Настоящее изобретение относится к способу модулирования c-Jun N-терминальных киназ (JNK) и способу лечения субъекта, страдающего заболеванием или состоянием, которое может быть облегчено посредством модуляции JNK гетероциклическими соединениями. Настоящее изобретение также относится к новым гетероциклическим соединениям и фармацевтическим композициям, содержащим таковые соединения.
N-терминальные киназы c-Jun (JNK) являются членами семейства митоген-активируемых протеинкиназ совместно с p38 и внеклеточными сигнал-регулируемыми киназами (ERK). Были идентифицированы три различных гена (jnk1, jnk2 и jnk3), кодирующие 10 сплайс-вариантов (Y.T. Ip и R.J. Davis, Curr. Opin. Cell Biol. (1998) 10:205-19). JNK1 и JNK2 экспрессируются в широком наборе тканей, тогда как JNK3 экспрессируется преимущественно в нейронах и, в меньшей степени, в сердце и яичках (D.D. Yang и др., Nature (1997) 389:865-70). Члены семейства JNK активируются провоспалительными цитокинами, такими как фактор некроза опухолей α (TNF-α) и интерлейкин-1β (ИЛ-1β), равно как и связанными с окружающей средой нагрузками. Активация JNK опосредована их киназами вверх по пути экспрессии, МКК4 и МКК7 (киназы митоген-активируемых протеинкиназ) посредством двойного фосфорилирования Thr-183 и Tyr-185 (В. Derijard и др., Cell (1994) 76:1025-37). Было показано, что MKK4 и MMK7 могут быть активированы различными киназами вверх по пути экспрессии, включая MEKK1 и MEKK4 (киназы митоген-активируемых протеинкиназ), в зависимости от внешних стимулов и клеточного контекста (D.Boyle и др., Arthritis Rheum (2003) 48:2450-24). Специфичность сигналов JNK достигается в результате образования JNK-специфичного сигнального комплекса, содержащего несколько компонентов киназного каскада с помощью вспомогательных матричных белков, называемых JNK-взаимодействующими белками (J. Yasuda и др., Mol. Cell. Biol. (1999) 19:7245-54). Было показано, что JNK играют важную роль в воспалениях, функциях Т-клеток, апоптозе и выживании клеток посредством фосфорилирования специфичных субстратов, включая транскрипционные факторы, такие как c-Jun, компонент семейства активаторного белка-1 (AP1), и ATF2 (активирующий транскрипционный фактор 2), равно как и не транскрипционные факторы IRS-1 (субстрат инсулинового рецептора 1) и Bcl-2 (ген B-клеточной лимфомы 2) (A.M. Manning и R.J. Davis, Nat. Rev. Drug Discov. (2003) 2:554-65). Как полагают, сверхактивация JNK является важным механизмом при аутоиммунных, воспалительных, метаболических и неврологических заболеваниях, равно как и при раке.
Ревматоидный артрит (PA) является системным аутоиммунным заболеванием, характеризуемым хроническим воспалением суставов. Помимо опухания суставов и вызываемой воспалительным процессом боли, у большинства пациентов с PA в конце концов развиваются дегенеративные повреждения и деформации суставов. Ряд линий убедительных фармакологических и генетических данных в клеточных моделях и моделях на животных решительно свидетельствуют в пользу участия и важности активированных JNK в патогенезе РА. Прежде всего, аномальная активация JNK была обнаружена в пораженных артритом суставах у страдающих PA пациентов (G. Schett и др., Arthritis Rheum (2000) 43:2501-12) и пораженных артритом суставах грызунов в моделях артрита на животных (Z. Han и др., J. Clin. Invest. (2001) 108:73-81). Кроме того, ингибирование активации JNK с помощью селективных ингибиторов JNK блокирует выработку провоспалительных цитокинов и матричных металлопротеиназ (MMP) в синовиоцитах, макрофагах и лимфоцитах человека (Z. Han и др., (2001) см. выше). Стоит заметить, что введение селективных ингибиторов JNK крысам, страдающим адъювантным артритом (Z.Han и др., (2001) см. выше), или мышам, страдающим коллагениндуцированным артритом (Р. Gaillard и др., J Med Chem. (2005) 14:4596-607), эффективно защищает суставы от разрушение и существенно снижает опухание лапок посредством ингибирования экспрессии цитокина и коллагеназы. Более того, мыши с недостатком JNK2 были частично защищено от разрушения суставов, но не выказали заметного эффекта в отношении опухания лапок и воспаления в модели с пассивным коллагениндуцированным артритом. Эти исследования показывают, что JNK2 функционально дублирует JNK1 в отношении их роли в матричной деградации, воспалении и опухании лапок. Таким образом, комбинированное ингибирование активности как JNK1, так и JNK2 требуется для эффективной терапии PA (Z. Han и др., Arthritis Rheum. (2002) 46:818-23).
Астма является хроническим воспалительным заболеванием дыхательных путей, характеризуемым присутствием клеточного воспалительного процесса и бронхиальной гиперчувствительностью, связанной со структурными изменениями дыхательных путей (В.Bradley и др., J. Allergy Clin. Immunol. (1991) 88:661-74). Было показано, что это расстройство опосредуется различными типами клеток дыхательных путей, включая Т-лимфоциты, эозинофилы, мастоциты, нейтрофилы и клетки эпителия (J.Bousquet и др., Am. J. Respir. Crit. Care Med. (2000) 161:1720-45). JNK стали рассматриваться как перспективные профилактические средства против астмы на основании недавних доказывающих эту концепцию исследований в клеточных моделях астмы и моделях астмы на животных, использующих селективные ингибиторы JNK (K. Blease и др., Expert Opin. Emerg. Drugs (2003) 8:71-81). Было показано, что ингибиторы JNK в существенной степени блокируют выработку RANTES (цитокин, регулируемый при активации, экспрессируемый и секрцируемый нормальными Т-клетками) в активированных гладких клетках дыхательных путей (K. Kujime и др., J. Immunol. (2000) 164:3222-28). Еще важнее то, что ингибиторы JNK демонстрировали хорошую эффективность в моделях на мышах и крысах с хроническими заболеваниями с точки зрения их способность к понижению клеточной инфильтрации, воспаления, гиперчувствительности, пролиферации гладких мышц, и выработки иммуноглобулина IgE (P. Nath и др., Eur. J. Pharmacol. (2005) 506:273-83, Р. Eynott и др., Br. J. Pharmacol. (2003) 140:1373-80). Эти наблюдения свидетельствуют о важной роли JNK при аллергических воспалениях, процессах ремоделирования дыхательных путей, связанных с гиперчувствительностью. Таким образом, ожидается, что блокада активности JNK будут полезны для лечения астмы.
Диабет типа 2 является наиболее серьезным и распространенным метаболическим заболеванием, характеризуемым резистентностью к инсулину и нарушенной секрецией инсулина, которые являются результатом хронического низкоуровневого воспаления и аномального липидного метаболизма, связанного с окислительной нагрузкой. Было показано, что активность JNK аномально повышена в различных тканях-мишенях диабета в условиях ожирения и диабета (J. Hirosumi и др., Nature (2002) 420:333-36, Н. Kaneto, Expert. Opin. Ther. Targets (2005) 9:581-92). Активация JNK-механизма провоспалительными цитокинами и окислительными нагрузками приводит к отрицательной регуляции инсулиновых сигналов посредством фосфорилирования субстрата инсулинового рецептора 1 (IRS-1) по Ser307 и вносит, таким образом, вклад в резистентность к инсулину и переносимость глюкозы (J. Hirosumi и др., Nature (2002) см. выше, Y. Lee и др., J. Biol. Chem. (2003) 278:2896-902, Y. Nakatani и др., J. Biol. Chem. (2004) 279:45803-09). Убедительные генетические доказательства были получены в изящных исследованиях в модели на животных с использованием мышей линии jnk-/-, скрещенных с мышами или с генетически обусловленным ожирением (ob/ob), или с ожирением, обусловленным питанием. Потеря функций JNK1(JNK1-/-), но не JNK2 (jnk2-/-) защищала страдающих ожирением мышей от увеличения массы тела, повышенных стационарных уровнях глюкозы в крови и пониженных уровнях инсулина в плазме (J.Hirosumi и др., Nature (2002) см. выше). Более того, полезнее эффекты наблюдались в генетической диабетической модели (мыши db/db) в результате введения или низкомолекулярного ингибитора JNK, CC105 (В. Bennett и др., Curr. Opin. Pharmacol. (2003) 3:420-25) или JNK-ингибирующего пептида I (JIP), производного от домена связывания JNK JNK-взаимодействующего белка-1 (ЛР-1) (Н. Kaneto и др., Nat. Med. (2004) 10:1128-32), включая существенно пониженный уровень глюкозы в крови и повышенные уровни инсулина в плазме. Что еще более интересно, другое недавнее исследование (A.Jaeschke и др., Proc. Natl. Acad. Sci. USA. (2005) 102:6931-35) продемонстрировало, что JNK2 играет важную роль при диабете типа 1, вызванном аутоиммунным разрушением вырабатывающих инсулин β-клеток. Страдающие диабетом, но не ожирением мыши с недостатком экспрессии JNK2 демонстрировали снижение деструктивного инсулита и замедленное развитие заболевания в сторону диабета, что, возможно, явилось следствием смещенной поляризации в направлении фенотипа Th2. Рассматриваемые совместно, эти исследования демонстрируют полезность ингибиторов JNK в лечении ожирения/диабета типа 2.
Нейродегенеративные заболевания, такие как болезнь Альцгеймера (БА), болезнь Паркинсона (БП) и инсульт характеризуются потерей синаптических связей, нейрональной атрофией и смертью. Было показано, что механизм JNK, ведущий к активации c-Jun играет причинную роль в апоптозе изолированных первичных эмбриональных нейронов и многих нейронных клеточных линий вследствие индукции разнообразными стимулирующими факторами (D. Bozyczko-Coyne и др., Curr. Drug Targets CNS Neurol. Disord. (2002) 1:31-49). Сверхактивация JNK наблюдалась в человеческом мозге у пациентов с БА (J. Pei и др., J. Alzheimers Dis. (2001) 3:41-48) или участках мозга грызунов, производных от моделей нейродегенеративных заболеваний на животных (М.Saporito и др., J. Neurochem. (2000) 75:1200-08). Например, повышенные уровни фосфо-JNK были обнаружены во взятом после смерти мозге пациентов, страдающих БА. Введение JNK-ингибирующего пептида (пептид JIP-1) в модели БА, вызванной введением β-амилоидного пептида, на грызунах предотвратило ухудшение синаптической пластичности. В модели БП на животных (модель МРТР (1-метил-4-фенил-1,2,3,6-тетрагидропиридина)) повышенные уровни фосфо-МКК4 и фосфо-JNK сопутствовали, как наблюдалось, гибели нейронных клеток. Аденовирусный перенос гена JNK-ингибирующего пептида (пептид JIP-1) в стриатум мышей ослабил поведенческие нарушения посредством ингибирования опосредованной МРТР активации JNK, c-Jun и каспазы, блокируй таким образом гибель нейронных клеток в черной субстанции (X. Xia и др., Proc. Natl. Acad. Sci. USA. (2001) 98:10433-38). Кроме того, в модели на животных ишемического инсульта, вызванного глутаматной эндотоксичностью, мыши с недостатком JNK3, но не JNK1 или JNK2, оказались резистентны к опосредованным каиновой кислотой (агонистом глутаматного рецептора) приступам или нейронной гибели (D.D. Yang и др., Nature (1997) 389:865-70). Эти данные свидетельствуют о том, что JNK3 в существенной степени ответственна за глутаматную эндотоксичность, важный компонент ишемических состояний. Рассматриваемые вместе, эти данные свидетельствуют о том, что JNK являются привлекательной мишенью для многих заболеваний центральной нервной системы (ЦНС), связанных с нейрональной клеточной гибелью.
Неконтролируемый клеточный рост, пролиферация и миграция совместно с дерегулированным ангиогенезом приводят к образованию злокачественных опухолей. Хотя связанный с JNK механизм передачи сигнала и не играет, может быть, исключительную роль в апоптозе, недавно было показано, что постоянная активация JNK, приводящая к активации AP1, вносит вклад в выживание клеток некоторых типов рака, таких как глиальные опухоли и BCL-ABL-трансформированные B-лимфобласты (М. Antonyak и др., Oncogene (2002) 21:5038-46, P. Hess и др., Nat. Genet. (2002) 32:201-05). В случае глиальных опухолей повышенная активность JNK/AP1 наблюдалась в большинстве образцов первичной опухоли мозга. Для трансформированных B-лимфобластов было показано, что BCL-ABL активирует JNK-механизм, который, в свою очередь, обеспечивает повышенную регуляцию экспрессии антиапоптотического гена bcl-2. Стоит заметить, что многолекарственная резистентность и гиперпролиферация, наблюдавшиеся у резистентным к лечению пациентов с острым миелогенным лейкозом (ОМЛ), была причинно связана с постоянной активностью JNK, присутствовавшей в соответствующих образцах ОМЛ (L. Cripe и др., Leukemia (2002) 16:799-812). Активация JNK в лейкозных клетках приводит к индуцированной экспрессии эффлюксных насосов, таких как mdr1 и MRP1 (белки многолекарственной резистентности), ответственных за многолекарственную резистентность. Кроме того, гены с улучшенной выживаемостью в условиях окислительной нагрузки, включающие глутатион-S-трансферазу и π и γ-глутамилцистеинсинтазу, также подвергаются повышенной регуляции активированным JNK-механизмом.
В соответствии с вышеизложенным, модуляторы JNK полезны в лечении разнообразных заболеваний и/или состояний.
В одном из вариантов осуществления настоящего изобретения его объектом является соединение формулы I:
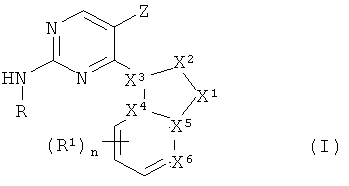
в котором
X1 является азотом, N-R3, CHR3, C-R3 или кислородом,
X2 является азотом, NH, N-CH3, CH, CH2, CHCH3 или C-CH3,
Х3 является азотом, углеродом или CH, причем Х1, Х2 и Х3 не являются все одновременно азотом,
каждый из Х4, X5 независимо является углеродом, CH или азотом,
X6 является азотом или C-R1,
причем не менее двух и не более четырех из X1, X2, X3, X4, X5 и X6 являются азотом, а связи между X1 и X2, X2 и X3, X3 и X4, X4 и X5, X5 и X1, а также X5 и X6 могут независимо являться одинарными или двойными, или же образовывать ароматический цикл, при том условии, что в результате получается химически устойчивая структура,
R является -(циклогексил)-R2 или -(фенил)-R8, где циклогексил и фенил необязательно замещены метилом, фтором, хлором или гидроксигруппой,
R1 является галоидом, нитрогруппой, -CN, -CH2CN, -OH, -NH2, -COOH или -Y1R4,
Y1 является -O-, -NH-, -S-, -SO-, -SO2-, -NHSO2-, -NHC(O)-, -C(O)NH-, -C(O)O- или связью,
R4 является (C1-C6)алкилом, ацилом, фенилом или бензилом, каждый из которых замещен 0-3 гидроксигруппами или галоидами,
n принимает значения 0, 1, или 2,
R3 является Н или (C1-C6)алкилом,
R2 является водородом, -OH, -SO2NH2, =O, -CN или -Y2-Y3-Y4-R5, где
Y2 является -C(O)-, -C(O)NRa-, -SO2-, -O-, -NH- или связью,
Y3 является (C1-C6)алкиленом или связью,
Y4 является -O-, -NRa-, -S-, -SO2-, -C(O)-, -C(O)NRa-, -NRaC(O)-, -NRaC(O)O-, -NRaC(O)NRa-, -NHSO2-, -SO2NH- или связью,
R5 является (C1-C6)алкилом, (C3-C10)циклоалкилом, гетероциклилом, причем R5 необязательно замещен -OH или -NHRa, где каждый из Ra независимо является водородом или (C1-C6)алкилом,
R8 является водородом, (C1-C6)алкилом, -OR3, -SO2NH2, -NHSO2R3, -COOR3, -SO2R3, -NH2, -CONRaR3, -NHC(O)R3, -CF3, -NO2, галоидом или -CN,
Z является водородом, галоидом, (C1-C12)алкилом или NH2,
или его фармацевтически приемлемая соль.
Предпочтительно,
X1 является азотом, N-R3, C-R3 или кислородом,
X2 является азотом, NH, N-CH3, CH или C-CH3,
Х3 является азотом или углеродом, причем Х1, Х2 и Х3 не являются азотом одновременно, а
каждый из X4, X5 независимо является углеродом или азотом.
Объектом настоящего изобретения также являются фармацевтические композиции, способы применения и способы получения вышеупомянутых соединений.
Соединения и композиции по настоящему изобретению применимы в лечении и/или профилактике опосредованного c-Jun N-терминальной киназой расстройства, такого как аутоиммунные расстройства, воспалительные расстройства, метаболические расстройства, неврологические расстройства и рак. В некоторых вариантах осуществления настоящего изобретения соединения и композиции по настоящему изобретению применимы в лечении и/или профилактике ревматоидного артрита, астмы, диабета типа II, болезни Альцгеймера, болезни Паркинсона и/или инсульта.
Определения
Если иное не оговорено специально, нижеследующие термины, употребляемые в настоящей заявке, включая описание изобретения и формулу изобретения, отвечают нижеприведенным определениям. Следует понимать, что формы единственного числа, как они употребляются в описании и формуле изобретения, включают и ссылку на множественное число, если иное не следует однозначно из контекста.
Термин «алкил» означает одновалентный линейный или разветвленный насыщенный углеводородный остаток, состоящий исключительно из атомов углерода и водорода и содержащий от одного до двенадцати атомов углерода. Термин «низший алкил» относится к алкильной группе, содержащей от одного до шести атомов углерода, т.е. к (C1-C6)алкилу. Примеры алкильных групп включают, не ограничиваясь перечисленным, метил, этил, пропил, изопропил, изобутил, втор-бутил, трет-бутил, пентил, н-гексил, октил, додецил. Термин «разветвленный алкил» относится к алкильному остатку, содержащему хотя бы одно разветвление, например, к изопропилу, изобутилу, трет-бутилу. Аналогично, термин «низшая алкоксигруппа» относится к остатку вида -OR, a термин «ацил» относится к остатку вида -C(O)R, где R является низшим алкилом.
Термин «алкилен» означает линейный насыщенный двухвалентный углеводородный остаток, содержащий от одного до шести атомов углерода или разветвленный насыщенный двухвалентный углеводородный радикал, содержащий от трех до шести атомов углерода, например, метилен, этилен, 2,2-диметилэтилен, пропилен, 2-метилпропилен, бутилен, пентилен.
Термин «алкилендиоксигруппа» означает двухвалентный остаток формулы -O-R-O-, где R является алкиленом, как он определен в контексте.
Термин «арил» означает одновалентный циклический ароматический углеводородный остаток, состоящий из моно-, би- или трициклической ароматической системы. Арильная группа может быть необязательно замещена, как это определено в контексте. Примеры арильных остатков включают, не ограничиваясь перечисленным, необязательно замещенные фенил, нафтил, фенантрил, флуоренил, инденил, пенталенил, азуленил, оксидифенил, бифенил, метилендифенил, аминодифенил, дифенилсульфидил, дифенилсульфонил, дифенилизопропилиденил, бензодиоксанил, бензофуранил, бензодиоксилил, бензопиранил, бензоксазинил, бензоксазинолил, бензопиперадинил, пинзопиперазинил, бензопирролидинил, бензоморфолинил, метилендиоксифенил, этилендиоксиденил, включая их частично гидрированные производные.
Термин «циклоалкил» означает одновалентный насыщенный карбоциклический остаток, представляющий собой моно- или бициклическую систему, содержащую от трех до десяти атомов углерода. Предпочтительно, циклоалкил является моноциклическим (C3-C7)циклоалкилом. Циклоалкил может быть необязательно замещен одним или более заместителями, где каждый из заместителей независимо является гидроксигруппой, алкилом, алкоксигруппой, галоидом, галоидалкилом, аминогруппой, моноалкиламиногруппой или диалкиламиногруппой, если иное не оговорено специально. Примеры циклоалкильных остатков включают, не ограничиваясь перечисленным, циклопропил, циклобутил, циклопентил, циклогексил и циклогептил, включая их частично ненасыщенные производные.
Термин «циклоалкилалкил» означает остаток формулы -Ra-Rb, где Ra является алкиленом, a Rb является циклоалкилом, как они определены в контексте.
Термин «гетероалкил» означает алкильный остаток, как он определен в контексте, включающий разветвленный (C4-C7)алкил, в котором один два или три атома водорода заменены заместителем, независимо выбранным из группы, включающей -ORa, -NRbRc, и -S(O)nRd (где n является целым числом от 0 до 2), при том условии, что гетероалкильный радикал присоединен через атом углерода, где Ra является водородом, ацилом, алкилом, циклоалкилом или циклоалкилалкилом, Rb и Rc независимо друг от друга являются водородом, ацилом, алкилом, циклоалкилом или циклоалкилалкилом, в том случае, если n равно 0, Rd является водородом, алкилом, циклоалкилом или циклоалкилалкилом, в том случае n равно 1, Rd является алкилом, циклоалкилом или циклоалкилалкилом, а в том случае, если n равно 2, Rd является алкилом, циклоалкилом, циклоалкилалкилом, аминогруппой, ациламиногруппой, моноалкиламиногруппой или диалкиламиногруппой. Характерные примеры включают, не ограничиваясь перечисленным, 2-гидроксиэтил, 3-гидроксипропил, 2-гидрокси-1-гидроксиметилэтил, 2,3-дигидроксипропил, 1-гидроксиметилэтил, 3-гидроксибутил, 2,3-дигидроксибутил, 2-гидрокси-1-метилпропил, 2-аминоэтил, 3-аминопропил, 2-метилсульфонилэтил, аминосульфонилметил, аминосульфонилэтил, аминосульфонилпропил, метиламиносульфонилметил, метиламиносульфонилэтил, метиламиносульфонилпропил.
Термин «гетероарил» означает моноциклический или бициклический остаток, содержащий от 5 до 12 кольцевых атомов и хотя бы один ароматический цикл, включающий один, два или три кольцевых гетероатома, выбранных из азота, кислорода или серы, тогда как остальные кольцевые атомы являются атомами углерода, при том условии, что точка присоединения гетероарильного радикала находится в ароматическом цикле. Гетероарильный цикл может быть необязательно замещен, как это определено в контексте. Примеры гетероарильных остатков включают, не ограничиваясь перечисленным, необязательно замещенные имидазолил, оксазолил, изоксазолил, тиазолил, изотиазолил, оксадиазолил, тиадиазолил, пиразинил, тиенил, тиофенил, фуранил, пиранил, пиридинил, пирролил, пиразолил, пиримидил, пиридазинил, хинолинил, изохинолинил, бензофурил, бензофуранил, бензотиофенил, бензотиопиранил, бензимидазолил, бензоксазолил, бензооксадиазолил, бензотиазолил, бензотиадиазолил, бензопиранил, индолил, изоиндолил, индазолил, триазолил, триазинил, хиноксалинил, пуринил, хиназолинил, хинолизинил, нафтиридинил, птеридинил, карбазолил, азепинил, диазепинил и акридинил, включая их частично гидрированные производные.
Термины «галоид», «галоген» и «галогенид» употребляются в контексте взаимозаменяемо и относятся к заместителям фтору, хлору, брому или иоду.
Термин «галоидалкил», означает алкил, как он определен в контексте, в котором один или более атом водорода заменены одинаковыми или разными галоидами. Примеры галоидалкилов включают -CH2Cl, -CH2CF3, -CH2CCl3, перфторалкил (например, -CF3).
Термин «гетероциклил» означает одновалентный насыщенный остаток, состоящий из от одного до трех циклов, включающих один, два, три или четыре гетероатома (выбранных из азота, кислорода или серы). Предпочтительным является моноциклический гетероциклил, содержащий от 3 до 8 кольцевых атомов. Гетероциклильный цикл может быть необязательно замещен, как это определено в контексте. Примеры гетероциклильных остатков включают, не ограничиваясь перечисленным, необязательно замещенные пиперидинил, пиперазинил, гомопиперазинил, азепинил, пирролидинил, пиразолидинил, имидазолинил, имидазолидинил, пиридинил, пиридазинил, пиримидинил, оксазолидинил, изоксазолидинил, морфолинил, тиазолидинил, изотиазолидинил, хинуклидинил, хинолинил, изохинолинил, бензимидазолил, тиадиазолидинил, бензотиазолидинил, бензоазолидинил, дигидрофурил, тетрагидрофурил, дигидропиранил, тетрагидропиранил, тиаморфолинил, тиаморфолинилсульфоксид, тиаморфолинилсульфон, дигидрохинолинил, дигидроизохинолинил, тетрагидрохинолинил и тетрагидроизохинолинил.
Термин «необязательно замещенный», употребляемый в сочетании с терминами «арил», «фенил», «гетероарил» или «гетероциклил», означает арил, фенил, гетероарил или гетероциклил, необязательно независимо замещенный одним или более заместителями, предпочтительно, одним - четырьмя, наиболее предпочтительно, одним - тремя заместителями, выбранными из (C1-C6)алкила, (C1-C6)гетероалкила, оксогруппы (т.е. =O), галоидалкила, -(CH2)mCOX1, -(CH2)mSO2X2, (C1-C6)алкоксигруппы, галоида, (C1-C6)алкилтиогруппы, (C1-C6)алкилсульфонильной группы, -SO2NR4R5, цианогруппы, нитрогруппы и -NR6R7, где m, X1, X2, R4 и R5 отвечают приведенным в контексте определениям.
Термин «уходящая группа» означает такую группу, с которой это понятие общепринято ассоциируют в синтетической органической химии, т.е. атом или группу, замещаемые в условиях реакции замещения. Примеры уходящих групп включают, не ограничиваясь перечисленным, галоид, алкан- или ариленсульфонилоксигруппу, такую как метансульфонилоксигруппа, этансульфонилоксигруппа, тиометил, бензолсульфонилоксигруппа, тозилоксигруппа и тиенилоксигруппа, дигалофосфиоилоксигруппу, необязательно замещенную бензилоксигруппу, изопропилоксигрупу и ацилоксигруппу.
Термин «необязательный» или «необязательно» означает, что описанное вслед за ним событие или обстоятельство может, но не должно непременно, иметь место и что описание включает как случаи, когда событие или обстоятельство имеет место, так и случаи, когда она места не имеет.
Термин «заболевание» или «болезненное состояние» означает любое заболевание, состояние, симптом, расстройство или показание.
Термин «инертный органический растворитель» или «инертный растворитель» означает, что растворитель инертен в описываемых в связи с ним условиях, и включает, например, бензол, толуол, ацетонитрил, тетрагидрофуран, N,N-диметилформамид, хлороформ, хлористый метилен или дихлорметан, дихлорэтан, диэтиловый эфир, этилацетат, ацетон, метилэтилкетон, метанол, этанол, пропанол, изопропанол, трет-бутанол, диоксан, пиридин. Если иное не оговорено специально, растворители, используемые во взаимодействиях по настоящему изобретению, являются инертными растворителями.
Термин «фармацевтически приемлемый» означает, что соответствующее вещество применимо в изготовлении фармацевтической композиции, которая является в целом безопасной, нетоксичной и не является нежелательной ни в биологическом, ни в ином смысле, включая применимость в ветеринарии, равно как и в медицине человека.
Термин «фармацевтически приемлемые соли» соединения означает соли, которые являются фармацевтически приемлемыми, как это определено в контексте, и которые обладают искомой фармакологической активностью родительского соединения. Подобные соли включают кислотно-аддитивные соли, образованные с неорганическими кислотами, такими как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота, или образованные с органическими кислотами, такими как уксусная кислота, бензолсульфоксилота, бензойная кислота, камфорсульфокислота, лимонная кислота, этансульфокислота, фумаровая кислота, глюкогептоновая кислота, глюконовая кислота, глутаминовая кислота, гликолевая кислота, гидроксинафтойная кислота, 2-гидроксиэтансульфокислота, молочная кислота, малеиновая кислота, яблочная кислота, малоновая кислота, миндальная кислота, метансульфокислота, муконовая кислота, 2-нафталинсульфокислота, пропионовая кислота, салициловая кислота, янтарная кислота, винная кислота, n-толуолсульфокислота, триметилуксусная кислота, или же соли, образуемые в том случае, когда присутствующий в родительском соединении кислый протон либо замещается ионом металла, например, ионом щелочного металла, ионом щелочноземельного металла или ионом алюминия, или координируется с органическим или неорганическим основанием. Приемлемые органические основания включают диэтаноламин, этаноламин, N-метилглюкамин, триэтаноламин, трометамин. Приемлемые неорганические основания включают гидроксид алюминия, гидроксид кальция, гидроксид калия, карбонат натрия, гидроксид натрия.
Предпочтительными фармацевтически приемлемыми солями являются соли, образованные с уксусной кислотой, соляной кислотой, серной кислотой, метансульфокислотой, малеиновой кислотой, фосфорной кислотой, винной кислотой, лимонной кислотой, натрием, калием, кальцием, цинком и магнием.
Термин «защитная группа» или «защищающая группа» означает группу, селективно блокирующую один реакционный центр в полифункциональном соединении таким образом, что химическое взаимодействие может быть осуществлено селективно по другому, незащищенному, реакционному центру, в том смысле, какой общепринято ассоциируют с этим в синтетической химии. Некоторые способы по настоящему изобретению основываются на использовании защитных групп с целью блокирования реакционноспособных атомов азота и/или кислорода, присутствующих в реагентах. Например, термины «защитная группа для аминогруппы» или «защитная группа для азота» употребляются в контексте взаимозаменяемо и относятся к органическим группам, предназначенным для защиты атома азота от нежелательны взаимодействий в ходе синтетических операций. Примеры защитных групп для атома азота включают, не ограничиваясь перечисленным, трифторацетил, ацетамидную группу, бензил (Bn), бензилоксикарбонильную группу (карбобензилоксигруппу, CBZ), n-метоксибензилоксикарбонильную группу, n-нитробензилоксикарбонильую группу, трет-бутоксикарбонильную группу (BOC). Специалистам в соответствующей области должно быть понятно, как выбирать группу с точки зрения легкости ее удаления и способности противодействовать соответствующим реакциям.
Термин «субъект» означает млекопитающих и не млекопитающих. Под млекопитающими понимаются любые члены класса млекопитающих, включая, но не ограничиваясь перечисленным, людей, не являющихся человеком приматов, таких как шимпанзе и другие виды приматов и обезьян, сельскохозяйственных животных, включая скот, лошадей, овец, коз и свиней, домашних животных, таких как кролики, собаки и кошки, лабораторных животных, включая грызунов, таких как крысы, мыши и морские свинки. Примеры не млекопитающих включают, не ограничиваясь перечисленным, птиц. Термин «субъект» не обозначает конкретный возраст или пол.
Термин «терапевтически эффективное количество» означает такое количество соединения, которое при введении субъекту с целью лечения болезненного состояния является достаточным для осуществления лечения такового болезненного состояния. «Терапевтически эффективное количество» будет варьироваться в зависимости от соединения, подвергаемого лечению болезненного состояния, тяжести подвергаемого лечению заболевания, возраста и сравнительного состояния здоровья субъекта, пути и формы введения, суждения лечащего врача или ветеринара и иных факторов.
Термины «определенные выше» и «определенные в контексте», когда они ссылаются на переменную величину, включают в качестве ссылки широкое определение переменной величины, равно как и предпочтительные, более предпочтительные и наиболее предпочтительные определения, если таковые имеются. Термин «лечить» или «лечение» болезненного состояния включает
(1) профилактику болезненного состояния, т.е. обеспечение того, чтобы клинические симптомы болезненного состояния не развивались у субъекта, который может быть уязвим или предрасположен в отношении болезненного состояния, но еще не испытывает и не проявляет симптомов такового болезненного состояния.
(2) ингибирование болезненного состояния, т.е. остановку развития болезненного состояния или его клинических симптомов, или
(3) облегчение болезненного состояния, т.е. обеспечение временного или постоянной регрессии болезненного состояния или его клинических симптомов.
Термин «обрабатывать», «вводить в соприкосновение» или «вводить во взаимодействие» означает, когда он относится к химическому взаимодействию, добавление или смешение двух или более реагентов в подходящих условиях с целью получения показанного и/или искомого продукта. Следует понимать, что взаимодействие, приводящее к получению показанного и/или искомого продукта, не обязательно является непосредственным результатом объединения двух исходно добавленных реагентов, т.е. может иметь место одно или более промежуточное соединение, получаемое из смеси, которое в конечном итоге приводит к образованию показанного и/или искомого продукта.
Номенклатура и структуры
Как правило, употребляемая в настоящей заявке номенклатура основана на AUTONOM™ v.4.0, компьютеризированной системе Института Бейльштейна для генерации систематической номенклатуры ИЮПАК. Приведенные в контексте химические структуры получены с помощью программы ISIS® версии 2.2. Любая некомпенсированная валентность, присутствующая на атоме углерода, кислорода или азота в приведенных в контексте структурах, означает присутствие атома водорода.
В тех случаях, когда в химической структуре присутствует хиральный атом, следует понимать, что структура включает в свой объем все стереоизомеры, связанные с этим хиральным атомом углерода.
Все упоминаемые в контексте патенты и публикации включены в настоящее описание в качестве ссылки во всей своей полноте и составляют его часть.
Общий способ
В одном из вариантов осуществления настоящего изобретения его объектом являются соединения формулы I
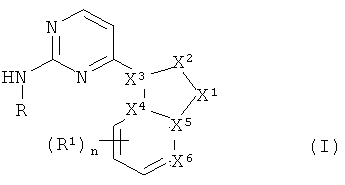
в которых
X1 является азотом, N-R3, CHR3, C-R3 или кислородом,
X2 является азотом, NH, N-CH3, CH или C-CH3,
X3 является азотом или углеродом, причем X1, X2 и X3 не являются все одновременно азотом,
каждый из X4, X5 независимо является углеродом или азотом,
Х6 является азотом или C-R1,
причем не менее двух и не более четырех из X1, X2, X3, X4, X5 и X6 являются азотом, а связи между X1 и X2, X2 и X3, X3 и X4, X4 и X5, X5 и X1, а также X5 и X6 могут независимо являться одинарными или двойными, или же образовывать ароматический цикл, при том условии, что в результате получается химически устойчивая структура («химически устойчивая структура» образуется в том случае, когда не превышены нормальные валентности и порядки связей, например, в химически устойчивой структуре не будет двойных связей между Х3-Х4, Х4-Х5 и Х5-Х1 одновременно),
R является -(циклогексил)-R2 или -(фенил)-R8, где циклогексил и фенил необязательно замещены метилом, фтором, хлором или гидроксигруппой,
R1 является галоидом, нитрогруппой, -CN, -CH2CN, -OH, -NH2, -COOH или -Y1R4,
Y1 является -O-, -NH-, -S-, -SO-, -SO2-, -NHSO2-, -NHC(O)-, -C(O)NH-, -C(O)O- или связью,
R4 является низшим алкилом, ацилом, фенилом или бензилом, каждый из которых замещен 0-3 гидроксигруппами или галоидами,
n принимает значения 0, 1, или 2,
R3 является Н или низшим алкилом,
R2 является водородом, -OH, -SO2NH2, =O, -CN или -Y2-Y3-Y4-R5, где
Y2 является -C(O)-, -C(O)NRa-, -SO2-, -O-, -NH- или связью,
Y3 является низшим алкиленом или связью,
Y4 является -O-, -NRa-, -S-, -SO2-, -C(O)-, -C(O)NRa-, -NRaC(O)-, -NRaC(O)O-, -NRaC(O)NRa-, -NHSO2-, -SO2NH- или связью,
R5 является низшим алкилом, циклоалкилом, гетероциклилом, причем R5 необязательно замещен -OH или -NHRa, где каждый из Ra независимо является водородом или низшим алкилом,
R8 является водородом, низшим алкилом, -OR3, -SO2NH2, -NHSO2R3, -COOR3, -SO2R3, -NH2, -CONRaR3, -NHC(O)R3, -CF3, -NO2, галоидом или -CN,
Z является водородом, галоидом, алкилом или NH2,
или его фармацевтически приемлемая соль.
В некоторых вариантах осуществления настоящего изобретения каждый из Х4, Х5 и Х6 является углеродом. В других вариантах осуществления настоящего изобретения R является -(циклогексил)-R2. В дальнейших вариантах осуществления настоящего изобретения X3 является азотом, а X1 является углеродом. В дополнительных вариантах осуществления настоящего изобретения R2 является OH. В некоторых вариантах осуществления настоящего изобретения R1 является бензилоксигруппой, а n равно 1. В еще некоторых вариантах осуществления настоящего изобретения R1 является -CN.
В некоторых вариантах осуществления настоящего изобретения Х2 является азотом. В других вариантах осуществления настоящего изобретения R2 является -Y2-Y3-Y4-R5, где Y2 является -C(O)NRa-, Ra является водородом, a Y3 является низшим алкиленом. В других вариантах осуществления настоящего изобретения Y4 является -SO2-. В дальнейших вариантах осуществления настоящего изобретения R5 является низшим алкилом.
В некоторых вариантах осуществления настоящего изобретения Y4 является -S-. В некоторых вариантах осуществления настоящего изобретения R5 является низшим алкилом.
В других вариантах осуществления настоящего изобретения Y4 является связью, а R5 является низшим алкилом.
В еще одном варианте осуществления настоящего изобретения его объектом является способ лечения воспаления, включающий введение эффективного количества соединения по настоящему изобретению нуждающемуся в том субъекту.
В еще одном варианте осуществления настоящего изобретения его объектом является фармацевтическая композиция, включающая соединение по настоящему изобретению и фармацевтически приемлемый наполнитель.
Следует понимать, что комбинации различных описанных в контексте групп могут образовывать другие варианты осуществления настоящего изобретения. Таким образом, в объем настоящего изобретения входит множество различных соединений.
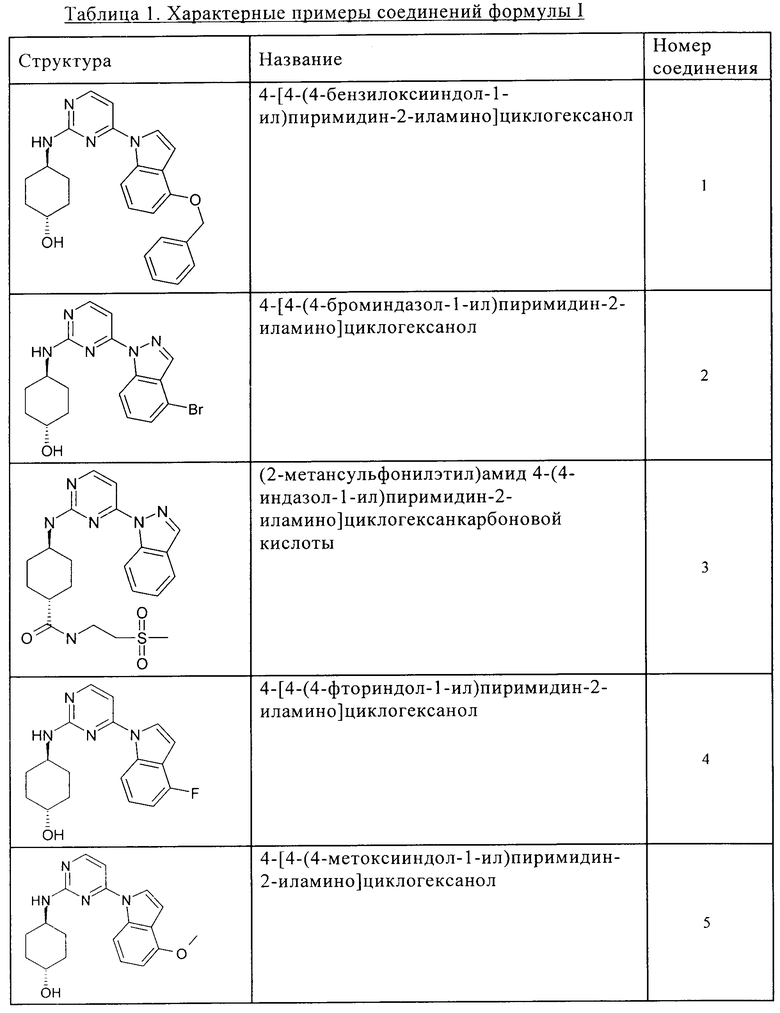
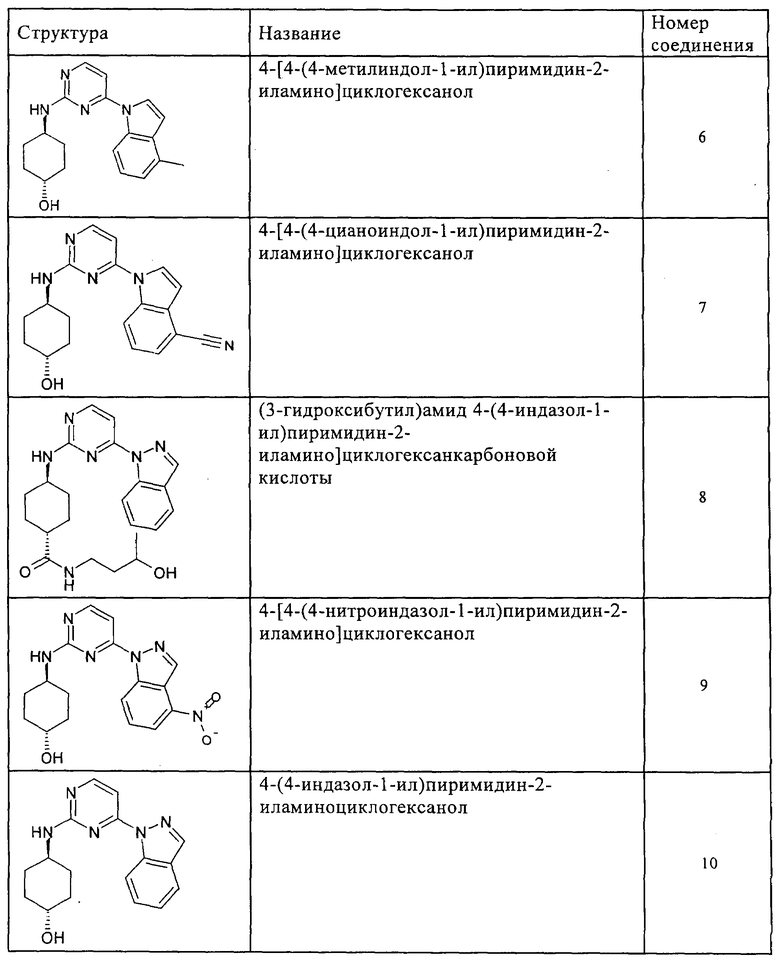
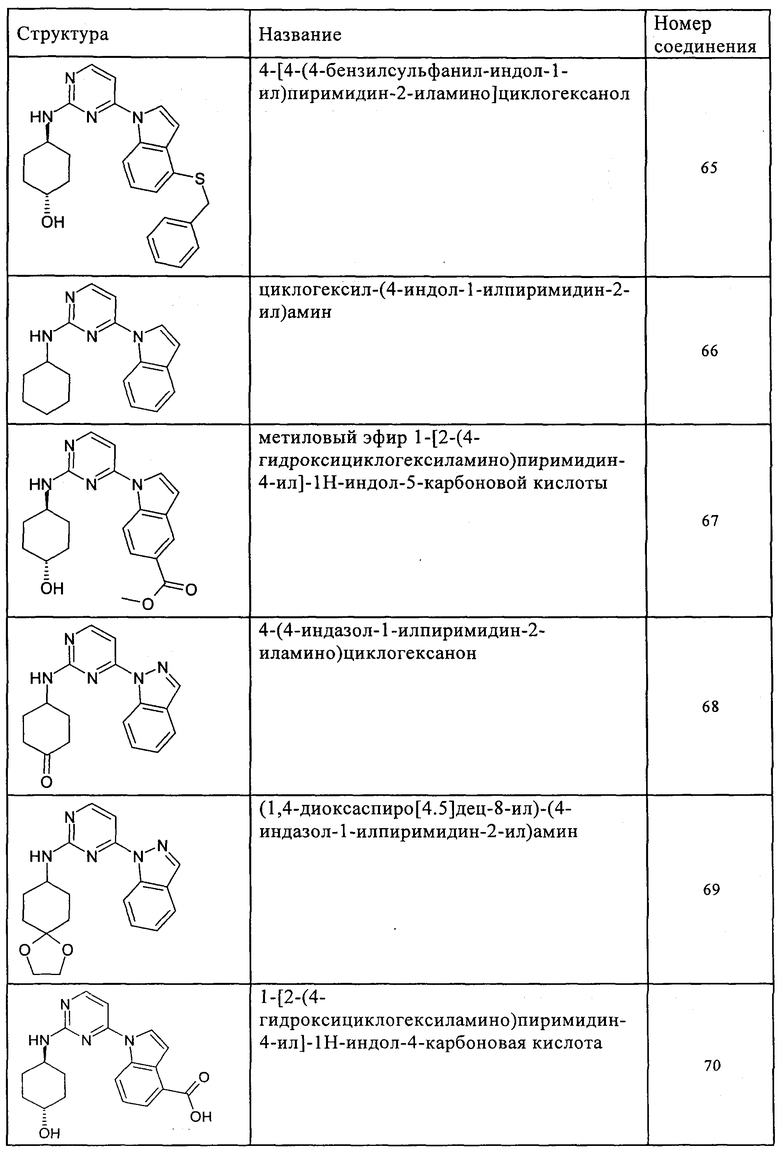
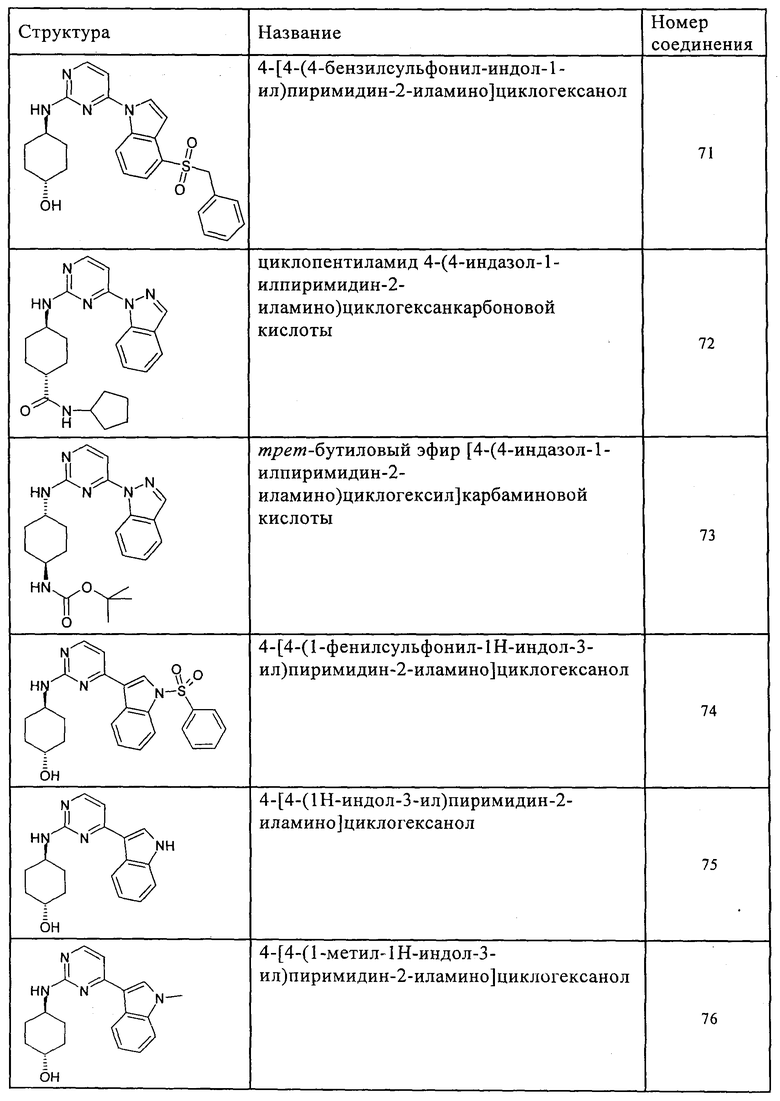
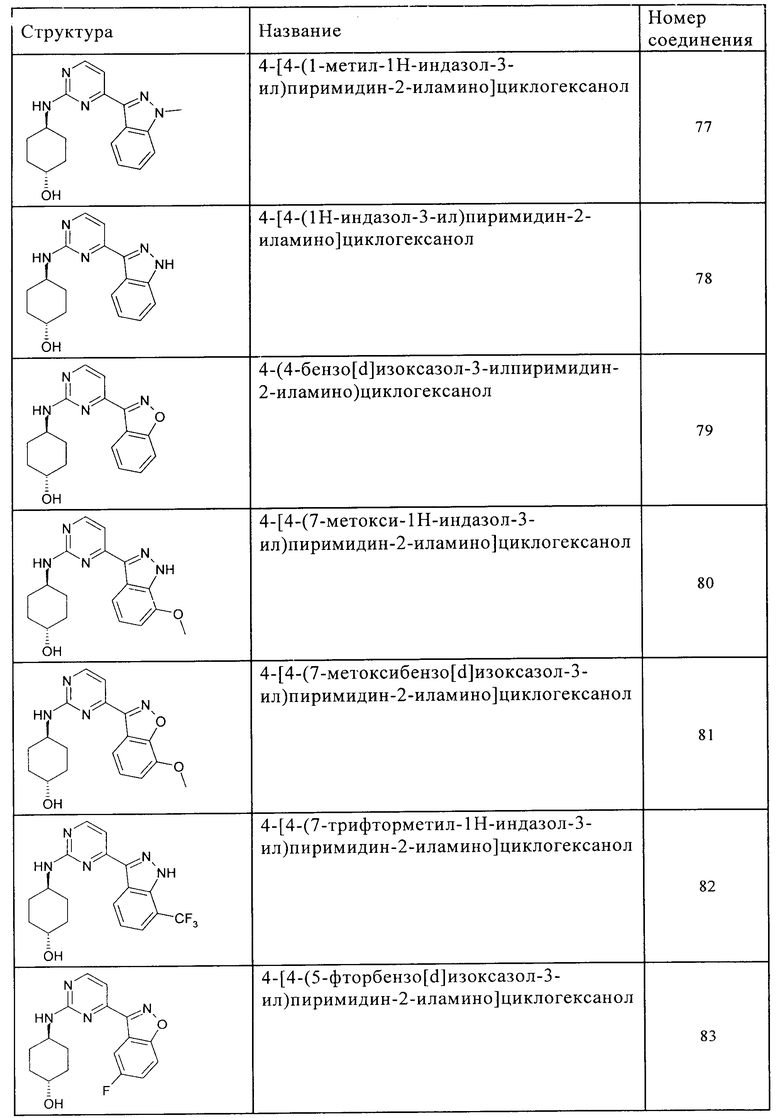
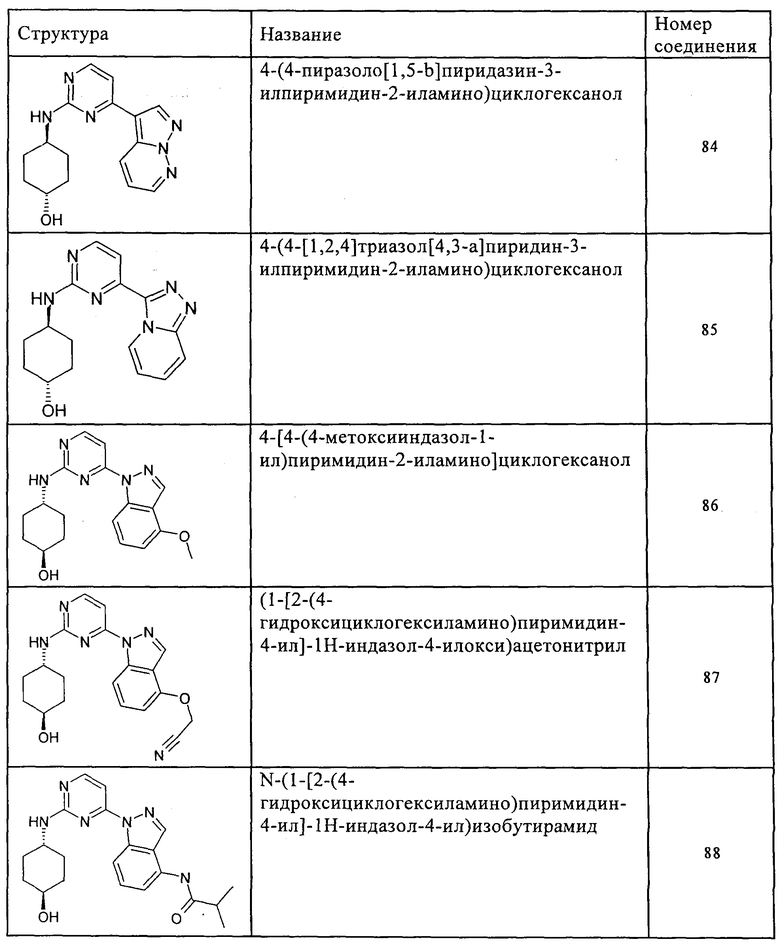
Характерные примеры соединений по настоящему изобретению представлены ниже в таблице 1.
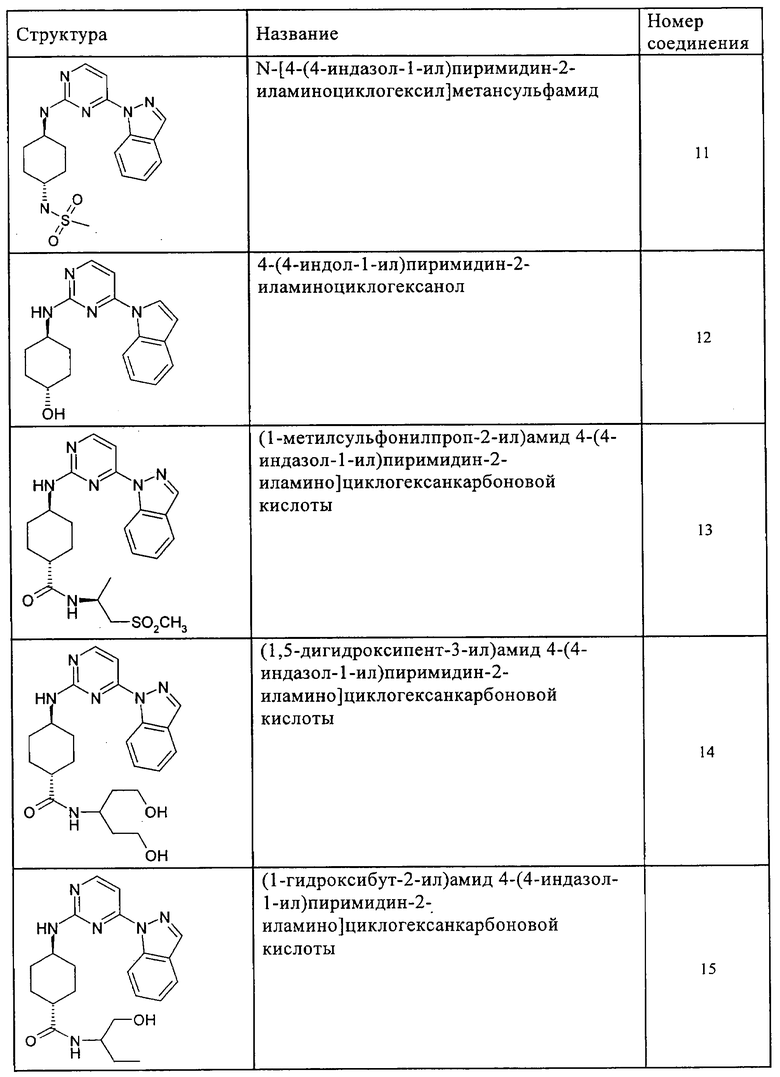
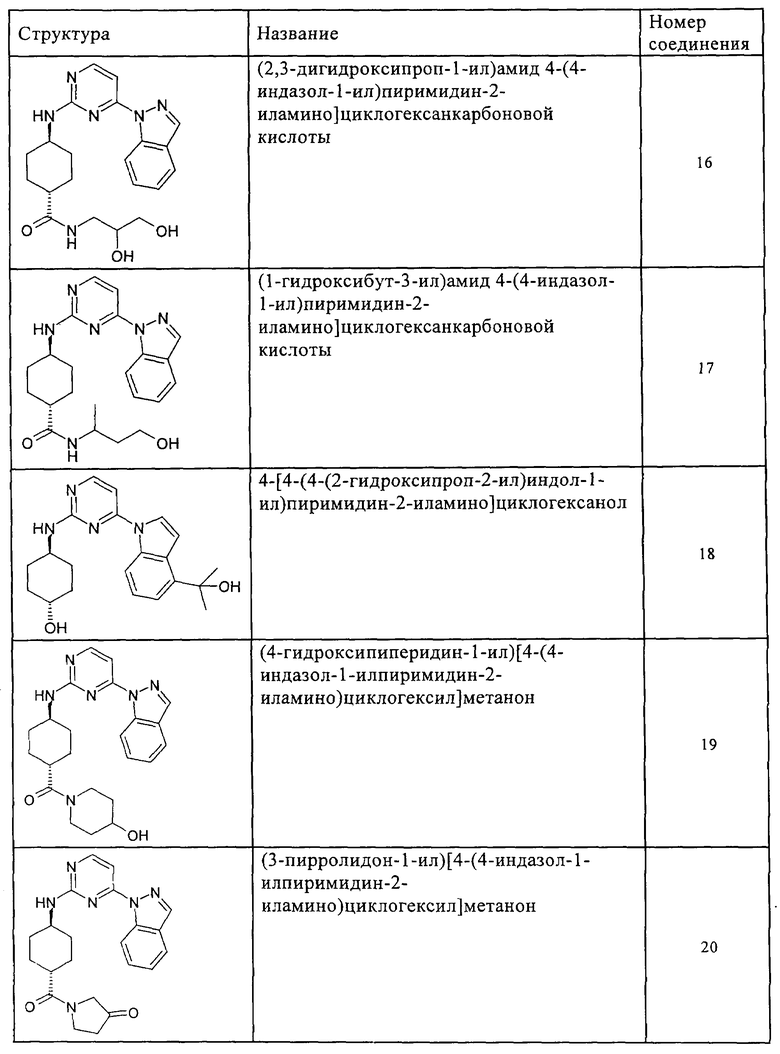

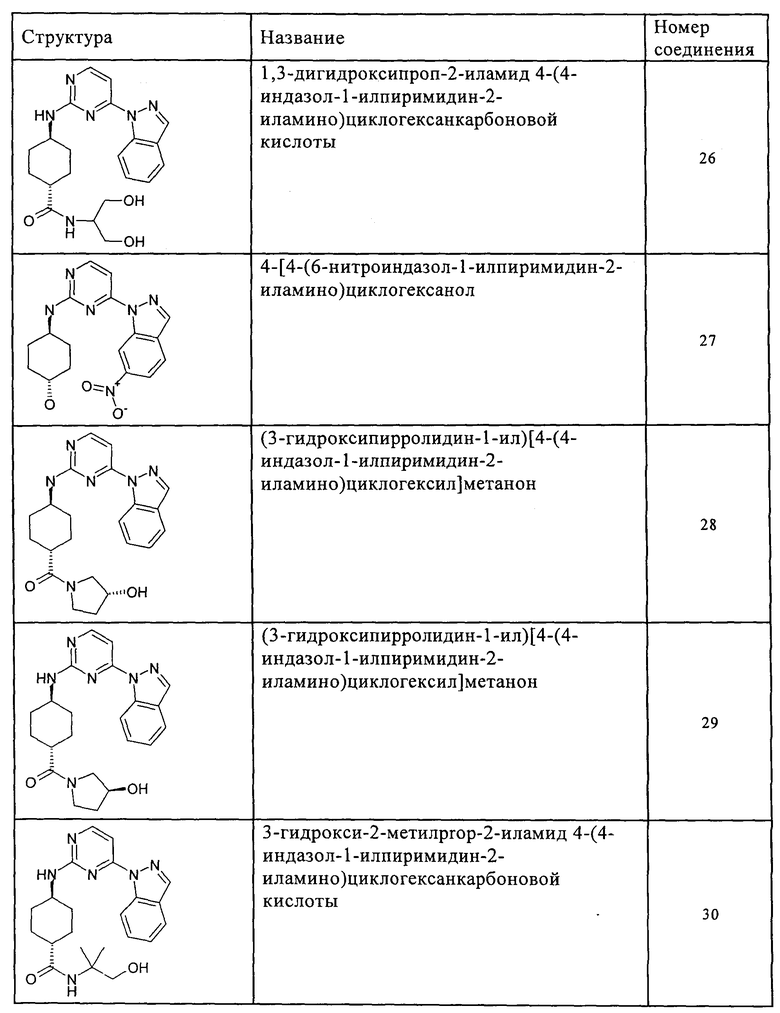

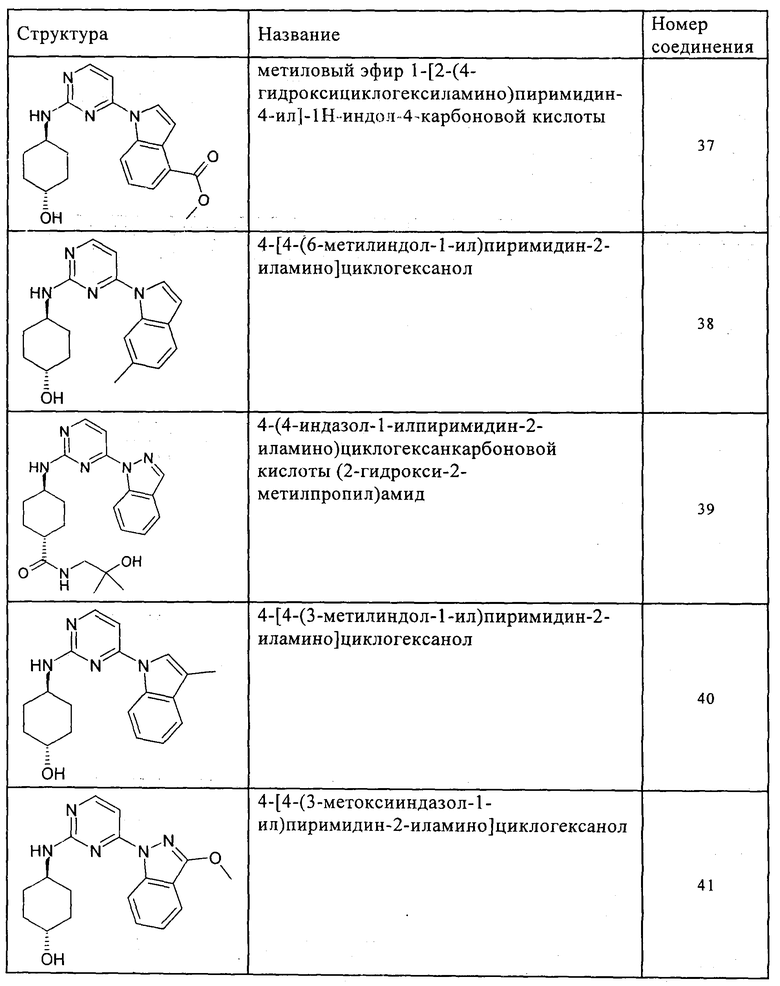
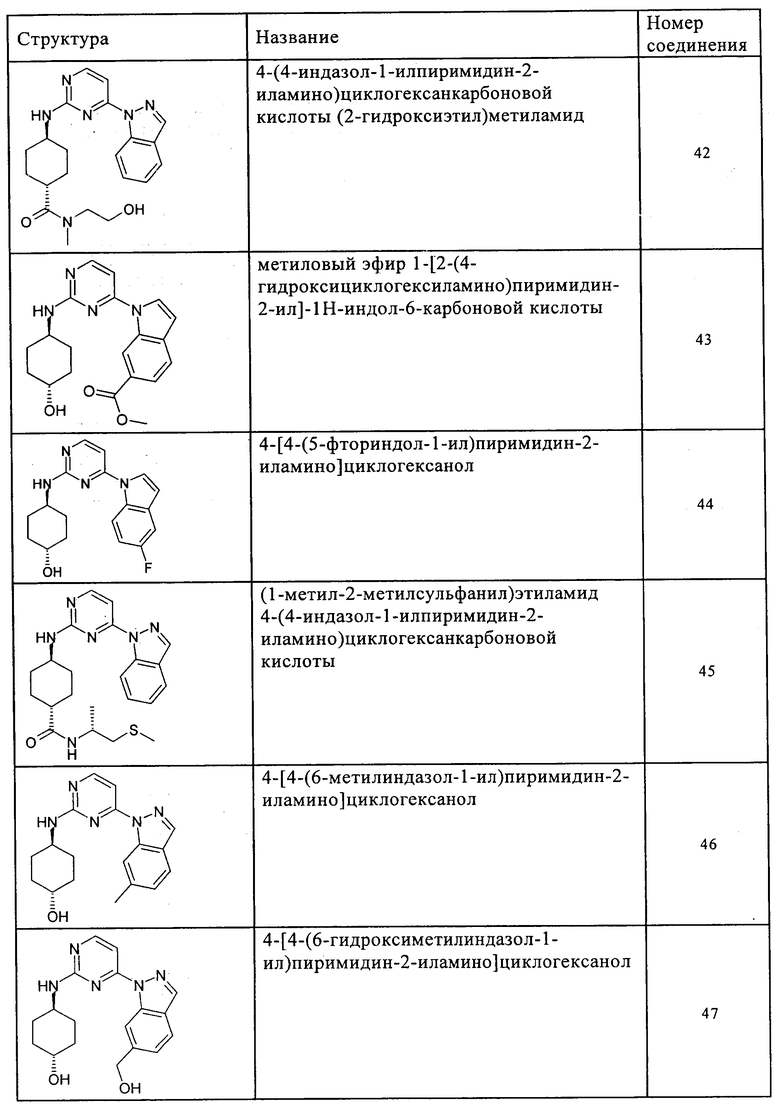
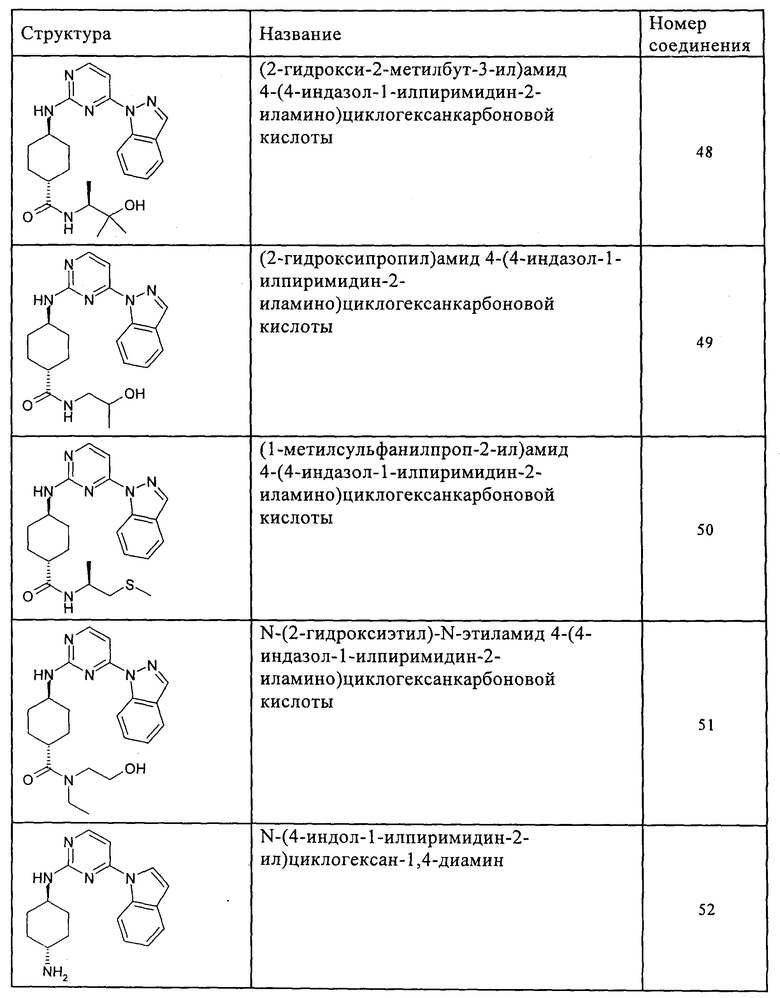
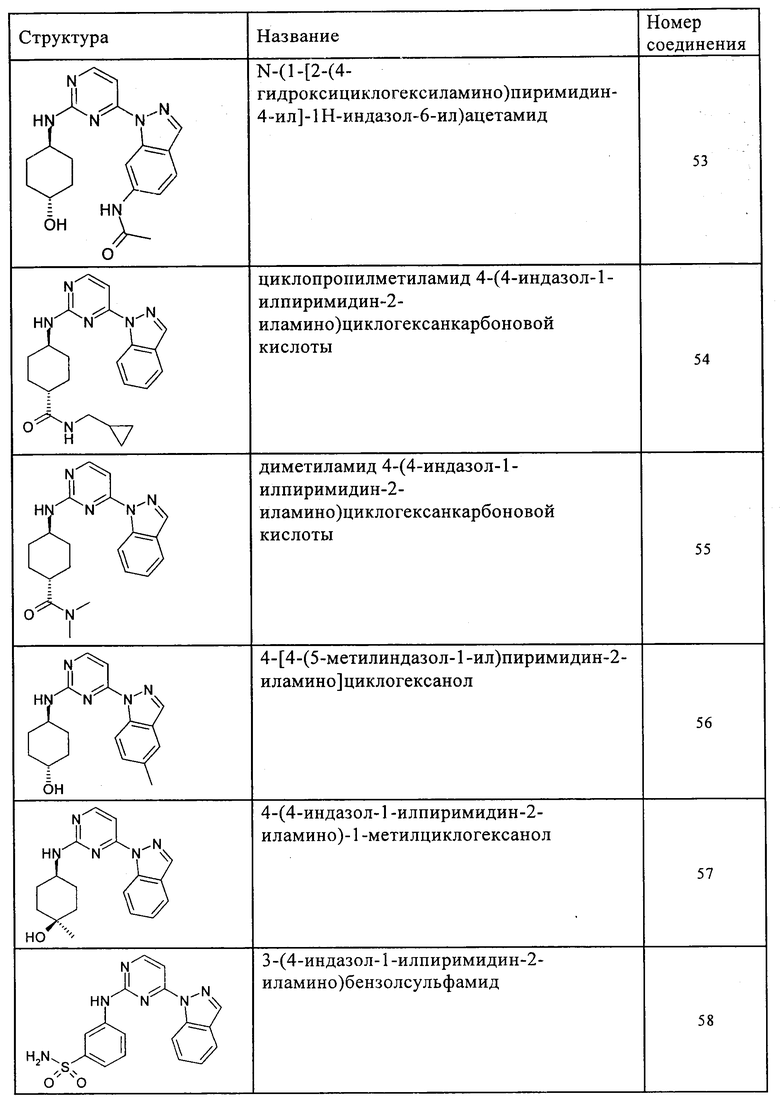
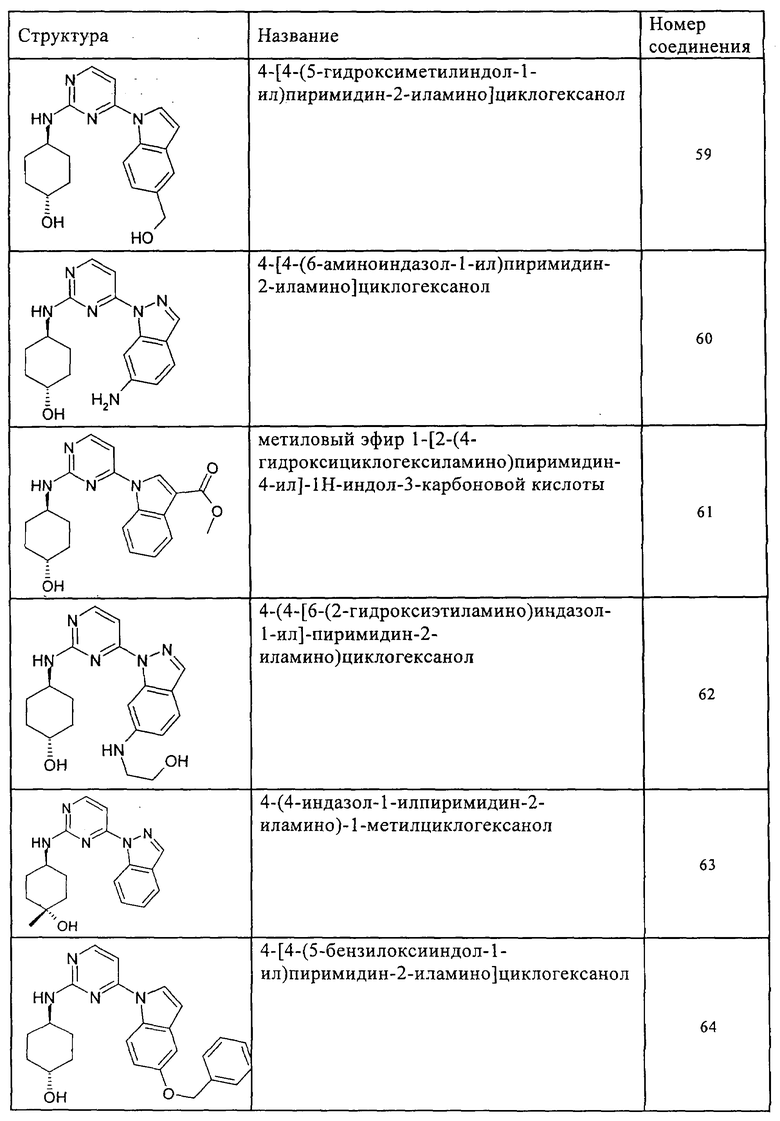
Синтез
Соединения по настоящему изобретению могут быть получены с помощью разнообразных способов, описанных в показательных примерах, представленных в нижеприведенном разделе «Примеры». Исходные вещества и реагенты, применяемые при получении этих соединений, как правило, либо являются доступными от коммерческих поставщиков, таких как Aldrich Chemical Co., либо могут быть получены с помощью способов, известных специалистам в соответствующей области, следуя методикам, приведенным в справочных изданиях, таких как Fieser and Fieser's Reagents for Organic Synthesis (Реагенты для органического синтеза), Wiley & Sons: New York, 1991, тт.1-15, Rodd's Chemistry of Carbon Compounds (Химия углеродных соединений), Elsevier Science Publishers, 1989, тт.1-5 и приложения, а также Organic Reactions (органические реакции), Wiley & Sons: New York, 1991, тт.1-40. Нижеследующие схемы реакций синтеза лишь иллюстрируют некоторые способы, с помощью которых соединения по настоящему изобретению могут быть получены. Специалистом в соответствующей области на основании содержащегося в настоящей заявке описания могут быть осуществлены и предложены различные модификации этих схем реакций синтеза.
Исходные вещества и промежуточные вещества в данных схемах реакций синтеза могут быть при необходимости выделены и очищены с помощью общепринятых способов, включающих фильтрование, перегонку, кристаллизацию, хроматографию, но не ограничивающихся ими. Подобные вещества могут быть охарактеризованы с помощью общепринятых средств, включающих физические константы и спектральные данные.
Если иное не оговорено специально, описанные в контексте взаимодействия предпочтительно осуществляют в инертной атмосфере, при атмосферном давлении, в диапазоне температур взаимодействия от, приблизительно, -78°C до, приблизительно, 230°C, а наиболее предпочтительно и легко - при комнатной температуре (или температуре внешней среды), например, около 20°C.
В нижеприведенных схемах, если не оговорено иначе, R1, X1, X2, X3 и им подобные отвечают вышеприведенным определениям, тогда как
А является хлором или SRb,
A1 является хлором, (S=O)Me, SO2Me или SO2Bu,
Ra является алкилом, циклоалкилом,
Rb является метилом или бутилом,
Z является азотом или углеродом,
Х является хлором, бромом или иодом,
Re и Rf независимо являются водородом, гидроксиалкилом, алкоксигруппой, алкилом, циклоалкилом, гетероалкилом, алкилсульфонильной группой, алкилсульфинильной группой,
Rg является низшим алкилом,
Rh является алкилом или циклоалкилом,
Ri является алкилом, циклоалкилом или гетероциклилом.

Стадия А: 1. иодид 1,3-диметилимидазолия, NaH, 1,4-диоксан, кипячение с обратным холодильником, 2. трет-бутилкарбазат, AcoH, MeOH, кипячение с обратным холодильником или NH2OH, EtOH, кипячение с обратным холодильником, 3. 1,8-диазабицикло[5.4.0]ундец-7-ен (ДБУ), тетрагидрофуран (ТГФ), микроволновой нагрев до температуры 150°C.
Стадия Б: NaH, N,N-диметилформамид (ДМФ) или гексаметилдисилазид лития (LiГМДC) или натрия (NaГМДC), ацетонитрил/ТГФ, низкая температура, или N,N-диизопропилэтиламин (ДИПЭА), ДМФ, 100°C.
Стадия В: тетракис(трифенилфосфин)палладия(0), Na2CO3, ацетонитрил, вода.
Стадия Г: 1. EtOH, кипячение с обратным холодильником, 2. диацетат иодбензола, дихлорметан (ДХМ).
Стадия Д: КОН, вода, ДХМ.
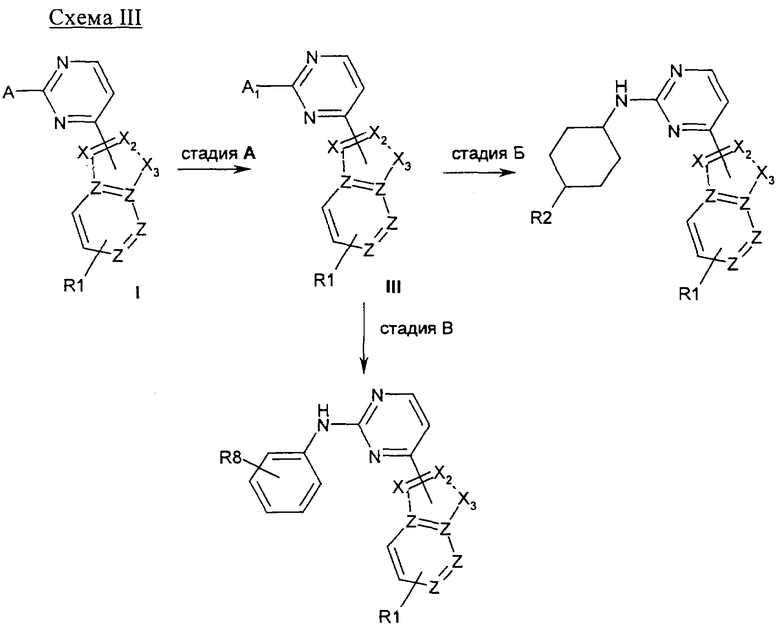
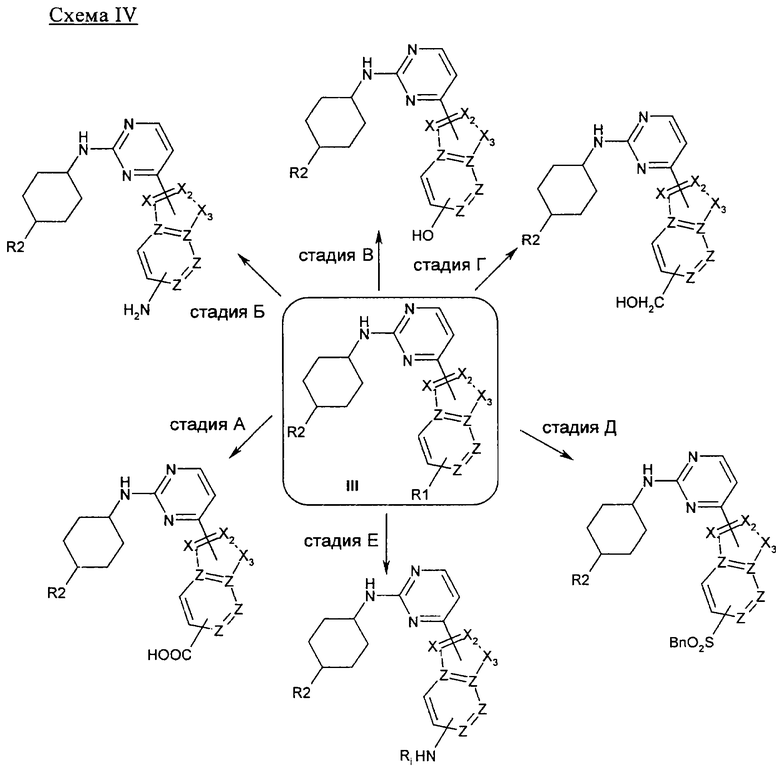
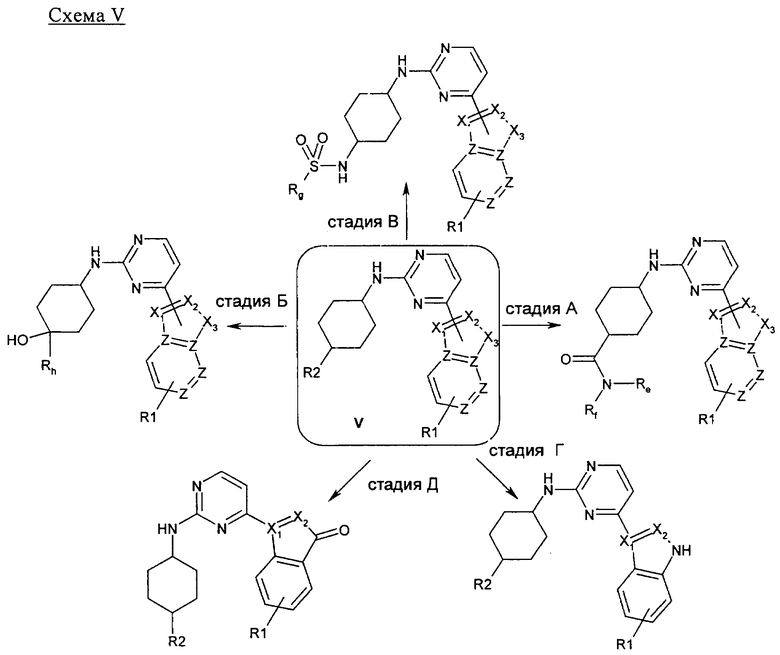
На стадии А 4-хлор-2-метилсульфанилпиримидин или 4-хлор-2-бутилсульфанилпиримидин взаимодействует при кипячении с обратным холодильником в присутствии сильного основания, такого как гидрид натрия, и иодида 1,3-диметилимидазолия с различным образом замещенным о-фторбензальдегидом в полярном апротонном растворителе, таком как 1,4-диоксан, что приводит к соответствующему кетону. В результате обработки этого продукта трет-бутилкарбазатом в присутствии уксусной кислоты в полярном апротонном растворителе, таком как метанол, при кипячении с обратным холодильником приводит к соответствующему гидразону, который при нагревании в условиях микроволнового облучения в присутствии основания, такого как ДБУ, в полярном растворителе, таком как ТГФ, циклизуется, что приводит к соответствующему производному индазола. Если вместо трет-бутилкарбазата используют гидроксиламин, получают соответствующее производное бензоксазола. На стадии Б 4-хлорпиримидин, несущий в положении 2-меркаптоалкильный или хлорный остаток, подвергается реакции ароматического нуклеофильного замещения (SNAr) с различным образом замещенным индолом, индазолом, азаиндолом или индолином в присутствии основания, такого как NaH, LiГМДC, NaГМДC или ДИПЭА, в полярном растворителе, таком как ДМФ, ацетонитрил, ТГФ, 1,4-диоксан или смесь перечисленного, при температуре в диапазоне от -10°C до 100°C, что приводит к соответствующим N1-арилированному индолу, N1- или N2-арилированному индазолу, N1-арилированному азаиндолу и N1-арилированному индолину. На стадии В 2,4-дихлорпиримидин подвергают реакции сочетания по Бухвальду с различным образом замещенной 3-индолбороновой кислотой, защищенной по положению N1, в присутствии катализатора, такого как тетракис(трифенилфосфин)палладий(0), и неорганического основания, такого как Na2CO3, в смеси полярных растворителей, такой как ацетонитрил/вода, что приводит к соответствующему 2-хлор-4-арилированному производному пиримидина. На стадии Г 2-метилсульфанилпиримидин-4-карбальдегид взаимодействует с замещенным или незамещенным пиридин-2-илгидразином в полярном протонном растворителе, таком как этанол, при кипячении с обратным холодильником, что приводит к соответствующему гидразону, который циклизуется в присутствии диацетата иодбензола в аполярном растворителе, таком как ДХМ, приводя к образованию соответствующего различным образом замещенного [1,2,4]триазоло[4,3-а]пиридина. На стадии Д, 4-этинил-2-метилсульфанилпиримидин взаимодействует с иодидом 1-аминопиридазиния-1 (J. Med. Chem. 2004, 47, 4716-30) в присутствии воды и неорганического основания, такого как КОН, в аполярном растворителе, таком как ДХМ, что приводит к соответствующему производному 3-(2-метилсульфанилпиримидин-4-ил)пиразоло[1,5-b]пиридазина.
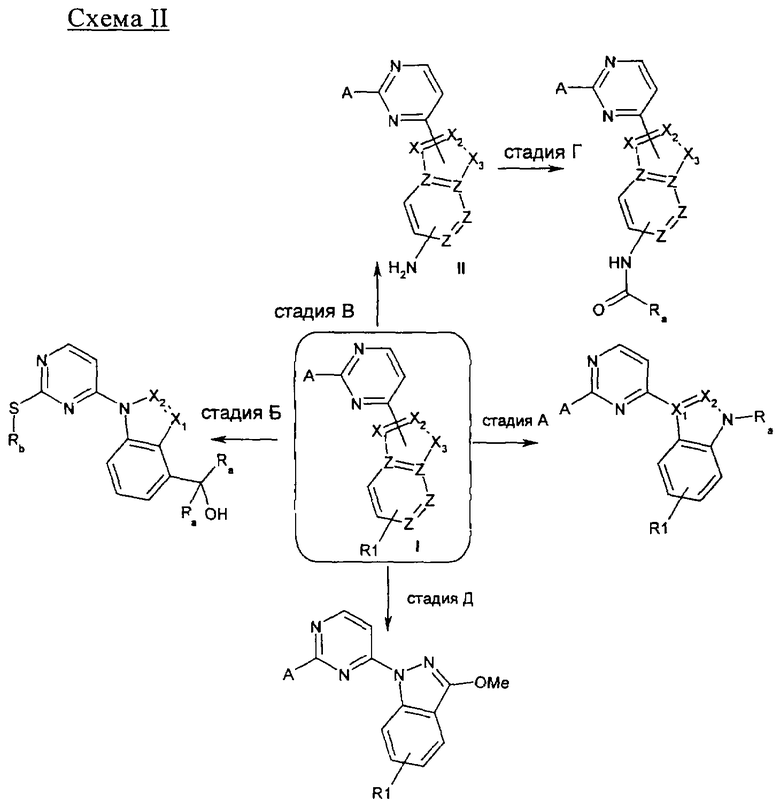
Если X3 является NH или NSO2Ph - стадия A: NaH, RaX, 1-метил-2-пирролидинон (НМП) или 1. NaOH, MeOH/ТГФ, 2. NaH, RaX, НМП.
Если R1 является COOMe - стадия Б: RaMgCl, ТГФ, низкая температура.
Если R1 является NO2 - стадия В: Fe(0), NH4Cl, EtOH, вода, нагревание, стадия Г: RaCOCl, триэтиламин (ТЭА), ТГФ.
Если X1 является C=O - стадия Д: NaH, RaX, ДМФ, 0°C.
На стадии А соединение общей структуры I, в котором X3 является NH, может быть проалкилировано посредством обработки сильным основанием, таким как NaH, и подходящим алкил- или циклоалкилгалогенидом, таким как MeI, в полярном растворителе, таком как НМП. Если X3 является фенилсульфамидным остатком, соединение общей структуры I может быть сначала лишено защиты посредством обработки сильным неорганическим основанием, таким как NaOH, в смеси полярных растворителей, такой как MeOH/ТГФ, а затем проалкилировано, как это описано выше. На стадии Б соединение общей структуры I, в котором R1 является остатком метилового сложного эфира, может быть подвергнуто двойному присоединению с помощью реактива Гриньяра, такого как MeMgCl, при низкой температуре в полярном растворителе, таком как ТГФ, с целью получения соответствующего третичного спирта. На стадии В соединение общей структуры I, в котором R1 является NO2, может быть восстановлено до соответствующего производного анилина посредством нагревания в присутствии восстановителя, такого как порошок Fe(0) от компании Fisher Scientific, хлорида аммония и воды в полярном растворителе, таком как этанол. Производное анилина общей формулы II может быть затем ацилировано с помощью подходящего ацилхлорида в присутствии основания, такого как триэтиламин, в полярном растворителе, таком как ТГФ, с целью получения соответствующего вторичного амида, как это описано на стадии Г. На стадии Д соединение общей структуры I, в котором X1 является C=O, может быть функционализировано группой O-алкил посредством обработки алкилирующим агентом, таким как MeI, после депротонирования под действием сильного основания, такого как NaH, в полярном растворителе, таком как ДМФ, при низкой температуре.
Стадия A: N-хлорсукцинимид (NCS), НМП или 3-хлорпероксибензойная кислота (м-ХПБК) или MeReO3/H2O2, ДХМ.
Стадия Б: R2C6H10NH2, НМП, нагревание или R2C6H10NH2, ДИПЭА, НМП, нагревание.
Стадия В: R8C6H4NH2, пара-толуолсульфокислота (n-TCK), i-PrOH, 150°C, микроволновой нагрев.
На стадии А соединение общей формулы I, в котором А является тиометильным остатком, может быть прохлорировано в присутствии N-хлорсукцинимида в полярном растворителе, таком как НМП, при высокой температуре. Альтернативно, соединение общей формулы I может быть окислено до соответствующего сульфона или сульфоксида с помощью 3-хлорпероксибензойной кислоты или смеси метилтриоксорения и перекиси водорода в качестве окислителя в аполярном растворителе, таком как ДХМ или хлороформ, при температуре в диапазоне между 0°C и комнатной температурой. Соответствующее хлорпроизводное, сульфон или сульфоксид III может быть подвергнуто SNAr-реакции с различным образом замещенным или незамещенным циклогексиламином в качестве нуклеофила, в присутствии или в отсутствие основания, такого как диизопропилэтиламин, в аполярном растворителе, таком как НМП, при температуре в диапазоне между 90 и 150°C, как это описано на стадии Б. Альтернативно, сульфоксид общей структуры III может быть введен во взаимодействие с различным образом замещенным или незамещенным анилином в присутствии n-толуолсульфокислоты в полярном протонном растворителе, таком как изопропанол, в условиях микроволнового облучения при температуре 150°C, как это описано на стадии В.
Если R1 является COOMe, COOEt - стадия A: LiOH или NaOH, ТГФ/MeOH.
Если R1 является NO2 - стадия Б: Fe(0), NH4Cl, EtOH, вода, нагревание.
Если R1 является OBn - стадия В: Н2, Pd/C, EtOH.
Если R1 является COOMe - стадия Г: литийалюминийгидрид (ЛАГ) или LiEt3BH, ТГФ, -10°C или комнатная температура (КТ).
Если R1 является SBn - стадия Д: м-ХПБК, хлороформ.
Если R1 является NH2 - стадия Е: RiX, Na2CO3, ДМФ, 80°C.
На стадии А соединение общей формулы IV, в котором R1 является остатком метилового или этилового сложного эфира, может быть гидролизовано до соответствующей карбоновой кислоты в присутствии сильного неорганического основания, такого как гидроксид натрия или лития, в смеси полярных растворителей, таких как ТГФ и MeOH. На стадии Б соединение общей формулы IV, в котором R1 является нитрогруппой, может быть восстановлено до соответствующего производного анилина посредством нагревания в присутствии хлорида аммония и воды в полярном протонном растворителе, таком как этанол, с помощью восстановителя, такого как порошок железа от компании Fisher Scientific. На стадии В соединение общей формулы IV, в котором R1 является бензилоксигруппой, может быть восстановлено до соответствующего производного фенола с помощью водорода в качестве восстановителя в присутствии палладия на угле в качестве катализатора в полярном протонном растворителе, таком как этанол. На стадии Г соединение общей формулы IV, в котором R1 является остатком метилового или этилового сложного эфира, может быть восстановлено до соответствующего производного бензилового спирта с помощью алюмогидрида лития или триметилборгидрида лития в качестве восстановителя в полярном растворителе, таком как ТГФ, при температуре в диапазоне между -10°C и комнатной температурой. На стадии Д соединение общей формулы IV, в котором R1 является меркаптобензильным остатком, может быть окислено до соответствующего сульфона с помощью 3-хлорпероксибензойной кислоты в качестве окислителя в аполярном растворите, таком как хлороформ. На стадии Е соединение общей формулы IV, в котором R1 является аминогруппой, может быть проалкилировано до соответствующего вторичного или третичного амина при нагревании в присутствии неорганического основания, такого как карбонат натрия, в полярном растворителе, таком как ДМФ, с помощью алкил-, циклоалкил- или гетероциклилгалогенида в качестве алкилирующего агента.
Если R2 является COOEt - стадия А: 1. NaOH, ТГФ, 2. ReRfNH, гексафторфосфат бензотриазол-1-илокситрис(диметиламино)фосфония (БОФ), ДИПЭА, ТГФ.
Если R2 является O(CH2)2O - стадия Б: 1. HCl, ТГФ, нагревание, 2. RhMgCl, ТГФ, -78°C.
Если R2 является NH2 - стадия В: (RgSO2)O.
Если X3 является NHSO2Ph - стадия Г: NaOH, MeOH.
Если X1 является C-OMe - стадия Д: триметилсилилиодид (ТМСИ), CHCl3.
На стадии А соединение общей формулы V, в котором R2 является остатком этилового или метилового сложного эфира, может быть гидролизовано до соответствующей карбоновой кислоты, в присутствии сильного неорганического основания, такого как гидроксид натрия, в полярном растворителе, таком как ТГФ. Затем может быть осуществлена реакция сочетания карбоновой кислоты с гидроксиалкил-, алкокси-, алкил-, циклоалкил-, гетероалкил-, алкилсульфонил- или алкилсульфиниламином в присутствии агента для сочетания, такого как БОФ, и основания, такого как ДИПЭА, в полярном растворителе, таком как ТГФ, с целью получения соответствующего амида. На стадии Б соединение общей формулы V, в котором R2 является 1,3-диоксолановым остатком, может быть гидролизовано до соответствующего кетона посредством нагревания в присутствии сильной неорганической кислоты, такой как HCl, в полярном растворителе, таком как ТГФ. Получаемый таким образом кетон может быть подвергнут реакции присоединения с реактивом Гриньяра при низкой температуре в полярном растворителе, таком как ТГФ, с целью получения соответствующего третичного спирта. На стадии В соединение общей формулы V, в котором R2 является аминогруппой, может быть сульфонилировано в присутствии ангидрида сульфокислоты в полярном растворителе, таком как НМП, что приводит к соответствующему сульфамиду. На стадии Г соединение общей формулы V, в котором X3 является фенилсульфамидным остатком, может быть гидролизовано до соответствующего производного индола, индазола или бензоксазола в присутствии неорганического основания, такого как гидроксид натрия, в полярном растворителе, таком как метанол. На стадии Д соединение общей формулы V, в котором X1 является остатком простого метилового эфира, может быть окислено до соответствующего кетона по реакции с триметилсилилиодидом в аполярном растворителе.
После этого продукты могут быть подвергнуты очистке, например, посредством экстракции, кристаллизации, препаративной высокоэффективной жидкостной хроматографии (ВЭЖХ), флэш-хроматографии, тонкослойной хроматографии.
Соединения по настоящему изобретению являются модуляторами JNK; ожидается, что в этом качестве они будут эффективны в лечении широкого спектра опосредованных JNK расстройств. Примеры опосредуемых JNK расстройств включают, не ограничиваясь, однако, перечисленным, аутоиммунное расстройство, воспалительное расстройство, метаболическое расстройство, неврологическое расстройство и рак. В соответствии с этим, соединения по настоящему изобретению могут быть задействованы для лечения одного или более подобных расстройств. В некоторых вариантах осуществления настоящего изобретения соединения по настоящему изобретению могут применяться для лечения опосредованного JNK расстройства, такого как ревматоидный артрит, астма, диабет типа II, болезнь Альцгеймера, болезнь Паркинсона или инсульт.
Введение и фармацевтическая композиция
Объектом настоящего изобретения являются фармацевтические композиции, содержащие хотя бы одно соединение по настоящему изобретению или его индивидуальный изомер, рацемическую или нерацемическую смесь изомеров или фармацевтически приемлемую соль или сольват совместно с хотя бы одним фармацевтически приемлемым носителем, а также, необязательно, другими терапевтическими и/или профилактическими ингредиентами.
Как правило, соединения по настоящему изобретению должны вводиться в терапевтически эффективном количестве одним из способов введения, принятых для средств, служащих сходным целям. Подходящие диапазоны дозировок составляют, как правило, 1-500 мг в сутки, предпочтительно 1-100 мг в сутки, наиболее же предпочтительно 1-30 мг в сутки, в зависимости от различных факторов, таких как тяжесть подвергаемого лечению заболевания, возраст и сравнительное состояние здоровья субъекта, действенность применяемого соединения, путь и форма введения, показания, обуславливающие введение, а также предпочтения и опыт лечащего врача. Специалист, обладающий базовыми знаниями в соответствующей области, сможет без недолжного экспериментирования, на основании своих знаний и описания в настоящей заявке, установить терапевтически эффективное количество соединений по настоящему изобретению для конкретного заболевания.
Соединения по настоящему изобретению могут вводиться в виде фармацевтических препаратов, включая подходящие для перорального (включая трансбуккальное и сублингвальное), перректального, интраназального, местного, внутрилегочного, интравагинального или парэнтерального (включая внутримышечное, внутриартериальное, интратекальное, подкожное и внутривенное) введения или в форме подходящей для введения посредством ингаляции или вдувания. Предпочтительным способом введения является, как правило, пероральный с применением удобного режима суточной дозировки, который может подстраиваться в зависимости от степени недуга.
Соединение или соединения по настоящему изобретению совместно с одним или более общепринятыми адъювантами, носителями или разбавителями может быть помещен в форму фармацевтических композиций или стандартных дозировок. Фармацевтические композиции и формы стандартных дозировок могут состоять из стандартных ингредиентов в стандартных пропорциях, с добавлением дополнительных активных соединений или действующих начал или без таковых, причем формы стандартной дозировки могут содержать любое подходящее эффективное количество активного ингредиента, соразмерное с предполагаемым к применению диапазоном суточных дозировок. Фармацевтические композиции могут применяться в виде твердых объектов, таких как таблетки или заполненные капсулы, полутвердых объектов, порошков, препаратов с замедленным высвобождением или жидкостей, таких как растворы, суспензии, эмульсии, эликсиры или заполненные капсулы для перорального применения, или в форме суппозиториев для перректального или интравагинального введения, или в форме стерильных растворов для инъекций для парэнтерального применения. Препараты, содержащие около одного (1) мг активного ингредиента или, в более широком случае, от, приблизительно, 0,01 до, приблизительно, ста (100) мг, на таблетку, являются, таким образом, подходящими характерными примерами форм стандартной дозировки.
Соединения по настоящему изобретению могут быть задействованы в широком диапазоне форм дозировки для перорального введения. Фармацевтические композиции и формы дозировки могут включать соединение или соединения по настоящему изобретению или их фармацевтически приемлемые соли в качестве активного компонента. Фармацевтически приемлемые носители могут быть как твердыми, так и жидкими. Твердые препараты включают порошки, таблетки, пилюли, капсулы, ожжет быть одним или более соединениями, которые также могут выступать в качестве разбавителей, ароматизаторов, солюбилизаторов, скользящих веществ, суспендирующих веществ, связующих веществ, консервантов, веществ, способствующих распадению таблеток или вещества-оболочки. В порошках носитель является, как правило, тонкоизмельченным твердым веществом в смеси с тонкоизмельченным активным компонентом. В таблетках активный компонент, как правило, смешан с носителем, характеризуемым необходимой связующей способностью, в подходящих пропорциях и спрессован до желаемой формы и размера. Порошки и таблетки предпочтительно содержат от, приблизительно, одного (1) до, приблизительно, семидесяти (70) процентов активного соединения. Подходящие носители включают, не ограничиваясь перечисленным, карбонат магния, стеарат магния, тальк, сахар, лактозу, пектин, декстрин, крахмал, желатин, трагакант, метилцеллюлозу, натрий-карбоксиметилцеллюлозу, воск с низкой температурой плавления, масло какао. Подразумевается, что термин «изготовление» включает препарат активного соединения с образующим оболочку веществом в качестве носителя, представляющий капсулу, в которой активный компонент, с носителями или без них, окружен носителем, находящимся с ним в контакте. Аналогичным образом, сюда включены облатки и лепешечки. Таблетки, порошки, капсулы, пилюли, облатки и лепешечки могут быть пригодны для перорального введения в качестве твердых форм.
Другие подходящие для перорального введения формы включают препараты в жидкой форме, включая эмульсии, сиропы, эликсиры, водные растворы, водные суспензии или препараты в твердой форме, предназначенные для преобразования в препараты в жидкой форме незадолго перед применением. Эмульсии могут быть приготовлены в растворах, например, в водных растворах пропиленгликоля, или могут содержать эмульгаторы, например, такие как лецитин, моноолеат сорбита или камедь акации. Водные растворы могут быть получены посредством растворения активного компонента в воде и добавления подходящих красителей, ароматизаторов, стабилизаторов и загустителей. Водные суспензии могут быть получены посредством диспергирования тонкоизмельченного активного компонента в воде с вязким веществом, таким как природные или синтетические смолы, камеди, метилцеллюлоза, натрий-карбоксиметилцеллюлоза и другие хорошо известные суспендирующие средства. Препараты в твердой форме включают растворы, суспензии и эмульсии и могут содержать, помимо активного компонента, красители, ароматизаторы, стабилизаторы, буферы, искусственные и природные подсластители, диспергирующие средства, загустители, солюбилизаторы.
Соединения по настоящему изобретению могут быть задействованы в препаратах для парэнтерального введения (например, посредством инъекции, например инъекции болюса или непрерывного вливания) и могут находиться в форме стандартной дозировки в ампулах, заранее заполненных шприцах, вливаниях малого объема или в контейнерах с многократной дозой с добавлением консерванта. Композиции могут принимать такие формы, как суспензии растворы или эмульсии в масляных или водных носителях, например, растворы в водном полиэтиленгликоле. Примеры масляных или неводных носителей, разбавителей, растворителей или основ включают пропиленгликоль, полиэтиленгликоль, растительные масла (например, оливковое масло) и пригодные для инъекций органические сложные эфиры (например, этилолеат), и могут содержать вспомогательные средства, такие как консерванты, смачивающие вещества, эмульгаторы или суспендирующие вещества, стабилизаторы и/или диспергирующие вещества. Альтернативно, активные ингредиент может находиться в форме порошка, полученного посредством асептического выделения стерильного твердого вещества или посредством лиофилизации из раствора, для соединения перед употреблением с подходящим носителем, например, стерильной водой, свободной от пирогенов.
Соединения по настоящему изобретению могут быть задействованы в препаратах для местного введения в эпидермис в виде мазей, кремов или лосьонов, или же в виде трансдермального пластыря. Мази и кремы могут, например, представлять собой препараты с водной или масляной основой с добавлением подходящих загустителей и/или желирующих веществ. Лосьоны могут представлять собой препараты с водной или масляной основой и, как правило, будут также содержать один или более эмульгатор, стабилизаторы, диспергирующее вещество, суспендирующее вещество, загуститель или краситель. Препараты, подходящие для местного применения во рту включают лепешечки, содержащие активные средства в ароматизированной основе, обычно - сахарозе и камеди акации или трагаканта, пастилки, содержащие активный ингредиент в инертной основе, такой как желатин и глицерин или сахароза и камедь акации, а также полоскания для рта, содержащие активный ингредиент в подходящем жидком носителе.
Соединения по настоящему изобретению могут быть задействованы в препаратах для введения в виде суппозиториев. Сначала расплавляют воск с низкой температурой плавления, такой как смесь глицеридов жирных кислот, или масло какао, а затем гомогенно диспергируют активный компонент посредством, например, перемешивания. После этого расплавленную гомогенную смесь выливают в формы соответствующих размеров и дают ей остыть и затвердеть.
Соединения по настоящему изобретению могут быть задействованы в препаратах для интравагинального введения. Это могут быть пессарии, тампоны, кремы, гели, пасты, пены или спреи, содержащие, помимо активного ингредиента, такие носители, которые считаются подходящими в соответствующей области.
Рассматриваемые соединения могут быть задействованы в препаратах для интраназального введения. Растворы и суспензии вводят непосредственно в носовую полость общепринятыми способами, например, с помощью капельницы, пипетки или спрея. Препараты могут находиться в форме одной или многократных доз. В последнем случае с капельницей или пипеткой это может быть достигнуто в результате введения пациентом подходящего заранее заданного объема раствора или суспензии. В случае спрея это может быть достигнуто посредством, например, измерительного распылительного насоса для спрея.
Рассматриваемые соединения могут быть задействованы в препаратах для аэрозольного введения, в частности, в дыхательные пути, включая интраназальное введение. Соединение будет, как правило, характеризоваться малым размером частиц порядка, например, пяти (5) микрон или менее. Подобный размер частиц может быть получен известными в соответствующей области способами, например, посредством тонкого измельчения. Активный ингредиент находится в упаковке под давлением совместно с подходящим газом-вытеснителем, таким как хлорфторуглероды (ХФУ), например, дихлордифторметан, трихлорфторметан или дихлортетрафторэтан, или же двуокись углерода или иной подходящий газ. Удобно также, если аэрозоль содержит поверхностно-активное вещество, такое как лецитин. Доза лекарственного средства может контролироваться с помощью измерительного клапана. Альтернативно, активные ингредиенты могут находиться в форме сухого порошка, например, порошкообразной смеси соединения в подходящей порошкообразной основе, такой как лактоза, крахмал, производные крахмала, такие как гидроксипропилметилцеллюлоза и полливинилпирролидин (ПВП). Порошкообразный носитель будет образовывать в носовой полости гель. Порошкообразная композиция может находиться в форме стандартной дозировки, например, в капсулах, или патронах из, например, желатина, или в блистерных упаковках, из которых порошок может быть введен с помощью ингалятора.
При необходимости препараты могут быть изготовлены с кишечнорастворимыми покрытиями, предназначенными для замедленного или контролируемого высвобождения активного ингредиента. Например, соединения по настоящему изобретению могут быть задействованы в устройствах для трансдермальной или подкожной доставки лекарства. Эти системы доставки имеют преимущества в тех случаях, когда необходимо замедленное высвобождение соединения и когда податливость пациента режиму лечения играет решающую роль. Соединения в системах трансдермальной доставки часто прикрепляют на прилипающую к коже твердую основу. Рассматриваемые соединения могут также сочетаться с веществом, способствующим проникновению, например, азоном (1-додецилазациклогептан-2-оном). Системы доставки с замедленным высвобождением вводят подкожно в подкожный слой хирургическим путем или посредством инъекции. Подкожные имплантаты инкапсулируют соединения в липидо-растворимой мембране, например, кремнийорганическом каучуке, или в подверженном биодеградации полимере, например, полимолочной кислоте.
Фармацевтические препараты предпочтительно находятся в форме стандартной дозировки. В подобной форме препарат разделен на стандартные дозы, содержащие соответствующие количества активного компонента. Форма стандартной дозировки может быть расфасованным препаратом, где упаковка содержит дискретные количества препарата, таким как расфасованные таблетки, капсулы, а также порошки в пузырьках или ампулах. Форма стандартной дозировки может также являться капсулой, таблеткой, облаткой ли лепешечкой самой по себе, а также может являться подходящим количеством любой из этих расфасованных форм.
Другие подходящие фармацевтические носители и их препараты описаны в справочнике Remington: The Science and Practice of Pharmacy (Наука и практика фармации) 1995, под ред. Е.W.Martin, Mack Publishing Company, 19-e издание, Истон, Пеннсильвания. Характерные примеры фармацевтических препаратов, содержащих соединение по настоящему изобретению, описаны ниже.
Дополнительные объекты, преимущества и новые свойства настоящего изобретения должны быть ясны специалистам в соответствующей области после рассмотрения нижеприведенных примеров изобретения, которые не рассматриваются как ограничивающие его.
Примеры
Список сокращений
Синтез 1: Синтез 3-(2-Метилсульфанилпиримидин-4-ил)-1H-индазола
Синтез 3-(2-метилсульфанилпиримидин-4-ил)-1H-индазол осуществляют согласно способу, показанному на схеме 1.
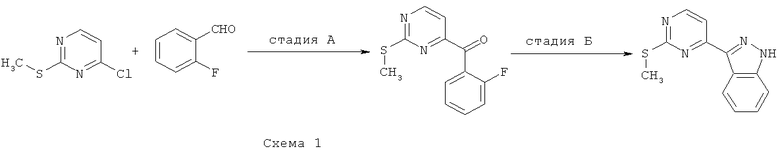
Стадия А: синтез (2-фторфенил)(2-метилсульфанилпиримидин-4-ил)-метанона
Добавляют гидрид натрия (60% дисперсия в минеральном масле, 598 мг, 14,94 ммоля) к перемешиваемому раствору 4-хлор-2-метилтиопиримидина (1,45 мл, 12,45 ммоля), 2-фторбензальдегида (1,57 мл, 14,94 ммоля) и иодида 1,3-диметилимидазолия (517 мг, 4,15 ммоля) (получаемого так, как это описано в Org. Synth. (1986) 64:9) в 1,4-диоксане (20 мл). Получаемую таким образом смесь кипятят с обратным холодильником в течение 1 часа, затем охлаждают ее и распределяют между фазами EtOAc и воды. Органический слой отделяют, сушат над Na2SO4, фильтруют и выпаривают при пониженном давлении. Сырой остаток подвергают очистке с помощью флэш-хроматографии (гексан/EtOAc, 95/5), что приводит к 840 мг (2-фторфенил)(2-метилсульфанилпиримидин-4-ил)метанона.
Аналогичным образом, используя соответствующие исходные вещества, получают следующие соединения:
(2-Фтор-3-метоксифенил)(2-метилсульфанилпиримидин-4-ил)метанон и (2,5-дифторфенил)(2-метилсульфанилпиримидин-4-ил)метанон.
Стадия Б: синтез 3-(2-метилсульфанилпиримидин-4-ил)-1H-индазола
Добавляют трет-бутилкарбазат (899 мг, 6,8 ммоля) и уксусную кислоту (0,5 мл) к смеси (2-фторфенил)(2-метилсульфанилпиримидин-4-ил)метанон (840 мг, 3,4 ммоля) в MeOH. Получаемую таким образом смесь кипятят с обратным холодильником в течение 16 часов, затем охлаждают ее до КТ и распределяют между фазами EtOAc и водного раствора гидрокарбоната натрия. Органический слой отделяют, сушат над Na2SO4, фильтруют и выпаривают при пониженном давлении. Остаток помещают в запаянную трубку с ДБУ (0,79 мл, 5,3 ммоля) и ТГФ. Получаемую таким образом смесь нагревают до температуры 150°C в микроволновом реакторе в течение 30 минут. После этого реакционную смесь выпаривают при пониженном давлении и подвергают сырой остаток очистке с помощью флэш-хроматографии (гексан/EtOAc, 4/1), что приводит к 525 мг 3-(2-метилсульфанилпиримидин-4-ил)-1H-индазола.
Аналогичным способом, используя соответствующие исходные вещества, получают следующие соединения:
7-Метокси-3-(2-метилсульфанилпиримидин-4-ил)-1H-индазол и 3-(2-Метилсульфанилпиримидин-4-ил)-7-трифторметил-1H-индазол.
Синтез 2: Синтез 1-Метил-3-(2-метилсульфанилпиримидин-4-ил)-1H-индазола
Синтез 1-метил-3-(2-метилсульфанилпиримидин-4-ил)-1H-индазол осуществляют согласно способу, показанному на схеме 2.
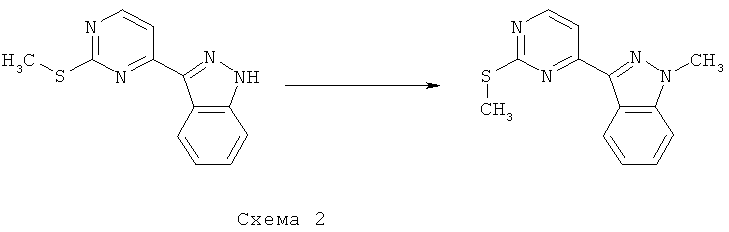
Добавляют гидрид натрия (60% суспензия в минеральном масле, 99 мг, 2,47 ммоля) к перемешиваемому раствору 3-(2-метилсульфанилпиримидин-4-ил)-1H-индазола (520 мг, 2,15 ммоля) в НМП при температуре 0°C. Смесь перемешивают в течение 15 минут, а затем добавляют метилиодид (0,14 мл, 2,25 ммоля) и перемешивают получаемую таким образом смесь в течение 2 часов. После этого реакционную смесь выливают в воду и дважды экстрагируют EtOAc. Объединенные органические фракции сушат над Na2SO4, фильтруют и выпаривают при пониженном давлении. Остаток подвергают очистке с помощью флэш-хроматографии (гексан/EtOAc, 9/1), что приводит к 415 мг 1-метил-3-(2-метилсульфанилпиримидин-4-ил)-1H-индазола.
Синтез 3: синтез 7-метокси-3-(2-метилсульфанилпиримидин-4-ил)-бензо[d]изоксазола
Синтез 7-метокси-3-(2-метилсульфанилпиримидин-4-ил)-бензо[d]изоксазола осуществляют согласно способу, показанному на схеме 3.
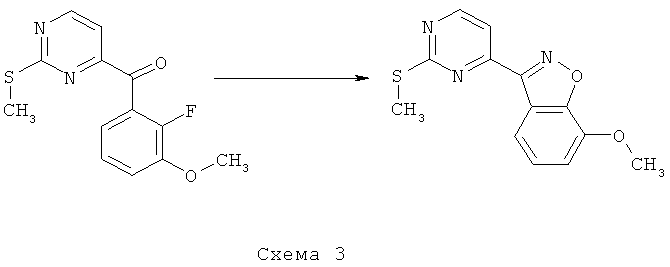
Смесь (2-фтор-3-метоксифенил)(2-метилсульфанилпиримидин-4-ил)метанона (1 г, 3,59 ммоля) и гидроксиламина (50% в воде, 1,5 мл) в этаноле кипятят с обратным холодильником в течение ночи. Получаемую таким образом смесь охлаждают до КТ, разбавляют EtOAc, а затем концентрируют при пониженном давлении. Остаток растворяют в ТГФ, добавляют ДБУ (0,84 мл, 5,6 ммоля), получаемую таким образом смесь нагревают до температуры 150°C в микроволновом реакторе в течение 30 минут. После этого реакционную смесь концентрируют при пониженном давлении и остаток подвергают очистке с помощью флэш-хроматографии (гексан/EtOAc, 95/5), что приводит к 458 мг 7-метокси-3-(2-метилсульфанилпиримидин-4-ил)бензо[d]изоксазола.
Аналогичным способом, используя соответствующие исходные вещества, получают следующие соединения:
3-(2-Метилсульфанилпиримидин-4-ил)-бензо[d]изоксазол и 5-Фтор-3-(2-метилсульфанилпиримидин-4-ил)бензо[d]изоксазол.
Синтез 4: Синтез 3-бром-1-(2-метилсульфанилпиримидин-4-ил)-1H-индазола
Синтез 3-бром-1-(2-метилсульфанилпиримидин-4-ил)-1H-индазол осуществляют согласно способу, показанному на схеме 4.
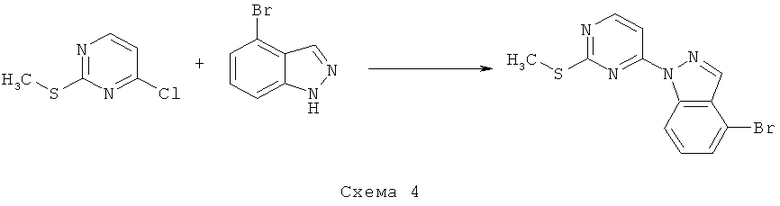
К раствору 4-бром-1H-индазола (8,831 г, 44,82 ммоля) в ДМФ (100 мл) добавляют NaH (60% суспензия в минеральном масле, 74,7 ммоля, 2,988 г), а затем 4-хлор-2-метилсульфанилпиримидин (4,34 мл, 37,35 ммоля). Получаемую таким образом смесь оставляют перемешиваться при КТ в течение 2 часов, твердый осадок собирают посредством фильтрования, промывают и сушат при пониженном давлении, что приводит к 10,7 г (89% выход) 3-бром-1-(2-метилсульфанилпиримидин-4-ил)-1H-индазола.
Аналогичным способом, используя соответствующие исходные вещества, получают следующие соединения:
1-(2-Метилсульфанилпиримидин-4-ил)-1H-индазол,
6-Метил-1-(2-метилсульфанилпиримидин-4-ил)-1H-индазол,
5-Метил-1-(2-метилсульфанилпиримидин-4-ил)-1H-индазол,
3-Метил-1-(2-метилсульфанилпиримидин-4-ил)-1H-индол,
6-Фтор-1-(2-метилсульфанилпиримидин-4-ил)-1H-индол,
4-Бензилокси-1-(2-метилсульфанилпиримидин-4-ил)-1H-индол,
5-Бензилокси-1-(2-метилсульфанилпиримидин-4-ил)-1H-индол,
1-(2-Метилсульфанилпиримидин-4-ил)-1H-индол,
1-(2-Метилсульфанилпиримидин-4-ил)-1H-индазол,
1-(2-Метилсульфанилпиримидин-4-ил)-6-нитро-1H-индазол,
1-(2-Метилсульфанилпиримидин-4-ил)-1H-пирроло[3,2-b]пиридин (твердое кристаллическое вещество белого цвета), масс-спектр (MC)=243 [M+H]+, температура плавления (tпл.)=120,1-123,0°C, и
1-(2-Метилсульфанилпиримидин-4-ил)-1H-пирроло[3,2-с]пиридин (твердое вещество желто-коричневого цвета), MC=243 [M+H]+, tпл.=167,7-169,2°C.
Синтез 5: синтез 2-бутилсульфанил-4-хлорпиримидина
Синтез 2-бутилсульфанил-4-хлорпиримидине осуществляют согласно способу, показанному на схеме 5.
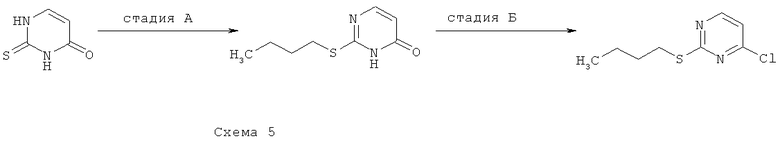
Стадия А: синтез 2-бутилсульфанил-3H-пиримидин-4-она
Добавляют 2-тиоксо-2,3-дигидро-1H-пиримидин-4-он (12,8 г) к раствору NaOH (8,0 г) в воде (70 мл). После полного растворения твердых веществ добавляют к раствору бутилиодид (12,4 мл) и перемешивают получаемую таким образом смесь при КТ в течение ночи. Затем добавляют вторую аликвоту бутилиодида (1,2 мл) и реакционную смесь перемешивают в течение 24 часов. Добавляют третью аликвоту бутилиодида (1,2 мл) и получаемую таким образом смесь перемешивают в течение 6 суток. Добавляют ледяную уксусную кислоту (5,5 мл) и перемешивают реакционную смесь в течение 30 минут, после чего выдерживают ее при температуре 4°C в течение ночи. Образующийся осадок собирают посредством фильтрования и сушат при пониженном давлении, что приводит к 12,3 г 2-бутилсульфанил-3H-пиримидин-4-ону.
Стадия Б: синтез 2-бутилсульфанил-4-хлорпиримидина
Смесь 2-бутилсульфанил-3H-пиримидин-4-она (3,0 г) и оксихлорида фосфора (15 мл) кипятят с обратным холодильником в течение 3 часов, затем охлаждают ее до КТ и выпаривают при пониженном давлении. Остаток выливают в смесь льда с водой и экстрагируют получаемую таким образом смесь ДХМ. Объединенные органические фракции промывают разбавленным водным раствором NaHCO3, сушат над Na2SO4, фильтруют и выпаривают при пониженном давлении, что приводит к 1,2 г 2-бутилсульфанил-4-хлорпиримидину.
Синтез 6: синтез 1-(2-метилсульфанилпиримидин-4-ил)-4-нитро-1H-индолу
Синтез 1-(2-метилсульфанилпиримидин-4-ил)-4-нитро-1H-индола осуществляют согласно способу, показанному на схеме 6.
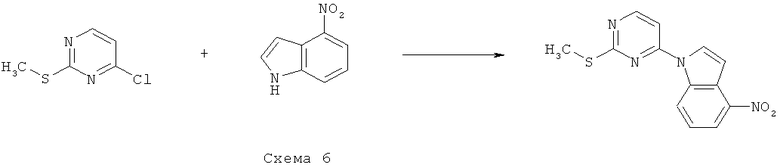
Раствор бис(триметилсилил)амида натрия (1 М в ТГФ, 26,42 мл) медленно добавляют к охлаждаемому (на бане из сухого льда с ацетоном) раствору 4-нитроиндола (4,2 г, 25,9 ммоля) в смеси ацетонитрил/ТГФ (2/1, 75 мл) и перемешивают получаемую таким образом смесь в течение 1 часа. Затем добавляют 4-хлор-4-метилтиопиримидин (4,202 г, 26,2 ммоля), реакционную смесь нагревают до КТ и перемешивают в течение 21 часа. Получаемую таким образом смесь выпаривают при пониженном давлении и растирают твердый остаток со смесью гексан/EtOAc/Et2O (5/1/1). Твердое вещество собирают посредством фильтрования, промывают и сушат при пониженном давлении, что приводит к 2,56 г 1-(2-метилсульфанилпиримидин-4-ил)-4-нитро-1H-индола, который используют без дополнительной очистки.
Аналогичным способом, используя соответствующие исходные вещества, получают следующие соединения:
Метиловый эфир 1-(2-метилсульфанилпиримидин-4-ил)-1H-индол-4-карбоновой кислоты,
4-Метокси-1-(2-метилсульфанилпиримидин-4-ил)-1H-индол,
6-Метил-1-(2-метилсульфанилпиримидин-4-ил)-1H-индол
4-Метил-1-(2-метилсульфанилпиримидин-4-ил)-1H-индол,
4-Фтор-1-(2-метилсульфанилпиримидин-4-ил)-1H-индол,
5-Фтор-1-(2-метилсульфанилпиримидин-4-ил)-1H-индол,
Метиловый эфир 1-(2-метилсульфанилпиримидин-4-ил)-1H-индол-3-карбоновой кислоты,
Метиловый эфир 1-(2-метилсульфанилпиримидин-4-ил)-1H-индол-5-карбоновой кислоты и
5-Метокси-1-(2-метилсульфанилпиримидин-4-ил)-1H-индол.
Аналогичным способом, используя соответствующие исходные вещества и LiГМДC в качестве основания, получают следующие соединения:
1-(2-Метилсульфанилпиримидин-4-ил)-1H-индол-4-карбонитрил,
Метиловый эфир 1-(2-Метилсульфанилпиримидин-4-ил)-1H-индол-6-карбоновой кислоты и
5-Метил-1-(2-метилсульфанилпиримидин-4-ил)-1H-индол.
Синтез 7: синтез гидрохлорида диметиламида 4-аминоциклогексанкарбоновой кислоты
Синтез гидрохлорида диметиламида 4-аминоциклогексанкарбоновой кислоты осуществляют согласно способу, показанному на схеме 7.
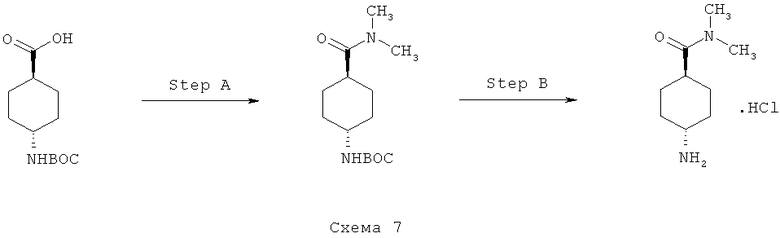
Стадия А: синтез трет-бутилового эфира (4-диметилкарбамоилциклогексил)карбаминовой кислоты
Смесь 4-трет-бутоксикарбониламиноциклогексанкарбоновой кислоты (2,43 г, 10 ммоля), ЭДКИ (6,52 г, 34 ммоля) и ГОБТ (4,59 г, 34 ммоля) в НМП (20 мл) перемешивают при КТ в течение 3 часов. Добавляют раствор диметиламина (2,0 М в ТГФ, 15 мл, 30 ммоля) и перемешивают получаемую таким образом смесь при КТ в течение 64 часов. Затем добавляют воду и EtOAc, органический слой отделяют и дважды промывают его насыщенным водным раствором K2CO3, водным раствором HCl (1 М), насыщенным водным раствором K2CO3 и соляным раствором. Органический слой сушат над Na2SO4, фильтруют и выпаривают при пониженном давлении, что приводит к трет-бутиловому эфиру (4-диметилкарбамоилциклогексил)карбаминовой кислоты (1,6 г, выход 59%) в виде твердого вещества белого цвета.
Стадия 2: синтез гидрохлорида диметиламида 4-аминоциклогексанкарбоновой кислоты
Добавляют HCl (концентрированная, 7 мл) к раствору трет-бутилового эфира (4-диметилкарбамоилциклогексил)карбаминовой кислоты (1,5 г) в 1,4-диоксане (20 мл) и перемешивают получаемую таким образом смесь при КТ в течение 5 часов. Добавляют толуол и выпаривают реакционную смесь при пониженном давлении. Маслянистый остаток растирают с EtOAc, что приводит к гидрохлориду диметиламида 4-аминоциклогексанкарбоновой кислоты (0,9 г).
Транс-(4-аминоциклогексил)морфолин-4-илметанон получают аналогичным способом, используя соответствующие исходные вещества.
Синтез 8: синтез этилового эфира транс-4-аминоциклогексанкарбоновой кислоты
Синтез этилового эфира транс-4-аминоциклогексанкарбоновой кислоты осуществляют согласно способу, показанному на схеме 8.
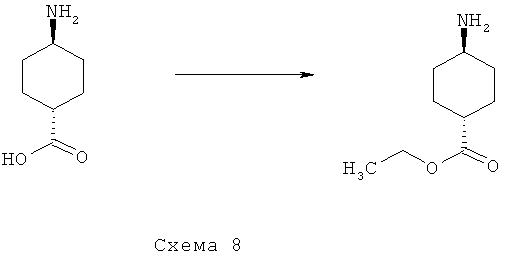
Смесь транс-4-аминоциклогексанкарбоновой кислоты (15 г, 83,8 ммоля), серной кислоты (9 мл) и EtOH (400 мл) перемешивают при КТ в течение ночи. Добавляют насыщенный водный раствор NaHCO3 и NaHCO3 (твердый) до достижения рН смеси 7. Растворитель затем отгоняют при пониженном давлении. Добавляют водный раствор K2CO3 (50%), экстрагируют получаемую таким образом смесь EtOAc, органические фракции сушат над Na2SO4, фильтруют и выпаривают при пониженном давлении, что приводит к этиловому эфиру транс-4-аминоциклогексанкарбоновой кислоты (12,81 г) в виде маслянистого вещества светло-желтого цвета.
Синтез 9: синтез 2-[1-(2-метилсульфанилпиримидин-4-ил)-1H-индол-4-ил]пропан-2-ола
Синтез 2-[1-(2-метилсульфанилпиримидин-4-ил)-1H-индол-4-ил]пропан-2-ола осуществляют согласно способу, показанному на схеме 9.
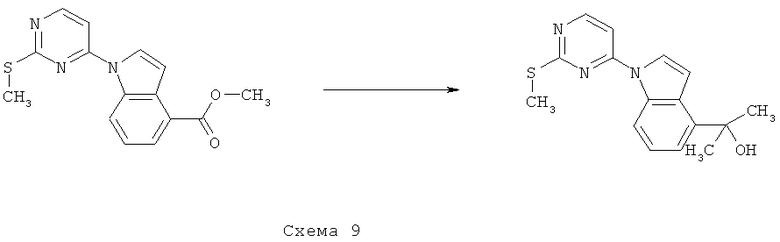
Суспензию 1-(2-метилсульфанилпиримидин-4-ил)-1H-индол-4-карбоновой кислоты метиловый эфир (500 мг) в ТГФ (6 мл) добавляют к раствору метилмагнийбромида (3,0 М в Et2O, 3,51 мл) и ТГФ (2 мл), охлаждают до температуры 0°C, и получаемую таким образом смесь перемешивают в атмосфере N2 в течение 5 часов. Затем добавляют вторую аликвоту метилмагнийбромида (3,0 М в Et2O, 1,80 мл) и перемешивают реакционную смесь при КТ в атмосфере N2 в течение 30 минут. Затем получаемую таким образом смесь охлаждают до температуры 0°C, выливают в холодную смесь вода/HCl (1 М, 1:1, 75 мл) и экстрагируют EtOAc. Объединенные органические фракции промывают один раз водой и дважды насыщенным водным раствором NaHCO3, сушат над Na2SO4, фильтруют и выпаривают при пониженном давлении, что приводит к 2-[1-(2-метилсульфанилпиримидин-4-ил)-1H-индол-4-ил]пропан-2-олу (0,48 г).
Синтез 10: синтез [6-(2,4-дифторфенокси)-1H-пиразоло[3,4-d]пиримидин-3-ил]изопропиламина
Синтез [6-(2,4-дифторфенокси)-1H-пиразоло[3,4-d]пиримидин-3-ил]-изопропиламина осуществляют согласно способу, показанному на схеме 10.

Стадия А: синтез (R)-2-boc-аминопопан-1-ола
(R)-2-Аминопропан-1-ол (30,0 г, 0,3994 моля) растворяют в MeOH (800 мл) и охлаждают раствор на ледяной бане в течение 30 минут. К перемешиваемой реакционной смеси добавляют порциями раствор ди-трет-бутилдикарбоната (87,17 г, 0,3994 моля) в MeOH (250 мл) добавляют. Удаляют ледяную баню и продолжают перемешивание в течение 3 часов. Реакционную смесь выпаривают досуха при пониженном давлении, что приводит к (R)-2-boc-аминопропан-1-олу, который используют без дополнительной очистки.
Стадия Б: синтез (R)-2-Вос-амино-1-метансульфонилпропан-1-ола
ДХМ (400 мл) и триэтиламин (83,9 мл, 0,60 моля) добавляют в колбу, содержащую (R)-2-boc-аминопропан-1-ол. Реакционную смесь охлаждают на ледяной бане в течение 30 минут и добавляют по каплям в продолжение пяти минут хлорангидрид метансульфокислоты (37,0 мл, 0,478 моля). Получаемую таким образом смесь перемешивают в течение 1 часа, удаляют ледяную баню и перемешивают смесь в течение 18 часов при КТ. Получаемую таким образом смесь промывают водным раствором NaOH (10%), органический слой сушат над MgSO4, фильтруют, и концентрируют при пониженном давлении, что приводит к (R)-2-boc-амино-1-метансульфонилпропан-1-олу (91,93 г).
Стадия В: синтез (R)-2-Вос-амино-1-метансульфанилпропана
Смесь (R)-2-boc-амино-1-метансульфонилпропан-1-ола (91,93 г, 363 ммоля), ТГФ (350 мл) и тиометоксида натрия (30,0 г, 406 ммолей) перемешивают при КТ в течение 18 часов. Смесь концентрируют при пониженном давлении и распределяют остаток между фазами Et2O и воды. Органический слой промывают соляным раствором, сушат над MgSO4, фильтруют, и концентрируют при пониженном давлении, что приводит к (R)-2-boc-амино-1-метансульфанилпропану (70,8 г).
Стадия Г: синтез (R)-1-метил-2-метилсульфанилэтиламин
Метанол (400 мл) охлаждают на ледяной бане в течение 30 минут и добавляют по каплям в продолжение 45 минут хлорангидрид уксусной кислоты (100 мл). Получаемый таким образом раствор добавляют к (R)-2-Вос-амино-1-метансульфанилпропану (70,47 г, 343 ммоля) при перемешивании при температуре ледяной бани. Реакционную смесь перемешивают в течение 15 минут по завершении добавления, удаляют ледяную баню и продолжают перемешивание в течение 2 часов. Реакционную смесь концентрируют до твердого состояния при пониженном давлении, затем переводят в ТГФ (700 мл). Получаемую таким образом смесь кипятят с обратным холодильником до растворения всех твердых веществ, затем охлаждают до КТ, после чего охлаждают до температуры 0°C в течение 30 минут. Получаемый таким образом осадок собирают посредством фильтрования, промывают холодным ТГФ и сушат при пониженном давлении, что приводит к (R)-1-метил-2-метилсульфанилэтиламину (23,57 г). MC=106 [M+H]+.
(S)-1-Метил-2-метилсульфанилэтиламин получают по вышеописанному способу, используя соответствующие исходные вещества.
Синтез 11: синтез (R)-2-Метансульфонил-1-метилэтиламина
Синтез (R)-2-метансульфонил-1-метилэтиламина осуществляют согласно способу, показанному на схеме 11.

Стадия А: синтез (R)-2-бензилоксикарбониламинопропилового эфира метансульфокислоты
Раствор бензилового эфира хлормуравьиной кислоты (10,0 мл) в ДХМ (25 мл) добавляют к перемешиваемому охлаждаемому раствору R-(-)-2-амино-1-пропанола (5,0 г) и триэтиламина (6,8 г) в ДХМ (75 мл) со скоростью, позволяющему поддерживать температуру ниже -5°C. Охлаждение и перемешивание продолжают в течение 1 часа, а затем добавляют раствор хлорангидрида метансульфокислоты (7,7 г) в ДХМ (25 мл) со скоростью, позволяющему поддерживать температуру ниже -5°C. Реакционной смеси дают нагреться до 5°C, затем дважды промывают водой (150 мл). Органический слой фильтруют над безводным сульфатом натрия и отгоняют растворитель посредством вакуумной перегонки, что приводит к 19,2 г твердого вещества белого цвета. Порцию этого вещества (15 г) растворяют в ДХМ (100 мл), и перегоняют получаемый таким образом раствор. Поддерживают объем посредством добавления метил-трет-бутилового эфира. После начала кристаллизации смесь перегоняют до объема приблизительно 75 мл и останавливают нагревание. Получаемую таким образом смесь разбавляют до объема приблизительно 150 мл посредством добавления гексана по каплям. После охлаждения собирают твердое кристаллическое вещество белого цвета, промывают его гексаном/метил-трет-бутиловым эфиром (1:1) и сушат, что приводит к (R)-2-бензилоксикарбониламинопропиловому эфиру метансульфокислоты (11,1 г).
Стадия Б: синтез бензилового эфира ((R)-2-метансульфонил-1-метилэтил)карбаминовой кислоты
К перемешиваемой смеси (R)-2-бензилоксикарбониламинопропилового эфира метансульфокислоты (11,1 г) в ацетонитриле (72 мл) добавляют водный раствор метилмеркаптида натрия (15%, 35 мл). Получаемую таким образом смесь нагревают до температуры приблизительно 55°C в течение 4-5 часов, затем охлаждают ее и отделяют нижний водный слой. Органический слой встряхивают с водой (10 мл) и снова отделяют нижний слой. Добавляют муравьиную кислоту (15 мл) к органическому слою, получаемую таким образом смесь перемешивают, и добавляют по каплям водный раствор H2O2 (30%, 12 г). После начального выделения тепла смесь охлаждают, а затем нагревают до температуры приблизительно 65°C. По прошествии 3 часов к смеси добавляют вторую аликвоту H2O2 (30%, 2 мл) и муравьиной кислоты (6 мл). Аналогичное добавление двух реагентов производят по прошествии 4 часов. По прошествии 6 часов реакционную смесь охлаждают и медленно разбавляют водой (около 200 мл). Во время добавления кристаллизуется твердое вещество белого цвета. Смесь охлаждают на ледяной бане, собирают твердое вещество посредством фильтрования, промывают водой и сушат при пониженном давлении, что приводит к бензиловому эфиру ((R)-2-метансульфонил-1-метилэтил)карбаминовой кислоты (9,2 г).
Стадия В: синтез (R)-2-метансульфонил-1-метилэтиламина
Смесь бензилового эфира ((R)-2-метансульфонил-1-метил-этил)карбаминовой кислоты (2,7 г), гидроксида палладия на угле (20%, 0,3 г) и изопропанола (30 мл) нагревают до температуры приблизительно 65°C в атмосфере азота. Добавляют к смеси в продолжение около 1 часа раствор формиата калия (2,7 г) в воде (4 мл). По прошествии 6 часов смесь охлаждают и фильтруют. Фильтрат перегоняют под вакуумом, получая вязкое маслянистое вещество, которое растворяют в изопропаноле (30 мл). Получаемый таким образом раствор фильтруют и выпаривают при пониженном давлении, что приводит к (R)-2-метансульфонил-1-метилэтиламину (1,2 г) в виде вязкого маслянистого вещества.
Синтез 12: синтез 3-триметилсиланилокси-1-(2-триметилсиланилокси-этил)пропиламина
Синтез 3-триметилсиланилокси-1-(2-триметилсиланилоксиэтил)-пропиламина осуществляют согласно способу, показанному на схеме 12.

Стадия А: синтез диметил-3-аминоглутаконата
Смесь диметилацетон-1,3-дикарбоксилата (24,55 кг), гидрокарбоната аммония (22,9 кг) и MeOH (81,8 кг) перемешивают при КТ в течение ночи. Твердые вещества удаляют посредством фильтрования, а фильтрат концентрируют при пониженном давлении. Добавляют изопропанол (27,2 кг) добавляют и отгоняют образующуюся в реакции воду посредством азеотропной перегонки. Отгоняют остаток растворителя, что приводит к диметил-3-аминоглутаконату (24,705 кг, выход 99%) в виде маслянистого вещества.
Стадия Б: синтез диметилового эфира 3-аминопентандикарбоновой кислоты
Получают разбавленный спиртовой раствор серной кислоты посредством добавления серной кислоты (20,69 кг) к изопропанолу (51,7 кг) при температуре 0-5°C. Получаемый таким образом раствору медленно добавляют к раствору трет-бутиламиноборана (9,19 кг) в ТГФ (69,3 кг), поддерживая температуру ниже -5°C. Медленно добавляют к получаемой таким образом смеси диметил-3-аминоглутаконат (18,28 кг), поддерживая температуру ниже 0°C. Получаемую таким образом реакционную смесь перемешивают при температуре между 0°C и 15°C в течение ночи, затем разлагают остатки реагентов посредством медленного добавления воды (166 кг), поддерживая при этом температуру ниже 10°C, а также рН между 5 и 7 посредством добавления концентрированного водного раствора NaOH. Затем доводят рН смеси до приблизительно 9-10 посредством добавления концентрированного водного раствора NaOH в конце процесса разложения остатков реагентов. Отделяют фазы и экстрагируют водную фазу ДХМ. Объединенные органические фракции промывают соляным раствором (20,6 кг), фильтруют через хлопковый патрон и концентрируют при пониженном давлении, что приводит к диметиловый эфир 3-аминопентандикарбоновой кислоты (17,87 кг, выход 97%).
Стадия В: синтез 3-аминопентан-1,5-диола
Раствор диметилового эфира 3-аминопентандикарбоновой кислоты (17,67 кг) в ТГФ (101,5 кг) добавляют к раствору ЛАГ (1,0 M в ТГФ, 90,2 кг) при температуре реактора в диапазоне 10-30°C и перемешивают получаемую таким образом смесь при температуре 25°C в течение 14-20 часов. Разлагают остатки реагентов посредством медленного добавления воды и ТГФ (1:1). Твердые вещества отфильтровывают и промывают смесью i-PrOH/ТГФ/ДЭА/пропиламин. Фильтрат выпаривают при пониженном давлении, что приводит к 3-аминопентан-1,5-диолу (9,55 кг, выход 72%) в виде густого маслянистого вещества.
Стадия Г: синтез 3-триметилсиланилокси-1-(2-триметилсиланилоксиэтил)пропиламина
Добавляют в реактор суспензию 3-аминопентан-1,5-диола (18,29 кг) в i-PrOH (16,91 кг) и выпаривают растворитель при пониженном давлении. Затем добавляют гексаметилдисилазан (31,985 кг) в ТГФ (55,3 кг) и триметилсилилихлорид (35,57 кг) в ТГФ (172,03 кг) и кипятят получаемую таким образом смесь с обратным холодильником в течение ночи. Реакционную смесь охлаждают, растворитель выпаривают при пониженном давлении и очищают остаток посредством перегонки (97-99°C), что приводит к 3-триметилсиланилокси-1-(2-триметилсиланилоксиэтил)пропиламину (24,36 кг).
Синтез 13: Синтез 3-(2-Метилсульфанилпиримидин-4-ил)[1,2,4]триазоло[4,3-a]пиридина
Синтез 3-(2-метилсульфанилпиримидин-4-ил)[1,2,4]триазоло[4,3-а]пиридина осуществляют согласно способу, показанному на схеме 13.
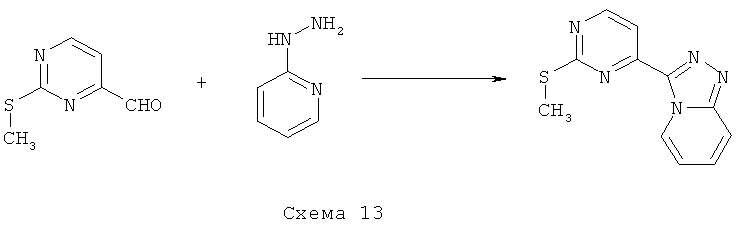
Смесь 2-метилсульфанилпиримидин-4-карбальдегида (1,00 г) (US 6218537) и 2-гидразинопиридина (0,71 г) в EtOH (20 мл) кипятят с обратным холодильником в течение 24 часов, затем охлаждают до КТ и собирают получаемое таким образом твердое вещество посредством фильтрования, что приводит к N-[1-(2-метилсульфанилпиримидин-4-ил)мет-(E)-илиден]-N'-пиридин-2-илгидразину (1,26 г). Смесь этого вещества и ДХМ (20 мл) аккуратно нагревают и добавляют вторую аликвоту ДХМ (10 мл) и хлороформа (5 мл) для облегчения солюбилизации. Добавляют диацетат иодбензола (1,65 г) и перемешивают получаемую таким образом смесь при КТ в атмосфере N2 в течение 17 часов. После этого реакционную смесь выпаривают при пониженном давлении и растирают сырой остаток с горячим Et2O. По охлаждении смеси до КТ твердое вещество собирают посредством фильтрования, что приводит к 3-(2-метилсульфанилпиримидин-4-ил)[1,2,4]триазоло[4,3-а]пиридину (1,12 г).
Синтез 14: синтез 1-(2-метилсульфанилпиримидин-4-ил)-2,3-дигидро-1H-индола
Синтез 1-(2-метилсульфанилпиримидин-4-ил)-2,3-дигидро-1H-индола осуществляют согласно способу, показанному на схеме 14.

Смесь 4-хлор-2-метилтиопиримидина (4,02 г, 25 ммоля), 2,3-дигидро-1H-индола (4,0 мл, 36 ммоля) и N,N-диизопропилэтиламина (4 мл, 22 ммоля) в ДМФ (10 мл) нагревают до температуры 100°C в атмосфере N2 в течение 16 часов. Затвердевшую реакционную смесь охлаждают, разбавляют EtOAc и фильтруют. Собранные твердые вещества растворяют в ДХМ и MeOH и перекристаллизовывают из смеси ДХМ/EtOAc, что приводит к 1-(2-метилсульфанилпиримидин-4-ил)-2,3-дигидро-1H-индолу (2,32 г, выход 38%) в виде твердого вещества светло-желтого цвета. MC=244 [М+H]+.
Пример 1: синтез транс-4-[4-(1-Метил-1H-индазол-3-ил)пиримидин-2-иламино]циклогексанола
Синтез транс-4-[4-(1-метил-1H-индазол-3-ил)пиримидин-2-иламино]циклогексанола осуществляют согласно способу, показанному на схеме 15.

Добавляют N-хлорсукцинимид (239 мг, 1,77 ммоля) к раствору 1-метил-3-(2-метилсульфанилпиримидин-4-ил)-1H-индазола (413 мг, 1,6 ммоля) в НМП и перемешивают получаемую таким образом смесь при температуре 85°C в течение 15 минут. Затем добавляют транс-аминоциклогексанол и перемешивают реакционную смесь в течение 1 часа, затем выливают ее в воду и дважды экстрагируют EtOAc. Объединенные органические фракции сушат над Na2SO4, фильтруют, и выпаривают при пониженном давлении. Сырой остаток очищают с помощью препаративной ТСХ, что приводит к транс-4-[4-(1-метил-1H-индазол-3-ил)пиримидин-2-иламино]циклогексанолу. MC=324 [M+H]+, Тпл.=180-183,3°C.
Транс-4-[4-(4-броминдазол-1-ил)пиримидин-2-иламино]циклогексанол (твердое вещество белого цвета) получают аналогичным образом, используя подходящее исходное вещество. MC=388 [M+H]+, tпл.=200,5-201,9°C.
Пример 2: синтез транс-4-[4-(7-метокси-1H-индазол-3-ил)пиримидин-2-иламино]циклогексанола
Синтез транс-4-[4-(7-метокси-1H-индазол-3-ил)пиримидин-2-иламино]циклогексанола осуществляют согласно способу, показанному на схеме 16.
Добавляют 3-хлорпероксибензойную кислоту (77%, 407 мг, 1,82 ммоля) к раствору 7-метокси-3-(2-метилсульфанилпиримидин-4-ил)-1H-индазола (225 мг, 0,8 ммоля) в ДХМ при температуре 0°C и перемешивают получаемую таким образом смесь в течение 3 часов. После этого реакционную смесь разбавляют ДХМ, промывают водным раствором NaHCO3 (5%), сушат над Na2SO4, фильтруют и выпаривают при пониженном давлении, что приводит к 3-(2-метансульфонилпиримидин-4-ил)-7-метокси-1H-индазолу, который используют без дополнительной очистки. К раствору этого вещества в НМП добавляют транс-аминоциклогексанол (190 мг), и получаемую таким образом смесь нагревают в запаянной трубке до температуры 120°C в течение 1,5 часов. Реакционную смесь охлаждают до КТ, добавляют воду и перемешивают получаемую таким образом суспензию в течение 30 минут. Твердый осадок собирают посредством фильтрования, промывают водой и сушат при пониженном давлении в течение ночи, что приводит к транс-4-[4-(7-метокси-1H-индазол-3-ил)пиримидин-2-иламино]циклогексанолу. MC=341 [M+H]+, tпл.=243,3-244,9°C.
Аналогичным способом, используя соответствующие исходные вещества, получают следующие соединения:
транс-4-[4-(1H-индазол-3-ил)пиримидин-2-иламино]циклогексанол, MC=310 [M+H]+,
транс-4-(4-бензо[d]изоксазол-3-илпиримидин-2-иламино)циклогексанол, MC=311 [M+H]+,
метиловый эфир транс-1-[2-(4-гидроксициклогексиламино)пиримидин-4-ил]-1H-индол-4-карбоновой кислоты (твердое вещество желтого цвета), MC=367 [M+H]+, tпл.=206,7-208,3°C,
циклогексил(4-индол-1-илпиримидин-2-ил)амин (кристаллическое вещество грязно-белого цвета), MC=293 [M+H]+, tпл.=192,5-193,0°C,
транс-4-(4-индол-1-илпиримидин-2-иламино)циклогексанол (твердое вещество грязно-белого цвета), MC=309 [M+H]+, tпл.=202,4-204,4°C,
транс-N-(4-индол-1-илпиримидин-2-ил)циклогексан-1,4-диамин (твердое вещество грязно-белого цвета), MC=308 [M+Н]+, tпл.=270,1-273,3°C,
транс-4-(4-индазол-2-илпиримидин-2-иламино)циклогексанол (белые иглы), MC=310 [M+H]+, tпл.=214,7-216,0°C,
транс-4-(4-индазол-1-илпиримидин-2-иламино)циклогексанол (твердое вещество белого цвета), MC=310 [M+H]+, tпл.=232,6-233,7°C,
транс-4-[4-(6-нитроиндазол-1-ил)пиримидин-2-иламино]циклогексанол (желтый порошок), MC=355 [M+H]+, tпл.=221,8-223,0°C,
транс-4-(4-[1,2,4]триазоло[4,3-а]пиридин-3-илпиримидин-2-иламино)циклогексанол (твердое вещество грязно-белого цвета), MC=311 [M+H]+, tпл.=253,4-255,7°C, and
транс-4-(4-пиразоло[1,5-b]пиридазин-3-илпиримидин-2-иламино)циклогексанол (белые иглы), (исходное вещество 3-(2-метилсульфанилпиримидин-4-ил)-пиразоло[1,5-b]пиридазин получают согласно методике, описанной в J. Med. Chem. 2004, 47, 4716-30), MC=311 [M+H]+, tпл.=214,9-215,6°C,
транс-4-(4-пирроло[3,2-b]пиридин-1-илпиримидин-2-иламино)циклогексанол (твердое вещество белого цвета), MC=310 [M+H]+, tпл.=198,9-200,3°C,
транс-4-(4-пирроло[3,2-с]пиридин-1-илпиримидин-2-иламино)циклогексанол (кристаллическое твердое вещество белого цвета), MC=310 [M+H]+, tпл.=229,9-230,2°C и
транс-4-[4-(2,3-дигидро-индол-1-ил)пиримидин-2-иламино]циклогексанол (твердое вещество белого цвета), MC=311 [M+H]+, tпл.=230,5-231,2°C.
Пример 3: синтез транс-4-[4-(7-трифторметил-1H-индазол-3-ил)пиримидин-2-иламино]циклогексанола
Синтез транс-4-[4-(7-трифторметил-1H-индазол-3-ил)пиримидин-2-иламино]циклогексанола осуществляют согласно способу, показанному на схеме 17.
Добавляют 3-хлорпербензойную кислоту (77%, 318 мг, 0,64 ммоля) к раствору 3-(2-метилсульфанилпиримидин-4-ил)-7-трифторметил-1H-индазола (200 мг, 0,64 ммоля) в ДХМ при температуре 0°C и перемешивают получаемую таким образом смесь в течение 2 часов. После этого реакционную смесь разбавляют ДХМ, промывают водным раствором NaHCO3 (5%), сушат над Na2SO4, фильтруют и выпаривают при пониженном давлении, что приводит к 3-(2-метансульфонилпиримидин-4-ил)-7-трифторметил-1H-индазолу, который используют без дополнительной очистки. К раствору этого вещества в НМП добавляют транс-аминоциклогексанол (148 мг), а затем ДИПЭА (0,11 мл, 0,64 ммоля), получаемую таким образом смесь перемешивают при температуре 120°C в течение 1 часа, затем охлаждают до КТ и добавляют воду. Получаемую таким образом суспензию перемешивают в течение 30 минут, собирают твердый осадок посредством фильтрования, промывают его водой и сушат при пониженном давлении в течение ночи. Твердое вещество промывают горячим EtOAc, фильтруют и сушат при пониженном давлении, что приводит к транс-4-[4-(7-метокси-1H-индазол-3-ил)пиримидин-2-иламино]циклогексанолу (75 мг). MC=378 [M+H]+, tпл.=265,5-270°C.
Аналогичным способом, используя соответствующие исходные вещества, получают следующие соединения:
транс-4-[4-(5-фторбензо[d]изоксазол-3-ил)пиримидин-2-иламино]циклогексанол, MC=329 [M+H]+, tпл.=172-173,9°C,
(1,4-диоксаспиро[4,5]дец-8-ил)(4-индазол-1-илпиримидин-2-ил)амин (твердое вещество белого цвета), MC=352 [M+H]+, tпл.=207,5-208,8°C,
транс-4-[4-(6-метоксииндазол-1-ил)пиримидин-2-иламино]циклогексанол (розовый порошок), MC=340 [M+H]+, tпл.=166,8-167,3°C,
транс-1-[2-(4-гидрокси-циклогексиламино)пиримидин-4-ил]-1H-индазол-6-карбонитрил (белый порошок), MC=335 [M+H]+, tпл.=212,0-212,6°C и
метиловый эфир транс-1-[2-(4-гидроксициклогексиламино)пиримидин-4-ил]-1H-индазол-6-карбоновой кислоты (светло-желтый порошок), MC=368 [M+H]+, tпл.=214,9-216,3°C.
Пример 4: синтез транс-(4-гидроксициклогексил)[4-(4-нитроиндазол-1-ил)пиримидин-2-ил]амина
Синтез транс-(4-гидроксициклогексил)[4-(4-нитроиндазол-1-ил)пиримидин-2-ил]амина осуществляют согласно способу, показанному на схеме 18.
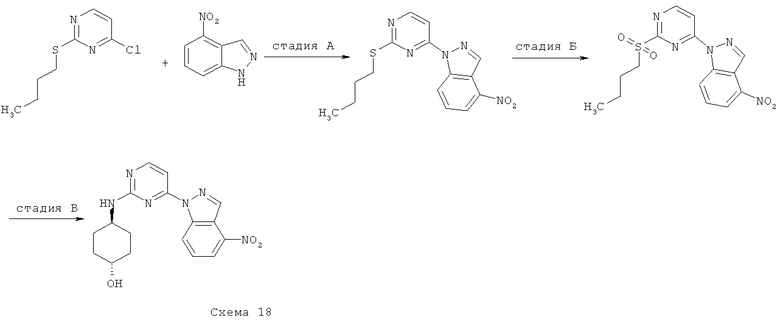
Стадия А: синтез 1-(2-бутилсульфанилпиримидин-4-ил)-4-нитро-1H-индазола
В колбу, содержащую 2-бутилсульфанил-4-хлорпиримидин (202 мг) добавляют 4-нитро-1H-индазол (228 мг), а затем NaH (60% дисперсия в минеральном масле, 84 мг) и НМП (4 мл). Получаемую таким образом смесь перемешивают при КТ в течение ночи, добавляют воду и экстрагируют смесь EtOAc. Объединенные органические фракции промывают водой, сушат над Na2SO4, фильтруют и выпаривают при пониженном давлении. Сырой остаток подвергают очистке с помощью флэш-хроматографии (гексан/EtOAc, 80:20), что приводит к 1-(2-бутилсульфанилпиримидин-4-ил)-4-нитро-1H-индазолу (156 мг).
Аналогичным способом, используя соответствующие исходные вещества, получают следующие соединения:
1-(2-бутилсульфанилпиримидин-4-ил)-1H-индазол-6-карбонитрил (исходное вещество, 1H-индазол-6-карбонитрил, получают, следуя способу, описанному в J. Med. Chem. 2000, 43(23): 4398-415),
1-(2-бутилсульфанилпиримидин-4-ил)-6-метокси-1H-индазол (исходное вещество, 6-метокси-1H-индазол, получают, следуя способу, описанному в Annalen der Chemie 1980, 908-27) и
метиловый эфир 1-(2-бутилсульфанилпиримидин-4-ил)-1H-индазол-6-карбоновой кислоты (исходное вещество, метиловый эфир 1H-индазол-6-карбоновой кислоты, получают, следуя способу, описанному в J. Med. Chem. 2000, 43, 47).
Стадия Б: синтез 1-[2-(бутан-1-сульфонил)пиримидин-4-ил]-4-нитро-1H-индазола
К раствору 1-(2-бутилсульфанилпиримидин-4-ил)-4-нитро-1H-индазола (660 мг) в ДХМ, охлажденному до температуры 0°C, добавляют метилтриоксорений (30 мг), а затем водный раствор H2O2 (30%, 1,3 мл). Реакционную смесь перемешивают при КТ в течение 2 часов, затем выливают ее в смесь воды и ДХМ. Органический слой отделяют и экстрагируют водный слой ДХМ. Объединенные органические фракции дважды промывают водным раствором Na2SO3 и один раз соляным раствором, сушат над Na2SO4, фильтруют и выпаривают при пониженном давлении, что приводит к 1-[2-(бутан-1-сульфонил)пиримидин-4-ил]-4-нитро-1H-индазолу (680 мг).
Стадия В: синтез транс-(4-гидроксициклогексил)[4-(4-нитроиндазол-1-ил)пиримидин-2-ил]амина
Смесь 1-[2-(бутан-1-сульфонил)пиримидин-4-ил]-4-нитро-1H-индазола (480 мг) и транс-аминоциклогексанола (500 мг) нагревают в неразбавленном виде до температуры 120°C. После образования осадка добавляют НМП (0,8 мл). Реакционную смесь перемешивают в течение 2 часов, затем охлаждают до КТ и выливают в смесь воды и ДХМ. Твердый осадок собирают посредством фильтрования и сушат при пониженном давлении, что приводит к транс-(4- гидроксициклогексил)[4-(4-нитроиндазол-1-ил)пиримидин-2-ил]амину (160 мг) в виде порошка желтого цвета. MC=355 [M+H]+ tпл.=253,9-254,8°C.
Пример 5: синтез (3-гидроксибутил)амида транс-4-(4-инл,азол-1-илпиримидин-2-иламино)циклогексанкарбоновой кислоты
Синтез (3-гидроксибутил)амида транс-4-(4-индазол-1-илпиримидин-2-иламино)циклогексапкарбоновой кислоты осуществляют согласно способу, показанному на схеме 19.
Смесь транс-4-(4-индазол-1-илпиримидин-2-иламино)циклогексанкарбоновой кислоты (100 мг, 0,30 ммоля), 2-амино-2-бутанола (39,2 мг, 0,44 ммоля), БОФ (194,6 мг, 0,44 ммоля) и ДИПЭА (77,4 мг, 0,60 ммоля) в ТГФ (8 мл) перемешивают при КТ в течение ночи. К получаемой таким образом суспензии добавляют воду (20 мл): твердые вещества сначала растворяется, а затем выпадает в осадок с добавлением дополнительного количества воды (100 мл). Осадок собирают посредством фильтрования, промывают водой и Et2O и сушат при пониженном давлении, что приводит к (3-гидрокси-бутил)амиду транс-4-(4-индазол-1-илпиримидин-2-иламино)циклогексанкарбоновой кислоты (78 мг) в виде твердого вещества белого цвета. MC=409 [M+H]+.
Аналогичным способом, используя соответствующие исходные вещества, получают следующие соединения:
(2-метансульфонилэтил)амид транс-4-(4-индазол-1-илпиримидин-2-иламино)циклогексанкарбоновой кислоты (твердое вещество светло-коричневого цвета), MC=443 [M+H]+, tпл.=229,6-231,2°C,
((R)-2-метансульфонил-1-метилэтил)амид транс-4-(4-индазол-1-илпиримидин-2-иламино)циклогексанкарбоновой кислоты (белый порошок), MC=457 [M+H]+, tпл.=289,0-291,0°C,
[3-гидрокси-1-(2-гидроксиэтил)пропил]амид транс-4-(4-индазол-1-илпиримидин-2-иламино)циклогексанкарбоновой кислоты (твердое вещество белого цвета), MC=439 [M+H]+, tпл.=215,0-217,8°C,
(1-гидроксиметилпропил)амид транс-4-(4-индазол-1-илпиримидин-2-иламино)циклогексанкарбоновой кислоты (белый порошок), MC=409 [M+H]+, tпл.=222,0-224,7°C,
(2,3-дигидроксипропил)амид транс-4-(4-индазол-1-илпиримидин-2-иламино)циклогексанкарбоновой кислоты (белый порошок), MC=411 [M+H]+, tпл.=243,3-245,1°C,
(3-гидрокси-1-метилпропил)амид транс-4-(4-индазол-1-илпиримидин-2-иламино)циклогексанкарбоновой кислоты (твердое вещество белого цвета), MC=409 [M+H]+, tпл.=231,9-234,4°C,
транс-(4-гидроксипиперидин-1-ил)[4-(4-индазол-1-илпиримидин-2-иламино)циклогексил]метанон (твердое вещество светло-коричневого цвета), MC=421 [M+H]+, tпл.=125,5-128,8°C,
транс-1-[4-(4-индазол-1-илпиримидин-2-иламино)циклогексанкарбонил]-пирролидин-3-он (светло-коричневый порошок) (исходный амин получают с помощью стандартного снятия защитной трет-бутилоксикарбонильной группы из коммерчески доступного трет-бутилового эфира 3-оксопирролидин-1-карбоновой кислоты), MC=405 [M+H]+, tпл.=172,6-174,8°C,
изопропиламид транс-4-(4-индазол-1-илпиримидин-2-иламино)циклогексанкарбоновой кислоты (твердое вещество белого цвета), MC=379 [М+H]+, tпл.=270,0-271,0°C,
((S)-2-гидрокси-1-метилэтил)амид транс-4-(4-индазол-1-илпиримидин-2-иламино)циклогексанкарбоновой кислоты (белый порошок), MC=395 [M+H]+, tпл.=254,6-256,9°C,
(2-метоксиэтил)амид транс-4-(4-индазол-1-илпиримидин-2-иламино)циклогексанкарбоновой кислоты (белый порошок), MC=395 [M+H]+, tпл.=216,9-219,1oC,
(2-гидрокси-1-гидроксиметилэтил)амид транс-4-(4-индазол-1-илпиримидин-2-иламино)циклогексанкарбоновой кислоты (белый порошок), MC=411 [M+H]+, tпл.=239,1-240,9°C,
транс-((R)-3-гидроксипирролидин-1-ил)[4-(4-индазол-1-илпиримидин-2-иламино)циклогексил]метанон (белый порошок), MC=407 [M+H]+, tпл.=203,3-204,0°C,
транс-((S)-3-гидроксипирролидин-1-ил)[4-(4-индазол-1-илпиримидин-2-иламино)циклогексил]метанон (твердое вещество белого цвета), MC=407 [М+H]+, tпл.=201,7-202,7°C,
(2-гидрокси-1,1-диметилэтил)амид транс-4-(4-индазол-1-илпиримидин-2-иламино)циклогексанкарбоновой кислоты (белый порошок), MC=409 [M+H]+, tпл.=256,5-259,5°C,
(2-гидрокси-2-метилпропил)амид транс-4-(4-индазол-1-илпиримидин-2-иламино)циклогексанкарбоновой кислоты (твердое вещество белого цвета) (исходный амид получают, следуя J. Med. Chem. 1998, 41, 3347), MC=409 [M+H]+, tпл.=224,2-225,9°C,
(2-гидроксиэтил)метиламид транс-4-(4-индазол-1-илпиримидин-2-иламино)циклогексанкарбоновой кислоты (твердое вещество белого цвета), MC=395 [M+H]+, tпл.=168,1-169,6°C,
((R)-1-метил-2-метилсульфанилэтил)амид транс-4-(4-индазол-1-илпиримидин-2-иламино)циклогексанкарбоновой кислоты (твердое вещество белого цвета), MC=425 [M+H]+, tпл.=215,1-218,8°C,
((S)-2-гидрокси-1,2-диметилпропил)амид транс-4-(4-индазол-1-илпиримидин-2-иламино)циклогексанкарбоновой кислоты (светло-коричневый порошок) (исходный амин синтезируют так, как это описано в международной заявке на изобретение WO 2002064594), MC=423 [M+H]+, tпл.=214,2-215,2°C,
(2-гидроксипропил)амид транс-4-(4-индазол-1-илпиримидин-2-иламино)циклогексанкарбоновой кислоты (грязно-белый порошок), MC=395 [M+H]+, tпл.=234,0-236,6°C,
((S)-1-метил-2-метилсульфанилэтил)амид транс-4-(4-индазол-1-илпиримидин-2-иламино)циклогексанкарбоновой кислоты (твердое вещество белого цвета), MC=425 [M+H]+, tпл.=221,5-222,5°C,
этил(2-гидроксиэтил)амид транс-4-(4-индазол-1-илпиримидин-2-иламино)циклогексанкарбоновой кислоты (твердое вещество белого цвета), MC=409 [M+H]+, tпл.=111,5-113,0°C,
циклопропилметиламид транс-4-(4-индазол-1-илпиримидин-2-иламино)циклогексанкарбоновой кислоты (твердое вещество белого цвета), MC=391 [M+H]+, tпл.=248,5-252,5°C и
циклопентиламид транс-4-(4-индазол-1-илпиримидин-2-иламино)циклогексанкарбоновой кислоты (белый порошок), MC=405 [M+H]+, tпл.=272,2-274,4°C.
Пример 6: синтез транс-[4-(4-индазол-1-илпиримидин-2-иламино)циклогексил]морфолин-4-илметанона
Синтез транс-[4-(4-индазол-1-илпиримидин-2-иламино)циклогексил]морфолин-4-илметанона осуществляют согласно способу, показанному на схеме 20.
В запаиваемую трубку (5 мл) помещают транс-(4-аминоциклогексил)морфолин-4-илметанон (89,3 мг, 0,36 ммоля), ДИПЭА (0,19 мл, 1,10 ммоля) и НМП (1 мл). Во вторую запаиваемую трубку (10 мл) помещают 1-(2-метансульфонилпиримидин-4-ил)-1H-индазол (100 мг, 0,36 ммоля) и НМП (1 мл). Обе смеси нагревают до температуры 120°C до полного растворения твердых веществ. Затем раствор 1-(2-метансульфонилпиримидин-4-ил)-1H-индазола переносят с помощью катетера в сосуд, содержащий смесь аминов и нагревают получаемую таким образом смесь до температуры 120°C в течение 3 часов. После этого реакционную смесь охлаждают до КТ и перемешивают в течение 2 суток, затем распределяют между фазами водного раствора NH4Cl (насыщенного) и ДХМ. Органический слой отделяют и экстрагируют водный слой ДХМ. Объединенные органические фракции сушат над Na2SO4, фильтруют и выпаривают при пониженном давлении, что приводит к маслянистому веществу янтарного цвета. Это сырое вещество перекристаллизовывают из Et2O, декантируют надосадочный раствор и промывают получаемые таким образом кристаллы Et2O. Кристаллическое вещество перекристаллизовывают из ДХМ, что приводит к 57 мг бледно-бежевого кристаллического вещества, которое подвергают очистке с помощью флэш-хроматографии (EtOAc/гексан, градиент от 50:50 к 70:30), что приводит к транс-[4-(4-индазол-1-илпиримидин-2-иламино)циклогексил]морфолин-4-ил-метанону (34 мг) в виде порошка грязно-белого цвета. MC=407 [M+H]+, tпл.=191,0-192,0°C.
Аналогичным способом, используя соответствующие исходные вещества, получают следующие соединения:
транс-4-[4-(6-метилиндазол-1-ил)пиримидин-2-иламино]циклогексанол (желтый порошок), MC=324 [M+H]+, tпл.=211,0-212,5°C,
диметиламид транс-4-(4-индазол-1-илпиримидин-2-иламино)циклогексанкарбоновой кислоты (белый порошок), MC=365 [M+H]+, tпл.=223,0-225,0°C и
транс-4-[4-(5-метилиндазол-1-ил)пиримидин-2-иламино]циклогексанол (грязно-белый порошок), MC=324 [M+H]+, tпл.=221,5-223,5°C.
Пример 7: синтез транс-1-[2-(4-гидрокси-циклогексиламино)пиримидин-4-ил]-1H-индол-4-карбоновой кислоты
Синтез транс-1-[2-(4-гидроксициклогексиламино)пиримидин-4-ил]-1H-индол-4-карбоновой кислоты осуществляют согласно способу, показанному на схеме 21.

Добавляют ЛАГ (327 мг, 13,6 ммоля) к суспензии метилового эфира 1-[2-(4-гидроксициклогексиламино)пиримидин-4-ил]-1H-индол-4-карбоновой кислоты (1,0 г, 2,7 ммоля) в смеси ТГФ (4 мл), воды (4 мл) и MeOH (1 мл). Реакционную смесь перемешивают при КТ в течение 16 часов, а затем выпаривают при пониженном давлении, что приводит к твердому веществу бежевого цвета. Добавляют к остатку водный раствор HCl (1 М, 14 мл), перемешивают получаемую таким образом смесь и выпаривают ее при пониженном давлении. Остаток растирают с MeOH, что приводит к транс-1-[2-(4-гидроксициклогексиламино)пиримидин-4-ил]-1H-индол-4-карбоновой кислоте (700 мг) в виде твердого вещества грязно-белого цвета. MC=353 [M+H]+, tпл.=259,0-261,0°C.
Пример 8: Синтез транс-4-[4-(4-аминоиндол-1-ил)пиримидин-2-иламино]циклогексанола
Синтез транс-4-[4-(4-аминоиндол-1-ил)пиримидин-2-иламино]циклогексанола осуществляют согласно способу, показанному на схеме 22.

Стадия А: синтез 1-(2-метансульфинилпиримидин-4-ил)-4-нитро-1H-индола
Суспензию 3-хлорпероксибензойной кислоты (77%, 3,11 г) в CHCl3 (40 мл) медленно добавляют при температуре 0°C в атмосфере N2 к смеси 1-(2-метилсульфанилпиримидин-4-ил)-4-нитро-1H-индола (3,93 г, 14,0 ммоля) в CHCl3 (50 мл). Получаемую таким образом смесь оставляют нагреться до КТ и перемешивают в течение ночи. После этого реакционную смесь разбавляют ДХМ (200 мл) и промывают водным раствором NaHCO3 (насыщенным). Получаемый таким образом осадок желтого цвета собирают посредством фильтрования и сушат при пониженном давлении, что приводит к 1-(2-метансульфинилпиримидин-4-ил)-4-нитро-1H-индолу (2,36 г). Маточные растворы сушат над Na2SO4, фильтруют и выпаривают при пониженном давлении, что приводит ко второй аликвоте (690 мг).
Стадия Б: синтез тронс-4-[4-(4-нитроиндол-1-ил)пиримидин-2-иламино]циклогексанола
Круглодонную колбу с 1-(2-метансульфинилпиримидин-4-ил)-4-нитро-1H-индолом (3,02 г) и транс-аминоциклогексанолом (2,87 г) помещают на масляную баню при температуре 75°C и добавляют НМП (5 мл). Получаемую таким образом смесь нагревают до температуры 105°C в течение 20 минут, а затем перемешивают при КТ в течение ночи. Получаемое таким образом твердое вещество трижды растирают со смесью гексан/Et2O (1:1, 50 мл), удаляют надосадочный раствор и выпаривают оставшийся растворитель при пониженном давлении. Твердый остаток суспендируют в воде (70 мл) при перемешивании, затем собирают посредством фильтрования и сушат при пониженном давлении, что приводит к транс-4-[4-(4-нитроиндол-1-ил)пиримидин-2-иламино]циклогексанолу (2,47 г),, который используют без дополнительной очистки.
Аналогичным образом, используя соответствующее твердое вещество, получают следующие соединения:
транс-4-[4-(4-метоксииндол-1-ил)пиримидин-2-иламино]циклогексанол (твердое вещество грязно-белого цвета), MC=339 [M+H]+, tпл.=208,0-209,8°C,
транс-4-[4-(3-метилиндол-1-ил)пиримидин-2-иламино]циклогексанол (твердое вещество белого цвета), MC=323 [M+H]+, tпл.=213,9-215,7°C,
транс-4-[4-(6-фториндол-1-ил)пиримидин-2-иламино]циклогексанол (твердое вещество белого цвета), MC=327 [M+H]+, tпл.=188,0-190,0°C,
транс-4-[4-(4-бензилоксииндол-1-ил)пиримидин-2-иламино]циклогексанол (твердое вещество светло-желтого цвета), MC=415 [M+H]+, tпл.=171,1-173,2°C,
транс-4-[4-(5-бензилоксииндол-1-ил)пиримидин-2-иламино]циклогексанол (твердое вещество грязно-белого цвета), MC=415 [M+H]+, tпл.=185,1-186,7°C,
транс-4-[4-(6-метилиндол-1-ил)пиримидин-2-иламино]циклогексанол (твердое вещество белого цвета), MC=323 [M+H]+, tпл.=207,0-208,0°C,
транс-4-[4-(4-метилиндол-1-ил)пиримидин-2-иламино]циклогексанол (твердое вещество грязно-белого цвета), MC=323 [M+H]+, tпл.=205,5-205,9°C,
транс-4-[4-(4-фториндол-1-ил)пиримидин-2-иламино]циклогексанол (твердое вещество белого цвета), MC=327 [M+H]+, tпл.=210,5-212,0°C,
транс-4-[4-(5-фториндол-1-ил)пиримидин-2-иламино]циклогексанол (твердое вещество белого цвета), MC=327 [M+H]+, tпл.=217,2-217,8°C,
метиловый эфир транс-1-[2-(4-гидроксициклогексиламино)пиримидин-4-ил]-1H-индол-3-карбоновой кислоты (твердое вещество грязно-белого цвета), MC=367 [M+H]+, tпл.=177,5-178,8°C,
метиловый эфир транс-1-[2-(4-гидроксициклогексиламино)пиримидин-4-ил]-1H-индол-5-карбоновой кислоты (твердое вещество белого цвета), MC=367 [M+H]+, tпл.=238,5-239,2°C,
транс-4-[4-(5-метоксииндол-1-ил)пиримидин-2-иламино]циклогексанол (грязно-белый порошок), MC=339 [M+H]+, tпл.=203,6-204,0°C,
транс-1-[2-(4-гидроксициклогексиламино)пиримидин-4-ил]-1H-индол-4-карбонитрил (твердое вещество грязно-белого цвета), MC=334 [M+H]+, tпл.=234,9-235,3°C,
метиловый эфир транс-1-[2-(4-гидроксициклогексиламино)пиримидин-4-ил]-1H-индол-6-карбоновой кислоты (твердое вещество светло-желтого цвета), MC=367 [M+H]+, tпл.=232,2-234,5°C,
транс-4-[4-(5-метилиндол-1-ил)пиримидин-2-иламино]циклогексанол (твердое вещество грязно-белого цвета), MC=323 [M+H]+, tпл.=202,5-203,5°C и
транс-4-{4-[4-(1-гидрокси-1-метилэтил)индол-1-ил]пиримидин-2-иламино}-циклогексанол (твердое вещество грязно-белого цвета), MC=367 [M+H]+, tпл.=168,0-169,0°C.
Стадия В: синтез транс-4-[4-(4-аминоиндол-1-ил)пиримидин-2-иламино]циклогексанола
Смесь транс-4-[4-(4-нитроиндол-1-ил)пиримидин-2-иламино]циклогексанола (2,44 г), EtOH (35 мл) и воды (11 мл) нагревают до температуры 80°C. К горячей суспензии добавляют хлорид аммония (1,46 г), а затем железо(0) (от компании Fisher, 1,55 г) и нагревают получаемую таким образом до температуры 80°C в течение 2 часов, а затем до температуры 50°C в течение еще 24 часов. Реакционную смесь фильтруют через слой целита, осадок на фильтре промывают EtOAc/MeOH (3:2) и выпаривают фильтрат при пониженном давлении. Сырой остаток подвергают очистке с помощью флэш-хроматографии (MeOH/ДХМ, градиент от 2,5:97,5 до 10:90), что приводит к транс-4-[4-(4-аминоиндол-1-ил)пиримидин-2-иламино]циклогексанолу в виде твердого вещества светло-желтого цвета. MC=324 [M+H]+, tпл.=218,9-220,1°C.
Транс-4-[4-(6-аминоиндазол-1-ил)пиримидин-2-иламино]циклогексанол получают аналогичным образом, используя подходящее исходное вещество. MC=325 [M+H]+, tпл.=236,5-237,5°C.
Пример 9: синтез транс-1-[2-(4-гидроксициклогексиламино)пиримидин-4-ил]-1H-индол-5-ола
Синтетическую методику, описанную в данном примере, осуществляют согласно способу, показанному на схеме 23.
Смесь 4-[4-(5-бензилоксииндол-1-ил)пиримидин-2-иламино]циклогексанола (70 мг) и Pd/C (15%, 38 мг) в EtOH перемешивают в атмосфере Н2 (при давлении резервуара) при КТ в течение 4 суток. После этого реакционную смесь фильтруют через слой целита, осадок на фильтре промывают EtOH и выпаривают фильтрат при пониженном давлении. Остаток подвергают очистке с помощью флэш-хроматографии (ДХМ/MeOH, 95:5), что приводит к транс-1-[2-(4-гидрокси-циклогексиламино)пиримидин-4-ил]-1H-индол-5-олу (51 мг) в виде твердого вещества грязно-белого цвета. MC=325 [M+H]+, tпл.=246,5-248,0°C.
Пример 10: синтез транс-4-[4-(4-гидроксиметилиндол-1-ил)пиримидин-2-иламино]циклогексанола
Синтез транс-4-[4-(4-гидроксиметилиндол-1-ил)пиримидин-2-иламино]циклогексанола осуществляют согласно способу, показанному на схеме 24.
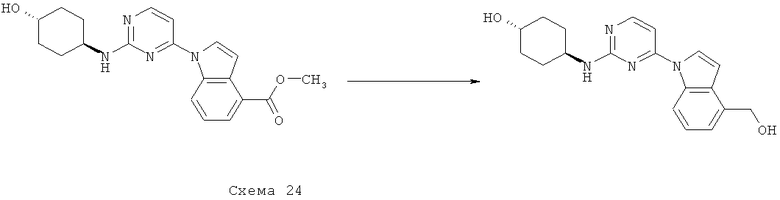
Раствор ЛАГ (1M в ТГФ, 0,57 мл) медленно добавляют при температуре -10°C в атмосфере аргона к раствору метилового эфира 1-[2-(4-гидроксициклогексиламино)пиримидин-4-ил]-1H-индол-4-карбоновой кислоты (100 мг) в ТГФ (0,3 мл). Получаемую таким образом смесь перемешивают при температуре -10°C в течение 30 минут и при КТ в течение ночи. После этого реакционную смесь охлаждают до температуры 0°C и добавляют воду (20 мкл), а затем водный раствор NaOH (15%, 20 мкл) и вторую аликвоту вода (70 мкл). Получаемую таким образом смесь перемешивают в течение 30 минут, а затем фильтруют через слой целита. Растирают неорганические соли, добавляют к фильтрату надосадочный раствор и выпаривают раствор при пониженном давлении, что приводит к транс-4-[4-(4-гидроксиметилиндол-1-ил)пиримидин-2-иламино]циклогексанолу (32,5 г, выход 35%) в виде твердого вещества белого цвета. MC=339 [M+H]+, tпл.=212,0-213,3°C.
Транс-4-[4-(5-гидроксиметилиндол-1-ил)пиримидин-2-иламино]циклогексанол (твердое вещество белого цвета) получают аналогичным способом, используя соответствующие исходные вещества. MC=339 [M+H]+, tпл.=232,0-233,9°C.
Пример 11: синтез транс-4-(4-индазол-1-илпиримидин-2-иламино)циклогексанкарбоновой кислоты
Синтез транс-4-(4-индазол-1-илпиримидин-2-иламино)циклогексанкарбоновой кислоты осуществляют согласно способу, показанному на схеме 25.
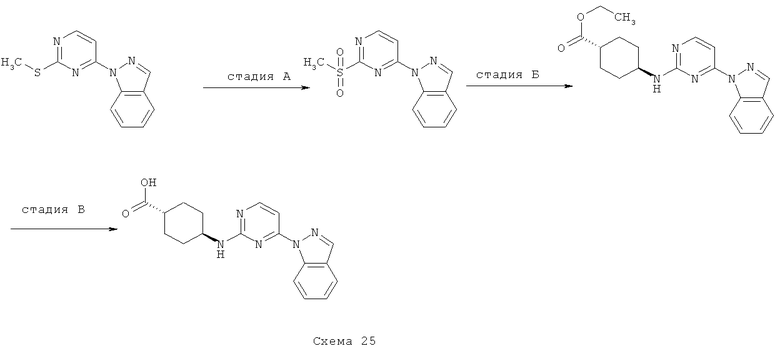
Стадия А: синтез 1-(2-метансульфонилпиримидин-4-ил)-1H-индазола
Добавляют 3-хлорпероксибензойную кислоту (77%, 14,97 г, 86,7 ммоля) к раствору 1-(2-метилсульфанилпиримидин-4-ил)-1H-индазола (10 г, 41,3 ммоля) в CHCl3 (50 мл), охлаждаемому на бане из льда с ацетоном, и перемешивают получаемую таким образом смесь в течение ночи. Затем реакционную смесь промывают водным раствором бисульфита натрия (10%). Получаемое таким образом твердое вещество отфильтровывают и отделяют фазы фильтрата. Водный слой экстрагируют ДХМ, объединенные органические фракции промывают водным раствором NaOH, водным раствором NaHCO3 (насыщенным), сушат над MgSO4, фильтруют и выпаривают при пониженном давлении, что приводит к 1-(2-метансульфонилпиримидин-4-ил)-1H-индазолу (6,4 г) в виде твердого вещества оранжевого цвета, используемого без дальнейшей очистки. MC=275 [M+H]+, tпл.=177,0-181,1°C.
Аналогичным способом, используя соответствующие исходные вещества, получают следующие соединения:
1-(2-метансульфонилпиримидин-4-ил)-6-метил-1H-индазол и
1-(2-метансульфонилпиримидин-4-ил)-5-метил-1H-индазол.
Стадия Б: синтез этилового эфира транс-4-(4-индазол-1-илпиримидин-2-иламино)циклогексанкарбоновой кислоты
1-(2-Метансульфонилпиримидин-4-ил)-1H-индазол (6,4 г) суспендируют в ТГФ и нагревают смесь до температуры 60°C до полного растворения твердых веществ. Добавляют этиловый эфир транс-4-аминоциклогексанкарбоновой кислоты (7,99 г, 46,6 ммоля), а затем ТЭА (9,7 мл, 69,9 ммоля) и перемешивают получаемую таким образом смесь в течение ночи. Затем реакционную смесь концентрируют при пониженном давлении до начала выпадения твердых веществ в осадок, после чего нагревают до температуры 60°C в течение 24 часов. Добавляют ТГФ (4 мл) и перемешивают реакционную смесь в течение 64 часов при температуре 60°C. Получаемую таким образом смесь экстрагируют EtOAc, объединенные органические фракции промывают водой, сушат над MgSO4, фильтруют и выпаривают при пониженном давлении. Сырой остаток подвергают очистке с помощью флэш-хроматографии (ДХМ/MeOH, 98:2) и растирают получаемое таким образом твердое вещество коричневого цвета с Et2O, что приводит к этиловому эфиру транс-4-(4-индазол-1-илпиримидин-2-иламино)циклогексанкарбоновой кислоты (2,883 г) в виде твердого вещества белого цвета.
Стадия В: синтез транс-4-(4-индазол-1-илпиримидин-2-иламино)циклогексанкарбоновой кислоты
Смесь этилового эфира транс-4-(4-индазол-1-илпиримидин-2-иламино)циклогексанкарбоновой кислоты (2,88 г, 7,9 ммоля), водного раствора NaOH (2 М, 65 мл) и ТГФ (65 мл) перемешивают в течение ночи. Добавляют MeOH (40 мл) и перемешивают реакционную смесь в течение еще 105 минут. Добавляют водный раствор HCl (1М) до достижения рН 3 и собирают получаемое таким образом твердое вещество посредством фильтрования, что приводит к транс-4-(4-индазол-1-илпиримидин-2-иламино)циклогексанкарбоновой кислоте (2,739 г) без дополнительной очистки.
Пример 12: синтез транс-N-[4-(4-индазол-1-илпиримидин-2-иламино)циклогексил]метансульфамида
Синтез транс-N-[4-(4-индазол-1-илпиримидин-2-иламино)циклогексил]-метансульфамида осуществляют согласно способу, показанному на схеме 26.
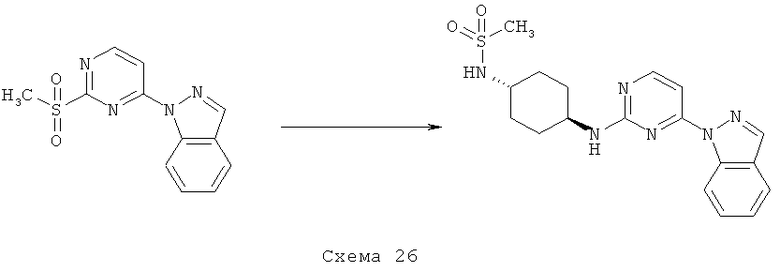
Смесь транс-циклогексан-1,4-диамина (0,62 г, 5,5 ммоля) и НМП (10 мл) нагревают до температуры 120°C в течение 5 минут, затем добавляют по каплям 1-(2-метансульфонилпиримидин-4-ил)-1H-индазол (300 мг) в НМП (10 мл) и перемешивают получаемую таким образом смесь в течение 15 минут. Медленно добавляют ангидрид метансульфокислоты (1,88 г, 10,8 ммоля) и перемешивают реакционную смесь в течение 30 минут. Растворитель выпаривают посредством перегонки под вакуумом, к остатку добавляют EtOAc и промывают получаемый таким образом раствор водой. Органический слой сушат над Na2SO4, фильтруют и выпаривают при пониженном давлении. Сырой остаток очищают посредством растирания с горячей смесью ДХМ/MeOH (90:10), что приводит к транс-N-[4-(4-индазол-1-илпиримидин-2-иламино)циклогексил]метансульфамиду (38,0 мг) в виде твердого вещества белого цвета. MC=387 [M+H]+, tпл.=269,6-270,4°C.
Пример 13: Синтез 4-(4-индазол-1-илпиримидин-2-иламино)-1-метилциклогексанола
Синтез 4-(4-индазол-1-илпиримидин-2-иламино)-1-метилциклогексанола осуществляют согласно способу, показанному на схеме 27.
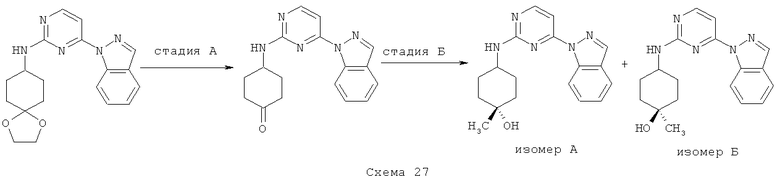
Стадия А: синтез 4-(4-индазол-1-илпиримидин-2-иламино)циклогексанона
Добавляют водный раствор HCl (3M, 40 мл) к (1,4-диоксаспиро[4,5]дец-8-ил)(4-индазол-1-илпиримидин-2-ил)амину (1,89 г, 5 ммоля) в ТГФ (40 мл) и нагревают получаемую таким образом смесь до температуры 80°C в течение 1 часа. Реакционную смесь охлаждают, выливают в смесь льда и водного раствора NaOH (1 M, 120 мл) и фильтруют получаемую таким образом суспензию. Собранные твердые вещества промывают водой, переводят в смесь ДХМ/MeOH и выпаривают часть растворителя при пониженном давлении. Получаемую таким образом суспензию фильтруют, собранные твердые вещества промывают гексанами и сушат при пониженном давлении, что приводит к 4-(4-индазол-1-илпиримидин-2-иламино)циклогексанону (1,57 г, выход 95%) в виде порошка белого цвета. MC=308 [M+H]+, tпл.=211,8-213,2°C.
Стадия Б: синтез 4-(4-индазол-1-илпиримидин-2-иламино)-1-метил-циклогексанола
Раствор метилмагнийхлорида (3 М в ТГФ, 2,4 мл, 7,2 ммоля) медленно добавляют к суспензии 4-(4-индазол-1-илпиримидин-2-иламино)циклогексанона (600 мг, 2 ммоля) в ТГФ (100 мл) при температуре -78°C в атмосфере N2. Реакционную смесь перемешивают в течение 15 минут, добавляют вторую аликвоту метилмагнийхлорида (3 М в ТГФ, 1,0 мл, 3 ммоля) и перемешивают получаемую таким образом смесь при температуре -78°C в течение 30 минут. После этого реакционную смесь нагревают до КТ и перемешивают в течение 4 часов, затем выливают в смесь льда и водного раствора NH4Cl (насыщенного). Получаемую таким образом смесь экстрагируют EtOAc, объединенные органические фракции промывают соляным раствором, сушат над MgSO4, фильтруют, и выпаривают при пониженном давлении, что приводит к твердому веществу белого цвета (609 мг, выход 96%). Это сырое вещество дважды подвергают очистке с помощью флэш-хроматографии (MeOH/ДХМ, градиент от 3:97 к 5:95,), что приводит к 4-(4-индазол-1-илпиримидин-2-иламино)-1-метил-циклогексанолу (123 мг, 19% выход менее полярного изомера в виде твердого вещества белого цвета, и 360 мг, 57% выход более полярного изомера в виде твердого вещества белого цвета). Оба изомера перекристаллизовывают из смеси ДХМ/гексаны (приблизительно 1:1), что приводит менее полярному изомеру А в виде твердого вещества белого цвета (100 мг) и более полярному изомеру Б в виде сыпучего твердого кристаллического вещества (290 мг).
Изомер А: MC=324 [М+H]+, tпл.=211,9-213,3°C.
Изомер Б: MC=324 [M+H]+, tпл.=168,1-170,1°C.
Пример 14: синтез транс-4-[4-(4-фенилметансульфонилиндол-1-ил)пиримидин-2-иламино]циклогексанола
Синтез транс-4-[4-(4-фенилметансульфонилиндол-1-ил)пиримидин-2-иламино]циклогексанола осуществляют согласно способу, показанному на схеме 28
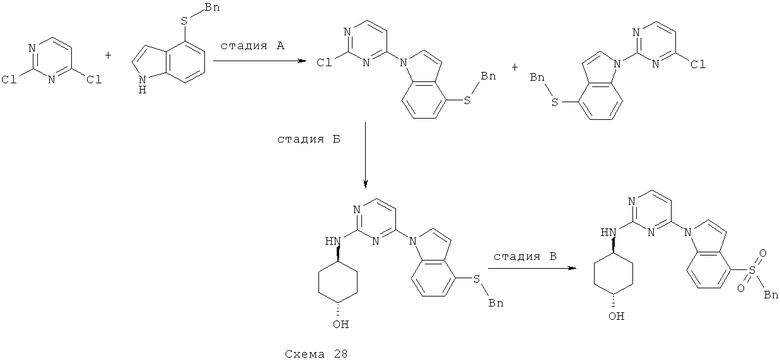
Стадия А: синтез 4-бензилсульфанил-1-(2-хлорпиримидин-4-ил)-1H-индола и 4-бензилсульфанил-1-(4-хлорпиримидин-2-ил)-1H-индола
Медленно добавляют 4-бензилсульфанил-1H-индол (5,00 г, 21 ммоля) (Can. J. Chem. 1962, 40, 511) к суспензии NaH (60% дисперсия в минеральном масле, 1,37 г, 34 ммоля) в ДМФ (40 мл) при температуре 0°C. Реакционную смесь перемешивают в течение 15 минут до прекращения выделения газа, после этого добавляют порциями 2,4-дихлорпиримидин (3,03 г, 20 ммоля), а затем дополнительную аликвоту ДМФ (10 мл). Получаемую таким образом смесь медленно нагревают до КТ, затем выливают в измельченный лед и воду и экстрагируют EtOAc. Органический слой дважды промывают смесью воды и соляного раствора, сушат над Na2SO4, фильтруют и выпаривают при пониженном давлении. Сырой остаток подвергают очистке с помощью флэш-хроматографии (EtOAc/гексаны, градиент от 10:90 к 50:50), что приводит к 4-бензилсульфанил-1-(4-хлорпиримидин-2-ил)-1H-индолу (979 мг, 14% выход) в виде воскообразного твердого вещества бледно-желтого цвета и смеси 4-бензилсульфанил-1-(2-хлорпиримидин-4-ил)-1H-индола и дизамещенного продукта (691 мг) после осаждения из смеси ДХМ/EtOAc. Данную смесь заново подвергают очистке с помощью флэш-хроматографии (EtOAc/гексаны, 20:80), что приводит к 4-бензилсульфанил-1-(2-хлорпиримидин-4-ил)-1H-индолу (270 мг, выход 4%) в виде твердого вещества грязно-белого цвета. MC=352 [M+H]+, tпл.=159,0-161,2°C.
Стадия Б: синтез транс-4-[4-(4-бензилсульфанилиндол-1-ил)пиримидин-2-иламино]циклогексанола
Добавляют транс-4-аминоциклогексанол (1,37 г, 12 ммоля) к раствору 4-бензилсульфанил-1-(2-хлорпиримидин-4-ил)-1H-индола и дизамещенного продукта (673 мг, 2 ммоля) в НМП (10 мл) и нагревают получаемую таким образом смесь до температуры 110°C в течение 4 часов. Реакционную смесь охлаждают и распределяют между фазами воды и EtOAc, органический слой дважды промывают водой, сушат над Na2SO4, фильтруют, и выпаривают при пониженном давлении. Сырой остаток подвергают очистке с помощью флэш-хроматографии (EtOAc/гексаны, градиент от 10:90 к 20:80), что приводит после перекристаллизации из смеси ДХМ/гексаны/EtOAc к транс-4-[4-(4-бензилсульфанил-индол-1-ил)пиримидин-2-иламино]циклогексанолу (309 мг, 38% выход) в виде твердого вещества белого цвета. Дополнительную аликвоту продукта выделяют из маточного раствора. MC=431 [M+H]+, tпл.=150,2-150,9°C.
Стадия В: синтез транс-4-[4-(4-фенилметансульфонилиндол-1-ил)пиримидин-2-иламино]циклогексанола
Добавляют 3-хлорпероксибензойную кислоту (0,55 г, 2,5 ммоля) к транс-4-[4-(4-бензилсульфанилиндол-1-ил)пиримидин-2-иламино]циклогексанолу (397 мг, 0,9 ммоля) в хлороформе (25 мл) и перемешивают получаемую таким образом смесь при КТ в течение 1 часа. Добавляют вторую аликвоту 3-хлорпероксибензойной кислоты (150 мг), реакционную смесь нагревают в течение короткого времени, затем охлаждают и концентрируют при пониженном давлении. Остаток распределяют между фазами водного раствора NaHCO3 и EtOAc. Органический слой отделяют, дважды промывают водным раствором NaHCO3 и водой, сушат над Na2SO4, фильтруют, и выпаривают при пониженном давлении на силикагель. Остаток подвергают очистке с помощью флэш-хроматографии (MeOH/ДХМ, 5:95), что приводит к транс-4-[4-(4-фенилметансульфонилиндол-1-ил)пиримидин-2-иламино]циклогексанолу (30 мг, 7% выход) в виде твердого вещества светло-желтого цвета. MC=463 [M+H]+, tпл.=132,1-134,5°C.
Пример 15: синтез транс-4-[4-(1H-индол-3-ил)пиримидин-2-иламино]циклогексанол
Синтез транс-4-[4-(1H-индол-3-ил)пиримидин-2-иламино]циклогексанола осуществляют согласно способу, показанному на схеме 29.
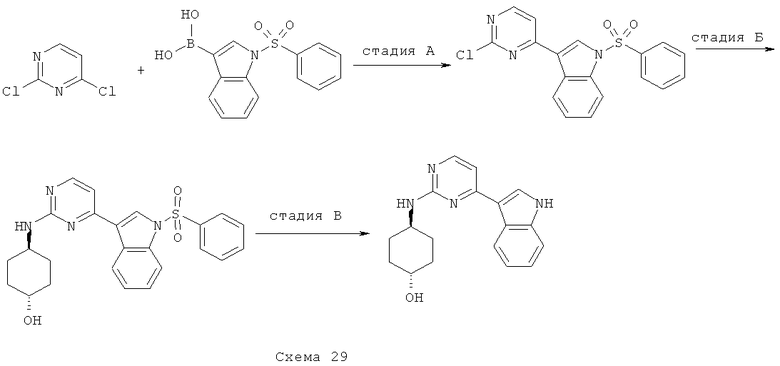
Стадия А: синтез 1-бензолсульфонил-3-(2-хлорпиримидин-4-ил)-1H-индола
Раствор 2,4-дихлорпиримидина (2,42 г, 16 ммоля), 1-(фенилсульфонил)-1H-индол-3-илбороновой кислоты (3,89 г, 13 ммоля) и карбоната натрия (5,03 г, 47 ммоля) в смеси ацетонитрил/вода (2:1, 60 мл) обезгаживают с помощью аргона, а затем добавляют тетракис(трифенилфосфин)палладий (300 мг, 1,6 моля). Получаемую таким образом смесь кипятят с обратным холодильником в течение 2 часов, в ходе чего образуется суспензия белого цвета. Осадок охлаждают и фильтруют через тампон из целита/силикагеля. Осадок на фильтре промывают EtOAc, органический слой отделяют, промывают соляным раствором, сушат над Na2SO4, фильтруют, и выпаривают при пониженном давлении. Остаток перекристаллизовывают из смеси EtOAc/гексаны, что приводит к 1-бензолсульфонил-3-(2-хлорпиримидин-4-ил)-1H-индолу в виде твердого вещества светло-розового цвета. Твердый фильтрат из реакционной смеси растворяют в ДХМ, сушат над Na2SO4, фильтруют и объединяют с предшествующей порцией продукта. Собранные твердые вещества растворяют в ДХМ, растворитель выпаривают при пониженном давлении, добавляют EtOAc с целью образования суспензии, продукт фильтруют и промывают гексанами, что приводит к 1-бензолсульфонил-3-(2-хлорпиримидин-4-ил)-1H-индолу (2,0 г, 33% выход) в виде твердого вещества светло-розового цвета. Дополнительные 0,45 г продукта получают из маточных растворов.
Стадия Б: синтез транс-4-[4-(1-бензолсульфонил-1H-индол-3-ил)пиримидин-2-иламино]циклогексанола
Раствор 1-бензолсульфонил-3-(2-хлорпиримидин-4-ил)-1H-индола (0,45 г, 1 ммоля) и транс-4-аминоциклогексанола (0,50 г, 4 ммоля) в НМП (8 мл) нагревают до температуры 120°C в течение 3 часов в атмосфере N2. Смесь затем охлаждают и распределяют между фазами воды и EtOAc, органический слой отделяют, дважды промывают соляным раствором, сушат над Na2SO4, фильтруют и выпаривают при пониженном давлении. Сырой остаток подвергают очистке с помощью флэш-хроматографии (MeOH/ДХМ, 5:95), что приводит к 405 мг пены желтого цвета. Этого вещества растворяют в EtOAc, дважды промывают водой, сушат над Na2SO4, фильтруют и выпаривают при пониженном давлении. Твердый остаток растирают с гексанами и сушат под высоким вакуумом, что приводит к транс-4-[4-(1-бензолсульфонил-1H-индол-3-ил)пиримидин-2-иламино]циклогексанолу (365 мг, выход 19%) в виде аморфного твердого вещества светло-желтого цвете. MC=449 [M+H]+, tпл.=132,1-133,3°C.
Стадия В: синтез транс-4-[4-(1H-индол-3-ил)пиримидин-2-иламино]циклогексанола
Добавляют NaOH (200 мг, 5 ммоля) к раствору транс-4-[4-(1-бензолсульфонил-1H-индол-3-ил)пиримидин-2-иламино]циклогексанола (296 мг, 0,66 ммоля) в MeOH (15 мл) и перемешивают получаемую таким образом смесь при КТ в течение 6 часов. Реакционную смесь концентрируют при пониженном давлении, остаток нейтрализуют посредством добавления водного раствора HCl (1 М), а затем распределяют между фазами воды и EtOAc. Органический слой отделяют, промывают водой, сушат над Na2SO4, фильтруют и выпаривают при пониженном давлении, что приводит к сырому твердому веществу желтого цвета. Это сырое вещество подвергают очистке с помощью флэш-хроматографии (MeOH/ДХМ, 5:95), что приводит к транс-4-[4-(1H-индол-3-ил)пиримидин-2-иламино]циклогексанолу (115 мг, выход 57%) в виде порошка белого цвета. MC=309 [M+H]+, tпл.=258,1-259,9°C.
Пример 16: синтез транс-1-[2-(4-гидроксициклогексиламино)-пиримидин-4-ил]-1,2-дигидроиндазол-3-она
Синтез транс-1-[2-(4-гидроксициклогексиламино)пиримидин-4-ил]-1,2-дигидроиндазол-3-она осуществляют согласно способу, показанному на схеме 30.
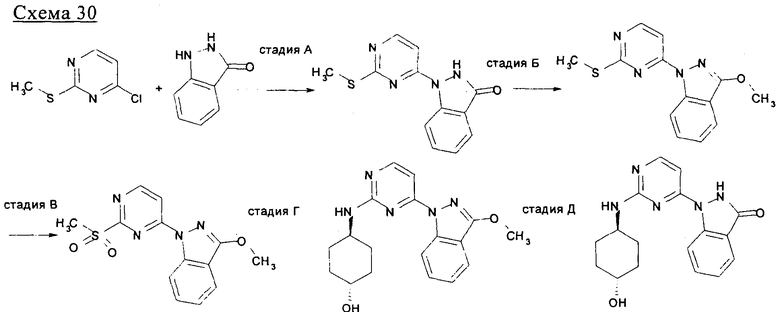
Стадия А: синтез 1-(2-метилсульфанилпиримидин-4-ил)-1,2-дигидроиндазол-3-она
3-Индазолинон (5,07 г, 38 ммоля) добавляют порциями при температуре 0°C к суспензии NaH (~60% дисперсия в минеральном масле, 2,18 г, 55 ммоля) в ДМФ (50 мл) и перемешивают получаемую таким образом смесь в течение 20 минут, до прекращения выделения газа. Добавляют 4-Хлор-2-метилтиопиримидин (6,10 г, 38 ммоля) и нагревают реакционную смесь до КТ, а затем нагревают до температуры 100°C в течение 3 часов. Получаемую таким образом смесь охлаждают, выливают в воду (150 мл), нейтрализуют водным раствором HCl (1 М) и добавляют EtOAc. Получаемую таким образом нерастворимую суспензию фильтруют и сушат, что приводит к 1-(2-метилсульфанилпиримидин-4-ил)-1,2-дигидроиндазол-3-ону (7,05 г, 72% выход) в виде твердого вещества грязно-белого цвета. MC=259 [M+H]+, tпл.=277,0-280,9°C.
Стадия Б: синтез 3-метокси-1-(2-метилсульфанилпиримидин-4-ил)-1H-индазола
Добавляют NaH (~60% дисперсия в минеральном масле, 0,75 г, 19 ммоля) к 1-(2-метилсульфанилпиримидин-4-ил)-1,2-дигидроиндазол-3-ону (2,70 г, 10 ммоля) в ДМФ (30 мл) при температуре 0°C и перемешивают получаемую таким образом смесь до прекращения выделения газа. Добавляют иодметан (0,90 мл, 14 ммоля), смесь перемешивают в течение 30 минут, а затем выливают в воду (100 мл) и экстрагируют EtOAc (100 мл). Объединенные органические фракции дважды промывают водой (100 мл), сушат над Na2SO4, фильтруют, и выпаривают при пониженном давлении. Сырой остаток подвергают очистке с помощью флэш-хроматографии (EtOAc/гексаны, градиент от 20:80 к 50:50) и перекристаллизовывают из гексанов, что приводит к 3-метокси-1-(2-метилсульфанилпиримидин-4-ил)-1H-индазолу (286 мг, 10% выход) в виде твердого вещества белого цвета. MC=272,9 [M+H]+.
Стадия В: синтез 1-(2-метансульфонилпиримидин-4-ил)-3-метокси-1H-индазола
Добавляют 3-хлорпероксибензойную кислоту (приблизительно 77%, 2,86 г, 18 ммоля) к раствору 3-метокси-1-(2-метилсульфанилпиримидин-4-ил)-1H-индазола (1,34 г, 5 ммоля) в хлороформе (30 мл) и перемешивают реакционную смесь при КТ в течение 20 минут. Получаемую таким образом смесь выливают в водный раствор NaHCO3 и перемешивают в течение 1 часа, а затем экстрагируют ДХМ. Объединенные органические фракции дважды промывают водным раствором NaHCO3 и водой, сушат над Na2SO4, фильтруют и выпаривают при пониженном давлении. Сырой остаток подвергают очистке с помощью флэш-хроматографии (EtOAc/гексаны, градиент от 50:50 к 100:0) и перекристаллизовывают из смеси ДХМ/гексаны/EtOAc, что приводит к двум порциям 1-(2-метансульфонилпиримидин-4-ил)-3-метокси-1H-индазола в виде твердого вещества грязно-белого цвета (927 мг, выход 62%). Дополнительное количество продукта выделяют из маточного раствора в виде твердого вещества желтого цвета (0,44 г, выход 29%). MC=305 [M+H]+, tпл.=206,3-207,8°C.
Стадия Г: синтез транс-4-[4-(3-метоксииндазол-1-ил)пиримидин-2-иламино]циклогексанола
Добавляют транс-4-Аминоциклогексанол (870 мг, 8 ммоля) к 1-(2-метансульфонилпиримидин-4-ил)-3-метокси-1H-индазолу (440 мг, 1 ммоля) в НМП (15 мл) и нагревают получаемую таким образом смесь до температуры 120°C в течение 3 часов. Затем реакционную смесь охлаждают и распределяют между фазами EtOAc и воды. Органический слой отделяют, промывают водой, сушат над Na2SO4, фильтруют, и концентрируют при пониженном давлении, что приводит к сырому твердому веществу желтого цвета. Это сырое вещество подвергают очистке с помощью флэш-хроматографии (MeOH/ДХМ, 5:95) и перекристаллизовывают из смеси ДХМ/гексаны, что приводит к транс-4-[4-(3-метоксииндазол-1-ил)пиримидин-2-иламино]циклогексанолу (181 мг (37% выход) в виде твердого вещества белого цвета. MC=340 [M+H]+, tпл.=202,3-205,1°C.
Стадия Д: синтез транс-1-[2-(4-гидроксициклогексиламино)пиримидин-4-ил]-1,2-дигидро-индазол-3-она
Триметилсилилиодид (0,80 мл, 5,6 ммоля) добавляют в атмосфере N2 к суспензии транс-4-[4-(3-метоксииндазол-1-ил)пиримидин-2-иламино]циклогексанола (251 мг, 0,7 ммоля) в хлороформе (10 мл). Реакционная смесь сразу становится гомогенной, но в течение 1 часа образует суспензию, и получаемую таким образом смесь перемешивают в течение 3 суток. Добавляют дополнительную аликвоту триметилсилилиодида (2 мл, 14 ммоля), кипятят получаемую таким образом смесь с обратным холодильником в течение ночи и перемешивают при КТ в течение суток. После этого реакционную смесь выливают в смесь льда с водой и экстрагируют ДХМ. Нерастворимые твердые вещества отфильтровывают и растворяют в смеси MeOH и ДХМ, объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении на силикагель. Остаток подвергают очистке с помощью флэш-хроматографии (MeOH/ДХМ, градиент от 3:97 к 5:95), что приводит к транс-1-[2-(4-гидроксициклогексиламино)пиримидин-4-ил]-1,2-дигидроиндазол-3-ону (71 мг, 30% выход) в виде твердого вещества грязно-белого цвета. MC=326 [M+H]+, tпл.=263,5-264,4°C.
Пример 17: синтез транс-4-[4-(1-Метил-1H-индол-3-ил)пиримидин-2-иламино]циклогексанола
Описанную в данном примере синтетическую методику осуществляют согласно способу, показанному на схеме 31.
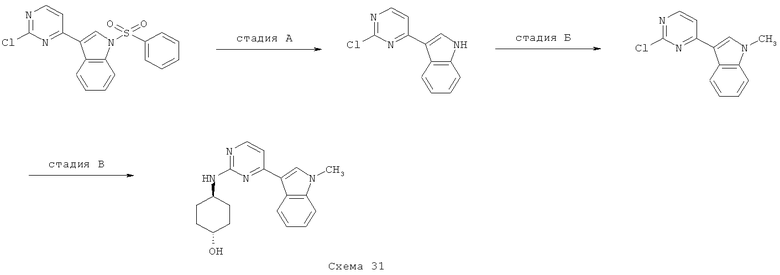
Стадия А: синтез 3-(2-хлорпиримидин-4-ил)-1H-индола
Суспензию свежеизмельченного NaOH (740 мг) в MeOH (20 мл) добавляют при температуре 0°C к суспензии 1-бензолсульфонил-3-(2-хлорпиримидин-4-ил)-1H-индола (0,91 г, 2 ммоля) в смеси MeOH/ТГФ (1:1, 40 мл). Реакционную смесь нагревают до КТ и перемешивают в течение 2 часов до достижения гомогенности. Получаемую таким образом смесь нейтрализуют водным раствором HCl (1 М) и концентрируют при пониженном давлении. Остаток распределяют между фазами воды и EtOAc, органический слой отделяют, промывают водой, сушат над Na2SO4, фильтруют, и концентрируют при пониженном давлении, что приводит к твердому веществу ярко-желтого цвета (0,63 г). Данное сырое вещество подвергают очистке с помощью флэш-хроматографии (1:1 EtOAc/гексаны), что приводит к 3-(2-хлорпиримидин-4-ил)-1H-индолу (502 мг, выход 89%) в виде твердого вещества желтого цвета.
Стадия Б: синтез 3-(2-хлорпиримидин-4-ил)-1-метил-1H-индола
Добавляют NaH (60% дисперсия в минеральном масле, 0,16 г, 7 ммоля) при температуре 0°C к 3-(2-хлорпиримидин-4-ил)-1H-индолу (0,24 г, 1 ммоля) в ДМФ (8 мл). Получаемую таким образом смесь перемешивают в течение 10 минут, затем добавляют иодметан (0,20 мл, 3,2 ммоля), нагревают реакционную смесь до КТ и выливают в смесь льда с водой. Получаемую таким образом смесь экстрагируют EtOAc, объединенные органические фракции дважды промывают водой, сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении, что приводит к 3-(2-хлорпиримидин-4-ил)-1-метил-1H-индолу (270 мг) в виде твердого вещества желтого цвета.
Стадия В: синтез транс-4-[4-(1-метил-1H-индол-3-ил)пиримидин-2-иламино]циклогексанола
Добавляют транс-4-аминоциклогексанол (0,45 г, 4 ммоля) к 3-(2-хлорпиримидин-4-ил)-1-метил-1H-индолу (0,23 г, 1 ммоля) в НМП (8 мл) и нагревают получаемую таким образом смесь до температуры 120°C в течение 3 часов. Добавляют вторую аликвоту транс-4-аминоциклогексанола (0,16 г, 1 ммоля) и нагревают получаемую таким образом смесь до температуры 130°C. Реакционную смесь охлаждают и распределяют между фазами воды и EtOAc, органический слой отделяют, дважды промывают его водой, сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Сырой остаток перекристаллизовывают из смеси ДХМ/EtOAc/гексаны, что приводит к транс-4-[4-(1-метил-1H-индол-3-ил)пиримидин-2-иламино]циклогексанолу (148 мг, 46% выход) в виде твердого вещества светло-желтого цвета. MC=323 [M+H]+, tпл.=194,5-195,3°C.
Пример 18: синтез транс-4-{4-[6-(2-гидроксиэтиламино)индазол-1-ил]-пиримидин-2-иламино}циклогексанола
Синтез транс-4-{4-[6-(2-гидрокси-этиламино)индазол-1-ил]-пиримидин-2-иламино}циклогексанола осуществляют согласно способу, показанному на схеме 32.
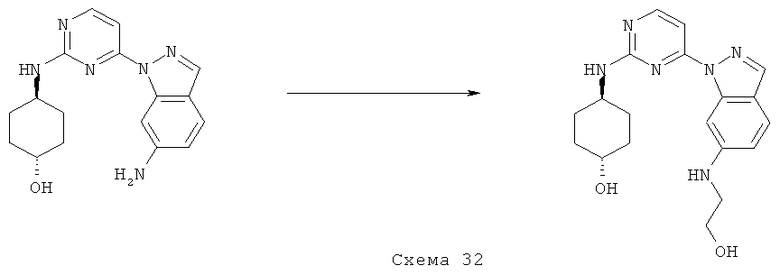
Схема 32
Добавляют 2-бромэтанол (42 мкл, 0,6 ммоля) к смеси транс-4-[4-(6-аминоиндазол-1-ил)пиримидин-2-иламино]циклогексанола (100 мг, 0,30 ммоля) и гидрокарбоната натрия (25 мг, 0,30 ммоля) в ацетонитриле (1 мл) и нагревают получаемую таким образом смесь до температуры 80°C в течение 2 часов. Добавляют ДМФ (0,5 мл) и нагревают получаемую таким образом смесь до температуры 80°C в течение ночи. Затем добавляют вторую аликвоту 2-бромэтанола (20 мкл) и продолжают нагревание в течение 3 суток. Получаемую таким образом смесь разбавляют MeOH и добавляют Et2O. Выпадающий из раствора смолистый остаток подвергают очистке с помощью препаративной TCX, что приводит к транс-4-{4-[6-(2-гидроксиэтиламино)индазол-1-ил]-пиримидин-2-иламино}циклогексанолу (29 мг) в виде твердого вещества желтого цвета. MC=369 [M+H]+.
Пример 19: синтез транс-N-{1-[2-(4-гидроксициклогексиламино)-пиримидин-4-ил]-1Н-индазол-6-ил}ацетамида
Синтез транс-N-{1-[2-(4-гидроксициклогексиламино)пиримидин-4-ил]-1Н-индазол-6-ил}ацетамида осуществляют согласно способу, показанному на схеме 33.
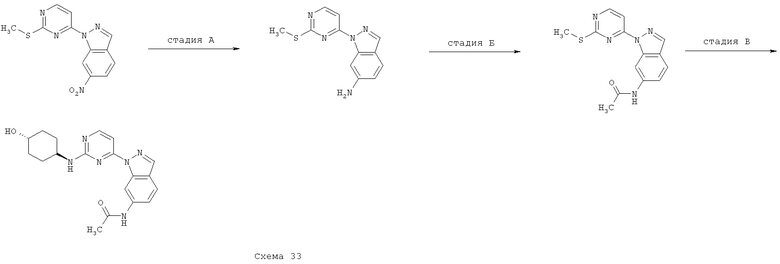
Стадия А: синтез 1-(2-метилсульфанилпиримидин-4-ил)-1H-индазол-6-иламина
Восстанавливают 1-(2-метилсульфанилпиримидин-4-ил)-6-нитро-1H-индазол, следуя методике, описанной в примере 8.
Стадия Б: синтез N-[1-(2-метилсульфанилпиримидин-4-ил)-1H-индазол-6-ил]ацетамида
Хлорангидрид уксусной кислоты (10 капель) добавляют по каплям при КТ к 1-(2-метилсульфанилпиримидин-4-ил)-1H-индазол-6-иламину (0,13 г, 0,5 ммоля) и триэтиламину (15 капель) в ТГФ (5 мл) и перемешивают получаемую таким образом смесь в течение 30 минут. Реакционную смесь разбавляют ДХМ и MeOH, дважды промывают водным раствором NaHCO3 и один раз соляным раствором, сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Сырой остаток обрабатывают Et2O и гексаном и собирают осадок посредством фильтрования, что приводит к N-[1-(2-метилсульфанилпиримидин-4-ил)-1H-индазол-6-ил]ацетамиду (146 мг) в виде твердого вещества грязно-белого цвета.
Стадия В: синтез транс-N-{1-[2-(4-гидроксициклогексиламино)-пиримидин-4-ил]-1Н-индазол-6-ил}ацетамида
Добавляют 3-хлорпероксибензойную кислоту (77%, 200 мг, 0,87 ммоля) к суспензии N-[1-(2-метилсульфанилпиримидин-4-ил)-1H-индазол-6-ил]ацетамида (0,43 ммоля, 0,13 г) в хлороформе (5 мл) и перемешивают получаемую таким образом смесь при КТ в течение ночи. Реакционную смесь разбавляют ДХМ/MeOH (4:1, 25 мл), промывают водным раствором NaOH (0,1 М) и соляным раствором, сушат над MgSO4, фильтруют, и концентрируют при пониженном давлении. Остаток растирают с Et2O, что приводит к N-[1-(2-метансульфонилпиримидин-4-ил)-1H-индазол-6-ил]ацетамиду (120 мг) в виде твердого вещества грязно-белого цвета. Порцию этого вещества (100 мг) смешивают с транс-4-аминоциклогексанолом (0,12 г) и нагревают смесь до температуры 130°C в течение 85 минут. Остаток обрабатывают смесью ДХМ/MeOH и выпаривают растворитель при пониженном давлении. Остаток обрабатывают MeOH (5 мл) и Et2O, декантируют надосадочную жидкость и обрабатывают образующееся смолообразное твердое вещество второй аликвотой MeOH (5 мл) и EtOAc (15 мл). Получаемый таким образом мелкий осадок собирают посредством фильтрования, что приводит к транс-N-{1-[2-(4-гидроксициклогексиламино)пиримидин-4-ил]-1Н-индазол-6-ил}ацетамиду (72 мг, 65% выход) в виде твердого вещества грязно-белого цвета. MC=367 [M+H]+, tпл.=288,0-289,5°C.
Пример 20: синтез транс-4-[4-(6-гидроксиметилиндазол-1-ил)пиримидин-2-иламино]циклогексанола
Синтез транс-4-[4-(6-гидроксиметилиндазол-1-ил)пиримидин-2-иламино]циклогексанола осуществляют согласно способу, показанному на схеме 34.

Схема 34
Раствор триэтилборгидрида лития (1 М в ТГФ, 0,8 мл) медленно добавляют при КТ к метиловому эфиру транс-1-[2-(4-гидроксициклогексиламино)-пиримидин-4-ил]-1H-индазол-6-карбоновой кислоты (100 мг) в ТГФ (3 мл). Получаемую таким образом смесь перемешивают в течение 30 минут и добавляют смесь AcOH/EtOH (1:1,2 мл). Реакционную смесь перемешивают в течение 30 минут, а затем выливают в водный раствор HCl (0,1 М). Получаемую таким образом смесь подщелачивают до рН 8 водным раствором NaOH и водным раствором NaHCO3, а затем экстрагируют ДХМ. Объединенные органические фракции сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Сырой остаток подвергают очистке с помощью препаративной TCX (ДХМ/MeOH, 95:5), что приводит к транс-4-[4-(6-гидроксиметил-индазол-1-ил)пиримидин-2-иламино]циклогексанолу (22 мг) в виде твердого вещества белого цвета. MC=340 [M+H]+, tпл.=216,0-217,7°C.
Пример 21: синтез 3-(4-индазол-1-илпиримидин-2-иламино)бензолсульфамида
Синтез 3-(4-индазол-1-илпиримидин-2-иламино)бензолсульфамида осуществляют согласно способу, показанному на схеме 35.
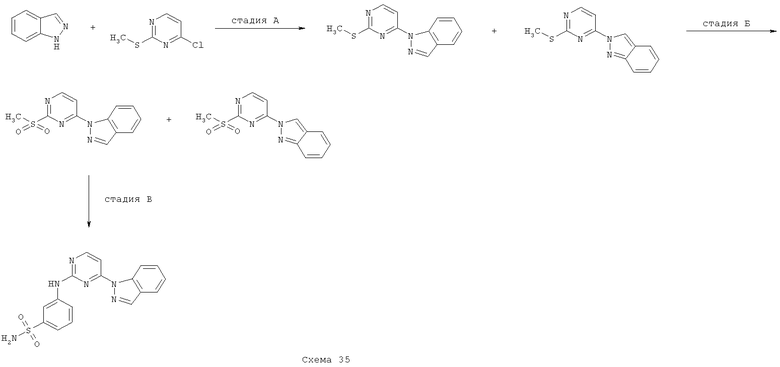
Стадия А: синтез 1-(2-метилсульфанилпиримидин-4-ил)-1H-индазола и 2-(2-метилсульфанилпиримидин-4-ил)-2H-индазола
Раствор 1H-индазола (2,36 г, 20,0 ммоля) в безводном ДМФ (100 мл) обрабатывают NaH (60% дисперсия в минеральном масле, 1,60 г, 40,0 ммоля) при КТ в течение 10 минут. Затем добавляют 4-хлор-2-метилтиопиримидин (2,55 мл, 22,0 ммоля) одной порцией при КТ и перемешивают получаемую таким образом смесь при температуре 70°C в течение 2,5 часов. Реакционную смесь охлаждают до КТ, разлагают остатки реагентов водным раствором HCl (1 М) и экстрагируют смесь EtOAc. Слои разделяют, органический слой промывают соляным раствором, сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Сырой продукт абсорбируют силикагелем и подвергают очистке с помощью флэш-хроматографии (этилацетат/гексан, градиент от 5:95 к 25:75), что приводит к смеси 1-(2-метилсульфанилпиримидин-4-ил)-1H-индазола и 2-(2-метилсульфанилпиримидин-4-ил)-2H-индазола (3,77 г, выход 78%) в виде твердого вещества бежевого цвета.
Стадия Б: синтез 1-(2-метансульфонилпиримидин-4-ил)-1H-индазола и 2-(2-метансульфонилпиримидин-4-ил)-2H-индазола
Смесь 1-(2-метилсульфанилпиримидин-4-ил)-1H-индазола и 2-(2-метилсульфанилпиримидин-4-ил)-2H-индазола (3,77 г, 15,55 ммоля) в хлороформе (80 мл) обрабатывают 3-хлорпероксибнзойной кислотой (80%, 7,37 г, 34,21 ммоля). Реакционную смесь перемешивают при температуре 50°C в течение 1,5 часов, а затем разлагают остатки реагентов водным раствором NaOH (1 М). Получаемую таким образом смесь экстрагируют ДХМ, разделяют слои, промывают органический слой соляным раствором, сушат его над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Сырой продукт абсорбируют силикагелем и подвергают очистке с помощью флэш-хроматографии (ДХМ/этилацетат, градиент от 0:100 к 5:95), что приводит к 1-(2-метилсульфанилпиримидин-4-ил)-1H-индазолу (менее полярный изомер) (1,92 г, выход 45%) и 2-(2-метилсульфанилпиримидин-4-ил)-2H-индазолу (более полярный изомер) (1,04 г, выход 24%).
Стадия В: синтез 3-(4-индазол-1-илпиримидин-2-иламино)бензолсульфамида
Раствор 1-(2-метилсульфонилпиримидин-4-ил)-1H-индазола (100 мг, 0,36 ммоля) и n-толуолсульфокислоты (139 мг, 0,73 ммоля) в i-PrOH (4 мл) обрабатывают 3-аминобензолсульфамидом (251 мг, 1,46 ммоля) в микроволновом реакторе при температуре 150°C в течение 20 минут. Смесь распределяют между фазами воды и этилацетата, органический слой отделяют, промывают его соляным раствором, сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Сырой остаток абсорбируют силикагелем и подвергают очистке с помощью флэш-хроматографии (EtOAc/гексан, градиент от 30:70 к 50:50), что приводит к указанному в заглавии соединению. Данный остаток растирают с водным раствором NaOH (1 М), твердое вещество собирают посредством фильтрования, промывают водой и сушат под вакуумом, что приводит к 3-(4-индазол-1-илпиримидин-2-иламино)бензолсульфамиду (50 мг, выход 37%). MC=367 [M+H]+.
Трет-бутиловый эфир 4-(4-индазол-1-илпиримидин-2-иламино)циклогексил]карбаминовой кислоты (твердое вещество белого цвета) получают аналогичным образом, используя подходящие исходные вещества. MC=409,2 [M+H]+.
Пример 22: Препараты
Фармацевтические препараты для введения различными путями приготавливают так, как это показано в нижеследующих таблицах. Термин «активный ингредиент» или «активное соединение», как он употребляется в таблице, означает одно или более соединение формулы I.
Ингредиент спешивают и распределяют по капсулам, каждая из которых содержит приблизительно 100 мг, т.е. одна капсула приблизительно составляет общую суточную дозировку.
Ингредиенты объединяют и гранулируют с помощью растворителя, такого как метанол. Затем препарат сушат и формуют в виде таблеток (содержащих приблизительно 20 мг активного соединения) с помощью соответствующего устройства для изготовления таблеток.
Ингредиенты смешивают с целью получения суспензии для перорального введения.
Активный ингредиент растворяют в порции воды для инъекций. Затем добавляют при перемешивании достаточное для достижения изотоничности количество хлорида натрия. Раствор доводят до конечной массы остатком воды для инъекций, фильтруют через 0,2-микронный мембранный фильтр и запаковывают в условиях стерильной атмосферы.
Ингредиенты совместно расплавляют и смешивают на паровой бане, после чего выливают в формы, содержащие 2,5 г общей массы.
Все ингредиенты, кроме воды, объединяют и нагревают при перемешивании до температуры приблизительно 60°C. После этого добавляют при энергичном перемешивании при температуре приблизительно 60°C существенное количество воды с целью эмульгации ингредиентов, после чего добавляют воду в количестве, требуемом для достижения массы приблизительно 100 г.
Препараты в виде назального спрея
Несколько водных суспензий, содержащих от, приблизительно, 0,025-0,5 процентов изготавливают в виде препаратов, применяемых в назальном спрее. Препараты необязательно содержат неактивные ингредиенты, такие как, например, микрокристаллическую целлюлозу, натрий-карбоксиметилцеллюлозу, декстрозу. Для получения нужного рН может быть добавлена соляная кислота. Препараты в виде назального спрея могут вводиться с помощью мерного насоса для назального спрея, обеспечивая, как правило, введение приблизительно 50-100 мкл препарата за одно приведение в действие. Типичное расписание дозировки составляет 2-4 распыления каждые 4-12 часов.
Пример 23: анализ JNK in vitro
Активность JNK измеряют посредством фосфорилирования химерного белка GST-ATF2 (19-96) (глутатион-S-трансфераза - активирующий транскрипционный фактор 2) [γ-33Р]АТФ (аденозинтрифосфатом). Ферментативную реакцию осуществляют при концентрациях АТФ и субстрата, равных Km в конечном объеме 40 мкл в буфере, содержащем 25 мМ 4-(2-гидроксиэтил)-1-пиперазинэтансульфокислоты (HEPES), рН 7,5, 2 мМ дитиотреитола, 150 мМ NaCl, 20 мМ MgCl2, 0,001% поверхностно-активного вещества Tween® 20, 0,1% бычьего сывороточного альбумина и 10% диметилсульфоксида (ДМСО). Анализ человеческой JNK2α2 содержит 1 нМ фермента, 1 мкМ ATF2, 8 мкМ АТФ с 1 мкКи [γ-33Р]АТФ. Анализ человеческой JNK1α1 содержит 2 нМ фермента, 1 мкМ ATF2, 6 мкМ АТФ с 1 мкКи [γ-33Р]АТФ. Анализ человеческой JNK3 (Upstate Biotech # 14-501М) содержит 2 нМ фермента, 1 мкМ ATF2, 4 мкМ АТФ с 1 мкКи [γ-33Р]АТФ. Ферментативный анализ осуществляют в присутствии нескольких концентрации соединения или в его отсутствие. JNK и соединение предварительно инкубируют в течение 10 минут, а затем инициируют ферментативную реакцию посредством добавления АТФ и субстрата. Реакционную смесь инкубируют при температуре 30°C в течение 30 минут. По окончании инкубирования реакцию останавливают посредством переноса 25 мкл реакционной смеси к 150 мкл 10% суспензии глутатионсефарозы® (Amersham # 27-4574-01), содержащей 135 мМ этилендиаминтетрауксусной кислоты (ЭДТА). Продукт реакции осаждают на аффинной смоле и шестикратно промывают на фильтровальном планшете (Millipore, MABVNOB50) забуференным фосфатом физиологическим раствором в течение с целью удаления свободного радиоактивно меченого нуклеотида. Количество внедренного в ATF2 33Р измеряют с помощью сцинтилляционного счетчика для микропланшетов (Packard Topcount). Ингибирующую способность соединения в отношении JNK измеряют величинами IC50 (концентрации, обеспечивающей пятидесятипроцентное ингибирование), полученными из десяти концентрационных зависимостей ингибирования, аппроксимированных трехпараметрической моделью: % ингибирования = максимум/(1+(IC50/[концентрация ингибирования])наклон). Данные анализируют с помощью программы Microsoft Excel с целью определения параметров. Результаты приведены ниже в таблице 2.
Пример 24: анализ in vivo стимулированной TNFα выработки интерлейкина ИЛ-6
Самкам крыс линии Wistar-Han, полученных от Charles River Laboratories дают акклиматизироваться в течение одной недели пред использованием и достичь массы тела 95-130 г. Крысам вводят испытуемое соединение через пероральный зонд за 30 минут до внутрибрюшинного введения 0,5 мкг рекомбинантного крысиного TNF-α (Biosource). Кровь собирают посредством сердечной пункции через 90 минут после введения TNF-α. Плазму подготавливают с помощью литий-гепариновых разделительных пробирок (BD microtainer) и замораживают при температуре -80°C до анализа. Уровни ИЛ-6 определяют с помощью специфичного для крыс набора для твердофазного иммуноферментного анализа ИЛ-6 (Biosource). Определяют процент ингибирования и величины ED50 (рассчитываемые как доза соединения, при которой выработка ИЛ-6 составляла 50% от контрольного значения). Результаты представлены в нижеприведенной таблице 3.
Пример 25: колагениндуцированный артрит грызунов
Самкам крыс линии Lewis, полученным от Harlan Laboratories в возрасте 7-8 недель позволяли акклиматизироваться в течение одной недели перед использованием и достичь приблизительной массы тела 120-140 г. В день 0 исследования крысам внутрикожно вводили в несколько точек на спине эмульсию 100 мкг бычьего коллагена типа II (Chondrex) в неполном адъюванте Фрейнда (НФА, общий объем 0,1 мл в 2-3 точки). Появление артрита наблюдали, как правило, через 12-14 суток после введения; осуществляли, однако, повторную инъекцию 100 мкг коллагена/НФА около 7-10 суток (общий объем до 0,1 мл) в основание хвоста или другую точку на спине с целью синхронизации стимуляции болезни. Дозировка соединений может быть профилактической (начиная от времени инъекции или за 1-2 суток до того) или терапевтической (начиная после инъекции и совпадая с начальными стадиями заболевания 1-2 - см. клиническую шкалу ниже). Состояние животных оценивали с точки зрения появления и развития заболевания в течение следующих 21 суток.
Крыс оценивают с помощью шкалы (описанной ниже), измеряя объем лап с помощью плетизмометрии для каждой лапы, или же измеряя толщину лап и суставов штангенциркулем. Измерения для базовой линии осуществляют в день 0 и заново начинают измерения при первых признаках опухания до трех раз в неделю до конца эксперимента. Для каждой лапы используют следующую шкалу оценок:
1 = опухание и/или покраснение лапы или одного пальца
2 = опухоль двух или более суставов
3 = сильная опухоль лапы, включающая более двух суставов
4 = тяжелый артрит всей лапы и пальцев.
Артритный индекс для каждой крысы определяют посредством сложения четырех оценок для отдельных лап, что дает максимальную оценку 16. C целью последовательного измерения начала и развития заболевания объем задних лап также определяют с помощью плетизмометрии.
В конце исследования задние лапы (и другие ткани) собирают для измерения массы, гистологических, клеточных и/или молекулярных анализов. Кроме того, собирают кровь с помощью сердечной пункции, подготавливают плазму с помощью литий-гепариновых разделительных пробирок (BD microtainer) и замораживают при температуре -70°C до анализа. Уровни воспалительных цитокинов (например, TNF-α, ИЛ-1 и ИЛ-6) из плазмы или из гомогенизированных суставных тканей определяют с помощью специфичного для крыс набора для твердофазного иммуноферментного анализа (R&D). Уровень ингибирования заболевания или защиты от него измеряют как составной показатель изменений в клинической оценке, объемах лап и гистопатологии по сравнению с контрольными животными.
Пример 26: анализ выработки ИЛ-8 в клетках стимулированной TNFα-хондросаркомы человека SW1353
Клетки SW1353 покупают у Американской коллекции Тканевых Культур и выдерживают в среде для роста, содержащую модифицированную Дюльбекко среду Игла (Invitrogen) с 10% околоплодной телячьей сыворотки (Invitrogen), аскорбиновыми кислотами (Sigma) и пенициллином (Invitrogen) в культуральных условиях 37°C при 5% CO2. Клетки помещают на планшеты с плотностью 1,0×104 клеток на ячейку в 100 мкл среды за 48 часов до обработки соединением. Сразу после обработки соединением среду заменяют 160 мкл свежей среды. Исходное соединение (10 мМ) разбавляют средой для роста и добавляют в каждую ячейку в виде 10× концентрированного раствора в объеме 20 мкл, смешивают и оставляют предварительно инкубироваться с клетками в течение 30 минут. Соединение-носитель (ДМСО) поддерживают при конечной концентрации 1% во всех образцах. По прошествии 30 минут клетки активируют 10 нг/мл TNF-α (Roche Biochem). TNF-α добавляют в виде 10× концентрированного раствора в среде для роста и добавляют в объеме 20 мкл на ячейку. Клеточные планшеты культивируют в течение 5 часов. Клеточную среду собирают и хранят при температуре -20°C. Аликвоты среды анализируют с помощью твердофазного иммуноферментного анализа сэндвичевого типа на присутствие ИЛ-8 согласно инструкции изготовителя (BD Bioscience). Величины IC50 рассчитывают как концентрацию соединения, при которой выработка ИЛ-8 снизилась на 50% от контрольной величины, с помощью процедуры Xlflt3 в программе Microsoft Excel. Некоторые соединения характеризуются в данном анализе величинами IC50 в диапазоне 0,1-20 мкМ.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПИРИМИДИНГИДРАЗИДНЫЕ СОЕДИНЕНИЯ КАК ИНГИБИТОРЫ PGDS | 2008 |
|
RU2464262C2 |
| ПРОИЗВОДНЫЕ 4-ФЕНИЛПИРИМИДИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2000 |
|
RU2243221C2 |
| ПИРИМИДИНАМИДНЫЕ СОЕДИНЕНИЯ КАК ИНГИБИТОРЫ PGDS | 2006 |
|
RU2420519C2 |
| ПРОИЗВОДНЫЕ ПИРИДИНА И ПИРИМИДИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2003 |
|
RU2293731C2 |
| МОДУЛЯТОРЫ АДЕНОЗИНОВЫХ РЕЦЕПТОРОВ | 2001 |
|
RU2277911C2 |
| ИНГИБИТОРЫ JNK | 2009 |
|
RU2504545C2 |
| ИНГИБИТОРЫ JAK | 2010 |
|
RU2538204C2 |
| 2,6-ЗАМЕЩЕННЫЕ-4-МОНОЗАМЕЩЕННЫЙ АМИНО-ПИРИМИДИНЫ КАК АНТАГОНИСТЫ РЕЦЕПТОРА ПРОСТАГЛАНДИНА D2 | 2005 |
|
RU2417990C2 |
| ЗАМЕЩЕННЫЕ ДИАМИНОКАРБОКСАМИДНЫЕ И ДИАМИНОКАРБОНИТРИЛЬНЫЕ ПРОИЗВОДНЫЕ ПИРИМИДИНОВ, ИХ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ С ИХ ПОМОЩЬЮ | 2012 |
|
RU2697712C2 |
| ЗАМЕЩЕННЫЕ ДИАМИНОКАРБОКСАМИДНЫЕ И ДИАМИНОКАРБОНИТРИЛЬНЫЕ ПРОИЗВОДНЫЕ ПИРИМИДИНОВ, ИХ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ С ИХ ПОМОЩЬЮ | 2012 |
|
RU2625309C2 |
Настоящее изобретение относится к области органической химии, а именно к новым производным пиримидина общей формулы (I), или к его фармацевтически приемлемым солям, где X1 обозначает N, N-R3, C-R3 или O; X2 обозначает N, CH или C-CH3; X3 обозначает N или C, где Х1, X2 и X3 все одновременно не обозначают N; X4, X5 каждый независимо обозначает C или N; X6 обозначает N или C-R1; где по меньшей мере два и не более четырех из X1, X2, X3, X4, X5 и X6 обозначают N; и где связи между X1 и X2, X2 и X3, X3 и X4, X4 и X5, X5 и X1, а также X5 и X6 могут каждая независимо быть одинарной или двойной, или же образовывать ароматический цикл, при условии, что образуется химически устойчивая структура; R обозначает (циклогексил)-R2; R1 обозначает галоид, нитрогруппу, -CN, -CH2CN, -OH, -NH2, -COOH или -Y1R4; Y1 обозначает -O-, -SO2-, -NHSO2-, -С(O)O- или связь; R4 обозначает (C1-C6)алкил, фенил или бензил, каждый из которых замещен 0-3 раза гидроксигруппой или галогеном; n равно 0, 1 или 2; R3 обозначает H или (C1-C6)алкил; R2 обозначает H, -OH, =O или -Y2-Y3-Y4-R5, где Y2 обозначает -C(O)-, -C(O)NRa, -NH- или связь; Y3 обозначает (C1-C6)алкилен или связь; Y4 обозначает -NRa-,
-S-, -SO2-, -NRaC(O)-, -NHSO2- или связь; R5 обозначает (C1-C6)алкил, (C3-C10)циклоалкил, 5-6-членный гетероциклил, содержащий 1-2 гетероатома, выбранных из N и O, где R5 является необязательно замещенным -OH или -NHRa; где каждый Ra независимо обозначает водород или (C1-C6)алкил; Z обозначает водород. Также изобретение относится к фармацевтической композиции на основе соединения формулы (I). Технический результат: получены новые производные пиримидина, полезные для терапевтического и/или профилактического лечения расстройства, опосредованного c-Jun N-терминальной киназой. 2 н. и 12 з.п. ф-лы, 3 табл., 26 пр.
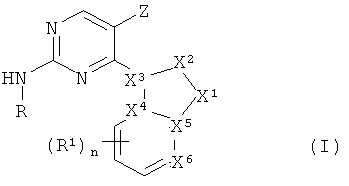
1. Соединение формулы I
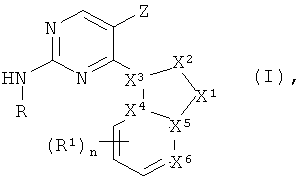
в котором
X1 обозначает N, N-R3, C-R3 или O;
X2 обозначает N, CH или C-CH3;
X3 обозначает N или C, где X1, X2 и X3 все одновременно не обозначают N;
X4, X5 каждый независимо обозначает C или N;
X6 обозначает N или C-R1;
где по меньшей мере два и не более четырех из X1, X2, X3, X4, X5 и X6 обозначают N; и где связи между X1 и X2, X2 и X3, X3 и X4, X4 и X5, X5 и X1, а также X5 и X6 могут каждая независимо быть одинарной или двойной, или же образовывать ароматический цикл, при условии, что образуется химически устойчивая структура;
R обозначает -(циклогексил)-R2;
R1 обозначает галоид, нитрогруппу, -CN, -CH2CN, -OH, -NH2, -COOH или -Y1R4;
Y1 обозначает -O-, -SO2-, -NHSO2-, -C(O)O- или связь;
R4 обозначает (C1-C6)алкил, фенил или бензил, каждый из которых замещен 0-3 раза гидроксигруппой или галогеном;
n равно 0, 1 или 2;
R3 обозначает H или (C1-C6)алкил;
R2 обозначает H, -OH, =O или -Y2-Y3-Y4-R5, где
Y2 обозначает -C(O)-, -C(O)NRa-, -NH- или связь;
Y3 обозначает (C1-C6)алкилен или связь;
Y4 обозначает -NRa-, -S-, -SO2-, -NRaC(O)-, -HHSO2- или связь;
R5 обозначает (C1-C6)алкил, (C3-C10)циклоалкил, 5-6-членный гетероциклил, содержащий 1-2 гетероатома, выбранных из N и О, где R5 является необязательно замещенным -OH или -NHRa; где каждый Ra независимо обозначает водород или (C1-C6)алкил;
Z обозначает водород;
или его фармацевтически приемлемая соль.
2. Соединение по п.1, в котором X3 обозначает N, и X1 обозначает CR3.
3. Соединение по п.1, в котором R2 обозначает OH.
4. Соединение по п.1, в котором R1 обозначает бензилоксигруппу, и n равно 1.
5. Соединение по п.1, в котором X2 обозначает N.
6. Соединение по п.1, в котором R2 обозначает -Y2-Y3-Y4-R5, где Y2 обозначает -C(O)NRa-, Ra обозначает H, а Y3 обозначает (C1-C6)алкилен.
7. Соединение по п.1, в котором Y4 обозначает -SO2-.
8. Соединение по п.1, в котором R5 обозначает (C1-C6)алкил.
9. Соединение по п.1, в котором Y4 обозначает -S-.
10. Соединение по п.1, в котором R5 обозначает (C1-C6)алкил.
11. Соединение по п.1, в котором Y4 обозначает связь, и R5 обозначает (C1-C6)алкил.
12. Соединение по п.1, в котором R1 обозначает -CN.
13. Соединение по п.1, выбранное из группы, включающей:
4-[4-(4-бензилоксииндол-1-ил)пиримидин-2-иламино]циклогексанол,
4-[4-(4-броминдол-1-ил)пиримидин-2-иламино]циклогексанол,
4-[4-(4-фториндол-1-ил)пиримидин-2-иламино]циклогексанол,
4-[4-(4-метоксииндол-1-ил)пиримидин-2-иламино]циклогексанол,
4-[4-(4-метилиндол-1-ил)пиримидин-2-иламино]циклогексанол,
4-[4-(4-цианоиндол-1-ил)пиримидин-2-иламино]циклогексанол,
4-[4-(4-(2-гидроксипроп-2-ил)индол-1-ил)пиримидин-2-иламино]-циклогексанол,
4-[4-(4-гидроксиметилиндол-1-ил)пиримидин-2-иламино]циклогексанол,
4-[4-(4-аминоиндол-1-ил)пиримидин-2-иламино]циклогексанол,
метиловый эфир 1-[2-(4-гидроксициклогексиламино)пиримидин-4-ил]-1H-индол-4-карбоновой кислоты,
4-[4-(4-бензилсульфанилиндол-1-ил)пиримидин-2-иламино]циклогексанол,
1-[2-(4-гидроксициклогексиламино)пиримидин-4-ил]-1H-индол-4-карбоновая кислота,
4-[4-(4-бензилсульфонилиндол-1-ил)пиримидин-2-иламино]циклогексанол,
4-[4-(7-метокси-1H-индазол-3-ил)пиримидин-2-иламино]циклогексанол,
4-[4-(7-метоксибензо[d]изоксазол-3-ил)пиримидин-2-иламино]циклогексанол,
4-[4-(7-метил-1H-индазол-3-ил)пиримидин-2-иламино]циклогексанол,
4-(4-пиразоло[1,5-b]пиридазин-3-илпиримидин-2-иламино)циклогексанол,
4-[4-(4-метоксииндазол-1-ил)пиримидин-2-иламино]циклогексанол, и
N-(1-[2-(4-гидроксициклогексиламино)пиримидин-4-ил]-1H-индазол-4-ил)изобутирамид.
14. Фармацевтическая композиция, предназначенная для терапевтического
и/или профилактического лечения расстройства, опосредованного c-Jun N-терминальной киназой, где опосредованное c-Jun N-терминальной киназой расстройство представляет собой ревматоидный артрит, астму, диабет типа II, болезнь Альцгеймера, болезнь Паркинсона или инсульт, содержащая соединение по п.1 и фармацевтически приемлемый наполнитель.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Ручка для пера с резервуаром для чернил | 1927 |
|
SU5974A1 |
| МОДУЛЯТОРЫ АДЕНОЗИНОВЫХ РЕЦЕПТОРОВ | 2001 |
|
RU2277911C2 |

