Настоящее изобретение относится к реагентам и способам [18F]-фторирования биомолекул, в частности пептидов. Полученные соединения, меченные 18F, полезны в качестве радиофармацевтических препаратов, особенно для применения в позитронной эмиссионной томографии (ПЭТ).
Применение меченных радиоактивным изотопом биоактивных пептидов для диагностической визуализации приобретает все большее значение в ядерной медицине. Биологически активные молекулы, которые избирательно взаимодействуют с определенными типами клеток, полезны для доставки радиоактивности к тканям-мишеням. Например, меченные радиоактивным изотопом пептиды обладают значительным потенциалом для доставки радионуклидов к опухолям, инфарктным и инфицированным тканям с целью диагностической визуализации и радиотерапии. 18F, имеющий период полураспада 110 минут, является предпочтительным позитрон-излучающим нуклидом во многих исследованиях рецепторной визуализации. Таким образом, 18F-меченные биоактивные пептиды вследствие своей полезности в ПЭТ в отношении количественного обнаружения и определения широкого круга заболеваний имеют большой клинический потенциал.
Одна из проблем приготовления 18F-меченных пептидов заключается в том, что получение существующих реагентов для 18F-мечения является трудоемким. Эффективное мечение пептидов и белков фтором 18F достигается только путем использования подходящих простетических групп. Несколько таких простетических групп предложено в литературе, в том числе N-сукцинимидил-4-[18F]фторбензоатная, м-малеимидо-N-(n-[18F]фторбензил)-бензамидная, N-(n-[18F]фторфенил)малеимидная и 4-[18F]фторфенацил-бромидная. Почти во всех используемых в настоящее время способах 18F-мечения пептидов и белков применяют активные сложные эфиры. Наиболее широко применяемым реагентом для 18F-мечения является N-сукцинимидил-4-[18F]фторбензоат (SFB). Недостаток SFB заключается в том, что его получение включает 3 синтетические стадии (фторирование, гидролиз и образование активного сложного эфира) с последующей трудоемкой стадией очистки с помощью ВЭЖХ (высокоэффективная жидкостная хроматография); таким образом, получение SFB является трудным для автоматизации. Более того, присутствие фенильного кольца в реагенте для мечения существенно повышает гидрофобность 18F-меченного продукта, что может неблагоприятно повлиять на его профиль биораспределения. Следовательно, все еще существует потребность в альтернативных реагентах и способах 18F-мечения, которые делают возможным быстрое хемоселективное введение 18F, в частности в пептиды, в мягких условиях с получением 18F-меченных продуктов. Кроме того, существует потребность в таких способах, которые поддаются автоматизации с целью облегчения получения радиофармацевтических препаратов в клинических условиях.
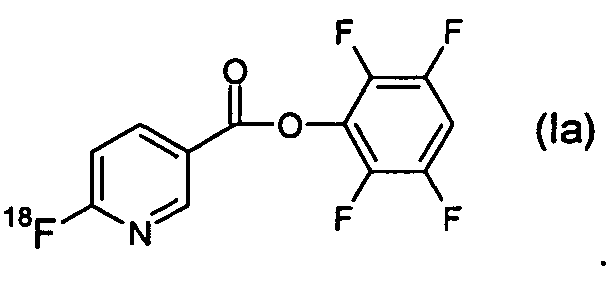
Согласно первому аспекту изобретения предложено соединение формулы (I):
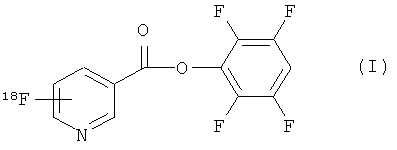
В одном из аспектов 18F-метка присоединена в орто-положении к пиридильному азоту, так что соединение формулы (I) имеет формулу (Ia):
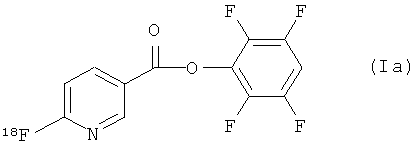
Соединения формулы (I) и (Ia) должны добавить 18F-мечению биомолекул существенные преимущества. Соединения формулы (I) и (Ia) можно метить фтором 18F за одну стадию, мечение происходит быстро при почти комнатной температуре, очистку можно осуществить с помощью системы на основе картриджа (например, колонки Oasis МСХ), что делает автоматизацию более доступной. Также известно, что пиридиновая система более гидрофильна, чем бензильный аналог, и поэтому ожидается, что будет добавлено положительное воздействие на профиль биораспределения 18F-продукта. Как показано ниже, было обнаружено, что сложный тетрафторфениловый эфир более стабилен во время 18F-мечения, чем другие активные сложные эфиры.
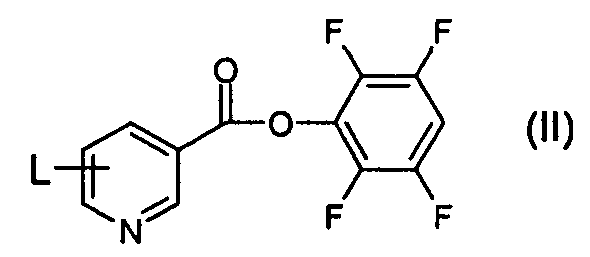
Соединения формулы (I) и (Ia) могут быть получены из соответствующего соединения формулы (II):
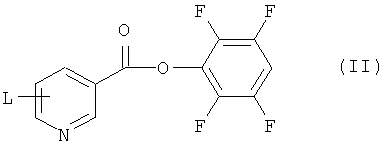
или его соли, где L представляет собой уходящую группу, выбранную из хлоро, бромо, йодо, нитро и три(C1-6алкил)аммония (например, триметиламмония). Такие соединения формулы (II) являются новыми и поэтому образуют дополнительный аспект изобретения.
В одном из аспектов L в соединении формулы (II) представляет собой три(C1-6алкил)аммоний (например, триметиламмоний) с соответствующим противоионом, выбранным из противоионов, полученных из неорганических кислот, например соляной, бромистоводородной, фосфорной, метафосфорной, хлорной, азотной и серной кислот, и противоионов, полученных из органических кислот, например винной, трифторуксусной, лимонной, яблочной, молочной, фумаровой, бензойной, гликолевой, глюконовой, янтарной, метансульфоновой, трифторметансульфоновой и лара-толуолсульфоновой кислот; с подходящим, выбранным из хлорида, бромида, перхлората, сульфоната, нитрата, фосфата и трифторметансульфоната; с более подходящим трифторметансульфонатным противоионом.
В одном из аспектов соединение формулы (II) представляет собой:
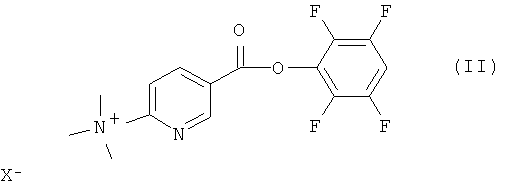
где X- представляет собой противоион, как он определен выше, и предпочтительно является трифторметансульфонатом.
Получение соединения формулы (I) из соответствующего соединения формулы (II) или его соли может быть осуществлено стандартными способами 18F-мечения. [18F]фторид легко получают из 18O-обогащенной воды с использованием (p, n)-ядерной реакции (Guillaume et al, Appl. Radiat. Isot. 42 (1991) 749-762) и обычно выделяют в виде соли, такой как Na18F, K18F, Cs18F, [18F]фторид тетраалкиламмония или 18F-фторид тетраалкилфосфония. Для увеличения реакционной способности [18F]фторида можно добавить катализатор фазового переноса, такой как аминополиэфир или краун-эфир, например 4,7,13,16,21,24-гексаокса-1,10-диазабицикло[8,8,8]гексакозан (Kryptofix 2.2.2); реакцию проводят в подходящем растворителе. В этих условиях получают реакционноспособные фторидные ионы. Возможно, для улучшения выходов фторидных продуктов можно использовать ловушку свободных радикалов, как описано в WO 2005/061415. Термин "ловушка свободных радикалов" определен как любой агент, который взаимодействует со свободными радикалами и инактивирует их. Подходящая для этой цели ловушка свободных радикалов может быть выбрана из 2,2,6,6-тетраметилпиперидин-N-оксида (TEMPO), 1,2-дифенилэтилена (DPE), аскорбата, парааминобензойной кислоты (РАВА), α-токоферола, гидрохинона, ди-трет-бутилфенола, β-каротина и гентизиновой кислоты.
Обработку соединения формулы (II) [18F]фторидом можно проводить в присутствии подходящего органического растворителя, такого как ацетонитрил, диметилформамид, диметилсульфоксид, диметилацетамид, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, сульфолан, N-метилпирролидинон, или в ионной жидкости, такой как имидазолиевое производное (например, гексафторфосфат 1-этил-3-метилимидазолия), пиридиниевое производное (например, тетрафторборат 1-бутил-4-метилпиридиния), фосфониевое соединение или тетраалкиламмониевое соединение, при непредельной температуре, например от 15°C до 50°C, предпочтительно примерно при температуре окружающей среды, такой как от 15°C до 30°C, например от 18°C до 25°C.
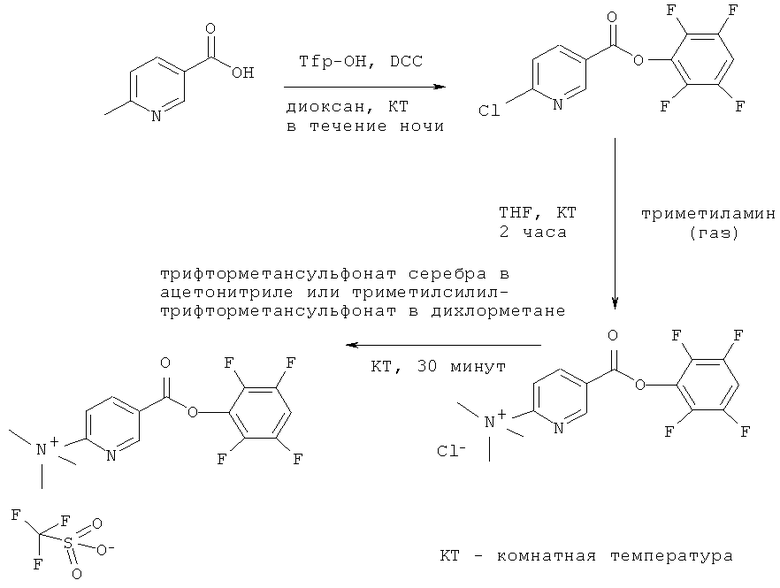
Соединения формулы (II) и их соли могут быть получены из имеющихся в продаже исходных веществ, таких как 6-хлорникотиновая кислота. Выходы для всего процесса в целом являются хорошими (больше 50%). Стадии включали этерификацию с тетрафторфенолом, активируемую, например, дициклогексилкарбодиимидом (DCC), образование триметиламмониевой соли путем обработки активного сложного эфира 6-хлорникотиновой кислоты насыщенным раствором триметиламина в тетрагидрофуране (THF) и образование трифторметансульфонатной (трифлатной) соли с помощью трифлата серебра.
После получения соединения формулы (I) его можно очистить стандартными способами, обычно с использованием твердофазной экстракции, например, на колонке Oasis МСХ™, из которой соединение формулы (I) можно элюировать с хорошей чистотой, используя подходящую смесь органического растворителя и воды.
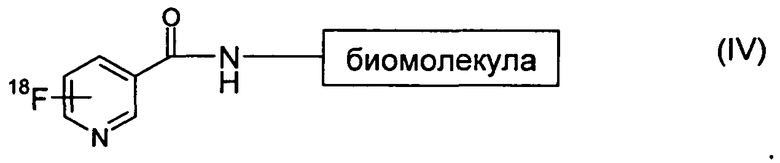
Согласно следующему аспекту изобретения предложен способ 18F-фторирования, включающий взаимодействие соединения формулы (I) с соединением формулы (III):

с образованием 18F-продукта формулы (IV):
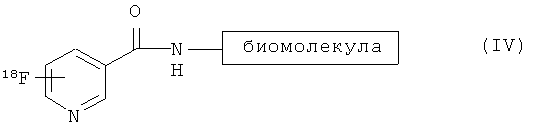
Взаимодействие соединения формулы (I) с соединением формулы (III) можно осуществлять в подходящем растворителе, например в водном буфере, при pH в диапазоне от 2 до 11, например от 3 до 11, и при непредельной температуре от 5 до 70°C, предпочтительно при температуре окружающей среды.
В формулах (III) и (IV) подходящие для мечения биомолекулы представляют собой пептиды, которые могут включать аналоги соматостатина, такие как октреотид, бомбезин, вазоактивный интестинальный пептид, аналоги хемотаксического пептида, α-меланоцитостимулирующий гормон, нейротензин, пептид Arg-Gly-Asp и его аналоги, проинсулинсвязывающий пептид человека, эндотелии, ангиотензин и формил-норлейцил-лейцил-фенилаланил-норлейцил-тирозил-лизин. Предпочтительными пептидами для мечения являются пептид Arg-Gly-Asp и его аналоги, такие как описанные в WO 01/77415 и WO 03/006491. Предпочтительные пептиды содержат фрагмент:
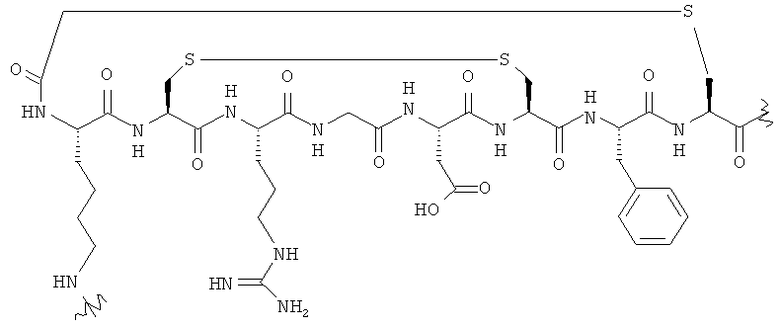
В одном конкретном аспекте биомолекула в формуле (III) или (IV) представляет собой пептид формулы (A):
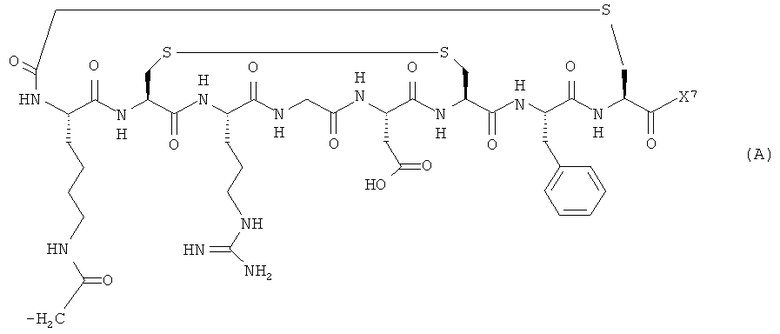
где X7 представляет собой либо -NH2, либо
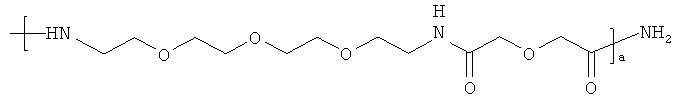
где a является целым числом от 1 до 10; предпочтительно a равно 1.
Специалисту будет очевидно, что способы по изобретению также можно применять для 18F-фторирования других биомолекул, таких как белки, гормоны, олигонуклеотиды и фрагменты антител, а также небольших лекарствоподобных молекул, с целью обеспечения множества ПЭТ индикаторов.
Соединения формулы (III) могут быть получены стандартными способами пептидного синтеза, например твердофазным пептидным синтезом, например, как описано в Atherton, Е. and Sheppard, R.C.; "Solid Phase Synthesis"; IRL Press: Oxford, 1989. Введение первичной аминной группы в соединение формулы (III) можно осуществлять путем взаимодействия с N- или C-концом пептида или с некоторыми другими функциональными группами, содержащимися в пептидной последовательности, модификация которых не влияет на характеристики вектора в отношении связывания. Первичную аминную группу предпочтительно вводят путем образования стабильной амидной связи в результате взаимодействия пептидной аминной функциональной группы с активированной кислотой и вводят либо во время, либо после пептидного синтеза. Если предшественником является кислота, то первичный амин можно ввести с использованием активирующих in situ агентов, таких как гексафторфосфат 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония (HBTU) или N-оксид гексафторфосфата N-[(диметиламино)-1Н-1,2,3-триазоло[4,5-b]пиридин-1-илметилен]-N-метилметанаминия (HATU).
Далее изобретение проиллюстрировано примерами.
ПРИМЕРЫ
Пример 1: Синтез 2,3,5,6-тетрафторфенилового эфира 6-фторникотиновой кислоты
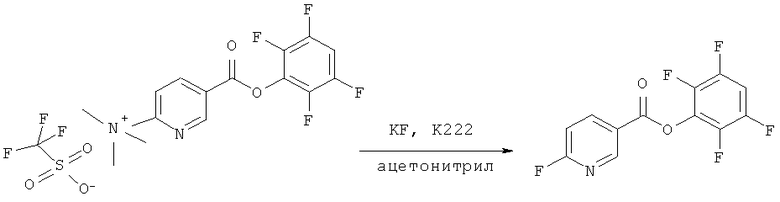
Активный сложный эфир 2,3,5,6-тетрафторфенола (Tfp) и 2-триметиламмоний-никотиновой кислоты синтезировали в три стадии, начиная с 6-хлорникотиновой кислоты (Sigma-Aldrich). Этерификация 6-хлорникотиновой кислоты (3 г, 19 ммоль) 2,3,5,6-тетрафторфенолом (3,25 мг, 19 ммоль), активируемая дициклогексилкарбодиимидом (DCC) (3,96 мг, 19 ммоль), в 50 мл диоксана с последующей кристаллизацией из гексана привела к Tfp эфиру 6-хлорникотиновой кислоты с выходом 73%. Образование триметиламмониевой соли путем обработки активного Tfp-эфира 6-хлорникотиновой кислоты (1 г, 3,27 ммоль) в насыщенном растворе триметиламина (непрерывное барботирование в течение двух часов) в тетрагидрофуране (THF) (15 мл) привело к получению триметиламмониевой соли с хлоридом в качестве противоиона с выходом 45%. Так как в тетрагидрофурановом растворе происходит осаждение соли, непрореагировавшее вещество можно было отфильтровать. Образования трифлатной соли можно добиться двумя путями: либо обработкой соответствующей хлоридной соли трифлатом серебра при молярном избытке 1,2 в ацетонитриле, либо обработкой триметилсилил-трифторметансульфонатом. Последний путь является предпочтительным, так как обработка является более простой и не требуется никакой препаративной хроматографии. Оба способа являются почти количественными.
Синтезированный предшественник (9,2 мг) сначала метили фтором 19F с помощью К222 (10 мг) и KF (1,1 мг) в ацетонитриле (0,7 мл). Реакцию 19F с Tfp эфиром изучали с помощью 1Н-ЯМР в ацетонитриле-d6 при 27°C для анализа кинетики реакции и образовавшихся примесей.
Сравнительный пример
N-гидроксисукцинимидный (NHS) эфир 2-триметиламмоний-никотиновой кислоты синтезировали из 6-хлорникотиновой кислоты и метили фтором 19F аналогично вышеописанным способам.
Результаты
Оба эфира синтезировали с хорошими выходами (больше 30% исходя из 6-хлорникотиновой кислоты), и они быстро реагировали с фторидом в ацетонитриле при комнатной температуре. NHS-эфир был более подвержен гидролизу, чем Tfp-эфир, и поэтому его далее не оценивали. Исследования взаимодействия Tfp-эфира с помощью 1Н ЯМР в течение 30 минут показали быстрое введение фторида при комнатной температуре, через 2,5 минуты 32% исходного вещества превратилось в желаемый фторированный продукт. В одной серии экспериментов 70% фторированного продукта получили менее чем за 20 минут. Вместе с желаемым продуктом были идентифицированы два производных никотиновой кислоты в качестве побочных продуктов.
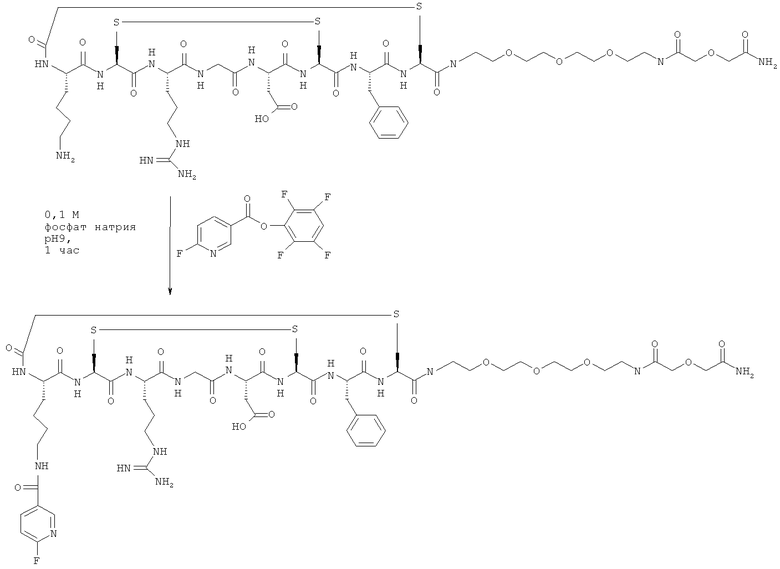
Пример 2: Взаимодействие 2,3.5.6-тетрафторфенилового эфира 6-фторникотиновой кислоты с функционализированным RGD пептидом
В результате взаимодействия фторированного продукта Примера 1 (1 мг) с соответствующим образом функционализированным RGD пептидом (5 мг, получен, как описано в WO 01/77415 и WO 03/006491) в растворе (1:1) ацетонитрила и 0,1 М фосфата натрия с рН 9 (всего 3 мл) получали желаемый продукт, который был проанализирован с помощью ЖХ-МС (жидкостная хроматография - масс-спектрометрия). Условия ЖХ-МС: Phenomenex Luna С18(2), 3 мкм, 2×50 мм, подвижная фаза А: вода/0,1% трифторуксусная кислота (TFA), подвижная фаза Б: ацетонитрил/0,1% TFA, поток 0,6 мл/мин, 10-30% Б за 5 мин. Время удерживания (Rt)=3,42 мин, М+Н+ (ожидаемое значение 1381,5; обнаруженное 1381,6).
Пример 3: Радиохимический синтез 2.3.5.6-тетрафторфенилового эфира 6-[18F]фторникотиновой кислоты
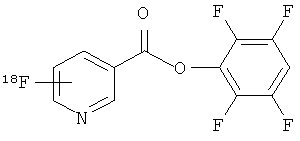
Водный [18F]фторид (до 150 МБк) азеотропно сушили в присутствии 15 мг Krytptofix 222 и 3 мг бикарбоната калия (КНСО3) при нагревании под N2 до 100°С в течение 9 минут. За этот период времени добавляли и выпаривали 2x1 мл ацетонитрила. После охлаждения до 40°С добавляли раствор метансульфоната триметил-[5-(2,3,5,6-тетрафторфеноксикарбонил)-пиридин-2-ил]аммония (7 мг в 1 мл ацетонитрила). Для осуществления мечения реакционный сосуд нагревали до 40°С в течение 10 минут. Неочищенную реакционную смесь подвергали радио-ВЭЖХ (высокоэффективная жидкостная хроматография) и радио-ТСХ (тонкослойная хроматография) с совместной инжекцией нерадиоактивного стандартного образца для подтверждения образования целевого F-соединения. Выходы продукта включения обычно составляли от 50 до 80% при анализе радио-ТСХ (n=3).
Радио-ТСХ: Предварительно покрытые силикагелевые пластины 60 F254 (Merck). Градиент: н-гексан/этилацетат 50:50. Для измерения распределения радиоактивности на пластинах ТСХ использовали устройство для быстрой визуализации Instant Imager (Packard Bioscience). Rf: 0,65.
Радио-ВЭЖХ: Аналитическую радио-ВЭЖХ осуществляли на системе Agilent (1100 series), оборудованной УФ детектором, последовательно соединенным с γ-детектором (Bioscan Flow-Count). Колонка Phenomenex Luna С18(2) (150 х 4,6 мм, 5 мкм), поток 1,0 мл/м с градиентом 20-80% Б за 20 мин (УФ детектирование при 214 и 254 нм объединяли с γ-детектором). Rt 14,4 мин.
Очистка
Неочищенную реакционную смесь, содержащую 2,3,5,6-тетрафторфениловый эфир -[18F]фторникотиновой кислоты в 2 мл ацетонитрила, разбавляли дистиллированной водой до 30% ацетонитрила. Водный раствор пропускали через картридж Oasis МСХ plus (подготовленный согласно рекомендациям производителей). Затем картридж ополаскивали 5 мл дистиллированной воды. После этого очищенный продукт элюировали из колонки 100% ацетонитрила с радиохимической чистотой выше 90%. Все остатки непрореагировавшего предшественника, представляющего собой метансульфонат триметил-[5-(2,3,5,6-тетрафторфеноксикарбонил)-пиридин-2-ил]аммония, оставались на картридже.
Пример 4: Конъюгация 2.3.5.6-тетраaторфенилового эфира 6-[18F]aторникотиновой кислоты с циклическим RGD-пептидом, имеющим свободную функциональную группу амино
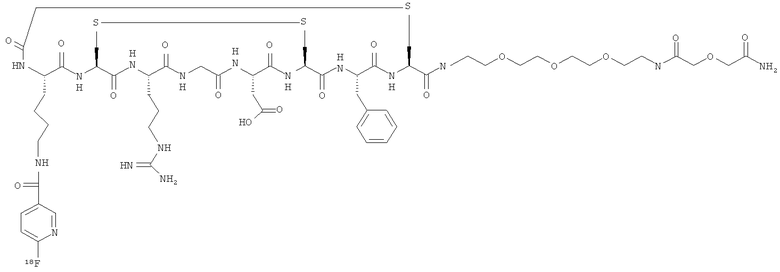
К раствору очищенного 2,3,5,6-тетрафторфенилового эфира 6-[18F]фторникотиновой кислоты в 1,5 мл раствора (1:1) ацетонитрил/вода добавляли 3 мг соответствующим образом функционализированного RGD пептида (молекулярная масса: 1258,47), растворенного в 1 мл раствора (1:1) ацетонитрила и 0,1 М NaHPO4. Полученную смесь с рН 9 нагревали до 40°C. Через 30 минут небольшую аликвоту смеси анализировали с помощью радио-ВЭЖХ. Радиохроматограмма показала превращение в желаемый продукт с выходом больше 65%. Продукт элюировали совместно с его 19F-стандартным образцом.
Радио-ВЭЖХ: Аналитическую радио-ВЭЖХ осуществляли на системе Agilent (1100 series), оборудованной УФ детектором, последовательно соединенным с γ-детектором (Bioscan Flow-Count). Колонка Phenomenex Luna С18(2) (150×4,6 мм, 5 мкм), поток 1,0 мл/м с градиентом 0-40% Б за 20 мин (УФ детектирование при 214 и 254 нм объединяли с γ-детектором). Rt 10,0 мин.
| название | год | авторы | номер документа |
|---|---|---|---|
| МЕЧЕННЫЕ РАДИОАКТИВНЫМ ИЗОТОПОМ КОНЪЮГАТЫ RGD-СОДЕРЖАЩИХ ПЕПТИДОВ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ С ПОМОЩЬЮ CLICK-ХИМИИ | 2005 |
|
RU2419627C2 |
| ДИАГНОСТИЧЕСКИЕ СОЕДИНЕНИЯ | 2005 |
|
RU2396272C9 |
| СПОСОБ ОБРАБОТКИ ФТОРИДА | 2008 |
|
RU2475448C2 |
| Молекула общей структуры Y-Nic-F, способы получения, предшественники для её получения, а также применение в качестве действующего вещества в составе потенциального радиофармацевтического лекарственного препарата | 2021 |
|
RU2811181C2 |
| Способ синтеза F-меченых биомолекул | 2012 |
|
RU2620598C2 |
| Раствор элюента | 2011 |
|
RU2608932C2 |
| НОВЫЕ ПРЕДШЕСТВЕННИКИ ПРОИЗВОДНЫХ ГЛУТАМАТА | 2012 |
|
RU2600981C2 |
| СПОСОБ ПОЛУЧЕНИЯ [F]ФТОРОРГАНИЧЕСКИХ СОЕДИНЕНИЙ В СПИРТОВЫХ РАСТВОРИТЕЛЯХ | 2005 |
|
RU2357947C2 |
| СПОСОБ ФТОРИРОВАНИЯ ДЛЯ СИНТЕЗА 2-[F]-ФТОР-2-ДЕЗОКСИ-D-ГЛЮКОЗЫ | 2005 |
|
RU2394040C2 |
| [F-18] МЕЧЕННАЯ L-ГЛЮТАМИНОВАЯ КИСЛОТА, [F-18] МЕЧЕННЫЙ L-ГЛЮТАМИН, ИХ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ, А ТАКЖЕ СПОСОБ ИХ ПОЛУЧЕНИЯ | 2006 |
|
RU2395489C2 |
Изобретение относится к соединениям формулы (I) и (II) и способу [18F]-фторирования биомолекул, в частности пептидов, с использованием соединения формулы (I). Полученные соединения, меченные 18F, полезны в качестве радиофармацевтических препаратов, особенно для применения в позитронной эмиссионной томографии (ПЭТ). 3 н. и 7 з.п. ф-лы, 4 пр.
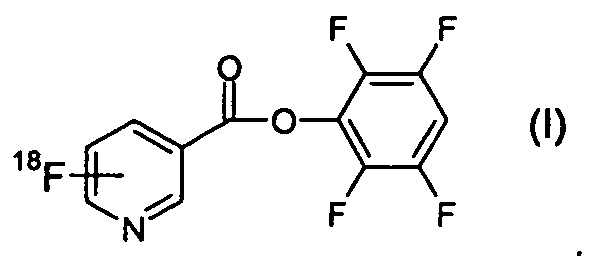
1. Соединение формулы (I):
2. Соединение по п.1 формулы (Ia):
3. Соединение формулы (II):
или его соль, где L представляет собой уходящую группу, выбранную из хлоро, бромо, йодо и три(C1-6алкил)аммония (например, триметиламмония).
4. Соединение формулы (II) или его соль по п.3, где L представляет собой три(C1-6алкил)аммоний (например, триметиламмоний).
5. Соединение формулы (II) по п.3 или 4, представляющее собой:
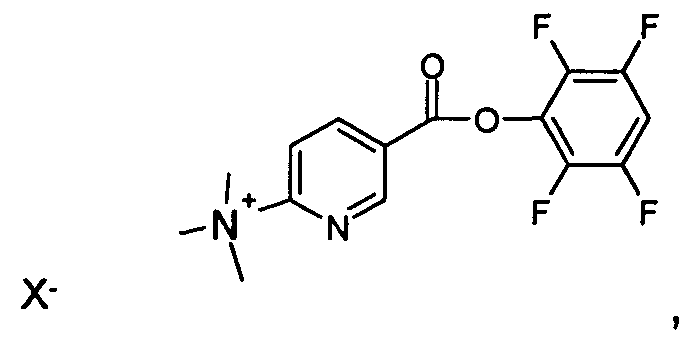
где X- представляет собой противоион и предпочтительно является трифторметансульфонатом.
6. Способ 18F-фторирования, включающий взаимодействие соединения формулы (I) или (Ia), как они определены в п.1 или 2, с соединением формулы (III):
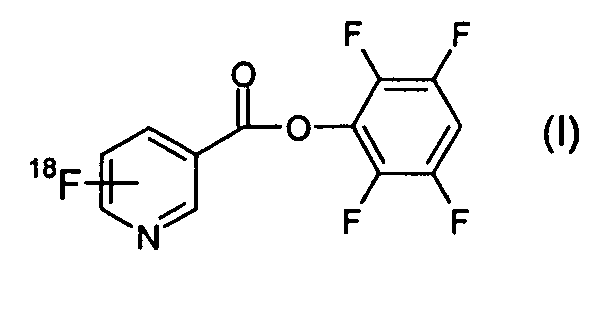

с образованием 18F-продукта формулы (IV):
7. Способ по п.6, где биомолекула представляет собой пептид.
8. Способ по п.6, где биомолекула представляет собой пептид, выбранный из аналогов соматостатина, таких как октреотид, бомбезин, вазоактивного интестинального пептида, аналогов хемотаксического пептида, α-меланоцитостимулирующего гормона, нейротензина, пептида Arg-Gly-Asp и его аналогов, проинсулинсвязывающего пептида человека, эндотелина, ангиотензина и формил-норлейцил-лейцил-фенилаланил-норлейцил-тирозил-лизина.
9. Способ по п.6, где биомолекула содержит фрагмент:
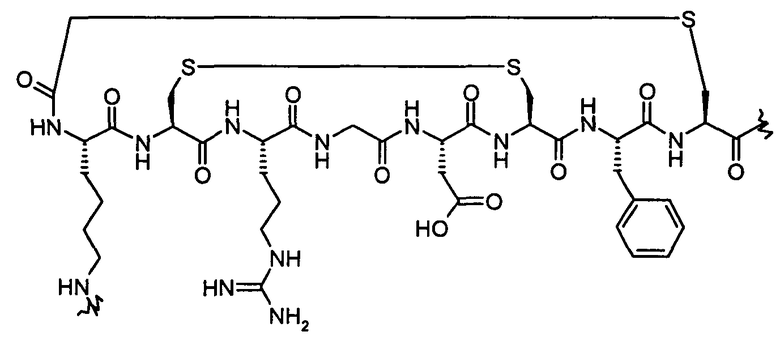
10. Способ по любому из пп.6-9, где биомолекула представляет собой пептид формулы (А):
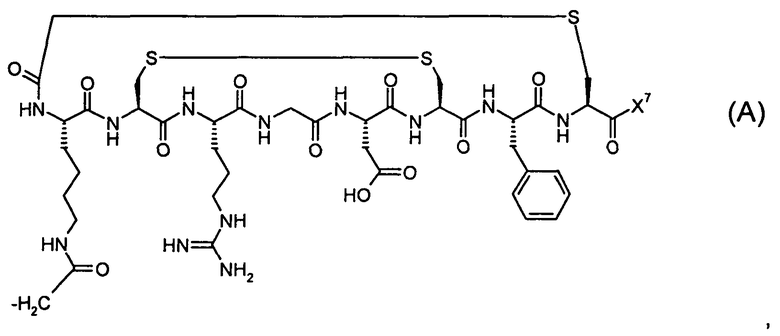
где X7 представляет собой либо -NH2, либо
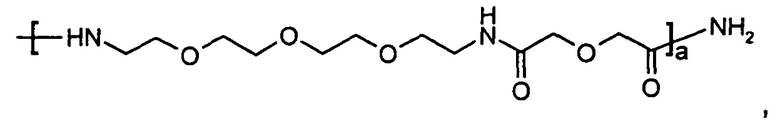
где а является целым числом от 1 до 10; предпочтительно а равно 1.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| C-Y SHIUE, "Synthesis of 18F-labelled N-(p-[18F]fluorophenyl)maleimide and its derivatives for labelling monoclonal antibody with 18F", JOURNAL OF LABELLED COMPOUNDS AND RADIOPHARMACEUTICALS, vol | |||
| Прибор для получения стереоскопических впечатлений от двух изображений различного масштаба | 1917 |
|
SU26A1 |
| СПОСОБ ИЗГОТОВЛЕНИЯ ЧЕРТЕЖЕЙ ДЛЯ ОДНООБРАЗНОЙ РАСКРОЙКИ ПРЕДМЕТОВ ОДЕЖДЫ | 1919 |
|
SU287A1 |
| FR 2926079 A1, 10.07.2009 | |||
| СОЕДИНЕНИЯ НА ОСНОВЕ ПЕПТИДОВ ДЛЯ НАПРАВЛЕННОЙ ДОСТАВКИ К РЕЦЕПТОРАМ ИНТЕГРИНОВ | 2002 |
|
RU2303042C2 |

