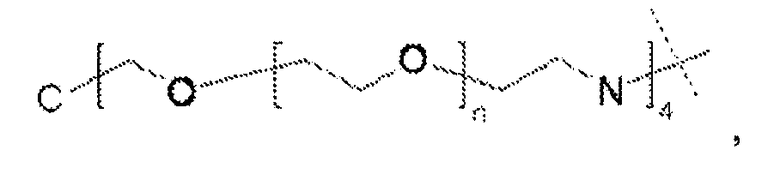
в которой n является целым числом от 5 до 50, а пунктирная линия указывает на присоединение к остальной части каркаса.
Предпочтительно, чтобы каркасная часть молекулы содержала гиперразветвленную часть Hyp.
Более предпочтительно, чтобы каркасная часть содержала гиперразветвленную часть Hyp со следующей формулой:
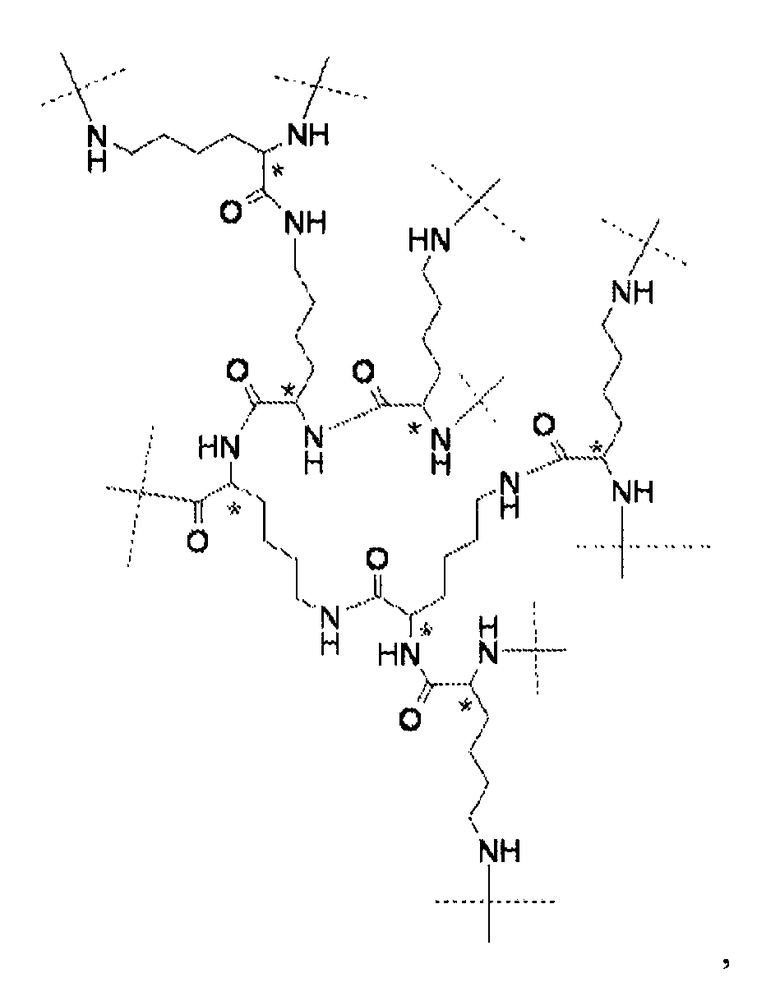
в которой пунктирная линия указывает на присоединение к остальной части каркасной молекулы, а атомы углерода, помеченные звездочкой, указывают S-конфигурацию.
Предпочтительно, чтобы каркасные части были присоединены по меньшей мере к одному спейсеру со следующей формулой:
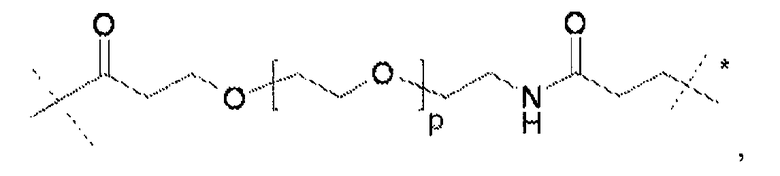
в которой пунктирная линия, помеченная звездочкой, указывает на связь между гидрогелем и азотом тиосукцинимидной группы,
в которой другая пунктирная линия указывает на присоединение к Hyp, и
в которой р является целым числом от 0 до 10.
Предпочтительно, чтобы каркасные части были присоединены по меньшей мере к одному спейсеру со следующей формулой:
в которой одна из пунктирных линий указывает присоединение к гиперразветвленной части Hyp, а вторая пунктирная линия указывает присоединение к остальной молекуле: и
в которой m является целым числом от 2 до 4.
Предпочтительно, чтобы каркасные части были соединены друг с другом через сшивающие части молекулы со следующей структурой:
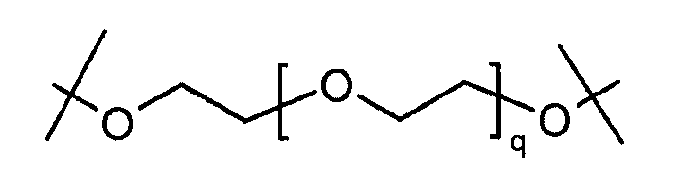
в которой
q является целым числом от 3 до 100;
на скорость гидролиза биодеградируемых связей между каркасными частями и сшивающими частям влияют или скорость гидролиза определяют число и тип соединенных атомов, прилегающих к ПЭГ-эфиру карбоксильной группы. Например, выбирая янтарную, адипиновую или глутаровую кислоту для образования эфиров с ПЭГ можно варьировать время половинной деградации биодеградируемого гидрогелевого носителя по изобретению.
Общее количество каркасных молекул можно измерить в растворе после полной деградации гидрогеля по изобретению, а в течение деградации фракции растворимых продуктов деградации каркаса можно отделить от нерастворимого гидрогеля по изобретению и исследовать без помех от других растворимых продуктов деградации, высвобождаемых из гидрогеля по изобретению. Гидрогель по изобретению можно отделить от избытка воды в буфере с физиологической осмоляльностью седиментацией или с помощью центрифугирования. Центрифугирование можно проводить таким образом, что супернатант будет составлять по меньшей мере 10% объема от набухшего гидрогеля по изобретению. После такой стадии седиментации или центрифугирования растворимые продукты деградации гидрогеля остаются в водном супернатанте, и водорастворимые продукты деградации, содержащие одну или несколько молекул каркаса детектируют, используя аликвоты этого супернатанта в соответствующих способах разделения и/или анализа.
Предпочтительно, чтобы водорастворимые продукты деградации можно было отделить от водонерастворимых продуктов деградации с помощью фильтрации через фильтры 0,45 мкм, после которой водорастворимые продукты деградации можно найти в проскоке. Водорастворимые продукты деградации также можно отделить от водонерастворимых продуктов деградации в помощью комбинации стадий центрифугирования и фильтрации.
Например, каркасные молекулы могут нести группы, УФ-спектр поглощения которых не совпадает с УФ-спектром поглощения других продуктов деградации. Такие группы с селективным поглощением в УФ-спектре могут быть структурными компонентами каркасной молекулы, как, например, амидные связи, или они могут быть введены в каркасную молекулу в результате присоединения к ее реакционно-способных функциональным группам ароматических кольцевых систем, таких как индольные группы.
В таких связанных с гидрогелем пролекарственных соединениях инсулина по изобретению желательно, чтобы высвобождение почти всего лекарственного соединения (>90%) происходило до высвобождения значительного количества каркасных молекул (<10%). Этого можно достичь, модифицируя время полужизни связанного с носителем пролекарственного соединения относительно кинетики деградации гидрогеля по изобретению.
Связанное с гидрогелем пролекарственное соединение инсулина по настоящему изобретению можно получить из гидрогеля по настоящему изобретению стандартными способами, известными в данной области. Для специалиста-практика в данной области будет очевидно, что существует несколько подходов. Например, пролекарственный линкер, указанный выше, к которому ковалентно присоединена биологически активная молекула, то есть инсулин, может вступать в реакцию с реакционно-способными функциональными группами гидрогеля по настоящему изобретению, уже несущими активную молекулу, то есть, инсулин, или без нее, частично или в целом.
В предпочтительном способе получения гидрогель синтезируют посредством химического лигирования. Гидрогель можно получить из двух макромолекулярных продуктов с комплементарными функциональными группами, которые участвуют в реакции, например, реакции конденсации или присоединения. Одним из этих исходных веществ является сшивающий реагент по меньшей мере с двумя идентичными функциональными группам, а другим исходным веществом является гомомультифункциональный каркасный реагент. Подходящие функциональные группы, присутствующие на сшивающем реагенте, включают концевые аминогруппы, карбоновые кислоты и производные, малеимид и другие альфа-бета-ненасыщенные акцепторы Михаэля, такие как винилсульфоновая, тиольная, гидроксильная группы. Подходящие функциональные группы, присутствующие на каркасном реагенте, включают, но не ограничены этим, аминогруппы, карбоновые кислоты и производные, малеимид и другие альфа-бета-ненасыщенные акцепторы Михаэля, такие как винилсульфоновая, тиольная, гидроксильная группы.
Если полимеризующиеся группы сшивающего реагента использовать в субстехиометрических количествах относительно полимеризующихся групп каркаса, то полученный гидрогель будет реакционно-способным гидрогелем со свободными реакционно-способными функциональными группами, присоединенными к каркасной структуре.
Необязательно, линкер пролекарственного соединения можно сначала конъюгировать с инсулином и полученный конъюгат инсулина с линкером пролекарственного соединения может затем вступать в реакцию с реакционно-способными функциональными группами гидрогеля. В качестве альтернативы, после активации одной из функциональных групп линкера пролекарственного соединения, конъюгат линкера с гидрогелем может вступать в контакт с инсулином на стадии второй реакции, и избыток инсулина можно удалить с помощью фильтрации после конъюгации инсулина с линкером пролекарственного соединения, связанным с гидрогелем.
Предпочтительным способом получения пролекарственного соединения по настоящему изобретению является следующий способ:
Предпочтительным исходным веществом для синтеза каркасного реагента является 4-лучевой ПЭГ-тетраамин или 8-лучевой ПЭГ-октаамин, причем молекулярная масса ПЭГ-реагента находится в диапазоне от 2000 до 10000 дальтон, наиболее предпочтительно - в диапазоне от 2000 до 5000 Да. К таким многолучевым производным ПЭГ остатки лизина присоединяются последовательно, образуя гиперразветвленный каркасный реагент. Очевидно, что остатки лизинов могут быть частично или полностью защищены защитными группами в ходе стадий присоединения, и что также конечный каркасный реагент может содержать защитные группы. Предпочтительным строительным блоком является бис-ВОС-лизин. В качестве альтернативы, вместо последовательного присоединения остатков лизина, сначала можно синтезировать дендритную полилизиновую часть молекулы и далее присоединить к 4-лучевому ПЭГ-тетраамину или 8-лучевому ПЭГ-октаамину. Желательно получить каркасный реагент, несущий 32 аминогруппы, соответственно, семь остатков лизина будут присоединены к каждому плечу 4-лучевого ПЭГ, или пять остатков лизина будет присоединены к каждому плечу 8-лучевого ПЭГ. В другом варианте осуществления изобретения многолучевым производным ПЭГ является тетра- или октакарбокси-ПЭГ. В этом случае дендритные части молекулы можно синтезировать из глутаровой или аспарагиновой кислоты, и полученный каркасный реагент будет нести 32 карбоксильных группы. Следует понимать, что все или часть функциональных групп каркасного реагента могут присутствовать в свободной форме, в виде солей или могут быть конъюгированы с защитными группами. Следует понимать, что исходя из практических причин число остатков лизина каркасного реагента на плечо ПЭГ будет составлять от шести до семи, более предпочтительно, приблизительно, семь.
Предпочтительный каркасный реагент приведен ниже:
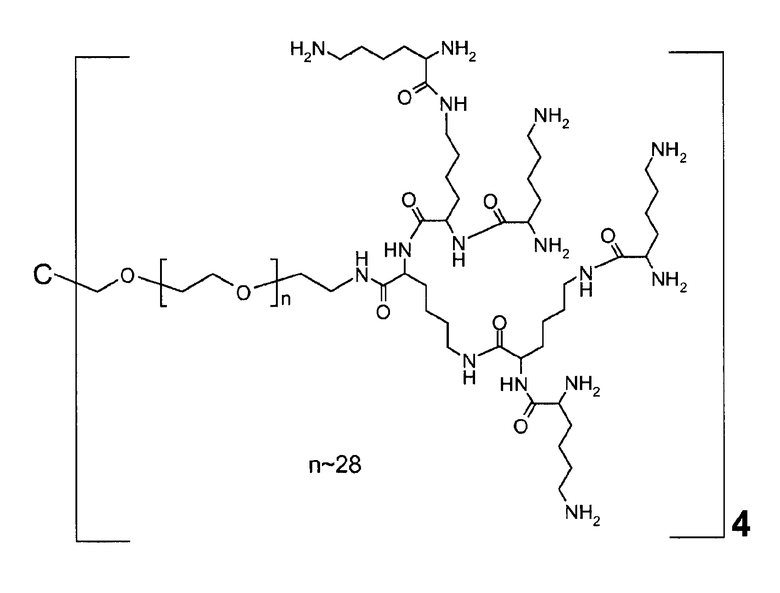
Синтез сшивающего реагента начинается с линейной цепи ПЭГ с молекулярной массой в диапазоне от 0,2 до 5 кДа, более предпочтительно, от 0,6 до 2 кДа, этерифицированной полуэфиром дикарбоновой кислоты, в основном, адипиновой кислоты или глутаровой кислоты. Предпочтительной защитной группой для образования полуэфира является бензильная группа. Полученные полуэфиры бис(дикарбоновой кислоты) и ПЭГ превращаются в более реакционно-способные карбоксильные соединения, как, например, ацилхлориды или активные эфиры, например, пентафторфенильные или N-гидроксисукцинимидные эфиры, наиболее предпочтительными являются N-гидроксисукцинимидные эфиры, из которых предпочтительная избранная структура приведена ниже.
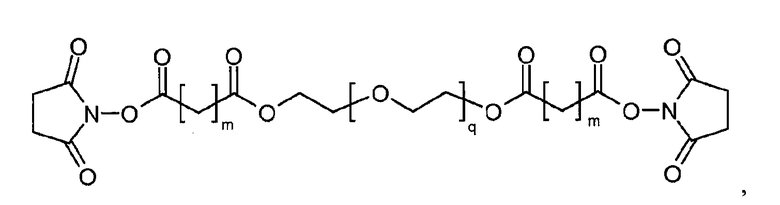
в которой каждое m независимо является целым числом в диапазоне от 2 до 4, а
q является целым числом от 3 до 100.
Более предпочтительной является следующая структура:

В качестве альтернативы, полуэфиры бис(дикарбоновой кислоты) и ПЭГ можно активировать в присутствии сшивающего реагента, такого как DCC или HOBt, или PyBOP.
В альтернативном варианте осуществления изобретения каркасный реагент несет карбоксильные группы, и соответствующий сшивающий реагент будет выбран из оканчивающихся аминогруппами цепей ПЭГ, содержащих сложноэфирные группы.
Каркасный реагент и сшивающий реагент можно полимеризовать с образованием гидрогеля по изобретению, используя полимеризацию в обращенной эмульсии. После выбора желаемого стехиометрического соотношения между полимеризуемыми группами каркаса и сшивки, каркасное соединение и сшивку растворяют в DMSO, и используют подходящий эмульгатор с соответственно выбранной величиной гидрофильно-липофильного баланса, предпочтительно, Арлацель P 135, для формирования обращенной эмульсии, используя механическую мешалку и контролируя скорость перемешивания. Полимеризацию индуцируют добавлением соответствующего основания, предпочтительно, N,N,N',N'-тетраметилэтилендиамина. После перемешивания в течение соответствующего периода времени реакцию останавливают добавлением кислоты, например уксусной кислоты, и воды. Шарики собирают, промывают и фракционируют по размеру с помощью механического просеивания. Необязательно, на этой стадии можно удалить защитные группы.
В альтернативном варианте осуществления настоящего изобретения многофункциональные части молекулы присоединяются к реакционно-способным функциональным группам полимеризуемого реакционно-способного гидрогеля для увеличения числа функциональных групп, что позволит повысить лекарственную емкость гидрогеля. Такими многофункциональными частями молекулы могут быть соответственно замещенные производные лизина, дилизина, трилизина, тетрализина, пентализина, гексализина, гептализина или олиголизина, низкомолекулярного PEI. Предпочтительной многофункциональной частью молекулы является лизин.
Кроме того, такой гидрогель по изобретению может быть функционализирован спейсером, несущим такую же функциональную группу, например, в гидрогель можно ввести аминогруппы посредством присоединения гетеробифункционального спейсера, например, соответственно активированного спейсера COOH-(EG)6-NH-fmoc (EG=этиленгликоль), и удаления fmoc-защитной группы.
После нагрузки функционализированного гидрогеля, содержащего малеимидные группы, конъюгатом инсулина и линкера, для предупреждения нежелательных побочных реакций все оставшиеся функциональные группы кэпируют соответствующим блокирующим реагентом, например, меркаптоэтанолом.
В предпочтительном варианте осуществления изобретения конъюгат инсулина с линкером, имеющий свободную тиольную группу соединенную с линкерной частью, вступает в реакцию с гидрогелем, несущим малеимидные функциональные группы, при температуре от комнатной до 4°С, более предпочтительно, при комнатной температуре, в буферном водном растворе с рН 2-5, предпочтительно, с рН 2,5-4,5, более предпочтительно, с рН 3,0-4,0. Далее соответствующий конъюгат инсулина, линкера и гидрогеля обрабатывают меркаптоэтанолом при температуре от комнатной до 4°С, более предпочтительно, при комнатной температуре, в буферном водном растворе с рН 2-5, предпочтительно, с рН 2,5-4,0, более предпочтительно, с рН 2,5-3,5.
В другом предпочтительном варианте осуществления изобретения конъюгат инсулина с линкером, несущим малеимидную группу соединенную с линкерной частью, вступает в реакцию с гидрогелем, имеющим тиольные функциональные группы, при температуре от комнатной до 4°С, более предпочтительно, при комнатной температуре, в буферном водном растворе с рН 2-5, предпочтительно, с рН 2,5-4,5, более предпочтительно, с рН 3,0-4,0. Далее, соответствующий конъюгат инсулина, линкера и гидрогеля обрабатывают низкомолекулярным соединением, содержащим малеимидную группу, предпочтительно, малеимид-содержащим соединением с массой от 100 до 300 Да, например, N-этил-малеимидом, при температуре от комнатной до 4°С, более предпочтительно, при комнатной температуре, в буферном водном растворе с рН 2-5, предпочтительно, с рН 2,5-4,0, более предпочтительно, с рН 2,5-3,5.
Другим аспектом настоящего изобретения является способ, включающий в себя стадии:
(а) контакта водной суспензии, содержащей микрочастицы гидрогеля с малеимидными функциональными группами, с раствором, содержащим инсулин-линкерный реагент, несущий тиольные группы, при температуре от комнатной до 4°С в буферном водном растворе с рН 2-5, дающего в результате конъюгат инсулина, линкера и гидрогеля;
(b) необязательно, обработки конъюгата инсулина, линкера и гидрогеля из стадии (а) тиол-содержащим соединением с массой от 34 до 500 кДа при температуре от комнатной до 4°С в буферном водном растворе с рН 2-5.
Еще одним аспектом настоящего изобретения является способ, включающий в себя стадии:
(а) контакта водной суспензии, содержащей микрочастицы гидрогеля с тиольными функциональными группами, с раствором, содержащим инсулин-линкерный реагент, несущий малеимидные группы, при температуре от комнатной до 4°С в буферном водном растворе с рН 2-5, дающего в результате конъюгат инсулина, линкера и гидрогеля;
(b) необязательно, обработки конъюгата инсулина, линкера и гидрогеля из стадии (а) малеимид-содержащим соединением с массой от 100 до 300 кДа при температуре от комнатной до 4°С в буферном водном растворе с рН 2-5.
Особенно предпочтительный способ получения пролекарственного соединения инсулина по настоящему изобретению включает стадии:
(а) реакции соединения с формулой C(A'-X1)4, в которой A'-X1 представляет А до его связывания с Hyp или предшественника Hyp, а Х1 является соответствующей функциональной группой, с соединением, имеющим формулу Hyp'-X2, в которой Hyp'-X2 представляет Hyp до его связывания с A или предшественника Нур, а Х2 является подходящей функциональной группой для участия в реакции с Х1;
(b) необязательно, участия соединения, полученного на стадии (а), в реакциях одной или нескольких дополнительных стадий с выходом соединения, имеющего формулу C(A-Hyp)4 по меньшей мере с четырьмя функциональными группами;
(с) реакции по меньшей мере четырех функциональных групп соединения, полученного на стадии (b), с сшивающим реагентом на основе полиэтиленгликоля, в которой реакционно-способные группы сшивающего реагента берутся в субстехиометрическом количестве относительно общего числа реакционно-способных групп C(A-Hyp)4, в результате которой образуется гидрогель;
(d) реакции оставшихся непрореагировавших функциональных групп (представляющих реакционно-способные функциональные группы каркаса, содержащиеся в гидрогеле по настоящему изобретению) в каркасе гидрогеля из стадии (с) с ковалентным конъюгатом инсулина и транзиентным линкером пролекарственного соединения; или сначала - реакцию непрореагировавших функциональных групп с транзиентным линкером пролекарственного соединения, и далее - с инсулином;
(е) необязательно, стадию кэпирования оставшихся непрореагировавших функциональных групп, дающую пролекарственное соединение по настоящему изобретению.
Более конкретно, гидрогели для пролекарственного соединения инсулина по настоящему изобретению синтезируют следующим образом:
Для массовой полимеризации каркасный реагент и сшивающий реагент смешивают в соотношении аминогрупп к реакционно-способным сложноэфирным группам от 2:1 до 1,05:1.
Оба реагента, каркасный и сшивающий, растворяют в DMSO, получая раствор с концентрацией от 5 до 50 г на 100 мл, предпочтительно, от 7,5 до 20 г на 100 мл и, наиболее предпочтительно, от 10 до 20 г на 100 мл.
Для осуществления полимеризации к раствору в DMSO, содержащему сшивающий реагент и каркасный реагент, добавляют от 2 до 10% (объемных) N,N,N',N'-тетраметилэтилендиамина (TMEDA), и смесь встряхивают в течение 1-20 сек и оставляют неподвижно. Смесь затвердевает меньшем чем за 1 мин.
Такой гидрогель по изобретению предпочтительно тонко измельчить механическими способами, такими как перемешивание, дробление, нарезка, прессование или помол, и, необязательно, просеивание. В случае полимеризации эмульсии реакционная смесь состоит из диспергированной фазы и дисперсионной фазы.
Для диспергированной фазы каркасный реагент и сшивающий реагент смешивают в соотношении аминогрупп к активным сложноэфирным группам от 2:1 до 1,05:1 и растворяют в DMSO, получая раствор с концентрацией от 5 до 50 г на 100 мл, предпочтительно, от 7,5 до 20 г на 100 мл и, наиболее предпочтительно, от 10 до 20 г на 100 мл.
Дисперсионной фазой является любой растворитель, который не смешивается с DMSO, является неосновным, апротонным и имеет вязкость ниже 10 Па*сек. Предпочтительно, чтобы растворитель, который не смешивается с DMSO, являлся неосновным, апротонным и имел вязкость ниже 2 Па*сек, и был не токсичным. Более предпочтительно, чтобы растворителем был насыщенный линейный или разветвленный углеводород с 5-10 атомами углерода. Более предпочтительно, чтобы растворителем был н-гептан.
Для образования эмульсии диспергированной фазы в дисперсионной фазе перед добавлением диспергированной фазы к дисперсионной фазе добавляют эмульгатор. Количество эмульгатора составляет от 2 до 50 мг на мл диспергированной фазы, более предпочтительно, от 5 до 20 мг на мл диспергированной фазы, наиболее предпочтительно, 10 мг на мл диспергированной фазы.
Величина HLB эмульгатора составляет от 3 до 8. Предпочтительно, чтобы эмульгатором был триэфир сорбита и жирной кислоты или конъюгат полигидроксижирной кислоты и полиэтиленгликоля. Более предпочтительно, чтобы эмульгатором был конъюгат полигидроксижирной кислоты и полиэтиленгликоля из линейного полиэтиленгликоля с молекулярной массой в диапазоне от 0,5 кДа до 5 кДа и звеньев полигидроксижирной кислоты с молекулярной массой в диапазоне от 0,5 кДа до 3 кДа на конце каждой цепи. Наиболее предпочтительным эмульгатором является полиэтиленгликоль-диполигидрокстеарат, Цитрол DPHS (Cithrol DPHS, бывшее название Arlacel P135, Croda International Plc).
Капли диспергированной фазы получают перемешиванием с помощью аксиально-поточной мешалки, имеющей геометрию, аналогичную мешалкам, таким как Isojet, Intermig, Propeller (EKATO Ruhr- und Mischtechnik GmbH, Germany), наиболее предпочтительно, аналогичную Isojet с диаметром от 50 до 90% от диаметра реакционной емкости. Предпочтительно начинать перемешивание до добавления диспергированной фазы. Скорость мешалки устанавливают от 0,6 до 1,7 м/сек. Диспергированную фазу добавляют при комнатной температуре, и концентрация диспергированной фазы составляет от 2% до 70%, предпочтительно, от 5 до 50%, более предпочтительно, от 10 до 40%, и наиболее предпочтительно, от 20 до 35% от общего объема реакции. Перед добавлением основания, инициирующего полимеризацию, смесь диспергированной фазы, эмульгатора и дисперсионной фазы перемешивают в течение 5-60 минут.
К смеси диспергированной и дисперсионной фазы добавляют от 5 до 10 эквивалентов основания (относительно каждой образуемой амидной связи). Основание является апротонным, ненуклеофильным и растворимым в дисперсионной фазе. Предпочтительно, чтобы основание было апротонным, ненуклеофильным, хорошо растворимым, как в дисперсионной фазе, так и в DMSO. Более предпочтительно, чтобы основание было апротонным, ненуклеофильным, хорошо растворимым, как в дисперсионной фазе, так и в DMSO, представляло собой аминооснование и не было токсичным. Наиболее предпочтительным основанием является N,N,N',N'-тетраметилэтилендиамин (TMEDA). Перемешивание в присутствии основания продолжают в течение 1-16 часов.
При перемешивании капельки диспергированной фазы затвердевают, становясь сшитыми шариками гидрогеля по изобретению, которые можно собрать и провести фракционирование по размеру на вибрационной непрерывной просеивающей машине с декой 75 мкл и 32 мкл, получая микрочастицы гидрогеля по изобретению.
Гидрогель для пролекарственного соединения инсулина по настоящему изобретению можно получить в форме микрочастиц. В предпочтительном варианте осуществления изобретения реакционно-способный гидрогель представляет собой предмет определенной формы, как, например, сетка или стент. Наиболее предпочтительно, чтобы гидрогель имел форму микрошариков, которые можно вводить с помощью подкожной или внутримышечной инъекции, используя стандартный шприц. Такие мягкие шарики могут иметь диаметр 1-500 микрометров.
Если микрочастицы ресуспендированы в изотоническом водном буфере, то предпочтительно, чтобы диаметр микрочастиц составлял от 10 до 100 микрометров, наиболее предпочтительно, от 20 до 100 мкм, наиболее предпочтительно, от 25 до 80 микрометров.
Предпочтительно, чтобы микрочастицы можно было вводить с помощью инъекций через иглу с внутренним диаметром меньше 0,6 мм, предпочтительно, через иглу с внутренним диаметром меньше 0,3 мм, более предпочтительно, через иглу с внутренним диаметром меньше 0,225 мм, еще более предпочтительно, через иглу с внутренним диаметром меньше 0,175 мм и, наиболее предпочтительно, через иглу с внутренним диаметром меньше 0,16 мм.
Следует понимать, что термины «может вводиться с помощью инъекций», «инъекционный» или «инъецируемость» относятся к комбинации факторов, таких как некоторая сила, прилагаемая к плунжеру шприца, содержащего биодеградируемый гидрогель по изобретению, набухший в жидкости при некоторой концентрации (масса/объем) и при некоторой температуре; игла данной внутреннего диаметра на конце такого шприца; и время, требуемое для вытеснения некоторого объема биодеградируемого гидрогеля по изобретению из шприца через иглу.
Для обеспечения инъецируемости объем 1 мл пролекарственных соединений инсулина по изобретению, набухший в воде до концентрации по меньшей мере 5% (масса/объем) и содержащийся в шприце с плунжером диаметром 4,7 мм, может вытесняться при комнатной температуре в течение 10 секунд при приложении силы меньше 50 ньютон.
Предпочтительно, чтобы инъецируемость достигалась у пролекарственного соединения инсулина по изобретению, набухшего в воде до концентрации приблизительно 10% (масса/объем).
В дополнительном варианте осуществления изобретения композиция характеризуется тем, что она является жидкой композицией, которая после инъекции образует депо-препарат.
В дополнительном варианте осуществления изобретения композиция характеризуется тем, что ее вводят с помощью инъекций, например, подкожных или внутримышечных.
В дополнительном варианте осуществления изобретения композиция предназначена для лечения или предупреждения заболевания или расстройства, ассоциированных с недостаточностью инсулина, лечение или предупреждение которых с использованием соединения инсулина является полезным, таких как гипергликемия, предиабет, нарушенная глюкозотолерантность, диабет I типа, диабет II типа, синдром Х. Обычно, такой болезнью или таким расстройством является диабет II типа.
В дополнительном варианте осуществления изобретения соединение инсулина выбрано из инсулина или аналогов инсулина человека или других биологических видов, и их производных и конъюгатов. Более предпочтительным является инсулин и аналоги инсулина человека, как, например, инсулин гларгин, инсулин детемир, инсулин лиспро, инсулин аспартат, инсулин глулизин. При введении максимальная пиковая концентрация соединения инсулина обычно достигается в течение первых 48 часов. В дополнительном варианте осуществления изобретения пиковая концентрация достигается в первые 24 часа после введения, например, в первые 12 часов после введения, например, в течение первых 6 часов после введения.
В дополнительном аспекте фармацевтическая композиция по настоящему изобретению дополнительно содержит соединение GLP-1, обычно, агонист GLP-1.
Такое соединение GLP-1 обычно выбрано из любого одного из:
[Seq ID No:1] Эксендин-4
HGEGTFTSDL SKQMEEEAVR LFIEWLKNGG PSSGAPPPS-NH2
[Seq ID No:2] Эксендин-3


[Seq ID No 13] GLP-1 (7-36)амид

Где аминокислота Хаа выбрана из P, F, Y.

Где аминокислота Хаа выбрана из T, α-аминомасляной кислоты, D-Ala, V, Gly.

Где R выбран из ацетила, пироглутамила, N-2-гидроксибензоила, N-транс-3-гексеноила.
[Seq ID No 20]
HXaaAEGTFTSDV SSYLEGQAAK EFIAWLVKGR-NH2
Где аминокислота Хаа является 6-аминогексаноилом.
В дополнительном аспекте настоящее изобретение относится к использованию соединения инсулина для получения фармацевтической композиции, содержащей соединение инсулина в концентрации, достаточной для поддержания терапевтически эффективного уровня соединения инсулина в плазме крови в течение по меньшей мере 3 дней, обычно, по меньшей мере 80 часов, например, в течение недели или более длительного периода времени, характеризуется тем, что она имеет фармакокинетический профиль in vivo по существу без выброса соединения инсулина, для лечения или предупреждения заболевания или расстройства, ассоциированных с недостаточностью инсулина, лечение или предупреждение которых с использованием соединения инсулина является полезным.
Такая концентрация будет варьировать от субъекта к субъекту и будет зависеть от терапевтического окна у отдельного субъекта, но для продолжительности терапевтического эффекта в течение по меньшей мере 3 дней, например, недели (то есть, примерно 7 дней), концентрация обычно составляет по меньшей мере около 10 мг/мл, например, превышает 10 мг/мл.
Предпочтительная композиция пролекарственного соединения инсулин-гидрогеля приведена в нижеследующих абзацах.
Композиция пролекарственного соединения инсулин-гидрогеля может представлять собой суспензионную композицию или сухую композицию. Предпочтительно, чтобы фармацевтическая композиция пролекарственного соединения инсулин-гидрогеля представляла собой сухую композицию. Подходящими способами сушки являются, например, сушка распылением и лиофилизация (сушка при замораживании). Предпочтительно высушивать фармацевтическую композицию пролекарственного соединения инсулин-гидрогеля с помощью лиофилизации.
Предпочтительно, чтобы композиция содержала дозу пролекарственного соединения инсулин-гидрогеля, достаточную для обеспечения терапевтически эффективного количества инсулина в течение по меньшей мере трех дней, при одном введении. Более предпочтительно, чтобы одного введения пролекарственного соединения инсулин-гидрогеля было достаточно на одну неделю.
Фармацевтическая композиция пролекарственного соединения инсулин-гидрогеля по настоящему изобретению содержит один или несколько вспомогательных веществ.
Вспомогательные вещества, используемые в парентеральных композициях можно классифицировать как буферные агенты, модификаторы изотоничности, консерванты, стабилизиторы, анти-адсорбционные агенты, агенты, защищающие от окисления, загустители/агенты, усиливающие вязкость, или другие вспомогательные агенты. В некоторых случаях эти ингредиенты может иметь двойные или тройные функции. Композиции пролекарственных соединений инсулин-гидрогеля по настоящему изобретению содержат одно или несколько вспомогательных веществ, выбранных из групп, состоящих из:
(i) Буферных агентов: физиологически совместимые буферы для поддержания рН в заданном диапазоне, такие как фосфат натрия, бикарбонат, сукцинат, гистидин, цитрат и ацетат, сульфат, нитрат, хлорид, пируват. Также можно использовать антациды, такие как Mg(OH)2 или ZnCO3. Буферную емкость можно подводить, чтобы соответствовать условиям, наиболее чувствительным к стабильности рН.
(ii) Модификаторов изотоничности: для минимизации боли, которая может возникнуть в результате повреждения клеток из-за разницы осмотического давления в месте депо-инъекции. Примерами являются глицерин и хлорид натрия. Эффективные концентрации можно определить с помощью осмометрии, используя принятую осмоляльность для сыворотки - 285-315 мОсмоль/кг.
(iii) Консервантов и/или противомикробных средств: для минимизации риска заражения пациентов в результате инъекции в мультидозовые парентеральные препараты необходимо добавлять консерванты в достаточной концентрации, и были установлены соответствующие нормативные требования. Типичные консерванты включают м-крезол, фенол, метилпарабен, этилпарабен, пропилпарабен, бутилпарабен, хлорбутанол, бензиловый спирт, нитрат фенилртути, тимеросал, сорбиновую кислоту, сорбат калия, бензойную кислоту, хлоркрезол и хлорид бензалкония.
(iv) Стабилизаторов: стабилизация достигается в результате повышения сил, стабилизирующих белок, дестабилизации денатурированного состояния или в результате прямого связывания вспомогательных веществ с белком. Стабилизаторами могут быть аминокислоты, такие как аланин, аргинин, аспарагиновая кислота, глицин, гистидин, лизин, пролин, сахара, такие как глюкоза, сахароза, трегалоза, многоатомные спирты, такие как глицерин, маннит, сорбит, соли, такие как фосфат калия, сульфат натрия, хелатирующие агенты, такие как EDTA, гексафосфат, лиганды, такие как бивалентные ионы металлов (цинка, кальция и т.п.), другие соли или органические молекулы, такие как фенольные производные. Кроме того, можно использовать олигомеры или полимеры, такие как циклодекстрины, декстран, дендримеры, ПЭГ или ПВП, или протамин или HSA.
(v) Анти-адсорбирующих агентов: для конкурентного покрытия внутренней поверхности или конкурентной адсорбции на внутренней поверхности контейнера, содержащего композицию, в основном используются ионные или неионные поверхностно-активные вещества или другие белки, или растворимые полимеры. Примерами являются полоксамер (Плюроник F68), ПЭГ-додецилэфир (Brij 35), полисорбат 20 и 80, декстран, полиэтиленгликоль, ПЭГ-полигистидин, BSA и HSA, и желатины. Выбор концентрации и типа вспомогательного вещества зависят от эффекта, которого следует избегать, но обычно монослой поверхностно-активного вещества образуется на поверхности раздела фаз немного выше значения критической концентрации мицеллообразования.
(vi) Лио- и/или криопротектантов: при лиофилизации или сушки распылением вспомогательные вещества могут противодействовать дестабилизирующим эффектам, вызванным разрушением водородных связей и удалением воды. Для этой цели можно использовать сахара и многоатомные спирты, но соответствующие положительные эффекты также наблюдались при использовании поверхностно-активных веществ, аминокислот, неводных растворителей и других пептидов. Трегалоза особенно эффективна для уменьшения агрегации, вызываемой влажностью, а также она увеличивает термостабильность белка, потенциально вызываемую экспозицией гидрофобных групп белка в воде. Также можно использовать маннит и сахарозу, либо в качестве единственного лио/криопротектанта, либо в комбинации друг с другом, причем известно, что высокое соотношение маннита к сахарозе увеличивает физическую стабильность лиофилизированного остатка. Маннит также можно комбинировать с трегалозой. Трегалозу также можно комбинировать с сорбитом, или использовать сорбит в качестве единственного протектанта. Также можно использовать крахмал или производные крахмала.
(vii) Агентов, защищающих от окисления: антиоксиданты, такие как аскорбиновая кислота, эктоин, метионин, глутатион, монотиолглицерин, морин, полиэтиленимин (PEI), пропилгаллат, витамин Е, хелатирующие агенты, такие как лимонная кислота, EDTA, гексафосфат, тиогликолевая кислота.
(viii) Загустителей/агентов, усиливающих вязкость: замедляют осаждение частиц в флаконе и шприце и используются для облегчения смешивания и ресуспендирования частиц, а также для более легкого введения суспензии с помощью инъекций (то есть, для приложения меньшей силы к плунжеру шприца). Подходящими загустителями или усилителями вязкости являются, например, карбомерные загустители, такие как Карбопол 940, Карбопол Ultrez 10, производные целлюлозы, такие как гидроксипропилметилцеллюлоза (гипромеллоза, HPMC) или диэтиламиноэтилцеллюлоза (DEAE или DEAE-C), коллоидный силикат магния (Veegum) или силикат натрия, гель гидроксиапатита, гель трикальцийфосфата, ксантаны, каррагинаны, такие как Satia gum UTC 30, алифатические полигидроксикислоты, такие как поли((D,L- или L-молочная кислота) (PLA) и полигликолевая кислота (PGA), а также их сополимеры (PLGA), терполимеры D,L-лактида, гликолид и капролактон, полоксамеры, гидрофильные полиоксиэтиленовые блоки и гидрофобные полиоксипропиленовые блоки для создания триблока полиоксиэтилен-полиоксипропилен-полиоксиэтилен (например, Плюроника®), сополимер простых и сложных полиэфиров, например, сополимер полиэтиленгликоль-терефталата и полибутилен-терефталата, изобутират ацетата сахарозы (SAIB), декстран или его производные, комбинации декстранов и ПЭГ, полидиметилсилоксан, коллаген, хитозан, поливиниловый спирт (PVA) и производные, полиалкилимиды, поли(сополимер акриламида и диаллилдиметиламмония (DADMA)), поливинилпирролидон (PVP), гликозаминогликаны (GAG), такие как дерматан сульфат, хондроитин сульфат, кератан сульфат, гепарин, гепаран сульфат, гиалуронан, АВА-триблок или АВ-блоксополимеры, состоящие из гидрофобных А-блоков, таких как полилактид (PLA) или сополимер лактида и гликолида (PLGA), и гидрофильных В-блоков, таких как полиэтиленгликоль (ПЭГ) или поливинилпирролидон. Такие блок-сополимеры, а также вышеуказанные полоксамеры могут проявлять поведение, обратное термическому гелеобразованию (жидкое состояние при комнатной температуре для облегчения введения соединения и гель выше температуры перехода жидкости в гель при температуре тела после инъекции).
(ix) Агента, усиливающего распределение или диффузию: модифицирует проницаемость соединительной ткани в результате гидролиза компонентов внеклеточного матрикса в интерстициальном пространстве, например (но не ограниченных этим), гиалуроновой кислоты, полисахарида, найденного в межклеточном пространстве соединительной ткани. Агент, усиливающий распределение, такой как, но не ограниченный этим, гиалуронидаза, временно снижает вязкость внеклеточного матрикса и усиливает диффузию лекарственных соединений, вводимых с помощью инъекций.
(x) Других вспомогательных агентов, таких как смачивающие агенты, модификаторы вязкости, антибиотики, гиалуронидаза. Кислоты и основания, такие как гидрохлористая кислота и гидроксид натрия, являются вспомогательными агентами, необходимыми для подведения рН при изготовлении соединения.
Предпочтительно, чтобы композиция пролекарственного соединения инсулин-гидрогеля содержала одно или несколько веществ из загустителя и/или агента, модифицирующего вязкость.
Термин «вспомогательное вещество» предпочтительно относится к разбавителю, адъюванту или носителю, с которым вводится терапевтическое соединение. Такие фармацевтические вспомогательные вещества могут быть стерильными жидкостями, такими как вода и масла, включая нефтяные масла, и масла животного, растительного или синтетического происхождения, включающие, но не ограниченные этим, арахисовое масло, соевое масло, минеральное масло, кунжутное масло и т.п. Вода является предпочтительным вспомогательным веществом при пероральном введении фармацевтической композиции. Физиологический раствор и водный раствор декстрозы являются предпочтительными вспомогательными веществами при внутривенном введении фармацевтической композиции. Физиологический раствор, водные растворы глюкозы и глицерина предпочтительно использовать в качестве жидких вспомогательных веществ для инъекционных растворов. Подходящие фармацевтические вспомогательные вещества включают крахмал, глюкозу, лактозу, сахарозу, желатин, солод, рис, муку, мел, силикагель, стеарат натрия, глицерина моностеарат, тальк, хлорид натрия, сухое обезжиренное молоко, глицерин, пропилен, гликоль, воду, этанол и т.п. Композиция, при желании, может также содержать минорные количества смачивающих или эмульгирующих агентов или рН-буферных агентов. Эти композиций могут представлять собой растворы, суспензии, эмульсии, таблетки, пилюли, капсулы, порошки, составы с замедленным высвобождением и т.п. Композиция может быть составлена в виде суппозитория с традиционными связующими веществами и вспомогательными веществами, такими как триглицериды. Пероральный состав может включать стандартные вспомогательные вещества, такие как фармацевтического качества маннит, лактоза, крахмал, стеарат магния, сахаринат натрия, целлюлоза, карбонат магния и т.п. Примеры подходящих фармацевтических вспомогательных веществ описаны в «Remington's Pharmaceutical Sciences» под авторством E.W. Martin. Такие композиции будут содержать терапевтически эффективное количество терапевтического соединения, предпочтительно, в очищенной форме, совместно с соответствующим количеством вспомогательного вещества, обеспечивающим форму для надлежащего введения пациенту. Состав должен соответствовать пути введения.
В общем варианте осуществления изобретения фармацевтическая композиция по настоящему изобретению либо в сухой форме, либо в виде суспензии или в виде другой лекарственной формы может представлять собой однодозовую или мультидозовую композицию.
В одном варианте осуществления настоящего изобретения сухая композиция пролекарственного соединения инсулин-гидрогеля представлена в виде однодозовой композиции, означающей, что контейнер, в котором она поставляется, содержит одну фармацевтическую дозу.
Поэтому, в другом аспекте настоящего изобретения композиция представлена в качестве однодозовой композиции.
Предпочтительно, чтобы суспензионная композиция была мультидозовой композицией, означающей, что она содержит более одной терапевтической дозы. Предпочтительно, чтобы мультидозовая композиция содержала по меньшей мере 2 дозы. Такую мультидозовую композицию инсулин-гидрогеля можно использовать либо у различных пациентов, нуждающихся в этом, либо она может быть предназначена для использования одним пациентом, в случае чего после применения первой дозы, оставшиеся дозы хранятся до необходимости в них.
В другом аспекте настоящего изобретения композиция содержится в контейнере. Предпочтительно, чтобы контейнером был двухкамерный шприц. В частности, сухая композиция по настоящему изобретению находится в первой камере двухкамерного шприца, а раствор для восстановления находится во второй камере двухкамерного шприца.
Перед введением сухой композиции пролекарственного соединения инсулин-гидрогеля пациенту, нуждающемуся в этом, восстанавливают сухую композицию. Восстановление можно проводить в контейнере, в котором находится сухая композиция пролекарственного соединения инсулин-гидрогеля, например, в флаконе, шприце, двухкамерном шприце, ампуле и картридже. Восстановление проводят, добавляя определенное количество раствора для восстановления к сухой композиции. Растворами для восстановления являются стерильные жидкости, такие как вода или буфер, которые могут содержать дополнительные добавки, такие как консерванты и/или противомикробные агенты. Если композиция пролекарственного соединения инсулин-гидрогеля представлена в однодозовой форме, то раствор для восстановления может содержать один или несколько из консервантов и/или противомикробных агентов. Предпочтительно, чтобы раствором для восстановления была стерильная вода. Если композиция пролекарственного соединения инсулин-гидрогеля является мультидозовой композицией, предпочтительно, чтобы раствор для восстановления содержал один или несколько из консервантов и/или противомикробных агентов, таких как, например, бензиловый спирт и крезол.
Дополнительный аспект настоящего изобретения относится к способу введения восстановленной композиции пролекарственного соединения инсулин-гидрогеля. Композицию пролекарственного соединения инсулин-гидрогеля можно вводить с помощью инъекций или инфузий, включая интрадермальные, подкожные, внутримышечные, внутривенные, внутрикостные и внутрибрюшинные.
Дополнительным аспектом является способ получения восстановленной композиции, содержащей терапевтически эффективное количество пролекарственного соединения инсулина, и, необязательно, одно или несколько фармацевтически приемлемых вспомогательных веществ, в котором инсулин транзиентно соединен с гидрогелем, причем способ включает в себя стадию:
• контакта композиции по настоящему изобретению с раствором для восстановления.
Другим аспектом является композиция для восстановления, содержащая терапевтически эффективное количество пролекарственного соединения инсулин-гидрогеля и, необязательно, одно или несколько фармацевтически приемлемых вспомогательных веществ, в котором инсулин транзиентно присоединен к гидрогелю, получаемому вышеуказанным способом.
Другим аспектом по настоящему изобретению является способ изготовления сухой композиции пролекарственного соединения инсулин-гидрогеля. В одном варианте осуществления изобретения такая суспензионная композиция изготовлена в результате:
(i) смешивания пролекарственного соединения инсулин-гидрогеля с одним или несколькими вспомогательными веществами,
(ii) переноса количества, равного одной или нескольким дозам в подходящий контейнер,
(iii) высушивания композиции в указанном контейнере, и
(iv) герметизации контейнера.
Подходящими контейнерами являются флаконы, шприцы, двухкамерные шприцы, ампулы и картриджи.
Другим аспектом является набор. Если устройством для введения является просто гиподермальный шприц, то в этом случае набор может включать шприц, иглу и контейнер, содержащий сухую композицию пролекарственного соединения инсулин-гидрогеля для применения с использованием шприца, и второй контейнер, содержащий раствор для восстановления. В более предпочтительных вариантах осуществления изобретения инъекционное устройство отличается от простого гиподермального шприца, а отдельный контейнер с восстановленным пролекарственным соединением инсулин-гидрогеля адаптирован под инъекционное устройство таким образом, чтобы при использовании жидкая композиция в контейнере была соединена с выходным отверстием инъекционного устройства с возможностью переноса текучей среды. Примеры устройств для введения включают, но не ограничены этим, гиподермальные шприцы и шприц-ручки. Особенно предпочтительными устройствами для введения являются шприц-ручки, в случае которых контейнером является картридж, предпочтительно, одноразовый картридж.
Предпочтительный набор включает в себя иглу и контейнер, содержащий композицию по настоящему изобретению и, необязательно, дополнительно содержащий раствор для восстановления, причем контейнер адаптирован для использования с иглой. Предпочтительно, чтобы контейнером был двухкамерный шприц.
В другом аспекте изобретение относится к картриджу, содержащему композицию пролекарственного соединения инсулин-гидрогеля, описанную выше, для использования с шприц-ручкой. Картридж может содержать одноразовую дозу или многоразовую дозу инсулина.
В одном варианте осуществления настоящего изобретения суспензионная композиция пролекарственного соединения инсулин-гидрогеля содержит не только пролекарственное соединение инсулин-гидрогеля и одно или несколько вспомогательных веществ, но также содержит другие биологически активные агенты, либо в свободной форме, либо в виде пролекарственных соединений. Предпочтительно, чтобы эти один или несколько биологически активных агентов были пролекарственными соединениями, более предпочтительно, пролекарственными соединениями с гидрогелем. Такие биологически активные агенты включают, но не ограничены этим, соединения из следующих классов:
(i) Сульфонилмочевины, такие как, например, хлорпропамид, толазамид, толбутамид, глибурид, глипизид, глимепирид и т.п.,
(ii) Меглитиниды, такие как, например, репаглинид,
(iii) Глюкагон-подобный пептид-1 (GLP-1) и его миметики, глюкозоинсулинотропный пептид (GIP) и его миметики, эксендин и его миметики, и ингибиторы дипептилпротеазы (DPPIV),
(iv) Бигуаниды, такие как, например, метформин,
(v) Тиазолидиндионы, такие как, например, розиглитазон, пиоглитазон, троглитазон, изаглитазон (известный как МСС-555), 2-[2-[(2R)-4-гексил-3,4-дигидро-3-оксо-2H-1,4-бензоксазин-2-ил]этокси]-бензолуксусная кислота и т.п.,
(vi) GW2570 и т.п.,
(vii) Модуляторы ретиноид-Х-рецепторов (RXR), такие как, например, таргретин, 9-цис-ретиноевая кислота и т.п.,
(viii) Другие инсулин-сенсибилизирующие агенты, такие как, например, INS-1, ингибиторы PTP-1B, ингибиторы GSK3, ингибиторы гликоген-фосфорилазы, ингибиторы фруктозо-1,6-бисфосфатазы и т.п.,
(ix) Инсулины, включая регулярный или короткого действия, средней продолжительности действия и длительного действия, ингаляционный инсулин и аналоги инсулина, такие как молекулы инсулина с минимальными отличиями в природной аминокислотной последовательности,
(x) Низкомолекулярные миметики инсулина, включающие, но не ограниченные этим, L-783281, TE-17411 и т.п.,
(xi) Ингибиторы Na-глюкозного ко-транспортера, такие как T-1095, T-1095 A, флоризин и т.п.,
(xii) Агонисты амилина, которые включают, не ограничиваясь этим, прамлинтид и т.п.,
(xiii) Антагонисты глюкагона, такие как AY-279955 и т.п.
Кроме антидиабетических агентов, биологически активными соединениями могут быть агенты против ожирения, такие как орлистат, ингибитор панкреатической липазы, который предупреждает расщепление и всасывание жиров, или сибутрамин, соединение, подавляющее аппетит и ингибирующее обратный захват серотонина, норэпинефрина, допамина в мозге, ростовые факторы, повышающие мобилизацию жиров (например, гормон роста, IGF-1, рилизинг-фактор гормона роста), оксинтомодулин и модуляторы грелина. Другие потенциальные биологически активные агенты против ожирения включают, но не ограничены этим, соединения, подавляющие аппетит, действующие через адренергические механизмы, такие как бензфетамин, фенметразин, фентермин, диэтилпропион, мазиндол, сибутрамин, фенилпропаноламин или эфедрин; агенты, подавляющие аппетит, действующие через серотонергические механизмы, такие как квипазин, флуоксетин, сертралин, фенфлурамин или дексфенфлурамин; агенты, подавляющие аппетит, действующие через допаминовые механизмы, например, апоморфин; агенты, подавляющие аппетит, действующие через гистаминергические механизмы, такие как (например, гистаминовые миметики, модуляторы Н3-рецептора); усилители энергозатрат, такие как бета-3 адренергические агонисты и стимуляторы функций разобщающих белков; лептин и лептиновые миметики (например, метрелептин); антагонисты нейропептида Y; модуляторы рецепторов меланокортина-1, 3 и 4; агонисты холецистокинина; миметики и аналоги глюкагон-подобного пептида-1 (GLP-1) (например, Эксендин); андрогены (например, дегидроэпиандростерон и производные, такие как этиохоландион), тестостерон, анаболические стероиды (например, оксандролон) и стероидные гормоны; антагонисты галаниновых рецепторов; цитокины, такие как цилиарный нейротропный фактор; ингибиторы амилазы; агонисты/миметики энтеростатина; антагонисты орексина/гипокретина; антагонисты урокортина; агонисты бомбезина; модуляторы протеинкиназы А; миметики кортикотропин-рилизинг фактора; миметики кокаин- и амфетамин-регулируемого транскрипта; миметики пептида, родственного гену кальцитонина; и ингибиторы синтетазы жирных кислот.
В альтернативном варианте осуществления изобретения композиция пролекарственного соединения инсулин-гидрогеля по настоящему изобретению объединена со вторым биологически активным соединением таким образом, что пролекарственное соединение инсулин-гидрогеля вводят нуждающемуся в нем пациенту первым, после чего вводят второе биологически активное соединение. В качестве альтернативы, композицию инсулин-гидрогеля вводят нуждающемуся в ней пациенту после введения другого соединения этому же пациенту.
Кроме антидиабетических агентов, биологически активными соединениями могут быть агенты против ожирения, такие как орлистат, ингибитор панкреатической липазы, который предупреждает расщепление и всасывание жиров, или сибутрамин, соединение, подавляющее аппетит и ингибирующее обратный захват серотонина, норэпинефрина, допамина в мозге, ростовые факторы, повышающие мобилизацию жиров (например, гормон роста, IGF-1, рилизинг-фактор гормона роста), оксинтомодулин и модуляторы грелина. Другие потенциальные биологически активные агенты против ожирения включают, но не ограничены этим, соединения, подавляющие аппетит, действующие через адренергические механизмы, такие как бензфетамин, фенметразин, фентермин, диэтилпропион, мазиндол, сибутрамин, фенилпропаноламин или эфедрин; агенты, подавляющие аппетит, действующие через серотонергические механизмы, такие как квипазин, флуоксетин, сертралин, фенфлурамин или дексфенфлурамин; агенты, подавляющие аппетит, действующие через допаминовые механизмы, например, апоморфин; агенты, подавляющие аппетит, действующие через гистаминергические механизмы, такие как (например, гистаминовые миметики, модуляторы Н3-рецептора); усилители энергозатрат, такие как бета-3 адренергические агонисты и стимуляторы функций разобщающих белков; лептин и лептиновые миметики (например, метрелептин); антагонисты нейропептида Y; модуляторы рецепторов меланокортина-1, 3 и 4; агонисты холецистокинина; миметики и аналоги глюкагон-подобного пептида-1 (GLP-1) (например, Эксендин); андрогены (например, дегидроэпиандростерон и производные, такие как этиохоландион), тестостерон, анаболические стероиды (например, оксандролон) и стероидные гормоны; антагонисты галаниновых рецепторов; цитокины, такие как цилиарный нейротропный фактор; ингибиторы амилазы; агонисты/миметики энтеростатина; антагонисты орексина/гипокретина; антагонисты урокортина; агонисты бомбезина; модуляторы протеинкиназы А; миметики кортикотропин-рилизинг фактора; миметики кокаин- и амфетамин-регулируемого транскрипта; миметики пептида, родственного гену кальцитонина; и ингибиторы синтетазы жирных кислот.
В альтернативном варианте осуществления изобретения композиция пролекарственного соединения инсулин-гидрогеля по настоящему изобретению объединена со вторым биологически активным соединением таким образом, что пролекарственное соединение инсулин-гидрогеля вводят нуждающемуся в нем пациенту первым, после чего вводят второе биологически активное соединение. В качестве альтернативы, композицию инсулин-гидрогеля вводят нуждающемуся в ней пациенту после введения другого соединения этому же пациенту.
Пациенты, которые нуждаются в лечении композициями инсулина длительного действия, описанного в настоящем изобретении, имеют высокий риск развития сопутствующих заболеваний. Соответственно, можно использовать комбинацию инсулина длительного действия по настоящему изобретению с соответствующими биологически активными соединениями, например, для предупреждения, задержки развития или лечения заболеваний и расстройств, выбранных из группы, состоящей из гипертонии (в том числе, но не ограниченной этим, изолированной систолической гипертонии и семейной дислипидемической гипертонии), застойной сердечной недостаточности, гипертрофии левого желудочка, периферической артериальной болезни, диабетической ретинопатии, макулярной дегенерации, катаракты, диабетической нефропатии, гломерулосклероза, хронической почечной недостаточности, диабетической нейропатии, синдрома Х, предменструального синдрома, коронарной болезни сердца, стенокардии, тромбоза, атеросклероза, инфаркта миокарда, транзиторных ишемических приступов, инсульта, рестеноза сосудов, гипергликемии, гиперинсулинемии, гиперлипидемии, гипертриглицеридемии, инсулинорезистентности, нарушенного метаболизма глюкозы, состояния нарушенной глюкозотолерантности, состояния нарушенного уровня глюкозы натощак, ожирения, эректильной дисфункции, заболеваний кожи и соединительной ткани, диабетической стопы и язвенного колита, эндотелиальной дисфункции и нарушенной податливости сосудов.
Предупредить, задержать развитие или осуществлять лечение выбранных из вышеуказанной группы заболеваний и расстройств, можно, используя комбинации композиции инсулина длительного действия по настоящему изобретению по меньшей мере с одним биологически активным соединением, выбранным из классов лекарственных соединений, используемых для лечения указанных состояний, включая антагонисты АТ1-рецепторов; ингибиторы ангиотензин-конвертирующего фермента (АСЕ); ингибиторы ренинна; блокаторы бета-адренергических рецепторов; блокаторы альфа-адренергических рецепторов; блокаторы кальциевых каналов; ингибиторы альдостеронсинтетазы; антагонисты альдостероновых рецепторов; ингибиторы нейтральной эндопептидазы (NEP); двойные ингибиторы ангиотензин-конвертирующего фермента/нейтральной эндопептидазы (ACE/NEP); антагонисты эндотелиновых рецепторов; диуретики; статины; нитраты; антикоагулянты; натрийуретические пептиды; соединения дигиталиса; модуляторы PPAR.
Соответственно, в дополнительном аспекте настоящее изобретение относится к применению соединения инсулина для получения фармакокинетической композиции, содержащей соединение инсулина с концентрацией по меньшей мере 10 мг/мл, характеризующейся тем, что она имеет фармакокинетический профиль по существу без выброса соединения инсулина, для лечения или предупреждения заболевания или расстройства, ассоциированных с недостаточностью инсулина, при которых лечение или предупреждение с использованием соединения инсулина является полезным.
В одном варианте осуществления настоящего изобретения концентрация соединения инсулина составляет по меньшей мере 11 мг/мл, например, от 11 мг/мл до 35 мг/мл, более предпочтительно, от 15 мг/мл до 25 мг/мл, еще более предпочтительно, около 20 мг/мл и, еще более предпочтительно, около 24 мг/мл.
Объем, вводимый, например с помощью шприца, субъекту, такому как человек, предпочтительно, составляет меньше 1,5 мл, обычно - 1,0 мл или меньше.
В дополнительном варианте осуществления заболевание или расстройство, ассоциированные с недостаточностью инсулина, при которых лечение или предупреждение с использованием соединения инсулина является благотворным, выбраны из гипергликемии, предиабета, нарушенной глюкозотолерантности, диабета I типа, диабета II типа, синдрома Х. Обычно, такой болезнью или таким расстройством является диабет II типа.
Еще один вариант осуществления изобретения касается лечения или предупреждения гипергликемии, предиабета, нарушенной глюкозотолерантности, диабета I типа, диабета II типа, синдрома Х у млекопитающего, например, у человека.
В дополнительном варианте осуществления изобретения у человека поставлен диагноз предиабета, нарушенной глюкозотолерантности, ожирения, диабета I типа, диабета II типа, синдрома Х.
В еще одном варианте осуществления настоящего изобретения фармакокинетический профиль измеряют в плазме крови млекопитающего, например, плазме крови человека.
В дополнительном варианте осуществления настоящего изобретения композиция характеризуется тем, что имеет соотношение пиковой концентрации к остаточной концентрации меньше 2, например, меньше 1,75, меньше 1,5 или меньше 1,25.
В еще одном варианте осуществления настоящего изобретения композиция отличается постоянным высвобождением соединения структурно интактного инсулина в течение всего интервала времени между введениями.
В дополнительном варианте осуществления изобретения полный интервал времени между введениями составляет по меньшей мере примерно 80 часов, например, примерно 110 часов, обычно, неделю.
В еще одном варианте осуществления настоящего изобретения соединение инсулина является пролекарственным соединением. Такое пролекарственное соединение обычно можно выбрать из описанных выше соединений, представленных формулой D-L.
В дополнительном варианте осуществления изобретения соединение инсулина полностью содержится в депо-препарате, обычно полимерном геле, таком как гидрогель, например, высоко гидратированном полимерном матриксе. Обычно, высоко гидратированный полимерный матрикс минимизирует межмолекулярные контакты молекул инсулина.
В еще одном варианте осуществления настоящего изобретения соединение инсулина ковалентно связано в депо-препарате, обычно, полимерном геле, таком как гидрогель, например, высоко гидратированном полимерном матриксе.
В дополнительном варианте осуществления изобретения композиция характеризуется тем, что она является жидкой композицией, которая после инъекции образует депо-препарат.
В еще одном дополнительном варианте осуществления изобретения композицию вводят с помощью инъекции, например, подкожной или внутримышечной.
В дополнительном варианте осуществления изобретения соединение инсулина выбрано из инсулина или аналогов инсулина человека или других биологических видов, и их производных и конъюгатов. Более предпочтительным является инсулин и аналоги инсулина человека, как, например, инсулин гларгин, инсулин детемир, инсулин лиспро, инсулин аспартат, инсулин глулизин.
В еще одном дополнительном варианте осуществления изобретения пиковая концентрация достигается в первые 24 часа после введения, например, в первые 12 часов после введения, например, в течение первых 6 часов после введения.
В еще одном варианте осуществления изобретения композиция дополнительно содержит соединение GLP-1.
Еще одним дополнительным вариантом осуществления изобретения является комбинация с соединением GLP-1. Обычно соединение инсулина вводят первым или наоборот, и соединение инсулина и соединение GLP-1 можно вводить одновременно или последовательно.
В дополнительном аспекте настоящее изобретение относится к способу лечения или предупреждения заболевания или расстройства, с недостаточностью инсулина, при которых лечение или предупреждение с использованием соединения инсулина является полезным, таких как гипергликемия, предиабет, нарушенная глюкозотолерантность, диабет I типа, диабет II типа, синдром Х, у млекопитающего, нуждающегося в таком лечении или предупреждении, путем введения терапевтически эффективного количества соединения инсулина в концентрации, достаточной для поддержания терапевтически эффективного уровня соединения инсулина в плазме крови в течение по меньшей мере 3 дней, обычно, по меньшей мере 80 часов, например, в течение недели или большего периода времени, которое характеризуется тем, что имеет фармакокинетический профиль in vivo по существу без выброса соединения инсулина.
В дополнительном аспекте настоящее изобретение относится к способу лечения или предупреждения заболевания или расстройства, с недостаточностью инсулина, при которых лечение или предупреждение с использованием соединения инсулина является полезным, таких как гипергликемия, предиабет, нарушенная глюкозотолерантность, диабет I типа, диабет II типа, синдром Х, у млекопитающего, нуждающегося в таком лечении или предупреждении, путем введения терапевтически эффективного количества соединения инсулина в концентрации по меньшей мере 10 мг/мл, которое характеризуется тем, что имеет фармакокинетический профиль по существу без выброса соединения инсулина.
Вариант осуществления изобретения относится к лечению или предупреждению гипергликемии, предиабета, нарушенной глюкозотолерантности, диабета I типа, диабета II типа, синдрома Х у млекопитающего, например, человека. Обычно у человека поставлен диагноз предиабета, нарушенной глюкозотолерантности, ожирения, диабета I типа, диабета II типа, синдрома Х.
В дополнительном варианте осуществления изобретения концентрация соединения инсулина составляет по меньшей мере 11 мг/мл, например, от 11 мг/мл до 35 мг/мл.
В еще одном варианте осуществления настоящего изобретения фармакокинетический профиль измеряют в плазме крови млекопитающего, например, плазме крови человека.
В дополнительном варианте осуществления настоящего изобретения композиция характеризуется тем, что имеет соотношение пиковой концентрации к остаточной концентрации меньше 2, например, меньше 1,75, меньше 1,5 или меньше 1,25.
В еще одном варианте осуществления настоящего изобретения композиция характеризуется постоянным высвобождением соединения структурно интактного инсулина в течение всего интервала времени между введениями.
В дополнительном варианте осуществления изобретения полный интервал времени между введениями составляет по меньшей мере примерно 80 часов, например, примерно 110 часов, обычно, неделю.
В еще одном варианте осуществления настоящего изобретения соединение инсулина является пролекарственным соединением, таким как любое одно из описанных в настоящем документе пролекарственных соединений.
В дополнительном варианте осуществления изобретения соединение инсулина полностью содержится в депо-препарате, обычно полимерном геле, таком как гидрогель, например, высоко гидратированном полимерном матриксе.
В еще одном варианте осуществления настоящего изобретения соединение инсулина ковалентно связано в депо-препарате, обычно, полимерном геле, таком как гидрогель, например, высоко гидратированном полимерном матриксе.
В дополнительном варианте осуществления изобретения композиция характеризуется тем, что она является жидкой композицией, которая после инъекции образует депо-препарат.
В еще одном дополнительном варианте осуществления изобретения композицию вводят с помощью инъекции, например, подкожной или внутримышечной.
В дополнительном варианте осуществления изобретения соединение инсулина выбрано из инсулина или аналогов инсулина человека или других биологических видов, и их производных и конъюгатов. Более предпочтительным является инсулин и аналоги инсулина человека, как, например, инсулин гларгин, инсулин детемир, инсулин лиспро, инсулин аспартат, инсулин глулизин. В еще одном дополнительном варианте осуществления изобретения пиковая концентрация достигается в первые 24 часа после введения, например, в первые 12 часов после введения, например, в течение первых 6 часов после введения.
В дополнительном варианте осуществления изобретения композиция дополнительно содержит соединение GLP-1.
Еще одним дополнительным вариантом осуществления изобретения является комбинация с соединением GLP-1. Обычно соединение инсулина вводят первым или наоборот, и соединение инсулина и соединение GLP-1 можно вводит одновременно или последовательно.
В дополнительном аспекте настоящее изобретение относится к набору, содержащему фармацевтическую композицию по любому одному из вариантов осуществления, описанных в настоящем документе, и контейнер для введения композиции. Обычно, контейнером является шприц.
Вариант набора дополнительно содержит соединение GLP-1.
Фиг.1а: UPLC-хроматограмма конъюгата инсулина и линкера 12a.
Фиг.1b: UPLC-хроматограмма конъюгата инсулина и линкера 12b.
На фигуре 2 показана средняя концентрация инсулина в плазме у животных 1-10 после однократного подкожного введения дозы тестируемого соединения 11а, содержащей 6 мг инсулина, здоровым крысам в течение 2-недельного периода. (Планки погрешностей указаны как ± среднеквадратичное отклонение, определенное для всех 10 животных, значения в точке t0 определяли за 3 дня до введения соединения).
Фигура 3: Средняя концентрация инсулина в плазме у животных 1-8 после однократного подкожного введения дозы тестируемого соединения 11da, содержащей 3 мг инсулина, здоровым крысам в течение 13 дней. (Планки погрешностей указаны как ± среднеквадратичное отклонение, определенное для всех 8 животных, значения в точке t0 определяли за 1 день до введения соединения).
Фигура 4: Концентрация инсулина в плазме (серые квадраты) и уровень глюкозы в крови (черные круги) после однократного подкожного введения дозы тестируемого соединения 11da, содержащей 6,4 мг инсулина, диабетическим крысам (n=7). (Планки погрешностей указаны как ± среднеквадратичное отклонение, определенное для всех 7 животных, значения в точке t0 определяли за 4 дня до введения соединения).
Фигура 5: Средняя концентрация инсулина в плазме после однократного подкожного введения дозы 8 мг/кг тестируемого соединения 11db здоровым крысам в течение первых 24 часов после дозировки (анализ выброса инсулина). 8 крыс распределяли по 2 группам, и образцы крови для фармакокинетического анализа собирали, чередуя обе группы. Ни в одной из групп не наблюдался заметный эффект выброса инсулина. (Планки погрешностей указаны как ± среднеквадратичное отклонение, определенное для всех животных в группе, значения в точке t0 определяли за 1 день до введения соединения).
Фигура 6: Концентрация инсулина в плазме (серые квадраты) и уровень глюкозы в крови (черные круги) в течение 4-недельного периода времени после 3 еженедельных подкожных доз 8 мг/кг тестируемого соединения 11da диабетическим крысам (n=8). (Планки погрешностей указаны как ± среднеквадратичное отклонение, определенное для всех 8 животных, значения в точке t0 определяли за 3 дня до введения соединения).
Фигура 7: Средняя концентрация инсулина в плазме у животных 1-8 (для животных 1-4 и животных 5-8 через 0,3, 1 час, 2 и 4 часа, соответственно) после однократной подкожной инъекции 12 мг/кг инсулин, в составе тестируемого соединения 11dc здоровым крысам в течение 13 дней. (Планки погрешностей указаны как ± среднеквадратичное отклонение, определенное для всех 8 животных, значения в точке t0 определяли за 4 дня до введения соединения).
Фигура 8: Наложение высвобождения инсулина и деградации гидрогеля для соединения инсулин-линкер-гидрогеля 11а. Приведена зависимость содержания инсулина в соединении инсулин-линкер-гидрогеля (треугольники) и высвобождения каркасных частей молекулы (круги) при инкубации инсулин-линкер-гидрогеля при рН 7,4 и 37°C от времени инкубации.
На фигуре 9 показан график зависимости потока от силы при использовании иглы 30G. Данные указаны: черные квадраты = этиленгликоль; черные треугольники = вода; черные точки = пролекарственное соединение инсулина и гидрогеля.
Примеры
Материалы и методы
Рекомбинантный инсулин человека был получен от Biocon Ltd., Bangalore, India.
Амино(4-лучевой ПЭГ), 5 кДа, был получен от JenKem Technology, Beijing, P. R. China.
NHS-эфир N-(3-малеимидопропил)-21-амино-4,7,10,13,16,19-гексаокса-генеэйкозановой кислоты (Mal-PEG6-NHS) был получен от Celares GmbH, Berlin, Germany.
Если не указано иное, то 2-хлортритилхлоридная смола, HATU, N-циклогексил-карбодиимид-N'-метилполистирол и аминокислоты были получены от Merck Biosciences GmbH, Schwalbach/Ts, Germany. Соединение Fmoc(NMe)-Asp(OtBu)-OH получено от Bachem AG, Bubendorf, Switzerland. S-Тритил-6-меркаптогексановая кислота была приобретена у Polypeptide, Strasbourg, France. Если не указано иное, то используемые аминокислоты имели L-конфигурацию.
Все другие химические соединения были от Sigma-ALDRICH Chemie GmbH, Taufkirchen, Germany.
Твердофазный синтез проводили на 2-хлортритилхлоридной (ТСР) смоле с нагрузкой 1,3 ммоль/г. В качестве реакционных сосудов использовали шприцы с полипропиленовыми фритами (фильтрующими элементами).
Связывание первой аминокислоты со смолой проводили по инструкции изготовителя.
Удаление защитной Fmoc-группы:
Для удаления Fmoc-защитной группы смолу промывали смесью пиперидина/DBU/DMF в объемном соотношении 2/2/96 (две промывки по 10 минут каждая), и промывали DMF (10 раз).
Удаление защитной Fmoc-группы со смол, нагруженных Fmoc-Aib:
Fmoc-группу с иммобилизованной Fmoc-Aib-OH удаляли, перемешивая смолу в смеси DMF/пиперидина в объемном соотношении 4/1 при 50°C в течение 20 минут (2 раза).
Протокол отщепления синтезированного продукта от 2-хлортритилхлоридной смолы:
После завершения синтеза смолу промывали DCM, высушивали под вакуумом и два раза по 30 минут обрабатывали смесью DCM/HFIP с объемным соотношением 6/4. Объединяли элюат, летучие соединения удаляли под током азота, и очистку полученного неочищенного продукта проводили с помощью обращенно-фазовой ВЭЖХ. ВЭЖХ-фракции, содержащие продукт, объединяли и лиофилизировали.
Продукты, содержащие амины, полученные в виде солей трифторуксусной кислоты (TFA) конвертировали в соответствующие соли гидрохлористой кислоты HCl), используя ионообменную смолу (Discovery DSC-SAX, Supelco, USA). Эту стадию проводили в том случае, если ожидали, что остаточная TFA, например, будет нарушать последующую реакцию присоединения.
Очистка с помощью обращенно-фазовой ВЭЖХ:
Обращенно-фазовую ВЭЖХ проводили на колонке C18 ReproSil-Pur 300 ODS-3 5 мкм (100×20 мм или 100×40 мм) (Dr. Maisch, Ammerbuch, Germany), соединенной с ВЭЖХ-системой Waters 600 и детектором Waters 2487. Использовали линейный градиенты раствора А (0,1% TFA в H2O) и раствора B (0,1% TFA в ацетонитриле). ВЭЖХ-фракции, содержащие продукт, лиофилизировали.
Флэш-хроматография
Очистку с помощью флэш-хроматографии проводили на системе Isolera One от Biotage AB, Sweden, используя силиконовые картриджи Biotage KP-Sil и н-гептан и этилацетат в качестве элюентов. Продукты детектировали при 254 нм.
В случае гидрогелевых шариков, в качестве реакционных сосудов или на стадиях промывки использовали шприцы, оборудованные полипропиленовыми фритами.
Методы анализа
Аналитическую ультра-производительную жидкостную хроматографию (UPLC) проводили на системе Waters Acquity, оборудованной колонкой Waters BEH300 C18 (2,1×50 мм, размер частиц 1,7 мкм) соединенной с масс-спектрометром LTQ Orbitrap Discovery от Thermo Scientific.
МС продуктов ПЭГ показал серии звеньев (CH2CH2O)n вследствие полидисперсности исходного ПЭГ. Для упрощения интерпретации в примерах приводили только один одиночный репрезентативный m/z-сигнал. МС конъюгатов инсулина приведены для представительных изотопов и относятся к четырехпротонным аддуктам [M+4H]4+.
Если не указано иное, то гель-фильтрацию (SEC) проводили на системе Amersham Bioscience AEKTAbasic, оборудованной колонкой Superdex 200 5/150 GL (Amersham Bioscience/GE Healthcare) с 0,45-микронным входным фильтром. В качестве подвижной фазы использовали 20 мМ фосфат натрия, 140 мМ NaCl, pH 7,4.
Пример 1
Синтез каркасного реагента 1g
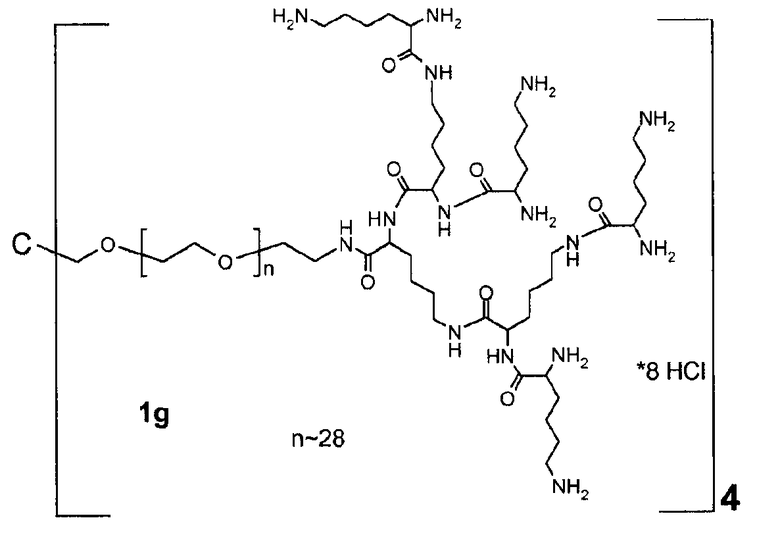
Каркасный реагент 1g синтезировали из амино(4-лучевого)-ПЭГ5000 1а по следующей схеме:
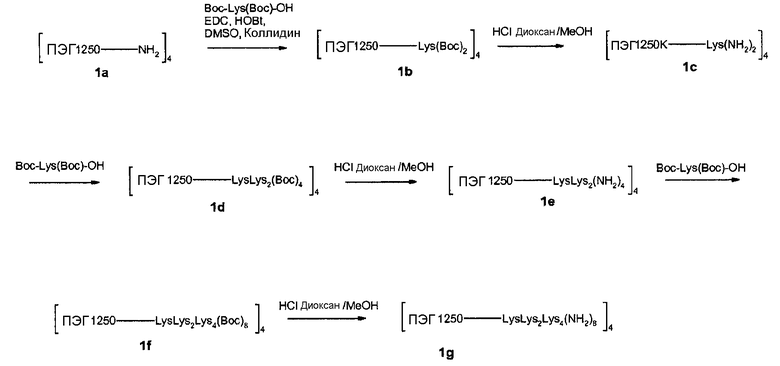
Для синтеза соединения 1b, 5,20 г (1,00 ммоль) HCl соли амино(4-лучевого)-ПЭГ5000 1а (MW приблизительно 5200 г/моль) растворяли в 20 мл безводного DMSO. Добавляли Boc-Lys(Boc)-OH (2,17 г, 6,25 ммоль) в 5 мл безводного DMSO, EDC•HCl (1,15 г, 6,00 ммоль), HOBt•H2O (0,96 г, 6,25 ммоль) и коллидин (5,20 мл, 40 ммоль). Реакционную смесь перемешивали в течение 30 минут при комнатной температуре.
Реакционную смесь разводили в 1200 мл дихлорметана и 2 раза промывали 600 мл 0,1н H2SO4, один раз маточным раствором, 2 раза 0,1 M NaOH и 4 раза смесью маточного раствора и воды в соотношении 1/1 (объемном). Водные фазы повторно экстрагировали 500 мл DCM. Органические фазы высушивали над Na2SO4, отфильтровывали и выпаривали, получая 6,3 г неочищенного продукта 1b в виде бесцветного масла. Соединение 1b очищали с помощью обращенно-фазовой ВЭЖХ.
Выход составлял 3,85 г (59%) бесцветного стекловидного продукта 1b.
МС: m/z 1294,4 = [M+5H]5+ (рассчитанное значение = 1294,6).
Соединение 1с получали, перемешивая 3,40 г соединения 1b (0,521 ммоль) в 5 мл метанола и 9 мл 4н HCl в диоксане при комнатной температуре в течение 15 минут. Летучие соединения удаляли под вакуумом. Продукт использовали в следующей стадии без дополнительной очистки.
МС: m/z 1151,9 = [M+5H]5+ (рассчитанное значение = 1152,0).
Для синтеза соединения 1d, 3,26 г соединения 1c (0,54 ммоль) растворяли в 15 мл безводного DMSO. Добавляли 2,99 г Boc-Lys(Boc)-OH (8,64 ммоль) в 15 мл безводного DMSO, 1,55 г EDC•HCl (8,1 ммоль), 1,24 г HOBt•H2O (8,1 ммоль) и 5,62 мл коллидина (43 ммоль). Реакционную смесь перемешивали в течение 30 минут при комнатной температуре.
Реакционную смесь разводили в 800 мл DCM и 2 раза промывали 400 мл 0,1н H2SO4, один раз маточным раствором, 2 раза 0,1 M NaOH и 4 раза смесью маточного раствора и воды в соотношении 1/1 (объемном). Водные фазы повторно экстрагировали 800 мл DCM. Органические фазы высушивали над Na2SO4, отфильтровывали и выпаривали, получая стекловидный продукт.
Продукт растворяли в DCM и высаживали холодным (-18°С) диэтилэфиром. Эту процедуру повторяли дважды, и высушивали осадок под вакуумом.
Выход составлял 4,01 г (89%) бесцветного стекловидного продукта 1d, который использовали в следующей стадии без дополнительной очистки.
МС: m/z 1405,4 = [M+6H]6+ (рассчитанное значение = 1405,4).
Соединение 1е получали, перемешивая раствор соединения 1d (3,96 г, 0,47 ммоль) в 7 мл метанола и 20 мл 4н HCl в диоксане при комнатной температуре в течение 15 минут. Летучие соединения удаляли под вакуумом. Продукт использовали в следующей стадии без дополнительной очистки.
МС: m/z 969,6 = [M+5H]7+ (рассчитанное значение = 969,7).
Для синтеза соединения 1f, 3,55 г соединения 1е (0,48 ммоль) растворяли в 20 мл безводного DMSO. Добавляли 5,32 г Boc-Lys(Boc)-OH (15,4 ммоль) в 18,8 мл безводного DMSO, 2,76 г EDC•HCl (14,4 ммоль), 2,20 г HOBt•H2O (14,4 ммоль) и 10,0 мл коллидина (76,8 ммоль). Реакционную смесь перемешивали в течение 60 минут при комнатной температуре.
Реакционную смесь разводили в 800 мл DCM и 2 раза промывали 400 мл 0,1н H2SO4, один раз маточным раствором, 2 раза 0,1 M NaOH и 4 раза смесью маточного раствора и воды в соотношении 1/1 (объемном). Водные фазы повторно экстрагировали 800 мл DCM. Органические фазы высушивали над Na2SO4, отфильтровывали и выпаривали, что давало неочищенный продукт 1f в виде бесцветного масла.
Продукт растворяли в DCM и высаживали холодным (-18°С) диэтилэфиром. Эту процедуру повторяли дважды, и высушивали осадок под вакуумом.
Выход составлял 4,72 г (82%) бесцветного стекловидного продукта 1f, который использовали в следующей стадии без дополнительной очистки.
МС: m/z 1505,3 = [M+6H]8+ (рассчитанное значение = 1505,4).
Каркасный реагент 1g получали, перемешивая раствор соединения 1f (MW приблизительно 12035 г/моль, 4,72 г, 0,39 ммоль) в 20 мл метанола и 40 мл 4н HCl в диоксане при комнатной температуре в течение 30 минут. Летучие соединения удаляли под вакуумом.
Выход составлял 3,91 г (100%) стекловидного каркасного продукта 1g.
МС: m/z 977,2 = [M+6H]9+ (рассчитанное значение = 977,4).
Альтернативный путь синтеза соединения 1g
Для синтеза соединения 1b, к суспензии 4-лучевого-ПЭГ5000- тетраамина (1a) (50,0 г, 10,0 ммоль) в 250 мл безводного iPrOH добавляли boc-Lys(boc)-OSu (26,6 г, 60,0 ммоль) и DIEA (20,9 мл, 120 ммоль) при 45°С, и смесь перемешивали в течение 30 минут.
Далее добавляли изопропиламин (2,48 мл, 30,0 ммоль). Через 5 минут раствор разводили 1000 мл MTBE и оставляли на ночь при -20°С без перемешивания. Декантировали приблизительно 500 мл супернатанта и отбрасывали. Добавляли 300 мл холодного MTBE, и после встряхивания в течение 1 минуты продукт собирали фильтрованием через стеклянный фильтр и промывали 500 мл холодного MTBE. Продукт высушивали под вакуумом в течение 16 часов.
Выход составлял 65,6 г (74%) соединения 1b в виде белого комковатого осадка.
МС: m/z 937,4 = [M+7H]7+ (рассчитанное значение = 937,6).
Соединение 1с получали перемешиванием соединения 1b из предшествующей стадии (48,8 g, 7,44 ммоль) в 156 мл 2-пропанола при 40°С. Смесь 196 мл 2-пропанола и 78,3 мл ацетилхлорида добавляли в течение 1-2 минут при перемешивании. Раствор при температуре 40°С перемешивали в течение 30 мин и охлаждали до -30°С в течение ночи без перемешивания. Добавляли 100 мл холодного MTBE, суспензию встряхивали в течение 1 минуты и охлаждали в течение 1 ч при -30°С. Продукт собирали фильтрованием через стеклянный фильтр и промывали 200 мл холодного MTBE. Продукт высушивали под вакуумом в течение 16 часов.
Выход составлял 38,9 г (86%) соединения 1с в виде белого порошка.
МС: m/z 960,1 = [M+6H]6+ (рассчитанное значение = 960,2).
Для синтеза соединения 1d к суспензии соединения 1с из предшествующей стадии (19,0 г, 3,14 ммоль) в 80 мл 2-пропанола добавляли Boc-Lys(boc)-OSu (16,7 г, 37,7 ммоль) и DIEA (13,1 мл, 75,4 ммоль) при 45°С, и смесь перемешивали в течение 30 минут при 45°С. Далее добавляли н-пропиламин (1,56 мл, 18,9 ммоль). Через 5 минут осадок из раствора высаживали 600 мл холодного MTBE и центрифугировали (3000 об/мин, 1 мин.). Осадок высушивали под вакуумом в течение 1 часа и растворяли в 400 мл THF. Добавляли 200 мл диэтилэфира, и продукт охлаждали при -30°С в течение 16 часов без перемешивания. Суспензию отфильтровывали через стеклянный фильтр и промывали 300 мл холодного MTBE. Продукт высушивали под вакуумом в течение 16 часов.
Выход составлял 21,0 г (80%) соединения 1d в виде белого осадка.
МС: m/z 1405,4 = [M+6H]6+ (рассчитанное значение = 1405,4).
Соединение 1е получали, растворяя соединение 1d из предшествующей стадии (15,6 г, 1,86 ммоль) в 3н HCl в метаноле (81 мл, 243 ммоль) и перемешивая в течение 90 минут при 40°С. Добавляли 200 мл MeOH и 700 мл iPrOH, и смесь оставляли на 2 часа при -30°С. Для полноты кристаллизации добавляли 100 мл МТВЕ, и суспензию оставляли на ночь при -30°С. Добавляли 250 мл холодного МТВЕ, и суспензию встряхивали 1 минуту, отфильтровывали через стеклянный фильтр и промывали 100 мл холодного МТВЕ. Продукт высушивали под вакуумом.
Выход составлял 13,2 г (96%) соединения 1е в виде белого порошка.
МС: m/z 679,1 = [M+10H]10+ (рассчитанное значение = 679,1).
Для синтеза соединения 1f к суспензии соединения 1е из предшествующей стадии (8,22 г, 1,12 ммоль) в 165 мл 2-пропанола добавляли boc-Lys(boc)-OSu (11,9 г, 26,8 ммоль) и DIEA (9,34 мл, 53,6 ммоль) при 45°С, и смесь перемешивали в течение 30 минут при 45°С. Далее добавляли н-пропиламин (1,47 мл, 17,9 ммоль). Через 5 минут раствор охлаждали при -18°С в течение 2 часов, затем добавляли 165 мл холодного МТВЕ, суспензию встряхивали в течение 1 минуты и отфильтровывали через стеклянный фильтр. Далее осадок на фильтре промывали 4 раза по 200 мл смесью холодного МТВЕ/iPrOH в соотношении 4:1 и один раз 200 мл холодного МТВЕ. Продукт высушивали под вакуумом в течение 16 часов.
Выход составлял 12,8 г (90%) соединения 1f в виде бледно-желтого комковатого осадка.
МС: m/z 1505,3 = [M+8H]8+ (рассчитанное значение = 1505,4).
Каркасный реагент 1g получали, растворяя 4-лучевой ПЭГ(5кДа)(-LysLys2Lys4(boc)8)4 (1f) (15,5 г, 1,29 ммоль) в 30 мл MeOH и охлаждая до 0°С. 4н HCl в диоксане (120 мл, 480 ммоль, охлажденную до 0°С) добавляли в течение 3 минут, и удаляли ледяную баню. Через 20 минут добавляли 3н HCl в метаноле (200 мл, 600 ммоль, охлажденную до 0°С) в течение 15 минут, и раствор перемешивали в течение 10 минут при комнатной температуре. Продукт из раствора высаживали 480 мл холодного МТВЕ и центрифугировали при 3000 об/мин 1 минуту. Осадок высушивали под вакуумом в течение 1 часа и повторно растворяли в 90 мл МеОН, осаждали 240 мл холодного МТВЕ, и суспензию центрифугировали при 3000 об/мин 1 минуту. Продукт 1g высушивали под вакуумом.
Выход составлял 11,5 г (89%) в виде бледно-желтых хлопьев.
МС: m/z 1104,9 = [M+8H]8+ (рассчитанное значение = 1104,9).
Пример 2
Синтез сшивающего реагента 2d
Сшивающий реагент 2d получали из монобензилового эфира адипиновой кислоты (English, Arthur R. et al., Journal of Medicinal Chemistry, 1990, 33(1), 344-347) и ПЭГ2000 по следующей схеме:
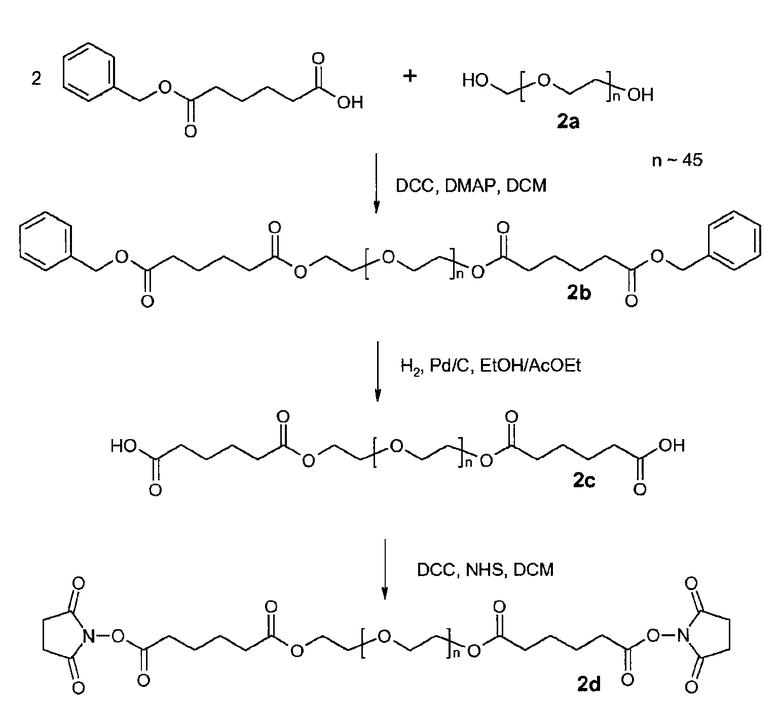
Раствор ПЭГ 2000 (2а) (11,0 г, 5,5 ммоль) и полуэфира бензиладипата (4,8 г, 20,6 ммоль) в дихлорметане (90,0 мл) охлаждали до 0°С. Добавляли дициклогексилкарбодиимид (4,47 г, 21,7 ммоль), после чего добавляли каталитическое количество DMAP (5 мг), раствор перемешивали, и реакцию оставляли на ночь (12 ч) при комнатной температуре. Колбу оставляли при +4°C на 5 часов. Осадок отфильтровывали, и растворитель полностью удаляли отгонкой под вакуумом. Осадок растворяли в 1000 мл смеси диэтилэфира/этилацетата в соотношении 1/1 (по объему) и оставляли при комнатной температуре на 2 часа, в течение которых выпадало небольшое количество осадка в виде хлопьев. Осадок удаляли фильтрованием через слой Celite®. Раствор хранили в хорошо закрытой колбе при -30°С в морозильной камере в течение 12 часов до завершения кристаллизации. Кристаллический продукт отфильтровывали через стеклянный фильтр и промывали охлажденным диэтилэфиром (-30°C). Осадок на фильтре высушивали под вакуумом. Выход составлял 11,6 г (86%) соединения 2b в виде бесцветного твердого вещества. Продукт использовали в следующей стадии без дополнительной очистки.
MS: m/z 813,1 = [M+3H]3+ (рассчитанное значение = 813,3)
В стеклянном автоклаве объемом 500 мл соединение ПЭГ2000-бис(адипиновая кислота)-бис(бензиловый эфир) 2b (13,3 g, 5,5 ммоль) растворяли в этилацетате (180 мл) и добавляли 10% палладия на угле (0,4 г). Раствор гидрировали при давлении 6 бар (6×105 Па) и 40°С до остановки поглощения водорода (5-12 ч.). Катализатор удалили на слое Celite®, а растворитель выпаривали под вакуумом. Выход (количественный) составлял 12,3 г соединения 2 с в виде желтоватого масла. Продукт использовали в следующей стадии без дополнительной очистки.
МС: m/z 753,1 = [M+3H]3+ (рассчитанное значение = 753,2).
Раствор ПЭГ2000-бис(полуэфира адипиновой кислоты) 2 с (9,43 г, 4,18 ммоль), N-гидроксисукцинимида (1,92 г, 16,7 ммоль) и дициклогексилкарбодиимида (3,44 г, 16,7 ммоль) в 75 мл безводного DCM перемешивали в течение ночи при комнатной температуре. Реакционную смесь охлаждали до 0°C, и осадок отфильтровывали. DCM выпаривали и осадок перекристаллизовали из THF.
Выход составлял 8,73 г (85%) сшивающего реагента 2d в виде бесцветного твердого вещества.
МС: m/z 817,8 = [M+3H]3+ (рассчитанная молекулярная масса = 817,9 г/моль).
Пример 3
Получение шариков гидрогеля (3) и (3а), содержащих свободные аминогруппы
Раствор 275 мг соединения 1g и 866 мг соединения 2d в 14 мл DMSO добавили к раствору 100 мг Арлацеля P135 (Croda International Plc) в 60 мл гептана. Смесь перемешивали при скорости 700 об/мин специальной металлической мешалкой в течение 10 минут при 25°С, в результате чего получали суспензию. Для осуществления полимеризации добавляли 1,0 мл N,N,N',N'-тетраметилэтилендиамина. Через 2 часа скорость перемешивания уменьшали до 400 об/мин, и смесь перемешивали в течение дополнительных 16 ч. Добавляли 1,5 мл уксусной кислоты, и через 10 минут добавляли 50 мл воды. Через 5 минут останавливали мешалку и сливали водную фазу.
Для фракционирования шариков по размеру суспензию гидрогеля и воды просеивали влажным способом на стальных ситах 75, 50, 40, 32 и 20 мкм. Фракции шариков, остающихся на ситах 32, 40 и 50 мкм объединяли и промывали 3 раза водой, 10 раз этанолом и высушивали в течение 16 часов при давлении 0,1 мбар, получая соединение 3 в виде белого порошка.
3а получали, как описано для 3, за исключением использования 1200 мг соединения 1g, 3840 мг соединения 2d, 28,6 мл DMSO, 425 мг Арацеля P135, 100 мл гептана и 4,3 мл TMEDA. Для завершения добавляли 6,6 мл уксусной кислоты и через 10 минут 50 мл воды и 50 мл насыщенного водного раствора хлорида натрия.
Содержание аминогрупп в гидрогеле определяли конъюгацией fmoc-аминокислот со свободными аминогруппами на гидрогеле и последующим определением fmoc-групп, описанным Gude, M., J. Ryf, et al. (2002) Letters in Peptide Science 9(4): 203-206.
Установленное содержание аминогрупп в соединениях 3 и 3а составляло от 0,11 до 0,16 ммоль/г.
Пример 4
Получение шариков гидрогеля с функционально-активными малеимидными группами (4), а также (4а) и (4аа), и определение степени замещения малеимидными группами
Раствор 600 мг Mal-PEG6-NHS (1,0 ммоль) в 4,5 мл смеси ацетонитрила с водой с соотношением 2/1 (объемным) добавляли к 200 мг сухих шариков гидрогеля 3. Добавляли 500 мкл натрий-фосфатного буфера (pH 7,4, 0,5 M), и суспензию интенсивно перемешивали в течение 30 минут при комнатной температуре. Шарики 4 промывали пять раз: смесью ацетонитрил/вода (в объемном соотношении 2/1), метанолом и смесью ацетонитрил/вода/TFA (в объемном соотношении 1/1/0,001).
Соединение 4а синтезировали как описано выше за исключением того, что использовали соединение 3а вместо соединения 3.
В качестве альтернативы, шарики гидрогеля 3а предварительно промывали смесью DMSO/DIEA (в объемном соотношении 99/1), промывали DMSO и инкубировали 45 минут с раствором Мал-ПЭГ6-NHS (в количестве 2,0 эквивалента относительно теоретического количества аминогрупп на гидрогеле) в DMSO. Шарики 4aa промывали два раз DMSO и три раза сукцинатным буфером с рН 3,0 (20 мМ, 1 мМ EDTA, 0,01% Tween-20). Образец инкубировали в фосфатном буфере с рН 6,0 (50 мМ, 50 мМ этаноламин, 0,01% Tween-20) в течение 1 часа при комнатной температуре и промывали пять раз натрий-сукцинатным буфером рН 3,0 (20 мМ, 1 мМ EDTA, 0,01% Tween-20).
Для определения содержания малеимидных групп аликвоту шариков гидрогеля 4, 4a или 4aa, соответственно, лиофилизировали с избытком меркаптоэтанола (в 50 мМ натрий-фосфатном буфере, 30 минут при комнатной температуре), и поглощение меркаптоэтанола определяли с помощью теста Эллмана (Ellman, G. L. et al., Biochem. Pharmacol., 1961, 7, 88-95). Установленное содержание малеимидных групп составляло от 0,11 до 0,13 ммоль/г сухого гидрогеля.
Пример 5
Синтез линкерного реагента 5d
Линкерный реагент 5d синтезировали по следующей схеме:
Синтез промежуточного линкерного реагента 5а:
4-Метокситритилхлорид (3 г, 9,71 ммоль) растворяли в DCM (20 мл) и по каплям добавляли к раствору этилендиамина (6,5 мл, 97,1 ммоль) в (20 мл). Через два часа раствор выливали в диэтиловый эфир (300 мл) и три раза промывали 50 мл смеси маточного раствора/0,1 M NaOH в объемном соотношении 30/1 и один раз маточным раствором (50 мл). Органическую фазу высушивали над Na2SO4, и летучие соединения удаляли под пониженным давлением, получая Mmt-защищенное промежуточное соединение (3,18 г, 9,56 ммоль).
Mmt-защищенное промежуточное соединение (3,18 г, 9,56 ммоль) растворяли в безводном DCM (30 мл). Добавляли 6-(тритилмеркапто)-гексановую кислоту (4,48 g, 11,47 ммоль), PyBOP (5,67 г, 11,47 ммоль) и DIEA (5,0 мл, 28,68 ммоль), и смесь интенсивно перемешивали в течение 30 минут при комнатной температуре. Раствор разбавляли диэтиловым эфиром (250 мл) и три раза промывали 50 мл смеси маточного раствора/0,1 M NaOH в объемном соотношении 30/1 и один раз маточным раствором (50 мл). Органическую фазу высушивали над Na2SO4, и летучие соединения удаляли под пониженным давлением. Соединение 5а очищали с помощью флэш-хроматографии.
Выход составлял 5,69 г (8,09 ммоль).
МС: m/z 705,4 = [M+H]+ (рассчитанное значение = 705,0).
Синтез промежуточного линкерного реагента 5b:
К раствору 5a (3,19 g, 4,53 ммоль) в безводном THF (50 мл) добавляли BH3•THF (1 M раствор, 8,5 мл, 8,5 ммоль), и раствор перемешивали в течение 16 часов при комнатной температуре. Добавляли дополнительное количество BH3•THF (1 M раствор, 14 мл, 14 ммоль) и перемешивали в течение 16 часов при комнатной температуре. Реакцию останавливали добавление метанола (8,5 мл), добавляли N,N-диметилэтилендиамин (3 мл, 27,2 ммоль), и раствор нагревали с обратным холодильником и перемешивали в течение 3 часов. Смесь разбавляли этилацетатом (300 мл) при комнатной температуре, промывали насыщенным водным раствором Na2CO3 (2 раза по 100 мл) и насыщенным водным раствором NaHCO3 (2 раза по 100 мл). Органическую фазу высушивали над Na2SO4, и летучие соединения удаляли под пониженным давлением, получая неочищенное промежуточное аминосоединение (3,22 г).
Промежуточное аминосоединение растворяли в DCM (5 мл), добавляли Boc2O (2,97 г, 13,69 ммоль), растворенное в DCM (5 мл), и DIEA (3,95 мл, 22,65 ммоль), и смесь интенсивно перемешивали при комнатной температуре в течение 30 минут. Смесь очищали с помощью флэш-хроматографии, получая неочищенное Boc- и Mmt-защищенное промежуточное соединение (3 г).
МС: m/z 791,4 = [M+H]+, 519,3 = [M-Mmt+H]+ (рассчитанное значение = 791,1).
0,4 M водный раствор HCl (48 мл) добавили к раствору Boc- и Mmt-защищенному промежуточному соединению в ацетонитриле (45 мл). Смесь разбавили ацетонитрилом (10 мл) и перемешивали в течение 1 часа при комнатной температуре. Далее рН реакционной смеси подводили до 5,5 добавлением 5 М раствора NaOH, ацетонитрил удаляли при пониженном давлении, и водный раствор экстрагировали DCM (4 раза по 100 мл). Объединенные органические фазы высушивали над Na2SO4, и летучие соединения удаляли под пониженным давлением. Неочищенное соединение 5b использовали без дополнительной очистки.
Выход составлял 2,52 г (3,19 ммоль).
МС: m/z 519,3 = [M+H]+ (рассчитанная MW = 518,8 г/моль).
Синтез промежуточного линкерного реагента 5с:
Соединение 5b (780 мг, 0,98 ммоль, чистота ~65%) и NaCNBH3 (128 мг, 1,97 ммоль) растворяли в безводном метаноле (13 мл). Добавляли раствор 2,4-диметоксибезальдегида (195 мг, 1,17 ммоль) в DCM (2 мл) и смесь перемешивали в течение 2 часов при комнатной температуре. Растворители упаривали при пониженном давлении, и неочищенный продукт растворяли в DCM и промывали насыщенным раствором NaCO3. Водную фазу три раза экстрагировали DCM, и объединенные органические фазы промывали маточным раствором, высушивали над MgSO4 и концентрировали при пониженном давлении. Соединение 5с очищали с помощью флэш-хроматографии, используя DCM и MeOH в качестве элюентов.
Выход составлял 343 мг (0,512 ммоль).
МС: m/z 669,37 = [M+H]+ (рассчитанное значение = 669,95).
Синтез линкерного реагента 5d:
Защиту с TCP-смолы, нагруженной Fmoc-Aib (980 мг, ~0,9 ммоль), удаляли в DMF/пиперидине, промывали DMF (5 раз) и DCM (6 раз) и высушивали в вакууме. Смолу обрабатывали раствором п-нитрофенилхлорформиата (364 мг, 1,81 ммоль) и коллидина (398 мкл, 3,0 ммоль) в безводном THF (6 мл), и качали в течение 30 минут. Раствор реагентов удаляли фильтрованием, и смолу промывали THF (5 раз) перед добавлением раствора амина 5c (490 мг, 0,7 ммоль) и DIEA (1,23 мл, 7,1 ммоль) в безводном THF (6 мл). После качания в течение 18 часов при комнатной температуре раствор реагентов удаляли фильтрованием, и смолу промывали DCM (5 раз). Линкерный реагент отщепляли от смолы и очищали с помощью обращенно-фазовой ВЭЖХ. рН фракций продукта подводили до рН 6 добавлением насыщенного водного раствора NaHCO3 и концентрировали при пониженном давлении. Полученную взвесь разделяли между насыщенным водным раствором NaCl и DCM, и водную фазу экстрагировали DCM. Объединенные органические фракции концентрировали до полного высушивания, что позволяло получить линкерный реагент 5d.
Выход составлял 230 мг, (0,29 ммоль).
МС: m/z 798,41 = [M+H]+ (рассчитанное значение = 798,1).
Пример 6
Синтез линкерного реагента 6с
Линкерный реагент 6с синтезировали по следующей схеме:
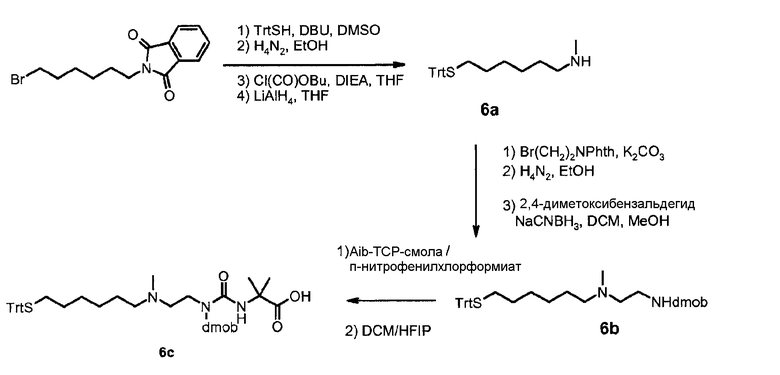
Синтез амина 6а:
Трифенилметантиол (11,90 г, 43,08 ммоль) ресуспендировали в DMSO (40 мл). Добавили DBU (7,41 мл, 49,55 ммоль) и 6-бромгексилфталимид (13,32 г, 42,94 ммоль), и смесь оставляли для прохождения реакции на 15 минут. Реакционную смесь разделяли в смеси этилацетата (700 мл) и 0,1 M HCl (200 мл). Водную фазу экстрагировали этилацетатом (3 раза по 50 мл), и объединенные органические фракции промывали насыщенным раствором NaCO3 (80 мл) и маточным раствором (80 мл), высушивали над MgSO4, фильтровали и концентрировали. Неочищенное желтое масло перекристаллизовали из н-гептана/этилацетата. Промежуточный продукт 6-(S-тритил-)меркаптогексилфталимид получили в виде белого твердого вещества (13,3 г, 26,4 ммоль, 62%).
6-(S-тритил-)меркаптогексилфталимид (14,27 г, 28,2 ммоль) ресуспендировали в этаноле (250 мл). Добавляли гидразин-гидрат (3,45 мл, 70,5 ммоль), и смесь нагревали с обратным холодильником в течение 2 часов. Смесь фильтровали, и фильтрат концентрировали в вакууме. К оставшемуся маслу добавляли хлороформ (180 мл), и полученную суспензию перемешивали при комнатной температуре в течение 1,5 ч. Смесь фильтровали, и фильтрат экстрагировали водой (60 мл) и маточным раствором (60 мл), высушивали над MgSO4 и концентрировали, получая неочищенный 6-(тритилмеркапто)-гексиламин (10,10 г, 26,87 ммоль, 95%).
МС: m/z 376,22 = [M+H]+ (рассчитанное значение = 376,20).
К охлажденному раствору (0°С) 6-(тритилмеркапто)-гексиламина (2,44 г, 6,49 ммоль) в THF (50 мл) добавляли DIEA (1,41 мл, 8,11 ммоль) и н-бутилхлорформиат (908 мкл, 7,14 ммоль, в 1 мл THF). Через 30 минут добавляли LiAlH4 (1 M в THF, 9,74 мл, 9,47 ммоль), и смесь нагревали с обратным холодильником в течение 90 минут. Добавление воды, 3,75 M водного раствора NaOH и воды приводило к образованию осадка, который удаляли из смеси фильтрованием. Фильтрат концентрировали в вакууме, получая соединение 6a.
Выход составлял 2,41 г (6,20 ммоль).
МС: m/z 390,22 = [M+H]+ (рассчитанное значение = 390,22).
Синтез промежуточного линкерного реагента 6b:
К раствору 6a (2,1 г, 5,31 ммоль) добавили 2-бромэтилфталимид (1,96 г, 7,7 ммоль) и K2CO3 (1,09 г, 7,9 ммоль), и смесь нагревали с обратным холодильником в течение 6 часов. После фильтрования и концентрирования, неочищенную смесь разделяли в этилацетате и насыщенном водном растворе NaHCO3. Неочищенный промежуточный продукт (2-(N-метил-N-(6-тритилмеркаптогексил-)амино-)этил)фталимид очищали с помощью флэш-хроматографии.
Выход составлял 1,23 г (2,18 ммоль).
МС: m/z: 563,27 = [M+H]+ (рассчитанное значение = 563,27).
К раствору (2-(N-метил-N-(6-тритилмеркаптогексил-)амино-)этил)фталимида (672 мг, 1,19 ммоль) в этаноле (12 мл) добавляли гидразина моногидрат (208 мкл, 4,17 ммоль), и смесь нагревали с обратным холодильником в течение 1 часа. Реакционную смесь фильтровали, концентрировали и N-(2-аминоэтил-)-N-метил-N-(6-тритилмеркаптогексил-)амин очищали с помощью обращенно-фазовой ВЭЖХ.
Выход составлял 624 мг (0,944 ммоль).
МС: m/z 433,27 = [M+H]+ (рассчитанное значение = 433,26).
К раствору N-(2-аминоэтил-)-N-метил-N-(6-тритилмеркаптогексил-)амина (151 мг, 0,229 ммоль) и NaCNBH3 (30 мг, 0,463 ммоль) в безводном MeOH (6 мл) добавляли раствор 2,4-диметоксибезальдегида в безводном CH2Cl2 (0,6 мкл). После перемешивания в течение 1 ч при комнатной температуре, реакционную смесь концентрировали, растворяли в 2 мл смеси вода/ацетонитрил в объемном соотношении 1/9 и соединение 6b очищали с помощью обращенно-фазовой ВЭЖХ.
Выход составлял 177 мг (0,219 ммоль).
МС: m/z 583,33 = [M+H]+ (рассчитанное значение = 583,33).
Синтез линкерного реагента 6с
Линкерный реагент 6с получали на смоле, нагруженной Fmoc-Aib (704 мг, ~0,6 ммоль), как описано для 5d, за исключением того, что использовали амин 6b (в виде TFA-соли, 430 мг, 0,53 ммоль) вместо 5c.
Выход составлял 285 мг (0,330 ммоль).
МС: m/z 712,37 = [M+H]+ (рассчитанное значение = 712,37).
Пример 7
Синтез линкерного реагента 7f
Линкерный реагент 7f синтезировали по следующей схеме:
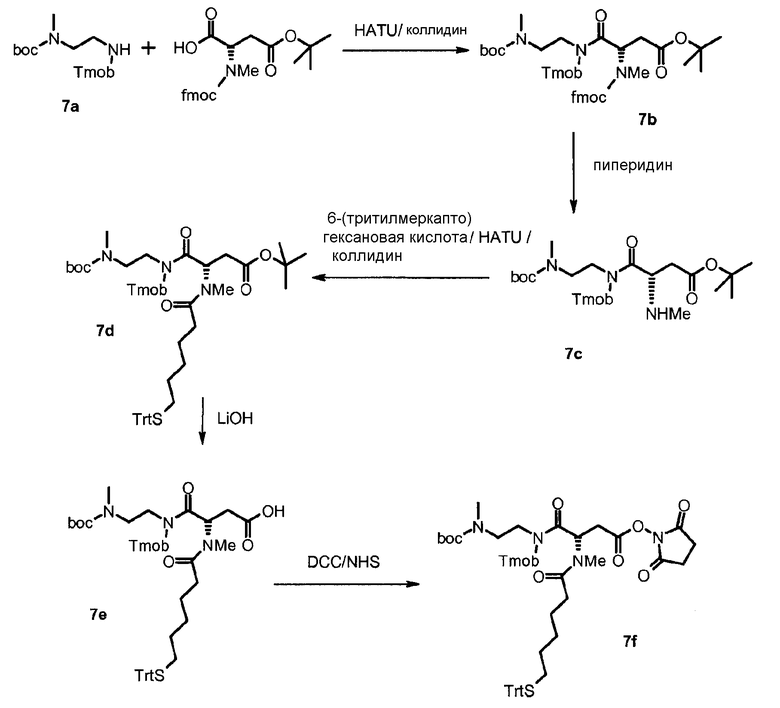
К охлажденному (0°С) раствору N-метил-N-boc-этилендиамина (0,5 мл, 2,79 ммоль) и NaCNBH3 (140 мг, 2,23 ммоль) в MeOH (10 мл) и уксусной кислоте (0,5 мл) добавляли раствор 2,4,6-триметоксибензальдегида (0,547 мг, 2,79 ммоль) в EtOH (10 мл). Смесь перемешивали при комнатной температуре в течение 2 часов, закисляли 2 М HCl (1 мл) и нейтрализовали насыщенным водным раствором Na2CO3 (50 мл). Выпаривание всех летучих соединений, экстракция с помощью DCM полученной водной взвеси и концентрирование органических фракций давали N-метил-N-boc-N'-tmob-этилендиамин (7a) в виде неочищенного масла, которое очищали с помощью обращенно-фазовой ВЭЖХ.
Выход составлял 593 мг (1,52 ммоль).
МС: m/z 377,35 = [M+Na]+ (рассчитанное значение = 377,14).
N-Fmoc-N-Me-Asp(OtBu)-OH (225 мг, 0,529 ммоль) растворяли в DMF (3 мл) и добавляли соединение 7a (300 мг, 0,847 ммоль), HATU (201 мг, 0,529 ммоль) и коллидин (0,48 мл, 3,70 ммоль). Смесь перемешивали при комнатной температуре в течение 2 часов, получая соединение 7b. Для снятия fmoc-защиты добавляли пиперидин (0,22 мл, 2,16 ммоль), и продолжали перемешивание в течение 1 часа. Добавляли уксусную кислоту (1 мл), и соединение 7с очищали с помощью обращенно-фазовой ВЭЖХ.
Выход составлял 285 мг (0,436 ммоль в виде TFA-соли).
МС: m/z 562,54 = [M+Na]+ (рассчитанное значение = 562,67).
6-Тритилмеркаптогексановую кислоту (0,847 г, 2,17 ммоль) растворяли в безводном DMF (7 мл). Добавляли HATU (0,825 г, 2,17 ммоль) и коллидин (0,8 мл, 6,1 ммоль) и соединение 7c (0,78 г, 1,44 ммоль). Реакционную смесь перемешивали в течение 60 минут при комнатной температуре, закисляли AcOH (1 мл) и очищали с помощью обращенно-фазовой ВЭЖХ. Фракции продукта нейтрализовали насыщенным водным раствором NaHCO3 и концентрировали. Оставшуюся водную фазу экстрагировали DCM, и соединение 7d выделяли после упаривания растворителя.
Выход составлял 1,4 г (94%).
МС: m/z 934,7 = [M+Na]+ (рассчитанное значение = 934,5).
К раствору соединения 7d (1,40 мг, 1,53 ммоль) в MeOH (12 мл) и H2O (2 мл) добавляли LiOH (250 мг, 10,4 ммоль), и реакционную смесь перемешивали в течение 14 часов при 70°С. Смесь закисляли AcOH (0,8 мл), и соединение 7e очищали с помощью обращенно-фазовой ВЭЖХ. Фракции продукта нейтрализовали насыщенным водным раствором NaHCO3 и концентрировали. Водную фазу экстрагировали DCM, и соединение 7е выделяли после упаривания растворителя.
Выход составлял 780 мг (60%).
МС: m/z 878,8 = [M+Na]+ (рассчитанное значение = 878,40).
К раствору 7e (170 мг, 0,198 ммоль) в безводном DCM (4 мл) добавляли DCC (123 мг, 0,59 ммоль) и N-гидроксисукцинимид (114 мг, 0,99 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Смесь фильтровали, и фильтрат закисляли добавлением 0,5 мл AcOH, и проводили очистку соединения 7f с помощью обращенно-фазовой ВЭЖХ. Фракции продукта нейтрализовали насыщенным водным раствором NaHCO3 и концентрировали. Оставшуюся водную фазу экстрагировали DCM, и соединение 7f выделяли после упаривания растворителя.
Выход составлял 154 мг (0,161 ммоль).
МС: m/z 953,4 = [M+H]+ (рассчитанное значение = 953,43).
В альтернативном варианте линкерный реагент 7f синтезировали по следующей схеме:
Альтернативная схема реакции:
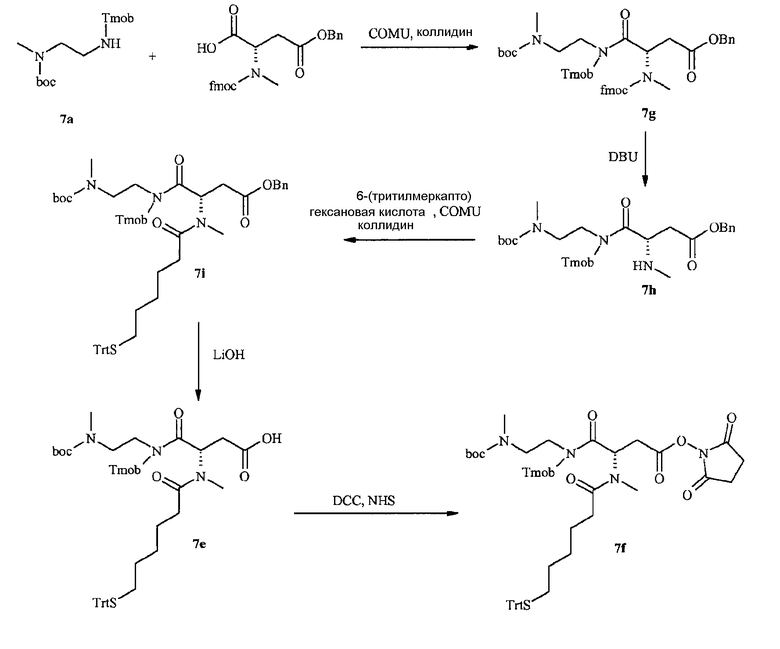
К раствору N-метил-N-boc-этилендиамина (2 г, 11,48 ммоль) и NaCNBH3 (819 мг, 12,63 ммоль) в MeOH (20 мл) добавляли частями 2,4,6-триметоксибензальдегид (2,08 мг, 10,61 ммоль). Смесь перемешивали при комнатной температуре в течение 90 минут, закисляли 3 M HCl (4 мл) и дополнительно перемешивали в течение 15 минут. Реакционную смесь добавляли к насыщенному раствору NaHCO3 (200 мл) и 5-кратно экстрагировали CH2Cl2. Объединенные органические фазы высушивали над Na2SO4, и растворители упаривали в вакууме. Полученное соединение N-метил-N-boc-N'-tmob-этилендиамин (7a) полностью высушивали в высоком вакууме и использовали в следующей реакционной стадии без дополнительной очистки.
Выход составлял 3,76 г, (11,48 ммоль, 89% чистоты, соотношение соединения 7a и продукта с двумя Tmob-защитными группами составляло 8:1).
МС: m/z 355,22 = [M+H]+ (рассчитанное значение = 354,21).
К раствору соединения 7a (2 г, 5,65 ммоль) в CH2Cl2 (24 мл) добавляли COMU (4,84 г, 11,3 ммоль), N-Fmoc-N-Me-Asp(OBn)-OH (2,08 г, 4,52 ммоль) и коллидин (2,65 мл, 20,34 ммоль). Реакционную смесь перемешивали в течение 3 часов при комнатной температуре, разбавляли CH2Cl2 (250 мл) и промывали 0,1 M H2SO4 (3 раза по 100 мл) и маточным раствором (3 раза по 100 мл). Водные фазы повторно экстрагировали CH2Cl2 (100 мл). Объединенные органические фазы высушивали над Na2SO4, фильтровали и остаток концентрировали до объема 24 мл. Соединение 7g очищали с помощью флэш-хроматографии.
Выход составлял 5,31 г (148%, 6,66 ммоль).
МС: m/z 796,38 = [M+H]+ (рассчитанное значение = 795,37).
К раствору соединения 7g [5,31 г, максимум 4,51 ммоль относительно N-Fmoc-N-Me-Asp(OBn)-OH] в THF (60 мл) добавили DBU (1,8 мл, 3% от объема смеси). Раствор перемешивали в течение 12 минут при комнатной температуре, разбавляли CH2Cl2 (400 мл) и промывали 0,1 M H2SO4 (3 раза по 150 мл) и маточным раствором (3 раза по 150 мл). Водные фазы повторно экстрагировали CH2Cl2 (100 мл). Объединенные органические фазы высушивали над Na2SO4 и фильтровали. Соединение 7h выделяли после упаривания растворителя и использовали в следующей реакции без дополнительной очистки.
МС: m/z 574,31 = [M+H]+ (рассчитанное значение = 573,30).
Соединение 7h (5,31 г, 4,51 ммоль, неочищенное) растворяли в ацетонитриле (26 мл) и добавляли COMU (3,87 г, 9,04 ммоль), 6-тритилмеркаптогексановую кислоту (2,12 г, 5,42 ммоль) и коллидин (2,35 мл, 18,08 ммоль). Реакционную смесь перемешивали в течение 4 часов при комнатной температуре, разбавляли CH2Cl2 (400 мл) и промывали 0,1 M H2SO4 (3 раза по 100 мл) и маточным раствором (3 раза по 100 мл). Водные фазы повторно экстрагировали CH2Cl2 (100 мл). Объединенные органические фазы высушивали над Na2SO4, фильтровали и соединение 7i выделяли после упаривания растворителя. Продукт 7i очищали с использованием флэш-хроматографии.
Выход составлял 2,63 г (62%, 94% чистоты).
МС: m/z 856,41 = [M+H]+ (рассчитанное значение = 855,41).
К раствору соединения 7i (2,63 г, 2,78 ммоль) в iPrOH (33 мл) и H2O (11 мл) добавляли LiOH (267 мг, 11,12 ммоль), и реакционную смесь перемешивали в течение 70 минут при комнатной температуре. Смесь разбавляли CH2Cl2 (200 мл) и промывали 0,1 M H2SO4 (3 раза по 50 мл) и маточным раствором (3 раза по 50 мл). Водные фазы повторно экстрагировали CH2Cl2 (100 мл). Объединенные органические фазы высушивали над Na2SO4, фильтровали и соединение 7е выделяли после упаривания растворителя. Соединение 7j очищали с использованием флэш-хроматографии.
Выход составлял 2,1 г (88%).
МС: m/z 878,4 = [M+Na]+ (рассчитанное значение = 878,40).
К раствору соединения 7e (170 мг, 0,198 ммоль) в безводном DCM (4 мл) добавляли DCC (123 мг, 0,59 ммоль) и каталитическое количество DMAP. Через 5 минут добавляли N-гидроксисукцинимид (114 мг, 0,99 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь фильтровали, растворитель удаляли в вакууме, и остаток растворяли в 90%-ом ацетонитриле и 0,1%-й TFA (3,4 мл). Неочищенную смесь очищали с помощью обращенно-фазовой ВЭЖХ. Фракции продукта нейтрализовали 0,5 М фосфатным буфером (рН 7,4) и концентрировали. Оставшуюся водную фазу экстрагировали DCM, и соединение 7f выделяли после упаривания растворителя.
Выход составлял 154 мг (81%)
МС: m/z 953,4 = [M+H]+ (рассчитанное значение = 953,43).
Пример 8
Синтез N αA1 -конъюгатов инсулин-линкера, 8b и 8c
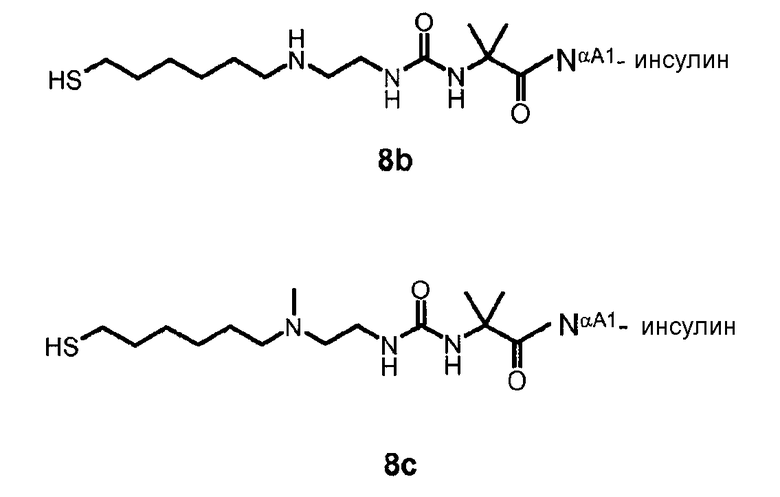
Синтез защищенного конъюгата инсулин-линкера, 8а
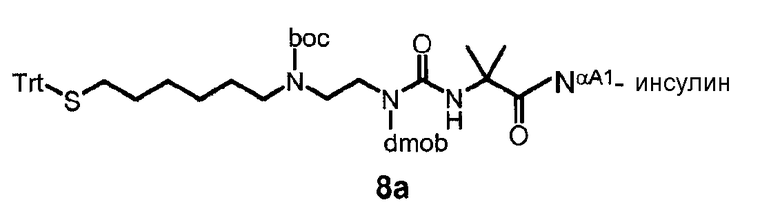
Линкерный реагент 5d растворяли в DCM (20 мг/мл) и активировали с помощью N-циклогексилкарбодиимид-N'-метилполистирольной смолы (1,9 ммоль/г, 10 экв.) в течение 1 часа. Раствор активированного линкерного реагента добавляли к раствору инсулина (1,2 экв.) и DIEA (3,5 экв.) в DMSO (100 мг инсулина/мл), и смесь встряхивали при комнатной температуре в течение 45 минут. Раствор закисляли уксусной кислотой, DCM упаривали при пониженном давлении, и NαA1-конъюгированный защищенный конъюгат инсулин-линкера 8а очищали с помощью обращенно-фазовой ВЭЖХ.
Лиофилизированный продукт 8а обрабатывали смесью HFIP/TFA/вода/триэтилсилан в объемном соотношении 90/10/2/2 (2 мл на 100 мг соединения 8a) в течение 45 минут при комнатной температуре. Реакционную смесь разбавляли водой, и все летучие вещества удаляли под током азота. NαA1-конъюгированный конъюгат инсулин-линкера 8b очищали с помощью обращенно-фазовой ВЭЖХ.
8b:
Выход составлял 139 мг (0,023 ммоль) из 62 мг (0,078 ммоль) линкера 5d
МС: m/z 1524,45 = [M+4H]4+ (рассчитанное значение = 1524,75).
NαA1-конъюгированный конъюгат инсулин-линкера 8с синтезировали как описано для 8b за исключением того, что вместо соединения 5d использовали соединение 6c (72 мг, 0,101 ммоль).
8c:
Выход составлял 237 мг (0,039 ммоль)
МС: m/z 1528,23 = [M+4H]4+ (рассчитанное значение = 1528,28).
Пример 9
Синтез N αВ1 -конъюгата инсулин-линкера, 9
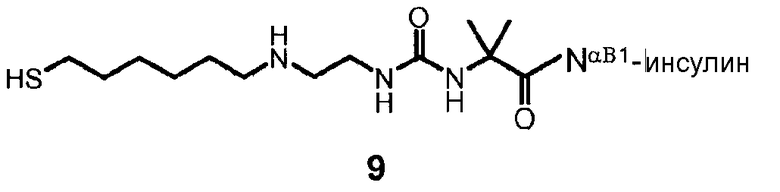
Инсулин с двумя защитными группами, N α-boc-GlyA1-N ε-boc-LysB29-инсулин, получали как описано ранее (J. Markussen, J. Halstrøm, F. C. Wiberg, L. Schaffer, J. Biol. Chem. 1991, 266, 18814-18818).
Линкерный реагент 5d (0,04 ммоль) растворяли в DCM (0,5 мл) и активировали с помощью N-циклогексилкарбодиимид-N'-метилполистирольной смолы (0,205 ммоль) при комнатной температуре в течение 2 часов. Полученный раствор активированного линкерного реагента добавляли к раствору инсулина с двумя защитными Boc-группами (24 мг, 0,004 ммоль) и DIEA (5 мкл, 0,0229 ммоль) и встряхивали при комнатной температуре в течение 1 часа. Реакционную смесь закисляли 100 мкл уксусной кислоты, и защищенный конъюгат инсулин-линкера очищали с помощью обращенно-фазовой ВЭЖХ.
Выход составлял 5 мг (0,00075 ммоль).
МС: m/z 1660,27 = [M+4H]4+ (рассчитанное значение = 1660,43).
Лиофилизированный защищенный конъюгат инсулин-линкера обрабатывали смесью HFIP/TFA/вода/TES в объемном соотношении 90/10/2/2 при комнатной температуре в течение 45 минут. Реакционную смесь разбавляли 0,5 мл воды, и все летучие вещества удаляли под током азота. NαВ1-конъюгированный конъюгат инсулин-линкера 9 очищали с помощью обращенно-фазовой ВЭЖХ.
Выход составлял 4 мг (0,0007 ммоль).
МС: m/z 1524,46 = [M+4H]4+ (рассчитанное значение = 1524,75).
Пример 10
Синтез N εB29 -конъюгата инсулин-линкера 10
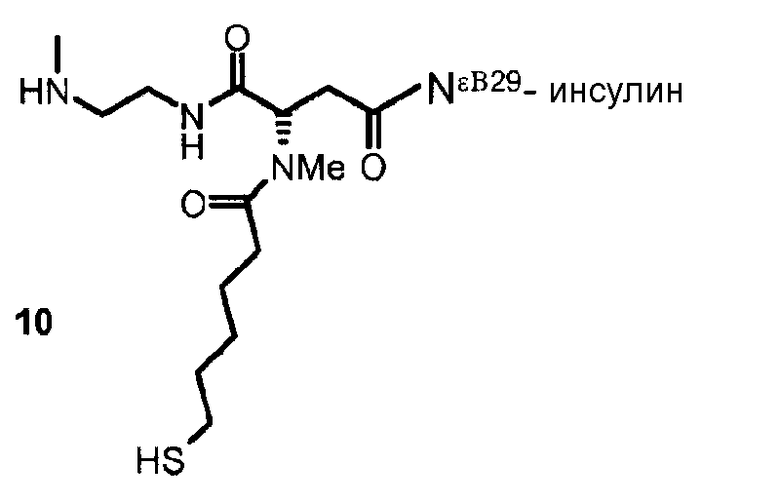
Инсулин (644 мг, 0,111 ммоль) растворяли в 6,5 мл DMSO. Добавляли 3 мл охлажденного (4°С) 0,5 М натрий-боратного буфера (pH 8,5) и соединение 7f (70 мг, 0,073 ммоль) в 2,5 мл DMSO, и смесь перемешивали в течение 5 минут при комнатной температуре. Добавляли 400 мкл АсОН, и защищенный конъюгат инсулина очищали с помощью обращенно-фазовой ВЭЖХ.
Выход составлял 172 мг (0,025 ммоль).
МС: m/z 1662,27 = [M+4H]4+ (рассчитанное значение = 1662,48).
Защитные группы удаляли, обрабатывая фракции лиофилизированного продукта 6 мл смеси HFIP/TFA/TES/вода в объемном соотношении 90/10/2/2 в течение 1 часа при комнатной температуре. NεB29-конъюгированный конъюгат инсулин-линкера 10 очищали с помощью обращенно-фазовой ВЭЖХ.
Выход составлял 143 мг (0,023 ммоль).
МС: m/z 1531,46 = [M+4H]4+ (рассчитанное значение = 1531,71).
Пример 11
Получение инсулин-линкер-гидрогеля 11a, 11b, 11с, 11d, 11da, 11db и 11dc
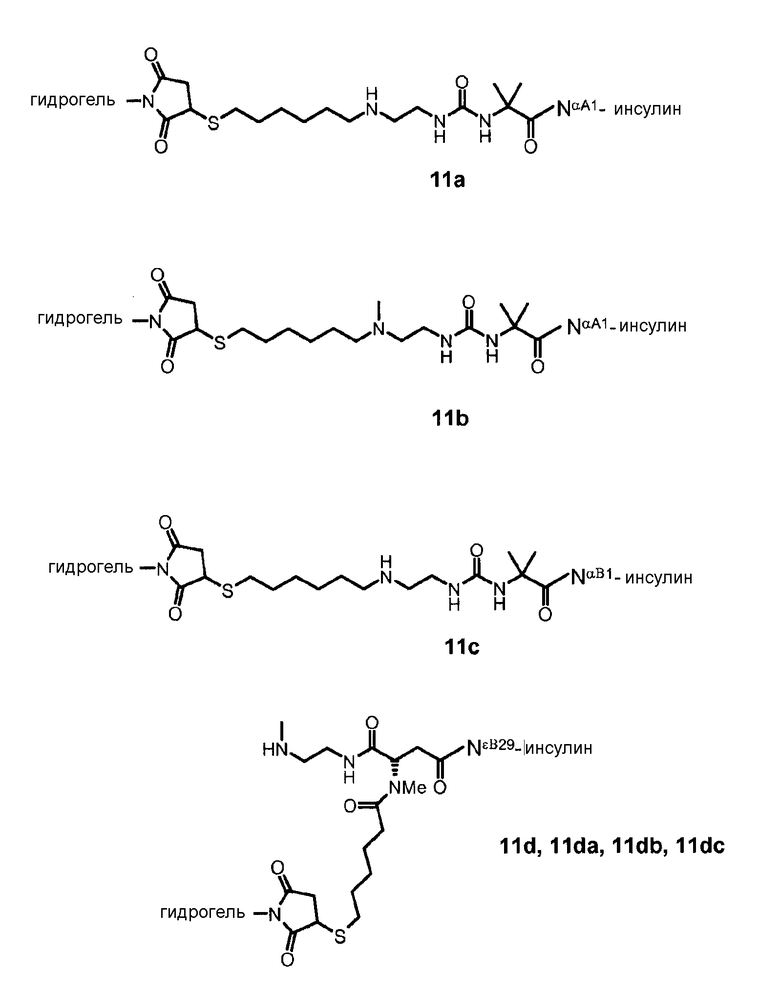
Сухим гидрогелем с функциональными малеимидными группами (соединение 4, 82 мг, 10,3 мкмоль малеимидных групп) заполняли шприц, оборудованный фрит-фильтром. Добавляли раствор соединения инсулин-линкер-тиола 8b (27,8 мг, 4,6 мкмоль) в 1,0 мл смеси ацетонитрил/вода/TFA в объемном соотношении 1/1/0,001, и суспензию инкубировали 5 минут при комнатной температуре. Добавляли ацетатный буфер (0,4 мл, pH 4,8, 1,0 M), и образец инкубировали при комнатной температуре в течение 1 часа. Расход тиольных групп контролировали с помощью теста Эллмана. Гидрогель промывали 10 раз смесью ацетонитрил/вода/TFA в объемном соотношении 1/0,43/0,001 и 2 раза смесью 1,0 М саркозин, рН 7,4/ацетонитрил/0,5 М фосфатный буфер рН 7,4/вода в соотношении 1/1/0,2/0,25. В конце гидрогель ресуспендировали в растворе саркозина и инкубировали 2 часа при комнатной температуре.
Соединение инсулин-линкер-гидрогель 11а промывали 10 раз смесью ацетонитрил/вода/TFA в соотношении 1/1/0,001 и хранили при +4°С.
Содержание инсулина определяли полным гидролизом аликвоты соединения инсулин-линкер-гидрогель при восстановительных условиях и рН 12, и последующую оценку количества А- и В-цепей инсулина проводили с помощью обращенно-фазовой ВЭЖХ.
Содержание инсулина в 11a: 175 мг инсулина/г инсулин-линкер-гидрогеля
Количество инсулина в суспензии соединения 11а в 10 мМ натрий-ацетатном буфере, рН 5, и 135 мМ хлориде натрия: 12 мг инсулина на 1 мл суспензии 11а.
Соединения 11b, 11с и 11d получали как описано выше за исключением того, что вместо соединения 8b использовали, соответственно, соединения 8c, 9 и 10.
Соединения 11da получали как описано выше за исключением того, что вместо соединений 8b и 4 использовали, соответственно, соединения 10 и 4а.
Соединение 11db получали следующим образом: суспензией гидрогеля с функциональными малеимидными группами 4а в HCl рН 2,5, 0,01% Tween-20 (5,0 мл, 119 мкмоль малеимидных групп) заполняли шприц с фильтром. К суспензии добавляли раствор инсулин-линкер-тиола (соединения 10) (166 мг, 24,4 мкмоль) в 8,0 мл HCl pH 2,5, 0,01% Tween-20, и суспензию инкубировали в течение 5 минут при комнатной температуре. Добавляли натрий-сукцинатный буфер (3,9 мл, pH 4,0, 150 мМ; 1 мМ EDTA, 0,01% Tween-20), получая рН 3,6, и образец инкубировали при комнатной температуре в течение 90 минут. Расход тиольных групп контролировали с помощью теста Эллмана. Гидрогель промывали 10 раз натрий-сукцинатным буфером (pH 3,0, 50 мМ; 1 мМ EDTA, 0,01% Tween-20) и 3 раза натрий-сукцинатным буфером (pH 3,0, 50 мМ; 1 мМ EDTA, 0,01% Tween-20), содержащим 200 мМ ацетилцистеин. В конце гидрогель ресуспендировали в буфере, содержащем ацетилцистеин, и инкубировали 1 час при комнатной температуре.
Соединение инсулин-линкер-гидрогеля 11db промывали 10 раз сукцинатным буфером (pH 3,0, 50 мМ; 1 мМ EDTA, 0,01% Tween-20) и 8 раз натрий-ацетатным буфером (pH 5,0, 10 мМ; 130 мМ NaCl, 0,01% Tween-20).
Содержание инсулина в 11db: 6,12 мг инсулина на мл суспензии инсулин-линкер-гидрогеля.
Соединение 11dc получали следующим образом: суспензию гидрогеля с функциональными малеимидными группами (4а) в HCl, рН 2,5, 0,01% Tween-20 (58,3 мл, 958 мкмоль малеимидных групп) добавляли в реактор для твердофазного синтеза. К соединению 4а добавляли раствор инсулин-линкер-тиола (соединения 10) (117 мл, 460 мкмоль) в 2,5 HCl, 0,01% Tween-20. Суспензию инкубировали 5 минут при комнатной температуре. Добавляли сукцинатный буфер (4,8 мл, pH 4,0, 150 мМ; 1 мМ EDTA, 0,01% Tween-20), получая рН 3,6, и образец инкубировали при комнатной температуре в течение 90 минут.
Расход тиольных групп контролировали с помощью теста Эллмана. Гидрогель промывали 10 раз сукцинатным буфером (pH 3,0, 50 мМ; 1 мМ EDTA, 0,01% Tween-20) и 2 раза натрий-сукцинатным буфером (pH 3,0, 50 мМ; 1 мМ EDTA, 0,01% Tween-20), содержащим 10 мМ меркаптоэтанол. В конце гидрогель ресуспендировали в буфере, содержащем меркаптоэтанол, и инкубировали 3 часа при комнатной температуре.
Соединение инсулин-линкер-гидрогеля (11dc) промывали 10 раз сукцинатным буфером (pH 3,0, 50 мМ; 1 мМ EDTA, 0,01% Tween-20) и 6 раз сукцинатным/Tris-буфером (pH 5,0, 10 мМ; 85 г/л трегалозы, 0,01% Tween-20).
Содержание инсулина в 11dc: 18,7 мг инсулина на мл суспензии инсулин-линкер-гидрогеля.
В альтернативном варианте вместо соединения 4а можно использовать микрочастицы малеимидного производного гидрогеля (4аа).
Пример 12
Кинетика высвобождения инсулина in vitro
Соединения инсулин-линкер-гидрогеля 11а, 11b, 11с и 11d (приблизительно содержащие 1 мг инсулина), соответственно, ресуспендировали в 2 мл буфера, содержащего 60 мМ фосфат натрия, 3 мМ EDTA, 0,01% Tween-20, pH 7,4, и инкубировали при 37°C. Суспензии центрифугировали через определенные промежутки времени, и анализ супернатанта проводили с помощью обращенно-фазовой ВЭЖХ с детекцией при 215 нм, а также используя ESI-MS. Значения УФ-поглощения, соответствующие высвобожденному инсулину, объединяли и строили график зависимости от времени инкубации.
Для определения соответствующего времени полувысвобождения инсулина использовали программное обеспечение для аппроксимации кривых.
Для соединений 11а, 11b, 11с и 11d было, соответственно, определено следующее время полужизни: 16 дней, 10 дней, 30 дней и 14 дней.
В альтернативном варианте инсулин-линкер-гидрогель (соединение 11db) переносили в шприцы, оборудованные фильтрами, ресуспендировали в 6 мл 60 мМ фосфата натрия, 3 мМ EDTA, 0,01% Tween-20, pH 7,4, и инкубировали при 37°C. В определенные интервалы времени обменивали супернатант, и количество высвобожденного инсулина определяли с помощью обращенно-фазовой ВЭЖХ при 215 нм. Строили график зависимости количества высвобожденного инсулина от времени инкубации. Определенное с помощью программного обеспечения для аппроксимации кривых время полужизни для соединения 11db составило 15 дней.
В альтернативном варианте соединением инсулин-линкер-гидрогеля 11db заполняли хроматографическую колонку и помещали в термостататируемый инкубатор (37°C). Через колонку пропускали натрий-фосфатный буфер (pH 7,4, 60 мМ; 3 мМ EDTA, 0,01% Tween-20) с постоянной скоростью потока 0,25 мл/час (номинальной) и собирали буфер вне инкубатора. В определенных временных точках раствор анализировали с помощью обращенно-фазовой ВЭЖХ при 215 нм. Строили график зависимости количества высвобожденного инсулина от времени инкубации. Определенное с помощью программного обеспечения для аппроксимации кривых время полужизни для соединения 11db составило 13 дней.
Пример 13
Синтез LysB29-конъюгата линкера и инсулина (12a) и LysB28-конъюгата линкера и инсулина лиспро (12b):
Синтез LysB29-конъюгата линкера и инсулина (12a)

1,2 г (0,206 ммоль, 0,85 экв.) инсулина растворяли в DMSO при комнатной температуре (RT). Через 30 минут раствор охлаждали до 0°С, при этом добавляли боратный буфер (0,5 M, pH 8,5, 21,6 мл) за 4,40 минуты. Температуру раствора поддерживали 25-28°С. Соединение 7f (0,239 ммоль, 1 экв.) растворяли в 40 мл DMSO и независимо от времени добавляли за 3 минуты. Удаляли ледяную баню, и реакционную смесь перемешивали 5 минут при комнатной температуре. Реакцию останавливали добавлением 70 мл MeCN/H2O (1:1, 0,1% TFA) и 400 мкл AcOH. Соединение 12a очищали с помощью обращенно-фазовой ВЭЖХ (растворитель А: Н2О с 0,1% TFA, растворитель B: MeCN с 0,1% TFA, градиент: 30-80% B за 14 минут, скорость потока: 40 мл/мин).
Избирательность по UPLC-анализу (до очистки с использованием обращенно-фазовой ВЭЖХ) была следующей: 0,70% соединения 7f было присоединено к GlyA1 инсулина, а 76,2% соединения 7f было присоединено к LysB29 инсулина (смотри фиг.1а).
Выход составлял 862 мг (TFA-соль, 60%).
МС: [M+H]1/4 + = 1662,25 г/моль (рассчитанная масса (MW+H)1/4 = 1662,35 г/моль).
Синтез LysB28-конъюгата линкера и инсулина лиспро (12b)

0,347 г (0,059 ммоль, 0,85 экв.) инсулина лиспро растворяли в 6 мл DMSO при комнатной температуре. Через 30 минут раствор охлаждали до 0°С, при этом добавляли боратный буфер (0,5 M, pH 8,5, 5,64 мл) за 1,40 минуты. Поддерживали температуру раствора 25-30°С. Далее независимо от времени в течение 2 минут добавляли раствор 67 мг (0,070 ммоль, 1 экв.) соединения 7f, растворенного в 8 мл DMSO. Удаляли ледяную баню, и реакционную смесь перемешивали 5 минут при комнатной температуре. Реакцию останавливали добавлением 20 мл MeCN/H2O (1:1, 0,1% TFA) и 1 мл AcOH. Соединение 12b очищали с помощью обращенно-фазовой ВЭЖХ (растворитель А: Н2О с 0,1% TFA, растворитель B: MeCN с 0,1% TFA, градиент: 30-80% B за 14 минут, скорость потока: 40 мл/мин).
Избирательность по UPLC-анализу (до очистки с использованием обращенно-фазовой ВЭЖХ) была следующей: 1,3% соединения 7f было присоединено к GlyA1 инсулина лиспро, а 76,7% соединения 7f было присоединено к LysB28 инсулина лиспро (смотри фиг.1b).
Выход составлял 305 мг (TFA-соль, 72%).
МС: [M+H]1/4 + = 1662,25 г/моль (рассчитанная масса (MW+H)1/4 = 1662,35 г/моль).
Пример 14
Фармакокинетические исследования на крысах
Фармакокинетику соединения 11а определяли, измеряя концентрацию инсулина в плазме крови после подкожного введения крысам однократной дозы исследуемого соединения.
Одну группу, состоящую из 10 самцов крыс Wistar (масса 200-250 г), использовали для исследования уровня инсулина в плазме в течение 14 дней. Каждое из животных получало однократную подкожную инъекцию 500 мкл суспензии соединения 11а в ацетатном буфере (рН 5), содержащей 6 мг инсулина (12 мг инсулина/мл). У каждого животного в определенные моменты времени забирали сублингвально по 200 мкл крови, получая 100 мкл Li-гепариновой плазмы. Образцы собирали перед введением препарата и через 4 часа, 1, 2, 3, 4, 7, 9, 11 и 14 дней после инъекции. Образцы плазмы замораживали в пределах 15 минут после забора крови и хранили при -80°С до проведения анализа.
Концентрацию инсулина в плазме измеряли, используя ультра-чувствительный набор ELISA для определения количества инсулина (Mercodia) и следуя протоколу изготовителя. Перед проведением измерений образцы плазмы разводили в буфере для проведения ELISA (1:5 и 1:10 с калибратором 0). Концентрацию инсулина вычисляли из калибровочной кривой, которую получали измерением стандартов инсулина в дупликатах и аппроксимацией значений с использованием линейной регрессии. Концентрацию инсулина устанавливали как среднее для двух независимых серий разведения, скорректированное на соответствующий фактор разведения, и строили зависимость от времени. Показанные на фиг.2 средние концентрации инсулина в плазме для каждой временной точки получали, вычисляя среднее для всех используемых в эксперименте животных.
Наблюдаемое в течение 14 дней высвобождение инсулина было постоянным и не имело выбросов.
Пример 15:
Фармакокинетические исследования на крысах
Фармакокинетику соединения 11da определяли, измеряя концентрацию инсулина в плазме крови в течение 13 дней после подкожного введения крысам однократной дозы исследуемого соединения.
8 крыс Wistar (масса приблизительно 250 г) получали однократную подкожную инъекцию 500 мкл суспензии тестируемого соединения 11da в ацетатном буфере (рН 5), содержащей 3 мг инсулина (приблизительно 12 мг/кг). У каждого животного в определенные моменты времени забирали по 200 мкл крови из хвостовой вены, получая 100 мкл Li-гепариновой плазмы. Образцы собирали за 1 день до введения препарата и через 4 часа, 1 день, 2 дня, 3 дня, 6 дней, 7 дней, 8 дней, 10 дней и 13 дней после введения тестируемого соединения. Образцы плазмы замораживали и хранили при -80°С до проведения анализа. Концентрацию инсулина в образцах плазмы измеряли, используя ультра-чувствительный набор ELISA для определения инсулина человека (DRG Instruments GmbH, Germany) и следуя протоколу изготовителя. Фоновые образцы (калибратор 0) включали в калибровочную кривую и вычитали из величин, полученных для образцов, а калибровочную кривую аппроксимировали, используя полиномиальное уравнение 3-го порядка. Перед проведением измерений образцы плазмы обрабатывали на вортексе и разбавляли в реакционных пробирках (1:5 и 1:10 с калибратором 0). Для анализа измеряли оптическую плотность OD при 450 нм на планшетном спектрофотометре (Tecan Ultra) без коррекции на референсную длину волны. Все результаты содержания инсулина в плазме до 13-го дня для всех исследуемых животных показаны на фигуре 3.
После однократной подкожной инъекции 500 мкл соединения 11d, содержащей 3 мг инсулина, средний уровень инсулина в плазме поднялся до максимума примерно 500 рМ в 1-й день. Как ожидалось, концентрация инсулина в плазме далее постепенно снижалась в течение 2 недель. Соотношение пиковой концентрации к остаточной концентрации инсулина в плазме в течение первой недели исследования приблизительно составляло 1,7.
Пример 16:
Фармакокинетические и фармакодинамические исследования на крысах
Количество и биологическую активность высвобождаемого инсулина изучали, анализируя концентрацию инсулина в плазме и эффект снижения концентрации глюкозы в крови в экспериментальном фармакокинетическом/фармакодинамическом исследовании, используя диабетических крыс Sprague-Dawley (SD).
Для этого у 8 крыс вызывали диабет, используя стрептозотоцин (STZ), и в данное исследование включали всех животных с содержанием глюкозы выше 350 мг/дл в нулевой день. У 7 из 8 крыс SD сформировался диабет, и они получали однократную подкожную инъекцию 500 мкл тестируемого соединения 11da в ацетатном буфере (рН 5), содержащую 6,4 мг инсулина. У каждого животного в определенные моменты времени забирали по 200 мкл крови из хвостовой вены, получая 100 мкл Li-гепариновой плазмы. Образцы собирали за 4 дня до введения препарата и через 2 часа, 1 день, 2 дня, 3 дня, 6 дней, 7 дней, 8 дней, 10 дней и 13 дней после введения тестируемого соединения. Образцы плазмы замораживали и хранили при -80°С до проведения анализа. Уровень глюкозы в крови определяли с помощью устройства AccuChek Comfort из хвостовой вены 3 раза перед инъекцией соединения и через 2 часа, 1 день, 2 дня, 3 дня, 6 дней, 7 дней, 8 дней, 10 дней, 13 дней, 15 дней, 17 дней, 20 дней, 22 дня и 24 дня после введения тестируемого соединения. Содержание инсулина в образцах плазмы измеряли, используя ультра-чувствительный набор ELISA для определения инсулина человека (DRG Instruments GmbH, Germany) и следуя протоколу изготовителя. Фоновые образцы (калибратор 0) включали в калибровочную кривую и вычитали из величин, полученных для образцов, а калибровочную кривую аппроксимировали, используя полиномиальное уравнение 3-го порядка. Перед проведением измерений образцы плазмы обрабатывали на вортексе и разбавляли в реакционных пробирках (1:5 и 1:10 с калибратором 0). Для анализа измеряли оптическую плотность OD при 450 нм на планшетном спектрофотометре (Tecan Ultra) без коррекции на референсную длину волны. Содержание инсулина в плазме контролировали в течение 2 недель, а содержание глюкозы в крови в течение 3-недельного периода, как показано на фигуре 4.
После однократной подкожной инъекции инсулин-гидрогеля 11da уровень глюкозы в крови был значительно снижен в течение 10 дней, при этом значения были ниже 100 мг/мл без каких-либо симптомов гипогликемии. В результате более высокой дозы (6,4 мг) инсулина на животное максимальная концентрация инсулина в плазме приблизительно составляла 800 рМ в 1-й день и постепенно снижалась в течение 2 недель до приблизительно 300 рМ. Одновременно, уровень глюкозы в крови начинал повышаться через 10 дней и возвращался к уровню до введения соединения через 3 недели.
Пример 17:
24-часовое фармакокинетическое исследование (исследование выброса инсулина) на крысах
Для доказательства того что инсулин высвобождается из инсулин-линкер-гидрогеля без выброса, у здоровых крыс наблюдали за концентрацией инсулина в плазме в течение 24 часов.
8 крыс Sprague-Dawley (масса 200-250 г) разделяли на 2 группы, и каждая получала однократную подкожную инъекцию 2 мл тестируемого соединения 11db в ацетатном буфере (рН 5) на кг массы тела. Тестируемое соединение имело концентрацию 4 мг/мл инсулина, так что каждое животное получало по 8 мг инсулина на кг массы тела. У каждого животного в определенные моменты времени забирали по 200 мкл крови из хвостовой вены, получая 100 мкл Li-гепариновой плазмы. Образцы в группе А собирали перед введением дозы соединения и через 5 минут, 30 минут, 2 часа, 4 часа и 8 часов после введения тестируемого соединения, а в группе В - перед введением дозы соединения и через 15 минут, 1 час, 3 часа, 6 часа и 24 часа после введения тестируемого соединения. Образцы плазмы замораживали и хранили при -80°С до проведения анализа. Содержание инсулина в образцах плазмы измеряли, используя ультра-чувствительный набор ELISA для определения инсулина человека (DRG Instruments GmbH, Germany) и следуя протоколу изготовителя. Фоновые образцы (калибратор 0) включали в калибровочную кривую и вычитали из величин, полученных для образцов, а калибровочную кривую аппроксимировали, используя полиномиальное уравнение 3-го порядка. Перед проведением измерений образцы плазмы обрабатывали на вортексе и разбавляли в реакционных пробирках (1:5 и 1:10 с калибратором 0). Для анализа измеряли оптическую плотность OD при 450 нм на планшетном спектрофотометре (Tecan Ultra) без коррекции на референсную длину волны. Результат приведен на фигуре 5 и ясно показывает, что инсулин высвобождается без какого-либо выброса.
Пример 18:
Фармакокинетические и фармакодинамические исследования многократного дозирования на крысах
Фармакокинетику и фармакодинамику после 3 еженедельных доз соединения 11da определяли, измеряя у диабетических крыс концентрацию инсулина в плазме и уровень глюкозы в крови в течение 4 недель.
Использовали 8 крыс Sprague-Dawley со средней массой 239 г. Диабет вызывали, используя стрептозотоцин (STZ), и всех животных с содержанием глюкозы выше 350 мг/дл в нулевой день включали в данное исследование. У 8 из 8 животных, получавших STZ, сформировался диабет, и они получили по 3 еженедельных подкожных инъекции в 0-й, 7-й и 14-й день по 2 мл тестируемого соединения 11da в ацетатном буфере (рН 5) на кг массы тела. Поскольку тестируемое соединение имело концентрацию 4 мг/мл инсулина, то вводимая доза составляла 8 мг инсулина на кг массы тела. У каждого животного в определенные моменты времени забирали по 200 мкл крови из хвостовой вены, получая 100 мкл Li-гепариновой плазмы. Образцы собирали за 3 дня до введения соединения и до 28-го дня после введения тестируемого соединения. Образцы плазмы замораживали и хранили при -80°С до проведения анализа. Уровень глюкозы в крови измеряли с помощью устройства AccuChek Comfort из хвостовой вены 3 раза перед инъекцией соединения и до 30-го дня после введения тестируемого соединения. Содержание инсулина в образцах плазмы измеряли, используя ультра-чувствительный набор ELISA для определения инсулина человека (DRG Instruments GmbH, Germany) и следуя протоколу изготовителя. Фоновые образцы (калибратор 0) включали в калибровочную кривую и вычитали из величин, полученных для образцов, а калибровочную кривую аппроксимировали, используя полиномиальное уравнение 3-го порядка. Перед проведением измерений образцы плазмы обрабатывали на вортексе и разбавляли в реакционных пробирках (1:5 и 1:10 с калибратором 0). Для анализа измеряли оптическую плотность OD при 450 нм на планшетном спектрофотометре (Tecan Ultra) без коррекции на референсную длину волны. Уровень инсулина в плазме и содержание глюкозы в крови контролировали в течение 4-недельного периода, и все эти значения показаны на фигуре 6.
Форма кривых указывает на то, что высвобождаемый инсулин был биологически активным, постоянно снижая содержание глюкозы в крови до показателей примерно 100 мг/дл после 3-й инъекции, которое оставалось низким около недели. Одновременно максимальная концентрация инсулина непрерывно повышалась, начиная с 200 рМ после первой дозы, до 300 рМ после второй дозы и 400 рМ после введения третьей дозы, и впоследствии снижалась снова в течение 2 недель до значений ниже 100 рМ.
Пример 19:
Фармакокинетические исследования на крысах
Фармакокинетику соединения 11dc определяли, измеряя концентрацию инсулина в плазме крови в течение 13 дней у здоровых крыс.
8 крыс Wistar (масса приблизительно 230 г) получали однократную подкожную инъекцию 2 мл/кг тестируемого соединения 11dc в сукцинатном буфере (рН 5) (10 мМ сукцинатный/tris-буфер, 85 г/л трегалозы, 0,01% Tween-20, pH 5,0), содержащую 3 мг инсулина (дозировка 12 мг/кг). У каждого животного в определенные моменты времени забирали по 200 мкл крови из хвостовой вены, получая 100 мкл Li-гепариновой плазмы. Образцы собирали за 4 дня до введения препарата и через 0,3 часа (4 животных), 1 час (4 животных), 2 часа (4 животных), 4 часа (4 животных), 1 день, 2 дня, 3 дня, 6 дней, 8 дней, 10 дней и 13 дней после введения тестируемого соединения. Образцы плазмы замораживали и хранили при -80°С до проведения анализа. Содержание инсулина в образцах плазмы измеряли, используя ультра-чувствительный набор ELISA для определения инсулина человека (DRG Instruments GmbH, Germany) и следуя протоколу изготовителя. Фоновые образцы (калибратор 0) включали в калибровочную кривую и вычитали из величин, полученных для образцов, а калибровочную кривую аппроксимировали, используя полиномиальное уравнение 3-го порядка. Перед проведением измерений образцы плазмы обрабатывали на вортексе и разбавляли в реакционных пробирках (1:5 и 1:10 с калибратором 0). Для анализа измеряли оптическую плотность OD при 450 нм на планшетном спектрофотометре (Tecan Ultra) без коррекции на референсную длину волны. Результаты содержания инсулина в плазме до 13-го дня для всех исследуемых животных показаны на фигуре 7.
После однократной подкожной инъекции 12 мг/кг соединения 11dc средний уровень инсулина в плазме поднялся в 1-й день до максимального значения примерно 500 рМ. Как ожидалось, концентрация инсулина в плазме впоследствии постепенно снижалась в течение 2 недель. Соотношение пиковой концентрации к остаточной концентрации инсулина в плазме в течение первой недели составляло приблизительно 1,4.
Пример 20
Высвобождение инсулина и деградация гидрогеля при рН 7,4 в режиме реального времени
Соединением инсулин-линкер-гидрогеля (11а) (730 мкл, содержащим 3,19 мг инсулина) в ацетатном буфере (рН 5,0, 10 мМ, 130 мМ NaCl, 0,01% (масса/объем) tween-20) заполняли пробирку для приготовления образцов, промывали 3 раза буфером для высвобождения инсулина с рН 7,4 (60 мМ фосфат натрия, 3 мМ EDTA, 0,01% (масса/объем) Tween-20) и объем подводили до 1,00 мл. Аликвотой суспензии (0,5 мл, 1,59 мг инсулина) заполняли хроматографическую колонку и помещали в термостатируемый инкубатор (37°C). Через колонку пропускали буфер для высвобождения инсулина (рН 7,4) с постоянной скоростью потока 0,25 мл/час (номинальной) и собирали буфер вне инкубатора. В определенных временных точках раствор анализировали с помощью обращенно-фазовой ВЭЖХ (215 нм). Строили график зависимости количества высвобожденного инсулина от времени инкубации, а для определения соответствующего половинного времени высвобождения использовали программное обеспечение для аппроксимации кривых. Было определено, что половинное время высвобождения инсулина составляет 9,4 дня.
Через 39 дней инкубации при 37°C суспензию гидрогеля переносили в пробирку для приготовления образцов, оставшийся гидрогель смывали с колонки буфером для высвобождения с рН 7,4, и объем образца подводили до 1,00 мл. Две аликвоты (по 300 мкл каждая) переносили в стерильные пробирки для образцов, объем подводили до 1,5 мл, и инкубировали при 37°C. Образцы отбирали через определенные интервалы времени и анализировали с помощью гель-фильтрации. УФ-поглощение, соответствующее водорастворимым продуктам деградации, высвобождаемым из гидрогеля, содержащим одну или несколько каркасных молекул (соответствующих реакционно-способным функциональным группам) объединяли и откладывали на графике относительно времени инкубации (см. фиг.8).
Пример 21
Инъецируемость пролекарственного соединения инсулин-линкер-гидрогеля
Использовали 5 мл пролекарственного соединения инсулин-линкер-гидрогеля 11dc (размер частиц 32-75 мкм, 18 мг инсулина/мл) в буфере янтарная кислота/tris с рН 5,0 (10 мМ, 40 г/л маннита, 10 г/л дигидрата трегалозы, 0,05% TWEEN-20). Суспензией пролекарственного соединения инсулин-линкер-гидрогеля заполняли шприц объемом 1 мл (длина 57 мм) через иглу 20G. Иглу 20G заменяли на иглу 30G и помещали шприц в держатель для шприцев (Aqua Computer GmbH&Co. KG), и измерения начинали со скоростью поршня 172 мм/мин (равно 50 мкл/с) (Стенд для силовых испытаний: Multitest 1-d; программное обеспечение для записи данных: EvaluatEmperor Lite, Version 1.16-015; Динамометр: BFG 200 N (все от компании Mecmesin Ltd., UK). Эксперименты с увеличивающейся скоростью поршня, показанные в таблице ниже, проводили с новым образцом пролекарственного соединения инсулин-линкер-гидрогеля. Эксперименты с водой и этиленгликолем проводили соответственно. Для всех экспериментов использовали одинаковые иглы 30G. На фиг.9 показана зависимость силы относительно скорости потока при использовании иглы 30G.
Н (вода)
Н 11dc
Сокращения:
AcOH - уксусная кислота
AcOEt - этилацетат
Aib - 2-аминоизомасляная кислота
Bn - бензил
Boc - т-бутилоксикарбонил
COMU (1-циано-2-этокси-2-оксоэтилиденаминоокси)диметиламино-морфолино-карбения гексафторфосфат
DBU - 1,3-диазабицикло[5.4.0]ундецен
DCC - N,N-дициклогексилкарбодиимид
DCM - дихлорметан
DIEA - диизопропилэтиламин
DMAP - диметиламинопиридин
DMF - N,N-диметилформамид
Dmob - 2,4-диметоксибензил
DMSO -диметилсульфоксид
EDC - 1-этил-3-(3-диметиламинопропил)карбодиимид
EDTA - этилендиаминтетрауксусная кислота
экв. - стехиометрический эквивалент
ESI-MS - масс-спектрометрия с ионизацией методом распыления в электромагнитном поле (электроспрей)
EtOH - этанол
Fmoc - 9-флуоренилметоксикарбонил
HATU O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат
HFIP - гексафторизопропанол
ВЭЖХ - высокоэффективная жидкостная хроматография
HOBt - N-гидроксибензотриазол
iPrOH - 2-пропанол
LCMS - масс-спектрометрия, совмещенная с жидкостной хроматографией
Mal - 3-малеимидопропил
Mal-PEG6-NHS - NHS-эфир N-(3-малеимидопропил)-21-амино-4,7,10,13,16,19-гексаокса-генеэйкозановой кислоты
Me - метил
MeCN - ацетонитрил
MeOH - метанол
Mmt - 4-метокситритил
МС - масс-спектр/масс-спектрометрия
MTBE - метил-трет-бутиловый эфир
MW - молекулярная масса
n.d. - не определено
NHS - N-гидроксисукцинимид
OD - оптическая плотность
OBu - бутилокси
OtBu - трет-бутилокси
ПЭГ - полиэтиленгликоль
Phth - фталь-
PyBOP - бензотриазол-1-ил-окси-трис-пирролидино-фосфоний гексафторфосфат
Обращенно-фазовая ВЭЖХ - Обращенно-фазовая высокоэффективная жидкостная хроматография
об/мин - оборотов в минуту
RT - комнатная температура
SEC - гель-фильтрация
Su - сукцинимидил
TCP - 2-хлортритилхлоридная смола
TES - триэтилсилан
TFA - трифторуксусная кислота
THF - тетрагидрофуран
TMEDA - N,N,N',N'-тетраметилэтилендиамин
Tmob - 2,4,6-триметоксибензил
Trt - трифенилметил, тритил
UPLC - ультра-производительная жидкостная хроматография
УФ - ультрафиолет
VIS - визуальный
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОЛЕКАРСТВА, СОДЕРЖАЩИЕ КОНЪЮГАТ ИНСУЛИНА И ЛИНКЕРА | 2010 |
|
RU2574667C2 |
| ПРОЛЕКАРСТВА, СОДЕРЖАЩИЕ КОНЪЮГАТ ЭКСЕНДИН-ЛИНКЕР | 2011 |
|
RU2593774C2 |
| ДИАГНОСТИКА, ПРОФИЛАКТИКА И ЛЕЧЕНИЕ БОЛЕЗНЕЙ СУСТАВОВ | 2013 |
|
RU2682676C2 |
| ПРОЛЕКАРСТВА НА ОСНОВЕ ГИДРОГЕЛЯ | 2013 |
|
RU2647729C2 |
| VEGF-НЕЙТРАЛИЗУЮЩИЕ ПРОЛЕКАРСТВА ДЛЯ ЛЕЧЕНИЯ ОФТАЛЬМОЛОГИЧЕСКИХ ЗАБОЛЕВАНИЙ | 2013 |
|
RU2673881C2 |
| БИОРАЗЛАГАЕМЫЕ НЕРАСТВОРИМЫЕ В ВОДЕ ГИДРОГЕЛИ НА ОСНОВЕ ПОЛИЭТИЛЕНГЛИКОЛЯ | 2010 |
|
RU2554854C9 |
| ПРОЛЕКАРСТВЕННЫЕ КОМПОЗИЦИИ НА ОСНОВЕ ГИДРОГЕЛЯ ПОПЕРЕЧНО-СШИТОЙ ГИАЛУРОНОВОЙ КИСЛОТЫ И СПОСОБЫ | 2018 |
|
RU2812787C2 |
| ПРОЛЕКАРСТВО, СОДЕРЖАЩЕЕ САМОРАСЩЕПЛЯЕМЫЙ ЛИНКЕР | 2014 |
|
RU2676324C2 |
| ПРОЛЕКАРСТВО, СОДЕРЖАЩЕЕ САМОРАСЩЕПЛЯЕМЫЙ ЛИНКЕР | 2018 |
|
RU2798085C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ СВЯЗАННЫХ С ГОРМОНОМ РОСТА ЗАБОЛЕВАНИЙ У ЧЕЛОВЕКА | 2014 |
|
RU2689336C2 |
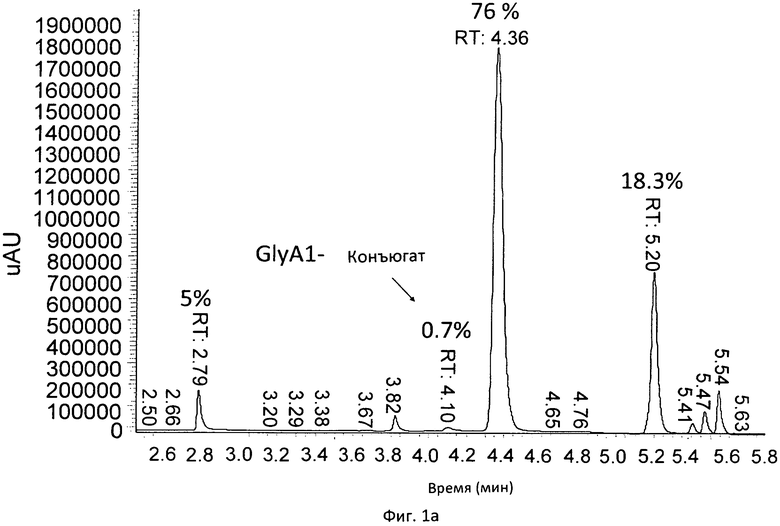
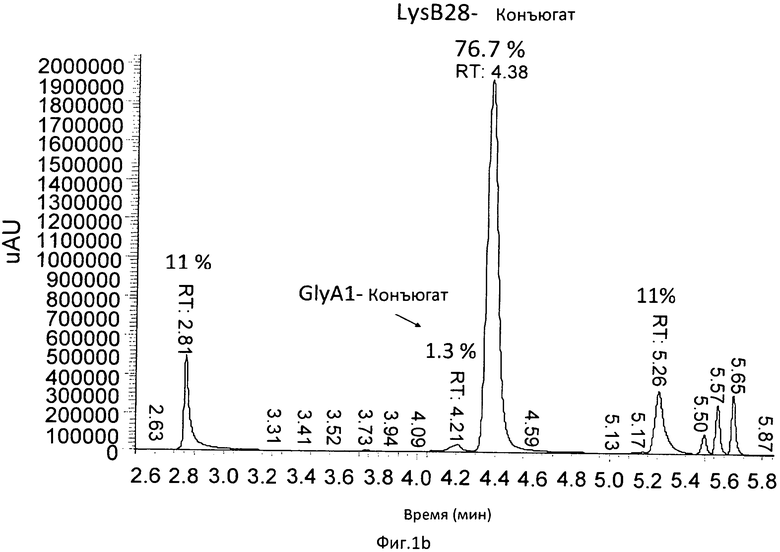
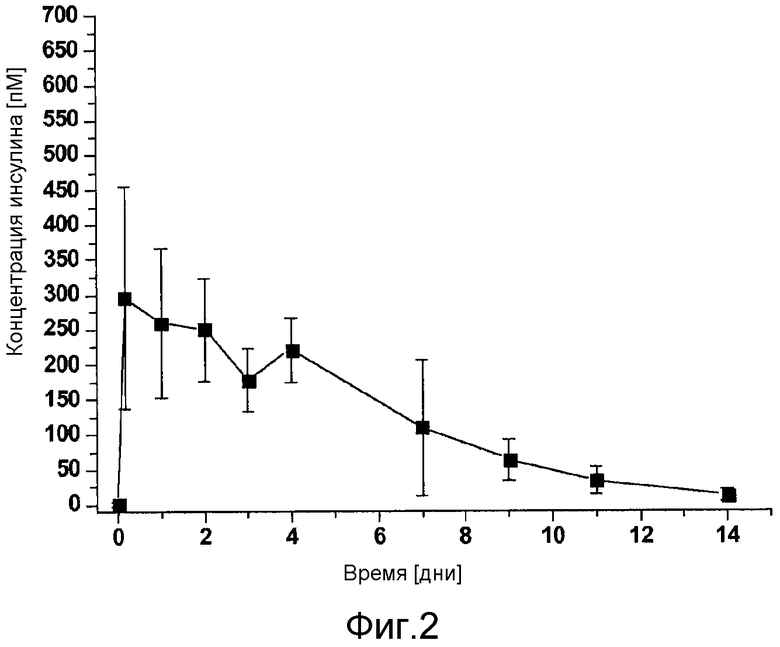
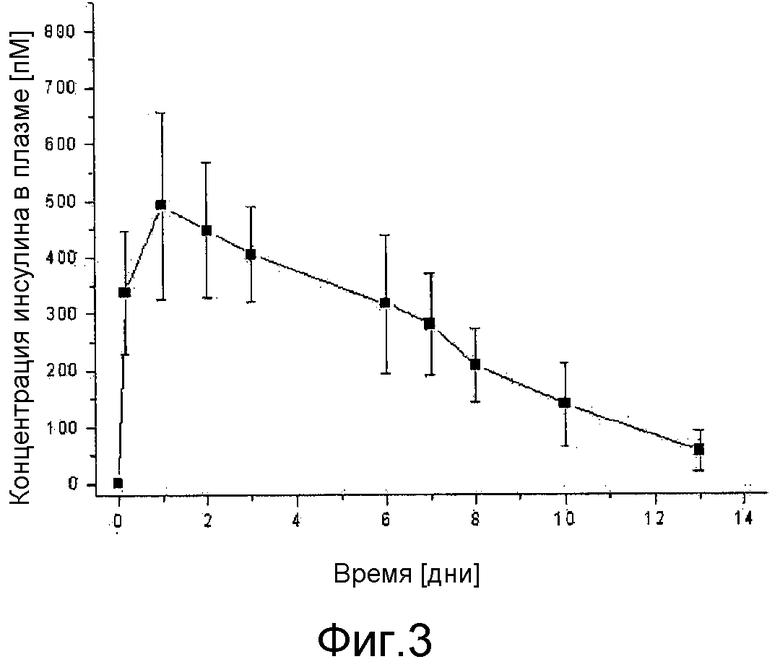
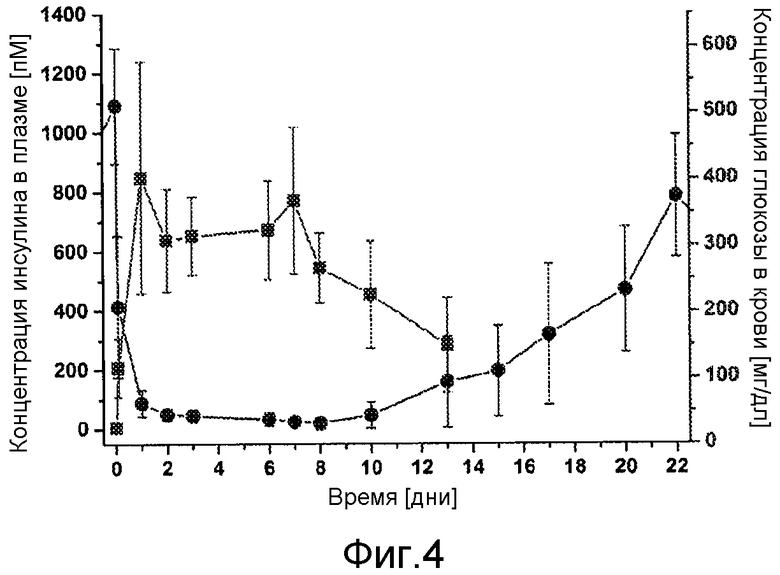
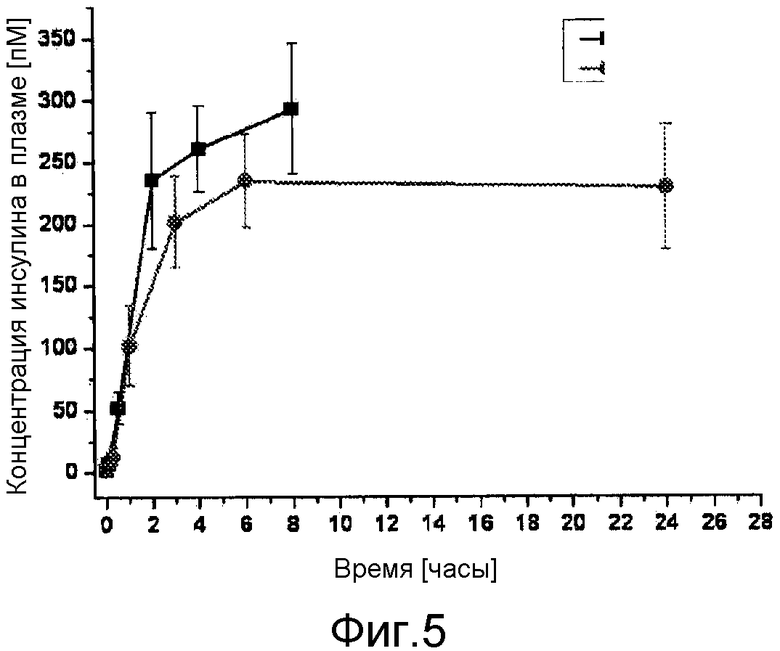
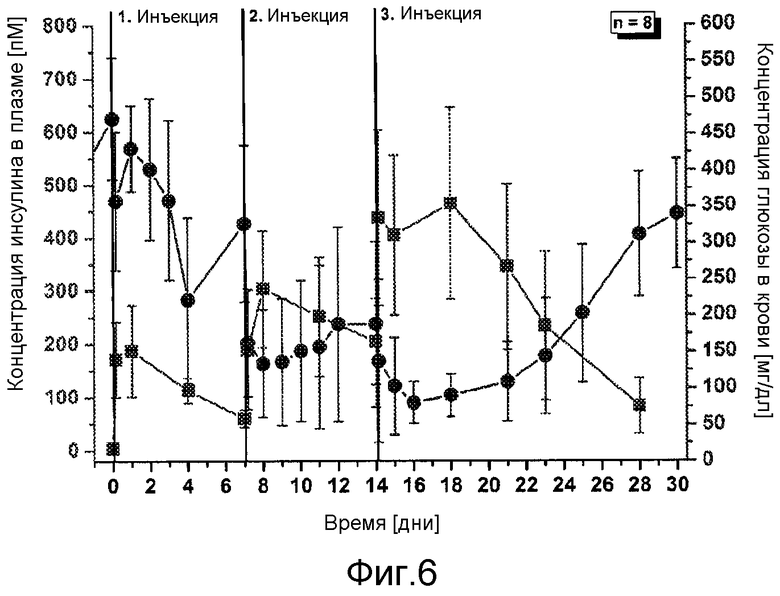
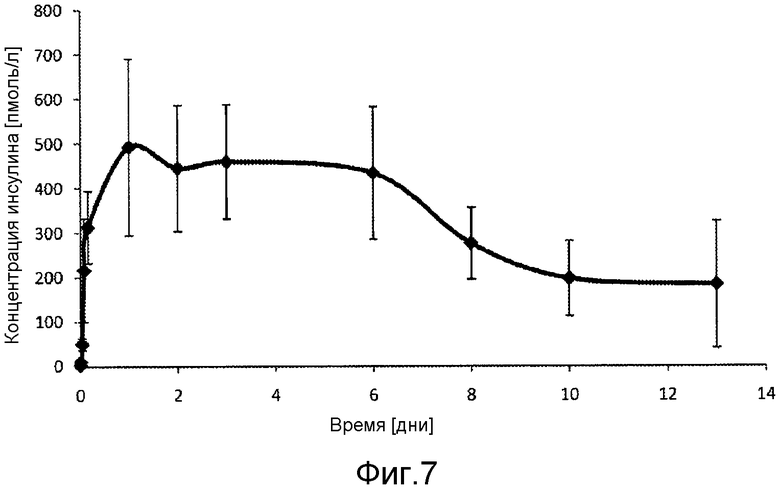
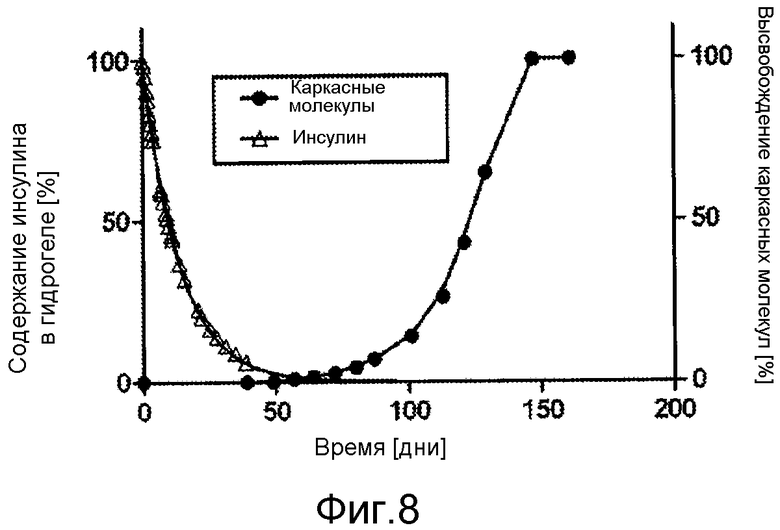
Настоящее изобретение относится к фармацевтической композиции, содержащей соединение инсулина в концентрации, достаточной для поддержания терапевтически эффективного уровня соединения инсулина в плазме крови в течение по меньшей мере 3 дней. Соединение инсулина относится к пролекарственному соединению, представляющему собой конъюгат инсулина с линкером, соединенный с гидрогелевым носителем. Также описана суспензия, содержащая фармацевтическую композицию конъюгата инсулина, способ получения суспензии, набор, включающий фармацевтическую композицию конъюгата инсулина и контейнер для введения композиции. Фармацевтическая композиция конъгата инсулина по изобретению характеризуется тем, что имеет фармакокинетический профиль in vivo по существу без выброса соединения инсулина. 6 н. и 20 з.п. ф-лы, 9 ил., 21 пр.
1. Фармацевтическая композиция, содержащая соединение инсулина в концентрации по меньшей мере 10 мг/мл, характеризующаяся тем, что она имеет фармакокинетический профиль in vivo по существу без выброса соединения инсулина, при этом соединение инсулина полностью содержится в депо-препарате, и соединение инсулина ковалентно связано в депо-препарате, где соединение инсулина представляет собой пролекарственное соединение или его фармацевтически приемлемую соль, представляющее собой конъюгат инсулина с линкером D-L, в котором
D представляет собой молекулу инсулина; а
- L является биологически неактивной линкерной молекулой -L1, представленной формулой (I),
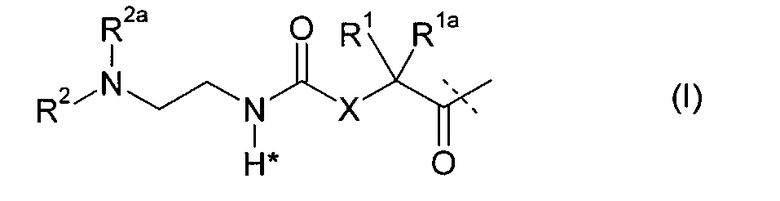
в которой пунктирная линия указывает присоединение одной из аминогрупп инсулина с образованием амидной связи;
X является C(R3R3a); или N(R3);
R1a, R3a независимо выбраны из группы, состоящей из Н, NH(R2b), N(R2b)C(O)R4 и С1-4алкила;
R1, R2, R2a, R2b, R3, R4 независимо выбраны из группы, состоящей из Н и С1-4алкила,
в которой L1 замещена одним заместителем L2-Z и необязательно дополнительно замещена, при условии, что атом водорода, обозначенный звездочкой в формуле (I) не замещен заместителем, и в которой
L2 является одиночной химической связью или L2 представляет собой С1-20алкильную цепь, которая необязательно прерывается одной или несколькими группами, независимо выбранными из -О- и С(О)N(R3aa); необязательно замещена одной или несколькими группами, независимо выбранными из ОН и С(О)N(R3aaR3aaa), где R3aa, R3aaa независимо выбраны из группы, состоящей из Н и С1-4алкила; а
Z является гидрогелем, содержащим каркасные части с четвертичным углеродом формулы С(A-Hyp)4, где каждая А независимо выбрана из формулы -(СН2)n1(OCH2CH2)nX1-, где n1 равняется 1 или 2; n представляет собой целое число в диапазоне от 5 до 50; а Х1 является химической функциональной группой, ковалентно соединяющей А и Hyp, где Hyp состоит из 5 - 32 лизинов.
2. Фармацевтическая композиция по п. 1, содержащая соединение инсулина в концентрации по меньшей мере 11 мг/мл, для введения в разовой дозе по меньшей мере 10 мг соединения инсулина.
3. Композиция по п. 1, в которой концентрация соединения инсулина составляет по меньшей мере 11 мг/мл, например от 11 мг/мл до 35 мг/мл.
4. Композиция по п. 1, характеризующаяся тем, что имеет соотношение пиковой концентрации к остаточной меньше 2, например меньше 1,75, меньше 1,5 или меньше 1,25.
5. Композиция по п. 1, характеризующаяся постоянным высвобождением соединения структурно интактного инсулина в течение всего интервала времени между введениями.
6. Композиция по п. 5, для которой полный интервал времени между введениями составляет по меньшей мере примерно 80 часов, например по меньшей мере примерно 110 часов, обычно по меньшей мере неделю.
7. Композиция по п. 1, характеризующаяся тем, что ее вводят с помощью инъекции, например, подкожной или внутримышечной.
8. Композиция по п. 1, в которой соединение инсулина выбрано из инсулина человека, инсулина гларгин, инсулина детемир, инсулина лизпро, инсулина аспарт, инсулина глулизин или их пролекарственных соединений.
9. Композиция по п. 1, для которой пиковая концентрация достигается в первые 24 часа после введения, например в первые 12 часов после введения, например в течение первых 6 часов после введения.
10. Фармацевтическая композиция по п. 1, причем фармацевтическая композиция является сухой.
11. Фармацевтическая композиция по п. 10, причем фармацевтическая композиция высушена с помощью лиофилизации.
12. Композиция по п. 10, причем композиция содержит дозу пролекарственного соединения инсулина с гидрогелем, достаточную для обеспечения терапевтически эффективного количества инсулина в течение по меньшей мере трех дней, при одном введении.
13. Композиция по п. 1, причем она является однодозовой композицией.
14. Композиция по п. 1, причем она является многодозовой композицией.
15. Фармацевтическая композиция по любому из пп. 1-13, находящаяся в контейнере.
16. Фармацевтическая композиция по п. 15, причем контейнер представляет собой двухкамерный шприц.
17. Применение соединения инсулина для изготовления фармацевтической композиции по любому из пп. 1-16 для лечения или предупреждения заболевания или расстройства, ассоциированных с недостаточностью инсулина, лечение или предупреждение которых с использованием соединения инсулина является полезным, таких как гипергликемия, предиабет, нарушенная глюкозотолерантность, диабет I типа, диабет II типа, синдром X.
18. Суспензия, содержащая фармацевтическую композицию по любому из пп. 1-16.
19. Способ получения суспензии по п. 18, включающий стадии восстановления сухой фармацевтической композиции по п. 10 или 11 путем добавления раствора для восстановления.
20. Набор, включающий фармацевтическую композицию по любому из пп. 1-14 и контейнер для введения композиции.
21. Набор, включающий иглу и контейнер, содержащий раствор для восстановления, и сухую композицию по п. 10 или 11 для применения с иглой.
22. Набор по п. 21, в котором контейнером является двухкамерный шприц и в котором одна из двух камер двухкамерного шприца содержит раствор для восстановления.
23. Набор по п. 22, в котором композицию можно вводить с помощью инъекции через иглу.
24. Набор по п. 23, в котором внутренний диаметр иглы меньше 300 мкм.
25. Набор по п. 23, в котором внутренний диаметр иглы меньше 225 мкм.
26. Набор по п. 23, в котором внутренний диаметр иглы меньше 175 мкм.
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Колосоуборка | 1923 |
|
SU2009A1 |
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| ЭНЦИКЛОПЕДИЯ ЛЕКАРСТВ, Регистр Лекарственных Средств, Москва, РЛС, 2000, стр.368-373 | |||

