Изобретение относится к области химии биополимеров-аминоглюканов и может быть использовано в фармации, медицине, ветеринарии, космецевтике.
Известны разработки, публикации, посвященные изучению процесса пероксидного гидролиза хитозана с целью получения его низкомолекулярных гомологов. Определены закономерности развития процесса деструкции хитозана в гетерогенных системах. Установлена зависимость степени деструкции аминогликана от технологических параметров процесса. Эксперементально определена следующая зависимость DD=ln(M0/M)=f{[H2O2]}·τ}, где DD - степень деструкции, M0 и M - средневязкостная молекулярная масса (ММ) соответственно исходного и деструктированного кислоторастворимого хитозана, τ - время в мин, позволяет установить условия деструкции для заданной ММ целевого хитозана при любых значениях ММ исходного полимера. Показано, что степень деацетилирования исходного хитозана в процессе его пероксидной деструкции практически не изменяется (Шийчук А.В. Универсальный показатель глубины деструкции полимера. Логарифм отношения молекулярных масс. Укр. Хим. журн. 1991. Т.57. 11. С.1229; Gamzazade A.I., Slimak V.M., Sklar A.M., Shtikova E.V., Pavlova S.V., Rogojin S.V. // Acta Polymerica. 1985. V.36. 8. P.420). Деструкция жесткоцепного аминогликана хитозана в гетерогенной системе приводит к снижению ММ и расширению молекулярно-массового распределения (ММР) ввиду возрастания содержания олигомерной фракции гомологов. Данные, полученные методом экслюзионной хроматографии показывают, что при изменении отношения M0/М от ~6,7 (DD=1,9) до ~21,1 (DD=3,1) в условиях гетерогенного пероксидного гидролиза-деполимеризации хитозана наблюдается расширение ММР, а содержание в продуктах гидролиза олигомерных фракций, в том числе мономера, возрастает до 30%. Для хитозана, деструктированного в гетерогенных условиях со средневязкостной ММ ~80-100 кДа, характерно широкое ММР - от 150 кДа до олигомерной фракции; для варианта ММ хитозана ≤25 кДа ММР включает фракции от ~50 кДа до мономера.
Наиболее близким к предлагаемому способу получения водорастворимых олигомерных гомологов хитозана является метод пероксидного гидролиза аминогликана с ММ ~350-600 кДа в гетерогенной системе, применяющийся для получения гомологов хитозана с пониженной молекулярной массой в диапазоне от ~80 кДа до олигомерных структур со степенью полимеризации 1-10. Этот метод включен в описание патента на способ получения водорастворимых форм хитозана (патент РФ 2215749, С08В 37/08, A61K 32/722, 2003). Сущностью заявляемого способа является получение образцов смесей гомологов хитозана с диапазоном молекулярных масс от ~100 кДа до олигомерной группы, включая мономер (глюкозамин). Конечной целью изобретения являлась подготовка препаратов с расширенным спектром биологического действия для использования в медицине, ветеринарии и косметике.
Гомологи хитозана использовались так же для проведения гидролиза и солеобразования с органическими (янтарная, L-глютаминовая) и неорганическими (хлористоводородная, ортофосфорная) кислотами, а также с ангидридами органических кислот. Кроме указанных методов обработки высокомолекулярного хитозана проводились способы аморфизации полимера с целью повышения его реакционной способности и гидрофильности путем переосаждения из растворов в кислотах в условиях повышения рН. С этой же целью использован метод гомогенизации суспензии хитозана в водных растворах органических кислот в режиме создания кавитационных механических полей со сдвиговым воздействием на рабочий объем.
Описание способов деполимеризации хитозана в гетерогенной системе с участием в качестве окислителя гликозидных центров (межзвеньевых связей) пероксида водорода приводится в двух вариантах:
а) Гидролиз хитозана с ММ ~600 кДа в системе, содержащей 0,15% H2O2 при 80°С в течение 25 мин. В результате образуется смесь гомологов хитозана нерастворимых в воде с Mср ~80 кДа.
б) Гидролиз хитозана с ММ ~300 кДа в системе, содержащей 0,2% H2O2 при 80°С в течение 34 мин. Образующаяся смесь гомологов хитозана характеризуется Мср ~20-25 кДа (нерастворимы в воде).
Олигомерную фракцию гомологов хитозана получали в условиях кислотного гидролиза аминогликанов в среде водного раствора ортофосфорной кислоты.
а) Гидролиз хитозана с ММ ~350 кДа. Ортофосфорная кислота использовалась при соотношении с хитозаном 0,6:1. Процесс проводили при 70°С в течение 4 часов. После нейтрализации смеси (Ca(OH)2), фазового разделения, лиофильной сушки и лиофилизации выделена смесь водорастворимых олигомеров хитозана (степень полимеризации=1-10); выход 13,4%.
б) Гидролиз хитозана с ММ ~25 кДа. Соотношение введенной в процесс гидролиза ортофосфорной кислоты и хитозана (водная среда) идентично методу (а). Температурный режим и длительность процесса идентичны методу (а). После обработки методом, указанным в (а) выделена олигомерная водорастворимая фракция гомологов хитозана (степень полимеризации=1-10) с выходом 20%.
Недостатками известной методики получения водорастворимых олигомерных и кислоторастворимых низкомолекулярных гомологов хитозана является усложнение процесса кислотного гидролиза (ортофосфорная кислота); стадии - гидролиз, нейтрализация кислоты, лиофилизация целевого продукта, а также низкий выход олигомерной фракции (степень полимеризации=1-10) до 20%.
Варианты известной методики пероксидного гидролиза хитозана имеют ограниченную возможность ввиду образования только гидрофобных фракций гомологов хитозана с Мср ~20-80 кДа. Возможность получения фракций водорастворимых олигомеров методом пероксидной деполимеризации хитозана не осуществлена.
Задачей предлагаемого изобретения является разработка способа пероксидной деполимеризации хитозана для получения фракций водорастворимых олигомеров.
Технический результат заключается в возможности количественно устанавливать степень превращения исходного высокомолекулярного аминогликана в олигомерные и низкомолекулярные структуры его гомологов.
Технический результат достигается тем, что в способе получения водорастворимых олигомерных гомологов хитозана в гетерогенной системе путем деполимеризацей высокомолекулярного хитозана перекисью водорода, согласно изобретению, процесс деполимеризации хитозана проводится в двухфазной системе, твердой фазой которой является активированный хитозан с Мср=450-650 кДа и средним размером частиц 0,05-0,20 мм, жидкая фаза - водный раствор H2O2 с концентрацией H 2 O 2 в реакционной системе 1-7%, длительностью реакции 120-180 - мин и температуре 70°С, далее проводят фазовое разделение образующихся гомологов хитозана путем фильтрации через бумажную или текстильную поверхность, полученный фильтрат содержит водорастворимые олигомеры хитозана.
В результате деполимеризации образуются две фракции низкомолекулярных гомологов хитозана. Фракция гомологов хитозана, растворимых в воде (нейтральная среда), представляет собой смесь олигомеров со степенью полимеризации в диапазоне 3-8; фракция, не растворимая в воде, но растворимая в 1-2% водной
СН3СООН, является смесью низкомолекулярных гомологов аминогликана со средневязкостной ММ до 25-46 кДа. Определена возможность регулирования процесса пероксидного гидролиза хитозана путем варьирования технологических параметров: концентрации Н2O2 в реакционной системе и длительности реакции. В качестве постоянных значений технологических параметров этого процесса приняты показатели: температура, соотношение твердой и жидкой фазы в системе на начальный период взаимодействия, размер частиц хитозана.
Таким образом, разработанный метод гидролиза позволяет количественно устанавливать степень превращения исходного высокомолекулярного аминогликана в олигомерные и низкомолекулярные структуры его гомологов. По разработанным показателям выход целевой фракции гомологов хитозана водорастворимых олигомеров со степенью полимеризации 3-8 достигает более 53% (масс).
Фазовое разделение образующихся гомологов хитозана осуществляется фильтрацией через бумажную или текстильную поверхность и дополнительной экстракцией путем обработки соответствующими растворителями. Реальной является возможность дополнительного фракционирования смесей водорастворимых олигомерных гомологов хитозана путем обработки выделенной первичной соответствующей фракции водными растворами этилового спирта (экстракция и седиментация при пониженных температурах). Этим путем достигается выделение смесей, состоящих из 2-3 олигомеров.
Оценка средней степени полимеризации фракции олигомеров хитозана производится по данным химического метода определения содержания редуцирующих звеньев в тестируемом образце полимер-гомолога. Количественное блокирование альдегидных групп (полуацетальных гидроксилов) осуществляется действием гидразида изоникотиновой кислоты в водной среде с применением йодометрии. Для характеристики гидрофобной фракции гомологов хитозана, образующихся по предлагаемому методу, использовался вискозиметрический способ, включающий расчет средневязкостной Мср по уравнению Марка-Куна-Хаувинка: [η]=KMα, где K=3,5·10-4, α=0,76.
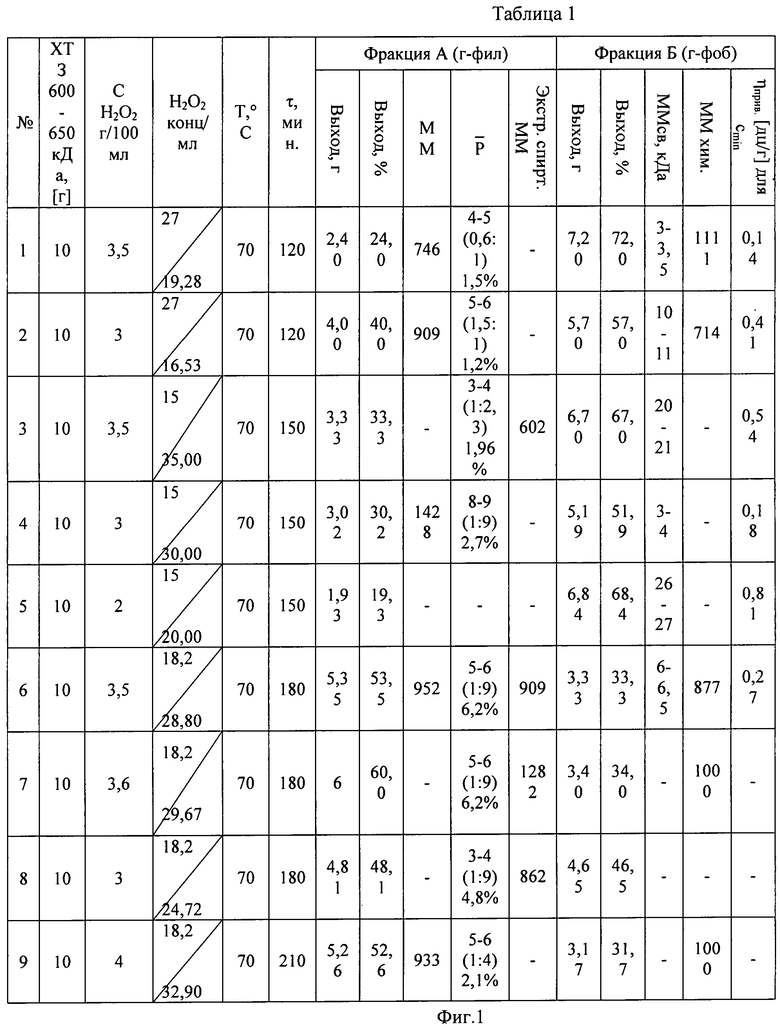
Данные по условиям пероксидного гидролиза хитозана и результатам процесса приведены в таблице (фиг.1).
Пример
10 г (0,06 моль) Хитозана (ХТЗ) (СДА>90%; [η]=29 дл/г, в 2% СН3СООН. Мср=600-650 кДа), гранулометрически подготовленного путем истирания со средним размером частиц 0,05-0,20 мм и 100 мл дистиллированной воды поместили в трехгорлую колбу, снабженную перемешивающим устройством и обратным воздушным холодильником. Образовавшуюся при перемешивании суспензию термостатировали при 40-50°С в течение 30 мин 5,46 г (0,16 моль) пероксида водорода в виде 50 мл водного раствора, добавляли в водную суспензию хитозана и продолжали перемешивание при 70°С в течение 180 мин. Концентрация H 2 O 2 3,6% масс. Жидкостной модуль реакционной системы 15; соотношение реагентов 1:2,6 моль (избыток H2O2) (табл.1). На заключительной стадии обработки визуально отмечено возрастание объема твердых частиц полимера. Образовавшуюся реакционную смесь охладили до 25-30°С и провели фазовое разделение путем фильтрации на воронке Бюхнера с бумажной фильтрующей поверхностью (-0,3-(-0,4) атм). Остаток на фильтре - мягкие, слегка окрашенные в желтый цвет частицы полимергомолога хитозана, не растворимые в воде, промывали дистиллированной водой в объеме 150-200 мл до исчерпывающей экстракции водорастворимой фракции полимерного гидролизата. Выделенную твердую фазу в виде набухших гранул сушили на воздухе при комнатной температуре в течение 48-50 часов. Остаточную влажность (5-6%) в составе полученного полимергомолога определяли путем дополнительного извлечения летучих продуктов из состава полимера в вакууме (-0,6 атм; 50°С). Выделен твердый порошкообразный продукт, окрашенный в слабо желтый цвет, растворимый в 1-2% водной уксусной кислоте, но практически не растворимый в воде. Для последующих аналитических испытаний проводили дополнительную экстракцию низкомолекулярных фракций из состава полимера путем обработки дистиллированной водой в колбе с магнитной мешалкой при 40-45°С в течение 30-45 мин. Выход гидрофобной фракций полимергомологов хитозана - 3,4 г (34% от массы исходного хитозана).
Фильтрат, полученный при фазовом разделении реакционной массы, образовавшегося после пероксидного гидролиза хитозана, помещали в стеклянный кристаллизатор (диаметр 17 см). Удаление летучих продуктов реакции производили в открытой системе при 40-45°С в течение 48-50 часов. Наблюдали образование бесцветного твердого вещества «по визуальной оценке» с аморфной структурой. Полученную смесь гидрофильных олигомеров хитозана экстрагировали 95% этанолом (40-45°С); остаток после экстракции высушивали под вакуумным колоколом (15 мм рт.ст.) при температуре окружающей среды. Выделен бесцветный порошкообразный продукт с признаками кристаллоидной структуры (наблюдение под микроскопом при 16-кратном увеличении). Полученная смесь олигомеров легко растворима в воде при 20-30°С. Для растворов установлена характерно выраженная редуцирующая способность, что подтверждалось реакцией образования серебряного зеркала при добавлении в раствор аммиачного AgNO3. Выход олигомеров составляет 6 г (60% от массы исходного хитозана).
Определение степени полимеризации и Мср синтезированного олигохитозана по редуцирующим звеньям
А. 0,5 г смеси олигомеров хитозана и 0,17 г (0,0012 моль) изониазида (фармакопейный) растворили в 40 мл дист, воды, раствор поместили в плоскодонную колбу с обратным воздушным холодильником. Раствор нагревали при 60-70°С в течение 90 мин. Полученный раствор слабожелтоватого цвета поместили в колбу Вюрца, присоединенную к системе с вакуумом -0,6-(-0,7) атм и вакуумировали при 60-70°С до образования гелеобразного остатка, который экстрагировали 95% этанолом в целях исчерпывающего экстрагирования избытка изониазида. Образовавшуюся при этом двухфазную систему разделили обработкой на фильтре из пористого стекла с бумажной прокладкой (вакуум -0,3 атм). Твердый остаток на поверхности фильтра - смесь гидразонов изониазида на основе олигохитозана. Фильтрат поместили в колбу Вюрца (100 мл) и отогнали этанол при 60-70°С (-0,4 атм). Остаток после удаления этанола (избыток изониазида) растворили в 100 мл дист. воды. Концентрацию изониазида в растворе определяли йодометрическим методом (ГФ XI). 2 г NaHCO3 и 50 мл 0,1 н раствора I2 смешивали с полученным раствором изониазида. Смесь термостатировали при 30°С в условиях светозащиты в течение 30 мин. В полученную смесь при охлаждении ее добавили 20 мл водного раствора HCl 1:2 (нейтрализация бикарбоната натрия). Остаточный I2 титровали 0,1 н раствором тиосульфата натрия. На титрование израсходовано 34 мл раствора Na2S2O3 (K=1, индикатор крахмал). Таким образом, 16 мл йода вступило во взаимодействие с избытком изониазида. Учитывая, что на 0,003428 г изониазида, расходуется 1 мл 0,1 н йода, количество изониазида, вступившего во взаимодействие с олигохитозаном - 0,054 г (0,0039 моль). Количество моль изониазида соответствует количеству редуцирующих звеньев в синтезированном олигохитозане. Мср. полученного олигохитозана составляет 1282. Рассчитанная молекулярная масса олигомера хитозана со степенью полимеризации (Р) равной 7, составляет 1177.
Б. Описание вискозиметрического метода определения Мср. 1,0 г гидрофобной фракции пероксидного гидролиза хитозана растворили в 100 мл 2% водной
СН3СООН. Уточненную концентрацию полученного раствора определяли по среднему значению сухого остатка (1,0 г/дл). Образовавшийся раствор фильтровали.
Для вискозиметрической оценки раствора хитозана в 2% СН3СООН использовали вискозиметр Оствальда (диаметр капилляра 0,9 мм, емкость 10 мл), помещенный в водный термостат с температурой 20°С. Время истечения растворителя и растворов с различной концентрацией (C1-C4) определяли трехкратным тестированием после термостатирования в течение 10 мин. Для этого использовались концентрации растворов, полученные разбавлением исходного раствора 2% уксусной кислотой: C1=0,13, С2=0,05, С3=0,04, С4=0,032 г/дл. Графическим методом определена характеристическая вязкость раствора хитозана ([η] 0,54 дл/г). Для расчета Мср использовали уравнение Марка-Куна-Хаувинка; Мср=20-21 кДа.
Определение степени деацетилирования (СДА)
Приготовили 100 мл водного раствора, содержащего 0,2 г олигомерной фракции хитозана. Потенциометрическое титрование 50 мл полученного раствора производили 0,1Н раством NaOH с использованием рН метра рН-150М с универсальным электродом ЭСКЛ 08М.1 при фиксированной температуре 20°С. По полученным данным строили график. Проведен анализ графической зависимости рН=f (D), где D - количество ммоль NaOH. Результаты анализа полученного графика и соответствующий расчет показывают, что на титрование солевой формы олигохитозана (0,10 г) израсходовано 5,6 ммоль NaOH, что соответствует степени деацетилирования олигохитозана 0,91-0,92 т(91-92%).
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАКОЛОГИЧЕСКОЕ СРЕДСТВО, ОБЛАДАЮЩЕЕ ПРОТИВОТУБЕРКУЛЕЗНОЙ АКТИВНОСТЬЮ | 2011 |
|
RU2469711C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИМЕРНОГО КОНЪЮГАТА ГИДРАЗОНА ИЗОНИКОТИНОВОЙ КИСЛОТЫ | 2010 |
|
RU2454226C1 |
| ПОЛИМЕРНЫЙ КОМПЛЕКС ХИТОЗАНА С ТЕТРАХЛОРИДОМ ПЛАТИНЫ | 2009 |
|
RU2426547C1 |
| СПОСОБ ПОЛУЧЕНИЯ НИЗКОМОЛЕКУЛЯРНОГО ХИТОЗАНА | 2010 |
|
RU2417088C1 |
| Способ получения низкомолекулярного хитозана и олигомеров хитозана | 2016 |
|
RU2627870C1 |
| СПОСОБ ПОЛУЧЕНИЯ ВОДОРАСТВОРИМЫХ ФОРМ ХИТОЗАНА | 2001 |
|
RU2215749C2 |
| СПОСОБ ПОЛУЧЕНИЯ НИЗКОМОЛЕКУЛЯРНОГО ОЛИГОМЕРНОГО ХИТОЗАНА И ЕГО ПРОИЗВОДНЫХ | 2018 |
|
RU2703437C1 |
| СПОСОБ ПОЛУЧЕНИЯ НИЗКОМОЛЕКУЛЯРНОГО ХИТОЗАНА В БЕССОЛЕВОЙ СРЕДЕ ПУТЕМ ФЕРМЕНТАТИВНОЙ ДЕПОЛИМЕРИЗАЦИИ | 2009 |
|
RU2425844C2 |
| АНТИАГРЕГАНТНОЕ СРЕДСТВО | 2017 |
|
RU2647366C1 |
| КОМПОЗИЦИИ ЧАСТИЧНО ДЕАЦЕТИЛИРОВАННЫХ ПРОИЗВОДНЫХ ХИТИНА | 2006 |
|
RU2421467C2 |
Изобретение относится к области химии биополимеров и может быть использовано в медицине, ветеринарии и космецевтике. Способ предусматривает деполимеризацию высокомолекулярного хитозана перекисью водорода. Процесс деполимеризации хитозана проводят в двухфазной системе. Твердой фазой является активированный хитозан с Мср=450-650 кДа и средним размером частиц 0,05-0,20 мм. Жидкая фаза - водный раствор H2O2 с концентрацией H2O2 в реакционной системе 1-7%. Реакцию ведут в течение 120-180 минут при температуре 70°С. Далее осуществляют фазовое разделение образующихся гомологов хитозана путем фильтрации через бумажную или текстильную поверхность полученной реакционной смеси. Образовавшийся фильтрат содержит водорастворимые олигомеры хитозана. Изобретение позволяет количественно регулировать степень превращения исходного высокомолекулярного хитозана в олигомерные и низкомолекулярные структуры его гомологов. 1 табл., 1 пр.
Способ получения водорастворимых олигомерных гомологов хитозана в гетерогенной системе путем деполимеризации высокомолекулярного хитозана перекисью водорода, отличающийся тем, что процесс деполимеризации хитозана проводится в двухфазной системе, твердой фазой которой является активированный хитозан с Мср=450-650 кДа и средним размером частиц 0,05-0,20 мм, жидкая фаза - водный раствор H2O2 с концентрацией Н2О2 в реакционной системе 1-7%, длительностью реакции 120-180 мин и температурой 70°С, далее проводят фазовое разделение образующихся гомологов хитозана путем фильтрации через бумажную или текстильную поверхность полученной реакционной смеси, образовавшийся фильтрат содержит водорастворимые олигомеры хитозана.
| СПОСОБ ПОЛУЧЕНИЯ ВОДОРАСТВОРИМЫХ ФОРМ ХИТОЗАНА | 2001 |
|
RU2215749C2 |
| CN 0101643516 А, 10.02.2010 | |||
| Кипятильник для воды | 1921 |
|
SU5A1 |

