Данное изобретение относится к новым соединениям - фторзамещенным (3R,4R,5S)-5-гуанидино-4-ациламино-3-(пентан-3-илокси)циклогексен-1-карбоновым кислотам и их эфирам-ингибиторам активности нейраминидазы.
Многие микроорганизмы, содержащие нейраминидазу, патогенны по отношению к человеку и животным, таким как птицы, лошади, свиньи, тюлени. Такие патогенные микроорганизмы включают вирус гриппа. Нейраминидаза связана с патогенностью вируса гриппа.
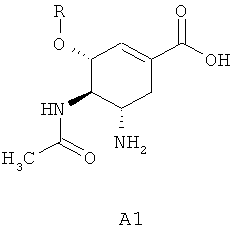
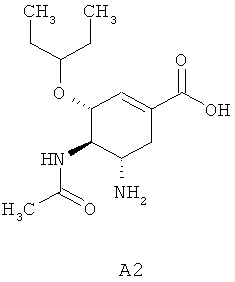
В этой связи новые фторзамещенные (3R,4R,5S)-5-гуанидино-4-ациламино-3-(пентан-3-илокси)циклогексен-1-карбоновые кислоты и их эфиры представляют интерес в качестве лекарственных субстанций для создания новых препаратов, предназначенных для профилактики и лечения гриппа. Известны ингибиторы нейраминидазы - (3R,4R,5S)-5-амино-3-алкилокси-4-ацетиламиноциклогексен-1-карбоновые кислоты A1, причем, наиболее активной из них является (3R,4R,5S)-5-амино-4-ацетиламино-3-(пентан-3-илокси)циклогексен-1-карбоновая кислота A2, которая, как показывают рентгеноструктурные данные для комплекса кислоты с нейраминидазой вируса гриппа, эффективно связывается с активным центром фермента (Oseltamivir Carboxylate) [С.U. Kim, W. Lew, M.A. Williams, et al. J. Am. Chem. Soc. 1997, 119, 681-690].
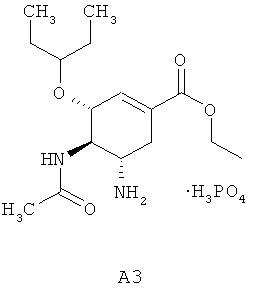
Этиловый эфир озельтамивир карбоксилата A3, известный как озельтамивир фосфат или тамифлю (Oseltamivir Phosphate, Tamiflu) [J.С. Rohloff, К.M. Kent, M.J. Postich, et al. J. Org. Chem. 1998, 63, 4545.], является лекарственным предшественником озельтамивир карбоксилата A2.
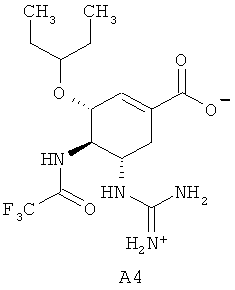
Известна также (3R,4R,5S)-5-гуанидино-4-трифторацетиламино-3-(пентан-3-илокси)циклогексен-1-карбоновая кислота A4, обладающая активностью по отношению к нейраминидазе H5N1 вируса гриппа [Q.-S. Du, R.-B. Huang, Y.-T. Wei, Z.-W. Pa, L.-Q. Du, K.-C. Chou. Fragment-Based Quantitative Structure-Activity Relationship (FB-QSAR) for Fragment-Based Drug Design. J. Comput. Chem. 2008, 30(2), 295-304].
Поиск высокоэффективных противогриппозных лекарственных препаратов, в том числе обладающих повышенной активностью по отношению к резистентным вирусам гриппа, по-прежнему является одним из основных направлений создания новых фармакологических средств для лечения гриппа. В этой связи актуальным является разработка новых противогриппозных субстанций, фармацевтических композиций и лекарственных препаратов, а также способов их получения и применения.
Ниже приведены определения терминов, которые использованы в описании этого изобретения.
«Алкенил» означает алифатическую линейную или разветвленную углеводородную группу, содержащую от 2 до 7 атомов углерода и включающую по крайней мере одну углерод-углеродную двойную связь.
Разветвленная означает, что к линейной алкенильной цепи присоединены один или несколько низших алкильных групп, таких как метил, этил или пропил. Алкильная группа может иметь один или несколько заместителей, например, таких как галоген, алкенилокси, циклоалкил, циано, гидрокси, алкокси, карбокси, алкинилокси, аралкокси, арилокси, арилоксикарбонил, алкилтио, гетероаралкилокси, гетероциклил, гетероциклилалкилокси, алкоксикарбонил, аралкоксикарбонил, гетроаралкилоксикарбонил или Rk aRk+1 aN-, Rk aRk+1 aNC(=O)-, Rk aRk+1 aNC(=S)-, Rk aRk+1 aNSO2-, где Rk a и Rk+1 a независимо друг от друга представляют собой «заместители амино группы», значение которых определено в данном разделе, например, атом водорода, алкил, арил, аралкил, гетероаралкил, гетероциклил или гетероарил, или Rk a и Rk+1 a вместе с атомом N, с которым они связаны, образуют через Rk a и Rk+1 a 4-7 членный гетероциклил или гетеоцикленил. Предпочтительными алкильными группами являются метил, трифторметил, циклопропилметил, циклопентилметил, этил, н-пропил, изо-пропил, н-бутил, трет-бутил, н-пентил, 3-пентил, метоксиэтил, карбоксиметил, метоксикарбнилметил, бензилоксикарбонилметил и пиридилметилоксикарбнилметил. Предпочтительными алкенильными группами являются этенил, пропенил, н-бутенил, изо-бутенил, 3-метилбут-2-енил, н-пентенил, и циклогексилбутенил.
«Алкил» означает алифатическую углеводородную линейную или разветвленную группу с 1-12 атомами углерода в цепи. Разветвленная означает, что алкильная цепь имеет один или несколько «низших алкильных» заместителей. Алкил может иметь один или несколько одинаковых или различных заместителей («алкильных заместителей») включая галоген, алкенилокси, циклоалкил, арил, гетероарил, гетероциклил, ароил, циано, гидрокси, алкокси, карбокси, алкинилокси, аралкокси, арилокси, арилоксикарбнил, алкилтио, гетероарилтио, аралкилтио, арилсульфонил, алкилсульфонилгетероаралкилокси, аннелированный гетероарилциклоалкенил, аннелированный гетероарилциклоалкил, аннелированный гетероарилгетероцикленил, аннелированный гетероарилгетероциклил, аннелированный арилциклоалкенил, аннелированный арилциклоалкил, аннелированный арилгетероцикленил, аннелированный арилгетероциклил, алкоксикарбонил, аралкоксикарбонил, гетероаралкилоксикарбонил или Rk aRk+1 aN-, Rk aRk+1 aNC(=O)-, Rk aRk+1 aNC(=S)-, Rk aRk+1 aNSO2-, где Rk a и Rk+1 a независимо друг от друга представляют собой «заместители амино группы», значение которых определено в данном разделе, например, водород, алкил, арил, аралкил, гетероаралкил, гетероциклил или гетероарил, или Rk a и Rk+1 a вместе с атомом N, с которым они связаны, образуют через Rk a и Rk+1 a 4-7 членный гетероциклил или гетероцикленил. Предпочтительными алкильными группами являются метил, трифторметил, циклопропилметил, циклопентилметил, этил, н-пропил, изо-пропил, н-бутил, трет-бутил, н-пентил, 3-пентил, метоксиэтил, карбоксиметил, метоксикарбонилметил, этоксикарбонилметил, бензилоксикарбонилметил метоксикарбонилметил и пиридилметилоксикарбнилметил. Предпочтительными «алкильными заместителями» являются циклоалкил, арил, гетероарил, гетероциклил, гидрокси, алкокси, алкоксикарбонил, аралкокси, арилокси, алкилтио, гетероарилтио, аралкилтио, алкилсульфонил, арилсульфонил, алкоксикарбонил, аралкоксикарбонил, гетероаралкилоксикарбонил или Rk aRk+1 aN-, Rk aRk+1 aNC(=O)-, аннелированный арилгетероцикленил, аннелированный арилгетероциклил.
«Алкинил» означает алифатическую линейную или разветвленную углеводородную группу, содержащую от 2 до 12 атомов углерода и включающую по крайней мере одну углерод-углеродную тройную связь.
Разветвленная означает, что к линейной алкинильной цепи присоединены один или несколько низших алкильных групп, таких как метил, этил или пропил. Алкильная группа может иметь один или несколько заместителей, например, таких как галоген, алкенилокси, циклоалкил, циано, гидрокси, алкокси, лкинилокси, аралкокси, арилокси, арилоксикарбонил, алкилтио, гетероаралкилокси, гетероциклил, гетероциклилалкилокси, алкоксикарбонил, аралкоксикарбонил, гетроаралкилоксикарбонил или Rk aRk+1 aN-, Rk aRk+1 aNC(=O)-, Rk aRk+1 aNSO2-, где Rk a и Rk+1 a независимо друг от друга представляют собой «заместители амино группы», значение которых определено в данном разделе, например, атом водорода, алкил, арил, аралкил, гетероаралкил, гетероциклил или гетероарил, или Rk a и Rk+1 a вместе с атомом N, с которым они связаны, образуют через Rk a и Rk+1 a 4-7 членный гетероциклил или гетероцикленил. Предпочтительными алкильными группами являются метил, трифторметил, циклопропилметил, циклопентилметил, этил, н-пропил, изо-пропил, н-бутил, трет-бутил, н-пентил, 3-пентил, метоксиэтил, карбоксиметил, метоксикарбнилметил, бензилоксикарбонилметил и пиридилметилоксикарбнилметил. Предпочтительными алкенильными группами являются этенил, пропенил, н-бутенил, изо-бутенил, 3-метилбут-2-енил, н-пентенил, бута-1,3-диин и гекса-1,3,5-триин.
«Гидрат» означает стехиометрическую или нестехиометрическую композицию соединения или его соли с водой.
«Лекарственное начало» (лекарственная субстанция лекарственное вещество, drug-substance) означает физиологически активное вещество синтетического или иного (биотехнологического, растительного, животного, микробного и прочего) происхождения, обладающее фармакологической активностью и являющееся активным началом фармацевтической композиции, используемой для производства и изготовления лекарственного препарата (средства).
«Лекарственное средство (препарат)» - вещество (или смесь веществ в виде фармацевтической композиции), в виде таблеток капсул инъекций, мазей и др. готовых форм предназначенное для восстановления, исправления или изменения физиологических функций у человека и животных, а также для лечения и профилактики болезней, диагностики, анестезии, контрацепции, косметологии и прочего.
Нейраминидаза (сиалидаза, ацилнейраминил гидролаза и ЕС 3.2.1.18) является обычным для животных и ряда микроорганизмов ферментом. Она представляет собой гликогидролазу, которая отщепляет альфакетосидикально связанные сиаловые кислоты от гликопротеинов, гликолипидов и олигосахаридов. Многие из микроорганизмов, содержащих нейраминидазу, патогенны по отношению к человеку и другим животным, включая птицу, лошадей, свиней и тюленей. Такие патогенные организмы включают вирус гриппа. Нейраминидаза связана с патогенностью вируса гриппа. Предположительно она содействует элюированию вновь синтезированных вирионов из инфицированных клеток и движению вируса (благодаря ее гидролазной активности) через слизь респираторного тракта.
«Фармацевтическая композиция» обозначает композицию, включающую в себя соединение формулы 1 и, по крайней мере, один из компонентов, выбранных из группы, состоящей из фармацевтически приемлимых и фармакологически совместимых наполнителей, растворителей, разбавителей, носителей, вспомогательных, распределяющих и воспринимающих средств, средств доставки, таких как консерванты, стабилизаторы, наполнители, измельчители, увлажнители, эмульгаторы, суспендирующие агенты, загустители, подсластители, отдушки, ароматизаторы, антибактериальные агенты, фунгициды, лубриканты, регуляторы пролонгированной доставки, выбор и соотношение которых зависит от природы и способа назначения и дозировки. Примерами суспендирующих агентов являются этоксилированный изостеариловый спирт, полиоксиэтилен, сорбитол и сорбитовый эфир, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакант, а также смеси этих веществ. Защита от действия микроорганизмов может быть обеспечена с помощью разнообразных антибактериальных и противогрибковых агентов, например, таких как, парабены, хлорбутанол, сорбиновая кислота и подобные им соединения. Композиция может включать также изотонические агенты, например, сахара, хлористый натрий и им подобные. Пролонгированное действие композиции может быть обеспечено с помощью агентов, замедляющих абсорбцию активного начала, например, моностеарат алюминия и желатин. Примерами подходящих носителей, растворителей, разбавителей и средств доставки являются вода, этанол, полиспирты, а также их смеси, растительные масла (такие, как оливковое масло) и инъекционные органические сложные эфиры (такие, как этилолеат). Примерами наполнителей являются лактоза, молочный сахар, цитрат натрия, карбонат кальция, фосфат кальция и им подобные. Примерами измельчителей и распределяющих средств являются крахмал, алгиновая кислота и ее соли, силикаты. Примерами лубрикантов являются стеарат магния, лаурилсульфат натрия, тальк, а также полиэтиленгликоль с высоким молекулярным весом. Фармацевтическая композиция для перорального, сублингвального, трансдермального, внутримышечного, внутривенного, подкожного, местного или ректального введения активного начала, одного или в комбинации с другим активным началом, может быть введена животным и людям в стандартной форме введения, в виде смеси с традиционными фармацевтическими носителями. Пригодные стандартные формы введения включают пероральные формы, такие как таблетки, желатиновые капсулы, пилюли, порошки, гранулы, жевательные резинки и пероральные растворы или суспензии, сублингвальные и трансбуккальные формы введения, аэрозоли, имплантаты, местные, трансдермальные, подкожные, внутримышечные, внутривенные, интраназальные или внутриглазные формы введения и ректальные формы введения.
«Фармацевтически приемлемая соль» означает относительно нетоксичные органические и неорганические соли кислот и оснований, заявленных в настоящем изобретении. Эти соли могут быть получены in situ в процессе синтеза, выделения или очистки соединений или приготовлены специально. В частности, соли оснований могут быть получены специально, исходя из очищенного свободного основания заявленного соединения и подходящей органической или неорганической кислоты. Примерами полученных таким образом солей являются гидрохлориды, гидробромиды, сульфаты, бисульфаты, фосфаты, нитраты, ацетаты, оксалаты, валериаты, олеаты, пальмитаты, стеараты, лаураты, бораты, бензоаты, лактаты, тозилаты, цитраты, малеаты, фумараты, сукцинаты, тартраты, мезилаты, малонаты, салицилаты, пропионаты, этансульфонаты, бензолсульфонаты, сульфаматы и им подобные (Подробное описание свойств таких солей дано в Berge S.M., et al., "Pharmaceutical Salts" J. Pharm. Sci. 1977, 66: 1-19). Соли заявленных кислот также могут быть специально получены реакцией очищенной кислоты с подходящим основанием, при этом могут быть синтезированы соли металлов и аминов. К металлическим относятся соли натрия, калия, кальция, бария, цинка, магния, лития и алюминия, наиболее желательными из которых являются соли натрия и калия. Подходящими неорганическими основаниями, из которых могут быть получены соли металлов, являются гидроксид, карбонат, бикарбонат и гидрид натрия, гидроксид и бикарбонат калия, поташ, гидроксид лития, гидроксид кальция, гидроксид магния, гидроксид цинка. В качестве органических оснований, из которых могут быть получены соли заявленных кислот, выбраны амины и аминокислоты, обладающие достаточной основностью, чтобы образовать устойчивую соль, и пригодные для использования в медицинских целях (в частности, они должны обладать низкой токсичностью). К таким аминам относятся аммиак, метиламин, диметиламин, триметиламин, этиламин, диэтиламин, триэтиламин, бензиламин, дибензиламин, дициклогексиламин, пиперазин, этилпиперидин, трис(гидроксиметил)аминометан и подобные им. Кроме того, для солеобразования могут быть использованы гидроокиси тетраалкиламмония, например, такие как, холин, тетраметиламмоний, тетраэтиламмоний и им подобные. В качестве аминокислот могут быть использованы основные аминокислоты - лизин, орнитин и аргинин.
Авторы обнаружили новые ингибиторы нейраминидазы, представляющие собой неизвестные ранее фторзамещенные (3R,4R,5S)-4-ациламино-5-гуанидино-3-(пентан-3-илокси)циклогексен-1-карбоновые кислоты и их эфиры общей формулы 1 и их фармацевтически приемлемые соли и/или гидраты.
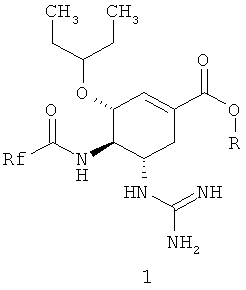
где R представляет собой водород, C1-C5алкил, C2-C5алкенил или C2-C5алкинил, необязательно замещенные C3-C6циклоалкилом, фенилом, пиридилом, C1-C3алкокси; Rf представляет собой CH2F или CHF2 радикал.
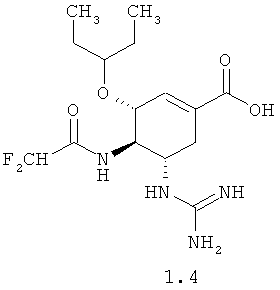
Новые соединения общей формулы 1, как установили авторы, неожиданно оказались более эффективными ингибиторами нейраминидазы вирусов гриппа, чем известные аналоги озельтамивир карбоксилат A2 и (3R,4R,5S)-5-гуанидино-4-трифторациламино-3-(пентан-3-илокси)-циклогексен-1-карбоновая кислота формулы A4, в том числе более активными по отношению к нейраминидазе озельтамивир резистентного вируса гриппа А / Владивосток / 16/09 (H1N1). Так, например, озельтамивир карбоксилат A2 и (3R,4R,5S)-5-гуанидино-4-(2-трифторацетиламино)-3-(пентан-3-илокси)-циклогексен-1-карбоновая кислота A.4, показали нейраминидазную активностью по отношению к вирусу A / Калифорния / 04/09 ИК50=0,8 nM (для A2) и ИК50=80 nM (для A4) соответственно. На нейраминидазе озельтамивир резистентного вируса A / Владивосток / 16/09 кислоты A2 и A.4 имеют слабую активность (ИК50=830 nM для A2 и ИК50>1000 nM для A.4). В то же время неизвестная ранее (3R,4R,5S)-5-гуанидино-4-фторацетиламино-3-(пентан-3-илокси)циклогексен-1-карбоновая кислота 1.1 имеет нейраминидазную активность по отношению к вирусу A / Калифорния / 04/09, соответствующую ИК50=0,2 nM (т.е. в 4 раза активнее A2 и в 400 раз активнее A4), а по отношению к вирусу A / Владивосток / 16/09 - новая кислота 1.1 имеет ИК50=4 nM, т.е. в 207 раза активнее A2 и, совершенно неожиданно, более чем в 250 раз активнее своего известного 4-(2-трифторацетильного) аналога. Более высокую нейраминидазную активность, чем аналоги A2 и A4 имеет и (3R,4R,5S)-5-гуанидино-4-дифторацетиламино-3-(пентан-3-илокси)циклогексен-1-карбоновая кислота 1.4: по отношению к вирусу A / Калифорния / 04/09, соответствующую ИК50=0,3 nM (т.е. в 2,7 раза активнее A2 и в 267 раз активнее A4); по отношению к вирусу A / Владивосток / 16/09 - новая кислота 1.4 имеет ИК50=7 nM (т.е. в 118 раза активнее A2 и более чем в 140 раз активнее A4).
Согласно данному изобретению предпочтительными являются соединения общей формулы 1, в которых R представляет собой водород, метил или этил.
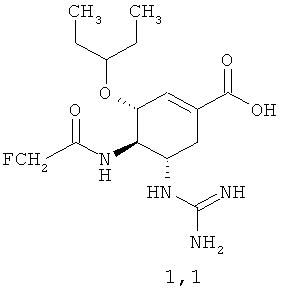
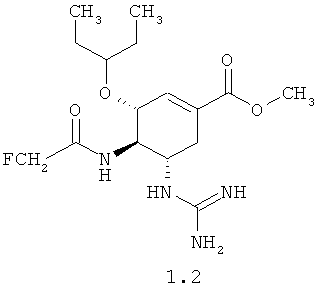
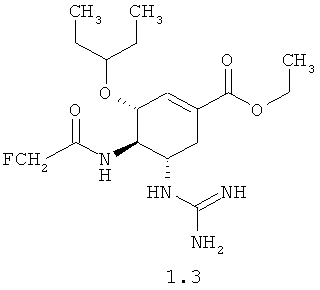
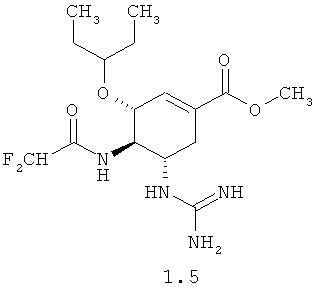
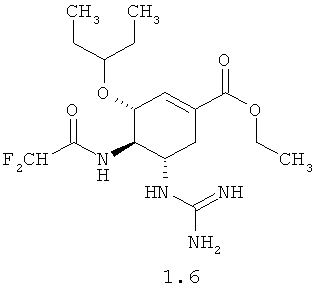
Согласно данному изобретению более предпочтительными соединениями общей формулы 1 являются: (3R,4R,5S)-5-гуанидино-4-фторацетиламино-3-(пентан-3-илокси)циклогексен-1-карбоновая кислота 1.1 и ее метиловый эфир 1.2, и этиловый 1.3 эфир, (3R,4R,5S)-5-гуанидино-4-дифторацетиламино-3-(пентан-3-илокси)циклогексен-1-карбоновая кислота 1.4 и ее метиловый эфир 1.5, и этиловый 1.6 эфир.
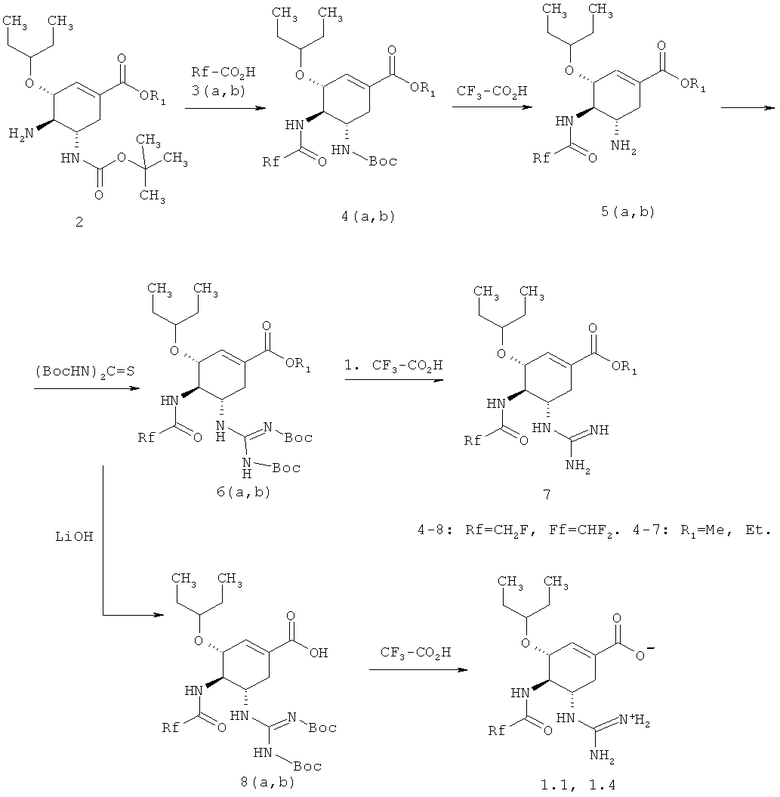
Ингибиторы нейраминидазы вирусов гриппа общей формулы 1 получают, исходя из алкил 4-амино-5-(трет-бутоксикарбониламино)-3-(пентан-3-илокси)циклогексен-1-карбоксилата общей формулы 2 по нижеследующей схеме.
Новые ингибиторы нейраминидазы вирусов гриппа, представляющие собой неизвестные ранее (3R,4R,5S)-4-(2-фторацетиламино)- и (3R,4R,5S)-4-(2,2-дифторацетиламино)-5-гуанидино-3-(пентан-3-илокси)циклогексен-1-карбоновые кислоты и их эфиры общей формулы 1 и их фармацевтически приемлемые соли проявили также высокую противогриппозную активность на животных моделях гриппозной пневмонии.
Антинейраминидазную активность соединений определяли по методу, описанному в [Who Collaborating Centre for Reference & Research on Influenza, Australia, Standard Operating Procedure WHO - 025. Reviewed by: Aeron Hurt, Senior Scientist Review Date: 13/3/2009].
Согласно данному изобретению новые соединения общей формулы 1 представляют собой лекарственное начало для приготовления фармацевтической композиции и готовых лекарственных форм для профилактики и лечения гриппа у теплокровных животных и людей.
Предметом данного изобретения является фармацевтическая композиция, включающая в качестве лекарственного начала соединения общей формулы 1 или их фармацевтически приемлемые соли в терапевтически эффективном количестве.
Фармацевтические композиции могут включать фармацевтически приемлемые эксципиенты. Под фармацевтически приемлемым экспициентами подразумеваются применяемые в сфере фармацевтики разбавители, вспомогательные агенты и/или носители. Фармацевтическая композиция наряду с соединением общей формулы 1 или их фармацевтически приемлемой солью и/или гидратом по настоящему изобретению может включать и другие активные субстанции, в том числе, обладающие противогриппозной активностью, при условии, что они не вызывают нежелательных эффектов.
При необходимости использования фармацевтической композиции по настоящему изобретению в клинической практике она может смешиваться с традиционными фармацевтическими носителями.
Носители, используемые в фармацевтических композиций по настоящему изобретению, представляют собой носители, которые применяются в сфере фармацевтики для получения распространенных форм, в том числе: в пероральных формах используются связующие вещества, смазывающие агенты, дезинтеграторы, растворители, разбавители, стабилизаторы, суспендирующие агенты, бесцветные агенты, корригенты вкуса; в формах для инъекций используются антисептические агенты, солюбилизаторы, стабилизаторы; в местных формах используются основы, разбавители, смазывающие агенты, антисептические агенты.
Предметом данного изобретения является способ получения фармацевтической композиции смешением с инертным наполнителем и/или растворителем, по крайней мере, одного лекарственного начала (субстанции) общей формулы 1 или его фармацевтически приемлемой соли в терапевтически эффективном количестве.
Предметом данного изобретения является также лекарственное средство, обладающее противогриппозной активностью, в форме таблеток, капсул или инъекций, помещенных в фармацевтически приемлемую упаковку, предназначенное для профилактики лечения гриппа у людей и теплокровных животных, включающее в свой состав противогриппозное лекарственное начало (субстанцию) общей формулы 1 или фармацевтическую композицию, включающую новое лекарственное начало общей формулы 1 в терапевтически эффективном количестве.
Предметом данного изобретения являются также терапевтические коктейли для лечения гриппа, включающие в качестве одного из компонентов новое лекарственное средство или новую фармацевтическую композицию, содержащих в качестве активного компонента, по крайней мере, одно соединение общей формулы 1 или его фармацевтически приемлемую соль и/или гидрат.
Терапевтический коктейль для лечения гриппа, наряду с лекарственным средством по данному изобретению, может включать другие известные препараты, предназначенные для лечения гриппа, или препараты, усиливающие иммунную систему пациента.
В соответствии с данным изобретением способ профилактики и лечения гриппа у животных и людей заключается во введении пациенту нового лекарственного средства, новой фармацевтической композиции или нового терапевтического коктейля.
Лекарственные средства могут вводиться через ингалятор, перорально или парентерально (например, внутривенно, подкожно, внутрибрюшинно или местно). Клиническая дозировка средства общей формулы 1 у пациентов может корректироваться в зависимости от терапевтической эффективности и биодоступности активных ингредиентов в организме, скорости их обмена и выведения из организма, а также в зависимости от возраста, пола и стадии заболевания пациента, при этом суточная доза у взрослых обычно составляет 10~500 мг, предпочтительно - 50~300 мг. Поэтому во время приготовления из фармацевтической композиции лекарственного средства по настоящему изобретению в виде единиц дозировки необходимо учитывать вышеназванную эффективную дозировку, при этом каждая единица дозировки препарата должна содержать 10~500 мг средства общей формулы 1 предпочтительно - 50~300 мг. В соответствии с указаниями врача или фармацевта данные препараты могут приниматься несколько раз в течение определенных промежутков времени (предпочтительно - от одного до шести раз).
Предметом данного изобретения является также способ ингибирования активности нейраминидазы в естественных условиях, в том числе нейраминидазы гриппа, включающий стадию контактирования соединения общей формулы 1 и нейраминидазы.
Данное изобретение иллюстрируется, но не ограничивается следующими примерами.
Пример 1. Получение (3R,4R,5S)-этил 4-(2,2-дифторацетамидо)-5-гуанидино-3-(пентан-3-илокси)циклогексен-1-карбоксилата мезилата 1.6·CH3SO3H. (3R,4R,5S)-Этил 4-амино-5-(трет-бутоксикарбониламино)-3-(пентан-3-илокси)циклогексен-1-карбоксилат (2 г, 0.0054 моль, 1 экв), 1H-бензо[d][1,2,3]триазол-1-ол (0.867 г, 0.0065 моль, 1.2 экв), N1-((этилимино)метилен)-N2,N2-диметилэтан-1,2-диамин гидрохлорид (1.239 г, 0.0065 моль, 1.2 экв) и диизопропилэтиламин (2.212 г, 0.0178 моль, 3.3 экв) растворяют в 20 мл ТГФ и по каплям добавляют 2,2-дифторуксусную кислоту 3b (0.624 г, 0.0065 моль, 1.2 экв). Реакционную массу перемешивают при комнатной температуре в течение четырех часов. Затем растворители отгоняют в вакууме, оставшееся масло растворяют в этилацетате, промывают 5% раствором NaHCOa, сушат над Na2S04, фильтруют и сушат в вакууме. Выход (3R,4R,5S)-этил 5-(трет-бутоксикарбониламино)-4-(2,2-дифторацетамидо)-3-(пентан-3-илокси)циклогексен-1-карбоксилата 4b составляет 89% (2.15 г). LCMS (M+H): Найдено 449; Вычислено 448.51. Полученный продукт 4b (2.15 г, 0.0048 моль) растворяют в 20 мл 10%-ного раствора трифторуксусной кислоты в хлористом метилене и перемешивают при комнатной температуре в течение 12 часов. Затем растворители отгоняют в вакууме, оставшееся масло растворяют в этилацетате, промывают 5% раствором NaHCO3, сушат над Na2SO4, фильтруют и сушат в вакууме. Выход продукта составляет 96% (1.605 г). Дополнительную очистку проводят методом колоночной хроматографии - элюент этилацетат/ТГФ или перекристалицацией из гексана. Получают (3R,4R,5S)-этил 5-амино-4-(2,2-дифторацетамидо)-3-(пентан-3-илоки)циклогексен-1-карбоксилат 5b, который растворяют в хлористом метилене и добавляют эквивалентное количество метансульфоновой кислоты. Через 10 минут растворитель отгоняют, полученный продукт промывают гексаном и сушат в вакууме. Выход мезилата 5b·CH3SO3H 90%. LCMS (M+H): Найдено 349; Вычислено 348.39. 1H ЯМР (DMSO-d6), 400 МГц: 8.91 (д, J=13,2 Гц, 1H), 7.83 (уш, 3H), 6.74 (с, 1H), 6.22 (т, J=54 Гц, 1H), 4.31 (д, J=8.4 Гц, 1H), 4.16 (к, J=7,2 Гц, 2H), 3.45 (м, 1H), 3.15 (д.д, J1=11.2 Гц, J2=8,8 Гц, 1H), 2.59 (д.д., J1=18 Гц, J2=6 Гц, 1H), 2.38 (м, 1H), 2.31 (с, 3H), 1.66 (м, 1H), 1.57 (м, 1H), 1.47 (м, 1H), 1.39 (м, 1H), 1.22 (т, J=7.6 Гц, 3H), 0.891 (т, J=7.2 Гц, 3H), 0.842 (т, J=7.2 Гц, 3H). (3R,4R,5S)-Этил 5-амино-4-(2,2-дифторацетамидо)-3-(пентан-3-илоки)циклогексен-1-карбоксилата 5b (1.6 г; 0.004598 моль; 1 экв) растворяют в 16 мл ДМФА и охлаждают в ледяной бане. Затем добавляют триэтиламин (2.57 г; 0.0253 моль; 5.5 экв), N,N'-ди-boc-тиомочевину (0.00505 моль; 1.35 г; 1.1 экв) и хлорид ртути(II) (0.0055 моль; 1.49 г; 1.2 экв). Полученную смесь перемешивают при охлаждении в ледяной бане в течение 1.5 часов. По окончании реакции осадок отфильтровывают через селит, ДМФА отгоняют в вакууме, полученное масло растворяют в этилацетате, промывают 5% раствором NaHCO3, сушат над Na2SO4, фильтруют и сушат в вакууме. Получают 1.82 г (67%) (3R,4R,5S)-этил 5-((Z)-2,3-бис(трет-бутоксикарбонил)гуанидино)-4-(2,2-дифторацетамидо)-3-(пентан-3-илокси)циклогксен-1-карбоксилата 6b. LCMS (M+H): Найдено 591; Вычислено 590.67. (3R,4R,5S)-этил 5-((Z)-2,3-бис(трет-бутоксикарбонил)гуанидино)-4-(2,2-дифторацетамидо)-3-(пентан-3-илокси)циклогексен-1-карбоксилат 6b (1.82 г; 0.00308 моль) растворяют в 20 мл 10%-ного раствора трифторуксусной кислоты в хлористом метилене и перемешивают при комнатной температуре в течение 12 часов. Затем растворители отгоняют в вакууме, оставшееся масло растворяют в этилацетате, промывают 5% раствором NaHCO3, сушат над Na2SO4, фильтруют и сушат в вакууме. Получают 1.1 г (92%) (3R,4R,5S)-этил 4-(2,2-дифторацетамидо)-5-гуанидино-3-(пентан-3-илокси)циклогексен-1-карбоксилата 1.6 Полученный продукт растворяют в хлористом метилене и добавляют эквивалентное количество метансульфоновой кислоты. Через 10 минут растворитель отгоняют, полученный продукт промывают гексаном и сушат в вакууме. Получают (3R,4R,5S)-этил 4-(2,2-дифторацетамидо)-5-гуанидино-3-(пентан-3-илокси)циклогексен-1-карбоксилата мезилата 1.6·CH3SO3H, выход 95%. LCMS (M+H): Найдено 391; Вычислено 390.43. 1H ЯМР (DMSO-d6), 400 МГц: 8.67 (д, J=8,4 Гц, 1H), 7.63 (д, J=9.6 Гц, 1H), 6.65 (с, 1H), 6.27 (т, J=53.6 Гц, 1H), 4.14 (к, J=7,2 Гц, 2H), 4.12 (м, 7Н), 3.54 (м, 1H), 3.41 (м, 1H), 2.6 (м, 1H), 2.36 (с, 3H), 1.66 (м, 1H), 1.44 (м, 4Н), 1.22 (т, J=7.2 Гц, 3H), 0.856 (т, J=7.6 Гц, 3H), 0.795 (т, J=7.6 Гц, 3H).
Используя в качестве исходного соединения 2 (3R,4R,5S)-аллил-4-амино-5-(трет-бутоксикарбониламино)-3-(пентан-3-илокси)циклогексен-1-карбоксилат и (3R,4R,5S)-проп-2-инил-4-амино-5-(трет-бутоксикарбониламино)-3-(пентан-3-илокси)циклогексен-1-карбоксилат аналогично получали аллиловый эфир (3R,4R,5S)-5-гуанидино-4-(2,2-дифторацетамидо)-3-(пентан-3-илокси)циклогексен-1-карбоновой кислоты 1.7 (LCMS (M+H): найдено 354; вычислено 353.44, по отношению к вирусу A / Калифорния / 04/09 ИК50<1 nM) и проп-2-иниловый эфир (3R,4R,5S)-5-гуанидино-4-(2,2,-дифторацетамидо)-3-(пентан-3-илокси)циклогексен-1-карбоновой кислоты 1.8 (LCMS (M+H): найдено 352; вычислено 351.42, по отношению к вирусу A / Калифорния / 04/09 ИК50<1 nM).
Используя в качестве исходного соединения соответствующий (3R,4R,5S)-4-амино-5-(трет-бутоксикарбониламин)-3-(пентан-3-илокси)циклогексен-1-карбоксилат 2 получали:
2-циклогексилэтиловый эфир (3R,4R,5S)-5-гуанидино-4-(2,2-дифторацетамидо)-3-(пентан-3-илокси)циклогексен-1-карбоновой кислоты 1.9 (LCMS (M+H): найдено 460; вычислено 459.55, по отношению к вирусу A / Калифорния / 04/09 ИК50<1 nM);
2-фенилэтиловый эфир (3R,4R,5S)-5-гуанидино-4-(2,2-дифторацетамидо)-3-(пентан-3-илокси)циклогексен-1-карбоновой кислоты 1.10 (LCMS (M+H): найдено 454; вычислено 453.51, по отношению к вирусу A / Калифорния / 04/09 ИК50<1 nM);
2-пиридин-3-илэтиловый эфир (3R,4R,5S)-5-гуанидино-4-(2,2-дифторацетамидо)-3-(пентан-3-илокси)циклогексен-1-карбоновой кислоты 1.11 (LCMS (M+H): найдено 455; вычислено 454.49, по отношению к вирусу A / Калифорния / 04/09 ИК50<1 nM);
2-метоксиэтиловый эфир (3R,4R,5S)-5-гуанидино-4-(2,2-дифторацетамидо)-3-(пентан-3-илокси)циклогексен-1-карбоновой кислоты 1.12 (LCMS (M+H): найдено 422; вычислено 421.46, по отношению к вирусу A / Калифорния / 04/09 ИК50<1 nM).
Пример 2. (3R,4R,5S)-этил 4-(2-фторацетамидо)-5-гуанидино-3-(пентан-3-илокси)циклогексен-1-карбоксилата мезилат 1.3·CH3SO3H получают по методике, приведенной в примере 1 с использованием монофторуксусной кислоты в качестве ацилирующего агента. LCMS (M+H): Найдено 373; Вычислено 372.44.
Пример 3. (3R,4R,5S)-4-(2,2-дифторацетамидо)-5-гуанидино-3-(пентан-3-илокси)циклогексен-1-карбоновая кислота 1.4. К раствору 250 мг (3R,4R,5S)-этил 5-((Z)-2,3-бис(трет-бутоксикарбонил)гуанидино)-4-(2,2-дифторацетамидо)-3-(пентан-3-илокси)циклогексен-1-карбоксилата 6b в 5 мл диоксана добавляют 2,5 мл 5%-го раствора гидроксида лития и перемешивают реакционную массу при комнатной температуре в течение 45 минут. Затем гидроксид лития пассивируют добавляя 300 мкл уксусной кислоты, растворители отгоняют в вакууме. Полученный осадок экстрагируют изопропиловым спиртом, экстракт сушат над Na2SO4 и в вакууме. Получают 200 мг (84%) (3R,4R,5S)-5-((Z)-2,3-бис(трет-бутоксикарбонил)гуанидино)-4-(2,2-дифторацетамидо)-3-(пентан-3-илокси)циклогексен-1-карбоксильной кислоты 8b. LCMS (M+H): Найдено 563; Вычислено 562.62. Растворяют 200 мг полученной кислоты 8b в 2 мл 10%-ного раствора трифторуксусной кислоты в хлористом метилене и перемешивают при комнатной температуре в течение 12 часов. Затем растворители отгоняют в вакууме. (3R,4R,5S)-4-(2,2-дифторацетамидо)-5-гуанидино-3-(пентан-3-илокси)циклогексен-1-карбоновую кислоту 1.4 выделяют методом HPLC. LCMS (M+H): Найдено 363; Вычислено 362.28. 1H ЯМР (DMSO-d6), 400 МГц: 8.68 (д, J=8,4 Гц, 1H), 7.6д (д, J=10 Гц, 1H), 7.26 (уш, 2H), 6.91 (уш, 2H), 6.63 (с, 1H), 6.27 (т, J=53.6 Гц, 1H), 4.18 (д, J=8 Гц, 2H), 4.09 (м, 1H), 3.54 (к, J=10 Гц, 1H), 3.39 (м, 2H), 2.57 (д.д, J1=18 Гц, J2=6 Гц, 1H), 2.31 (м, 1H), 1.44 (м, 4Н), 0.85 (т, J=8 Гц, 3H), 0.795 (т, J=7.6 Гц, 3H).
Пример 4. (3R,4R,5S)-4-(2-фторацетамидо)-5-гуанидино-3-(пентан-3-илокси)циклогексен-1-карбоновую кислоту 1.1 получают аналогично примеру 3. LCMS (M+H): Найдено 345; Вычислено 345. 1H ЯМР (DMSO-d6), 400 МГц: 8.16 (д, J=10 Гц, 1H), 7.56 (д, J=9.6 Гц, 1H), 6.64 (с, 1H), 4.80 (д, J=47.3 Гц, 2H), 4.21 (д, J=8,4 Гц, 1H), 3.89 (к, J=10.4 Гц, 1H), 3.71 (м, 1H), 2.67 (м, 1H), 2.25 (м, 1H), 1.42 (м, 4Н), 0.85 (т, J=7.2 Гц, 3H), 0.78 (т, J=7.2 Гц, 3H).
Пример 5. (3R,4R,5S)-Метил 4-(2-фторацетамидо)-5-гуанидино-3-(пентан-3-илокси)циклогексен-1-карбоксилата мезилат 1.2·CH3SO3H был получен по методике, приведенной в примере 1 с использованием в качестве исходного (3R,4R,5S)-метил 4-амино-5-(трет-бутоксикарбониламино)-3-(пентан-3-илокси)циклогексен-1-карбоксилата и монофторуксусной кислоты в качестве ацилирующего агента. LCMS (M+H): Найдено 359; Вычислено 358.42.
Пример 6. (3R,4R,5S)-Метил 4-(2,2-дифторацетамидо)-5-гуанидино-3-(пентан-3-илокси)циклогексен-1-карбоксилата мезилат 1.5·CH3SO3H был получен по методике, приведенной в примере 1 с использованием в качестве исходного (3R,4R,5S)-метил 4-амино-5-(трет-бутоксикарбониламино)-3-(пентан-3-илокси)циклогексен-1 карбоксилата и дифторуксусной кислоты в качестве ацилирующего агента. LCMS (M+H): Найдено 373; Вычислено 372.44.
Пример 7. Получение фармацевтической композиции в форме таблеток. Смешивают 1600 мг крахмала, 1600 мг измельченной лактозы, 400 мг талька и 1000 мг (3R,4R,5S)-4-(2-фторацетамидо)-5-гуанидино-3-(пентан-3-илокси)циклогексен-1-карбоновой кислоты 1.1. Полученный брусок измельчают в гранулы и просеивают через сита, собирая гранулы размером 14-16 меш. Полученные гранулы таблетируют в подходящую форму таблетки весом 560 м г каждая.
Пример 8. Получение фармацевтической композиции в форме капсул. Тщательно смешивают (3R,4R,5S)-4-(2-фторацетамидо)-5-гуанидино-3-(пентан-3-илокси)циклогексен-1-карбоновой кислоты 1.1 с порошком лактозы в соотношении 2:1. Полученную порошкообразную смесь упаковывают по 300 мг в желатиновые капсулы подходящего размера.
Пример 9. Получение фармацевтической композиции для внутримышечных, внутрибрюшинных или подкожных инъекций. Смешивают 500 мг (3R,4R,5S)-4-(2-фторацетамидо)-5-гуанидино-3-(пентан-3-илокси)циклогексен-1-карбоновой кислоты 1.1 с 300 мг хлорбутанола, 2 мл пропиленгликоля и 100 мл инъекционной воды. Полученный раствор фильтруют и помещают по 1 мл в ампулы, которые запаивают.
Пример 10. Определение активности соединений общей формулы 1 по отношению к нейраминидазе вирусов гриппа. В предварительных экспериментах были определены рабочие разведения для штаммов аллантоисных вирусов гриппа A / California / 07/09 (H1N1) и А / Аичи / 2/69 (H3N2), а также для полученного в культуре клеток озельтамивир резистентного вируса гриппа А / Владивосток / 16/09 (H1N1). Для этого было приготовлено по 60 мкл каждого двухкратно разведеного вируса на реакционной буферной смеси (РБС, 50 мМ MES, 5 мМ CaCl2, pH 6,5) в 96-луночном планшете с круглым дном. Из этого планшета по 50 мкл двухкратно разведеного вируса было перенесено в 96-луночные планшеты с плоским дном для измерения флуоресценции (FluoroNunc, черные, кат. No.237105), далее к ним был добавлен равный объем субстратного буфера (СБ, 12,5 мМ 2'-(4-метилумбеллиферил)-α-D-N-ацетилнейраминовой кислоты, Sigma, на 40 мМ ацетатного буфера pH=5,8). В качестве контроля были использованы лунки, к которым вместо вируса было добавлено по 50 мкл РБС. После инкубации планшета при 37° в течение 1 часа при слабом покачивании к каждой лунке планшета было добавлено по 100 мкл стоп-раствора (2.225 mL 0.824 M NaOH в 11.0 mL этанола), после чего было проведено измерение флуоресценции на приборе Varioskan Flach (Thermo Scientific) при λex=360 нм и λem=448 нм. Для дальнейшей работы были выбраны те разведения вирусов, которые соответствовали середине линейного участка кривой зависимости разведения вируса от величины флуоресценции. Для определения антинейраминидазной активности соединений в 96-луночные планшеты с плоским дном для измерения флуоресценции (FluoroNunc, черные, кат. No.237105) в лунки рядов от B до H было добавлено по 50 мкл приготовленных на РБС разведении соединений общей формулы 1 и аналогов формулы A2 и формулы A4 (концентрации 0,03; 0,3; 3; 30; 300; 3000; 30000 нм - каждая концентрация на ряд). В качестве вирусного контроля были использованы лунки ряда А, к которым было добавлено по 50 мкл РБС. Далее к соответствующим лункам было добавлено 50 мкл выбранных рабочих разведении каждого из вирусов на РБС. В качестве контроля были использованы лунки, к которым вместо вируса был добавлен такой же объем РБС. После перемешивания и инкубации при комнатной температуре в течение 45 минут ко всем лункам был добавлен равный объем СБ. После перемешивания и инкубации планшета при 37°C в течение 1 часа к каждой лунке планшета было добавлено по 100 мкл стоп-раствора. Измерение флуоресценции было проведено на приборе Varioskan Flach (Thermo Scientific) при λex=360 нм и λem=448 нм. Все определения проводились не менее чем в двух повторах (две лунки планшета). Процент ингибирования нейраминидазной активности изучаемым веществом общей формулы 1 определяли по формуле: процент ингибирования = 100 - (УЕФ опыта - УЕФ контроля / УЕФ вирусного контроля в отсутствии соединения - УЕФ контроля). Концентрация препарата, уменьшающая значение величины УЕФ на 50%, принималась за ингибирующую концентрацию 50 (ИК50).
Пример 11. Изучение противогриппозной активности соединений общей формулы 1 (1.1., 1.3., 1.4 и 1.6) на модели гриппозной пневмонии мышей. Предварительно взвешенные мыши (самки нелинейные, средний вес 12-15 г) инфицировались интраназально под легким эфирным наркозом вирусом гриппа А / Аичи / 2/69 (H3N2) (10 ЛД50 в 50 мкл). В предварительном опыте было проведено определение ЛД50 путем титрования аллантоисного вируса на таких же мышах, которые затем использовались в основном опыте. Была использована следующая схема лечения соединениями: за 24 часа до инфицирования, за 1 час до инфицирования, через 24 часа и далее 1 раз в день в течение 5 дней. Для перорального введения использовали одноразовый инсулиновый шприц со специальной иглой (лаваж), исследовался эффект следующих доз: 25 мг/кг/день соединений в объеме 100 мкл. В качестве препарата сравнения был использован Тамифлю в дозах от 5 мг/кг/день до 30 мг/кг/день. В группе «вирусного контроля», в группах «леченых соединениями» общей формулы 1 и Тамифлю формулы A3 было по 10 мышей в каждой. За лечеными и контрольными животными велось ежедневное наблюдение, в первые 5 дней после инфицирования мышей взвешивали каждый день, далее - через день. Химиотерапевтическую активность соединений на модели гриппозной пневмонии мышей оценивали по показателю защиты от смертельной вирусной инфекции, и снижению веса в группах животных, леченых препаратом по сравнению с контрольной группой. Уменьшение или увеличение веса рассчитывалось отдельно для каждой мыши и выражалось в процентах. При этом за 100% принимался вес животного перед инфицированием. Для всех мышей одной группы определялось среднее значение процента потери или увеличения веса.
В предварительном опыте определяют дозу вируса, содержащую 10 ЛД50 в объеме 100 мкл. Всех животных в опыте заражают этой дозой вируса. Эффективность действия соединений общей формулы 1 на модели гриппозной пневмонии мышей оценивалась по количеству животных, выживших после инфицирования вирусом, средней продолжительности жизни и изменению веса инфицированных животных.
Было установлено, что на 7-й день наблюдения все мыши, зараженные вирусом и не прошедшие лечение указанными соединениями (группа «вирусного контроля»), погибли.
Проведенные эксперименты показали, что к последнему дню гибели животных из группы «вирусного контроля» лечение указанными соединениями общей формулы 1 и Тамифлю животных из групп «леченных соединениями» позволило предотвратить их гибель полностью.
Противогриппозная эффективность изученных препаратов общей формулы 1 и Тамифлю формулы A3 выражается в снижении темпов потери веса в группах леченных мышей по сравнению с группой «вирусного контроля». Потеря веса животного является одним из клинических признаков проявления гриппозной пневмонии. Большее снижение веса животного свидетельствует о более тяжелом протекании заболевания. Взвешивание мышей проводилось на 1, 2, 3, 4, 5 дни после инфицирования, а далее через день до 15 дня наблюдения. Было установлено, что в группе вирусного контроля животные больше всего теряли вес на 5 день после инфицирования (около 10%). В отличие от вирусного контроля в группах животных, проходящих лечение всеми указанными соединениями общей формулы 1 и Тамифлю, в среднем не наблюдалось потери веса. Начиная с 9 дня, все животные в группах «леченных соединениями» активно и стабильно набирали в весе.
Таким образом, показана высокая эффективность лечения гриппозной пневмонии мышей соединениями формулы 1.1., 1.3., 1.4 и 1.6.
Данное изобретение относится к новым ингибиторам активности нейраминидазы и их использованию для профилактики и лечения инфекции гриппа, а именно к новым фторзамещенным 4-ациламино-5-гуанидино-3-(пентан-3-илокси)никлогексен-1-карбоновым кислотам, их эфирам общей формулы 1 и их фармацевтически приемлемым солям и/или гидратам. В формуле 1
R представляет собой водород, C1-C5алкил, C2-C5алкенил или C2-C5алкинил, необязательно замещенные C3-C6циклоалкилом, фенилом, пиридилом, C1-C3алкокси; Rf представляет собой CH2F или CHF2. Изобретение также относится к фармацевтической композиции, способу ингибирования нейраминидазы, в частности нейраминидазы вируса гриппа, и способу профилактики и лечения гриппа и вирусных заболеваний, обусловленных вирусом гриппа, таких как пневмония. 5 н. и 5 з.п. ф-лы.
1. Фторзамещенные (3R,4R,5S)-5-гуанидино-4-ациламино-3-(пентан-3-илокси)циклогексен-1-карбоновые кислоты и их эфиры формулы 1 и их фармацевтически приемлемые соли и/или гидраты,
где R представляет собой водород, C1-C5алкил, C2-C5алкенил или C2-C5алкинил, необязательно замещенные C3-C6циклоалкилом, фенилом, пиридилом, C1-C3алкокси; Rf представляет собой CH2F или CHF2.
2. Соединения по п.1, представляющие собой соединения общей формулы 1, в которых R представляет собой водород, метил или этил.
3. Соединения по п.1, представляющие собой
(3R,4R,5S)-5-гуанидино-4-фторацетиламино-3-(пентан-3-илокси)циклогексен-1-карбоновая кислота,
метиловый эфир (3R,4R,5S)-5-гуанидино-4-фторацетиламино-3-(пентан-3-илокси)циклогексен-1-карбоновой кислоты,
этиловый эфир (3R,4R,5S)-5-гуанидино-4-фторацетиламино-3-(пентан-3-илокси)циклогексен-1-карбоновой кислоты,
(3R,4R,5S)-5-гуанидино-4-дифторацетиламино-3-(пентан-3-илокси)циклогексен-1-карбоновая кислота,
метиловый эфир (3R,4R,5S)-5-гуанидино-4-дифторацетиламино-3-(пентан-3-илокси)циклогексен-1-карбоновой кислоты,
этиловый эфир (3R,4R,5S)-5-гуанидино-4-дифторацетиламино-3-(пентан-3-илокси)циклогексен-1-карбоновой кислоты.
4. Лекарственное начало, обладающее активностью в отношении нейраминидазы, представляющее собой соединение по любому из пп.1-3.
5. Фармацевтическая композиция, обладающая противовирусной активностью и предназначенная для лечения гриппа и сопутствующих заболеваний, вызванных вирусом гриппа, включающая лекарственное начало по п.4 в терапевтически эффективном количестве.
6. Фармацевтическая композиция по п.5 в форме таблеток, капсул или инъекций, помещенных в фармацевтически приемлемую упаковку.
7. Фармацевтическая композиция по любому из п.5 или 6, для лечения пневмонии, обусловленной вирусом гриппа, в эффективном количестве.
8. Способ ингибирования активности нейраминидазы, заключающийся в контактировании нейраминидазы с соединением по любому из пп.1-3.
9. Способ по п.8, в котором в качестве нейраминидазы используют нейраминидазу вируса гриппа.
10. Способ профилактики и лечения гриппа и заболеваний, обусловленных вирусом гриппа, заключающийся во введении терапевтически эффективного количества лекарственного начала по п.4, или фармацевтической композиции по любому из пп.5-7.
| RU 2181357 C2, 20.07.1999 | |||
| ZHENG, MINQUIE et al | |||
| QSAR analyses on avian influenza virus neuraminidase inhibitors using CoMFA, CoMSIA, and HQSAR | |||
| Journal of Computer-Aided Molecular Design (English), 2006, 20(9), 549-566 | |||
| DU, QI-SHI; et al., Fragment-Based Quantitative Structure-Activity Relationship (FB-QSAR) for Fragment-Based Drug Design | |||
| Journal |

