Область изобретения
Настоящее изобретение относится к твердой пероральной лекарственной форме, включающей терапевтический агент, например LCZ696. Указанную фармацевтическую композицию получают сухим способом, таким как прямое прессование или уплотнение, при этом получают твердую пероральную лекарственную форму.
Предпосылки создания настоящего изобретения
Ангиотензин II представляет собой гормон, который вызывает сокращение кровеносных сосудов что может привести к развитию гипертензии и увеличить нагрузку на сердце. Указанный гормон взаимодействует со специфическими рецепторами на поверхности клеток-мишеней. К настоящему времени идентифицировано два подтипа рецепторов ангиотензина II, например, AT1 и АТ2. В последнее время большое внимание уделяется поиску соединений, которые связываются с рецептором AT1. Блокаторы рецепторов ангиотензина (ARB, антагонисты ангиотензина II) предотвращают связывание ангиотензина II с его рецепторами на стенках кровеносных сосудов, таким образом, снижая кровяное давление. Следовательно, благодаря ингибированию рецептора AT1, указанные антагонисты можно использовать в качестве антигипертензивных агентов или для лечения застойной сердечной недостаточности, наряду с другими показаниями.
Нейтральная эндопептидаза (ЕС 3.4.24.11, энкефалиназа, атриопептидаза, NEP) является цинксодержащей металлопротеазой, которая расщепляет множество пептидных субстратов по аминогруппе гидрофобных остатков (см. Pharmacol. Rev., т.45, с.87 (1993)). Субстраты указанного фермента включают, не ограничиваясь только ими, атриальный натрийуретический пептид (ANP, также известный как ANF), мозговой натрийуретический пептид (BNP), мет- и лей-энкефалин, брадикинин, нейрокинин А, эндотелин-1 и вещество P. ANP является высокоэффективным сосудосуживающим средством и натрийуретическим агентом (см. J. Hypertens., т.19, с.1923 (2001)). Вливание ANP нормальным субъектам приводит к воспроизводимому значительному усилению натрийуреза и диуреза, включая повышение фракционной экскрекции натрия, скорости мочеотделения и скорости клубочковой фильтрации в почках (см. J. Clin. Pharmacol., т.27, p.927 (1987)). Однако ANP характеризуется коротким периодом полураспада в кровотоке, и было установлено, что в мембранах коркового вещества почки NEP является главным ферментом, ответственным за деградацию указанного пептида (см. Peptides, т.9, с.173 (1988)). В связи с этим, ингибиторы NEP (ингибиторы нейтральной эндопептидазы, NEPi) повышают уровень ANP в плазме и, следовательно, могут индуцировать натрийуретическое и диуретическое действия.
Хотя соединения, такие как блокаторы рецептора ангиотензина и ингибиторы нейтральной эндопептидазы, можно использовать для контроля гипертензии, первичная артериальная гипертензия представляет собой полигенное заболевание и не во всех случаях адекватно контролируется лечением в режиме монотерапии. Повышенное кровяное давление в 2000 г. было зарегистрировано приблизительно у 333 миллионов взрослых пациентов в экономически развитых странах и приблизительно у 65 миллионов американцев (у каждого третьего взрослого пациента, см. Lancet, т.365, с.217 (2005) и Hypertension, т.44, с.398 (2004)). Продолжительная и неконтролируемая сосудистая гипертензия в конечном счете приводит к множеству патологических изменений в органах-мишенях, таких как сердце и почки. Стабильная артериальная гипертензия также приводит к увеличению числа инсультов.
Соединение двойного действия или комбинация, прежде всего надмолекулярный комплекс двух активных агентов с различными механизмами действия, или связанное пролекарство или, особенно надмолекулярный комплекс двух активных агентов с различными механизмами действия, а именно антагониста рецептора ангиотензина и ингибитора нейтральной эндопептидазы, описаны в заявках U.S. №№60/735093, поданной 9 ноября 2005 г., 60/735541, поданной 10 ноября 2005 г., 60/789332, поданной 4 апреля 2006 г., и 60/822086, поданной 11 августа 2006 г., а также в международной заявке WO 2007/056546, которые включены в полном объеме в настоящее описание в качестве ссылок. Указанные надмолекулярные комплексы можно использовать для лечения пациентов с различными сердечно-сосудистыми и/или почечными заболеваниями.
Особенно пригодным терапевтическим агентом является надмолекулярный комплекс - полупентагидрат тринатриевой соли [3-((1S,3R)-1-бифенил-4-илметил-3-этоксикарбонил-1-бутилкарбамоил)пропионат-(S)-3'-метил-2'-(пентаноил{2''-(тетразол-5-илат)бифенил-4'-илметил}амино)масляной кислоты], также известный как LCZ696.
Существует необходимость получения такого надмолекулярного комплекса в виде фармацевтических композиций, особенно твердых пероральных лекарственных форм, которые оказывают терапевтически благоприятное действие соединений на пациента, нуждающегося в указанном лечении. В одном объекте настоящего изобретения предлагается пример твердой пероральной лекарственной формы, которую может проглатывать пациент.
Начиная со второй половины девятнадцатого века использование таблеток получило широкое распространение, и большинство твердых пероральных лекарственных форм начали выпускать в виде таблеток. Главные причины популярности таблеток заключаются в простом способе получения, низкой стоимости лекарственной формы и высокой производительности при ее производстве. Другие причины включают стабильность полученного лекарственного средства, удобство упаковки, транспортировки и распространения. С точки зрения пациента таблетки характеризуются простотой введения, точной дозировкой, компактностью, транспортабельностью и слабо выраженным вкусом. В связи с этим, в другом объекте настоящего изобретения предлагается получение композиции терапевтического агента в виде таблетки.
Состав, включающий соединения двойного действия, таких как надмолекулярные комплексы, является нетривиальным, так как обычные методики получения лекарственных форм могут оказывать отрицательное действие на лекарственное соединение, что приводит, например к повышенной аморфности и/или диссоциации компонентов соединения двойного действия. В основном, при получении таких составов следует исключить действие на терапевтическое соединение влаги, избыточного тепла и/или высокого сдвига. Это может вызвать ряд проблем и трудностей при получении составов лекарственных форм.
Краткое описание сущности изобретения
В настоящем изобретении предлагаются твердые пероральные лекарственные формы для фармацевтических соединений, содержащие терапевтический агент, особенно надмолекулярный комплекс. В одном объекте настоящего изобретения предлагаемым надмолекулярным комплексом является соединение двойного действия. Соединение двойного действия или комбинация представляет собой надмолекулярный комплекс двух активных агентов с различными механизмами действия, или связанное пролекарство, или, особенно надмолекулярный комплекс двух активных агентов с различными механизмами действия, а именно антагониста рецептора ангиотензина и ингибитора нейтральной эндопептидазы. В другом объекте настоящего изобретения предлагаемым надмолекулярным комплексом является полупентагидрат тринатриевой соли [3-((1S,3R)-1-бифенил-4-илметил-3-этоксикарбонил-1-бутилкарбамоил)пропионат-(S)-3'-метил-2'-(пентаноил{2''-(тетразол-5-илат)бифенил-4'-илметил}амино)масляной кислоты]. Было установлено, что профиль высвобождения указанной композиции в значительной степени отличается от профиля высвобождения двух отдельных компонентов, валсартана и этилового эфира N-(3-карбокси-1-оксопропил)-(4S)-пара-фенилфенилметил)-4-амино-2R-метилмасляной кислоты. В частности, состав характеризуется улучшенным действием и, в связи с этим более высокой биодоступностью по сравнению с валсартаном. Указанные неожиданные преимущества открывают возможность получения фармацевтических композиций новых терапевтических агентов в более низких дозах. Фармацевтические составы, содержащие терапевтический агент, особенно надмолекулярный комплекс, можно получать сухим способом, таким как прямое прессование или ротационное прессование. В связи с этим, в другом объекте настоящего изобретения предлагается твердая пероральная лекарственная форма, полученная при смешивании терапевтического агента, по крайней мере, с одним фармацевтически приемлемым эксципиентом, и при последующем прямом прессовании смеси в пригодном оборудовании, таком как пресс для таблетирования, или при уплотнении смеси в пригодном оборудовании, таком как ротационный пресс.
Краткое описание фигур
Прилагаемые фигуры, которые включены в настоящее описание и являются его частью, иллюстрируют типичные варианты осуществления настоящего изобретения.
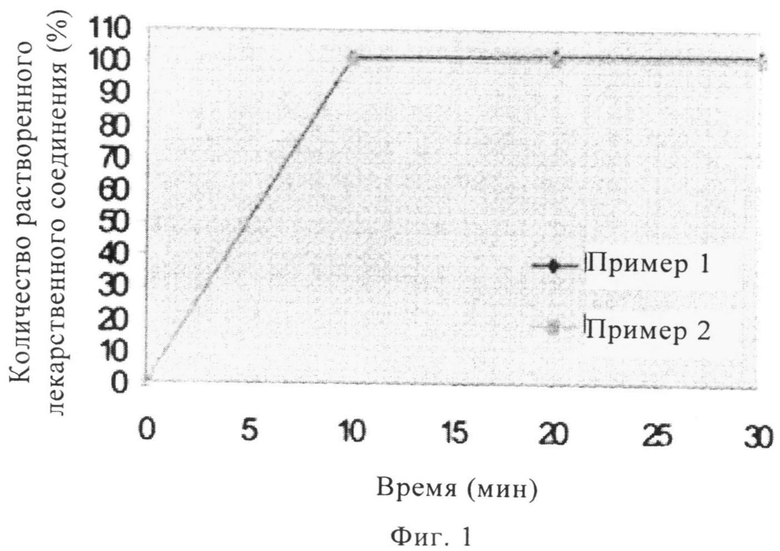
На фиг.1 представлен график профилей растворения in vitro таблеток массой 5 мг и 50 мг, включающих надмолекулярный комплекс полупентагидрата тринатриевой соли [3-((1S,3R)-1-бифенил-4-илметил-3-этоксикарбонил-1-бутилкарбамоил)пропионат-(S)-3'-метил-2'-(пентаноил{2''-(тетразол-5-илат)бифенил-4'-илметил}амино)масляной кислоты], полученных методом прямого прессования.
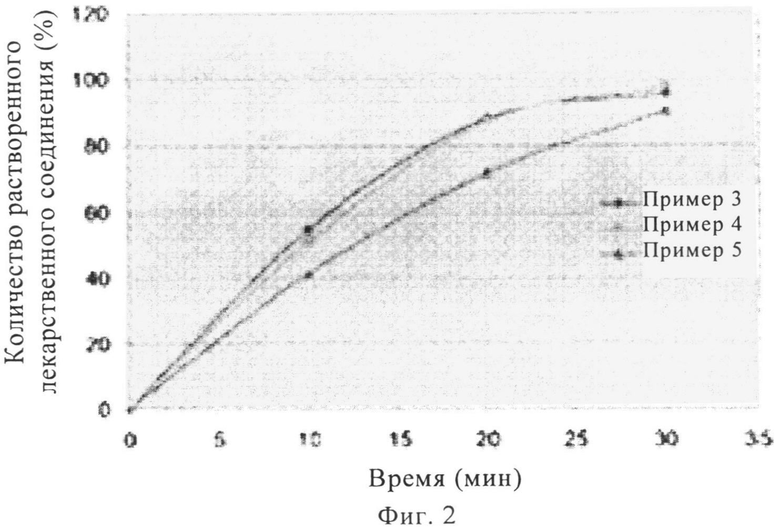
На фиг.2 представлен график профилей растворения при pH 6,8 in vitro таблеток массой 100, 200 и 400 мг с покрытием, включающих надмолекулярный комплекс полупентагидрата тринатриевой соли [3-((1S,3R)-1-бифенил-4-илметил-3-этоксикарбонил-1-бутилкарбамоил)пропионат-(S)-3'-метил-2'-(пентаноил{2''-(тетразол-5-илат)бифенил-4'-илметил}амино)масляной кислоты], полученных методом ротационного прессования.
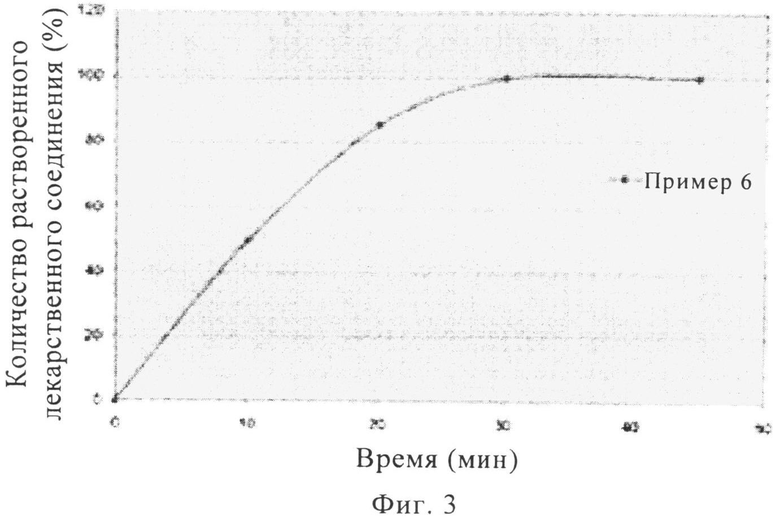
На фиг.3 представлен график профилей растворения при pH 4,5 in vitro таблеток массой 400 мг с покрытием, включающих надмолекулярный комплекс полупентагидрата тринатриевой соли [3-((1S,3R)-1-бифенил-4-илметил-3-этоксикарбонил-1-бутилкарбамоил)пропионат-(S)-3'-метил-2'-(пентаноил{2''-(тетразол-5-илат)бифенил-4'-илметил}амино)масляной кислоты], полученных методом ротационного прессования.
Подробное описание изобретения
Настоящее изобретение относится к фармацевтическим композициям, включающим терапевтический агент. Фармацевтические композиции по настоящему изобретению получают прямым прессованием или предпочтительно способом ротационного прессования, с получением фармацевтически приемлемых таблеток.
Использованный в данном контексте термин «терапевтический агент» обозначает надмолекулярный комплекс - полупентагидрат тринатриевой соли [3-((1S,3R)-1-бифенил-4-илметил-3-этоксикарбонил-1-бутилкарбамоил)пропионат-(S)-3'-метил-2'-(пентаноил{2''-(тетразол-5-илат)бифенил-4'-илметил}амино)масляной кислоты], структура которого схематически показана ниже:
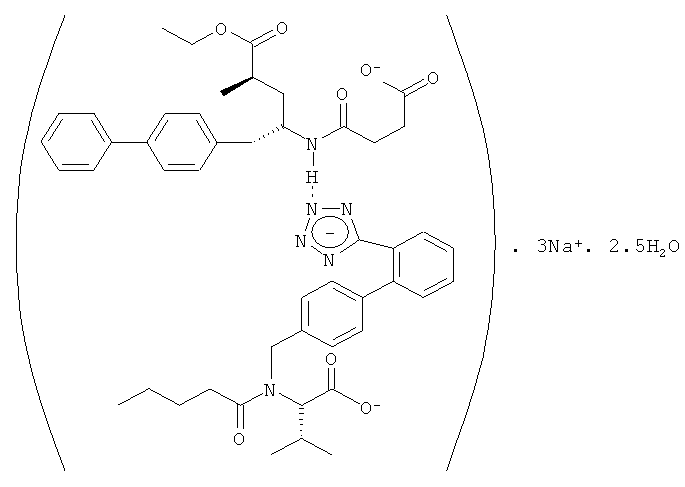
Указанный выше терапевтический агент включает антагонист рецептора ангиотензина, ингибитор нейтральной эндопептидазы (NEPi) и катион, т.е. Na. Получение указанного терапевтического агента и его характеристики подробно описаны в заявке WO 2007/056546. Указанный терапевтический агент представляет собой «соединение двойного действия», которое характеризуется одновременно двумя различными типами действия, т.е. одно действие представляет собой блокаду рецептора ангиотензина, обусловленную присутствием в соединении молекулярного фрагмента ARB, а другое действие заключается в ингибировании нейтральной эндопептидазы в результате присутствия в соединении молекулярного фрагмента NEPi. Терапевтический агент присутствует в фармацевтической композиции в количестве в диапазоне от приблизительно 4 мас.% до приблизительно 90 мас.%, например, от 4 мас.% до 60 мас.% в расчете на массу композиции.
Использованный в данном контексте термин «надмолекулярный комплекс» обозначает взаимодействие между двумя фармацевтически активными агентами, катионами и любыми другими присутствующими компонентами, такими как растворитель, особенно вода, за счет их нековалентного межмолекулярного связывания. Указанное взаимодействие приводит к ассоциации присутствующих в надмолекулярном комплексе компонентов, т.е. образуется комплекс в отличие от физической смеси компонентов.
Использованный в данном контексте термин «фармацевтическая композиция» обозначает, например смесь, содержащую терапевтически эффективное количество терапевтического соединения в фармацевтически приемлемом носителе, предназначенную для введения млекопитающему, например, человеку для лечения заболеваний, зависимых от киназ. Прежде всего, пригодной фармацевтической композицией по настоящему изобретению является фармацевтически приемлемая таблетка.
Использованный в данном контексте термин «фармацевтически приемлемые» обозначает такие соединения, материалы, композиции и/или лекарственные формы, которые по мнению специалистов в области медицины пригодны для контактирования с тканями млекопитающих, особенно человека, при этом не наблюдается избыточная токсичность, раздражение, аллергическая ответная реакция и другие осложнения, которые соизмеримы с допустимым соотношением польза/риск. В отношении фармацевтически приемлемой таблетки указанный термин включает также приемлемый профиль растворения in vitro.
Терапевтический агент присутствует в фармацевтической композиции в терапевтически эффективном количестве, которое зависит от скорости абсорбции, инактивации и скорости выведения терапевтического агента, а также от других факторов, известных специалисту в данной области техники. Кроме того, следует отметить, что дозировки также изменяются в зависимости от тяжести состояния, подлежащего лечению. Следует также понимать, что для любого конкретного реципиента в течение времени необходимо подбирать специальные курсы лечения в соответствии с индивидуальными потребностями пациента и мнением врача, осуществляющего введение или назначающего введение фармацевтических композиций. Терапевтическое соединение вводят в виде однократной дозы или дозу разделяют на ряд более низких доз, предназначенных для введения через различные интервалы времени. Таким образом, пригодное терапевтически эффективное количество определяется специалистом в данной области медицины.
Например, стандартная лекарственная форма терапевтического агента составляет величину в диапазоне от приблизительно 1 мг до приблизительно 1000 мг, например, от 40 мг до 400 мг (например, 100 мг, 200 мг, 400 мг) в сут. В другом варианте, используют более низкие дозы, например, дозы от 0,5 мг до 100 мг, от 0,5 мг до 50 мг, или от 0,5 мг до 20 мг в сут. В данном случае неожиданно было установлено, что компонент валсартана при доставке в форме соединения двойного действия, такого как надмолекулярный комплекс, характеризуется повышенным действием и, в связи с этим, более высокой биодоступностью по сравнению с введением валсартана в отдельности. Следовательно, можно снизить дозу компонента валсартана. Более конкретно, обычные дозы валсартана в композиции Diovan® составляют 80 мг, 160 мг и 320 мг. С учетом того, что соединение двойного действия, такое как надмолекулярный комплекс, включает компоненты - валсартан и этиловый эфир N-(3-карбокси-1-оксипропил)-(4S)-пара-фенилфенилметил)-4-амино-2R-метилмасляной кислоты, характеризующиеся достаточно близкими значениями молекулярной массы, в соотношении 1:1, то на основе их действия нельзя предсказать, что дозы 100 мг, 200 мг и 400 мг соединения двойного действия будут соответствовать однократным дозам 80 мг, 160 мг и 320 мг, соответственно, валсартана в составе композиции Diovan®.
Использованный в данном контексте термин «немедленное высвобождение» относится к быстрому высвобождению большей части терапевтического соединения, например более приблизительно 50%, приблизительно 55%, приблизительно 60%, приблизительно 65%, приблизительно 70%, приблизительно 75%, приблизительно 80%, или более приблизительно 90% в относительно короткий период времени, например в течение 1 ч, 40 мин, 30 мин или 20 мин после перорального введения. Прежде всего, пригодные условия для немедленного высвобождения обозначают высвобождение, по крайней мере, приблизительно 80% терапевтического соединения в течение 30 мин после перорального введения. Конкретные условия немедленного высвобождения конкретного терапевтического соединения известны или определяются специалистом в данной области техники. Профиль немедленного высвобождения определяют при проведении испытания на растворимость in vitro.
Использованный в данном контексте термин «растворение» относится к процессу диспергирования твердого вещества, в данном контексте активных ингредиентов, в молекулярной форме в среде. Скорость растворения активных ингредиентов фиксированной фармацевтической пероральной комбинации по настоящему изобретению определяется количеством лекарственного соединения, которое переходит в раствор в единицу времени в стандартных условиях: граница раздела фаз жидкость/поверхность твердого вещества, температура и состав растворителей. Скорость растворения определяют стандартными способами, известными специалисту в данной области техники, см. утвержденную методику, описанную в фармакопее США, раздел <711>, а также в европейской фармакопее, раздел 2.9.3, и в японской фармакопее. Согласно настоящему изобретению испытание для определения растворимости индивидуальных активных ингредиентов проводят по методике, описанной в фармакопее США, раздел <711>, при pH 6,8 при перемешивании лопастной мешалкой при 50 об/мин, или в другом варианте при pH 4,5 при перемешивании лопастной мешалкой при 75 об/мин. Растворяющей средой предпочтительно является буферный раствор, обычно фосфатный буферный раствор, прежде всего буферный раствор, описанный в примере «Определение растворимости».
Соответственно, в настоящем изобретении предпочтительно предлагаются твердые пероральные лекарственные формы, предназначенные для доставки терапевтически эффективного количества валсартана в форме свободной кислоты или фармацевтически приемлемой соли указанного соединения, причем пероральные лекарственные формы характеризуются таким профилем растворения in vitro, определенным методом, описанным в фармакопее США, при перемешивании лопастной мешалкой при приблизительно 50 об/мин в 900 мл фосфатного буферного раствора (0,05 М, pH 6,8) при 37±0,5°C, что при этом через 10 мин высвобождается в среднем от приблизительно 10 мас.% до приблизительно 100 мас.% валсартана в форме свободной кислоты или его фармацевтически приемлемой соли, через 20 мин высвобождается в среднем от приблизительно 30 мас.% до приблизительно 100 мас.% валсартана в форме свободной кислоты или его фармацевтически приемлемой соли, через 30 мин высвобождается в среднем от приблизительно 40 мас.% до приблизительно 100 мас.% валсартана в форме свободной кислоты или его фармацевтически приемлемой соли.
Кроме того, в настоящем изобретении предпочтительно предлагаются твердые пероральные лекарственные формы, предназначенные для доставки терапевтически эффективного количества валсартана в форме свободной кислоты или фармацевтически приемлемой соли указанного соединения, причем пероральная лекарственная форма характеризуется таким профилем растворения in vitro, определенным методом, описанным в фармакопее США, при перемешивании лопастной мешалкой при приблизительно 75 об/мин в 1000 мл фосфатного буферного раствора при pH 4,5 и 37±0,5°C, что при этом через 10 мин высвобождается в среднем от приблизительно 20 мас.% до приблизительно 100 мас.% валсартана в форме свободной кислоты или его фармацевтически приемлемой соли, через 20 мин высвобождается в среднем от приблизительно 30 мас.% до приблизительно 100 мас.% валсартана в форме свободной кислоты или его фармацевтически приемлемой соли, через 30 мин высвобождается в среднем от приблизительно 40 мас.% до приблизительно 100 мас.% валсартана в форме свободной кислоты или его фармацевтически приемлемой соли.
В одном варианте осуществления настоящего изобретения терапевтический агент присутствует в стандартной лекарственной форме в количестве приблизительно 100 мг, и пероральная лекарственная форма характеризуется таким профилем растворения in vitro, что при этом через 10 мин высвобождается в среднем приблизительно 50 мас.% валсартана в форме свободной кислоты, через 20 мин высвобождается в среднем приблизительно 85 мас.% валсартана в форме свободной кислоты, через 30 мин высвобождается в среднем приблизительно 95 мас.% валсартана в форме свободной кислоты. В другом варианте терапевтический агент присутствует в стандартной лекарственной форме в количестве приблизительно 200 мг, и пероральная лекарственная форма характеризуется таким профилем растворения in vitro, что при этом через 10 мин высвобождается в среднем приблизительно 50 мас.% валсартана в форме свободной кислоты, через 20 мин высвобождается в среднем приблизительно 85 мас.% валсартана в форме свободной кислоты, через 30 мин высвобождается в среднем приблизительно 95 мас.% валсартана в форме свободной кислоты. В еще одном варианте терапевтический агент присутствует в стандартной лекарственной форме в количестве приблизительно 400 мг, и пероральная лекарственная форма характеризуется таким профилем растворения in vitro, что при этом через 10 мин высвобождается в среднем приблизительно 40 мас.% валсартана в форме свободной кислоты, через 20 мин высвобождается в среднем приблизительно 70 мас.% валсартана в форме свободной кислоты, через 30 мин высвобождается приблизительно 90 мас.% валсартана в форме свободной кислоты.
Точная доза терапевтического агента и состав конкретной композиции, предназначенной для введения, зависят от ряда факторов, например, состояния, подлежащего лечению, требуемой продолжительности лечения и скорости высвобождения активного агента. Например, требуемое количество активного агента и скорость его высвобождения определяют на основе известных методик испытаний in vitro или in vivo, по продолжительности периода, в течение которого концентрация конкретного активного агента в плазме крови сохраняется на приемлемом для терапевтического действия уровне.
Неожиданно было установлено, что фармакокинетический/фармакодинамический (PK/PD) профиль, наблюдаемый для твердой пероральной лекарственной формы валсартана в форме свободной кислоты по настоящему изобретению, значительно отличается от профиля композиции, содержащей только валсартан, в частности состава Diovan®. Указанный отличительный уникальный фармакокинетический/фармакодинамический профиль валсартана в форме свободной кислоты, наблюдаемый после введения терапевтического агента, характеризуется в значительной степени повышенной пероральной биодоступностью (приблизительно в 1,4-1,6 раза, прежде всего, в 1,6 раза выше) и более быстрым началом действия (tмакс составляет 1,8±0,3 ч) по сравнению с профилем, наблюдаемым после введения валсартана в составе композиции Diovan® tмакс приблизительно составляет 2,6 ч). В целом, указанный фармакокинетический/фармакодинамический профиль лекарственной формы терапевтического агента по настоящему изобретению представляет перспективы для дальнейшей разработки улучшенного способа лечения сердечно-сосудистых заболеваний.
Соответственно, в настоящем изобретении предлагаются твердые пероральные лекарственные формы, предназначенные для доставки терапевтически эффективного количества валсартана в форме свободной кислоты или фармацевтически приемлемой соли указанного соединения, и среды носителя, причем указанная пероральная лекарственная форма обеспечивает быструю скорость абсорбции валсартана в форме свободной кислоты (tмакс составляет от 1 до 2,2 ч) после введения однократной дозы указанной лекарственной формы. Более конкретно, наблюдаемое tмакс составляет от 1,4 до 2,0 ч. Указанная величина в значительной степени отличается от скорости абсорбции валсартана после введения композиции Diovan®, при этом наблюдаемое tмакс составляет от 2,5 до 4,0 ч, более конкретно, от 2,8 до 3,0 ч. Например, в настоящем изобретении предлагается твердая пероральная лекарственная форма, включающая приблизительно 200 мг полупентагидрата тринатриевой соли [3-((1S,3R)-1-бифенил-4-илметил-3-этоксикарбонил-1-бутилкарбамоил)пропионат-(S)-3'-метил-2'-(пентаноил{2''-(тетразол-5-илат)бифенил-4'-илметил}амино)масляной кислоты], или соответствующее количество валсартана в форме свободной кислоты, и среду носителя, причем указанная лекарственная форма характеризуется величиной tмакс валсартана в форме свободной кислоты от 1,5 до 1,9 ч после введения однократной дозы указанной лекарственной формы.
Кроме того, в настоящем изобретении предлагается твердая пероральная лекарственная форма для доставки терапевтически эффективного количества валсартана в форме свободной кислоты, или фармацевтически приемлемой соли указанного соединения, и среда носителя, причем указанная пероральная лекарственная форма обеспечивает нормированное относительно дозы среднее содержание активного вещества в плазме (AUC0-24) от 230 до 400 нг·ч/мл/мг-экв. после введения однократной дозы указанной лекарственной формы. Более конкретно, нормированное относительно дозы среднее геометрическое содержание активного вещества в плазме (AUC0-24) составляет от 270 до 320 нг·ч/мл/мг-экв. Соответствующее содержание при введении валсартана форме композиции Diovan® составляет значительно более низкую величину. В связи с этим, в настоящем изобретении предлагается твердая пероральная лекарственная форма для доставки терапевтически эффективного количества валсартана в форме свободной кислоты или фармацевтически приемлемой соли указанного соединения, и среды носителя, причем относительная средняя биодоступность валсартана составляет от 140% до 185%, например, от 150% до 165% по сравнению с биодоступностью валсартана после введения композиции Diovan®. Например, в настоящем изобретении предлагается твердая пероральная лекарственная форма, включающая приблизительно 200 мг полупентагидрата тринатриевой соли [3-((1S,3R)-1-бифенил-4-илметил-3-этоксикарбонил-1-бутилкарбамоил)пропионат-(S)-3'-метил-2'-(пентаноил{2''-(тетразол-5-илат)бифенил-4'-илметил}амино)масляной кислоты], или соответствующее количество валсартана в форме свободной кислоты, и среду носителя, причем указанная лекарственная форма обеспечивает среднее содержание активного соединения в плазме (AUC0-24) от 16000 до 18000 нг·ч/мл, например, 16970 нг·ч/мл после введения однократной дозы указанной лекарственной формы.
Таким образом, твердая пероральная лекарственная форма по настоящему изобретению обеспечивает не только более высокую скорость абсорбции, но и более высокую степень абсорбции по сравнению с композицией Diovan®. Следует ожидать, что указанные фармакокинетические свойства будут обеспечивать терапевтические преимущества по сравнению с введением валсартана в отдельности.
Упомянутая выше твердая пероральная лекарственная форма включает части
валсартана или его соли, и
(2R,4S)-5-бифенил-4-ил-5-(3-карбоксипропиониламино)-2-метилпентановой кислоты или этилового эфира (2R,4S)-5-бифенил-4-ил-5-(3-карбоксипропиониламино)-2-метилпентановой кислоты или их соль.
Термин «части» обозначает, что компонент присутствует сам по себе или, предпочтительно, в форме надмолекулярного комплекса. Наиболее предпочтительно, твердая пероральная лекарственная форма включает полупентагидрат тринатриевой соли [3-((1S,3R)-1-бифенил-4-илметил-3-этоксикарбонил-1-бутилкарбамоил)пропионат-(S)-3'-метил-2'-(пентаноил{2''-(тетразол-5-илат)бифенил-4'-илметил}амино)масляной кислоты], т.е. оба фрагмента присутствуют в одной молекуле двойного действия или в надмолекулярном комплексе.
Использованный в данном контексте термин «эксципиент» относится к фармацевтически приемлемому инертному ингредиенту, который используют при получении твердых пероральных лекарственных форм. Примеры классов эксципиентов включают, но не ограничиваясь только ими, связующие, дезинтегрирующие агенты, замасливатели, регуляторы сыпучести, стабилизаторы, наполнители и разбавители. Эксципиенты повышают технологические характеристики фармацевтической композиции, т.е. упрощают процесс прямого прессования композиции за счет повышения текучести и/или когезионной способности.
Использованный в данном контексте термин «прямое прессование» относится к общему процессу прямого прессования ингредиентов (т.е. терапевтического агента и эксципиентов) в фармацевтическую композицию без изменения физических и химических свойств терапевтического агента. Терапевтический агент, наряду с фармацевтически приемлемыми эксципиентами, в форме порошков смешивают в устройстве с низким сдвигом, например, в смесителе с двойным корпусом. Затем полученной после смешивания композицией заполняют матрицу и непосредственно прессуют пуансоном. Указанную стадию прессования завершают с использованием, например, пресса для таблетирования. Пригодные для прямого прессования эксципиенты включают, но не ограничиваясь только ими, наполнители, связующие, замасливатели и регуляторы сыпучести. Прямое прессование, прежде всего, можно использовать для твердых пероральных лекарственных форм, включающих от 0,5 мг до 200 мг терапевтического агента.
Использованный в данном контексте термин «уплотнение» относится к общему процессу сухой грануляции для получения таблетки (например, при агрегировании или ротационном прессовании). Терапевтические агенты и фармацевтически приемлемые эксципиенты перерабатывают и получают агрегаты (при агрегировании) или ленты (при ротационном прессовании). В процессе ротационного прессования происходит уплотнение порошка за счет удаления воздуха. Уплотненный материал затем измельчают на гранулы одинакового размера и затем прессуют. Пригодные для ротационного прессования эксципиенты включают, но не ограничиваясь только ими, наполнители, связующие, замасливатели, дезинтегрирующие агенты и регуляторы сыпучести. Ротационное прессование, прежде всего, можно использовать для твердых пероральных лекарственных форм, включающих от 50 мг до 800 мг терапевтического агента.
Было установлено, что преимущество ротационного прессования, прежде всего, заключается в обеспечении включения более высоких доз терапевтического агента в таблетку пригодного размера без ухудшения качества лекарственного соединения. Прежде всего, сводится к минимуму или предотвращается образование избыточного количества аморфной формы, а также разделение компонентов соединения двойного действия.
Твердая пероральная лекарственная форма по настоящему изобретению включает стандартные добавки для указанной лекарственной формы. Используют вспомогательные вещества для таблетирования, обычно применяемые при получении таблеток, которые подробно описаны в соответствующей литературе, см., прежде всего, книгу Fiedler, «Lexicon der Hilfstoffe», А изд., ECV, Aulendorf (1996), которая включена в настоящее описание в качестве ссылки. Указанные вещества включают, но не ограничиваясь только ими, дезинтегрирующие агенты, связующие, замасливатели, регуляторы сыпучести, стабилизаторы, наполнители или разбавители, ПАВ и т.п.
Примеры фармацевтически приемлемых дезинтегрирующих агентов включают, но не ограничиваясь только ими, крахмалы, глины, целлюлозы, альгинаты, камеди, сшитые полимеры, например, сшитый поливинилпирролидон или кросповидон, например, продукт POLYPLASDONE XL фирмы International Specialty Products (Wayne, NJ), сшитую натриевую соль карбоксиметилцеллюлозы или натриевую соль кроскармеллозы, например, продукт AC-DI-SOL фирмы FMC, а также сшитую кальциевую соль карбоксиметилцеллюлозы, соевые полисахариды и гуаровую камедь, наиболее предпочтительно сшитый поливинилпирролидон или кросповидон. Дезинтегрирующий агент присутствует в количестве от приблизительно 0 до приблизительно 65 мас.%, например, от приблизительно 1 мас.% до приблизительно 40 мас.% (например, от приблизительно 0,05 мас.% до приблизительно 10 мас.%) в расчете на общую массу композиции (до необязательного нанесения покрытия).
Примеры фармацевтически приемлемых связующих включают, но не ограничиваясь только ими, крахмалы, целлюлозы и их производные, например, микрокристаллическую целлюлозу, например, продукт AVICEL РН фирмы FMC (Филадельфия, РА), гидроксипропилцеллюлозу, прежде всего, гидроксипропилцеллюлозу с низкой степенью замещения, например, гидроксипропилцеллюлозу с содержанием гидроксипропильных групп от 5 мас.% до 16 мас.% и ММ от приблизительно 80000 до 1150000, более предпочтительно от 140000 до 850000, например, продукт LH21, гидроксиэтилцеллюлозу и гидроксипропилметилцеллюлозу METHOCEL фирмы Dow Chemical Corp.(Midland, MI), сахарозу, декстрозу, кукурузный сироп, полисахариды и желатин, наиболее предпочтительно целлюлозу, такую как гидроксипропилцеллюлоза, прежде всего, гидроксипропилцеллюлоза с низкой степенью замещения. Если используют способы прямого прессования, связующее присутствует в количестве от приблизительно 1 мас.% до приблизительно 60 мас.%, например, от 5 мас.% до приблизительно 40 мас.% в расчете на общую массу композиции, прежде всего, от 10 мас.% до приблизительно 40 мас.% в расчете на общую массу композиции (до необязательного нанесения покрытия), или в случае ротационного прессования количество связующего составляет от приблизительно 5 мас.% до приблизительно 30 мас.% в расчете на общую массу композиции (до необязательного нанесения покрытия). Хотя некоторые из указанных эксципиентов можно использовать также в качестве дезинтегрирующих агентов, в настоящем изобретении указанные соединения использованы исключительно в качестве связующих.
Примеры фармацевтически приемлемых наполнителей включают, но не ограничиваясь только ими, кондитерский сахар, прессованный сахар, декстраты, декстрин, декстрозу, лактозу, маннит, микрокристаллическую целлюлозу, прежде всего, продукт Cellulose MK GR, порошкообразную целлюлозу, сорбит, а также сахарозу, прежде всего, микрокристаллическую целлюлозу. Наполнитель присутствует в количестве от приблизительно 4 мас.% до приблизительно 60 мас.%), например, от приблизительно 20 мас.% до приблизительно 40 мас.% в расчете на общую массу композиции (до необязательного нанесения покрытия).
Примеры фармацевтически приемлемых замасливателей и фармацевтически приемлемых регуляторов сыпучести включают, но не ограничиваясь только ими, коллоидный диоксид кремния, трисиликат магния, крахмалы, тальк, трикальций фосфат, стеарат магния, стеарат алюминия, стеарат кальция, карбонат магния, оксид магния, полиэтиленгликоль, порошкообразную целлюлозу, бегенат глицерина, стеариновую кислоту, гидрированное касторовое масло, моностеарат глицерина и стеарилфумарат натрия. Регулятор сыпучести присутствует в количестве от 0 до 10 мас.%, например, вплоть до 2 мас.%, например, приблизительно 0,1 мас.% (до необязательного нанесения покрытия). Замасливатель присутствует в количестве от 0 до 5 мас.%, например, от 0,5 мас.% до приблизительно 5 мас.% (до необязательного нанесения покрытия).
Предпочтительные твердые пероральные лекарственные формы характеризуются относительно низким содержанием добавок при высоком содержании активного агента. Указанные свойства позволяют получать стандартные лекарственные формы небольшого размера. Общее количество добавок в данной стандартной лекарственной форме составляет приблизительно 65 мас.% или менее в расчете на общую массу твердой пероральной лекарственной формы (до необязательного нанесения покрытия), более предпочтительно приблизительно 55 мас.% или менее. Предпочтительно, содержание добавок составляет величину в диапазоне от приблизительно 35 мас.%) до 55 мас.%), более предпочтительно от 40 мас.%о до 45 мас.%.
Абсолютные количества каждой добавки и количества относительно других добавок аналогичным образом зависят от требуемых свойств твердой пероральной лекарственной формы и определяются специалистом в данной области техники при проведении стандартных экспериментов. Например, можно выбрать твердую пероральную лекарственную форму с ускоренным и/или замедленным высвобождением активного агента, с количественно контролируемым или не контролируемым высвобождением активного агента.
Таким образом, если требуется ускоренное высвобождение, например, высвобождение 90 мас.% в течение 10 мин, более предпочтительно в течение 5 мин, используют дезинтегрирующий агент, такой как сшитый поливинилпирролидон, например, продукты, с зарегистрированными торговыми названиями Polyplasdone® XL или Kollidon® CL, прежде всего, характеризующиеся молекулярной массой более 1000000, более предпочтительно характеризующиеся распределением частиц по размеру менее 400 мкм или менее 74 мкм, или реакционноспособные добавки (шипучие смеси), которые вызывают быстрое распадение таблетки в присутствии воды, например, так называемые шипучие таблетки, которые содержат кислоту в твердой форме, обычно лимонную кислоту, или которые действуют в воде за счет высвобождения входящего в их состав химически связанного диоксида углерода, например, содержат гидрокарбонат натрия или карбонат натрия.
И наоборот, если требуется замедленное высвобождение, используют методику нанесения покрытия на пеллеты, матричные системы на основе воска, полимерные матричные таблетки или стандартные полимерные покрытия.
Количественный контроль высвобождения активного агента осуществляют по известным стандартным методикам. Указанные лекарственные формы известны и включают пероральные осмотические системы (OROS), таблетки с покрытиями, матричные таблетки, таблетки с прессованным покрытием, многослойные таблетки и т.п.
При получении твердой пероральной лекарственной формы, в которой активный агент полностью состоит из соединения двойного действия, полупентагидрата тринатриевой соли [3-((1S,3R)-1-бифенил-4-илметил-3-этоксикарбонил-1-бутилкарбамоил)пропионат-(S)-3'-метил-2'-(пентаноил{2''-(тетразол-5-илат)бифенил-4'-илметил}амино)масляной кислоты], предпочтительными добавками являются микрокристаллическая целлюлоза, гидроксипропилцеллюлоза, кросповидон, стеарат магния, стеарат кальция или стеарат алюминия, безводный коллоидный диоксид кремния и тальк. Количество используемой добавки зависит от требуемого количества активного агента. Стеарат, например, стеарат магния, предпочтительно используют в количестве от 1,0 мас.% до 6,0 мас.%), например, от 1,5 мас.% до 4,0 мас.% (до необязательного нанесения покрытия). Тогда как диоксид кремния предпочтительно используют в количестве от 0,1 мас.% до 2 мас.%. Микрокристаллическая целлюлоза предпочтительно присутствует в количестве от 10 мас.% до 30 мас.%, например, 20-21 мас.%. Кросповидон предпочтительно присутствует в количестве от 1 мас.% до 20 мас.%, более предпочтительно от 5 мас.% до 15 мас.%), например, 8-10 мас.%
Твердые пероральные лекарственные формы по настоящему изобретению могут быть в виде драже, в этом случае на твердую пероральную лекарственную форму наносят покрытие, обычно сахар, шеллак или другое стандартное пленочное покрытие. В данной области техники известно множество способов нанесения покрытий, например, нанесение покрытий при распылении в псевдоожиженном слое, например, известными способами с использованием приборов фирм Aeromatic, Glatt, Wurster или Huttlin, способом Accela Cota в перфорированном поддоне, или методом «погружения меча». В указанных способах применяют стандартные добавки, обычно используемые для получения лекарственных средств.
В другом объекте настоящего изобретения предлагается способ получения описанной выше твердой пероральной лекарственной формы. Указанную твердую пероральную лекарственную форму получают при обработке пригодных количеств определенной выше конечной композиции, при этом получают стандартные лекарственные формы.
В одном варианте предлагается способ получения описанных выше твердых пероральных лекарственных форм, который включает стадии:
(a) смешивания соединения двойного действия, по крайней мере, с одним фармацевтически приемлемым эксципиентом, с получением смеси,
(b) прямое прессование указанной смеси в твердую пероральную лекарственную форму.
В другом предпочтительном варианте осуществления настоящего изобретения предлагается способ получения твердой пероральной лекарственной формы по настоящему изобретению, включающей повышенные дозы соединения двойного действия. Указанную твердую пероральную лекарственную форму получают способом, который включает стадии смешивания соединения двойного действия, по крайней мере, с одним фармацевтически приемлемым эксципиентом, с получением смеси, уплотнение указанной смеси, например методом ротационного прессования, необязательное смешивание с дополнительными фармацевтически приемлемыми эксципиентами, и необязательное прессование конечной смеси, с получением твердой пероральной лекарственной формы.
Более конкретно, указанный способ включает стадии:
(a) просеивания соединения двойного действия и фармацевтически приемлемых эксципиентов, с получением просеянного материала,
(b) смешивания просеянного материала, с получением смешанного материала,
(c) уплотнения смешанного материала, например методом ротационного прессования, с получением уплотненного материала,
(d) измельчения уплотненного материала, с получением измельченного материала, называемого гранулятом,
(e) необязательного смешивания измельченного материала с внешней фазой, т.е. с фармацевтически приемлемыми эксципиентами, с получением конечной смеси,
(f) необязательного прессования конечной смеси, с получением таблетки, и
(g) необязательного нанесения пленочного покрытия, с получением таблетки с пленочным покрытием.
Указанный способ проводят в отсутствии воды, т.е. он представляет собой способ сухого прессования при относительной влажности предпочтительно <55% и предпочтительно при комнатной температуре (20-25°C), но температуру можно повысить вплоть до 65°C, например, вплоть до 40°C. Указанные условия являются предпочтительными и позволяют исключить распад на индивидуальные компоненты или образование аморфной формы терапевтического агента.
При уплотнении требуется уплотнение сухих измельченных компонентов. Уплотнение можно проводить с использованием метода агрегации или, предпочтительно, метода ротационного прессования. В приборе для ротационного прессования в основном используют два ролика, вращающихся во встречном направлении. Гидравлический плунжер сближает вращающиеся ролики друг с другом, при этом происходит прессование измельченных частиц, которые подаются в ротационный пресс с помощью системы шнеков. Предпочтительно используют силу прессования от 20 кН до 60 кН, более предпочтительно от 20 кН до 40 кН. Неожиданно было установлено, что при прессовании в указанном диапазоне силы прессования можно получать композицию терапевтического агента без распада терапевтического агента на индивидуальные компоненты или без образования его аморфной формы. Конкретная оптимальная сила прессования зависит от содержания активного агента в данной композиции и, в связи с этим, зависит также от количества и природы присутствующих добавок.
Неожиданно было установлено, что, несмотря на относительно слабые нековалентные силы, связывающие компоненты терапевтического агента, описанные выше методики позволяют обрабатывать композиции без нарушения целостности терапевтического агента и обеспечивают надежные способы получения пригодных твердых пероральных лекарственных форм.
Следующие примеры приведены для иллюстрации настоящего изобретения и не ограничивают его объем. В примерах описан способ осуществления настоящего изобретения на практике.
Количества ингредиентов фармацевтической композиции, представленные в массовых процентах и использованные в каждом примере, приведены в соответствующих таблицах.
Примеры 1 и 2
В указанном примере в качестве терапевтического агента использовали полупентагидрат тринатриевой соли [3-((1S,3R)-1-бифенил-4-илметил-3-этоксикарбонил-1-бутилкарбамоил)пропионат-(S)-3'-метил-2'-(пентаноил{2''-(тетразол-5-илат)бифенил-4'-илметил}амино)масляной кислоты]. В табл.1 представлены композиции, полученные в примерах 1 и 2 и включающие 5 мг и 50 мг терапевтического агента, соответственно.
Терапевтический агент сначала просеивали через сито с размером ячеек 40 меш. К терапевтическому агенту добавляли микрокристаллическую целлюлозу и кросповидон, смесь просеивали через сито с размером ячеек 20 меш. Затем смесь перемешивали в бункерном смесителе (приблизительно 100 вращений). Затем в бункерный смеситель добавляли гидроксипропилцеллюлозу с низкой степенью замещения и коллоидный диоксид кремния и смесь перемешивали (еще 100 вращений). Затем в смесь добавляли тальк и снова перемешивали, затем добавляли стеарат магния. Затем порошкообразную смесь прессовали в таблетку массой приблизительно 115 мг (пример 1) и приблизительно 575 мг (пример 2). На фиг.1 представлены профили растворения при pH 6,8 для таблеток, полученных в указанных примерах.
Примеры 3-6
В указанном примере в качестве терапевтического агента использовали полупентагидрат тринатриевой соли [3-((1S,3R)-1-бифенил-4-илметил-3-этоксикарбонил-1-бутилкарбамоил)пропионат-(S)-3'-метил-2'-(пентаноил{2''-(тетразол-5-илат)бифенил-4'-илметил}амино)масляной кислоты]. В табл.2 и табл.3 представлены композиции, полученные в примерах 3-6 и включающие 40 мг, 100 мг, 200 мг и 400 мг терапевтического агента, соответственно.
Стеарат магния, тальк, коллоидный диоксид кремния и микрокристаллическую целлюлозу сначала просеивали через сито с размером ячеек 30 меш. Указанную выше смесь, терапевтический агент, кросповидон и гидроксипропилцеллюлозу с низкой степенью замещения затем смешивали в бункерном смесителе (приблизительно 120 вращений). Затем полученную смесь просеивали через сито с размером ячеек 30 меш. Просеянную смесь затем перемешивали в бункерном смесителе (приблизительно 120 вращений). Смесь прессовали в ротационном прессе ВЕРЕХ 50 при силе прессования 30 кН. После прессования смесь измельчали на вибрационной мельнице Frewitt и просеивали через сито с размером ячеек 18 меш, при этом получали конечную внутреннюю фазу или гранулят. Гранулят смешивали с кросповидоном и тальком (эксципиенты внешней фазы), просеивали через сито с размером ячеек 30 меш, смешивали в бункерном смесителе (приблизительно 50 вращений). Затем полученную смесь смешивали со стеаратом магния (эксципиент внешней фазы), просеивали через сито с размером ячеек 30 меш, смешивали в бункерном смесителе (приблизительно 50 вращений). Затем полученную конечную смесь прессовали в таблетку в прессе Fette 3090. Наносили полимерное покрытие с красителем Opadry, при этом получали таблетки с покрытием. Профили растворения при pH 6,8 для таблеток, полученных в примерах 3-5, представлены на фиг.2, а профиль растворения при pH 4,5 для таблеток, полученных в примере 6, показан на фиг.3.
Пример
Определение растворимости
Исследовали растворимость таблеток, описанных в примерах, в 900 мл фосфатного буферного раствора при pH 6,8 при перемешивании лопастной мешалкой при 50 об/мин.
Прибор включает следующие элементы: закрытый сосуд из стекла или другого инертного прозрачного материала, мотор, и в качестве перемешивающего элемента лопасть и вал. Сосуд частично погружен в пригодную водяную баню любого стандартного размера или снабжен нагревающей рубашкой. Водяная баня или нагревающая рубашка обеспечивают температуру в сосуде во время испытания при 37±0,5° и равномерную циркуляцию нагревающей жидкости. Ни один из элементов прибора, включая окружающую среду, в которой он находится, кроме перемешивающего равномерно вращающегося элемента, не является источником интенсивного движения, встряхивания или вибрации. Прибор, который позволяет наблюдать за образцом и перемешивающим элементом во время испытания, характеризуется следующими размерами и емкостью: высота составляет от 160 мм до 210 мм, а внутренний диаметр составляет от 98 мм до 106 мм. Стороны прибора загнуты в верхней части. Для снижения испарения можно использовать подходящую крышку. Вал расположен таким образом, чтобы его ось не отклонялась более чем на 2 мм в каждой точке от вертикальной оси сосуда, и вращается равномерно без отклонений. Вертикальная центральная линия лопасти проходит через ось вала таким образом, что нижняя часть лопасти совпадает с нижней частью вала. Конструкция лопасти аналогична конструкции, представленной на фиг.2 в разделе <711> фармакопеи США. Во время испытаний поддерживается расстояние 25±2 мм между лопастью и дном сосуда. Металлические или изготовленные из пригодного инертного материала лопасть и вал составляют единое целое. Можно использовать пригодную состоящую из двух съемных элементов конструкцию, при условии, что указанные элементы надежно закреплены во время испытаний. Лопасть мешалки и вал можно покрывать пригодным инертным покрытием. Лекарственную форму погружают на дно сосуда, затем включают вращение мешалки. Чтобы исключить всплывание лекарственной формы, к ней можно прикрепить небольшое количество нереакционноспособного материала, например нескольких витков проволоки. Можно использовать другие пригодные балластные материалы.
Водный буферный раствор (1 л), pH которого доводили до 6,8±0,05 (0,05 М фосфатный буферный раствор, названный в данном контексте «растворяющая среда», полученный при растворении в воде 6,805 г дигидрофосфата калия и 0,896 г гидроксида натрия, объем доводили водой до 1000 мл, pH доводили до 6,80±0,05 с использованием 0,2 М гидроксида натрия или 1 М фосфорной кислоты), помещали в сосуд прибора, прибор собирали, устанавливали температуру растворяющей среды при 37±0,5°C и термометр удаляли. Одну лекарственную форму (например, таблетку или капсулу) помещали в прибор, при этом следует исключить образование пузырьков воздуха на поверхности стандартной лекарственной формы, и сразу же включали прибор со скоростью 50+2 об/мин. В течение определенного периода времени (например, 10, 20, 30, 45, 60, 90 и 120 мин) или в каждый указанный момент времени отбирали образец (>1 мл) из средней зоны между поверхностью растворяющей среды и верхней частью вращающейся лопасти, на расстоянии не менее 1 см от стенки сосуда. (Примечание: отобранные для анализа аликвотные части заменяли эквивалентным объемом свежей растворяющей среды при 37° или, если установлено, что необходимости в возмещении растворяющей среды не существует, расчеты проводили с поправкой на изменение объема. Сосуд в течение испытаний закрывали и измеряли температуру исследуемой смеси через определенные промежутки времени). Образец фильтровали через пригодный фильтр, например, через фильтр PVDF (Millipore) с диаметром пор 0,45 мкм и первые образцы (от 2 до 3 мл) фильтрата отбрасывали. Анализ проводили методом ЖХВР с детекцией в УФ. Испытания повторяли по крайней мере 6 раз с использованием дополнительных стандартных лекарственных форм.
Соответствующие профили растворения представлены на фиг.1 и фиг.2. Для таблеток, полученных в примере 1 и примере 2, наблюдается высвобождение более 90 мас.% терапевтического агента в течение менее 10 мин, а для таблеток, полученных в примерах 3-5, наблюдается высвобождение более 70 мас.% терапевтического агента в течение менее 20 мин.
Описанный выше способ можно также использовать для анализа полученных в примерах таблеток при pH 4,5 со следующими изменениями.
Фосфатный буферный раствор, pH 4,5, получали при растворении 13,61 г дигидрофосфата калия в 750 мл воды, при необходимости доводили pH с использованием 0,1 М гидроксида натрия или 0,1 М соляной кислоты, затем объем раствора доводили водой до 1000,0 мл.
Условия определения растворимости при pH 4,5
Условия
На фиг.3 представлен профиль растворения таблеток, полученных в примере 6, при pH 4,5. Для таблеток, полученных в примере 6, наблюдается высвобождение более 80 мас.% терапевтического агента в течение менее 20 мин.
Примеры
Определение фармакокинетических параметров
1) Дозы от 5 до 80 мг
Проводили двухфазные клинические испытания с плацебо-контролем с использованием параллельных групп при увеличении однократной дозы (5 мг, 20 мг, 80 мг препарата LCZ696 и 40 мг Diovan®, коммерческая композиция валсартана) и выбором доз на основании рекомендаций по исследованию новых лекарственных препаратов Комитета по контролю за пищевыми продуктами и лекарственными средствами (FDA). Препарат LCZ696 вводили в виде таблеток, содержащих 5 мг и 50 мг активного агента, получение которых описано выше, а для введения дозы 20 мг и 80 мг использовали несколько таблеток массой 5 мг и/или 50 мг.
Для сравнения действия валсартана в виде препарата LCZ696 и в виде препарата Diovan® в различных дозах содержание валсартана в плазме (AUC) и Cмакс нормировали относительно реального количества введенного безводного не содержащего соли валсартана (нормировали в мг-экв.). Значения AUC0-24 рассчитывали в виде нг·ч/мл/мг-экв. валсартана и сравнивали геометрические средние значения. Средняя относительная биодоступность валсартана при введении препарата LCZ696 значительно превышала среднюю относительную биодоступность валсартана при введении препарата Diovan®, при этом соотношение геометрических средних для трех групп, которым вводили препарат LCZ696, изменялось от 107% до 249%. Содержание валсартана в плазме после ведения препарата LCZ696 линейно возрастало с увеличением дозы, а так как для 3 групп не наблюдалось статистически значимых отклонений нормированного по отношению к дозе содержания валсартана, то для суммарной оценки относительной биодоступности валсартана в виде препарата LCZ696 по сравнению с препаратом Diovan® (40 мг) данные для всех 3 групп объединяли (n равно 24). Значения содержания в плазме (AUC) и Cмакс, нормированные относительно мг-экв. безводного не содержащего соли валсартана, представлены в табл.4.
Скорость и степень абсорбции валсартана после ведения препарата LCZ696 повышаются по сравнению с препаратом Diovan®. После введения препарата LCZ696 наблюдается более высокое нормированное в отношении дозы Смакс валсартана по сравнению со значением Cмакс валсартана после введения препарата Diovan® (соотношение средних геометрических для AUC составляет 161%, 90% доверительный коэффициент: 140-185%). Кроме того, после введения препарата LCZ696 наблюдается тенденция к снижению tмакс (среднее значение 1,3-1,8 ч) по сравнению с tмакс (среднее значение 2,4-3,0 ч) после введения препарата Diovan®.
2) Дозы от 50 до 1200 мг
Для оценки безопасности, переносимости и фармакокинетических свойств препарата LCZ696 проводили перекрестные рандомизированные периодические клинические испытания двойным слепым методом с контролем плацебо при увеличении однократной и многократных доз с использованием параллельных групп с участием здоровых добровольцев. В ходе указанных испытаний в течение 14 дней вводили следующие дозы: для групп с однократной дозой использовали дозы 200 мг, 600 мг, 900 мг и 1200 мг, для групп с многократной дозой использовали дозы 50 мг, 200 мг, 600 мг и 900 мг.
Оценки средних значений tмакс и t1/2 для всех исследуемых соединений были сопоставимы с полученными ранее данными при проведении описанных выше исследований с использованием более низких однократных доз.
Для всех исследуемых соединений наблюдается минимальное накопление после введения 50-900 мг препарата LCZ696 один раз в сут в течение 14 дней.
Следует понимать, что хотя настоящее изобретение подробно описано в данном контексте, представленное выше описание предназначено для иллюстрации настоящего изобретения и не ограничивает его объем, который определен в пунктах следующей формулы изобретения. Другие объекты, преимущества и модификации включены в объем формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ГАЛЕНОВЫЕ КОМПОЗИЦИИ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ | 2017 |
|
RU2773029C2 |
| ЛЕЧЕНИЕ ГИПЕРТЕНЗИИ И/ИЛИ ПРЕДОТВРАЩЕНИЕ И ЛЕЧЕНИЕ СЕРДЕЧНОЙ НЕДОСТАТОЧНОСТИ У МЛЕКОПИТАЮЩЕГО, ПОЛУЧАЮЩЕГО ТЕРАПИЮ АНТИКОАГУЛЯНТАМИ | 2011 |
|
RU2564941C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМБИНАЦИИ АНТАГОНИСТА РЕЦЕПТОРА АНГИОТЕНЗИНА И ИНГИБИТОРА NEP | 2006 |
|
RU2503668C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМБИНАЦИИ АНТАГОНИСТА РЕЦЕПТОРА АНГИОТЕНЗИНА И ИНГИБИТОРА NEP | 2006 |
|
RU2459809C2 |
| ИНГИБИТОРЫ NEP ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, ХАРАКТЕРИЗУЮЩИХСЯ УВЕЛИЧЕНИЕМ ИЛИ РЕМОДЕЛИРОВАНИЕМ ПРЕДСЕРДИЯ | 2018 |
|
RU2809222C2 |
| ИНГИБИТОРЫ NEP ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, ХАРАКТЕРИЗУЮЩИХСЯ УВЕЛИЧЕНИЕМ ИЛИ РЕМОДЕЛИРОВАНИЕМ ПРЕДСЕРДИЯ | 2013 |
|
RU2667643C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2001 |
|
RU2333757C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ВАЛСАРТАНА | 2008 |
|
RU2487710C2 |
| ГАЛЕНОВЫЙ СОСТАВ АЛИСКИРЕНА И ГИДРОХЛОРТИАЗИДА | 2007 |
|
RU2491058C2 |
| СОСТАВ С МОДИФИЦИРОВАННЫМ ВЫСВОБОЖДЕНИЕМ, СОДЕРЖАЩИЙ 1-[(3-ГИДРОКСИАДАМАНТ-1-ИЛАМИНО)АЦЕТИЛ]ПИРРОЛИДИН-2(S)-КАРБОНИТРИЛ | 2006 |
|
RU2423124C2 |
Изобретение относится к твердой пероральной лекарственной форме, предназначенной для доставки терапевтически эффективного количества валсартана в виде свободной кислоты или его фармацевтически приемлемой соли. Лекарственная форма включает соединение двойного действия в количестве от 4 до 90% в расчете на массу композиции и фармацевтически приемлемые эксципиенты. Соединением двойного действия является полупентагидрат тринатриевой соли [3-((1S,3R)-1-бифенил-4-илметил-3-этоксикарбонил-1-бутилкарбамоил)пропионат-(S)-3'-метил-2'-(пентаноил{2''-(тетразол-5-илат)бифенил-4'-илметил}амино)масляной кислоты]. Указанное соединение двойного действия присутствует в количестве 40, 50, 100, 200 или 400 мг в одной стандартной лекарственной форме. Также изобретение относится к способу получения твердой пероральной лекарственной формы. Лекарственная форма по изобретению характеризуется таким профилем растворения in vitro, что через 30 мин высвобождается в среднем от приблизительно 10 до приблизительно 100 мас.% валсартана в форме свободной кислоты или его фармацевтически приемлемой соли. Указанные профиль растворения и скорость абсорбции валсартана обеспечивают общую более низкую дозу валсартана. 5 н. и 10 з.п. ф-лы, 3 ил., 6 табл., 6 пр.
1. Твердая пероральная лекарственная форма, предназначенная для доставки терапевтически эффективного количества валсартана в форме свободной кислоты или его фармацевтически приемлемой соли, включающая:
(a) соединение двойного действия в количестве от приблизительно 4 до приблизительно 90% в расчете на массу композиции; и
(b) по крайней мере, один фармацевтически приемлемый эксципиент, где соединением двойного действия является полупентагидрат тринатриевой соли [3-((1S,3R)-1-бифенил-4-илметил-3-этоксикарбонил-1-бутилкарбамоил)пропионат-(S)-3'-метил-2'-(пентаноил{2''-(тетразол-5-илат)бифенил-4'-илметил}амино)масляной кислоты] и где соединение двойного действия присутствует в количестве 40, 50, 100, 200 или 400 мг в одной стандартной лекарственной форме; и
где твердая пероральная лекарственная форма характеризуется таким профилем растворения in vitro, что через 30 мин высвобождается в среднем от приблизительно 10 до приблизительно 100 мас.% валсартана в форме свободной кислоты или его фармацевтически приемлемой соли.
2. Твердая пероральная лекарственная форма по п.1, где указанная пероральная дозированная форма включает полупентагидрат тринатриевой соли [3-((1S,3R)-1-бифенил-4-илметил-3-этоксикарбонил-1-бутилкарбамоил)пропионат-(S)-3'-метил-2'-(пентаноил{2''-(тетразол-5-илат)бифенил-4'-илметил}амино)масляной кислоты] в количестве от 4 до 60% в расчете на массу композиции.
3. Твердая пероральная лекарственная форма по п.1, где указанная твердая пероральная лекарственная форма представляет собой таблетку.
4. Твердая пероральная лекарственная форма по п.3, где указанная таблетка представляет собой состав немедленного высвобождения.
5. Твердая пероральная лекарственная форма по п.4, где указанная таблетка представляет собой ротационно спрессованную таблетку.
6. Твердая пероральная лекарственная форма по п.1, где фармацевтически приемлемые эксципиенты включают (i) микрокристаллическую целлюлозу, (ii) гидроксипропилцеллюлозу, (iii) кросповидон, (iv) стерат Mg, Ca или Al, (v) безводный коллоидный диоксид кремния и (vi) тальк.
7. Твердая пероральная лекарственная форма по п.6, где стеарат Mg используют в количестве от 1,0 мас.% до 6 мас.%, безводный коллоидный диоксид кремния используют в количестве от 0,1 мас.% до 2 мас.%, микрокристаллическая целлюлоза присутствует в количестве от 10 мас.% до 30 мас.%, и кросповидон присутствует в количестве от 1 мас.% до 20 мас.%.
8. Способ получения твердой пероральной лекарственной формы по п.1 или 2, включающий стадии:
(a) смешивания полупентагидрата тринатриевой соли [3-((1S,3R)-1-бифенил-4-илметил-3-этоксикарбонил-1-бутилкарбамоил)пропионат-(S)-3'-метил-2'-(пентаноил{2''-(тетразол-5-илат)бифенил-4'-илметил}амино)масляной кислоты], по крайней мере, с одним фармацевтически приемлемым эксципиентом с получением смеси;
(b) прямого прессования указанной смеси в твердую пероральную лекарственную форму.
9. Способ получения твердой пероральной лекарственной формы по п.1 или 2, включающий стадии:
(a) смешивания полупентагидрата тринатриевой соли [3-((1S,3R)-1-бифенил-4-илметил-3-этоксикарбонил-1-бутилкарбамоил)пропионат-(S)-3'-метил-2'-(пентаноил{2''-(тетразол-5-илат)бифенил-4'-илметил}амино)масляной кислоты], по крайней мере, с одним фармацевтически приемлемым эксципиентом с получением смеси;
(b) уплотнения указанной смеси ротационным прессованием;
(c) необязательного смешивания с дополнительными фармацевтически приемлемыми эксципиентами; и
(d) необязательного прессования конечной смеси в твердую пероральную лекарственную форму.
10. Способ получения твердой пероральной лекарственной формы по п.9, включающий стадии:
(a) просеивания соединения двойного действия и фармацевтически приемлемых ингредиентов с получением просеянного материала;
(b) смешивания просеянного материала с получением смешанного материала;
(c) уплотнения смешанного материала, например, методом ротационного прессования, с получением уплотненного материала;
(d) измельчения уплотненного материала с получением измельченного материала, называемого гранулятом;
(e) необязательного смешивания измельченного материала с дополнительными фармацевтически приемлемыми эксципиентами с получением конечной смеси;
(f) необязательного прессования конечной смеси с получением таблетки; и
(g) необязательного нанесения пленочного покрытия с получением таблетки с пленочным покрытием.
11. Твердая пероральная лекарственная форма, полученная способом по любому из пп.8-10, которая представлена в форме таблетки.
12. Твердая пероральная лекарственная форма по п.1 или 2, где указанная твердая пероральная лекарственная форма характеризуется таким профилем растворения in vitro, что через 10 мин высвобождается в среднем по меньшей мере приблизительно 40 мас.% валсартана в форме свободной кислоты или его фармацевтически приемлемой соли.
13. Твердая пероральная лекарственная форма по любому из пп.1 или 2, где (i) соединение двойного действия присутствует в количестве приблизительно 100 мг в одной стандартной лекарственной форме; и где твердая пероральная лекарственная форма характеризуется таким профилем растворения in vitro, что через 10 мин высвобождается в среднем приблизительно 50 мас.% валсартана в форме свободной кислоты, через 20 мин высвобождается в среднем приблизительно 85 мас.% валсартана в форме свободной кислоты, через 30 мин высвобождается в среднем приблизительно 95 мас.% валсартана в форме свободной кислоты, или
(ii) соединение двойного действия присутствует в количестве приблизительно 200 мг в одной стандартной лекарственной форме; и где твердая пероральная лекарственная форма характеризуется таким профилем растворения in vitro, что через 10 мин высвобождается в среднем приблизительно 50 мас.% валсартана в форме свободной кислоты, через 20 мин высвобождается в среднем приблизительно 85 мас.% валсартана в форме свободной кислоты, через 30 мин высвобождается в среднем приблизительно 95 мас.% валсартана в форме свободной кислоты, или
(iii) соединение двойного действия присутствует в количестве приблизительно 400 мг в одной стандартной лекарственной форме; и где твердая пероральная лекарственная форма характеризуется таким профилем растворения in vitro, что через 10 мин высвобождается в среднем приблизительно 40 мас.% валсартана в форме свободной кислоты, через 20 мин высвобождается в среднем приблизительно 70 мас.% валсартана в форме свободной кислоты, через 30 мин высвобождается в среднем приблизительно 90 мас.% валсартана в форме свободной кислоты.
14. Твердая пероральная лекарственная форма по п.1 или 2, предназначенная для доставки, после введения, терапевтически эффективного количества валсартана в форме свободной кислоты или его фармацевтически приемлемой соли, причем после введения однократной дозы указанной лекарственной формы скорость абсорбции валсартана в форме свободной кислоты tмакс составляет от 1 до 2,2 ч и/или после введения однократной дозы указанной лекарственной формы нормированное относительно дозы среднее содержание в плазме (AUC0-24) составляет от 230 до 400 нг·ч/мл/мг-экв.
15. Применение твердой пероральной лекарственной формы по п.14 для повышения скорости абсорбции и/или содержания валсартана в форме свободной кислоты в плазме, причем указанный способ заключается во введении указанной твердой пероральной лекарственной формы пациенту, нуждающемуся в этом.
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| US 6248729 B1, 19.06.2001 | |||
| WO 00/38676 A1, 06.07.2000 | |||
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |

