Изобретение относится к медицине и, в частности к созданию противовирусных препаратов на основе пептидов, гомологичных функциональным доменам вирусных белков.
Актуальность поиска средств для терапии гриппа (семейство Orthomyxoviridae, род Influenzavirus) диктуется развитием пандемической ситуации в мире и опасностью появления в человеческой популяции высокопатогенных штаммов вируса гриппа, в частности, подтипа H5N1. В настоящее время смертность от этой инфекции составляет порядка 70%, при этом заболевание, как и тяжелые формы гриппа другой этиологии, характеризуется острейшим началом и крайне тяжелым течением: высокой (более 40°С) температурой и длительной лихорадкой с резко выраженными симптомами интоксикации (сильной головной болью, ломотой, бессонницей, бредом, анорексией, тошнотой, рвотой, менингиальными симптомами, иногда энцефалитическим синдромом). Вариантом молниеносной формы может быть стремительное развитие геморрагического токсического отека легких и смертельный исход от дыхательной и сердечнососудистой недостаточности [Int. J. Tuberc. Lung Dis. 2007 11(7): 710-21].
Вирионы гриппа представляют собой сферические или нитевидные частицы диаметром 80-100 нм, покрытые липидной оболочкой с интегрированными молекулами поверхностных гликопротеидов (НА и NA) и ионного канала М2, ассоциированной со слоем мембранного белка M1. Сердцевина вирионов представлена 8 сегментами однонитевой РНК отрицательной полярности (вРНК), ассоциированной с тремя белками полимеразного комплекса (PA, PB1, РВ2) и молекулами нуклеопротеина (NP).
После проникновения в клетку и «раздевания» вириона сегменты генома попадают в цитоплазму, где вирусная полимераза осуществляет транскрипцию и репликацию вРНК. В результате образуются молекулы вирусспецифической матричной РНК (мРНК) и комплементарной РНК (кРНК) соответственно. На матрице кРНК впоследствии синтезируется цепь вРНК, входящая затем в состав вирионов потомства. мРНК транслируется при помощи клеточных механизмов с образованием структурных и неструктурных белков, первые из которых образуют вирионы потомства, а последние играют важные регуляторные функции в ходе жизненного цикла вируса.
Несмотря на наличие в жизненном цикле вируса множества компонентов, представляющих потенциальную мишень для специфической химиотерапии, на сегодняшний день лишь два из них используются в этом качестве.
Во-первых, это белок М2 вируса гриппа, играющий роль ионного канала в вирусной мембране, который блокируют препараты адамантанового ряда - ремантадин (а-Метил-1-адамантил-метиламина гидрохлорид) и амантадин (1-аминоадамантан) [Antiviral Res. 1998, 37, 83-95].
Другой мишенью для лекарственного вмешательства является вирусная нейраминидаза - фермент, необходимый для нормального почкования вирусных частиц и проявления инфекционных свойств вируса гриппа, против которой эффективны нейраминидазные ингибиторы занамивир (5-(ацетиламино)-4-[(аминоиминометил)-амино]-2,6-ангидро-3,4,5-тридезокси-D-глицеро-D-галактонон-2-еноновая кислота), озелтамивир (Тамифлю™) [J Antimicrob Chemother. 1999 Nov; 44 Suppl B: 17-22] и перамивир ((1S,2S,3S,4R)-3-[(1S)-1-ацетамидо-2-этил-бутил]-4-(диаминометилиденамино)-2-гидрокси-циклопентан-1-карбоновая кислота) [Ann Pharmacother. 2010 44(7-8): 1240-9].
Обе группы соединений имеют свои недостатки.
В отношении группы производных адамантана можно отметить сравнительно высокую токсичность, узкий спектр действия (препараты активны против гриппа А, но не против гриппа В) и быстрое формирование устойчивости вируса к препаратам.
Для ингибиторов нейраминидазы характерны формирование резистентности вирусов и высокая стоимость синтеза, что делает эти препараты менее доступными для широкого использования.
Ограниченность выбора противовирусных препаратов является причиной низкой эффективности лечения острых и хронических вирусных инфекций [Nature Rev. Microbiol. 2004, 2, 704-720; Nature Rev. Drug. Discov. 2007, Vol.6, p.1001-1018.].
Многочисленные патогенетические и симптоматические препараты, применяемые при лечении гриппа, направлены на корректировку только реактивных процессов в организме, и не устраняют причину заболевания.
Многочисленные химические соединения, не дошедшие до клинических испытаний, относятся к различным химическим классам, направлены на различные мишени в жизненном цикле вируса и демонстрируют различный уровень противогриппозной активности в клеточных культурах и в редких случаях лабораторных животных (Antiviral Research 88 (2010) 227-235; PNAS 2010 vol.107 3157-3162; Bioorganic & Medicinal Chemistry 18 (2010) 5379-5390; Antiviral Research 88 (2010) 10-18; Virology Journal 2009, 6:197; J. Med. Chem. 2009, 52, 7368-7371).
Известен препарат нуклеозин, имеющий высокую степень сродства к белку NP вируса гриппа и эффективно ингибирующий его репликацию. Однако все изоляты вируса, вызвавшего пандемию 2009 г., оказались устойчивы к этому препарату (Nature Biotechnology 28 6:600-607).
Известен препарат Рибавирин, представляющий собой нуклеозидный аналог, эффективный против 85% РНК-геномных вирусов млекопитающих. Применение его ограничено побочными последствиями, преимущественно анемией, проявляющимися вследствие его сходства с субстратами клеточных ферментов.
Близкими аналогами изобретения являются как рибавирин, так и разработанный японскими исследователями препарат Т-705 (фавипиравир), представляющий собой высокоэффективный противовирусный аналог нуклеозида, ингибирующий вирусные полимеразы (Antiviral Research 82 (2009) 95-102).
Несмотря на высокую эффективность, его химическая природа как нуклеотидного аналога предполагает, по аналогии с Рибавирином, побочные последствия при широком клиническом применении.
В последние годы в связи с разработкой фундаментальных подходов в области молекулярного дизайна появились принципиально новые возможности в конструировании лекарственных препаратов, максимально направленных на конкретную молекулярную мишень, характерную для вируса и отсутствующую в клетке хозяина [Nature Rev. Drug. Discov. 2007, Vol.6, p.1001-1018]. На основании анализа аминокислотных последовательностей функционально значимых вирусных белков разрабатываются их олигопептидные ингибиторы. Так, известны примеры разработки пептидов, угнетающих репродукцию а) вируса иммунодефицита человека [Antimicrob Agents Chemother. 2010, 54(3): 1343-6; Retrovirology 2008, 5:93], б) респираторно-синцитиального вируса [Antimicrob Agents Chemother. 2003, p.3470-3477], в) цитомегаловируса человека [J. Virol, 2003, p.8336-8344], г) вирусов лихорадки денге и Западного Нила [Virology Journal 2005, 2:49], д) вируса гриппа [J. Virol, 2007, р.7801-7804; J. Virol, 2006, p.11960-11967; Intl. J. Biol. Sci. 2009, 5(6): 543-548], и др.
Для профилактики и лечения различных заболеваний предпочтение отдается препаратам, обладающим возможно более широким спектром фармакологической активности. Большие возможности в разработке лекарственных препаратов связаны с конструированием пептидов, гомологичных функционально значимым доменам вирус - специфических белков [Protein Engineering, 2003, 16(4), pp.311-317; Virology Journal 2005, 2:49; USPTO Application #: 20100041604].
Для разработки лекарственных препаратов, направленных на конкретные молекулярные мишени, необходимо понимание структуры и функции отдельных доменов вирусных белков, играющих ключевую роль в жизненном цикле вируса. В связи с этим необходимо в каждом случае осуществлять выбор вирус-специфических белков - перспективных лекарственных мишеней. Применительно к вирусу гриппа среди таких мишеней особого внимания заслуживают каталитическая субъединица РНК - зависимой РНК - полимеразы как фермент, катализирующий синтез вирусной РНК, и полифункциональный неструктурный белок NS1, в значительной степени определяющий патогенность вирусов гриппа. Полимераза вируса гриппа, как и белок NS1 [J Virol. 2006; 80: 3957-65], являются, с одной стороны, консервативными белками среди всех типов и подтипов вируса, с другой - жизненно необходимыми компонентами в цикле вирусной репликации в клетке. Третичная структура каталитической субъединицы РНК - зависимой РНК - полимеразы вирусов гриппа типа А недавно была расшифрована (Nature 2008, 454: 1123-6; Nucl Acids Res., 2007, 1-10).
Структурная самосборка функционально активного полимеразного комплекса осуществляется путем взаимодействия межмолекулярных интерфейс - доменов - участков молекул, вступающих в контакт и обеспечивающих связь между субъединицами комплекса [Nucl. Acids Res. 2007, Vol.35, p.3774-3783]. Картирование функциональных доменов белков осуществляется с помощью гомологических полипептидов, структура которых соответствует предполагаемым функционально значимым доменам.
Первичная структура субъединицы РВ 1 представлена на Рис.1 «Первичная структура каталитической субъединицы РВ 1 РНК - зависимой РНК - полимеразы вирусов гриппа H5N1». Аминокислоты 531-540, входящие в состав активного противовирусного пептида, выделены рамкой.
Известны работы [J. Virol. 81: 7801-7804; PLoS ONE 4(10): e7517], где продемонстрирована высокая противовирусная активность олигопептидов, гомологичных функционально значимым последовательностям субъединицы РВ1, конкурирующих за сайты связывания ее с другими субъединицами полимеразного комплекса. Каждый раз создание биологически активных олигопептидов требует учета всех факторов, влияющих на заданную активность препарата.
При разработке пептидных противовирусных соединений существенное значение имеет проблема их проникновения через заряженную мембрану клетки.
Относительно низкомолекулярные соединения проникают в цитоплазму сравнительно легко в силу малых размеров молекулы, однако в силу тех же причин они не в состоянии обеспечить высокую избирательность в противовирусной активности. Олигопептиды размером 12-25 аминокислот, будучи высоко избирательны в отношении мишени, плохо проникают через мембрану клетки.
Для решения этой задачи применяются различные модификации собственно противовирусных пептидов. Так, вирусная полимераза эффективно ингибируется 25-аминокислотным пептидом MDVNPTLLFLKVPAQNAISTTFPYT (аминокислоты 1-25 субъединицы РВ1), однако для доставки его через клеточную мембрану приходится модифицировать его, добавляя мембранотропный остаток YGRKKRRQRRRPP белка Tat вируса иммунодефицита человека [J Virol 81: 7801-7804]. То же можно сказать о пептиде, имеющем активность против вирусов гриппа А и В [PLoS ONE 4(10): е7517]. Ингибирование полимеразной активности непосредственно гомологичными пептидами достигается при помощи специальной процедуры трансфекции клеток плазмидами, кодирующими эти пептиды [PLoS ONE 4(10): е7517].
Целью данного изобретения являлось создание синтетического пептидного препарата, обладающего двумя важными качествами: противовирусной активностью в отношении вируса гриппа и способностью проникать через клеточную мембрану без дополнительных модификаций. Поставленная задача решена путем синтеза низкомолекулярного аспарагин- богатого пептидного ингибитора вирусной репликации, соответствующего последовательности белка РВ1 (531-540), т.е. аминокислотного состава KNNMINNDLG, направленного на самосборку полимеразного комплекса вируса. Установлено, что при невысокой токсичности и относительно малой молекулярной массе этот полипептид обладает высокой селективностью в отношении вирусов гриппа разных подтипов - H1 и Н5. Благодаря специфическому аминокислотному составу этот пептид проявляет противовирусные свойства в отношении вируса гриппа без дополнительных химических модификаций или специальных приемов для облегчения его проникновения в клетку.
Использование такого пептида, богатого аспарагином, для ингибирования репликации вируса гриппа неизвестно. Его действие на вирус гриппа ранее никогда не проверяли.
Противовирусные свойства пептида РВ1 (531-540) в отношении вирусов гриппа изучались в культуре клеток MDCK на модели экспериментальной гриппозной инфекции, вызванной вирусами гриппа человека A(H1N1) и гриппа птиц A(H5N2).
Испытание РВ1 (531-540) в опытах по угнетению репликации вирусов гриппа выявило, что этот пептид эффективно снижает инфекционную активность вируса гриппа человека A(H1N1) (EC50 составляет 28,6 мкг/мл) и проявляет несколько меньшую активность в отношении вируса гриппа птиц A(H5N2) (EC50 составляет 35,2 мкг/мл). В качестве сравнения использованы последовательности РВ1 (6-25) и РВ1 (391-400), синтезированные в процессе поиска оптимального для лечения гриппа препарата.
Результаты испытаний представлены в таблице 1. Как видно из представленных данных, изученные пептиды проявляли гораздо меньший уровень токсичности по сравнению с существующим противовирусным препаратом ремантадином (в 8-20 раз), что выражалось в более высоких значениях 50% цитотоксических концентраций (CTD50). На основании полученных результатов были рассчитаны 50% эффективная концентрация (EC50) для каждого из препаратов, а также индекс селективности - отношение CTD50 к ЕС50. Противовирусная активность пептидов сравнения РВ1 (6-25) и РВ1 (391-400) была невысока, что выражалось в низких значениях индекса селективности (4, 7 и 2, 6, соответственно), и была ниже, чем у препарата сравнения - ремантадина, тогда как противовирусная активность пептида РВ 1(531-540) (индекс селективности 43,0) была выше, чем у всех трех препаратов сравнения в отношении ремантадин-устойчивого штамма H1N1. В отношении вируса гриппа птиц A(H5N2) пептид РВ1 (531-540) проявил меньшую активность, чем ремантадин (индексы селективности 34,8 и 150,0, соответственно), однако был существенно активнее, чем два другие пептида РВ1 (6-25) и РВ1 (391-400) (индекс селективности у обоих 3,9).
Представленные данные убедительно подтверждают вывод, что в опытах РВ1 (531-540) проявляет низкую токсичность и высокую противовирусную активность в отношении вирусов гриппа человека и птиц. Его способность ингибировать репликацию вируса H1N1 оказалась примерно в 4 раза выше, чем у препарата сравнения - ремантадина. Кроме того, этот пептид обладал более широким спектром противогриппозной активности по сравнению с ремантадином, который проявлял ингибирующие свойства против гриппа птиц A(H5N2), но не против вируса гриппа человека A(H1N1).
Преимущества синтезированного и заявляемого пептида пред ремантадином делают его пригодным для использования в качестве фармпрепарата для борьбы с гриппозной инфекцией.
Изобретение иллюстрируется следующими примерами.
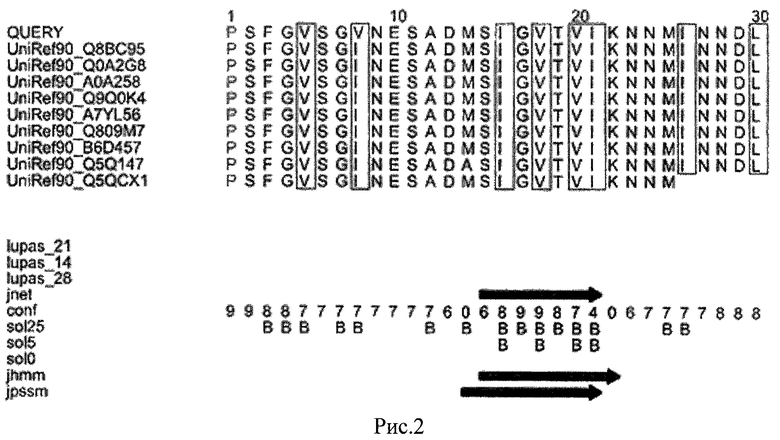
Пример 1. Для получения дополнительных данных о строении идентифицированного пептида - ингибитора репликации вирусов гриппа типа А исследовали свойства участка 510-539 из последовательности белка PB1 вируса А/Санкт-Петербург/04/2009 (H1N1)v. Данная последовательность гомологична структуре этого же домена для всех пандемических изолятов вирусов гриппа H1N1v-2009 и вирусов гриппа птиц H5N1. Анализ структуры проводили при помощи программы Jpred. Результаты анализа представлены на рис.2 («Анализ вторичной структуры домена 510-539 белка РВ1 вирусов гриппа типа A H1N1. Оценка конформации полипептида в отношении β-структуры (обозначена как В). Практически вся последовательность полипептида имеет структуру β-цепи, по центру она приближается к 100%, а далее носит такой же, но менее устойчивый характер»).
Полученные данные свидетельствуют, что домен белка РВ1 - 510-539 может быть отнесен к бета - структурным доменам, для которых характерны внутри - и межмолекулярные взаимодействия, описанные для доменов такого типа для других компонентов полимеразного комплекса вирусов гриппа типа A [PLoS ONE 4(10): е7517].
В связи с тем, что пептид РВ1 - 510-539 может быть сконструирован в виде более короткого β-структурного блокатора функционально значимого домена субъединицы вирусной полимеразы РВ1, он был выбран для практического применения, т.е. препарата, позволяющего получить технический эффект. Анализ аминокислотного состава такого пептида РВ1 (531-540) показал, что он обогащен аспарагином, имеет структуру в виде β-цепи или релаксированной спирали, и может представлять собой частный случай класса олигопептидов общей формулы XNNYYNNZY, где Х - положительно заряженная аминокислота, Y - гидрофобная аминокислота, Z - отрицательно заряженная аминокислота.
Пример 2. Синтез пептидов
Синтез пептидов проводили твердофазным методом на 2-Cl-тритилхлоридной смоле (емкостью 1,55 ммоль/г) по Fmoc-/t-Bu стратегии с использованием как стандартных методик последовательного наращивания цепи, так и конвергентного подхода.
В реакциях последовательного наращивания пептидной цепи в качестве конденсирующих агентов использовали N,N'-диизопропилкарбодиимид (DIC) и 1-гидрокси-бензотриазол (HOBt) (20% мольные избытки относительно активируемых аминокислот), реакции проводили в диметилформамиде (DMF). В реакциях использовали 2-3-кратные избытки защищенных аминокислот, продолжительность протекания реакций варьировалась от 1 до 48 часов. Для фрагментных конденсаций наряду с указанными реагентами использовались также 1-гидрокси-7-аза-бензотриазол (HOAt) и N-метилпирролидон (NMP).
Полноту протекания реакций оценивали с помощью теста Кайзера на свободные аминогруппы. В случае завершения реакции (отрицательный тест Кайзера) для последующих операций смолу промывали 6-8 раз DMF от компонентов реакционного раствора. В случае положительного теста реакцию повторяли, увеличивая избыток реагентов и время протекания реакций.
Также полноту протекания конденсации контролировали с помощью индикатора бромфенолового синего. В реакционную смесь добавляли 3 капли 1% раствора бромфенолового синего в диметилацетамиде (DMA). По мере протекания реакции окраска раствора изменялась с синей на желтую.
Для временной защиты α-аминогрупп использовалась 9-флуоренилметилоксикарбонильная (Fmoc) - группа, удаление которой проводили 2-3-кратной обработкой смолы 20% раствором диэтиламина в диметилформамиде в течение 5-20 минут. В некоторых случаях смолу обрабатывали смесью DMF: диэтиламин: DBU (1,8-диазобицикло [5.4.0] ундец-7-ен) в объемном соотношении 48:1:1 в течение 10 минут. Для удаления остаточных количеств диэтиламина или DBU смолу промывали 6-8 раз DMF.
В качестве постоянных защитных групп использовались: трет-бултилоксикарбонильная (ВОС) - для ε-аминогруппы лизина, 2,2,4,6,7-пентаметилдигидробензофуран-5-сульфонильная (Pbf) - для гуанидиновой группы аргинина, трифенилметильная (Trt) - для амидных групп глутамина и аспарагина, трет-бутильная (tBu) - для ОН-групп треонина, тирозина и серина, (O-tBu) для β- и γ - карбоксильных групп аспарагиновой и глутаминовой кислот.
Защищенные фрагменты для конвергентного синтеза получали следующим образом: для удаления DMF смолу дважды промывали дихлорметаном (DCM), 10-15 раз по 1,5-2 минуты обрабатывали 1% раствором трифторуксусной кислоты (TFA) в DCM; полученные растворы объединяли в колбе, содержащей 10% раствор пиридина в метаноле, смесь концентрировали на ротационном испарителе. К остатку добавляли воду, после чего защищенный пептидный фрагмент отфильтровывали. Чистота полученных в указанных условиях фрагментов по данным ВЭЖХ составила более 95%.
Для финального деблокирования целевого соединения, а также отщепления его от полимерного носителя, смолу в течение двух часов обрабатывали раствором TFA, содержащим этандитиол (EDT), воду и триизопропилсилан (TIS) в следующих объемных соотношениях 92,5:2,5:2,5:2,5. Полученный раствор концентрировали на ротационном испарителе. После добавления к остатку метилтретбутилового (МТВЕ) или диэтилового эфиров продукт отфильтровывали. Полноту деблокирования боковых функциональных групп контролировали с помощью аналитической обращеннофазной ВЭЖХ, при необходимости указанную процедуру повторяли.
В случае окисления метионин содержащие пептиды после финального деблокирования восстанавливали по методу, описанному в [Chan W.C., White P.D., Fmoc solid phase peptide synthesis, Oxford University press, 2004]: к раствору пептида в TFA добавляли EDT и триметилбромсилан (TMSBr) в количестве 15,7 и 13 мкл/мл соответственно, раствор выдерживали 15-30 минут при перемешивании, далее концентрировали и выделяли продукт с использованием МТВЕ, как описано выше.
Очистку полученных пептидов проводили методом препаративной обращеннофазной ВЭЖХ в градиентном режиме в системе ацетонитрил-вода-TFA. Фракции, содержащие целевой компонент, после объединения и концентрирования в вакууме подвергались лиофилизации. Чистота полученных соединений составила не менее 95%, их структура подтверждена масс-спектрометрически (MALDI-TOF).
Синтез фрагмента РВК391-400) - пептида сравнения
H-Lys-Ile-Arg-Pro-Leu-Leu-Val-Glu-Gly-Thr-OH
1. В твердофазном реакторе суспендировали 0.5 г (0.775 ммоль) 2-хлортритилхлоридной смолы в 10 мл DCM, выдерживали в течение 2 мин и отфильтровывали смолу.
2. К смоле добавляли раствор 140 мг (0.35 ммоль) С-концевой защищенной аминокислоты (FmocThr(t-Bu)OH) и 512 мкл (3.1 ммоль, 4 эквивалента по отношению к смоле) DIPEA в 3 мл DCM, перемешивали в течение двух часов при комнатной температуре. Смолу отфильтровывали и промывали 2×8 мл DCM в течение двух минут.
3. Далее в реактор добавляли 10 мл смеси ОСМ/метанол/DIPEA в соотношении 17:2:1, перемешивали в течение 10 минут и отфильтровывали, данную операцию проводили дважды, после чего смолу промывали DMF порциями по 10 мл (3×2 мин).
4. Деблокирование Fmoc-группы осуществляли описанным ранее способом.
5. К смоле добавляли охлажденный раствор 0.7 ммоль (2-кратный избыток) следующей защищенной аминокислоты, 129 мг (0.84 ммоль) HOBt и 131 мкл (0.84 ммоль) DIC в 3 мл DMF, перемешивали в течение двух часов при комнатной температуре, отфильтровывали смолу. Полноту протекания конденсации контролировали по изменению окраски индикатора бромфенолового синего. Промывали смолу порциями по 10 мл DMF (7×2 мин).
Операции 4, 5 повторяли до присоединения FmocLys(Boc)-1. Стадию финального деблокирования проводили по описанной ранее схеме. Получено 103 мг(26.1%).
Синтез фрагмента РВ1 (531-540)
H-Lys-Asn-Asn-Met-Ile-Asn-Asn-Asp-Leu-Gly-OH
1. В твердофазном реакторе суспендировали 1 г (1.55 ммоль) 2-хлортритилхлоридной смолы в 10 мл DCM, выдерживали в течение 2 мин и отфильтровывали смолу.
2. К смоле добавляли раствор 297 мг (1 ммоль) С-концевой защищенной аминокислоты (FmocGlyOH) и 1.02 мл (6.2 ммоль, 4 эквивалента по отношению к смоле) DIPEA в 5 мл DCM, перемешивали в течение двух часов при комнатной температуре. Смолу отфильтровывали и промывали 2×10 мл DCM в течение двух минут.
3. Далее в реактор добавляли 10 мл смеси DCM/метанол/DIPEA в соотношении 17:2:1, перемешивали в течение 10 минут и отфильтровывали, данную операцию проводили дважды, после чего смолу промывали DMF порциями по 10 мл (3×2 мин).
4. Деблокирование Fmoc-группы осуществляли описанным ранее способом.
5. К смоле добавляли охлажденный раствор 2 ммоль (2-кратный избыток) следующей защищенной аминокислоты, 337 мг (2.2 ммоль) HOBt и 343 мкл (2.2 ммоль) DIC в 5 мл DMF, перемешивали в течение двух часов при комнатной температуре, отфильтровывали смолу. Полноту протекания конденсации контролировали по изменению окраски индикатора бромфенолового синего. Промывали смолу порциями по 10 мл DMF (7×2 мин).
Операции 4, 5 повторяли до присоединения FmocLys(Boc)-1. Стадию финального деблокирования проводили по описанной ранее схеме. Получено 376 мг (33.2%).
Синтез фрагмента РВ1 (6-25) - пептида сравнения
H-Thr-Leu-Leu-Phe-Leu-Lys-Val-Pro-Ala-Gln-Asn-Ala-lle-Ser-Thr-Thr-Phe-Pro-Tyr-Thr-OH
Синтез данного пептида проводили конвергентно, последовательность была разделена на 2 фрагмента по пролину в положении 8.
В твердофазный реактор поместили 250 мг смолы, содержащей 0,25 ммоль фрагмента 9-20 (данный фрагмент был синтезирован по стандартной описанной выше схеме). К смоле добавили растворенные в 10 мл NMP 85 мг (0,625 ммоль) HOAt, 620 мг (0,495 ммоль) защищенного фрагмента 1-8 и 110 мкл (0,7 ммоль) DIC. Реакционную смесь перемешивали в течение 2 недель, контролируя полноту протекания реакции ранее описанными методами. Стадию финального деблокирования проводили по описанной выше схеме. Получено 154 мг (27,7%).
Пример 3. Определение биологической активности пептидов
Токсичность синтезированных пептидов. Навески пептидов (5 мг) растворяли в 2,5 мл среды для клеточных культур Игла MEM. Из полученных растворов готовили серии двукратных разведении препарата для определения противовирусной активности. Клетки MDCK инкубировали с пептидами в различных концентрациях в течение 48 часов при 37°С. По окончании инкубации количество выживших клеток определяли при помощи микротетразолиевого теста. На основании полученных данных вычисляли 50% цитотоксическую дозу каждого пептида. Для клеток MDCK она оказалась равной >500, 375 и >250 мкг/мл для пептидов РВ1 (531-540), РВ1 (391-400) и РВ1(6-25), соответственно, т.е. все пептиды проявляют низкую токсичность в опытах in vitro. В то же время 50% цитотоксическая доза ремантадина составила 60 мкг/мл, что говорит о большей его токсичности по сравнению с исследованными пептидами.
Противовирусная активность пептидов. Изучаемые препараты были тестированы в отношении вирусов гриппа человека A/Puerto Rico/8/34 (H1N1) и птиц A/Mallard/Pennsylvania/10218/84 (H5N2). Зараженные клетки культивировали в присутствии исследуемого пептида и препарата сравнения - ремантадина - при 37°С в атмосфере 5% СО2. Срок культивирования вируса составлял 24 часа. По окончании срока инкубации уровень репродукции вируса в лунках панели оценивали по реакции гемагглютинации (РГА) эритроцитов. За титр вируса принимали величину, противоположную десятичному логарифму наибольшего разведения вируса, способного вызвать положительную реакцию гемагглютинации и выражали в логарифмах 50% экспериментальной инфекционной дозы вируса (lg ЭИД50). На основании полученных данных рассчитывали 50% эффективную дозу препаратов (ED50) и индекс селективности (отношение CTD50/ED50). Противовирусная активность пептидов сравнения РВ1(6-25) и РВ1(391-400) была невысока, что выражалось в низких значениях индекса селективности (4, 7 и 2, 6, соответственно), и была ниже, чем у препарата сравнения - ремантадина, тогда как противовирусная активность пептида РВ 1(531-540) (индекс селективности 43,0) была выше, чем у всех трех препаратов сравнения в отношении ремантадин-устойчивого штамма H1N1. В отношении вируса гриппа птиц A(H5N2) пептид РВ 1(531-540) проявил меньшую активность, чем ремантадин (индексы селективности 34,8 и 150,0, соответственно), однако был существенно активнее, чем два другие пептида РВ 1(6-25) и РВ 1(391-400) (индекс селективности у обоих 3, 9).
Полученные данные свидетельствуют, что из трех изученных пептидов, пептид РВ1 (531-540) обладает наиболее выраженной селективностью в отношении вирусов гриппа человека и птиц, что делает этот пептид пригодным для лечения вирусной гриппозной инфекции, т.е. заявляемый пептид обладает техническим эффектом.
| название | год | авторы | номер документа |
|---|---|---|---|
| Композиция на основе пептида, подавляющего репликацию вируса гриппа А | 2018 |
|
RU2695336C1 |
| 1-[ω-АРИЛОКСИАЛКИЛ(БЕНЗИЛ)]ЗАМЕЩЕННЫЕ 2-АМИНОБЕНЗИМИДАЗОЛЫ, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ В ОТНОШЕНИИ ВИРУСА ГРИППА | 2012 |
|
RU2570906C2 |
| УНИВЕРСАЛЬНАЯ ВАКЦИНА ПРОТИВ ВИРУСА ГРИППА ПТИЦ | 2007 |
|
RU2358981C2 |
| Штамм A/goose/Kalmykia/813/16 H5N8 вируса гриппа птиц Influenza virus avicum типа А подтипа H5 для контроля антигенной и иммуногенной активности вакцин против гриппа птиц и для изготовления биопрепаратов для диагностики и специфической профилактики гриппа птиц типа А подтипа Н5 | 2017 |
|
RU2647566C1 |
| ПРИМЕНЕНИЕ 1,7,7-ТРИМЕТИЛБИЦИКЛО[2.2.1]ГЕПТАН-2-ИЛИДЕН-АМИНОЭТАНОЛА В КАЧЕСТВЕ ИНГИБИТОРА РЕПРОДУКЦИИ ВИРУСА ГРИППА | 2013 |
|
RU2530554C1 |
| ИМИНОПРОИЗВОДНЫЕ КАМФОРЫ - ЭФФЕКТИВНЫЕ ИНГИБИТОРЫ РЕПРОДУКЦИИ ВИРУСА ГРИППА (штамм A/California/07/09 (H1N1)pdm09) | 2014 |
|
RU2554934C1 |
| ФАРМАЦЕВТИЧЕСКИЕ СОЛИ АМИНОБИЦИКЛО[2.2.1]ГЕПТАНОВ КАК ИНГИБИТОРЫ ТРАНСКРИПЦИОННОГО ФАКТОРА NF-KB С ПРОТИВОВИРУСНОЙ АКТИВНОСТЬЮ (ВАРИАНТЫ) И ИХ ПРИМЕНЕНИЕ | 2009 |
|
RU2448692C2 |
| Рекомбинантный штамм вируса гриппа A/PR8-NS124-Luc и способ оценки поствакцинальных нейтрализующих антител с использованием биолюминесцентной детекции | 2019 |
|
RU2759054C2 |
| 6,8-Дифтор-2-(4-(трифторметил)фенил)хроман-4-он в качестве ингибитора репродукции вирусов гриппа А и В и способ его получения | 2023 |
|
RU2826250C1 |
| ПРИМЕНЕНИЕ ФУЛЛЕРЕНОЛОВ С60(OH)18-24 И С60(OH)30-38 В КАЧЕСТВЕ ПРОТИВОВИРУСНЫХ ПРЕПАРАТОВ | 2011 |
|
RU2472496C2 |
Изобретение относится к области молекулярной биологии, фармакологии и медицины. Предложен выделенный из олигопептида РВ1 (531-540) олигопептид KNNMINNDLG, активный как против гриппа человека H1N1 (EC50=28,6 мкг/мл), так и против гриппа птиц H5N2 (EC50=35,3 мкг/мл). Изобретение может быть использовано для вакцинации против вируса гриппа подтипов H1 и Н5. 1 з.п. ф-лы, 1 табл, 2 ил, 3 пр.
1. Олигопептид последовательности 531-540 белка полимеразного комплекса РВ1 вируса гриппа, имеющий аминокислотный состав KNNMINNDLG, характеризующийся тем, что обладает высокой противовирусной активностью ЕС50 в отношении вирусов гриппа человека A(H1N1), составляющей 28,6 мкг/мл, и птиц A(H5N2), составляющей 35,3 мкг/мл при низкой токсичности пептида CTD50, равной 1229 мкг/мл.
2. Олигопептид по п.1, отличающийся тем, что обладает противовирусной активностью в отношении ремантадин - устойчивого вируса гриппа человека A(H1N1), превосходящей референс - препарат ремантадин в 4 раза.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Прибор для получения стереоскопических впечатлений от двух изображений различного масштаба | 1917 |
|
SU26A1 |
| КРЕМ ПРОТИВОВОСПАЛИТЕЛЬНОГО И ПРОТИВОЗУДНОГО ДЕЙСТВИЯ ДЛЯ ЛЕЧЕНИЯ ДЕРМАТОЛОГИЧЕСКИХ ЗАБОЛЕВАНИЙ | 2002 |
|
RU2233152C1 |

