Область техники, к которой относится изобретение
Изобретение относится к фармацевтической промышленности и касается новых производных мишень-специфичного макролидного антибиотика олигомицина А и способа их получения.
Уровень техники
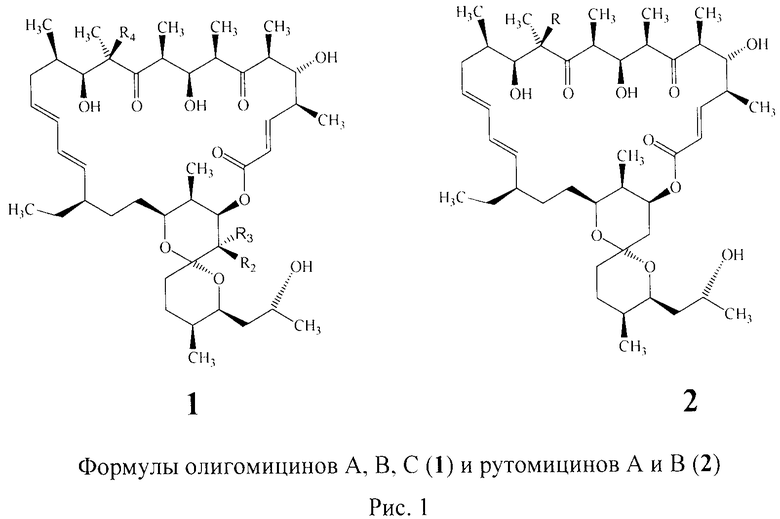
Макролидные антибиотики являются высокоэффективными природными препаратами, проявляющими противогрибковый и антибактериальный эффекты. Важнейшими представителями этой группы являются олигомицин А (формула 1, R1=СН3, R2=СН3, R3=Н, R4=ОН), олигомицин В (формула 1 R1=С=О, R2=Н, R3=СН3, R4=ОН), олигомицин С (формула 1, R1=СН2, R2=Н, R3=СН3, R4=Н)2, R4=Н), рутамицин А (формула 2, R4=OH), рутамицин В (формула 2, R=H) (Рис.1).
Олигомицины и рутамицины представляют собой 26-членные α,β-ненасыщенные лактоны, сочлененные с бициклической спирокетальной структурой.
Механизм действия макролидных антибиотиков группы олигомицинов основан на нарушении связи F0 и F1 факторов митохондриальной АТФ-синтазы и ингибировании процессов фосфорилирования в митохондриях. Олигомицин специфичен в отношении АТФ-синтазы и в микромолярных концентрациях блокирует транспорт протонов через субъединицу F0. Ферментный комплекс F0F1 АТФ-синтаза рассматривается как молекулярная мишень новых лекарств для терапии опухолей и инфекций (Shchepina et al. Oligomycin, inhibitor of the FO part of H+-ATP-synthase, suppresses the TNE-incklced apoptosis, Oncogene (2002) 21, 8149-8157). Благодаря специфическому подавлению митохондриальной АТФ-синтазы олигомицины проявляют антибактериальное, противогрибковое и противоопухолевое действие. Антибиотики группы олигомицинов наряду с ценными свойствами имеют ряд недостатков, основными из которых являются: высокая токсичность и низкая растворимость.
Химическая модификация макролидных антибиотиков группы олигомицинов высокоактивных ингибиторов АТФ-синтазы направлена на получение новых производных, обладающих селективным противоопухолевым или антиинфекционным действием и имеющих преимущества перед исходными антибиотиками. В то же время химическая модификация других макролидов (эритромицин) привела к высокоактивным избирательно действующим антибактериальным препаратам, доминирующим в настоящее время на фармацевтическом рынке. Одним из важных синтетических подходов в химии макролидов является осуществление полных синтезов некоторых антибиотиков этой группы и получение их аналогов (Evans et al., Total synthesis of oligomycin C.J. Org. Chem. 1998. 63. 4572-4576; Panek. Nareshkumar. Total synthesis of rutamycin В and oligomycin C.J. Org. Chem., 2001, 66, 2747 2756; White et al., Total synthesis of rutamycin B, a macrolide antibiotic from Streptomyces aureofaciens. J. Org. Chem., 2001, 66, 5217 5231). В рамках этих синтезов были разработаны синтетические подходы к синтезу макролактонных циклов и гликозилированию полученных агликонов различными, в том числе неприродными, сахарами. Наиболее полно эти подходы были использованы при синтезе антибиотиков семейства апоптолидинов и их производных (Wehlan et al. Apoptolidin A: total synthesis and partially glycosylated analogues. Chemistry - Eur. 7., 2006, 12, 7378-7397; Ghidu et al., Synthesis and evaluation of the cytotoxicity of apoptolidinones A and D. J. Org. Chem. 2008, 73, 4949-4955).
Получение полусинтетических олигомицинов затруднено из-за сложности структуры молекулы - наличием большого числа асимметрических атомов, функциональных групп и лабильностью антибиотика, неустойчивого уже в слабокислых и щелочных условиях. Серьезную проблему представляет не только выбор селективных методов модификации, но и строгая идентификация полученных производных. Наиболее доступными для модификации представляются кислородсодержащие группы (гидрокси- и карбонильные группы. Описано частичное ацетилирование олигомицина А, которое привело к образованию 5,9,33-три-О-ацетил и 5,9,13,33-тетраацетил производных. Эти производные не проявили ингибирующей активности при тестировании на спорах Aspergillus niger (Szilagyi et al., Structure elucidation of two acetylated derivatives of oligomycin A, Spectroscopy Lett., 1995, 28, 699-707). Олигомицин А содержит несколько кето-групп, реакционная способность которых изучена (Ramirez et al., Effects of borohydride-treated oligomycins on processes of energy transduction in mitochondria. Eur. J. Biochem., 1982, 121, 2275-2279). Авторы показали, что постепенное прибавление боргидрида натрия в этанольный раствор рутамицина и олигомицинов восстанавливает кето-группы по С-7 и С-11 положениям макролидного агликона до соответствующих гидроксильных групп без дальнейшего изменения лактонного цикла. Восстановленные соединения, как и исходные антибиотики, ингибируют АДФ-зависимое дыхание и выброс протона при гидролизе АТФ, но не влияют на другие связанные с дыханием процессы в митохондриях печени крыс.
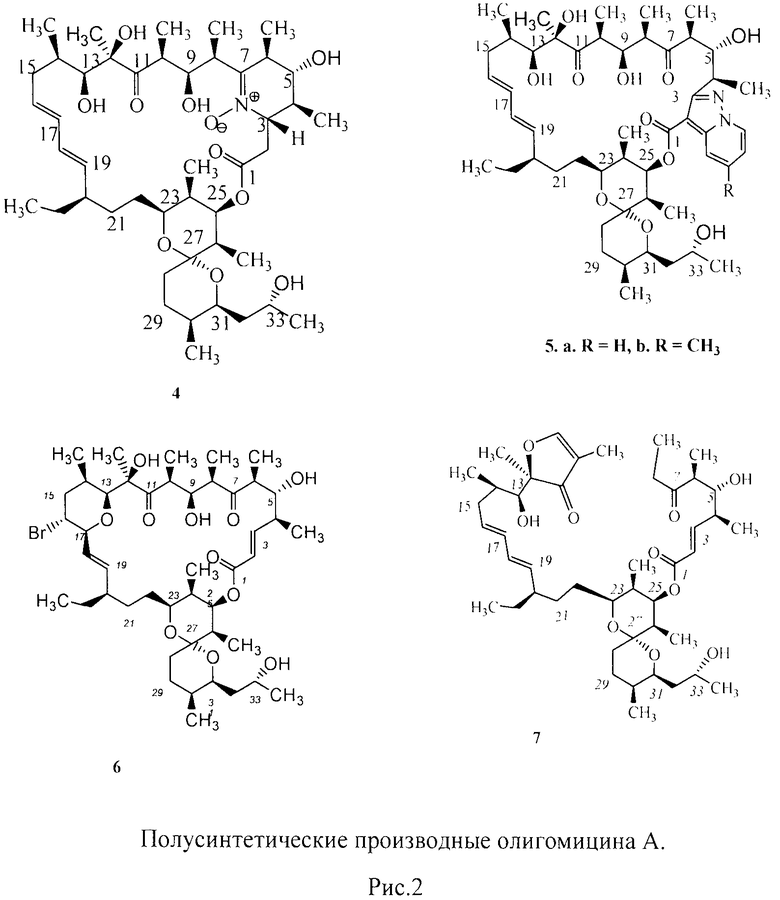
Описаны способы химической модификации олигомицина А, селективно затрагивающие 7-кето-группу и С-2, С-3-двойную связь. Взаимодействие олигомицина А с гидроксиламином привело к образованию 6-членного нитрона, аннелированного с исходным антибиотиком по положениям 3, 4, 5, 6, 7. Реакция с 1-аминопиридиниум йодидом дала пиразоло[1,5-а]пиридины, конъюгированные с олигомицином А по положениям С-2 и С-3, с последующим спонтанным окислением продукта присоединения по двойной связи С2 С3. Полученные производные менее токсичны, чем исходный антибиотик и, в отличие от последнего, не проявляют противогрибковую активность.
Структуры полученных соединений подтверждены методами ЯМР и масс-спектрометрии, включая соединения, меченные изотопом 15N (Lysenkova et al. The first examples of chemical modification of oligomycin A. J. Antibiot. (Tokyo), 2010, 63, 17 22). Олигомицин А может бромироваться N-бромсукцинимидом в нейтральных условиях, образуя монобромпроизводное (Lysenkova et al. Synthesis and properties of a novel brominated oligomycin A derivative. J. Antibiot. (Tokyo), 2012. doi: 10.1038/ja.2012.4. Детальное ЯМР-исследование показало, что полученное полусинтетическое производное имеет структуру 6-членного пирана, аннелированного с макроциклом (рис.2). При проведении реакций в присутствии оснований из олигомицина А образуется устойчивое соединение с расщепленным макроциклом (Lysenkova et al., A novel acyclic oligomycin A derivative formed via retro-aldol rearrangement of oligomycin A.J.Antibiot. (Tokyo), 2012, в печати).
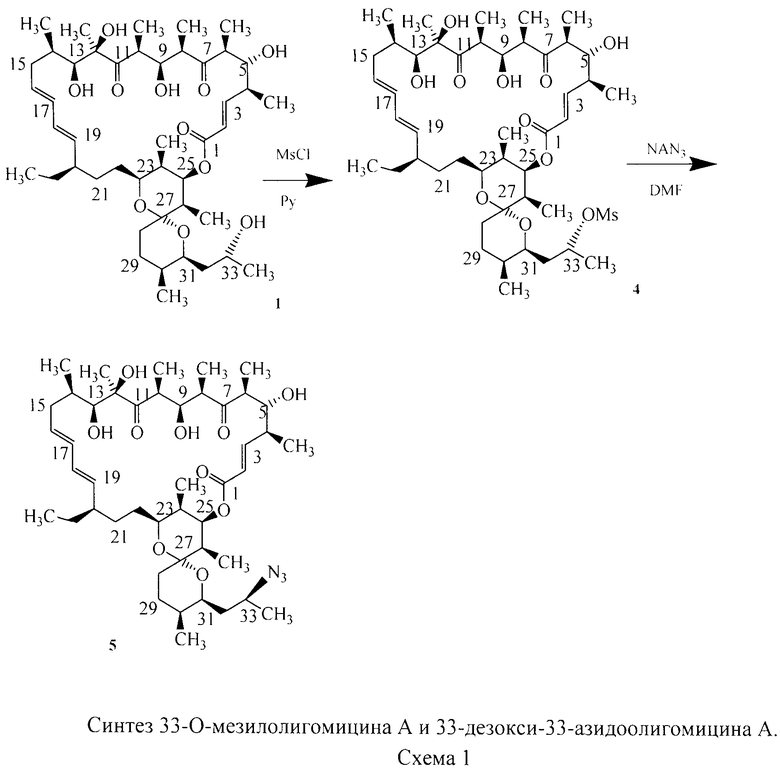
Предварительные исследования показывают, что изменения структуры макролактонного цикла в разной степени снижают активность описанных выше полусинтетических антибиотиков в отношении АТФ-синтазы, однако замена гидроксильной группой по С-33 положению олигомицина А (1) (Схема 1) на другие функциональные группы - метансульфонильную (4) и азидо (5), не препятствовала взаимодействию антибиотика с мишенью АТФ-синтазой. Цитотоксичность полученных соединений 4 и 5 близка к таковой исходного антибиотика [Патент на изобретение №2454420 от 27.06.2012? Бюл.№18] (рис.3).
Раскрытие изобретения
По сравнению с полусинтетическими производными, описанными выше, в соединениях по настоящему изобретению азидогруппа по 33-азидоолигомицина А трансформирована в 1,4-дизамещенные 1,2,3-триазолы.
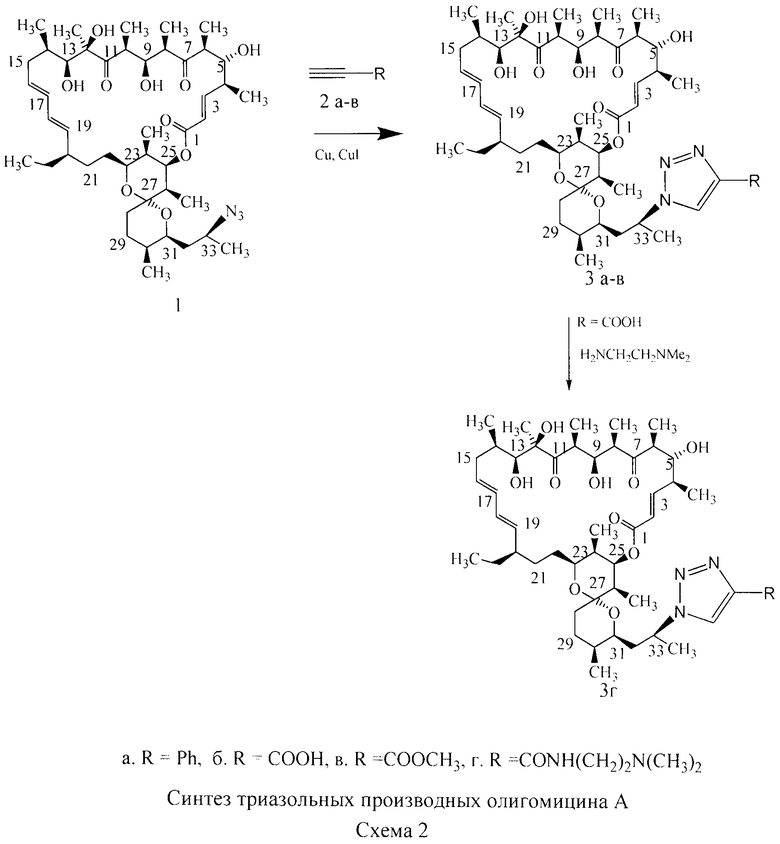
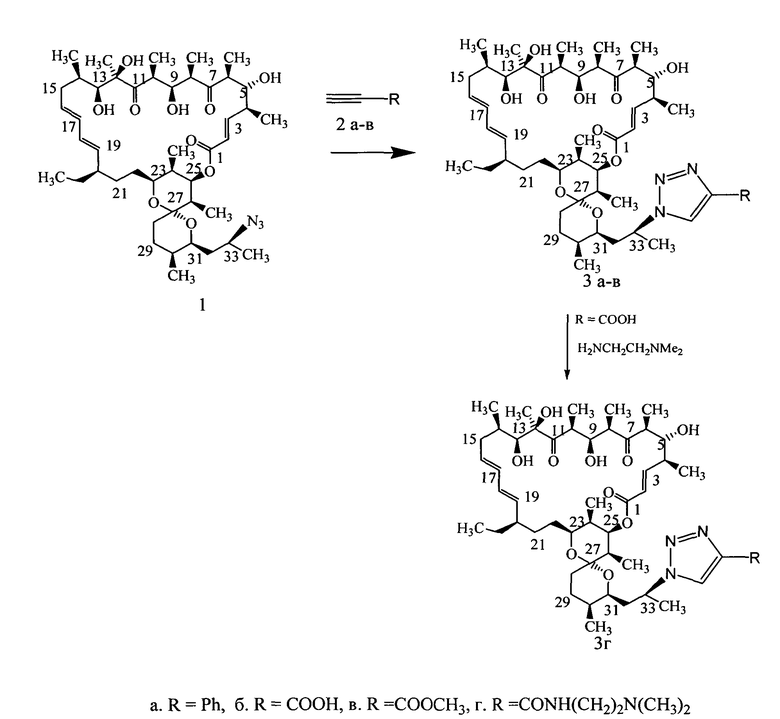
Изобретение включает способ получения новых полусинтетических аналогов антибиотика олигомицина А (формула 3), заключающийся в региоселективном [3+2]-диполярном циклоприсоединении 33-азидогруппы к монозамещенным алкинам (Схема 2). Циклоприсоединение 33-дезокси-33-азидоолигомицина (формула 1) к алкинам (формула 2) проводили в смеси трет-бутанол:вода (1:1) как в присутствии катализатора (солей одновалентной меди), так и без катализатора.
Примеры получения идентификации производных олигомицина А по настоящему изобретению и изучение их антибактериальной и противоопухолевой активности
Пример 1. 33-Дезокси-33-(4-фенил-триазол-1-ил)олигомицин (формула 3а).
Азид олигомицина (1) (0,05 г, 0,06 ммоль) помещали в круглодонную колбу, растворяли в 5 мл смеси т-бутанол:вода (1:1), приливали фенилацетилен (0,6 ммоль), присыпали аскорбат натрия 0,018 г (0,06 ммоль), Cu 0,06 г (0,95 ммоль) и CuI 0,05 г (0,26 ммоль) в виде порошка и перемешивали при 60-65°С в течение 18 часов, реакционную смесь продували аргоном. Контроль реакции осуществляли методом тонкослойной хроматографии на пластинках Merck в системе гексан:ацетон (1:1). По окончании реакции продукт экстрагировали этилацетатом из разбавленной водой реакционной смеси дважды по 15 мл, экстракт фильтровали, высушивали сульфатом натрия, растворитель удаляли на роторном испарителе. Фенилтризол олигомицина очищали методом колоночной хроматографии на силикагеле в системе гексан:ацетон (4:1). Получали 0,022 г фенилтриазола олигомицина в виде аморфного белого порошка. Выход 39%. Брутто формула C53H79N3O10, мол. вес 917,5765, в масс-спектре наблюдали ион 940,5684 (M+Na)+.
УФ (МеОН) λ max (ε) 205 (60000), 225 пл. (54000), 232 (56000), 241 пл. (45000).
ИК ν (см-1) 3466, 2971, 2931, 2870, 1699, 1643, 1456, 1384, 1278, 1226, 1191, 1097, 1049.
[α]D24 -65° (с - 0.184, СН2ОН).
RT 16,053 (A - H2O; В - MeCN), A-20%, В-80%.
Пример 2. 33-Дезокси-33-(4-карбокситриазол-1-ил)олигомицин (формула 3б). Азид олигомицина (1) (0,05 г, 0,06 ммоль) помещали в круглодонную колбу, растворяли в 5 мл смеси т-бутанол:толуол (1:1), добавляли ацетиленкарбоновую кислоту 0,2 мл (0,2 ммоль), реакционную смесь перемешивали при 45°С в течение 24 часов, через каждые 6 часов добавляли ацетиленкарбоновую кислоту по 0,05 мл (0,05 ммоль). Контроль реакции осуществляли методом тонкослойной хроматографии на пластинках Merck в системе хлороформ:метанол:уксусная кислота (объемные соотношения 10:2:0,1). Реакционную смесь упаривали от толуола, продукт реакции экстрагировали этилацетатом из разбавленной водой реакционной смеси дважды по 15 мл, экстракт промывали 1% раствором NaHCO3 от избытка ацетиленкарбоновой кислоты, фильтровали, высушивали сульфатом натрия, растворитель удаляли на роторном испарителе. Карбокситриазол олигомицина очищали методом колоночной хроматографии на силикагеле в системе хлороформ:метанол:уксусная кислота (объемные соотношения 20:2:0,1), чистые фракции промывали водой, сушили сульфатом натрия, концентрировали в вакууме, осаждали карбокситриазол олигомицина гексаном. Получали 0,027 г карбокситриазола олигомицина в виде аморфного белого порошка. Выход 50%. Брутто формула C48H75N3O12, мол. вес 885,5350, в масс-спектре наблюдали ион 908,5281 (M+Na)+.
УФ (МеОН) λ max (ε) 213-223 (58571), 232 пл. (51428), 241 пл. (31428).
ИК ν (см-1) 3462, 2972, 2936, 2810, 1704, 1644, 1548, 1456, 1385, 1280, 1226, 1193, 1135, 1033, 1048, 986, 922, 880.
[α]D24 -91° (с - 0.176, СН3ОН).
RT 10,30 (А - H3PO4 0,01 М pH 2,6; В - MeCN), А-30%, В-70.
Пример 3. 33-Дезокси-33-(4-метоксикарбонилтриазол-1-ил)олигомицин (формула 3в).
Азид олигомицина (1) (0,06 г, 0,075 ммоль) помещали в круглодонную колбу, растворяли в 5 мл смеси т-бутанол:вода (объемные соотношения 1:1), приливали метиловый эфир ацетиленкарбоновой кислоты 0,03 мл (0,37 ммоль), присыпали Cu 0,06 г (0,95 ммоль) в виде порошка и перемешивали при комнатной температуре в течение 6 часов. Контроль реакции осуществляли методом тонкослойной хроматографии на пластинках Merck в системе толуол:этилацетат (2:1). По окончании реакции продукт экстрагировали этилацетатом из разбавленной водой реакционной смеси дважды по 15 мл, экстракт фильтровали, высушивали сульфатом натрия, растворитель удаляли на роторном испарителе. Метиловый эфир карбокситриазола олигомицина очищали методом колоночной хроматографии на силикагеле, в системе толуол:этилацетат (25:1). Получали 0,034 г (0,037 ммоль) метилового эфира карбокситриазола олигомицина в виде аморфного белого порошка. Выход 52%.
Брутто формула C49H77N3O12, мол. вес 899,5072, в масс-спектре наблюдали ион 922,5406 (М+Na)+.
УФ (МеОН) λ max (ε) 223 (86364), 232 пл. (73636), 239 пл. (43536).
ИК ν (см-) 3486, 2972, 2934, 1704, 1456. 1385, 1278, 1226, 1099, 1046, 987.
[α]D24 -66° (с - 0.18, СН3ОН).
RT 14,274 (A - H3PO4 0,01 М pH 2,6; В - MeCN), А-30%, В-70%.
Пример 4. 33-Дезокси-33-[(4-диметиламиноэтиламидокарбонил)триазол-1-ил] олигомицин А (формула 3г).
Карбокситриазол олигомицина (36) (0,05 г, 0,056 ммоль) помещали в круглодонную колбу, растворяли в 5 мл диметилформамида, присыпали PyBOP 0,05 г (0,1 ммоль), добавляли N,N-диметилэтилендиамин 0,012 мл (0,1 ммоль) и перемешивали 24 часа. Контроль реакции осуществляли методом тонкослойной хроматографии на пластинках Merck в системе хлороформ:метанол:уксусная кислота (10:3:0,2).
Реакционную смесь разбавляли водой, добавляли NaCl, экстрагировали этилацетатом дважды по 15 мл, экстракт промывали 1% раствором NaHCO3 и водой, высушивали сульфатом натрия, растворитель удаляли на роторном испарителе. Амид карбокситриазола олигомицина очищали методом колоночной хроматографии на силикагеле в системе хлороформ:метанол:уксусная кислота (10:0.1), чистые фракции промывали 1% раствором NaHCO3, концентрировали в вакууме, затем амид карбокситриазола очищали на сефадексе LH-20 в метаноле. Получали 0,008 г амида карбокситриазола олигомицина в виде аморфного белого порошка. Выход 15%.
Брутто формула C52H85O11, мол. вес 955,6245, в масс-спектре наблюдали ион 956.6328 (М+Н)+.
Пример 5. ЯМР-спектры олигомицина А и его триазольных производных (δC, δH, м.д.).
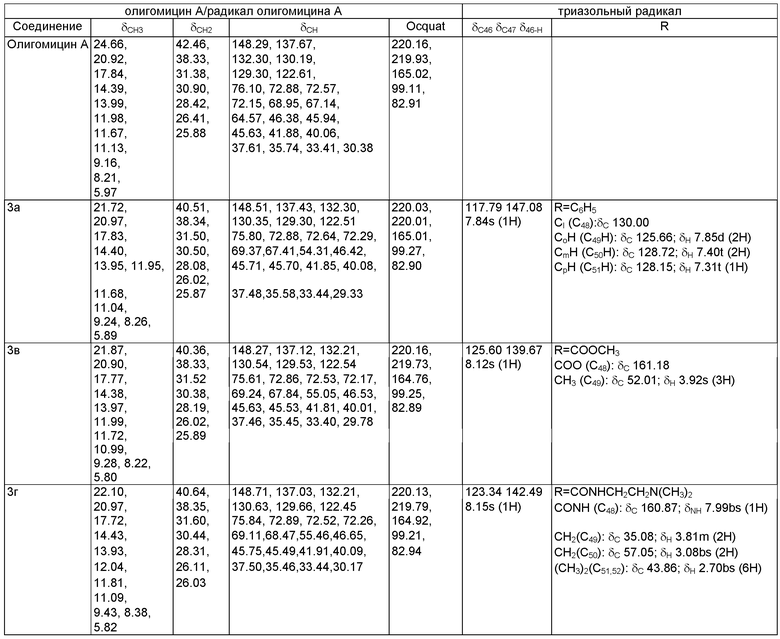
Пример 6.
Сравнение способности олигомицина А и новых производных ингибировать FoFi-АТФ-синтазу оценивали по подавлению роста штамма Streptomyces fradiae, чувствительного к очень низким (в пикомолярном диапазоне) концентрациям олигомицина А.
Тестирование чувствительности бактерий к ингибированию FoFi-АТФ-синтазы проводили методом наложения дисков и заключалось оно в определении зоны подавления роста штамма Streptomyces fradiae, засеянного газоном на агаризованной среде, вокруг бумажных дисков, содержащих олигомицин А и азид олигомицина А в различных концентрациях. Для приготовления газона споровую суспензию смешивали с агаризованной средой МГ-2 (0,7% агара) в соотношении 2×106-1×107 спор на чашку Петри и засевали чашки с агаризованной средой МГ-1 (2% агара). После застывания агаризованной споровой суспензии на чашки наносили бумажные диски, содержащие тестируемые соединения. Газон выращивали 40-48 часов при 28°С. Среды с общим названием МГ содержат мальтэкстракт, дрожжевой экстракт, NaCl, MgSO4, K2HPO4, FeSO4, KNO3, глюкозу.
Результаты тестирования представлены в таблице 1.
Таким образом, в тест-системе Streptomyces fradiae азид олигомицина и соединение 3г ~ в 1000 раз менее активны, чем олигомицин А, 3а ~ в 100 раз менее активен, активность 3в ~ в 10000 раз ниже.
Пример 7.
Изучение способности олигомицина А и новых производных вызывать гибель опухолевых клеток проводили по следующей методике. Адгезионные клетки (НСТ116, аденокарцинома толстой кишки человека) открепляли от подложки версеном-трипсином, тщательно пипетировали для образования одноклеточной суспензии, вносили 50 мкл суспензии в 450 мкл физиологического буфера, пипетировали и подсчитывали в камере Горяева. Клетки лейкоза человека (линия K562), растущие в суспензии, вносили 50 мкл суспензии в 450 мкл физиологического буфера, пипетировали и считали в камере Горяева. Использовали культуры в логарифмической фазе роста. В лунки 96-луночного планшета вносили по 190 мл клеточной взвеси и оставляли в СО2-инкубаторе на 24 часа. Приготовляли маточные растворы исследуемых препаратов (растворитель ДМСО) с концентрацией 10 мМ, из которых проводили серийные разведения препаратов в среде для культивирования клеток (модифицированная Дульбекко среда Игла с добавлением 5% эмбриональной сыворотки теленка, 2 мМ L-глутамина, 100 ЕД/мл пенициллина, 100 мкг/мл стрептомицина). Для контроля воздействия растворителя использовали растворы ДМСО в тех же разведениях, что и исследуемые растворы. Объемами растворителя и изменением общего объема среды в лунке пренебрегали. Получали следующие концентрации препаратов в лунке: 0,1 мкМ; 0,2 мкМ; 0,4 мкМ; 0,8 мкМ; 1,6 мкМ; 3,2 мкМ, 6,4 мкМ; 12,5 мкМ; 25 мкМ; 50 мкМ. Каждую концентрацию исследуемого препарата изучали в трех повторностях. Планшеты с внесенными соединениями помещали в СО2-инкубатор при 37°С на 72 часа. В лунки вносили по 10 мкл водного раствора МТТ (5 мг/мл). Инкубировали 1-4 часа до развития интенсивной темно-фиолетовой окраски формазана внутри клеток. Среду отбирали, не затрагивая клеток. В лунки добавляли по 100 мкл ДМСО, пипетировали до гомогенности и встряхивали на шейкере 2 мин. Измеряли оптическую плотность (ОП) полученных растворов при 540 нм, строили кривые выживаемости клеток. При их построении за 100% принимали ОП540 контрольных лунок, к которой относили ОП лунок с той или иной концентрацией исследуемого препарата. В качестве критической отмечали концентрацию выживаемости 50% клеток (IC50).
В таблице 2 приведены результаты исследования цитотоксичности олигомицина А и новых производных для клеток лейкоза (линия К562) и рака толстой кишки (линия НСТ116).
- Показатель не определяли.
Таким образом, предложенный новый способ модификации антибиотика олигомицина А позволяет получать новые триазольные производные олигомицина А, обладающие преимуществами перед исходным антибиотиком олигомицином А и его полусинтетическим аналогом 33-азидоолигомицином, а именно, высокой цитотоксичностью для опухолевых клеток и хорошей растворимостью.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЦИТОТОКСИЧЕСКИЕ ПОЛУСИНТЕТИЧЕСКИЕ ПРОИЗВОДНЫЕ МАКРОЛИДНОГО АНТИБИОТИКА ОЛИГОМИЦИНА А И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2011 |
|
RU2454420C1 |
| Способ получения 2,3,16,17,18,19-гексагидроолигомицина А и его применение для ингибирования роста дрожжей рода Candida | 2016 |
|
RU2623087C1 |
| НЕИММУНОСУПРЕССОРНЫЙ ЦИКЛОСПОРИН ДЛЯ ЛЕЧЕНИЯ ВРОЖДЕННОЙ МИОПАТИИ УЛЬРИХА | 2008 |
|
RU2462262C2 |
| НИЗКОМОЛЕКУЛЯРНЫЕ КОНЪЮГАТЫ ПРОТИВООПУХОЛЕВЫХ АГЕНТОВ И ВЫСОКОСЕЛЕКТИВНЫХ ЛИГАНДОВ АСИАЛОГЛИКОПРОТЕИНОВОГО РЕЦЕПТОРА ДЛЯ ТЕРАПИИ ОНКОЛОГИЧЕСКИХ ПАТОЛОГИЙ ПЕЧЕНИ | 2017 |
|
RU2696096C2 |
| N''-ЗАМЕЩЕННЫЕ 9a-N-(N'-КАРБАМОИЛ-ГАММА-АМИНОПРОПИЛЬНЫЕ) И 9a-N-(N'-ТИОКАРБАМОИЛ-ГАММА-АМИНОПРОПИЛЬНЫЕ), ПРОИЗВОДНЫЕ 9-ДЕЗОКСО-9-ДИГИДРО-9a-АЗА-9a-ГОМОЭРИТРОМИЦИНА A И 5-O-ДЕЗОЗАМИНИЛ-9-ДЕЗОКСО-9-ДИГИДРО-9a-АЗА-9a-ГОМОЭРИТРОНОЛИДА A | 2003 |
|
RU2328503C2 |
| СПОСОБ ДИАГНОСТИКИ НАСЛЕДСТВЕННОЙ ОПТИЧЕСКОЙ НЕЙРОПАТИИ | 2014 |
|
RU2566271C9 |
| ЛЕКАРСТВЕННОЕ СРЕДСТВО ДЛЯ ЛЕЧЕНИЯ ЭНДОМЕТРИОЗА | 2022 |
|
RU2818816C1 |
| N-СУЛЬФАТИРОВАННЫЕ ОЛИГОСАХАРИДЫ, АКТИВИРУЮЩИЕ РЕЦЕПТОРЫ FGF, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ В ТЕРАПИИ | 2010 |
|
RU2559629C2 |
| ЛЕКАРСТВЕННОЕ СРЕДСТВО ДЛЯ ЛЕЧЕНИЯ ЭНДОМЕТРИОЗА | 2024 |
|
RU2839308C1 |
| ПРОИЗВОДНОЕ ЦИНКОВОГО МЕТАЛЛОКОМПЛЕКСА ХЛОРИНА-e И ЕГО ПРИМЕНЕНИЕ | 2018 |
|
RU2691754C1 |
Изобретение относится к фармацевтической промышленности и касается новых производных противоопухолевого антибиотика олигомицина А и способа их получения региоселективным [3+2]диполярным циклоприсоединением азидогруппы 33-дезокси-33-азидоолигомицина А(1) к монозамещенным алкинам. Новые производные антибиотика олигомицина А, соответствующие формуле:
где R представляет собой 1,4-дизамещенные 1,2,3-триазолы, а именно а. - (фенил-триазол-1-ил), б. - (4-карбокси-триазол-1-ил), в. - (4-4-метоксикарбонил-триазол-1-ил), г. - (4-диметиламиноэтиламидокарбокситриазол-1-ил), обладают выраженной противоопухолевой активностью и более высокой растворимостью по сравнению с исходным олигомицином А. 3 н.п. ф-лы., 4 ил., 3 табл.
1. Способ получения полусинтетических производных антибиотика олигомицина А (3а-г), заключающийся в региоселективном [3+2]-диполярном циклоприсоединении азидогруппы 33-дезокси-33-азидоолигомицина А (1) к монозамещенным алкинам 2а-в как в присутствии катализатора (смеси порошка меди и солей галогенидов одновалентной меди), так и в его отсутствие, при получении соединения 3г проводят амидирование карбокситриазола олигомицина (3б) N,N-диметилэтилендиамином.
2. Производные антибиотика олигомицина А 3а-г, соответствующие общей формуле:
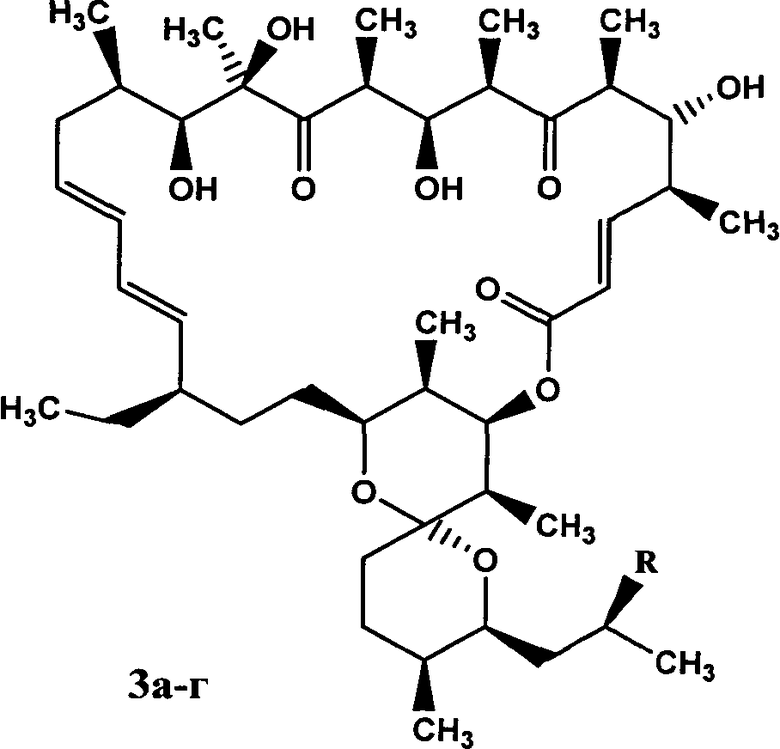
где R представляет собой 1,4-дизамещенные 1,2,3-триазолы: а. - (фенил-триазол-1-ил), б. - (4-карбокси-триазол-1-ил), в. - (4-4-метоксикарбонил-триазол-1-ил), г. - (4-диметиламиноэтиламидокарбокситриазол-1-ил).
3. Применение соединений формулы 3а-г из п.1 в качестве противоопухолевых средств.
| JP 0009208587 12.08.1997 |

