Данное изобретение относится к новому ингибитору РНК-зависимой РНК - полимеразы (RdRp) различных РНК вирусов - 3-гидрокси-6-фтор-хиноксалин-2-карбоксамиду к фармацевтической композиции и способам их получения и применения. РНК - вирус представляет собой вирус, который имеет РНК (рибонуклеиновая кислота) в качестве своего генетического материала. К наиболее распространенным заболеваниям, вызываемым РНК вирусами, относятся грипп, эбола, тяжелый острый респираторный синдром (ОРВИ, SARS), коронавирус SARS-CoV-2, желтая лихорадка, и др. [P. Poltronieri ct al. RNA Viruses: RNA Roles in Pathogenesis, Complication and Viral Load. Curr. Genomics. 2015, 16(5), 327-335.].
Грипп, является инфекционным заболеванием, вызываемым вирусом гриппа. Симптомы могут быть от слабых до тяжелых. Осложнения гриппа могут включать вирусную пневмонию, вторичную бактериальную пневмонию, синусовые инфекции и ухудшение предыдущих проблем со здоровьем, таких как астма или сердечная недостаточность. Ежегодные вспышки гриппа распространяются по всему миру, что приводит к от трех до пяти миллионов случаев тяжелых заболеваний и от 290000 до 650000 смертей. Более крупные вспышки, известные как пандемии, встречаются реже. В 20-м веке произошли три пандемии гриппа: испанский грипп в 1918 году (17-100 миллионов смертей), азиатский грипп в 1957 году (два миллиона смертей) и гонконгский грипп в 1968 году (один миллион смертей). Всемирная организация здравоохранения объявила вспышку нового типа гриппа А / H1N1 пандемией в июне 2009 г. Грипп может также поражать других животных, включая свиней, лошадей и птиц [https://en.wikipedia.org/wiki/Influenza].
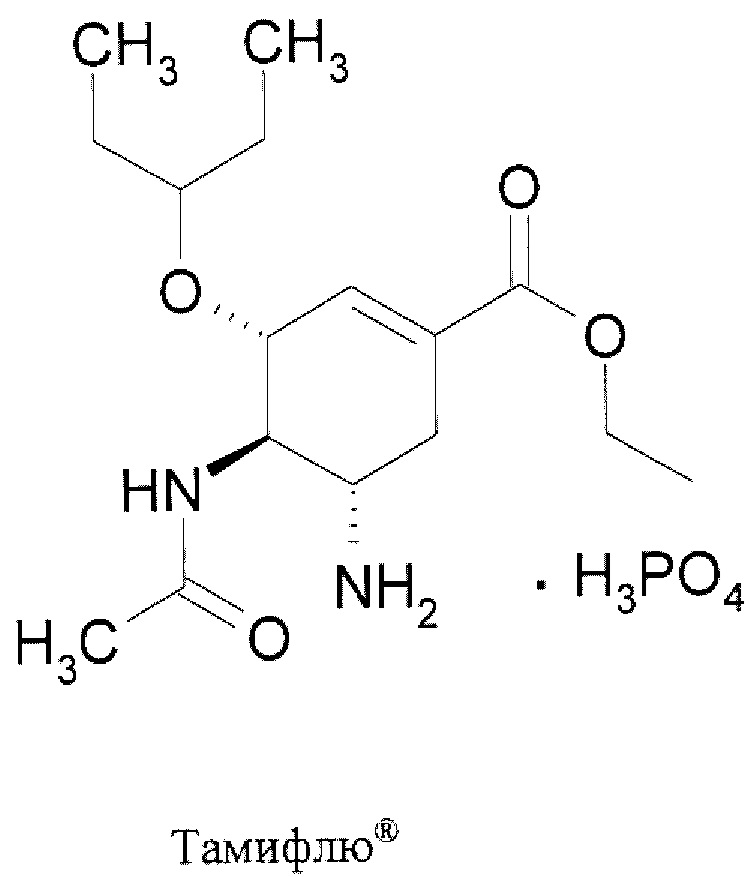
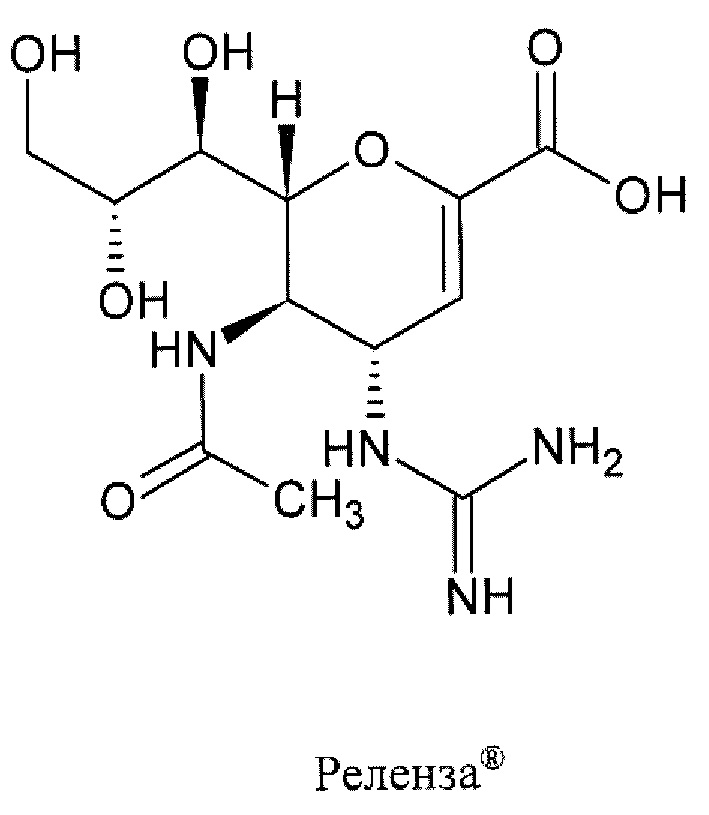
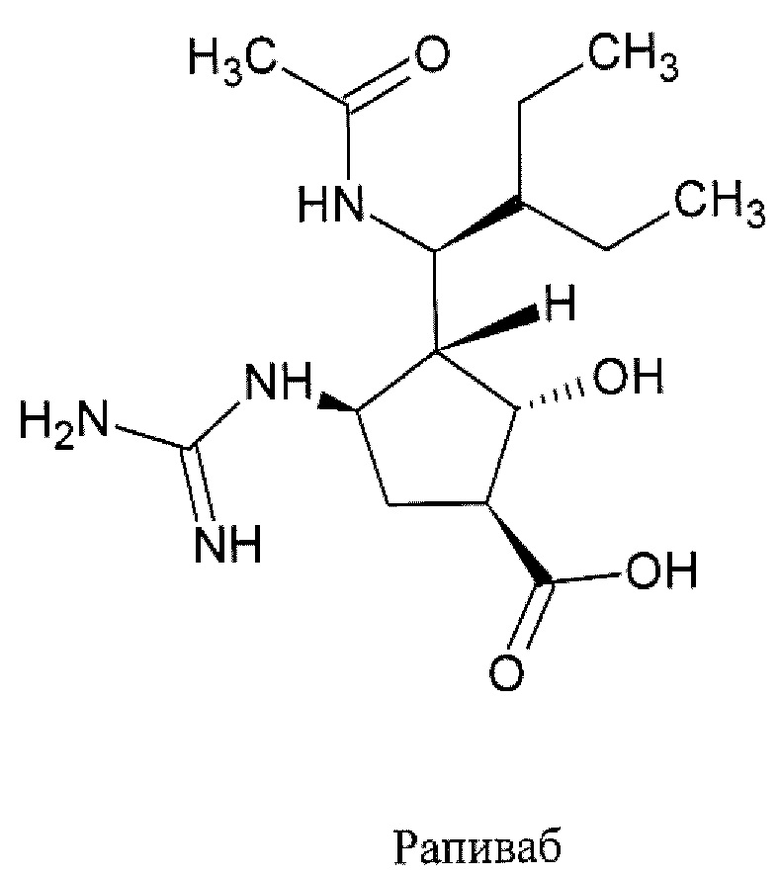
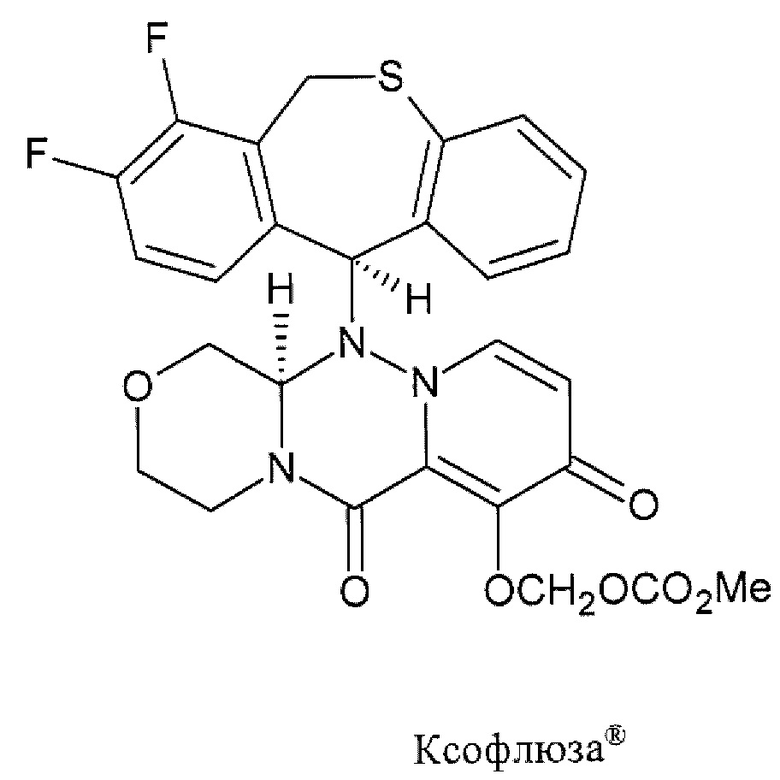
Центр по контролю и профилактике заболеваний (CDC) США рекомендует для лечения гриппа в использовать четыре противогриппозных препарата, в том числе: осельтамивир фосфат (торговым названием Тамифлю (Tamiflu®)), занамивир (торговое наименование Реленза (Relenza)®), перамивир (торговое наименование Рапиваб (Rapivab)®) и балоксавир марбоксил (торговое наименование Ксофлюза (Xofluza)®) [https://www.cdc.gov/flu/treatment/whatyoushould.htm]. Известные противогрппозные препараты.
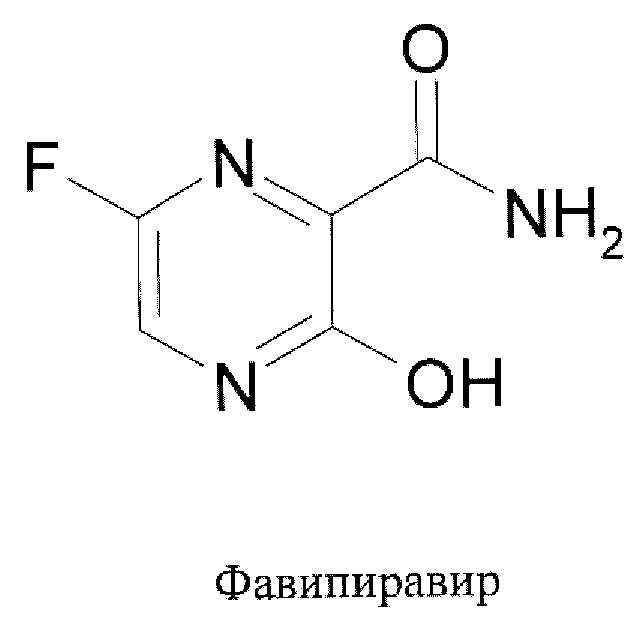
Известен также противогриппозный препарат Фавипиравир (Т-705, Авиган, Фавилавир), запатентованный в 1999 году японской компанией Тояма Ксмикал Ко [RU 2224520]. Фавипиравир используется в Японии для лечения гриппа, в том численовых высокопатогенного A H5N1 штамма птичьего гриппа [R.W. Sidwell at al. Antimicrob. Agents Chemother. 2007, 51(3): 845-851].
Фавипиравир проявляет антивирусную активность в отношении многих других РНК-вирусов, такие как аренавирусы, буньявирусы и филовирусы, которые, как известно, вызывают смертельную геморрагическая лихорадку. [Y. Furuta et al. Review Favipiravir (T-705), a broad spectrum inhibitor of viral RNA polymerase. Proc. Jpn. Acad., Scr. B 93, 2017, 449-463].
Фавипиравир успешно прошел испытание лечения прогрессирующей инфекции у мышей, вызванной вирусом Эбола [L. Oestereich et al. Ebola virus infection with T-705 (favipiravir) in a small animal model. Antiviral Research 2014, 105, 17-21.] и является обнадеживающим драг-кандидатом, но пока он не одобрен ВОЗ.
Вирусная Эбола (EVD), также известная как геморрагическая лихорадка Эбола (EIIF) или просто лихорадка Эбола, представляет собой вирусную геморрагическую лихорадку людей и других приматов, вызванную эболавирусами. Заболевание имеет высокий риск смерти, убивая от 25% до 90% инфицированных, в среднем около 50%. Вспышки EVD периодически возникают в тропических регионах Африки к югу от Сахары. В период с 1976 по 2013 год ВОЗ сообщает о 24 вспышках, в которых зарегистрировано 2387 случаев заболевания с 1590 смертельными исходами. Крупнейшей вспышкой на сегодняшний день была эпидемия в Западной Африке, которая произошла с декабря 2013 года по январь 2016 года, с 28646 случаями заболевания и 11323 смертельными исходами. Вакцина против Эболы была одобрена в США в декабре 2019 года. По состоянию на 2019 год не было утвержденного лечение Эболы [https://en.wikipedia.org/wiki/Ebola_virus_disease].
В феврале 2020 года Фавипиравир был успешно опробован в начальном рандомизированном исследовании в Китае в качестве противовирусной терапии коронавируса SARS-CoV-2 (COVID-19). Фавипиравир получил краткосрочное одобрение в Китае 16 февраля 2020 года в качестве эффективного противовирусного средства против COVID-19 в течение пяти лет. В настоящее время он производится в Китае под названием Фавилавир [https://de.wikipedia.org/wiki/Favipiravir]. На 25 апреля 2020 г в мире было зарегистрировано (за 4 месяца) 2837155 заболевших COV1D-19, 808975 выздоровевших пациентов и 197698 умерших пациентов.
Следует отметить, что Фавипиравир имеет по крайней мере три существенных недостатка: низкую микромолярную активность, необходимость использования при терапии РНК вирусных заболеваний высоких доз фавипиравира (суточная доза от 1200 мг до ≥3200 мг [https://researchgate.net/publication/340000976_Experimental_Treatment_with_Favipiravir_for_COVID-19_An_Open-Label_Control_Study] и серьезные побочные эффекты [L. Voytko. Japanese Flu Drug 'Effective' Against Coronavirus In Clinical Trials, Chinese Officials Say. Mar 18, 2020. https://www.forbes.com/sites/lisettevoytko/2020/03/18/japanese-flu-drug-effective-against-coronaviras-in-clinical-trials-chinese-officials-say/#7e5300b56bad].
По нашему мнению все эти недостатки связаны с его драматической химической лабильностью [J. Huchting et al. Prodrugs of the Phosphoribosylated Forms of Hydroxypyrazinecarboxamide Pseudobase T-705 and Its De-Fluoro Analogue T-1105 as Potent Influenza Vims Inhibitors. J. Med, Chem. 2018, 61, 6193-6210], обусловленной наличием фтора, который легко может вступать в реакции нуклеофильного замещения. Даже в чистой воде и водных средах происходит нуклеофильное замещение фтора на гидроксил с последующим разложением пиразинового гетероцикла T-705 [J. Huchting et al. Synthesis of T705-ribonucleoside and T-705-ribonucleotide and studies of chemical stability. ChemMedChem 2017, 12, 652-659]. Кроме того, высоко реакционноспособный атома фтора Фавипиравира может легко замещаться О-, N- и S-нуклеофилами, например, цистеином или лизином в белках [S.B. Cohen, R.L. Halcomb. Synthesis and characterization of an anomeric sulfur analogue of CMP-sialic acid. J. Org. Chem. 2000, 65, 6145-52], a неселективное или плохо селективное ковалентное связывание таких агентов с биомолекулами может привести к серьезному повреждение клеток и тканей или активация врожденной иммунной системы в ответ на образование модифицированных белков, которые считаются чужеродными, с последующей деградацией в лизосомах. Кроме того, многие необратимые ингибиторы обладают мутагенным действием, и генотоксическими эффектами [J. Kazius et al. Derivation and validation of toxicophores for mutagenicity prediction. J. Med. Chem. 2005, 48, 312-20].
Учитывая, что РНК вирусы, и особенно короновирусы, представляет собой серьезную угрозу для общественного здравоохранения (в глобальном масштабе ежегодные эпидемии приводят к 3-5 миллионам случаев тяжелой болезни, миллионам госпитализаций и до 650000 смертей во всем мире) представляется целесообразным поиск новых противо-РНК вирусных препаратов, обладающих улучшенными характеристиками.
Ниже приведены определения терминов, которые использованы в описании этого изобретения.
Термин «пролекарство» относится к соединениям по изобретению, которые расщепляются химически или метаболически и становятся, путем сольволиза или в физиологических условиях, соединением по настоящему изобретению, которое фармацевтически активно в естественных условиях. Пролекарства часто имеют более высокую растворимость, тканевую совместимость, доставку или замедленное высвобождение у млекопитающих (Bungard, Н., Desing of products, pp. 7-9, 21-24, Elsevier, Amsterdam 1985). Пролекарства включают кислотные производные, хорошо известные специалистам в данной области техники, такие как, например, сложные эфиры, полученные реакцией исходного кислотного соединения с подходящим спиртом, или амиды, полученные реакцией соединения исходной кислоты с подходящим амином. Примеры пролекарств включают, но не ограничиваются ими, ацетат, формиат, бензоат или другие ацилированные производные спиртов или аминов функциональных групп в соединениях по настоящему изобретению.
Термин «активный компонент» (лекарственное вещество) относится к физиологически активному веществу синтетического или иного (биотехнологического, растительного, животного, бактерицидного и так далее) происхождения, обладающему фармакологической активностью, которое является активным ингредиентом фармацевтической композиции.
Термин «лекарственный препарат» означает вещество (или смесь веществ в виде фармацевтической композиции) в виде таблеток, капсул, инъекций, мазей и др. готовых форм, предназначенное для восстановления, исправления или изменения физиологических функций у человека и животных, а также для лечения и профилактики болезней, диагностики, анестезии, контрацепции, косметологии и прочего.
Термин «фармацевтическая композиция» обозначает композицию, включающую в себя соединение формулы 1 и, по крайней мере, один из компонентов, выбранных из группы, состоящей из фармацевтически приемлемых и фармакологически совместимых наполнителей, растворителей, разбавителей, носителей, вспомогательных, распределяющих и воспринимающих средств, средств доставки, таких как консерванты, стабилизаторы, наполнители, измельчители, увлажнители, эмульгаторы, суспендирующие агенты, загустители, подсластители, отдушки, ароматизаторы, антибактериальные агенты, фунгициды, лубриканты, регуляторы пролонгированной доставки, выбор и соотношение которых зависит от природы и способа назначения и дозировки. Примерами суспендирующих агентов являются этоксилированный изостеариловый спирт, полиоксиэтилен, сорбитол и сорбитовый эфир, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакант, а также смеси этих веществ. Защита от действия микроорганизмов может быть обеспечена с помощью разнообразных антибактериальных и противогрибковых агентов, например, таких как парабены, хлорбутанол, сорбиновая кислота и подобные им соединения. Композиция может включать также изотонические агенты, например, сахара, хлористый натрий и им подобные. Пролонгированное действие композиции может быть обеспечено с помощью агентов, замедляющих абсорбцию активного компонента, например, моностеарат алюминия и желатин. Примерами подходящих носителей, растворителей, разбавителей и средств доставки являются вода, этанол, полиспирты, а также их смеси, растительные масла (такие, как оливковое масло) и инъекционные органические сложные эфиры (такие, как этилолеат). Примерами наполнителей являются лактоза, молочный сахар, цитрат натрия, карбонат кальция, фосфат кальция и им подобные. Примерами измельчителей и распределяющих средств являются крахмал, альгиновая кислота и ее соли, силикаты. Примерами лубрикантов являются стеарат магния, лаурилсульфат натрия, тальк, а также полиэтиленгликоль с высоким молекулярным весом. Фармацевтическая композиция для перорального, сублингвального, транедермального, внутримышечного, внутривенного, подкожного, местного или ректального введения активного компонента, одного или в комбинации с другим активным компонентом, может быть введена животным и людям в стандартной форме введения, в виде смеси с традиционными фармацевтическими носителями. Пригодные стандартные формы введения включают пероральные формы, такие как таблетки, желатиновые капсулы, пилюли, порошки, гранулы, жевательные резинки и пероральные растворы или суспензии, сублингвальные и трансбуккальные формы введения, аэрозоли, имплантаты, местные, транедермальные, подкожные, внутримышечные, внутривенные, интраназальные или внутриглазные формы введения и ректальные формы введения.
Термин «инертный наполнитель», используемый в данном описании, относится к соединению, которое используют для получения фармацевтической композиции, и, как правило, безопасному, нетоксичному и ни биологически, ни иным образом нежелательному, и включает в себя вспомогательные вещества, которые являются приемлемыми для применения в ветеринарии, а также фармакологически приемлемыми для человеческого использования. Соединения по данному изобретению могут быть введены отдельно, но обычно их будут вводить в смеси с одним или более фармацевтически приемлемыми эксципиентами, разбавителями или носителями, выбранными с учетом предполагаемого пути введения и стандартной фармацевтической практики.
Термин «фармацевтически приемлемая соль» означает относительно нетоксичные органические и неорганические соли кислот и оснований, заявленных в настоящем изобретении. Эти соли могут быть получены in situ в процессе синтеза, выделения или очистки соединений или приготовлены специально. В частности, соли оснований могут быть получены специально, исходя из очищенного свободного основания заявленного соединения и подходящей органической или неорганической кислоты. Примерами полученных таким образом солей являются гидрохлориды, гидробромиды, сульфаты, бисульфаты, фосфаты, нитраты, ацетаты, океалаты, валериаты, олеаты, пальмитаты, стеараты, лаураты, бораты, бензоаты, лактаты, тозилаты, цитраты, малеаты, фумараты, сукцинаты, тартраты, мезилаты, малонаты, салицилаты, пропиоиаты, этансульфонаты, бензолсульфонаты, сульфаматы и им подобные (Подробное описание свойств таких солей дано в Berge S.M., et al., "Pharmaceutical Salts" J. Pharm. Sci. 1977, 66: 1-19). Соли заявленных кислот также могут быть специально получены реакцией очищенной кислоты с подходящим основанием, при этом могут быть синтезированы соли металлов и аминов. К металлическим относятся соли натрия, калия, кальция, бария, цинка, магния, лития и алюминия, наиболее желательными из которых являются соли натрия и калия. Подходящими неорганическими основаниями, из которых могут быть получены соли металлов, являются гидроксид, карбонат, бикарбонат и гидрид натрия, гидроксид и бикарбонат калия, поташ, гидроксид лития, гидроксид кальция, гидроксид магния, гидроксид цинка. В качестве органических оснований, из которых могут быть получены соли заявленных кислот, выбраны амины и аминокислоты, обладающие достаточной основностью, чтобы образовать устойчивую соль, и пригодные для использования в медицинских целях (в частности, они должны обладать низкой токсичностью). К таким аминам относятся аммиак, метиламин, диметиламин, триметиламин, этиламин, диэтиламин, триэтиламии, бензиламин, дибензиламин, дициклогексиламин, пиперазин, этилпиперидин, трис(гидроксиметил)аминометан и подобные им. Кроме того, для солсобразования могут быть использованы гидроокиси тетраалкиламмопия, например, такие как холин, тетраметиламмоний, тетраэтиламмоний и им подобные. В качестве аминокислот могут быть использованы основные аминокислоты - лизин, орнитин и аргинин.
Термин «кристаллическая форма» означает структуру вещества, характеризующуюся упаковкой образующих ее молекул в один из видов кристаллической решетки.
Термин «поликристаллическая форма» означает структуру вещества, имеющую поликристаллическое строение, т.е. состоящую из множества мелких монокристаллов, т.е. кристаллитов определенной кристаллической формы.
Термин «терапевтически эффективное количество», используемый здесь, означает количество субстанции, пролекарства или лекарства, необходимое для уменьшения симптомов заболевания у субъекта. Доза субстанции, пролекарства или лекарства будет соответствовать индивидуальным требованиям в каждом конкретном случае. Эта доза может варьироваться в широких пределах в зависимости от многочисленных факторов, таких как тяжесть заболевания, подлежащего лечению, возраста и общего состояния здоровья пациента, других лекарственных средств, с помощью которых пациент проходит лечение, способа и формы введения и опыта лечащего врача. Для перорального введения суточная доза составляет приблизительно от 0,01 до 10 г, включая все значения между ними, в день в монотерапии и/или в комбинированной терапии. Предпочтительная суточная доза составляет примерно от 0,1 до 7 г в день. Как правило, лечение начинают с большой начальной «нагрузочной дозы», чтобы быстро уменьшить или устранить вирус, сопровождающей убывающую дозу до уровня, достаточного для предотвращения всплеска инфекции.
Термин «сольват» означает комплекс или агрегат, образуемый одной или более молекулами растворенного вещества, т.е. соединением согласно изобретению или его фармацевтически приемлемой солью и одной или более молекулами растворителя. Такие сольваты являются типичными твердыми кристаллами, имеющими, по существу, фиксированное молярное отношение растворенного вещества и растворителя. Репрезентативные растворители включают в себя, не ограничиваясь перечисленными, воду, этанол, изопропанол, уксусную кислоту и пр. Когда растворителем является вода, образуемый сольват представляет собой гидрат.
Термин «субъект» означает млекопитающее, которое включает, но не ограничивается ими, крупный рогатый скот, свиней, овец, куриц, индеек, буйволов, лам, страусов, собак, кошек и человека, предпочтительно субъектом является человек. Предполагается, что в способе лечения субъекта может быть любое из пролекарств общей формулы 1, его стереоизомер, изотопно-обогащенный аналог, его фармацевтически приемлемая соль, гидрат, сольват, кристаллическая и полиморфная форма, либо в сочетании их с другим соединением, в том числе с ингибитором РНК-зависимой РНК - полимеразы (RdRp) различных РНК вирусов.
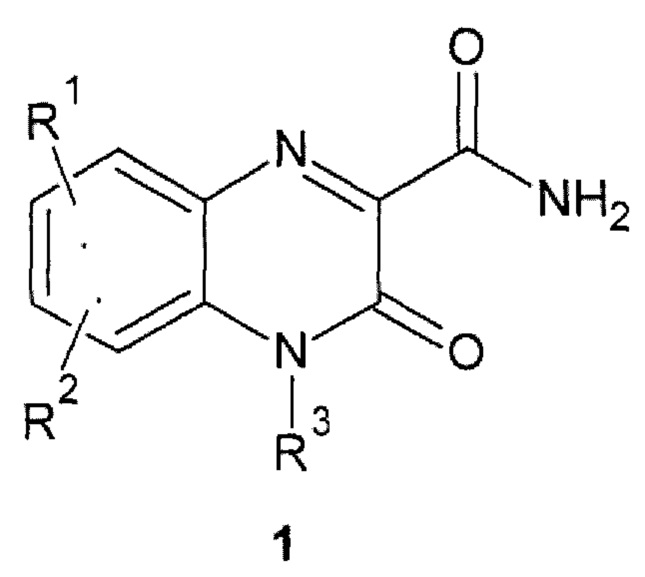
Авторы обнаружили новое противо-РНК вирусное, в том числе противокороновирусное средство, представляющее собой замещенный хиноксалин общей формулы 1, его стереоизомер, фармацевтически приемлемую соль, аморфную, поликристаллическую или кристаллическую форму.
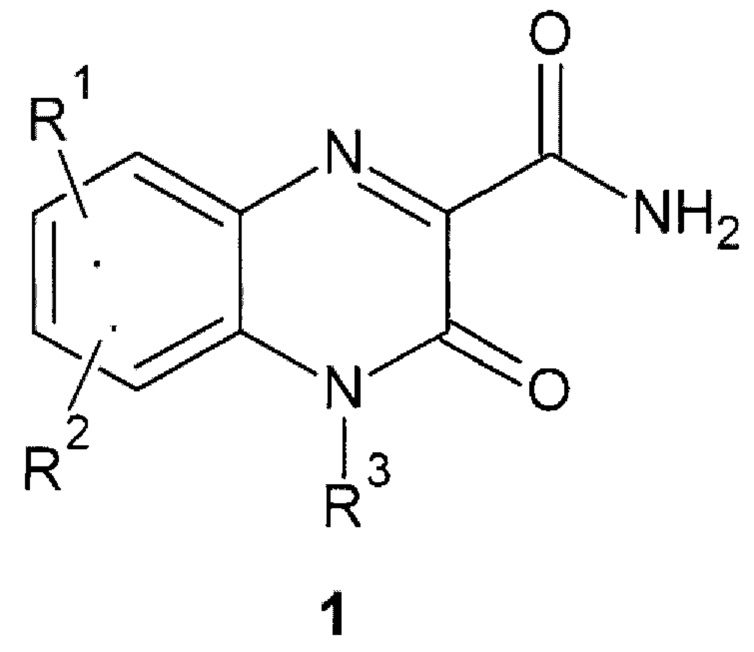
где: R1 и R2 представляют собой необязательно одинаковые атомы водорода или галогена; R3 представляют собой атом водорода или фрагмент общей формулы 2 или его стереоизомер.
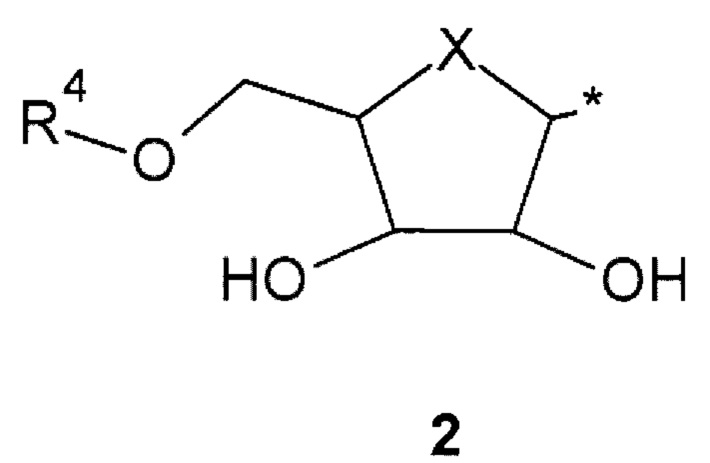
где: X представляет собой атом кислорода или С=СН2 группу; * - место присоединения фрагмента 2;
R4 представляет собой атом водорода или заместитель общей формулы 3 или его стереоизомер
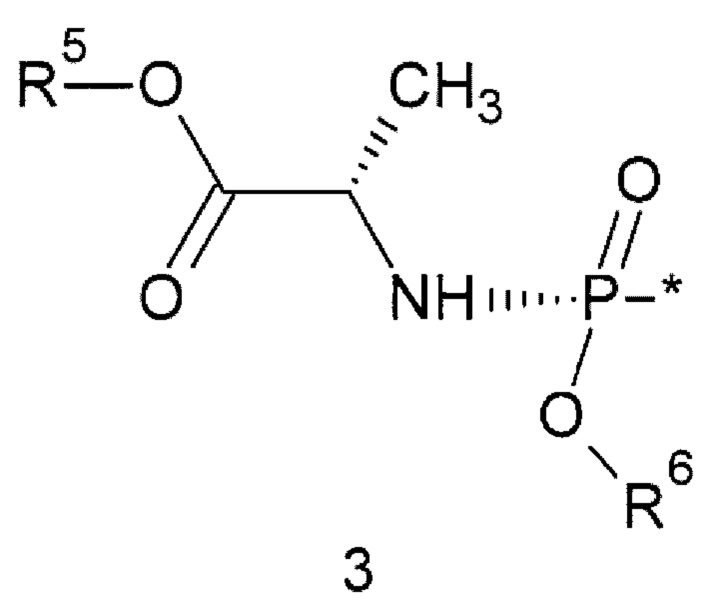
где: * - место присоединения заместителя 3;
R5 представляет собой C1-С6алкил или С3-С6циклоалкил; R6 представляет собой необязательно замещенный арил, предпочтительно фенил.
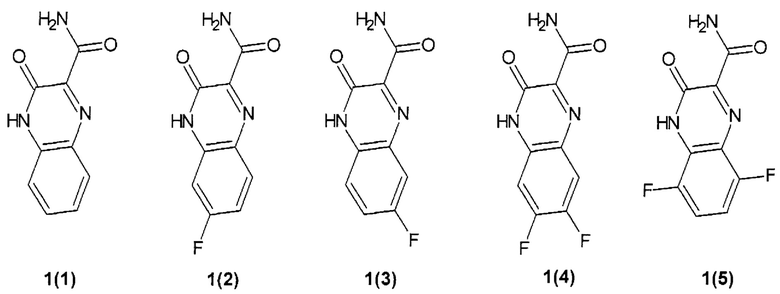
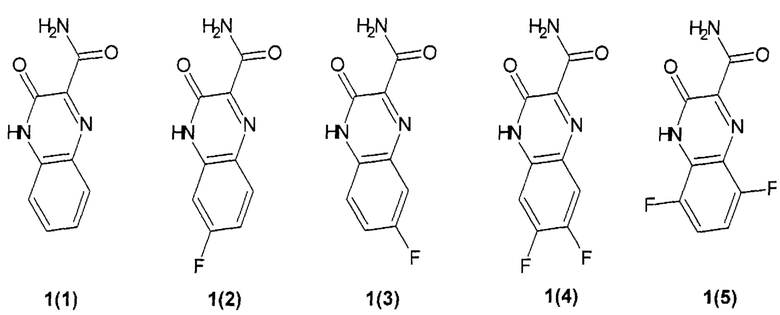
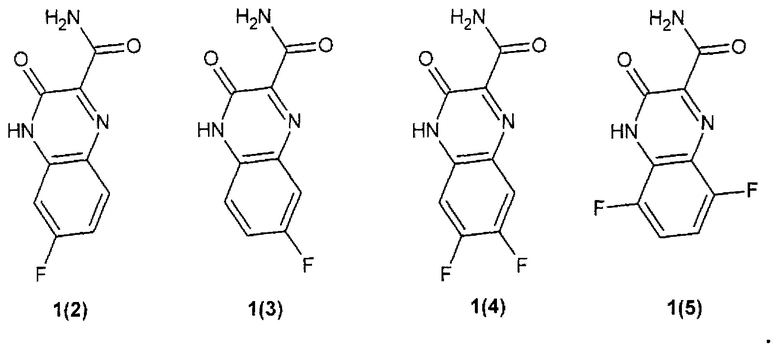
Предпочтительным являются замещенный хиноксалин выбранный из ряда:
3-оксо-3,4-дигидрохиноксалин-2-карбоксамид {1(1)},
6-фтор-3-оксо-3,4-дигидрохиноксалин-2-карбоксамид {1(2)},
7-фтор-3-оксо-3,4-дигидрохиноксалин-2-карбоксамид {1(3)},
6,7-дифтор-3-оксо-3,4-дигидрохиноксалин-2-карбоксамид {1(4)},
5,8-дифтор-3-оксо-3,4-дигидрохиноксалин-2-карбоксамид {1(5)}.
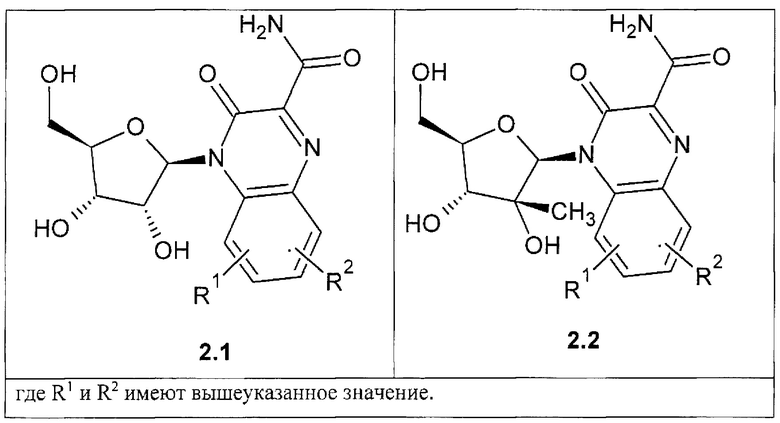
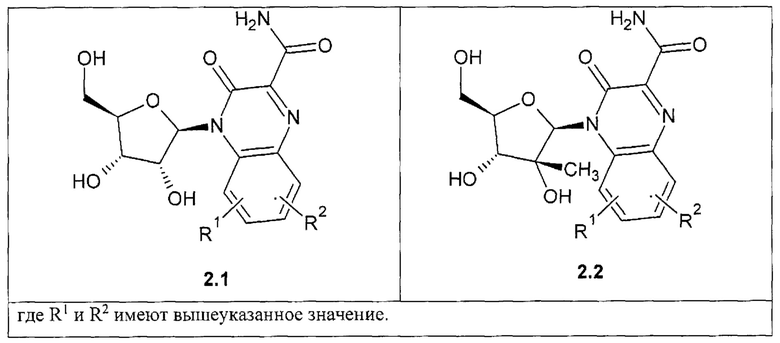
Предпочтительным являются также замещенный хиноксалин выбранный из ряда
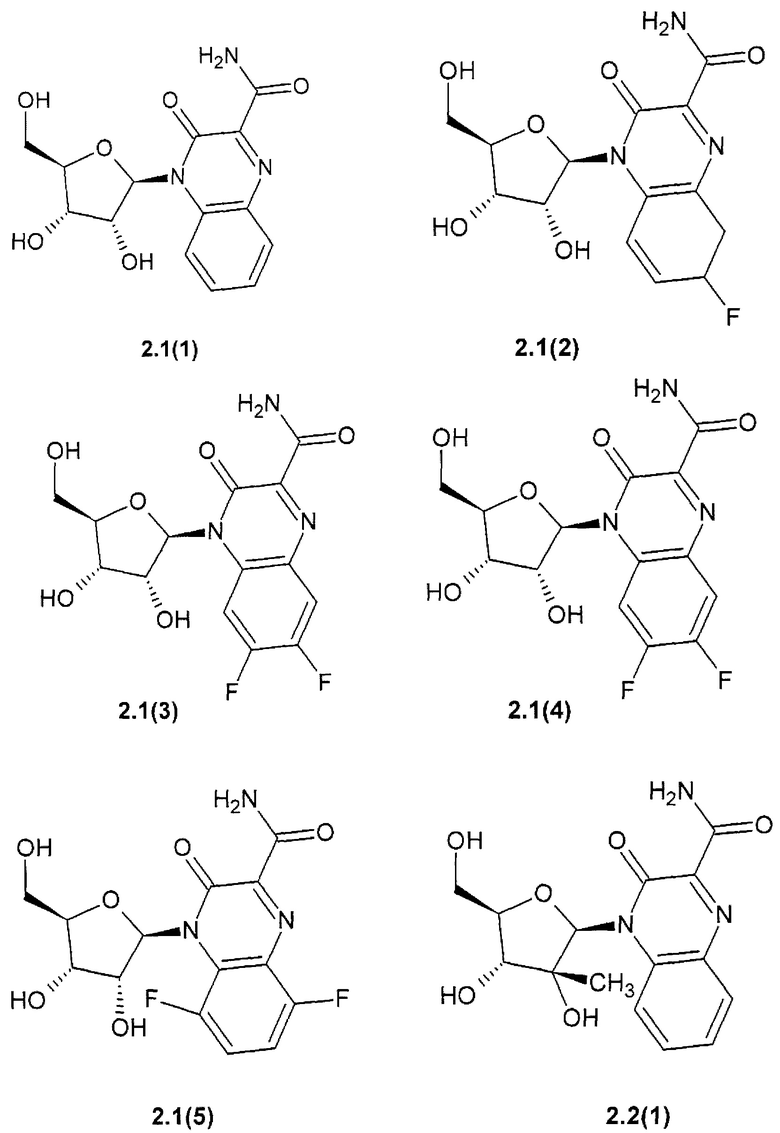
3-оксо-4-b-D-рибофуранозид-3,4-дигидрохиноксалин-2-карбоксамид общей формулы {2.1},
4-(2-С-метил-b-D-рибофуранозил)-3-оксо-3,4-дигидрохиноксалин-2-карбоксамид общей формулы {2.2}.
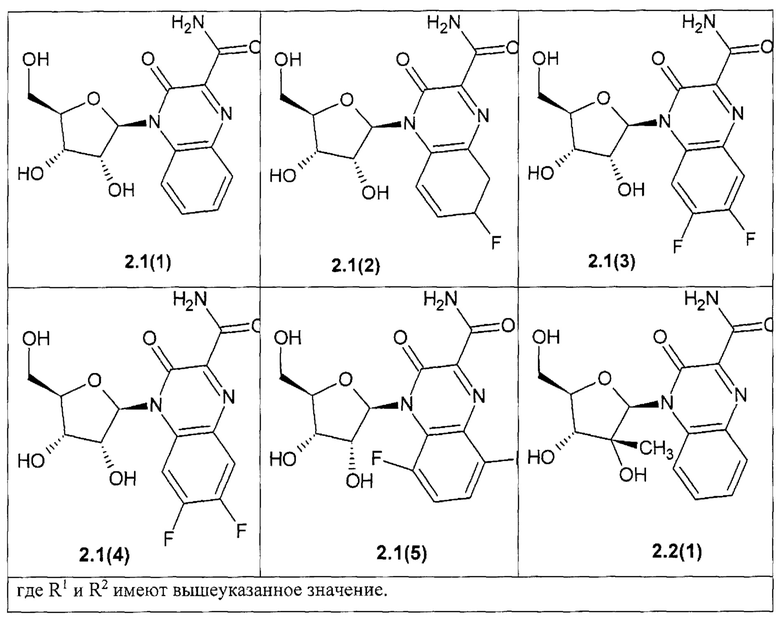
Более предпочтительным является замещенный хиноксалин выбранный из ряда:
3-оксо-4-b-D-рибофуранозид-3,4-дигидрохиноксалин-2-карбоксамид {2.1(1)},
6- фтор-3-оксо-4-b-D-рибофуранозид-3,4-дигидрохиноксалин-2-карбоксамид {2.1(2)},
7-фтор-3-оксо-4-b-D-рибофуранозид-3,4-дигидрохиноксалин-2-карбоксамид {2.1(3)},
6,7-фтор-3-оксо-4-b-D-рибофуранозид-3,4-дигидрохиноксалин-2-карбоксамид {2.1(4)},
5,8-фтор-3-оксо-4-b-D-рибофуранозид-3,4-дигидрохинокеалин-2-карбокеамид {2.1(5)},
4-(2-С-метил-b-D-рибофуранозил)-3-оксо-3,4-дигидрохиноксалин-2-карбоксамид {2.2(1)},
Предпочтительным являются также замещенный хиноксалин выбранный из ряда:
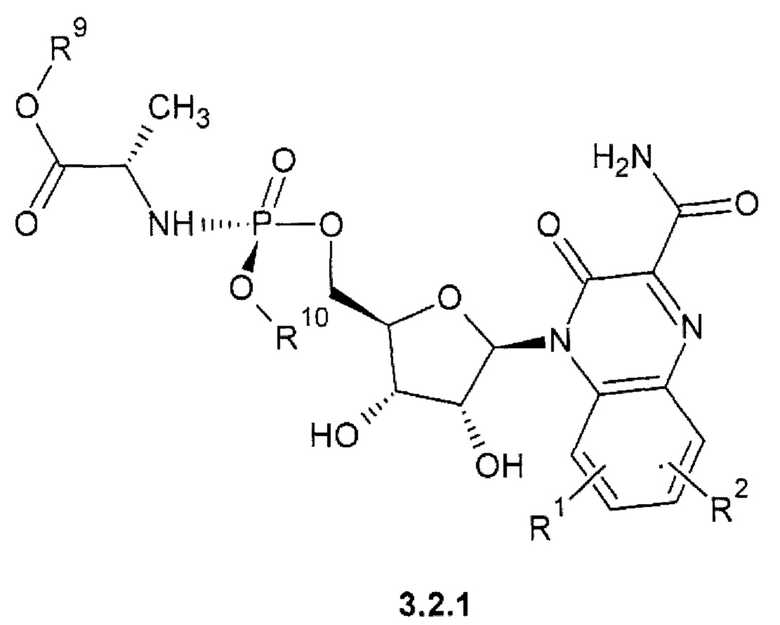
1-(С1-Сбалкил) (2S)-2-{[(S)-{[(2R,3S,4R,5R)-5-(3-карбамоил-2-оксохиноксалин-1(2H)-ил)-3,4-дигидрокситетрагидрофуран-2-ил]метокси}(арилокси)фосфорил]амино}пропаноат {3.2.1},
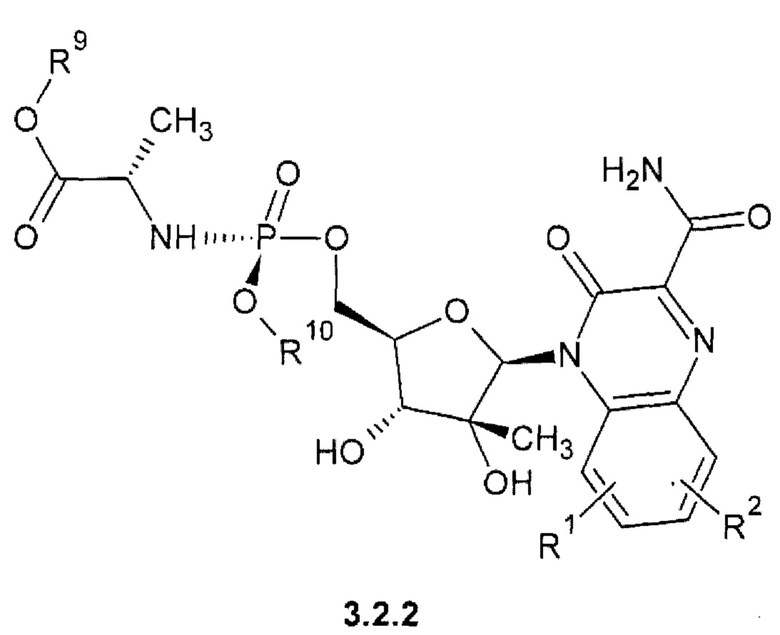
1-(С1-С6алкил) (2S)-2-{[(S)-{[(2R,3R,4R,5R)-5-(3-карбамоил-2-оксохиноксалин-1(2H)-ил)-3,4-дигидрокси-4-дифтор-3-гидрокситетрагидрофуран-2-ил]метокси}(арилокси)фосфорил]амиио}пропаноат {3.2.2} или их стереоизомеров.
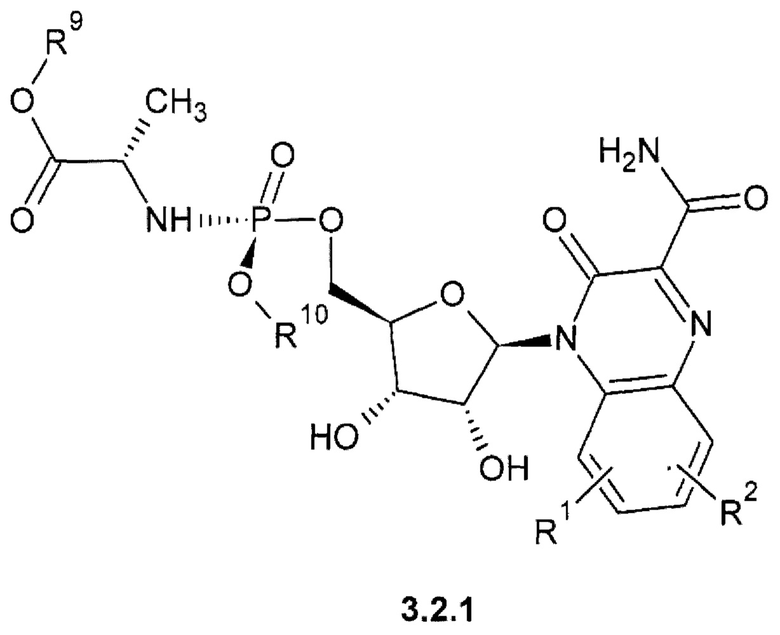
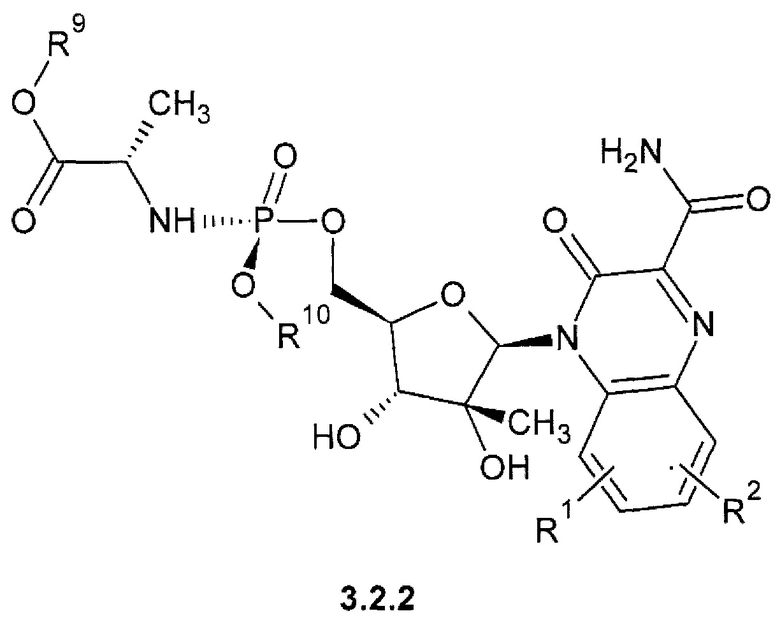
где R1, R2 имеют вышеуказанное значение. R9 и R10 соответственно имеют вышеуказанное значение для R5 и R6.
Более предпочтительным являются замещенный хиноксалин выбранный из ряда: 1-метилэтил (2S)-2-{[(S)-{[(2R,3S,4R,5R)-5-(3-карбамоил-2-оксохиноксалин-1(2H)-ил)-3,4-дигидрокситетрагидрофуран-2-ил]метокси}(фенокси)фосфорил]амино}пропаноат {3.2.1(1)},
1-метилэтил (2S)-2-{[(S)-{[(2R,3S,4R,5R)-5-(3-карбамоил-7-фтор-2-оксохиноксалин-1(2H)-ил)-3,4-дигидрокситетрагидрофуран-2-ил]метокси}(фенокси)фосфорил]амино}пропаноат {3.2.1(2)},
1-метилэтил (2S)-2-{[(S)-{[(2R,3S,4R,5R)-5-(3-карбамоил-6,7-дифтор-2-оксохиноксалин-1(2H)-ил)-3,4-дигидрокситетрагидрофуран-2-ил]метокси}(фенокси)фосфорил]амино}пропаноат {3.2.1(3)},
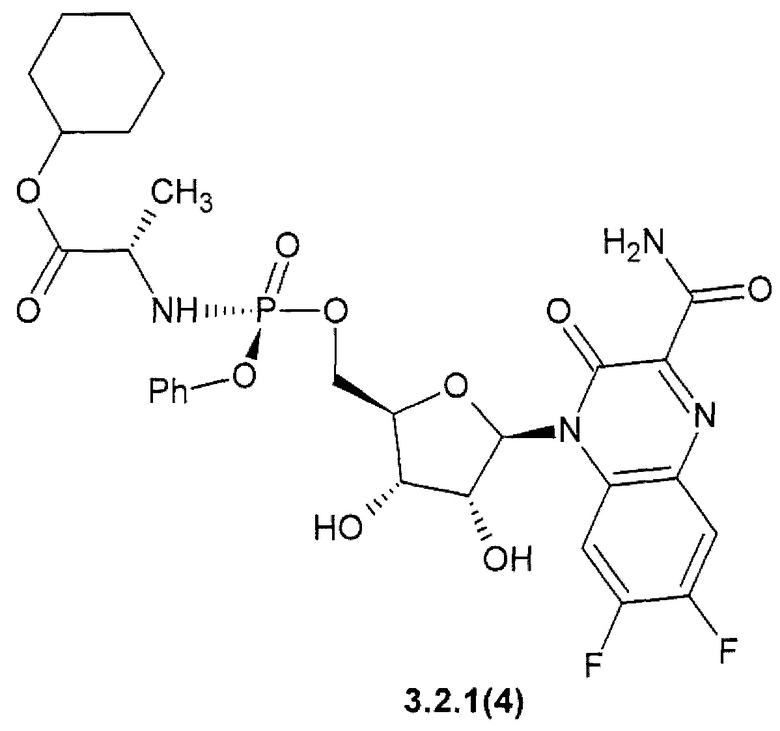
циклогексил (2S)-2-{[(S)-{[(2R,3S,4R,5R)-5-(3-карбамоил-6,7-дифтор-2-оксохиноксалин-1(2H)-ил)-3,4-дигидрокситетрагидрофуран-2-ил]метокси}(фенокси)фосфорил]амино}пропаноат {3.2.1(4)} или их стерео изомеров, фармацевтически приемлемых солей, аморфных, поликристаллических или кристаллических форм.
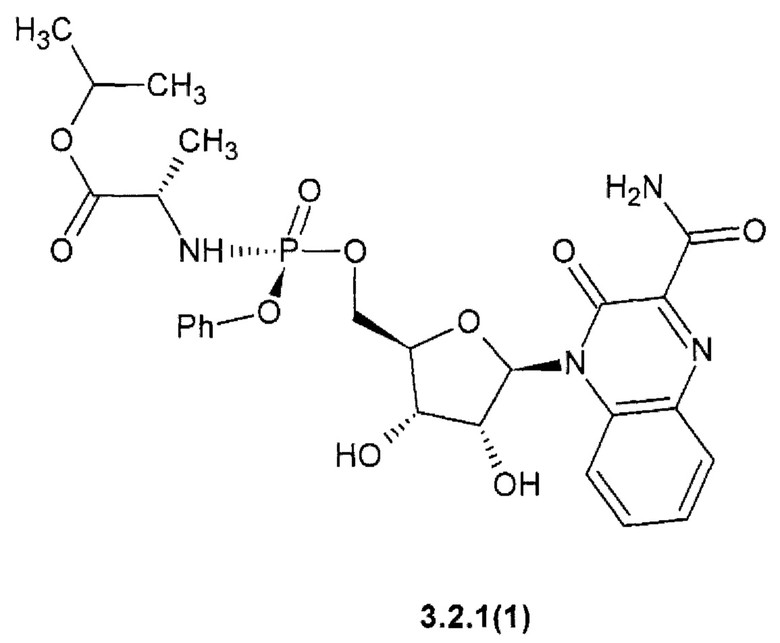
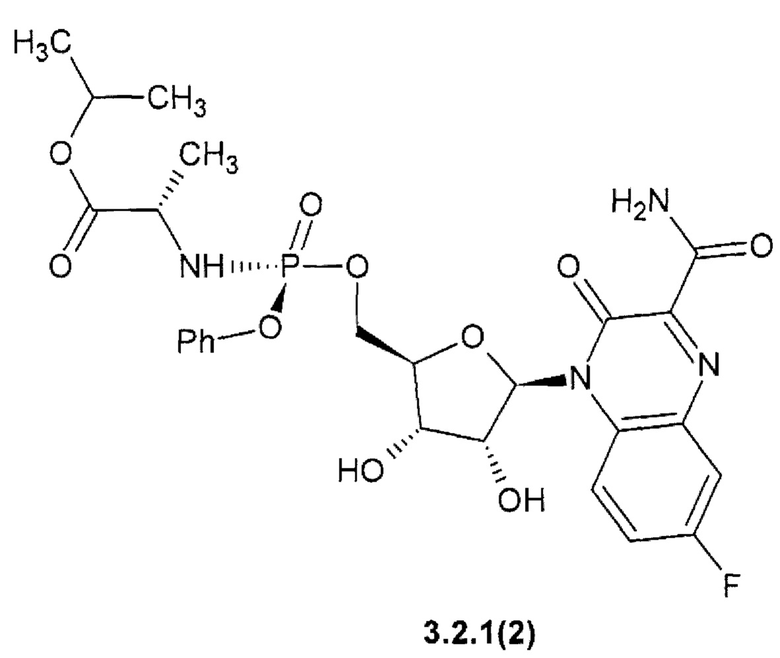
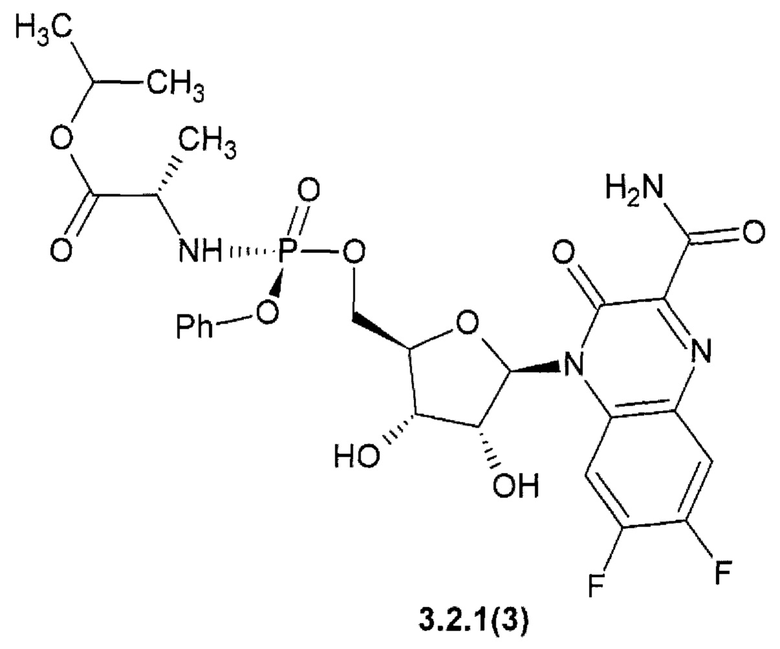
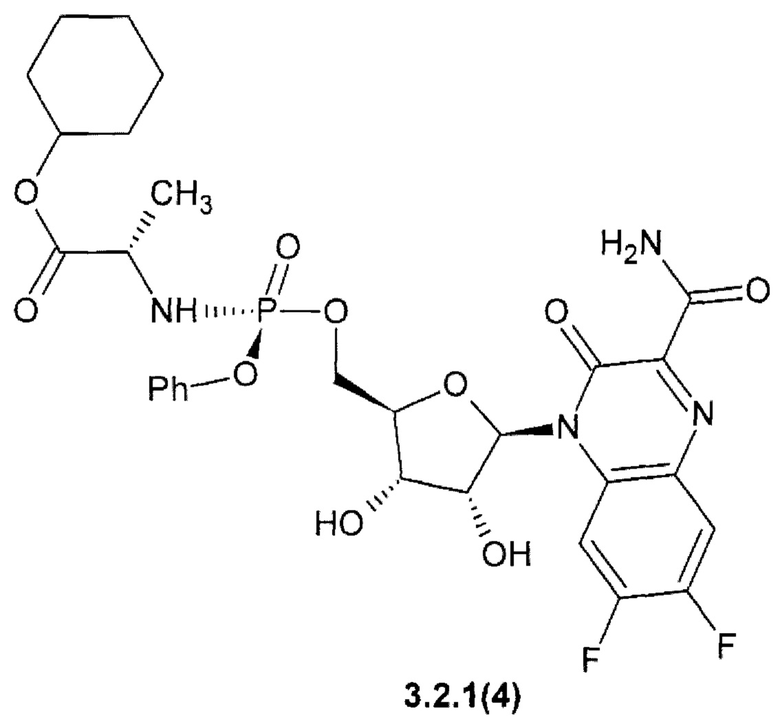
Новое противо-РНК вирусное, в том числе противокороновирусное средство, представляющее собой замещенный хиноксалин общей формулы 1 более активен, чем известный Фавипиравир, который активен против целого ряда РНК вирусов. Например, 7-фтор-3-оксо-3,4-дигидрохиноксалин-2-карбоксамид {1(3)} имеет активность EC50=2,5 мкМ против высокопатогенного штамм Ia/duck/MN/1525/81 (H5N1) РНК вируса гриппа, в то время, как Фавипиравир имеет к этому РНК вирусу EC50=4,5 мкМ [R.W. Sidwell at al. Antimicrob. Agents Chemother. 2007, 51(3): 845-851], т.е. последний в 1.8 раза менее активен, чем новый ингибитор по данному изобретению. Кроме того, Фавипиравир имеет более высокую цитотоксичность и (СС50 > 661 мкМ) [R.W. Sidwell at al. Antimicrob. Agents Chemother. 2007, 51(3): 845-851] и терапевтический индекс ТИ СС50/ЕС50 > 147, значительно уступающий 6-фтор-3-оксо-3,4-дигидрохиноксалин-2-карбоксамиду (1(2)} (СС50 > 1207 мкМ, ТИ > 480).
Исследование противовирусной активности 6,7-дифтор-3-оксо-3,4-дигидрохиноксалин-2-карбоксамида {1(4)} на in vitro модели вируса SARS-COV-2 (штамм BetaCoV/Hong Kong/VM20001061 /2020) продемонстрировало выраженную противо-РНК вирусную активность. В концентрации 100 мкМ 6,7-дифтор-3-оксо-3,4-дигидрохиноксалин-2-карбоксамид {1(4)} на 2 порядка снизил количество копий РНК SARS-Cov-2, а также полностью защитил клетки от цитопатического действия вируса. В тех же условиях Фавипиравир не оказал влияния на репродукцию вируса.
Кроме того, замещенные хиноксалины общей формулы 1 стабильны в интервале рН 2÷4, в то время как Фавипиравир при рН 2 и рН 4 не стабилен (Таблица 1).

Предметом данного изобретения является фармацевтическая композиция, в форме таблеток капсул или инъекций, обладающая свойством ингибитора РНК-зависимой РНК - полимеразы (RdRp) РНК вируса, содержащая в терапевтически эффективном количестве замещенный хиноксалин общей формулы 1 или его стереоизомер, фармацевтически приемлемую соль, аморфную, поликристаллическую или кристаллическую форму необязательно в комбинации с фармацевтически приемлемым носителем или вспомогательным веществом.
Фармацевтические композиции могут включать фармацевтически приемлемые эксцяпиенты. Под фармацевтически приемлемым экспициентами подразумеваются применяемые в сфере фармацевтики разбавители, вспомогательные агенты и/или носители. При необходимости использования фармацевтической композиции по настоящему изобретению в клинической практике она может смешиваться с традиционными фармацевтическими носителями.
Носители, используемые в фармацевтических композиций по настоящему изобретению, представляют собой носители, которые применяются в сфере фармацевтики для получения распространенных форм, в том числе: в пероральных формах используются связующие вещества, смазывающие агенты, дезинтеграторы, растворители, разбавители, стабилизаторы, суспендирующие агенты, бесцветные агенты, корригенты вкуса; в формах для инъекций используются антисептические агенты, солюбилизаторы, стабилизаторы; в местных формах используются основы, разбавители, смазывающие агенты, антисептические агенты.
Предметом данного изобретения является также лекарственное средство, обладающее противо-РНК вирусной активностью, в форме таблеток, капсул или инъекций, помещенных в фармацевтически приемлемую упаковку, предназначенное для профилактики лечения РНК вирусных инфекций у людей и теплокровных животных, включающее в свой состав замещенный хиноксалин общей формулы 1, его стереоизомер, фармацевтически приемлемую соль, аморфную, поликристаллическую или кристаллическую форму или фармацевтическую композицию, включающую замещенный хиноксалин общей формулы 1, его стереоизомер, фармацевтически приемлемую соль, аморфную, поликристаллическую или кристаллическую форму.
Предметом данного изобретения являются также терапевтические коктейли для лечения РНК вирусных инфекций, включающие в качестве одного из компонентов лекарственное средство или фармацевтическую композицию по настоящему изобретению, содержащих в качестве активного компонента замещенный хиноксалин общей формулы 1, его стереоизомер, фармацевтически приемлемую соль, аморфную, поликристаллическую или кристаллическую форму. Терапевтический коктейль для лечения РНК вирусных инфекций, наряду с лекарственным средством по данному изобретению, может включать другие известные препараты, предназначенные для лечения РНК вирусных инфекций, или препараты, усиливающие иммунную систему пациента, в том числе интерфероны.
В соответствии с данным изобретением способ профилактики и лечения РНК вирусных инфекций у животных и людей заключается во введении пациенту лекарственного средства, фармацевтической композиции или терапевтического коктейля по данному изобретению.
Лекарственные средства могут вводиться через ингалятор, перорально или парентерально (например, внутривенно, подкожно, внутрибрюшинно или местно). Клиническая дозировка замещенного хиноксалина общей формулы 1 или его стереоизомера, фармацевтически приемлемой соли, аморфной, поликристаллической или кристаллической формы у пациентов может корректироваться в зависимости от терапевтической эффективности и биодоступности активных ингредиентов в организме, скорости их обмена и выведения из организма, а также в зависимости от возраста, пола и стадии заболевания пациента, при этом суточная доза у взрослых обычно составляет 100÷4000 мг. Поэтому во время приготовления из фармацевтической композиции лекарственного средства по настоящему изобретению в виде единиц дозировки необходимо учитывать вышеназванную эффективную дозировку, при этом каждая единица дозировки препарата должна содержать 100÷4000 мг замещенного хиноксалина общей формулы 1 или его стереоизомера, фармацевтически приемлемой соли, аморфной, поликристаллической или кристаллической формы. В соответствии с указаниями врача или фармацевта данные препараты могут приниматься несколько раз в течение определенных промежутков времени (предпочтительно - от одного до шести раз).
Предметом данного изобретения является также замещенный хиноксалин общей формулы 1, его стереоизомер, фармацевтически приемлемая соль, аморфная, поликристаллическая или кристаллическая форма, исключая 3-оксо-3,4-дигидрохиноксалин-2-карбоксамид {1(1)}
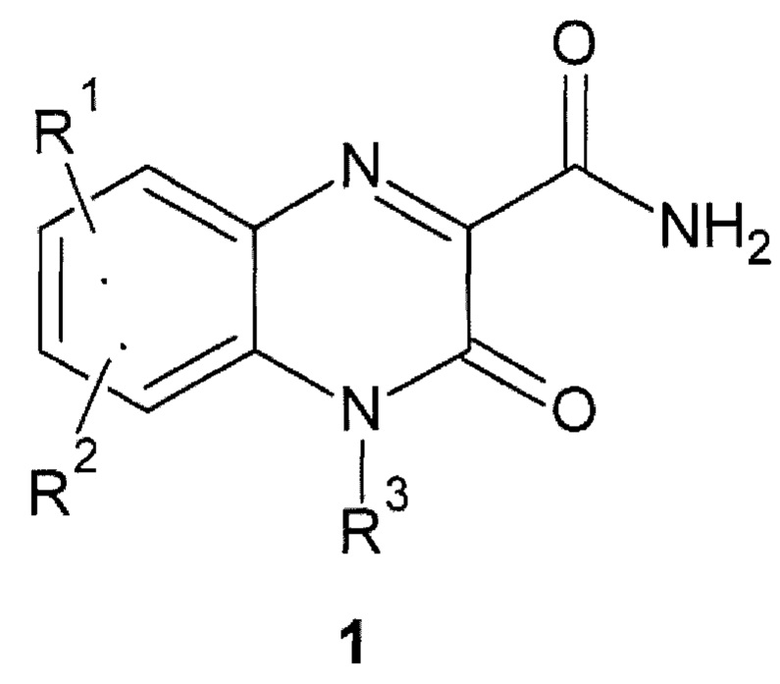
где: R1 и R2 представляют собой необязательно одинаковые атомы водорода или галогена, исключая R1=R2=Н; R3 представляют собой атом водорода или фрагмент общей формулы 2 или его стереоизомер.
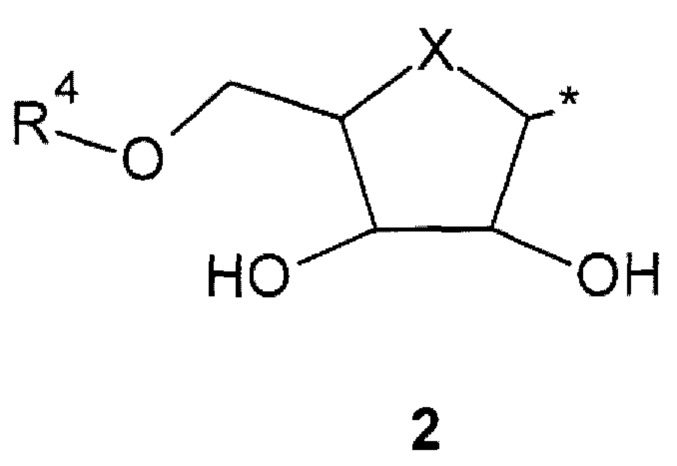
где: X представляет собой атом кислорода или С=СН2 группу; * - место присоединения фрагмента 2; R4 представляет собой атом водорода или заместитель общей формулы 3 или его стереоизомер
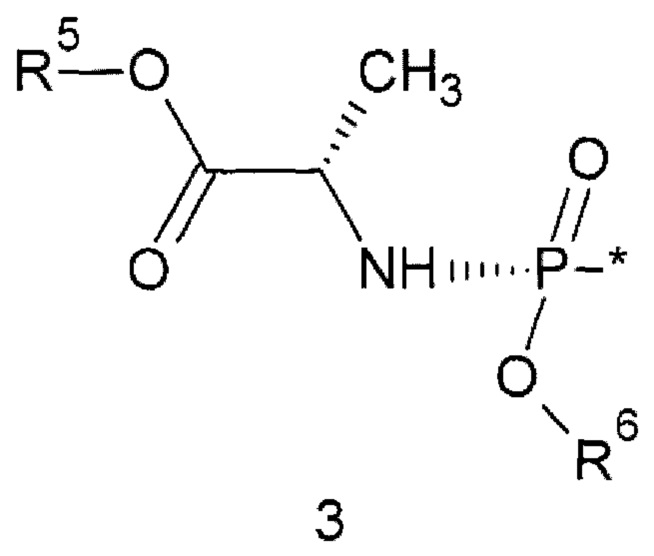
где: * - место присоединения заместителя 3; R5 представляет собой С1-С6алкил или С3-С6циклоалкил; R6 представляет собой необязательно замещенный арил, предпочтительно фенил.
Предпочтительным являются замещенный хиноксалин выбранный из ряда:
3-оксо-3,4-дигидрохиноксалин-2-карбоксамид (1(1)}, 6-фтор-3-оксо-3,4-дигидрохиноксалин-2-карбоксамид {1(2)},
7-фтор-3-оксо-3,4-дигидрохиноксалин-2-карбоксамид {1(3)},
6,7-дифтор-3-оксо-3,4-дигидрохиноксатин-2-карбоксамид {1(4)},
5,8- дифтор-3-оксо-3,4-дигидрохиноксалин-2-карбоксамид {1(5)}.
Предпочтительным являются также замещенный хиноксалин выбранный из ряда 3-оксо-4-β-D-рибофуранозид-3,4-дигидрохиноксалин-2-карбоксамид общей формулы {2.1},
4-(2-С-метил-β-D-рибофуранозил)-3-оксо-3,4-дигидрохиноксалин-2-карбоксамид общей формулы {2.2}.
Более предпочтительным является замещенный хиноксалин выбранное из ряда:
3-оксо-4-β-D-рибофуранозид-3,4-дигидрохиноксалин-2-карбоксамид {2.1(1)},
6-фтор-3-оксо-4-β-D-рибофуранозид-3,4-дигидрохиноксалин-2-карбоксамид {2.1(2)},
7-фтор-3-оксо-4-β-D-рибофуранозид-3,4-дигидрохиноксалин-2-карбоксамид {2.1(3)},
6,7-фтор-3-оксо-4-β-D-рибофуранозид-3,4-дигидрохиноксалин-2-карбоксамид {2.1(4)},
5,8-фтор-3-оксо-4-β-D-рибофуранозид-3,4-дигадрохиноксалин-2-карбоксамид {2.1(5)},
4-(2-С-метил-β-D-рибофуранозид)-3-оксо-3,4-дигидрохиноксалин-2-карбоксамид (2.2(1)},
Предпочтительным являются также замещенный хиноксалин выбранный из ряда:
1-(С1-Сбалки) (2S)-2-{[(S)-{[(2R,3,S,4R,5R)-5-(3-карбамоил-2-оксохиноксалин-1(2Н)-ил)-3,4-дигидрокситетрагидрофуран-2-ил]метокси}(арилокси)фосфорил]амино}пропаноат {3.2.1},
1-(С1-С6алки) (2S)-2-{[(S)-{[(2R,3R,4R,5R)-5-(3-карбамоил-2-оксохиноксалин-1(2H)-ил)-3,4-дигидрокси-4-дифтор-3-гидрокситетрагидрофуран-2-ил]метокси}(арилокси)фосфорил]амино}пропаноат {3.2.2} или их стереоизомеров.
где R1, R2 имеют вышеуказанное значение, R9 и R10 имеют соответствующее вышеуказанное значение для R5 и R6
Более предпочтительным являются замещенный хиноксалин выбранный из ряда: 1-метилэтил (2S)-2-{[(S)-{[(2R,3S,4R,5R)-5-(3-карбамоил-2-оксохиноксачин-1(2Н)-ил)-3,4-дигидрокситетрагидрофуран-2-ил]метокси}(фенокси)фосфорил]амино}пропаноат (3.2.1(1)},
1-метилэтил (2S)-2-{[(S)-{[(2R,3S,4R,5R)-5-(3-карбамоил-7-фтор-2-оксохиноксалин-1(2H)-ил)-3,4-дигидрокситетрагидрофуран-2-ил]метокси}(фенокси)фосфорил]амино}пропаноат (3.2.1(2)},
1-метилэтил (2S)-2-{[(S)-{[(2R,3S,4R,5R)-5-(3-карбамоил-6,7-дифтор-2-оксохиноксалин-1(2H)-ил)-3,4-дигидрокситетрагидрофуран-2-ил]метокси}(фенокси)фосфорил]амино}пропаноат (3.2.1(3)},
циклогексил (2S)-2-{[(S)-{[(2R,3S,4R,5R)-5-(3-карбамоил-6,7-дифтор-2-оксохиноксалин-1(2H)-ил)-3,4-дигидрокситетрагидрофуран-2-ил]метокси}(фенокси)фосфорил]амино}пропаноат (3.2.1(4)},
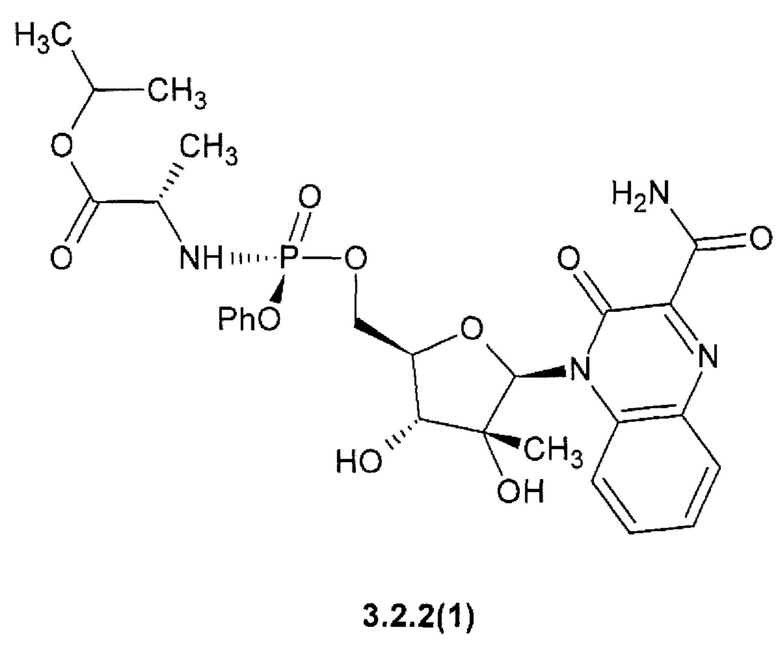
1-метилэтил (2S)-2-([(S)-{[(2R,3R,4R,5R)-5-(3-карбамоил-6,7-дифтор-2-оксохиноксалин-1(2H)-ил)-3,4-дигидрокси-4-метилтетрагидрофуран-2-2-ил]метокси}(фенокси)фосфорил]амино}пропаноат {3.2.2(1)} или их стереоизомеров, фармацевтически приемлемых солей, аморфных, поликристаллических или кристаллических форм.
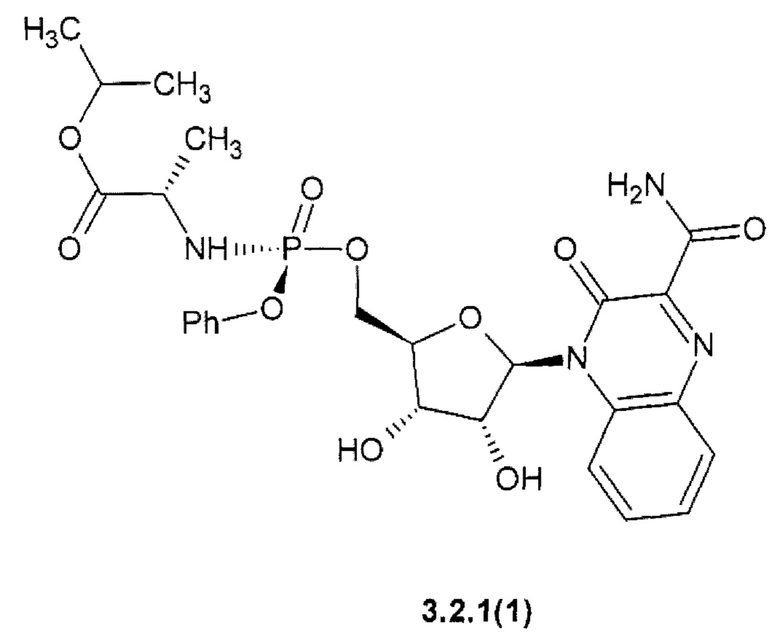
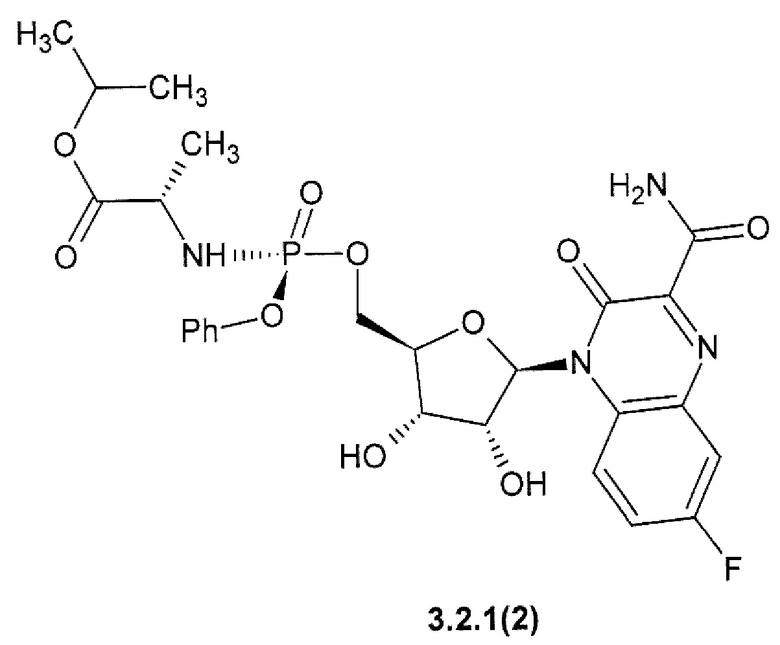
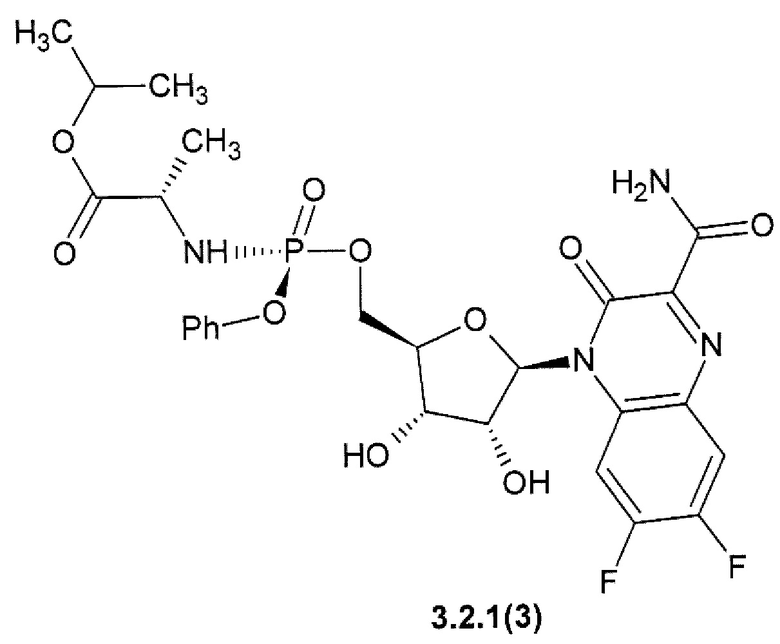
Предметом данного изобретения является способ получения 3-оксо-3,4-дигидрохиноксалин-2-карбоксамида общей формулы 1, исключая 3-оксо-3,4-дигидрохиноксалин-2-карбоксамид {1(1)}, взаимодействием С1-С4-алкил 3-оксо-3,4-дигидрохиноксалин-2-карбоксилата общей формулы В1
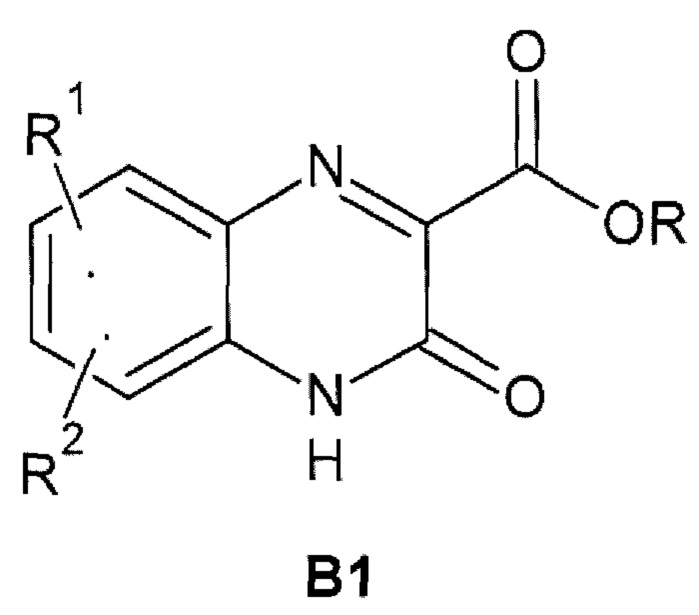
где R1 и R2 имеют вышеуказанное значение, исключая R1=R2=Н.
Предметом данного изобретения является способ получения 3-оксо-3,4-дигидрохиноксалин-2-карбоксамида общей формулы 2.1-2.7 или их стереоизомеров. В качестве исходных билдинг блоков использованы, с одной стороны, 3-оксо-3,4-дигидрохиноксалин-2-карбоксамйД общей формулы 1а или его триметилсилильное производное общей формулы 1b (Схема 1), а с другой стороны сахара 2.1-2.7 (Схема 2)
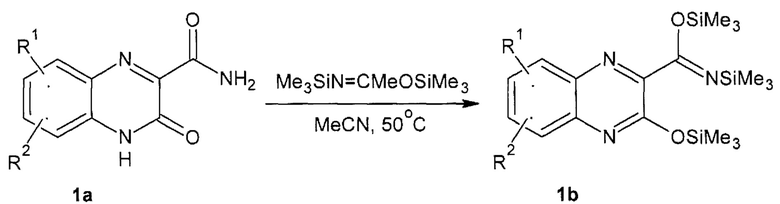
где: R1 и R2 имеют вышеуказанное значение
Схема 1. Замещенные 3-оксо-3,4-дигидрохиноксалин-2-карбоксамиды 1а и 1b.
Схема 2. Исходные билдинг-блоки 2.1, 2.2.
Способ получения замещенного 3-оксо-3,4-дигидрохиноксалин-2-карбоксамида общей формулы 2.1 и общей формулы 3.2.1 представлен на Схеме 3 и заключается во взаимодействии тетра-O-ацетил-β-D-рибофуранозы (В2.1) с триметилсилильным производным 3-оксо-3,4-дигидрохиноксалин-2-карбоксамида общей формулы 1b с последующим снятием ацетильных защитных групп с В2.1.1 и взаимодействием образующегося 2.1 с 2,2-димстоксипроиан в присуствии моногидрата n-толуолсульфокислоты, последующим взаимодействием образующегося В2.1.2 с (S)-изопропил 2-((S)-(перфторфенокси)(фенокси)фосфориламино)пропаноатом в присуствии t-BuMgCl с получением В2.1.3 и превращением последнего под действием TFA в целевой продукт общей формулы 3.2.1.
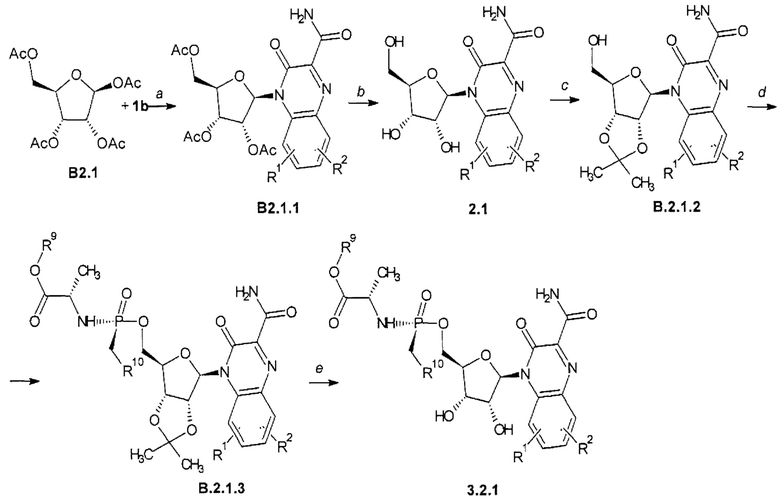
R1, R2, R9 и R10 имеют вышеуказанное значение.
Схема 3. Схема получения замещенного 3-оксо-3,4-дигидрохиноксалин-2-карбоксамида общей формулы 2.1 и 3.2.1. а - SnCl4, MeCN; b - NH3, MeOH; с - Ме2С(ОМе)2, TsOH, Ме2СО; d - t-BuMgCl, (S)-изопроиил 2-((S)-(перфторфенокси)(фенокси)фосфориламино)-пропаноат, THF, 0-10°С; е - TFA, H2O, MeOH.
Способ получения замещенного 3-оксо-3,4-дигидрохиноксалин-2-карбоксамида общей формулы 2.2 и общей формулы 3.2.2 представлен на Схеме 4 и заключается во взаимодействии (2S,3R,4R,5R)-2,4-бис(бензоилокси)-5-[(бензоилокси)метил]-3-метилоксалан-3-ил бензоат (В2.2) с триметилсилильным производным 3-оксо-3,4-дигидрохиноксалин-2-карбоксамида общей формулы 1b с последующим снятием защитных бензоильных групп с В2.2.1 и взаимодействием образующегося 2.2 с 2,2-диметоксипропан в присутствии моногидрата n-толуолсульфокислоты, последующим взаимодействием образующегося В2.2.2 с (S)-изопропил 2-((S)-(перфторфенокси)(фенокси)фосфориламино)-пропаноатом в присуствии t-BuMgCl с получением В2.2.3 и превращением последнего под действием TFA в целевой продукт общей формулы 3.2.2.
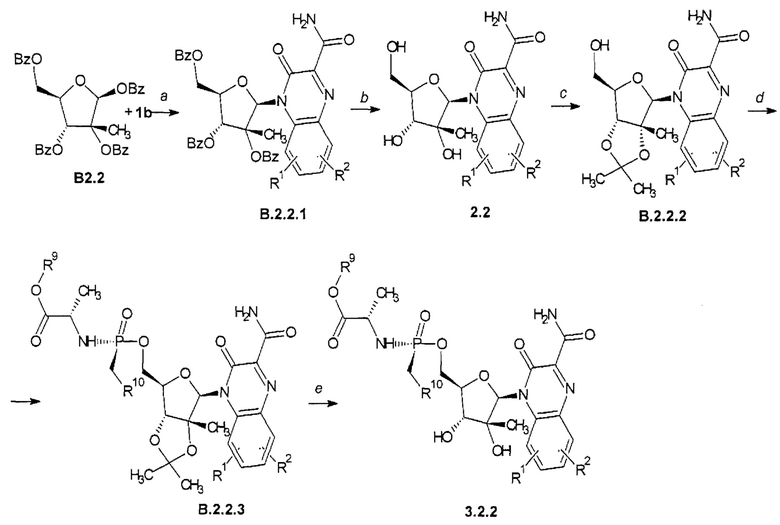
R1, R2, R9 и R10 имеют вышеуказанное значение.
Схема 4. Схема получения замещенного 3-оксо-3,4-дигидрохиноксалин-2-карбоксамида общей формулы 2.2 и 3.2.2. а - SnCl4, MeCN; b - NH3, MeOH; с - Me2C(OMe)2, TsOH, Me2CO; d - t-BuMgCl, (S)-изопропил 2-((S)-(перфторфенокси)(фенокси)фосфориламино)-пропаноат, THF, 0-10°С; e - TFA, H2O, MeOH.
Изобретение иллюстрируется следующими чертежами
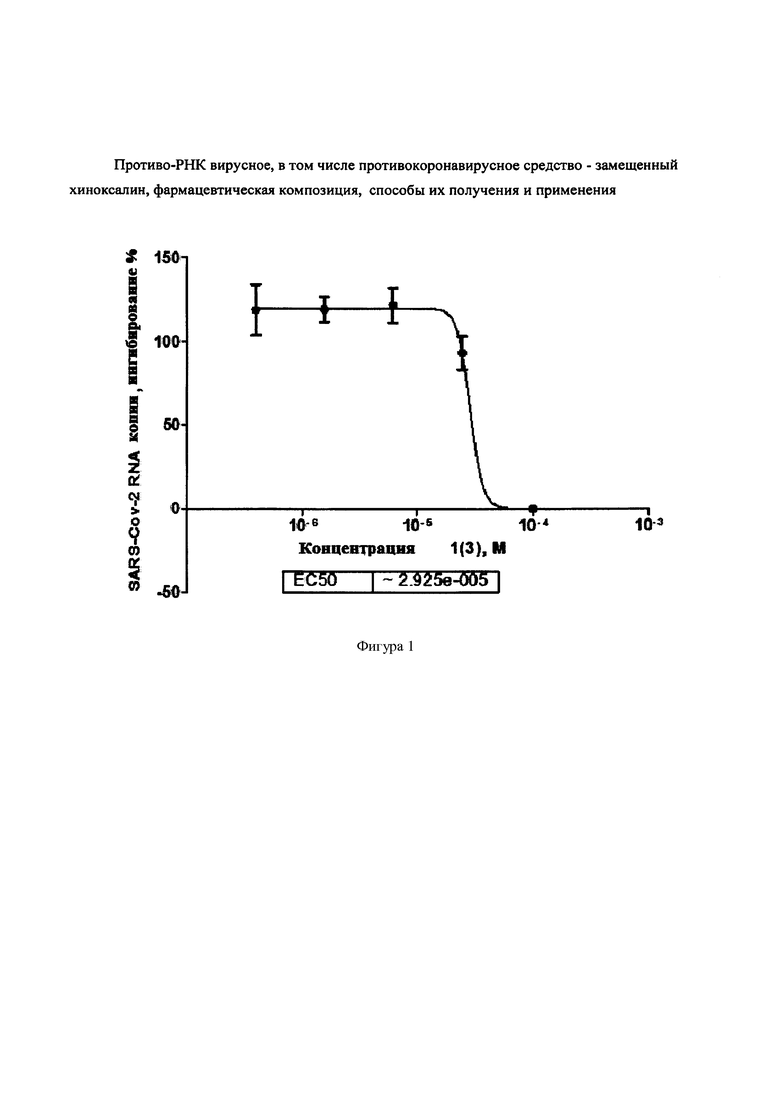
Фигура 1. Концентрационная зависимость ингибирования вируса SARS-Cov-2 6,7-дифтор-3-оксо-3,4-дигидрохиноксалин-2-карбоксамидом {1(4)}.
Данное изобретение иллюстрируется, но не ограничивается следующими примерами.
Пример 1. Получение 6-фтор-3-оксо-3,4-дигидрохиноксалин-2-карбоксамида формулы {1(2)}. Этил 6-фтор-3-оксо-3,4-дигидрохиноксалин-2-карбоксилата {B1(1): R=Et, R1=Н, R2=F} (0,236 г, 1 ммоль) перемешивали в закрытой пробирке с 2,5 мл 7N метанольного раствора NH3 при 50°С. в течение 15 ч. После охлаждения желтый осадок отфильтровывали, промывали эфиром и сушили в вакууме. Получают 1(2): 1Н NMR (DMSO-d6, 400 MHz) δ 12.77 (brs, 1H), 8.52 (brs, 1H), 7.84 (brs, 1H), 7.69 (dd, J1=8.8 Hz, J2=2.4 Hz, 1H), 7.54 (dt, J1=8.8 Hz, J2=2.4 Hz, 1H), 7.37 (dd, J1=8.8 Hz, J2=5.2 Hz, 1H).
Пример 2. Получение 7-фтор-3-оксо-3,4-дигидрохиноксалин-2-карбоксамида формулы {1(3)}. Получают, как в примере 1 исходя из этил 7-фтор-3-оксо-3,4-дигидрохшюксалин-2-карбоксилата {(В1(2): R=Et, R1=F, R2=Н} и аммиака. 1(3): 1Н NMR (DMSO-d6, 400 MHz) δ 12.77 (brs, 1H), 8.52 (brs, 1H), 7.84 (brs, 1H), 7.69 (dd, J1=8.8 Hz, J2=2.4 Hz, 1H), 7.54 (dt, J1=8.8 Hz, J2=2.4 Hz, 1H), 7.37 (dd, J1=8.8 Hz, J2=5.2 Hz, 1H).
Пример 3. Получение этил 6,7-дифтор-3-оксо-3,4-дигидрохиноксалин-2-карбоксилата {В1(3): R=Et, R1=R2=F}. К раствору 4,5-дифторбензол-1,2-диамина (10 ммоль) в EtOH (30 мл) добавляли диэтилкетомалонат (1,74 г, 10 ммоль) и кипятили с обратным холодильником в течение 2 часов. После охлаждения реакционную смесь концентрировали и обрабатывали этилацетатом. Осадок собирали фильтрацией, промывали этилацетатом, эфиром и сушили в вакууме, получая В1(3): 1Н NMR (DMSO-d6, 400 MHz) δ 12.98 (brs, 1Н), 8.01 (dd, J1=10.8 Hz, J2=8.0 Hz, 1H), 7.27 (dd, J1=10.8 Hz, J2=7.2 Hz, 1H), 4.37 (q, J=7.0 Hz, 2H), 1.31 (t, J=7.0 Hz, 3H).
Пример 4. Получение 6,7-дифтор-3-оксо-3,4-дигидрохиноксалин-2-карбоксамида {1(4)}. Получают, как в примере 1 исходя из этил 6,7-дифтор-3-оксо-3,4-дигидрохиноксалин-2-карбоксилата {В1(3)} и аммиака. 1(4): 1Н NMR (DMSO-d6, 400 MHz) δ 12.77 (brs, 1Н), 8.52 (brs, 1H), 7.84 (brs, 1H), 7.69 (dd, J1=8.8 Hz, J2=2.4 Hz, 1H), 7.54 (dt, J1=8.8 Hz, J2=2.4 Hz, 1H), 7.37 (dd, J1=8.8 Hz, J2=5.2 Hz, 1H).
Пример 5. Получение 3-оксо-4-β-D-рибофунарозид-3,4-дигадрохиноксалин-2-карбоксамида общей формулы {2.1} и 4-(2-С-метил-β-D-рибофуранозид)-3-оксо-3,4-дигидрохиноксалин-2-карбоксамид общей формулы {2.2}.
К суспензии соединения 1b (1 ммоль) в 10 мл сухого ацетонитрила под аргоном добавляют N,O-бис(триметилсилил)ацетамид (0.856 мл, 3.5 ммоль) и смесь перемешивают 2 часа при 50°С. Раствор охлаждают и добавляют соответствующую рибофуранозу (1.1 ммоль) и олова(IV) хлорид (0.176 мл, 1.5 ммоль). Смесь перемешивают под аргоном при 70°С в течение 15 часов. Раствор упаривают в вакууме, разбавляют дихлорметаном, промывают водой и насыщенным NaHCO3, сушат над Na2SO4 и упаривают в вакууме. Целевой продукт выделяют методом колоночной хроматографии на силикагеле (дихлорметан : метанол 19:1).
К полученному продукту добавляют ледяной 4N раствор аммиака в метаноле и смесь перемешивают в холодильнике в течение 12 ч. Раствор упаривают, остаток обрабатывают ацетонитрилом. Осадок отфильтровывают, промывают ацетонитрилом, эфиром и сушат в вакууме. Получают соответственно целевой продукт общей формулы 2.1 или 2.2, в том числе: 3-оксо-4-β-D-рибофунарозид-3,4-дигидрохиноксалин-2-карбоксамид {2.1(1)}, Мол. вес 321, LC-MS (ESI) 322 (М+Н)+; 6-фтор-3-оксо-4-β-D-рибофунарозид-3,4-дигидрохиноксалин-2-карбоксамид {2.1(2)}, Мол. вес 341, LC-MS (ESI) 342 (М+Н)+; 7-фтор-3-оксо-4-β-D-рибофуранозид-3,4-дигидрохиноксалин-2-карбоксамид {2.1(3)}, Мол. вес 341, LC-MS (ESI) 342 (М+Н)+; 6,7-фтор-3-оксо-4-β-D-рибофунарозид-3,4-дигидрохиноксалин-2-карбоксамид {2.1(4)}, Мол. вес 357, LC-MS (ESI) 348 (М+Н)+; 5,8-фтор-3-оксо-4-β-D-рибофунарозид-3,4-дигидрохинокеалин-2-карбоксамид {2.1(5)}, Мол. вес 357, LC-MS (ESI) 348 (М+Н)+; 4-(2-С-метил-β-D-рибофуранозид)-3-оксо-3,4-дигидрохиноксалин-2-карбоксамид {2.2(1)}, Мол. вес 335, LC-MS (ESI) 336 (М+Н)+.
Пример 6. Получение 1-(С1-С6алки) (2S)-2-{[(S)-{[(2R,3S,4R,5R)-5-(3-карбамоил-2-оксохиноксалин-1(2H)-ил)-3,4-дигидрокситетрагидрофуран-2-ил]метокси}(арилокси)-фосфорил]амино}пропаноата общей формулы {3.2.1} и 1-(С1-С6алки)(2S)-2-{[(S)-{[(2R,3R,4R,5R)-5-(3-карбамоил-2-оксохиноксалин-1(2H)-ил)-3,4-дигидрокси-4-дифтор-3-гидрокситетрагидрофуран-2-ил}метокси}(арилокси)фосфорил]амино}пропаноат общей формулы {3.2.2}.
К суспензии соединения 2.1 или 2.2 (1 ммоль) в 10 мл ацетона добавляют 2,2-диметоксипропан (1.23 мл, 10 ммоль) и n-толуолсульфокислоты моногидрат (0.228 г, 1.2 ммоль) и смесь перемешивают при комнатной температуре в течение 15 ч. Смесь упаривают в вакууме, растворяют в метаноле, обрабатывают Амберлитом А-26 в щелочной форме (предварительно промывают 2N NaOH и водой) и перемешивают в течение 2 ч. Смесь фильтруют, упаривают в вакууме, остаток промывают этилацетатом, сушат в вакууме и получают соответственно В.2.1.2 или В.2.2.2.
К раствору соединения В.2.1.2 или В.2.2.2 (1 ммоль) в 10 мл ТГФ под аргоном при температуре -10-0°С добавляют 1М раствор трет-бутилмагния хлорида в ТГФ (2.25 мл, 2.25 ммоль), поддерживая температуру смеси ниже 0°С. Реакционную массу перемешивают под аргоном при температуре около 0°С в течение 1 ч. Затем при температуре -10-0°С присыпают (S)-изопропил 2-((S)-(перфторфенокси)(фенокси)фосфориламино)пропаноат (0.63 г, 1.2 моль) и смесь перемешивают под аргоном при комнатной температуре в течение 15 ч. Реакционную массу разбавляют дихлорметаном, промывают 5%-м раствором лимонной кислоты с 10% NaCl и упаривают в вакууме. Остаток В.2.1.3 или В.2.2.3 растворяют в 8 мл TFA, добавляют 2 мл воды и перемешивают в течение 30 мин. Раствор упаривают в вакууме и выделяют целевой продукт общей формулы 3.2.1 или 3.2.2 посредством ВЭЖХ, в том числе: 1-метилэтил (2S)-2-{[(S)-{[(2R,3S,4R,5R)-5-(3-карбамоил-2-оксохиноксалин-1(2H)-ил)-3,4-дигидрокситетрагидрофуран-2-ил]метокси}(фенокси)-фосфорил]амино}пропаноат {3.2.1(1)}, Мол. вес 590, LC-MS (ESI) 591 (М+Н)+;
1-метилэтил (2S)-2-{[(S)-{[(2R,3S,4R,5R)-5-(3-карбамоил-7-фтор-2-оксохиноксалин-1(2H)-ил)-3,4-дигидрокситетрагидрофуран-2-ил]метокси}(фенокси)фосфорил]амино}пропаноат {3.2.1(2)}, Мол. вес 608, LC-MS (ESI) 681 (М+Н)+; 1-метилэтил (2S)-2-{[(S)-{[(2R,3S,4R,5R)-5-(3-карбамоил-6,7-дифтор-2-оксохиноксалин-1(2H)-yl)-3,4-дигидрокситетрагидрофуран-2-ил]метокси}(фенокси)фосфорил]амино}пропаноат {3.2.1(3)}, Мол. вес 626, LC-MS (ESI) 627 (М+Н)+; циклогексил (2S)-2-{[(S)-{[(2R,3S,4R,5R)-5-(3-карбамоил-6,7-дифтор-2-оксохиноксалин-1(2Н)-ип)-3,4-дигидрокситетрагидрофуран-2-ил]метокси}(фенокси)фосфорил]амино}пропаноат {3.2.1(4)}, Мол. вес 666, LC-MS (ESI) 667 (М+Н)+; 1-метилэтил (2S)-2-{[(S)-{[(2R,3R,4R,5R)-5-(3-карбамоил-6,7-дифтор-2-оксохииоксачин-1(2H)-yl)-3,4-диг-идрокси-4-метилтетрагидрофуран-2-2-ил]метокси}(фенокси)-фосфорил]амино}пропаноат {3.2.2(1)}, Мол. вес 604, LC-MS (ESI) 605 (М+Н)+.
Пример 7. Получение фармацевтической композиции в форме таблеток. Смешивают 1600 мг крахмала, 1600 мг измельченной лактозы, 400 мг талька и 1000 мг 6,7-дифтор-3-оксо-3,4-дигидрохиноксалин-2-карбоксамид {1(1), 1(3) или 1(4)}. Полученный брусок измельчают в гранулы и просеивают через сита, собирая гранулы размером 14-16 меш. Полученные гранулы таблетируют в подходящую форму таблетки весом 920 мг, каждая из которых содержит 200 мг 6,7-дифтор-3-оксо-3,4-дигидрохиноксалин-2-карбоксамид {1(1), 1(3) или 1(4)} весом 560 мг каждая.
Пример 8. Получение фармацевтической композиции в форме капсул. Тщательно смешивают 6,7-дифтор-3-оксо-3,4-дигидрохиноксалин-2-карбоксамид {3.2.1(1), 3.2.1(2) или 3.2.1 (3)}с порошком лактозы в соотношении 2:1. Полученную порошкообразную смесь упаковывают по 600 мг в желатиновые капсулы подходящего размера, каждая из которых содержит 200 мг 6,7-дифтор-3-оксо-3,4-дигидрохиноксалин-2-карбоксамид {3.2.1(1), 3.2.1(2) или 3.2.1(3)}.
Пример 9. Противо-РНК вирусная активность и цитотоксичность 6,7-дифтор-3-оксо-3,4-дигидрохиноксалин-2-карбоксамид {1(3)}. Противо-РНК вирусная активность против Ia/duck/MN/1525/81 штамма РНК вируса гриппа испытуемого соединения определялась в Институте противовирусных исследований университет штата Юта с использованием нейтрального красного эссея. Цитотоксичность испытуемых соединений определяют параллельно. 7-фтор-3-оксо-3,4-дигидрохиноксалин-2-карбоксамид {1(3)} имеет активность EC50=2,5 мкМ, цитотоксичность СС50>1207 мкМ и терапевтический интервал ТИ>480).
Пример 10. Противо-РНК вирусная активность и цитотоксичность. Протокол тестирования веществ против вируса SARS-CoV-2 описан в статье Ka-Tim Choy al. Remdesivir, lopinavir, emetine, and homoharringtonine inhibit SARS-CoV-2 replication in vitro. Antiviral Research 2020, 178, 10478; https://www.sciencedirect.com/science/ article/pii/S016635422030200X. Активность соединений определяли по отношению к вирусу SARS-CoV-2, штамм BetaCoV/Hong Kong/VM20001061/2020, выделенному из образцов пациентов, больных COVID-2019. Культуру VERO Е6 сеяли в 96-луночный планшет, инкубировали при 37°С и 5% CO2 в течение 24х часов. Готовили 100 мкМ сток-раствор исследуемого вещества в ДМСО, делали последовательное разведения в ДМСО с шагом 4 (чтобы получились 1000-кратные растворы в ДМСО). Соответствующее количество маточного раствора исследуемого вещества в ДМСО подвергали промежуточному разведению в среде для культивирования клеток, добавляли полученные растворы вещества к клеткам, финальная концентрация ДМСО составила 0.1%. Каждое вещество исследовалось в пяти концентрациях: 100 мкМ, 25 мкМ, 6.25 мкМ, 1.56 мкМ, 0.39 мкМ и 0 (пустой контроль) в 2х повторах (лунках). Через 2 часа после добавления веществ, добавляли вирус при MOI=0.02, и инкубировали при 37°С и 5% СО2 в течение 48 часов. Отбирали 200 мкл супернатанта из культуры вируса, выделяли нуклеиновые кислоты, использовали QRT-ПЦР для выявления вирусной нагрузки с использованием набора TaqMan™ Fast Virus 1-Step Master Mix. Получали, в частности для соединения 1(4) ЕС50=29,25 мкМ (Фигура 1). Цитотоксичность (СС50 > 100 мкМ) определяли в клетках с добавленными веществами и без вируса с помощью набора CellTiter-Glo (Promega).
| название | год | авторы | номер документа |
|---|---|---|---|
| Макрогетероциклические нуклеозидные производные и их аналоги, получение и применение | 2017 |
|
RU2731385C1 |
| Нуклеотиды, включающие N-[(S)-1-циклобутоксикарбонил]фосфорамидатный фрагмент, их аналоги и их применение | 2017 |
|
RU2659388C1 |
| Замещенный 3,4,12,12а-тетрагидро-1Н-[1,4]оксазино[3,4-c]пиридо[2,1-f] [1,2,4]триазин-6,8-дион, фармацевтическая композиция, способы их получения и применения | 2019 |
|
RU2720305C1 |
| Пролекарство ингибитора NS5B HCV полимеразы, способ его получения и применения | 2017 |
|
RU2644156C1 |
| Противовирусная композиция и способ ее применения | 2017 |
|
RU2650610C1 |
| Анелированные 9-гидрокси-1,8-диоксо-1,3,4,8-тетрагидро-2Н-пиридо[1,2-a]пиразин-7-карбоксамиды - ингибиторы интегразы ВИЧ, способы их получения и применения | 2019 |
|
RU2717101C1 |
| Антикоронавирусный терапевтический агент - замещенный 7-гидрокси-3,4,12,12а-тетрагидро-1H-[1,4]оксазино[3,4-c]пиридо[2,1-f][1,2,4]триазин-6,8-дион для профилактики и лечения COVID-19 | 2020 |
|
RU2745985C1 |
| Ингибитор вируса гепатита В (ВГВ) | 2017 |
|
RU2666727C1 |
| ЗАМЕЩЕННЫЕ (2R,3R,5R)-3-ГИДРОКСИ-(5-ПИРИМИДИН-1-ИЛ)ТЕТРАГИДРОФУРАН-2-ИЛМЕТИЛ АРИЛ ФОСФОРАМИДАТЫ | 2013 |
|
RU2553996C1 |
| Противокоронавирусное средство для комбинированной терапии COVID-19 (SARS-CoV-2). | 2020 |
|
RU2742116C1 |
Изобретение относится к замещенному хиноксалину общей формулы 1, где R1 и R2 представляют собой необязательно одинаковые атомы водорода или галогена, R3 представляет собой атом водорода, при условии, что R1, R2 и R3 одновременно не означают водород, или оба R1 и R2 не означают 6,7-дихлор, или если R1 означает водород, то R2 не означает 7-хлор. Изобретение также относится к фармацевтической композиции, обладающей свойством ингибитора РНК-зависимой РНК-полимеразы (RdRp) РНК вирусов, на основе указанного соединения формулы 1. Технический результат: получены новые соединения и фармацевтические композиции на их основе, которые могут найти применение в медицине в качестве средств, ингибирующих РНК-зависимую РНК-полимеразу (RdRp) РНК вирусов, где вирус представляет собой коронавирус SARS-CoV-2 и вирус гриппа. 3 н. и 2 з.п. ф-лы, 1 ил., 1 табл., 10 пр.
1. Замещенный хиноксалин общей формулы 1
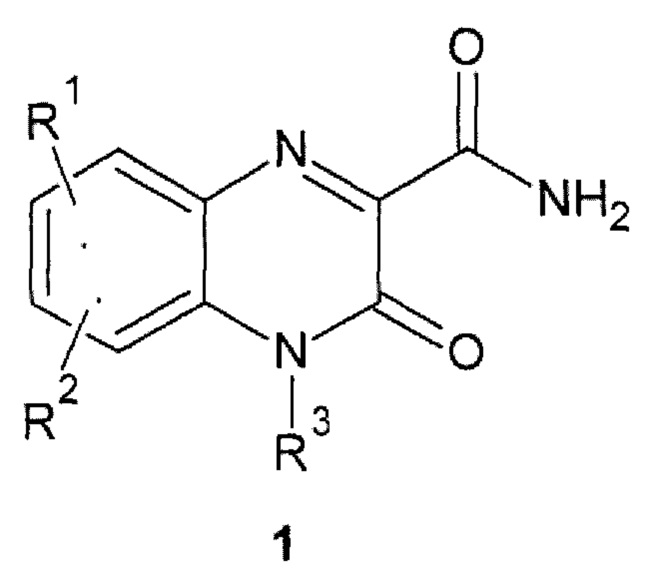 ,
,
где R1 и R2 представляют собой необязательно одинаковые атомы водорода или галогена, R3 представляет собой атом водорода,
при условии, что R1, R2 и R3 одновременно не означают водород, или оба R1 и R2 не означают 6,7-дихлор, или если R1 означает водород, то R2 не означает 7-хлор.
2. Замещенный хиноксалин по п. 1, выбранный из ряда
6-фтор-3-оксо-3,4-дигидрохиноксалин-2-карбоксамид {1 (2)},
7-фтор-3-оксо-3,4-дигидрохиноксалин-2-карбоксамид {1(3)},
6,7-дифтор-3-оксо-3,4-дигидрохиноксалин-2-карбоксамид {1(4)},
5,8-дифтор-3-оксо-3,4-дигидрохиноксалин-2-карбоксамид {1(5)}
3. Замещенный хиноксалин по п. 1 или 2, обладающий свойствами ингибитора РНК-зависимой РНК-полимеразы (RdRp) РНК вируса, где вирус представляет собой коронавирус SARS-CoV-2 и вирус гриппа.
4. Фармацевтическая композиция, обладающая свойством ингибитора РНК-зависимой РНК-полимеразы (RdRp) РНК вируса, где вирус представляет собой коронавирус SARS-CoV-2 и вирус гриппа, содержащая в терапевтически эффективном количестве замещенный хиноксалин общей формулы 1 по п. 1 или 2 в комбинации с фармацевтически приемлемым носителем или вспомогательным веществом.
5. Применение замещенного хиноксалина общей формулы 1 по п. 1 или 2 или фармацевтической композиции по п. 4 для ингибирования РНК-зависимой РНК-полимеразы (RdRp) РНК вируса, где вирус представляет собой коронавирус SARS-CoV-2 и вирус гриппа.
| АЗОТСОДЕРЖАЩИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ КАРБОКСАМИДА ИЛИ ИХ СОЛИ И ПРОТИВОВИРУСНЫЕ СРЕДСТВА, ВКЛЮЧАЮЩИЕ ИХ | 1999 |
|
RU2224520C2 |
| RU 2015104888 A, 27.08.2016 | |||
| WO 1992011245 A1, 09.07.1992 | |||
| P | |||
| Sanna et al | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Снеговая лыжа для самолетов | 1913 |
|
SU455A1 |

