Изобретение относится к биохимии, а именно к новому пептидному соединению, обладающему высоким сродством к альфа7 типу никотинового ацетилхолинового рецептора (нАХР) и имеющему следующую аминокислотную последовательность: GCCSRPPCALNNPRYC-NH2 (SEQ ID NO:1), а также его радиоактивному [125I]-меченному производному. Более конкретно данное изобретение относится к применению соединения SEQ ID NO:1 в биохимии и медицине.
Никотиновый ацетилхолиновый рецептор (нАХР), относящийся к тому же семейству лиганд-управляемых ионных каналов, что и глициновый, 5НТ3-, GABA-A и некоторые другие рецепторы, - один из наиболее изученных в структурно-функциональном плане нейрорецепторов (см., например, обзор [Hucho et al (1996) Eur. J. Biochem. 239: 539-557]). К настоящему времени получена криоэлектронно-микроскопическая структура нАХР из электрического органа ската Torpedo marmorata с разрешением 4 Å [Unwin (2005) J. Mol. Biol. 346: 967-989]. Благодаря многочисленным биохимическим исследованиям, а также упомянутой разрешенной структуре мы знаем, что нАХР из Torpedo состоит из 5-ти субъединиц (двух альфа1 и по одной бета1, гамма и дельта), располагающихся в мембране псевдосимметрично вокруг центральной оси, по которой проходит ионный канал. Два лигандсвязывающих участка холинергических агонистов и конкурентных антагонистов при этом располагаются в областях контакта больших N-концевых внеклеточных доменов двух альфа1 и соседних с ними гамма- и дельта-субъединиц рецептора примерно в их средней части (по отношению к мембранной поверхности). По своим фармакологическим и структурным характеристикам, а также представленности в электрическом органе (перерожденная мышечная ткань), нАХР Torpedo относят по современной классификации к холинорецепторам мышечного типа.
Позднее кроме мышечных рецепторов в различных нейрональных тканях были обнаружены новые «нейрональные» подтипы нАХР. К настоящему времени известно о существовании 9-ти нейрональных альфа-субъединиц (альфа2-альфа10) и 3-х бета-субъединиц (бета2-бета4), которые могут образовывать как гомоолигомерные формы нАХР (кроме альфа7, также и альфа8, и альфа9 подтипы), так и гетеромерные (состоящие из различных композиций альфа- и бета-субъединиц). Наиболее распространенными из выявленных в нервных тканях подтипов нейрональных нАХР являются альфа4бета2. альфа3альфа6бета2, альфа7 и другие. Пространственных структур ни для одного нейронального нАХР пока не получено, но и для этих рецепторов предполагается соответствующая 5-субъединичная структура, стехиометрия которой (для сложных гетеромерных подтипов) неизвестна. Позднее многие типы нейрональных нАХР были выявлены и в различных не-нейрональных тканях и органах (клетках иммунной системы, легких, коже) [Conti-Fine et al (2000) Eur. J. Pharmacol. 393: 279-294; Sciamanna et al (1997) J. Neurochem. 69: 2302-2311; Kumar et al (2005) J. Biol. Chem. 280: 25928-25935], где они могут быть вовлечены в развитие ряда патологических состояний [Grando (2008) J. Pharmacol. Sci. 106: 174-179]. К настоящему времени показано, что нарушение работы некоторых подтипов нАХР может вызывать или быть следствием ряда заболеваний, таких как мышечные дистрофии (миастении) [Vincent et al (2000) Eur. J. Biochem. 267: 6717-6728] и некоторые виды эпилепсии [Steinlein (2004) Prog. Brain Res. 145: 275-285]. В ряде работ была выявлена взаимосвязь между болезнями Альцгеймера и Паркинсона, а также шизофренией и нарушением в уровне определенных подтипов нейрональных нАХР [Olivera et al (2008) Channels 2: 143-152; O'Neill et al (2002) Curr. Drug Targets CNS Neurol. Disord. 1: 399-411].
Поиск новых высокоэффективных средств для диагностики и лечения этих заболеваний диктует необходимость создания новых лигандов, избирательно действующих на определенный подтип нАХР. Однако для создания подобных лигандов необходимо знать детальное устройство лигандсвязывающего участка. Общей пространственной структуры нАХР для этого недостаточно, и для этих целей сегодня используются другие возможности. В первую очередь речь идет об использовании природного структурного аналога лигандсвязывающего домена всех нАХР - ацетилхолинсвязывающего белка (АХСБ). Среди 4-х известных на сегодня АХСБ - наиболее структурно и фармакологически охарактеризованными являются белки из моллюсков Lymnaea stagnalis [Smit et al (2001) Nature 411: 261-268] и Aplysia californica [Hansen et al (2004) J. Biol. Chem. 279: 24197-24202]. К настоящему времени получены высокоразрешенные структуры комплексов этих белков с холинергическими лигандами самой разной природы [Celie et al (2004) Neuron 41: 907-914; Hansen et al (2005) EMBO J. 24: 3635-3646; Bourne et al (2005) EMBO J. 24: 1512-1522; Celie et al (2005) Nat. Struct. Mol. Biol. 12: 582-588; Ulens et al (2006) Proc. Natl. Acad. Sci. USA 103: 3615-3620; Dutertre et al (2007) EMBO J. 26: 3858-3867]. Эти структуры в свою очередь стали базой для многочисленных компьютерных моделей нАХР различных типов [Sgrignani et al (2009) J. Comp. Chem. 30: 2443-2454; Parthibanet al (2009) J. Biomol. Struct. Dyn. 26: 535-547; Perez et al (2009) Bioorg. Med. Chem. Lett. 19: 251-254], что позволяет вести целенаправленные поиски и конструирование новых соединений, высокоэффективных и селективных для определенных подтипов рецептора.
Особенностью нАХР является наличие большого количества лигандов самой разной природы от низкомолекулярных соединений до полипептидов и белков. Среди наиболее известных (и исторически самых первых) были полипептидные альфа-нейротоксины из яда змей - антагонисты некоторых типов холинорецепторов. Однако в последнее время при исследовании нАХР в дополнение к ним все чаще стали применять небольшие токсические пептиды, выделенные из яда морских улиток семейства Conus - так называемые альфа-конотоксины. Первые альфа-конотоксины были выделены в начале 80-х годов прошлого века [Gray et al. (1981) J. Biol. Chem. 256: 4734-4740], а в настоящее время благодаря быстро растущему числу новых пептидов количество используемых в исследованиях нАХР соединений на основе альфа-конотоксинов и их аналогов исчисляется уже сотнями (широкий спектр синтетических аналогов альфа-конотоксинов представлен в обзоре [Kasheverov et al. (2009) Curr. Pharm. Des. 15: 2430-2452]). Причиной такой популярности альфа-конотоксинов являются их два неоспоримых преимущества (по сравнению с теми же альфа-нейротоксинами из яда змей). Первое из них - это удивительное разнообразие в специфичности их разных представителей к разным типам нАХР, а второе - это небольшие размеры (в 5-6 раз меньшие, чем альфа-нейротоксины), что позволяет достаточно просто получать препаративные количества альфа-конотоксинов методами твердофазного пептидного синтеза.
Однако успешным структурно-функциональным исследованиям и нАХР с помощью альфа-конотоксинов во многом мешает их недостаточная эффективность и особенно селективность по отношению к разным подтипам холинорецепторов. Поэтому уже с момента выделения первых альфа-конотоксинов были начаты работы по созданию их различных аналогов и к настоящему времени в литературе описаны сотни подобных пептидных лигандов (структуры большей части представлены в обзорах [Kasheverov et al. (2009) Curr. Pharm. Des. 15: 2430-2452; Кашеверов и др. (2009) Успехи биол. химии 49: 275-318]).
Наиболее успешными при создании высокоэффективных и селективных к конкретному подтипу нАХР аналогов альфа-конотоксинов были работы, в которых в структуру природного пептида вводились единичные или множественные аминокислотные замены. Подобные работы проводились для альфа-конотоксинов MI, RgIA, Vc1.1, GID, ImI, PnIA, PnIB и MII [Jacobsen et al (1999) Biochemistry 38: 13310-13315; Ellison et al (2008) J. Mol. Biol. 377: 1216-1227; Halai et al (2009) J. Biol. Chem. 284: 20275-20284; Millard et al (2009) J. Biol. Chem, 284: 4944-4951; Servent et al (1998) J. Physiol. Paris 92: 107-111; Hogg et al (2003) J. Biol. Chem. 278: 26908-26914; Everhart et al (2004) Biochemistry 43: 2732-2737]. Для альфа-конотоксинов MII и ArIB эти работы привели к созданию нескольких исключительно активных и специфичных аналогов. Введением в последовательность первого пептида одиночных, двойных и даже тройных замен нескольких аминокислотных остатков (а.о.) на аланин удалось получить самый мощный на сегодня антагонист альфа6альфа3бета2бета3 нАХР - MII[А11] [McIntosh et al (2004) Mol. Pharmacol. 65: 944-952; Bordia et al (2007) Mol. Pharmacol. 72: 52-61], а также соединения MII[А9, А15] и MII[А4, A11, A15], более чем на три порядка различающиеся по сродству к родственным подтипам рецептора, содержащим альфа6- и альфа3-субъединицы [McIntosh et al (2004) Mol. Pharmacol. 65: 944-952; Azam et al (2008) J. Biol. Chem. 283: 11625-11632]. Перспективность введения аминокислотных замен была продемонстрирована и в случае с альфа-конотоксином ArIB, когда из «полиспецифичного» пептида были получены эффективные и высокоспецифичные к альфа7 нАХР лиганды - ArIB[L11, A16] и ArIB[L11, D16] [Whiteaker et al (2007) Biochemistry 46: 6628-6638]. Известно также получение аналогов альфа-конотоксинов с введенными в них заряженными а.о., в некоторых случаях это приводило к созданию более эффективных лигандов определенных типов нАХР и АХСБ [Groebe et al (1997) Biochemistry 36: 6469-6474; Ellison et al (2008) J. Mol. Biol. 377: 1216-1227; Kasheverov et al (2006) FEBS J. 273: 4470-4481; Celie et al (2005) Nat. Struct. Mol. Biol. 12: 582-588; Dutertre et al (2007) EMBO J. 26: 3858-3867].
Еще один аспект возможного практического применения альфа-конотоксинов в медицине - это создание на их основе маркеров определенных подтипов нАХР для количественного определения последних в органах и тканях для диагностики некоторых мышечных и нервных заболеваний. Одним из самых высокочувствительных методов детекции малопредставленных рецепторов является метод радиолигандного анализа. Известно применение радиоактивных форм альфа-конотоксинов MI и GI, которые служат эффективной заменой широко применяемому коммерческому [125I]-альфа-бунгаротоксину при работе с нАХР мышечного типа [Kasheverov et al (2006) FEBS J. 273: 4470-4481; Luo et al (2004) Biochemistry 43: 6656-6662; Myers et al (1991) Biochemistry 30: 9370-9377; Kasheverov et al (2001) Eur. J. Biochem. 268: 3664-3673; Sugiyama et al (1998) Mol. Pharmacol. 53: 787-794; Золотарев и др (2000) Биоорган. химия 26: 587-592]. Известно радиоактивное производное альфа-конотоксина ArIB[L11, A16] - мощного антагониста альфа7 нАХР [Whiteaker et al (2008) J. Pharmacol. Exp. Ther. 325: 910-919]. Известно использование [125I]иодированного аналога альфа-конотоксина MII для детекции подтипов нАХР, содержащих альфа6 и/или альфа3 субъединицы, на самых разных клеточных и тканевых препаратах и срезах (см, например, [Cui et al (2003) J. Neurosci. 23: 11045-11053; Whiteaker et al (2000) Mol. Pharmacol. 57: 913-925; Champtiaux et al (2002) J. Neurosci. 22: 1208-1217; Salminen et al (2005) Neuropharmacology 48: 696-705; Doura et al (2008) Brain Res. 1215: 40-52]).
Известны антиболевые агенты альфа-конотоксины Vc1.1 и RgIA [Sandall et al (2003) Biochemistry 42: 6904-6911], дающие надежду на возможное использование и других альфа-конотоксинов в качестве анальгетиков.
Известны наиболее близкие к заявляемому пептиду аналоги альфа-конотоксина PnIA - PnIA[L10], PnIA[L10, K14] [Celie et al (2005) Nat. Struct. Mol. Biol. 12: 582-588; Кашеверов и др (2009) Успехи биол. химии 49: 275-318] и PnIA[R5, L10] [Dutertre et al (2007) EMBO J. 26: 3858-3867]. Однако все указанные аналоги обладают как минимум на порядок меньшей активностью в конкуренции с радиоактивным альфа-бунгаротоксином при взаимодействии с альфа7 нАХР человека, чем патентуемое соединение.
Задачей изобретения является расширение ассортимента высокоспецифичных лигандов для никотиновых ацетилхолиновых рецепторов (нАХР) альфа7 типа.
Поставленная задача решается за счет структуры аналога альфа-конотоксина PnIA - пептида PnIA[R5, L10, R14], имеющего следующую аминокислотную последовательность: GCCSRPPCALNNPRYC-NH2 и содержащего две внутримолекулярные дисульфидные связи между Cys2 и Cys8, Cys3 и Cys16 и амидированный С-концевой цистеин 16. Пептид PnIA[R5, L10, R14] имеет соответствующую заявляемой структуре молекулярную массу 1747.7 Да.
Заявляемый пептид (SEQ ID NO:1) представляет собой новое соединение, ранее не описанное в научно-технической и патентной литературе.
Сущность изобретения заключается в проявлении пептидом PnIA[R5, L10, R14] высокого сродства к альфа7 нАХР человека. В конкуренции с радиоактивно меченным альфа-бунгаротоксином ((125-I)-Bgt) за связывание с этим рецептором PnIA[R5, L10, R14] показал сродство, выраженное в значениях IC50 (концентрация препарата, вызывающая 50% ингибирование специфического связывания радиолиганда с рецепторами) от 340 до 670 нМ. Сродство других аналогов альфа-конотоксина PnIA к альфа7 нАХР человека в аналогичном тесте было на порядок ниже: наиболее активный среди них PnIA[L10, K14] давал IC50=7200±700 нМ.
Другим аспектом изобретения является возможность получения из заявленного пептида PnIA[R5, L10, R14] его радиоактивного (125-I)-меченного производного - PnIA[R5, L10, R14, Y15(125-I)] (SEQ ID NO:1). Данное производное получают химической модификацией в присутствии хлорамина Т и выделяют в индивидуальном виде с помощью ВЭЖХ (высокоэффективная жидкостная хроматография). В качестве побочного продукта подобной реакции получают и выделяют в индивидуальном виде ди-(125-I)-меченное производное - PnIA[R5, L10, R14, Y15(125-I)2]. Молекулярные массы обоих соединений, полученных в одинаковых условиях с применением нерадиоактивного изотопа 127-I йода, подтверждены MALDI (матрично-активированная лазерная десорбция/ионизация) масс-спектрометрией и составили 1873.6 Да и 1999.5 Да для моно- и ди-(127-I)-меченных пептидов, соответственно.
Следующим аспектом изобретения является способность PnIA[R5, L10, R14, Y15(125-I)] (SEQ ID NO:1) с высоким сродством напрямую связываться с альфа7 нАХР человека и таким образом использоваться в качестве эффективного и специфического маркера данного типа рецептора. Константа диссоциации (KD) такого взаимодействия варьировала для разных фракций PnIA[R5, L10, R14, Y15(125-I)] в независимых экспериментах от 190 до 1600 нМ со средним значением по всем проведенным опытам KD≈1000±600 нМ. Данное соединение эффективно вытеснялось с альфа7 нАХР человека своей нерадиоактивной формой (PnIA[R5, L10, R14])-IC50=60±7 нМ; для наиболее близкого по структуре PnIA[L10, K14] аналога это значение составило 1800±600 нМ.
Техническим результатом заявляемого изобретения являются получение нового пептидного соединения (SEQ ID NO:1) и его радиоактивного производного (SEQ ID NO:1), высокая эффективность взаимодействия обоих пептидов с нАХР человека альфа7 типа и возможность их использования для детекции данного подтипа никотинового холинорецептора в ряде органов и тканей человека, а также для диагностики и лечения некоторых нейродегенеративных заболеваний, вызванных нарушением нормального функционирования этого рецептора.
Изобретение иллюстрируют рисунки:
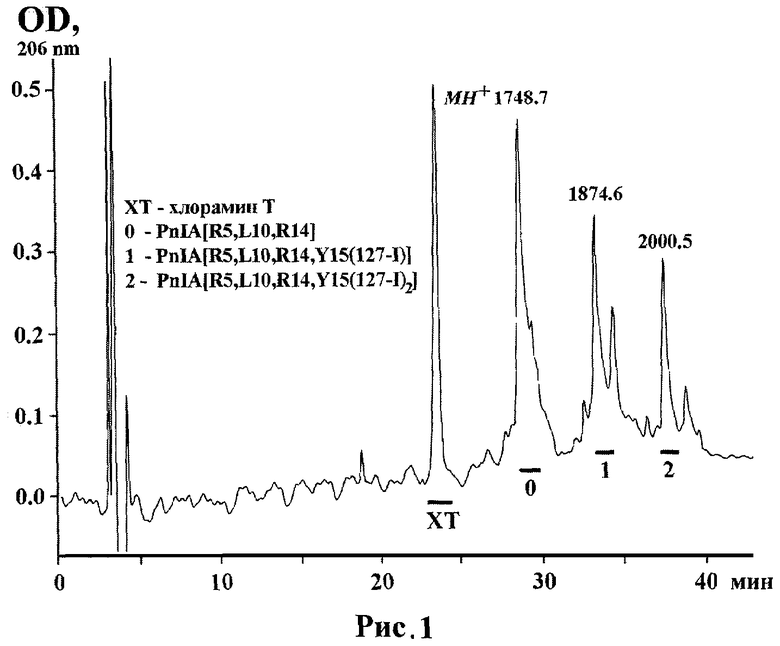
рис.1. ВЭЖХ хроматография продуктов разделения реакции [127-I]иодирования PnIA[R5, L10, R14] (SEQ ID NO:1);
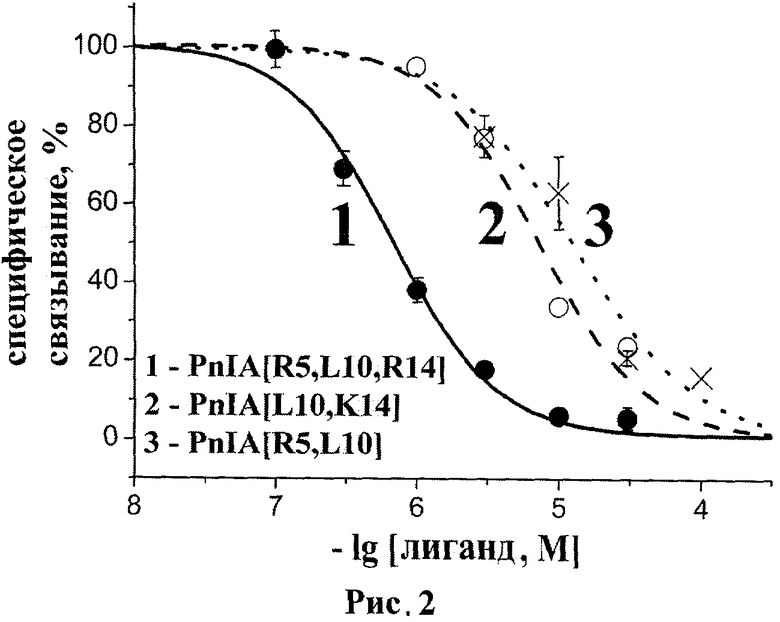
рис.2. Кривая ингибирования специфического связывания [125-I]-Bgt с альфа7 нАХР человека для пептида PnIA[R5, L10, R14] (SEQ ID NO:1) и его наиболее близких аналогов;
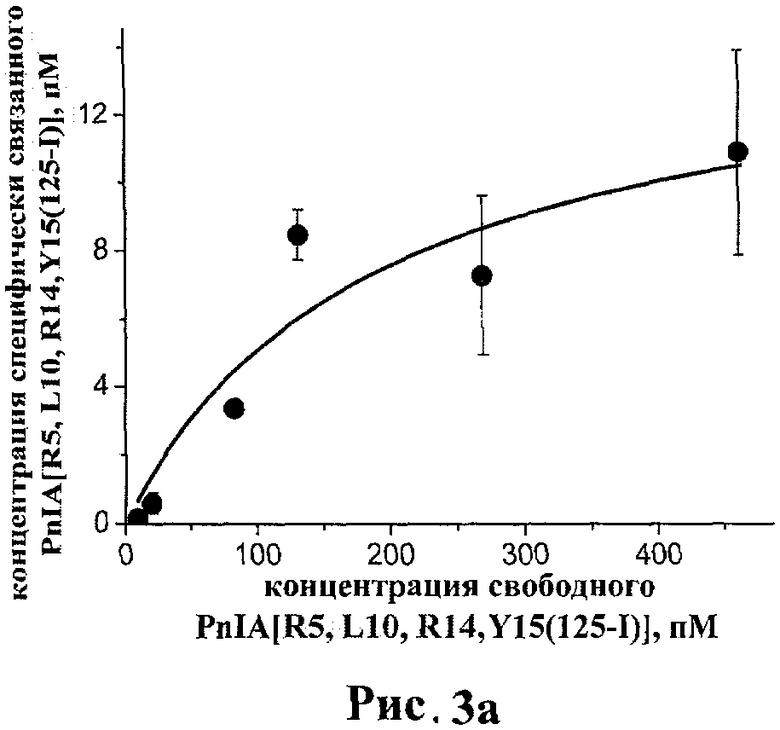
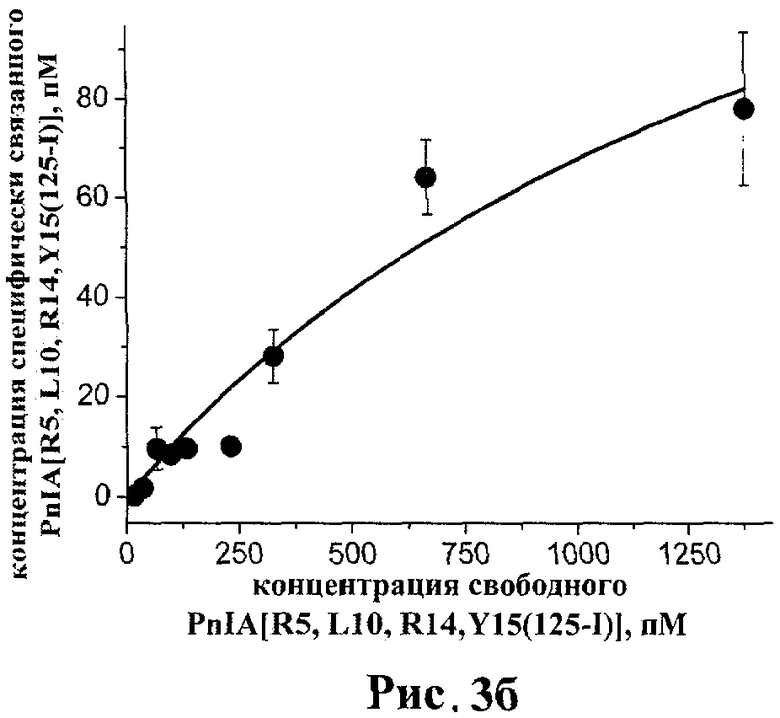
рис.3а, 3б. Кривые специфического связывания PnIA[R5, L10, R14, Y15(125-I)] с альфа7 нАХР человека, полученные из двух независимых экспериментов;
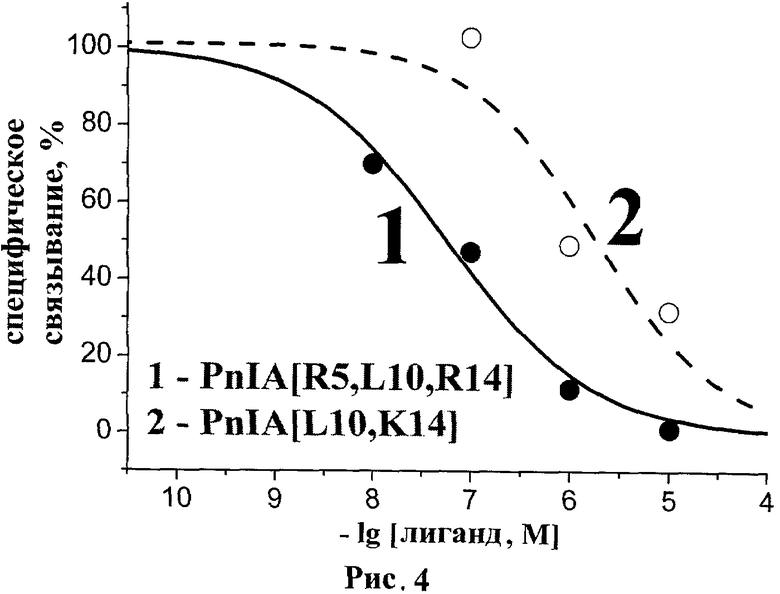
рис.4. Кривые ингибирования специфического связывания PnIA[R5, L10, R14, Y15(125-I)] с альфа7 нАХР человека для пептида PnIA[R5, L10, R14] (SEQ ID NO:1) и его наиболее близкого аналога.
Примеры осуществления изобретения.
Пример 1. Синтез PnIA[R5, L10, R14] (SEQ ID NO:1).
Присоединение первой аминокислоты. Получение Fmoc-Cys(Trt)-P(Fmoc=9-флуоренилметилоксикарбонил).
400 мг N-[(9Н-флуорен-9-илметокси)карбонил]-2,4-диметокси-4-(карбоксиметилокси)-бензгидриламин аминометил полимера (полимер Ринка) с содержанием гидроксильных групп 0.55 ммоль/г промывают диметилформамидом, затем в течение 20 мин обрабатывают 20%-ным раствором пиперидина в диметилформамиде. Полимер промывают последовательно 5 мл следующих растворителей: диметилформамидом - 3 раза по 2 мин, смесью диоксан-вода (2:1) - 2 раза по 5 мин, диметилформамидом - 5 раз по 2 мин. В 5 мл диметилформамида растворяют 644 мг (1.1 ммоль) Fmoc-Cys(Trt)-OH, 150 мг (1.1 ммоль) 1-гидроксибензотриазола и 172 мкл (1.1 ммоль) NN'-диизопропилкарбодиимида; раствор перемешивают 10 мин при 0°С и затем приливают к полимеру. Реакцию проводят 4 ч при периодическом перемешивании. По окончании реакции полимер отфильтровывают, промывают диметилформамидом и обрабатывают 5 мл смеси Ac2O-пиридин-CH2Cl2 (20:20:60) в течение 1 ч, после чего полимер промывают изопропанолом и диметилформамидом.
Пример одного синтетического цикла.
Получение Fmoc-Tyr(tBu)-Cys(Trt)-P:
а) Полученный на предыдущем этапе пептидил-полимер в течение 20 мин обрабатывают 20%-ным раствором пиперидина в диметилформамиде. Полимер промывают последовательно 5 мл следующих растворителей: диметилформамидом - 3 раза по 2 мин, смесью диоксан-вода (2:1) - 2 раза по 5 мин, диметилформамидом - 5 раз по 2 мин.
б) В 5 мл диметилформамида растворяют 505 мг (1.1 ммоль) Fmoc-Tyr(tBu)-OH, 150 мг (1.1 ммоль) 1-гидроксибензотриазола и 172 мкл (1.1 ммоль) NN'-диизопропилкарбодиимида; раствор перемешивают 10 мин при 0°С и затем приливают к суспендированному полимеру. Реакцию проводят 4 ч при периодическом перемешивании. По окончании реакции полимер отфильтровывают, промывают диметилформамидом и обрабатывают 5 мл смеси Ас2О-пиридин-диметилформамид (20:20:60) в течение 1 ч, после чего полимер последовательно промывают диметилформамидом, изопропанолом, диметилформамидом.
Синтез полипептидной цепи проводят вручную в стеклянном проточном реакторе (2×20 см) по следующему протоколу для каждого синтетического цикла (из расчета 10 мл растворителя на 400 мг исходного полимера); при проведении реакции конденсации (операция 6) используют объем реакционной смеси 7 мл:
1. ДМФА(диметилформамид) (5×2 мин);
2. 20% пиперидин в ДМФА (20 мин);
3. ДМФА (3×2 мин);
4. диоксан:вода, 2:1, (2×5 мин);
5. ДМФА (5×2 мин);
6. Реакция конденсации: 5 экв. активированной Fmoc-аминокислоты (4 ч);
7. ДМФА (3×2 мин);
8. ацилирование: Ас2О-пиридин-диметилформамид, 20:20:60, (1 ч.);
9. ДМФА (3×2 мин);
10. изопропанол (3×2 мин).
Для активации Fmoc-производных аминокислот DIPCDI/HOBT (N,N'-диизопропилкарбодиимид/1-гидроксибензотриазол)-методом к раствору 5 экв. (1.1 ммоль) Fmoc-защищенной аминокислоты и 150 мг (5 экв., 1.1 ммоль) HOBt в 3-4 мл ДМФА добавляют 170 мкл (5 экв., 1.1 ммоль) DIPCDI; раствор перемешивают 10 мин.
Полноту протекания реакции конденсации контролируют с помощью нингидринового или, в случае N-концевого пролина, изатинового тестов после операции 6 синтетического протокола.
Для синтеза были использованы следующие производные аминокислот: Fmoc-Gly-ОН, Fmoc-Cys(Trt)-OH (для присоединения остатков Cys3 и Cys16), Fmoc-Cys(tBu)-OH (для присоединения остатков Cys2 и Cys8), Fmoc-Ser(tBu)-OH, Fmoc-Arg(Pbf)-OH, Fmoc-Pro-ОН, Fmoc-Ala-OH, Fmoc-Asn-OH, Fmoc-Leu-OH, Fmoc-Tyr(tBu)-OH.
Отщепление продукта и замыкание дисульфидов.
Для реакции отщепления пептида от полимера и одновременного деблокирования защитных групп боковых цепей аминокислот отбирают 800 мг пептидил-полимера. К пептидил-полимеру приливают 15 мл смеси ТФУ(трифторуксусная кислота)-H2O в объемном соотношении 97.5:2.5, суспензию перемешивают в течение 2 ч; затем полученный раствор пептида отфильтровывают от полимера, промывают 5 мл ТФУ и избыток ТФУ упаривают при пониженном давлении. Пептид осаждают 100 мл этилового эфира, отфильтровывают и промывают эфиром (5×20 мл). Осадок растворяют в 5 мл 10% уксусной кислоты 20 мин, отфильтровывают и промывают 5 мл 10% уксусной кислоты. Полученный раствор пептида лиофилизируют и обессоливают на колонке (2.5×60 см) с Сефадексом G-10 в 0.1М уксусной кислоте. Очистку пептида проводят с помощью обращенно-фазовой ВЭЖХ в градиенте ацетонитрила (от 10% до 70% за 60 мин) в 0.1% ТФУ при расходе элюента 4 мл/мин; поглощение элюата регистрируют при длине волны 226 нм. Фракции, соответствующие основному пику на хроматограмме, собирают и лиофилизируют. Выход пептида GC(tBu)CSRPPC(tBu)ALNNPRYC-NH2 в расчете на C-концевую аминокислоту составляет 73%. Время удерживания пептида в условиях аналитической ВЭЖХ составляет 15.7 мин. Экспериментальная молекулярная масса 1863.6; теоретическая молекулярная масса 1863.2.
Пептид растворяют в 50% ацетонитриле в концентрации 0.5 мг/мл и устанавливают рН раствора до 8.5 с помощью диизопропилэтиламина. Реакционную смесь оставляют при перемешивании на 30 ч; полноту протекания реакции проверяют с помощью теста Эллмана. После завершения реакции рН раствора доводят до 5.0 с помощью уксусной кислоты и лиофилизируют. Пептид очищают с помощью препаративной обращенно-фазовой ВЭЖХ. Выход пептида GC(tBu)CSRPPC(tBu)ALNNPRYC-NH2, содержащего дисульфидную связь в положении Cys3/Cys16 в расчете на этап окисления, составляет 40%. Время удерживания пептида в условиях аналитической ВЭЖХ составляет 15.3 мин. Экспериментальная молекулярная масса - 1861.6, теоретическая молекулярная масса - 1861.3.
Для замыкания второй дисульфидной связи пептид и растворяют в ТФУ в концентрации 1 мг/мл; затем при перемешивании добавляют дифенилсульфоксид (10 экв.), метилтрихлорсилан (150 экв.) и анизол (100 экв.). Смесь оставляют при перемешивании на 15 мин при температуре 0°С; затем выливают в холодный диэтиловый эфир в объемном соотношении ТФУ-эфир 1:10. Пептид трижды экстрагируют водой. Затем водную фракцию отделяют на делительной воронке и лиофилизируют. Очистку пептида проводят с помощью обращенно-фазовой ВЭЖХ в градиенте ацетонитрила. Время удерживания пептида в условиях аналитической ВЭЖХ составляет 14.7 мин. Экспериментальная молекулярная масса - 1747.1, теоретическая молекулярная масса - 1747.4.
Пример 2. Синтез радиоактивного производного PnIA[R5, L10, R14, Y15(125-I)] (SEQ ID NO:1).
Отработка условий синтеза и подтверждение структуры с помощью MALDI масс-спектрометрии йодированных производных PnIA[R5, L10, R14] проводят с использованием нерадиоактивного изотопа 127-I: к 9.6 наномолям PnIA[R5, L10, R14], растворенных в 12.5 мкл 125 мМ натрий-фосфатного буфера (рН 7.2), добавляют 10 наномолей раствора йодида натрия (7.5 мкл в том же буфере) и 44 наномоля водного раствора хлорамина Т (1 мкл). Реакционную смесь перемешивают и оставляют на 25 мин при комнатной температуре. Продукты реакции разделяют на колонке Reprosil C18 AQ (5µ; 150×4 мм) в градиенте водного ацетонитрила (от 10% до 40% за 60 мин) в присутствии 0.1% ТФУ при скорости элюции 0.5 мл/мин (Рис.1). Предварительными контрольными заколами в тех же условиях служат 1) аликвота исходного пептида и 2) буферная смесь с йодидом натрия и хлорамином Т без пептида. В результате, два основных пика новых продуктов, присутствующих в реакционной смеси и отсутствующих в контрольных заколах, собирают и анализируют с помощью MALDI масс-спектрометрии. Молекулярные массы первого и второго пиков соответствуют моно- (1873.6 Да) и ди-(1999.5 Да)иодированным производным PnIA[R5, L10, R14] (Рис.1).
По тому же протоколу проводят получение радиоактивных производных PnIA[R5, L10, R14] с использованием раствора смеси [125-I]- и [127-I]йодида натрия в соотношении 1:333 - к 9.6 наномолям PnIA[R5, L10, R14], растворенных в 12.5 мкл 125 мМ натрий-фосфатного буфера (рН 7.2), добавляют 10 наномолей раствора смеси [125-I]- и [127-I]йодида натрия (7.5 мкл в том же буфере) и 44 наномоля водного раствора хлорамина Т (1 мкл). Реакционную смесь перемешивают и оставляют на 20 мин при комнатной температуре. Продукты реакции разделяют в выше приведенных условиях и собирают в коллектор по 0.5 мин на фракцию. Аликвоты фракций просчитывают на γ-счетчике и по результатам полученной радиограммы собирают основной продукт PnIA[R5, L10, R14, Y15(125-I)] (SEQ ID NO:1) с примерной удельной радиоактивностью 6 Кu/ммоль, соответствующий по времени выхода пику 1 на Рис.1, и дополнительное ди-(125-I)-производное с примерной удельной радиоактивностью 12 Ки/ммоль, соответствующее пику 2. Собранные радиоактивные фракции для удаления ацетонитрила упаривают на 50% объема в присутствии аликвот стоковых растворов Трис-HCl буфера, рН 7.5, и БСА (бычий сывороточный альбумин) из расчета их примерной конечной концентрации в упаренных фракциях 50 мМ Трис-HCl буфера и 0.1 мг/мл БСА.
Пример 3. Функциональная характеристика PnIA[R5, L10, R14] и PnIA[R5, L10, R14, Y15(125-I)] по связыванию с альфа7 нАХР человека.
Функциональную активность PnIA[R5, L10, R14] (SEQ ID NO:1) оценивают по его способности конкурировать с радиоактивным альфа-бунгаротоксином ((125-I)-Bgt) за связывание с альфа7 нАХР человека, трансфецированного в клеточной линии GH4C1 крысы (фирма EliLilly). Связывание различных концентраций пептида с клетками проводят в 50 мкл 20 мМ Трис-HCl буфера, рН 8.0, содержащего 1 мг/мл БСА, в течение 2.5 ч при комнатной температуре и постоянном перемешивании. Общее количество клеточного белка в реакционной смеси составляет 6.5 мкг, а конечная концентрация токсин-связывающих участков альфа7 нАХР - 0.4 нМ. После этого к смеси добавляют (125I)-Bgt в конечной концентрации 0.2 нМ на 5 мин. Удаление не связавшегося радиолиганда осуществляют быстрой фильтрацией (3 раза по 3 мл 20 мМ Трис-HCl буфера, рН 8.0, содержащего 0.1 мг/мл БСА) через стеклянные фильтры GF/C фирмы Whatman, замоченные предварительно в течение 3 ч в 0.25% полиэтиленимине. Перед нанесением на фильтр реакционной смеси его промывают 3 мл указанного выше буфера. Уровень неспецифической сорбции радиолиганда оценивают, проводя вышеизложенное связывание в присутствии 2 мкМ альфа-кобратоксина.
Анализ конкурентного связывания и построение кривых ингибирования осуществляют с использованием программы ORIGIN 7.5 (OriginLab Corporation) для односайтовой модели по уравнению: % связывания = 100/{1+([пептид]/IC50)n}, где IC50 - концентрация пептида, вызывающая 50% ингибирование специфического связывания радиолиганда с мишенями, а n - коэффициент Хилла.
На Рисунке 2 представлена полученная в одном из нескольких независимых экспериментов кривая ингибирования специфического связывания (125-I)-Bgt с альфа7 нАХР человека для патентуемого пептида PnIA[R5, L10, R14] (SEQ ID NO:1). В качестве сравнения представлены также аналогичные кривые для его наиболее близких по структуре PnIA[L10, K14] и PnIA[R5, L10] аналогов, полученные параллельно в одном из экспериментов.
Эффективность взаимодействия указанных соединений, выраженная в виде значений IC50, представлена в Таблице 1.
Представленные данные показывают высокое сродство патентуемого соединения PnIA[R5, L10, R14] (SEQ ID NO:1) к альфа7 нАХР человека.
Равновесное связывание PnIA[R5, L10, R14, Y15(125-I)] (SEQ ID NO:1) с альфа7 нАХР человека, трансфецированного в клеточной линии GH4C1 крысы, проводят в 50 мкл 20 мМ Трис-HCl буфера, рН 8.0, содержащего 1 мг/мл БСА, в течение 1.5 ч при комнатной температуре и постоянном перемешивании. Для получения параметров специфического связывания различные концентрации радиолиганда (общий диапазон по разным опытам составлял от 6 до 1600 нМ) инкубируют с 0.4 нМ токсинсвязывающих участков альфа7 нАХР. Уровень неспецифического связывания определяли преинкубацией в течение 1.5 ч клеток GH4C1 с 25 мкМ альфа-кобратоксина. Удаление не связавшегося с клетками PnIA[R5, L10, R14, Y15(125-I)] осуществляют быстрой фильтрацией (3 раза по 3 мл 20 мМ Трис-HCl буфера, рН 8.0, содержащего 0.1 мг/мл БСА) через стеклянные фильтры GF/C фирмы Whatman, замоченные предварительно в течение 3 ч в 0.25% полиэтиленимине. Перед нанесением на фильтр реакционной смеси его промывают 3 мл указанного выше буфера.
Анализ равновесного связывания осуществляют с использованием программы ORIGIN 7.5 (OriginLab Corporation) для односайтовой модели по уравнению: В(х)=Bmax/(1+KD/x), где В(x) - концентрация специфически связанного радиолиганда, x - свободная концентрация радиолиганда (полученная вычитанием из добавленного в реакционную смесь количества радиолиганда, связавшегося с фильтрами и сорбировавшегося на стенках пробирки), Bmax - максимальное количество специфически связанного радиолиганда, a KD - константа диссоциации.
Было проведено несколько независимых экспериментов по получению кривых связывания различных фракций PnIA[R5, L10, R14, Y15(125-I)] (SEQ ID NO:1) с альфа7 нАХР на клеточной линии GH4C1 крысы. Среднее значение константы диссоциации по всем опытам составляет KD≈1000±600 нМ, однако в зависимости от использованной фракции радиолиганда и диапазона его концентрации это значение варьируется от KD≈190±130 нМ до KD≈1600±800 нМ. Результаты экспериментов, в которых были получены эти крайние значения, представлены на рисунках 3а и 3б соответственно. В любом случае, все значения KD для PnIA[R5, L10, R14, Y15(125-I)] (SEQ ID NO:1) в прямом радиолигандном тесте близки к значениям IC50 для PnIA[R5, L10, R14] (SEQ ID NO:1) в конкурентном тесте (см. Табл.1), указывая на сохранение высокого сродства к альфа7 нАХР патентуемого радиоактивного производного.
Конкурентный радиолигандный анализ с использованием PnIA[R5, L10, R14, Y15(125-I)] (SEQ ID NO:1) в качестве радиолиганда проводят на клеточной линии GH4C1 крысы, трансфецирующей альфа7 нАХР человека в 50 мкл 20 мМ Трис-HCl буфера, рН 8.0, содержащего 1 мг/мл БСА. Инкубация с клетками (0.4 нМ токсинсвязывающих участков альфа7 нАХР) различных концентраций PnIA[R5, L10, R14] (SEQ ID NO:1) и для сравнения PnIA[L10, K14] аналога длится 100 мин при комнатной температуре и постоянном перемешивании, после чего к смеси добавляют 180 нМ PnIA[R5, L10, R14, Y15(125-I)] (SEQ ID NO:1) и дополнительно инкубируют 100 мин в тех же условиях. Уровень неспецифического связывания определяют в присутствии 25 мкМ альфа-кобратоксина. Удаление не связавшегося с клетками радиолиганда и анализ конкурентного связывания осуществляют аналогично опытам с применением (125-I)-Bgt (см. выше).
Полученные результаты радиолигандного анализа с использованием PnIA[R5, L10, R14, Y15(125-I)] (SEQ ID NO:1) в качестве радиолиганда представлены на рисунке 4. Соответствующие расчетные значения IC50 для PnIA[R5, L10, R14] (SEQ ID NO:1) и PnIA[L10, K14] составляют 60±7 и 1800±600 нМ соответственно.
Перечень последовательностей.
SEQ ID NO:1. Аминокислотная последовательность пептида PnIA[L5R, A10L, D14R].
Glyl-Cys2-Cys3-Ser4-Arg5-Pro6-Pro7-Cys8-Ala9-Leu10-Asnll-Asnl2-Prol3-Argl4-X15-Cysl6-NH2,
где Х=Tyr или (I-125)Tyr,
дисульфидные связи между Cys2 и Cys8, Cys3 и Cys16.
| название | год | авторы | номер документа |
|---|---|---|---|
| АНАЛОГ АЛЬФА-КОНОТОКСИНА PnIA, ОБЛАДАЮЩИЙ ВЫСОКИМ СРОДСТВОМ И СЕЛЕКТИВНОСТЬЮ К АЦЕТИЛХОЛИН-СВЯЗЫВАЮЩЕМУ БЕЛКУ ИЗ APLYSIA CALIFORNICA | 2011 |
|
RU2458068C1 |
| Аналог альфа-конотоксина RgIA для лечения боли | 2016 |
|
RU2731217C2 |
| ПЕПТИДЫ И ИХ ПРОИЗВОДНЫЕ, ВЗАИМОДЕЙСТВУЮЩИЕ С НИКОТИНОВЫМ АЦЕТИЛХОЛИНОВЫМ РЕЦЕПТОРОМ И ПРИГОДНЫЕ ДЛЯ ИСПОЛЬЗОВАНИЯ В КОСМЕТОЛОГИИ ПРОТИВ МИМИЧЕСКИХ И ВОЗРАСТНЫХ МОРЩИН | 2013 |
|
RU2524428C1 |
| ПЕПТИД АЗЕМИОПСИН, ИЗБИРАТЕЛЬНО ВЗАИМОДЕЙСТВУЮЩИЙ С НИКОТИНОВЫМИ ХОЛИНОРЕЦЕПТОРАМИ МЫШЕЧНОГО ТИПА И ПРИГОДНЫЙ ДЛЯ ИСПОЛЬЗОВАНИЯ В КАЧЕСТВЕ МЫШЕЧНОГО РЕЛАКСАНТА В МЕДИЦИНЕ И КОСМЕТОЛОГИИ | 2011 |
|
RU2473559C1 |
| ЛИГАНДЫ МЕЛАНОКОРТИНОВЫХ РЕЦЕПТОРОВ, МОДИФИЦИРОВАННЫЕ ГИДАНТОИНОМ | 2008 |
|
RU2450017C2 |
| АНАЛОГИ ГРЕЛИНА | 2006 |
|
RU2427587C2 |
| ПОЛИПЕПТИД, ИМЕЮЩИЙ АНТИБАКТЕРИАЛЬНУЮ АКТИВНОСТЬ И ПРИГОДНЫЙ ДЛЯ ПОЛУЧЕНИЯ ЛЕКАРСТВЕННЫХ СРЕДСТВ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ ЧЕЛОВЕКА | 2010 |
|
RU2434880C1 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ КОНЪЮГАТОВ СОМАТОСТАТИН-ДОФАМИН | 2009 |
|
RU2464039C2 |
| СПОСОБ МОДУЛЯЦИИ ПРОЛИФЕРАЦИИ КЛЕТОК МЕДУЛЛЯРНОЙ КАРЦИНОМЫ ЩИТОВИДНОЙ ЖЕЛЕЗЫ | 2002 |
|
RU2352578C2 |
| СПОСОБ МОДУЛЯЦИИ ПРОЛИФЕРАЦИИ КЛЕТОК МЕДУЛЛЯРНОЙ КАРЦИНОМЫ ЩИТОВИДНОЙ ЖЕЛЕЗЫ | 2002 |
|
RU2275934C2 |
Настоящее изобретение относится к области биохимии. Предложен аналог альфа-конотоксина PnIA, обладающий высоким сродством к нейрональному альфа7 типу никотинового ацетилхолинового рецептора и представленный последовательностью SEQ ID NO:1. Использование изобретения позволяет расширить ассортимент веществ, являющихся конкурентными антагонистами альфа7 типа никотинового ацетилхолинового рецептора. Изобретение может быть использовано в медицине для создания лекарственных средств при лечении болезней, связанных с нарушением работы нейронального альфа7 типа никотинового ацетилхолинового рецептора. 2 н.п. ф-лы, 5 ил., 1 табл., 3 пр.
1. Пептид, имеющий высокое сродство к альфа7 типу никотинового ацетилхолинового рецептора человека и представленный последовательностью SEQ ID NO: 1.
2. Применение пептида по п.1 для детекции альфа7 типа никотинового ацетилхолинового рецептора человека.
| Прибор для сигнализирования пропуска папирос при их укладке | 1926 |
|
SU5365A1 |
| Прибор для сигнализирования пропуска папирос при их укладке | 1926 |
|
SU5365A1 |

