Данное изобретение относится к фармацевтическим композициям, которые содержат кислую сернокислую соль (2-гидроксиэтокси)амида 6-(4-бром-2-хлорфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты (далее в данной заявке обозначается как "Агент"), в частности к композициям, которые доставляются перорально, содержащим Агент; к применению указанных композиций в качестве лекарственного средства; и к способам получения указанных композиций.
Агент раскрывается в международной патентной заявке WO 2007/076245 и представляет собой мощный ингибитор MEK. Агент представляет собой кислую сернокислую соль соединения со структурой формулы I:
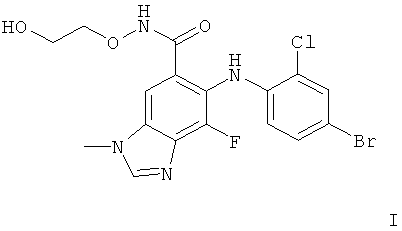
Агент обладает антипролиферативной активностью и, как можно ожидать, будет полезным в лечении заболеваний или медицинских состояний, которые опосредуются полностью или частично MEK, и, в частности, разных видов рака, таких как рак мозга, легких, чешуйчатых клеток, желчного или мочевого пузыря, желудка, поджелудочной железы, молочных желез, головы, шеи, ренального рака, рака почки, яичника, предстательной железы, колоректального рака, рака пищевода, тестикулярного, гинекологического рака, рака щитовидной железы или злокачественной меланомы. Агент может также использоваться в лечении неракового гиперпролиферативного заболевания, такого как доброкачественная гиперплазия кожи (например, псориаз), рестеноз или гипертрофия предстательной железы (например, доброкачественная гипертрофия предстательной железы (ВРН)), и для лечения других опосредованных MEK заболеваний, включая заболевание поджелудочной железы или почек (включая пролиферативный гломерулонефрит и индуцированное диабетом почечное заболевание), или для лечения боли у млекопитающего. Также предполагается, что Агент также будет полезным для предотвращения бластоцитарной имплантации у млекопитающего или для лечения заболевания, связанного с васкулогенезом или ангиогенезом у млекопитающего. Такие заболевания могут включать опухолевый ангиогенез, хроническое воспалительное заболевание, такое как ревматоидный артрит, атеросклероз, воспалительное заболевание кишечника, заболевания кожи, такие как псориаз, экзема, а также склеродерму, диабет, диабетическую ретинопатию, ретролентальну фиброплазию, возрастную дегенерацию желтого пятна, гемангиому, глиому, меланому, саркому Капоши и рак яичника, молочной железы, легкого, поджелудочной железы, предстательной железы, кишечника и эпидермоидный рак.
Форма свободного основания Агента (то есть (2-гидроксиэтокси)амида 6-(4-бром-2-хлорфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты) была классифицирована как соединение класса 4 BCS (в соответствии с Фармацевтической Системой Классификации, как определено в Руководстве для промышленности: Исключение in vivo исследований биодоступности и биоэквивалентности для дозированных твердых пероральных форм немедленного высвобождения на основе Биофармацевтической Системы Классификации), что свидетельствует о том, что она имеет низкую скорость солюбилизации/растворимости и низкую проницаемость. Такие соединения типично демонстрируют низкую и/или изменчивую биодоступность и, кроме того, биодоступность формы свободного основания Агента из традиционной композиции для таблеток является относительно слабой (~18% у собак).
Заявители раньше идентифицировали специфическую форму соли (2-гидроксиэтокси)амида 6-(4-бром-2-хлорфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты, которая демонстрирует уникальные фармацевтические свойства, что делает ее особенно приемлемой для применения в качестве лекарственного средства. Эта специфическая форма соли, в частности кислая сернокислая соль (1:1 лечебное средство: H2SO4) (2-гидроксиэтокси)амида 6-(4-бром-2-хлорфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты (выше и ниже в данной заявке обозначается как "Агент"), была раскрыта в WO 2007/076245. Эта соль является кристаллической и неожиданно была выявлена такой, которая обладает улучшенными фармацевтическими свойствами при сравнении с формой свободного основания Агента и другими солями (2-гидроксиэтокси)амида 6-(4-бром-2-хлорфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты. В частности, скорость растворения этой соли, а также ее биодоступность были выявлены как особенно высокие по сравнению с формой свободного основания Агента и другими солями (2-гидроксиэтокси)амида 6-(4-бром-2-хлорфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты.
Для того чтобы рецептировать фармацевтически активное соединение, такое как Агент, в виде приемлемой дозированной формы, активное соединение должно дополнительно обладать приемлемыми биофармацевтическими свойствами, такими как способность к солюбилизации и свойства растворения, а также, соответственно, обладать приемлемой стабильностью и свойствами, которые делают его приемлемым для обработки. В этой связи особая проблема возникает с Агентом. Форма свободного основания Агента является слабо основным соединением и имеет две основные группы со значениями 
Таким образом, существует потребность в фармацевтической композиции, которая содержит Агент (то есть кислую сернокислую соль), в частности в композициях, в которых стабильность Агента поддерживается во время процессов обработки и хранения, для обеспечения приемлемой абсорбции и/или в которых биодоступность Агента достигается во время дозирования.
Согласно первому аспекту данного изобретения обеспечивается фармацевтическая композиция, которая включает Агент и матриксный носитель, где матриксный носитель существенно состоит из одного или более фармацевтически приемлемых носителей, выбранных из следующих:
(a) d-альфа-токоферилполиэтиленгликоль 1000 сукцинат;
(b) полигликозилированные глицериды;
(c) полиэтиленгликоли (ПЭГи); и
(d) твердые жиры;
и где Агент является диспергированным в матриксном носителе.
Мы неожиданно обнаружили, что стабильность Агента может поддерживаться в композициях данного изобретения. Много материалов, которые являются приемлемыми для образования матриксного носителя, являются традиционно известными в области техники, такие как, например, эмульгаторы, солюбилизирующие агенты и энхансеры абсорбции, и используются для улучшения кинетик растворения и биодоступности плохо растворимых лечебных средств. Однако заявители неожиданно обнаружили, что такие наполнители могут также использоваться как инертные матриксные носители для стабилизации Агента в его форме кислой сернокислой соли во время фармацевтической обработки и долгосрочного хранения.
Согласно этому композиции данного изобретения обеспечивают средства стабилизации Агента в виде его кислой сернокислой формы во время обработки композиции и дальнейшего долгосрочного хранения и, как следствие, обеспечивают тот факт, что приемлемая абсорбция и/или биодоступность Агента достигается во время дозирования.
Дополнительное преимущество данного изобретения относится к процессу производства, который используется для получения приемлемых композиций в соответствии с данным изобретением. В то время как большинство традиционных процессов рецептирования, таких как те, что используются для рецептирования форм дозирования в виде таблеток, могут втягивать большое количество продолжительных и сложных этапов, что возможно будет приводить к нестабильности Агента, композиции данного изобретения могут быть получены путем относительно простых и способных к изменениям процессов.
Матриксный носитель
Матриксный носитель включает один или больше фармацевтически приемлемых носителей, упомянутых выше. Матриксный носитель может включать единственный фармацевтически приемлемый носитель, выбранный из групп, определенных выше, или, альтернативно, он может включать смесь. Фармацевтически приемлемый носитель является выбранным из любой одной из следующих групп:
(a) d-альфа-токоферил полиэтиленгликоль 1000 сукцинат;
(b) полигликозилированные глицериды;
(c) полиэтиленгликоли; и
(d) твердые жиры.
D-альфа-токоферил полиэтиленгликоль 1000 сукцинат (который также является известным как витамин Е TPGS) представляет собой водорастворимую производную естественного витамина Е и имеет двойную природу, подобную амфифильности, гидрофильности и липофильности. Витамин Е TPGS получают путем эстерификации кристаллического сукцината d-α-токофериловой кислоты с помощью полиэтиленгликоля (см. Фармакопею США 25 - Национальный Формуляр 20). Витамин Е TPGS является также известным благодаря его применение при фармацевтических назначениях как эмульгатора, солюбилизирующего агента и энхансера абсорбции, при этом WO 96/36316, US 5891845 и WO 00/76482 могут быть приведенные как примеры. См. также "Eastman Vitamin Е TPGS" Eastman Brochure, Eastman Chemical Co., Kingsport, Tenn. (November 2002) для дополнительной информации о применении витамина Е TPGS при таких назначениях.
Полигликозилированные глицериды представляют собой смеси глицеридов жирных кислот и эстеров полиоксиэтилена с жирными кислотами. В этих смесях жирные кислоты являются насыщенными или ненасыщенными, а глицериды является моно-, ди- или триглицеридами или их смесями в любых соотношениях. Примеры приемлемых полигликозилированных глицеридов включают, но не ограничены, каприлокапроил макроголглицериды (например, Лабразол), олеоил макроголглицериды (например, Лабрафил M1944 CS), линолеоил макроголглицериды (например, Лабрафил М2125 CS), лауроил макроголглицериды (например, лауроил макрогол-32 глицериды) и стеароил макроголглицериды, например, Гелуцир 50/13 (см. Pheur 6ое издание, 2008, для дополнительных подробностей относительно этих полигликозилированных глицеридов). В особой группе композиций полигликозилированные глицериды, которые содержатся в матриксном носителе, имеют значение гидрофильно/липофильного баланса (HLB) более 10. В дополнительной особой группе композиций полигликозилированные глицериды, которые содержатся в матриксном носителе, являются способными к диспергированию в воде. В дополнительной особой группе композиций полигликозилированные глицериды представляют собой лауроил макроголглицериды или стеароил макроголглицериды. Еще в одной дополнительной особой группе композиций полигликозилированные глицериды представляют собой лауроил макроголглицериды. Еще в одной дополнительной особой группе композиций полигликозилированные глицериды представляют собой лауроил макрогол-32 глицериды или Гелуцир 50/13. Еще в одной дополнительной особой группе композиций полигликозилированные глицериды представляют собой лауроил макрогол-32 глицериды. Лауроил макрогол-32 глицериды (коммерчески обеспечиваются как Гелуцир 44/14 или Акконон® С-44, ЕР) представляют собой насыщенный полигликозилированный глицерид, который состоит из моно-, ди- или триглицеридов и моно- и ди-жирных кислот полиэтиленгликоля (ПЭГ). Лауроил макрогол-32 глицериды являются полутвердыми/твердыми при комнатной температуре и имеют точку плавления при 44°С, их получают путем реакции гидрогенизировапного масла ядра кокосового ореха с полиэтиленгликолем 1500.
Полиэтиленгликоли USP (ПЭГи), которые альтернативно являются известными как макроголы (см. Pheur 6ое издание 2008), представляют собой гидрофильные полимеры оксиэтилена. ПЭГи, которые имеют средний молекулярный вес более 900 дальтон, в общем случае являются полутвердыми или твердыми при комнатной температуре. Приемлемый интервал среднего молекулярного веса для ПЭГов в данном изобретении составляет от 900 до 35000 дальтон. Приемлемые коммерчески доступные продукты включают, но не ограничены, ПЭГ 900, ПЭГ 1000, ПЭГ 1450, ПЭГ 2000, ПЭГ 6000 и ПЭГ 20000. В особой группе композиций ПЭГ(и), который(ые) присутствует(ют) в матриксном носителе, имеет интервал среднего молекулярного веса от 900 до 25000 дальтон. В дополнительной особой группе композиций этого воплощения этот ПЭГ имеет средний молекулярный вес приблизительно 6000 дальтон. Еще в одной дополнительной группе композиций этого воплощения ПЭГ имеет средний молекулярный вес приблизительно 20000 дальтон.
Твердые жиры представляют собой твердые смеси моноглицеридов, диглицеридов и триглицеридов, которые является практически не растворимыми в воде. Примеры приемлемых твердых жиров включают, но не ограничены, Гелуцир 33/01 (см. USP-NF 'твердый жир'), Гелуцир 39/01 (см. USP-NF и ЕР 'твердый жир') и Гелуцир 43/01 (см. ЕР 3е издание и USP24/NF19 'твердый жир').
В соответствии с одним воплощением изобретения матриксный носитель состоит из одного или более фармацевтически приемлемых носителей, выбранных из следующих:
(a) d-альфа-токоферил полиэтиленгликоль 1000 сукцинат;
(b) полигликозилированные глицериды; и
(c) полиэтиленгликоли (ПЭГи);
где Агент является диспергированным в матриксном носителе.
В дополнительном воплощении изобретения матриксный носитель представляет собой витамин Е TPGS.
Еще в одном дополнительном воплощении изобретения матриксный носитель представляет собой полигликозилированный глицерид. Является приемлемым, когда полигликозилированный глицерид представляет собой лауроил макрогол-32 глицериды или Гелуцир 50/13, в частности лауроил макрогол-32 глицериды.
В дополнительном воплощении изобретения матриксный носитель включает смесь витамина Е TPGS и, по крайней мере, одного полигликозилированного глицерида. Является приемлемым, когда, по крайней мере, один полигликозилированный глицерид, присутствующий в этом воплощении, представляет собой лауроил макрогол-32 глицериды, также является приемлемым, когда лауроил макрогол-32 глицериды являются присутствующими в количестве, которое составляет 1-60% по весу компонента матриксного носителя композиции, и приемлемым образом приблизительно 30-55% и еще более приемлемо приблизительно 50% по весу компонента матриксного носителя композиции. Желательно, когда лауроил макрогол-32 глицериды являются единственным полигликозилированным глицеридом, присутствующим в этом воплощении.
В дополнительном воплощении изобретения матриксный носитель включает смесь витамина Е TPGS и, по крайней мере, одного ПЭГа. Является приемлемым, когда, по крайней мере, один ПЭГ, который присутствует в этом воплощении, имеет средний молекулярный вес от 900 до 25000 дальтон, и является приемлемым, когда ПЭГ является присутствующим в количестве, которое составляет 1-30% по весу компонента матриксного носителя композиции, приемлемым образом приблизительно 5-15% и еще более приемлемо приблизительно 10% по весу компонента матриксного носителя композиции. Желательно, когда только один ПЭГ присутствует в этом воплощении. В особой группе композиций этого воплощения такой ПЭГ имеет средний молекулярный вес 6000 дальтон. Еще в одной дополнительной группе композиций этого воплощения ПЭГ имеет средний молекулярный вес 20000 дальтон. Еще в одной дополнительной группе композиций этого воплощения ПЭГ имеет средний молекулярный вес 1000 дальтон.
При этом является понятным, что термин 'приблизительно', как используется в данной заявке выше для соотношения наполнителей, таких как лауроил макрогол-32 глицериды или ПЭГ, в компоненте матриксного носителя композиции, относится к ±2% по весу компонента матриксного носителя.
Является приемлемым, когда композиция содержит от 40 до 99% по весу, в частности от приблизительно 60 до 95% по весу, в частности от приблизительно 65 до 95% по весу матриксного носителя.
В особой группе композиций данного изобретения композиция содержит приблизительно 90-95% по весу матриксного носителя, и в частности, приблизительно 95% по весу матриксного носителя.
В особой дополнительной группе композиций данного изобретения композиция содержит от приблизительно 85 до 90% по весу матриксного носителя, и в частности, приблизительно 90% по весу матриксного носителя.
Еще в одной дополнительной особой группе композиций данного изобретения композиция содержит от приблизительно 75 до 85% по весу матриксного носителя, и в частности приблизительно 80% по весу матриксного носителя.
Еще в одной дополнительной особой группе композиций данного изобретения композиция содержит от приблизительно 65 до 80% по весу матриксного носителя, и в частности приблизительно 70% по весу матриксного носителя.
При этом является понятным, что срок 'приблизительно', когда касается соотношения матриксного носителя в композиции, относится к ±2% от общего веса композиции. В качестве примера, если говорят, что композиция содержит приблизительно 70% по весу матриксного носителя, то это будет охватывать композиции, которые содержат от 68 до 72% по весу матриксного носителя.
Еще в одной дополнительной особой группе композиций данного изобретения композиция содержит 79-81%, например 79,83%, по весу матриксного носителя.
Агент
Типично Агент будет присутствовать в количестве, которое находится в интервале от 1 до 50%, приемлемым образом от приблизительно 1 до 35% и особенно от приблизительно 5 до 30% по весу композиции. В особой группе композиций Агент будет присутствовать в количестве приблизительно 5% по весу заключительной композиции. В дополнительной особой группе композиций Агент будет присутствовать в количестве приблизительно 10% от веса заключительной композиции. Еще в одной дополнительной особой группе композиций Агент будет присутствовать в количестве приблизительно 20% от веса заключительной композиции. Еще в одной дополнительной особой группе композиций Агент будет присутствовать в количестве приблизительно 30% от веса заключительной композиции. Еще в одной дополнительной особой группе композиций Агент будет присутствовать в количестве 19-21%, например 20,17%, от веса заключительной композиции.
При этом является понятным, что термин 'приблизительно', когда касается соотношения Агента, присутствующего в композиции, относится к ±2% от общего веса композиции.
Является приемлемым, когда единичная доза композиции в соответствии с изобретением может содержать от 0,01 мг до 500 мг Агента. Является приемлемым, когда каждая терапевтическая доза композиции будет содержать достаточное количество Агента для обеспечения суточной дозы Агента в одной или более единицах. Приемлемые количества Агента в единичных дозах в разных воплощениях включают, например, приблизительно 6,05, 12,1, 18,15, 30,25, 60,5, 72,6, 78,65, 84,7, 90,75, 96,8, 102,85, 108,9, 114,95, 121, 151,25, 181,5, 242, 302,5, 363, 423,5, 484 мг или более в зависимости от дозы, которая является необходимой, и от конкретной формы фармацевтической композиции. В отдельном воплощении единичная доза композиции содержит от 1 мг до 150 мг Агента, и в частности от 50 мг до 130 мг Агента, например приблизительно 72,6, 78,65, 84,7, 90,75, 96,8, 102,85, 108,9, 114,95 или 121 мг Агента, и особенно 72,6, 78,65, 84,7, 90,75 или 96,8 мг Агента. Термин 'приблизительно', как используется непосредственно выше в данной заявке, определяется как +/- 2 мг от указанного весового количества. В отдельном воплощении единичная доза композиции содержит 90,75 или 60,5 мг Агента. В отдельном воплощении единичная доза композиции содержит 90,75 мг Агента. В отдельном воплощении единичная доза композиции содержит 60,5 мг Агента.
Агент может использоваться в различных формах, которые все включаются в объем данного изобретения. Такие включают аморфные или кристаллические формы, безводные формы, а также сольваты и гидраты. В особой группе композиций Агент является кристаллическим и находится в безводной форме.
Мы обнаружили, что Агент может быть стабилизирован в приемлемом матриксном носителе данного изобретения. Как используется в данной заявке, термин "стабилизированный" означает, что активный ингредиент ((2-гидроксиэтокси)амид 6-(4-бром-2-хлорфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты), который присутствует в композиции после обработки и/или хранения, присутствует существенно в виде кислой сернокислой соли, то есть, в виде Агента, в отличие от формы свободного основания Агента. Специалист в данной области техники может легко оценить, что указание количества формы свободного основания Агента и количества Агента (то есть, формы кислой сернокислой соли) в композиции может быть получено при использовании методик, таких, как, например, XRPD и 19F ЯМР спектроскопия твердого тела, и может также подвергаться мониторингу путем анализа растворения.
Как используется в данной заявке, термин "диспергированный" описывает двухфазную систему, где одна фаза состоит из Агента, который распределяется во второй фазе, которая включает матриксный носитель, при этом Агент представляет собой диспергированную фазу, а матриксный носитель, который включает эту фазу, является беспрерывной фазой. В особой группе композиций Агент, который образует "диспергированную фазу", находится в форме тонко измельченных частиц, которые являются распределенными во "второй фазе", которая включает матриксный носитель. В особой группе композиций более 60% от веса общего количества Агента, который является присутствующим в композиции, находятся в диспергированном виде. Еще в одной особой группе композиций более 90% и преимущественно более 95% от веса общего количества Агента, который является присутствующим в композиции, находятся в диспергированном виде. Специалист в данной области техники сможет оценить, что указание соотношения лекарственного средства, которое присутствует в форме твердой дисперсии, может быть установлено при использовании методик, таких как дифференциальная сканирующая калориметрия (DSC), термический гравиметрический анализ (TGA), дифференциальная сканирующая калориметрия и 19F спектроскопия ЯМР твердого тела. Квалифицированный специалист в данной области техники сможет оценить, что кристаллическая структура лекарственного средства в композиции может быть определена при использовании методик, таких как, например, дифракция рентгеновских лучей.
В особой группе композиций данного изобретения размер частиц диспергированного Агента может варьировать от приблизительно 1 до 20 микрон. Является желательным, когда диспергированный Агент имеет распределение размера частиц, такое, что 90% частиц имеют диаметр менее чем 15 микрон.
В одном воплощении изобретения Агент является диспергированным в матриксном носителе и не содержит дополнительных растворителей или вспомогательных агентов. Композиции этого воплощения могут быть получены с особенно высокой загрузкой Агента, и это является желательным, поскольку дополнительные компоненты часто имеют недостатки, такие как потенциально повышенный риск токсичности и увеличенный размер дозированной формы. Оба эти недостатка могут осуществлять свой вклад в плохое соблюдение режима приема пациентом и в приемлемость лечения.
Согласно дополнительному аспекту данного изобретения обеспечивается фармацевтическая композиция, которая включает:
(i) Агент; и
(ii) матриксный носитель;
где матриксный носитель имеет любые определения, приведенные выше;
и где Агент является диспергированным в матриксном носителе, а композиция является полутвердой или твердой при комнатной температуре.
Как используется в данной заявке, термин "полутвердый" описывает компонент или композицию, которые имеют жесткость и вязкость, которые являются промежуточными между твердым веществом и жидкостью. Полутвердые вещества не имеют такой текучести, как порошок, и не являются жидкими при комнатной температуре (то есть они имеют точку плавления, которая выше комнатной температуры). Как используется в данной заявке, термин "затвердение" означает образование твердого или полутвердого вещества. Под комнатной температурой понимают значение температуры в интервале от 18 до 23°С.
В соответствии с дополнительным аспектом данного изобретения обеспечивается фармацевтическая композиция, которая включает:
(i) Агент; и
(ii) матриксный носитель, который существенно состоит из витамина Е TPGS;
где Агент является диспергированным в витамине Е TPGS, а композиция является полутвердой или твердой при комнатной температуре.
В соответствии с дополнительным аспектом данного изобретения обеспечивается фармацевтическая композиция, которая включает:
(i) Агент; и
(ii) матриксный носитель, который существенно состоит из полигликозилированного глицерида;
где Агент является диспергированным в полигликозилированном глицериде, а композиция является полутвердой или твердой при комнатной температуре.
В соответствии с дополнительным аспектом данного изобретения обеспечивается фармацевтическая композиция, которая включает:
(i) Агент; и
(ii) матриксный носитель, который существенно состоит из витамина Е TPGS и лауроил макрогол-32 глицеридов;
где Агент является диспергированным в матриксном носителе, а композиция является полутвердой или твердой при комнатной температуре.
В соответствии с дополнительным аспектом данного изобретения обеспечивается фармацевтическая композиция, которая включает:
(i) Агент; и
(ii) матриксный носитель, который существенно состоит из витамина Е TPGS и ПЭГ;
где Агент является диспергированным в матриксном носителе, а композиция является полутвердой или твердой при комнатной температуре.
В отдельном воплощении обеспечивается фармацевтическая композиция, которая включает:
(i) от 15 до 30 (в частности, от 15 до 25) частей Агента; и
(ii) от 70 до 85 (в частности, от 75 до 85) частей матриксного носителя;
где обе части являются весовыми, а сумма частей (i)+(ii)=100, матриксный носитель имеет любое из значений, определенных в данной заявке выше, и Агент является диспергированным в матриксном носителе, а композиция является полутвердой или твердой при комнатной температуре.
В отдельном воплощении обеспечивается фармацевтическая композиция, которая включает:
(i) от 15 до 25 (в частности, от 18 до 22) частей Агента; и
(ii) от 75 до 85 (в частности, от 78 до 82) частей матриксного носителя;
где обе части являются весовыми, а сумма частей (i)+(ii)=100, матриксный носитель имеет любое из значений, определенных в данной заявке выше, и Агент является диспергированным в матриксном носителе, а композиция является полутвердой или твердой при комнатной температуре.
В отдельном воплощении обеспечивается фармацевтическая композиция, которая включает:
(i) от 25 до 40 (в частности, от 25 до 35) частей Агента; и
(ii) от 60 до 75 (в частности, от 65 до 75) частей матриксного носителя;
где обе части являются весовыми, а сумма частей (i)+(ii)=100, матриксный носитель имеет любое из значений, определенных в данной заявке выше, и Агент является диспергированным в матриксном носителе, а композиция является полутвердой или твердой при комнатной температуре.
В отдельном воплощении обеспечивается фармацевтическая композиция, которая включает:
(i) от 25 до 35 (в частности, от 28 до 32) частей Агента; и
(ii) от 65 до 75 (в частности, от 68 до 72) частей матриксного носителя;
где обе части являются весовыми, а сумма частей (i)+(ii)=100, матриксный носитель имеет любое из значений, определенных в данной заявке выше, и Агент является диспергированным в матриксном носителе, а композиция является полутвердой или твердой при комнатной температуре.
В отдельном воплощении обеспечивается фармацевтическая композиция, которая включает:
(i) от 15 до 25 (в частности, от 18 до 22) частей Агента; и
(ii) от 75 до 85 (в частности, от 78 до 82) частей витамина Е TPGS;
где обе части являются весовыми, а сумма частей (i)+(ii)=100;
и где Агент является диспергированным в витамине Е TPGS, а композиция является полутвердой или твердой при комнатной температуре.
В отдельном воплощении обеспечивается фармацевтическая композиция, которая включает:
(i) от 25 до 35 (в частности, от 28 до 32) частей Агента; и
(ii) от 65 до 75 (в частности, от 68 до 72) частей витамина Е TPGS;
где обе части являются весовыми, а сумма частей (i)+(ii)=100;
и где Агент является диспергированным в витамине Е TPGS, а композиция является полутвердой или твердой при комнатной температуре.
В отдельном воплощении обеспечивается фармацевтическая композиция, которая включает:
(i) 19-21, например, 20,17, частей Агента; и
(ii) 79-81, например, 79,83, частей витамина Е TPGS;
где обе части являются весовыми, а сумма частей (i)+(ii)=100;
и где Агент является диспергированным в витамине Е TPGS, а композиция является полутвердой или твердой при комнатной температуре.
В отдельном воплощении обеспечивается фармацевтическая композиция, которая включает:
(i) от 25 до 35 (в частности, от 28 до 32) частей Агента; и
(ii) от 65 до 75 (в частности, от 68 до 72) частей матриксного носителя, который состоит из смеси витамина Е TPGS и, по крайней мере, одного полигликозилированного глицерида;
где обе части являются весовыми, а сумма частей (i)+(ii)=100;
и где Агент является диспергированным в витамине Е TPGS и, по крайней мере, одном полигликозилированном глицериде, а композиция является полутвердой или твердой при комнатной температуре.
Композиция
Необязательно, в композицию в соответствии с данным изобретением могут включаться дополнительные наполнители при условии, что включение таких наполнителей не будет неблагоприятным образом влиять на стабильность формы соли Агента в композиции. В соответствии с этим любой квалифицированный специалист в данной области техники сможет оценить, что в некоторых воплощениях изобретения Агент, который присутствует в композиции в соответствии с изобретением, может быть диспергированным в смеси, которая состоит из матриксного носителя и дополнительных наполнителей, таких как те, что описанные в некоторых конкретных примерах, которые приведены в данной заявке ниже. Дополнительные наполнители, которые могут присутствовать, включают, например, консерванты, стабилизаторы, эмульгаторы, антиоксиданты, подсластители, вкусовые агенты, агенты для доведения значения рН, агенты, которые содействуют диспергированию (например, поверхностно-активные соединения, такие как, например, этоксилированное касторовое масло (Кремофор EL), этоксилированное гидрогенизированное касторовое масло (Кремофор RH40) или полисорбат 80) и модификаторы вязкости. Такие дополнительные наполнители является хорошо известными специалисту в данной области техники и являются описанными, например, в Руководстве по фармацевтическим наполнителям, 4е издание, Американская фармацевтическая ассоциация; Теория и практика промышленной фармацевтики, 3е издания, Lachman и др. 1986; Фармацевтические дозированные формы: Таблетки, том 1, 2е издание, Lieberman, Hebert А., и др., 1989; Современная фармацевтика, Banker, Gilbert и Rhodes, Christopher Т, 3е издания, 1995; и Remington's Pharmaceutical Sciences, 20е издание, 2000.
Является приемлемым, когда композиция в соответствии с данным изобретением находится в форме, адаптированной для перорального введения, например в форме капсульной композиции или жидкой дисперсии, приемлемой для перорального введения. Приемлемые композиции для капсул является хорошо известными и включают, например, твердые, жидкие или полутвердые композиции, которые содержатся в мягких или твердых желатиновых капсулах; капсулы из водорастворимых этеров целлюлозы (например, гипромеллозы) или крахмальные капсулы.
В соответствии с этим дополнительный аспект изобретения представляет собой фармацевтическую композицию, адаптированную для перорального введения, которая включает Агент и матриксный носитель, где матриксный носитель имеет любое значение, как определено в данной заявке выше; и где Агент является диспергированным в матриксном носителе.
Еще один дополнительный аспект изобретения представляет собой фармацевтическую капсульную композицию, которая включает Агент и матриксный носитель, где матриксный носитель имеет любое значение, как определено в данной заявке выше; и где Агент является диспергированным в матриксном носителе.
Композиции в соответствии с данным изобретением могут быть получены при использовании традиционных способов, которые является хорошо известными в области фармацевтики. Например, в одном конкретном воплощении компонент(ы) матриксного носителя нагревают до плавления, а Агент, размер частиц которого может быть снижен, например, путем измельчения или микронизации, постепенно вводят в расплавленную смесь при постоянном встряхивании/перемешивании для обеспечения гомогенного распределения. Расплавленная смесь может потом вноситься в твердые или мягкие капсулы, после чего ее оставляют для охлаждения с образованием вязкой жидкости, твердой или полутвердой массы внутри капсулы. Потом капсулу закрывают при использовании традиционных способов, известных в данной области техники, таких как, например, объединение с помощью кромки.
Альтернативно, композиции в соответствии с данным изобретением могут быть получены с помощью других известных способов, таких, как, например, экструзия расплава или грануляция расплава (см. A. Royce, J, Drug Dev. Ind. Pharm. 22 (1996) 9 IP-924, G. Verreck, Bull. tech. Gattefosse (2004) 85-95 и J. Breitenbach, Eur. J. Pharm. Biopharm. 54 (2002) 107-117 для деталей приемлемых способов производства).
Агент обладает антипролиферативной активностью, и в соответствии с этим композиции данного изобретения являются полезными для лечения состояний, таких как те, что описанные в международной патентной заявке WO 2007/076245, которая раскрывает Агент (то есть кислую сернокислую соль), а также в WO 03/077914, где представлена форма свободного основания Агента. Например, композиция в соответствии с данным изобретением является полезной для лечения многих общих видов рака человека, таких как злокачественная меланома, рак мозга, легких, чешуйчатых клеток, мочевого и желчного пузыря, желудка, поджелудочной железы, молочной железы, рак головы и шеи, ренальный рак, рак почки, яичника, предстательной железы, колоректальний рак, рак пищевода, рак яичка, гинекологический рак и рак щитовидной железы. Дополнительно можно ожидать, что композиции в соответствии с данным изобретением будут полезными для лечения заболеваний, которые втягивают избыточную пролиферацию клеток, таких как доброкачественная гиперплазия кожи, например псориаза, рестеноза или доброкачественной гипертрофии предстательной железы (ВРН). Другие примеры опосредованных MEK заболеваний, которые также могут лечиться при использовании Агента, включают заболевания поджелудочной железы или почек (включая пролиферативный гломерулонефрит и индуцированное диабетом почечное заболевание) или лечение боли у млекопитающего. Кроме того. Агент также может использоваться для предотвращения бластоцитарной имплантации у млекопитающего или для лечения заболевания, связанного с васкулогенезом или ангиогенезом у млекопитающего. Такие заболевания могут включать опухолевый ангиогенез, хроническое воспалительное заболевание, такое как ревматоидный артрит, атеросклероз, воспалительное заболевание кишечника, заболевания кожи, такие как псориаз, экзема, а также склеродерму, диабет, диабетическую ретинопатию, ретролентальну фиброплазию, возрастную дегенерацию желтого пятна, гемангиому, глиому, меланому, саркому Капоши и рак яичника, молочной железы, легких, поджелудочной железы, предстательной железы, кишечника и эпидермоидный рак.
Дополнительный аспект данного изобретения обеспечивает фармацевтическую композицию в соответствии с изобретением, как определено в данной заявке выше, для применения в качестве лекарственного средства.
Агент, который является присутствующим в композициях в соответствии с данным изобретением, обладает антипролиферативными свойствами, такими как противораковые свойства, которые, как предполагается, происходят от его активности в отношении ингибирования MEK. В соответствии с этим композиция данного изобретения, как предполагается, является полезной в лечении заболеваний или медицинских состояний, опосредованных полностью или частично MEK, то есть композиция в соответствии с данным изобретением может использоваться для получения ингибиторного эффекта в отношении MEK у теплокровного животного, которое нуждается в таком лечении. Таким образом, композиция в соответствии с данным изобретением обеспечивает способ лечения пролиферации злокачественных опухолей, который характеризуется ингибированием MEK, то есть композиция в соответствии с данным изобретением может использоваться для получения антипролиферативного эффекта, опосредованного полностью или частично ингибированием MEK. В соответствии с этим композиция данного изобретения, как можно ожидать, будет полезной в лечении рака путем обеспечения антипролиферативного эффекта, в частности в лечении чувствительных к MEK видов рака, таких, как виды рака, описанные в данной заявке выше.
В одном воплощении в соответствии с данным изобретением обеспечивается фармацевтическая композиция в соответствии с изобретением, как определено в данной заявке выше, для применения в получении антипролиферативного эффекта у теплокровного животного (преимущественно у человека). В другом воплощении обеспечивается фармацевтическая композиция в соответствии с изобретением, как определено в данной заявке выше, для применения в лечении рака. Еще в одном дополнительном воплощении обеспечивается фармацевтическая композиция в соответствии с изобретением для применения в предотвращении или лечении опухолей, которые являются чувствительными к ингибированию MEK.
Дополнительный аспект данного изобретения обеспечивает применение композиции в соответствии с изобретением, как определено в данной заявке выше, в производстве лекарственного средства для применения в получении антипролиферативного эффекта у теплокровного животного (желательно у человека).
Дополнительный аспект данного изобретения обеспечивает применение композиции в соответствии с изобретением, как определено в данной заявке выше, в производстве лекарственного средства для применения в лечении рака.
Дополнительный аспект данного изобретения обеспечивает способ предотвращения неприемлемого снижения биодоступности Агента у пациента, который нуждается в применении Агента, который включает пероральное введение указанному пациенту фармацевтической композиции в соответствии с данным изобретением, как определено в данной заявке выше.
Дополнительный аспект данного изобретения обеспечивает применение фармацевтической композиции в соответствии с данным изобретением, как определено в данной заявке выше, в производстве лекарственного средства для предотвращения неприемлемого снижения биодоступности Агента.
Фармацевтические композиции в соответствии с данным изобретением могут вводиться самостоятельно как базисная терапия или могут вводиться дополнительного к одному или более других лечебных средств и/или способов лечения. Такое совместное лечение может быть достигнуто путем одновременного, последовательного или отдельного введения индивидууму компонентов лечения. В области медицинской онкологии является обычной практикой использовать комбинацию разных форм лечения для обработки каждого пациента с раком. В медицинской онкологии другой(другие) компонент(ы) такого совместного лечения в дополнение к композициям данного изобретения может представлять собой: хирургию, радиотерапию или химиотерапию. Такая химиотерапия может включать разные категории терапевтических агентов, таких как:
(i) другие антиангиогенные агенты, такие как те, что ингибируют эффекты фактора роста сосудистого эндотелия (например, антитело к фактору роста сосудистого эндотелия бевацизумаб [Avastin™]), и такие, которые работают с помощью разных механизмов из тех, что определены в данной заявке выше (например, линомид, ингибиторы функции интегрина αvβ3, ангиостатин, разоксин, талидомид, ингибиторы ММР-2 (матриксной металлопротеиназы 2), ингибиторы ММР-9 (матриксной металопротеиназы 9), и ингибиторы COX-II (циклооксигеназы II)), и включая агенты нацеливания на сосуды (например, комбрестатин фосфат и соединения, раскрытые в международных патентных заявках WO 00/40529, WO 00/41669, WO 01/92224, WO 02/04434 и WO 02/08213, а также агенты, которые повреждают сосуды, описанные в публикации международной патентной заявки WO 99/02166, полное раскрытие указанного документа введено в данную заявку в качестве ссылки (например, ацетилколхинол-O-фосфат));
(ii) цитостатические агенты, такие как антиэстрогены (например, тамоксифен, торемифен, ралоксифен, дролоксифен, иодоксифен), агенты, которые выявляют понижающую регуляцию эстрогенового рецептора (например, фулвестрант), прогестогены (например, мегестрол ацетат), ингибиторы ароматазы (например, анастрозол, летразол, воразол, эксеместан), антипрогестогены, антиандрогены (например, флутамид, нилутамид, бикалутамид, циротерон ацетат), LHRH агонисты и антагонисты (например, гозерелин ацетат, лупролид, бузерелин), ингибиторы 5α-редуктазы (например, финастерид), антиинвазивные агенты (например, ингибиторы металлопротеиназы, подобные маримастату, и ингибиторы функции рецептора урокиназного активатора плазминогена) и ингибиторы функции фактора роста (такие факторы роста включают, например, фактор роста тромбоцитов и фактор роста гепатоцитов), такие ингибиторы включают антитела к фактору роста, антитела к рецептору фактора роста, (например, анти-erbb2 антитело трастузумаб [Herceptin™] и анти-erbbl антитело цетуксимаб [С225]), ингибиторы фарнезилтрансферазы, ингибиторы тирозинкиназы, например ингибиторы семейства эпидермального фактора роста (например, EGFR семейство ингибиторов тирозинкиназы, например N-(4-фторфенил)-метокси-6-(3-морфолинопропокси)хиназолин-4-амин (гефитиниб), N-(3-этинилфенил)-6,7-бис(2-метоксиэтокси)хиназолин-4-амин (эрлотиниб, OSI-774) и 6-акриламидо-(4-фторфенил)-7-(3-морфолинопропокси)хиназолин-4-амин (CI 1033) и ингибиторы серин/треонинкиназы; и
(iii) антипролиферативные/антинеопластические лекарственные средства и их комбинации, как используется в медицинской онкологии, такие как антиметаболиты (например, антифолаты, подобные метотрексату, фторпиримидины, подобные 5-фторурацилу, тегафур, пурин и аналоги аденозина, цитозин арабинозид); противоопухолевые антибиотики (например, антрациклины, подобные адриамицину, бдеомицину, доксорубицину, дауномицину, эпирубицину и идарубицину, митомицину С, дактиномицину, митрамицину); производные платины (например, цисплатин, карбоплатин); алкилирующие агенты (например, азотистый иприт, мелфалан, хлорамбуцил, бусулфан, циклофосфамид, ифосфамид, нитрозомочевины, тиотепа); антимитотические агенты (например, алкалоиды барвинка, подобные винкристину, винбластину, виндезину, винорелбину, и таксоиды, подобные таксолу, таксотере); ингибиторы топоизомеразы (например, эпиподофилотоксины, подобные этопозиду и тенипозиду, амсакрину, топотекану, каптотецину, а также иринотекану); а также ферменты (например, аспарагиназа); и ингибиторы тимидилатсинтазы (например, ралтитрексед);
и дополнительные типы химиотерапевтических агентов включают:
(iv) модификаторы биологического ответа (например, интерферон);
(v) антитела (например, эдреколомаб);
(vi) антисмысловые терапии, например те, которые направлены на мишени, приведенные выше, такие как ISIS 2503, антисмысловой анти-ras;
(vii) подходы генной терапии, включая, например, подходы для замены абберантных генов, таких как аберрантный р53 или абберантный BRCA1 или BRCA2, GDEPT (терапия на основе направленного на ген ферментативного пролекарственного средства) подходы, такие как те, что используют цитозиндезаминазу, тимидинкиназу или бактериальный фермент нитроредуктазу, а также подходы для повышения толерантости пациента к химиотерапии или радиотерапии, такие как генная терапия мультилекарственной резистентности; и
(viii) иммунотерапевтические подходы, включая, например, ex vivo и in vivo подходы для повышения иммуногенности опухолевых клеток пациента, такие как трансфекция с помощью цитокинов, таких как интерлейкин 2, интерлейкин 4 или фактор стимуляции колоний гранулоцитов-макрофагов, подходы для снижения иммунологической толерантности Т-клеток, подходы при использовании трансфицированных иммунных клеток, таких как трансфицированные цитокинами дендритные клетки, подходы при использовании трансфицированных цитокинами опухолевых линий клеток и подходы при использовании антиидиотипических антител.
(ix) митотические ингибиторы, например винбластин;
(х) алкилирующие агенты, например цисплатин, карбоплатин и циклофосфаниид;
(xi) антиметаболиты, например 5-фторурацил, цитозин арабинозид и гидроксимочевина, или, например, один из преобладающих антиметаболитов, которые раскрыты в европейском патенте ЕР 0239362 В1 (опубликован 12 апреля 1991), такие как N-(5-[N-(3,4-дигидро-2-метил-4-оксохиназолин-6-илметил)-N-метиламино-2-теноил)-L-глутаминовая кислота;
(xii) ингибиторы факторов роста; ингибиторы клеточного цикла;
интеркалирующие антибиотики, например адриамицин и блеомицин; ферменты, например интерферон; и антигормоны, например антиэстрогены, такие как Nolvadex™ (тамоксифен), или, например, антиандрогены, такие как Casodex™ (4'-циано-3-(4-фторфенилсульфонил)-2-гидрокси-2-метил-3'-(трифторметил)пропионанилид).
В частности, фармацевтические композиции в соответствии с данным изобретением используются в сочетании с эффективным количеством одного или более веществ, выбранных из антиангиогенных агентов, ингибиторов сигнальной трансдукции и антипролиферативных агентов.
В отдельном воплощении антиангиогенные агенты, такие как ингибиторы ММР-2 (матриксной металлопротеиназы 2), ингибиторы ММР-9 (матриксной металлопротеиназы 9) и ингибиторы COX-II (циклогеназы II), могут использоваться в сочетании с фармацевтической композицией в соответствии с данным изобретением. Примеры полезных ингибиторов COX-II включают CELEBREX™ (алекоксиб), валдекоксиб и ролекоксиб. Примеры полезных ингибиторов матриксной металлопротеиназы являются описанными в WO 96/33172 (опубликована 24 октября 1996), WO 96/27583 (опубликована 7 марта 1996), публикации европейского патента ЕР 0818442 А2 (опубликован 14 января 1998), европейском патенте ЕР 1004578 В1 (выдан 25 февраля 2004), WO 98/07697 (опубликована 26 февраля 1998), WO 98/03516 (опубликована 29 января 1998), WO 98/34918 (опубликована 13 августа 1998), WO 98/34915 (опубликована 13 августа 1998), WO 98/33768 (опубликована 6 августа 1998), WO 98/30566 (опубликована 16 июля 1998), публикации европейского патента 606,046 (опубликован 13 июля 1994), публикации европейского патента 931,788 (опубликован 28 июля 1999), WO 90/05719 (опубликована 31 мая 1990), WO 99/52910 (опубликована 21 октября 1999), WO 99/52889 (опубликована 21 октября 1999), WO 99/29667 (опубликована 17 июня 1999), международной заявке РСТ WO 99/07675 (опубликована 18 февраля 1999), европейском патенте ЕР 0952148 В1 (выдан 12 мая 2004), патентной заявке Великобритании №9912961,1 (подана 3 июня 1999), временной заявке США №60/148,464 (подана 12 августа 1999), патенте США №5,863,949 (выдан 26 января 1999), патенте США №5,861,510 (выдан 19 января 1999) и публикации европейского патента 780,386 (опубликован 25 июня 1997), которые все введены в данную заявку в своей целостности в качестве ссылки. Желательные ингибиторы ММР-2 и ММР-9 является такими, которые обладают небольшой ингибирующей активностью или не имеют никакой ингибирующей активности в отношении ММР-1. Более желательными является те, которые селективно ингибируют ММР-2 и/или ММР-9 по отношению к другим матриксным металлопротеиназам (то есть ММР-1, ММР-3, ММР-4, ММР-5, ММР-6, ММР-7, ММР-8, ММР-10, ММР-11, Mmp-12 и Mmp-13).
Некоторые специфические примеры ингибиторов ММР, полезных в данном изобретении, представляют собой AG-3340, RO 32-3555 и RS 13-0830.
Доза Агента, который требуется в композиции в соответствии с данным изобретением для терапевтического или профилактического лечения конкретного заболевания или медицинского состояния (например, пролиферативного заболевания), будет с необходимостью варьировать в зависимости, например, от хозяина, которого подвергают лечению, и тяжести заболевания, которое подвергают лечению. Количество активного соединения, которое вводится, будет зависеть от субъекта, которого подвергают лечению, тяжести расстройства или состояния, скорости введения, характера соединения и от точки зрения врача, который выписывает лекарственное средство. Однако эффективная доза находится в интервале от приблизительно 0,01 до приблизительно 100 мг на кг веса тела в сутки, желательно от приблизительно 1 до приблизительно 35 мг/кг/сутки, в виде одной или разделенных доз. Для человека весом 70 кг это количество будет составлять от приблизительно 0,7 до 7000 мг/сутки, желательно от приблизительно 70 до приблизительно 2500 мг/сутки. В некоторых случаях равные дозировки, которые являются более низкими, чем нижняя граница указанного интервала, будут более адекватными, в то время как в других случаях более высокие дозы могут использоваться без вредного побочного эффекта, при условии, что такие более высокие дозы сначала разделяют на несколько маленьких доз для введения в течение суток. Единичная доза композиции, конечно, будет содержать, например, 1-500 мг активного ингредиента и желательно 5-150 мг активного ингредиента. Предполагается, что желательная суточная доза находится в интервале 0,03-6 мг/кг.
Изобретение иллюстрируется ниже неограничивающими примерами, где, если не указанное другое, то "Агент" представляет собой кислую сернокислую соль (2-гидроксиэтокси)амида6-(4-бром-2-хлорфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты.
Краткое описание фигур
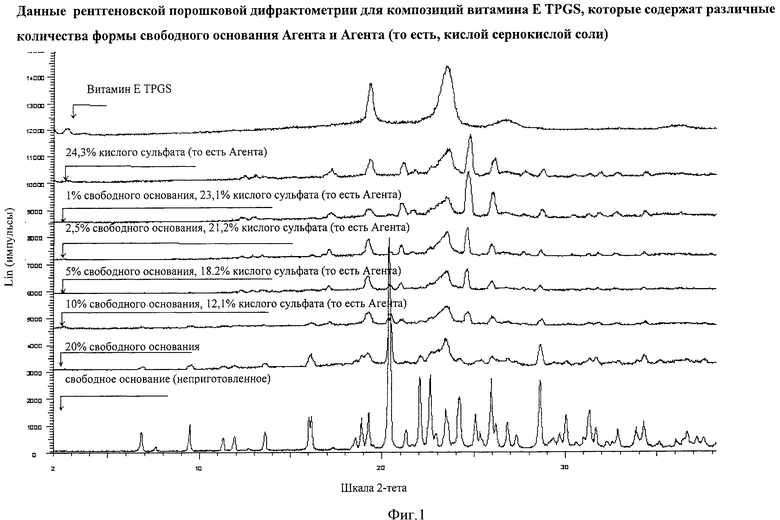
Фигура 1 демонстрирует результаты рентгеновской порошковой дифрактометрии для композиций, которые содержат разные количества формы свободного основания Агента и Агента (то есть кислой сернокислой соли), где ось Х демонстрирует 2-тета значение, а ось Y демонстрирует Lin (импульсы). Данные обеспечивают указание уровня определения формы свободного основания Агента в композиции при использовании рентгеновской порошковой дифрактометрии.
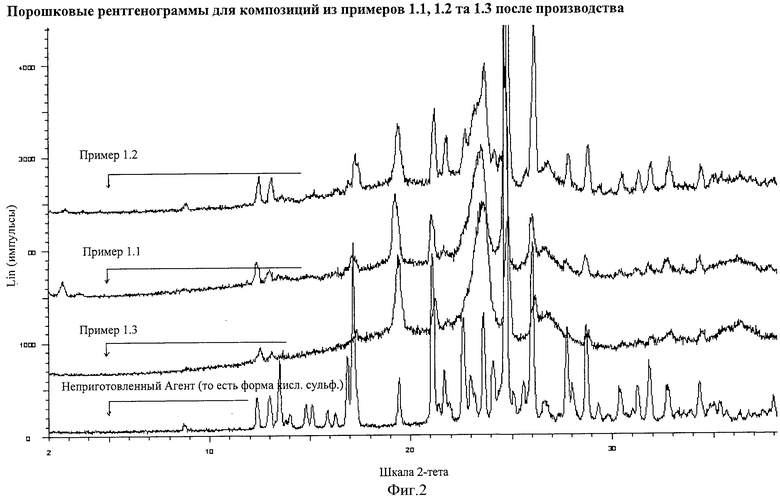
Фигура 2 демонстрирует порошковые рентгенограммы для композиций данного изобретения, полученных после производства, где ось Х демонстрирует 2-тета значение, а ось Y демонстрирует Lin (импульсы). Данные демонстрируют, что только Агент (то есть форма кислой сернокислой соли) подвергается определению в композициях.
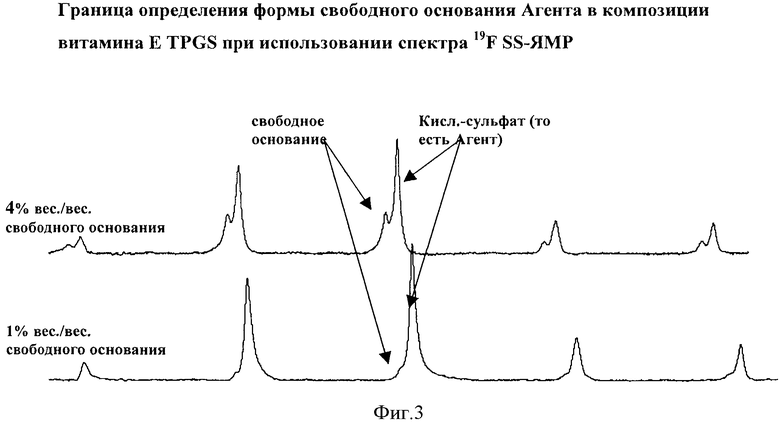
Фигура 3 демонстрирует 19F SS-ЯМР спектры, которые используются для определения приблизительной границы определения в композициях формы свободного основания Агента в витамине Е TPGS при использовании 19F SS-ЯМР.
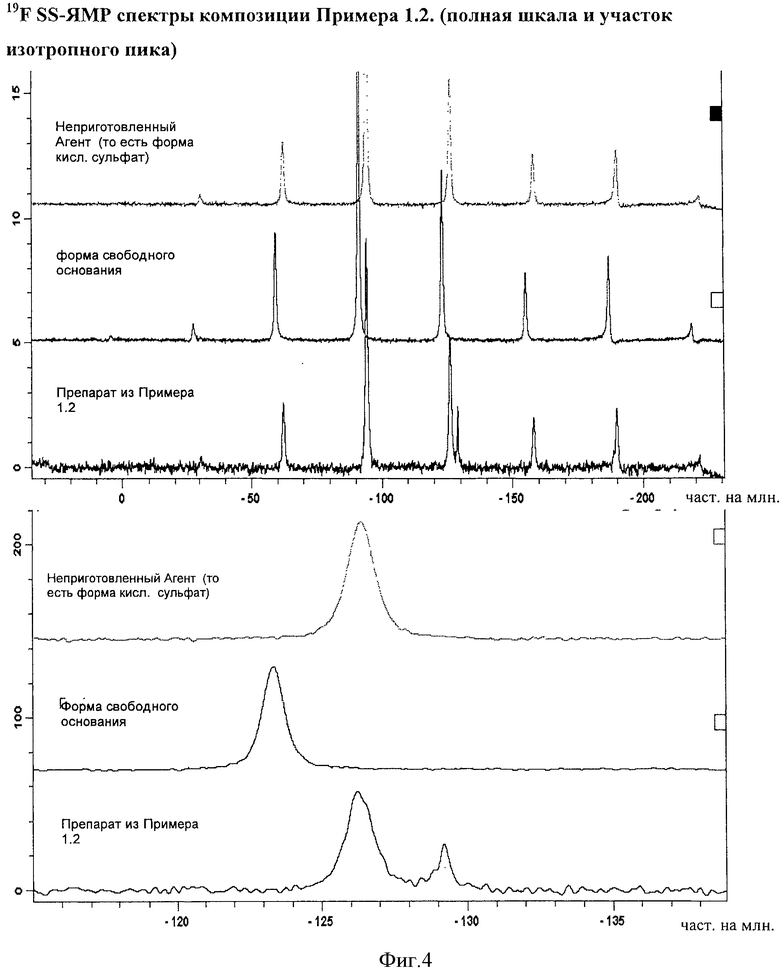
Фигура 4 демонстрирует 19F SS-ЯМР спектры композиции Примера 1.2. Спектры показывают отсутствие способного к определению уровня формы свободного основания Агента в композиции.
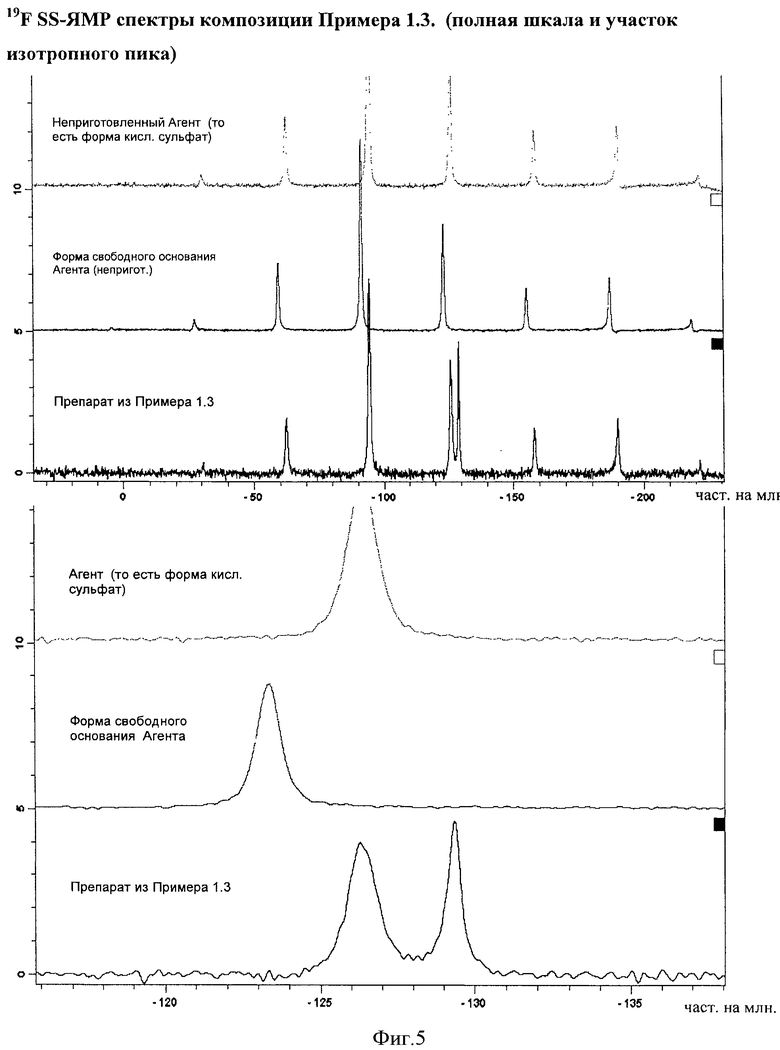
Фигура 5 демонстрирует 19F SS-ЯМР спектры композиции Примера 1.3. Спектры демонстрируют отсутствие способного к определению уровня формы свободного основания Агента в композиции.
Пример 1: Получение композиций в соответствии сданным изобретением
Композиции, представленные в Таблице 1, получали путем нагревания матриксного носителя до температуры 60-70°С при использовании термостата. Температуру поддерживали на протяжении приблизительно 2 часов для обеспечения полного расплавления всего материала. Потом постепенно прибавляли Агент и механически перемешивали в матриксном носителе при использовании магнитной мешалки или гомогенизатора высокого сдвига. Систему поддерживали при достаточно высокой температуре для того, чтобы поддержать смесь в расплавленном состоянии во время перемешивания, которое продолжали до получения визуально гомогенной смеси. Время перемешивания варьировало в зависимости от конкретной композиции, однако в общем случае находилось в интервале от 3 до 35 минут. Полученную смесь потом вносили в НРМС капсулы и оставляли для охлаждения до комнатной температуры. Капсулы запечатывали и в общем случае сохраняли в замороженном состоянии до использования.
Гелуцир 44/14 (71,49)
ПЭГ 20000 (11,98)
ПЭГ 6000 (11,98)
Пример 2: Исследование стабильности композиций данного изобретения с помощью рентгеновской порошковой дифрактометрии (XRPD)
Определение стабильности Агента (то есть кислой сернокислой соли) в композиции может обеспечиваться путем XRPD. Эта методика является способной к одновременному определению кристаллической формы свободного основания Агента и кристаллической формы кислой сернокислой соли Агента в композиции. Образцы композиций помещали на силиконовую пластинку основания и анализировали при использовании рентгеновского дифрактометра Siemen's D5000. Образцы подвергали сканированию на протяжении 4 секунд на 0,02° θ при использовании интервала от 2° до 40° 2θ в беспрерывном тета-тета режиме.
Приблизительную границу определения кристаллической формы свободного основания Агента в композиции в соответствии с данным изобретением определяли путем получения композиций с разными соотношениями количества кристаллической формы свободного основания Агента и кристаллического Агента (то есть формы кислой сернокислой соли). Эти композиции подвергали анализу с помощью XRPD. Фигура 1 демонстрирует, что форма свободного основания Агента определяется до уровня 2,5% вес./вес. свободного основания в витамине Е TPGS на основе композиции, которая номинально содержит 21,2% вес./вес. Агента.
XRPD порошковые рентгенограммы получали для каждой композиции, описанной в Примерах 1.1, 1.2 и 1.3, непосредственно после их производства. Эти рентгенограммы (показанные на Фигуре 2) демонстрировали только присутствие Агента (то есть формы кислой сернокислой соли).
Пример 3: Исследование стабильности композиций данного изобретения с помощью ЯМР спектроскопии твердого тела
Определение стабильности Агента в композициях в соответствии с данным изобретением может обеспечиваться при использовании 19F ЯМР спектроскопии твердого тела (19F SS-ЯМР). Эта методика является способной к одновременному определению кристаллической формы свободного основания Агента и кристаллической формы кислой сернокислой соли Агента в композиции. Форма свободного основания Агента и Агента (то есть кислой сернокислой соли) обеспечивает отличные и характерные пики фтора в спектре. Эти пики могут быть интегрированы нормальным образом для ЯМР сигналов, и соотношение пиков является пропорциональным соотношению двух присутствующих форм твердого состояния, то есть формы свободного основания Агента и Агента (то есть формы кислой сернокислой соли). Анализ композиций осуществляли путем размещения материала образца на 4 мм MAS (вращение образца под магическим углом) роторе. 19F ЯМР [376 МГц] спектр с 1Н расщеплением составного импульса [ТРРМ15] регистрировали на Avance 400 спектрометре при использовании 4 мм HFX (Bruker Biospin) зонда. Все образцы подвергали обработке при 12 кГц при использовании импульсной программы "aringdec" (анти-кольцо с расщеплением). Следует отметить, что силы трения, которые ассоциируются с методикой вращения образца под магическим углом, могут приводить к нагреванию образца приблизительно на 10°С-20°С выше температуры окружающего среды.
Приблизительную границу определения кристаллической формы свободного основания Агента в композиции в соответствии с данным изобретением определяли путем получения композиций с разными соотношениями количества кристаллической формы свободного основания Агента и кристаллического Агента (то есть формы кислой сернокислой соли). Эти композиции потом подвергали анализу с помощью 19F SS-ЯМР. Спектры ЯМР, изображенные на Фигуре 3, показывают, что форма свободного основания Агента определяется до уровня 1% вес./вес. свободного основания в витамине Е TPGS на основе композиции, которая также содержала 28,9% вес./вес. Агента (то есть формы кислой сернокислой соли).
Композиции, описанные в Примерах 1.2 и 1.3, анализировали с помощью 19F SS-ЯМР после производства, при этом не было выявлено никакого подтверждения присутствия формы свободного основания Агента, смотри Фигуру 4 и Фигуру 5. Наблюдали некоторое нагревание образцов при их анализе, который может приводить к появления изотропного пика при -129,5 част. на млн. Без намерения связывать это с какой-либо теорией, можно сказать, что пик может быть связан с (2-гидроксиэтокси)амидом 6-(4-бром-2-хлорфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты, растворенным в витамине Е TPGS, который расплавляется при нагревании образца.
Пример 4. Стабильность композиций при хранении
Исследование стабильности на композициях, описанных в Примерах 1.2 и 1.3, на протяжении периода времени до 12 месяцев показало, что они являются стабильными при повышенных температурах и высокой влажности, если они содержатся в белых бутылках из полиэтилена высокой плотности (HDPE) (индукционно запечатанные и содержат десикант). Никаких значительных изменений данных относительно стабильности для композиций Примера 1.2 и 1.3 не наблюдали через 12 месяцев хранения в HDPE бутылках при 25°С/60% относительной влажности (ОВ) и 30°С/65% 0 В, см. данные, приведенные в Таблице 1 и Таблице 2.
25°С/60% ОВ
30°С/65% ОВ
25°С/60% ОВ
30°С/65% ОВ
= 30% В, 10,5 мин = 36% В, 16,5 мин = 36% В, 30,5 мин = 90% В, 33 мин = 90% В, 34 мин = 30% В, 40 мин = 30% В. Параметры ВЭЖХ: скорость истечения = 1,2 мл/мин, температура колонки = 40°С, длина волны = 258 нм, объем, который вводится = 10 мкл.
Пример 5. Растворение композиций в соответствии с данным изобретением
Образ растворения in vitro совершенствовали для анализа поведения композиций, которые содержатся в НРМС капсулах. Растворение осуществляли при использовании композиций, приведенных ниже в Таблице 3.
Растворение капсул проводили согласно общей процедуре с использованием аппаратуры II Фармакопеи США (устройство с мешалкой). Образцы среды для растворения отбирали в разные моменты времени после добавления капсул и количественно оценивали концентрации (2-гидроксиэтокси)амида 6-(4-бром-2-хлорфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты путем сравнения их площади пика ответа ВЭЖХ с такими стандартного раствора, который готовили при уровне, эквивалентном 100% высвобождению соединения. В этом способе использовали сосуды из прозрачного стекла с заостренным концом для растворения, а спиральные держатели из нержавеющей стали использовали для укрепления капсул. Использовали 900 мл рН 2,0 735 мОсмол/л буферного раствора на основе фосфата при 27°С и скорость перемешивания 100 об./минуту.
В дополнение к композициям, описанным в Примерах 1.1-1.3 и 1.5-1.7, изготовляли несколько дополнительных композиций для сравнения при использовании смесей кристаллического свободного основания Агента и кристаллического Агента (то есть кислой сернокислой соли). Смесь этих двух форм диспергировали в витамине Е TPGS в соответствии со способами, аналогичными тем, которые описаны в Примере 1, и вносили их в НРМС капсулы.
Данные относительно растворения композиций сравнения показывают, что растворение снижается при повышении количества формы свободного основания Агента в композиции. Снижение растворения до 17% через 50 минут наблюдали в композиции, которая содержала 2% вес./вес. формы свободного основания Агента. Данные, полученные путем анализа свободного основания, которое содержится в композиции для сравнения, показали, что способ растворения обеспечивает получение показателя уровня формы свободного основания Агента, который присутствует в композициях. Результаты растворения для композиций, описанных в Примерах 1.1-1.3 и 1.5-1.7, показали, что достигали растворения на уровне 95% или более, это свидетельствует о том, что в этих композициях соединение является существенно присутствующим в форме своей кислой сернокислой соли (то есть, в виде Агента).
Пример 6. Получение дополнительных композиций в соответствии с данным изобретением
Композиции, представленные в Таблице 4, получали путем нагревания матриксного носителя в термостате, установленном на 70°С, на протяжении, по крайней мере, одного часа. Постепенно прибавляли Агент и механически перемешивали в матриксном носителе при использовании механической мешалки или гомогенизатора высокого сдвига. Систему поддерживали при достаточно высокой температуре для того, чтобы поддержать смесь в расплавленном состоянии во время перемешивания. Перемешивание осуществляли до получения визуально гомогенной смеси. Время перемешивания варьировало в зависимости от композиции, однако составляло, по крайней мере, 10 минут и могло составлять вплоть до 60 минут. Общий вес систем колебался в интервале от 3,75 г до 75 г (как указано в Таблице 4). Полученную смесь вводили в НРМС капсулы и оставляли для охлаждения до комнатной температуры и затвердения. Капсулы хранили или при комнатной температуре, или в замороженном состоянии до использования.
Твин 80 (30,00)
Кремофор EL (30,00)
ПлюроникР-68 (30,00)
ПЭГ 1000 (30,00)
ПЭГ 1000 (22,50)
ПЭГ 1000 (15,00)
ПЭГ 1000 (7,50)
Пример 7. Растворение композиций в среде для растворения с рН 6,5
In vitro способ растворения, который использует среду для растворения с рН 6,5, применяли для анализа поведения композиций, которые содержатся в НРМС капсулах. Способ растворения при рН 6,5 обеспечивал улучшенное установление отличия в присутствии формы свободного основания Агента в композициях по сравнению со способом, описанным в Примере 5. Растворение в двукратных и трехкратных повторах проводили на композициях, приведенных в Таблице 4, а также на композиции Примера 1.7.
Растворение капсул проводили в соответствии с общей процедурой при использовании аппаратуры II Фармакопеи США (устройство с мешалкой). Образцы среды для растворения отбирали в разные моменты времени после добавления капсул и количественно оценивали концентрации (2-гидроксиэтокси)амида 6-(4-бром-2-хлорфениламино)-7-фтор-3-метил-3H-бензоимидазол-5-карбоновой кислоты путем сравнения их площади пика ответа ВЭЖХ с такой стандартного раствора, который готовили при уровне, эквивалентном 100% высвобождению соединения. В этом способе использовали сосуды из прозрачного стекла с заостренным концом для растворения, а спиральные держатели из нержавеющей стали использовали для закрепления капсул. Использовали 1000 мл среды для растворения с рН 6,5 при 37°С и скорости перемешивания 50 об./минуту.
Среду для растворения с рН 6,5 готовили путем прибавления 1,7 г гранул гидроокиси натрия, 19,77 г водного дигидрофосфата натрия (или 17,19 г безводного дигидрофосфата натрия) и 30,93 г хлорида натрия к 5 литрам деионизированной воды. Значение рН потом доводили до 6,5 с помощью 1 М хлористоводородной кислоты или 1 М гидроокиси натрия.
В дополнение к композициям, описанным в Таблице 4, изготавливали несколько дополнительных композиций для сравнения при использовании смесей кристаллического свободного основания Агента и кристаллического Агента (то есть кислой сернокислой соли). Смесь этих двух форм диспергировали в витамине Е TPGS в соответствии со способом, аналогичным тем, что описаны в Примере 6, и вносили их в НРМС капсулы. Конкретные составы композиций для сравнения показаны в Таблице 5.
Данные относительно растворения для сравнительных композиций (Таблица 6) показывают, что растворение снижается при повышении количества формы свободного основания Агента в композиции. Снижение растворения до 90% через 60 минут наблюдали для композиции, которая содержит 0,4% вес./вес. формы свободного основания Агента. Кроме того, присутствие 0,02% вес./вес. свободного основания Агента вызывало 13% снижение растворения через 60 минут. Данные, полученные путем анализа свободного основания, которое содержится в сравнительных композициях, показали, что способ растворения при рН 6,5 обеспечивает хороший показатель уровня формы свободного основания Агента, который является присутствующей в композициях.
Результаты растворения для композиций, описанных в Примере 6, а также для композиции Примера 1.7 представлены в Таблицы 7. Значение, превышающее 96% растворение через 60 минут, достигалось для всех композиций, это свидетельствует о том, что в этих композициях Агент присутствует существенно в форме своей кислой сернокислой соли.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПЕРОРАЛЬНЫЕ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ТАКСАНЫ, И СПОСОБЫ ЛЕЧЕНИЯ С ИХ ПРИМЕНЕНИЕМ | 1999 |
|
RU2236226C2 |
| ТРАНСБУККАЛЬНАЯ СИСТЕМА ДОСТАВКИ | 2006 |
|
RU2406480C2 |
| НОВАЯ ГИДРОСУЛЬФАТНАЯ СОЛЬ | 2006 |
|
RU2418790C2 |
| ПОЛУТВЕРДЫЕ СИСТЕМЫ, СОДЕРЖАЩИЕ ПРОИЗВОДНЫЕ АЗЕТИДИНА | 2004 |
|
RU2343915C2 |
| НОВАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ НЕИОННЫЕ СУРФАКТАНТЫ | 2015 |
|
RU2720680C2 |
| ФАРМАЦЕВТИЧЕСКАЯ ДОЗИРОВАННАЯ ФОРМА ДЛЯ ПЕРОРАЛЬНОГО ВВЕДЕНИЯ ИНГИБИТОРА СЕМЕЙСТВА BCL-2 | 2010 |
|
RU2568599C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРЕДНАЗНАЧЕННАЯ ДЛЯ ПЕРОРАЛЬНОГО ВВЕДЕНИЯ ПРОИЗВОДНОГО ПИРАЗОЛ-3-КАРБОКСАМИДА | 2004 |
|
RU2321404C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ТАКСОИДОВ | 2011 |
|
RU2603833C2 |
| ФАРМАЦЕВТИЧЕСКАЯ ДОЗИРОВАННАЯ ФОРМА ДЛЯ ПЕРОРАЛЬНОГО ВВЕДЕНИЯ ИНГИБИТОРА СЕМЕЙСТВА BCL-2 | 2010 |
|
RU2711359C2 |
| ФАРМАЦЕВТИЧЕСКАЯ ЛЕКАРСТВЕННАЯ ФОРМА ИНГИБИТОРА ТИРОЗИНКИНАЗЫ ДЛЯ ПЕРОРАЛЬНОГО ВВЕДЕНИЯ | 2007 |
|
RU2468788C2 |
Изобретение относится к фармацевтическим композициям, которые содержат кислую сернокислую соль (2-гидроксиэтокси)амида 6-(4-бром-2-хлорфениламино)-7-фтор-3-метил-3Н-бензоимидазол-5-карбоновой кислоты и сольваты, их кристаллические формы и аморфные формы, к применению указанных композиций в качестве лекарственного средства; и к способам получения указанных композиций. 4 н. и 16 з.п. ф-лы, 7 табл., 7 пр., 5 ил.
1. Фармацевтическая композиция, которая включает кислую сернокислую соль (2-гидроксиэтокси)амида 6-(4-бром-2-хлорфениламино)-7-фтор-3-метил-3Н-бензоимидазол-5-карбоновой кислоты и матриксный носитель, где матриксный носитель существенно состоит из одного или более фармацевтически приемлемых носителей, выбранных из следующих групп:
(a) d-альфа-токоферил полиэтиленгликоль 1000 сукцинат;
(b) полигликозилированные глицериды;
(c) полиэтиленгликоли (ПЭГи); и
(d) твердые жиры;
и где кислая сернокислая соль (2-гидроксиэтокси)амида 6-(4-бром-2-хлорфениламино)-7-фтор-3-метил-3Н-бензоимидазол-5-карбоновой кислоты является диспергированной в матриксном носителе.
2. Фармацевтическая композиция по п.1, где матриксный носитель существенно состоит из одной или более групп, выбранных из следующих групп:
а. d-альфа-токоферилполиэтиленгликоль 1000 сукцинат;
b. полигликозилированные глицериды; и
с. полиэтиленгликоль.
3. Фармацевтическая композиция по п.1 или 2, где матриксный носитель существенно состоит из одного или обоих компонентов:
а. d-альфа-токоферил полиэтиленгликоль 1000 сукцината; и
b. полигликозилированных глицеридов.
4. Фармацевтическая композиция по п.1, где матриксный носитель представляет собой d-альфа-токоферил полиэтиленгликоль 1000 сукцинат или лауроил макрогол-32 глицериды.
5. Фармацевтическая композиция по п.1, где матриксный носитель представляет собой смесь d-альфа-токоферил полиэтиленгликоля 1000 сукцината и лауроил макрогол-32 глицеридов, и где лауроил макрогол-32 глицериды являются присутствующими в количестве, которое составляет приблизительно 30-55 вес.% компонента матриксного носителя композиции.
6. Фармацевтическая композиция по п.1, где матриксный носитель представляет собой d-альфа-токоферил полиэтиленгликоль 1000 сукцинат.
7. Фармацевтическая композиция по п.6, где d-альфа-токоферил полиэтиленгликоль 1000 сукцинат присутствует в количестве, которое составляет приблизительно от 65 до 95% от веса композиции.
8. Фармацевтическая композиция по п.1, где более 90% от общего весового количества кислой сернокислой соли (2-гидроксиэтокси)амида 6-(4-бром-2-хлорфениламино)-7-фтор-3-метил-3Н-бензоимидазол-5-карбоновой кислоты, которая является присутствующей в композиции, является диспергированным в матриксном носителе.
9. Фармацевтическая композиция по п.1, где композиция содержит от 5 до 30 вес.% кислой сернокислой соли (2-гидроксиэтокси)амида 6-(4-бром-2-хлорфениламино)-7-фтор-3-метил-3Н-бензоимидазол-5-карбоновой кислоты.
10. Фармацевтическая композиция по п.1, где композиция является полутвердой или твердой при комнатной температуре.
11. Фармацевтическая композиция по п.1, где кислая сернокислая соль (2-гидроксиэтокси)амида 6-(4-бром-2-хлорфениламино)-7-фтор-3-метил-3Н-бензоимидазол-5-карбоновой кислоты является диспергированной в форме тонко измельченных частиц, которые являются распределенными в фазе, которая включает матриксный носитель.
12. Фармацевтическая композиция по п.1, которая включает:
(i) от 15 до 25 частей кислой сернокислой соли (2-гидроксиэтокси)амида 6-(4-бром-2-хлорфениламино)-7-фтор-3-метил-3Н-бензоимидазол-5-карбоновой кислоты; и
(ii) от 75 до 85 частей витамина Е TPGS;
где обе части являются весовыми, а сумма частей (i)+(ii)=100;
и где кислая сернокислая соль (2-гидроксиэтокси)амида 6-(4-бром-2-хлорфениламино)-7-фтор-3-метил-3Н-бензоимидазол-5-карбоновой кислоты является диспергированной в витамине Е TPGS, и композиция является полутвердой или твердой при комнатной температуре.
13. Фармацевтическая композиция по п.1, которая включает:
(i) от 18 до 22 части кислой сернокислой соли (2-гидроксиэтокси)амида 6-(4-бром-2-хлорфениламино)-7-фтор-3-метил-3Н-бензоимидазол-5-карбоновой кислоты; и
(ii) от 78 до 82 части витамина Е TPGS;
где обе части являются весовыми, а сумма частей (i)+(ii)=100;
и где кислая сернокислая соль (2-гидроксиэтокси)амида 6-(4-бром-2-хлорфениламино)-7-фтор-3-метил-3Н-бензоимидазол-5-карбоновой кислоты является диспергированной в витамине Е TPGS, и композиция является полутвердой или твердой при комнатной температуре.
14. Фармацевтическая композиция по п.1, которая включает:
(i) 19-21 части кислой сернокислой соли (2-гидроксиэтокси)амида 6-(4-бром-2-хлорфениламино)-7-фтор-3-метил-3Н-бензоимидазол-5-карбоновой кислоты; и
(ii) 79-81 части витамина Е TPGS;
где обе части являются весовыми, а сумма частей (i)+(ii)=100; и где кислая сернокислая соль (2-гидроксиэтокси)амида 6-(4-бром-2-хлорфениламино)-7-фтор-3-метил-3Н-бензоимидазол-5-карбоновой кислоты является диспергированной в витамине Е TPGS, и композиция является полутвердой или твердой при комнатной температуре.
15. Фармацевтическая композиция по п.1, где композиция представляет собой композицию пероральных капсул.
16. Способ получения фармацевтической композиции по п.1, который включает этапы:
а. смешивание и расплавление компонентов матриксного носителя;
b. смешивание агента с матриксным носителем для получения гомогенной смеси; и
с. внесение продукта этапа (b) в капсулу и охлаждение смеси с образованием густой жидкой, полутвердой или твердой массы в капсуле.
17. Способ лечения теплокровного животного (желательно человека), которое страдает от состояния, которое может подвергаться лечению с помощью кислой сернокислой соли (2-гидроксиэтокси)амида 6-(4-бром-2-хлорфениламино)-7-фтор-3-метил-3Н-бензоимидазол-5-карбоновой кислоты, который включает введение фармацевтической композиции по любому из пп.1-15.
18. Способ лечения рака у теплокровного животного (желательно у человека), который включает введение фармацевтической композиции по любому из пп.1-15.
19. Фармацевтическая композиция по п.1 для применения в качестве лекарственного средства в лечении состояния, которое может подвергаться лечению с помощью кислой сернокислой соли (2-гидроксиэтокси)амида 6-(4-бром-2-хлорфениламино)-7-фтор-3-метил-3Н-бензоимидазол-5-карбоновой кислоты.
20. Фармацевтическая композиция по п.1 для применения в качестве лекарственного средства в лечении рака.
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| RU 2006105000 A1, 08.07.2004 | |||
| Некоторые особенности технологии получения капсул, подбора композиций желатиновых масс и наполнителей | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |

