Область техники, к которой относится изобретение
Настоящее изобретение относится к способу получения сахарида, включающего глюкозу, путем гликозилирования растительного волокнистого материала и выделения полученного сахарида.
Описание предшествующего уровня техники
Было предложено производить сахарид, включающий в основном глюкозу или ксилозу, из целлюлюзы или гемицеллюлозы разложением растительного материала, который представляет собой биомассу, такую как остатки после обжима сахарного тростника (багасса) или древесные стружки, и эффективно использовать полученный сахарид в качестве корма или топлива, и данный процесс был введен в действие на практике. В частности, технология, по которой моносахарид, полученный разложением растительных волокон, подвергали ферментации для изготовления спирта, а именно этанола в качестве топлива, привлекла внимание. До настоящего времени было предложено множество способов для получения сахарида, такого как глюкоза, разложением целлюлюзы или гемицеллюлозы (например, публикация японской патентной заявки № 8-299000 (JP-A-8-299000), публикация японской патентной заявки № 2006-149343 (JP-A-2006-149343), публикация японской патентной заявки № 2006-129735 (JP-A-2006-129735) и публикация японской патентной заявки № 2002-59118 (JP-A-2002-59118). Обычный способ включает фермент, гидролизующий целлюлозу, при использовании серной кислоты, а именно разбавленной серной кислоты или концентрированной серной кислоты или хлористоводородной кислоты (JP-A-8-299000). Также доступными являются способ, в котором использована целлюлаза (JP-A-2006-149343), способ, в котором использован твердый катализатор, такой как активированный уголь или цеолит (JP-A-2006-129735), и способ, в котором использована горячая вода под давлением (JP-A-2002-59118).
Однако проблема, связанная со способом, по которому целлюлоза разлагается при использовании кислоты, такой как серная кислота, состоит в том, что кислота, служащая в качестве катализатора, и произведенный сахарид трудны для выделения из гидролизной реакционной смеси, полученной в результате гидролиза. Причина состоит в том, что глюкоза, которая представляет собой основной компонент продукта гидролиза целлюлозы, и кислота, которая служит катализатором гидролиза, обе растворимы в воде. Удаление кислоты нейтрализацией или обменом ионов из гидролизной реакционной смеси представляет собой не только сложное и дорогое дело, но также трудно удалить кислоту полностью, и кислота часто остается в процессе ферментации для этанола. В результате этого, даже когда рН оптимизирован с точки зрения активности дрожжей в процессе ферментации для этанола, концентрация соли повышается, тем самым снижается активность дрожжей и уменьшается эффективность ферментации.
В частности, когда используют концентрированную серную кислоту, серная кислота очень трудна для удаления до такой степени, чтобы дрожжи не дезактивировались в процессе ферментации для этанола, и такое удаление требует значительной энергии. Напротив, когда используют разбавленную серную кислоту, серную кислоту относительно легко удалять. Однако существует необходимость разлагать целлюлозу в условиях высоких температур, что требует потребления энергии. Еще другая проблема, возникающая, когда используют концентрированную серную кислоту, возникает в том случае, когда реакцию проводят в течение длительного времени, при этом полученный сахарид дегидратируется и выход сахарида снижается. В результате этого, даже когда растительный волокнистый материал добавляют в реакционную систему в течение гидролиза, чтобы увеличить количество растительного волокнистого материала, подлежащего гидролизу, выход сахарида, относящегося к растительному волокнистому материалу, не повышается. Кроме того, кислота, такая как серная кислота и хлористоводородная кислота, очень трудна для того, чтобы отделять, собирать и повторно использовать. Таким образом, применение данных кислот в качестве катализатора для получения глюкозы является причиной повышенной стоимости биоэтанола.
В способе, в котором используют горячую воду под давлением, трудно регулировать условия и трудно производить глюкозу со стабильным выходом. Кроме того, по данному способу глюкоза еще и разлагается, тем самым снижая выход глюкозы. Более того, активность дрожжей снижается деградируемыми компонентами, и ферментация может быть подавлена. Другая проблема связана со стоимостью, так как реактор (аппарат для суперкритической обработки) является дорогим и имеет малый срок службы.
Сущность изобретения
Авторы изобретения провели всестороннее изучение гликозилирования целлюлозы и обнаружили, что кластерная кислота в псевдорасплавленном состоянии обладает отличной каталитической активностью в отношении гидролиза целлюлозы и может быть легко отделена от полученного сахарида. Патентные заявки, которые охватывают соответствующий способ, уже были зарегистрированы (японская патентная заявка № 2007-115407 и японская патентная заявка № 2007-230711). Согласно данному способу, в противоположность обычному способу с применением концентрированной серной или разбавленной серной кислоты, катализатор гидролиза может быть выделен и использован повторно, и эффективность энергии процесса, ведущего к выделению водного раствора сахарида и регенерации катализатора гидролиза от гидролиза целлюлозы, может быть повышена. Кроме того, в способе приведенных выше патентных заявок кластерная кислота в псевдорасплавленном состоянии действует в качестве катализатора гидролиза, а также действует в качестве растворителя реакционной среды.
Авторы данного изобретения далее провели исследование гликозилирования целлюлозы, используя кластерный кислотный катализатор, и удачно повысили обрабатываемое количество растительного волокнистого материала на единицу массы кластерного кислотного катализатора. Таким образом, настоящее изобретение основано на результатах, полученных в ходе данного исследования, и относится к способу гликозилирования и выделения растительного волокнистого материала с применением кластерного кислотного катализатора в псевдорасплавленном состоянии, в котором повышено обрабатываемое количество растительного волокнистого материала на единицу массы кластерного кислотного катализатора, понижено количество используемого кластерного кислотного катализатора и увеличена эффективность энергии.
Первый аспект изобретения относится к способу гидролиза растительного волокнистого материала и получения и выделения сахарида, включающего глюкозу. Данный способ включает процесс гидролиза при использовании кластерного кислотного катализатора в псевдорасплавленном состоянии, чтобы гидролизовать целлюлозу, содержащуюся в растительном волокнистом материале, и получить глюкозу. В процессе гидролиза кластерный кислотный катализатор и первое количество растительного волокнистого материала, которое повышает вязкость кластерного кислотного катализатора в псевдорасплавленном состоянии при добавлении к кластерному кислотному катализатору в псевдорасплавленном состоянии, нагревают и смешивают и затем добавляют второе количество растительного волокнистого материала, когда происходит снижение вязкости горячей смеси кластерного кислотного катализатора и первого количества растительного волокнистого материала. Первое количество и второе количество могут быть одинаковыми.
В японской патентной заявке № 2007-115407 и японской патентной заявке № 2007-230711 авторы раскрыли способ гликозилирования и выделения растительного волокнистого материала, в котором кластерную кислоту нагревают с получением псевдорасплавленного состояния и используют в качестве катализатора гидролиза для растительного волокнистого материала. В данном способе гликозилирования и выделения кластерная кислота в псевдорасплавленном состоянии действует в качестве катализатора гидролиза, а также в качестве растворителя реакционной среды для гидролиза. По этой причине соотношение в смеси кластерного кислотного катализатора и растительного волокнистого материала в процессе гидролиза определяется таким образом, чтобы гарантировать смешиваемость кластерного кислотного катализатора и растительного волокнистого материала. Иными словами, количество растительного волокнистого материала, которое может быть смешано в одном цикле с кластерным кислотным катализатором, ограничено, и обрабатываемое количество растительного волокнистого материала на единицу массы кластерного кислотного катализатора также ограничено.
Результаты исследования, проведенного авторами изобретения, показали, что в способе гликозилирования и выделения растительного волокнистого материала при использовании кластерного кислотного катализатора, где растительный волокнистый материал загружают во множество циклов, как описано в настоящем описании выше, когда кластерный кислотный катализатор и растительный волокнистый материал перемешивают при нагревании и растительный волокнистый материал гидролизуют, обрабатываемое количество растительного волокнистого материала на единицу массы кластерного кислотного катализатора повышается. Таким образом, сначала кластерный кислотный катализатор и количество растительного волокнистого материала, которое добавляют для повышения вязкости, нагревают и перемешивают, и гидролиз растительного волокнистого материала начинается. Затем, когда вязкость горячей смеси, в которой растительный волокнистый материал гидролизован, снижается, растительный волокнистый материал добавляют дополнительно. Таким образом, было обнаружено, что горячая смесь кластерного кислотного катализатора в псевдорасплавленном состоянии и растительного волокнистого материала имеет высокую вязкость на начальной стадии реакции гидролиза, но вязкость снижается по мере того, как происходит гидролиз растительного волокнистого материала. Кроме того, было обнаружено, что из-за сниженной вязкости горячей смеси, даже когда растительный волокнистый материал помещен снова в горячую смесь, горячая смесь еще может быть смешана и перемешана, и как в начале загруженный растительный волокнистый материал, так и дополнительно добавленный растительный волокнистый материал могут быть гидролизованы с одновременным обеспечением высокого выхода сахарида. Иными словами, согласно изобретению обработанное количество растительного волокнистого материала может быть увеличено за счет количества растительного волокнистого материала, которое добавлено дополнительно, по сравнению с традиционным количеством. В результате этого увеличивается обрабатываемое количество растительного волокнистого материала на единицу массы кластерного кислотного катализатора, и получен эффект снижения стоимости при производстве сахарида из-за снижения количества используемого кластерного кислотного катализатора. Кроме того, так как растительный волокнистый материал дополнительно добавлен в горячую смесь в каталитическом процессе, который включает кластерный кислотный катализатор в псевдорасплавленном состоянии, энергия, требуемая для нагревания, которое необходимо для получения псевдорасплавленного состояния кластерного кислотного катализатора, может быть снижена. Таким образом, эффективность энергии может быть увеличена. Степень снижения вязкости горячей смеси, при которой дополнительно добавляется растительный волокнистый материал, может быть определена в соответствии с количеством растительного волокнистого материала, который будет добавлен дополнительно. Таким образом, в случае, когда дополнительно должно быть добавлено небольшое количество, оно может быть загружено после относительно небольшого снижения вязкости, но в случае, когда должно быть добавлено большое количество, растительный волокнистый материал не добавляют до тех пор, пока реакция гидролиза происходит достаточно и вязкость снижается значительно. В любом случае предпочтительно, чтобы растительный волокнистый материал дополнительно добавляли таким образом, чтобы не превышать вязкость, достигнутую после первоначального добавления волокнистого материала.
Индикатор периода, когда следует еще добавлять второе количество растительного волокнистого материала, может, например, представлять собой время, когда вязкость горячей смеси уменьшается до или ниже 1500 сП (=1500 мПа.с).
Объемный показатель первого количества растительного волокнистого материала по отношению к кластерному кислотному катализатору может быть равен или быть больше 60%. Объемный показатель количества растительного волокнистого материала, который впоследствии добавляют к кластерному кислотному катализатору, может быть равен или быть больше 60%.
Согласно настоящему изобретению в способе гликозилирования и выделения растительного волокнистого материала с использованием кластерного кислотного катализатора в псевдорасплавленном состоянии возможно повышать гидролизуемое количество растительного волокнистого материала на единицу массы кластерного кислотного катализатора, снижать количество используемой кластерной кислоты и повышать эффективность энергии. Поэтому согласно настоящему изобретению стоимость и потребление энергии при производстве сахарида гидролизом растительного волокнистого материала могут быть снижены.
Второй аспект изобретения относится к способу гидролиза растительного волокнистого материала и получения и выделения сахарида, включающего глюкозу. Данный способ включает процесс гидролиза при использовании кластерного кислотного катализатора в псевдорасплавленном состоянии, чтобы гидролизовать целлюлозу, содержащуюся в растительном волокнистом материале, и получить глюкозу. В процессе гидролиза растительный волокнистый материал добавляют, когда вязкость смеси кластерного кислотного катализатора и растительного волокнистого материала становится первой заранее определенной величиной, и затем добавление растительного волокнистого материала останавливают, когда вязкость смеси становится второй заранее определенной величиной, которая больше, чем первая заранее определенная величина.
Краткое описание чертежей
Вышеизложенные и дополнительные объекты, особенности и преимущества настоящего изобретения станут очевидными из следующего описания конкретных вариантов осуществления со ссылкой на сопутствующие графики, где использована одинаковая нумерация, чтобы изображать одинаковые элементы, и где:
на фиг.1 показана структура Кеггина гетерополикислоты;
на фиг.2 представлен график, показывающий зависимость между показателем кристаллизационной воды в кластерном кислотном катализаторе и кажущейся температурой плавления;
на фиг.3 представлена схематическая диаграмма, показывающая вариант осуществления устройства для реакции загрузочного типа, которое может быть использовано в процессе гидролиза; и
на фиг.4 представлена схематическая диаграмма поточного реакционного аппарата, который использован в процессе гидролиза примера 3.
Подробное описание вариантов осуществления
Способ гликозилирования и выделения растительного волокнистого материала, который представляет собой вариант осуществления изобретения, будет описан ниже. Сначала будет описан процесс гидролиза, в котором целлюлоза, содержащаяся в растительном волокнистом материале, подвергается гидролизу и получается сахарид, включающий в основном глюкозу. В объяснении ниже, изобретение сфокусировано на процессе, в котором глюкоза получена в основном из целлюлозы, но процесс, в который включена гемицеллюлоза в дополнение к целлюлозе в растительном волокнистом материале, и процесс, в котором продукт включает другие моносахариды, такие как ксилоза, в дополнение к глюкозе, также охватываются объемом настоящего изобретения.
Растительный волокнистый материал особенно не ограничивают при условии, что он включает целлюлозу или гемицеллюлозу, и его примеры включают биомассу на основе целлюлозы, а именно широколиственные деревья, бамбук, хвойные деревья, кенаф, древесные отходы от мебели, рисовую солому, солому пшеницы, рисовые отходы и остатки от обжима сахарного тростника (багасса). Растительный волокнистый материал может представлять собой целлюлозу или гемицеллюлозу, которая отделена от биомассы, или может представлять собой целлюлозу или гемицеллюлозу, которая синтезирована искусственно. Такие волокнистые материалы обычно используются в размельченной форме, чтобы улучшить диспергируемость в реакционной системе. Способ измельчения может представлять собой обычно применяемый способ. С точки зрения облегчения смешивания с кластерным кислотным катализатором и реакционной смесью предпочтительно, чтобы растительный волокнистый материал был размельчен в порошок с диаметром от примерно нескольких микрон до 200 мкм.
Лигнин, содержащийся в волокнистом материале, может быть растворен, если необходимо, путем проведения заранее пульпирующей обработки. Количество остатка в течение гликозилирования и выделения может быть уменьшено путем растворения и удаления лигнина, можно препятствовать смешиванию полученного сахарида или кластерной кислоты с остатком, и может быть проингибировано снижение выхода сахарида или показателя регенерации кластерной кислоты. В случае, где проведена пульпирующая обработка, степень измельчения растительного волокнистого материала может быть сравнительно небольшой (грубое измельчение). Полученный эффект состоит в том, что работа, стоимость и энергия для измельчения волокнистого материала могут быть уменьшены. Пульпирующая обработка может быть выполнена, например, путем введения в контакт растительного волокнистого материала (например, от нескольких сантиметров до нескольких миллиметров) со щелочью или солью, такой как NaOH, KOH, Ca(OH)2, Na2SO3, NaHCO3, NaHSO3, Mg(HSO3)2, Ca(HSO3)2, их водным раствором, их смесью с раствором SO3 или газа, такого как NH3 с водяным паром. Специфические условия включают температуру реакционной смеси от 120 до 160°С и время реакции от нескольких десятков минут до примерно 1 ч.
Согласно настоящему изобретению кластерная кислота, используемая в качестве катализатор для гидролиза растительного волокнистого материала, означает кислоту, в которой конденсированы многие оксокислоты, то есть так называемую поликислоту. В большинстве поликислот множество кислородных атомов присоединены к центральному элементу. В результате этого поликислоты, как известно, находятся, главным образом, в состоянии окисления до максимальной степени окисления, показывают отличные свойства в качестве катализатора окисления и являются сильными кислотами. Например, сила кислоты, а именно фосфовольфрамовой кислоты (рКа=-13,16), которая представляет собой гетерополикислоту, больше, чем сила кислоты, а именно серной кислоты (рКа=-11,93). Таким образом, даже в мягких температурных условиях, таких как температура 50°С, например, возможно разложить целлюлозу или гемицеллюлозу для получения моносахарида, такого как глюкоза или ксилоза.
Кластерная кислота, используемая в изобретении, может представлять собой либо гомополикислоту, либо гетерополикислоту, но предпочтительной является гетерополикислота, так как она имеет высокую окисляющую способность и высокую силу кислоты. Гетерополикислота, которая может быть использована, особенно не ограничена. Например, гетерополикислота может быть изображена общей формулой HwAxByOz (A означает гетероатом, В означает полиатом, который служит в качестве скелета поликислоты, w означает коэффициент атомов водорода в структуре, х означает коэффициент гетероатомов в структуре, y означает коэффициент полиатомов в структуре, и z означает коэффициент атомов кислорода в структуре). Примеры полиатома В включают атомы, такие как W, Mo, V и Nb, которые могут образовывать поликислоту. Примеры гетероатома А включают атомы, такие как P, Si, Ge, As и B, которые могут образовывать гетерополикислоту. Число видов полиатомов и гетероатомов, которые содержатся в одной молекуле гетерополикислоты, может составлять один или несколько видов.
Из-за хорошего баланса силы кислоты и окисляющей способности предпочтительно использовать фосфовольфрамовую кислоту H3[PW12O40] или кремнефольфрамовую кислоту H4[SiW12O40], которые представляют собой вольфраматы. Фосфомолибденовая кислота H3[PMo12O40], которая находится в виде молибдата, также преимущественно может быть использована. Структура гетерополикислоты (фосфовольфрамовой кислоты) Кеггин-типа [Xn+M12O40: X означает P, Si, Ge, As и т.д., М означает Мо, W и т.д.] показана на фиг.1. Четырехгранник ХО4 присутствует в центре политетраэдра, составленного единицами МО6 октаэдра, и большое количество кристаллизационной воды присутствует вокруг данной структуры. Структура кластерной кислоты особенно не ограничена и может быть не только Кеггин-типа, но также, например, Доусон-типа. В данном случае, вода, которая гидратирована или координирована с кластерным кислотным катализатором в кристаллическом состоянии или кластерным кислотным катализатором в кластерном состоянии, составленном несколькими молекулами кластерного кислотного катализатора, описана обычно применяемым термином “кристаллизационная вода”. Кристаллизационная вода включает анион воды, который через водород связан с анионом, составляющим кластерный кислотный катализатор, координационную воду, которая координирована с катионом, кристаллическую воду, которая не координирована с катионом или анионом, а также воду, которая заключена в форме ОН групп. Кластерный кислотный катализатор в кластерном состоянии представляет собой совокупность, составленную одной или несколькими молекулами кластерных кислот, и отличается от кристалла. Кластерный кислотный катализатор в кластерном состоянии может представлять собой форму твердого состояния, псевдорасплавленного состояния и растворенного состояния в растворителе (коллоидное состояние).
В гидролизном процессе гликозилирования и способе выделения согласно настоящему изобретению, растительный волокнистый материал разделяют и добавляют циклами. Поэтому моносахарид, который получался из растительного волокнистого материала, который был загружен в начале гидролизного процесса, нагревают и непрерывно перемешивают с кластерным кислотным катализатором вместе с дополнительно загруженным растительным волокнистым материалом. Поэтому наличие реакции дегидратации моносахарида (гиперреакция) заторможено, тем самым создается возможность для того, чтобы повышать выход моносахарида. С этой точки зрения предпочтительно, чтобы катализатор в кластерном состоянии, который имеет силу кислоты, подходящую для гидролиза целлюлозы, был использован в качестве кластерного кислотного катализатора. Так как кластерная кислота в кластерном состоянии почти не вызывает гиперреакцию моносахарида, выход моносахарида не снижается даже при длительном нагревании кластерной кислоты с моносахаридом. Способ получения кластерного кислотного катализатора в кластерном состоянии не особенно ограничен. Конкретный способ его получения будет описан ниже.
Вышеописанный кластерный кислотный катализатор находится в твердом состоянии при нормальной температуре, но становится псевдорасплавленным при более высокой температуре. Псевдорасплавленное состояние, указанное в настоящем описании, означает состояние, при котором кластерная кислота плавится кажущимся образом, но не полностью плавится в жидкое состояние; псевдорасплавленное состояние напоминает коллоидное (растворенное) состояние, в котором кластерная кислота диспергирована в жидкости, и представляет собой состояние, в котором кластерная кислота показывает текучесть. Находится ли кластерная кислота в псевдорасплавленном состоянии, можно подтвердить визуальными наблюдениями или, в случае гомогенной системы, при помощи DTG (дифференциальная сканирующая калориметрия). Псевдорасплавленное состояние кластерной кислоты изменяется в зависимости от температуры и количества кристаллизационной воды в кластерном кислотном катализаторе (см. фиг.2). Более конкретно, в случае, где количество кристаллизационной воды, содержащейся в фосфовольфрамовой кислоте, которая представляет собой кластерную кислоту, является высоким, температура, при которой данная кислота показывает псевдорасплавленное состояние, снижается. Таким образом, кластерный кислотный катализатор, содержащий большое количество кристаллизационной воды, показывает каталитический эффект на реакцию гидролиза целлюлозы при температуре, более низкой, чем температура кластерного кислотного катализатора с относительно небольшим количеством кристаллизационной воды. Иными словами, контролированием количества кристаллизационной воды, заключенной в кластерном кислотном катализаторе в реакционной системе гидролизного процесса, можно вводить кластерный кислотный катализатор в псевдорасплавленное состояние при целевой температуре реакции гидролиза. Например, когда фосфовольфрамовую кислоту используют в качестве кластерного кислотного катализатора, можно контролировать температуру реакции гидролиза в пределах интервала от 40 до 110°С за счет изменения количества кристаллизационной воды в кластерной кислоте (см. фиг.2).
Фиг.2 иллюстрирует зависимость между показателем кристаллизационной воды в гетерополикислоте (фосфовольфрамовая кислота), которая является типичным кластерным кислотным катализатором, и температурой (кажущаяся температурой плавления), при которой псевдорасплавленное состояние показано в начале. Кластерный кислотный катализатор находится в твердом состоянии в области под кривой и в псевдорасплавленном состоянии в области выше кривой. Кроме того, на фиг.2 соотношение кристаллизационной воды (%) представляет собой величину, полученную с предположением, что стандартное количество кристаллизационной воды n (n=30) в кластерной кислоте (фосфовольфрамовая кислота) составляет 100%. Поскольку нет компонента кластерного кислотного катализатора, который термически разлагается и улетучивается даже при высокой температуре, такой как 800°С, количество кристаллизационной воды может быть определено пиролитическим методом (термогравиметрические (TG) определения).
Стандартное количество кристаллизационной воды, указанной в настоящем описании, представляет собой количество (число молекул) кристаллизационной воды, содержащейся в молекуле кластерной кислоты в твердом состоянии при комнатной температуре, и стандартное количество изменяется в зависимости от вида кластерной кислоты. Например, стандартное количество кристаллизационной воды равно примерно 30 в фосфовольфрамовой кислоте (H3[PW12O40].nH2O (n≈30)), примерно 24 в кремнефольфрамовой кислоте (H4[SiW12O40].nH2O (n≈24)), и примерно 30 в фосфомолибденовой кислоте (H3[PMo12O40].nH2O (n≈30)).
Количество кристаллизационной воды, содержащейся в кластерном кислотном катализаторе, можно регулировать контролированием количества воды, присутствующей в системе реакции гидролиза. В частности, когда требуется увеличить количество кристаллизационной воды, содержащейся в кластерном кислотном катализаторе, то есть снизить температуру реакции, воду можно внести в систему реакции гидролиза путем добавления воды к смеси, содержащей растительный волокнистый материал и кластерный кислотный катализатор, или путем подъема относительной влажности атмосферы реакционной системы. В результате этого кластерная кислота берет в добавленной воде воду в виде кристаллизационной и кажущаяся температура плавления кластерного кислотного катализатора снижается.
Напротив, когда требуется уменьшить количество кристаллизационной воды, содержащейся в кластерном кислотном катализаторе, то есть поднять температуру реакции, можно снизить количество кристаллизационной воды, содержащейся в кластерном кислотном катализаторе, путем удаления воды из системы реакции гидролиза, например, нагреванием реакционной смеси для испарения воды или добавлением десиканта к смеси, содержащей растительный волокнистый материал и кластерный кислотный катализатор. В результате этого кажущаяся температура плавления кластерного кислотного катализатора поднимается. Как описано выше, можно легко контролировать количество кристаллизационной воды, содержащейся в кластерной кислоте, а также можно легко регулировать температуру реакции, при которой гидролизуется целлюлоза, путем контроля количества кристаллизационной воды.
Как описано выше, кластерная кислота проявляет высокую каталитическую активность в отношении гидролиза целлюлозы даже при низкой температуре из-за высокой кислотной силы кластерной кислоты. Поскольку диаметр молекулы кластерной кислоты составляет примерно от 1 до 2 нм, обычно несколько больше, чем 1 нм, кластерная кислота легко смешивается с растительным волокнистым материалом, который представляет собой необработанный материал, и поэтому эффективно способствует гидролизу целлюлозы. Таким образом, можно гидролизовать целлюлозу в мягких температурных условиях с высокой эффективностью энергии и малой нагрузкой для окружающей среды. Кроме того, в противоположность обычному способу гидролиза целлюлозы, который использует кислоту, такую как серная кислота, способ согласно настоящему изобретению использует кластерную кислоту в качестве катализатора, эффективность выделения сахарида и катализатора является высокой, и они могут быть легко разделены. Поскольку кластерная кислота находится в твердом состоянии при определенной температуре, она может быть отделена от сахарида, который является продуктом. Поэтому отделенная кластерная кислота может быть регенерирована и получена обратно. Кроме того, поскольку кластерный кислотный катализатор в псевдорасплавленном состоянии также функционирует как катализатор реакции, количество растворителя, используемого в качестве растворителя реакции, может быть сильно уменьшено по сравнению с количеством обычного способа. Это означает, что разделение кластерной кислоты и сахарида, который является продуктом, и выделение кластерной кислоты может быть проведено с повышенной эффективностью. Таким образом, изобретение, в котором кластерная кислота использована в качестве катализатора гидролиза целлюлозы, может уменьшать себестоимость и снижать нагрузку на окружающую среду.
Согласно настоящему изобретению в процессе гидролиза, в котором растительный волокнистый материал гидролизуют и сахарид получают при использовании кластерного кислотного катализатора в псевдорасплавленном состоянии, кластерный кислотный катализатор и количество растительного волокнистого материала, который увеличивает вязкость кластерного кислотного катализатора в псевдорасплавленном состоянии при добавлении к кластерному кислотному катализатору в псевдорасплавленном состоянии, нагревают и смешивают, тем самым происходит гидролиз растительного волокнистого материала, и растительный волокнистый материал затем дополнительно загружают после того, как вязкость горячей смеси снижена.
В данном случае количество растительного волокнистого материала, которое повышает вязкость кластерного кислотного катализатора в псевдорасплавленном состоянии при добавлении к кластерному кислотному катализатору в псевдорасплавленном состоянии, как указано в настоящем описании, представляет собой количество, которое повышает вязкость, когда кластерный кислотный катализатор, используемый в процессе гидролиза, находится в псевдорасплавленном состоянии, и растительный волокнистый материал добавлен к кластерному кислотному катализатору в псевдорасплавленном состоянии в пределах вязкости кластерного кислотного катализатора в псевдорасплавленном состоянии перед добавлением растительного волокнистого материала. Конкретное количество растительного волокнистого материала различается в зависимости от свойств используемого растительного волокнистого материала (размер, вид, пористая структура и тому подобное) и температуры нагревания, состояния перемешивания (растирания) и распределения температуры в течение смешивания кластерного кислотного катализатора растительного волокнистого материала в псевдорасплавленном состоянии и растительного волокнистого материала. Поэтому конкретное количество может быть определено заранее. Обычно, в случае, где объемный показатель растительного волокнистого материала по отношению к кластерному кислотному катализатору, используемому в процессе гидролиза, равен или больше 60%, вязкость увеличивается, когда растительный волокнистый материал добавлен к кластерному кислотному катализатору в псевдорасплавленном состоянии. В частности, с точки зрения эффективности обработки растительного волокнистого материала предпочтительно, что объемный показатель растительного волокнистого материала по отношению к кластерному кислотному катализатору, используемому в процессе гидролиза, равен или больше 50%, более предпочтительно равен или больше 65%.
Последовательность, с которой кластерный кислотный катализатор и растительный волокнистый материал загружают в реакционный контейнер, не особенно ограничена. Например, кластерный кислотный катализатор может быть загружен и нагрет с получением псевдорасплавленного состояния, и затем может быть загружен растительный волокнистый материал. В альтернативном случае кластерный кислотный катализатор и растительный волокнистый материал могут быть загружены вместе и затем нагреты, чтобы вносить кластерный кислотный катализатор в псевдорасплавленном состоянии. В случае, где кластерный кислотный катализатор и растительный волокнистый материал нагревают после загрузки, кластерный кислотный катализатор и растительный волокнистый материал предпочтительно смешивают и перемешивают заранее, перед нагреванием. Степень контакта между кластерной кислотой и растительным волокнистым материалом может быть увеличена при смешивании до определенной степени, перед тем как кластерные кислоты вводят в псевдорасплавленное состояние.
Как представлено в настоящем описании, поскольку кластерный кислотный катализатор находится в псевдорасплавленном состоянии и функционирует как катализатор реакции в процессе гидролиза, согласно настоящему изобретению можно не использовать воду или органический растворитель в качестве растворителя реакции в процессе гидролиза, но вода или органический растворитель могут потребоваться в зависимости от формы (размер, состояние волокон и т.д.) растительного волокнистого материала, соотношения смеси и объемного соотношения кластерного кислотного катализатора и растительного волокнистого материала и тому подобное. Однако вода необходима для гидролизуемой целлюлозы в процессе гидролиза. Более конкретно, (n-1) молекул воды требуются, чтобы разложить глюкозу, в которой (n) глюкоз было полимеризовано в (n) глюкозы (n является природным числом). Поэтому в случае, где общая сумма количества кристаллизационной воды, необходимого для приведения кластерной кислоты в псевдорасплавленное состояние при температуре реакции, и количества воды, необходимого для гидролиза всего загруженного количества целлюлозы в глюкозу, не представлена в реакционной системе, для гидролиза целлюлозы используют кристаллизационную воду кластерного кислотного катализатора, при этом количество кристаллизационной воды кластерного кислотного катализатора снижается, и кластерная кислота твердеет. Таким образом, степень контакта между кислотным катализатором и растительным волокнистым материалом или вязкость смеси растительного волокнистого материала и кластерного кислотного катализатора повышается и требуется продолжительное время, чтобы перемешать смесь в достаточной степени.
Поэтому, чтобы обеспечить каталитическое действие кластерного кислотного катализатора и его функцию как растворителя реакционной смеси при температуре реакции в процессе гидролиза, то есть чтобы создавать возможность кластерному кислотному катализатору поддерживать псевдорасплавленное состояние, предпочтительно, чтобы количество воды в системе реакции удовлетворяло следующему условию. Таким образом, предпочтительно, чтобы количество воды в системе реакции было равно или было больше, чем общая сумма (А) количества кристаллизационной воды, необходимого для всего кластерного кислотного катализатора, присутствующего в системе реакции, чтобы быть в псевдорасплавленном состоянии при температуре реакции в процессе гидролиза, и (В) количества воды, необходимого для всего количества целлюлозы, присутствующей в системе реакции, предназначенной для гидролиза в глюкозу. Особенно предпочтительно добавлять общее количество из суммы (А) и (В). Из-за того что добавлена дополнительная вода, полученный сахарид и кластерная кислота растворяются в избытке воды, и процесс выделения сахарида и кластерной кислоты становится трудным. Перед дополнительной загрузкой растительного волокнистого материала, который загружают дополнительно, количество (В) воды представляет собой количество (В1) воды, которое необходимо, чтобы гидролизовать в глюкозу все количество целлюлозы, содержащейся в растительном волокнистом материале, который подвергнут гидролизу в начале, и после дополнительной загрузки растительного волокнистого материала, количество (В) воды представляет собой количество (В1+В2), которое необходимо, чтобы гидролизовать в глюкозу все количество целлюлозы, содержащейся в растительном волокнистом материале, который гидролизован дополнительно, и затем в добавленном растительном волокнистом материале. Что касается количества (В) воды, общее количество (В1+В2) может быть добавлено перед дополнительной загрузкой растительного волокнистого материала, или В1 и В2 могут быть добавлены по отдельности соответственно дополнительной загрузке растительного волокнистого материала.
В процессе гидролиза, количество воды в системе реакции уменьшается, и количество кристаллизационной воды кластерного кислотного катализатора также уменьшается. В результате этого, кластерный кислотный катализатор может стать твердым и степень контакта с растительным волокнистым материалом и способность к смешиванию реакционной системы может ухудшаться. Наличие таких проблем может быть устранено повышением температуры гидролиза таким образом, чтобы кластерный кислотный катализатор приводился в псевдорасплавленное состояние. Кроме того, предпочтительно, чтобы требуемое количество кристаллизационной воды кластерного кислотного катализатора могло быть обеспечено, даже когда относительная влажность реакционной системы снижена нагреванием в процессе гидролиза. В частности, может быть использован способ, по которому состояние давления насыщенного пара получают при температуре реакции гидролиза внутри предварительно герметизированного реакционного контейнера, так что атмосфера реакционной системы при заранее определенной температуре реакции находится под давлением насыщенного пара, температуру снижают для конденсации паров, одновременно поддерживая герметичность, и конденсированную воду добавляют к растительному волокнистому материалу и кластерному кислотному катализатору. Кроме того, в случае, где используют растительный волокнистый материал, содержащий влагу, предпочтительно, количество влаги, содержащейся в растительном волокнистом материале, должно быть принято во внимание как количество влаги, присутствующей в реакционной системе; это не является особенно необходимым в случае, где используют сухой растительный волокнистый материал.
Когда реакция гидролиза растительного волокнистого материала произошла, и вязкость горячей смеси уменьшилась, растительный волокнистый материал загружают дополнительно. Вязкость горячей смеси может быть измерена непосредственно вискозиметром (например, сдвиговым звуковым резонатором или тому подобное), расположенным внутри реакционного контейнера, или определена косвенно из вращающего момента лопасти для перемешивания, которая перемешивает горячую смесь, высоты жидкости, измеренной прибором для определения уровня жидкости, размещенным внутри реакционного контейнера, и зависимости между вращением и вращающим моментом лопасти для перемешивания. Вязкость горячей смеси в период, когда растительный волокнистый материал дополнительно загружен, не ограничена до определенной величины, при условии, что она меньше, чем вязкость горячей смеси, включающей кластерный кислотный катализатор в псевдорасплавленном состоянии и растительный волокнистый материал на начальной стадии реакции процесса гидролиза, и что горячая смесь может быть смешана, несмотря на дополнительную загрузку растительного волокнистого материала к горячей смеси. Вязкость горячей смеси, в которую растительный волокнистый материал должен быть дополнительно загружен, может быть приблизительно определена соответственно количеству растительного волокнистого материала, который будет дополнительно загружен. Таким образом, в случае, где дополнительно должно быть загружено небольшое количество, дополнительная загрузка может быть проведена, когда вязкость слегка уменьшилась, и где дополнительно должно быть загружено большое количество, дополнительную загрузку проводят после ожидания, пока реакция гидролиза протекает достаточно и вязкость снижается значительно. В любом случае, предпочтительно, чтобы дополнительная загрузка выполнялась таким образом, чтобы не превышать вязкость горячей смеси на начальной стадии реакции, когда волокнистый материал добавлен в начале. Обычно предпочтительно, чтобы растительный волокнистый материал дополнительно загружали после того, как вязкость горячей смеси уменьшилась до или ниже 1500 сП (=1500 мПа.с), предпочтительно была равна или ниже 1200 сП (=1200 мПа.с) и даже более предпочтительно была равна или ниже 1000 сП (=1000 мПа.с).
Количество растительного волокнистого материала, который загружают дополнительно, не особенно ограничено и может быть приблизительно определено при условии, что оно находится в интервале, в котором способность к смешиванию горячей смеси после дополнительной загрузки растительного волокнистого материала может быть гарантирована. С точки зрения эффективности обработки растительного волокнистого материала обычно предпочтительно, чтобы объемный показатель затем добавленного растительного волокнистого материала по отношению к кластерному кислотному катализатору, который используется в процессе гидролиза, был равен или больше 60%. Дополнительная загрузка растительного волокнистого материала может быть выполнена путем множества циклов. Таким образом, возможно повторять процесс, в котором растительный волокнистый материал дополнительно загружают, после того как вязкость горячей смеси уменьшилась от предыдущей дополнительной загрузки растительного волокнистого материала.
Дополнительную загрузку волокнистого материала в процессе гидролиза можно легко контролировать путем установки обратной связи, адресующей изменения вязкости горячей смеси, которые измеряют описанным выше вискозиметром, вращающим моментом лопасти для перемешивания, прибором для определения уровня жидкости или тому подобное, к механизму загрузки растительного волокнистого материала и дополнительной загрузки растительного волокнистого материала к горячей смеси, когда вязкость горячей смеси уменьшается. Более конкретно, например, в случае фиксированного реакционного аппарата (загрузочный тип), показанного на фиг.3, вязкость горячей смеси 4, расположенной в реакционном контейнере 1, может быть измерена сенсором вязкости 2 и сенсором уровня жидкости 3. Сенсор вязкости 2, который измеряет вязкость горячей смеси 4, предпочтительно размещен на донной поверхности реакционного контейнера 1 или в положении близко к поверхности дна на стороне. Кроме того, в случае, где множество сенсоров уровня жидкости 3 расположено на боковой поверхности реакционного контейнера 1, изменения в уровне жидкости горячей смеси 4 внутри реакционного контейнера 1 могут быть точно измерены. В данном случае, обеспечением множества сенсоров температуры 5 вместе с сенсорами уровня жидкости 3 можно проводить адекватное регулирование скорости вращения лопасти для перемешивания (см. фиг.3). Как описано выше, изменения в вязкости горячей смеси 4, которые измеряются сенсором вязкости 2 и сенсором уровня жидкости 3, предпочтительно взаимосвязаны с механизмом загрузки 9 растительного волокнистого материала. В расположении, показанном на фиг.3, обогреватель 7 и сенсор температуры 8 помещены на донной поверхности реакционного контейнера 1, и температура горячей смеси 4, находящейся в реакционном контейнере 1, может быть проконтролирована. Кроме того, конфигурация фиксированного реакционного аппарата (загрузочный тип) не ограничена конфигурацией, показанной на фиг.3. Например, как описано в настоящем описании выше, вязкость горячей смеси 4 может быть измерена косвенно из вращающего момента лопасти для перемешивания 6.
На фиг.4 показан вариант осуществления реакционного аппарата с потоком. В цилиндрическом реакционном контейнере 100 с устройством для перемешивания (лопасть для перемешивания 10), показанным на фиг.4, множество входов 11(1)-11(4) для загрузки растительного волокнистого материала и сенсоров вязкости 12(1)-12(4) расположено в направлении потока вниз по течению из загрузочного входа 13 для кластерного кислотного катализатора в псевдорасплавленном состоянии. Дополнительный период загрузки или дополнительно загруженное количество растительного волокнистого материала, предназначенного для загрузки загрузочных входов 11, могут быть определены из вязкости горячей смеси, которую измеряют сенсором вязкости 12 при условии нисходящего потока для положений, где расположены загрузочные входы 11. В данном случае реакцию можно легко контролировать в процессе гидролиза путем установления обратной связи, адресующей изменения в вязкости горячей смеси, которые измеряют сенсорами вязкости 12 вискозиметром, дополнительному загрузочному периоду или дополнительно загруженному количеству растительного волокнистого материала, который будет дополнительно загружен из загрузочных входов 11. Места расположения сенсоров вязкости 12 и загрузочных входов 11 особенно не ограничены. Например, сенсор вязкости 12(1) может быть расположен по соседству к и против потока из загрузочного входа 11(2), который расположен в нисходящем потоке из загрузочного входа 11(1) (см. фиг.4). Загрузочные входы 11(2) и 11(3) и сенсоры вязкости 12(2) и 12(3) расположены аналогично. В реакционном аппарате с потоком в случае, где использован растительный волокнистый материал, включающий лигнин, гетерополикислоту и полученный сахарид не удаляют сразу перед загрузочными входами 11, но остаток против потока предпочтительно удаляют, располагая фильтр, который может удалять лигнин.
Преимущество снижения температуры реакции в процессе гидролиза означает, что эффективность энергии может быть повышена. Селективность образования глюкозы при гидролизе целлюлозы, содержащейся в растительном волокнистом материале, изменяется в зависимости от процесса гидролиза. Эффективность реакции обычно поднимается по мере того, как поднимается температура реакции. Например, как описано в японской патентной заявке № 2007-115407, в реакции гидролиза целлюлозы, использующей фосфовольфрамовую кислоту с соотношением кристаллизационной воды 160%, показатель реакции R при температуре 50-90°C поднимается с повышением температуры и почти вся целлюлоза реагирует примерно при 80°С. Выход глюкозы показывает аналогичную тенденцию к повышению при температуре 50-60°С, достигает пика при 70°С и затем уменьшается. Таким образом, глюкозу получают с высокой селективностью при температуре 50-60°С, но при температуре 70-90°С также протекают реакции, другие чем образование глюкозы, а именно образование других сахаридов, таких как ксилоза, и образование продуктов разложения. Поэтому температура реакции гидролиза является важным фактором, который регулирует селективность показателя реакции целлюлозы и селективность получения глюкозы, и предпочтительно, чтобы температура реакции гидролиза была низкой ввиду эффективности энергии. Однако предпочтительно, чтобы температура реакции гидролиза была определена с учетом также показателя реакции целлюлозы и селективности получения глюкозы.
Как описано выше, температурные условия в процессе гидролиза могут быть соответственно определены при рассмотрении нескольких факторов (например, селективности реакции, эффективности энергии, показателя реакции целлюлозы и т.д.), но с точки зрения баланса эффективности энергии, показателя реакции целлюлозы и выхода глюкозы, обычно предпочтительно температура равна или ниже чем 140°С и особенно предпочтительно температура равна или ниже чем 120°С. В зависимости от формы растительного волокнистого материала можно также использовать температуру, равную или ниже 100°С. В данном случае глюкоза может быть получена с особенно высокой эффективностью энергии.
Давление в процессе гидролиза особенно не ограничено, но поскольку каталитическая активность кластерного кислотного катализатора в отношении реакции гидролиза целлюлозы является высокой, гидролиз целлюлозы может протекать с хорошей эффективностью даже в мягких условиях давления, таких как интервал от нормального давления (атмосферное давление) до 1 МПа.
Поскольку смесь, содержащая кластерный кислотный катализатор и растительный волокнистый материал, в процессе гидролиза имеет высокую вязкость, например, преимущественно может быть использована шаровая мельница с применением подогрева, но также может быть использовано обычное устройство для перемешивания.
Продолжительность процесса гидролиза особенно не ограничена, и может быть надлежащим образом установлена в соответствии с видом используемого растительного волокнистого материала, соотношения растительного волокнистого материала и кластерного кислотного катализатора, каталитической активности кластерного кислотного катализатора, температуры реакции и тому подобное.
Там, где температура реакционной системы снижается, после того как снижено завершение реакции, сахарид, полученный в процессе гидролиза, становится водным раствором сахарида, когда вода, которая растворяла сахарид, присутствует в смеси реакции гидролиза, содержащей кластерный кислотный катализатор, и где вода не присутствует, сахарид осаждается и содержится в твердом состоянии. Часть полученного сахарида может присутствовать в форме водного раствора, и баланс может содержаться в виде смеси в твердом состоянии. Поскольку кластерный кислотный катализатор также растворим в воде, в тех случаях, где достаточное количество воды содержится в смеси после процесса гидролиза, кластерный кислотный катализатор также растворен в воде.
Процесс выделения сахарида, в котором выделены сахарид (в основном включающий глюкозу), полученный в процессе гидролиза, и кластерный кислотный катализатор, будет описан ниже. В способе гликозилирования и выделения согласно настоящему изобретению способ выделения сахарида и кластерной кислоты не ограничен описанным ниже способом.
Реакционная смесь после процесса гидролиза (может быть также названа в настоящем описании как “смесь реакции гидролиза”) содержит по меньшей мере кластерный кислотный катализатор и полученный сахарид. В случае, где количество воды в процессе гидролиза представляет собой общую сумму из (А) и (В), сахарид из смеси реакции гидролиза осаждается. Одновременно кластерный кислотный катализатор также становится твердым, когда температура снижается. В зависимости от типа используемого растительного волокнистого материала остаток (не прореагировавшая целлюлоза или лигнин и т.д.) содержится в виде твердого компонента в смеси реакции гидролиза.
Кластерный кислотный катализатор проявляет растворимость в органических растворителях, в которых сахарид, в основном включающий глюкозу, нерастворим или имеет малую растворимость. Поэтому можно добавлять органический растворитель, который является слабым растворителем для сахарида и хорошим растворителем для кластерного кислотного катализатора, к смеси реакции гидролиза, проводить перемешивание, селективно растворять кластерный кислотный катализатор в органическом растворителе и затем отделять раствор в органическом растворителе, содержащий растворенные кластерные кислоты и твердый компонент, включающий сахарид, путем разделения твердого вещества-жидкости. В зависимости от используемого растительного волокнистого материала в твердом компоненте, включающем сахарид, может содержаться осадок или тому подобное. Способ разделения раствора в органическом растворителе и твердого компонента особенно не ограничен, и может быть использован обычный способ разделения твердого вещества-жидкости, такой как декантация или фильтрация.
Органический растворитель особенно не ограничен при условии, что он является хорошим растворителем для кластерного кислотного катализатора и слабым растворителем для сахарида, но для подавления растворения сахарида в органическом растворителе предпочтительно, чтобы растворимость сахарида в органическом растворителе была равна или меньше 0,06 г/100 мл и более предпочтительно была равна или меньше 0,6 г/100 мл. В данном случае, чтобы повысить показатель извлечения кластерного кислотного катализатора, предпочтительно, чтобы растворимость кластерной кислоты в органическом растворителе была равна или больше 20 г/100 мл, более предпочтительно была равна или больше 40 г/100 мл.
Конкретные примеры органического растворителя включают спирты, такие как этанол, метанол, н-пропанол и октанол, и простые эфиры, такие как диэтиловый эфир и диизопропиловый эфир. Преимущественно могут быть использованы спирты и простые эфиры и среди них, с точки зрения способности к растворению и точки кипения, предпочтительно этанол и диэтиловый эфир. Диэтиловый эфир не растворяет сахариды, такие как глюкоза, и обладает высокой способностью к растворению кластерных кислот. Поэтому простой диэтиловый эфир является одним из оптимальных растворителей для выделения сахаридов и кластерных кислотных катализаторов. Этанол также почти не растворяет сахариды, такие как глюкоза, и обладает высокой способностью к растворению кластерных кислот. Поэтому он также является одним из оптимальных растворителей. Диэтиловый эфир превосходит этанол в плане перегонки, но преимущество этанола состоит в том, что его легче получать, чем диэтиловый эфир.
Количество используемого органического растворителя отличается в зависимости от способности растворителя растворять сахарид и кластерный кислотный катализатор и количества влаги, содержащейся в смеси реакции гидролиза. Поэтому подходящее количество органического растворителя может быть определено соответствующим образом.
Обычно предпочтительно перемешивание смеси реакции гидролиза и органического растворителя выполняют внутри температурного интервала от комнатной температуры до 60°С, причем конкретная температура зависит от точки кипения органического растворителя. Способ перемешивания смеси реакции гидролиза и органического растворителя особенно не ограничен, и перемешивание может быть выполнено обычным способом. С точки зрения эффективности извлечения кластерной кислоты, перемешивание и размол на шаровой мельнице предпочтительно в качестве способа перемешивания.
Чтобы повысить показатель извлечения сахарида и кластерной кислоты и увеличить чистоту полученного сахарида, предпочтительно органический растворитель (органический растворитель, который является слабым растворителем для сахарида и хорошим растворителем для кластерного кислотного растворителя) добавляют и перемешивают с твердым компонентом, полученным путем приведенного выше разделения твердого вещества-жидкости, при этом проводят промывание органическим растворителем. По этой причине кластерный кислотный растворитель, который примешан к твердому компоненту, может быть удален и извлечен. Смесь, в которой органический растворитель добавлен к твердому компоненту, может быть разделена на твердый компонент и раствор в органическом растворителе, содержащий кластерную кислоту, путем разделения твердого вещества-жидкости как для смеси реакции гидролиза. Если необходимо, твердый компонент может быть промыт несколько раз органическим растворителем. Добавлением воды, а именно дистиллированной воды к твердому компоненту, полученному разделением твердого вещества-жидкости водой, перемешиванием и затем проведением разделения твердого вещества-жидкости (поскольку сахарид растворим в воде), можно отделить водный раствор сахарида от твердого компонента, содержащего осадок или тому подобное.
Удалением органического растворителя от жидкого компонента (раствор в органическом растворителе, включающий кластерный кислотный катализатор, растворенный в нем), полученного разделением твердого вещества-жидкости, можно разделить кластерный кислотный катализатор и органический растворитель и извлечь кластерный кислотный катализатор. Способ удаления органического растворителя особенно не ограничен. Примеры подходящих способов включают перегонку в вакууме, сушку вымораживанием и испарительную сушку. Среди них предпочтительна перегонка в вакууме при температуре, равной или меньше чем 50°С. Извлеченный кластерный кислотный катализатор может быть снова использован в качестве катализатора гидролиза для растительного волокнистого материала. Раствор в органическом растворителе, содержащий выделенную кластерную кислоту после промывания твердого компонента, полученного указанным выше разделением твердого вещества-жидкости, может быть снова использован для промывания органического компонента.
В зависимости от количества влаги в процессе гидролиза смесь реакции гидролиза может содержать водный раствор, содержащий сахарид и кластерную кислоту в нем. В данном случае твердый компонент, содержащий сахарид, и органический растворитель, содержащий растворенный в нем кластерный кислотный катализатор, могут быть выделены путем удаления влаги из смеси реакции гидролиза, чтобы осадить растворенный сахарид и кластерную кислоту, и последующего добавления органического растворителя, перемешивания и проведения разделения твердого вещества-жидкости. Особенно предпочтительно, чтобы количество влаги в смеси реакции гидролиза было установлено так, чтобы соотношение кристаллизационной воды во всем кластерном кислотном катализаторе, содержащемся в смеси реакции гидролиза, было менее чем 100%. В случае, где кластерный кислотный катализатор имеет большое количество кристаллизационной воды, обычно количество кристаллизационной воды, которое равно или больше, чем стандартное количество кристаллизационной воды, сахарид, который является продуктом, были растворены в избытке влаги, и показатель извлечения сахарида снижен подмешиванием сахарида к жидкой фазе, включающей раствор в органическом растворителе, содержащий кластерную кислоту. Снижением соотношения кристаллизационной воды в кластерном кислотном катализаторе до менее чем 100% можно препятствовать сахариду таким образом примешиваться к кластерному кислотному катализатору.
Способ, который может уменьшать количество влаги в смеси реакции гидролиза, может быть использован для снижения показателя кристаллизационной воды в кластерном кислотном катализаторе, содержащемся в смеси реакции гидролиза. Примеры такого способа включают способ, по которому деблокируют герметичное состояние реакционной системы и осуществляют нагревание для испарения влаги, содержащейся в смеси реакции гидролиза, и способ, по которому десикант или тому подобное добавляют к гидролизной смеси и влага, содержащаяся в гидролизной смеси, удаляется.
Способ получения кластерного кислотного катализатора в кластерном состоянии будет рассмотрен ниже. Превращение кластерного кислотного катализатора в кластерное состояние усиливают, например, путем перемешивания кластерной кислоты в псевдорасплавленном состоянии или добавления кластерной кислоты к растворителю и перемешивания при нагревании, или перемешивания кластерной кислоты вместе с растительным волокнистым материалом при нагревании и приведением кластерной кислоты к действию в качестве катализатора гидролиза. Следующие три конкретных способа могут использоваться для усиления превращения в кластерное состояние. (1) Способ, включающий процесс нагревания и смешивания кластерного кислотного катализатора и органического растворителя, который может растворять кластерный кислотный катализатор; (2) способ, по которому в процессе гидролиза, где растительный волокнистый материал гидролизуется с использованием кластерного кислотного катализатора, часть растительного волокнистого материала в количестве, которое может быть загружено в одну порцию, перемешивают при нагревании с кластерным кислотным катализатором в псевдорасплавленном состоянии и проводят гидролиз растительного волокнистого материала; и (3) способ нагревания и перемешивания кластерного кислотного катализатора в псевдорасплавленном состоянии. Данные способы (1)-(3) будут описаны ниже.
По способу (1), который включает процесс нагревания и смешивания кластерного кислотного катализатора и органического растворителя, который может растворять кластерный кислотный катализатор, температуру нагревания можно соответственно устанавливать согласно изменению состояния кластерной кислоты в растворителе, но обычно предпочтительно температура равна или выше 30°С. С точки зрения предотвращения перекристаллизации кластерного кислотного катализатора предпочтительно температура равна или ниже 65°С, в частности равна или ниже 55°С. Примеры органических растворителей, которые могут растворять кластерный кислотный катализатор, включают органические растворители, которые могут быть использованы в описанном выше процессе выделения сахарида. Среди них, с точки зрения способности к растворению кластерной кислоты и точки кипения органического растворителя, предпочтительны этанол и метанол. Смесевое соотношение органического растворителя и кластерного кислотного катализатора особенно не ограничено и может быть соответственно подобрано надлежащим образом к растворимости кластерного кислотного катализатора в органическом растворителе. Время нагревания и перемешивания может быть соответственно подобрано надлежащим образом к растворимости кластерного кислотного катализатора в используемом органическом растворителе и температуре нагревания, и обычно время нагревания и перемешивания составляет примерно от 10 мин до 60 мин или примерно от 30 мин до 60 мин. Способ смешивания особенно не ограничен, и можно применять хорошо известный способ.
Даже в случае, где использован необычный новый кластерный кислотный реагент, такое нагревание и перемешивание кластерного кислотного катализатора и органической кислоты может превращать кластерный кислотный катализатор в кластерное состояние и ингибировать реакцию дегидратации сахарида в процессе гидролиза. Кроме того, кластерообразование повторно используемого кластерного кислотного катализатора может быть усилено добавлением органического растворителя к смеси реакции гидролиза и перемешиванием в процессе выделения сахарида и последующим нагреванием и перемешиванием раствора в органическом растворителе, включающем кластерную кислоту, полученную разделением твердого вещества-жидкости.
Кластерный кислотный катализатор, подлежащий обработке, повышающей кластерообразование, может быть выделен удалением органического растворителя из смеси кластерного кислотного катализатора и органического растворителя после нагревания и перемешивания. В данном случае быстрым удалением органического растворителя можно легко поддерживать кластерное состояние кластерного кислотного катализатора. Более конкретно предпочтительно органический растворитель удаляют вакуумной перегонкой, сушкой вымораживанием или подобным. Органический растворитель может быть также удален нагреванием, но с точки зрения поддержания кластерного состояния кластерной кислоты предпочтительно органический растворитель удаляют при низкой температуре (более конкретно при температуре, равной или ниже 65°С), и можно говорить, что предпочтительны указанные выше вакуумная перегонка и сушка вымораживанием.
Кроме того, кластерообразование добавленного кластерного кислотного катализатора и повторно используемого кластерного кислотного катализатора может быть также усилено добавлением органического растворителя к смеси реакции гидролиза и перемешиванием в процессе выделения сахарида, затем добавлением кластерного кислотного катализатора в кристаллическом состоянии (необычный кластерный кислотный реагент или тому подобное) к раствору в органическом растворителе, содержащем кластерную кислоту, полученную разделением твердого вещества-жидкости, и перемешиванием при нагревании. В дополнение к повторному извлечению и неоднократному использованию кластерного кислотного катализатора, даже в случае, где выделенное количество кластерной кислоты снижено, можно проводить кластерообразующую обработку кластерного кислотного катализатора в кристаллическом состоянии путем добавления кластерного кислотного катализатора в кристаллическом состоянии и использования процесса выделения сахарида, тем самым восполняя потерю кластерного кислотного катализатора в процессе выделения сахарида.
В способе 2, по которому часть растительного волокнистого материала в количестве, которое может быть загружено одной порцией, перемешивают при нагревании с кластерным кислотным катализатором в псевдорасплавленном состоянии и проводят гидролиз растительного волокнистого материала в процессе гидролиза, при этом гидролизом одной части растительного волокнистого материала, который может быть загружен в одну порцию, можно снизить количество моносахарида, которое может быть дегидратировано кластерным кислотным катализатором на начальной стадии процесса гидролиза, и повысить кластерообразование кластерного кислотного катализатора. После того как кластерный кислотный катализатор принял кластерное состояние, дополнительно загружают оставшийся растительный волокнистый материал, тем самым создается возможность ингибировать гиперреакцию сахарида, полученного из дополнительно загруженного растительного волокнистого материала.
“Растительный волокнистый материал в количестве, которое может быть загружено одной порцией”, как указано в настоящем описании, представляет собой количество, которое способно придать смеси полностью гомогенно смешанное и тестообразное состояние, когда данное количество смешано с кластерным кислотным катализатором (количество, используемое в процессе гидролиза) в псевдорасплавленном состоянии, которое используют в процессе гидролиза. В данном случае растительный волокнистый материал не находится в смеси в сухом состоянии. Поскольку количество растительного волокнистого материала, которое может быть загружено одной порцией, изменяется в зависимости от типа месильной машины, данное количество не может быть определено однозначно, но обычно предпочтительно массовое отношение (растительный волокнистый материал:кластерный кислотный катализатор) растительного волокнистого материала в количестве, которое может быть загружено одной порцией, и кластерного кислотного катализатора в псевдорасплавленном состоянии, которое используют в процессе гидролиза, составляет от 1:2 до 1:6. Кроме того, “часть растительного волокнистого материала в количестве, которое может быть загружено одной порцией”, как указано в настоящем описании, представляет собой часть указанного выше “растительного волокнистого материала в количестве, которое может быть загружено одной порцией” и не ограничено определенным количеством. Обычно оно представляет собой очень небольшое количество, такое, что вязкость кластерного кислотного катализатора в псевдорасплавленном состоянии перед добавлением сохраняется даже после того, как данное количество растительного волокнистого материала добавлено к и перемешано с кластерным кислотным катализатором в псевдорасплавленном состоянии. В случае, где такое очень небольшое количество растительного волокнистого материала первоначально добавлено к кластерному кислотному катализатору, используемому в процессе гидролиза, может ожидаться эффект повышения активности реакции в целом, из-за такой, как говорится, жертвы. Конкретное количество “части растительного волокнистого материала в количестве, которое может быть загружено одной порцией” предпочтительно равно или меньше 10% масс., в частности равно или меньше 5% масс. растительного волокнистого материала в количестве, которое может быть загружено одной порцией.
Время гидролиза порции растительного волокнистого материала особенно не ограничено и может быть установлено определением снижения в вязкости гидролизной смеси в качестве индикатора. Обычно время гидролиза составляет примерно от 10 мин до 300 мин или от 60 мин до 300 мин. Другие условия, такие как время реакции и давление, могут быть аналогичными условиям процесса гидролиза.
Проведением гидролиза данной порции растительного волокнистого материала с кластерным кислотным катализатором можно превращать кластерный кислотный катализатор в кластерное состояние и ингибировать реакцию дегидратации сахарида в процессе гидролиза, хотя снижается количество моносахарида, дегидратированного кластерным кислотным катализатором, до минимума даже в случае, когда используют необычный кластерный кислотный реагент. Кроме того, поскольку кластерообразующая обработка кластерной кислоты может быть выполнена при использовании процесса гидролиза, повышение трудности производственного процесса может быть заторможено.
Способ (3) нагревания и перемешивания кластерного кислотного катализатора в псевдорасплавленном состоянии обычно представляет собой способ, по которому кластерный кислотный катализатор нагревают и приводят в псевдорасплавленное состояние, прежде чем растительный волокнистый материал и кластерный кислотный катализатор смешиваются в процессе гидролиза, и затем выполняют нагревание и перемешивание. Обычно кластерный кислотный катализатор изменяют до псевдорасплавленного состояния, нагревают и перемешивают в реакционном контейнере для применения в процессе гидролиза и выполняют кластерообразующую обработку, затем добавляют растительный волокнистый материал и осуществляют процесс гидролиза.
Температура нагревания не особенно ограничена при условии, что кластерная кислота может сохранять псевдорасплавленное состояние, и может быть соответственно установлена согласно типу кластерной кислоты и соотношению кристаллизационной воды. Чтобы выполнить кластерообразование кластерного кислотного катализатора с хорошей эффективностью, предпочтительно нагревание проводить при температуре, которая по меньшей мере на 10-30°С, более предпочтительно по меньшей мере на 10-20°С, еще более предпочтительно по меньшей мере на 5-10°С выше температуры, при которой кластерный кислотный катализатор первоначально переходит в псевдорасплавленное состояние.
Кластерный кислотный катализатор предпочтительно нагревают и перемешивают с водой в таком количестве, что соотношение кристаллизационной воды кластерного кислотного катализатора становится равным или выше 100%. Особенно предпочтительно кластерный кислотный катализатор нагревают и перемешивают с водой в таком количестве, что соотношение кристаллизационной воды кластерного кислотного катализатора становится равным или выше 100%, водой, которая необходима для гидролиза растительного волокнистого материала в последующем процессе гидролиза, и водой, обеспечивающей присутствие насыщенного водяного пара в мертвом объеме реактора. Из-за этого нагревание и перемешивание в присутствии воды усиливает переход кластерного кислотного катализатора в псевдорасплавленное состояние, тем самым усиливая кластерообразование.
Время нагревания и перемешивания может быть установлено определением снижения вязкости гидролизной смеси в качестве индикатора. Обычно, время нагревания и перемешивания может составлять примерно от 60 до 300 мин. Процесс нагревания и перемешивания кластерной кислоты в псевдорасплавленном состоянии может быть легко включен в качестве предварительного подготовительного процесса для процесса гидролиза при использовании кластерной кислоты в псевдорасплавленном состоянии в качестве катализатора гидролиза в уже существующем процессе. Кроме того, реакция дегидратации моносахарида в процессе гидролиза может быть ингибирована, даже когда используют необычный кластерный кислотный реагент.
Прошло ли кластерообразование кластерного кислотного катализатора, можно определить, например, инфракрасными (ИК) измерениями, спектроскопией комбинационного рассеяния, ядерным магнитным резонансом (ЯМР) и подобным.
Например, в ИК-измерениях определение может быть сделано наблюдением спектра воды (указанная выше кристаллизационная вода), которая координирована к кластерной кислоте, и сравнительной оценкой интенсивности пика абсорбции (в области 3200 см-1), полученного от молекулы Н2О, связанной в кристалле, и пика абсорбции (в области 3500 см-1), полученного от ОН группы, связанной с сильно кислым субстратом. Более конкретно, когда сравнивают ИК-спектр кластерного кислотного катализатора перед обработкой, повышающей кластерообразование, и ИК-спектр кластерного кислотного катализатора после обработки, повышающей кластерообразование, в случае, где интенсивность пика в области 3200 см-1, который получен от молекулы Н2О, связанной в кристалле кластерного кислотного катализатора после обработки, повышающей кластерообразование, меньше, чем интенсивность пика в аналогичной области кластерного кислотного катализатора перед обработкой, повышающей кластерообразование, и интенсивность пика в области 3500 см-1, полученного от ОН группы, связанной с сильно кислым субстратом кластерного кислотного катализатора после обработки, повышающей кластерообразование, больше, чем интенсивность пика в аналогичной области кластерного кислотного катализатора перед обработкой, повышающей кластерообразование, можно определить, что кластерообразование произошло. В ИК-измерениях пик абсорбции, полученный от молекулы Н2О, не ограничен до абсорбции пика абсорбции, полученного от ОН групп, связанных с сильно кислым субстратом, и обычно может наблюдаться как широкий пик.
Кроме того, в спектроскопии комбинационного рассеяния, например, где акцент сфокусирован на симметричных вытягивающих вибрациях WO6 октаэдра фосфовольфрамовой кислоты, резкий высокий пик рассеяния наблюдался в области 985 см-1 в кластерном кислотном катализаторе в кристаллическом состоянии перед кластерообразующей обработкой. Однако в кластерном кислотном катализаторе в кластерном состоянии после кластерообразующей обработки происходит сдвиг к более высокой частоте в область 1558 см-1, и интенсивность пика значительно снижается, то есть светочувствительность снижается. Такой сдвиг к более высокой частоте и снижение в светочувствительности вызваны описанными ниже структурными изменениями, обусловленными кластерообразованием кластерного кислотного катализатора. В WO6 октаэдре, поскольку радиус иона W равен только 0,074 нм, расстояние между W и O является экстремально сжатым, как показано на фиг.1. Когда поверхностная энергия стабилизирована кластерообразованием и форма изменена ближе к сферической форме, симметрия WO6 уменьшается, и расстояние между W и О становится еще короче. В результате этого уменьшение светочувствительности и увеличение силы связывания вызывают одновременное рассеяние и сдвиг к более высокой частоте. Это явление не свойственно фосфовольфрамовой кислоте и аналогично происходит в других кислотах. Следовательно, кластерное состояние кластерного кислотного катализатора может быть подтверждено наблюдением структурных изменений в кластерном кислотном катализаторе с помощью спектроскопии комбинационного рассеяния.
Примеры
Количественное определение D-(+)-глюкозы и D-(+)-ксилозы проводили методом высокоэффективной жидкостной хроматографии (ВЭЖХ) с последующим детектированием флуоресцентной метки. Кластерную кислоту идентифицировали и количественно определяли индуктивно связанной плазмой (ICP).
Пример 1
Дистиллированную воду помещали в заранее герметически закрытый реакционный контейнер (загрузочный тип; см. фиг.3), поднимали температуру до предварительно определенной температуры реакции (70°С), получали состояние давления насыщенного пара внутри контейнера, и водяной пар прилипал к внутренней поверхности контейнера. Затем 1 кг повторно использованной гетерополикислоты в кластерном состоянии (количество кристаллизационной воды измеряли заранее; фосфовольфрамовая кислота) и дистиллированную воду (35 г) в количестве, представляющем дефицит воды (воду компонента с давлением насыщенного пара при 70°С исключали) по отношению к общей сумме количества, необходимого для приведения кристаллизационной воды гетерополикислоты к 100%, и количество воды (55,6 г), необходимой для гидролиза целлюлозы и получения глюкозы, загружали в контейнер и нагревали и перемешивали. Когда температура внутри контейнера достигала 70°С, перемешивание дополнительно продолжали в течение 10 мин. Затем загружали 0,5 кг целлюлозы и проводили смешивание при нагревании при 70°С. Через 10 мин после того, как смешивание при нагревании было начато, вязкость составляла 3000 сП (=3000 мПа.с). Через 1 ч вязкость нагреваемой смеси снижалась до 700 сП (=700 мПа.с). Поэтому загружали 0,5 кг целлюлозы и воду (55,6 г) в количестве, необходимом для гидролиза целлюлозы в глюкозу, и продолжали перемешивание при нагревании при 70°С в течение 2 ч. Нагревание затем останавливали, контейнер открывали и смесь реакции гидролиза охлаждали до комнатной температуры, выпуская избыточный водяной пар.
Общее количество в 500 мл этанола, который дважды использовали для промывания, затем добавляли к смеси реакции гидролиза, расположенной внутри контейнера, перемешивание проводили в течение 30 мин с последующим фильтрованием, что давало выход первого фильтрата и первый отфильтрованный продукт. Получали первый фильтрат (раствор гетерополикислоты в этаноле). Общее количество в 500 мл этанола, который однажды использовали для промывания, затем добавляли к отфильтрованному продукту и перемешивание проводили в течение 30 мин с последующим фильтрованием, что давало выход второго фильтрата и второй отфильтрованный продукт. Общее количество в 500 мл новой порции этанола добавляли ко второму отфильтрованному продукту и перемешивание проводили в течение 30 мин с последующим фильтрованием, что давало выход третьего фильтрата и третий отфильтрованный продукт. Дистиллированную воду добавляли к полученному третьему отфильтрованному продукту и перемешивание проводили в течение 10 мин. Не было какого-либо остатка, который мог быть подтвержден как присутствующий в полученном водном растворе, но раствор все-таки фильтровали и получали водный раствор сахарида. Выход моносахаридов (общая сумма глюкозы, ксилозы, арабинозы, маннозы и галактозы) рассчитывали из полученного водного раствора сахарида. Результат составил 85,3%. Выход моносахаридов вычисляли следующим образом.
Выход моносахаридов (%): отношение (массовое отношение) общей суммы действительно выделенных моносахаридов к теоретическому количеству полученных моносахаридов, которые образуются, когда все количество загруженной целлюлозы превращено в моносахариды.
Пример 2
Дистиллированную воду помещали в заранее герметически закрытый реакционный контейнер (загрузочный тип; см. фиг.3), поднимали температуру до предварительно определенной температуры реакции (70°С), получали состояние давления насыщенного пара внутри контейнера, и водяной пар прилипал к внутренней поверхности контейнера. Затем 1,15 кг повторно использованной гетерополикислоты в кластерном состоянии (количество кристаллизационной воды измерено заранее; фосфовольфрамовая кислота) и дистиллированную воду (35 г) в количестве, представляющем дефицит воды (воду компонента с давлением насыщенного пара при 70°С исключали) по отношению к общей сумме количества, необходимого для приведения кристаллизационной воды гетерополикислоты к 100%, и количество воды (55,6 г), необходимое для гидролиза целлюлозы и получения глюкозы, загружали в контейнер и нагревали и перемешивали. Когда температура внутри контейнера достигала 70°С, перемешивание дополнительно продолжали в течение 10 мин. Затем загружали 0,5 кг древесных стружек, содержащих лигнин, и проводили перемешивание при нагревании при 70°С. Через 10 мин после того, как смешивание при нагревании было начато, вязкость составляла 3000 сП (=3000 мПа.с). Через 3 ч лигнин удаляли из полученной горячей смеси, используя синтерированный фильтр. Вязкость горячей смеси, из которой лигнин был удален, снижалась до 700 сП (=700 мПа.с). Поэтому загружали 0,5 кг древесных стружек и воду (35 г), необходимую для гидролиза целлюлозы, содержащейся в древесных стружках, в глюкозу и продолжали перемешивание при нагревании при 70°С в течение 3 ч.
Нагревание затем останавливали, контейнер открывали и смесь реакции гидролиза охлаждали до комнатной температуры, выпуская избыточный водяной пар. Выделение моносахаридов и гетерополикислоты затем проводили по способу примера 1. Дистиллированную воду добавляли к полученному третьему отфильтрованному продукту и перемешивание проводили в течение 10 мин. Было подтверждено, что осадок, содержащий лигнин, присутствует в полученном водном растворе, и водный раствор сахарида получали фильтрованием. Выход моносахаридов рассчитывали из полученного водного раствора сахарида. Результат составлял 80,2%. В данном случае выход моносахаридов рассчитывали при предположении, что лигнин, удаленный с помощью синтерированного фильтра, и лигнин, удаленный фильтрованием из водного раствора, составляет 30% масс. (то есть 300 г) от использованной древесины. В данном примере, когда лигнин удаляли синтерированным фильтром, гетерополикислота, которая адсорбировалась на лигнине, удалялась вместе с лигнином. По этой причине количество использованной гетерополикислоты было равно показателю 1,15, превышающему количество примера 1.
Пример 3
Использовали реактор поточного типа (см. фиг.4), в котором перемешивание может быть выполнено в трубопроводе с подогревом (постоянная температура 70°С) для главного потока гетерополикислоты (фосфовольфрамовая кислота) в псевдорасплавленном состоянии. Лопасть 10 для перемешивания в данном трубопроводе имеет структуру, которая эффективна только для перемешивания и практически не влияет на подачу содержимого в реакционный бак 100. Поэтому скорость подачи содержимого представляет собой общую сумму загрузочных скоростей компонентов (гетерополикислота, растительный волокнистый материал). В реакционном баке 100 загрузочный впуск 13 для гетерополикислоты в псевдорасплавленном состоянии находится на самом верху по течению и множество загрузочных впусков 11 (первый-четвертый загрузочный впуск) для растительного волокнистого материала предназначено нисходящему потоку из загрузочного впуска 13 для гетерополикислоты. Сенсоры вязкости 12 (первый-четвертый сенсоры вязкости) предназначены нисходящему потоку из загрузочного впуска 11 для растительного волокнистого материала (непосредственно в фасадной стороне загрузочного впуска, дающего нисходящий поток из данного загрузочного впуска, или непосредственно в фасадной стороне стенки реактора вниз по течению), и вязкость содержимого внутри реакционного бака 100, которую определяют сенсорами вязкости 12, используют для обратной связи, контролирующей количество растительного волокнистого материала, загруженного из загрузочных впусков 11 для растительного волокнистого материала.
Древесные стружки (содержащие лигнин) загружали через загрузочный впуск 11(1), тогда как в реакционный бак 100 загружали гетерополикислоту (повторно использованная, в кластерном состоянии) в псевдорасплавленном состоянии, в которую воду гидролиза добавляли заранее. В данном случае скорость загрузки древесных опилок составляла половину скорости загрузки гетерополикислоты. Скорость загрузки гетерополикислоты и скорость загрузки древесных стружек через первый загрузочный впуск 11(1) регулировали, чтобы получить вязкость в первом сенсоре вязкости 12(1) в 700 сП (=700 мПа.с). Скорость загрузки через второй-четвертый загрузочные впуски 11(2)-11(4) также регулировали, чтобы получить вязкость во втором-четвертом сенсорах вязкости 12(2)-12(4) в 700 сП (=700 мПа.с). Реакционную смесь, выпущенную нисходящим потоком реактора, охлаждали до комнатной температуры, в то время как избыточный водяной пар удаляли. В данном примере массовое соотношение гетерополикислоты и использованных древесных стружек составляло 1:1,2 (гетерополикислота:древесные стружки). Сахариды и гетерополикислоту затем извлекали из смеси реакции гидролиза по способу примера 1. Выход моносахаридов составлял 82,1%. В данном случае выход моносахаридов рассчитывали при предположении, что удаленный лигнин составляет 30% масс. от использованных древесных стружек.
Сравнительный пример 1
Дистиллированную воду помещали в заранее герметически закрытый реакционный контейнер (загрузочный тип), поднимали температуру до предварительно определенной температуры реакции (70°С), получали состояние давления насыщенного пара внутри контейнера, и водяной пар прилипал к внутренней поверхности контейнера. Затем 1 кг повторно использованной гетерополикислоты в кластерном состоянии (количество кристаллизационной воды измерено заранее; фосфовольфрамовая кислота) и дистиллированную воду (35 г) в количестве, представляющем дефицит воды (воду компонента с давлением насыщенного пара при 70°С исключали) по отношению к общей сумме количества, необходимого для приведения кристаллизационной воды гетерополикислоты к 100%, и количество воды (55,6 г), необходимое для гидролиза целлюлозы и получения глюкозы, загружали в контейнер и нагревали и перемешивали. Когда температура внутри контейнера достигала 70°С, перемешивание дополнительно продолжали в течение 5 мин. Затем загружали 0,5 кг целлюлозы и проводили перемешивание в течение 2 ч при нагревании при 70°С. Нагревание затем останавливали, контейнер открывали и проводили охлаждение до комнатной температуры при выпуске избыточного водяного пара. Моносахариды и гетерополикислоту затем извлекали из смеси реакции гидролиза по способу примера 1. Выход моносахаридов составлял 85,3%.
Результаты
Выход моносахаридов и массовое соотношение гетерополикислоты и растительного волокнистого материала, полученных в примерах 1-3 и сравнительном примере 1, показаны в таблице 1.
Как показано в таблице 1, в примерах 1-3, количество гетерополикислоты, используемое на единицу массы волокнистого материала, могло быть сильно снижено по отношению к количеству сравнительного примера 1, в то время как сохраняется выход моносахаридов. Кроме того, при сравнении примера 1 и сравнительного примера 1 (оба используют реактор загрузочного типа) видно, что в примере 1 обработанное количество волокнистого материала на единицу массы гетерополикислоты удвоено. Поэтому энергия, требуемая для подогрева, который необходим, чтобы привести гетерополикислоту в псевдорасплавленное состояние, могла быть снижена. Кроме того, в примере 3, в котором использован реакционный аппарат непрерывного действия, количество используемой гетерополикислоты могло быть уменьшено и энергия подогрева также могла быть снижена. В производственном реакционном аппарате, где воду в реакционную систему добавляют введением водяного пара, операции подогрева и добавления воды могут быть проведены одновременно, причем такой процесс предназначен исключительно для введения дистиллированной воды, исходя из энергии.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ГИДРОЛИЗА РАСТИТЕЛЬНОГО ВОЛОКНИСТОГО МАТЕРИАЛА ДЛЯ ПОЛУЧЕНИЯ И ВЫДЕЛЕНИЯ САХАРИДА, ВКЛЮЧАЮЩЕГО ГЛЮКОЗУ | 2009 |
|
RU2455365C1 |
| СПОСОБ ГИДРОЛИЗА РАСТИТЕЛЬНОГО ВОЛОКНИСТОГО МАТЕРИАЛА ДЛЯ ПОЛУЧЕНИЯ И ВЫДЕЛЕНИЯ САХАРИДА, ВКЛЮЧАЮЩЕГО ГЛЮКОЗУ | 2009 |
|
RU2453607C1 |
| СПОСОБ ПРЕОБРАЗОВАНИЯ РАСТИТЕЛЬНОГО ВОЛОКНИСТОГО МАТЕРИАЛА | 2008 |
|
RU2440419C2 |
| СПОСОБ ОСАХАРИВАНИЯ И СЕПАРИРОВАНИЯ ДЛЯ РАСТИТЕЛЬНЫХ ВОЛОКНИСТЫХ МАТЕРИАЛОВ | 2008 |
|
RU2451087C2 |
| СПОСОБ ПРЕДВАРИТЕЛЬНОЙ ОБРАБОТКИ ДЛЯ ОСАХАРИВАНИЯ РАСТИТЕЛЬНОГО ВОЛОКНИСТОГО МАТЕРИАЛА И СПОСОБ ОСАХАРИВАНИЯ | 2010 |
|
RU2486256C2 |
| МНОГОСТАДИЙНЫЙ ГИДРОЛИЗ ЦЕЛЛЮЛОЗЫ И БЫСТРОЕ ОХЛАЖДЕНИЕ С ИСПОЛЬЗОВАНИЕМ И БЕЗ ИСПОЛЬЗОВАНИЯ КИСЛОТЫ | 2012 |
|
RU2608999C2 |
| СПОСОБ ПОЛУЧЕНИЯ 5-ГИДРОКСИМЕТИЛФУРФУРОЛА | 2015 |
|
RU2583953C1 |
| УСТАНОВКА ДЛЯ ПРЕДВАРИТЕЛЬНОЙ ОБРАБОТКИ ЦЕЛЛЮЛОЗОСОДЕРЖАЩЕГО РАСТИТЕЛЬНОГО СЫРЬЯ | 2013 |
|
RU2577066C2 |
| СПОСОБ ПОЛУЧЕНИЯ ГЛЮКОЗНОГО ГИДРОЛИЗАТА ИЗ ДРЕВЕСИНЫ БЕРЕЗЫ | 2015 |
|
RU2600134C1 |
| СПОСОБ ПОЛУЧЕНИЯ МИКРОКРИСТАЛЛИЧЕСКОЙ ЦЕЛЛЮЛОЗЫ | 2013 |
|
RU2528261C1 |
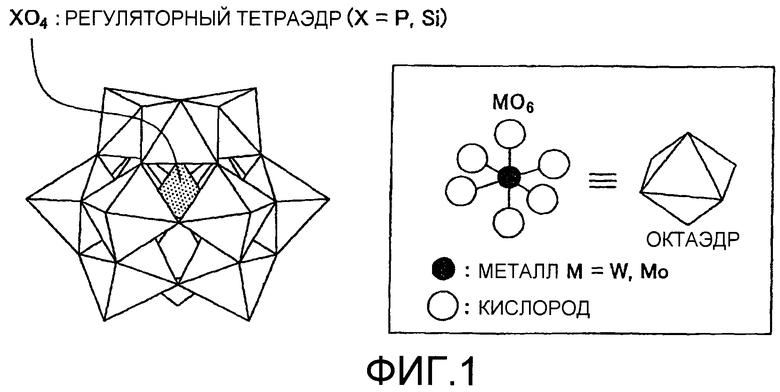
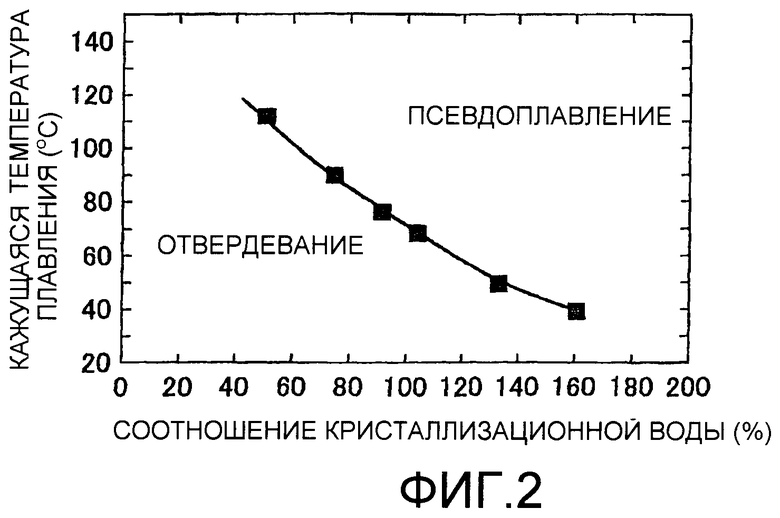
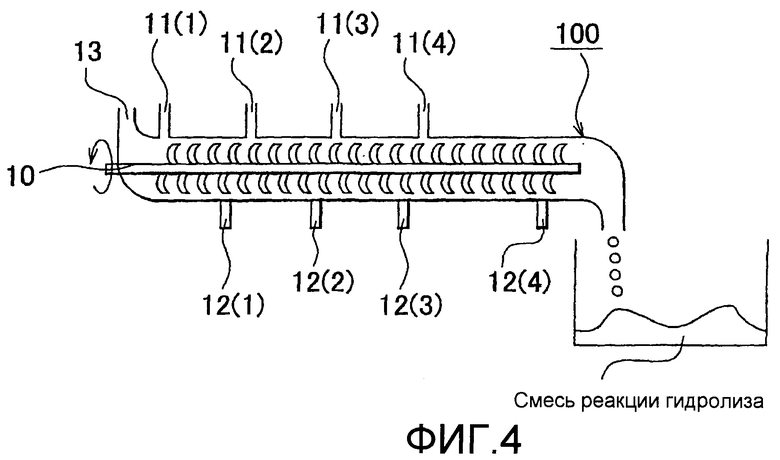
Изобретение относится к пищевой промышленности. Способ включает процесс гидролиза при использовании кластерного кислотного катализатора в псевдорасплавленном состоянии для гидролиза целлюлозы, содержащейся в растительном волокнистом материале, и получения глюкозы. В процессе гидролиза кластерный кислотный катализатор и первое количество растительного волокнистого материала, который повышает вязкость кластерного кислотного катализатора в псевдорасплавленном состоянии при добавлении к кластерному кислотному катализатору в псевдорасплавленном состоянии, нагревают и смешивают и второе количество растительного волокнистого материала затем добавляют, когда происходит снижение вязкости горячей смеси кластерного кислотного катализатора и первого количества растительного волокнистого материала. При этом в качестве кластерного кислотного катализатора используют гомополикислоты или гетерополикислоты. Предложенный способ позволяет повысить количество обрабатываемого растительного волокнистого материала на единицу массы кластерного кислотного катализатора. 2 з.п. ф-лы, 4 ил., 1 табл., 4 пр.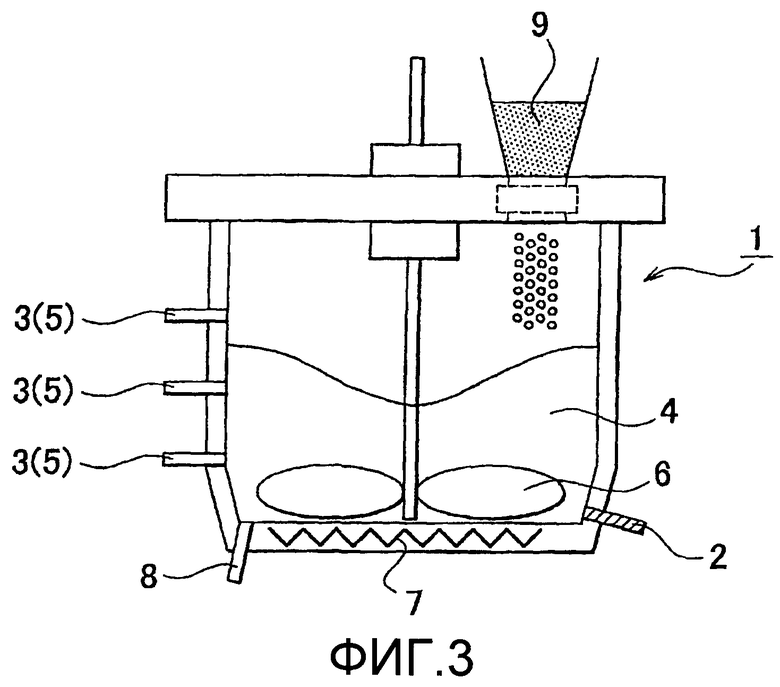
1. Способ гидролиза растительного волокнистого материала и получения и выделения сахарида, содержащего глюкозу, включающий:
процесс гидролиза при использовании гомополикислоты или гетерополикислоты в качестве кластерного кислотного катализатора в псевдорасплавленном состоянии для гидролиза целлюлозы, содержащейся в растительном волокнистом материале, и получения глюкозы, где в процессе гидролиза кластерный кислотный катализатор и первое количество растительного волокнистого материала, который повышает вязкость кластерного кислотного катализатора в псевдорасплавленном состоянии при добавлении к кластерному кислотному катализатору в псевдорасплавленном состоянии, нагревают и смешивают и второе количество растительного волокнистого материала затем добавляют, когда происходит снижение вязкости горячей смеси кластерного кислотного катализатора и первого количества растительного волокнистого материала.
2. Способ по п.1, где объемное отношение первого количества растительного волокнистого материала к количеству кластерного кислотного катализатора равно или больше 60%.
3. Способ по п.1 или 2, где объемное отношение второго количества растительного волокнистого материала к количеству кластерного кислотного катализатора равно или больше 60%.
| Способ обработки грубых растительных кормов | 1976 |
|
SU651652A3 |
| RU 2075384 С1, 20.03.1997 | |||
| Пневматический или гидравлический расширитель | 1932 |
|
SU34477A1 |
| ЕР 1860201 А1, 28.11.2007. | |||

