Область техники, к которой относится изобретение.
Настоящее изобретение относится к биохимии, а именно к новому пептидному соединению, способному блокировать прохождение болевого сигнала. Более конкретно данное изобретение относится к применению этого пептида для лечения боли различной этиологии.
Уровень техники.
Под хронической болью как правило подразумевают боль, продолжительность ощущения которой заведомо превосходит время, необходимое для излечения от причины ее вызвавшей. По этиологии хроническая боль многими специалистами разделяется на ноцицептивную и нейропатическую. Ноцицептивная хроническая боль вызвана активацией болевых рецепторов в органах и тканях, например, в результате воспалительной реакции. Нейропатическая хроническая боль вызвана нарушениями в работе нервной системы и может иметь как периферическое происхождение (соматические нервы, нервная система пищеварительного тракта) так и центральное (нарушения в работе спинного или головного мозга).
Хроническая боль является крайне актуальной проблемой современной медицины. По различным данным от 10,1% до 55,2% людей в мире страдают от различных проявлений хронической боли (Harstall С, Ospina М. How Prevalent Is Chronic Pain? June 2003 volume XI issue2 Pain Clinical Updates, International Association for the Study of Pain. Pages=1-4). Стоит отметить, что, по различным данным, вплоть до 67% обращений к хирургическому персоналу так или иначе связано с ощущением неспецифических болей в теле, которые можно отнести к хронической боли. В Великобритании, например, негативный экономический эффект, связанный с подобной хронической болью оценивается приблизительно в 100 млн. фунтов в год (Chronic Abdominal and Visceral Pain: Theory and Practice. Pankaj Jay Pasricha, William D. Willis, G.F. Gebhart). Несмотря на огромную значимость проблемы, не выработано единого подхода к лечению хронической боли: диапазон методов простирается от психологической или когнитивной терапии до применения опиоидных анальгетиков. При этом каждый из подходов имеет определенные недостатки, что подтверждает наличие неудовлетворенного спроса на анальгетик нового поколения для лечения хронической боли.
Весьма эффективны в снятии симптомов хронической боли опиоидные анальгетики. Однако их применение сопряжено с развитием привыкания и уменьшением эффективности при длительном применении. Вместе с тем феномен хронической боли заключается именно в длительности болевых ощущений. Таким образом, даже весьма эффективные опиоиды не являются оптимальным средством борьбы с хронической болью (Chronic Abdominal and Visceral Pain: Theory and Practice. Pankaj Jay Pasricha, William D. Willis, G.F. Gebhart).
Однако, относительно недавно для лечения хронической боли начал применяться пептидный антагонист кальциевых каналов «Ziconotide», представляющий собой омега-конотоксин. Основным преимуществом данного конотоксина перед опиоидными анальгетиаками является отсутствие многих побочных эффектов, таких как развитие зависимости, постепенное уменьшение анальгетического эффекта и бессонница. Вместе с тем, «Ziconotide» имеет ряд побочных эффектов, включая парадоксальные реакции - усиление болевых ощущений под действием препарата (Sanford, Mark. "Intrathecal ziconotide: a review of its use in patients with chronic pain refractory to other systemic or intrathecal analgesics." CNS drugs 27.11 (2013): 989-1002.).
К настоящему времени из яда морских улиток были выделены и исследованы несколько альфа-конотоксинов способных подавлять нейропатическую боль. Так недавно открытый представитель подгруппы 4/3 α-конотоксинов - RgIA, в отличие от родственных ImI и ImII, оказался высоко активным и селективным блокатором α9α10 нАХР [Ellison, М. et al (2006) Biochemistry, 45, 1511-1517.]. Особый интерес к этому пептиду и α-конотоксину Vc1.1 вызван их способностью подавлять нейропатическую боль на моделях in vivo [Vincler, М. et al Proc. Natl. Acad. Sci. USA, 103, 17880-17884]. Однако задействованность в антиболевом эффекте α9α10 нАХР в настоящее время активно дискутируется [Nevin, S.T., et al Mol. Pharmacol., 72, 1406-1410], в частности и потому, что природный прототип α-конотоксина Vc1.1 (Vela, несущий две пост-трансляционные модификации [Jakubowski, J.A., et al J. Mass Spectrom., 39, 548-557]), также является мощным антагонистом α9α10 нАХР, но не обладает антиболевой активностью.
Лекарственный препарат представляет собой синтетический вариант природного соединения - пептида RgIA, впервые обнаруженного в библиотеке кДНК ядовитых желез морского моллюска Conus regius. Исходный пептид RgIA является представителем семейства альфа-конотоксинов - коротких 10-15-членных пептидов, ингибирующих никотиновые ацетилхолиновые рецепторы (нАхР). Природным источником альфа-конотоксинов являются яды морских моллюсков рода Conus. RgIA имеет два принципиально различных типа молекулярных мишеней.
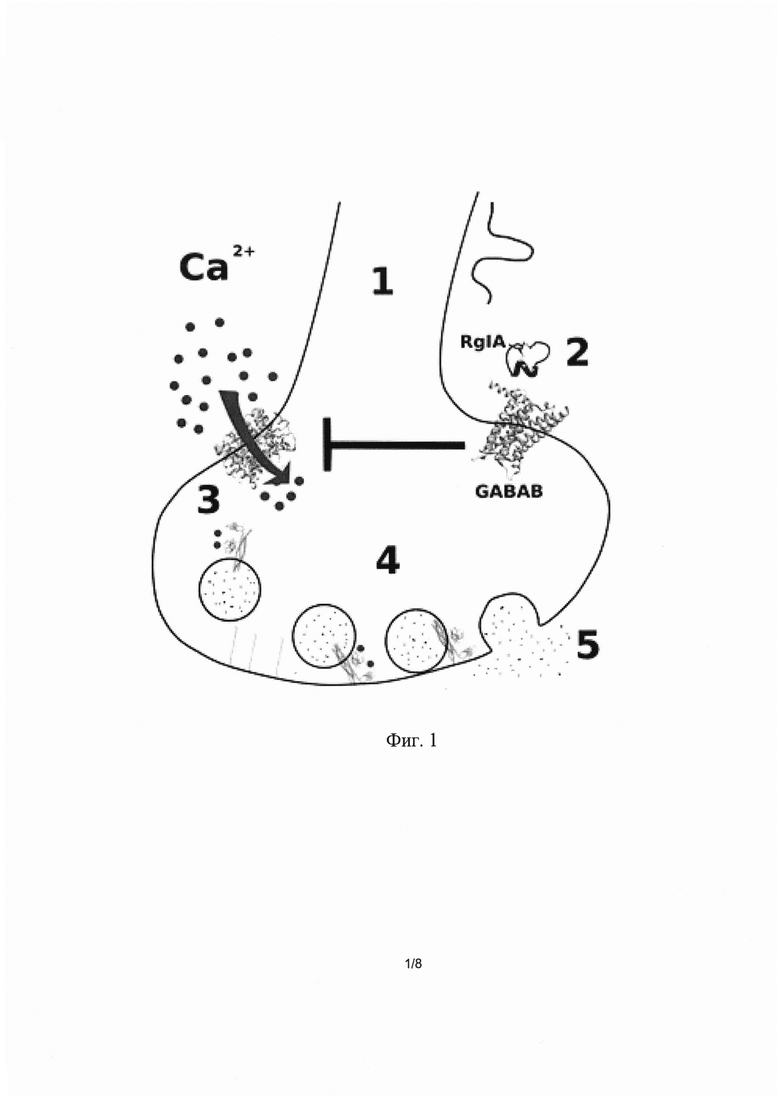
Первый тип молекулярных мишеней RgIA - потенциал чувствительные кальциевые каналы. Этот тип ионных каналов играет ключевую роль в высвобождении нейромедиаторов, ответственных за болевые ощущения. Блокирование потенциал-зависимых кальциевых каналов уменьшает выброс болевых нейромедиаторов из пресинаптических окончаний, что способствует достижению обезболивающего эффекта (Фиг. 1). Рассмотрим процесс поэтапно. На первом этапе (1) сигнал, кодирующий болевые ощущения, приходит в окончание эффекторного отростка (аксона) чувствительного нейрона. В нормальных условиях это приводит к активации потенциал-зависимых кальциевых каналов (3) и входу ионов кальция в цитоплазму нейрона. Ионы кальция связываются с белком синаптотагмином, который запускает процесс слияния мембран синаптических везикул с пресинаптической мембраной (4) и выброс нейромедиаторов в синаптическую щель (5). Для уменьшения эффективности передачи болевого сигнала достаточно снизить активность потенциалчувствительных кальциевых каналов. Одним из путей такого уменьшения активности является активация метаботропных рецепторов гамма-аминомасляной кислоты (GABAB). RgIA активирует GABAB, которые, в свою очередь, запускают сигнальный каскад, приводящий к уменьшению активности кальциевых каналов (2). В результате такого воздействия уменьшается количество «болевых» нейромедиаторов в синаптической щели и, как следствие, уменьшается интенсивность болевых ощущений.
Второй тип мишеней - нАхР альфа9/альфа10 подтипов, к которым сродство пептида наиболее высоко (100-200 нМ). Особенностью рецепторов данных подтипов является то, что они участвуют в проведении болевых сигналов различной этиологии, преимущественно нейропатической и воспалительной. В частности, модельные животные, нокаутные по гену альфа9 нАхР менее восприимчивы к механически вызванной гиперальгезии - одному из компонентов нейропатической боли, что безусловно подтверждает актуальность применения лигандов данного нАхР в качестве модуляторов болевых ощущений (doi: 10.1186/1744-8069-10-64).
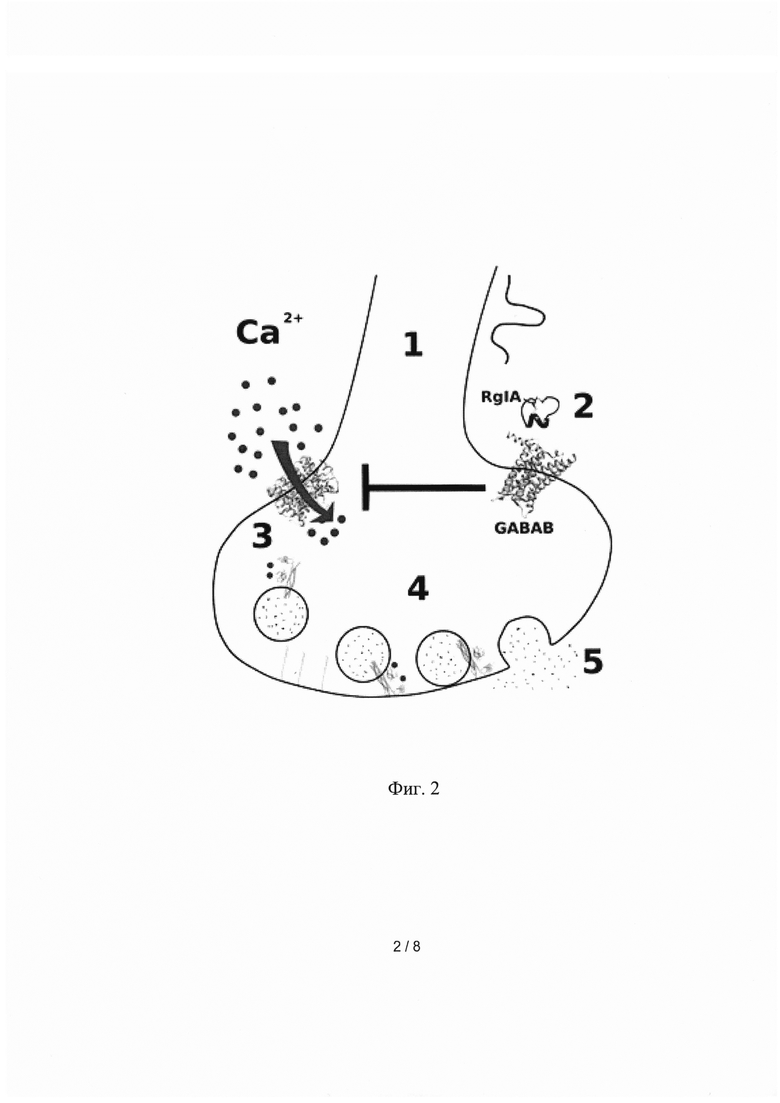
Особенностью данного механизма анальгезии является то, что нАхР альфа9/альфа10 подтипа сами являются лиганд-управляемыми кальциевыми каналами (Фиг. 2). Поэтому их активация приводит к входу ионов кальция в цитоплазму нейрона и стимуляции выброса «болевых» нейромедиаторов в синаптическую щель. RgIA непосредственно ингибирует нАхР и препятствует входу ионов кальция, что уменьшает выброс нейромедиаторов и передачу болевого сигнала.
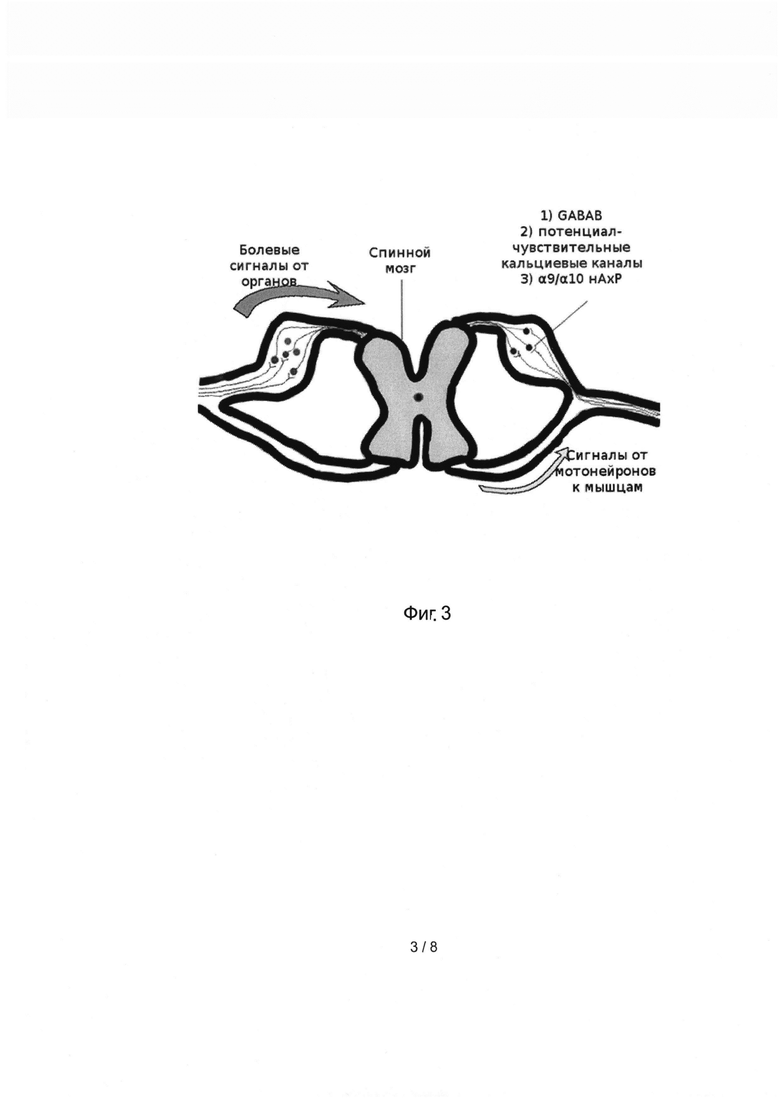
Оба типа мишеней - GABAB и альфа9/альфа10 нАхР - экспрессированы в нейронах ганглиев задних корешков спинного мозга (dorsal root ganglion, DRG). Нейроны этих ганглиев задействованы в передаче сенсорной информации от периферических органов в спинной мозг, в частности, сигналы, передаваемые по немиелинизированным волокнам класса С, передающим болевые сигналы (Фиг. 3). Болевые сигналы по волокнам класса «С» поступают в спинной мозг через задние корешки. При этом в ганглиях этих корешков происходит передача сигнала от чувствительного нейрона к вставочному нейрону. В синаптическом контакте данных нейронов экспрессированы интересующие нас молекулярные мишени: метаботропные рецепторы гамма-аминомасляной кислоты (GABAB), сопряженные с потенциал-чувствительными кальциевыми каналами, а также ионотропные ацетилхолиновые рецепторы α9/α10 типа.
Таким образом, при взаимодействии RgIA с рецепторами на поверхности нейронов заднекорешковых ганглиев происходит ингибирование проведения болевых сигналов в результате подавления выброса нейромедиаторов из пресинаптических окончаний чувствительных нейронов. Это приводит к тому, что эффективность передачи информации через такой синапс становится значительно ниже. Следовательно, в мозг поступают сигналы меньшей интенсивности, что интерпретируется, как уменьшение болевых ощущений.
Стоит отметить, что RgIA и его аналоги выгодно отличаются от различных низкомолекулярных анальгетиков, присутствующих на рынке. Будучи пептидными соединениями они плохо проникают через гематоэнцефалический барьер, что исключает развитие "центральных" побочных эффектов. С другой стороны, основная мишень их действия - рецепторы на поверхности нейронов заднекорешковых ганглиев в меньшей степени изолированы от системного кровотока, что позволяет использовать традиционные методы введения препарата вместо сложных систем с введением анальгетика в цереброспинальную жидкость.
В то же время, одним из препятствий для внедрения конотоксинов, как и других дисульфидсодержащих пептидов, в медицинскую практику в качестве лекарственных средств является нестабильность дисульфидных связей (doi: 10.1016/j.ijpharm.2015.04.041, doi:10.1089/ars.2009.3068). В результате реакций тиолдисульфидного обмена или восстановления, исходные молекулы теряют свою структуру и, как следствие, сродство к своим мишеням в организме, снижается или исчезает совсем физиологический эффект. Поэтому исследователи прилагают значительные усилия для создания на основе конотоксинов RgIA и Vc1.1, как терапевтически интересных, более стабильных молекул (doi: 10.1021/jm501126u, doi: 10.1002/anie.201409678, doi: 10.1002/bip.22699, doi: 10.1038/srep13264). Все известные попытки относятся либо к полной замене дисульфидной связи, либо к ее модификации. Основным недостатком полученных соединений является снижение их активности. Таким образом, весьма актуальной является задача разработки стабильных и активных аналогов конотоксинов для применения их в качестве анальгетиков нового поколения, лишенных недостатков уже существующих препаратов. Ближайшим аналогом объекта изобретения является природный альфа-конотоксин RgIA.
Раскрытие изобретения.
Настоящее изобретение решает задачу получения стабильной молекулы, которую можно использовать в лекарственных препаратах для лечения боли. Технический результат достигается за счет изменения природной структуры молекулы альфа-конотоксина RgIA путем модификации дисульфидной связи, которое заключается в ее частичном десульфурировании Объектом изобретения является пептид, состоящий из тех же аминокислотных остатков, что и природный RgIA, но содержащий две внутримолекулярные тиоэфирные (сульфидные) связи между остатками цистеина 2 и 8, 3 и 12. Аминокислотная последовательность пептида Rg2S (SEQ ID:1): H-Gly-Cys-Cys-Ser-Asp-Pro-Arg-Cys-Arg-Tyr-Arg-Cys-Arg-OH. Такое изменение структуры позволяет решить проблему устойчивости молекулы, поскольку сульфидная связь не восстанавливается в обычных условиях тиольными агентами (например, дитиотреитолом). Это в свою очередь приводит к жесткой структуре, которая не расщепляется ферментами. Например, под воздействием трипсина от пептида SEQ ID:1 отщепляется только С-концевой аргинин, остальная часть молекулы остается стабильной. В то же время, изменение структуры не повлияло на биологическую активность молекулы. Так в тесте «горячая пластина» было показано (Табл. 1), что, во-первых, пептид SEQ ID:1 проявляет большую активность по сравнению с природными конотоксинами VC1.1 и Rg1A, а, во-вторых, активность заявляемого пептида является дозозависимой.
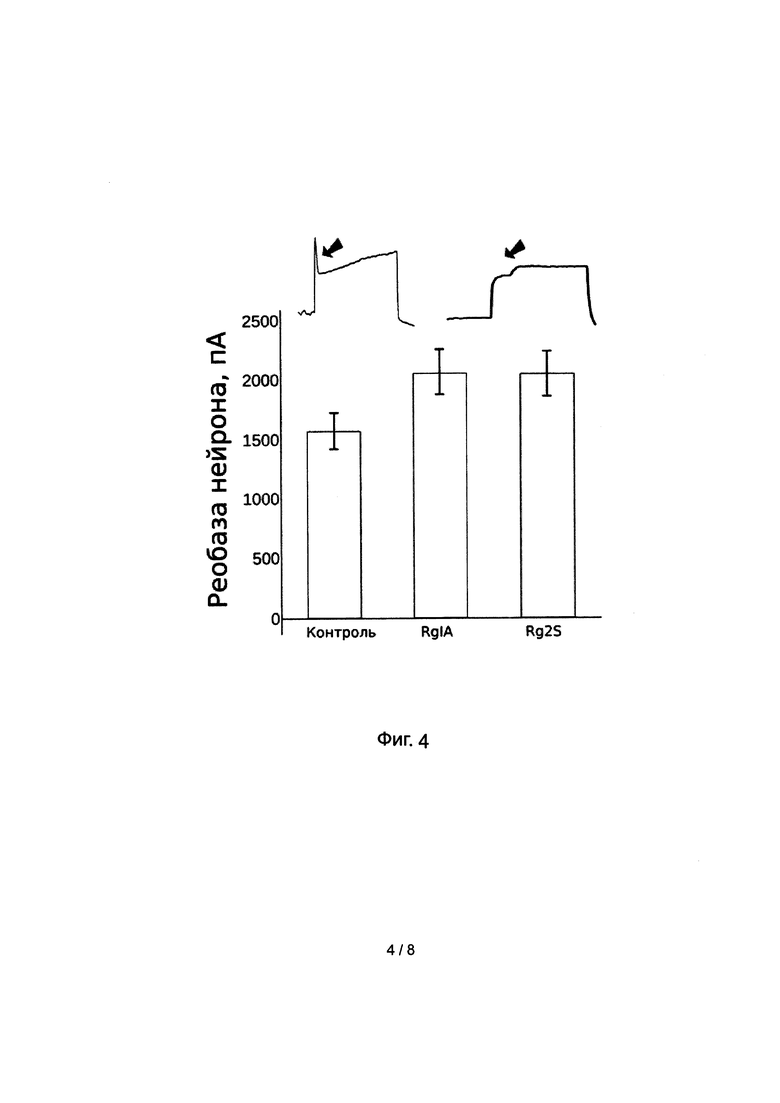
Эксперименты на природных мишенях конотоксина Rg1A показали, что активность пептида Rg2S сохранилась либо незначительно уменьшилась. Так, В опытах по электрофизиологическому тестированию возбудимости нейронов заднекорешковых ганглиев было установлено, что аппликация модифицированных альфа-конотоксинов RgIA вызывает уменьшение возбудимости нейронов (увеличение показателя реобазы Фиг. 4)
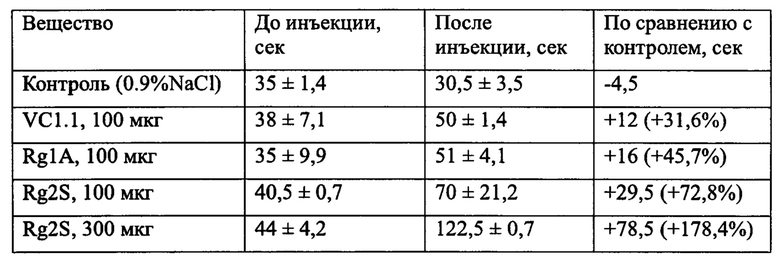
Талица 1. Время отдергивания и облизывания лапки мыши в тесте горячей пластины. Средние значения ± несмещенное стандартное отклонение.
Полноклеточная регистрация мембранного потенциала нейронов заднекорешковых ганглиев была проведена в режиме фиксации тока. Микропипетки электрода изготавливались с помощью автоматического пулера Narishige (сопротивление пипетки не менее 5 МОм) и заполнялись внутриклеточным раствором (140 мМ CsCl, 6 мМ CaCl2, 2 мМ MgCl2, 2 мМ MgATP, 0.4 мМ NaGTP, 10 мМ HEPES/CsOH, 20 мМ ВАРТА/КОН; рН 7.3). Измерения проводят при комнатной температуре (23-25°С) с помощью пэтч-кламп усилителя НЕКА. В данном опыте проводили последовательную подачу ступенек инъецируемого в клетку тока ступеньками возрастающей амплитуды с шагом 50 пикоА и длительностью 500 мс.В результате опыта определялась реобаза нейрона до и после воздействия модифицированного конотоксина RgIA в концентрации 1 мкрМ. Возрастание реобазы свидетельствует об ингибировании нейрона, а следовательно и о затруднении проведения болевого сигнала через данный нейрон. Как ранее было установлено в литературе (doi: 10.1136/gutjnl-2015-310971), подобные эффекты вероятнее всего опосредованы активацией метаботропных рецепторов гамма-аминомасляной кислоты.
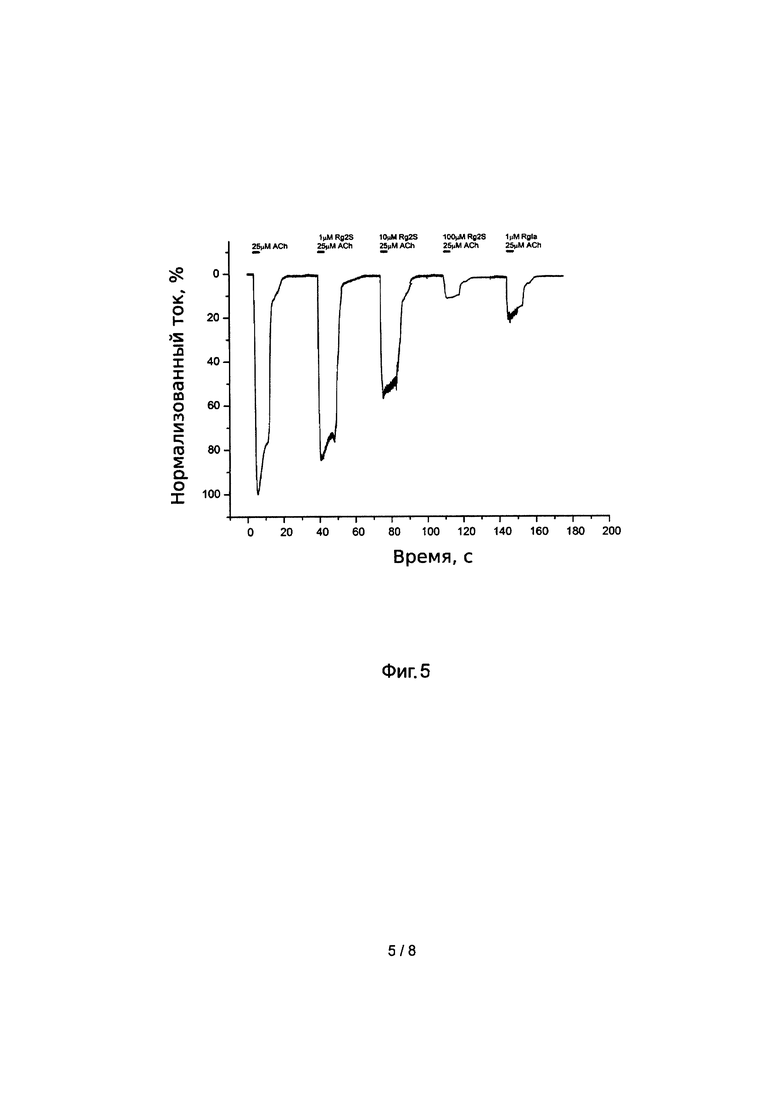
Для изучения активности пептида Rg2S на альфа9/альфа10 нАхР использовали ооциты Xenopus, экспрессирующие этот рецептор. Измерения проводили при помощи дифференциального усилителя TURBO ТЕС-03Х (DruMMond, Германия) методом двухэлектродной фиксации потенциала (TEVC, two-electrode voltage сlамр) при -60 мВ. Объем камеры, в которой находился ооцит, составлял 50 мкл. Регистрацию ионных токов ооцита проводили при аппликации 25 мкМ раствора ацетилхолина йодида (SigMa-Aldrich, США) приготовленного на основе буфера Barth's, содержащим ионы кальция. Объем вносимого лиганда - 100 мкл, промывка буфером - 1 мл. Между аппликациями лиганда выдерживали паузу 5 минут. Для изучения эффекта конотоксинов на α9α10 нАХР, ооциты преинкубировали с растворами RgIa (1 мкМ) и его аналога (1 мкМ, 10 мкМ, 100 мкМ) в течение 5 минут перед нанесением агониста.
Раствор аналога Rg2S-2 в концентрации 1-100 мкМ блокирует ионный ток α9α10 экспрессирующих ооцитов, вызванный 25 мкМ раствором ацетилхолина (Фиг. 5). В то же время, для RgIA блокирующий эффект был продемонстрирован при концентрации токсина равной 1 мкМ и менее, что говорит о меньшем сродстве аналога к данному подтипу нАхР.
Опыты по изучению активности пептида Rg2S (SEQ ID:1) показали, что он способен воздействовать на пути передачи болевых импульсов и подавлять боль путем взаимодействия с различными мишенями.
Изобретение иллюстрируют Фиг. 1-3:
Фиг 1. Механизм действия конотоксина RgIA через метаботропные рецепторы гама-аминомасляной кислоты.
Фиг 2. Механизм действия конотоксина RgIA через ионотропные ацетилхолиновые рецепторы (нАхР).
Фиг 3. Упрощенная схема передачи болевых сигналов с участием ганглиев задних корешков спинного мозга.
Фиг 4. Увеличение реобазы нейронов заднекорешковых ганглиев под воздействием модифицированного аналога альфа-конотоксина RgIA. В результате воздействия тестируемых пептидов возбудимость нейронов уменьшается, блокируется генерация потециала действия. Стрелка отмечает наличие потенциала действия в контроле (пик) и отсутствие после апликации Rg2S.
Фиг 5. Электрофизиологическое тестирование влияния аналога конотоксина RgIA на активность α9α10 нАхР.
Фиг 6. Реакционная смесь при окислении линейного предшественника пептида Rg2S.
Фиг 7. Аналитическая хроматограмма пептида Rg2S.
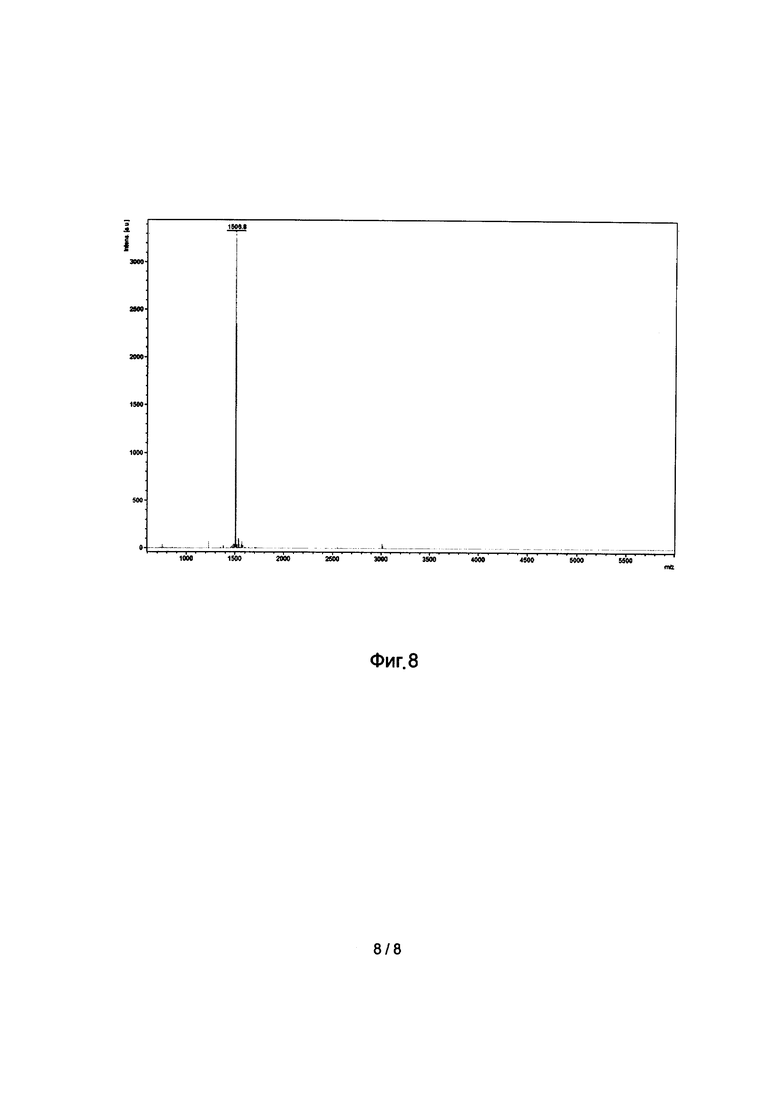
Фиг 8. Масс-спектр пептида Rg2S.
Примеры осуществления изобретения.
Пример 1. Химический синтез пептида SEQ ID NO: 1: H-Gly-Cys-Cys-Ser-Asp-Pro-Arg-Cys-Arg-Tyr-Arg-Cys-Arg-OH
Присоединение первой аминокислоты. Получение Fmoc-Arg(Pbf)-P (Fmoc=9-флуоренилметилоксикарбонил).
200 мг 2-хлоротритилхлоридного полимера с содержанием гидроксильных групп 1,0 ммоль/г промывают безводным хлористым метиленом. В 5 мл хлористого метилена растворяют 713 мг (1,1 ммоль) Fmoc-Arg(Pbf)-OH и 374 мкл (2,2 ммоль) диизопропилэтиламина, раствор перемешивают 5 мин и затем приливают к полимеру. Реакцию проводят 1 час при комнатной температуре и перемешивании. По окончании реакции полимер отфильтровывают и трижды промывают последовательно хлористым метиленом, этиловым спиртом, диметилформамидом.
Описание одного синтетического цикла наращивания полипептидной цепи.
Получение Fmoc-Cys(Trt)-Arg(Pbf)-P:
а) Полученный на предыдущем этапе пептидил-полимер в течение 20 мин обрабатывают 20%-ным раствором пиперидина в диметилформамиде. Полимер промывают последовательно 5 мл следующих растворителей: диметилформамидом - 3 раза по 2 мин., смесью диоксан-вода (2:1) - 2 раза по 5 мин., диметилформамидом - 5 раз по 2 мин.
б) В 5 мл диметилформамида растворяют 644 мг (1,1 ммоль) Fmoc- Cys(Trt)-OH, 150 мг (1,1 ммоль) 1-гидроксибензотриазола, и 172 мкл (1,1 ммоль) N,N'-диизопропилкарбодиимида, раствор перемешивают 10 мин при 0°С и затем приливают к суспендированному полимеру. Реакцию проводят 4 часа при периодическом перемешивании. По окончании реакции полимер отфильтровывают, промывают диметилформамидом, и обрабатывают 5 мл смеси Ac2O-пиридин-диметилформамид (20:20:60) в течение 1 ч., после чего полимер последовательно промывают диметилформамидом, изопропанолом, диметилформамидом.
Синтез полипептидной цепи проводят вручную в стеклянном проточном реакторе (2×20 см) по следующему протоколу для каждого синтетического цикла (из расчета 8-10 мл растворителя на 400 мг исходного полимера), при проведении реакции конденсации (операция 6) используют объем реакционной смеси 5-7 мл:
1. ДМФА (диметилформамид) (5×2 мин.);
2. 20% пиперидин в ДМФА (20 мин.);
3. ДМФА (3×2 мин.);
4. диоксан-вода, 2:1, (2×5 мин.);
5. ДМФА (5×2 мин.);
6. Реакция конденсации: 5 молярных эквивалентов активированной Fmoc-аминокислоты (4 ч.);
7. ДМФА (3×2 мин.);
8. ацилирование: Ас2О-пиридин-диметилформамид, 20:20:60, (1 ч.);
9. ДМФА (3×2 мин.);
10. изопропанол (3×2 мин.);
Для активации Fmoc-производных аминокислот с использованием смеси DIPCDI/HOBT (N,N'-диизопропилкарбодиимид/1-гидроксибензотриазол) к раствору 1,1 ммоль (5 эквивалентов) Fmoc-защищенной аминокислоты и 150 мг (1,1 ммоль, 5 эквивалентов) HOBt в 4 мл ДМФА добавляют 170 мкл (1,1 ммоль, 5 эквивалентов) DIPCDI, раствор перемешивают 10 мин.
Полноту протекания реакции конденсации контролируют с помощью нингидринового или, в случае N-концевого пролина, изатинового тестов после операции 6 синтетического протокола.
Для синтеза используют следующие производные аминокислот: Fmoc-Gly-OH, Fmoc-Ser(tBu)-OH, Fmoc-Cys(Trt)-OH, Fmoc-Pro-OH, Fmoc-Asp(OtBu)-OH, Fmoc-Arg(Pbf)-OH, Fmoc-Tyr(tBu)-OH.
Отщепление пептида от полимера.
Для реакции отщепления пептида от полимера и одновременного деблокирования защитных групп боковых цепей аминокислот отбирают 800 мг пептидил-полимера. К пептидил-полимеру приливают 15 мл смеси ТФУ (трифторуксусная кислота) -H2O в объемном соотношении 97.5:2.5, суспензию перемешивают в течение 2 ч., затем полученный раствор пептида отфильтровывают от полимера, промывают 5 мл ТФУ и избыток ТФУ упаривают при пониженном давлении. Пептид осаждают 100 мл этилового эфира, отфильтровывают и промывают эфиром (5×20 мл). Осадок растворяют в 5 мл 10% уксусной кислоты 20 мин., отфильтровывают и промывают 5 мл 10% уксусной кислоты. Полученный раствор пептида лиофилизируют и обессоливают на колонке (2,5×60 см) с Сефадексом G-10 в 0.1М уксусной кислоте. Очистку пептида проводят с помощью обращенно-фазной ВЭЖХ в градиенте ацетонитрила (от 10% до 35% за 75 мин.) в 0.1% ТФУ при расходе элюента 3 мл/мин, поглощение элюата регистрируют при длине волны 226 нм. Фракции, соответствующие основному пику на хроматограмме собирают и лиофилизируют. Молекулярная масса пептида, установленная методом масс-спектрометрии составила 1574,6 Да, теоретическая расчетная молекулярная масса - 1574,8 Да.
Получение сульфидных связей между цистеинами в линейном конотоксине RgIA.
В 80 мл смеси ацетонитрил\вода (1\1) при перемешивании растворяют 40 мг линейного пептида RgIA, затем добавляют 300 мкл диизопропилэтиламина (рН≥12). Реакцию проводят в открытой колбе при перемешивании двое суток. Затем добавляют уксусную кислоту до рН=5, раствор замораживают и лиофилизовывают.Из полученной смеси (Фиг. 6) выделяют основной пик (Фиг. 7). Молекулярная масса пептида, установленная методом масс-спектрометрии составила 1506,8 Да (Фиг. 8), теоретическая расчетная молекулярная масса - 1506,8 Да.
Пример 2. Активность пептида SEQ ID NO: 1 в тесте горячая пластина.
Подопытный самец белой мыши массой тела 20-25 г помещался на термостатируемую пластину (53,5±0,3 град С). Регистрировали время первого отдергивания и облизывания задней лапки, после чего мышь перемещали в отдельный контейнер. До инъекции препарата осуществляли три измерения. Интервал между измерениями составлял 7,5 минут. Исследуемое вещество в указанной дозе (см. Таблицу 1) в водном растворе с 0.9% хлорида натрия вводили этой же особи внутрибрюшинно в объеме не более 0.3 мл. Через 7,5 минут после инъекции проводили четыре измерения времени первого отдергивания и облизывания задней лапки с интервалом 7,5 минут. Рассчитывали среднее время отдергивания и облизывания задней лапки мыши до инъекции препарата и после инъекции.
Пример 3. Двухэлектродная фиксация потенциала мембраны ооцитов Xenopus laevis
Плазмиды, содержащие гены α9 и α10 субъединиц нАХР крысы (вектор pGEMNE) были линеаризованы эндонуклеазой рестрикции NheI (NEB, USA). Полученные линеаризованные плазмиды выделяют при помощи набора QIAquick PCR Purification Kit (Quiagen, Нидерланды). Затем, используя набор Т7 MMessage мМасhinе transcription kit (Амbion Inc., Austin, ТХ, USA), проводят синтез мРНК α9 и α10 субъединиц нАХР in vitro. Для очистки полученной мРНК был использован набор RNeasy MinElute Cleanup (Quiagen, Нидерланды).
Для последующей трансфекции отбирают ооциты Xenopus laevis IV и V стадии, полученные путем прямой диссекции брюшной полости самки с выделением яичников одной стороны. Лягушку помещают в холодный раствор парааминобензоата 0,5 г/л («Potaba», Германия) на 15-30 мин и затем переносят в лед. Охлажденными ножницами делают надрез около 10 мм длиной, откуда извлекают фрагмент яичника. Выделенные яичники обрабатывают коллагеназой типа А (4 мг/мл, Worthington, USA) в Barth's буфере, не содержащем кальция (88.0 мМ NaCl, 1.1 мМ KCl, 2.4 мМ NaHCO3, 0.8 мМ MgSO4, 15.0 мМ HEPES/NaOH, рН 7.6) в течение 4 часов. Около 7 нг мРНК α9 и α10 субъединиц нАХР инъецируют в районе анимального полюса ооцитов при помощи вакуумного наноинжектора NanoJect-2 (Druммond, Германия) в объеме 23 нл на ооцит. Инъецированные ооциты, затем, инкубируют в течение 72-120 часов при постоянной температуре +17°С в буфере Barth's, содержащим ионы кальция (88.0 мМ NaCl, 1.1 мМ KCl, 2.4 мМ NaHCO3, 0.3 мМ Ca(NO3)2, 0.4 мМ CaCl2, 0.8 мМ MgSO4, 15.0 мМ HEPES/NaOH, рН 7.6) с добавлением 100 мкг/мл апмициллина и 40 мкг/мл гентамицина.
--->
Sequence listing
<110> Limited Liability Company «Syneuro»
<120> An analogue of alpha-conotoxin RgIA to treat pain.
<130> Rg2S
<150>
<151> 2016-11-24
<160> 1
<210> 1
<211> 13
<212> PRT
<213> Art
<220>
<223> Sulfide bond between Cys2/Cys8 and Cys3/Cys12
<400>1
Gly-Cys-Cys-Ser-Asp-Pro-Arg-Cys-Arg-Tyr-Arg-Cys-Arg
1 5 10
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| ПЕПТИД, ИМЕЮЩИЙ ВЫСОКОЕ СРОДСТВО К АЛЬФА7 ТИПУ НИКОТИНОВОГО АЦЕТИЛХОЛИНОВОГО РЕЦЕПТОРА ЧЕЛОВЕКА, И ЕГО ПРИМЕНЕНИЕ | 2011 |
|
RU2455359C1 |
| ПЕПТИД АЗЕМИОПСИН, ИЗБИРАТЕЛЬНО ВЗАИМОДЕЙСТВУЮЩИЙ С НИКОТИНОВЫМИ ХОЛИНОРЕЦЕПТОРАМИ МЫШЕЧНОГО ТИПА И ПРИГОДНЫЙ ДЛЯ ИСПОЛЬЗОВАНИЯ В КАЧЕСТВЕ МЫШЕЧНОГО РЕЛАКСАНТА В МЕДИЦИНЕ И КОСМЕТОЛОГИИ | 2011 |
|
RU2473559C1 |
| ПЕПТИДНЫЙ МОДУЛЯТОР ПУРИНЕРГИЧЕСКИХ РЕЦЕПТОРОВ | 2010 |
|
RU2422459C1 |
| ПЕПТИДЫ И ИХ ПРОИЗВОДНЫЕ, ВЗАИМОДЕЙСТВУЮЩИЕ С НИКОТИНОВЫМ АЦЕТИЛХОЛИНОВЫМ РЕЦЕПТОРОМ И ПРИГОДНЫЕ ДЛЯ ИСПОЛЬЗОВАНИЯ В КОСМЕТОЛОГИИ ПРОТИВ МИМИЧЕСКИХ И ВОЗРАСТНЫХ МОРЩИН | 2013 |
|
RU2524428C1 |
| АНАЛОГ АЛЬФА-КОНОТОКСИНА PnIA, ОБЛАДАЮЩИЙ ВЫСОКИМ СРОДСТВОМ И СЕЛЕКТИВНОСТЬЮ К АЦЕТИЛХОЛИН-СВЯЗЫВАЮЩЕМУ БЕЛКУ ИЗ APLYSIA CALIFORNICA | 2011 |
|
RU2458068C1 |
| ПОЛИПЕПТИД АКТИНИИ, ОБЛАДАЮЩИЙ АНАЛЬГЕТИЧЕСКИМ ДЕЙСТВИЕМ | 2008 |
|
RU2368621C1 |
| ПЕПТИДНЫЙ МОДУЛЯТОР ПУРИНЕРГИЧЕСКИХ РЕЦЕПТОРОВ | 2016 |
|
RU2650780C1 |
| СПОСОБ СТИМУЛЯЦИИ ХРОНИЧЕСКОЙ БОЛЬЮ ЗЛОКАЧЕСТВЕННОГО РОСТА В ЛЁГКИХ КРЫС | 2018 |
|
RU2676641C1 |
| ПЕПТИД, ОБЛАДАЮЩИЙ АКТИВНОСТЬЮ БЕЛКА LYNX1 (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ ТРЕВОЖНЫХ РАССТРОЙСТВ И ДЕПРЕССИИ ИЛИ КОРРЕКЦИИ КОГНИТИВНЫХ НАРУШЕНИЙ ПРИ НЕЙРОДЕГЕНЕРАТИВНЫХ ЗАБОЛЕВАНИЯХ, СОДЕРЖАЩАЯ УКАЗАННЫЙ ПЕПТИД, И СПОСОБ ЛЕЧЕНИЯ И КОРРЕКЦИИ УКАЗАННЫХ НАРУШЕНИЙ | 2020 |
|
RU2734649C1 |
| НЕЦИТОТОКСИЧЕСКОЕ ВЕЩЕСТВО, СПОСОБ РЕГУЛЯЦИИ ВЫДЕЛЕНИЯ НЕЙРОМЕДИАТОРА ИЛИ НЕЙРОМОДУЛЯТОРА ИЗ ПЕРВИЧНЫХ СЕНСОРНЫХ АФФЕРЕНТНЫХ КЛЕТОК И СПОСОБ РЕГУЛЯЦИИ ВЫДЕЛЕНИЯ НЕЙРОМЕДИАТОРА И НЕЙРОМОДУЛЯТОРА ИЗ ПЕРВИЧНЫХ НОЦИЦЕПТИВНЫХ АФФЕРЕНТНЫХ КЛЕТОК | 1996 |
|
RU2165976C2 |
Настоящее изобретение относится к биохимии, а именно к новому пептидному соединению, способному блокировать прохождение болевого сигнала. Объектом изобретения является аналог конотоксина RgIA, в котором дисульфидные связи модифицированы в тиоэфирные. Новый аналог показывает высокую биологическую и физиологическую активность. Изобретение может быть использовано при получении лекарственных препаратов для лечения боли различной этиологии. 2 н.п. ф-лы, 8 ил., 1 табл., 3 пр.
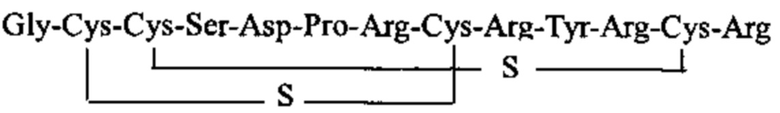
1. Соединение общей формулы (I):
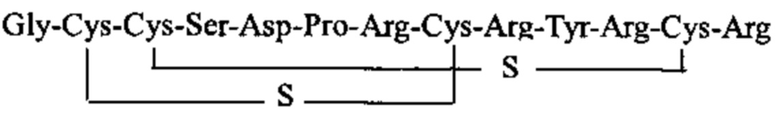 .
.
2. Применение соединения по п. 1 для лечения боли.
| WO 2016073949 A1, 12.05.2016 | |||
| ПЕПТИД, ИМЕЮЩИЙ ВЫСОКОЕ СРОДСТВО К АЛЬФА7 ТИПУ НИКОТИНОВОГО АЦЕТИЛХОЛИНОВОГО РЕЦЕПТОРА ЧЕЛОВЕКА, И ЕГО ПРИМЕНЕНИЕ | 2011 |
|
RU2455359C1 |
| АНАЛОГ АЛЬФА-КОНОТОКСИНА PnIA, ОБЛАДАЮЩИЙ ВЫСОКИМ СРОДСТВОМ И СЕЛЕКТИВНОСТЬЮ К АЦЕТИЛХОЛИН-СВЯЗЫВАЮЩЕМУ БЕЛКУ ИЗ APLYSIA CALIFORNICA | 2011 |
|
RU2458068C1 |
| US 20050256301 A1, 17.11.2005 | |||
| WO 2007118270 A1, 25.10.2007 | |||
| US 20120220539 A1, 30.08.2012. | |||

