Предпосылки изобретения
1. Область изобретения
Изобретение относится к композициям для перорального введения паклитаксела и родственных таксанов пациентам и способам лечения с применением таких композиций.
2. Описание предшествующего уровня знаний
Многие ценные фармакологически активные соединения не могут эффективно вводиться пациентам пероральным путем по причине их предварительного или несовместимого всасывания из желудочно-кишечного тракта. Поэтому такие фармацевтические средства вводятся преимущественно внутривенным путем врачом или другим медицинским профессионалом, что влечет за собой заметный дискомфорт и возможную местную травму у пациента и даже требует введения в условиях госпиталя с хирургическим вмешательством в случае некоторых в./в. инфузий.
Одним из важных классов цитотоксических агентов, которые обычно биологически недоступны при пероральном введении людям, являются таксаны, которые включают паклитаксел, его производные и аналоги. Паклитаксел (в настоящее время продается Bristol-Meyers Squibb Oncology Division под названием TAXOL®; ТАКСОЛ) представляет собой природный дитерпеновый продукт, выделенный из тиса тихоокеанского (Taxus brevifolia). Он является членом таксанового семейства терпенов. Он был впервые выделен в 1971 Wani et al. (J.Am.Chem. Soc., 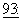 2325, 1971), которые охарактеризовали его структуру химическими методами и методом рентгеновской кристаллографии. Один из механизмов его активности имеет отношение к способности паклитаксела связывать тубулин, подавляя таким образом рост клеток злокачественной опухоли. Schiff et al., Proc. Natl. Acad. Sci. USA,
2325, 1971), которые охарактеризовали его структуру химическими методами и методом рентгеновской кристаллографии. Один из механизмов его активности имеет отношение к способности паклитаксела связывать тубулин, подавляя таким образом рост клеток злокачественной опухоли. Schiff et al., Proc. Natl. Acad. Sci. USA, 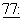 1561-1565 (1980); Schiff et al., Nature, 277: 665-667 (1979); Kumar, J.Biol.Chem.,
1561-1565 (1980); Schiff et al., Nature, 277: 665-667 (1979); Kumar, J.Biol.Chem., 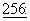 10435-10441 (1981).
10435-10441 (1981).
Паклитаксел принят к клиническому применению в лечении рефракторного рака яичника в Соединенных Штатах (Markman et al., Yale Journal of Biology and Medicine, 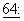 583, 1991; McGuire et al., Ann. Intern. Med.,
583, 1991; McGuire et al., Ann. Intern. Med., 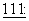 273, 1989). Он является эффективным при химиотерапии некоторых типов новообразований, включая таковые молочной железы (Holmes et al., J. Nat. Cancer Inst.,
273, 1989). Он является эффективным при химиотерапии некоторых типов новообразований, включая таковые молочной железы (Holmes et al., J. Nat. Cancer Inst., 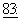 1797, 1991), а также принят для лечения рака молочной железы. Он является потенциальным кандидатом для лечения новообразований кожи (Einzig et al., Proc. Am. Soc. Clin. Oncol.,
1797, 1991), а также принят для лечения рака молочной железы. Он является потенциальным кандидатом для лечения новообразований кожи (Einzig et al., Proc. Am. Soc. Clin. Oncol., 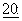 46), рака легких и карцином головы и шеи (Forastire et al., Sem. Oncol.,
46), рака легких и карцином головы и шеи (Forastire et al., Sem. Oncol., 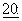 56, 1990). Данное соединение также обладает возможным потенциалом в лечении поликистоза почек (Woo et al., Nature, 368:750, 1994) и малярии.
56, 1990). Данное соединение также обладает возможным потенциалом в лечении поликистоза почек (Woo et al., Nature, 368:750, 1994) и малярии.
Паклитаксел лишь слабо растворим в воде, и это создает значительные проблемы при разработке подходящих инъекционных и инфузионных композиций, полезных для противораковой химиотерапии. Разработаны некоторые композиции паклитаксела для в./в. инфузии, с использованием CREMOPHOR EL™ (полиэтоксилированное касторовое масло) в качестве лекарственного носителя ввиду нерастворимости паклитаксела в воде. Например, паклитаксел, использованный в клиническом испытании под эгидой NCI, входил в состав композиции на основе 50% CREMOPHOR EL™ и 50% дегидратированного этилового спирта. Однако CREMOPHOR EL™ при внутривенном введении сам по себе является токсичным и вызывает у собак вазодилатацию, одышку, летаргию, гипотензию и смерть. Также предполагают, что он по меньшей мере частично отвечает за реакции аллергического типа, наблюдаемые во время введения паклитаксела, хотя имеются некоторые доказательства того, что паклитаксел сам по себе может провоцировать острые реакции даже в отсутствие Cremophor.
При попытках увеличить растворимость паклитаксела и разработать более безопасные клинические композиции исследования были направлены на синтез аналогов паклитаксела, в которых положения 2' и/или 7 замещены группами, которые могут увеличить растворимость в воде. Такие попытки привели к получению соединений - пролекарств, которые являются более растворимыми в воде, чем исходное соединение, и которые при активации проявляют цитотоксические свойства. Одна важная группа таких пролекарств включает 2’-oниeвыe соли паклитаксела и доцетаксела, в частности, соли 2’-мeтилпиpидиниймeзилaтa (2'-МРМ).
При пероральном введении паклитаксел очень слабо всасывается (менее 1%); см. Eiseman et al., Second NCI Workshop on Taxol and Taxus (Sept. 1992); Suffness et al., в Taxol Science and Application (CRC Press 1995). Eiseman et al. указывают на то, что паклитаксел при пероральном введении обладает биодоступностью, равной 0%, a Suffness et al. сообщают, что пероральное дозирование паклитаксела кажется невозможным, поскольку при пероральном введении противоопухолевая активность не была доказана при дозах вплоть до 160 мг/кг/сутки. По этой причине, согласно предшествующему уровню техники в данной области, паклитаксел не вводили пациентам перорально, и, конечно, не использовали таким образом в курсе лечения заболеваний, поддающихся лечению паклитакселом.
Доцетаксел (N-дебензоил-N-трет-бутоксикарбонил-N-деацетилпаклитаксел) стал коммерчески доступным под названием TAXOTERE® (ТАКСОТЕР; Rhone-Poulenc-Rorer S.A.) в парентеральной форме для лечения рака груди. К настоящему времени в научной литературе не приведено ни одной ссылки на пероральное всасывание доцетаксела у животных или пациентов.
Предполагается, что в некоторых случаях слабая или отсутствующая биодоступность лекарственного средства, такого как паклитаксел, после перорального введения является результатом активности мультилекарственного транспортера, мембраносвязанного Р-гликопротеина, который функционирует как энергонезависимый транспортер или насос утечки для понижения внутриклеточного накопления лекарственного средства путем вытеснения ксенобиотиков из клетки. Данный Р-гликопротеин идентифицирован в нормальных тканях секреторного эндотелия, таких как билиарная выстилка, ресничный край проксимального канальца в почке и поверхность просвета кишечника, и в васкулярных эндотелиальных клетках, образующих гематоэнцефалический барьер, барьеры плаценты и яичек.
Считается, что Р-гликопротеиновый насос утечки предотвращает проникновение некоторых фармацевтических соединений через слизистые клетки тонкого кишечника и, таким образом, от всасывания в системную циркуляцию. Показано, что некоторое количество нецитотоксических фармакологических средств ингибируются Р-гликопротеином, включая, среди прочих, циклоспорин А (также известный как циклоспорин), верапамил, тамоксифен, хинидин и фенотиазины. Многие из данных исследований направлены на достижение большего накопления внутривенно введенных цитотоксических лекарственных средств внутри опухолевых клеток. В действительности, проводились клинические испытания, чтобы изучить воздействия циклоспорина на фармакокинетику и токсичность паклитаксела (Fischer et al.,. Proc. An. Soc. Clin. Oncol., 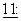 143, 1994); доксорубицина (Bartlett et al., J. Clin. Oncol.,
143, 1994); доксорубицина (Bartlett et al., J. Clin. Oncol., 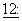 835-842, 1994) и этопозида (Lum et al., J. Clin. Oncol.,
835-842, 1994) и этопозида (Lum et al., J. Clin. Oncol., 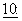 1635-42, 1992), причем все из них являются противораковыми средствами, известными как зависимые от мультилекарственной резистентности (MDR). Данные испытания показали, что пациенты, получавшие циклоспорин внутривенно перед или одновременно с противораковыми лекарственными средствами, обладали более высоким содержанием данных лекарственных средств в крови, преимущественно за счет сниженного клиренса организма, и проявляли ожидаемую токсичность при существенно более низких уровнях дозы. Эти результаты указывают на то, что сопутствующее введение циклоспорина подавляло MDR-действие Р-гликопротеина, приводя к большему внутриклеточному накоплению терапевтических средств. Для общей дискуссии по фармакологическим применениям ингибиторов Р-гликопротеина для клинического использования см. Lum et al., Drug Resist. Clin. Onc. Hemat.,
1635-42, 1992), причем все из них являются противораковыми средствами, известными как зависимые от мультилекарственной резистентности (MDR). Данные испытания показали, что пациенты, получавшие циклоспорин внутривенно перед или одновременно с противораковыми лекарственными средствами, обладали более высоким содержанием данных лекарственных средств в крови, преимущественно за счет сниженного клиренса организма, и проявляли ожидаемую токсичность при существенно более низких уровнях дозы. Эти результаты указывают на то, что сопутствующее введение циклоспорина подавляло MDR-действие Р-гликопротеина, приводя к большему внутриклеточному накоплению терапевтических средств. Для общей дискуссии по фармакологическим применениям ингибиторов Р-гликопротеина для клинического использования см. Lum et al., Drug Resist. Clin. Onc. Hemat., 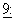 319-336 (1995); Schinkel et al., Eur. J. Cancer,
319-336 (1995); Schinkel et al., Eur. J. Cancer, 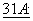 1295-1298 (1995).
1295-1298 (1995).
В описанных выше исследованиях, относящихся к использованию циклоспорина для повышения содержания фармацевтических средств в крови, активные противоопухолевые средства и циклоспорин вводили внутривенно. В данных публикациях не было сделано предположения о том, что циклоспорин мог бы вводиться перорально, чтобы существенно повысить биодоступность перорально вводимых противораковых лекарственных средств и других фармацевтических средств, которые сами по себе слабо всасываются в кишечнике, не вызывая высокотоксичных побочных эффектов. Ни в одном из опубликованных исследований не приводится какого-либо регламента для применения эффективного перорального введения людям лекарственных средств с пониженной биодоступностью, таких как паклитаксел, например, указание соответственных пределов доз и временного распорядка введения конкретных целевых лекарственных средств, и средств, повышающих биодоступность, являющихся наиболее применимыми для улучшения перорального всасывания каждого целевого лекарственного средства или класса лекарственных средств.
В опубликованной заявке РСТ WO 95/20980 (опубликовано 10 августа 1995) Benet et al. описывают способ, нацеленный на повышение биодоступности перорально вводимых гидрофобных фармацевтических соединений. Данный способ включает пероральное введение пациентам таких соединений одновременно с биоусилителем (bioenhancer), включающим ингибитор фермента цитохрома Р450 3А или ингибитор опосредованного Р-гликопротеином мембранного транспортера. Однако, Benet et al. по существу не предоставили средств для определения, какое именно усиливающее биодоступность средство будет улучшать доступность конкретных “целевых” фармацевтических соединений, и также они не привели конкретных значений доз, схем и регламентов для введения усиливающего или целевого средства. В действительности, хотя в заявке Benet et al. перечислено множество потенциальных усилителей (ингибиторов Р450 3А) и целевых лекарственных средств (субстратов Р450 3А), единственным сочетанием усилителя и целевого средства, подтвержденного в заявке каким-либо экспериментальным доказательством, является кетоконазол как усилитель и циклоспорин А как целевое лекарственное средство.
При описании основных характеристик соединений, которые можно использовать в качестве биоусилителей, понижающих активность Р-гликопротеинового транспортера, Benet et al. отмечают, что они представляют собой гидрофобные соединения, которые в основном, но не обязательно, содержат два лежащих в одной плоскости ароматических кольца, положительно заряженную группу азота или карбонильную группу - класс, включающий огромное количество соединений, большинство из которых не будут обладать требуемой усиливающей всасывание активностью в случае конкретного целевого средства. Более того, классы целевых средств, описанных Benet et al., включают большое число фармацевтических средств, перечисленных в Physician, s Desk Reference. Эти критерии включения не имеют смысла для практических медиков, нуждающихся в безопасных, практичных и эффективных способах перорального введения конкретных фармацевтических средств.
В общем, Benet et al. не предоставляют руководства, которому могут следовать специалисты в медицинской и фармацевтических областях, чтобы определить подходящие комбинации биоусилитель/целевое лекарственное средство или составить конкретные регламенты и схемы лечения, которые будут делать целевые средства терапевтически эффективными при пероральном введении пациентам. Benet et al. также не предоставляют чего бы то ни было, касающегося возможного перорального введения людям паклитаксела и других таксанов при наличии терапевтической эффективности и приемлемой токсичности.
В опубликованной заявке РСТ WO 97/15269, которая соответствует патентной заявке США с серийным No 08/733142 (предшествует настоящей заявке) и которая находится в общем владении с настоящей заявкой, раскрыто, что различные терапевтически эффективные фармацевтические “целевые средства”, проявляющие низкую пероральную биодоступность, могут стать биодоступными с обеспечением терапевтических уровней содержания активного агента в крови путем совместного перорального введения конкретных средств, повышающих биодоступность. Предпочтительные примеры таких целевых средств, описанных в WO 97/15269, включают циклоспорины, например, циклоспорин A, D и G. Предпочтительные примеры целевых средств включают таксановый класс средств против новообразований, в частности, паклитаксел. Также описаны терапевтические регламенты и величины доз для совместного введения целевых средств и усиливающих средств. Все раскрытия опубликованной заявки WO 97/15269 включены в данное описание в качестве ссылки.
Ни заявка общего владения WO 97/15269, ни какие-либо сообщения предшествующего уровня в данной области не описывают классы пероральных составов или композиций, содержащих активное целевое средство, например, паклитаксел, которые в основном адаптированы для совместного введения с пероральным средством, повышающим биодоступность с целью добиться терапевтических уровней содержания целевого средства в крови, что прежде считалось невозможным при пероральном введении.
Сущность изобретения
Настоящее изобретение относится к пероральным фармацевтическим композициям, содержащим таксановые противоопухолевые средства, например, паклитаксел или доцетаксел, которые при введении млекопитающему предпочтительно совместно с пероральным средством, повышающим биодоступность, обеспечивают существенное всасывание таксанового средства из желудочно-кишечного тракта в кровяное русло с достижением терапевтически значимых уровней содержания активного лекарственного средства.
Композиции по изобретению включают несущее средство, в состав которого входит носитель, в котором растворено или диспергировано таксановое средство. Несущее средство может также включать снижающий вязкость сорастворитель, который приводит несущее средство в более текучее состояние при температуре тела или по меньшей мере уменьшает точку плавления несущего средства ниже температуры тела, и может также обеспечивать повышенную растворимость таксана.
Носитель, используемый в новых композициях, предпочтительно представляет собой неионное поверхностно-активное вещество или эмульгатор, характеризующийся значением коэффициента гидрофильно-липофильного баланса (HLB), равным по меньшей мере приблизительно 10. Снижающий вязкость сорастворитель выбран, например, из органических растворителей, подходящих для перорального введения, растительных масел, гидрогенированного или полиоксиэтилированного касторового масла, сложных эфиров цитрата и насыщенных полигликолизированных глицеридов. Определенные насыщенные полигликолизированные глицериды могут также служить в композициях по изобретению в качестве носителей.
Новые фармацевтические композиции содержат приблизительно 2-500 мг/мл или мг/г таксана и предпочтительно приблизительно 2-50 мг/мл или мг/г таксана. Терапевтически неактивное несущее средство включает по меньшей мере 30% по массе носителя и приблизительно 0-70% сорастворителя и может также содержать обычные фармацевтические добавки и эксципиенты, как ароматизаторы и красители и тому подобное.
Другой аспект изобретения относится к способам лечения млекопитающих, страдающих заболеваниями, подлежащими воздействию таксанов, путем введения таким пациентам пероральных фармацевтических композиций по изобретению, предпочтительно при совместном введении средства, повышающего биодоступность.
Подробное описание изобретения
Пероральные фармацевтические композиции по изобретению содержат по меньшей мере два компонента: активный агент, включающий таксан, предпочтительно, противоопухолевое средство паклитаксел или доцетаксел, и терапевтически неактивное несущее средство, включающее фармацевтически приемлемый носитель для указанного таксана.
Чтобы получить композиции для перорального введения, которые представляют собой жидкость или по меньшей мере текучую форму при температуре тела (приблизительно 37°С), что в основном требуется для пероральной биодоступности, в некоторых случаях необходимо добавлять к несущему средству дополнительный компонент: снижающий вязкость сорастворитель, который уменьшает вязкость и увеличивает текучесть несущего средства при температуре тела, и также может увеличивать количество активного агента, которое может быть растворено или диспергировано в несущем средстве, по сравнению с использованием одного носителя.
Новые композиции могут включать более чем один таксан в качестве активного ингредиента и более чем один носитель и/или сорастворитель в качестве неактивных несущих компонентов. Несущее средство включает по меньшей мере 30% по массе носителя, предпочтительно 30-90% по массе. Предпочтительные для использования по изобретению носители представляют собой неионные поверхностно-активные вещества или эмульгаторы, характеризующиеся значениями HLB, по меньшей мере, приблизительно 10. Обнаружено, что такие неионные поверхностно-активные вещества или эмульгаторы не только являются совместимыми носителями для липофильных таксанов (которые лишь слаборастворимы в воде), но также способствуют всасыванию активного ингредиента из желудочно-кишечного тракта в кровяное русло.
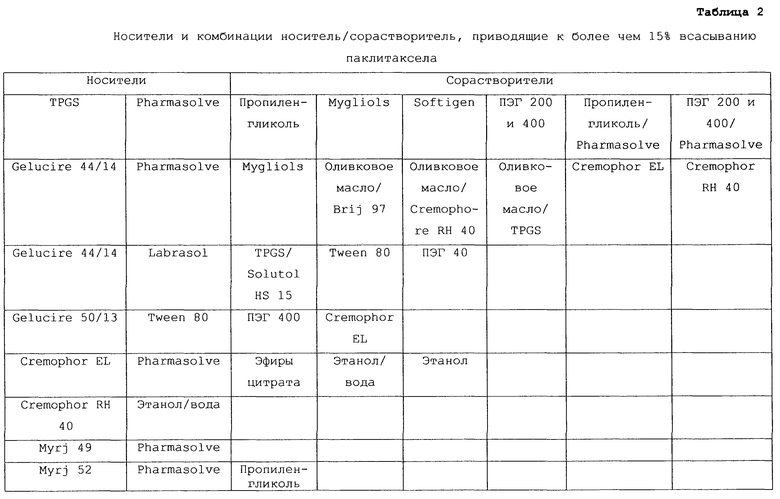
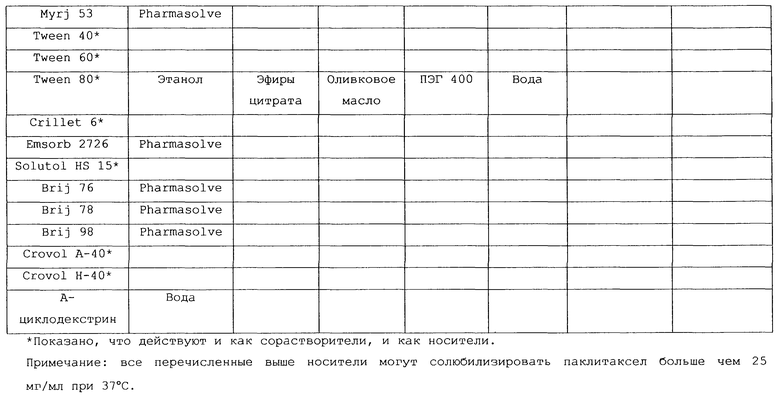
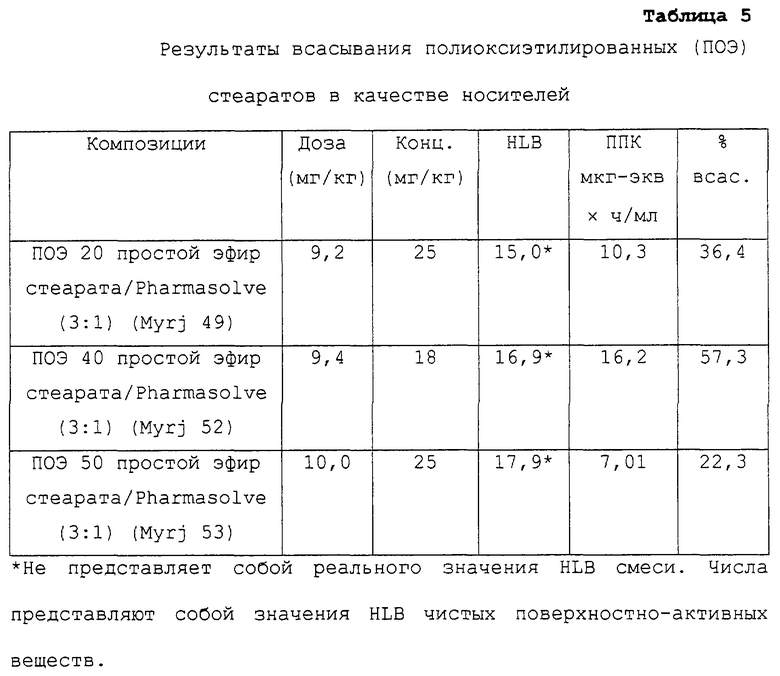
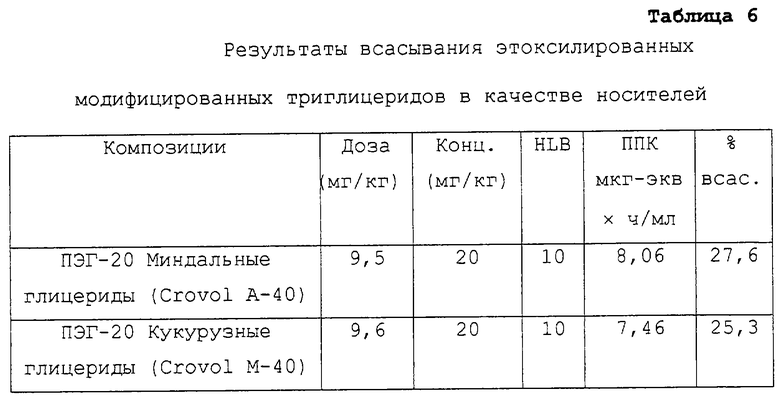
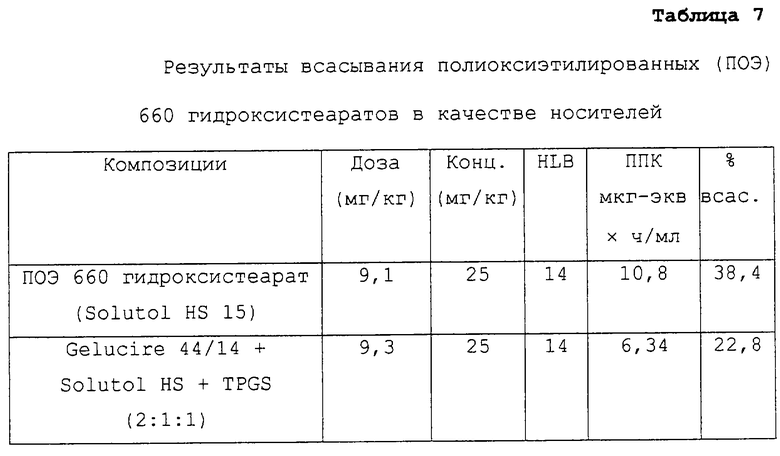
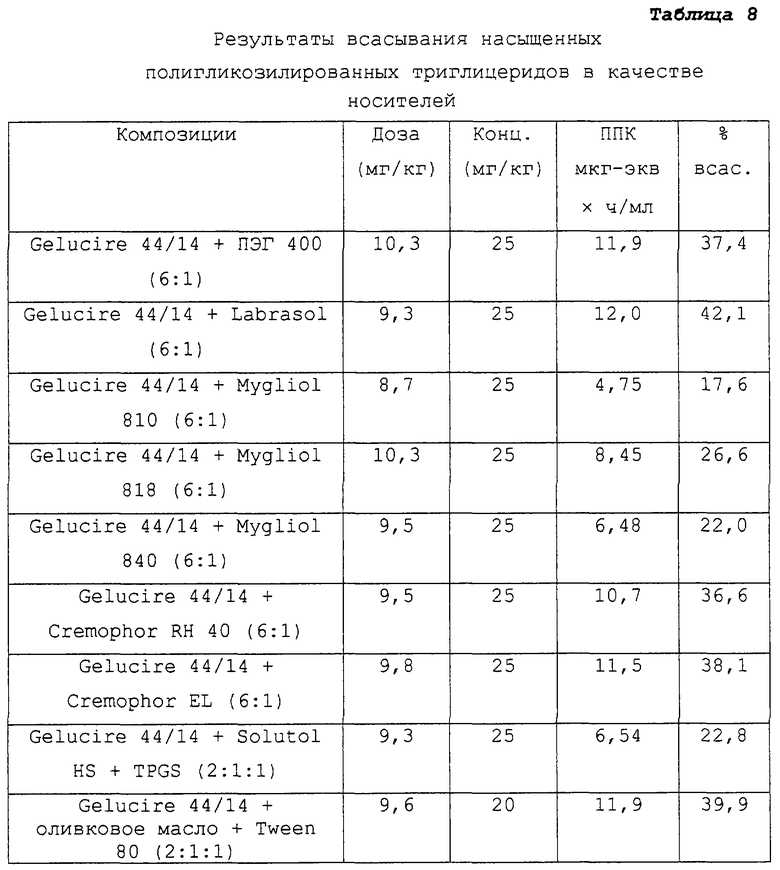
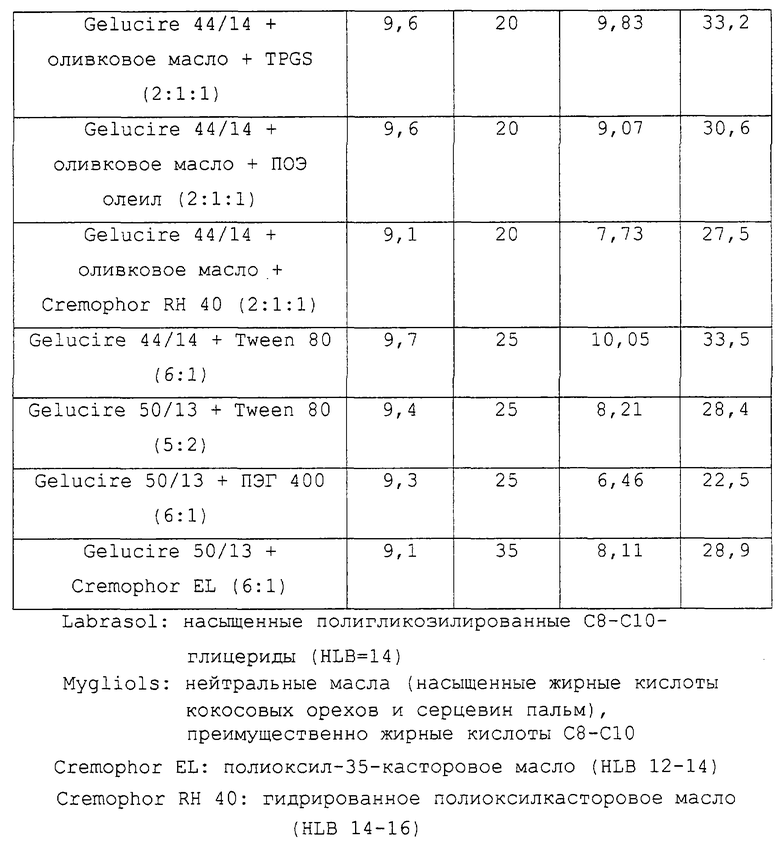
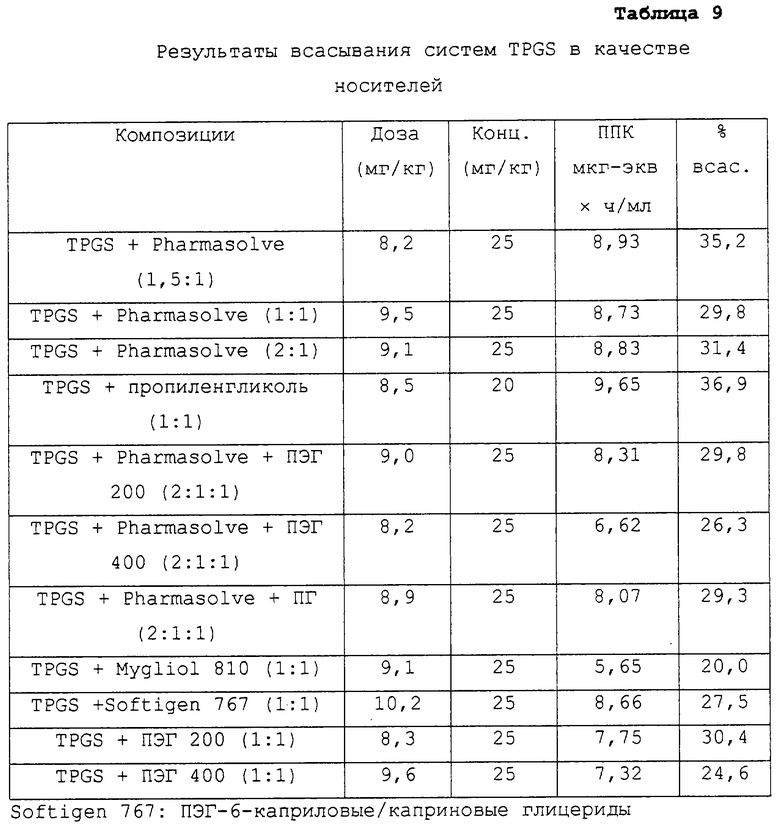
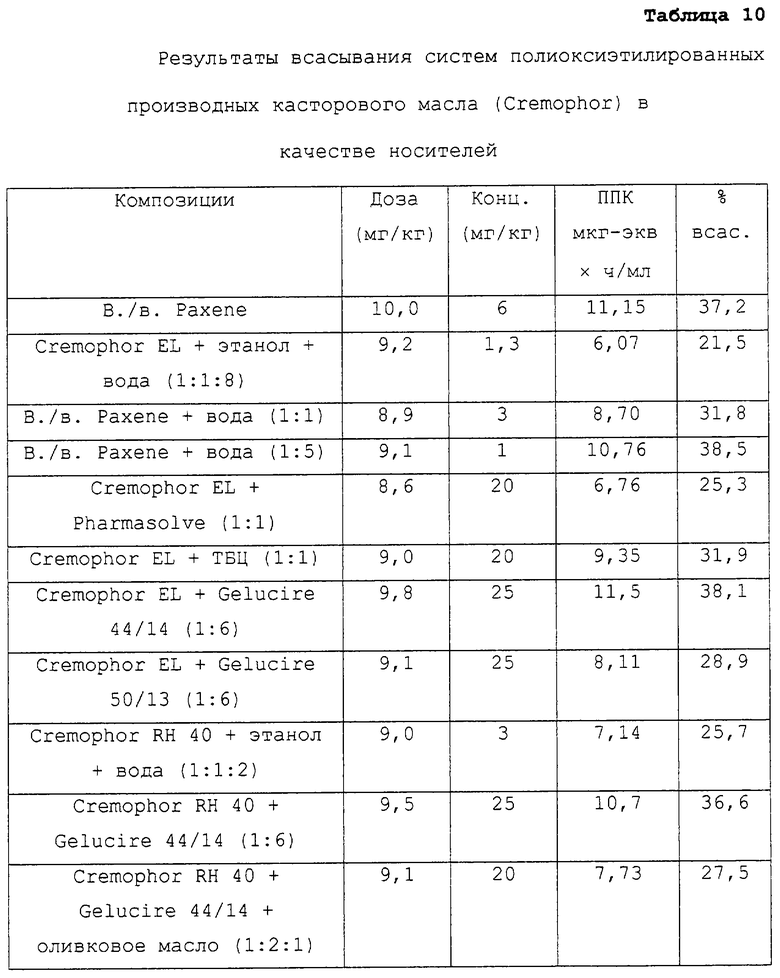
Предпочтительные для использования по изобретению носители включают, например, витамин Е TPGS (сукцинат d-α-токоферилполиэтиленгликоля 1000; Eastman Chemical Company Co., Kingsport, TN); насыщенные полигликолизированные глицериды, такие как продукты GELUCIRE™ и LABRASOL™ (Gattefosse Corp., Westwood, NJ), которые включают глицериды С8–С18-жирных кислот; модифицированные касторовые масла CREMOPHOR™ EL или RH40 (BASF, Mt Olive, NJ); полиоксиэтилированный сложный эфир стеарата MYRJ™ (ICI Americas) и полиоксиэтилированные сорбитановые сложные эфиры TWEEN™ (ICI Americas) и CRILLET™ (Croda Inc., Parsipanny, NJ); полиоксиэтилированные жирные простые эфиры BRIJ™ (ICI Americas); модифицированные (полиэтиленгликолем) глицериды миндального и кукурузного масел CROVOL™ (Croda Inc.); сорбитановые сложные эфиры диизостеарата EMSORB™ (Henkel Corp., Ambler, PA); полиоксиэтилированные гидроксистеараты SOLUTOL™ (BASF); и β-циклодекстрин. Только такие члены данных семейств поверхностно-активных веществ, которые характеризуются значениями HLB приблизительно 10 или выше, могут использоваться в качестве носителей в представленных композициях.
Предпочтительные снижающие вязкость сорастворители включают, например, PHARMASOLVE™ (N-метил-2-пирролидон, International Specialty Products, Wayne, NJ); глицериновые или пропиленгликолевые эфиры каприловой и каприновой кислот MIGLYOL™ (Huels AG, Marl, Germany); полиоксиэтилированные гидроксистеараты (например, SOLUTOL™ HS 15); полиоксиэтилированные сорбитановые сложные эфиры TWEEN™; полиэтиленгликолевые эфиры каприловой и каприновой кислот (Huels AG); модифицированные касторовые масла (как CREMOPHOR™ EL или RH 40); растительные масла, такие как оливковое масло, полиоксиэтилированные простые жирные эфиры или модифицированные касторовые масла, определенные насыщенные полигликолизированные глицериды (такие как LABRASOL™); сложные эфиры цитрата, такие как трибутилцитрат, триэтилцитрат и ацетилтриэтилцитрат; пропиленгликоль, один или в комбинации с PHARMASOLVE™; этанол; вода; и полиэтиленгликоли низкой молекулярной массы, как ПЭГ 200 и 400.
Несущее средство содержит приблизительно 0-70% по массе сорастворителя и предпочтительно приблизительно 10-50% по массе.
Следует отметить, что некоторые из материалов, установленных в качестве носителей, обнаруживают свойство быть эффективными сорастворителями или сами по себе, или в комбинации с другими средствами, снижающими вязкость, для определенных других носителей. В общем, любой растворитель, в котором паклитаксел или другие таксаны по меньшей мере среднерастворимы при температуре тела или осторожном нагревании, может использоваться в качестве сорастворителя в несущем средстве новых композиций. Предпочтительными сорастворителями являются те, в которых можно растворить по меньшей мере 25 мг/мл паклитаксела или другого таксана приблизительно при 20-25°С.
Концентрация активного таксанового ингредиента или ингредиентов в композиции может варьировать в зависимости от растворимости активного агента в носителе(-лях) или системе носитель(-ли)/сорастворитель(-и) и требуемой общей дозы таксана, которую следует вводить пациенту перорально. Концентрация таксана может колебаться в пределах приблизительно от 2 до приблизительно 500 мг/мл или мг/г в несущем средстве и предпочтительно приблизительно от 2 до приблизительно 50 мг/мл или мг/г.
Композиции по изобретению можно приготовить обычным способом, известным специалистам в области фармацевтики для приготовления жидких или других текучих пероральных составов, содержащих поверхностно-активные носители и липофильные активные ингредиенты. Поскольку большинство предпочтительных носителей при комнатной температуре являются очень вязкими и в некоторых случаях сохраняют относительно высокую вязкость даже после добавления сорастворителя в малом соотношении, при приготовлении новых композиций, как правило, предпочтительно смешивать используемые носители и сорастворители, добавлять таксановый активный ингредиент и нагревать при помешивании полученную в результате смесь, например, до приблизительно 40°С. Данный способ обеспечивает получение чистых растворов. Однако некоторые сорастворители, в частности PHARMASOLVE™, снижают вязкость носителя и усиливают растворимость таксана до такой степени, что композицию можно приготовить, перемешивая при комнатной температуре без нагревания.
Желательно, чтобы вязкость конечной композиции не превышала 40000 сантипуаз при температуре тела (приблизительно 37°С).
Пероральные композиции по изобретению могут существовать в форме истинных растворов, эмульсий или даже суспензий, но предпочтительными являются растворы активного таксанового ингредиента в носителе или системе носитель/сорастворитель.
Настоящее изобретение также охватывает способы лечения людей, страдающих от злокачественных опухолей, опухолей, саркомы Калоши, злокачественных новообразований, неконтролируемых тканевых или клеточных пролифераций, вторичных по отношению к повреждению ткани, и от любых других патологических состояний, которые поддаются воздействию таксанов, таких как паклитаксел и доцетаксел, и/или пролекарств и производных указанных выше соединений, новыми фармацевтическими композициями для перорального введения. В числе типов карциномы, которые можно лечить перорально паклитакселом, доцетакселом, другими таксанами и их пролекарствами и производными особенно эффективно, находятся гепатоцеллюлярная карцинома и печеночные метастазы, раки желудочно-кишечного тракта, поджелудочной железы, предстательной железы и легких и саркома Капоши. Примерами не злокачественных патологических состояний, которые можно эффективно лечить данными активными агентами, вводимыми перорально по настоящему изобретению, являются неконтролируемые тканевые или клеточные пролиферации, вторичные по отношению к повреждению ткани, поликистоз почек, воспалительные заболевания (например, артрит) и малярия, включая инфекцию малярийными паразитами, устойчивыми к хлорохину и пириметамину (Pouvelle et al., J. Cin. Invest., 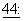 413-417, 1994).
413-417, 1994).
Хотя некоторые из пероральных фармацевтических композиций по изобретению могут обеспечить терапевтические уровни содержания таксанового активного ингредиента в крови при введении только их, предпочтительным способом по изобретению для лечения млекопитающих (в частности, человека), страдающих от патологических состояний, поддающихся воздействию таксанов, является введение пероральных композиций, содержащих таксановое целевое средство, одновременно с введением по меньшей мере одной дозы средства, повышающего биодоступность.
Предпочтительное осуществление способа по изобретению для перорального введения людям паклитаксела, его производных, аналогов и пролекарств и других таксанов включает пероральное введение человеку перорального средства, усиливающего всасывание или биодоступность, одновременно с или предварительно, или как одновременно с, так и перед пероральным введением для повышения всасывания интактного целевого средства в кровяное русло.
Усиливающие средства перорального введения, которые могут использоваться при выполнении предпочтительного осуществления изобретения, включают следующие неограничивающие примеры:
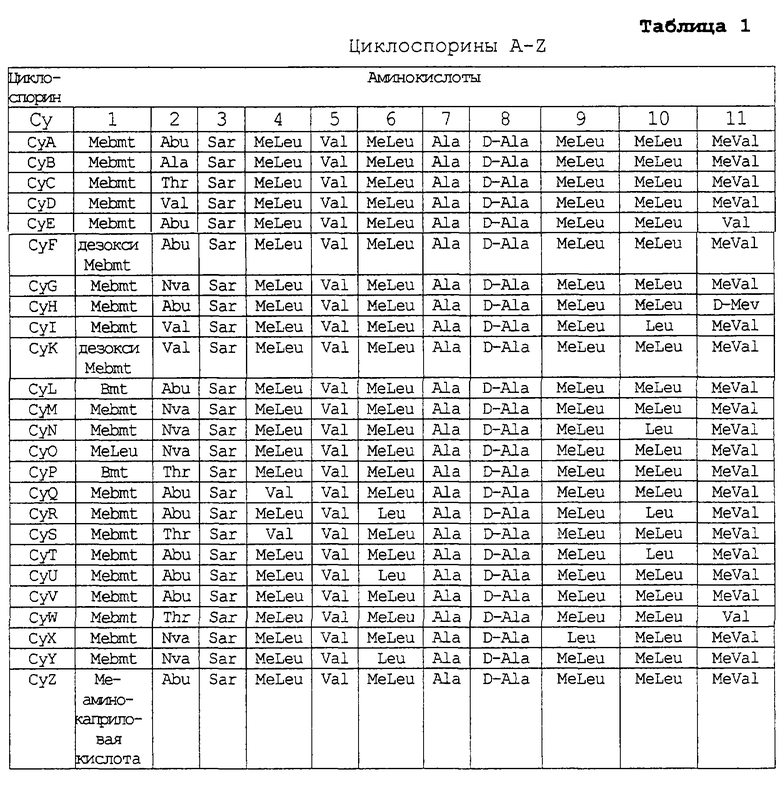
Циклоспорины, включая циклоспорины от А до Z, но особенно циклоспорин А (циклоспорин), циклоспорин F, циклоспорин D, дигидроциклоспорин А, дигидроциклоспорин С, ацетилциклоспорин A, PSS-833, SDZ-NIM 8111 (оба от Sandoz Pharmaceutical Corp). Структуры циклоспоринов A-Z описаны в таблице 1.
1 SDZ-NIM 811 представляет собой (Me-Ile-4)-циклоспорин, антивирусный, неиммунодепрессивный циклоспорин.
Циклоспорины представляют собой группу неполярных циклических олигопептидов (некоторые из которых обладают иммунодепрессивной активностью), продуцируемых организмами рода Topycladium, включая, например, Topycladium inflatum Gams (ранее называемый Trichoderma polysporum), Topycladium terricola и другие виды несовершенных грибков. Главный компонент, циклоспорин А (циклоспорин или СsА) был идентифицирован вместе с некоторыми другими минорными метаболитами, например, циклоспоринами от В до Z, некоторые из которых проявляют существенно меньшую иммунодепрессивную активность, нежели циклоспорин А. Также было получено много синтетических и полусинтетических аналогов. В целом см. Jegorov et al., Phytochemistry, 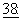 403-407 (1995). Настоящее изобретение охватывает природные, полусинтетические и синтетические аналоги циклоспоринов.
403-407 (1995). Настоящее изобретение охватывает природные, полусинтетические и синтетические аналоги циклоспоринов.
Циклоспорины представляют собой нейтральные, липофильные, циклические ундекапептиды с молекулярной массой приблизительно 1200. Они применяются внутривенно или перорально в качестве иммунодепрессантов, в первую очередь, при трансплантации органов и некоторых других состояниях. Циклоспорины, в частности, циклоспорин (циклоспорин А), являются известными ингибиторами Р-гликопротеинового насоса утечки и других насосов-транспортеров, так же как и некоторых ферментов деградации Р450, но до сих пор ни одного эффективного регламента для клинического применения данного свойства не было доведено до уровня клинического и коммерческого осуществления или одобрения распорядительными органами.
Величина дозировки усиливающего средства, подлежащего совместному введению с целевым средством по изобретению, составляют от приблизительно 0,1 до приблизительно 20 мг/кг массы тела пациента. “Совместное введение” усиливающего средства включает: по сути одновременное введение с целевым средством (или менее чем за 0,5 ч, менее чем через 0,5 ч, или совместно); введение заранее, от приблизительно 0,5 до приблизительно 72 ч, до введения целевого средства; или и то, и другое, то есть, одна или более дозы одного и того же или разных усиливающих средств, которые дают по меньшей мере за 0,5 ч до, и одна доза, вводимая по сути одновременно (или вместе, или немедленно до или после) с целевым средством. В дополнение, “совместное введение” охватывает введение более чем одной дозы целевого средства за период длительностью 72 ч после дозы усиливающего средства, иными словами, усиливающее средство(-а) не обязательно вводить снова до или вместе с каждым введением целевого средства, но оно может вводиться с перерывами в течение курса лечения.
Величина дозировки перорально вводимых таксановых целевых средств будет варьировать от соединения к соединению, в зависимости от их терапевтического индекса, требований к условиям лечения, состояния субъекта и так далее. Способ по изобретению дает возможность перорально вводить паклитаксел и другие таксаны в пределах от приблизительно 20 мг/м2 до приблизительно 1000 мг/м2 (из расчета на площадь поверхности тела пациента) или приблизительно 0,5-30 мг/кг (из расчета на массу тела пациента) в виде одной или разделенных (2-3) суточных доз, и поддерживать содержание паклитаксела в плазме крови человека в пределах 50-500 нг/мл в течение длительного периода времени (например, 8-12 ч) после каждой пероральной дозы. Данные уровни по меньшей мере сравнимы с таковыми, получаемыми при 96-часовой в./в. терапии инфузией паклитаксела (которая вызывает у пациента большие неудобства, дискомфорт, потерю полноценного времени, риск инфекций и т.д.). Более того, такие концентрации паклитаксела в плазме являются более чем достаточными для того, чтобы обеспечить требуемые виды фармакологической активности целевого лекарственного средства, например, ингибирование распада тубулина (которое происходит при уровнях приблизительно 0,1 мкМ, или приблизительно 85 нг/мл) и ингибирование изопренилирования белка (которое происходит при уровнях приблизительно 0,03 мкМ, или приблизительно 25 нг/мл), которые напрямую связаны с его противоопухолевыми эффектами, осуществляемыми путем ингибирования функций онкогенов и других белков сигнальной трансдукции, играющих основную роль в регуляции роста клеток.
Два или более усиливающих средств и/или два или более разных таксановых целевых средства могут вводиться вместе, альтернативно или с перерывами во всех различных аспектах способа по изобретению.
Как отмечалось выше, один паклитаксел, введенный перорально (например, в твердой дозированной форме или в жидком несущем средстве, не содержащем пероральный носитель, способствующий всасыванию), проявляет биодоступность, близкую к нулю. Чтобы считаться биодоступной перорально фармацевтичекой композицией, содержащей паклитаксел или другие таксаны для целей настоящего изобретения, композиция должна отвечать следующему критерию: когда данная композиция вводится перорально млекопитающему (например, лабораторной крысе или пациенту), т.е., поглощается субъектом через один час после введения эффективной пероральной дозы перорального средства, повышающего биодоступность, количество активного ингредиента, поступившего в кровоток, составляет по меньшей мере 15% от количества, поступившего в кровоток в случае введения субъекту той же дозы паклитаксела внутривенно в стандартном несущем средстве для внутривенного введения, например, в несущем средстве CREMOPHOR™ EL/этанол. Близкая процентная доля всасывания получается при сравнении соответствующих значений ППК (площадь под кривой) кривой уровня содержания таксана в крови в зависимости от времени, построенной после перорального введения, и соответствующей кривой, построенной после внутривенного введения.
Предпочтительным для использования средством, повышающим биодоступность, при выполнении экспериментального определения того, удовлетворяет ли данная пероральная композиция критерию 15% от в./в. поступления, является циклоспорин А, например, в единичной пероральной дозе СsА, равной 5 мг/кг.
Новые фармацевтические композиции могут вводиться в любых известных фармацевтических дозированных формах. Например, композиции могут быть инкапсулированы в мягкие или твердые желатиновые капсулы и могут вводиться в форме жидкого препарата. Каждая дозированная форма кроме существенных компонентов композиции (по меньшей мере один носитель и один таксановый активный ингредиент, и в некоторых случаях по меньшей мере один сорастворитель) может включать обычные фармацевтические эксципиенты, разбавители, подсластители, ароматизаторы, красители и любые другие инертные ингредиенты, обычно входящие в состав дозированных форм, предназначенных для перорального введения (см. например, Remington's Pharmaceutical Sciences, 17th Ed., 1985).
Точные количества каждого из целевых лекарственных средств, включенных в пероральные дозированные формы, будут варьировать в зависимости от возраста, веса, заболевания и состояния пациента. Например, дозированные формы паклитаксела или другого таксана могут содержать достаточные количества целевого средства, чтобы обеспечить суточную дозу приблизительно 20-1000 мг/м2 (из расчета на площадь поверхности тела пациента или млекопитающего) или приблизительно 0,5-30 мг/кг (из расчета на массу тела пациента или млекопитающего) в виде одной или разделенных (2-3) суточных доз. Предпочтительные количества дозировки составляют приблизительно 50-200 мг/м2 или приблизительно 2-6 мг/кг.
Регламент дозирования для способа лечения по настоящему изобретению, например, лечения заболеваний, поддающихся воздействию паклитаксела, пероральными дозированными формами паклитаксела, вводимыми совместно с усиливающими средствами, также может быть установлен с учетом характеристик пациента и состояния заболевания. Предпочтительные регламенты дозирования для введения паклитаксела перорально представляют собой: (а) ежесуточное введение пациенту при такой необходимости 1-3 равноразделенных доз, составляющих приблизительно 20-1000 мг/м2 (из расчета на площадь поверхности тела) и предпочтительно приблизительно 50-200 мг/м2, при том, что описанное ежесуточное введение продолжается в течение 1-4 последующих дней каждые 2-3 недели, или (b) введение в течение одного дня каждой недели. Предшествующий регламент сравним с использованием 96-часовой инфузии паклитаксела каждые 2-3 недели, которая считается некоторыми наиболее предпочтительным режимом в./в. лечения.
Пероральное введение таксанов по настоящему изобретению может существенно снизить токсические побочные эффекты во многих случаях, по сравнению с применяемой в настоящее время в./в. терапией. По сравнению с внезапным и быстрым возникновением высоких концентраций в крови, что обычно происходит в случае в./в. инфузии, всасывание активного агента через стенку кишечника (которому способствует усиливающее средство) обеспечивает более постепенное достижение данного уровня в крови и стабильное, стационарное поддержание такого уровня в идеальном или близком к нему пределу в течение длительного периода времени.
В дальнейшем осуществлении настоящего изобретения пероральные композиции по изобретению могут вводиться в двухкомпонентной системе лекарственного средства. Так, например, это могут быть определенные носители, входящие в объем настоящего изобретения, которые желательно использовать в несущих средствах для определенных таксановых средств вследствие их способности солюбилизировать таксан и способствовать его пероральному всасыванию, но носитель может быть химически или физически не совместимым с требуемыми дополнительными ингредиентами, такими как ароматизаторы и красители. В таких случаях активный ингредиент может вводиться пациенту в качестве первой части лекарственного средства в относительно малом объеме любого подходящего жидкого солюбилизирующего несущего средства (такого как вода, CREMOPHOR™ или этанол), которое может быть подслащено, ароматизировано или окрашено, как требуется, чтобы замаскировать неприятный вкус несущего средства и сделать его более приятным. За введением активного ингредиента может следовать введение второй части лекарственного средства: больший объем жидкости, например, от 1 до 8 унций (30-240 мл), содержащий по меньшей мере один носитель или систему носитель/сорастворитель по изобретению. Обнаружено, что введение второго, “преследующего” состава через короткое время после таксанового активного ингредиента может задержать преципитацию таксана, которая может произойти в противном случае после его поступления в желудочный сок, и способствовать пероральному всасыванию на уровне, сравнимом с тем, который наблюдается, когда таксан смешивается с носителем и вводится одновременно с ним.
Иллюстративные примеры “преследующих” композиций, которые могут использоваться в двухкомпонентном пероральном таксановом лекарственном средстве, включают:
a) 2-20% (по массе) витамина Е TPGS+вода по необходимости;
b) 2-25% витамина Е TPGS+2 - 25% PHARMSOLVE™+вода по необходимости;
c) 2-20% витамина Е TPGS+2 - 25% пропиленгликоля+вода по необходимости.
В соответствии с еще одним аспектом изобретения пероральные композиции по изобретению могут содержать не только один или более таксановых активных ингредиентов, но также один или более средств, повышающих биодоступность в комбинированной дозированной форме. Например, такая комбинированная дозированная форма может содержать от приблизительно 0,1 до приблизительно 20 мг/кг (из расчета на среднюю массу тела пациента) одного или более циклоспоринов А, D, С, F и G, дигидро-СsА, дигидро-СsС и ацетил-СsА вместе от приблизительно 20 до приблизительно 1000 мг/м2 (из расчета на среднюю площадь поверхности тела пациента), и, предпочтительно, приблизительно 50-200 мг/м2 паклитаксела, доцетаксела, другого таксана или производных паклитаксела или доцетаксела.
Композиции и способы по настоящему изобретению предоставляют много преимуществ по сравнению с предшествующими в данной области внутривенными композициями, содержащими паклитаксел и другие таксаны, и предшествующими в данной области внутривенными режимами введения. Помимо вопросов сниженной токсичности, удобства и комфорта пациента, простоты введения и сниженной стоимости, которые обсуждались выше, изобретение дает возможность вводить пациентам сильнодействующие таксановые противоопухолевые средства с сильно сниженной вероятностью развития аллергических реакций гиперчувствительности, которые являются обычными при в./в. введении. Таким образом, может отпасть необходимость в режимах премедикации Н-1- и Н-2-блокаторами и стероидами.
Настоящее изобретение также делает возможным вводить таксаны, например, паклитаксел, сравнительно не частыми ежесуточными дозами (например, приблизительно двух раз в сутки) и согласно регламентам, которые, с другой стороны, не могли бы быть возможными или применяться на практике при внутривенном пути введения. Применение усилителя биодоступности (например, циклоспорин А) способствует пероральному всасыванию паклитаксела первой дозы, и если вторая доза паклитаксела должна быть введена позже в тот же день, дополнительное использование циклоспорина А даже может быть ненужным. Таким образом, паклитаксел может вводиться с перерывами в виде единичной дозы по фиксированному регламенту (еженедельно, раз в две недели, и т.д.) или постоянно в течение периода последовательных дней (например, 4 дней) каждые 2-4 недели с целью поддержания уровней его содержания в пределах безопасного и эффективного “окна”.
Последующие примеры иллюстрируют различные аспекты изобретения. Однако эти примеры никоим образом не ограничивают данное изобретение или перечисления конкретных активных ингредиентов, носителей, совместных солюбилизаторов, усиливающих средств, величин дозировки, тестовых процедур или других параметров, которые должны использоваться исключительно для осуществления изобретения.
ПРИМЕР 1
Модель скрининга на животных
Группы по три самца крысы в каждой не получали корма в течение 16-18 ч перед введением дозы радиоактивно меченного 3H паклитаксела. Каждая группа животных получала одну пероральную дозу циклоспорина А (5 мг/кг) перед введением дозы экспериментального перорального состава паклитаксела. По прошествии одного часа после введения дозы циклоспорина, каждая группа получала приблизительно 9 мг/кг паклитаксела перорально в виде композиции по изобретению. Каждая из групп получала разный пероральный состав.
Образцы крови отбирали у каждого животного через 0,5, 1, 2, 3, 4, 6, 8, 12 и 24 ч после введения дозы паклитаксела.
Образцы крови сжигали и измеряли их общую радиоактивность.
Используя уровни общей радиоактивности крови (соответствующие концентрации в крови 3H-паклитаксела), строили график их зависимости от времени после введения дозы. Данные по каждой группе крыс компилировали как ППК, Сmax и Тmax.
Процентную долю всасывания 3H-паклитаксела для каждой группы животных рассчитывали путем сравнения среднего значения ППК в каждой группе с соответствующим средним ППК контрольной группы крыс, которым вводили 3H-паклитаксел (9 мг/кг) внутривенно в форме PAXENE™ (Baker Norton Pharmaceuticals, Miami, PL), которая включает CREMOPHOR™ EL, этанол и лимонную кислоту.
В таблице 2 перечислены все носители и комбинации носитель/сорастворитель, которые были составлены в содержащие паклитаксел пероральные композиции по изобретению, тестированы на крысах по описанной выше процедуре и, как было показано, обеспечивали у экспериментальных животных значения процента всасывания на уровне 15% или выше по сравнению с паклитакселом в./в.
ПРИМЕР 2
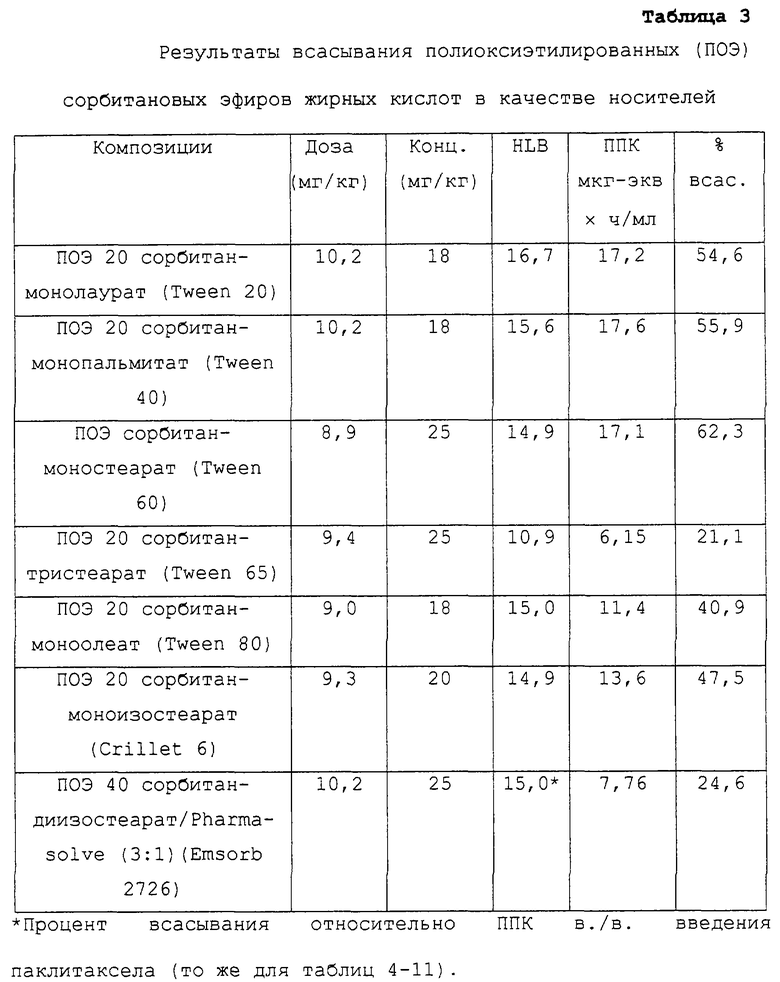
Полиоксиэтилированные (ПОЭ) сорбитановые эфиры жирных кислот в качестве носителей
В таблице 3 перечислены несущие составы, включающие определенные ПОЭ сорбитановые эфиры жирных кислот в качестве носителей для перорального паклитаксела как таковые или в комбинации с сорастворителем. Для составов, где в несущем средстве присутствует более чем один компонент, приведены соотношения масс компонентов. Каждый из данных составов тестировали на моделях животных, описанных в примере 1, и было обнаружено, что процент всасывания паклитаксела после перорального введения был выше (в некоторых случаях намного выше), чем 15% от всасывания введенной внутривенно приблизительно сравнимой дозы паклитаксела. В таблице 3 показаны общая доза паклитаксела, включенная в каждое несущее средство, как действительно введенная экспериментальным животным, концентрация паклитаксела в композиции, значение HLB носителя, среднее значение ППК для группы крыс, получавших состав и процент всасывания паклитаксела по сравнению с крысами, получавшими в./в. введение.
ПРИМЕР 3
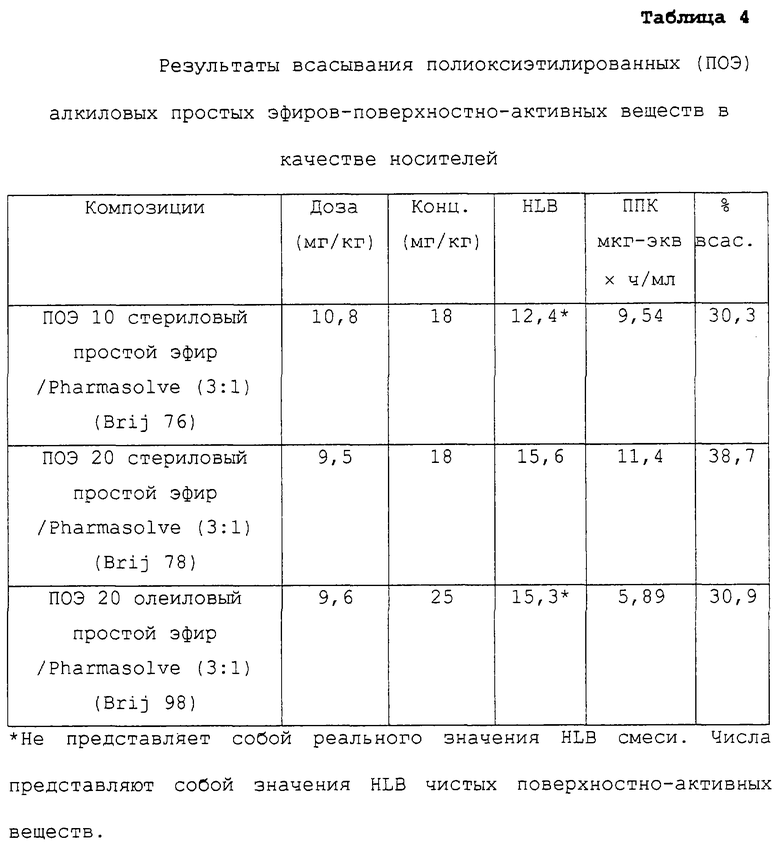
ПОЭ алкиловые простые эфиры в качестве носителей
Таблица 4 относится к композициям несущих средств, содержащих в качестве носителей ПОЭ алкиловые простые эфиры. Указанные данные представляют собой данные, описанные в предшествующем примере в соответствии с таблицей 3.
ПРИМЕР 4
ПОЭ стеараты в качестве носителей
Таблица 5 относится к композициям несущих средств, содержащих в качестве носителей ПОЭ стеараты. Указанные данные представляют собой данные, описанные в примере 2 в соответствии с таблицей 3.
ПРИМЕР 5
Этоксилированные модифицированные триглицериды в качестве носителей
Таблица 6 относится к композициям несущих средств, содержащих в качестве носителей этоксилированные модифицированные триглицериды. Указанные данные представляют собой данные, описанные в примере 2 в соответствии с таблицей 3.
ПРИМЕР 6
ПОЭ 660 гидроксистеараты в качестве носителей
Таблица 7 относится к композициям несущих средств, содержащих в качестве носителей ПОЭ 660 гидроксистеараты. Указанные данные представляют собой данные, описанные в примере 2 в соответствии с таблицей 3.
ПРИМЕР 7
Насыщенные полигликозилированные триглицериды в качестве носителей
Таблица 8 относится к композициям несущих средств, содержащих в качестве носителей насыщенные полигликозилированные триглицериды. Указанные данные представляют собой данные, описанные в примере 2 в соответствии с таблицей 3.
ПРИМЕР 8
Системы витамина Е TPGS в качестве носителей
Таблица 9 относится к композициям несущих средств, содержащих в качестве носителей системы витамина Е TPGS. Указанные данные представляют собой данные, описанные в примере 2 в соответствии с таблицей 3.
ПРИМЕР 9
ПОЭ и гидрированные производные касторового касла в качестве носителей
Таблица 10 относится к композициям несущих средств, содержащих в качестве носителей ПОЭ и гидрированных производных касторового масла. Указанные данные представляют собой данные, описанные в примере 2 в соответствии с таблицей 3.
ПРИМЕР 10
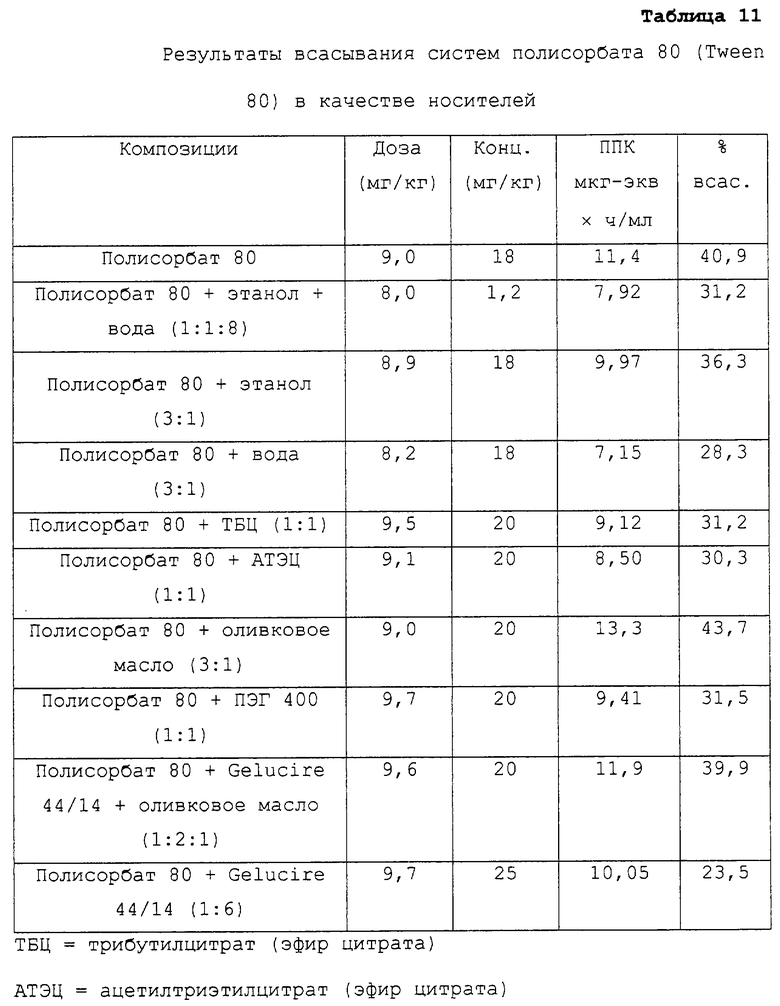
Носители на основе полисорбата 80
Таблица 11 относится к композициям несущих средств, содержащих полисорбат 80 и по меньшей мере один носитель.
Указанные данные представляют собой данные, описанные в примере 2 в соответствии с таблицей 3.
Таким образом, показано, что предоставленные композиции и способы достигают различных целей изобретения и хорошо адаптированы для соответствия условиям практического применения.
Поскольку возможны варианты осуществления описанного выше изобретения и поскольку в описанных выше вариантах осуществления могут быть сделаны различные изменения, должно быть очевидно, что все рассмотренные вопросы следует интерпретировать как иллюстративные, но не в смысле ограничения.
То новое, что заявлено и требует защиты патентной грамотой, описано в следующих пунктах формулы изобретения.
Фармацевтические композиции для перорального введения млекопитающим включают таксан или производное таксана (например, паклитаксел или доцетаксел) в качестве активного ингредиента и несущее средство, включающее по меньшей мере 30% по массе носителя для таксана, причем указанный носитель имеет значение HLB по меньшей мере приблизительно 10. Композиции также могут включать 0-70% снижающего вязкость сорастворителя. Композиции могут входить в состав обычных пероральных фармацевтических дозированных форм или существовать в форме двухкомпонентного лекарственного средства, где первая часть представляет собой таксан в солюбилизирующем несущем средстве, и вторая часть представляет собой носитель для таксана, для того чтобы способствовать пероральному всасыванию. Также описаны способы лечения поддающихся воздействию таксанами патологических состояний, задействующие новые композиции, где композиции могут вводиться сами по себе или в ассоциации с пероральным средством, повышающим биодоступность. Композиция обеспечивает повышенную биодоступность таксанов и терапевтический уровень содержания таксанов в крови. 5 н. и 110 з.п. ф-лы, 11 табл.
| WO 9853811 А1, 03.12.1998 | |||
| WO 9715269 А2, 01.05.1997 | |||
| УСТРОЙСТВО ПОДАВЛЕНИЯ НАВЕДЕННЫХ ЗВУКОВЫХ ПОМЕХ ДЛЯ ВИДЕОДИСПЛЕЙНОГО ПРИБОРА | 1996 |
|
RU2122770C1 |

