Настоящее изобретение относится к новой фармацевтической композиции для пероральной доставки фармацевтических соединений, прежде всего полиаминокислот, включая пептиды, или в другом варианте пептидомиметики.
Прежде всего, настоящее изобретение относится к новой пероральной фармацевтической композиции, включающей полиаминокислоты, предназначенные для лечения нарушения, вызванного аномальной резорбцией костной ткани и/или для лечения артрита и других заболеваний.
Предпосылки создания настоящего изобретения
Гормоны
Полиаминокислоты, которые используют или предлагают для использования в фармацевтических или ветеринарных целях, включают, без ограничения перечисленным, соединения из синтетического, природного или рекомбинантного источников: полипептидные гормоны, такие как кальцитонины, например кальцитонин лосося, гормон роста, включая гормон роста человека (hGH), рекомбинантный гормон роста человека (rhGH), бычий гормон роста и свиной гормон роста, факторы, высвобождающие гормон роста и гипофизарный гормон.
Паратироидный гормон (РТН) является полноцепным пептидом, содержащим 84 аминокислотных остатка, и включает, например, гормон человека, hPTH (1-84), или любой полипептид, белок, фрагмент белка или модифицированный фрагмент, т.е. РТН-родственные пептиды и аналоги РТН, способные проявлять активность hPTH (1-84) в контролируемом метаболизме кальция и фосфатов для формирования костной ткани в организме человека. Фрагменты РТН обычно включают по крайней мере первые 28 N-концевых остатков и включают, например, РТН (1-28), РТН (1-31), РТН (1-34), РТН (1-37), РТН (1-38) и РТН (1-41) и их аналоги, например PTS893. РТН является индивидуальным соединением или комбинацией двух или более РТН.
Предпочтительным фрагментом РТН является РТН (1-34).
Указанные паратироидные гормоны выпускаются в промышленности или их получают рекомбинантным способом, пептидным синтезом или экстракцией из биологических жидкостей человека по известным методикам.
РТН обычно вводят в эффективном количестве для стимуляции образования новой костной ткани, т.е. в терапевтически эффективном количестве. Это количество зависит от возраста, массы, пола и состояния субъекта, нуждающегося в таком лечении, природы и тяжести заболевания и т.п. Однако однократная доза может включать меньшую дозу по сравнению с назначенной дозой, если вводить несколько лекарственных композиций, т.е. можно вводить общее эффективное количество, которое составляет сумму однократных доз. Однократная доза РТН может иногда включать большую дозу по сравнению с эффективным количеством, если композиция является средством с замедленным высвобождением фармакологически активного агента. Общее количество используемого РТН определяют известными способами. Однако в общем случае удовлетворительные результаты получают при систематическом введении суточной дозы от приблизительно 0,001 мкг/кг до приблизительно 10 мг/кг массы тела животного, предпочтительно от 1 мкг/кг до приблизительно 6 мкг/кг массы тела.
Предпочтительным фармакологически активным агентом является фармакологически активный пептид, прежде всего кальцитонин. Известный класс фармакологически активных агентов, к которому относятся кальцитонины, используют в различных областях фармацевтики и обычно применяют для лечения, например, болезни Педжета, гиперкальцемии и постклимактерического остеопороза. Кальцитонин, например кальцитонин угря (Asu-17) или человека, является длинноцепочечным полипептидным гормоном, секретируемым парафолликулярными клетками щитовидной железы млекопитающих, ультимобранхиальными железами рыб и птиц. Различные кальцитонины, включая кальцитонин лосося, свиньи и угря, являются коммерческими препаратами и обычно применяются для лечения, например, заболевания Педжета, злокачественной гиперкальцемии и остеопороза. Кальцитонин означает любой кальцитонин, включая природный, синтетический или рекомбинантный, а также производные кальцитонина, такие как кальцитонин угря 1,7-Asu. Композиции включают индивидуальный кальцитонин или любые комбинации двух и более кальцитонинов. Предпочтительным является синтетический кальцитонин лосося.
Кальцитонин является коммерческим препаратом или его получают с использованием известных методик.
Количество фармакологически активного агента обычно означает эффективное количество, обеспечивающее достижение требуемой цели, например терапевтически эффективное количество. Однако однократная доза может включать меньшую дозу по сравнению с назначенной дозой, если вводить несколько лекарственных композиций, т.е. можно вводить общее эффективное количество, которое составляет общую дозу. Однократная доза активного агента может иногда включать большую дозу по сравнению с эффективным количеством, если композиция является средством с замедленным высвобождением фармакологически активного агента. Общее количество используемого активного агента определяют известными способами. Однако поскольку композиции обеспечивают более эффективную доставку активного агента по сравнению с известными композициями, субъекту можно вводить меньшее количество активного агента по сравнению и известными стандартными лекарственными формами или системами доставки, при этом достигается аналогичный уровень лекарственного агента в крови и/или терапевтический эффект.
Пригодная доза кальцитонина зависит, например, от количества вводимого кальцитонина и тяжести состояния, подлежащего лечению. Однако в общем случае удовлетворительные результаты получают при систематическом интраназальном введении или введении в виде инъекций в суточной дозе от приблизительно 0,5 мкг/кг до приблизительно 10 мкг/кг массы тела животного в сутки, предпочтительно от 1 мкг/кг до приблизительно 6 мкг/кг массы тела.
Гормон роста человека (hGH) (или соматотропный гормон или соматотропин) представляет собой полипептидный гормон, секретируемый передней долей гипофиза, который обеспечивает рост тела, прежде всего за счет стимуляции высвобождения соматомедина, и влияет на метаболизм белков, углеводов и липидов.
Термин hGH включает различные природные или синтетические соединения, регулирующие рост животных или растений, такие как гипофизарный гормон роста у позвоночных и ауксин у растений.
Заболевания костной ткани
Известно множество заболеваний костной ткани. Первый класс заболеваний относится к нарушениям, вызванным резорбцией костной ткани. Примеры таких нарушений включают остеопороз, остеолиз и заболевание Педжета.
Ко второму классу нарушений относятся артриты. Примерами таких нарушений являются остеоартриты.
Новые составы
В последнее время предпринималось множество попыток улучшить абсорбцию полиаминокислот, таких как пептиды и белки, например гормоны. В основном предполагается, что пептиды и белки необходимо защищать от воздействия среды желудочно-кишечного тракта, в которой содержится большое количество пептидаз и происходит деградация пептидов и белков. Установлено, что нанесение энтеросолюбильного покрытия и добавление ингибиторов пептидаз в состав фармацевтических композиций эффективно повышает абсорбцию полиаминокислот, например белков и пептидов при пероральном введении.
Однако такие подходы сами по себе не обеспечивают достаточной защиты для достижения удовлетворительного уровня концентрации пептидов и белков в плазме, таким образом существует необходимость в разработке альтернативных способов доставки лекарственных средств, включающих пептиды и белки, и одновременно способов их защиты от химической и ферментативной деградации для обеспечения терапевтического эффекта.
Разработка таких способов прежде всего необходима в случае кальцитонинов, для которых пероральный способ является предпочтительным способом введения, поскольку этот способ является наиболее простым, удобным и практически безболезненным, что повышает согласие пациентов с курсом лечения по сравнению с другими курсами лечения.
Краткое изложение сущности изобретения
Настоящее изобретение относится к фармацевтической композиции для эффективной доставки лекарственных средств в фармацевтически эффективном количестве, прежде всего полиаминокислот, таких как пептиды, пептидомиметики и белки, например гормоны, при пероральном введении субъекту для достижения требуемого терапевтического эффекта.
Настоящее изобретение также относится к пероральной фармацевтической композиции, включающей в качестве активного ингредиента полиаминокислоту, например пептид или белок, при этом время распадаемости фармацевтической композиции и/или скорость растворения являются достаточно высокими, чтобы активный ингредиент оказывал терапевтический эффект.
В одном объекте настоящего изобретения предлагаются фармацевтические композиции, включающие в качестве активного ингредиента пептид или белок, при этом время распадаемости фармацевтической композиции, например таблетки, составляет до 10 мин.
Настоящее изобретение также относится к фармацевтической композиции, например таблетке или капсуле, время растворения которых составляет до 30 мин, например до 20 мин, обычно до 10 мин.
Прежде всего, настоящее изобретение относится к фармацевтическим композициям, включающим в качестве активного ингредиента кальцитонин в смеси с агентом для доставки 5-CNAC, и полученная фармацевтическая композиция обеспечивает улучшенную пероральную биодоступность, например удовлетворительную или оптимальную пероральную биодоступность кальцитонина в качестве активного ингредиента.
Термин «биодоступность», использованный в настоящем описании, означает часть дозы, которая достигает кровотока после введения указанной лекарственной формы. Более подробно указанный термин означает отношение количества лекарственного средства, «абсорбированного» из исследуемого состава, к количеству, «абсорбированному» после введения стандартного состава. Часто термин «стандартный состав», используемый для оценки биодоступности, означает водный раствор лекарственного средства, который вводят внутривенно.
Абсорбированное количество лекарственного средства рассматривают как меру способности состава доставлять лекарственное средство к участку воздействия, которая зависит от распадаемости и растворения лекарственной формы и скорости биотрансформации по отношению к скорости абсорбции.
Лекарственные формы, включающие одинаковое количество активного лекарственного средства, могут значительно различаться по биодоступности лекарственного средства и, следовательно, по способности проявлять ожидаемые фармакодинамические и терапевтические свойства лекарственного средства.
В настоящем изобретении неожиданно было установлено, что более быстрая распадаемость фармацевтических композиций по настоящему изобретению у субъекта, например, в желудке обеспечивает улучшенные абсорбционные свойства активных пептидов и белков, где пептиды и белки в значительной степени расщепляются в присутствии пепсина или других ферментов.
Настоящее изобретение также относится к фармацевтической композиции для доставки пептида или белка при пероральном введении без необходимости нанесения энтеросолюбильного покрытия или включения ингибитора пептидаз. Таким образом, в одном варианте композиции по настоящему изобретению не содержат энтеросолюбильное покрытие или ингибиторы пептидаз, или не используют оба варианта.
Включающие кальцитонин композиции по настоящему изобретению можно использовать для лечения нарушений, связанных с аномальной резорбцией костной ткани или артрита, как описано в данном контексте.
В одном варианте настоящего изобретения предлагается пероральная фармацевтическая композиция в твердой фазе, включающая:
1) полиаминокислоту,
2) агент для доставки и необязательно,
3) разбавитель,
где время распадаемости композиции составляет не более 10 мин, а растворение >80% происходит в течение 20 мин, прежде всего время распадаемости составляет не более 6 мин, а растворение >90% происходит в течение 20 мин.
Прежде всего, композиция по настоящему изобретению характеризуется временем распадаемости не более 2 мин.
В еще одном варианте композиция по настоящему изобретению дополнительно включает дезинтегрирующий агент, прежде всего выбранный из группы, включающей любой из супердезинтегрирующих агентов, таких как кросповидон или повидон, и/или другой агент, снижающий время распадаемости, например, при получении шипучих таблеток или других лекарственных форм.
В другом варианте настоящего изобретения фармацевтическая композиция характеризуется растворением в желудочной среде >80% в течение не более 20 мин.
Настоящее изобретение также относится к фармацевтической композиции в форме таблетки, прежде всего прессованной таблетки, твердость которой составляет от 3 кПа до 20 кПа, прежде всего от 5 кПа до 15 кПа, более предпочтительно от 5 кПа до 7 кПа.
В одном варианте композиция по настоящему изобретению включает полипептидный гормон, прежде всего кальцитонин, более предпочтительно кальцитонин лосося.
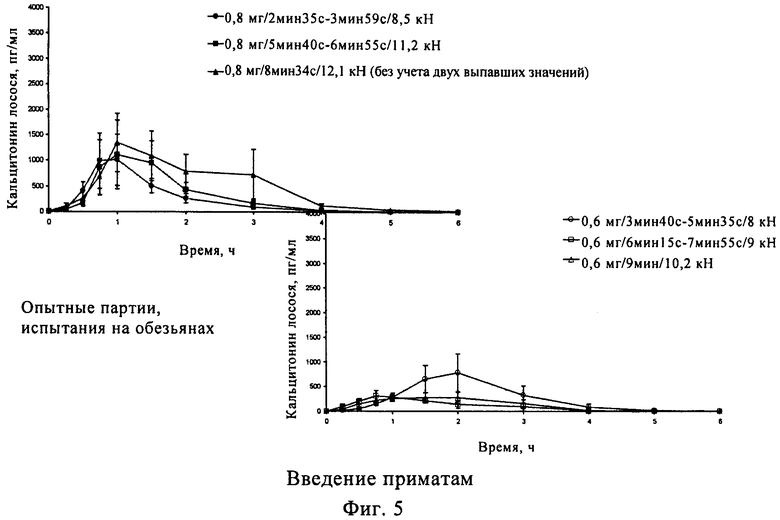
Прежде всего кальцитонин содержится в терапевтически эффективном количестве в свободной форме или в форме соли, что обеспечивает достижение максимальной концентрации в плазме крови (Сmах) не менее 400 пг/мл, прежде всего не менее 800 пг/мл, более предпочтительно не менее 1000 пг/мл, и/или снижение уровня кальция в плазме до величины >20% в течение 6 ч, как показано на модели приматов, прежде всего обезьян.
В еще одном варианте настоящего изобретения композиция включает терапевтически эффективное количество кальцитонина в свободной форме или в форме соли в количестве от 0,15 мг до 2,5 мг, прежде всего от 0,15 мг до 0,4 мг.
Композиция по настоящему изобретению может также включать агент для доставки 5-CNAC и/или кросповидон и/или повидон в качестве дезинтегрирующего агента. Композиция также может включать один или более загустителей, стабилизаторов и сухих связующих агентов.
В одном варианте настоящего изобретения предлагается фармацевтическая композиция в форме таблетки массой 500 мг.
В отдельном варианте настоящего изобретения фармацевтическая композиция включает:
где общее количество составляет 100%.
Настоящее изобретение также относится к фармацевтической композиции, включающей:
а) композицию по настоящему изобретению, описанную в данном контексте и
б) совместный агент, которым является ингибитор резорбции костной ткани или ингибитор катепсина К.
В еще одном варианте изобретение относится к способу получения пероральной фармацевтической композиции, который включает следующие стадии:
а) смешивание полиаминокислоты, носителя и дезинтегрирующего агента, при этом получают первую смесь,
б) необязательно добавление сухого связующего в первую смесь, при этом получают вторую смесь,
в) необязательно добавление стабилизатора во вторую смесь, при этом получают третью смесь,
г) прессование третьей смеси в таблетки, твердость которых составляет от 5 кПа до 20 кПа.
В еще одном варианте изобретение относится к применению фармацевтической композиции по настоящему изобретению, описанной в данном контексте, для получения лекарственного средства, предназначенного для лечения заболеваний, вызванных аномальной резорбцией костной ткани, таких как, например, остеопороз, артрит или остеоартрит.
Настоящее изобретение также относится к способу определения абсорбционных свойств композиции по настоящему изобретению, описанной в данном контексте, который заключается в том, что:
а) определяют время распадаемости,
б) коррелируют время распадаемости и время растворения.
В другом варианте настоящее изобретение относится к способу предварительного определения максимума концентрации активного ингредиента (Сmах) в плазме пациента, которого лечат при пероральном введении фармацевтической композиции, включающей указанный активный ингредиент, предпочтительно кальцитонин, более предпочтительно кальцитонин лосося, и агент для доставки. Указанный способ включает такое сочетание времени распадаемости фармацевтической композиции и/или времени растворения активного ингредиента, которое обеспечивает благоприятное микроокружение в желудочно-кишечном тракте для растворения активного ингредиента в кишечнике, чтобы оптимизировать абсорбцию активного ингредиента и достижение терапевтически эффективной максимальной концентрации активного ингредиента в плазме крови, прежде всего максимальная концентрация в плазме составляет не менее 400 пг/мл.
Прежде всего, благоприятное микроокружение в желудочно-кишечном тракте для растворения активного ингредиента в кишечнике обеспечивается при добавлении в композицию 5-CNAC.
В одном варианте предлагается способ предварительного определения максимальной концентрации активного ингредиента в плазме (Сmах) у пациента, которого лечат при пероральном введении фармацевтической композиции, и пероральную фармацевтическую композицию получают в форме таблетки, а время распадаемости регулируют при изменении твердости таблетки, прежде всего получают таблетки, твердость которых составляет от 3 кПа до 20 кПа и/или время распадаемости которых составляет менее 10 мин, прежде всего менее 1 мин.
В еще одном варианте настоящее изобретение относится к применению для получения пероральной фармацевтической композиции, характеризующейся временем распадаемости и/или растворения не более 10 мин, и включающей
1) полиаминокислоту,
2) агент для доставки,
3) дезинтегрирующий агент.
Прежде всего, для обеспечения благоприятного микроокружения для растворения кальцитонина лосося в желудочно-кишечном тракте используют 5-CNAC.
Подробное описание вариантов осуществления изобретения
Настоящее изобретение относится к пероральной фармацевтической композиции, включающей полиаминокислоту в качестве активного ингредиента, например пептид или белок, а время распадаемости фармацевтической композиции является достаточным, чтобы обеспечивать адекватное терапевтическое действие активного ингредиента.
Настоящее изобретение также относится к пероральной фармацевтической композиции, включающей в качестве активного ингредиента полиаминокислоту, например пептид или белок, а скорость растворения фармацевтической композиции является достаточной, чтобы обеспечивать адекватное терапевтическое действие активного ингредиента.
Настоящее изобретение относится к пероральной фармацевтической композиции, включающей в качестве активного ингредиента полиаминокислоту, например пептид или белок, в которой величины и времена распадаемости и скорости растворения фармацевтической композиции являются достаточными, чтобы обеспечивать адекватное терапевтическое действие активного ингредиента.
Учитывая, что скорость распадаемости является высокой, т.е. составляет миллисекунды, предполагается, что такая быстрая раепадаемость не компенсируется растворением. Однако неожиданно было установлено, что достаточно высокий терапевтический уровень активного ингредиента достигается за относительно короткий промежуток времени, который компенсирует биохимическое разложение (например, в желудочно-кишечном тракте) активного ингредиента.
Следует отметить, что в результате увеличения концентрации терапевтически активного ингредиента в плазме исключается необходимость добавлять в композицию значительное количество активного ингредиента по сравнению с композициями, не обладающими свойствами, описанными в данном контексте. Такие свойства обеспечивают не только преимущество за счет снижения стоимости полученного лекарственного средства, но и снижают риск образования нежелательных или даже токсичных метаболитов активного ингредиента в организме у субъекта.
Кроме того, композиции по настоящему изобретению можно использовать в способе контроля терапевтического уровня активного ингредиента, например концентрации активного ингредиента в плазме. Другими словами, если существует линейная или практически линейная зависимость между распадаемостью, растворением и/или концентрацией активного ингредиента в плазме, то можно предварительно определить требуемую концентрацию в плазме в любое данное время при выборе конкретного состава с определенным временем распадаемости и/или растворения.
С этой целью настоящее изобретение также включает библиотеку композиций с различным временем распадаемости и/или растворения, как описано в данном контексте. Одна группа композиций включает таблетки, характеризующиеся различной твердостью, например от 3 кПа до 20 кПа, предпочтительно от 5 кПа до 20 кПа, более предпочтительно от 5 кПа до 15 кПа, наиболее предпочтительно от 5 кПа до 7 кПа. В подгруппе этой группы каждая таблетка с определенной твердостью может также включать, например, различное количество активного ингредиента, носителя, разбавителя, замасливателя, глиданта или дезинтегрирующего агента.
Настоящее изобретение также включает библиотеку композиций, в которых отсутствие замасливателя может приводить к более быстрому началу распадаемости и растворения.
В одном объекте настоящего изобретения предлагается пероральная фармацевтическая композиция, для которой время растворения или время распадаемости составляет до 10 мин, или оба параметра составляют до 10 мин.
Настоящее изобретение относится к композициям, степень растворения которых составляет от 20% до 100% по данным анализа USP II с использованием лопастной мешалки при растворении в 0,1 н. НСl и 0,01% твин-80 в течение определенного периода времени.
Прежде всего композиции по настоящему изобретению характеризуются степенью растворения от 20% до 100%, например 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90% и 100% в течение периода времени от 0 до 60 мин, например, 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 25, 30, 35, 40, 45, 50, 55 и 60 мин.
В предпочтительном объекте настоящего изобретения время растворения и степень растворения, как описано выше, соответствуют композициям, время распадаемости которых составляет менее 10 мин.
В одном варианте воплощения настоящего изобретения композиция характеризуется временем распадаемости не более 10 мин и степенью растворения >80% в течение не более 20 мин, прежде всего временем распадаемости не более 6 мин и степенью растворения >90% в течение не более 20 мин, прежде всего в желудочной среде.
В еще одном варианте композиция характеризуется временем распадаемости не более 10 мин и степенью растворения >80% в течение 20 мин, прежде всего временем распадаемости не более 6 мин и степенью растворения >90% в течение 20 мин, прежде всего в желудочной среде.
Специалисту в данной области представляется очевидным, что на время распадаемости или растворения твердой фазы перорального состава влияют различные параметры, включающие:
- лекарственную форму (капсулу или таблетку),
- конкретный активный агент,
- конкретные дополнительные ингредиенты, например агент для доставки, дезинтегрирующий агент, глидант, замасливатель, разбавитель,
- количество (соотношение) ингредиентов,
- размеры частиц,
- твердость таблетки.
Таким образом, невозможно установить универсальный набор параметров, определяющих все композиции, характеризующиеся определенным временем распадаемости и/или растворения, исключив все остальные композиции. Тем не менее, специалист в данной области может получить композиции, характеризующиеся указанными временем растворения и распадаемости. Чтобы исключить сомнения, в настоящее описание включена методика измерения времени распадаемости и растворения, а также способы получения композиций с определенным временем распадаемости и растворения.
Время растворения соединения может напрямую влиять на концентрацию активного ингредиента в плазме в любое заданное время.
Настоящее изобретение включает фармацевтическую композицию в твердой фазе, включающую:
полиаминокислоту,
агент для доставки и
при необходимости разбавитель,
и композиция характеризуется временем распадаемости не более 10 мин.
Прежде всего настоящее изобретение включает:
фармацевтическую композицию для пероральной доставки полиаминокислот, включающую:
1) полиаминокислоту,
2) агент для доставки,
3) разбавитель,
и композиция характеризуется временем распадаемости не более 10 мин. Настоящее изобретение также относится к
твердой фармацевтической композиции для пероральной доставки полиаминокислот, включающей:
1) полиаминокислоту,
2) агент для доставки
3) дезинтегрирующий агент
4) разбавитель
и композиция характеризуется временем распадаемости не более 10 мин.
Твердую композицию можно получать в форме таблетки. Таблетки можно получать прессованием, как описано в данном контексте.
Полиаминокислотой является любое лекарственное средство на основе полиаминокилоты, например, включающее белок или его фрагмент. Полиаминокислота означает любую полиаминокислоту, описанную выше в разделе «Предпосылки создания настоящего изобретения». Отдельный класс фармацевтических композиций включает в качестве полиаминокислоты гормон, например полипептидный гормон, такой как кальцитонин, например кальцитонин лосося, гормон роста, включая гормоны роста человека (hGH), рекомбинантные гормоны роста человека (rhGH), свиные и бычьи гормоны роста, факторы, высвобождающие гормоны роста, и гипофизарные гормоны.
Предпочтительным активным ингредиентом является полиаминокислота.
Вопреки общепринятому мнению, неожиданно было установлено, что более быстрая распадаемость фармацевтических композиций по настоящему изобретению, например, в желудке у субъекта обеспечивает лучшие абсорбционные характеристики активных пептидов и белков, где основное расщепление пептидов и белков происходит под действием пепсина и других ферментов.
Предпочтительный класс фармацевтических композиций включает в качестве активного ингредиента кальцитонин лосося. Полиаминокислота находится в свободной форме или в форме соли.
Содержание полиаминокислоты, например кальцитонина, в композиции предпочтительно составляет от 0,03 мас.% до 1 мас.%, предпочтительно от 0,05 мас.% до 1 мас.%, более предпочтительно от 0,03 мас.% до 0,5 мас.% в расчете на общую массу фармацевтической композиции. Прежде всего, содержание полиаминокислоты, например кальцитонина, в композиции составляет от 0,05 мас.% до 0,5 мас.%, например от 0,1 мас.% до 0,2 мас.%. Например, если масса конечной фармацевтической композиции составляет 500 мг, то содержание полиаминокислоты, например кальцитонина, составляет от 0,25 мг до 5 мг.
Агентом для доставки является любой агент для доставки, пригодный для доставки полиаминокислоты пероральным способом. Агенты для доставки, использованные для получения составов, например пероральных составов, включают любые агенты, пригодные для доставки конкретного фармакологичеки активного агента. Пригодные агенты для доставки включают любые модифицированные аминокислоты, описанные в упомянутом выше патенте US 5866536 или любые модифицированные аминокислоты, описанные в упомянутом патенте US 5773647, а также любые их комбинации. Содержание упомянутых патентов US 5773647 и 5866536 включено в данный контекст в качестве ссылки.
Кроме того, в качестве агента для доставки можно использовать двунатриевую соль любой упомянутой модифицированной аминокислоты, а также их этанольные сольваты и гидраты. Пригодные соединения включают соединения формулы I:
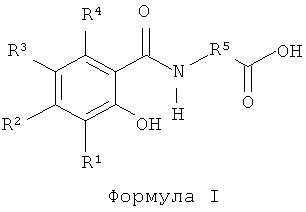
где R1, R2, R3 и R4 независимо означают водород, -ОН, -NR6R7, галоген, С1-С4 алкил или С1-С4 алкокси,
R5 означает замещенный или незамещенный С2-С16 алкилен, замещенный или незамещенный С2-С16 алкенилен, замещенный или незамещенный С1-С12 алкил(арилен) или замещенный или незамещенный арил (C1-С12 алкилен), а
R6 и R7 независимо означают водород, кислород или С1-С4 алкил
или их гидраты и спиртовые сольваты.
Соединения формулы I, а также их двунатрисвые соли и спиртовые сольваты и гидраты, а также способы их получения описаны в заявке WO 00/059863.
Кроме того, в качестве агента для доставки можно использовать двунатриевую соль любой упомянутой выше модифицированной аминокислоты, а также ее этанольный сольват или гидрат.
Двунатриевую соль получают из этанольного сольвата при упаривании или высушивании этанольного сольвата по известной методике, при этом получают безводную двунатриевую соль. Высушивание обычно проводят при температуре от приблизительно 80 до приблизительно 120°С, предпочтительно от приблизительно 85 до приблизительно 90°С, наиболее предпочтительно при приблизительно 85°С. Стадию высушивания обычно проводят при давлении 26 мм рт.ст или более. Безводная двунатриевая соль обычно содержит приблизительно менее 5 мас.% этанола, предпочтительно менее приблизительно 2 мас.% этанола в расчете на 100% безводной двунатриевой соли. Двунатриевую соль агента для доставки также получают при суспендировании агента для доставки в воде и добавлении двух молярных эквивалентов водного раствора гидроксида натрия, алкоксида натрия и т.п.
Пригодные алкоксиды натрия включают, без ограничения перечисленным, метоксид натрия, этоксид натрия и их смеси. Еще один способ получения двунатриевой соли включает взаимодействие агента для доставки с одним молярным эквивалентом гидроксида натрия, при этом получают двунатриевую соль. Двунатриевую соль выделяют в виде твердого вещества при концентрировании раствора двунатриевой соли до состояния густой пасты при перегонке в вакууме. Пасту высушивают в вакуумном шкафу, при этом получают двунатриевую соль агента для доставки в виде твердого вещества. Твердое вещество также получают при высушивании водного раствора двунатриевой соли в сушилке с распылением. Агенты для доставки получают по известным методикам, например, как описано выше, а также как описано в US 5773647 и 5866536. Этанольные сольваты, как описано в заявке WO 00/059863, включают, без ограничения перечисленным, молекулярные или ионные комплексы молекул или ионов этанола с молекулами или ионами двунатриевой соли агента для доставки. Обычно этанольные сольваты включают приблизительно одну молекулу или один ион этанола на каждую молекулу двунатриевой соли агента для доставки. Этанольный сольват двунатриевой соли агента для доставки получают при растворении агента для доставки в этаноле.
Обычно один грамм агента для доставки растворяют в приблизительно от 1 до приблизительно 50 мл этанола, в общем случае в приблизительно от 2 до приблизительно 10 мл этанола. Агент для доставки/раствор в этаноле затем взаимодействует с молярным избытком натриевой соли, например мононатриевой соли, в отношении к агенту для доставки, т.е. на каждый моль агента для доставки добавляют более одного моля катионов натрия, при этом получают этанольный сольват. Пригодные мононатриевые соли включают, без ограничения перечисленным, гидроксид натрия, алкоксид натрия, например метоксид натрия и этоксид натрия, или любую их комбинацию.
Предпочтительно по крайней мере два молярных эквивалента мононатриевой соли добавляют в раствор этанола, т.е. на каждый моль агента для доставки добавляют приблизительно по крайней мере два моля катионов натрия. В общем случае реакцию проводят при температуре кипения смеси или менее, например, при КТ. Этанольный сольват затем выделяют по известным методикам, таким как концентрирование полученной суспензии при перегонке в атмосферных условиях, охлаждение концентрата и фильтрование твердого вещества. Полученное твердое вещество затем высушивают в вакууме и получают этанольный сольват. Гидраты двунатриевой соли агентов для доставки получают при высушивании этанольного сольвата с образованием безводной двунатриевой соли, как описано выше, и при гидратации безводной двунатриевой соли. Предпочтительно образуется моногидрат двунатриевой соли. Поскольку безводная двунатриевая соль является чрезвычайно гигроскопичной. гидрат образуется под действием влаги из атмосферы.
В общем случае стадию гидратации проводят при температуре от КТ до приблизительно 50°С, предпочтительно от КТ до 30°С и при относительной влажности окружающей среды по крайней мере 50%. В другом варианте безводную двунатриевую соль гидратируют паром.
Предпочтительные агенты для доставки выбирают из группы, включающей N-(5-хлорсалицилоил)-8-аминокаприловую кислоту (5-CNAC), N-(10-[2-гидроксибензоил]амино)каприновую кислоту (SNAD), N-(8-[2-гидроксибензоил]амино)каприловую кислоту (SNAC) и их моно- и дисоли, например мононатриевая или двунатриевая соли, этанольные сольваты солей и моногидраты солей и любые комбинации вышеперечисленных соединений, такие как, например, этанольные сольваты натриевых солей и моногидраты натриевых солей в любой комбинации. Также можно использовать другие соли, например соли калия, лития и кальция. Агенты для доставки 5-CNAC, SNAD и SNAC характеризуются высокой растворимостью в воде, прежде всего в щелочной среде кишечника и практически полностью, т.е. более 90% абсорбируются в желудочно-кишечном тракте, например в двенадцатиперстной кишке, после проглатывания в виде тонкоизмельченных или крупных частиц. Напротив, агенты для доставки могут образовывать осадок в кислотной среде, например в желудке. Предпочтительно агент для доставки получают в виде тонкоизмельченных частиц.
Неожиданно было также установлено, что определенный агент для доставки может влиять на время растворения активного ингредиента. Например, если носителем является 5-CNAC, превращение нерастворимой формы, например твердой формы натриевой соли или свободной кислоты 5-CNAC, в конкретной окружающей среде, например в кишечнике, в растворимую форму 5-CNAC, например образование раствора 5-CNAC, происходит по механизму, который способствует увеличению скорости растворения активного ингредиента, например скорость растворения составляет не более 10 мин.
Таким образом, можно предположить, что нерастворимая форма агента для доставки, которая переходит в растворимую при контактировании с окружающей средой желудочно-кишечного тракта (например, двенадцатиперстной кишки), обеспечивает высокую скорость растворения активного ингредиента.
Следовательно, агент для доставки, такой как 5-CNAC или его соли, может обеспечивать удовлетворительное или оптимальное микроокружение для удовлетворительной или оптимальной скорости растворения и/или абсорбции полиаминокислоты в качестве активного ингредиента.
Прежде всего, двунатриевая соль 5-CNAC обеспечивает удовлетворительное или оптимальное микроокружение для абсорбции кальцитонина лосося. Абсорбцию кальцитонина лосося оценивают, например, по концентрации в плазме.
В отдельном предпочтительном классе фармацевтических композиций в качестве агента для доставки используют 5-CNAC. 5-CNAC находится в свободной форме или в форме соли и необязательно содержит частицы различного размера в широком диапазоне, например средний размер частиц составляет от 50 до 5 мкм.
Предпочтительно агент для доставки находится в тонкоизмельченной форме.
Средний размер частиц тонкоизмельченного агента для доставки, например 5-CNAC, измеряют, например, измельчая крупные частицы 5-CNAC и периодически сравнивая полученные частицы с частицами известного размера до образования частиц с требуемым средним размером. Процесс измельчения 5-CNAC описан в заявке WO 2005/014031, включенной в данное описание в качестве ссылки, см. например, стр.10 и пример 1, в котором описано получение частиц 5-CNAC различного размера.
Содержание агента для доставки предпочтительно составляет от 5 мас.% до 80 мас.%, предпочтительно от 10 мас.% до 70 мас.%, более предпочтительно от 20 мас.% до 60 мас.%, еще более предпочтительно от 40 мас.% до 60 мас.% в расчете на общую массу фармацевтической композиции, например 50 мас.%. Если конечная масса фармацевтической композиции составляет 500 мг, то количество агента для доставки в этой композиции составляет от 2,5 до 400 мг.
Кроме того, если агентом для доставки является 5-CNAC или его соль, содержание солевой формы предпочтительно составляет более 90 мас.% в расчете на общую массу 5-CNAC в композиции, прежде всего в отношении двунатриевой соли 5-CNAC.
Предпочтительным агентом для доставки является двунатриевая соль 5-CNAC.
Соотношение активного ингредиента и агента для доставки предпочтительно составляет от 1:25 до 1:400, предпочтительно от 1:50 до 1:300, более предпочтительно от 1:100 до 1:200, наиболее предпочтительно соотношение в случае композиции sCT/5-CNAC составляет 0,5-1 мг sCT на 200 мг - 300 мг двунатриевой соли 5-CNAC.
Дезинтегрирующий агент выбирают из группы, включающей любой супердезинтегрирующий агент, например синтетический полимер, набухающий при поглощении воды, прежде всего кросповидоны и повидоны. Более конкретные примеры дезинтегрирующих агентов включают кросповидон, повидон, Explotab или AC-Di-Sol. В предпочтительном классе фармацевтических композиций в качестве дезинтегрирующего агента используют кросповидон. Кросповидон представляет собой синтетический сшитый гомополимер N-винил-2-пирролидона, химическое название этенил-2-пирролидинон, с молекулярной массой 1000000 или более.
Супердезинтегрирующие агенты представляют собой вещества, которые поглощают воду и в значительной степени набухают за счет капиллярного эффекта или гидратации. Эти соединения более эффективны по сравнению с обычными дезинтегрирующими агентами вследствие способности к поглощению воды и набуханию.
Для снижения времени распадаемости можно использовать также другие агенты, шипучие смеси/или другие средства.
Содержание дезинтегрирующего агента предпочтительно составляет от 0,02 мас.% до 10 мас.%, предпочтительно от 0,2 мас.% до 10 мас.%, более предпочтительно от 1,0 мас.% до 8 мас.%, например от 3 мас.% до 7 мас.% в расчете на общую массу фармацевтической композиции, например 5 мас.%. Если масса полученной фармацевтической композиции составляет 500 мг, то содержание дезинтегрирующего агента составляет от 0,1 мг до 50 мг.
Коммерческие кросповидоны включают Polyplasdone XL, Polyplasdone XL-10, PolyplasdoneINF-10 фирмы ISP, Kollidon CL фирмы BASF. Предпочтительным кросповидоном является Polyplasdone XL. Повидон является синтетическим полимером, содержащим линейные группы 1-винил-2-пирролидинона с молекулярной массой обычно от 2500 до 3000000. Коммерческие повидоны включают Kollidon K-30, Kollidon K-90F фирмы BASF, а также Plasdone K-30 и PlasdoneK-29/32 фирмы ISP. В другом варианте эти соединения получают по известным методикам.
В качестве разбавителя используют, например, Avicel PH 102 или 101. Содержание разбавителя в фармацевтической композиции составляет до 90 мас.% в расчете на общую массу композиции, или разбавитель используется для компенсации любого различия между требуемой и реальной массой конечной фармацевтической композиции, которое составляет до 600 мг, например 500 мг. Предпочтительно содержание связующего агента составляет от 20 до 70 мас.% в расчете на общую массу композиции, например от 40 до 60 мас.%, например 50 мас.%. Если масса конечной фармацевтической композиции составляет 500 мг, то содержание связующего агента, например, составляет от 100 мг до 350 мг.
В предпочтительном варианте настоящего изобретения разбавитель представляет собой микрокристаллическую целлюлозу.
Добавление разбавителя снижает время распадаемости таблетки.
Время растворения активного агента не обязательно зависит от наличия разбавителя.
Добавлением глиданта или замасливателя в таблетку может увеличивать скорость растворения активного ингредиента, как известно, из-за гидрофобности замасливателя, например стеарата магния, стеарилфумарата натрия, стеарата кальция и т.п.
Распадаемость и растворение
Термины «распадаемость» и «растворение» определены в фармакопее USP, разделы <701> и <711>, включенные в данный контекст в качестве ссылок.
Термин «время растворения» в данном контексте означает время, требуемое для высвобождения данного количества (или части) лекарственного средства из твердой лекарственной формы в раствор. Время растворения измеряют in vivo в условиях, аналогичных условиям in vivo, и в которых количество лекарственного средства в растворе определяют в зависимости от времени.
Например, растворение определяют согласно инструкциям USP XXIII, лопастным методом с использованием прибора для испытаний 2 при 50 об/мин.
Термин «время распадаемости» в данном контексте означает время, необходимое для распада лекарственного средства (т.е. капсулы или таблетки) на первичные частицы в строго определенных условиях испытаний. Лабораторные испытания in vitro проводят в условиях, близких к условиям in vivo.
Например, если композиция представляет собой таблетку, время распадаемости означает время, необходимое для распада таблетки на гранулы определенного размера. Время распадаемости определяется такими факторами, как вид и содержание связующего в таблетке, степень прессования, использованная для прессования ингредиентов таблетки.
Время распадаемости фармацевтической композиции по настоящему изобретению составляет не более 10 мин, например не более 9 мин. Предпочтительно время распадаемости составляет до 8 мин, например 6 мин, например, оно может составлять менее 8 мин или 7 мин. Еще один класс фармацевтических композиций характеризуется временем распадаемости до 5 мин, например от 1 мин до 4 мин, например 2 мин. Другой класс фармацевтических композиций характеризуется временем распадаемости менее 2 мин, например 1 мин или менее.
В еще одном объекте настоящего изобретения композиции характеризуются временем растворения до 10 мин.
В основном время распадаемости композиций по настоящему изобретению составляет 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10 мин или любую часть от указанного времени, например 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 120, 130, 140, 150, 160, 170, 180 с и т.п.
Распадаемость в данном контексте означает физический процесс, в ходе которого таблетка распадается на мелкие частицы, например, диаметром менее 0,065 см. Данный процесс регистрируют визуально в отношении к физически целостной таблетке. Обычно распадаемость определяют по времени, необходимому для прохождения 100% частиц дисперсии через цилиндр, снабженный отверстиями, например, длиной 7,75 см с внутренним диаметром 21,5 мм и толщиной стенок приблизительно 2 мм, на водяной бане при 37°С (±2°С), как описано в инструкции USP <701>.
Время распадаемости композиции может быть связано со временем растворения. Время растворения означает период, в который активный ингредиент растворяется в жидкой среде. Растворение определяют методом УФ или ЖХВР и определяют приблизительное время, необходимое для полного высвобождения лекарственного средства.
Активные ингредиенты в дезинтегрированной композиции, например в таблетке, не обязательно присутствуют в растворе и способны абсорбироваться. Продолжительное время распадаемости несовместимо с быстрой абсорбцией лекарственного средства, а быстрая распадаемость сама по себе не означает быструю абсорбцию.
Обычно существует определенное соотношение между распадаемостью и растворением. Для композиций по настоящему изобретению можно установить линейное соотношение между временем растворения и временем распадаемости. Например, меньшее время распадаемости соответствует более быстрому растворению, в то время как большее время распадаемости соответствует более медленному растворению. Более подробно время распадаемости 6 мин и менее соответствует растворению >90% в течение 20 мин, время распадаемости 9 мин соответствует растворению ~30% в течение 20 мин.
В одном специфическом варианте осуществления настоящего изобретения время растворения композиции линейно зависит от времени распадаемости композиции. В еще одном объекте настоящего изобретения время распадаемости композиции можно использовать для предсказания времени растворения композиции. И наоборот, если известно время растворения композиции, то можно рассчитать время распадаемости данной композиции.
Соотношение между временем распадаемости и временем растворения прежде всего можно эффективно использовать, если композиция находится в форме таблетки. В данном случае время распадаемости соответствует времени распадаемости таблетки в желудке. Таким образом, факторы, влияющие на время распадаемости таблетки, например твердость таблетки, также можно использовать для предсказания времени растворения активного ингредиента.
Степень растворения может отражаться на степени абсорбции. Следовательно, характерным параметром растворения является терапевтически эффективное количество активного вещества или веществ в плазме крови.
В фармакокинетических исследованиях с использованием обезьян состав, содержащий 0,8 мг кальцитонина, обеспечивает максимальную концентрацию в плазме (Сmах) не менее 400 пг/мл и/или снижение уровня кальция в плазме >20% в течение 6 ч.
Таким образом, предполагается, что в результате изменения времени распадаемости композиции по настоящему изобретению можно оптимизировать и/или при необходимости изменить скорость и/или количество абсорбированного вещества. Например, если композиция находится в форме таблетки, биодоступность можно регулировать, например, при изменении твердости таблетки. Таким образом, ингредиенты таблетки (экципиенты/носители) и степень прессования, использованные при формировании таблетки, могут влиять на биодоступность активного ингредиента в составе композиции по настоящему изобретению.
Другой класс соединений по настоящему изобретению включает фармацевтическую композицию в форме прессованной таблетки. В этом случае твердость таблетки предпочтительно составляет от 5 до 10 кПа.
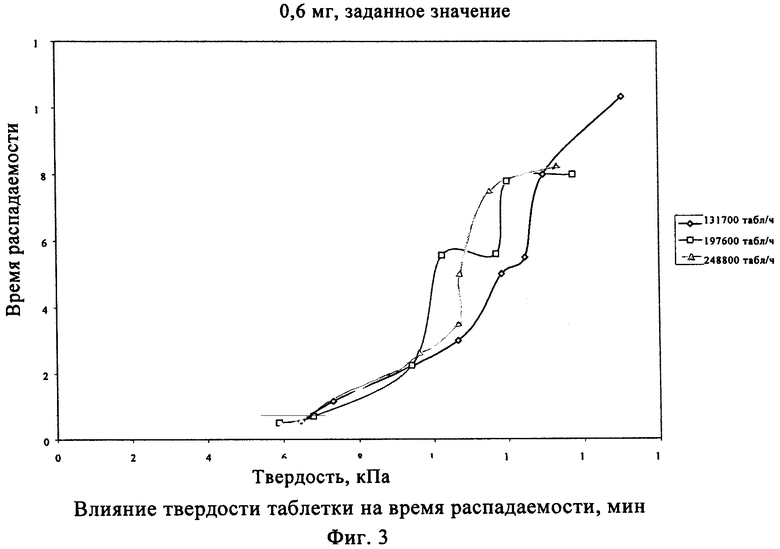
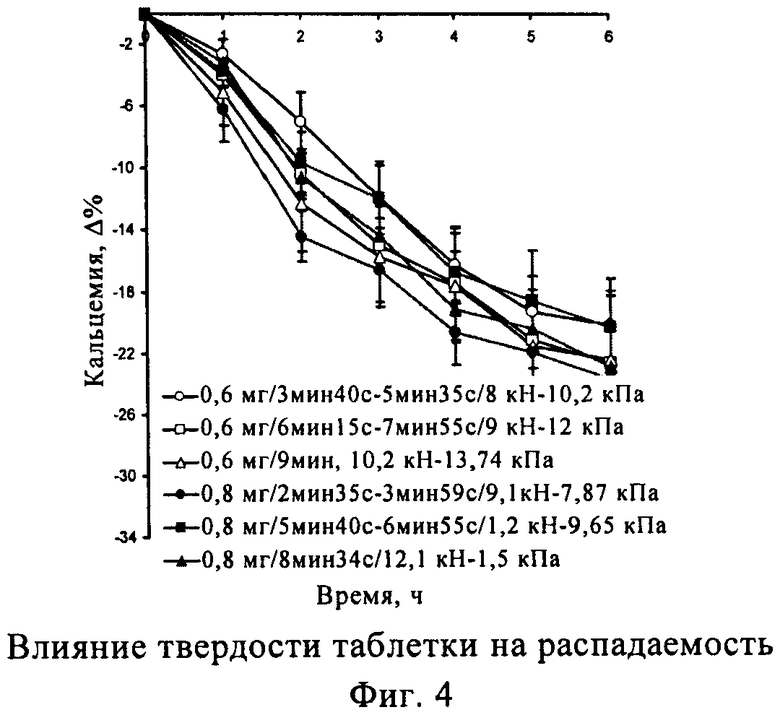
Твердость таблетки данного класса можно использовать для дополнительного определения времени распадаемости фармацевтической композиции. В настоящем изобретении установлено, что при использовании одной и той же фармацевтической композиции твердость таблетки находится в линейной зависимости от времени распадаемости. Таким образом, в еще одном объекте настоящего изобретения время распадаемости фармацевтической композиции зависит от твердости таблетки. Более подробно определенное время распадаемости достигается при изменении величины твердости прессованной таблетки.
Твердость таблетки
Таблетка предпочтительной массы, предпочтительного состава, твердость которой составляет от 5 до 20 кПа, обычно характеризуется временем распадаемости менее 6 мин.
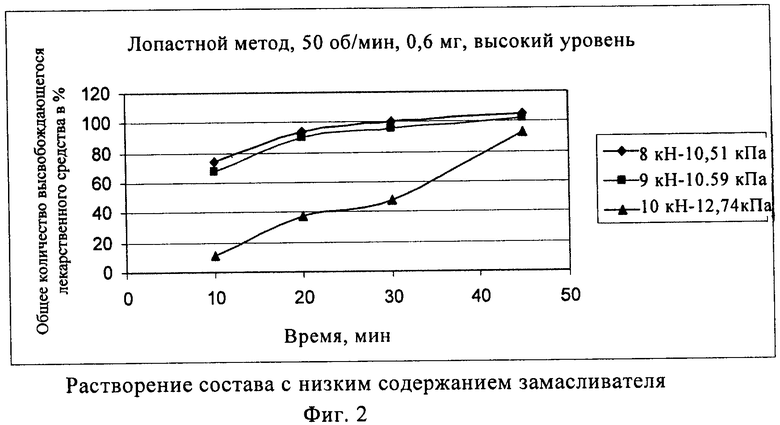
Твердость таблетки напрямую связана со временем распадаемости таблетки, как указано в приведенных ниже табл.1 и 2 и фиг.3 и 4.
где СО - относительное стандартное отклонение.
Таким образом, сила прессования, используемая при получении таблетки, может определять время распадаемости фармацевтической композиции.
Дополнительные ингредиенты
Еще один класс фармацевтических композиций также включает глидант и/или сухой связующий агент.
Один класс фармацевтических композиций дополнительно включает глидант.
Глидантом является, например, продукт cab-o-sil.
Содержание глиданта составляет до 1,5 мас.%, например от 0,02 до 0,5 мас.% в расчете на общую массу композиции, например 0,3 мас.%. Если конечная масса фармацевтической композиции составляет 500 мг, содержание глиданта составляет до 7,5 мг.
Еще один класс фармацевтических композиций содержит замасливатель. Замасливателем является, например, стеарат магния.
Содержание замасливателя составляет, например, от 0,5 до 1,5 мас.% в расчете на общую массу композиции, например от 0,75 до 1,25 мас.%, например 1 мас.%. Если конечная масса фармацевтической композиции составляет 500 мг, содержание замасливателя составляет от 2,5 мг до 7,5 мг (см. фиг.1 и 2).
Кроме упомянутых выше специфических ингредиентов для получения композиции по настоящему изобретению, характеризующейся указанным временем распадаемости и/или растворения, могут использоваться другие методы, например, описанные в заявке WO 94/26778 и в патентах US 5359030, 5438040,5681811, 6191105, 6309633, 6380405, 6436990, 6458776, а также заявке WO 97/33531 и патентах US 5912014, 608618 и 6479692 (включенных в данное описание в качестве ссылок).
Способы
Настоящее изобретение относится к способам получения составов и композиций, описанных в данном контексте.
Прежде всего, настоящее изобретение относится к способу получения таблеток, время распадаемости которых составляет не более 10 мин, и указанный способ включает следующие стадии:
а) смешивание полиаминокислоты, агента для доставки и дезинтегрирующего агента, при этом получают смесь,
б) добавление в полученную смесь разбавителя и перемешивание,
в) прессование продукта.
Необязательно способ также включает следующие стадии:
1. Просеивание смеси, полученной на стадии а).
2. Добавление дезинтегрирующего агента и перемешивание после проведения стадии б).
3. Добавление замасливателя и/или глиданта перед проведением стадии с).
Типичный способ получения показан ниже на схеме 1.
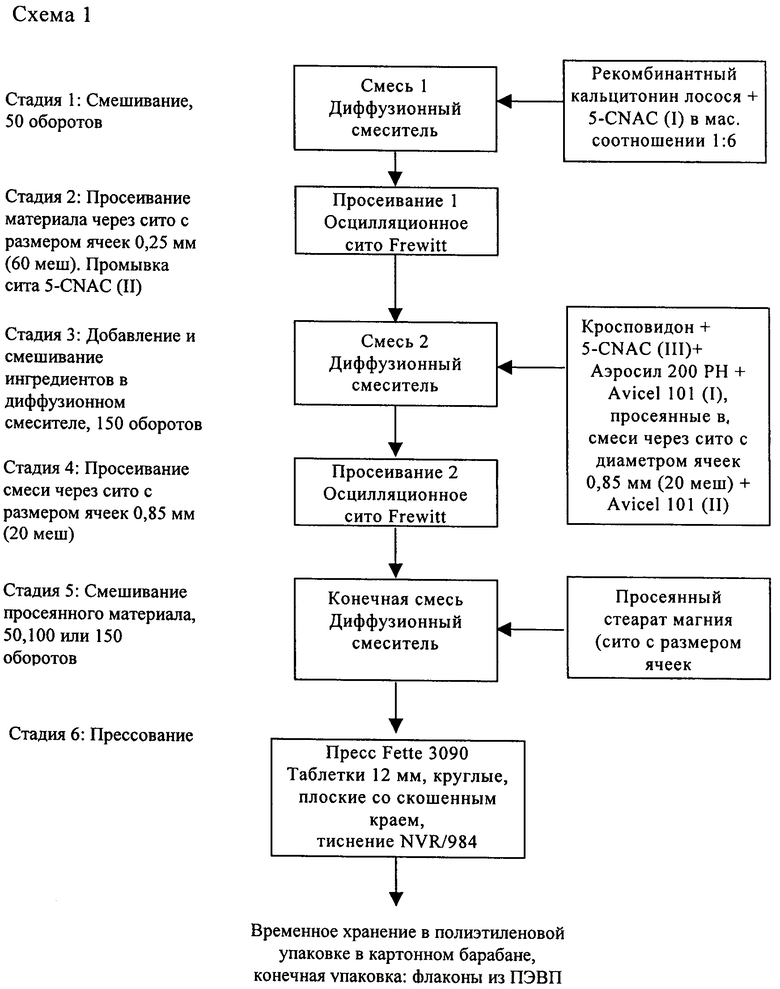
В одном варианте описанного выше способа, после прессования смеси в таблетки получают таблетки, твердость которых составляет от 5 до 20 Па, время распадаемости которых составляет менее 10 мин.
Как указано выше, если полиаминокислотой является кальцитонин, состав по настоящему изобретению можно использовать для лечения нарушения резорбции костной ткани, такого как остеоартрит, остеолиз или болезнь Педжета, или артрита, например остеоартрита. Таким образом, предлагается способ профилактики и/или лечения нарушения резорбции костной ткани и/или артрита у пациентов, нуждающихся в таком лечении, который заключается во введении указанному пациенту терапевтически эффективного количества фармацевтической композиции по настоящему изобретению, при этом полиаминокислотой является кальцитонин, например кальцитонин лосося, в свободной форме или в форме соли, а время распадаемости фармацевтической композиции составляет до 10 мин.
Кроме того фармацевтическую композицию по настоящему изобретению, включающую требуемую полиаминокислоту, например кальцитонин, можно использовать в следующих случаях:
1. Способ подавления резорбции и нормализации обмена веществ в околохрящевых костях у пациента, нуждающегося в таком лечении, включающий введение указанному пациенту терапевтически эффективного количества фармацевтической композиции по настоящему изобретению.
2. Способ поддерживания и стимуляции роста хряща при прямом или косвенном воздействии на хондроциты пациента, нуждающегося в таком лечении, включающий введение указанному пациенту терапевтически эффективного количества фармацевтической композиции по настоящему изобретению.
3. Способ ингибирования активности фосфолипазы А2 и/или коллагеназы у пациента, нуждающегося в таком лечении, включающий введение указанному пациенту терапевтически эффективного количества фармацевтической композиции по настоящему изобретению.
4. Способ стимуляции синтеза глюкозаминогликанов и/или протеогликанов у пациента, нуждающегося в таком лечении, включающий введение указанному пациенту терапевтически эффективного количества фармацевтической композиции по настоящему изобретению.
5. Способ воздействия на неоднородную плотность или неподвижность околохрящевой кости пациента, нуждающегося в таком лечении, включающий введение указанному пациенту терапевтически эффективного количества фармацевтической композиции по настоящему изобретению.
6. Способ воздействия на воспалительный процесс, приводящий к ослаблению боли при движении и связанных с воспалением симптомов (окружность коленного сустава, угол сгибания коленного сустава, неподвижность при опухании) у пациента, нуждающегося в таком лечении, включающий введение указанному пациенту терапевтически эффективного количества фармацевтической композиции по настоящему изобретению.
7. Способ снижения дегенеративных изменений в суставе у пациента, нуждающегося в таком лечении, включающий введение указанному пациенту терапевтически эффективного количества фармацевтической композиции по настоящему изобретению.
Комбинации
В еще одном объекте настоящего изобретения фармацевтическую композицию по настоящему изобретению можно вводить совместно, например включать в состав второе лекарственное средство, где указанным вторым лекарственным средством является, например, второй ингибитор резорбции костной ткани, средство, способствующее образованию костной ткани, или обезболивающее средство.
Если фармацевтическую композицию по настоящему изобретению вводят совместно со вторым, третьим или четвертым лекарственным средством, каждое соединение независимо можно вводить одновременно, в отдельности или последовательно в отношении к композиции по настоящему изобретению.
Пригодное второе лекарственное средство включает кальцитонин различного происхождения, например кальцитонин лосося, угря Asul-7 или человека, аналоги и производные кальцитонина, стероидные гормоны, например эстроген, частичные агонисты эстрогена или комбинацию эстроген-гестаген, селективный модулятор рецептора эстрогена (SERM), например ралоксифен, лазофоксифен, TSE-424, FC1271, тиболон (Livial О), витамин D или его аналоги РТН, фрагменты РТН, производные РТН, например РТН (1-84), РТН (1-34), РТH (1-36), РТН (1-38), РТН (1-31) NH2 или PTS 893, бисфосфонаты (например, алендронат, ризедронат, золедроновая кислота, ибандронат), ингибиторы протеаз, например ингибитор катепсина, предпочтительно ингибитор катепсин К, вещества, высвобождающие РТН, селективные молекулы андрогенного рецептора (SARM), ингибиторы ММР (ингибиторы металлопротеаз), соединения стронция, ингибиторы СОХ-2, например люмиракоксиб (Prexige (E)), целекоксиб (Celebrex0), рофекоксиб(Viхх (D), валдекоксиб (BextraS), эторикоксиб (ArcoxiaG) или смеси ингибиторов СОХ-1 и СОХ-2, например диклофенак.
Таким образом, в соответствии с указанным объектом в настоящем изобретении предлагается фармацевтическая комбинация, включающая:
а) первый агент, включающий фармацевтическую композицию, включающую кальцитонин, например кальцитонин лосося, кальцитонин угря Asul-7 или кальцитонин человека в свободной форме или в форме соли, предпочтительно в фармацевтически приемлемой пероральной форме, агент для доставки и дезинтегрирующий агент, и время распадаемости указанной фармацевтической композиции составляет до 10 мин, и
б) совместный агент, который подавляет резорбцию костной ткани, или является агентом, способствующим образованию костной ткани или
обезболивающим средством, например, как описано выше.
Термин «фармацевтическая комбинация», использованный в данном контексте, означает продукт, который получают при смешивании или комбинировании нескольких активных ингредиентов, и включает фиксированные и нефиксированные комбинации активных ингредиентов.
Термин «фиксированная комбинация» означает, что активные ингредиенты, например кальцитонин лосося, и совместный агент вводят пациенту одновременно в виде единой лекарственной формы или дозы.
Термин «нефиксированная комбинация» означает, что активные ингредиенты, например кальцитонин лосося, и совместный агент вводят пациенту в составе различных лекарственных форм совместно, одновременно или последовательно без ограничений по времени, и после указанного введения достигается терапевтически эффективные уровни двух соединений в организме пациента.
Предпочтительно кальцитонин, например кальцитонин лосося в свободной форме или в форме фармацевтически приемлемой соли, вводят совместно с ингибитором протеаз, например ингибитором катепсина, например ингибитором катепсина К.
В указанном выше объекте настоящего изобретения предлагается также набор компонентов для профилактики и/или лечения нарушения резорбции костной ткани и/или артрита, включающий:
а) первый агент, кальцитонин, например кальцитонин лосося, кальцитонин угря (Asu 1-7) или человека в свободной форме или в форме соли, в составе фармацевтической композиции, включающей:
1) поламинокислоту,
2) агент для доставки,
3) дезинтегрирующий агент и
время распадаемости которой составляет до 10 мин, и
б) совместный агент, подавляющий резорбцию костной ткани, или который является лекарственным средством, способствующим образованию костной ткани или обезболивающим агентом, например, как описано выше.
Предлагаются также способы совместного введения согласно каждому из описанных выше методов (1)-(7), и способы включают совместное введение терапевтически эффективного количества фармацевтической композиции по настоящему изобретению, например фармацевтической композиции, включающей кальцитонин, например кальцитонин лосося в свободной форме или в форме соли, в составе фармацевтической композиции, и время распадаемости которой составляет до 10 мин, и второго лекарственного средства, являющегося ингибитором резорбции костной ткани, агентом, способствующим образованию костной ткани, или обезболивающим лекарственным средством в свободной форме или в форме соли.
Термин «совместное введение», использованный в данном контексте, означает введение выбранных терапевтических агентов одному пациенту и включает курсы лечения, при которых агенты необязательно вводят одним и тем же способом или одновременно.
Дозировки
Если фармакологически активным ингредиентом является кальцитонин лосося, пригодная доза зависит, например, от субъекта, природы и тяжести заболевания, подлежащего лечению. Однако в общем случае удовлетворительные результаты получают при систематическом введении суточной дозы от приблизительно 0,5 мгк/кг до приблизительно 10 мкг/кг массы тела животного, предпочтительно от 1 мкг/кг до 6 мкг/кг массы тела. Фармацевтически приемлемые неактивные экципиенты, используемые в составе фармацевтических композиций кальцитонина, например в составе пероральных фармацевтических композиций кальцитонина, необязательно включают полимеры и неактивные соединения, которые, например, упрощают обработку фармацевтической композиции или получение твердой пероральной лекарственной формы по настоящему изобретению, или способствуют высвобождению твердой пероральной композиции в желудочно-кишечном тракте.
В настоящем изобретении предлагается конкретный диапазон доз кальцитонина, прежде всего кальцитонина в свободной форме или в форме соли, например кальцитонина лосося, характеризующийся эффективностью и достаточно высокой переносимостью, т.е являющийся безопасным для пациента.
Предпочтительный диапазон доз составляет от 0,15 мг до 2,5 мг, прежде всего от 0,4 мг до 2,5 мг кальцитонина лосося для пациента, например человека, например среднего человека массой 70 кг. Более предпочтительно доза составляет приблизительно 1 мг, например, от 0,8 мг до 1,2 мг. Предпочтительны также дозы не более 1 мг, но не менее 0,4 мг. Более предпочтительно доза составляет приблизительно 1 мг, например, 1 мг. Наиболее предпочтительна доза от 0,5 мг до 1,1 мг, прежде всего от 0,6 мг до 0,8 мг, еще более предпочтительна доза от 0,15 мг до 0,4 мг, прежде всего доза 0,15 мг. Дозы вводят один раз в сут пациенту, нуждающемуся в лечении.
Фармацевтические композиции по настоящему изобретению используют в следующих случаях:
- Способ профилактики и/или лечения остеоартрита у пациента, нуждающегося в таком лечении, включающий введение указанному пациенту фармацевтической композиции, включающей от 0,4 до 2,5 мг, предпочтительно от 0,8 до 1,2 мг, наиболее предпочтительно приблизительно 1 мг кальцитонина, например кальцитонина лосося, время распадаемости которой составляет до 10 мин.
- Фармацевтическая композиция, включающая от 0,4 до 2,5 мг, предпочтительно от 0,8 до 1,2 мг, наиболее предпочтительно приблизительно 1 мг кальцитонина, например кальцитонина лосося.
- Применение кальцитонина, например кальцитонина лосося, для получения лекарственного средства, предназначенного для лечения и/или профилактики нарушения резорбции костной ткани и/или артрита, и указанное лекарственное средство включает кальцитонин в количестве от 0,4 до 2,5 мг, предпочтительно от 0,8 до 1,2 мг, наиболее предпочтительно приблизительно 1 мг кальцитонина, например кальцитонина лосося, при этом время распадаемости указанной фармацевтической композиции составляет до 10 мин.
Такой пероральной формой является, например, фармацевтическая композиция для перорального введения кальцитонина лосося, включающая:
(A) терапевтически эффективное количество указанного кальцитонина лосося,
(B) по крайней мере один усилитель абсорбции, улучшающий биодоступность указанного кальцитонина лосося, и время распадаемости указанной композиции составляет до 10 мин.
Примеры усилителей включают 5-CNAC, SNAC и жирные кислоты, такие как капрат Na и капралат Na.
Фармацевтические композиции, на примере которых установлена применимость кальцитонина для лечения остеоартрита, можно получать в виде капсул, включая мягкие капсулы, таблеток, микротаблеток, суппозитория или других твердых пероральных форм, которые получают по известным методикам, при этом время распадаемости композиции составляет до 10 мин.
В предпочтительном варианте настоящего изобретения время распадаемости композиции составляет до 10 мин, или по данным испытаний в соответствующих условиях более 90% продукта растворяется в течение 20 мин.
Типичный способ получения твердых фармацевтических композиций по настоящему изобретению заключается в том, что композицию получают при измельчении агента для доставки или агента для доставки в смеси с любой комбинацией дополнительных ингредиентов настоящей композиции, при этом получают тонкоизмельченные частицы. Тонкоизмельченный агент для доставки или агент для доставки в смеси с тонкоизмельченными дополнительными ингредиентами по настоящему изобретению затем перерабатывают пригодными способами, например перемешиванием смеси активного агента или нескольких активных агентов, агента для доставки, кросповидона или повидона и/или других ингредиентов, и полученной смесью заполняют капсулы, или смесь прессуют с последующим таблетированием. Кроме того, твердую дисперсию получают по известным методикам с последующей переработкой в таблетки или капсулы.
В данном контексте и формуле изобретения термин «включает, «содержит» и другие варианты означают «включают без ограничения перечисленным» и не исключают другие фрагменты, добавки, компоненты, ингредиенты или стадии.
В данном контексте и формуле изобретения единственное число включает множественное, если не указано иное. Прежде всего, если использовано единственное число, то подразумевается как единственное, так и множественное, если из контекста не следует иное.
Признаки, целые числа, характеристики, соединения, химические фрагменты или группы, описанные в связи с конкретным объектом, вариантом или примером по настоящему изобретению, применимы к любому другому объекту, варианту или примеру, описанным в данном контексте, при условии, что они применимы в каждом конкретном случае.
Примеры
Приведенные ниже примеры представлены для иллюстрации настоящего изобретения и представляются очевидными для специалиста в данной области. Данные примеры не ограничивают объем настоящего изобретения.
Пример 1
Фармацевтическая композиция 1
На первой стадии смешивали кальцитонин лосося, 5-CNAC и кросповидон. На второй стадии Avicel PH 102 просеивали, добавляли в смесь и перемешивали. На конечной стадии добавляли стеарат магния и перемешивали. Полученную смесь прессовали в таблетки массой 500 мг и их эффективность оценивали на макаках-резус. Полученные результаты приведены на фиг.5.
Пример 2
Альтернативная фармацевтическая композиция (3 партии)
Получали композицию, как описано в примере 1, включающую:
Однако в отличие от примера 1 на первой стадии смешивали кальцитонин лосося и Avicel PH 102. Затем на второй стадии в полученную смесь добавляли 5-CNAC и кросповидон. На последней стадии добавляли стеарат магния.
Полученную смесь прессовали, используя 3 различных уровня прессования, при этом получали 3 различные партии таблеток, характеризующихся различной твердостью и тремя различными временами распадаемости:
1) время распадаемости 1 мин 10 с,
2) время раепадаемости 5 мин 40 с,
3) время распадаемости 8 мин 51 с.
Пример 3
Альтернативная фармацевтическая композиция
Получали смесь, аналогичную описанной в примере 1, кроме того, добавляли продукт Cab-o-sil, при этом получали композицию, включающую:
На первой стадии смешивали кальцитонин лосося, 5-CNAC и кросповидон. На второй стадии добавляли просеянные Avicel и Cab-o-sil. На последней стадии добавляли стеарат магния. Полученную смесь прессовали в таблетки массой 500 мг. Добавление Cab-o-sil улучшает профиль прессования таблетки.
Пример 4
Альтернативная фармацевтическая композиция
Композицию получали, как описано в примере 3, включающую:
Пример 5
Альтернативная фармацевтическая композиция
Масса (а+b), указанная для двунатриевой соли 5-CNAC, соответствует общей массе 200 мг 5-CNAC в виде свободной кислоты.
Масса (а+b), указанная для Avicel РН 101 (I) и (II), соответствует общей массе Avicel РН 101.
Пример 6
Альтернативная фармацевтическая композиция
Масса (a+b+с), указанная для двунатриевой соли 5-CNAC, соответствует общей массе 200 мг 5-CNAC в виде свободной кислоты.
Масса (а+b), указанная для Avicel РН 101 (I) и (II), соответствует общей массе Avicel РН 101.
Для получения описанных выше составов использовали аналогичный способ, как описано в примере 1. Однако альтернативный слособ получения композиций, прежде всего описанных в примерах 5 и 6, включает:
1. Взвешивание 0,25 г sCT DS
2. Смешивание с 5-CNAC (партия I)
3. Просеивание смеси, полученной на стадии 2, через сито #60 (0,25 мм)
4. Промывку сита (после стадии 3) 5-CNAC (партия II)
5. Просеивание Аэросила 200 РН и Avicel PH 101 (партия I) через сито #20 (0,85 мм)
6. Добавление Avicel PH 101 (партия II), просеянного материала, полученного на стадии 5, 5-CNAC (партия III), просеянного материала, полученного на стадии 4, и кросповидона в диффузионный смеситель и перемешивание (150 оборотов)
7. Просеивание смеси через сито #20 (0,85 мм)
8. Просеивание стеарата магния через сито #20 (0,85 мм) и добавление в смесь, полученную на стадии 7
9. Смешивание с замасливателем (50 оборотов)
10. Прессование смеси в круглые таблетки диаметром 12 мм, FFBE с тиснением.
Использовали оборудование, описанное в примере 1.
Пример 7
Испытания на приматах
Таблетки получали по описанной выше методике и испытывали на макаках-резус:
Макаки-резус не получали корм в течение ночи до введения лекарственного средства и в течение всего периода испытаний животных в полном сознании фиксировали в положении сидя. Одну таблетку из каждой партии вводили каждой обезьяне через желудочный зонд, а затем вводили 10 мл воды. Кровь у макак-резус отбирали непосредственно перед введением и через 0,25, 0,5, 0,75, 1, 1, 5, 2, 3, 4, 5 и 6 ч после введения лекарственного средства. Содержание кальцитонина лосося в плазме для каждой дозы и для каждого животного определяли методом радиоиммунологического анализа.
Рассчитанные для каждой макаки значения содержания кальцитонина лосося в плазме приматов (SCt) для одной партии и одного периода времени, средней концентрации SCt в плазме для всех обезьян для одной партии и одного периода времени, стандартного отклонения концентрации SCt в плазме для одной серии и одного периода времени и стандартного отклонения (СО) от среднего значения концентрации SCt в плазме для всех макак для одной партии и одного периода времени приведены на фиг.5.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРИМЕНЕНИЕ КАЛЬЦИТОНИНА ДЛЯ ЛЕЧЕНИЯ РЕВМАТОИДНОГО АРТРИТА | 2006 |
|
RU2453330C2 |
| ПРИМЕНЕНИЕ КАЛЬЦИТОНИНА ПРИ ОСТЕОАРТРИТЕ | 2004 |
|
RU2368390C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ПЕРОРАЛЬНОГО ВВЕДЕНИЯ ФАРМАКОЛОГИЧЕСКИ ДЕЙСТВУЮЩИХ ВЕЩЕСТВ | 2001 |
|
RU2287999C2 |
| ПЕРОРАЛЬНОЕ ВВЕДЕНИЕ ПАРАТИРЕОИДНОГО ГОРМОНА И КАЛЬЦИТОНИНА | 2002 |
|
RU2300392C2 |
| 5-CNAC В КАЧЕСТВЕ АГЕНТА ДЛЯ ПЕРОРАЛЬНОЙ ДОСТАВКИ ФРАГМЕНТОВ ПАРАТИРОИДНОГО ГОРМОНА | 2002 |
|
RU2322256C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ чГР, ДЛЯ ПЕРОРАЛЬНОГО ВВЕДЕНИЯ | 2007 |
|
RU2493868C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ДИНАТРИЕВОЙ СОЛИ N-(5-ХЛОРСАЛИЦИЛОИЛ)-8-АМИНОКАПРИЛОВОЙ КИСЛОТЫ | 2006 |
|
RU2507196C2 |
| ПЕРОРАЛЬНОЕ ВВЕДЕНИЕ КАЛЬЦИТОНИНА | 2003 |
|
RU2355417C2 |
| ПЕПТИДНЫЕ АНАЛОГИ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ И РАССТРОЙСТВ | 2012 |
|
RU2616511C2 |
| МИМЕТИК КАЛЬЦИТОНИНА ДЛЯ ЛЕЧЕНИЯ БОЛЕЗНЕЙ И НАРУШЕНИЙ | 2014 |
|
RU2689551C1 |

Группа изобретений относится к области медицины, в частности к пероральной фармацевтической композиции в прессованной таблетированной форме, включающей паратироидный гормон (РТН), 5-CNAC или ее соль, дезинтегрирующий агент и растворитель, при этом время распадаемости композиции составляет не более 6 мин, а растворимость более 90% происходит в течение 20 мин, а также к способу получения указанной фармацевтической композиции. Группа изобретений обеспечивает эффективную доставку РТН, таблетка быстро распадается и/или растворяется и активный ингредиент оказывает терапевтическое воздействие. 2 н. и 8 з.п. ф-лы, 7 пр., 5 ил., 2 табл.
1. Пероральная фармацевтическая композиция в прессованной таблетированной форме, включающая: паратироидный гормон (PTH);
N-(5-хлорсалицилоил)-8-аминокаприловую кислоту (5-CNAC) или ее соль; дезинтегрирующий агент и растворитель; при этом время распадаемости композиции составляет не более 6 мин, а растворимость >90% происходит в течение 20 мин.
2. Композиция по п.1, у которой время распадаемости составляет не более 2 мин.
3. Композиция по п.1, дополнительно включающая агент, снижающий время распадаемости, например, при получении шипучих составов.
4. Фармацевтическая композиция по п.1 в форме прессованной таблетки, твердость которой составляет от 3 кПа до 20 кПа.
5. Фармацевтическая композиция по п.4, где твердость прессованной таблетки составляет от 5 кПа до 15 кПа.
6.Фармацевтическая композиция по п.4, в которой время распадаемости таблетки составляет менее 1 мин и твердость от 5 кПа до 7 кПа.
7. Композиция по п.1, где агентом для доставки является динатриевая соль 5-CNAC.
8. Композиция по п.1, где дезинтегрирующим агентом является кросповидон и/или повидон.
9. Композиция по п.1, в которой PTH представляет собой PTH 1-34.
10. Способ получения пероральной фармацевтической композиции по п.1, который включает следующие стадии;
1) смешивание РТН, агента доставки и дезинтегрирующего агента с образованием смеси,
2) прессование смеси в таблетки, твердость которых составляет от 5 кПа до 20 кПа.
| US 20020123459 A1, 05.09.2002 | |||
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИРБЕСАРТАН | 1996 |
|
RU2181590C2 |
| Белая Ж.Е., Рожинская Л.Я | |||
| Возможности применения паратиреоидного гормона для лечения остеопороза | |||
| Остеопороз и остеопатии, №3, 2004. | |||

