2-ола,Область изобретения
Изобретение относится к новым 2,6,9-замещенным пуриновым производным и их биологическому применению. В частности, настоящее изобретение относится к пуриновым производным, обладающим антипролиферативными свойствами, которые пригодны для лечения пролиферативных заболеваний, таких как рак, лейкемия, псориаз и тому подобное.
Предпосылки создания изобретения
Инициация, развитие и завершение клеточного цикла млекопитающих регулируются различными циклинзависимыми киназными (CDK) комплексами, которые являются определяющими для клеточного роста. Такие комплексы включают по меньшей мере каталитическую (сама CDK) и регуляторную (циклин) субъединицу. Некоторые из более важных комплексов для регуляции клеточного цикла включают циклин А (CDK1 - известный также как cdc2 и CDK2), циклин В1-В3 (CDK1), циклин D1-D3 (CDK2, CDK4, CDK5, CDK6), циклин Е (CDK2). Каждый из таких комплексов участвует в конкретной фазе клеточного цикла. Однако не все представители семейства CDK участвуют исключительно в контроле клеточного цикла. Например, CDK 7, 8 и 9 вовлечены в регуляцию транскрипции, тогда как CDK5 играет свою роль в функции нейронных и секреторных клеток.
Активность CDK регулируется посттрансляционно путем временных связей с другими белками и путем изменений их внутриклеточной локализации. Развитие опухоли тесно связано с генетическим изменением и нарушением регуляции CDK и их регуляторов, что говорит о том, что ингибиторы CDK могут быть полезными противораковыми терапевтическими средствами. Разумеется, первоначальные результаты говорят о том, что трансформированные и нормальные клетки отличаются по их потребности в отношении, например, циклина А/CDK2, и что возможна разработка новых противоопухолевых средств, не обладающих общей токсичностью в отношении организма хозяина, наблюдаемой у общепринятых цитотоксических и цитостатических лекарственных средств. Хотя ингибирование связанных с клеточным циклом CDK явно важно, например, для применения в онкологии, это не может быть аргументом для ингибирования регулирующих РНК-полимеразу CDK. С другой стороны, ингибирование функции CDK9/циклина Т до недавнего времени связывали с предотвращением репликации ВИЧ, и открытие новой биологии CDK, таким образом, продолжает открывать новые терапевтические показания для ингибиторов CDK (Sausville, E.A. Trends Molec. Med. 2002, 8, S32-S37).
Функция CDK состоит в фосфорилировании и, таким образом, в активации или дезактивации определенных белков, включая, например, белки ретинобластомы, ламины, гистон Н1 и компоненты митотического веретена. Каталитическая стадия, опосредуемая CDK, включает реакцию фосфопереноса от АТФ к макромолекулярному субстрату фермента. Несколько групп соединений (рассмотренные, например, у Fischer, P.M. Curr. Opin. Drug Discovery Dev. 2001, 4, 623-634), как обнаружено, обладают антипролиферативными свойствами в силу CDK-специфического антагонизма АТФ.
В WO 98/05335 (CV Therapeutics Inc) описаны 2,6,9-трехзамещенные пуриновые производные, которые являются селективными ингибиторами киназ клеточного цикла. Такие соединения полезны при лечении аутоиммунных заболеваний, например, ревматоидного артрита, волчанки, диабета I типа, рассеянного склероза; лечении рака, сердечно-сосудистых заболеваний, таких как рестеноз, реакции организма хозяина против трансплантата, подагры, поликистозной болезни почек и других пролиферативных заболеваний, патогенез которых связан с аномальной клеточной пролиферацией.
В WO 99/07705 (The Regents of the university of California) описаны пуриновые аналоги, которые ингибируют, среди прочего, протеинкиназы, G-белки и полимеразы. Более конкретно, настоящее изобретение относится к способам применения таких пуриновых аналогов для лечения заболеваний с пролиферацией клеток и нейродегенеративных заболеваний.
В WO 97/20842 (CNRS) также описаны пуриновые производные, проявляющие антипролиферативные свойства, которые полезны при лечении рака, псориаза и нейродегенеративных нарушений. Дополнительные пуриновые производные описаны в WO 03/002565, WO 04/016613 и WO 04/016612.
Данным изобретением была предпринята попытка получения новых 2,6,9-замещенных пуриновых производных, особенно таких, которые обладают антипролиферативными свойствами.
Сущность изобретения
В первом аспекте настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли
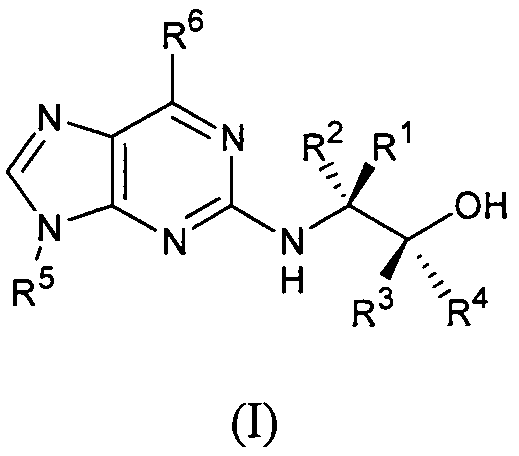
где:
R1 и R2, каждый независимо, представляет собой Н, алкил или галогеналкил;
R3 и R4, каждый независимо, представляет собой Н, алкил, галогеналкил или арил;
R5 представляет собой алкил или циклоалкил, или циклоалкилалкил, каждый из которых может быть необязательно замещен одной или более ОН группами;
R6 выбран из циклопропиламино, циклопропилметиламино, циклобутиламино, циклобутилметиламино и
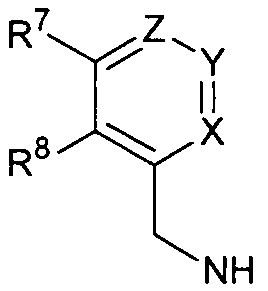
где один из Х, Y и Z представляет собой N, и остальные представляют собой CR9;
R7, R8 и каждый R9 независимо представляют собой Н, алкил или галогеналкил; где по меньшей мере один из R7, R8 и каждый R9 не является Н.
Во втором аспекте настоящее изобретение относится к соединению формулы II или его фармацевтически приемлемой соли,
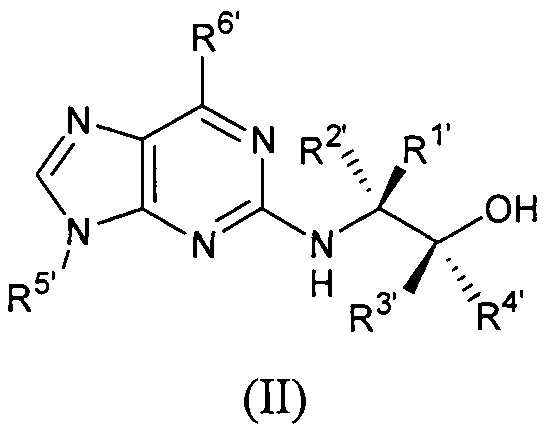
где:
по меньшей мере один из R1', R2', R3' и R4' является галогеналкилом, и остальные, каждый независимо, представляет собой Н, алкил или галогеналкил;
R5' представляет собой алкил или циклоалкил, или циклоалкилалкил, каждый из которых может быть необязательно замещен одной или более OH группами;
R6' выбран из циклопропиламино, циклопропилметиламино, циклобутиламино, циклобутилметиламино и
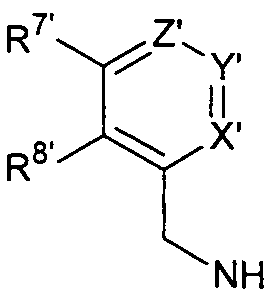
где Х', Y' и Z', каждый независимо, представляет собой CR9', или один из Х', Y' и Z' является N, и остальные представляют собой CR9'; и
R7', R8' и каждый R9', каждый независимо, представляет собой Н, галоген, алкил или галогеналкил.
Третий аспект настоящего изобретения относится к фармацевтической композиции, содержащей соединение настоящего изобретения и фармацевтически приемлемый носитель, разбавитель или эксципиент.
Четвертый аспект настоящего изобретения относится к применению соединения настоящего изобретения для получения лекарственного средства для лечения одного или более следующих заболеваний:
пролиферативного заболевания;
вирусного заболевания;
удара;
облысения;
расстройства ЦНС;
нейродегенеративного расстройства и
различных типов диабета.
Пятый аспект настоящего изобретения относится к применению соединения настоящего изобретения в качестве антимитотического средства.
Шестой аспект настоящего изобретения относится к применению соединения настоящего изобретения для ингибирования протеинкиназы.
Седьмой аспект настоящего изобретения относится к способу лечения пролиферативного заболевания, где указанный способ включает введение млекопитающему терапевтически эффективного количества соединения настоящего изобретения.
Восьмой аспект настоящего изобретения относится к применению соединения настоящего изобретения для анализа по идентификации дополнительных кандидатов на соединения, которые влияют на активность одного или нескольких ферментов CDK.
Подробное описание изобретения
Как указано выше, первый аспект настоящего изобретения относится к соединению формулы (I), как определено выше.
Как использовано в данном описании, термин «алкил» включает алкильные группы как с прямой насыщенной цепью, так и с разветвленной. Предпочтительно, алкильная группа является С1-20алкильной группой, более предпочтительно, С1-15, еще более предпочтительно С1-12алкильной группой, и наиболее предпочтительно С1-6алкильной группой, более предпочтительно, С1-3алкильной группой. Особенно предпочтительные алкильные группы включают, например, метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, пентил и гексил.
Как использовано в данном описании, термин «циклоалкил» относится к циклической алкильной группе. Предпочтительно, циклоалкильная группа является С3-12циклоалкильной группой.
Как использовано в данном описании, термин «циклоалкилалкил» относится к группе, имеющей как циклоалкильную, так и функциональную алкильную группу.
Предпочтительно, один из R1 и R2 представляет собой Н, и другой является алкилом.
Более предпочтительно, один из R1 и R2 представляет собой Н, и другой является метилом, этилом или изопропилом.
В одном предпочтительном варианте осуществления R1 представляет собой этил, и R2 представляет собой Н.
В одном из предпочтительных вариантов осуществления настоящего изобретения R3 и R4, каждый независимо, представляет собой Н, алкил, галогеналкил или арил; и по меньшей мере один из R3 и R4 не является Н.
В одном из предпочтительных вариантов осуществления настоящего изобретения один из R3 и R4 представляет собой Н, и другой является алкилом или галогеналкилом.
В более предпочтительном варианте осуществления R3 представляет собой Н, и R5 представляет собой алкил или галогеналкил.
Более предпочтительно, R3 представляет собой Н, и R4 представляет собой метил.
В одном из предпочтительных вариантов осуществления настоящего изобретения R6 представляет собой
В одном из предпочтительных вариантов осуществления настоящего изобретения Y является N. Предпочтительно для данного варианта осуществления, Х является СН, Z представляет собой C-алкил, R7 является Н, и R8 представляет собой алкил. Более предпочтительно для данного варианта осуществления, Х является СН, Z представляет собой С-Ме, R7 является Н, и R8 представляет собой Ме. В альтернативном предпочтительном варианте осуществления Х является СН, Z представляет собой С-Ме, R7 и R8, оба являются Н. В еще одном альтернативном предпочтительном варианте осуществления Х является СН, Z представляет собой С-CF3, и R7 и R8, оба являются Н.
В одном из предпочтительных вариантов осуществления Х является N. Предпочтительно для данного варианта осуществления, Y является С-Ме, Z является СН, и R7 и R8, оба являются Н. В еще одном альтернативном предпочтительном варианте осуществления Y и Z представляют собой СН, R7 является Н, и R8 является Ме.
В одном из предпочтительных вариантов осуществления настоящего изобретения Z является N. Предпочтительно для данного варианта осуществления, Х является СН, Y представляет собой С-Ме, R7 является Ме, и R8 является Н.
В другом предпочтительном варианте осуществления настоящего изобретения R6 представляет собой циклопропиламино, циклопропилметиламино, циклобутиламино или циклобутилметиламино.
В другом предпочтительном варианте осуществления настоящего изобретения R5 является изопропилом.
В одном из самых предпочтительных вариантов осуществления соединение настоящего изобретения выбирают из следующего:
Другой аспект настоящего изобретения относится к соединению формулы II или его фармацевтически приемлемой соли
где:
по меньшей мере один из R1', R2', R3' и R4' является галогеналкилом, и остальные, каждый независимо, представляет собой Н, алкил или галогеналкил;
R5' представляет собой алкил или циклоалкил, или циклоалкилалкил, каждый из которых может быть необязательно замещен одной или более OH группами;
R6' выбран из циклопропиламино, циклопропилметиламино, циклобутиламино, циклобутилметиламино и
где Х', Y' и Z', каждый независимо, представляет собой CR9', или один из Х', Y' и Z' является N, и остальные представляют собой CR9'; и
R7', R8' и каждый R9' независимо представляют собой Н, галоген, алкил или галогеналкил.
Предпочтительно, в данном аспекте настоящего изобретения R5' представляет собой изопропил.
Предпочтительно, в данном аспекте настоящего изобретения R6' представляет собой
В одном из предпочтительных вариантов осуществления Y' является N, Х' и Z' представляют собой СН, и R7' и R8', оба являются Н.
В другом предпочтительном варианте осуществления настоящего изобретения один из R1' и R2' является Н, и другой является алкилом, или R1' и R2', оба являются Н.
В другом предпочтительном варианте осуществления настоящего изобретения один из R3' и R4' является Н, и другой является CF3.
В одном из самых предпочтительных вариантов осуществления соединение настоящего изобретения выбирают из следующего:
В дополнительном аспекте настоящее изобретение относится к соединению, выбранному из следующего:
В одном из особенно предпочтительных вариантов осуществления соединение настоящего изобретения выбирают из следующего:
В одном особенно предпочтительном варианте осуществления настоящего изобретения соединение выбирают из соединений [1], [3], [4], [6], [7], [8], [9], [10] и [11]. Более предпочтительно, данное соединение выбирают из соединений [1], [3], [4], [6] и [11], еще более предпочтительно, когда данным соединением является соединение [1] или [11].
Предпочтительно, соединение настоящего изобретения проявляет по меньшей мере 3-кратное повышение активности по сравнению с селициклибом, более предпочтительно, по меньшей мере 4-кратное или 5-кратное повышение активности, еще более предпочтительно, по меньшей мере 8-кратное или 10-кратное повышение активности.
В одном особенно предпочтительном варианте осуществления настоящего изобретения данным соединением является (2R,3S-3-(6-((4,6-диметилпиридин-3-илметиламино)-9-изопропил-9Н-пурин-2-иламино)пентан-2-ол [1] или его фармацевтически приемлемые соль или сложный эфир.
Преимущественно, соединение [1] проявляет неожиданно высокую активность в анализах на клеточную токсичность в ряде различных клеточных линий по сравнению со структурно родственными соединениями, известными специалистам в данной области. Дополнительные детали данных анализов представлены в сопровождающих примерах (см., в частности, таблицу 8).
Кроме того, эксперименты показали, что в отличие от структурно родственных соединений, известных специалистам в данной области, соединение [1] не ингибирует значительно CYP3A4. А также, указанные анализы описаны более подробно в сопровождающих примерах (см., в частности, таблицу 5). Разумеется, соединение [1], как видно, не ингибирует CYP3A4 до концентраций более 20 мкМ, что составляет ~60-кратную его клеточную IC50. Так как значение IC50 для ингибирования CYP3A4 для ингибирования соединением [1] значительно выше его клеточной IC50 (см. таблицу 8), это показывает, что при цитотоксических концентрациях в данном случае не должно быть воздействия на активность CYP3A4. Это важно, так как CYP3A4 участвует в метаболизме большого числа лекарственных средств. Если CYP3A4 ингибируется одним лекарственным средством, это может привести к неожиданной токсичности из-за сниженного метаболизма субстратов CYP3A4, приводя, тем самым, к заметно повышенным уровням таких средств.
Подобным образом, дополнительные эксперименты показали, что в отличие от его структурно родственных аналогов соединение [1] не является субстратом для шести испытанных изоформ CYP (см., в частности, таблицу 6). Это различие вполне соответствует наблюдаемому различию в ингибировании CYP, обсужденному выше. Обычный механизм, приводящий к ингибированию CYP, проявляется, если данное соединение является также субстратом для данной CYP.
Соответственно, соединение [1] не является ни субстратом для CYP3A4, ни ингибитором CYP3A4, что придает важное, неожиданное полезное свойство, по сравнению с его структурно родственными аналогами.
Фармацевтические композиции
Второй аспект настоящего изобретения относится к фармацевтической композиции, содержащей соединение настоящего изобретения в смеси с фармацевтически приемлемым разбавителем, эксципиентом или носителем, или их смесью. Хотя соединения настоящего изобретения (включая их фармацевтически приемлемые соли, сложные эфиры и фармацевтически приемлемые сольваты) можно вводить отдельно, они будут обычно применяться в смеси с фармацевтическим носителем, эксципиентом или разбавителем, особенно для лечения людей. Фармацевтические композиции могут быть предназначены для применения на людях или животных в медицине или ветеринарии.
Примеры таких подходящих эксципиентов для различных форм фармацевтических композиций, описанных в данном описании, можно найти в «Handbook of Pharmaceutical Excipients», 2nd Edition, (1994), edited by A. Wade and P.J. Weller.
Приемлемые носители или разбавители для терапевтического применения хорошо известны специалистам в фармацевтической области и описаны, например, в Remington's Pharmaceutical Sciences, Mack Publishing Co. (A.R. Gennaro edit. 1985).
Примеры подходящих носителей включают лактозу, крахмал, глюкозу, метил, целлюлозу, стеарат магния, манит, сорбит и тому подобное. Примеры подходящих разбавителей включают этанол, глицерин и воду.
Выбор фармацевтического носителя, эксципиента или разбавителя может быть сделан в соответствии с предназначенным путем введения и единичной фармацевтической практикой. Фармацевтические композиции могут содержать в качестве носителя, эксципиента или разбавителя или в дополнение к ним любые подходящие связующее(ие) вещество(а), лубрикант(ы), суспендирующее(ие) вещество(а), вещество(а) для покрытия и солюбилизирующее(ие) вещество(а).
Примеры подходящих связующих веществ включают крахмал, желатин, природные сахара, такие как глюкоза, безводная лактоза, свободно текучая лактоза, бета-лактоза, сахаристые вещества из кукурузы, природные и синтетические камеди, такие как камедь акации, трагакант или альгинат натрия, карбоксиметилцеллюлозу и полиэтиленгликоль.
Примеры подходящих лубрикантов включают олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия и тому подобное.
В фармацевтической композиции могут быть представлены консерванты, стабилизаторы, красители и маскирующие улучшающие вкус и запах вещества. Примеры консервантов включают бензоат натрия, сорбиновую кислоту и сложные эфиры п-гидроксибензойной кислоты. Могут также использоваться антиоксиданты и суспендирующие средства.
Соли/сложные эфиры
Соединения настоящего изобретения могут быть представлены в виде солей или сложных эфиров, в частности, фармацевтически приемлемых солей или сложных эфиров.
Фармацевтически приемлемые соли соединений настоящего изобретения включают их подходящие аддитивные соли кислот или оснований. Обзор подходящих фармацевтических солей можно найти у Berge et al., J. Pharm. Sci., 66, 1-19 (1977). Соли образуются, например, с сильными неорганическими кислотами, такими как минеральные кислоты, например, серная кислота, фосфорная кислота или галогенводородная кислота; с сильными органическими карбоновыми кислотами, такими как алканкарбоновые кислоты с 1-4 атомами углерода, которые являются незамещенными или замещены (например, галогеном), такими как уксусная кислота; с насыщенными или ненасыщенными дикарбоновыми кислотами, например, щавелевой, малоновой, янтарной, малеиновой, фумаровой, фталевой или тетрафталевой; с гидроксикарбоновыми кислотами, например, аскорбиновой, гликолевой, молочной, яблочной, винной или лимонной; с аминокислотами, например, аспарагиновой или глутаминовой; с бензойной кислотой или с органическими сульфоновыми кислотами, такими как (С1-С4)алкил- или арилсульфоновыми кислотами, которые являются незамещенными или замещены (например, галогеном), такими как метан- или п-толуолсульфоновая кислота. Сложные эфиры образуются или с использованием органических кислот, или спиртов/гидроксидов, в зависимости от функциональной группы, которую нужно этерифицировать. Органические кислоты включают карбоновые кислоты, такие как алканкарбоновые кислоты из 1-12 атомов углерода, которые являются незамещенными или замещены (например, галогеном), такие как уксусная кислота; с насыщенной или ненасыщенной дикарбоновой кислотой, например, щавелевой, малоновой, янтарной, малеиновой, фумаровой, фталевой или тетрафталевой; с гидроксикарбоновыми кислотами, например, аскорбиновой, гликолевой, молочной, яблочной, винной или лимонной кислотой; с аминокислотами, например, аспарагиновой или глутаминовой кислотой; с бензойной кислотой; или с органическими сульфоновыми кислотами, такими как (С1-С4)алкил- или арилсульфоновые кислоты, которые являются незамещенными или замещены (например, галогеном), такими как метан- или п-толуолсульфоновой кислотой. Подходящие гидроксиды включают неорганические гидроксиды, такие как гидроксид натрия, гидроксид калия, гидроксид кальция, гидроксид алюминия. Спирты включают алкановые спирты из 1-12 атомов углерода, которые могут быть незамещенными или замещены, например, галогеном.
Энантиомеры/таутомеры
Во всех аспектах настоящего изобретения, обсужденных выше, настоящее изобретение включает, где это приемлемо, все энантиомеры и таутомеры соединений настоящего изобретения. Специалист в данной области сможет распознать соединения, которые обладают оптическими свойствами (один или более хиральных атомов углерода) или таутомерными характеристиками. Соответствующие энантиомеры и/или таутомеры могут быть выделены или получены методами, известными специалистам в данной области.
Стерео и геометрические изомеры
Некоторые из соединений настоящего изобретения могут существовать в виде стереоизомеров и/или геометрических изомеров, например, они могут иметь один или более асимметрических и/или геометрических центров, и поэтому могут существовать в двух или более стереоизомерных и/или геометрических формах. Данным изобретением предполагается использование всех отдельных стереоизомеров и геометрических изомеров таких ингибирующих агентов и их смесей. Термины, использованные в формуле изобретения, охватывают все такие формы, при условии, что указанные формы сохраняют соответствующую функциональную активность (хотя необязательно в такой же степени).
Настоящее изобретение включает также все подходящие изотопные вариации данного вещества или его фармацевтически приемлемой соли. Изотопная вариация средства настоящего изобретения или его фармацевтически приемлемой соли определяется как вариация, в которой по меньшей мере один атом заменен атомом, имеющим тот же самый атомный номер, но атомную массу, отличающуюся от атомной массы, обычно обнаруживаемой в природе. Примеры изотопов, которые могут включаться в данное вещество и его фармацевтически приемлемые соли, включают изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора и хлора, такие как 2Н, 3Н, 13С, 14С, 15N, 17O, 18O, 31P, 32P, 35S, 18F и 36Cl, соответственно. Некоторые изотопные вариации данного средства и их фармацевтически приемлемых солей, например, которые включают радиоактивный изотоп, такой как 3Н или 14С, могут применяться в лекарственном средстве и/или для анализа распределения субстрата в тканях. Тритированные, т.е. 3Н, и с углеродом-14, т.е. 14С, изотопы особенно предпочтительны из-за их легкого получения и детектирования. Кроме того, замена изотопами, такими как дейтерий, т.е. 2Н, могут давать некоторые терапевтические преимущества в результате большей метаболической стабильности, например, повышенного периода полувыведения in vivo или сниженных требований к дозе, и следовательно могут быть предпочтительными в некоторых обстоятельствах. Изотопные вариации соединения настоящего изобретения и фармацевтически приемлемых солей настоящего изобретения обычно можно получить по общепринятым методикам с использованием соответствующих изотопных вариантов подходящих реагентов.
Сольваты
Настоящее изобретение включает также сольватированные формы соединений настоящего изобретения. Термины, использованные в формуле изобретения, охватывают все такие формы.
Полиморфы
Настоящее изобретение, кроме того, относится к соединениям настоящего изобретения в их различных кристаллических формах, полиморфных формах и (без)водных формах. В фармацевтической промышленности хорошо установлено, что химические соединения могут быть выделены в любой из таких форм путем небольшого изменения метода очистки и/или выделения из растворителей, используемых в синтетическом препарате таких соединений.
Пролекарства
Настоящее изобретение, кроме того, включает соединения настоящего изобретения в форме пролекарства. Такие пролекарства обычно являются соединениями настоящего изобретения, у которых одна или более соответствующих групп модифицированы таким образом, что модификация может быть обратима при введении человеку или млекопитающему. Такая обратимость обычно производится с помощью фермента, присутствующего у такого субъекта, хотя возможно введение второго средства вместе с таким пролекарством, чтобы произвести это превращение in vivo. Примеры таких модификаций включают сложный эфир (например, любой из тех, которые описаны выше), причем превращение может быть осуществлено эстеразой и т.д. Другие такие системы хорошо известны специалистам в данной области.
Введение
Фармацевтическая композиция настоящего изобретения может быть адаптирована для перорального, ректального, вагинального, парентерального, внутримышечного, внутрибрюшинного, интратекального, интрабронхиального, подкожного, внутрикожного, внутривенного, назального, защечного или подъязычного путей введения.
Для перорального введения, в частности, используют прессованные таблетки, пилюли, таблетки, пилюли с гелем, капли и капсулы. Предпочтительно, данные композиции содержат от 1 до 250 мг, и более предпочтительно, от 10 до 100 мг активного ингредиента на дозу.
Другие формы для введения включают растворы или эмульсии, которые можно вводить путем инъекции внутривенно, внутриартериально, интратекально, подкожно, внутрикожно, внутрибрюшинно или внутримышечно, и которые готовят из стерильных или стерилизованных растворов. Фармацевтические композиции настоящего изобретения также могут быть в форме суппозиториев, пессариев, суспензий, эмульсий, лосьонов, мазей, кремов, гелей, препаратов аэрозолей, растворов и присыпок.
Альтернативные средства трансдермального введения представлены использованием кожного пластыря. Например, активный ингредиент может быть включен в крем, состоящий из водной эмульсии полиэтиленгликолей или вазелинового масла. Активный ингредиент может быть также включен в концентрации от 1 до 10% по массе в мазь, состоящую из основы из отбеленного пчелиного воска или белого вазелина вместе с такими стабилизаторами и консервантами, которые могут быть необходимы.
Инъекционные формы могут содержать от 10 до 1000 мг, предпочтительно от 10 до 250 мг активного ингредиента на дозу.
Композиции могут быть изготовлены в единичной дозированной форме, например, в форме отдельных порций, содержащих единичную дозу, или в виде множественных или разделенных доз единичной дозы.
Дозировка
Специалист в данной области может легко определить соответствующую дозу одной из настоящих композиций для введения пациенту без ненужного экспериментирования. Обычно врач определяет действующую дозу, которая будет наиболее подходящей для конкретного пациента, и она будет зависеть от ряда факторов, включая активность конкретного применяемого соединения, метаболическую стабильность и продолжительность действия данного соединения, возраст, массу тела, общее состояние здоровья, пол, диету, способ и время введения, скорость выведения, комбинацию лекарственных средств, тяжесть конкретного состояния и индивидуальную применяемую терапию. Дозировки, описанные в данном описании, являются примерами для обычного случая. Разумеется, могут быть индивидуальные примеры, когда показаны более высокие или более низкие интервалы доз, и такие случаи охватываются объемом настоящего изобретения.
В зависимости от потребности средство можно вводить в дозе от 0,01 до 30 мг/кг массы тела, такой как от 0,1 до 10 мг/кг, более предпочтительно, от 0,1 до 1 мг/кг массы тела.
В конкретном примере варианта осуществления одна или более доз от 10 мг до 150 мг/сутки будут вводиться пациенту для лечения злокачественного новообразования.
Терапевтическое применение
Соединения настоящего изобретения, как обнаружено, обладают антипролиферативной активностью, и поэтому, как полагают, будут полезны при лечении пролиферативных заболеваний, таких как различные типы рака, лейкемии и другие заболевания, связанные с нерегулируемой клеточной пролиферацией, таких как псориаз и рестеноз.
Как определено в данном описании, антипролиферативный эффект в соответствии с объемом настоящего изобретения может быть продемонстрирован способностью ингибировать пролиферацию клеток анализом in vitro на целых клетках, например, с использованием любой из клеточных линий А549, HeLa, HT-29, MCF7, Saos-2, CCRF-CEM, H460, HL-60 и K-562, или путем демонстрации ингибирования киназы при соответствующем анализе. Такие анализы, включая способы их выполнения, описаны более подробно в сопровождающих примерах. Применением таких анализов можно определить, является ли соединение антипролиферативным в контексте настоящего изобретения.
Один предпочтительный вариант осуществления настоящего изобретения поэтому относится к применению одного или более соединений настоящего изобретения для получения лекарственного средства для лечения пролиферативного заболевания.
В соответствии с описанием фраза «получение лекарственного средства» включает применение соединения настоящего изобретения непосредственно в качестве лекарственного средства в дополнение к его применению в программе отбора на терапевтические средства в дальнейшем или на любой стадии производства такого лекарственного средства.
Термин «пролиферативное заболевание» использован в данном описании в широком смысле, так что включает любое заболевание, которое требует контроля над клеточным циклом, например, сердечно-сосудистые заболевания, такие как рестеноз и кардиомиопатия, аутоиммунные заболевания, такие как гломерулонефрит и ревматоидный артрит, дерматологические заболевания, такие как псориаз, грибковые, паразитарные заболевания, такие как малярия, эмфизема и облысение. При данных заболеваниях соединения настоящего изобретения могут вызывать апоптоз или поддерживать стаз в желаемых клетках, когда необходимо. Предпочтительно, пролиферативное заболевание представляет собой рак или лейкемию.
В другом предпочтительном варианте осуществления пролиферативным заболеванием является псориаз.
Соединения настоящего изобретения могут ингибировать любой из этапов или любых стадий в клеточном цикле, например, образование ядерной оболочки, выход из фазы покоя клеточного цикла (G0), G1 развитие, разрыхление хромосомы, разрыв ядерной оболочки, СТАРТ, инициация репликации ДНК, развитие репликации ДНК, терминация репликации ДНК, дупликация центросомы, развитие G2, активация митотических или мейтических функций, конденсация хромосомы, разделение центросомы, нуклеация микротрубочек, образование и функционирование веретена, взаимодействие с моторными белками микротрубочек, хроматидное разделение и расщепление, инактивация митотических функций, образование сократительного кольца и функций цитокинеза. В частности, соединения настоящего изобретения могут влиять на некоторые функции генов, такие как связывание хроматина, образование комплексов репликации, лицензирование репликации, фосфорилирование или другая активность вторичной модификации, протеолитическое разложение, связывание с микротрубочками, связывание актина, связывание септина, активность нуклеации организующего микротрубочки центра и связывание компонентов сигнальных путей клеточного цикла.
Дополнительный аспект настоящего изобретения относится к способу лечения пролиферативного заболевания, где указанный способ включает введение млекопитающему терапевтически эффективного количества соединения настоящего изобретения.
В предпочтительном варианте осуществления данного аспекта пролиферативным заболеванием является рак или лейкемия.
В еще более предпочтительном варианте осуществления данного аспекта данное соединение вводят в количестве, достаточном для ингибирования по меньшей мере одного фермента CDK.
Предпочтительно, соединение настоящего изобретения вводят в количестве, достаточном для ингибирования по меньшей мере одной из CDK1, CDK2, CDK3, CDK4, CDK6, CDK7, CDK8 и/или CDK9.
Более предпочтительно, соединение настоящего изобретения применяют в количестве, достаточном для ингибирования по меньшей мере одной из CDK2 и/или CDK4.
Еще более предпочтительно, ферментом CDK является CDK2.
В одном из предпочтительных вариантов осуществления данного аспекта соединение вводят перорально.
Другой аспект настоящего изобретения относится к применению соединения настоящего изобретения в качестве антимитотического средства.
Еще один аспект настоящего изобретения относится к применению соединения настоящего изобретения для лечения нейродегенеративного нарушения.
Предпочтительно нейродегенеративным нарушением является апоптоз нейронов.
Другой аспект изобретения относится к применению соединения настоящего изобретения в качестве противовирусного средства.
Таким образом, еще один аспект настоящего изобретения относится к применению соединения настоящего изобретения для получения лекарственного средства для лечения вирусного заболевания, такого как заболевание, вызванное человеческим цитомегаловирусом (HCMV), вирусом простого герпеса типа 1 (HSV- 1), вирусом иммунодефицита человека типа 1 (HIV-1) и вирусом ветряной оспы и опоясывающего лишая (VZV).
В более предпочтительном варианте осуществления изобретения соединение настоящего изобретения применяют в количестве, достаточном для ингибирования одной или более CDK клетки-хозяина, участвующих в вирусной репликации, т.е. CDK2, CDK7, CDK8 и CDK9. [Wang D., De la Fuente C., Deng L., Zilberman I., Eadie C., Healey M., Stein D., Denny T., Harrison L.E., Meijer L., Kashanchi F. Inhibition of human immunodeficiency virus type 1 transcription by chemical cyclin-dependent kinase inhibitors. J. Virol. 2001; 75: 7266-7279].
Как определено выше, противовирусное действие в объеме настоящего изобретения может быть продемонстрировано способностью ингибировать CDK2, CDK7, CDK8 или CDK9.
В особенно предпочтительном варианте осуществления настоящее изобретение относится к применению одного или более соединений настоящего изобретения для лечения вирусного заболевания, которое является зависимым или чувствительным к CDK. Зависимые от CDK заболевания связаны с уровнем активности выше нормального у одного или более CDK ферментов. Такие заболевания предпочтительно связаны с аномальным уровнем активности CDK2, CDK7, CDK8 и/или CDK9. Чувствительное к CDK заболевание является заболеванием, при котором отклонение от нормы уровня CDK не является основной причиной, но является вытекающим следствием главного метаболического отклонения от нормы. При такой программе действий можно сказать, что CDK2, CDK7, CDK8 и/или CDK9 являются частью чувствительного метаболического пути, и ингибиторы CDK поэтому могут быть активны при лечении таких заболеваний.
Соединения настоящего изобретения пригодны также для получения лекарственных средств для лечения различных глазных заболеваний. Предпочтительно, глазным заболеванием является глаукома, экссудативная, возрастная дегенерация желтого пятна (AMD) или пролиферативная диабетическая ретинопатия (PDR).
Болезненное состояние, называемое глаукомой, характеризуется стойкой потерей зрительной функции из-за необратимого повреждения оптического нерва. Несколько морфологически или функционально отличающихся типов глаукомы обычно характеризуются повышенным внутриглазным давлением (IOP), которое, как считают, причинно связано с патологическим течением заболевания. Глазная гипертензия является состоянием, при котором внутриглазное давление повышено, но явной потери зрительной функции не произошло; таких пациентов рассматривают как имеющих высокий риск возможного развития потери зрения, связанной с глаукомой. Ингибиторы GSK-3 полезны для лечения глазных заболеваний, таких как глаукома. Было показано, что компонент Wnt сигнального пути, связанный с ожогом белок (FRP), дифференциально экспрессируется в ряде линий клеток глаукоматозной трабекулярной сетчатой структуры и может разрывать сигнальный каскад, вызывая повышение сопротивления оттоку и развитие повышенного IOP. Hellberg M.R. et al. (US20040186159) показали, что через взаимодействие GSK-3 с компонентами Wnt сигнального пути ингибирование GSK-3 фармакологическими средствами можно преодолеть опосредованный FRP антагонизм Wnt сигнальному пути, вызванный повышенными уровнями FRP, и противодействовать повышению сопротивления оттока, которое является результатом повышения продукции FRP у индивидуумов с глаукомой.
CTGF является секретируемым цитокином, который, как известно, повышает продукцию внеклеточного матрикса (ECM), главным образом путем депонирования коллагена I и фибринонектина. Сверхэкспрессию CTGF ранее связывали в качестве главного причинного фактора с патологическими состояниями, такими как склеродерма, фибропролиферативные заболевания, рубцевание и т.д., при которых присутствует сверхнакопление компонентов ECM. Сверхнакопление веществ внеклеточного матрикса в области трабекулярной сетчатой структуры (TM) также является признаком многих форм глаукомы; такое повышение, как полагают, приводит к повышенному сопротивлению оттоку внутриглазной жидкости и поэтому к повышенному внутриглазному давлению. Fleenor D.L. et al. (US20050234075) показали, что ингибиторы GSK-3 и ингибиторы CDK могут ингибировать как базовую, так и вызванную TGF-бета-2 экспрессию CTGF в человеческих ТМ клетках, поэтому соединения настоящего изобретения могут быть применены при лечении глаукомы.
Соединения настоящего изобретения пригодны также для лечения AMD и PDR. Экссудативная возрастная дистрофия желтого пятна (AMD) и пролиферативная диабетическая ретинопатия (PDR) являются главными причинами возникающей слепоты в развитых странах и характеризуются патологической поздней сегментной неоваскуляризацией в глазу. Провоцирующая причина как AMD, так и PDR еще неизвестна, однако, выработка различных проангиогенных факторов роста, по-видимому, является общим стимулом. Растворимые факторы роста, такие как сосудистый эндотелиальный фактор роста (VEGF), фактор роста, полученный из тромбоцитов (PDGF), основной фактор роста фибробластов (bFGF или FGF-2), инсулиноподобный фактор роста 1 (IGF-1), ангиопоэтины и т.д., были обнаружены в тканях и жидкостях, взятых от пациентов с патологическим ангиогенезом глаз. Ингибирование или блокада активности данных факторов роста и их других внутриклеточных ферментов, таких как aurora-киназы, как показано, обладают антиангиогенным действием. Таким образом, соединения настоящего изобретения могут быть применены при лечении глазных заболеваний, характеризующихся неоваскуляризацией.
Другой аспект настоящего изобретения относится к применению соединений настоящего изобретения или их фармацевтически приемлемых солей для получения лекарственного средства для лечения диабета.
В особенно предпочтительном варианте осуществления диабетом является диабет II типа.
GSK-3 является одной из нескольких протеинкиназ, которая фосфорилирует гликогенсинтазу (GS). Стимуляция синтеза гликогена инсулином в скелетных мышцах происходит в результате дефосфорилирования и активации GS. Действие GSK-3 на GS, таким образом, приводит к дезактивации последней и, таким образом, ингибированию превращения глюкозы в гликоген в мышцах.
Диабет типа II (инсулин независимый сахарный диабет) является многофакторным заболеванием. Гипергликемия связана с инсулиновой резистентностью в печени, мышцах и других тканях в сочетании с нарушенной секрецией инсулина. Скелетные мышцы являются главным сайтом стимулируемого инсулином усвоения глюкозы, здесь она или удаляется из кровотока, или превращается в гликоген. Отложение гликогена в мышцах является главной детерминантой гомеостаза глюкозы, и при диабете II типа происходит дефектное депонирование гликогена в мышцах. Существуют данные, что повышение активности GSK3 важно при диабете типа II [Chen, Y.H.; Hansen, L.; Chen, M.x.; Bjorbaek, C.; Vestergaard, H.; Hansen, T.; Cohen, P.T.; Pedersen, O. Diabetes, 1994, 43, 1234]. Кроме того, было показано, что при диабете II типа происходит сверхэкспрессия GSK3 в клетках мышц, и что существует обратная корреляция между активностью GSK3 в скелетных мышцах и действием инсулина [Nikoulina, S.E.; Ciaraldi, T.P.; Mudaliar, S.; Mohideen, P.; Carter, L.; Henry, R.R. Diabetes, 2000, 49, 263].
Ингибирование GSK3 поэтому имеет терапевтическое значение при лечении диабета, в частности, II типа, и диабетической нейропатии.
Примечательно, что GSK3, как известно, фосфорилирует многие субстраты, помимо GS, и, таким образом, участвует в регуляции многих биохимических путей. Например, высокая экспрессия GSK происходит в центральной и периферической нервных системах.
В еще одном аспекте настоящее изобретение относится к применению соединений настоящего изобретения или его фармацевтически приемлемых солей для получения лекарственного средства для лечения заболеваний ЦНС, например, нейродегенеративных заболеваний.
Предпочтительно, заболеванием ЦНС является болезнь Альцгеймера.
Тау-белок является субстратом GSK-3, который связан с этиологией болезни Альцгеймера. В здоровых нервных клетках тау сокомпонуется с тубулином в микротрубочки. Однако при болезни Альцгеймера тау образует большие сплетения нитей, что разрушает структуру микротрубочек в нервной клетке, нарушая, тем самым, транспорт питательных веществ, а также передачу сигналов нервных клеток.
Без желания быть связанными теорией, полагают, что ингибиторы GSK3 могут быть способны предотвращать и/или исправлять аномальное фосфорилирование связанного с микротрубочками белка тау, что является инвариантным признаком болезни Альцгеймера и ряда других нейродегенеративных заболеваний, таких как прогрессивный супрануклеарный паралич, кортикобазальная дегенерация и болезнь Пика. Мутации в гене тау вызывают врожденные формы фронтотемпорального слабоумия, дополнительно акцентируя на значении дисфункции тау-белка для нейродегенеративного процесса [Goedert, M. Curr. Opin. Gen. Dev., 2001, 11, 343].
Еще один аспект настоящего изобретения относится к применению соединений настоящего изобретения или их фармацевтически приемлемых солей для получения лекарственного средства для лечения биполярного расстройства.
Еще один аспект настоящего изобретения относится к применению соединений настоящего изобретения или их фармацевтически приемлемых солей для получения лекарственного средства для лечения удара.
Снижение апоптоза нейронов является важной терапевтической целью в контексте травмы головы, удара, эпилепсии и болезни двигательных нейронов [Mattson, M.P. Nat. Rev. Mol. Cell. Biol., 2000, 1, 120]. Поэтому GSK3 в качестве проапоптозного фактора в нейронных клетках делает эту протеинкиназу привлекательной терапевтической мишенью для создания ингибирующих лекарственных средств для лечения таких заболеваний.
Еще один аспект настоящего изобретения относится к применению соединений настоящего изобретения или его фармацевтически приемлемых солей для получения лекарственного средства для лечения облысения.
Рост волос регулируется Wnt сигнальным путем, в частности Wnt3. На модельных системах кожи в виде тканевой культуры экспрессия не разрушаемых мутантов β-катенина приводит к заметному повышению популяции предполагаемых стволовых клеток, которые обладают большим пролиферативным потенциалом [Zhu, A.J.; Watt, F.M. Development, 1999, 126, 2285]. Данная популяция стволовых клеток экспрессирует не связанный с кадхерином β-катенин на более высоком уровне [DasGupta, R.; Fuchs, E. Development, 1999, 126, 4557], что может способствовать их высокому пролиферативному потенциалу. Кроме того, у трансгенных мышей со сверхэкспрессией усеченного β-катенина в коже по-новому происходит морфогенез волосяных фолликул, что в норме устанавливается только во время эмбриогенеза. Эктопическое применение ингибиторов GSK3 поэтому может быть терапевтически полезно при лечении облысения и для восстановления роста волос после вызванного химиотерапией облысения.
Дополнительный аспект настоящего изобретения относится к способу лечения зависимого от GSK3 заболевания, где указанный способ включает введение пациенту, нуждающемуся в этом, соединения настоящего изобретения или его фармацевтически приемлемой соли, как определено выше, в количестве, достаточном для ингибирования GSK3.
Предпочтительно, соединение настоящего изобретения или его фармацевтически приемлемую соль вводят в количестве, достаточном для ингибирования GSK3.
В одном из вариантов осуществления соединение настоящего изобретения вводят в количестве, достаточном для ингибирования по меньшей мере одного фермента PLK.
Полоподобные киназы (PLK) образуют семейство серин/треониновых протеинкиназ. Митотические мутанты Drosophila melanogaster в поло локусе проявляют аномалии веретена [Sunkel et al., J. Cell Sci., 1988, 89, 25] и поло, как обнаружено, кодирует митотическую киназу [Llamazares et al., Genes Dev., 1991, 5, 2153]. У людей существуют три близкородственные PLK [Glover et al., Genes Dev., 1998, 12, 3777]. Они содержат высоко гомологичный аминоконцевой каталитический киназный домен, и их карбоксильные концы содержат две или три консервативные области, боксы поло. Функция боксов поло остается не полностью понятной, но они задействованы в направлении PLK в субклеточные компартменты [Lee et al., Proc. Natl. Acad. Sci. USA, 1998, 95, 9301; Leung et al., Nat. Struct. Biol., 2002, 9, 719], опосредовании взаимодействий с другими белками [Kauselmann et al., EMBO J., 1999, 18, 5528] или могут образовывать часть ауторегуляторного домена [Nigg, Curr. Opin. Cell Biol., 1998, 10, 776]. Кроме того, зависимая от бокса поло активность PLK1 необходима для правильного перехода метафаза/анафаза и цитокинеза [Yuan et al., Cancer Res., 2002, 62, 4186; Seong et al., J. Biol. Chem., 2002, 277, 32282].
Исследования показали, что человеческие PLK регулируют некоторые фундаментальные аспекты митоза [Lane et al., J. Cell. Biol., 1996, 135, 1701; Cogswell et al., Cell Growth Differ., 2000, 11, 615]. В частности, активность PLK1, как полагают, необходима для функционального созревания центросом в поздней G2/ранней профазе и последующего образования биполярного веретена. Уменьшение клеточной PLK1 методом малых интерферирующих РНК (миРНК) также подтвердило, что этот белок необходим для многих митотических процессов и завершения цитокинеза [Liu et al., Proc. Natl. Acad. Sci. USA, 2002, 99, 8672].
В более предпочтительном варианте осуществления настоящего изобретения соединение настоящего изобретения вводят в количестве, достаточном для ингибирования PLK1.
Из трех человеческих PLK PLK1 лучше всего охарактеризована; она регулирует ряд действий цикла деления клетки, включая начало митоза [Toyoshima-Moritomo et al., Nature, 2001, 410, 215; Roshak et al., Cell Signaling, 2000, 12, 405], активацию контрольной точки повреждения ДНК [Smits et al., Nat. Cell Biol., 2000, 2, 672; van Vugt et al., J. Biol Chem., 2001, 276, 41656], регуляцию активирующего анафазу комплекса [Sumara et al., Mol. Cell, 2002, 9, 515; Golan et al., J. Biol. Chem., 277, 15552; Kotani et al., Mol. Cell, 1998, 1, 371], фосфорилирование протеасомы [Feng et al., Cell Growth Differ., 2001, 12 29] и удвоение и созревание центросомы [Dai et al., Oncogene, 2002, 21, 6195].
В частности, инициация митоза требует активации вызывающего М-фазу фактора (MPF), комплекса между циклинзависимой киназой CDK1 и циклинами В-типа [Nurse, Nature, 1990, 344, 503]. Последний накапливается во время S и G2 фаз клеточного цикла и вызывает подавляющее фосфорилирование комплекса MPF киназами WEE1, MIK1 и MYT1. В конце фазы G2 соответствующее дефосфорилирование с помощью фосфатазы с двойной специфичностью CDC25C запускает активацию MPF [Nigg, Nat. Rev. Mol. Cell Biol., 2001, 2, 21]. В интерфазе циклин В локализуется в цитоплазме [Hagting et al., EMBO J., 1998, 17, 4127], затем он становится фосфорилированным во время профазы, и это явление вызывает ядерную транслокацию [Hagting et al., Curr. Biol., 1999, 9, 680; Yang et al., J. Biol. Chem., 2001, 276, 3604]. Ядерное накопление активной MPF во время профазы, как полагают, важно для инициации событий М-профазы [Takizawa et al., Curr. Opin. Cell Biol., 2000, 12, 658]. Однако ядерная MPF остается неактивной с помощью WEE1, если ей не препятствует CDC25C. Сама фосфатаза CDC25C, локализованная в цитоплазме во время интерфазы, накапливается в ядре в профазе [Seki et al., Mol. Biol. Cell, 1992, 3, 1373; Heald et al., Cell, 1993, 74, 463; Dalal et al., Mol. Cell. Biol., 1999, 19, 4465]. Поступление в ядро как циклина В [Toyoshima-Moritomo et al., Nature, 2001, 410, 215], так и CDC25C [Toyoshima-Moritomo et al., EMBO Rep., 2002, 3, 341] стимулируется фосфорилированием PLK1 [Roshak et al., Cell Signalling, 2000, 12, 405]. Данная киназа является важным регулятором инициации M-фазы.
В одном особенно предпочтительном варианте осуществления соединения настоящего изобретения являются АТФ-антагонистическими ингибиторами PLK1.
В данном контексте АТФ-антагонизм относится к способности подавляющего соединения снижать или предотвращать каталитическую активность PLK, т.е. фосфоперенос от АТФ к макромолекулярному субстрату PLK, в силу обратимого или необратимого связывания в активном сайте фермента таким образом, что ослабляется или аннулируется связывание АТФ.
В другом предпочтительном варианте осуществления соединение настоящего изобретения применяют в количестве, достаточном для ингибирования PLK2 и/или PLK3.
PLK2 (известная также как SNK) и PLK3 (известная также как PRK и FNK) млекопитающих, как первоначально было показано, являются прямыми ранними продуктами генов. Киназная активность PLK3, по-видимому, достигает максимума во время фазы S и G2. Она активируется также во время активации в контрольной точке повреждения ДНК и при тяжелом окислительном стрессе. PLK3 играет также важную роль в регуляции динамики микротрубочек и функции центросомы в клетке, и разрегулированная экспрессия PLK3 приводит к остановке клеточного цикла и апоптозу [Wang et al., Mol. Cell. Biol., 2002, 22, 3450]. PLK2 является по меньшей мере вполне понятным гомологом трех PLK. Как PLK2, так и PLK3 может иметь дополнительные важные постмитотические функции [Kauselmann et al., EMBO J., 1999, 18, 5528].
Другой аспект настоящего изобретения относится к применению соединения настоящего изобретения для ингибирования протеинкиназы.
В предпочтительном варианте осуществления в данном аспекте протеинкиназа является циклинзависимой киназой. Предпочтительно, протеинкиназа является CDK1, CDK2, CDK3, CDK4, CDK6, CDK7, CDK8 или CDK9, более предпочтительно, CDK2.
Дополнительный аспект настоящего изобретения относится к способу ингибирования протеинкиназы, где указанный способ включает контактирование указанной протеинкиназы с соединением настоящего изобретения.
В предпочтительном варианте осуществления в данном аспекте протеинкиназа является циклинзависимой киназой, еще более предпочтительно, CDK2.
Анализы
Другой аспект настоящего изобретения относится к применению соединения, как определено выше, в анализе по идентификации соединений-кандидатов, которые влияют на активность одной или более циклинзависимых киназ, aurora-киназ, GSK и/или ферментов PLK.
Предпочтительно анализ дает возможность идентификации соединений-кандидатов, которые способны ингибировать одну или более циклинзависимых киназ, aurora-киназ и GSK и/или ферментов PLK.
Более предпочтительно, анализом является анализ на конкурентное связывание.
Более предпочтительно, соединение-кандидат получают общепринятой SAR модификацией соединения настоящего изобретения.
Как использовано в данном описании, термин «общепринятая SAR модификация» относится к стандартным методам, известным специалистам в данной области, для изменения данного соединения путем химического получения его производных.
Таким образом, в одном из аспектов идентифицированное соединение может действовать в качестве модели (например, матрицы) для получения других соединений. Соединения, используемые в таком испытании, могут быть свободными в растворе, фиксированными на твердой подложке, размещенными на поверхности клетки или расположены внутриклеточно. Устранение активности или образование связывающих комплексов между соединением и средством, которое нужно испытать, может быть оценено количественно.
Анализ по настоящему изобретению может быть отбором, при котором испытывают ряд средств. В одном из аспектов способ анализа по настоящему изобретению является высоко производительным отбором.
По настоящему изобретению предполагается также применение анализов скринирования лекарственных средств по конкуренции, при которых нейтрализующие антитела, способные к связыванию соединения, специфически конкурируют с испытуемым соединением за связывание с соединением.
Другая методика отбора обеспечивает отбор с высокой производительностью (HTS) средств, обладающих соответствующей аффинностью связывания с данными веществами, и основана на методе, подробно описанном в WO 84/03564.
Подразумевается, что методы анализа настоящего изобретения будут полезны как при скрининге небольшого объема, так и при крупномасштабном скрининге испытуемых соединений, а также при количественных анализах.
Предпочтительно, анализ по конкурентному связыванию включает контактирование соединения настоящего изобретения с циклинзависимой киназой, aurora-киназой, GSK или PLK ферментом в присутствии известного субстрата указанного CDK фермента и детектирование какого-либо изменения во взаимодействии между указанной киназой и указанным известным субстратом.
Дополнительный аспект настоящего изобретения представляет собой способ детектирования связывания лиганда с циклинзависимой киназой, aurora-киназой, GSK или PLK ферментом, где указанный способ включает стадии:
(i) контактирования лиганда с циклинзависимой киназой, aurora-киназой, GSK или PLK ферментом в присутствии известного субстрата указанной киназы;
(ii) детектирования любого изменения во взаимодействии между указанной киназой и указанным известным субстратом;
и где указанный лиганд является соединением настоящего изобретения.
Один из аспектов настоящего изобретения относится к способу, включающему стадии:
(а) осуществления способа анализа, описанного выше;
(b) определения одного или более лигандов, способных к связыванию со связывающим лиганд доменом; и
(с) получения некоторого количества указанного одного или более лигандов.
Другой аспект настоящего изобретения представляет собой способ, включающий стадии:
(а) осуществления способа анализа, описанного выше;
(b) определения одного или более лигандов, способных к связыванию со связывающим лиганд доменом; и
(с) получения фармацевтической композиции, содержащей указанный один или более лигандов.
Еще один аспект настоящего изобретения представляет собой способ, включающий стадии:
(а) осуществления способа анализа, описанного выше;
(b) определения одного или более лигандов, способных к связыванию со связывающим лиганд доменом; и
(с) модификации указанного одного или более лигандов, способных к связыванию со связывающим лиганд доменом;
(d) осуществления способа анализа, описанного выше;
(e) необязательно, получения фармацевтической композиции, содержащей указанный один или более лигандов.
Настоящее изобретение относится также к лиганду, идентифицированному способом, описанным выше.
Еще один аспект настоящего изобретения относится к фармацевтической композиции, содержащей лиганд, идентифицированный способом, описанным выше.
Еще один аспект настоящего изобретения относится к применению лиганда, идентифицированного способом, описанным выше, для получения фармацевтической композиции для применения при лечении пролиферативных заболеваний.
Представленные выше способы можно использовать для отбора лиганда, используемого в качестве ингибитора одного или более CDK ферментов.
Настоящее изобретение далее описывается с помощью следующих примеров и со ссылками на следующую фигуру, где:
на фиг.1 представлена понижающая регуляция Mcl-1 соединениями настоящего изобретения. Клетки Н460 обрабатывали в течение 24 часов различными концентрациями соединений и анализировали путем Вестерн-блоттинга на изменения уровня Mcl-1;
на фиг.2 показано действие некоторых из последующих соединений на уровни Mcl-1 в клетках Н460. Клетки обрабатывали рядом концентраций каждого лекарственного средства и анализировали через 5 часов.
Примеры
Общие сведения
Химические вещества и растворители приобретали из коммерческих источников и использовали по получении, если не указано иное. ТГФ и Et2O сушили при температуре кипения с обратным холодильником с бензофеноном натрия в атмосфере N2 и отделяли путем перегонки. Толуол сушили при температуре кипения с обратным холодильником над натрием в атмосфере N2. CH2Cl2 сушили при температуре кипения с обратным холодильником над СаН2 в атмосфере азота. Используемым микроволновым генератором была модель CEM “Discover” с конструкцией в виде одной круглой полости, которая фокусирует микроволновое излучение на пробирке для образца. ТСХ (тонкослойную хроматографию) выполняли, используя стеклянные пластины, покрытые силикагелем G60 (0,25 см). Проявленные пластины сушили на воздухе и анализировали под УФ лампой (254/365 нм). Безводный MgSO4 использовали в качестве стандартного средства для сушки органических растворов, если не указано иное. Колоночную флэш-хроматографию выполняли, используя силикагель Fluorochem (35-70 мкм). Температуру плавления (т.п.) определяли с помощью капиллярного аппарата для определения температуры плавления Electrothermal 9100 и не корректировали. Сокращение (разл.) означает температуру разложения. Спектры 1Н-ЯМР регистрировали на спектрофотометре Bruker Avance 300 (300,1 МГц) или Varian Gemini 2000 (300 МГц) с использованием дейтерированного растворителя в качестве фиксатора и остаточного растворителя в качестве внутреннего стандарта во всех случаях. Спектры 13С-ЯМР с использованием последовательности PENDANT регистрировали на спектрофотометре Bruker Avance 300 (75,5 МГц). Все другие спектры 13С регистрировали на спектрофотометре Varian Gemini 2000 (75,5 МГц), используя сложный импульс разъединения 1Н. Константы связи (J) оценивали с точностью 0,1 Гц. Использовали следующие сокращения: с - синглет; д - дуплет; т - триплет; кв. - квартет; квин. - квинтет; м - мультиплет и ушир. - широкий. Элементные микроанализы выполняли Mrs S. Williamson, Scholl of Chemistry, Purdie Building, University of St, Andrews, UK. Полученные результаты находились в пределах 0,4% от рассчитанных значений. Масс-спектры с электрораспылением (ESI) регистрировали на масс-спектрофотометре Micromass LCT, в комбинации с ВЭЖХ Waters 2975. Аналитическую ОФ-ВЭЖХ выполняли, используя автоматический инжектор образца Dionex ASI-100, соединенный с насосом Dionex P580. Для аналитических целей использовали колонку Phenomenex (150×4,60 мм, Synergi 4 мкм гидро-RP 80 Å), выдержанную при температуре 25°C. Установку ВЭЖХ регулировали, используя программное обеспечение Chromeleon. Линейное градиентное элюирование проводили с использованием систем Н2О/MeCN (содержащих 0,1% CF3COOH) при скорости потока 1 мл/мин. Чистоту оценивали путем интегрирования хроматограмм (λ=254 нм).
Синтез
(2R,3S)-3-Аминопентан-2-ол получали одним или другим из двух путей, отличающихся защитной группой, используемой для амина.
Путь 1 с использованием тритила в качестве защитной группы
(S)-2-(тритиламино)бутан-1-ола
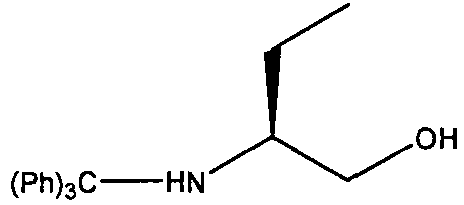
К перемешиваемому раствору (S)-(+)-2-(тритиламино)бутан-1-ола (10 г, 112,18 ммоль) в дихлорметане (DCM, 250 мл) в атмосфере аргона при комнатной температуре добавляли диизопропилэтиламин (DIEA, 19,4 мл, 112,18 ммоль), с последующим добавлением тритилхлорида (31,2 г, 112,18 ммоль). Реакционную смесь перемешивали при этой температуре в течение 48 часов, пока ТСХ (гексан:эфир:МеОН; 55:40:5) не показала, что реакция подошла к завершению. Растворитель выпаривали в вакууме и остаток забирали в этилацетат. Органический раствор промывали водой (2×), сушили над сульфатом натрия. Растворитель удаляли, с получением (S)-2-(тритиламино)бутан-1-ола в виде светло-желтого масла; выход: 33 г (89%).
1H-ЯМР (CDCl3, 250 МГц): δ 0,72 (3H, т, J=7,5 Гц, -NHCH(CH2 CH 3)CH2OH), 1,15-1,10 (м, 2H, -NHCH(CH 2CH3)CH2OH), 2,05 (1H, с, ушир., NH), 2,24 (1H, с, ушир., OH), 2,62-2,54 (м, 1H, -NHCH(CH2CH3)CH2OH), 3,17-3,08 (1H, м, -NHCH(CH2CH3)CHHOH), 3,35-3,29 (1H, м, NHCH(CH2CH3)CHHOH), 7,37-7,2 (12H, м, ArH), 7,65-7,58 (3H, м, ArH); δC (250 МГц, CDCl3) 146,86 (C), 129,43 (6×CH), 127,90 (6×CH), 126,48 (3×CH), 71,27 (C), 62,72 (CH2), 48,91 (CH), 24,55 (CH2), 10,47 (CH3).
(S)-2-(Тритиламино)бутиральдегид
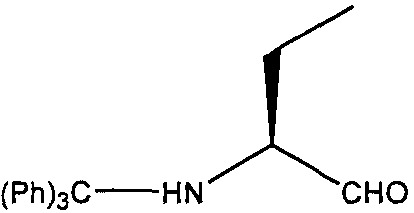
К перемешиваемому раствору сухого диметилсульфоксида (2,4 мл, 2,8 экв., 33,82 ммоль) в сухом дихлорметане (30 мл) в атмосфере аргона при -78ºC по каплям добавляли оксалилхлорид (2М раствор в DCM, 8,45 мл, 1,40 экв. 16,9 ммоль). Реакционную смесь перемешивали при -78ºС в течение 1 часа, затем по каплям при перемешивании добавляли (S)-2-(тритиламино)бутан-1-ол (4 г, 1 экв, 12,07 ммоль) в DCM (30 мл). Реакционную смесь перемешивали при этой температуре в течение 2 часов, затем добавляли раствор триэтиламина (TEA, 8,4 мл, 5 экв., 60,27 ммоль) в DCM и раствору давали нагреться до комнатной температуры за 1 час. Реакционную смесь разбавляли дополнительно DCM (100 мл) и промывали водой (250 мл). Водную фазу экстрагировали DCM (3×50 мл) и объединенную органическую фазу промывали насыщенным раствором соли (50 мл), сушили (Na2SO4) и выпаривали в вакууме. Остаток очищали флэш-хроматографией на диоксиде кремния (этилацетат:гексан 1:4), с получением (S)-2-(тритиламино)бутиральдегида в виде светло-желтого масла; выход: 3,64 г (91%).
1H-ЯМР (CDCl3, 250 МГц): δ 0,95 (3H, т, J=7,5 Гц, -NHCH(CH2 CH 3)CHO), 1,72-1,52 [2H, м, NHCH(CH 2CH3)CHO], 2,76 (1H, с, ушир., -NH), 3,41-3,36 [1H, м, NHCH(CH2CH3)CHO], 7,35-7,17 (12H, м, ArH), 7,67-7,51 (3H, м, ArH), 9,05 (1H, с, NHCH(CH2CH3)CHO). δC (250 МГц, CDCl3) 202,95 (CO), 146,23 (C), 129,23 (6×CH), 127,96 (6×CH), 126,85 (3×CH), 71,13 (C), 62,62 (CH), 24,78 (CH2), 10,48 (CH3).
(2R,3S)-3-(Тритиламино)пентан-2-ол
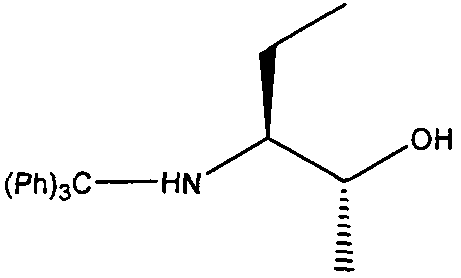
К перемешиваемой суспензии CuBr·SMe2 (3 г, 14,6 ммоль) в безводном эфире (100 мл) в атмосфере аргона при -78ºС по каплям добавляли метиллитий (1,6М в эфире, 16,5 мл, 4,0 экв., 26,5 ммоль), и раствору давали нагреться до комнатной температуры в течение 1 часа. Смесь снова охлаждали до -78ºС и по каплям добавляли раствор (S)-2-(тритиламино)бутиральдегида (2,2 г, 6,62 ммоль) в эфире (25 мл) при перемешивании. Реакционную смесь перемешивали при этой температуре в течение 2 часов, затем давали нагреться до комнатной температуры в течение 1 часа. Добавляли насыщенный водный раствор NH4Cl (50 мл) и два слоя разделяли. Органическую фазу промывали насыщенным раствором соли (50 мл), сушили (MgSO4) и выпаривали в вакууме. Остаток очищали колоночной флэш-хроматографией на силикагеле при элюировании смесью гексан:этилацетат (80:20), с получением (2R,3S)-3-(тритиламино)пентан-2-ола в виде светло-желтого масла; выход: 1,5 г (66%). (75% de 2R,3S: 25% de 2S,3S).
1H-ЯМР (d6-ДМСО, 250 МГц): δ 0, 0,47+0,55 (2×т, J=7,50+7,26 Гц -NHCH(CH2 CH 3)CH(CH3)OH), 0,99-1,12 (м, 5H, -NHCH(CH 2CH3)CH(CH 3)OH), 2,01 (1H, м, -NHCH(CH2CH3)CH(CH3)OH), 3,22-3,43 (м, 1H, -NHCH(CH2CH3)CH(CH3)OH), 4,41 [1H, д, J=3,3, NHCH(CH2CH3)CH(CH3)OH], 7,14-7,56 (15H, м, ArH). δC (250 МГц, CDCl3) 146,88 (C), 128,97 (6×CH), 127,83 (6×CH), 126,43 (3×CH), 71,03 (C), 68,13 (CH), 58,77 (CH), 23,09 (CH2), 17,88 (CH3), 10,47 (CH3).
(2R,3S)-3-Аминопентан-2-ол
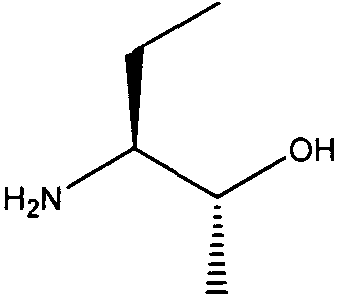
К перемешиваемому раствору (2R,3S)-3-(тритиламино)пентан-2-ола (1,64 г. 4,75 ммоль) в дихлорметане (20 мл) в атмосфере аргона при комнатной температуре по каплям добавляли трифторуксусную кислоту (10 мл) и раствор перемешивали при этой температуре в течение 1 часа. Растворитель выпаривали в вакууме и остаток осаждали из эфира (15 мл) гексаном (150 мл) при перемешивании, с получением желтого масла. Растворитель декантировали из масла, масло промывали гексаном (30 мл) и сушили в вакууме, с получением (2R,3S)-3-аминопентан-2-ола в виде светло-желтого масла; выход: 0,30 г (98%). (75% de 2R,3S: 25% de 2S,3S).
1H-ЯМР (d6-ДМСО, 250 МГц): δ 0,913+0,923 (2×т, 3H, J=7,50+7,50 Гц, NH2CH(CH2 CH 3)CH(CH3)OH), 1,11+1,18 (3H, 2×д, J=6,48+6,48 Гц, NH2CH(CH2CH3)CH(CH 3)OH), 1,41-1,65 (2H, м, NH2CH(CH 2CH3)CH(CH3)OH), 2,76+2,93 [2×1H, м, NH2 CH(CH2CH3)CH(CH3)OH], 3,61-3,69+3,80-3,90 [2×1H, м, NH2CH(CH2CH3)CH(CH3)OH], 7,73 (2H, с, ушир., NH 2).
При пути 2 амин защищали путем дибензилирования.
(S)-2-(Дибензиламино)бутан-1-ол
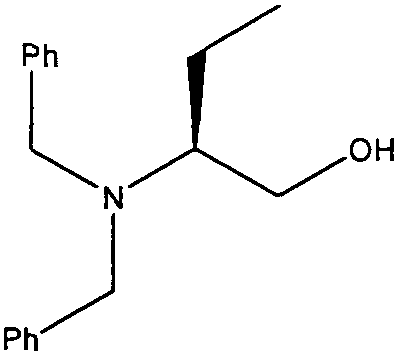
К перемешиваемому раствору (S)-(+)-2-аминобутан-1-ола (5 г, 56,18 ммоль) в сухом ацетонитриле (100 мл) добавляли сухой порошковый карбонат калия (31 г, 224,72 ммоль), с последующим добавлением бензилбромида (19 г, 111,11 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 24 часов. Растворитель удаляли в вакууме и остаток забирали в этилацетат (100 мл) и воду (100 мл). Органическую фазу снова промывали водой, сушили (Na2SO4) и концентрировали, с получением чистого продукта в виде светло-желтого масла (14,5 г, 97,3%).
δH (250 МГц, CDCl3) 0,98 (3H, т, J=7,5, CHCH2 CH 3), 1,38-1,2 (1H, м, CHCHHCH3), 1,94-1,78 (1H, м, CHHCH3), 2,83-2,71 (1H, м, CHCHHCH3), 3,22 (1H, с, ушир., OH), 3,65-3,4 (2H, м, CH 2OH), 3,47 (2H, д, J=17,5, 2×CHHPh), 3,94 (2H, д, J=17,5, 2×CHHPh), 7,46-7,26 (10H, м, 2×C6H5); δC (250 МГц, CDCl3) 139,42 (2×C), 129,1 (2×CH), 128,52 (2×CH), 127,25 (2×CH), 61,97 (CH), 60,67 (CH2), 53,23 (CH2), 17,92 (CH2), 11,83 (CH3); m/z 270,2 (M+H).
(S)-2-(Дибензиламино)бутаналь
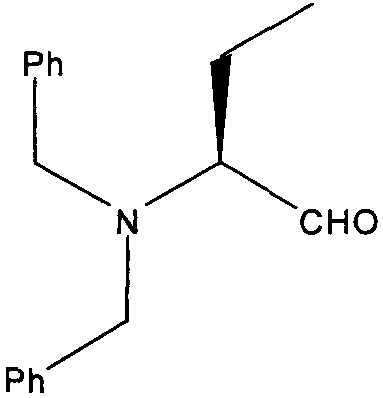
2М раствор оксалилхлорида в дихлорметане (3,18 мл, 6,36 ммоль) охлаждали до -78°С и разбавляли сухим дихлорметаном (20 мл) в атмосфере сухого азота. К охлажденному перемешиваемому раствору по каплям добавляли раствор диметилсульфоксида (1 г, 12,72 ммоль) в безводном дихлорметане. Реакционную смесь перемешивали в течение еще 1 часа после завершения добавления. Добавляли раствор (S)-2-(дибензиламино)бутан-1-ола (1,43 г, 5,3 ммоль) в дихлорметане в течение 5 минут. Через 10 минут добавляли диизопропилэтиламин (2,73 г, 21,2 ммоль). Реакционной смеси давали нагреться до комнатной температуры и оставляли перемешиваться на 1 час. Ее охлаждали до 0°С и добавляли смесь этилацетат/вода (50 мл:50 мл). Органический слой промывали водой (50 мл), насыщенным раствором соли (50 мл), сушили (MgSO4) и концентрировали. Продукт очищали колоночной флэш-хроматографией (этилацет:гексан 1:4), с получением чистого продукта (1,28 г, 90,5%).
δH (250 МГц, CDCl3) 0,88 (3H, т, J=7,5, CHCH2CH 3), 1,77-1,54 (2H, м, CH 2CH3), 2,99 (1H, т, J=7,5, CHCH2CH3), 3,74-3,57 (4H, м, 2×CH 2Ph), 7,31-7,11 (10H, м, 2×C6H5) 9,64 (1H, с, CHO); δC (250 МГц, CDCl3) 203,9 (CO), 139,33 (2×C), 128,99 (4×CH), 128,45 (4×CH), 127,3 (2×CH), 68,46 (CH), 54,85 (CH2), 17,44 (CH2), 11,83 (CH3); m/z 268,2 (M+H).
(2R,3S)-2-(Дибензиламино)бутан-2-ол
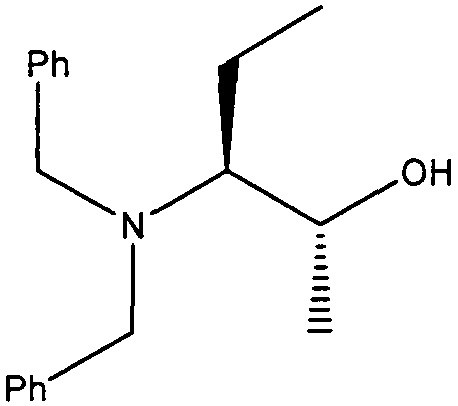
К перемешиваемой суспензии CuBr·SMe2 (1.54 г, 7.5 ммоль) в безводном эфире (100 мл) в атмосфере аргона при -78ºС по каплям добавляли метиллитий (1,6М в эфире, 9.4 мл, 15 ммоль). После того, как добавление завершалось, реакционной смеси давали нагреться до комнатной температуры. Реакционную смесь снова охлаждали до -78°С и по каплям добавляли раствор (S)-2-(дибензиламино)бутаналя (1 г, 3,75 ммоль) в эфире (20 мл). После добавления перемешивание продолжали в течение 2 часов. Затем реакционную смесь гасили насыщенным водным раствором NH4Cl (10 мл). Реакционную смесь экстрагировали эфиром (2×30 мл) и объединенную органическую фазу промывали насыщенным раствором соли (20 мл), сушили (MgSO4) и выпаривали в вакууме. Остаток очищали колоночной градиентной флэш-хроматографией на силикагеле при элюировании смесью гексан:этилацетат (100:0→80:20), с получением продукта в виде светло-желтого масла (0,95 г, 89%) в виде единственного изомера.
δH (250 МГц, CDCl3) 1,05 (3H, т, J=7,5, CHCH2CH 3), 1,25 [3H, д, J=7,5, CH(CH 3)OH], 1,6-1,49 (1H, м, CHHCH3), 1,88-1,73 (1H, м, CHHCH3), 2,41 (1H, с, ушир., OH), 2,66-2,59 (1H, м, CHCH2CH3), 3,85-3,65 (4H, м, 2×CH 2Ph), 4,05-3,9 (1H, м, CHOH), 7,41-7,25 (10H, м, ArH). δC (250 МГц, CDCl3) 140,05 (2×C), 128,98 (4×CH), 128,37 (4×CH), 127,3 (2×CH), 66,81 (CH), 63,65 (CH), 55,41 (CH2), 20,63 (CH3), 18,44 (CH2), 12,5 (CH3).
Пример 1
2-хлор-4,6-диметилникотинонитрил
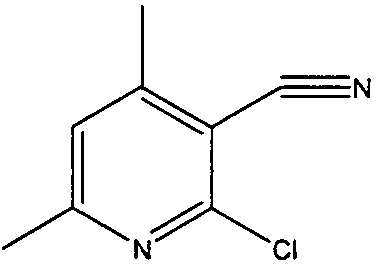
4,6-Диметил-2-оксо-1,2-дигидропиридин-3-карбонитрил (5 г, 34 ммоль) добавляли к оксихлориду фосфора (20 мл). Реакционную смесь перемешивали при кипячении с обратным холодильником в течение 2 часов, затем наблюдали, что она завершилась. Летучие вещества удаляли и остаток растирали с бензином. Полученное твердое вещество отфильтровывали, промывали гексаном и сушили, с получением чистого белого твердого вещества (5,1 г, 90%).
δH (250 МГц, CDCl3) 2,55 (3H, с, CH3), 2,57 (3H, с, CH3), 7,09 (1H, с, ArH); δС (250 МГц, CDCl3) 162,64 (C), 154,39 (C), 152,26 (C), 123,22 (CH), 114,28 (C), 108,31 (C), 24,5 (CH3), 20,54 (CH3); m/z 189 (M+Na).
Трет-бутиловый эфир 4,6-диметилпиридин-3-илметилкарбаминовой кислоты
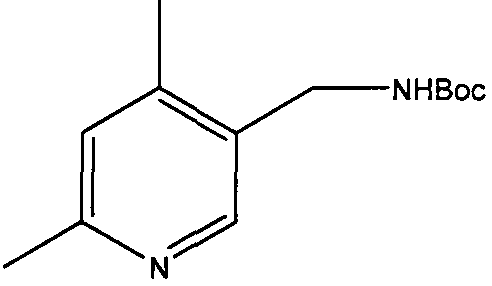
2-Хлор-4,6-диметилникотинонитрил (5 г, 30,1 ммоль) растворяли в смеси 10% уксусная кислота/этанол (30 мл). Добавляли катализатор 10% палладий на углероде (0,5 г) и реакционную смесь перемешивали в атмосфере водорода в течение 24 часов при 60°С. Смесь фильтровали через рыхлый слой целита. Летучие вещества удаляли и неочищенный остаток растворяли в дихлорметане (30 мл). К перемешиваемому раствору затем добавляли триэтиламин (5 мл), с последующим добавлением ди-трет-бутилдикарбоната (6,5 г, 30 ммоль). Через 3 часа растворитель удаляли и остаток растворяли в этилацетате. Полученный раствор промывали водой (50 мл), насыщенным раствором бикарбоната (50 мл), сушили и выпаривали. Неочищенный продукт очищали колоночной флэш-хроматографией на силикагеле (этилацетат:гексан 1:2), с получением 1,4 г чистого указанного в заголовке соединения (20% выход).
δH (250 МГц, CDCl3) 1,43 (9H, с, 3×CH3), 2,19 (3H, с, CH3), 2,38 (3H, с, CH3), 4,19 (2H, с, ушир., ArCH 2NH), 6,84 (1H, с, ArH), 8,15 (1H, с, ArH); δC (250 МГц, CDCl3) 157,41 (CO), 155,63 (C), 148,93 (CH), 145,91 (C), 129,51 (C), 124,76 (CH), 79,44 (C), 46,12 (CH2), 28,32 (3×CH3), 23,74 (CH3), 18,97 (CH3); m/z 237,2 (M+H).
(4,6-Диметилпиридин-3-илметил)-(2-фтор-9Н-пурин-6-ил)амин
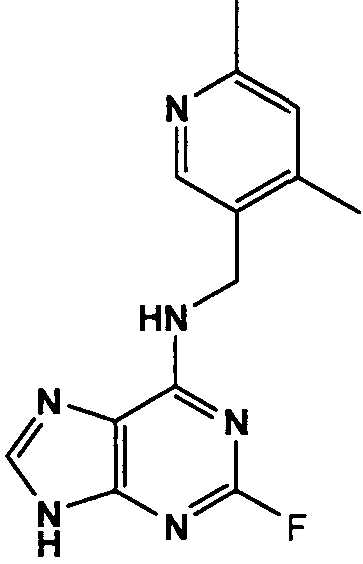
К перемешиваемому раствору 6-хлор-2-фторпурина (0,83 г, 4,9 ммоль) в н-BuOH (50 мл) в атмосфере аргона при 0°С добавляли DIEA (2,5 мл, 14,7 ммоль), с последующим добавлением (4,6-диметилпиридин-3-ил)метанамина (1 г, 7,35 ммоль). Реакционную смесь перемешивали при этой температуре в течение 1 часа, затем давали вернуться к комнатной температуре и перемешивали в течение 4 часов, при этом наблюдалось, что реакция еще не завершена, и поэтому реакционную смесь нагревали до 100°С и оставляли при этой температуре в течение 2 часов. Растворитель выпаривали в вакууме и остаток очищали градиентной колоночной флэш-хроматографией на силикагеле при элюировании CHCl3:MeOH (100:0→90:10), с получением продукта в виде белого твердого вещества; выход: 0,86 г (65%).
δH (250 МГц, CDCl3) 2,35 (3H, с, CH3), 2,39 (3H, с, CH3), 4,61 (2H, с, ушир., NHCH 2), 7,07 (1H, с, ArH), 8,13 (1H, с, ArH), 8,33 (1H, с, ArH), 8,69 (1H, с, ушир., NH); δc (250 МГц, CDCl3) 161,2 (C), 158,57 (C), 156,08 (C), 150 (C), 148,08 (CH), 148,14 (CH), 147,9 (CH), 145,93 (C), 129,92 (C), 129,76 (C), 124,37 (CH), 41,7 (CH2), 23,17 (CH3), 18,14 (CH3); m/z 273,2 (M+H).
(4,6-Диметилпиридин-3-илметил)-(2-фтор-9-изопропил-9Н-пурин-6-ил)амин
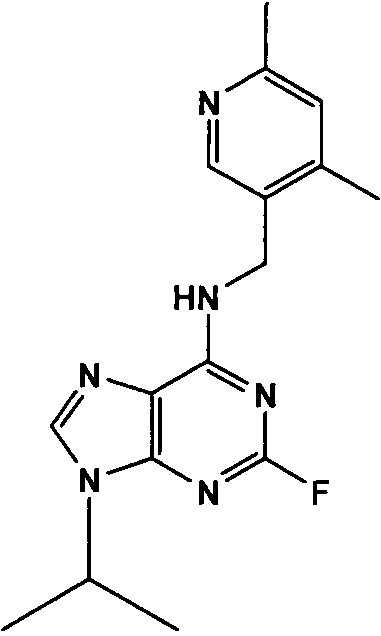
К перемешиваемому раствору (4,6-диметилпиридин-3-илметил)-(2-фтор-9Н-пурин-6-ил)амина (0,6 г, 1,9 ммоль) в ДМФА (10 мл) в атмосфере аргона при к.т. добавляли K2CO3 (порошковый, безводный, 1,77 г, 5 экв., 13 ммоль), с последующим добавлением 2-бромпропана (1,8 мл, 10 экв., 19 ммоль). Реакционную смесь перемешивали при к.т. в течение 24 часов, пока ТСХ (CHCl3:MeOH; 90:10) не показала, что реакция пришла к завершению. Растворитель выпаривали в вакууме и остаток распределяли между водой (50 мл) и этилацетатом (50 мл), водную фазу отделяли и дополнительно экстрагировали EtOAc (2×50 мл). Объединенную органическую фазу промывали насыщенным раствором соли (50 мл), сушили (MgSO4), выпаривали в вакууме и остаток очищали градиентной колоночной хроматографией на силикагеле при элюировании CHCl3:MeOH (100:1→95:5), с получением продукта в виде желтой пленки (0,4 г, 59%).
δH (250 МГц, CDCl3) 1,52 [6H, д, J=7,5 CHCCH 3)2], 2,27 (3H, с, CH3), 2,45 (3H, с, CH3), 4,73-4,62 (3H, м, NHCH 2 и CH[CH3]2), 6,91 (1H, с, ArH), 7,12 (1H, NH), 7,47 (1H, с, ArH), 8,32 (1H, с, ArH); δC (250 МГц, CDCl3) 160,77 (C), 157,89 (C), 157,43 (C), 156,12 (C), 155,79 (C), 149,14 (CH), 137,7 (CH), 128,7 (C), 129,76 (C), 124,83 (CH), 47,2 (CH), 40,14 (CH2), 23,9 (CH3), 22,47 (2×CH3), 18,54 (CH3); m/z 315,3 (M+H).
(2R,3S-3-(6-((4,6-диметилпиридин-3-илметиламино)-9-изопропил-9Н-пурин-2-иламино)пентан-2-ол [1]
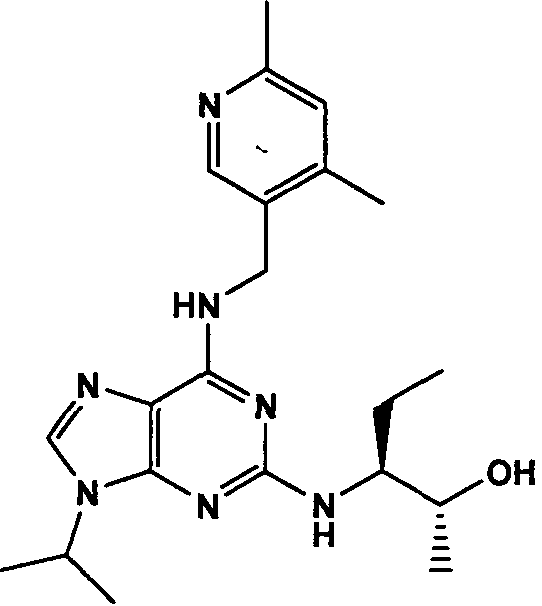
К перемешиваемому раствору (4,6-диметилпиридин-3-илметил)-(2-фтор-9-изопропил-9Н-пурин-6-ил)амина (300 мг, 0,84 ммоль) в н-BuOH/ДМСО (5 мл, 4:1) при комнатной температуре в атмосфере аргона добавляли DIEA (1,7 мл, 10 экв., 8,4 ммоль), с последующим добавлением (2R,3S)-3-аминопентан-2-ола (0,5 г, 4,8 ммоль). Колбу снабжали конденсатором и реакционную смесь помещали на предварительно нагретую масляную баню при 140°С и перемешивали при этой температуре в течение 72 часов. Реакционной смеси давали охладиться до комнатной температуры и растворитель выпаривали в вакууме. Остаток распределяли между этилацетатом (50 мл) и водой (50 мл), водную фазу экстрагировали EtOAc (2×25 мл) и объединенную органическую фазу промывали насыщенным раствором соли (50 мл), сушили (MgSO4) и выпаривали в вакууме. Остаток очищали градиентной колоночной хроматографией на силикагеле при элюировании CHCl3:MeOH (100:1→95:5), с получением 55 мг чистого соединения, указанного в заголовке (12%).
δH (250 МГц, CDCl3) 0,95 (3H, т, J=7,5, CHCH2CH3), 1,06 (3H, д, J=7,5, CHCH3OH), 1,48 [6H, д, J=7,5 CHCCH 3)2], 2,24 (3H, с, CH3), 2,4 (3H, с, CH3), 3,92-3,82 (2H, м, NHCH2), 4,67-4,45 (3H, м, CHEtCHMeOH), 6,15 (1H, с, ушир., NH), 6,87 (1H, с, ArH), 7,37 (1H, ArH), 8,31 (1H, с, ArH); δC (250 МГц, CDCl3) 160,11 (C), 157,68 (C), 154,57 (C), 149,42(CH), 146,38 (C), 134,54 (CH), 129,24 (C), 124,84 (CH), 71,52 (CH), 59,65(CH), 46,47 (CH), 40,33 (CH2), 24,94 (CH2), 23,89 (CH3), 23,52 (2×CH3), 17,37 (CH3), 12,57 (CH3); m/z 398,3 (M+H).
Пример 2
2-Фтор-N-((6-метилпиридин-3-ил)метил)-9Н-пурин-6-амин
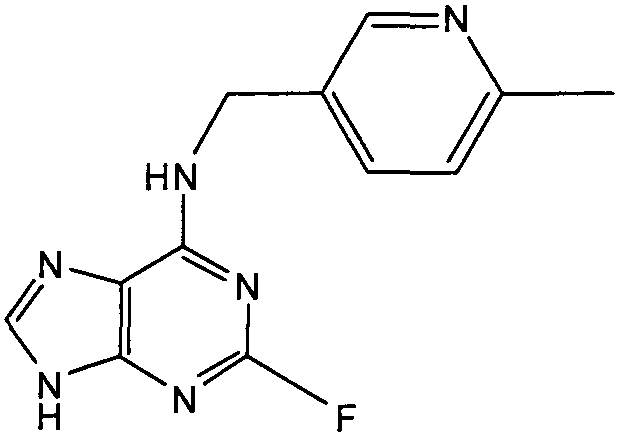
К перемешиваемому раствору 6-хлор-2-фторпурина (0,4 г, 2,3 ммоль) в н-BuOH (50 мл) в атмосфере аргона при 0°С добавляли DIEA (2,5 мл, 14,7 ммоль), с последующим добавлением (6-метилпиридин-3-ил)метиламина (0,36 г, 2,95 ммоль). Реакционную смесь перемешивали при этой температуре в течение 1 часа, затем давали вернуться к комнатной температуре и перемешивали в течение 4 часов, наблюдали, что реакция все еще не завершена, поэтому реакционную смесь нагревали до 100°С и оставляли при этой температуре на 8 часов. Растворитель выпаривали в вакууме и остаток очищали градиентной колоночной хроматографией на силикагеле при элюировании CHCl3:MeOH (100:0→90:10), с получением продукта в виде в виде белого твердого вещества; выход: 0,38 г (65%).
δH CDCl3, 250 МГц) 2,44 (3H, с, CH3), 3,66-3,57 (2H, м, NHCH 2), 4,63 (1H, с, ушир., NH), 7,25 (1H, д, J=7,5, ArH), 7,71 (1H, дд, J=2,5, 7,5, ArH), 8,14 (1H, с, ArH), 8,49 (1H, с, ArH), 9,07 (1H, с, ушир., NH); δС (CDCl3, 250 МГц) 159,12 (C), 158,62 (C), 157,61 (C), 155,56 (C), 147,44 (CH), 146,99 (CH), 136,32 (C), 123,05 (2×CH), 119,42 (C), 41,64 (CH2), 18,47 (CH3); m/z 259,2 (M+H).
2-Фтор-9-изопропил-N-((6-метилпиридин-3-ил)метил)-9Н-пурин-6- амин
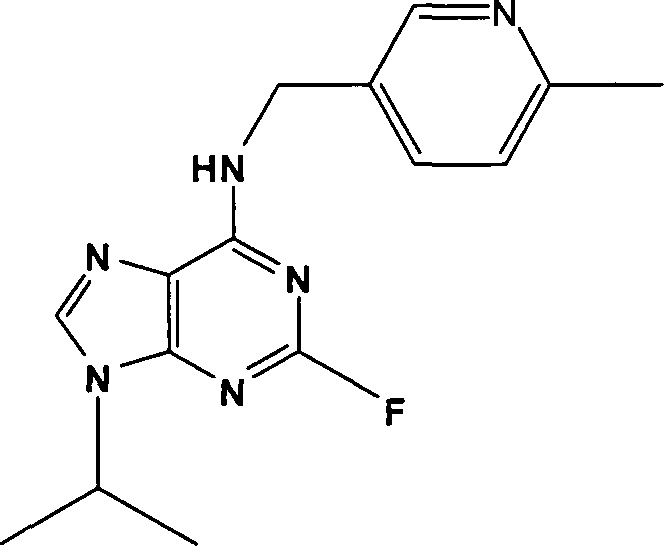
К перемешиваемому раствору 2-фтор-N-(6-метилпиридин-3-ил)метил-9Н-пурин-6-амина (0,3 г, 1,17 ммоль) в диметилформамиде (10 мл) при комнатной температуре в атмосфере аргона добавляли порошковый безводный К2СО3 (0,8 г, 5 экв., 5,85 ммоль), с последующим добавлением 2-бромпропана (1,15 мл, 11,7 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 24 часов, пока смесь DCM:эфир:МеОН (55:40:5) не показала, что реакция подошла к завершению. Растворитель выпаривали в вакууме и остаток распределяли между EtOAc (100 мл) и водой (100 мл). Водную фазу дополнительно экстрагировали EtOAc (2×50 мл) и объединенную органическую фазу промывали насыщенным раствором соли (50 мл), сушили (MgSO4) и выпаривали в вакууме. Остаток очищали колоночной флэш-хроматографией на силикагеле при элюировании CHCl3:MeOH (98:2), с получением указанного в заголовке соединения в виде слегка желтой пленки (195 мг, 55%).
δH (CDCl3, 250 МГц) 1,52 (6H, д, J=7,5, CH[CH3]2), 2,5 (3H, с, CH3), 4,76-4,6 (3H, м, NHCH 2 и CHMe2), 7,06 (1H, д, J=2,5, ArH), 7,35 (1H, с, ушир., NH), 7,56 (2H, с, ушир., ArH), 8,47 (1H, с, ушир., ArH); δC (CDCl3, 250 МГц) 157,6 (C), 156,32 (C), 156 (C), 148,47 (CH), 137,72 (CH), 136,08 (CH), 130,83 (C), 123,11 (CH), 118,2 (C), 47,38 (CH), 43,2 (CH2), 23,99 (CH3), 22,5 (2×CH3); m/z 301,2 (M+H).
2R,3S-3-(9-изопропил-6-((6-метилпиридин-3-ил)метиламино)-9Н-пурин-2-иламино)пентан-2-ол [2]
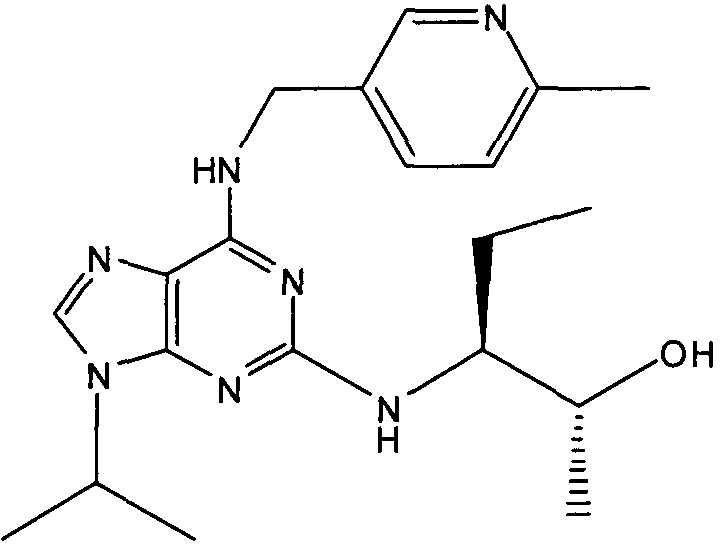
К перемешиваемому раствору 2-фтор-9-изопропил-N-(6-метилпиридин-3-илметил)-9Н-пурин-6-амина (180 мг, 0,59 ммоль) в н-BuOH/ДМСО (5 мл, 4:1) при комнатной температуре в атмосфере аргона добавляли DIEA (1 мл, 10 экв., 5.6 ммоль), с последующим добавлением (2R,3S)-3-аминопентан-2-ола (0,34 г, 6 ммоль). Колбу снабжали конденсатором и реакционную смесь помещали на предварительно нагретую масляную баню при 140°С и перемешивали при этой температуре в течение 72 часов. Реакционной смеси давали охладиться до комнатной температуры и растворитель выпаривали в вакууме. Остаток распределяли между EtOAc (50 мл) и водой (50 мл), водную фазу дополнительно экстрагировали EtOAc (2×25 мл) и объединенную органическую фазу промывали насыщенным раствором соли (50 мл), сушили (MgSO4) и выпаривали в вакууме. Остаток очищали градиентной колоночной флэш-хроматографией на силикагеле при элюировании CHCl3:MeOH (100:1→95:5), с получением указанного в заголовке соединения (40 мг, 19%).
δH (CDCl3, 250 МГц) 0,95 (3H, т, J=7,5, CHCH2CH 3), 1,14 (3H, д, J=5, CHCH 3OH), 154 (6H, д, J=7,5, CH[CH 3]2), 1,62-1,43 (2H, м, CHCH2CH3), 2,44 (3H, с, ArCH3), 3,93 (1H, м, CHMe2), 4,77-4,58 (1H, м, CHCH3OH), 4,8-4,6 (2H, м, NHCH 2Ar), 5,8 (1H, с, ушир., NH), 6,82 (1H, с, ушир., NH), 7,09 (1H, д, J=10, ArH), 7,31-7,23 (2H, м, ArH), 8,49 (1H, с, ушир., ArH); δC (CDCl3, 250 МГц) 157,71 (C), 156,28 (C), 155,95 (C), 148,58 (CH), 137,73 (CH), 129,01 (C), 128,52 (C), 128,42 (CH), 1231,14 (CH), 68,84 (CH), 50,45 (CH2), 47,25 (CH), 23,27 (CH3), 22,53 (2×CH3), 20,9 (CH2), 19,46 (CH3), 10,45 (CH3); m/z 384,3 (M+H).
Пример 3
(3-Хлорбензил)-(2-фтор-9Н-пурин-6-ил)амин
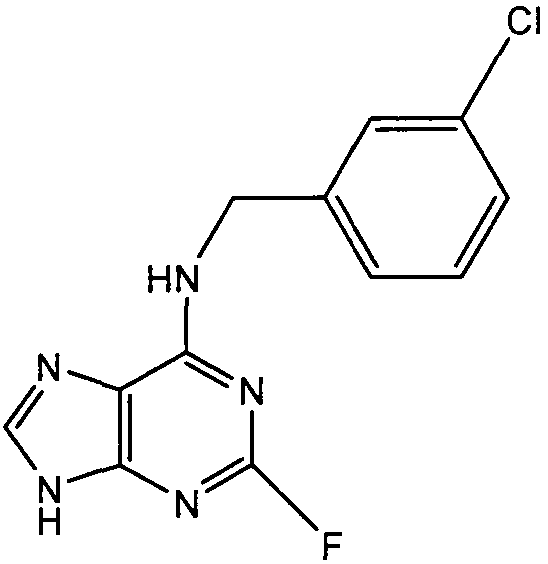
К перемешиваемому раствору 6-хлор-2-фторпурина (1 г, 1 экв., 5,9 ммоль) в н-BuOH (50 мл) в атмосфере аргона добавляли DIEA (2,6 мл, 2,5 экв., 14,75 ммоль), с последующим добавлением 3-хлорбензиламина (1,25 г, 1,5 экв., 8,85 ммоль). Реакционную смесь нагревали до 100°С и выдерживали при этой температуре в течение 3 часов, затем реакция завершилась. Растворитель выпаривали в вакууме и остаток очищали градиентной колоночной хроматографией на силикагеле при элюировании смесью DCM:эфир:МеОН (55:45:0→55:43:2), с получением указанного в заголовке соединения в виде белого твердого вещества; выход: 1,15 г (70%).
δH (250 МГц, d6-ДМСО) 4,78-4,6 (2H, м, NHCH 2Ar), 7,41-7,28 (4H, м, ArH), 7,57 (1H, с, ушир., ArH), 8,15 (1H, с, ушир., NH), 8,88 (1H, с, ушир., NH); δС (250 МГц, d6-ДМСО) 142,3 (C), 133,02 (C), 132,92 (C), 130,25 (CH), 130,17 (C), 127,82 (CH), 127,27 (CH), 127,03 (CH), 126,76 (C), 125,92 (CH), 116,4 (C), 115 (C), 42,67(CH2); m/z 278 (M+H).
(3-Хлорбензил)-(2-фтор-9-изопропил-9Н-пурин-6-ил)амин
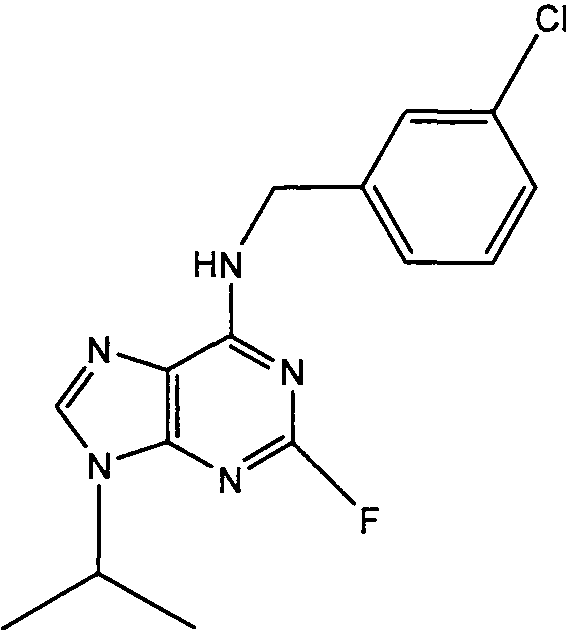
К перемешиваемому раствору (3-хлорбензил)-(2-фтор-9Н-пурин-6-ил)амина (0,6 г, 2,16 ммоль) в диметилформамиде (10 мл) при комнатной температуре в атмосфере аргона добавляли порошковый безводный карбонат калия (1,47 г, 5 экв., 10,8 ммоль), с последующим добавлением 2-бромпропана (2,2 мл, 10 экв., 21,6 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 24 часов, пока элюирование смесью DCM:эфир:МеОН (55:40:5) не показало, что реакция подошла к завершению. Растворитель выпаривали в вакууме и остаток распределяли между этилацетатом (50 мл) и водой (50 мл). Водную фазу дополнительно экстрагировали этилацетатом (2×50 мл) и объединенную органическую фазу промывали насыщенным раствором соли (50 мл), сушили (MgSO4) и выпаривали в вакууме. Остаток очищали колоночной флэш-хроматографией на силикагеле при элюировании CHCl3, с получением указанного в заголовке соединения в виде слегка желтой смолы: выход 0,45 г (65%).
δH (250 МГц, CDCl3) 1,57 (6H, д, J=7,5, CH[CH3]2), 4,81-4,63 (3H, м, NHCH 2Ar и CHMe2), 5,98 (1H, с, ушир., NH), 7,29-7,19 (3H, м, ArH), 7,34 (1H, с, ушир., ArH), 7,42 (1H, д, J=7,5, ArH), 7,51 (1H, с, ушир., NH); δC (250 МГц, CDCl3) 161,93 (C), 156,37 (C), 156,05 (C), 141,33 (C), 140,42 (C), 137,75 (CH), 134,49 (C), 129,92 (CH), 127,69 (CH), 125,81 (CH), 125,64 (CH), 47,43 (CH), 43,82 (CH2), 22,56 (2×CH3); m/z 320,3 (M+H).
2R,3S-3-(6-(3-Хлорбензиламино-9-изопропил-9Н-пурин-2-иламино)пентан-2-ол [3]
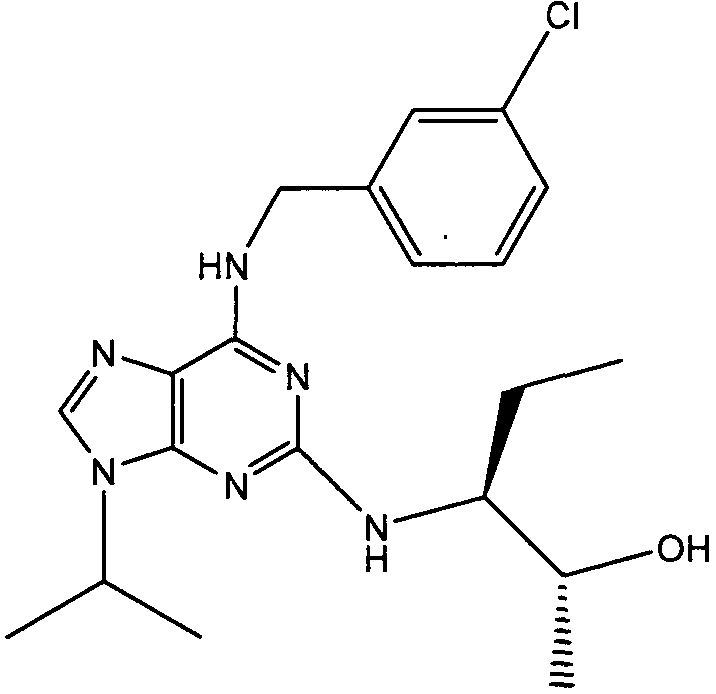
К перемешиваемому раствору (3-хлорбензил)-(2-фтор-9-изопропил-9Н-пурин-6-ил)амина (0,3 г, 0,93 ммоль) в н-BuOH/ДМСО (5 мл, 4:1) при комнатной температуре в атмосфере аргона добавляли DIEA (0,6 мл, 10 экв., 10 ммоль), с последующим добавлением (2R,3S)-3-аминопентан-2-ола (0,5 г, 4,8 ммоль). Реакционную смесь помещали на предварительно нагретую масляную баню при 140°С и перемешивали при этой температуре в течение 72 часов, пока ТСХ со смесью хлороформ:метанол (95:5) не показала, что реакция подошла к завершению. Реакционной смеси давали остыть до комнатной температуры и растворитель выпаривали в вакууме. Остаток распределяли между дихлорметаном (100 мл) и водой (100 мл), водную фазу дополнительно экстрагировали дихлорметаном (3×50 мл) и объединенную органическую фазу промывали насыщенным раствором соли (50 мл), сушили (MgSO4) и выпаривали в вакууме. Остаток очищали градиентной колоночной флэш-хроматографией на силикагеле при элюировании смесью DCM:эфир:MeOH (60:40:0→60:40:2), с получением указанного в заголовке соединения в виде бесцветной пленки; выход 90 мг, (22,5%).
δH (250 МГц, CDCl3) 0,97 (3H, т, J=7,5, CHCH2CH 3), 1,08 (3H, д, J=7,5, CHCH 3OH), 1,45 (6H, д, J=7,5, CH[CH 3]2), 1,6-1,35 (2H, м, CHCH 2CH3), 3,93-3,8 (2H, м, CHEt и NH), 4,55-4,45 [1H, м, CH(CH3)OH], 4,74-4,64 (3H, м, NHCH 2Ar и CHMe2), 5,5 (1H, с, ушир., OH), 6,53 (1H, с, ушир., NH), 7,19-7,13 (3H, м, ArH), 7,22 (1H, с, ArH), 7,32 (1H, с, ArH); δC (250 МГц, CDCl3) 175,06 (C), 160,17 (C), 154,75 (C), 141,22 (C), 134,57 (CH), 134,29 (C), 129,73 (CH), 127,75 (CH), 127,30 (CH), 125,7 (CH), 114,56 (C), 71,53 (CH), 59,58 (CH), 46,44(CH), 43,78 (CH2), 23,96 (2×CH3), 18,65 (CH3), 18,32 (CH3), 12,56 (CH3); m/z 403,2 (M+H).
Пример 4
(3-Фторбензил-2-фтор-9Н-пурин-6-ил)амин
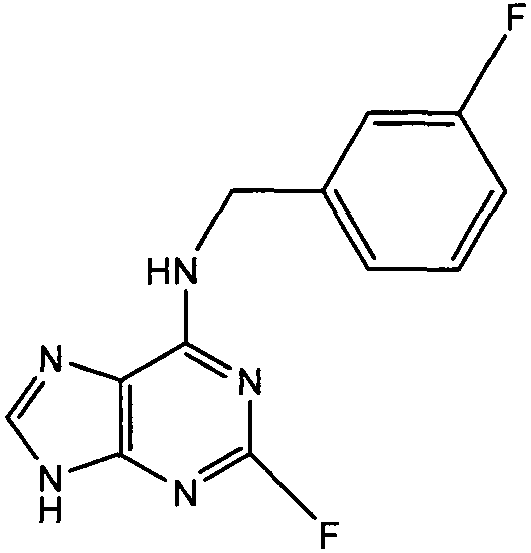
К перемешиваемому раствору 6-хлор-2-фторпурина (1 г, 5,9 ммоль) в н-BuOH (30 мл) в атмосфере аргона добавляли DIEA (2,6 мл, 2,5 экв., 14,75 ммоль), с последующим добавлением 3- торбензиламина (1,1 г, 1,5 экв., 8,85 ммоль). Реакционную смесь нагревали до 100°С и выдерживали при этой температуре в течение 3 часов, затем реакция завершилась. Растворитель выпаривали в вакууме и остаток очищали градиентной колоночной флэш-хроматографией на силикагеле при элюировании смесью DCM:эфир:МеОН (55:45:0→55:43:2), с получением указанного в заголовке соединения в виде белого твердого вещества; выход: 1,1 г (71,7%).
δH (250 МГц, d6-ДМСО) 4,79-4,66 (2H, с, ушир., NHCH 2Ar), 7,41-7,28 (4H, м, ArH), 7,57 (1H, с, ушир., ArH), 8,15 (1H, с, ушир., NH), 8,87 (1H, с, ушир., NH); δC (250 МГц, d6-ДМСО) 142,5 (C), 133,02 (C), 132,92 (C), 130,24 (CH), 130,17 (C), 128,41 (CH), 127,75 (C), 127,04 (CH), 126,76 (CH), 125,92 (CH), 116,2 (C), 115 (C), 42,68 (CH2); m/z 262,1 (M+H).
(3-Фторбензил-(2-фтор-9-изопропил-9Н-пурин-6-ил)амин
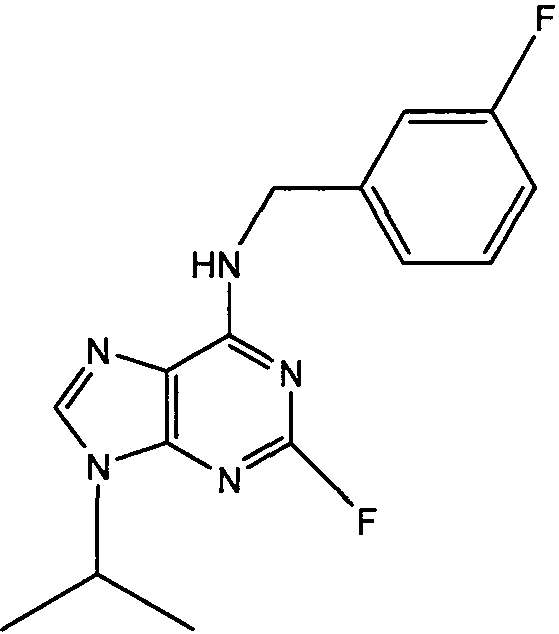
К перемешиваемому раствору (3-фторбензил)-(2-фтор-9Н-пурин-6-ил)амина (0,6 г, 2,24 ммоль) в диметилформамиде (10 мл) при комнатной температуре в атмосфере аргона добавляли порошковый безводный карбонат калия (1,52 г, 5 экв., 11,2 ммоль), с последующим добавлением 2-бромпропана (2,3 мл, 10 экв., 22,4 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 24 часов, пока ТСХ со смесью DCM:эфир:МеОН (55:40:5) не показала, что реакция подошла к завершению. Растворитель выпаривали в вакууме и остаток распределяли между этилацетатом (100 мл) и водой (100 мл). Водную фазу дополнительно экстрагировали этилацетатом (2×50 мл) и объединенную органическую фазу промывали насыщенным раствором соли (50 мл), сушили (MgSO4) и выпаривали в вакууме. Остаток очищали колоночной флэш-хроматографией на силикагеле при элюировании хлороформом, с получением указанного в заголовке соединения в виде слегка желтой смолы: выход 0,37 г (54%).
δH (250 МГц, CDCl3) 1,55 (6H, д, J=7,5, CH[CH3]2), 4,85-4,65 (3H, м, NHCH 2Ar и CHMe2), 5,96 (1H, с, ушир., NH), 7,31-6,94 (4H, м, ArH), 7,59 (1H, с, ушир., ArH); δC (250 МГц, CDCl3) 164,93 (C), 161,01 (C), 156,04 (C), 139,77 (C), 137,76 (CH), 130,13 (CH), 129,92 (C), 123,19 (CH), 114,59 (CH), 114,26 (CH), 47,22 (CH), 43,93 (CH2), 22,56 (2×CH3); m/z 304,2 (M+H).
2R,3S-3-[6-(3-Фторбензиламино)-9-изопропил-9Н-пурин-2-иламино]пентан-2-ол [4]
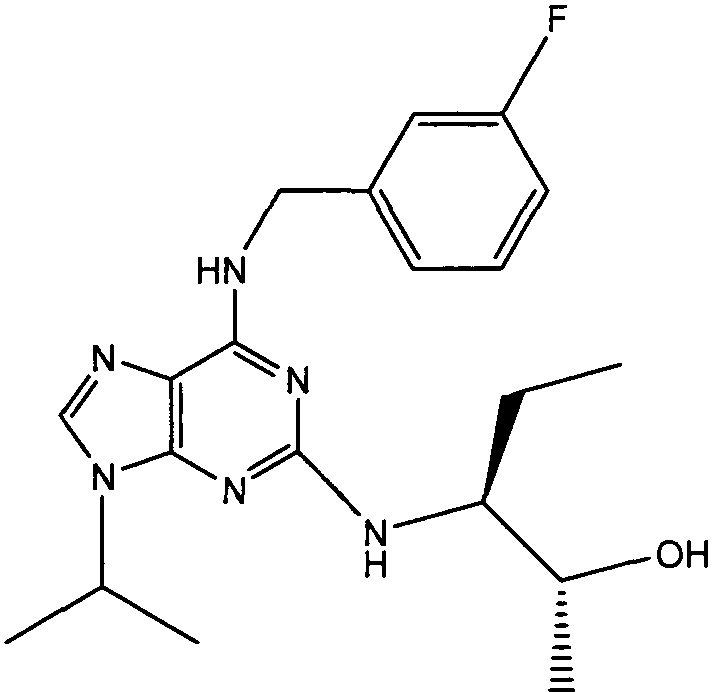
К перемешиваемому раствору (3-фторбензил)-(2-фтор-9-изопропил-9Н-пурин-6-ил)амина (0,3 г, 0,99 ммоль) в н-BuOH/ДМСО (5 мл, 4:1) при комнатной температуре в атмосфере аргона добавляли DIEA (0,6 мл, 10 экв., 10 ммоль), с последующим добавлением (2R,3S)-3-аминопентан-2-ола (0,5 г, 4,8 ммоль). Реакционную смесь помещали на предварительно нагретую масляную баню при 140°С и перемешивали при этой температуре в течение 72 часов, пока ТСХ со смесью хлороформ:метанол (95:5) не показала, что реакция подошла к завершению. Реакционной смеси давали остыть до комнатной температуры и растворитель выпаривали в вакууме. Остаток распределяли между дихлорметаном (100 мл) и водой (200 мл), водную фазу дополнительно экстрагировали дихлорметаном (3×25 мл) и объединенную органическую фазу промывали насыщенным раствором соли (50 мл), сушили (MgSO4) и выпаривали в вакууме. Остаток очищали градиентной колоночной флэш-хроматографией на силикагеле при элюировании смесью DCM:эфир:MeOH (60:40:0→60:40:2), с получением указанного в заголовке продукта в виде бесцветной пленки; выход: 80 мг (20%).
δH (250 МГц, CDCl3) 1,05 (3H, т, J=7,5, CHCH2CH 3), 1,15 (3H, д, J=7,5, CHCH 3OH), 1,55 (6H, д, J=7,5, CH[CH 3]2), 1,65-1,4 (2H, м, CHCH 2CH3), 4-3,91 (2H, м, CHEt и CHMe2), 4,66-4,55 (1H, м, CHMeOH), 4,8 (2H, д, J=5, NHCH 2Ar), 6,41 (1H, с, ушир., NH), 7,33-6,92 (4H, м, ArH), 7,46 (1H, с, ушир., ArH); δC (250 МГц, CDCl3) 165,2 (C), 164,92 (C), 160,18 (C), 154,77 (C), 141,67 (C), 141,56 (C), 134,59 (CH), 130,08 (CH), 123,14 (CH), 114,71 (CH), 113,95 (CH), 71,61 (CH), 59,65 (CH), 46,48 (CH), 43,94 (CH2), 25,02 (CH2), 22,52 (2×CH3), 17,24 (CH3), 10,59 (CH3); m/z 387,3 (M+H).
Пример 5
(9-Циклопропилметил-2-фтор-9Н-пурин-6-ил)пиридин-3-илметиламин
К перемешиваемому раствору 2-фтор-6-[(пиридин-3-илметил)амино]пурина (1 г, 4,10 ммоль) в ДМФА (12 мл) в атмосфере аргона при к.т. добавляли К2СО3 (порошковый безводный, 2,84 г, 5 экв., 20,52 ммоль), с последующим добавлением (бромэтил)циклопропана (5,53 мл, 10 экв., 41 ммоль). Реакционную смесь перемешивали при к.т. в течение 24 часов, пока ТСХ (CHCl3:MeOH; 90:10) не показала, что реакция подошла к завершению. Растворитель выпаривали в вакууме и остаток распределяли между водой (50 мл) и EtOAc (50 мл); водную фазу отделяли и дополнительно экстрагировали EtOAc (2×50 мл). Объединенную органическую фазу промывали насыщенным раствором соли (50 мл), сушили (MgSO4) и выпаривали в вакууме, и остаток очищали градиентной колоночной флэш-хроматографией на силикагеле при элюировании CHCl3:MeOH (100:0→95:5), с получением продукта в виде бесцветной смолы; выход: 0,8 г (68%).
δH (250 МГц, CDCl3) 0,24-0,15 (2H, м, CHH и CHH от Cp), 0,48-0,38 (2H, м, CHH и CHH от Cp), 1,11-0,95 (1H, м, CH от Cp), 2,56 (1H, с, ушир., NH), 3,69 (2H, д, J=7,5, NCH 2Cp), 4,58 (2H, с, ушир., NHCH 2Ar), 7,04-6,96 (1H, м, ArH), 7,51-7,45 (2H, м, ArH), 8,26-8,24 (1H, м, ArH), 8,37 (1H, с, ушир., ArH); δC (250 МГц, CDCl3) 156,32 (C), 156,01 (C), 149,29 (CH), 148,97 (CH), 139,85 (CH), 139,8 (C), 135,54 (CH), 133,81 (C), 123,51 (CH), 117,86 (C), 48,52 (CH2), 42,09 (CH2), 11,06 (CH3), 5,28 (2×CH2); m/z 299,2 (M+H).
2R,3S-3-(9-(Циклопропилметил)-6-(пиридин-3-илметиламино)-9Н-пурин-2-иламино)пентан-2-ол [5]
К перемешиваемому раствору (9-циклопропилметил-2-фтор-9Н-пурин-6-ил)пиридин-3-илметиламина (300 мг, 1 ммоль) в н-BuOH/ДМСО (5 мл, 4:1) при комнатной температуре в атмосфере аргона добавляли DIEA (1,9 мл, 10 экв., 10,5 ммоль), с последующим добавлением (2R,3S)-3-аминопентан-2-ола (500 мг, 4,8 экв., 4,8 ммоль). Колбу снабжали обратным холодильником, реакционную смесь помещали на предварительно нагретую масляную баню при 140°С и перемешивали при этой температуре в течение 72 часов. Реакционной смеси давали остыть до комнатной температуры и растворитель выпаривали в вакууме. Остаток распределяли между этилацетатом (50 мл) и водой (50 мл), водную фазу дополнительно экстрагировали этилацетатом (2×25 мл) и объединенную органическую фазу промывали насыщенным раствором соли (50 мл), сушили (MgSO4) и выпаривали в вакууме. Остаток очищали градиентной колоночной флэш-хроматографией на силикагеле при элюировании CHCl3:MeOH (100:0→95:5), с получением указанного в заголовке продукта в виде бесцветной пленки. Выход (85 мг, 22%).
δH (250 МГц, CDCl3) 0,45-0,35 (2H, м, CHH и CHH от Cp), 0,7-0,6 (2H, м, CHH и CHH от Cp), 1,02 (3H, т, J=7,5, CHCH2CH 3), 1,16 (3H, д, J=7,5, CHCH 3OH), 1,35-1,2 (1H, м, CHCHHCH3), 1,68-1,38 (2H, м, CHCHHCH3 и CH от Cp), 3,9-3,8 (2H, м, CHOH и CHEt), 4,05 (2H, д, J=7,5, NCH 2Cp), 4,76 (2H, с, ушир., NHCH 2Ar), 4,87 (1H, д, J=5, OH), 5,77 (1H, с, ушир., NH), 6,67 (1H, с, ушир., NH), 7,22-7,17 (1H, м, ArH), 7,53 (1H, с, ArH), 7,65 (1H, дд, J=2,5, 7,5, ArH), 8,5 (1H, д, J=5, ArH), 8,61 (1H, с, ArH), 1,11-0,95 (1H, м, CH от Cp), 2,56 (1H, с, ушир., NH), 3,69 (2H, д, J=7,5, NCH 2Cp), 4,58 (2H, с, ушир., NHCH 2Ar), 7,04-6,96 (1H, м, ArH), 7,51-7,45 (2H, м, ArH), 8,26-8,24 (1H, м, ArH), 8,37 (1H, с, ушир., ArH); δC (250 МГц, CDCl3) 160,3 (C), 154,71 (C), 151,05 (C), 151,05 (C), 149,28 (CH), 148,64 (CH), 136,85 (CH), 135,30 (CH), 134,56 (C), 129,2 (C), 123,37 (CH), 122,1 (C), 114,21 (C), 77,27 (CH), 59,5 (CH), 48,02 (CH2), 41,99 (CH2), 24,84 (CH2), 17,4 (CH3), 11,53 (CH3), 11,01 (CH3), 4,2 (CH2); m/z 382,3 (M+H).
Пример 6
6-Хлор-2-фтор-9-изопропил-9Н-пурин
Смесь 2-фтор-6-хлорпурина (2 г, 11,7 ммоль) и порошковый карбонат калия (4 г, 28 ммоль) энергично перемешивали в 30 мл ДМФ. Изопропилйодид (6 мл, 60 ммоль) очень медленно добавляли в течение 2 часов. Реакционную смесь перемешивали в течение еще 5 часов. ДМФА удаляли и неочищенный продукт забирали в этилацетат, промывали водой (50 мл), насыщенным раствором соли (50 мл), сушили (MgSO4) и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (30% этилацетат в гексане), с получением чистого продукта в виде твердого белого вещества (1,1 г, 44%).
δH (CD3OD, 250 МГц) 1,65 [6H, д, J=7,5, CHC(CH 3)2], 4,92 [1H, м, CH(CH3)2], 8,66 (1H, с, ArH); δС (CD3OD, 250 МГц) 154,7 (C), 153,88 (C), 152,2 (C), 147,65 (CH), 132,44 (C), 50,66 (CH), 22,72 (2×CH3); m/z 215,2 (M+H).
N-Циклопропил-2-фтор-9-изопропил-9Н-пурин-6-амин
6-Хлор-2-фтор-9-изопропил-9Н-пурин (0,3 г, 1,4 ммоль), диизопропилэтиламин (0,2 г, 1,55 ммоль) и циклопропиламин (0,2 г, 2,66 ммоль) перемешивали вместе в этаноле (30 мл) при комнатной температуре в течение 6 часов. Летучие вещества удаляли и остаток забирали в этилацетат, промывали водой (50 мл), насыщенным раствором соли (50 мл), сушили (MgSO4) и концентрировали. Неочищенный продукт очищали колоночной флэш-хроматографией на силикагеле (этилацетат:гексан 3:2), с получением чистого продукта (232 мг, 70,5%).
δH (CDCl3, 500 МГц) 0,63-0,55 (2H, м, CHHCHH от Cp), 0,85- 0,78 (2H, м, CHHCHH от Cp), 1,54 (6H, д, J=7,5, CH[CH 3]2), 2,97 (1H, с, ушир., CH от Cp), 4,73-4,62 (1H, м, CHMe2), 6,72 (1H, с, ушир., NH) 7,7 (1H, с, ArH); δC (CDCl3, 500 МГц) 159,29 (C), 157,63 (C), 156,8 (C), 156,64 (C), 137,69 (CH), 47,35 (CH), 22,6 (2×CH3), 21,06 (CH), 7,28 (2×CH2); m/z 236,2 (M+H).
2R,3S-3-(6-Циклопропиламино)-9-изопропил-9Н-пурин-2-иламино)пентан-2-ол [6]
К перемешиваемому раствору N-циклопропил-2-фтор-9-изопропил-9Н-пурин-6-амина (232 мг, 0,99 ммоль) в N-метилпирролидиноне (10 мл) при комнатной температуре в атмосфере аргона добавляли DIEA (0,2 мл, 10,96 экв., 1,14 ммоль), с последующим добавлением (2R,3S)-3-аминопентан-2-ола (540 мг, 5 экв., 5,25 ммоль). Реакционную смесь помещали на предварительно нагретую масляную баню при 140°С и перемешивали при этой температуре в течение 24 часов. Реакционной смеси давали остыть до комнатной температуры и добавляли избыток воды, вследствие чего продукт выделялся в виде масла. Добавляли этилацетат и органический слой тщательно промывали водой (3×50 мл), сушили (MgSO4) и выпаривали в вакууме. Остаток очищали колоночной флэш-хроматографией на силикагеле при элюировании этилацетатом, с получением указанного в заголовке продукта в виде бледно-коричневого твердого вещества (46 мг, 15%).
δH (CDCl3, 500 МГц) 0,58-0,49 (2H, м, CHHCHH от Cp), 0,83-0,81 (2H, м, CHHCHH от Cp), 1,03 (3H, т, J=7,5, CHCH2CH 3), 1,13 (3H, д, J=7,5, CHCH 3OH), 1,55 (6H, д, J=7,5, CH[CH 3]2), 1,57-1,45 (2H, м, CHCH 2CH3), 2,91 (1H, с, ушир., CH от Cp), 3,95 (2H, с, ушир., CHMe2 и CHEt), 4,6-4,57 (1H, м, CHMeOH), 4,84 (1H, с, ушир., NH), 6,25 (1H, с, ушир., NH), 7,49 (1H, с, ArH); δC (CDCl3, 500 МГц) 160,16 (C), 155,91 (C), 150,24 (C), 134,57 (CH), 114,63 (C), 71,57 (CH), 59,88 (CH), 46,27 (CH), 25,15 (CH2), 22,59 (CH3), 17,22 (CH3), 11,61 (CH3), 7,34 (CH2), 47,35 (CH), 22,6 (2×CH3), 21,06 (CH), 7,28(2×CH2); m/z 319,3 (M+H).
Пример 7
N-(Циклопропилметил)-2-фтор-9-изопропил-9Н-пурин-6-амин
6-Хлор-2-фтор-9-изопропил-9Н-пурин (0,3 г, 1,4 ммоль), диизопропилэтиламин (0,2 г, 1,55 ммоль) и циклопропилметиламин (0,24 г, 2,7 ммоль) перемешивали вместе с этанолом (30 мл) при комнатной температуре в течение 6 часов. Летучие вещества удаляли и остаток забирали в этилацетат, промывали водой (50 мл), насыщенным раствором соли (50 мл), сушили (MgSO4) и концентрировали. Неочищенный продукт очищали колоночной флэш-хроматографией на силикагеле (этилацетат:гексан 3:2), с получением чистого продукта (290 мг, 83%) в виде бесцветной смолы.
δH (CDCl3, 500 МГц) 0,29-0,27 (2H, м, CHHCHH от Cp), 0,56-0,54 (2H, м, CHHCHH от Cp), 1,13 (1H, с, ушир., CH от Cp), 1,58 (6H, д, J=7,5, CH[CH3]2), 3,44 (2H, с, ушир., NHCH 2Cp) 2,97 (1H, с, ушир., CH от Cp), 4,74-4,72 (1H, м, CHMe2), 6,22 (1H, с, ушир., NH), 7,76 (1H, с, ArH); δС (CDCl3, 500 МГц) 160,09 (C), 156,14 (C), 147,4 (C), 137,38 (CH), 118,26 (C), 47,14 (CH), 45,78 (CH2), 22,46 (2×CH3), 10,06 (CH), 3,5 (2×CH2).
2R,3S-3-(6-(Циклопропилметиламино)-9-изопропил-9Н-пурин-2-иламино)пентан-2-ол [7]
К перемешиваемому раствору N-(циклопропилметил)-2-фтор-9-изопропил-9Н-пурин-6-амина (290 мг, 1,1 ммоль) в N-метилпирролидиноне (10 мл) при комнатной температуре в атмосфере аргона добавляли DIEA (1,42 г, 1,11 ммоль), с последующим добавлением (2R,3S)-3-аминопентан-2-ола (566 мг, 5 экв., 5,5 ммоль). Реакционную смесь помещали на предварительно нагретую масляную баню при 140°С и перемешивали при этой температуре в течение 24 часов. Реакционной смеси давали остыть до комнатной температуры и добавляли избыток воды, вследствие чего продукт выделялся в виде масла. Добавляли этилацетат и органический слой тщательно промывали водой (4×50 мл). Органический слой промывали насыщенным раствором соли (50 мл), сушили (MgSO4) и выпаривали в вакууме. Остаток очищали колоночной флэш-хроматографией на силикагеле при элюировании этилацетатом, с получением указанного в заголовке продукта в виде бледно-коричневого твердого вещества (120 мг, 31%).
δH (CDCl3, 500 МГц) 0,22-0,19 (2H, м, CHHCHH от Cp), 0,36-0,34 (2H, м, CHHCHH от Cp), 0,8 (3H, т, J=7,5, CHCH2CH 3), 0,89 (3H, д, J=7,5, CHCH 3OH), 1,13 (1H, с, ушир., CH от Cp), 1,33 (6H, д, J=7,5, CH[CH 3]2), 3,24 (2H, с, ушир., NHCH 2Cp), 3,7 (2H, с, ушир., CHEt и CHMe2), 6,52 (1H, с, ушир., NH), 7,26 (1H, с, ArH); δC (CDCl3, 500 МГц) 160,2 (C), 154,89 (C), 134,35 (CH), 71,72 (CH), 59,76 (CH), 46,238 (CH), 25,23 (CH2), 22,59 (CH3), 17,15 (CH3), 11,61 (CH3), 3,48 (2×CH2); m/z 333,3 (M+H).
Пример 8
N-Циклобутил-2-фтор-9-изопропил-9Н-пурин-6-амин
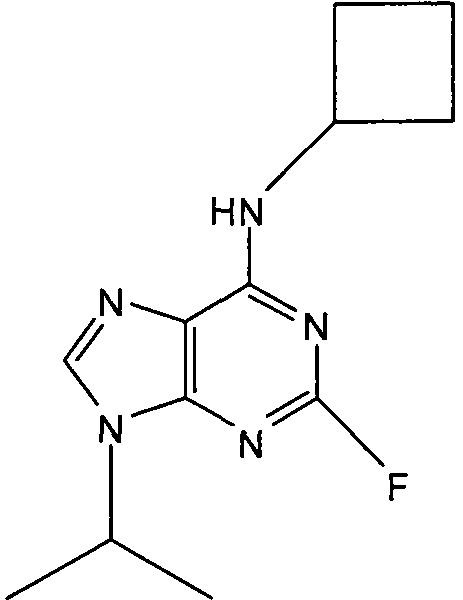
6-Хлор-2-фтор-9-изопропил-9Н-пурин (0,3 г, 1,4 ммоль), диизопропилэтиламин (0,2 г, 1,55 ммоль) и циклобутиламин (0,2 г, 2,8 ммоль) перемешивали вместе с этанолом (30 мл) при комнатной температуре в течение 6 часов. Летучие вещества удаляли и остаток забирали в этилацетат, промывали водой (50 мл), насыщенным раствором соли (50 мл), сушили (MgSO4) и концентрировали. Неочищенный продукт очищали колоночной флэш-хроматографией на силикагеле (этилацетат:гексан 3:2), с получением чистого продукта (237 мг, 68%).
δH (CDCl3, 500 МГц) 1,56 (6H, д, J=7,5, CH[CH 3]2), 1,76-1,74 (2H, м, CH2 циклобутила), 1,98 -1,94 (2H, м, CH2 циклобутила), 2,45 (2H, с, ушир., CH2 циклобутила)) 4,72-4,7 (2H, м, CH циклобутила и CHMe2), 6,35 (1H, с, ушир., NH), 7,75 (1H, с, ArH); δC (CDCl3, 500 МГц) 160,08 (C), 158,43 (C), 155,34 (C), 155,18 (C), 150,09 (C), 137,44 (CH), 47,26 (CH), 31,56 (2×CH2), 22,38 (2×CH3), 15,09 (CH2); m/z 250,2 (M+H).
2R,3S-3-(6-(Циклобутиламино)-9-изопропил-9Н-пурин-2-иламино)пентан-2-ол [8]
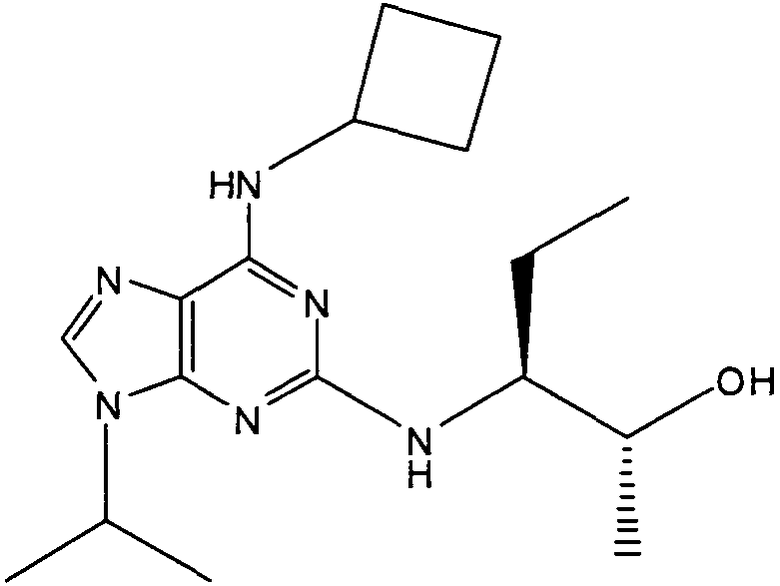
К перемешиваемому раствору N-циклобутил-2-фтор-9-изопропил-9Н-пурин-6-амина (237 мг, 0,95 ммоль) в N-метилпирролидиноне (10 мл) при комнатной температуре в атмосфере аргона добавляли DIEA (1,28 г, 10 ммоль), с последующим добавлением (2R,3S)-3-аминопентан-2-ола (566 мг, 5 экв., 5,5 ммоль). Реакционную смесь помещали на предварительно нагретую масляную баню при 140°С и перемешивали при этой температуре в течение 24 часов. Реакционной смеси давали остыть до комнатной температуры и добавляли избыток воды, вследствие чего продукт выделялся в виде масла. Добавляли этилацетат и органический слой тщательно промывали водой (4×50 мл). Органическую фазу промывали насыщенным раствором соли (50 мл), сушили (MgSO4) и выпаривали в вакууме. Остаток очищали колоночной флэш-хроматографией на силикагеле при элюировании этилацетатом, с получением указанного в заголовке продукта в виде бледно-коричневого твердого вещества (120 мг, 31%).
δH (CDCl3, 500 МГц), 0,97 (3H, т, J=7,5, CHCH2CH 3), 1,07 (3H, д, J=7,5, CHCH 3OH), 1,47 (6H, д, J=7,5, CH[CH 3]2), 1,53-1,42 (2H, м, CHCH 2CH3), 1,71-1,68 (2H, м, CH2 циклобутила), 1,89-1,87 (2H, м, CH2 циклобутила), 2,39-2,37 (2H, м, CH2 циклобутила), 3,89 (1H, д, J=5, NH циклобутила)), 4,53-4,5 (3H, м, CHMe2 и CHEt и CH циклобутила), 5,62 (1H, м, CHMeOH), 6,15 (1H, с, ушир., NH), 7,43 (1H, с, ArH); δC (CDCl3, 500 МГц) 161,16 (C), 154,76 (C), 152,2 (C), 134,43 (CH), 114,67 (C), 71,7 (CH), 59,74 (CH), 46,36 (CH), 31,68 (CH2), 25,25 (2×CH2), 22,59 (CH3), 17,11 (CH3), 15,04 (CH2), 11,61 (CH3); m/z 333,3 (M+H).
Пример 9
(2-фтор-9-Н-пурин-6-ил)пиридин-4-илметиламин
К перемешиваемому раствору 6-хлор-2-фторпурина (1 г, 5,8 ммоль) в н-BuOH (50 мл) в атмосфере аргона при 0°С добавляли диизопропилэтиламин (3 мл, 17,4 ммоль), с последующим добавлением 4-(аминометил)пиридина (0,9 мл, 1,5 экв., 8,7 ммоль). Реакционную смесь перемешивали при этой температуре в течение 1 часа, затем давали вернуться к комнатной температуре и перемешивали в течение 4 часов, пока ТСХ (CHCl3:MeOH; 90:10) не показала, что реакция подошла к завершению. Растворитель выпаривали в вакууме и остаток очищали градиентной колоночной хроматографией на силикагеле при элюировании CHCl3:MeOH (100:0→90:10), с получением продукта в виде в виде белого твердого вещества; выход: 0,85 г (60%). Т.пл. 200-202°С.
1H-ЯМР (d6-ДМСО, 250 МГц): δ 4,67 (2H, д, J=5, -HNCH 2-Pyr), 7,34-7,28 (2H, м, Pyr-H), 8,48-8,42 (2H, м, Pyr-H), 8,54 (1H, с, -N=CH-NH-), 8,84 (1H, с, ушир., -HNCH2-Pyr), 13,13 (1H, с, ушир., -N=CH-NH-); m/z 245 ([M+H]+.
(2-Фтор-9-изопропил-9Н-пурин-6-ил)пиридин-4-илметиламин
К перемешиваемому раствору (2-фтор-9Н-пурин-6-ил)пиридин-4-илметиламина (0,6 г, 2,46 ммоль) в ДМФА (10 мл) в атмосфере аргона при к.т. добавляли К2СО3 (порошковый безводный, 1,7 г, 5 экв., 12,3 ммоль), с последующим добавлением 2-бромпропана (2,3 мл, 10 экв., 24,6 ммоль). Реакционную смесь перемешивали при к.т. в течение 24 часов, пока ТСХ (CHCl3:MeOH; 90:10) не показала, что реакция подошла к завершению. Растворитель выпаривали в вакууме и остаток распределяли между водой (200 мл) и EtOAc (50 мл). Водную фазу отделяли и дополнительно экстрагировали EtOAc (2×50 мл). Объединенную органическую фазу промывали насыщенным раствором соли (50 мл), сушили (MgSO4) и выпаривали в вакууме, остаток очищали градиентной колоночной хроматографией на силикагеле при элюировании смесью хлороформ:метанол (100:0→95:5), с получением продукта в виде белого твердого вещества; выход: 0,40 г (57%). Т.пл. 170-173°С.
1H-ЯМР (d6-ДМСО, 250 МГц): δ 1,49 (6H, д, J=7,5, -CH(CH 3)2), 4,63 (3H, м, -CH(CH3)2+-HNCH 2-Pyr), 7,30, 8,47 (4H, 2×м, Pyr-H), 8,28 (с, 1H, -N=CH-N-), 8,97 (1H, с, ушир., -HNCH2-Pyr); m/z: 287 [M+H].
2R,3S-3-(9-изопропил-6(пиридин-4-илметиламино)-9Н-пурин-2-иламино)пентан-2-ол [9]
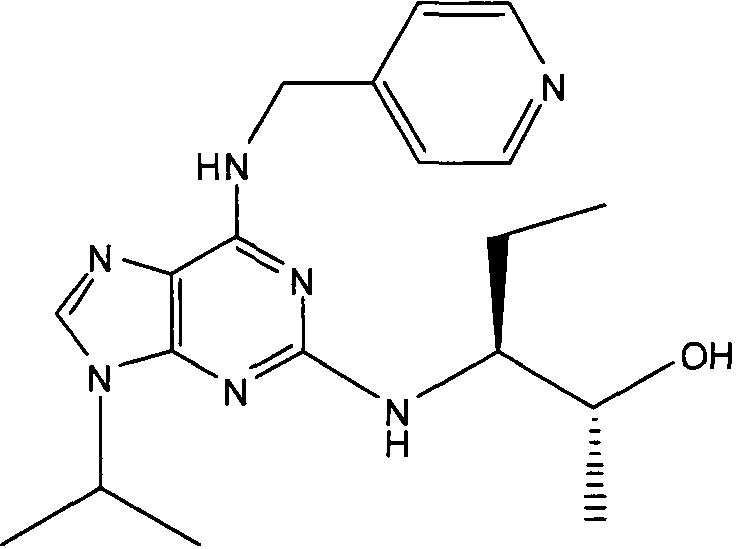
К перемешиваемому раствору (2-фтор-9-изопропил-9Н-пурин-6-ил)пиридин-4-илметиламина (300 мг, 1,05 ммоль) в н-BuOH/ДМСО (5 мл, 4:1) при комнатной температуре в атмосфере аргона добавляли DIEA (2 мл, 10 экв., 10,5 ммоль), с последующим добавлением (2R,3S)-3-аминопентан-2-ола (600 мг, 5,5 экв., 5,8 ммоль). Колбу снабжали обратным холодильником, реакционную смесь помещали на предварительно нагретую масляную баню при 140°С и перемешивали при этой температуре в течение 72 часов. Реакционной смеси давали охладиться до комнатной температуры и растворитель выпаривали в вакууме. Остаток распределяли между EtOAc (50 мл) и водой (50 мл), водную фазу дополнительно экстрагировали EtOAc (2×25 мл) и объединенную органическую фазу промывали насыщенным раствором соли (50 мл), сушили (MgSO4) и выпаривали в вакууме. Остаток очищали градиентной колоночной флэш-хроматографией на силикагеле при элюировании CHCl3:MeOH (100:0→95:5), с получением продукта в виде бесцветной пленки. Выход (225 мг, 58%);
δH (CDCl3, 500 МГц) 0,92 (3H, т, J=7,5, CHCH2CH 3), 1,05 (3H, д, J=7,5, CHCH 3OH), 1,4 (6H, д, J=7,5, CH[CH3]2), 1,47-1,36 (2H, м, CHCH 2CH3), 3,88-3,85 (2H, м, CHEt и NH), 4,53-4,5 (1H, м, CHCH3OH), 4,72 (2H, с, ушир., NHCH 2Ar), 6,5 (1H, с, ушир., NH), 7,2 (2H, с, ушир., ArH), 7,45-7,42 (1H, м, ArH), 8,46-8,43 (2H, м, ArH); δC (CDCl3, 500 МГц) 154,66 (C), 149,86 (2×CH), 149,69 (C), 148,26 (C), 134,84 (CH), 122,22 (2×CH), 71,51 (CH), 59,57 (CH), 46,58 (CH), 43,45 (CH2), 24,9 (CH2), 22,59 (2×CH3), 17,27 (CH3), 11,53 (CH3); m/z 370,2 (M+H).
Пример 10
2,6-Диметилизоникотинонитрил
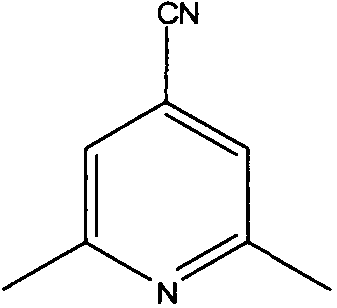
К перемешиваемому количеству 2,6-лутидин-1-оксида (12,3 г, 100 ммоль) медленно добавляли диметилсульфат (12,6 г, 100 ммоль) с такой скоростью, чтобы температура реакционной смеси сохранялась на уровне 80°С во время добавления. Когда добавление завершилось (примерно один час), раствор перемешивали при этой же температуре в течение еще 2 часов. Соль кристаллизовалась при охлаждении, и ее перекристаллизовывали из безводного ацетона, что давало белые призмы; т.пл. 96-97°С. Выход 18 г (73%). К раствору полученного 1-метокси-2,6-диметилпиридинийметилсульфата (11,65 г, 50 ммоль) в воде (50 мл) в атмосфере азота добавляли раствор цианида калия (10 г, 150 ммоль), растворенного в 50 мл воды. Раствор оставляли стоять при комнатной температуре в течение 2 дней, в это время нитрил, который выделялся из раствора в виде длинных белых игл, удаляли фильтрованием, получая 2,8 г чистого продукта (42%).
δH (CDCl3, 500 МГц) 2,44 (6H, с, 2×CH3), 4,2 (2H, д, J=5, NHCH 2), 6,78 (2H, с, ArH); δC (CDCl3, 500 МГц) 157,83 (2×C), 122,71 (C), 118,2 (2×CH), 24,28 (2×CH3).
трет-Бутил(2,6-диметилпиридин-4-ил)метилкарбамат
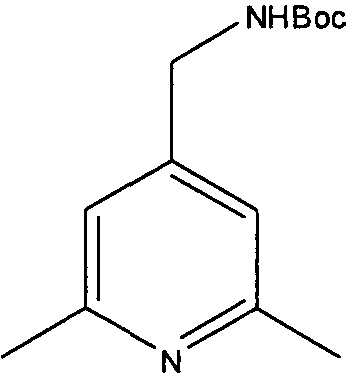
2,6-диметилизоникотинонитрил (2 г, 15,15 ммоль) растворяли в смеси 10% уксусная кислота/этанол (30 мл). Добавляли катализатор 10% палладий на углероде (0,5 г) и реакционную смесь перемешивали в атмосфере водорода в течение 24 часов при 60°С. Смесь фильтровали через рыхлый слой целлита. Летучие вещества удаляли и неочищенный остаток растворяли в дихлорметане (30 мл). К перемешиваемому раствору затем добавляли триэтиламин (5 мл), с последующим добавлением ди-трет-бутилдикарбоната (3,3 г, 15,15 ммоль). Через 3 часа растворитель удаляли и остаток растворяли в этилацетате. Полученный раствор промывали водой (50 мл), насыщенным раствором бикарбоната (50 мл), сушили и выпаривали. Неочищенный продукт очищали колоночной флэш-хроматографией на силикагеле (этилацетат:гексан 1:2), с получением 0,6 г чистого указанного в заголовке соединения (17,24% выход).
δH (CDCl3, 500 МГц) 1,42 (9H, с, 3×CH3), 2,44 (6H, с, 2×CH3), 4,2 (2H, д, J=5, NHCH 2), 5,25 (1H, с, ушир., NH), 6,81 (2H, с, ArH); δC (CDCl3, 500 МГц) 157,83 (C), 156,01 (CO), 148,71 (C), 118,58 (2×CH), 79,77 (C), 43,41 (CH2), 28,34 (3×CH3), 24,28 (2×CH3); m/z 237,2 (M+H).
N-((2,6-Диметилпиридин-4-ил)метил)-2-фтор-9Н-пурин-6-амин
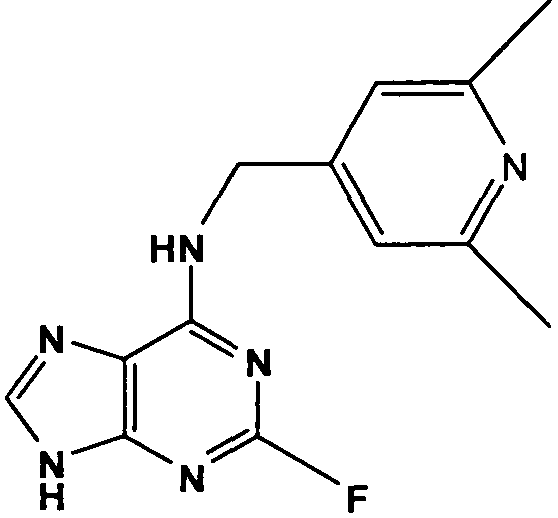
К перемешиваемому раствору 6-хлор-2-фторпурина (0,4 г, 2,9 ммоль) в н-BuOH (50 мл) в атмосфере аргона при 0°С добавляли DIEA (2,5 мл, 14,7 ммоль), с последующим добавлением (2,6-диметилпиридин-4-ил)метанамина (0,54 г, 4 ммоль). Реакционную смесь перемешивали при этой температуре в течение 1 часа, затем давали вернуться к комнатной температуре и перемешивали в течение 4 часов, при этом наблюдалось, что реакция еще не завершена, и поэтому реакционную смесь нагревали до 100°С и оставляли при этой температуре на 8 часов. Растворитель выпаривали в вакууме и остаток очищали градиентной колоночной флэш-хроматографией на силикагеле при элюировании CHCl3:MeOH (100:0→90:10), с получением продукта в виде белого твердого вещества; выход: 0,5 г (63%).
δH (CDCl3, 250 МГц) 2,48 (6H, с, 2×CH3), 3,65-3,54 (2H, м, NHCH 2), 4,61 (1H, с, ушир., NH), 7,32 (2H, с, ArH), 7,84 (1H, с, ArH),), 9,05 (1H, с, ушир., NH); δC (CDCl3, 250 МГц) 159,2(C), 156,62 (C), 156,61 (C), 155,46 (C), 146,92 (CH), 136,3 (C), 122,05 (2×CH), 119,48 (C), 41,6 (CH2), 18,06 (2×CH3).
N-((2,6-Диметилпиридин-4-ил)метил)-2-фтор-9-изопропил-9Н-пурин-6-амин
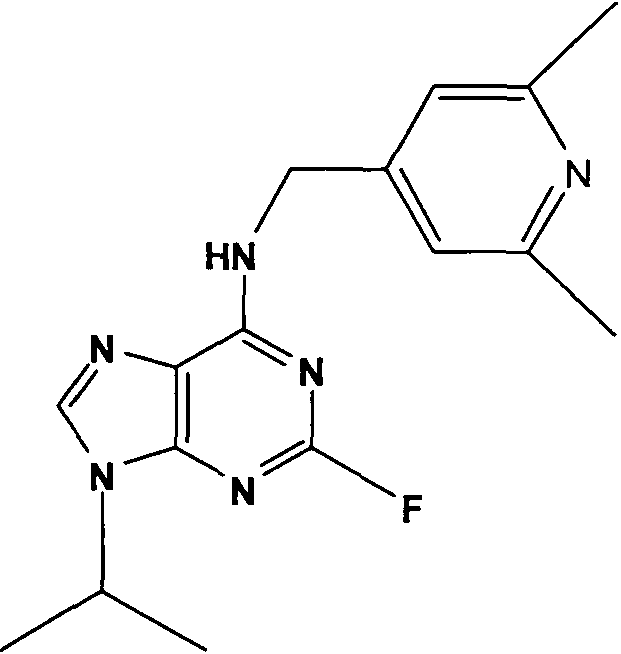
К перемешиваемому раствору 2-фтор-N-(2,6-диметилпиридин-4-ил)метил-9Н-пурин-6-амина (0,4 г, 1,48 ммоль) в диметилформамиде (10 мл) при комнатной температуре в атмосфере аргона добавляли порошковый безводный K2CO3 (1 г, 5 экв., 7,4 ммоль), с последующим добавлением 2-бромпропана (1,2 мл, 12,2 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 24 часов, пока ТСХ со смесью DCM:эфир:МеОН (55:40:5) не показала, что реакция пришла к завершению. Растворитель выпаривали в вакууме и остаток распределяли между EtOAc (50 мл) и водой (50 мл). Водную фазу дополнительно экстрагировали EtOAc (2×50 мл) и объединенную органическую фазу промывали насыщенным раствором соли (50 мл), сушили (MgSO4) и выпаривали в вакууме. Остаток очищали колоночной хроматографией на силикагеле при элюировании CHCl3:MeOH (98:2), с получением указанного в заголовке соединения в виде бесцветной пленки (250 мг, 54%).
δH (CDCl3, 500 МГц) 1,46 (6H, д, J=7,5, CH[CH3]2), 2,38 (3H, с, ArCH 3), 2,39 (3H, с, ArCH 3), 4,73-4,6 (3H, м, NHCH 2 и CHMe2), 6,85 (2H, с, ArH), 7,35 (1H, с, ушир., NH), 7,5 (1H, с, ушир., ArH); δc (CDCl3, 500 МГц) 157,8 (C), 158,6 (C), 154,32 (C), 151 (C), 148,42 (CH), 122,2 (2×CH), 121,08 (C), 47,52 (CH), 43,6 (CH2), 23,68 (2×CH3), 22,3 (2×CH3); m/z 315,2 (M+H).
2R,3S-3-(9-изопропил-6-(2,6-диметилпиридин-4-илметиламино)-9Н-пурин-2-иламино)пентан-2-ол [10]
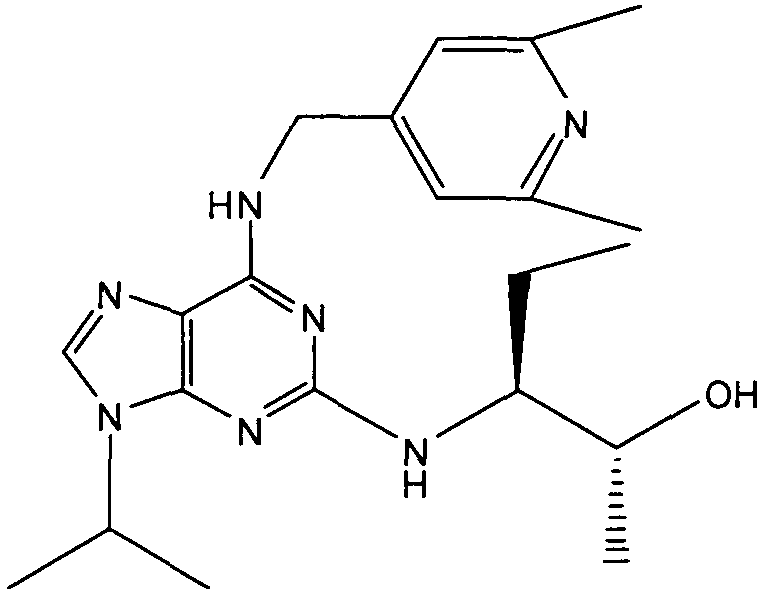
К перемешиваемому раствору 2-фтор-9-изопропил-N-(2,6-диметилпиридин-4-илметил)-9Н-пурин-6-амина (250 мг, 0,8 ммоль) в н-BuOH/ДМСО (5 мл, 4:1) при комнатной температуре в атмосфере аргона добавляли DIEA (1,4 мл, 10 экв., 8 ммоль), с последующим добавлением (2R,3S)-3-аминопентан-2-ола (0,35 г, 3,4 ммоль). Колбу снабжали обратным холодильником, реакционную смесь помещали на предварительно нагретую масляную баню при 140°С и перемешивали при этой температуре в течение 72 часов. Реакционной смеси давали остыть до комнатной температуры и растворитель выпаривали в вакууме. Остаток распределяли между EtOAc (50 мл) и водой (50 мл), водную фазу дополнительно экстрагировали EtOAc (2×25 мл) и объединенную органическую фазу промывали насыщенным раствором соли (50 мл), сушили (MgSO4) и выпаривали в вакууме. Остаток очищали градиентной колоночной флэш-хроматографией на силикагеле при элюировании CHCl3:MeOH (100:0→95:5), с получением указанного в заголовке соединения в виде бесцветного масла (56 мг, 18%).
δH (CDCl3, 500 МГц) 0,95 (3H, т, J=7,5, CHCH2CH 3), 1,05 (3H, д, J=5, CHCH 3OH), 1,43 (6H, д, J=10, CH[CH 3]2), 1,55-1,4 (2H, м, CHCH 2CH3), 2,43 (3H, с, ArCH3), 2,44 (3H, с, ArCH3), 3,9-3,8 (2H, м, CHEt и CHMe2), 4,57-4,46 (1H, м, CHCH3OH), 4,63-4,58 (2H, м, NHCH 2Ar), 6,28 (1H, с, ушир., NH), 6,8 (2H, с, ушир., ArH), 7,45 (1H, с, ArH); δC (CDCl3, 250 МГц) 160,08 (C), 157,76 (C), 155,36 (C), 148,97 (C), 135,36 (C), 134,75 (CH), 118,95 (2×CH), 71,52 (CH), 59,59 (CH), 46,65 (CH), 44,92 (CH2), 24,91 (CH2), 24,01 (2×CH3), 22,5 (2×CH3), 17,27 (CH3), 11,51 (CH3); m/z 398,3 (M+H).
Пример 11
2-Фтор-N-((6-трифторметил)пиридин-3-ил)метил)-9Н-пурин-6-амин
К перемешиваемому раствору 6-хлор-2-фторпурина (344 г, 2 ммоль) и [6-(трифторметил)пиридин-3-ил]метанамина (0,4 г, 2,27 ммоль) в н-BuOH (50 мл) в атмосфере аргона при 0°С добавляли DIEA (2,5 мл, 14,7 ммоль). Реакционную смесь перемешивали при этой температуре в течение 1 часа, затем давали вернуться к комнатной температуре и перемешивали в течение 4 часов, при этом наблюдалось, что реакция еще не завершена, и поэтому реакционную смесь нагревали до 100°С и оставляли при этой температуре на 5 часов. Растворитель выпаривали в вакууме и остаток очищали градиентной колоночной хроматографией на силикагеле при элюировании CHCl3:MeOH (100:0→90:10), с получением продукта в виде белого твердого вещества; выход: 0,46 г (75%).
δH (CDCl3, 500 МГц) 3,62-3,57 (2H, м, NHCH 2), 4,69 (1H, с, ушир., NH), 7,2-7,1 (2H, с, ушир., ArH и NH), 7,69 (1H, д, J=7,5, ArH), 8,27 (1H, с, ArH), 8,71 (1H, с, ArH); δC (CDCl3, 500 МГц) 158,69 (C), 158,18 (C), 157,67 (C), 154,56 (C), 147,48(CH), 146,99 (CH), 144,32 (C), 133,87 (CH), 121,67 (CH), 118,88 (C), 43,48 (CH2); m/z 313,1 (M+H).
2-Фтор-9-изопропил-N-((6-(трифторметил)пиридин-3-ил)метил)-9Н-пурин-6-амин
К перемешиваемому раствору 2-фтор-N-[(6-(трифторметил)пиридин-3-ил)метил-9Н-пурин-6-амина (0,4 г, 1,27 ммоль) в диметилформамиде (10 мл) при комнатной температуре в атмосфере аргона добавляли порошковый безводный K2CO3 (0,86 г, 5 экв., 6,54 ммоль), с последующим добавлением 2-бромпропана (1 мл, 10,3 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 24 часов, пока ТСХ со смесью DCM:эфир:МеОН (55:40:5) не показала, что реакция пришла к завершению. Растворитель выпаривали в вакууме и остаток распределяли между EtOAc (100 мл) и водой (100 мл). Водную фазу дополнительно экстрагировали EtOAc (2×50 мл) и объединенную органическую фазу промывали насыщенным раствором соли (50 мл), сушили (MgSO4) и выпаривали в вакууме. Остаток очищали колоночной хроматографией на силикагеле при элюировании CHCl3:MeOH (98:2), с получением указанного в заголовке соединения в виде бесцветной пленки (350 мг, 77%).
δH (CDCl3, 500 МГц) 1,5 (6H, д, J=7,5, CH[CH3]2), 4,7-4,6 (3H, м, NHCH 2 и CHMe2), 7,16 (1H, д, J=2,5, ArH), 7,25 (1H, с, ушир., NH), 7,66 (2H, с, ушир., ArH), 8,67 (1H, с, ушир., ArH); δC (CDCl3, 500 МГц) 158,8 (C), 156,72 (C), 156,3 (C), 147,42 (CH), 137,92 (CH), 138,08 (CH), 131,73 (C), 123,31 (CH), 119,25 (C), 48,38 (CH), 44,1 (CH2), 22,72 (2×CH3); m/z 355,1 (M+H).
2R,3S-3-(9-Изопропил-6-((6-трифторметил)пиридин-3-ил)метиламино)-9Н-пурин-2-иламино)пентан-2-ол [11]
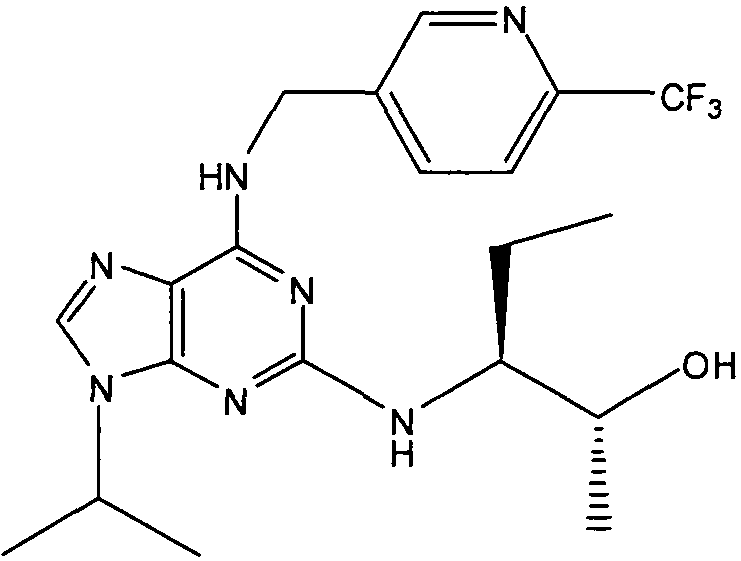
К перемешиваемому раствору 2-фтор-9-изопропил-N-(6-(трифторметилпиридин-3-илметил)-9Н-пурин-6-амина (200 мг, 0,56 ммоль) в н-BuOH/ДМСО (5 мл, 4:1) при комнатной температуре в атмосфере аргона добавляли DIEA (1,4 мл, 10 экв., 8 ммоль), с последующим добавлением (2R,3S)-3-аминопентан-2-ола (0,25 г, 2,4 ммоль). Колбу снабжали обратным холодильником, реакционную смесь помещали на предварительно нагретую масляную баню при 140°С и перемешивали при этой температуре в течение 72 часов. Реакционной смеси давали остыть до комнатной температуры и растворитель выпаривали в вакууме. Остаток распределяли между EtOAc (50 мл) и водой (50 мл), водную фазу дополнительно экстрагировали EtOAc (2×25 мл) и объединенную органическую фазу промывали насыщенным раствором соли (50 мл), сушили (MgSO4) и выпаривали в вакууме. Остаток очищали градиентной колоночной флэш-хроматографией на силикагеле при элюировании CHCl3:MeOH (100:0→95:5), с получением указанного в заголовке соединения в виде бесцветного масла (30 мг, 12%).
δH (CDCl3, 500 МГц) 0,85 (3H, т, J=7,5, CHCH2CH 3), 1,12 (3H, д, J=5, CHCH 3OH), 1,52 (6H, д, J=7,5, CH[CH 3]2), 1,61-1,41 (2H, м, CHCH 2CH3), 3,82 (1H, м, CHMe2), 4,75-4,57 (1H, м, CHCH3OH), 4,82-4,64 (2H, м, NHCH 2Ar), 5,8 (1H, с, ушир., NH), 6,85 (1H, с, ушир., NH), 7 (1H, д, J=10, ArH), 7,31-7,23 (2H, м, ArH), 8,45 (1H, с, ушир., ArH); δC (CDCl3, 250 МГц) 158,61 (C), 157,35 (C), 156,97 (C), 147,62 (CH), 137,43 (CH), 129,22 (C), 128,72 (C), 128,92 (CH), 123,14 (CH), 118,32 (C), 69,64 (CH), 60,91 (CH2), 57,68 (CH), 23,55 (CH2), 22,53 (2×CH3), 12,12 (CH3); m/z 438,3 (M+H).
Пример 12
2-Фтор-N-((6-метилпиридин-2-ил)метил)-9Н-пурин-6-амин
К перемешиваемому раствору 6-хлор-2-фторпурина (0,4 г, 2,3 ммоль) в н-BuOH (50 мл) в атмосфере аргона при 0°С добавляли DIEA (2,5 мл, 14,7 ммоль), с последующим добавлением (6-метилпиридин-2-ил)метанамина (0,36 г, 2,95 ммоль). Реакционную смесь перемешивали при этой температуре в течение 1 часа, затем давали ей вернуться к комнатной температуре и перемешивали в течение 4 часов, при этом наблюдалось, что реакция еще не завершена, и поэтому реакционную смесь нагревали до 100°С и оставляли при этой температуре на 8 часов. Растворитель выпаривали в вакууме и остаток очищали градиентной колоночной флэш-хроматографией на силикагеле при элюировании CHCl3:MeOH (100:0→90:10), с получением продукта в виде белого твердого вещества; выход: 0,35 г (60%).
δH (CDCl3, 500 МГц) 2,44 (3H, с, CH3), 4,83-4,54 (3H, м, NH и NHCH2), 7,1-7,05 (2H, м, ArH), 7,61-7,55 (1H, м, ArH), 8,13-8,09 (1H, м, ArH), 8,61(1H, с, ушир., NH); δC (CDCl3, 500 МГц) 159,41 (C), 158,78 (C), 158,53 (C), 157,19 (C), 155,38 (C), 147,69 (CH), 137,03 (C), 136,93 (CH), 121,34 (CH), 117,76 (CH), 117,31 (C), 46,47 (CH2), 23,9 (CH3); m/z 259,2 (M+H).
2-Фтор-9-изопропил-N-((6-метилпиридин-2-ил)метил)-9Н-пурин-6-амин
К перемешиваемому раствору 2-фтор-N-[(6-метилпиридин-2- ил)метил-9Н-пурин-6-амина (0,3 г, 1,17 ммоль) в диметилформамиде (10 мл) при комнатной температуре в атмосфере аргона добавляли порошковый безводный K2CO3 (0,8 г, 5 экв., 5,85 ммоль), с последующим добавлением 2-бромпропана (1,15 мл, 11,7 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 24 часов, пока ТСХ со смесью DCM:эфир:МеОН (55:40:5) не показала, что реакция пришла к завершению. Растворитель выпаривали в вакууме и остаток распределяли между EtOAc (100 мл) и водой (100 мл). Водную фазу дополнительно экстрагировали EtOAc (2×50 мл) и объединенную органическую фазу промывали насыщенным раствором соли (50 мл), сушили (MgSO4) и выпаривали в вакууме. Остаток очищали колоночной хроматографией на силикагеле при элюировании CHCl3:MeOH (98:2), с получением указанного в заголовке соединения в виде слегка желтой пленки (180 мг, 51%).
δH (CDCl3, 500 МГц) 1,6 (6H, д, J=7,5, CH[CH3]2), 2,54 (3H, с, CH3), 4,86-4,73 (3H, м, NHCH 2 и CHMe2), 7,06 (1H, д, J=10, ArH), 7,14 (1H, д, J=10, ArH), 7,53 (1H, т, J=10, ArH), 7,8 (1H, с, ArH); δC (CDCl3, 500 МГц) 158,42 (C), 158,04 (C), 156,16 (C), 156 (C), 155,49 (C), 150,12 (C), 137,67 (CH), 136,93 (CH), 121,89 (CH), 118,1 (C), 118,53 (CH), 47,15 (CH), 45,52 (CH2), 24,38 (CH3), 22,58 (2×CH3); m/z 301,2 (M+H); 19F ЯМР δ -50,22.
2R,3S-3-(9-Изопропил-6-((6-метилпиридин-2-ил)метиламино)-9Н-пурин-2-иламино)пентан-2-ол [12]
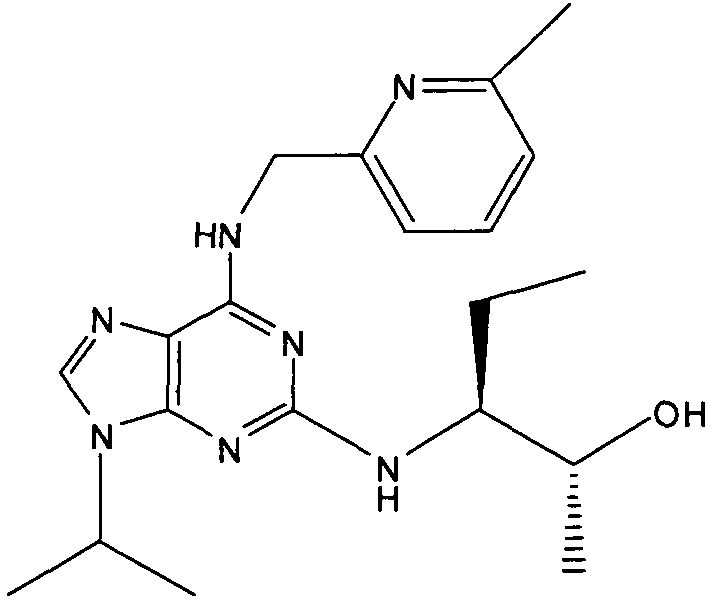
К перемешиваемому раствору 2-фтор-9-изопропил-N-(6-метилпиридин-2-илметил)-9Н-пурин-6-амина (180 мг, 0,59 ммоль) в н-BuOH/ДМСО (5 мл, 4:1) при комнатной температуре в атмосфере аргона добавляли DIEA (1 мл, 10 экв., 5,6 ммоль), с последующим добавлением (2R,3S)-3-аминопентан-2-ола (0,34 г, 6 ммоль). Колбу снабжали обратным холодильником, реакционную смесь помещали на предварительно нагретую масляную баню при 140°С и перемешивали при этой температуре в течение 72 часов. Реакционной смеси давали остыть до комнатной температуры и растворитель выпаривали в вакууме. Остаток распределяли между EtOAc (50 мл) и водой (50 мл), водную фазу дополнительно экстрагировали EtOAc (2×25 мл) и объединенную органическую фазу промывали насыщенным раствором соли (50 мл), сушили (MgSO4) и выпаривали в вакууме. Остаток очищали градиентной колоночной флэш-хроматографией на силикагеле при элюировании CHCl3:MeOH (100:0→95:5), с получением указанного в заголовке соединения в виде слегка желтого масла (28 мг, 13%).
δH (CDCl3, 500 МГц) 1,05 (3H, т, J=7,5, CHCH2CH 3), 1,13 (3H, д, J=5, CHCH 3OH), 1,43 (6H, д, J=7,5, CH[CH 3]2), 1,6-1,4 (2H, м, CHCH2CH3), 1,75 (1H, с, ушир., NH), 2,32 (3H, с, ArCH3), 3,95 (1H, с, ушир., CHMe2), 4,57-4,53 (1H, м, CHCH3OH), 4,8-4,6 (2H, м, NHCH 2Ar), 5,8 (1H, с, ушир., NH), 6,82 (1H, с, ушир., NH), 6,95 (1H, д, J=5, ArH), 7,05 (1H, д, J=5, ArH), 7,45-7,4 (2H, м, ArH); δC (CDCl3, 500 МГц) 160,17 (C), 157,9 (C), 156,72 (C), 154,8 (C), 150,12 (C), 136,8 (CH), 134,55 (CH), 121,64 (CH), 118,58 (CH), 114,87(C), 71,59 (CH), 59,66 (CH), 46,41 (CH), 44,79 (CH2), 24,12 (CH2), 23,41 (CH3), 21,59 (CH3), 16,18 (CH3), 10,6 (CH3); m/z 384,3 (M+H).
Пример 13
2-Фтор-N-((3-метилпиридин-2-ил)метил)-9Н-пурин-6-амин
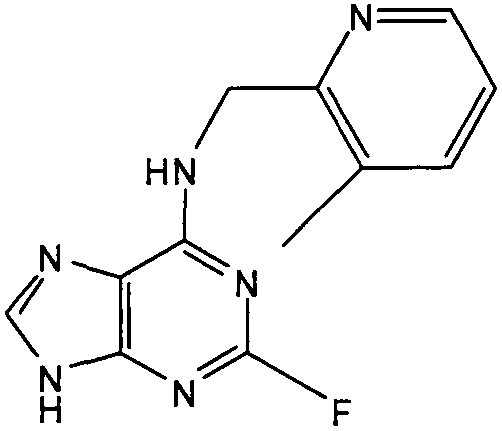
К перемешиваемому раствору 6-хлор-2-фторпурина (0,4 г, 2,3 ммоль) в н-BuOH (50 мл) в атмосфере аргона при 0°С добавляли DIEA (2,5 мл, 14,7 ммоль), с последующим добавлением (3-метилпиридин-2-ил)метанамина (0,36 г, 2,95 ммоль). Реакционную смесь перемешивали при этой температуре в течение 1 часа, затем давали вернуться к комнатной температуре и перемешивали в течение 4 часов, при этом наблюдалось, что реакция еще не завершена, и поэтому реакционную смесь нагревали до 100°С и оставляли при этой температуре на 8 часов. Растворитель выпаривали в вакууме и остаток очищали градиентной колоночной хроматографией на силикагеле при элюировании CHCl3:MeOH (100:0→90:10), с получением продукта в виде белого твердого вещества; выход: 0,3 г (51%).
δH (CDCl3, 500 МГц) 2,35 (3H, с, CH3), 4,71 (3H, м, NH и NHCH2), 7,24 (1H, с, ушир., ArH), 7,63 (1H, с, ушир., ArH), 8,28 (1H, с, ушир., ArH), 8,38 (1H, с, ушир., ArH); δC (CDCl3, 500 МГц) 161,03 (C), 158,49 (C), 154,77 (C), 152,16 (C), 144,86(CH), 137,53 (CH), 137,14 (CH), 130,67 (C), 121,58 (CH), 118,73 (C), 42,87 (CH2), 18,16 (CH3); m/z 259,3 (M+H).
2-Фтор-9-изопропил-N-((3-метилпиридин-2-ил)метил)-9Н-пурин-6-амин
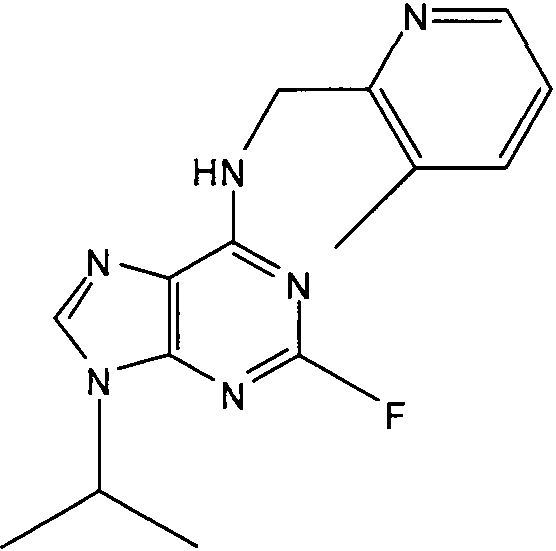
К перемешиваемому раствору 2-фтор-N-(3-метилпиридин-2-ил)метил-9Н-пурин-6-амина (0,3 г, 1,17 ммоль) в диметилформамиде (10 мл) при комнатной температуре в атмосфере аргона добавляли порошковый безводный K2CO3 (0,8 г, 5 экв., 5,85 ммоль), с последующим добавлением 2-бромпропана (1,15 мл, 11,7 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 24 часов, пока ТСХ со смесью DCM:эфир:МеОН (55:40:5) не показала, что реакция пришла к завершению. Растворитель выпаривали в вакууме и остаток распределяли между EtOAc (50 мл) и водой (50 мл). Водную фазу дополнительно экстрагировали EtOAc (2×50 мл) и объединенную органическую фазу промывали насыщенным раствором соли (50 мл), сушили (MgSO4) и выпаривали в вакууме. Остаток очищали колоночной хроматографией на силикагеле при элюировании CHCl3:MeOH (98:2), с получением указанного в заголовке соединения в виде слегка желтой пленки (170 мг, 48%).
δH (CDCl3, 500 МГц) 1,51 (6H, д, J=7,5, CH[CH3]2), 2,28 (3H, с, CH3), 4,68-4,65 (3H, м, NHCH 2 и CHMe2), 7,08-7,05 (1H, м, ArH), 7,4 (1H, д, J=5, ArH), 7,72 (1H, с, ArH), 7,93 (1H, с, ушир., NH), 8,34 (1H, д, J=5, ArH); δC (CDCl3, 500 МГц) 160,09 (C), 158,43 (C), 155,97 (C), 154,06 (C), 145,85 (CH), 137,73 (CH), 137,54 (CH), 130,67 (C), 122,28 (CH), 118,73 (C), 47,28 (CH), 42,87 (CH2), 22,43 (2×CH3), 17,66 (CH3); 19F ЯМР δ -50,20; m/z 301,2 (M+H).
2R,3S-3-(9-Изопропил-6-((3-метилпиридин-2-ил)метиламино)-9Н-пурин-2-иламино)пентан-2-ол [13]
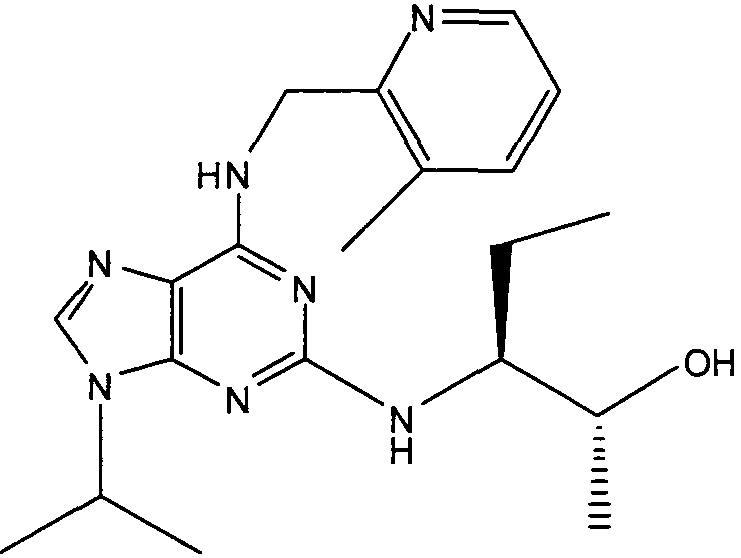
К перемешиваемому раствору 2-фтор-9-изопропил-N-(3-метилпиридин-2-илметил)-9Н-пурин-6-амина (170 мг, 0,56 ммоль) в н-BuOH/ДМСО (5 мл, 4:1) при комнатной температуре в атмосфере аргона добавляли DIEA (1 мл, 10 экв., 5,6 ммоль), с последующим добавлением (2R,3S)-3-аминопентан-2-ола (0,34 г, 6 ммоль). Колбу снабжали обратным холодильником, реакционную смесь помещали на предварительно нагретую масляную баню при 140°С и перемешивали при этой температуре в течение 72 часов. Реакционной смеси давали остыть до комнатной температуры и растворитель выпаривали в вакууме. Остаток распределяли между EtOAc (50 мл) и водой (50 мл), водную фазу дополнительно экстрагировали EtOAc (2×25 мл) и объединенную органическую фазу промывали насыщенным раствором соли (50 мл), сушили (MgSO4) и выпаривали в вакууме. Остаток очищали градиентной колоночной флэш-хроматографией на силикагеле при элюировании CHCl3:MeOH (100:0→95:5), с получением указанного в заголовке соединения в виде слегка желтого масла (28 мг, 13%).
δH (CDCl3, 500 МГц) 1 (3H, т, J=7,5, CHCH2CH 3), 1,1 (3H, д, J=5, CHCH 3OH), 1,44 (6H, д, J=7,5, CH[CH 3]2), 1,6-1,4 (2H, м, CHCH2CH3), 1,75 (1H, с, ушир., NH), 2,3 (3H, с, ArCH3), 3,92 (1H, с, ушир., CHMe2), 4,56-4,52 (1H, м, CHCH3OH), 4,7 (2H, с, ушир., NHCH2Ar), 6,1 (1H, с, ушир., NH), 7,06 (1H, дд, J=2,5, 5, ArH), 7,41 (1H, дд, J=2,5, 5, ArH), 7,47 (1H, с, ArH), 8,37 (1H, д, J=5, ArH); δC (CDCl3, 500 МГц) 160,21 (C), 154,68 (C), 153,65 (C), 146,07 (CH), 137,53 (CH), 134,53 (CH), 130,59 (C), 122,31 (C), 122,13 (CH), 115,14 (C), 71,73 (CH), 59,93 (CH), 46,43 (CH), 43,11 (CH2), 25,33 (CH2), 22,63 (CH3), 17,61 (CH3), 17,25 (CH3), 11,67 (CH3); m/z 384,3 (M+H).
Пример 14
(R)-1-(9-изопропил-6-(пиридин-3-илметиламино)-9Н-пурин-2-иламино)пропан-2-ол [14]
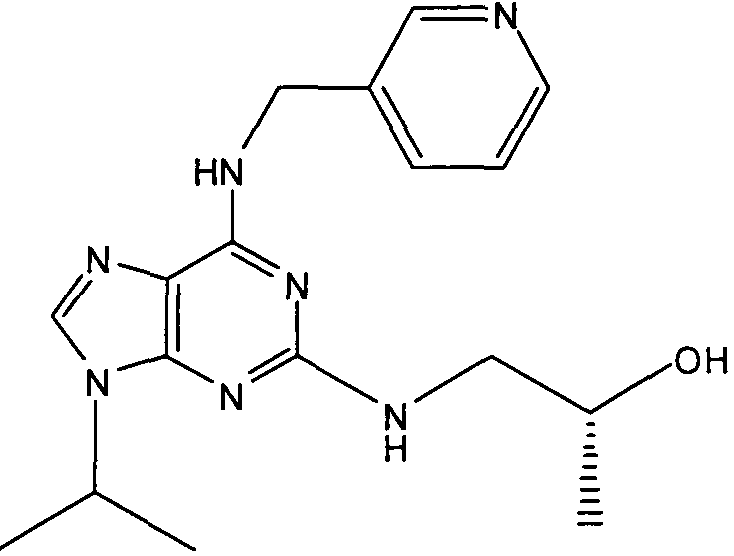
К перемешиваемому раствору 2-фтор-9-изопропил-6-[(пиридин-3-илметил)амино]пурина (300 мг, 1,05 ммоль) в н-BuOH/ДМСО (5 мл, 4:1) при комнатной температуре в атмосфере аргона добавляли DIEA (2 мл, 10 экв., 10,5 ммоль), с последующим добавлением (R)-1-аминопропан-2-ола (0,395 г, 5,25 ммоль). Колбу снабжали обратным холодильником, реакционную смесь помещали на предварительно нагретую масляную баню при 140°С и перемешивали при этой температуре в течение 72 часов. Реакционной смеси давали остыть до комнатной температуры и растворитель выпаривали в вакууме. Остаток распределяли между этилацетатом (50 мл) и водой (50 мл), водную фазу дополнительно экстрагировали этилацетатом (2×25 мл) и объединенную органическую фазу промывали насыщенным раствором соли (50 мл), сушили (MgSO4) и выпаривали в вакууме. Остаток очищали градиентной колоночной флэш-хроматографией на силикагеле при элюировании CHCl3:MeOH (100:0→95:5), с получением чистого продукта в виде бесцветного масла (38 мг, 10,6%).
δH (250 МГц, CDCl3) 1,15 (3H, д, J=7,5, CHCH 3OH), 1,45 (6H, д, J=7,5, CH[CH 3]2), 3,3-3,2 (1H, м, NHCHHCHMeOH)), 3,45-3,36 (1H, м, NHCHHCHMeOH), 3,96-3,9 (1H, м, CHMe2), 4,58-4,47 (1H, м, CHMeOH), 4,69 (2H, д, J=5, NHCH 2Ar), 5,23 (1H, т, J=5, NHCH2CHMeOH), 6,32 (1H, с, ушир., NHCH2Ar), 7,16-7,11 (1H, м, ArH), 7,4 (1H, с, ArH), 7,6 (1H, д, J=7,5, ArH), 8,4 (1H, д, J=2,5, ArH), 8,55 (1H, с, ArH); δС (250 МГц, CDCl3 160,07 (C), 154,95 (C), 154,7 (C), 151,1 (C), 149,27 (CH), 148,54 (CH), 135,3 (CH), 134,73 (CH), 123,39 (CH), 114,61 (C), 68,83 (CH), 50,09 (CH2), 46,49 (CH), 41,95 (CH2), 22,49 (2×CH3), 20,85 (CH3); m/z 342,2 (M+H).
Пример 15
1,1,1-Трифтор-3-(9-изопропил-6-(пиридин-3-илметиламино)-9Н-пурин-2-иламино)пропан-2-ол [15]
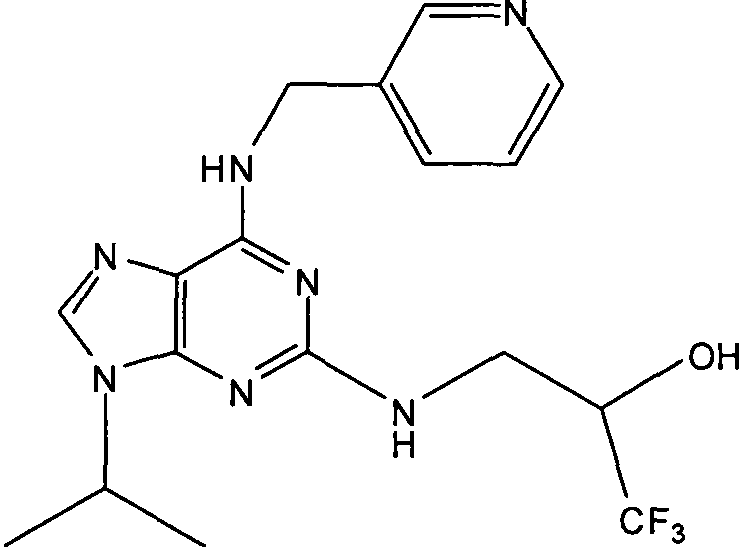
К перемешиваемому раствору (2-фтор-9-изопропил-9Н-пурин-6-ил)пиридин-3-илметиламина (300 мг, 1,05 ммоль) в н-BuOH/ДМСО (5 мл, 4:1) при комнатной температуре в атмосфере аргона добавляли DIEA (1,4 мл, 10 экв., 8 ммоль), с последующим добавлением 3-амино-1,1,1-трифторпропан-2-ола (1 г, 7,8 ммоль) [полученный по реакции аммиака с 2-(трифторметил)оксирана]. Колбу снабжали обратным холодильником, реакционную смесь помещали на предварительно нагретую масляную баню при 140°С и перемешивали при этой температуре в течение 72 часов. Реакционной смеси давали остыть до комнатной температуры и растворитель выпаривали в вакууме. Остаток распределяли между EtOAc (50 мл) и водой (50 мл), водную фазу дополнительно экстрагировали EtOAc (2×25 мл) и объединенную органическую фазу промывали насыщенным раствором соли (50 мл), сушили (MgSO4) и выпаривали в вакууме. Остаток очищали градиентной колоночной флэш-хроматографией на силикагеле при элюировании CHCl3:MeOH (100:0→95:5), с получением указанного в заголовке соединения в виде бесцветного масла (62 мг, 15%).
δH (CDCl3, 500 МГц) 1,53 (6H, д, J=7,5, CH[CH 3]2), 3,64-3,6 (1H, м, NHCHHCOH[CF3][CH3]), 3,76-3,73 (1H, м, NHCHHCOH[CF3][CH3]), 4,18-4,16 (1H, м, CHMe2), 4,62-4,56 (1H, м, CHOHCF3), 4,72 (3H, с, ушир., NHCH 2Ar и NHCH2Ar), 7,22-7,2 (1H, м, ArH), 7,52 (1H, с, ArH), 7,66 (1H, д, J=10, ArH), 8,47 (1H, д, J=5, ArH), 8,58 (1H, с, ArH); δC (CDCl3, 500 МГц) 159,77 (C), 154,68 (C), 149,04 (CH), 148,45 (CH), 135,44 (CH), 135,03 (CH), 134,44 (C), 125,91 (C), 123,49 (CH), 114,78 (C), 71,57 (CH), 46,76 (CH), 42,94 (CH2), 32,2 (CH2), 22,47 (2×CH3); 19FЯМР δ -78,48; m/z 396,2 (M+H).
Пример 16
1,1,1-Трифтор-3-нитропентан-2-ол
1-Нитропропан (3,25 г, 36,6 ммоль), трифторацетальдегидэтилгемиацеталь (5,85 г, 36,6 ммоль, 90% чистоты) и порошковый К2СО3 (0,34 г, 2,5 ммоль) смешивали и перемешивали при 60°С в течение 3 часов, затем при комнатной температуре в течение 3 дней. Добавляли насыщенный раствор соли (10 мл) и 1н. раствор HCl (10 мл), и нижний органический слой отделяли. Водный слой экстрагировали эфиром (2×30 мл) и объединенные органические слои промывали насыщенным раствором соли, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией на силикагеле с градиентом элюирования смесью CH2Cl2:гексан (50:50), CH2Cl2:гексан (75:25), CH2Cl2 (100%) и МеОН:CH2Cl2 (5:95), с получением продукта в виде воскового белого твердого вещества (4,4 г, 64%).
δH (CDCl3, 500 МГц) 1,04-1,00 (3H, м, CHCH2CH 3), 2,16-1,99 (2H, м, CHCH 2CH3), 4,37 (1H, с, ушир., OH), 4,71-4,59 (2H, м, 2×CH); δC (CDCl3, 500 МГц) дкв. (126,79, 126,70; 124,55, 124,46; 122,30, 122,21; 120,06, 119,96, CF3), два пика (88,34, 87,60 CH NO2), восемь пиков (71,13, 70,87, 70,81, 70,62, 70,56, 70,36, 70,30, 70,05 CHOH), два пика (23,57 и 21,98 CH2), два пика (9,77 и 9,66 CH3); 71,5 (CH), 54,03 (CH), 25,5 (CH2), 11,05 (CH3).
19F ЯМР δ -76,2 и -77,5.
3-Амино-1,1,1-трифторпентан-2-ол
1,1,1-трифтор-3-нитропентан-2-ол (3 г, 16 ммоль) растворяли в метаноле (40 мл). Добавляли никелевый катализатор Ренея и реакционную смесь энергично перемешивали в атмосфере водорода в течение 24 часов. Катализатор отфильтровывали и фильтрат концентрировали в вакууме, с получением относительно чистого амина (2,35 г, 94%).
δH (CDCl3, 500 МГц) 1,05 (3H, т, J=5, CHCH2CH 3), 1,55-1,45 (1H, м, CHCHHCH3), 1,8-1,7 (1H, м, CHCHHCH3), 2,95-2,9 (1H, м, CHEt), 3,95-3,85 (1H, м, CHOHCF3); δc (CDCl3, 500 МГц) 124,5 (C), 71,5 (CH), 54,03 (CH), 25,5 (CH2), 11,05 (CH3).
1,1,1-Трифтор-3-(9-изопропил-6-(пиридин-3-илметиламино)-9Н-пурин-2-иламино)пентан-2-ол [16]
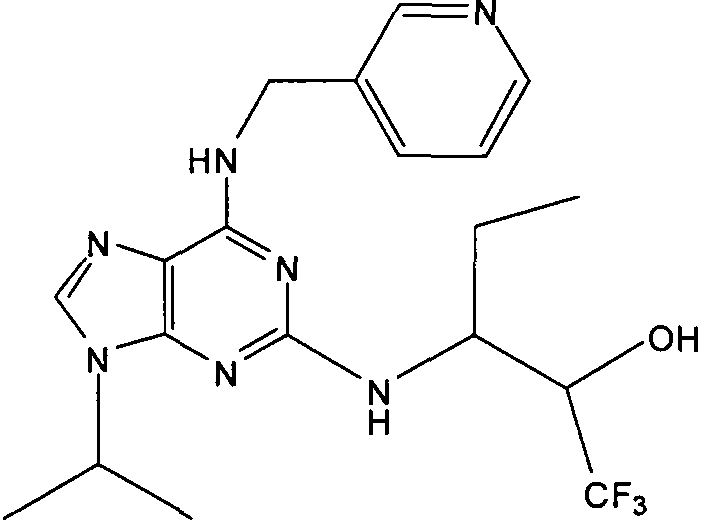
К перемешиваемому раствору (2-фтор-9-изопропил-9Н-пурин-6- ил)пиридин-3-илметиламина (300 мг, 1,05 ммоль) в н-BuOH/ДМСО (5 мл, 4:1) при комнатной температуре в атмосфере аргона добавляли DIEA (1,4 мл, 10 экв., 8 ммоль), с последующим добавлением 3-амино-1,1,1-трифторпентан-2-ола (1 г, 6,4 ммоль) [полученный по реакции аммиака с 2-(трифторметил)оксирана]. Колбу снабжали обратным холодильником, реакционную смесь помещали на предварительно нагретую масляную баню при 140°С и перемешивали при этой температуре в течение 72 часов. Реакционной смеси давали остыть до комнатной температуры и растворитель выпаривали в вакууме. Остаток распределяли между EtOAc (50 мл) и водой (50 мл), водную фазу дополнительно экстрагировали EtOAc (2×25 мл) и объединенную органическую фазу промывали насыщенным раствором соли (50 мл), сушили (MgSO4) и выпаривали в вакууме. Остаток очищали градиентной колоночной флэш-хроматографией на силикагеле при элюировании CHCl3:MeOH (100:0→95:5), с получением указанного в заголовке соединения в виде бесцветной пленки (42 мг, 9,4%).
δH (CDCl3, 500 МГц) 1,04 (3H, т, J=7,5, CHCH2CH 3). 1,56 (6H, д, J=7,5, CH[CH 3]2), 1,8-1,73 (2H, м, CHCH 2CH3), 4,13-4,07 (1H, м, CHMe2), 4,22-4,2 (1H, м, CHEt) 4,57-4,55 (1H, м, CHOHCF3), 4,71 (2H, с, ушир., NHCH 2Ar), 5,08 (1H, с, ушир., NH), 7,2 (1H, дд, J=5, 10 ArH), 7,51 (1H, с, ArH), 7,64 (1H, д, J=10, ArH), 8,46 (1H, д, J=5, ArH), 8,55 (1H, с, ArH); δC (CDCl3, 500 МГц) 159,28 (C), 154,72 (C), 149,07 (CH), 148,47 (CH), 135,44 (CH), 134,83 (C), 134,48 (CH), 124,12 (C), 123,44 (CH), 114,62 (C), 73,1 (CH), 55,88 (CH), 46,70 (CH), 41,78 (CH2), 23,68 (CH2) 22,36 (2×CH3), 11,55 (CH3);
19FЯМР δ -74,83; m/z 424,2 (M+H).
Пример 17
1,1,1-Трифтор-3-(9-изопропил-6-((6-(трифторметил)пиридин-3-ил)метиламино)-9Н-2-иламино)пентан-2-ол [17]
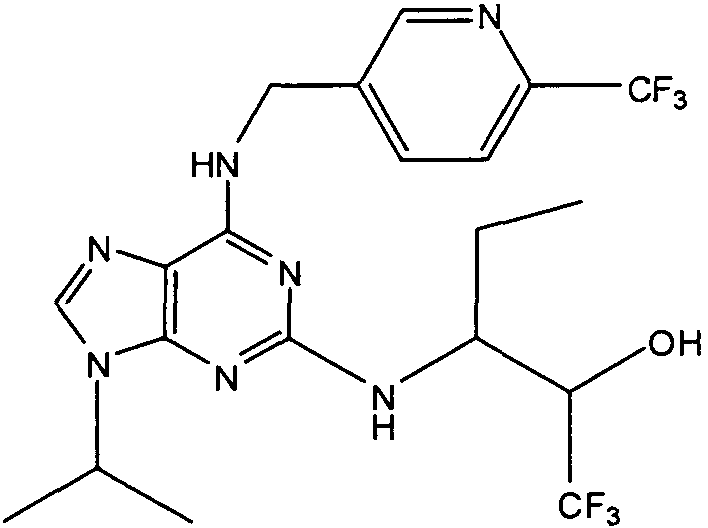
К перемешиваемому раствору 2-фтор-9-изопропил-N-(6-(трифторметилпиридин-3-илметил)-9Н-пурин-6-амина (200 мг, 0,56 ммоль) в н-BuOH/ДМСО (5 мл, 4:1) при комнатной температуре в атмосфере аргона добавляли DIEA (1,4 мл, 10 экв., 8 ммоль), с последующим добавлением 3-амино-1,1,1-трифторпентан-2-ола (0,377 г, 2,4 ммоль). Колбу снабжали обратным холодильником, реакционную смесь помещали на предварительно нагретую масляную баню при 140°С и перемешивали при этой температуре в течение 72 часов. Спустя указанное время, реакция проходила только на 30%. Проводили добавление аминоспирта еще в течение 4 дней, для завершения конверсии. Реакционной смеси давали остыть до комнатной температуры и растворитель выпаривали в вакууме. Остаток распределяли между EtOAc (50 мл) и водой (50 мл), водную фазу дополнительно экстрагировали EtOAc (2×25 мл) и объединенную органическую фазу промывали насыщенным раствором соли (50 мл), сушили (MgSO4) и выпаривали в вакууме. Остаток очищали градиентной колоночной флэш-хроматографией на силикагеле при элюировании CHCl3:MeOH (100:0→95:5), с получением указанного в заголовке соединения в виде коричневого порошка (30 мг, 11%).
δH (CDCl3, 500 МГц) 0,95 (3H, т, J=7,5, CHCH2CH 3), 1,56 (6H, д, J=7,5, CH[CH 3]2), 1,721-1,61 (2H, м, CHCH 2CH3), 3,22-3,05 (1H, м, CHEt), 3,88 (1H, м, CHMe2), 4,75-4,57 (1H, м, CHCF3OH), 4,92-4,76 (2H, м, NHCH 2Ar), 5,3 (1H, с, ушир., NH), 6,83 (1H, с, ушир., NH), 7,55 (1H, д, J=10, ArH), 7,61-7,53 (2H, м, ArH), 8,85 (1H, с, ArH); δC (CDCl3, 250 МГц) 157,67 (C), 156,65 (C), 156,9 (C), 146,62 (CH), 139,55 (CH), 128,22 (C), 126,84 (C), 125,97 (CH), 123,14 (CH), 120,32 (C), 71,12 (CH), 61,56 (CH2), 56,43 (CH), 25,74 (CH2), 22,42 (2×CH3), 11,56 (CH3); 19FЯМР δ -67,87, -74,30; ESMS 492 (M+1).
Пример 18
1,1,1,3,3,3-Гексафтор-2-((9-изопропил-6-(пиридин-3-илметиламино)-9Н-пурин-2-иламино)метил)пропан-2-ол [18]
К перемешиваемому раствору 2-фтор-9-изопропил-6-[(пиридин-3-илметил)амино]пурина (50 мг, 0,175 ммоль) в н-BuOH/ДМСО (2,5 мл, 4:1) при комнатной температуре и в атмосфере аргона добавляли диизопропилэтиламин (135 мг, 1,05 ммоль), с последующим добавлением 2-(аминометил)-1,1,1,3,3,3-гексафторпропан-2-ола, полученного по реакции 30% раствора гидроксида аммония с 2,2-бис[(трифторметил)оксираном]. Реакционную смесь помещали на предварительно нагретую масляную баню при 140°С и перемешивали при этой температуре в течение 3 дней. За ходом реакции следили с помощью ЖХМС, и она проходила только на 30%. Добавляли вновь полученный 2-(аминометил)-1,1,1,3,3,3-гексафторпропан-2-ол, и реакция продолжалась. Такое добавление проводили еще в течение трех последовательных дней до достижения полного завершения. После удаления растворителя остаток очищали колоночной флэш-хроматографией, с получением указанного в заголовке соединения в виде бледно-коричневого порошка (19 мг, 23%).
δH (CDCl3, 500 МГц) 1,57 (6H, д, J=7,5, CH[CH 3]2), 3,48-3,44 (1H, м, NHCHHCOH[CF3]2, 3,86-3,81 (1H, м, NHCHHCOH[CF3]2, 4,62-4,555 (1H, м, CHMe2), 4,78-4,74 (2H, м, NHCH 2ArH), 5,35 (1H, с, ушир., OH), 6,36 (1H, с, ушир., NH), 7,27-7,22 (2H, м, ArH), 7,7-7,67 (1H, м, ArH), 8,5 (1H, д, J=5, ArH), 8,66 (1H, с, ArH); δC (CDCl3, 500 МГц) 159,99 (C), 154,63 (C), 149,53 (C), 149,05 (C), 148,6 (CH), 137,79 (C), 135,41 (C), 135,38 (CH), 135 (CH), 123,44 (CH), 75,03 (C), 47,62 (CH2), 46,84 (CH), 42,05 (CH2), 22,1 (2×CH3); m/z 464,2 (M+H).
Анализы по киназам
Всего оценивали 21 соединение в анализах in vitro рекомбинантных киназ и на опухолевых клетках и сравнивали с селициклибом. Большинство таких ингибиторов cdk, как обнаружено, являются более сильными, чем селициклиб.
Чтобы оценить активность данных соединений в отношении киназ in vitro, их скринировали в отношении CDK2 и CDK9. Анализы по киназам выполняли в 96-луночных планшетах с использованием рекомбинантных CDK-циклинов, полученных в Cyclacel, Ltd., Dundee, UK. Анализы CDK2 и CDK9 выполняли в общем объеме, равном 25 мкл, в буфере для анализов (25 мМ b-глицерофосфата, 20 мМ MOPS, 5 мМ EGTA, 1 мМ DTT и 1 мМ NaVO3, рН 7,4), куда добавляли 2-4 мкг активного фермента с соответствующими субстратами (очищенный гистон Н1 для CDK2/циклина Е и CDK2/циклина А, биотинил-Ahx-(YSPTSPS)4 для CDK9/циклина Т1). Реакцию инициировали добавлением смеси Mg/АТФ (15 мМ MgCl2+100 мкМ АТФ с 30-50 kBq на лунку [-32P]-АТФ), и смеси инкубировали в течение 15 мин (CDK2/циклин Е), 30 мин (CDK2/циклин А) или 45 мин (CDK9/циклин Т1), как необходимо, при 30°С). Реакции с CDK останавливали добавлением 25 мкл 75 мМ фосфорной кислоты с последующим фильтрованием через фильтровальные пластины Р81 (Whatman Polyfiltronics, Kent, UK). В отношении CDK9 реакцию останавливали добавлением 25 мкл 75 мМ фосфорной кислоты, затем 5 мкл раствора авидина 10 мг/мл в каждую лунку и дополнительно инкубировали 2 мин с последующим фильтрованием, как при анализе CDK2. После промывания 3 раза 75 мМ ортофосфорной кислотой планшеты сушили, добавляли сцинтилляционную жидкость и включенную радиоактивность оценивали на сцинтилляционном счетчике (TopCount, Packard Instruments, Pangbourne, Berks, UK). Соединения для анализов киназ готовили в виде 10 мМ исходных стандартизованных растворов в ДМСО и разводили 10% раствором ДМСО в буфере для анализов. Данные анализировали с использованием программного обеспечения для корректировки кривых (Xlfit, Версия 4.00, ID Business Solutions Ltd, Guildford, Surrey, UK) для определения IC50 (концентрация соединения, которая ингибирует активность киназы на 50%). Среднее по двум показателям можно найти в таблицах 2 и 4. Результаты показывают, что многие из соединений являются более активными ингибиторами CDK2 и CDK9, чем родственное соединение селициклиб.
Определение IC 50 на линии опухолевых клеток NSCLC H460 с использованием анализа на цитотоксичность с Alamar Blue
Чтобы определить активность соединений в отношении клеток, цитотоксичность каждого соединения определяли в отношении линии клеток немелкоклеточного рака легких (NSCLC) Н460. Стандартные методы определения цитотоксичности осуществляли следующим образом: клетки Н460 засевали в 96-луночные планшеты в соответствии с их временем удвоения (3000 клеток на лунку) в среде RPMI, содержащей 10% FCS, и инкубировали в течение ночи при 37°С и с 5% СО2. Среду удаляли и добавляли 100 мкл свежей среды, содержащей концентрации соответствующего соединения, и клетки инкубировали в течение 72 часов при 37°С и 5% СО2. 10% стандартизованный раствор alamar blue (Roche, Lewes, United Kingdom) готовили в среде и по 100 мкл добавляли к клеткам, которые инкубировали в течение 2 часов. Поглощение измеряли на полиадресном (multi-label) счетчике Wallac Victor 2 1420 при 544-595 нм. Среднее по трем независимым экспериментам представлено в таблице 3.
Два соединения имеют значения IC50 менее 1 мкМ (соединение [1] и соединение [11]).
Сравнение способа действия соединений настоящего изобретения и селициклиба
Ранее было показано, что селициклиб вызывает апоптоз путем его воздействия на транскрипцию. Таким образом, ингибирует CDK7 и CDK9, которые ответственны за фосфорилирование РНК-полимеразы II, которое необходимо для инициации и продолжения транскрипции. В результате ингибирования транскрипции уровни ряда белков с коротким периодом полужизни снижаются, таких как Mcl-1, запуская, тем самым, апоптоз.
Чтобы подтвердить то, что такие соединения вызывают также понижающую регуляцию Mcl-1, клетки Н460 засевали в количестве 5×105 клеток на 10 см2 чашку с 10 мл среды RPMI, содержащую 10% FCS, и инкубировали в течение ночи при 37°С и при 5% СО2. Затем к клеткам добавляли 1 мл 11× концентрированного соединения и инкубировали в течение 5 или 24 часов. Среду удаляли и прикрепленные клетки промывали 5 мл PBS. Клетки лизировали на чашках добавлением 100 мкл буфера для лизиса (50 мМ HEPES) рН 7,20 мМ NaCl, 1 мМ DTT, коктейля ингибиторов протеаз (1:1000) и ингибиторы фосфатазы (10 мМ пирофосфата натрия, 10 мМ фторида натрия и 1 мМ ортованадата натрия). Лизаты быстро замораживали в жидком азоте и хранили при -70°С. Замороженные лизаты оттаивали и обрабатывали ультразвуком импульсами 2×10 секунд на льду. Концентрацию белка в каждом лизате определяли с использованием набора для определения белка BCA (Pierce) согласно инструкциям производителя. Лизат (30 мкг) смешивали с 1× буфером для загрузки на гель, содержащим 10% β-меркаптоэтанол, и разделяли в 4-12% бис-трис полиакриламидном геле с использованием денатурирующих электрофоретических условий (Invitrogen, Glasgow, United Kingdom). Белки переносили на нитроцеллюлозные мембраны (Shleicher & Schuell, Dassel, Germany) с использованием влажного электрофоретического переноса. Мембраны окрашивали пунцовым S для подтверждения одинаковой нагрузки перед блокированием в 5% растворе нежирного молока в PBS с 0,05% твин 20 (PBSTM) в течение 2 часов. Мембраны инкубировали в течение ночи при 4°С с первыми антителами (кроличьи поликлональные антитела против Mcl-1, Santa Cruz), разводили 1:1000 PBSTM. Мембраны промывали 2×5 минут с последующим промыванием 2×10 минут PBS и 0,05% твин 20 (PBST) и инкубировали в течение 1 часа в PBSTM, содержащем вторые антитела, конъюгированные с пероксидазой хрена. Мембраны промывали, как указано выше, и инкубировали с усиливающим хемилюминесценцию раствором (Amersham) и экспонировали на рентгеновской пленке (Amersham).
Как и ожидалось, селициклиб проявляет очень умеренные изменения, так как наивысшая концентрация, использованная в данном эксперименте, близка к значению IC50 для данного соединения. Соединение [1] явно превосходит селициклиб по его способности снижать уровни Mcl-1 со значительными эффектами, наблюдаемыми при 1,5 мкМ через 24 часа. Это находится в согласии с его повышенной активностью.
Во втором эксперименте исследовали действие дополнительных соединений на уровни Mcl-1. В данном случае клетки Н460 обрабатывали 0,5; 1,5; 4,5 и 13,5 мкМ в течение 5 часов, на данном сроке клетки собирали для Вестерн-блоттинг анализа. Результаты представлены на фиг.2.
Результаты показывают, что соединение [1], как видно, является наиболее эффективным соединением из данной группы для понижающей регуляции уровней Mcl-1 и обладает активностью, превосходящей активность селициклиба. Однако ясно, что все такие соединения вызывают снижение Mcl-1 при концентрациях, эквивалентных примерно 2-3-кратному значению IC50.
Ингибирование цитохрома Р450 (определение IC 50 5 изоформ)
Цель
Чтобы установить, ингибирует ли соединение [1] активность пяти изоформ CYP путем анализа метаболизма специфических субстратов CYP. Сравнительные анализы выполняли с использованием соединений предшествующего уровня техники [А1], [А2] и [А3], представленных ниже:
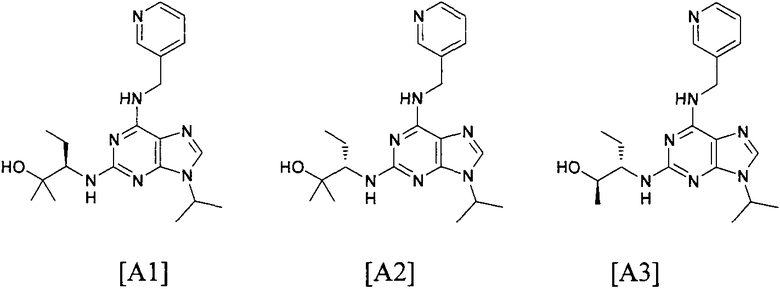
Методика эксперимента
Соединения [А1], [А2] и [А3] синтезировали в соответствии с методами, представленными в WO 2004/016612 (Cyclacel Limited).
Ингибирование CYP1A
Микросомы печени человека (0,25 мг/мл) и NADPH (1 мМ) инкубировали с шестью концентрациями испытуемого соединения (0,05; 0,25; 0,5; 2,5; 5; 25 мкМ в ДМСО; конечная концентрация ДМСО=0,35%) в присутствии оцениваемого субстрата этоксирезоруфина (0,5 мкМ) в течение 5 мин при 37°С. Селективный ингибитор CYP1A, альфа-нафтофлавон, скринировали вместе с испытуемыми соединениями в качестве положительного контроля.
Ингибирование CYP2C9
Микросомы печени человека (0,25 мг/мл) и NADPH (1 мМ) инкубировали с шестью концентрациями испытуемого соединения (0,05; 0,25; 0,5; 2,5; 5; 25 мкМ в ДМСО; конечная концентрация ДМСО=0,25%) в присутствии оцениваемого субстрата толбутамида (120 мкМ) в течение 60 мин при 37°С. Селективный ингибитор CYP2C9, сульфафеназол, скринировали вместе с испытуемыми соединениями в качестве положительного контроля.
Ингибирование CYP2C19
Микросомы печени человека (0,25 мг/мл) и NADPH (1 мМ) инкубировали с шестью концентрациями испытуемого соединения (0,05; 0,25; 0,5; 2,5; 5; 25 мкМ в ДМСО; конечная концентрация ДМСО=0,25%) в присутствии оцениваемого субстрата мефенитоина (25 мкМ) в течение 60 мин при 37°С. Селективный ингибитор CYP2C19, транилципромин, скринировали вместе с испытуемыми соединениями в качестве положительного контроля.
Ингибирование CYP2D6
Микросомы печени человека (0,25 мг/мл) и NADPH (1 мМ) инкубировали с шестью концентрациями испытуемого соединения (0,05; 0,25; 0,5; 2,5; 5; 25 мкМ в ДМСО; конечная концентрация ДМСО=0,25%) в присутствии оцениваемого субстрата декстрометорфана (5 мкМ) в течение 30 мин при 37°С. Селективный ингибитор CYP2D6, хинидин, скринировали вместе с испытуемыми соединениями в качестве положительного контроля.
Ингибирование CYP3A4
Микросомы печени человека (0,25 мг/мл) и NADPH (1 мМ) инкубировали с шестью концентрациями испытуемого соединения (0,05; 0,25; 0,5; 2,5; 5; 25 мкМ в ДМСО; конечная концентрация ДМСО=0,26%) в присутствии оцениваемого субстрата мидазолама (2,5 мкМ) в течение 5 мин при 37°С. Селективный ингибитор CYP3A4, кетоконазол, скринировали вместе с испытуемыми соединениями в качестве положительного контроля.
Для инкубации с CYP1A реакции прерывали добавлением метанола, и образование метаболита, резоруфина, отслеживали по флуоресценции (волна возбуждения=535 нм, волна излучения=595 нм). Для инкубации с CYP2C9, CYP2C19, CYP2D6 и CYP3A4 реакции прерывали добавлением содержащего метанол внутреннего стандарта. Образцы затем центрифугировали и надосадочные жидкости объединяли для одновременного анализа 4-гидрокситолбутамида, 4-гидроксимефенитоина, декстрометорфана и 1-гидроксимидазолама плюс внутренний стандарт с помощью ЖХ-МС/МС с использованием характерных условий для ЖХ-МС/МС. В конечный образец перед анализом добавляли муравьиную кислоту в деионизированной воде (конечная концентрация=0,1%). Снижение образования метаболитов в сравнении с контролем носителя использовали для расчета значения IC50 (концентрация испытуемого соединения, которая дает 50% ингибирование).
Результаты
IC50 (мкМ) для каждого соединения в отношении пяти изоформ CYP представлены в таблице 5.
Данные показывают, что три соединения являются сильными ингибиторами CYP3A4, тогда как соединение [1] не является таковым. Так как значение IC50 соединения [1] значительно выше его IC50 для клеток (см., таблицу 7), это показывает, что цитотоксические концентрации там не должны оказывать воздействия на активность CYP3A4. Это является важным, так как CYP3A4 участвует в метаболизме большого числа лекарственных препаратов. Если CYP3A4 ингибируется одним лекарственным средством, это может привести к неожиданной токсичности из-за сниженного метаболизма субстратов CYP3A4, приводя, тем самым, к явно повышенным уровням этих средств.
Идентификация субстрата цитохрома Р450
Цель
Установить, какие из главных изоформ цитохрома Р450 участвуют в метаболизме четырех испытуемых соединений.
Методика эксперимента
Препараты человеческого фермента CYP450, экспрессированные кДНК, со-экспрессированные с человеческой NADPH-цитохром-Р450-редуктазой (Bactosomes™), были поставлены Cypex Ltd. Bactosomes™ (конечная концентрация CYP1A2 100 пмоль/мл, CYP2C8 50 пмоль/мл, CYP2C9 25 пмоль/мл, CYP2C19 100 пмоль/мл, CYP2D6 50 пмоль/мл и CYP3A4 25 пмоль/мл), 0,1М фосфатный буфер рН 7,4 и испытуемое соединение (конечная концентрация субстрата=5 мкМ; конечная концентрация ДМСО=0,25%) предварительно инкубировали при 37°С перед добавлением NADPH (конечная концентрация=1 мМ) для инициации реакции. Инкубацию выполняли, используя контрольные бактосомы (при отсутствии Р450 ферментов), чтобы выявить любое неферментативное разложение. Конечный объем инкубации составил 25 мкл. Соединения, о которых известно, что они специфически метаболизируются каждой изоформой CYP450, использовали в качестве контрольных соединений.
Каждое соединение инкубировали отдельно в течение 0, 5, 15, 30 и 45 мин с каждой из изоформ CYP. Реакции останавливали добавлением содержащего метанол внутреннего стандарта в соответствующие моменты времени. Планшеты для инкубации центрифугировали при 2500 об/мин в течение 20 мин при 4°С для осаждения белка. После осаждения белка надосадочные жидкости объединяли в кассеты, включающие до 4 соединений, и анализировали, используя характерные условия ЖХ-МС/МС.
Данные анализа
Отношения площади пика ln (которое было скорректировано на любую потерю при инкубации с контрольными бактосомами) помещали на график зависимости от времени, и определяли градиент линии.
Константа скорости элиминации (k)=(-градиент)
Период полужизни(t1/2)(мин)=0,693/k
Результаты
Период полужизни (минуты) для каждого соединения в присутствии каждого из шести CYP представлены в таблице 6.
Данные показывают, что в данной системе Bactosomes™ соединение [1] не является субстратом для шести испытанных изоформ CYP. Существует большое различие с другими тремя соединениями, так как они все являются субстратами CYP3A4, и два также являются субстратами CYP1A2. Это различие хорошо соответствует различию ингибирования CYP, обсужденному в таблице 4. Общий механизм, приводящий к ингибированию CYP, существует, если соединение является также субстратом для данного CYP. Как можно видеть, соединение [1] не является ни субстратом, ни ингибитором CYP3A4, тогда как другие три соединения являются ими.
Анализы по киназам
Чтобы оценить активность данных соединений в отношении киназ in vitro, их скринировали в отношении CDK2 и CDK9. Анализы по киназам выполняли в 96-луночных планшетах с использованием рекомбинантных CDK-циклинов, полученных в Cyclacel, Ltd., Dundee, Uk. Анализы с CDK2 и CDK9 выполняли в общем объеме, равном 25 мкл в буфере для анализов (25 мМ b-глицерофосфата, 20 мМ MOPS, 5 мМ EGTA, 1 мМ DTT и 1 мМ NaVO3, рН 7,4), куда добавляли 2-4 мкг активного фермента с соответствующими субстратами (очищенный гистон Н1 для CDK2/циклина Е и CDK2/циклина А, биотинил-Ahx-(YSPTSPS)4 для CDK9/циклина Т1). Реакцию инициировали добавлением смеси Mg/АТФ (15 мМ MgCl2+100 мкМ АТФ с 30-50 kBq на лунку [-32P]-АТФ), и смеси инкубировали в течение 15 мин (CDK2/циклин Е), 30 мин (CDK2/циклин А) или 45 мин (CDK9/циклин Т1), как необходимо, при 30°С). Реакции с CDK останавливали добавлением 25 мкл 75 мМ фосфорной кислоты с последующим фильтрованием через фильтровальные пластины Р81 (Whatman Polyfiltronics, Kent, UK). В отношении CDK9 реакцию останавливали добавлением 25 мкл 75 мМ фосфорной кислоты, затем 5 мкл раствора авидина 10 мг/мл в каждую лунку и дополнительно инкубировали 2 мин с последующим фильтрованием, как при анализе с CDK2. После промывания 3 раза 75 мМ ортофосфорной кислотой планшеты сушили, добавляли сцинтилляционную жидкость и включенную радиоактивность оценивали на сцинтилляционном счетчике (TopCount, Packard Instrument, Panbourne, Berks, UK). Соединения для анализов с киназами готовили в виде 10 мМ исходных стандартизованных растворов в ДМСО и разводили 10% раствором ДМСО в буфере для анализов. Данные анализировали с использованием программного обеспечения для корректировки кривых (Xlfit, Версия 4.00, ID Business Solutions Ltd, Guildford, Surrey, UK) для определения IC50 (концентрация соединения, которая ингибирует активность киназы на 50%). Среднее по двум показателям можно найти в таблицах 6 и 8.
Результаты
Результаты в таблице 7 показывают, что соединение предшествующего уровня техники [А3] и соединение [1] являются наиболее активными ингибиторами киназ.
Определение IC 50 в линиях опухолевых клеток с использованием анализа на цитотоксичность с Alamar Blue
Чтобы определить активность данных соединений в отношении клеток, цитотоксичность каждого соединения определяют в отношении ряда клеточных линий. Стандартные способы определения цитотоксичности выполняли следующим образом: клетки засевали в 96-луночные планшеты в соответствии со временем их удвоения (2-5000 клеток на лунку) на среду RPMI или DMEM, содержащую 10% FCS, и инкубировали в течение ночи при 37°С и 5% СО2. Среду удаляли и добавляли 100 мкл свежей среды, содержащей повышающиеся концентрации соответствующего соединения, и клетки инкубировали в течение 72 часов при 37°С и 5% СО2. 10% стандартизованный раствор alamar blue (Roche, Lewes, United Kingdom) готовили в среде и по 100 мкл добавляли к клеткам, которые инкубировали в течение 2 часов. Поглощение измеряли на полиадресном (multi-label) счетчике Wallac Victor 2 1420 при 544-595 нм.
Результаты
Результаты анализа на клеточную цитотоксичность в отношении 21 линии клеток представлены в таблице 8. Соединение [1] значительно более активно, чем соединения [А1], [А2] или [А3] предшествующего уровня техники.
Различные модификации и варианты настоящего изобретения, не отходя от существа и объема изобретения, будут очевидны специалистам в данной области. Хотя настоящее изобретение было описано в связи с конкретными предпочтительными вариантами осуществления, будет понятно, что изобретение, которое заявлено, не должно неоправданно ограничиваться такими конкретными вариантами осуществления. Разумеется, различные модификации описанных способов осуществления настоящего изобретения, которые очевидны специалистам в данной области, как подразумевается, охватываются данным изобретением.
Выбранные соединения настоящего изобретения
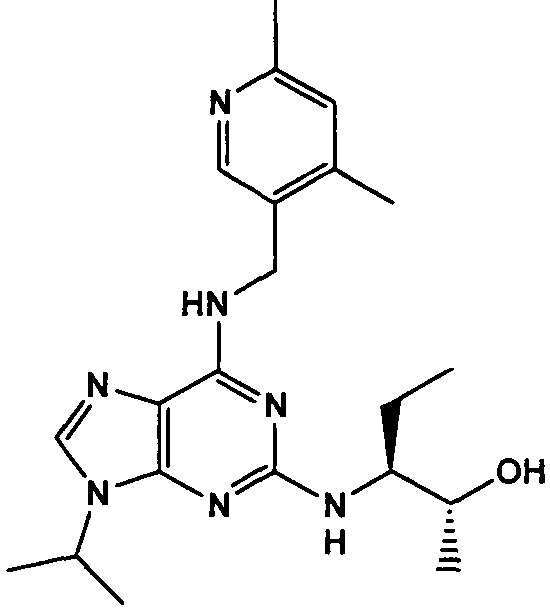
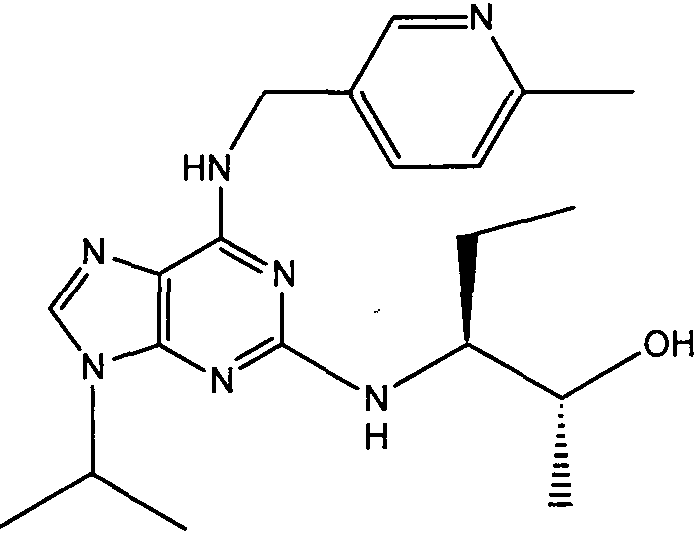
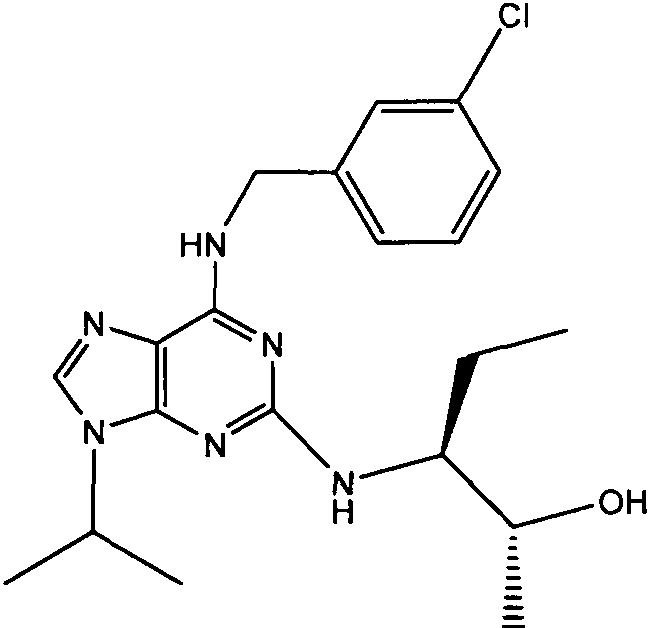
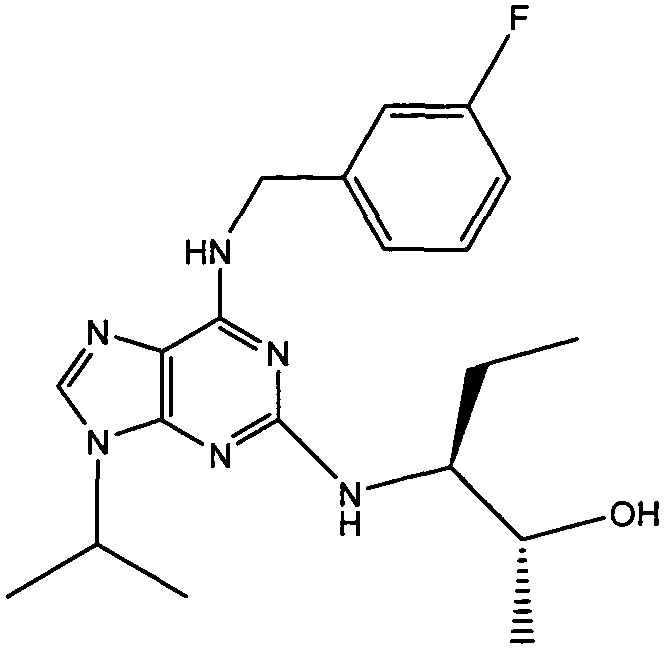
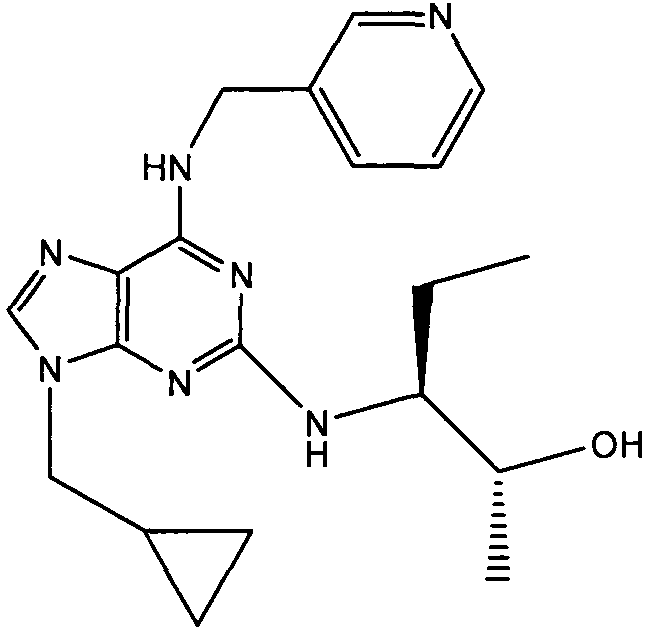
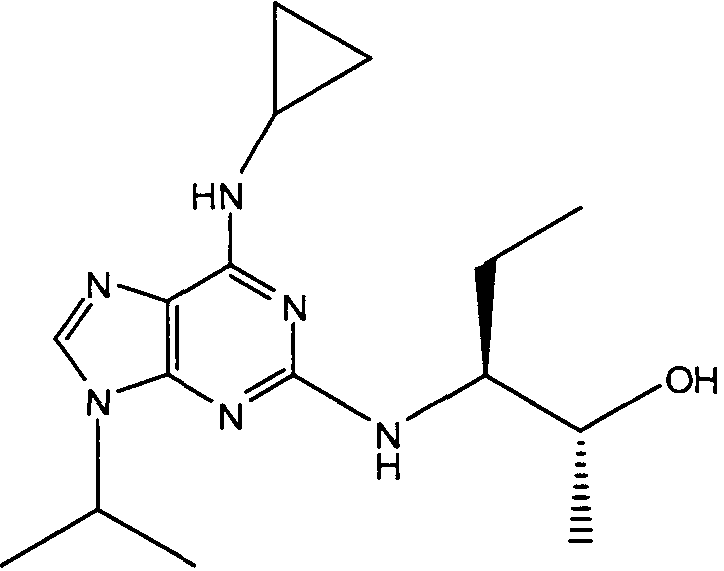
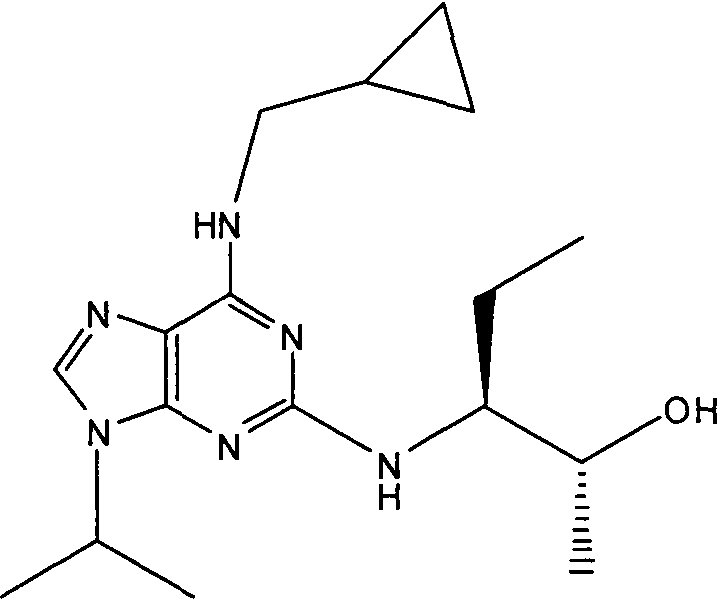
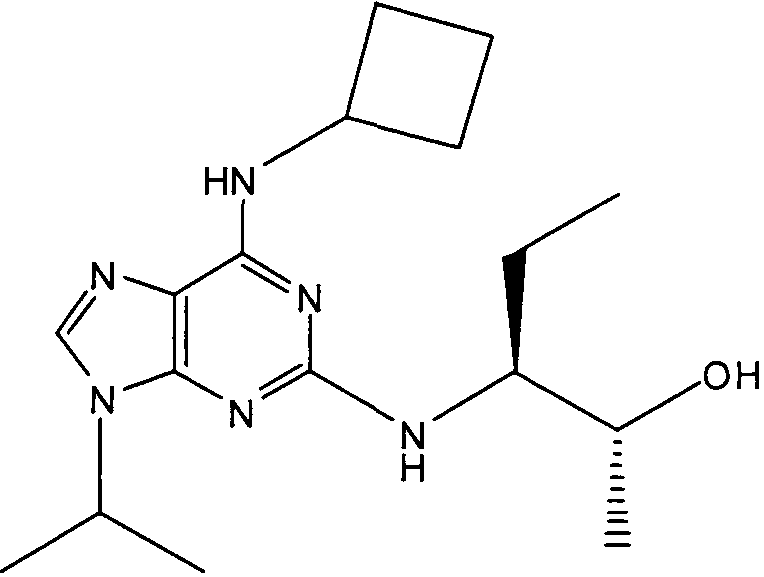
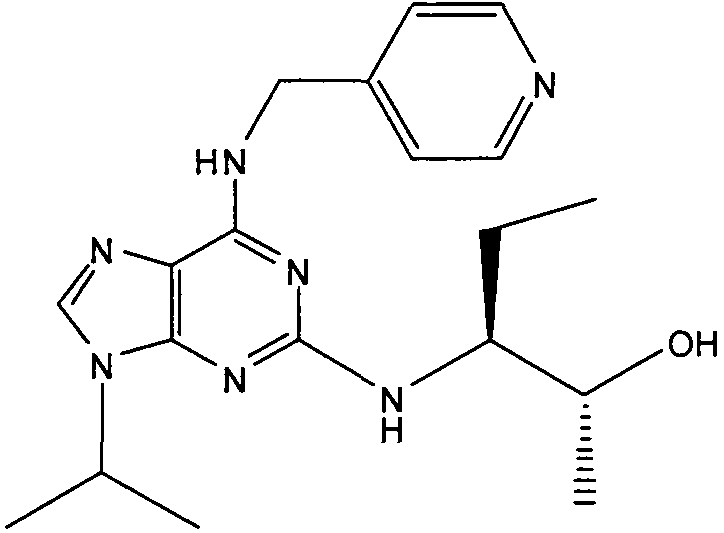
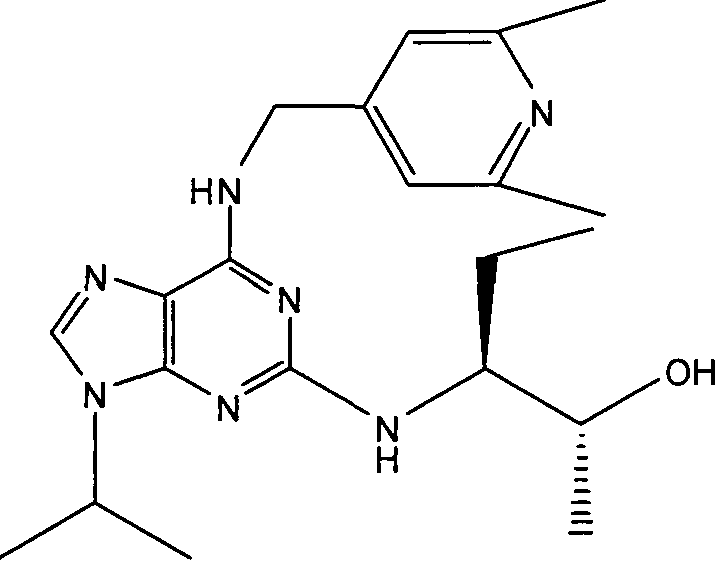
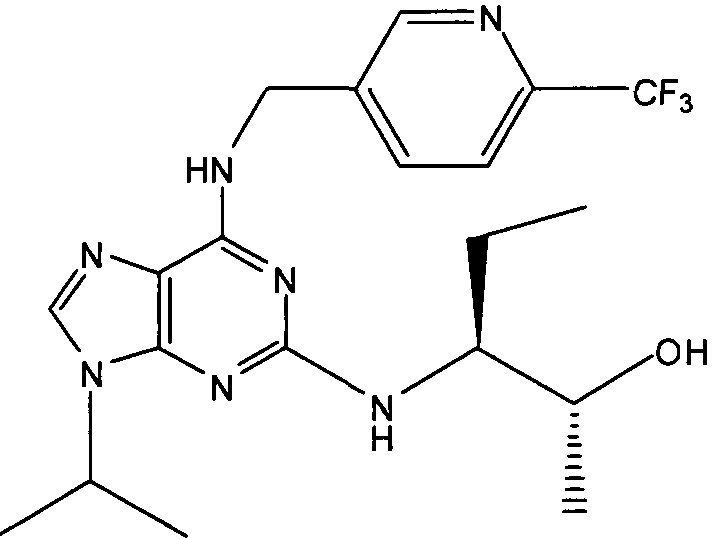
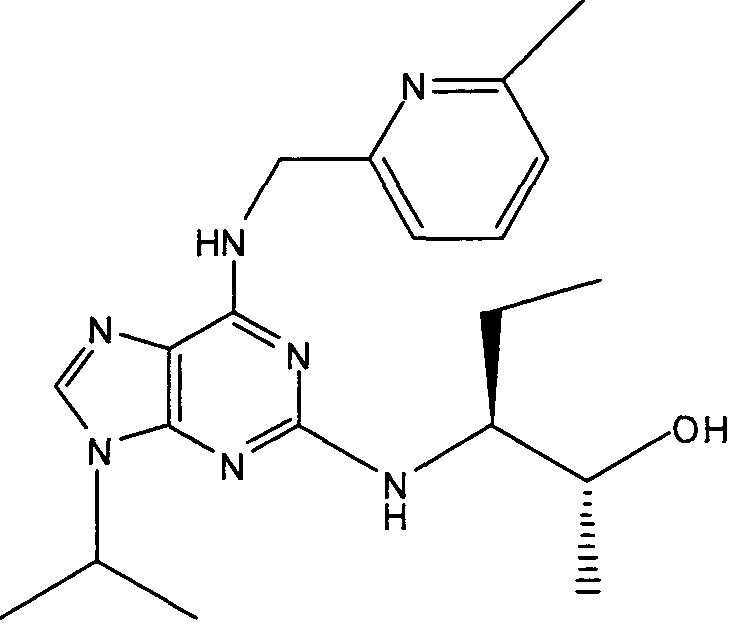
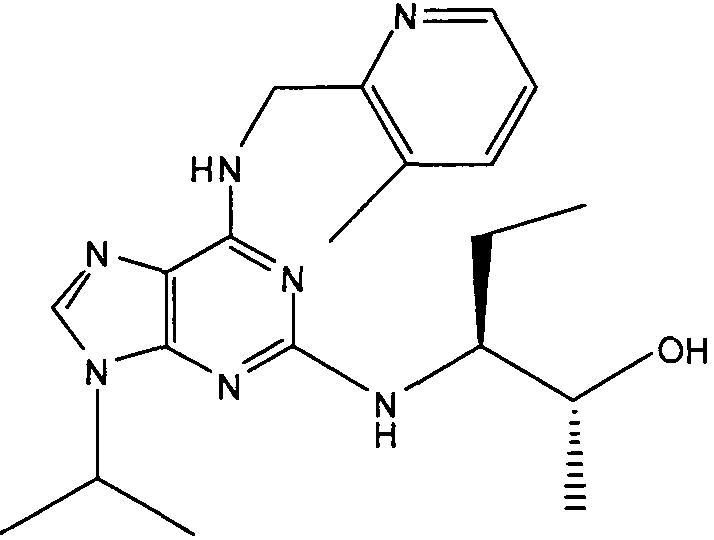
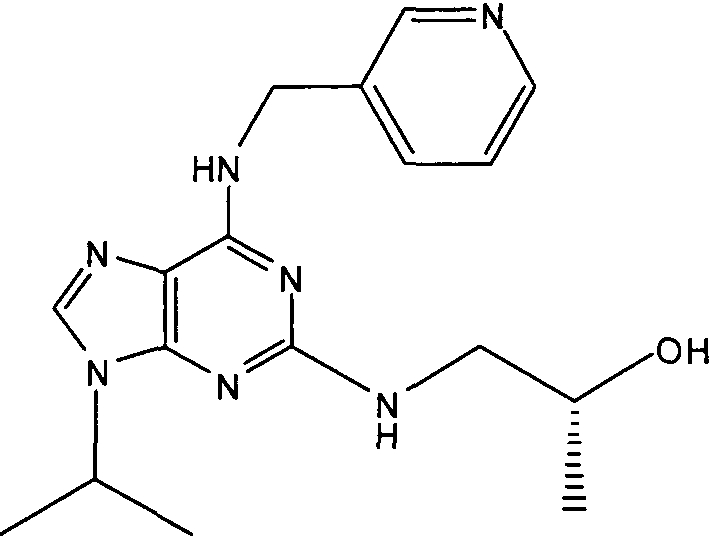
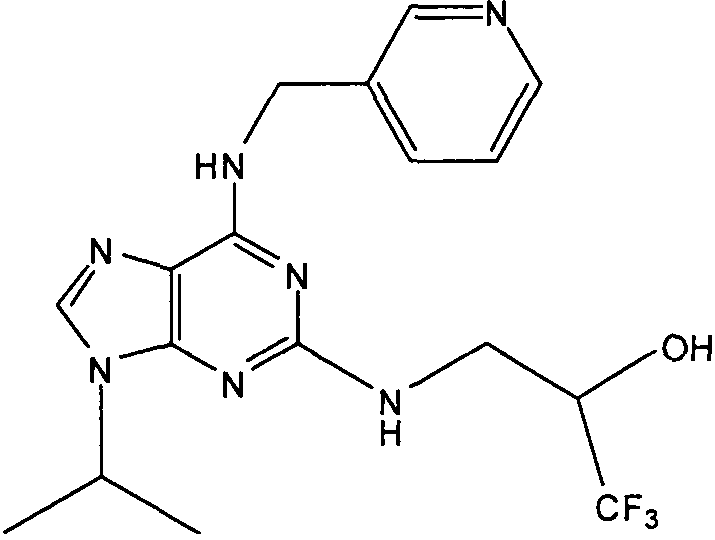
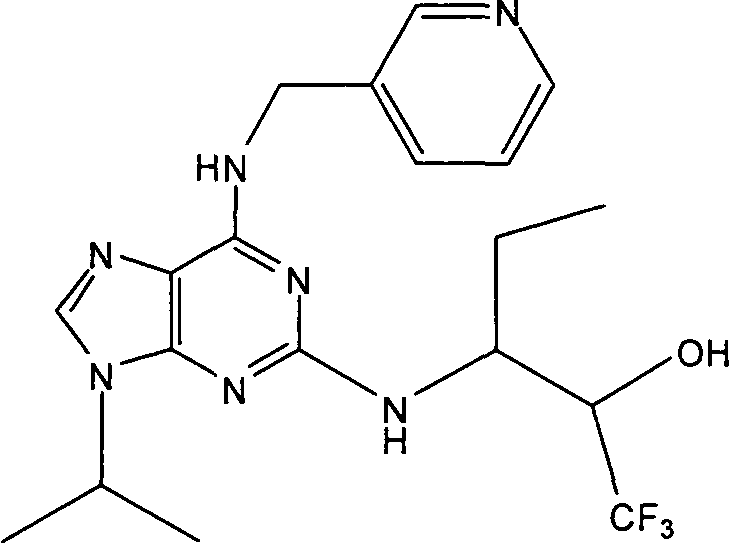
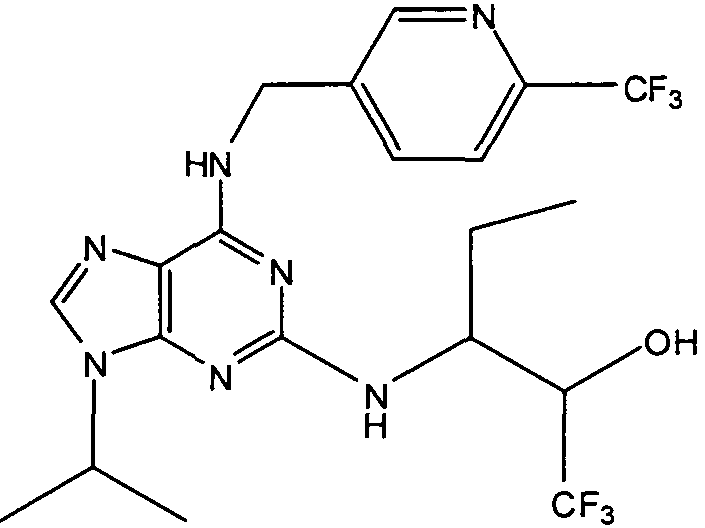
Сводная таблица по анализам с киназой in vitro при выборе соединений второго поколения и селициклиба. Значения IC50 выражены в мкМ. Соединения анализировали в две партии, и в обоих случаях селициклиб проходил анализ в качестве контроля, представляя селициклиб 1 и 2, которые дали сходные результаты
NA - не анализировали
Сводная таблица значений IC50 для клеток представленных соединений, которые определяли на клетках Н460. В последней колонке показано, насколько многие из данных соединений более активны по сравнению с селициклибом
Сводная таблица по анализам с киназой при скрининге соединений [17] и [18] (мкМ)
IC
50
(М) для соединений [А1], [А2] и [А3] предшествующего уровня техники и соединения [1] настоящего изобретения в отношении пяти изоформ CYP
Период полужизни (мин) для соединений [А1], [А2] и [А3] предшествующего уровня техники и соединения [1] настоящего изобретения в присутствии каждого из шести CYP
Активность в отношении ингибирования киназ (IC
50
, М) соединений [А1], [А2] и [А3] предшествующего уровня техники и соединения [1] настоящего изобретения
Результаты анализа на клеточную цитотоксичность для соединений [А1], [А2] и [А3] предшествующего уровня техники и соединения [1] настоящего изобретения в отношении различных линий клеток
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКИЕ СОЕДИНЕНИЯ | 2005 |
|
RU2422449C2 |
| АМИНОПРОИЗВОДНЫЕ ОКСО- ИЛИ ГИДРОКСИЗАМЕЩЕННЫХ ГИДРАЗИНОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ИНГИБИРОВАНИЯ РЕТРОВИРУСНОЙ ПРОТЕАЗЫ | 1993 |
|
RU2126794C1 |
| ПРОТИВОВОСПАЛИТЕЛЬНЫЕ СРЕДСТВА | 2004 |
|
RU2365585C2 |
| ИНГИБИТОРЫ ФОСФОИНОЗИТИД-3-КИНАЗЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2007 |
|
RU2468027C2 |
| ЛЕЧЕНИЕ ИНФЕКЦИЙ H. PYLORI С ПРИМЕНЕНИЕМ ИНГИБИТОРОВ MTAN | 2015 |
|
RU2663803C2 |
| ПРОИЗВОДНОЕ ИНДАНОНА, ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ ИЛИ ЭНАНТИОМЕР, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПРОФИЛАКТИКИ ИЛИ ЛЕЧЕНИЯ ВИРУСНОГО ЗАБОЛЕВАНИЯ | 2012 |
|
RU2574591C2 |
| ЗАМЕЩЕННЫЕ ДИАМИНОКАРБОКСАМИДНЫЕ И ДИАМИНОКАРБОНИТРИЛЬНЫЕ ПРОИЗВОДНЫЕ ПИРИМИДИНОВ, ИХ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ С ИХ ПОМОЩЬЮ | 2012 |
|
RU2697712C2 |
| ПРОИЗВОДНЫЕ 1,3-ДИОКСОИНДЕНА, ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ АНТИВИРУСНАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ИХ В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 2012 |
|
RU2566761C2 |
| ПРОИЗВОДНЫЕ 2-АМИНО-2-ФЕНИЛАЛКАНОЛА, ИХ ПОЛУЧЕНИЕ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2009 |
|
RU2486174C2 |
| ТЕТРАГИДРОФУРО(3,2-b)ПИРРОЛ-3-ОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ КАТЕПСИНА К | 2007 |
|
RU2456290C2 |
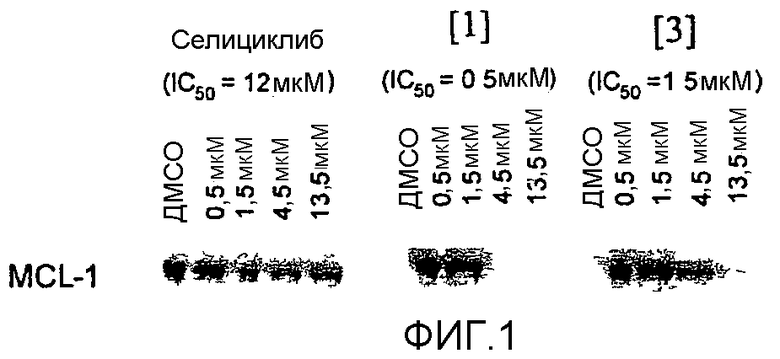
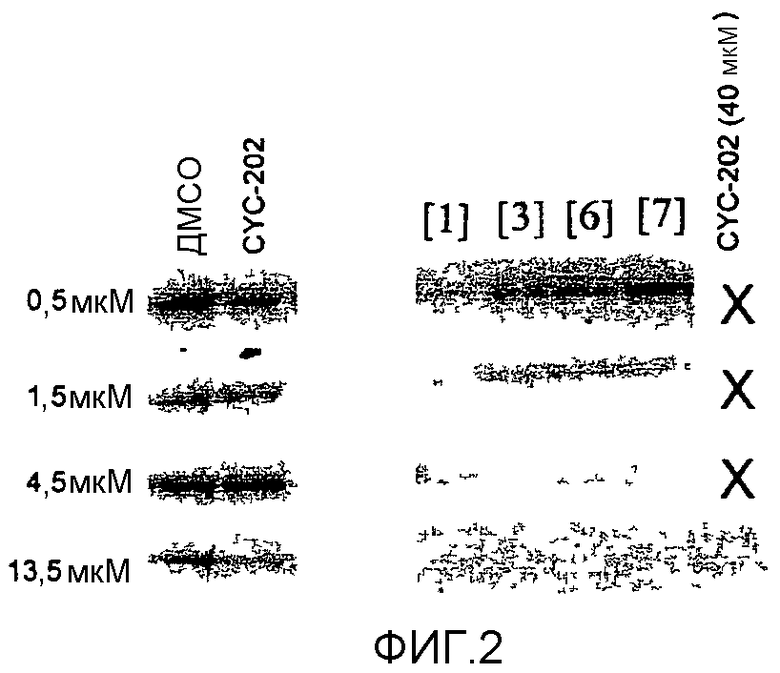
Изобретение относится к новым производным пурина, обладающим свойствами ингибитора фермента CDK, выбранного из CDK1, CDK2, CDK3, CDK4, CDK6, CDK7, CDK8 и CDK9. В формуле (I):
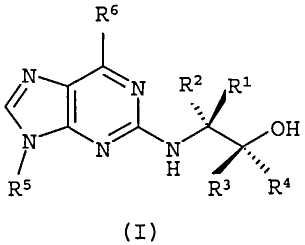
R1 и R2, каждый независимо, представляет собой Н, C1-6-алкил или С1-6-галогеналкил; R3 и R4, каждый независимо, представляет собой Н, С1-6-алкил или С1-6-галогеналкил; R5 представляет собой C1-6-алкил или С3-12-циклоалкил, или С3-12-циклоалкил-С1-6-алкил, каждый из которых может быть необязательно замещен одной или более ОН группами; R6 представляет собой  , где Y представляет собой N, X и Z представляют собой CR9; R7, R8 и каждый R9 независимо представляют собой Н, C1-6-алкил или C1-6-галогеналкил; где по меньшей мере один из R7, R8 и R9 не является Н. Изобретение также относится к фармацевтической композиции, содержащей указанные соединения, к применению соединений для лечения облысения, удара, пролиферативного заболевания, такого как рак, лейкемия, гломерулонефрит, ревматоидный артрит, псориаз, вирусного заболевания, такого как заболевание, вызванное человеческим цитомегаловирусом, вирусом простого герпеса типа 1, вирусом иммунодефицита человека типа 1, нейродегенеративного заболевания, заболевания ЦНС, такого как болезнь Альцгеймера. 13 н. и 17 з.п. ф-лы, 8 табл., 18 пр., 2 ил.
, где Y представляет собой N, X и Z представляют собой CR9; R7, R8 и каждый R9 независимо представляют собой Н, C1-6-алкил или C1-6-галогеналкил; где по меньшей мере один из R7, R8 и R9 не является Н. Изобретение также относится к фармацевтической композиции, содержащей указанные соединения, к применению соединений для лечения облысения, удара, пролиферативного заболевания, такого как рак, лейкемия, гломерулонефрит, ревматоидный артрит, псориаз, вирусного заболевания, такого как заболевание, вызванное человеческим цитомегаловирусом, вирусом простого герпеса типа 1, вирусом иммунодефицита человека типа 1, нейродегенеративного заболевания, заболевания ЦНС, такого как болезнь Альцгеймера. 13 н. и 17 з.п. ф-лы, 8 табл., 18 пр., 2 ил.
1. Соединение формулы (I) или его фармацевтически приемлемая соль
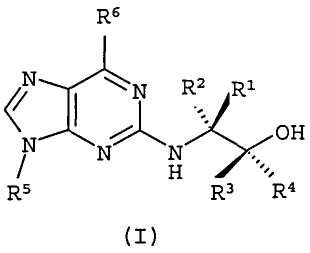 \
\
где R1 и R2, каждый независимо, представляет собой Н, C1-6-алкил или C1-6-галогеналкил;
R3 и R4, каждый независимо, представляет собой Н, С1-6-алкил или C1-6-галогеналкил;
R5 представляет собой C1-6-алкил или С3-12-циклоалкил, или С3-12-циклоалкил-С1-6-алкил, каждый из которых может быть необязательно замещен одной или более ОН группами;
R6 представляет собой
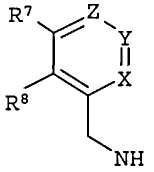
где Y представляет собой N, X и Z представляют собой CR9;
R7, R8 и каждый R9 независимо представляют собой Н, C1-6-алкил или С1-6-галогеналкил; где по меньшей мере один из R7, R8 и R9 не является Н.
2. Соединение по п.1, где один из R1 и R2 является Н, и другой является С1-6-алкилом.
3. Соединение по п.1, где один из R1 и R2 является Н, и другой является метилом, этилом или изопропилом.
4. Соединение по п.1, где R1 является этилом, и R2 является Н.
5. Соединение по п.1, где R3 и R4, каждый независимо, представляет собой Н, C1-6-алкил или C1-6-галогеналкил, и где по меньшей мере один из R3 и R4 не является Н.
6. Соединение по п.1, где один из R3 и R4 является Н, и другой является С1-6-алкилом или C1-6-галогеналкилом.
7. Соединение по п.1, где R3 является Н, и R4 является C1-6-алкилом или C1-6-галогеналкилом.
8. Соединение по п.1, где R3 является Н, и R4 является метилом.
9. Соединение по п.1, где X является СН, Z является С-Ме, и R7 является Н, и R8 является Me.
10. Соединение по п.1, где X является СН, Z является С-Ме, и R7 и R8, оба являются Н.
11. Соединение по п.1, где X является СН, Z является C-CF3, и R7 и R8, оба являются Н.
12. Соединение по п.1, где R5 является изопропилом.
13. Соединение по п.1, которое выбирают из следующего:
14. Соединение (2R,3S-3-(6-((4,6-диметилпиридин-3-илметиламино)-9-изопропил-9Н-пурин-2-иламино)пентан-2-ол или его фармацевтически приемлемые соль или сложный эфир.
15. Фармацевтическая композиция, обладающая свойствами ингибитора фермента CDK, выбранного из CDK1, CDK2, CDK3, CDK4, CDK6, CDK7, CDK8 и CDK9, содержащая терапевтически эффективное количество соединения по п.1 в смеси с фармацевтически приемлемым разбавителем, эксципиентом или носителем, или их смесью.
16. Применение соединения по п.1 для получения лекарственного средства для лечения пролиферативного заболевания, опосредованного активностью фермента CDK, выбранного из CDK1, CDK2, CDK3, CDK4, CDK6, CDK7, CDK8 и CDK9.
17. Применение по п.16, где указанным пролиферативным заболеванием является рак или лейкемия.
18. Применение по п.16, где пролиферативным заболеванием является гломерулонефрит, ревматоидный артрит или псориаз.
19. Применение соединения по п.1 для получения лекарственного средства для лечения вирусного заболевания, опосредованного активностью фермента CDK, выбранного из CDK1, CDK2, CDK3, CDK4, CDK6, CDK7, CDK8 и CDK9, где вирусное заболевание выбирают из заболеваний, вызванных человеческим цитомегаловирусом (HCMV), вирусом простого герпеса типа 1 (HSV-1), вирусом иммунодефицита человека типа 1 (HIV-1) и вирусом ветряной оспы и опоясывающего лишая (VZV).
20. Применение соединения по п.1 для получения лекарственного средства для лечения заболевания ЦНС, опосредованного активностью фермента CDK, выбранного из CDK1, CDK2, CDK3, CDK4, CDK6, CDK7, CDK8 и CDK9, где заболеванием ЦНС является болезнь Альцгеймера.
21. Применение соединения по п.1 для получения лекарственного средства для лечения облысения.
22. Применение соединения по п.1 для получения лекарственного средства для лечения удара.
23. Применение по п.16, где данное соединение применяют в количестве, достаточном для ингибирования по меньшей мере одного фермента CDK.
24. Применение соединения по п.1 для лечения нейродегенеративного заболевания, опосредованного активностью фермента CDK, выбранного из CDK1, CDK2, CDK3, CDK4, CDK6, CDK7, CDK8 и CDK9.
25. Применение по п.24 для лечения апоптоза нейронов.
26. Применение соединения по п.1 для ингибирования протеинкиназы, которая представляет собой циклинзависимую киназу, выбранную из CDK1, CDK2, CDK3, CDK4, CDK6, CDK7, CDK8 и CDK9.
27. Способ лечения пролиферативного заболевания, опосредованного активностью фермента CDK, выбранного из CDK1, CDK2, CDK3, CDK4, CDK6, CDK7, CDK8 и CDK9, включающий введение млекопитающему терапевтически эффективного количества соединения по п.1.
28. Способ по п.27, где указанное соединение применяют в количестве, достаточном для ингибирования по меньшей мере одного фермента CDK.
29. Соединение по п.1 для применения в медицине для лечения заболевания, опосредованного активностью фермента CDK, выбранного из CDK1, CDK2, CDK3, CDK4, CDK6, CDK7, CDK8 и CDK9.
30. Соединение по п.1 для лечения заболевания, опосредованного активностью фермента CDK, выбранного из CDK1, CDK2, CDK3, CDK4, CDK6, CDK7, CDK8 и CDK9, где указанное заболевание выбрано из пролиферативного заболевания, вирусного заболевания, нейродегенеративного заболевания, заболевания ЦНС, облысения и удара.
| Буровой резец | 1985 |
|
SU1399446A1 |
| УСТРОЙСТВО И СПОСОБ СВЯЗИ, МОНИТОРИНГА И КОНТРОЛЯ ЖЕЛЕЗНОДОРОЖНОГО ДВИЖЕНИЯ | 2005 |
|
RU2392155C2 |
| US 20030191086 А1, 09.10.2003 | |||
| US 20030229105 А1, 11.12.2003 | |||
| 2,6,9-ТРИЗАМЕЩЕННОЕ ПРОИЗВОДНОЕ ПУРИНА, ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ КАТИОННАЯ СОЛЬ, СПОСОБ ИНГИБИРОВАНИЯ ПРОЛИФЕРАЦИИ КЛЕТОК, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1997 |
|
RU2220968C2 |
| WO 1997020842 А1, 12.06.1997 | |||
| HAESSLEIN J.-L | |||
| "Recent advances in cyclin-dependent kinase inhibition | |||
| Purine-based derivatives as anti-cancer agents | |||
| Roles and perspectives for the future", Current topics in medicinal chemistry, vol.2(9), pp.1037-1050. | |||

