1. Область техники, к которой относится изобретение
Изобретение относится к способу получения сахарида, включающего глюкозу, путем осахаривания растительного волокнистого материала, и выделения полученного сахарида.
2. Уровень техники
Было предложено получать сахарид, включающий главным образом глюкозу или ксилозу, из целлюлозы или гемицеллюлозы путем разложения растительного материала, который представляет собой биомассу, такого как остатки выжатого сахарного тростника (жмыха) или древесные стружки, и эффективно использовать полученный сахарид в качестве пищевого продукта или топлива, и этот способ был внедрен в практику. В частности, привлекала внимание технология, с помощью которой моносахарид, полученный разложением растительных волокон, сбраживают для получения спирта, такого как этанол, в качестве топлива. К настоящему времени были предложены многочисленные способы получения сахарида, такого как глюкоза, разложением целлюлозы или гемицеллюлозы (например, Публикация Японской Патентной Заявки № 8-299000 (JP-A-8-299000), Публикация Японской Патентной Заявки № 2006-149343 (JP-A-2006-149343), Публикация Японской Патентной Заявки № 2006-129735 (JP-A-2006-129735), и Публикация Японской Патентной Заявки № 2002-59118 (JP-A-2002-59118)), и типичный способ включает гидролиз целлюлозы с использованием серной кислоты, такой как разбавленная серная кислота или концентрированная серная кислота, или соляной кислоты (JP-A-8-299000). Имеется также способ, в котором используют фермент целлюлазу (JP-A-2006-149343), способ, в котором применяют твердый катализатор, такой как активированный уголь или цеолит (JP-A-2006-129735), и способ, в котором используют горячую воду под давлением (JP-A-2002-59118).
Однако со способом, в котором целлюлозу подвергают разложению с использованием такой кислоты, как серная кислота, связана проблема трудности выделения кислоты, служащей в качестве катализатора, и полученного сахарида из гидролитической реакционной смеси, образованной при гидролизе. Это обусловлено тем, что как глюкоза, которая является основным компонентом продуктов гидролиза целлюлозы, так и кислота, которая служит в качестве катализатора гидролиза, растворимы в воде. Трудным и дорогостоящим оказывается не только удаление кислоты из гидролитической реакционной смеси путем нейтрализации или ионного обмена, но также затруднительно удалить кислоту полностью, и часто кислота остается в процессе ферментации для получения этанола. В результате, даже когда значение рН оптимизируют в отношении активности дрожжей в (последующем) процессе спиртового брожения, возрастает концентрация соли, тем самым снижая активность дрожжей и уменьшая эффективность сбраживания.
В частности, когда используют концентрированную серную кислоту, весьма затруднительно удалить серную кислоту до такой степени, чтобы не дезактивировать дрожжи в процессе брожения для получения этанола, и для такого удаления требуется значительное количество энергии. Напротив, когда применяют разбавленную серную кислоту, удаление кислоты становится относительно простым. Однако необходимо проводить разложение целлюлозы в условиях высокой температуры, что связано с повышенным энергопотреблением. Кроме того, такие кислоты, как серная кислота и соляная кислота, очень трудно отделять, собирать и повторно использовать. Таким образом, применение этих кислот в качестве катализатора для получения глюкозы обусловливает повышение стоимости биоэтанола.
При использовании способа, в котором применяют горячую воду под давлением, затруднительно регулировать условия и трудно получать глюкозу со стабильным выходом. В дополнение, в этом способе даже происходит разложение глюкозы, тем самым снижая выход глюкозы. Более того, продукты разложения снижают активность дрожжей, и ферментация может быть подавлена. Еще одна проблема сопряжена с затратами, поскольку реактор (устройство для обработки в суперкритических условиях) является дорогостоящим и имеет малый ресурс прочности.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Авторы настоящего изобретения провели исчерпывающее исследование осахаривания целлюлозы и обнаружили, что комплексная кислота в псевдорасплавленном состоянии имеет превосходную каталитическую активность в отношении гидролиза целлюлозы и может быть без труда отделена от полученного сахарида. Патентные заявки, которые относятся к соответствующему способу, уже были поданы (Японская Патентная Заявка № 2007-115407 и Японская Патентная Заявка № 2007-230711). Согласно данному способу, в отличие от традиционного способа с использованием концентрированной серной кислоты или разбавленной серной кислоты, катализатор гидролиза может быть извлечен и повторно использован, и может быть повышен энергетический коэффициент полезного действия процесса, имеющего результатом выделение водного раствора сахарида и отделение катализатора гидролиза от продукта, полученного гидролизом целлюлозы. Кроме того, вышеупомянутые патентные заявки также предлагают способ разделения сахарида, полученного гидролизом растительного волокнистого материала, и комплексного кислотного катализатора. Более конкретно, предложен способ, в котором после гидролиза к реакционной смеси, включающей образованный сахарид и комплексный кислотный катализатор, добавляют органический растворитель, в результате чего комплексная кислота растворяется, и сахарид вместе с остатком в виде твердой фракции отделяют от комплексной кислоты и органического растворителя.
Авторы настоящего изобретения далее продолжили изучение осахаривания целлюлозы с использованием комплексного кислотного катализатора и успешно повысили селективность комплексного кислотного катализатора в отношении реакции осахаривания растительного волокнистого материала. Таким образом, изобретение основывается на результатах, полученных в ходе этого исследования, и представляет способ осахаривания и отделения растительного волокнистого материала с использованием комплексного кислотного катализатора в псевдорасплавленном состоянии, в котором подавляют развитие реакции дегидратации (последующей реакции) сахарида комплексным кислотным катализатором, обеспечивают протекание реакции гидролиза целлюлозы с высокой селективностью и повышают выход сахарида.
Первый аспект изобретения относится к способу осахаривания и отделения растительного волокнистого материала для получения и выделения сахарида, включающего глюкозу. Способ включает процесс гидролиза с использованием комплексного кислотного катализатора в псевдорасплавленном состоянии для гидролиза целлюлозы, содержащейся в растительном волокнистом материале, и получения глюкозы, причем комплексный кислотный катализатор подвергают обработке, усиливающей агрегацию, при которой повышают агрегацию комплексного кислотного катализатора в кристаллическом состоянии. При применении способа осахаривания и отделения согласно изобретению подавляют реакцию дегидратации (последующей реакции) сахарида, включающего глюкозу, который получен гидролизом растительного волокнистого материала, и повышают выход сахарида.
Подверганием комплексного кислотного катализатора обработке, усиливающей агрегацию, в тот момент времени, когда в процесс гидролиза растительный волокнистый материал загружается в количестве, которое может быть загружено в одном цикле для всей реакционной системы, то есть в момент времени, в который начинается основное действие процесса гидролиза, можно эффективно ингибировать последующее реагирование моносахарида, образованного в процессе гидролиза.
Агрегация комплексного кислотного катализатора в результате обработки, усиливающей агрегацию, может быть подтверждена несколькими методами, например, с помощью инфракрасного (IR, ИК) спектра. Более конкретно, когда комплексный кислотный катализатор кристаллизуется, комплексный кислотный катализатор захватывает воду в виде кристаллизационной воды, и имеет полосу поглощения в области 3200 см-1, но когда кристаллы разрушаются и переходят в агрегированное состояние, полоса поглощения располагается в области 3500 см-1. Поэтому, когда сравнивают ИК-спектр комплексного кислотного катализатора перед обработкой, усиливающей агрегацию, и ИК-спектр комплексного кислотного катализатора после обработки, усиливающей агрегацию, то агрегация комплексного кислотного катализатора в результате обработки, усиливающей агрегацию, может быть подтверждена в случае, где интенсивность полосы в области 3200 см-1, которая обусловлена молекулой Н2О, которая сэндвичеобразно размещена между кристаллами комплексного кислотного катализатора, после обработки, усиливающей агрегацию, является меньшей, чем интенсивность полосы комплексного кислотного катализатора до обработки, усиливающей агрегацию, и интенсивность полосы в области 3500 см-1, которая обусловлена ОН-группой, связанной с сильной кислотой комплексного кислотного катализатора, после обработки, усиливающей агрегацию, является большей, чем интенсивность полосы комплексного кислотного катализатора до обработки, усиливающей агрегацию.
Конкретный способ обработки, усиливающей агрегацию, включает процесс нагревания и перемешивания комплексного кислотного катализатора и органического растворителя, который может растворять комплексный кислотный катализатор, и процесс удаления органического растворителя после процесса нагревания и перемешивания. В этом случае нагревание и перемешивание комплексного кислотного катализатора и органического растворителя может быть проведено при температуре, равной или меньшей 65°С.
В случае, где способ согласно изобретению включает процесс отделения сахарида добавлением органического растворителя, в котором может быть растворен комплексный кислотный катализатор, к реакционной смеси после процесса гидролиза и твердофазно-жидкостного разделения полученной смеси на жидкостную фракцию, включающую комплексный кислотный катализатор и органический растворитель, и твердую фракцию, включающую сахарид, конкретный способ обработки, усиливающей агрегацию, включает процесс добавления комплексного кислотного катализатора в кристаллическом состоянии в количестве, которое компенсирует потерю комплексного кислотного катализатора в процессе отделения сахарида, к раствору комплексной кислоты в органическом растворителе, который получен в процессе отделения сахарида и образован растворением комплексного кислотного катализатора в органическом растворителе и затем выполнения нагревания и перемешивания.
Еще один способ обработки, усиливающей агрегацию, включает нагревание и перемешивание части количества растительного волокнистого материала, которое может быть загружено в одном цикле, вместе с комплексным кислотным катализатором в псевдорасплавленном состоянии, и проведение гидролиза растительного волокнистого материала в процессе гидролиза. В этом случае, при обработке, усиливающей агрегацию, количество растительного волокнистого материала, который нагревают и перемешивают вместе с комплексным кислотным катализатором в псевдорасплавленном состоянии, является равным или меньшим, чем 10% по весу от количества растительного волокнистого материала, который может быть загружен в одном цикле. Более того, нагреванию и перемешиванию вместе с комплексным кислотным катализатором в псевдорасплавленном состоянии растительный волокнистый материал может быть подвергнут в количестве, которое не изменяет вязкости комплексного кислотного катализатора в псевдорасплавленном состоянии.
Еще один дополнительный способ обработки, усиливающей агрегацию, включает нагревание и перемешивание комплексного кислотного катализатора в псевдорасплавленном состоянии. В этом случае нагревание и перемешивание проводят при температуре, которая является по меньшей мере на величину от 5 до 10°С более высокой, чем температура, при которой состояние комплексного кислотного катализатора начинает изменяться на псевдорасплавленное состояние. Нагревание и перемешивание комплексного кислотного катализатора может быть проведено с водой в таком количестве, чтобы доля кристаллизационной воды в комплексном кислотном катализаторе стала равной или превышающей 100%.
В соответствии с изобретением, при осахаривании и отделении растительного волокнистого материала с использованием комплексного кислотного катализатора в псевдорасплавленном состоянии может быть подавлено развитие реакции дегидратации (последующей реакции) моносахарида комплексным кислотным катализатором. Поэтому, в соответствии с изобретением, обеспечивают протекание реакции гидролиза целлюлозы с высокой селективностью и может быть повышен выход реакции гидролиза. Кроме того, может быть повышена скорость реакции гидролиза.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Вышеуказанные и дополнительные цели, признаки и преимущества изобретения станут очевидными из нижеследующего описания примерных вариантов осуществления с привлечением сопроводительных чертежей, в которых подобные номера позиций использованы для представления сходных элементов и в которых:
Фиг.1 показывает кеггиновскую структуру гетерополикислоты;
Фиг.2 представляет график, показывающий взаимосвязь между долей кристаллизационной воды в комплексном кислотном катализаторе и кажущейся температурой плавления;
Фиг.3 показывает результаты измерений ИК-спектров в Примере 1, Примере 2 и Сравнительном Примере 1;
Фиг.4 показывает результаты измерений Рамановской спектроскопии в Примере 2 и Сравнительном Примере 1;
Фиг.5 показывает методику процесса гидролиза в примерах;
Фиг.6 показывает методику процесса выделения сахарида в примерах; и
Фиг.7 показывает методику извлечения гетерополикислоты в примерах.
ПОДРОБНОЕ ОПИСАНИЕ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
Способ осахаривания и отделения растительного волокнистого материала в соответствии с изобретением представляет собой способ гидролиза растительного волокнистого материала для получения и выделения сахарида, включающего главным образом глюкозу. Этот способ включает процесс гидролиза с использованием комплексного кислотного катализатора в псевдорасплавленном состоянии для гидролиза целлюлозы, содержащейся в растительном волокнистом материале, и получения глюкозы, причем комплексный кислотный катализатор подвергают обработке, усиливающей агрегацию, в результате которой усиливается агрегация комплексного кислотного катализатора в микрокристаллическом состоянии и/или поликристаллическом состоянии.
В вышеупомянутых патентных заявках (Японская Патентная Заявка № 2007-115407 и Японская Патентная Заявка № 2007-230711) авторы настоящего изобретения раскрыли способ осахаривания и отделения растительного волокнистого материала, в котором комплексную кислоту нагревают для достижения псевдорасплавленного состояния и используют в качестве катализатора гидролиза растительного волокнистого материала. Результаты исследования, проведенного авторами настоящего изобретения, демонстрируют, что в способе осахаривания и отделения растительного волокнистого материала с использованием комплексных кислот, в случае, где в качестве реагента применяют не бывший в употреблении новый комплексный кислотный реагент, после начальной стадии реакции гидролиза (более конкретно, в течение 10 минут с момента начала реакции при реакционной температуре 70°С) протекает реакция дегидратации (последующая реакция) моносахарида, такого как образованная глюкоза, но после этого дегидратация моносахарида не происходит (более конкретно, через 10 минут после момента начала реакции при реакционной температуре 70°С). Поскольку реакция дегидратации моносахарида снижает выход моносахарида, важно эту реакцию эффективно ингибировать. Для ингибирования реакции дегидратации сахарида может быть снижена реакционная температура процесса гидролиза, но такой подход ведет к увеличению продолжительности реакции и может снижать стабильность реакции.
Соответственно этому авторы настоящего изобретения провели анализ состояния гетерополикислот, которые являются показательными примерами комплексных кислот, с использованием ИК-спектроскопии (см. Фиг.3). Более конкретно, ИК-измерения были проведены в отношении следующих гетерополикислот (А), (В) и (С). (А): гетерополикислота, полученная растворением не использовавшегося ранее нового гетерополикислотного реагента в этаноле при комнатной температуре (от 20 до 25°С), затем выпариванием этанола и высушиванием (см. Сравнительный Пример 1); (В) гетерополикислота, полученная перемешиванием не использовавшегося ранее нового гетерополикислотного реагента и этанола при нагревании при температуре 60°С, снижением температуры до 45°С, вакуумированием внутренности резервуара смесителя, быстрым испарением этанола и высушиванием (см. Пример 1); и (С) гетерополикислота, полученная добавлением не использовавшегося ранее гетерополикислотного реагента к этанолу, содержащему гетерополикислоту, которая была использована в качестве катализатора гидролиза растительного волокнистого материала (отношение использованной гетерополикислоты к неиспользованной гетерополикислоте составляет 9:1), перемешиванием с нагреванием при температуре 50°С, вакуумированием внутренности резервуара смесителя, быстрым испарением этанола и высушиванием (см. Пример 2).
В результате ИК-измерения гетерополикислоты (А) подтвердили, что гетерополикислота содержала связанные в кристалле молекулы Н2О (полоса поглощения в области 3200 см-1, показанная на Фиг.3), тем самым демонстрируя, что гетерополикислота (А) содержала гетерополикислоту в кристаллическом состоянии. Далее, когда гетерополикислоту (А) использовали в качестве катализатора гидролиза растительного волокнистого материала, выход сахарида составлял 60%. Напротив, в ИК-измерениях гетерополикислот (В) и (С) наблюдался сдвиг полосы. Более конкретно, интенсивность полосы поглощения молекул Н2О, связанных в кристалле (полоса поглощения в области 3200 см-1, показанная на Фиг.3), снижалась, и увеличивалась интенсивность полосы поглощения ОН-групп, находящихся на сильнокислотном субстрате (полоса поглощения в области 3500 см-1, показанная на Фиг.3). Таким образом, было найдено, что изменялось состояние агрегации гетерополикислот, состоящее в количестве молекул гетерополикислот в процессе гидролиза растительного волокнистого материала, или вследствие нагревания и перемешивания в этаноле, который может растворять гетерополикислоты. Кроме того, когда в качестве катализаторов гидролиза растительного волокнистого материала использовали гетерополикислоты (В) и (С), выход сахарида составлял 83,5% с гетерополикислотой (В) и 86,5% с гетерополикислотой (С), тем самым демонстрируя значительное повышение выхода сахарида сравнительно с выходом в случае применения гетерополикислоты (А).
Вышеописанные результаты наводят на мысль, что последующее реагирование моносахарида происходит вследствие того, что гетерополикислота в кристаллическом состоянии, такая как гетерополикислота (А), проявляет значительную поляризацию и чрезмерно большую силу кислоты. Кроме того, можно предположить, что последующее реагирование моносахарида не происходит потому, что кислотность гетерополикислоты в агрегированном состоянии, таком как гетерополикислоты (В) и (С), является более пригодной, чем таковая гетерополикислоты в кристаллическом состоянии, и можно провести гидролиз растительного волокнистого материала селективно. Кислотность комплексной кислоты в кристаллическом состоянии является более высокой, чем кислотность гетерополикислоты в агрегированном состоянии, по-видимому, вследствие усиления поляризации, обусловленного кристаллизацией.
Изобретение основывается на вышеописанной информации. Таким образом, комплексный кислотный катализатор в кристаллическом состоянии имеет высокую кислотность и обусловливает реакцию дегидратации (последующую реакцию) моносахарида, тогда как комплексный кислотный катализатор в агрегированном состоянии не вызывает реакции дегидратации образованного моносахарида и стимулирует гидролиз растительного волокнистого материала с высокой селективностью. Таким образом, в соответствии с изобретением, повышение выхода сахарида сделано возможным путем подвергания комплексного кислотного катализатора обработке, усиливающей агрегацию. Поскольку обработка, усиливающая агрегацию, увеличивает скорость диффузии комплексного кислотного катализатора в гидролитической реакционной системе, может быть также получен эффект повышения скорости реакции гидролиза.
В соответствии с изобретением, комплексная кислота, используемая в качестве катализатора гидролиза растительного волокнистого материала, означает кислоту, в которой сконденсированы многочисленные кислородные кислоты, то есть так называемую поликислоту. Для большинства поликислот известно, что в поликислотах с центральным элементом связаны многочисленные атомы кислорода, и в результате поликислоты являются окисленными настолько, что степень окисления становится максимальной, и поликислоты демонстрируют превосходные свойства как катализатор окисления, и поликислоты являются сильными кислотами. Например, фосфорновольфрамовая кислота (рКа=-13,16), которая представляет собой гетерополикислоту, является более сильной кислотой, чем серная кислота (рКа=-11,93). Таким образом, даже при мягких температурных условиях, например, таких как температура 50°С, можно провести разложение целлюлозы или гемицеллюлозы с образованием моносахарида, такого как глюкоза или ксилоза.
Комплексная кислота, используемая в изобретении, может представлять собой либо гомополикислоту, либо гетерополикислоту, но предпочтительной является гетерополикислота, поскольку она имеет высокую окислительную способность и большую силу кислоты. Гетерополикислота, которая может быть использована, не является специально ограниченной. Например, гетерополикислота может быть представлена общей формулой HwAxByOz (А означает гетероатом, В означает многовалентный атом, который служит в качестве каркаса поликислоты, “w” означает число атомов водорода в структуре, “x” означает число гетероатомов в структуре, “y” означает число многовалентных атомов в структуре и “z” означает число атомов кислорода в структуре). Примеры многовалентного атома В включают такие атомы, как W, Mo, V и Nb, которые могут формировать поликислоту. Примеры гетероатома А включают такие атомы, как P, Si, Ge, As и В, которые могут формировать гетерополикислоту. В одиночной молекуле гетерополикислоты может содержаться один или более видов многоатомных атомов и гетероатомов.
Благодаря хорошему балансу силы кислоты и окислительной способности предпочтительно применяют фосфорновольфрамовую кислоту H3[PW12O40] или кремневольфрамовую кислоту H4[SiW12O40], которые представляют собой вольфраматы. Преимущественно может быть применена также фосфорномолибденовая кислота H3[PMo12O40], которая представляет собой молибдат.
Структура кеггиновского типа [Xn+M12O40: X = P, Si, Ge, As и т.д., M = Mo, W и т.д.] гетерополикислоты (фосфорновольфрамовой кислоты) показана на Фиг.1. В центре многогранника, составленного октаэдрическими структурными единицами МО6, находится тетраэдр ХО4, и вокруг этой структуры присутствует большое количество кристаллизационной воды. Структура комплексной кислоты не является специально ограниченной и может быть не только кеггиновского типа, но также, например, доусоновского типа. Здесь вода, которая гидратирует комплексный кислотный катализатор в кристаллическом состоянии или координирована с ним, или связана с комплексным кислотным катализатором в агрегированном состоянии, составленном несколькими молекулами комплексного кислотного катализатора, описана общеупотребительным термином «кристаллизационная вода». Кристаллизационная вода включает анион воды, который водородной связью соединен с анионом, составляющим комплексный кислотный катализатор, координационную воду, которая координационно связана с катионом, воду кристаллической решетки, которая не координирована ни с катионом, ни с анионом, и также воду, которая содержится в форме ОН-групп. Комплексный кислотный катализатор в агрегированном состоянии представляет собой агрегат, который составлен молекулами комплексных кислот числом от одной до нескольких и отличается от кристалла. Комплексный кислотный катализатор в агрегированном состоянии может находиться в твердом состоянии, псевдорасплавленном состоянии и в растворенном состоянии в растворителе (коллоидальном состоянии).
Вышеописанный комплексный кислотный катализатор при нормальной температуре находится в твердом состоянии, но его состояние становится псевдорасплавленным состоянием при нагревании до более высокой температуры. Псевдорасплавленное состояние, как упоминаемое здесь, означает состояние, в котором комплексная кислота по внешнему виду расплавляется, но она не полностью плавится до перехода в жидкое состояние; псевдорасплавленное состояние напоминает коллоидальное состояние (золь), в котором комплексная кислота диспергирована в жидкости, и представляет собой состояние, в котором комплексная кислота проявляет текучесть. Находится ли комплексная кислота в псевдорасплавленном состоянии, может быть подтверждено визуальными наблюдениями, или, в случае гомогенных систем, с помощью ДСК (дифференциальной сканирующей калориметрии).
Комплексная кислота, как описанная здесь выше, проявляет высокую каталитическую активность в гидролизе целлюлозы даже при низких температурах благодаря высокой кислотности комплексной кислоты. Поскольку диаметр молекулы комплексной кислоты составляет от около 1 до 2 нм, типично чуть больше 1 нм, комплексная кислота легко смешивается с растительным волокнистым материалом, который представляет собой сырьевой материал и поэтому эффективно стимулирует гидролиз целлюлозы. Таким образом, возможно гидролизовать целлюлозу при мягких температурных условиях с высоким энергетическим коэффициентом полезного действия и малым влиянием на окружающую среду.
В дополнение, в отличие от традиционного способа гидролиза целлюлозы, в котором применяют такую кислоту, как серная кислота, в способе согласно изобретению, в котором используют комплексную кислоту в качестве катализатора, высока эффективность разделения сахарида и катализатора, и они могут быть легко разделены. Поскольку комплексная кислота находится в твердом состоянии при определенной температуре, она может быть отделена от сахарида, который представляет собой продукт. Поэтому отделенная комплексная кислота может быть выделена и использована повторно. Кроме того, поскольку комплексный кислотный катализатор в псевдорасплавленном состоянии также действует как реакционный растворитель, количество растворителя, применяемого в качестве реакционного растворителя, может быть значительно сокращено сравнительно с таковым в традиционном способе. Это значит, что разделение комплексной кислоты и сахарида, который представляет собой продукт, и извлечение комплексной кислоты могут быть проведены с повышенной эффективностью. Таким образом, изобретение, в котором комплексную кислоту применяют в качестве катализатора гидролиза целлюлозы, может сократить расходы и снизить нагрузку на окружающую среду.
Произошла ли агрегация комплексного кислотного катализатора, может быть определено, например, с помощью ИК-измерений, Рамановской спектроскопии, ядерного магнитного резонанса (NMR, ЯМР) и тому подобного.
Например, в ИК-измерениях определение может быть сделано наблюдением спектра воды (вышеупомянутой кристаллизационной воды), которая координирована с комплексной кислотой, и сравнительной оценкой интенсивности полосы поглощения (в области 3200 см-1), которая обусловлена молекулой Н2О, связанной в кристалле, и полосы поглощения (в области 3500 см-1), которая обусловлена ОН-группой, связанной с сильнокислотным субстратом. Более конкретно, когда сравнивают ИК-спектр комплексного кислотного катализатора перед обработкой, усиливающей агрегацию, и ИК-спектр комплексного кислотного катализатора после обработки, усиливающей агрегацию, то может быть установлено, что агрегация произошла, в случае если интенсивность полосы в области 3200 см-1, которая обусловлена молекулой Н2О, связанной в кристалле комплексного кислотного катализатора, после обработки, усиливающей агрегацию, является меньшей, чем интенсивность полосы комплексного кислотного катализатора до обработки, усиливающей агрегацию, а интенсивность полосы в области 3500 см-1, которая обусловлена ОН-группой, связанной с сильнокислотным субстратом комплексного кислотного катализатора, после обработки, усиливающей агрегацию, является большей, чем интенсивность полосы комплексного кислотного катализатора до обработки, усиливающей агрегацию. В ИК-измерениях полоса поглощения, обусловленная молекулой Н2О, не перекрывается полосой поглощения, обусловленной ОН-группами, связанными с сильнокислотным субстратом, и в общем может наблюдаться в виде широкой полосы.
Кроме того, в Рамановской спектроскопии, например, где сосредоточивают внимание на симметричных валентных колебаниях октаэдрической структуры WO6 фосфорновольфрамовой кислоты, узкая линия комбинационного рассеяния наблюдается в области 985 см-1 для комплексного кислотного катализатора в кристаллическом состоянии до агрегационной обработки (см. Сравнительный Пример 1 на Фиг.4). Однако в комплексном кислотном катализаторе в агрегированном состоянии после агрегационной обработки происходит сдвиг в сторону более высокой частоты, в область 1558 см-1, и интенсивность линии значительно снижается, то есть снижается чувствительность (см. Пример 2 на Фиг.4). Такой сдвиг в область более высокой частоты и снижение чувствительности обусловливаются описанными ниже структурными изменениями, вызванными агрегацией комплексного кислотного катализатора. В октаэдрической структуре WO6, поскольку ионный радиус W составляет всего 0,074 нм, расстояние между атомами W и О является предельно малым, как показано на Фиг.1. Там, где поверхностная энергия стабилизирована агрегацией и компоновка деформирована ближе к сферической форме, симметрия фрагмента WO6 искажается, и расстояние между W и О даже сокращается. В результате снижение чувствительности и увеличение прочности связи обусловливает одновременное рассеяние и сдвиг в более высокочастотную область. Это явление не является присущим только фосфорновольфрамовой кислоте и подобным образом проявляется в других комплексных кислотах. Поэтому агрегированное состояние комплексного кислотного катализатора может быть подтверждено наблюдением структурных изменений в комплексном кислотном катализаторе с использованием Рамановской спектроскопии.
В соответствии с изобретением, конкретный способ обработки, усиливающей агрегацию, не является специально ограниченным, при условии, что комплексная кислота может быть переведена в вышеописанное агрегированное состояние. Конкретную обработку, усиливающую агрегацию, рекомендуемую согласно изобретению, проводят перед процессом гидролиза, в котором комплексную кислоту используют в качестве катализатора гидролиза растительного волокнистого материала, но, как здесь описано выше, комплексная кислота может быть отделена от полученного сахарида после процесса гидролиза, извлечена и повторно использована вновь в качестве катализатора гидролиза. Поэтому обработка, усиливающая агрегацию, может быть проведена в процессе гидролиза или процессе отделения сахарида перед повторным применением. Соответственно этому, ниже сначала будет описан каждый процесс способа осахаривания и отделения растительного волокнистого материала с использованием комплексного кислотного катализатора, и затем будет приведено разъяснение обработки, усиливающей агрегацию, комплексного кислотного катализатора.
В соответствии с изобретением, комплексный кислотный катализатор подвергают обработке, усиливающей агрегацию, в тот момент времени, когда в процесс гидролиза загружают растительный волокнистый материал в таком количестве, которое может быть загружено в одном цикле для всей реакционной системы, то есть в момент времени, в который начинается основная операция процесса гидролиза. В результате можно эффективно ингибировать последующую реакцию моносахарида, образованного в процессе гидролиза. Выражение «растительный волокнистый материал в количестве, которое может быть загружено в одном цикле», упоминаемое в настоящем описании, означает количество, которое позволяет привести смесь в полностью однородное и перемешанное состояние, когда это количество смешано с комплексным кислотным катализатором (количество, используемое в процессе гидролиза) в псевдорасплавленном состоянии, который применяют в процессе гидролиза. В этом случае растительный волокнистый материал в смеси находится не в сухом состоянии. Поскольку количество растительного волокнистого материала, которое может быть загружено в одном цикле, меняется в зависимости от типа месильной машины, это количество нельзя определить однозначно, но в основном является предпочтительным, чтобы весовое соотношение (растительный волокнистый материал: комплексный кислотный катализатор) растительного волокнистого материала в количестве, которое может быть загружено в одном цикле, и комплексного кислотного катализатора в псевдорасплавленном состоянии, который используют в процессе гидролиза, составляло от 1:2 до 1:6. В настоящем описании выражение «момент времени, в который количество растительного волокнистого материала, которое может быть загружено в одном цикле для всей реакционной системы, загружают в процесс гидролиза» означает момент времени, в который количество растительного волокнистого материала, которое смешивают с комплексным кислотным катализатором, который используют в процессе гидролиза, достигает «количества, которое может быть загружено в одном цикле» в процессе гидролиза.
Во-первых, будет описан процесс гидролиза, в котором целлюлозу, содержащуюся в растительном волокнистом материале, гидролизуют и получают сахарид, главным образом включающий глюкозу. В нижеприведенном разъяснении внимание сосредоточено на процессе, в котором глюкозу получают главным образом из целлюлозы, но в объем изобретения входит также процесс, в котором в растительный волокнистый материал, в дополнение к целлюлозе, включена гемицеллюлоза, и процесс, в котором продукт, в дополнение к глюкозе, включает другие моносахариды, такие как ксилоза.
Растительный волокнистый материал не является специально ограниченным, при условии, что он включает целлюлозу или гемицеллюлозу, и примеры его включают биомассу на основе целлюлозы, такую как широколиственные растения, бамбуки, хвойные породы деревьев, гибискус коноплевый, отходы древесины мебельного производства, рисовая солома, пшеничная солома, рисовая шелуха и выжатые остатки сахарного тростника (жмых). Растительный волокнистый материал может представлять собой целлюлозу или гемицеллюлозу, которая выделена из биомассы, или может быть целлюлозой или гемицеллюлозой, которая синтезирована искусственно. Такие волокнистые материалы обычно применяют в измельченной до порошкообразного состояния форме для улучшения диспергируемости в реакционной системе. Способ измельчения в порошок может представлять собой общеупотребительный способ. Из соображений облегчения смешения с комплексным кислотным катализатором и реагирования является предпочтительным, чтобы волокнистый материал был измельчен в порошок с диаметром частиц от около нескольких микрон до 200 мкм.
Далее, лигнин, содержащийся в волокнистом материале, может быть растворен, если необходимо, путем проведения предварительной варочной обработки. Растворением и удалением лигнина можно повысить вероятность контактирования между комплексным кислотным катализатором и целлюлозой в процессе гидролиза и в то же время снизить количество остатка, содержащегося в гидролитической реакционной смеси, и подавить снижение выхода сахарида или коэффициента извлечения комплексной кислоты, обусловленное смешением полученного сахарида или комплексной кислоты с остатком. В том случае, когда проводят варочную обработку, степень измельчения растительного волокнистого материала может быть сравнительно малой (грубое дробление). В результате этого можно достигнуть снижения трудозатрат, расходов и энергопотребления, требуемых для измельчения волокнистого материала в порошок. Варочная обработка может быть проведена, например, приведением растительного волокнистого материала (например, в кусках величиной от нескольких сантиметров до нескольких миллиметров) в контакт со щелочью или с солью, такими как NaOH, KOH, Ca(OH)2, Na2SO3, NaHCO3, NaHSO3, Mg(HSO3)2, Ca(HSO3)2, их водным раствором, их смесью с раствором SO2, или таким газом, как NH3, в атмосфере водяного пара. Конкретные условия включают реакционную температуру от 120 до 160°С и продолжительность реакции от нескольких десятков минут до около 1 часа.
Последовательность, в которой комплексный кислотный катализатор и растительный волокнистый материал загружают в реакционный контейнер, не является специально ограниченной. Например, комплексный кислотный катализатор может быть загружен в реакционный контейнер и нагрет для получения псевдорасплавленного состояния и затем может быть загружен растительный волокнистый материал. Альтернативно, комплексный кислотный катализатор и растительный волокнистый материал могут быть загружены совместно и затем нагреты для приведения комплексного кислотного катализатора в псевдорасплавленное состояние. В случае, когда комплексный кислотный катализатор и растительный волокнистый материал нагревают после загрузки, комплексный кислотный катализатор и растительный волокнистый материал предпочтительно смешивают и перемешивают заблаговременно, перед нагреванием. Степень контакта между комплексной кислотой и растительным волокнистым материалом может быть повышена проведением смешения до некоторой степени перед тем как комплексную кислоту переводят в псевдорасплавленное состояние. Как здесь описано выше, поскольку комплексный кислотный катализатор переходит в псевдорасплавленное состояние и действует как реакционный растворитель в процессе гидролиза, то в соответствии с изобретением можно не применять воду или органический растворитель в качестве реакционного растворителя в процессе гидролиза, но вода или органический растворитель могут понадобиться в зависимости от формы (размера, состояния волокон и т.д.) растительного волокнистого материала, соотношения и объемного соотношения в смеси комплексного кислотного катализатора и растительного волокнистого материала и тому подобного.
Псевдорасплавленное состояние комплексной кислоты изменяется в зависимости от температуры и количества кристаллизационной воды, содержащейся в комплексном кислотном катализаторе (см. Фиг.2). Более конкретно, когда количество кристаллизационной воды, содержащейся в фосфорновольфрамовой кислоте, которая представляет собой комплексную кислоту, является высоким, температура, при которой кислота демонстрирует псевдорасплавленное состояние, снижается. Таким образом, комплексный кислотный катализатор, содержащий большое количество кристаллизационной воды, проявляет каталитическое действие в реакции гидролиза целлюлозы при более низкой температуре, чем комплексный кислотный катализатор с относительно малым количеством кристаллизационной воды. Другими словами, регулированием количества кристаллизационной воды, содержащейся в комплексном кислотном катализаторе в реакционной системе процесса гидролиза, можно привести комплексный кислотный катализатор в псевдорасплавленное состояние при целевой температуре реакции гидролиза. Например, когда в качестве комплексного кислотного катализатора применяют фосфорновольфрамовую кислоту, можно регулировать температуру реакции гидролиза в пределах диапазона между 40 и 110°С путем изменения количества кристаллизационной воды в комплексной кислоте (см. Фиг.2).
Фиг.2 показывает взаимосвязь между долей кристаллизационной воды в гетерополикислоте (фосфорновольфрамовой кислоте), которая представляет собой типичный комплексный кислотный катализатор, и температурой (кажущейся температурой плавления), при которой состояние комплексного кислотного катализатора начинает изменяться до псевдорасплавленного состояния, и комплексный кислотный катализатор находится в твердом состоянии в области ниже кривой, и в псевдорасплавленном состоянии в области выше кривой. Кроме того, на Фиг.2 доля кристаллизационной воды (%) представляет собой значение, полученное при допущении, что стандартное количество кристаллизационной воды n (n=30) в комплексной кислоте (фосфорновольфрамовой кислоте) составляет 100%. Поскольку ни один компонент комплексного кислотного катализатора не подвергается термическому разложению и не улетучивается даже при высокой температуре, такой как 800°С, количество кристаллизационной воды может быть установлено пиролитическим методом (термогравиметрическими (TG) измерениями).
Стандартное количество кристаллизационной воды, как оно здесь упоминается, представляет собой количество (число молекул) кристаллизационной воды, содержащейся в одной молекуле комплексной кислоты в твердом состоянии при комнатной температуре, и стандартное количество варьирует в зависимости от типа комплексной кислоты. Например, стандартное количество кристаллизационной воды составляет около 30 в фосфорновольфрамовой кислоте (H3[PW12O40]·nH2O (n≈30)), около 24 в кремневольфрамовой кислоте (H4[SiW12O40]·nH2O (n≈24)) и около 30 в фосфорномолибденовой кислоте (H3[PMo12O40]·nH2O (n≈30)).
Количество кристаллизационной воды, содержащейся в комплексном кислотном катализаторе, можно регулировать путем корректирования количества воды, присутствующей в гидролитической реакционной системе. Более конкретно, когда желательно увеличивать количество кристаллизационной воды, содержащейся в комплексном кислотном катализаторе, то есть снижать реакционную температуру, то можно добавлять воду в гидролитическую реакционную систему, например, добавлением воды к смеси, содержащей растительный волокнистый материал и комплексный кислотный катализатор, или повышением относительной влажности атмосферы в реакционной системе. В результате комплексная кислота захватывает добавленную воду как кристаллизационную воду и кажущаяся температура плавления комплексного кислотного катализатора снижается.
Напротив, когда желательно уменьшать количество кристаллизационной воды, содержащейся в комплексном кислотном катализаторе, то есть повышать реакционную температуру, то можно снижать количество кристаллизационной воды, содержащейся в комплексном кислотном катализаторе, удалением воды из гидролитической реакционной системы, например нагреванием реакционной системы для испарения воды или добавлением поглотителя влаги к смеси, содержащей растительный волокнистый материал и комплексный кислотный катализатор. В результате кажущаяся температура плавления комплексного кислотного катализатора повышается. Как описано выше, можно без труда контролировать количество кристаллизационной воды, содержащейся в комплексной кислоте, и также возможно легко регулировать реакционную температуру, при которой гидролизуют целлюлозу, путем корректирования количества кристаллизационной воды.
Далее, является предпочтительным, чтобы желательное количество кристаллизационной воды в комплексном кислотном катализаторе могло быть обеспечено даже когда относительная влажность реакционной системы снижается при нагревании в процессе гидролиза. Более конкретно, может быть использован способ, в котором внутри реакционного резервуара, который был предварительно герметизирован, создают состояние давления насыщенных водяных паров при температуре реакции гидролиза таким образом, что атмосфера в реакционной системе находится в состоянии ниже давления насыщенных водяных паров при предварительно заданной реакционной температуре, снижают температуру для конденсации водяных паров, в то же время сохраняя герметичное состояние, и сконденсированную воду добавляют к растительному волокнистому материалу и комплексному кислотному катализатору. Кроме того, в случае, где используют растительный волокнистый материал, содержащий влагу, предпочтительно, чтобы количество влаги, содержащейся в растительном волокнистом материале, также учитывалось как количество влаги, присутствующей в реакционной системе; это не является специально необходимым в случае, где применяют сухой растительный волокнистый материал.
Преимущество снижения реакционной температуры в процессе гидролиза состоит в том, что может быть повышена эффективность использования энергии. Селективность получения глюкозы при гидролизе целлюлозы, содержащейся в растительном волокнистом материале, варьирует в зависимости от температуры в процессе гидролиза. Выход реакции в общем повышается по мере роста реакционной температуры. Например, как описано в Японской Патентной Заявке № 2007-115407, в реакции гидролиза целлюлозы с использованием фосфорновольфрамовой кислоты с 160%-ным количеством кристаллизационной воды степень R конверсии реакции при температуре от 50 до 90°С возрастает при повышении температуры и почти вся целлюлоза реагирует при температуре около 80°С. Выход η глюкозы проявляет подобную тенденцию к повышению при температуре от 50 до 60°С, достигает максимума при 70°С и затем снижается. Таким образом, глюкоза получается с высокой селективностью при температурах от 50 до 60°С, но при температурах от 70 до 90°С протекают также иные реакции, нежели образование глюкозы, такие как образование других сахаридов, таких как ксилоза, и формирование продуктов разложения. Поэтому реакционная температура при гидролизе представляет собой важный фактор, который определяет селективность в пределах степени конверсии целлюлозы и селективность образования глюкозы, и предпочтительно, чтобы температура реакции гидролиза была низкой в плане энергетического выхода. Однако предпочтительно, чтобы температуру реакции гидролиза определяли, принимая во внимание также степень конверсии целлюлозы и селективность образования глюкозы.
Чтобы гидролизовать целлюлозу в процессе гидролиза, необходима вода. Более конкретно, для разложения целлюлозы, полученной полимеризацией (n) фрагментов глюкозы, на (n) молекул глюкозы, необходимы (n-1) молекул воды (n представляет натуральное число). Поэтому в случае, где в реакционной системе не наличествует общее суммарное количество кристаллизационной воды, которое необходимо для приведения комплексной кислоты в псевдорасплавленное состояние при реакционной температуре, и количество воды, необходимое для гидролиза всего загруженного количества целлюлозы до глюкозы, для гидролиза целлюлозы расходуется кристаллизационная вода комплексного кислотного катализатора, и количество кристаллизационной воды комплексного кислотного катализатора сокращается, и комплексная кислота затвердевает. Таким образом, степень контакта между комплексным кислотным катализатором и растительным волокнистым материалом, или вязкость смеси растительного волокнистого материала и комплексного кислотного катализатора возрастает, и требуется продолжительное время для достаточного перемешивания смеси.
Поэтому, чтобы обеспечить функционирование комплексного кислотного катализатора как реакционного растворителя и катализатора при реакционной температуре в процессе гидролиза, то есть, чтобы иметь возможность поддерживать комплексный кислотный катализатор в псевдорасплавленном состоянии, является предпочтительным, чтобы количество воды в реакционной системе удовлетворяло следующим условиям. Так, предпочтительно, чтобы количество воды в реакционной системе было равным или большим, чем общая сумма (а) количества кристаллизационной воды, необходимого для поддержания всего комплексного кислотного катализатора, присутствующего в реакционной системе, в псевдорасплавленном состоянии при реакционной температуре в процессе гидролиза, и (b) количества воды, необходимого для гидролиза до глюкозы всего количества целлюлозы, присутствующей в реакционной системе. В особенности предпочтительно, чтобы добавлялась общая сумма (а) и (b). Это обусловлено тем, что, если добавляют избыточное количество воды, образованный сахарид и комплексная кислота растворяются в излишней воде, тем самым затрудняя процесс разделения сахарида и комплексной кислоты.
В процессе гидролиза имеет место ситуация, где количество воды в реакционной системе уменьшается и также сокращается количество кристаллизационной воды комплексного кислотного катализатора, вследствие чего комплексный кислотный катализатор становится твердым, и степень контакта с растительным волокнистым материалом и эффективность перемешивания реакционной системы ухудшаются. Возникновения такой проблемы можно избежать повышением температуры гидролиза так, чтобы комплексный кислотный катализатор перевести в псевдорасплавленное состояние.
Как описано выше, температурные условия в процессе гидролиза могут быть надлежащим образом определены с учетом нескольких факторов (например, селективности реакции, энергетического выхода, степени конверсии в реакции целлюлозы и тому подобных), но с позиции баланса между энергетическим выходом, степенью конверсии в реакции целлюлозы и выходом глюкозы обычно предпочтительна температура, равная и меньшая, чем 140°С, и в особенности предпочтительна температура, равная или меньшая 120°С. В зависимости от формы растительного волокнистого материала процесс гидролиза может быть также проведен при низкой температуре, равной или меньшей 100°С. В этом случае глюкоза может быть получена с особенно высоким энергетическим выходом.
Давление в процессе гидролиза не является специально ограниченным, но, поскольку каталитическая активность комплексного кислотного катализатора в отношении реакции гидролиза целлюлозы высока, гидролиз целлюлозы может проходить с хорошей эффективностью даже при мягких условиях давления, таких как диапазон от нормального давления (атмосферного давления) до 1 МПа.
Соотношение растительного волокнистого материала и комплексного кислотного катализатора варьирует в зависимости от свойств (например, размера и тому подобного) и типа используемого растительного волокнистого материала, и способа перемешивания или метода смешения, применяемого в процессе гидролиза. Поэтому, хотя это соотношение может быть надлежащим образом определено соответственно применяемым условиям, предпочтительное отношение комплексного кислотного катализатора к растительному волокнистому материалу (весовое отношение) предпочтительно варьирует в диапазоне от 2:1 до 6:1 и обычно может составлять от около 2:1 до 4:1. Поскольку смесь, включающая комплексный кислотный катализатор и растительный волокнистый материал в процессе гидролиза, имеет высокую вязкость, преимущественно может быть применена, например, шаровая мельница с нагреванием, но может быть также использовано типичное перемешивающее устройство.
Продолжительность процесса гидролиза не является специально ограниченной и может быть надлежащим образом отрегулирована согласно форме используемого растительного волокнистого материала, соотношению растительного волокнистого материала и комплексного кислотного катализатора, каталитической активности комплексного кислотного катализатора, реакционной температуре, реакционному давлению и тому подобным.
Когда температура в реакционной системе снижается после завершения гидролиза, сахарид, полученный в процессе гидролиза, образует водный раствор сахарида, когда вода, которая растворяет сахарид, присутствует в гидролитической реакционной смеси, включающей комплексный кислотный катализатор, и там, где вода не присутствует, сахарид выпадает в осадок и находится в твердом состоянии. Часть образованного сахарида может присутствовать в форме водного раствора и остальное количество может содержаться в форме смеси в твердом состоянии. Поскольку комплексный кислотный катализатор также растворим в воде, то там, где в смеси после процесса гидролиза содержится достаточное количество воды, комплексный кислотный катализатор также растворен в воде.
Ниже будет описан процесс выделения сахарида, в котором разделяют сахарид (включающий главным образом глюкозу), образованный в процессе гидролиза, и комплексный кислотный катализатор. В способе осахаривания и отделения согласно изобретению способ разделения сахарида и комплексной кислоты не ограничивается описанным ниже способом.
Реакционная смесь после процесса гидролиза (ниже может также называться «гидролитическая реакционная смесь») включает по меньшей мере комплексный кислотный катализатор и полученный сахарид. В случае, где количество воды в процессе гидролиза представляет собой общую сумму (а) и (b), сахарид выпадает в осадок в гидролитической реакционной смеси. Между тем комплексный кислотный катализатор также переходит в твердое состояние, когда температура снижается. В зависимости от типа используемого растительного волокнистого материала в гидролитической реакционной смеси в виде твердого компонента содержится остаток (непрореагировавшая целлюлоза или лигнин и т.д.).
Комплексный кислотный катализатор проявляет растворимость в органических растворителях, в которых сахарид, включающий главным образом глюкозу, нерастворим или имеет плохую растворимость. Поэтому к гидролитической реакционной смеси можно добавить органический растворитель, который является плохим растворителем для сахарида и хорошим растворителем для комплексного кислотного катализатора, провести перемешивание, избирательно растворить комплексный кислотный катализатор в органическом растворителе и затем с помощью твердофазно-жидкостного разделения отделить раствор в органическом растворителе, содержащий растворенные комплексные кислоты, от твердого компонента, включающего сахарид. В зависимости от используемого растительного волокнистого материала, в твердом компоненте, включающем сахарид, может содержаться остаток или тому подобное. Способ разделения раствора в органическом растворителе и твердого компонента не является специально ограниченным и может быть применен типичный способ твердофазно-жидкостного разделения, такой как декантация и фильтрование.
Органический растворитель не является специально ограниченным, при условии, что он является хорошим растворителем для комплексного кислотного катализатора и плохим растворителем для сахарида, но для подавления растворения сахарида в органическом растворителе предпочтительно, чтобы растворимость сахарида в органическом растворителе была равной или меньшей, чем 0,6 г/100 мл, и более предпочтительно равной или меньшей, чем 0,06 г/100 мл. В этом случае для повышения коэффициента извлечения комплексного кислотного катализатора предпочтительно, чтобы растворимость комплексной кислоты в органическом растворителе была равной или большей, чем 20 г/100 мл, более предпочтительно равной или большей, чем 40 г/100 мл.
Конкретные примеры органического растворителя включают спирты, такие как этанол, метанол, н-пропанол и октанол, и простые эфиры, такие как диэтиловый простой эфир и диизопропиловый простой эфир. Преимущественно могут быть применены спирты и простые эфиры и среди них, с позиции растворяющей способности и температуры кипения, предпочтительны этанол и диэтиловый простой эфир. Диэтиловый простой эфир не растворяет сахариды, такие как глюкоза, и имеет высокую способность растворять комплексные кислоты. Поэтому диэтиловый простой эфир представляет собой один из оптимальных растворителей для разделения сахаридов и комплексных кислотных катализаторов. Этанол также почти не растворяет сахариды, такие как глюкоза, и имеет высокую способность растворять комплексные кислоты. Поэтому он также является одним из оптимальных растворителей. Диэтиловый эфир превосходит этанол в отношении дистилляции, но преимущество этанола состоит в том, что его проще получать, чем диэтиловый простой эфир.
Количество используемого органического растворителя варьирует в зависимости от способности растворителя растворять сахарид и комплексный кислотный катализатор, и количества влаги, содержащейся в гидролитической реакционной смеси. Поэтому подходящее количество органического растворителя может быть определено надлежащим образом.
Обычно является предпочтительным, чтобы перемешивание гидролитической реакционной смеси и органического растворителя проводили при конкретной температуре в пределах температурного диапазона от комнатной температуры до 60°С, причем конкретная температура зависит от температуры кипения органического растворителя. Способ перемешивания гидролитической реакционной смеси и органического растворителя не является специально ограниченным и перемешивание может быть выполнено обычным способом. С позиции коэффициента извлечения комплексной кислоты, в качестве способа перемешивания предпочтительно перемешивание и измельчение в шаровой мельнице.
Для повышения коэффициента извлечения сахарида и комплексной кислоты и увеличения чистоты полученного сахарида предпочтительно, чтобы органический растворитель (органический растворитель, который является плохим растворителем для сахарида и хорошим растворителем для комплексного кислотного катализатора) добавляли к твердому компоненту, полученному путем вышеупомянутого твердофазно-жидкостного разделения, и перемешивали с ним, тем самым выполняя промывание органическим растворителем. Это обусловлено тем, что комплексный кислотный катализатор, который был примешан к твердому компоненту, может быть удален и извлечен. Смесь, в которой органический растворитель добавляют к твердому компоненту, может быть разделена на твердый компонент и раствор комплексной кислоты в органическом растворителе с помощью твердофазно-жидкостного разделения таким же образом, как в гидролитической реакционной смеси. Если необходимо, твердый компонент может быть промыт органическим растворителем несколько раз. Добавлением воды, такой как дистиллированная вода, к твердому компоненту, полученному твердофазно-жидкостным разделением, перемешиванием и затем проведением твердофазно-жидкостного разделения (поскольку сахарид растворим в воде), можно отделить водный раствор сахарида от твердого компонента, включающего остаток или тому подобное.
Удалением органического растворителя из жидкостного компонента (раствора в органическом растворителе, включающего растворенный в нем комплексный кислотный катализатор), полученного твердофазно-жидкостным разделением, можно разделить комплексный кислотный катализатор и органический растворитель и извлечь комплексный кислотный катализатор. Способ удаления органического растворителя не является специально ограниченным, за исключением дистилляции при атмосферном давлении. Примеры применимых способов включают вакуумную дистилляцию и лиофильную сушку вымораживанием. Среди них предпочтительна вакуумная дистилляция при температуре, равной и меньшей 50°С. Извлеченный комплексный кислотный катализатор может быть опять использован в качестве катализатора гидролиза растительного волокнистого материала. Раствор в органическом растворителе, включающий извлеченную комплексную кислоту, после промывания твердого компонента может быть вновь применен для промывания твердого компонента (см. Фиг.6).
В зависимости от количества влаги в процессе гидролиза, гидролитическая реакционная смесь может содержать водный раствор, включающий растворенные в нем сахарид и комплексную кислоту. В этом случае твердый компонент, включающий сахарид, и органический растворитель, включающий растворенный в нем комплексный кислотный катализатор, могут быть разделены удалением влаги из гидролитической реакционной смеси для осаждения растворенного сахарида и комплексной кислоты и затем добавлением органического растворителя, перемешиванием и проведением твердофазно-жидкостного разделения. Является в особенности предпочтительным, чтобы количество влаги в гидролитической реакционной смеси было отрегулировано так, что доля кристаллизационной воды во всем комплексном кислотном катализаторе, содержащемся в гидролитической реакционной смеси, составляет менее 100%. В случае, где комплексный кислотный катализатор имеет большое количество кристаллизационной воды, обычно такое количество кристаллизационной воды, которое является равным или большим, чем стандартное количество кристаллизационной воды, сахарид, который представляет собой продукт, растворяется в избыточной влаге, и коэффициент извлечения сахарида снижается вследствие примешивания сахарида к раствору в органическом растворителе, включающему комплексную кислоту. Снижением доли кристаллизационной воды в комплексном кислотном катализаторе до уровня менее 100% можно предотвратить такое примешивание сахарида к комплексному кислотному катализатору.
Для снижения доли кристаллизационной воды в комплексном кислотном катализаторе, содержащемся в гидролитической реакционной смеси, может быть применен способ, которым можно сократить влагосодержание гидролитической реакционной смеси. Примеры такого способа включают способ, которым реакционную систему переводят из замкнутого состояния в открытое и проводят нагревание для испарения влаги, содержащейся в гидролитической смеси, и способ, которым к гидролитической смеси добавляют поглотитель влаги или тому подобное, и удаляют влагу, содержащуюся в гидролитической смеси.
Ниже будет приведено разъяснение обработки, усиливающей агрегацию, комплексного кислотного катализатора. Как здесь описано выше, конкретную обработку, усиливающую агрегацию, которая рекомендуется в соответствии с изобретением, выполняют перед процессом гидролиза, в котором комплексную кислоту используют в качестве катализатора гидролиза растительного волокнистого материала, но в случае, где комплексную кислоту, извлеченную в процессе выделения сахарида, применяют повторно, обработка, усиливающая агрегацию, также может быть реализована в процессе гидролиза или процессе выделения сахарида. Преобразование комплексного кислотного катализатора в агрегированное состояние стимулируют, например, перемешиванием комплексной кислоты в псевдорасплавленном состоянии или добавлением комплексной кислоты к растворителю и перемешиванием при нагревании, или перемешиванием комплексной кислоты вместе с растительным волокнистым материалом при нагревании, что заставляет комплексную кислоту действовать в качестве катализатора гидролиза.
Для стимулирования конверсии в агрегированное состояние могут быть использованы следующие три конкретных способа.
(1) Способ нагревания и перемешивания комплексного кислотного катализатора и органического растворителя, который может растворять комплексный кислотный катализатор; (2) способ, реализуемый в процессе гидролиза, в котором растительный волокнистый материал гидролизуют с использованием комплексного кислотного катализатора, включающий нагревание и перемешивание части растительного волокнистого материала в количестве, которое может быть загружено в одном цикле, с комплексным кислотным катализатором в псевдорасплавленном состоянии и выполнение гидролиза растительного волокнистого материала; и (3) способ нагревания и перемешивания комплексного кислотного катализатора в псевдорасплавленном состоянии.
Эти способы (1)-(3) будут описаны ниже.
В способе (1) нагревания и перемешивания комплексного кислотного катализатора и органического растворителя, который может растворять комплексный кислотный катализатор, температура нагревания может быть надлежащим образом установлена соответственно вариации состояния комплексной кислоты в растворителе, но обычно предпочтительна температура, равная или превышающая 30°С. Из соображений предотвращения повторной кристаллизации комплексного кислотного катализатора предпочтительно, чтобы температура была равной или меньшей 65°С, в особенности равной или меньшей 55°С. Примеры органических растворителей, которые могут растворять комплексный кислотный катализатор, включают органические растворители, которые могут быть использованы в вышеописанном процессе выделения сахарида. Среди них, с позиции растворяющей способности и температуры кипения, предпочтительны этанол и метанол. Смесевое соотношение органического растворителя и комплексного кислотного катализатора не является специально ограниченным и может быть надлежащим образом выбрано соответственно растворимости комплексного кислотного катализатора в органическом растворителе. Продолжительность нагревания и перемешивания может быть надлежащим образом определена соответственно растворимости комплексного кислотного катализатора в применяемом органическом растворителе и температуре нагревания, и обычно продолжительность нагревания и перемешивания составляет от около 10 минут до 60 минут, или от около 30 минут до 60 минут. Способ смешения не является специально ограниченным и может быть применен общеизвестный способ.
Даже в случае, где используют не бывший в употреблении новый комплексный кислотный реагент, такое нагревание и перемешивание комплексного кислотного катализатора и органической кислоты может преобразовать состояние комплексного кислотного катализатора в агрегированное состояние и ингибировать реакцию дегидратации сахарида в процессе гидролиза. Кроме того, агрегирование повторно используемого комплексного кислотного катализатора может быть стимулировано добавлением органического растворителя к гидролитической реакционной смеси и перемешиванием в процессе выделения сахарида и затем нагреванием и перемешиванием раствора комплексной кислоты в органическом растворителе, полученного в результате твердофазно-жидкостного разделения.
Комплексный кислотный катализатор, подвергнутый обработке, усиливающей агрегацию, может быть выделен удалением органического растворителя из смеси комплексного кислотного катализатора и органического растворителя после нагревания и перемешивания. В этом случае быстрым удалением органического растворителя с использованием метода вакуумирования можно без труда сохранять комплексный кислотный катализатор в агрегированном состоянии. Более конкретно, предпочтительно, чтобы органический растворитель удаляли вакуумной дистилляцией, лиофильной сушкой вымораживанием или тому подобным путем. Органический растворитель может быть также удален нагреванием, но из соображений сохранения комплексной кислоты в агрегированном состоянии предпочтительно, чтобы органический растворитель удаляли при низкой температуре (более конкретно, при температуре, равной или меньшей 65°С), и можно сказать, что предпочтительны вышеупомянутые вакуумная дистилляция и лиофильная сушка вымораживанием.
Кроме того, агрегирование добавленного комплексного кислотного катализатора и используемого повторно комплексного кислотного катализатора может быть также стимулировано добавлением органического растворителя к гидролитической реакционной смеси и перемешиванием в процессе выделения сахарида, затем добавлением комплексного кислотного катализатора в кристаллическом состоянии (не использованного ранее комплексного кислотного реагента или тому подобного) к раствору комплексной кислоты в органическом растворителе, полученному твердофазно-жидкостным разделением, и перемешиванием при нагревании. В дополнение к повторяющимся извлечению и повторному использованию комплексного кислотного катализатора, даже в случае, где количество извлеченной комплексной кислоты сокращалось, можно выполнить агрегирующую обработку комплексного кислотного катализатора в кристаллическом состоянии добавлением комплексного кислотного катализатора в кристаллическом состоянии в количестве, которое компенсирует потерю комплексного кислотного катализатора в процессе выделения сахарида, и применением процесса отделения сахарида.
В способе (2), в котором часть растительного волокнистого материала в количестве, которое может быть загружено в одном цикле, перемешивают при нагревании с комплексным кислотным катализатором в псевдорасплавленном состоянии, и гидролиз растительного волокнистого материала выполняют в процессе гидролиза, гидролизом только части растительного волокнистого материала, который может быть загружен в одном цикле, можно сократить количество моносахарида, который может быть дегидратирован комплексным кислотным катализатором на начальной стадии процесса гидролиза, и стимулировать агрегирование комплексного кислотного катализатора. После перевода комплексного кислотного катализатора в агрегированное состояние дополнительно загружают остальной растительный волокнистый материал, тем самым делая возможным ингибирование последующей реакции сахарида, полученного из дополнительно загруженного растительного волокнистого материала.
Выражение «часть растительного волокнистого материала в количестве, которое может быть загружено в одном цикле», как оно здесь упоминается, означает часть вышеупомянутого «растительного волокнистого материала в количестве, которое может быть загружено в одном цикле», и не ограничивается конкретным количеством. Обычно это очень маленькое количество, такое, чтобы вязкость комплексного кислотного катализатора в псевдорасплавленном состоянии до добавления сохранялась даже после того, как это количество растительного волокнистого материала добавляют к комплексному кислотному катализатору в псевдорасплавленном состоянии и перемешивают с ним. Когда такое очень малое количество растительного волокнистого материала поначалу добавляют к комплексному кислотному катализатору, который используют в процессе гидролиза, можно ожидать, что за счет столь малого убытка в целом будет достигнут эффект повышения выхода реакции. Конкретное количество «части растительного волокнистого материала в количестве, которое может быть загружено в одном цикле» предпочтительно является равным или меньшим, чем 10% по весу, в особенности равным или меньшим, чем 5% по весу от растительного волокнистого материала в количестве, которое может быть загружено в одном цикле.
Продолжительность гидролиза части растительного волокнистого материала не является специально ограниченной и может быть установлена, принимая во внимание снижение вязкости гидролитической смеси в качестве индикатора. Обычно продолжительность гидролиза составляет от около 10 минут до 300 минут или от около 60 минут до 300 минут. Другие условия, такие как продолжительность реакции и давление, могут быть подобными таковым в процессе гидролиза.
Проведением гидролиза этой части растительного волокнистого материала с помощью комплексного кислотного катализатора можно преобразовать комплексный кислотный катализатор в агрегат и ингибировать реакцию дегидратации сахарида в процессе гидролиза, в то же время сокращая количество моносахарида, дегидратируемого комплексным кислотным катализатором, до минимума, даже в случае, где используют не бывший ранее в употреблении комплексный кислотный реагент. Кроме того, поскольку агрегирующая обработка комплексной кислоты может быть реализована с использованием процесса гидролиза, можно избежать усложнения производственного процесса.
Способ (3) нагревания и перемешивания комплексного кислотного катализатора в псевдорасплавленном состоянии типично представляет собой способ, которым комплексный кислотный катализатор нагревают и переводят в псевдорасплавленное состояние и затем перемешивают при нагревании перед тем, как растительный волокнистый материал и комплексный кислотный катализатор смешивают в процессе гидролиза. Обычно комплексный кислотный катализатор нагревают и перемешивают для получения псевдорасплавленного состояния в реакционном контейнере для применения в процессе гидролиза и проводят агрегационную обработку, а затем добавляют растительный волокнистый материал и выполняют процесс гидролиза.
Температура нагревания не является специально ограниченной, при условии, что комплексная кислота может поддерживаться в псевдорасплавленном состоянии, и может быть надлежащим образом установлена соответственно типу комплексной кислоты и доле кристаллизационной воды. Для выполнения агрегирования комплексного кислотного катализатора с хорошей эффективностью предпочтительно, чтобы нагревание проводили при температуре, которая по меньшей мере на величину от 10 до 30°С, более предпочтительно на величину по меньшей мере от 10 до 20°С, еще более предпочтительно по меньшей мере на величину от 5 до 10°С выше, чем температура, при которой состояние комплексного кислотного катализатора начитает изменяться на псевдорасплавленное состояние.
Комплексный кислотный катализатор предпочтительно нагревают и перемешивают с водой в таком количестве, что доля кристаллизационной воды в комплексном кислотном катализаторе становится равной или большей 100%. В особенности является предпочтительным, чтобы комплексный кислотный катализатор нагревали и перемешивали с водой в таком количестве, что доля кристаллизационной воды в комплексном кислотном катализаторе становится равной или большей, чем 100%, водой, которая необходима для гидролиза растительного волокнистого материала в последующем процессе гидролиза, и водой, обеспечивающей присутствие насыщенных водяных паров в мертвом пространстве реактора. Это обусловлено тем, что нагревание и перемешивание в присутствии воды стимулирует переход комплексного кислотного катализатора в псевдорасплавленное состояние, тем самым усиливая агрегирование.
Продолжительность нагревания и перемешивания может быть установлена, принимая во внимание снижение вязкости гидролитической смеси в качестве индикатора. Обычно продолжительность нагревания и перемешивания может составлять от 20 до 300 минут или от 60 до 300 минут. Процесс нагревания и перемешивания комплексной кислоты в псевдорасплавленном состоянии может быть без труда включен в уже существующий процесс как процесс предварительной подготовки для процесса гидролиза с использованием комплексной кислоты в псевдорасплавленном состоянии в качестве катализатора гидролиза. Кроме того, реакция дегидратации моносахарида в процессе гидролиза может быть ингибирована, даже когда используют не бывший в употреблении комплексный кислотный реагент.
ПРИМЕРЫ
Количественное определение D-(+)-глюкозы и D-(+)-ксилозы проводили с использованием высокоэффективной жидкостной хроматографии (HPLC, ВЭЖХ) методом детектирования после введения флуоресцентной метки. Идентификацию и количественное определение комплексной кислоты проводили методом спектрометрии с индуктивно-связанной плазмой (ICP).
ПРИМЕР 1. (Обработка, усиливающая агрегацию, Комплексного Кислотного Катализатора)
Общее количество в 1 кг не использованной ранее гетерополикислоты (фосфорновольфрамовой кислоты) в качестве реагента и 500 мл этанола перемешивали при нагревании, и перемешивание проводили в течение 1 часа при постоянной температуре 60°С. Затем температуру снижали до 45°С, внутренность контейнера с мешалкой эвакуировали (вакуумирование до давления около 20 кПа), быстро испаряли этанол и получали порошкообразную гетерополикислоту, подвергнутую обработке, усиливающей агрегацию.
Общее количество в 1,0 г гетерополикислоты, подвергнутой обработке, усиливающей агрегацию, растворяли в 0,5 мл этанола и раствор перемешивали при комнатной температуре. Затем этанол выпаривали и затем проводили ИК-измерения при следующих условиях. Результаты показаны на Фиг.3.
(Осахаривание Целлюлозы и Отделение) В закрытый реакционный контейнер заблаговременно помещали дистиллированную воду, повышали температуру до предварительно заданной реакционной температуры (70°С), создавали внутри контейнера состояние давления насыщенных водяных паров и обеспечивали возможность осаждения водяных паров на внутренней поверхности контейнера. Затем 1 кг гетерополикислоты, подвергнутой обработке, усиливающей агрегацию, (заблаговременно измеряли количество кристаллизационной воды), и дистиллированную воду (35 г) в количестве, представляющем недостающее количество воды (за исключением водного компонента с давлением насыщенных водяных паров при температуре 70°С) относительно общего суммарного количества, необходимого для доведения кристаллизационной воды в гетерополикислоте до 100%, и количества воды (55,6 г), необходимого для гидролиза всей целлюлозы и получения глюкозы, помещали в контейнер, нагревали и перемешивали. Когда температура внутри контейнера достигала 70°С, перемешивание дополнительно продолжали в течение 5 минут. Затем 0,5 кг целлюлозы загружали в контейнер, перемешивание проводили в течение 2 часов с нагреванием при температуре 70°С. Затем нагревание прекращали, контейнер открывали и гидролитическую реакционную смесь охлаждали до комнатной температуры, в то же время с выведением избыточных водяных паров.
Затем, как показано на Фиг.6, общее количество в 500 мл этанола, который был дважды использован для промывания, добавляли к гидролитической реакционной смеси, размещенной внутри контейнера, проводили перемешивание в течение 30 минут, с последующим фильтрованием, которое давали фильтрат 1 и отфильтрованный материал 1. Фильтрат 1 (раствор гетерополикислоты в этаноле) отделяли. Общее количество в 500 мл этанола, который был однажды использован для промывания, затем добавляли к отфильтрованному материалу 1 и проводили перемешивание в течение 30 минут, с последующим фильтрованием, которое давали фильтрат 2 и отфильтрованный материал 2. К отфильтрованному материалу 2 дополнительно добавляли всего 500 мл нового этанола и перемешивание проводили в течение 30 минут, с последующим фильтрованием, которое давали фильтрат 3 и отфильтрованный материал 3. К полученному отфильтрованному материалу 3 добавляли дистиллированную воду и проводили перемешивание в течение 10 минут. В полученном водном растворе не было обнаружено никакого остатка, но тем не менее раствор профильтровали и получали водный раствор сахарида. Выход моносахаридов (общая сумма глюкозы, ксилозы, арабинозы, маннозы и галактозы) рассчитывали из полученного водного раствора сахарида. Результат составил 83,5%. Как показано на Фиг.7, фильтраты 1-3, выделенные вышеописанным способом (раствор гетерополикислоты в этаноле), подвергали вакуумной дистилляции при температуре от 45 до 50°С, испаряли этанол и извлекали гетерополикислоту. Выход моносахаридов рассчитывали следующим образом.
Выход моносахаридов (%): отношение (весовое отношение) общей суммы реально извлеченных моносахаридов к теоретическому количеству полученных моносахаридов, которые получаются, когда все количество загруженной целлюлозы преобразовано в моносахариды.
ПРИМЕР 2. (Обработка, усиливающая агрегацию, Комплексного Кислотного Катализатора)
Гидролиз целлюлозы и разделение сахарида и гетерополикислоты проводили и раствор гетерополикислоты в этаноле отделяли таким же образом, как в Примере 1, за тем исключением, что применяли гетерополикислоту, которая не была подвергнута обработке, усиливающей агрегацию. Около 100 г не использованного ранее гетерополикислотного реагента добавляли к извлеченному раствору гетерополикислоты в этаноле, растворяли в нем (содержащем 900 г гетерополикислоты и 300 мл этанола) и проводили перемешивание при нагревании. После перемешивания в течение 20 минут при температуре 50°С проводили вакуумирование (давление снижали до около 20 кПа), испаряли этанол и получали порошкообразную гетерополикислоту, подвергнутую обработке, усиливающей агрегацию.
ИК-измерения выполняли таким же образом, как в Примере 1, в отношении гетерополикислоты, подвергнутой обработке, усиливающей агрегацию. Результаты показаны на Фиг.3.
Измерения Рамановского рассеяния на полученной порошкообразной гетерополикислоте, подвергнутой обработке, усиливающей агрегацию, проводили с использованием аргонового (Ar) лазера (длина волны 488 нм). Результаты показаны на Фиг.4.
(Осахаривание Целлюлозы и Отделение). Целлюлозу гидролизовали и сахарид и гетерополикислоту разделяли таким же образом, как в Примере 1, за тем исключением, что в контейнер помещали 1 кг гетерополикислоты, подвергнутой обработке, усиливающей агрегацию, вышеописанным способом (заблаговременно измеряли количество кристаллизационной воды) и дистиллированную воду (35 г) в количестве, представляющем недостающее количество воды (за исключением водного компонента с давлением насыщенных водяных паров при температуре 70°С) относительно общего суммарного количества, необходимого для доведения кристаллизационной воды в гетерополикислоте до 100%, и количества воды (55,6 г), необходимого для гидролиза всей целлюлозы и получения глюкозы. Выход моносахарида составил 86,5%.
ПРИМЕР 3. (Обработка, усиливающая агрегацию, Комплексного Кислотного Катализатора и Осахаривание Целлюлозы и Отделение)
В закрытый реакционный контейнер заблаговременно помещали дистиллированную воду, повышали температуру до предварительно заданной реакционной температуры (70°С), создавали внутри контейнера состояние давления насыщенных водяных паров и обеспечивали возможность осаждения водяных паров на внутренней поверхности контейнера. Затем 1 кг не использованной ранее гетерополикислоты (заблаговременно измеряли количество кристаллизационной воды) и дистиллированную воду (35 г) в количестве, представляющем недостающее количество воды (за исключением водного компонента с давлением насыщенных водяных паров при температуре 70°С) относительно общего суммарного количества, необходимого для доведения кристаллизационной воды в гетерополикислоте до 100%, и количества воды (55,6 г), необходимого для гидролиза всей целлюлозы и получения глюкозы, помещали в контейнер, нагревали и перемешивали. Когда температура внутри контейнера достигала 70°С, перемешивание дополнительно продолжали в течение 5 минут. Затем 0,05 кг целлюлозы [10% по весу от 0,5 кг количества для гидролитической обработки (количество, которое может быть загружено в одном цикле)] загрузили в контейнер и перемешивание проводили в течение 10 минут при температуре 70°С. Затем загружали остальную целлюлозу, 0,45 кг (90% по весу от количества для гидролитической обработки) и перемешивание продолжали дополнительно в течение 80 минут при температуре 70°С. Затем нагревание прекращали, контейнер открывали и гидролитическую реакционную смесь охлаждали до комнатной температуры, в то же время с выведением избыточных водяных паров. Затем сахарид и гетерополикислоту извлекали из гидролитической реакционной смеси таким же образом, как в Примере 1. Выход моносахарида был 82,1%.
ПРИМЕР 4. (Обработка, усиливающая агрегацию, Комплексного Кислотного Катализатора и Осахаривание Целлюлозы и Отделение)
В закрытый реакционный контейнер заблаговременно помещали дистиллированную воду, повышали температуру до предварительно заданной реакционной температуры (70°С), создавали внутри контейнера состояние давления насыщенных водяных паров и обеспечивали возможность осаждения водяных паров на внутренней поверхности контейнера. Затем 1 кг не использованной ранее гетерополикислоты (заблаговременно измеряли количество кристаллизационной воды) и дистиллированную воду (35 г) в количестве, представляющем недостающее количество воды (за исключением водного компонента с давлением насыщенных водяных паров при температуре 70°С) относительно общего суммарного количества, необходимого для доведения кристаллизационной воды в гетерополикислоте до 100%, и количества воды (55,6 г), необходимого для гидролиза всей целлюлозы и получения глюкозы, и дополнительно 50 г дистиллированной воды помещали в контейнер, нагревали и перемешивали. Когда температура внутри контейнера достигала 70°С, перемешивание дополнительно продолжали в течение 20 минут. Затем 0,5 кг целлюлозы загружали в контейнер и перемешивание проводили в течение 2 часов при температуре 70°С. Затем нагревание прекращали и гидролитическую реакционную смесь охлаждали до комнатной температуры. Затем сахарид и гетерополикислоту извлекали из гидролитической реакционной смеси таким же образом, как в Примере 1. Выход моносахарида был 75,1%.
СРАВНИТЕЛЬНЫЙ ПРИМЕР 1
Общее количество в 1,0 г не использованного ранее нового гетерополикислотного реагента растворяли в 0,5 мл этанола при комнатной температуре (от 20 до 25°С). Затем этанол испаряли, выполняли высушивание и проводили ИК-измерения таким же образом, как в Примере 1. Результаты показаны на Фиг.3. Измерения Рамановского рассеяния проводили таким же образом, как в Примере 2. Результаты показаны на Фиг.4.
Между тем в закрытый реакционный контейнер заблаговременно помещали дистиллированную воду, повышали температуру до предварительно заданной реакционной температуры (70°С), создавали внутри контейнера состояние давления насыщенных водяных паров и обеспечивали возможность осаждения водяных паров на внутренней поверхности контейнера. Затем 1 кг не использованной ранее новой гетерополикислоты (заблаговременно измеряли количество кристаллизационной воды) и дистиллированную воду (35 г) в количестве, представляющем недостающее количество воды (за исключением водного компонента с давлением насыщенных водяных паров при температуре 70°С) относительно общего суммарного количества, необходимого для доведения кристаллизационной воды в гетерополикислоте до 100%, и количества воды (55,6 г), необходимого для гидролиза всей целлюлозы и получения глюкозы, помещали в контейнер, нагревали и перемешивали. Когда температура внутри контейнера достигала 70°С, перемешивание дополнительно продолжали в течение 5 минут. Затем загружали в контейнер 0,5 кг целлюлозы и проводили перемешивание в течение 2 часов с нагреванием при температуре 70°С. Затем нагревание прекращали, контейнер открывали, и гидролитическую реакционную смесь охлаждали до комнатной температуры, в то же время с выведением избыточных водяных паров. Затем моносахариды и гетерополикислоту извлекали из гидролитической реакционной смеси таким же образом, как в Примере 1. Выход моносахаридов составил 60,0%.
РЕЗУЛЬТАТЫ. Выход моносахарида, полученного в Примерах 1-4 и в Сравнительном Примере 1, показан в Таблице 1.
Как показано на Фиг.3, когда ИК-спектр не использованного ранее нового гетерополикислотного реагента, применяемого в Сравнительном Примере 1, сравнивают с ИК-спектром гетерополикислоты, подвергнутой обработке, усиливающей агрегацию, которую использовали в Примере 1 и Примере 2, в гетерополикислоте, подвергнутой обработке, усиливающей агрегацию, которую использовали в Примере 1 и Примере 2, интенсивность полосы поглощения в области 3200 см-1, которая обусловлена молекулой Н2О, связанной в кристалле, снижается, интенсивность полосы поглощения в области 3500 см-1, которая обусловлена ОН-группой, координированной с сильной кислотой, возрастает и подтверждается усиление агрегации. Кроме того, как показано на Фиг.4, когда сравнивают Рамановские спектры гетерополикислоты, подвергнутой обработке, усиливающей агрегацию, которую использовали в Примере 2, и не использованного ранее нового гетерополикислотного реагента, применяемого в Сравнительном Примере 1, в области 985 см-1 наблюдается узкая интенсивная линия, но в комплексном кислотном катализаторе из Примера 2 происходит высокочастотный сдвиг в область 1558 см-1 и интенсивность линии значительно снижается, тем самым подтверждая, что агрегация усиливается.
Как показано в Таблице 1, в Примерах 1-4 выход моносахарида значительно повысился относительно выхода в Сравнительном Примере 1. Очевидно, что это обусловлено тем, что в Примерах 1-4 гетерополикислота была агрегирована в кристаллическом состоянии в результате обработки, усиливающей агрегацию, гетерополикислоты, благодаря чему кислотность гетерополикислоты была снижена и последующая реакция (реакция дегидратации) моносахарида в процессе гидролиза целлюлозы была ингибирована. В частности, выход моносахарида в Примерах 1-3 превышает 80%, и был достигнут эффект повышения выхода сахарида.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ГИДРОЛИЗА РАСТИТЕЛЬНОГО ВОЛОКНИСТОГО МАТЕРИАЛА ДЛЯ ПОЛУЧЕНИЯ И ВЫДЕЛЕНИЯ САХАРИДА, ВКЛЮЧАЮЩЕГО ГЛЮКОЗУ | 2009 |
|
RU2453607C1 |
| СПОСОБ ГИДРОЛИЗА РАСТИТЕЛЬНОГО ВОЛОКНИСТОГО МАТЕРИАЛА ДЛЯ ПОЛУЧЕНИЯ И ВЫДЕЛЕНИЯ САХАРИДА, ВКЛЮЧАЮЩЕГО ГЛЮКОЗУ | 2009 |
|
RU2461633C2 |
| СПОСОБ ОСАХАРИВАНИЯ И СЕПАРИРОВАНИЯ ДЛЯ РАСТИТЕЛЬНЫХ ВОЛОКНИСТЫХ МАТЕРИАЛОВ | 2008 |
|
RU2451087C2 |
| СПОСОБ ПРЕДВАРИТЕЛЬНОЙ ОБРАБОТКИ ДЛЯ ОСАХАРИВАНИЯ РАСТИТЕЛЬНОГО ВОЛОКНИСТОГО МАТЕРИАЛА И СПОСОБ ОСАХАРИВАНИЯ | 2010 |
|
RU2486256C2 |
| СПОСОБ ПРЕОБРАЗОВАНИЯ РАСТИТЕЛЬНОГО ВОЛОКНИСТОГО МАТЕРИАЛА | 2008 |
|
RU2440419C2 |
| УСТАНОВКА ДЛЯ ПРЕДВАРИТЕЛЬНОЙ ОБРАБОТКИ ЦЕЛЛЮЛОЗОСОДЕРЖАЩЕГО РАСТИТЕЛЬНОГО СЫРЬЯ | 2013 |
|
RU2577066C2 |
| СПОСОБ ПОЛУЧЕНИЯ БИОНЕФТИ | 2009 |
|
RU2501840C2 |
| СПОСОБ ПОЛУЧЕНИЯ 5-ГИДРОКСИМЕТИЛФУРФУРОЛА | 2015 |
|
RU2583953C1 |
| СПОСОБ ПОЛУЧЕНИЯ МИКРОКРИСТАЛЛИЧЕСКОЙ ЦЕЛЛЮЛОЗЫ | 2013 |
|
RU2528261C1 |
| ПОВЫШЕННЫЙ ВЫХОД ПРОДУКТА РАСТВОРИМЫХ С5-САХАРОВ | 2012 |
|
RU2609000C2 |




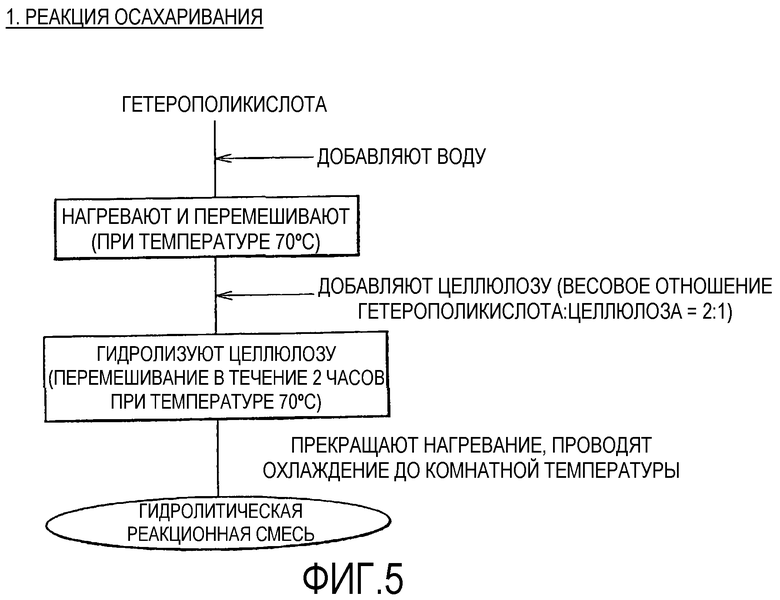

Изобретение относится к пищевой промышленности. Способ включает процесс гидролиза с использованием комплексного кислотного катализатора на основе гомополикислоты или гетерополикислоты в псевдорасплавленном состоянии для гидролиза целлюлозы, содержащейся в растительном волокнистом материале, и получения глюкозы. При этом катализатор на основе гомополикислоты или гетерополикислоты подвергают обработке, усиливающей агрегацию, в результате чего усиливается агрегирование катализатора на основе гомополикислоты или гетерополикислоты в кристаллическом состоянии. Предложенный способ позволяет значительно повысить выход сахарида. 14 з.п. ф-лы, 7 ил., 1 табл., 5 пр.
1. Способ гидролиза растительного волокнистого материала для получения и выделения сахарида, включающего глюкозу, включающий:
процесс гидролиза с использованием комплексного кислотного катализатора на основе гомополикислоты или гетерополикислоты, в псевдорасплавленном состоянии, для гидролиза целлюлозы, содержащейся в растительном волокнистом материале, и получения глюкозы,
в котором катализатор на основе гомополикислоты или гетерополикислоты подвергают обработке, усиливающей агрегацию, в результате чего усиливается агрегирование катализатора на основе гомополикислоты или гетерополикислоты в кристаллическом состоянии.
2. Способ по п.1, в котором температуру гидролиза регулируют путем регулирования количества кристаллизационной воды, содержащейся в катализаторе на основе гомополикислоты или гетерополикислоты в процессе гидролиза.
3. Способ по п.1, в котором катализатор на основе гомополикислоты или гетерополикислоты подвергают обработке, усиливающей агрегацию, в тот момент времени, когда в процесс гидролиза загружают количество растительного волокнистого материала, которое может быть загружено в одном цикле для всей реакционной системы.
4. Способ по п.1, в котором
когда сравнивают ИК-спектр катализатора на основе гомополикислоты или гетерополикислоты перед обработкой, усиливающей агрегацию, и ИК-спектр катализатора на основе гомополикислоты или гетерополикислоты после обработки, усиливающей агрегацию, то интенсивность полосы в области 3200 см-1, которая обусловлена молекулой H2O, которая сэндвичеобразно размещена между кристаллами катализатора на основе гомополикислоты или гетерополикислоты, после обработки, усиливающей агрегацию, является меньшей, чем интенсивность полосы катализатора на основе гомополикислоты или гетерополикислоты до обработки, усиливающей агрегацию, и интенсивность полосы в области 3500 см-1, которая обусловлена ОН-группой, связанной с сильной кислотой катализатора на основе гомополикислоты или гетерополикислоты, после обработки, усиливающей агрегацию, является большей, чем интенсивность полосы катализатора на основе гомополикислоты или гетерополикислоты до обработки, усиливающей агрегацию.
5. Способ по п.1, в котором
обработка, усиливающая агрегацию, включает процесс нагревания и перемешивания катализатора на основе гомополикислоты или гетерополикислоты и органического растворителя, который может растворять катализатор на основе гомополикислоты или гетерополикислоты, и процесс удаления органического растворителя после процесса нагревания и перемешивания.
6. Способ по п.5, в котором
при обработке, усиливающей агрегацию, катализатор на основе гомополикислоты или гетерополикислоты и органический растворитель нагревают и перемешивают при температуре, равной или меньшей 65°С.
7. Способ по п.5, дополнительно включающий:
процесс отделения сахарида добавлением органического растворителя, в котором может быть растворен катализатор на основе гомополикислоты или гетерополикислоты, к реакционной смеси после процесса гидролиза и твердофазно-жидкостное разделение полученной смеси на жидкостную фракцию, включающую катализатор на основе гомополикислоты или гетерополикислоты и органический растворитель, и твердую фракцию, включающую сахарид.
8. Способ по п.7, в котором
обработка, усиливающая агрегацию, включает процесс добавления катализатора на основе гомополикислоты или гетерополикислоты в кристаллическом состоянии в количестве, которое компенсирует потерю катализатора на основе гомополикислоты или гетерополикислоты в процессе отделения сахарида, к раствору катализатора на основе гомополикислоты или гетерополикислоты в органическом растворителе, который получен в процессе выделения сахарида и образован растворением катализатора на основе гомополикислоты или гетерополикислоты в органическом растворителе, и затем выполнение нагревания и перемешивания.
9. Способ по п.1, в котором
обработка, усиливающая агрегацию, включает нагревание и перемешивание части от количества растительного волокнистого материала, которое может быть загружено в одном цикле, вместе с катализатором на основе гомополикислоты или гетерополикислоты в псевдорасплавленном состоянии и выполнение гидролиза растительного волокнистого материала в процессе гидролиза.
10. Способ по п.9, в котором
при обработке, усиливающей агрегацию, количество растительного волокнистого материала, который нагревают и перемешивают вместе с катализатором на основе гомополикислоты или гетерополикислоты в псевдорасплавленном состоянии, является равным или меньшим, чем 10% по весу от количества растительного волокнистого материала, которое может быть загружено в одном цикле.
11. Способ по п.9, в котором
при обработке, усиливающей агрегацию, количество растительного волокнистого материала, который нагревают и перемешивают вместе с катализатором на основе гомополикислоты или гетерополикислоты в псевдорасплавленном состоянии, представляет собой количество, которое не изменяет вязкости катализатора на основе гомополикислоты или гетерополикислоты в псевдорасплавленном состоянии.
12. Способ по п.1, в котором
обработка, усиливающая агрегацию, включает нагревание и перемешивание катализатора на основе гомополикислоты или гетерополикислоты в псевдорасплавленном состоянии.
13. Способ по п.12, в котором
при обработке, усиливающей агрегацию, катализатор на основе гомополикислоты или гетерополикислоты нагревают и перемешивают при температуре, которая является более высокой по меньшей мере на величину от 5 до 10°С, чем температура, при которой катализатор на основе гомополикислоты или гетерополикислоты начинает переходить в псевдорасплавленное состояние.
14. Способ по п.12, в котором
при обработке, усиливающей агрегацию, катализатор на основе гомополикислоты или гетерополикислоты нагревают и перемешивают с водой в таком количестве, чтобы доля кристаллизационной воды в катализаторе на основе гомополикислоты или гетерополикислоты стала равной или большей 100%.
15. Способ по любому из пп.1-14, в котором катализатор на основе гомополикислоты или гетерополикислоты представляет собой гетерополикислоту, подвергнутую обработке, усиливающей агрегацию.
| JP 11343301 А, 14.12.1999 | |||
| ЕР 1860201 A1, 28.11.2007 | |||
| ГЕТЕРОГЕННЫЙ КАТАЛИЗАТОР ОКИСЛЕНИЯ НЕОРГАНИЧЕСКИХ И/ИЛИ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ НА ПОЛИМЕРНОМ НОСИТЕЛЕ | 2003 |
|
RU2255805C2 |

