Область техники, к которой относится изобретение
Настоящее изобретение относится к новым предшественникам, пригодным для введения радиоактивной метки 18F в производные глутамата, к способам получения таких соединений и к их промежуточным продуктам, к композициям, содержащим такие соединения, к наборам, содержащим такие соединения, или к композициям и к способам введения радиоактивной метки 18F в производные глутамата, при этом полученные меченные радиоактивным изотопом 18F производные глутамата пригодны для диагностической визуализации позитронной эмиссионной томографией (ПЭТ) пролиферативных заболеваний, например, опухоли у млекопитающих.
Предпосылки создания изобретения
Раннее диагностирование злокачественных опухолевых заболеваний играет важную роль для прогноза выживания онкологических больных. В случае такого диагноза важным вспомогательным средством являются неинвазивные диагностические способы визуализации. В последние годы в этом отношении особенно полезной оказалась в частности ПЭТ технология (позитронная эмиссионная томография). Чувствительность и специфичность ПЭТ технологии, по существу, зависит от применяемого сигнального вещества (радиоактивного индикатора) и его распределения в организме. В поисках пригодных радиоактивных индикаторов пытаются использовать определенные свойства опухолей, которые отличают опухолевую ткань от окружающей здоровой ткани. Предпочтительным коммерчески доступным используемым изотопом, применяемым для ПЭТ, является 18F. В случае 18F за счет его короткого периода полураспада менее 2 ч предъявляются особые требования в отношении получения пригодных радиоактивных индикаторов. С этим изотопом невозможны сложные длительные пути синтеза и методики очистки, так как в противном случае значительная доля радиоактивности изотопа уже исчезает, прежде чем радиоактивный индикатор сможет быть использован для диагностики. Поэтому зачастую невозможно применять установленные пути синтеза для нерадиоактивных фторирований в случае синтеза 18F радиоактивных индикаторов. Кроме того, высокая удельная активность 18F (примерно 80 ГБк/нмоль) приводит к очень незначительным количествам вещества, [18F]-фторида для синтеза радиоактивного индикатора, что снова обусловливает предельный избыток предшественника и делает непредсказуемым результат стратегии радиосинтеза, которая основывается на нерадиоактивной реакции фторирования
ПЭТ на основе ФДГ ([18F]-2-фтордеоксиглюкоза) представляет собой широко распространенное и часто применяемое вспомогательное средство при диагностике и дальнейшем клиническом наблюдении опухолевых патологий. Злокачественные опухоли конкурируют с организмом-хозяином за снабжение глюкозой в качестве питательного вещества. (Warburg О., Über den Stoffwechsel der Carcinomzelle [Обмен веществ раковых клеток], Biochem. Zeitschrift 1924; 152:309-339; Kellof G., Progress and Promise of FDG-PET Imaging for Cancer Patient Management and Oncologic Drug Development, Clin. Cancer Res. 2005; 11(8):2785-2807). При этом опухолевые клетки, по сравнению с окружающими клетками нормальной ткани, обычно обладают повышенным метаболизмом глюкозы. Это используют при применении фтордезоксиглюкозы (ФДГ), производного глюкозы, которое усиленно переносится в клетки, однако, там после фосфорилирования метаболически блокируется в виде ФДГ-6-фосфата («Эффект Варбурга»). Поэтому 18F-меченая ФДГ является эффективным радиоактивным индикатором для определения онкологических заболеваний у пациента с помощью технологии ПЭТ. В поисках новых радиоактивных индикаторов для ПЭТ в последнее время также все более использовали аминокислоты для 18F визуализации ПЭТ (например, (обзор): Eur J Nucl Med Mol Imaging. May 2002; 29(5):681-90). При этом некоторые из 18F-меченых аминокислот пригодны для измерения скоростной нормы синтеза белка, однако, большинство других производных пригодно для измерения прямого клеточного поглощения в опухоли. Известными 18F-мечеными аминокислотами являются, например, производные от тирозиновых аминокислот, фенилаланиновых аминокислот, пролиновых аминокислот, аспарагиновых аминокислот и искусственных аминокислот (например, J. Nucl. Med. 1991; 32:1338-1346, J. Nucl. Med. 1996; 37:320-325, J. Nucl. Med. 2001; 42:752-754 и J. Nucl. Med. 1999; 40:331-338).
В последнее время были опубликованы применение и синтез 18F/19F-меченых производных глутаминовой кислоты и производных глутамина (WO 2008052788, WO 2009141091). Соединения с многообещающими преклиническими результатами (WO 2008052788, J. Med. Chem. 2011; (54):406-410, J Nucl Med. 2010; 51 (Supplement 2):1535) были испытаны в первых клинических исследованиях. Было обнаружено хорошее поглощение опухоли для [18F]-4-фтор-глутаминовой кислоты. Тем не менее, было замечено некоторое дефторирование, что негативно воздействовало на отношение фон-сигнал опухоли. (J Nucl Med. 2010; 51 (дополнение 2):118). В первых клинических исследованиях были получены лучшие результаты с применением (S)-4-(3-[18F]фторпропил)-L-глутаминовой кислоты. Очень хорошие результаты были обнаружены при выявлении рака легкого (Koglin et al., Abstract Nr. 412, SNM 2011, San Antonio; Baek et al., Abstract Nr. 195, SNM 2011, San Antonio).
Общими уходящими группами для введения метки в положения алкила, описанными в литературных источниках, являются сульфонаты, такие как мезилат, тозилат, и трифлат или галогениды (Ernst Schering Res Found Workshop. 2007; (62):15-50 и Eur. J. Org. Chem. 2008, 2853-2873).
Были опубликованы новые уходящие группы с различными возможностями. Lu et al. описывает применение уходящих групп, которые уже содержат катализатор межфазного переноса для введения [18F]фторида (Lu et al. J. Org. Chem. 2009; (74):5290-5296). Эти уходящие группы содержат арилсульфонат и хелатообразующую единицу, которая присоединена к арильному кольцу через циклический эфир. Кроме того, было описано применение особых уходящих групп, которые поддерживают удаление предшественника на стадии очистки после введения радиоактивной метки (WO 2011006610). Описанные уходящие группы представляют собой сульфонаты, содержащие липофильную часть, чтобы позволить простую очистку.
Были описаны различные предшественники для синтеза 4-(3-[18F]фторпропил)-L-глутаминовой кислоты.
В заявках WO 2008052788 и WO 2009141091, предшественник представляет собой комбинацию известных амино- и карбоксильных защитных групп и уходящих групп, таких как хлора, брома, производных сульфоната, таких как тозилокси, который приводит к образованию пригодного 18F-меченого радиоактивным изотопом предшественника в виде масла. Документ WO 2010000409 относится к применению новых перфорированных предшественников, к введению в них радиоактивной метки 18F и к очистке полученного соединения. Эти способы также применяли для получения 4-(3-[18F]фторпропил)-L-глутаминовой кислоты.
Тем не менее, синтез соединения остается затруднительным. Одним важным фактором в производстве радиоактивного индикатора является предшественник, пригодный для введения радиоактивной метки 18F. В связи с наличием различных функциональных групп (карбоксильной группы, аминогруппы) введение защитных групп необходимо для осуществления введения радиоактивной метки без потери функциональных групп. Кроме того, присутствие уходящей группы требуется для того, чтобы способствовать нуклеофильному введению 18F-метки.
До сих пор не был описан твердый предшественник для синтеза 4-(3-[18F]фторпропил)-L-глутаминовой кислоты.
Задача, которая должна быть решена с помощью изобретения, и ее решение
Для стандартного клинического применения 4-(3-[18F]фторпропил)-L-глутаминовой кислоты, необходим надежный и устойчивый способ производства, соответствующий требованиям Надлежащей производственной практики (GMP) и обеспечивающий устойчивый раствор для инъекций (изотонический, с соответствующим pH) радиоактивного индикатора с низким содержанием примесей.
Несмотря на короткий период полураспада 18F (110 мин), способ должен обеспечивать меченый радиоактивный индикатор с высоким радиохимическим выходом в течение короткого времени синтеза (предпочтительно менее чем 60 мин). Обычно изготовление меченых радиоактивных индикаторов осуществляют на автоматизированных системах. Для стандартного применения часто применяют предварительно изготовленные наборы, содержащие (среди прочего) необходимое количество предшественника. Как правило, реагенты, используемые для изготовления меченых радиоактивных индикаторов - включая предшественник - нуждаются в достаточной стабильности для транспортировки и хранения.
Кроме того, также важной является физико-химическая природа предшественника: масляные или смолистые предшественники вызывают технические проблемы во время наполнения (например, в наборы). Или взвешивание точного количества предшественника является трудоемким и дорогостоящим, или взвешенное количество не является точным. Последнее может вызвать синтетические проблемы или привести к высокому содержанию примесей. Вследствие этого предпочтение отдается твердым предшественникам.
Согласно настоящему изобретению производные глутаминовой кислоты формулы Ia и IIa, а также Ib и IIb имеют два стереоцентра во 2 и 4 положениях. Способ получения этих соединений должен обеспечить высокую оптическую чистоту.
18F-меченые производные глутаминовой кислоты формулы IIIa-F18 и IVa-F18, а также IIIb-F18 и IVb-F18 также имеют два стереоцентра во 2 и 4 положениях. Способ получения этих соединений должен обеспечить то, что реакционные условия введения метки не приведут к значительной степени эпимеризации в одном или обоих стереоцентрах.
Поэтому для получения (S)-4-(3-[18F]фторпропил)-L-глутаминовой кислоты или (R)-4-(3-[18F]фторпропил)-L-глутаминовой кислоты желательно иметь предшественник, который является:
1. устойчивым
2. твердым и
3. меченым при достаточно мягких условиях, предотвращающих потерю стереохимической целостности.
В настоящем изобретении указанные выше задачи решают с помощью предоставления устойчивых (например, хранение при >-20°C), оптически чистых, твердых и достаточно реакционноспособных предшественников для изготовления меченных фтором производных глутамата.
Дистанционно управляемые синтезаторы для введения 18F метки могут быть адаптированы к этим предшественникам, чтобы обеспечить согласно стандарту GMP гибкое получение изотопных индикаторов.
Краткое изложение изобретения
Для синтеза (S)-4-(3-[18F]фторпропил)-L-глутаминовой кислоты были изобретены новые устойчивые и твердые предшественники формулы Ia для введения метки. Указанные выше задачи были решены с помощью ведения особой комбинации защитных групп и уходящих групп. В особенности, применение защитной группы тритила на аминофункции в комбинации с ароматическим кольцом, содержащим уходящую группу, обеспечивает твердые соединения. В полученные предшественники могут быть легко введены радиоактивные метки 18F, и с них может быть снята защита, чтобы получить (S)-4-(3-[18F]фторпропил)-L-глутаминовую кислоту (схема 1а). Новые предшественники формулы Ib, несущие заместитель на С-4 в "R" ориентации можно использовать для получения (R)-4-(3-[18F]фторпропил)-L-глутаминовой кислоты (схема 1b).
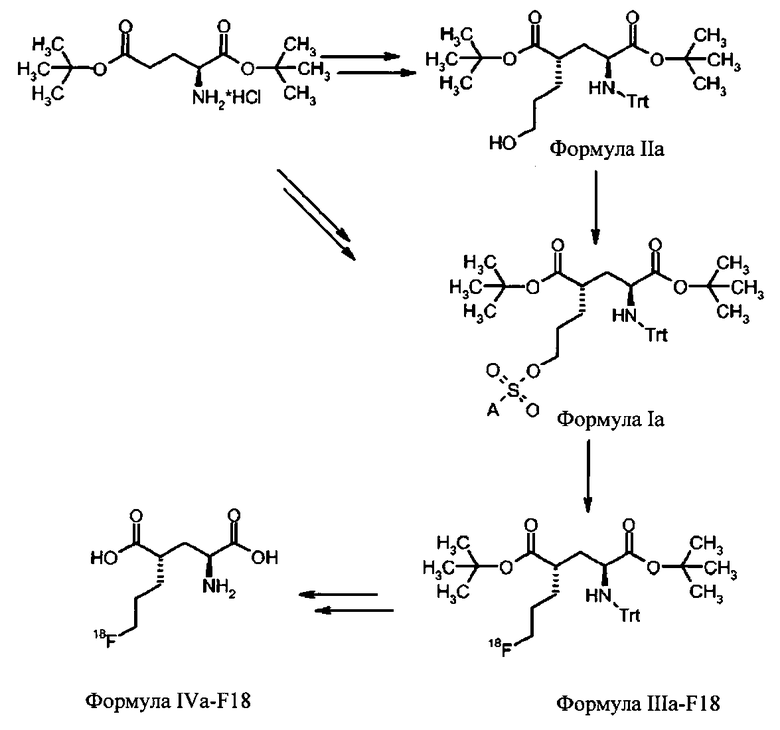
Схема 1a: синтез (S)-4-(3-[18F]фторпропил)-L-глутаминовой кислоты (IVa-F18) из соединений формулы Ia.
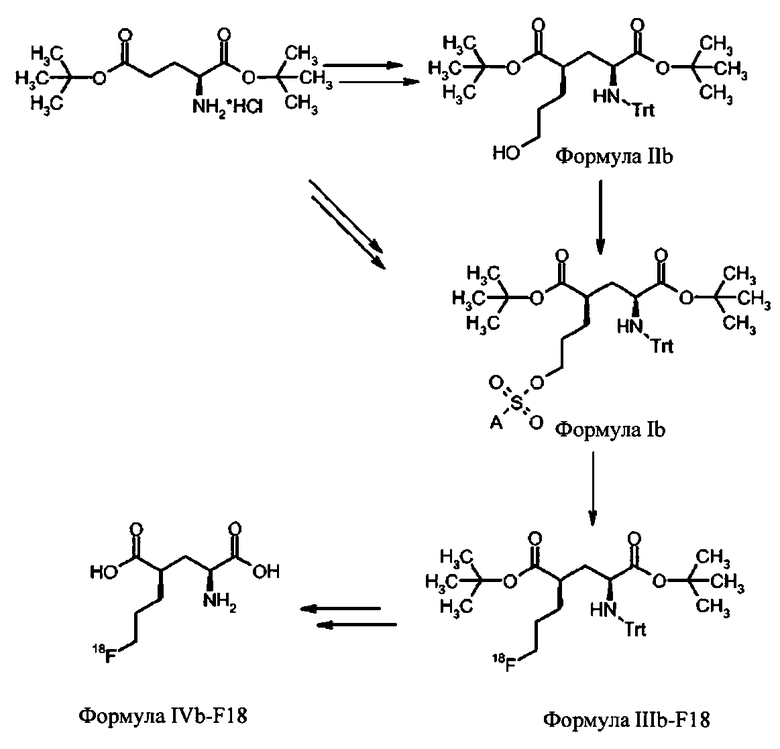
Схема 1b: Синтез (R)-4-(3-[18F]фторпропил)-L-глутаминовой кислоты (IVb-F18) из соединений формулы Ib.
Кроме того, настоящее изобретение обеспечивает способы получения меченных радиоактивным изотопом соединений формулы IV-F18, IVa-F18 и IVb-F18, используя раскрытые в данном описании соединения формулы I, Ia и Ib.
Подробное описание изобретения
В первом аспекте, изобретение относится к соединениям формулы I (предшественники)
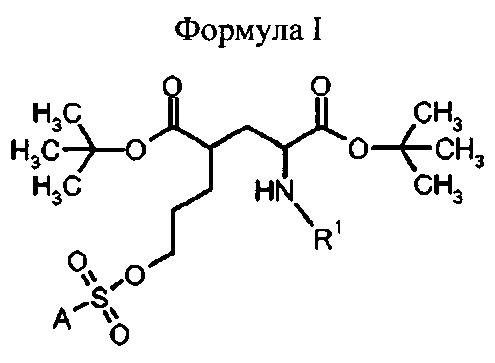
в которой
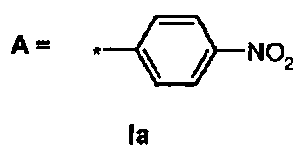
R1 представляет собой трифенилметил (тритил), А выбран из группы:
a) моноциклический арил,
b) бициклический арил,
c) биарил,
d) моноциклический гетероарил, и
e) бициклический гетероарил
по выбору, А несет один или несколько заместителей, выбранных из группы, включающей:
a) галоген,
b) нитро,
c) алкил,
d) трифторметил, и
e) Z,
при этом Z представляет собой
R1 представляет собой трифенилметил (тритил),
# указывает положение связи с А, и
к отдельным изомерам, таутомерам, диастереомерам, энантиомерам, стереоизомерам, их смесям и их приемлемым солям.
Предпочтительные признаки:
Предпочтительно, А выбран из группы:
a) фенил,
b) бифенил,
c) нафтил, и
d) хинолинил,
по выбору, А несет от 1 до 4 заместителей, выбранных из группы, включающей:
a) галоген,
b) нитро,
c) С1-С3-алкил,
d) трифторметил, и
e) Z.
Более предпочтительно, А выбран из группы:
a) фенил,
b) бифенил,
c) нафтил, и
d) хинолинил,
по выбору, А несет от 1 до 3 заместителей, выбранных из группы, включающей:
a) галоген,
b) нитро,
c) трифторметил, и
d) Z.
Еще более предпочтительно, А выбран из группы:
a) фенил,
b) бифенил,
c) нафтил, и
d) хинолинил,
по выбору, А несет от 1 до 3 заместителей, выбранных из группы, включающей:
a) хлор,
b) нитро,
c) трифторметил, и
d) Z.
Еще более предпочтительно, А выбран из группы:
a) фенил,
b) бифенил,
c) нафтил, и
d) хинолинил,
по выбору, А несет от 1 до 3 заместителей, выбранных из группы, включающей:
a) хлор,
b) нитро, и
c) трифторметил.
Еще более предпочтительно, А выбран из группы:
a) фенил,
b) бифенил,
c) нафтил, и
d) хинолинил,
по выбору, А несет от 1 до 3 заместителей, выбранных из хлора, и по выбору, А несет 1 заместитель, выбранный из группы, состоящей из:
a) нитро, и
b) трифторметил.
В предпочтительном варианте осуществления А представляет собой фенил, по выбору замещенный, как описано выше.
В другом предпочтительном варианте осуществления А представляет собой бифенил, по выбору замещенный, как описано выше.
В другом предпочтительном варианте осуществления А представляет собой нафтил, по выбору замещенный, как описано выше.
В другом предпочтительном варианте осуществления А представляет собой хинолинил, по выбору замещенный, как описано выше.
В более предпочтительном варианте осуществления А представляет собой нитрофенил.
В другом более предпочтительном варианте осуществления А представляет собой бифенил.
В другом более предпочтительном варианте осуществления А представляет собой хинолинил.
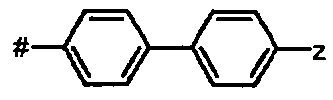
В другом более предпочтительном варианте осуществления А представляет собой бифенил-Z.
В более предпочтительном варианте осуществления А представляет собой нитро-(трифторметил)фенил.
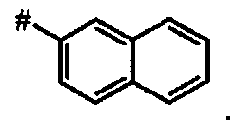
В более предпочтительном варианте осуществления А представляет собой нафтил.
В более предпочтительном варианте осуществления А представляет собой трихлорфенил.
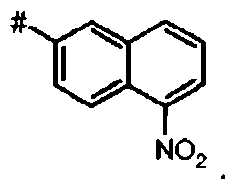
В более предпочтительном варианте осуществления А представляет собой нитронафтил.
В еще более предпочтительном варианте осуществления А представляет собой

В другом еще более предпочтительном варианте осуществления А представляет собой

В другом еще более предпочтительном варианте осуществления А представляет собой

В другом еще более предпочтительном варианте осуществления А представляет собой

В другом еще более предпочтительном варианте осуществления А представляет собой
В другом еще более предпочтительном варианте осуществления А представляет собой
В другом еще более предпочтительном варианте осуществления А представляет собой
В другом еще более предпочтительном варианте осуществления А представляет собой

В другом еще более предпочтительном варианте осуществления А представляет собой

В другом еще более предпочтительном варианте осуществления А представляет собой
В другом еще более предпочтительном варианте осуществления А представляет собой

# указывает положение связи с А в формуле I.
Галоген представляет собой хлор, фтор, йод или бром. Предпочтительно, галоген представляет собой хлор.
Алкил представляет собой разветвленный или неразветвленный C1-С6 алкил. Предпочтительно, алкил представляет собой метил, этил или пропил.
В предпочтительном варианте осуществления формула I относится к соединениям с (2S,4S)-конфигурацией (соединение формулы Ia) с диастереомерной и энантиомерной чистотой в >80%, предпочтительно >90%, более предпочтительно 95% и еще более предпочтительно >98%.
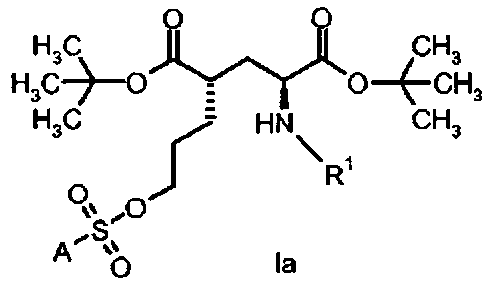
в которой А и R1 определены, как для формулы I выше.
В другом предпочтительном варианте осуществления формула I относится к соединениям с (2S,4R)-конфигурацией (соединение формулы Ib) со диастереомерной и энантиомерной чистотой в >80%, предпочтительно >90%, более предпочтительно 95% и еще более предпочтительно >98%.

в которой А и R1 определены, как для формулы I выше.
Предпочтительное соединение формулы I представляет собой ди-трет-бутил(4S)-4-(3-{[(4-нитрофенил)сульфонил]окси}пропил)-N-тритил-L-глутамат
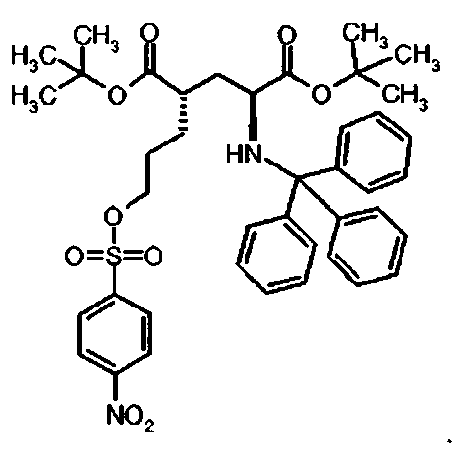
Предпочтительное соединение формулы I представляет собой ди-трет-бутил(4S)-4-(3-{[(3-нитрофенил)сульфонил]окси}пропил)-N-тритил-L-глутамат
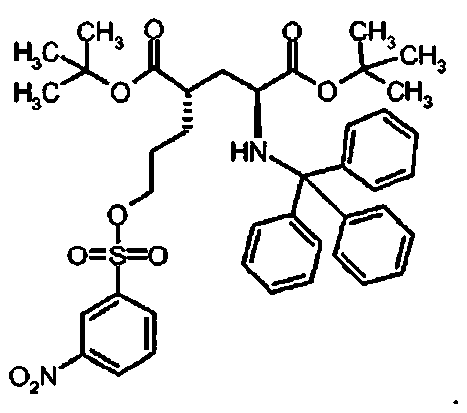
Предпочтительное соединение формулы I представляет собой ди-трет-бутил(4S)-4-{3-[(бифенил-4-илсульфонил)окси]пропил}-N-тритил-L-глутамат
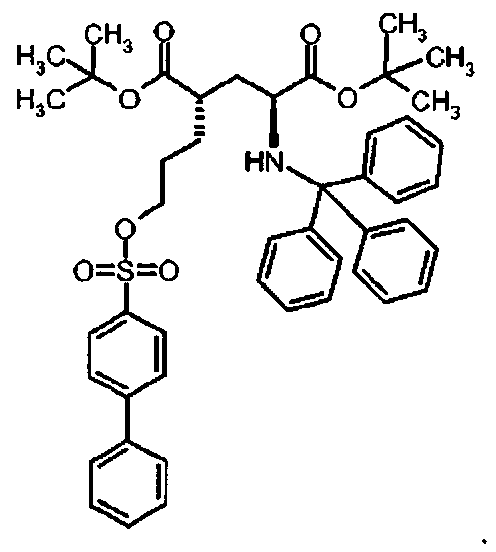
Предпочтительное соединение формулы I представляет собой ди-трет-бутил(4S)-4-{3-[(2-нафтилсульфонил)окси]пропил}-N-тритил-L-глутамат

Предпочтительное соединение формулы I представляет собой ди-трет-бутил(4S)-4-{3-[(1-нафтилсульфонил)окси]пропил}-N-тритил-L-глутамат
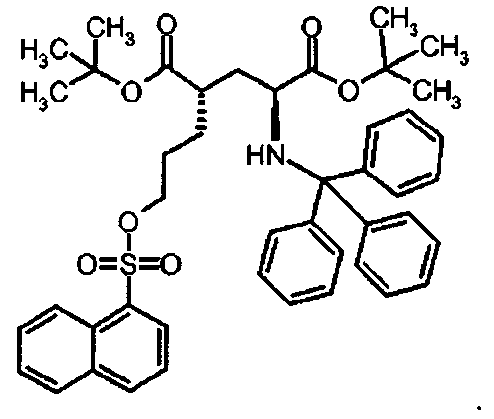
Предпочтительное соединение формулы I представляет собой ди-трет-бутил(4S)-4-{3-[(хинолин-8-илсульфонил)окси]пропил}-N-тритил-L-глутамат

Предпочтительное соединение формулы I представляет собой ди-трет-бутил(4S)-4-(3-{[(2,4,6-трихлорфенил)сульфонил]окси}пропил)-N-тритил-L-глутамат
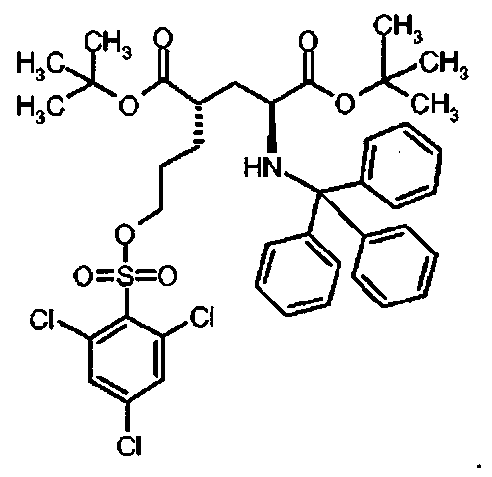
Предпочтительное соединение формулы I представляет собой тетра-трет-бутил(2S,4S,2′S,4′S)-2,2,-[бифенил-4,4′-диилбис(сульфонилоксипропан-3,1-диил)]бис[4-(тритиламино)пентандиоат]

Предпочтительное соединение формулы I представляет собой ди-трет-бутил(4S)-4-(3-{[(7-нитро-1-нафтил)сульфонил]окси}пропил)-N-тритил-L-глутамат

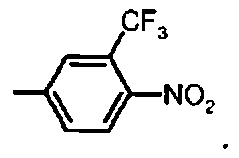
Предпочтительное соединение формулы I представляет собой ди-трет-бутил(4S)-4-[3-({[4-нитро-3-трифторметил)фенил]сульфонил}окси)пропил]-N-тритил-L-глутамат
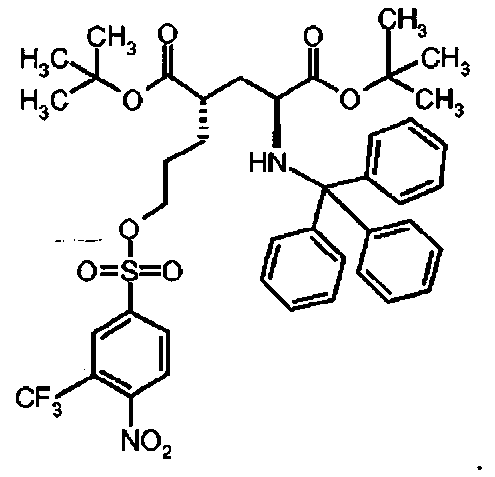
Предпочтительное соединение формулы I представляет собой ди-трет-бутил(4S)-4-(3-{[(4-метилфенил)сульфонил]окси}пропил)-N-тритил-L-глутамат.

Предпочтительное соединение формулы I представляет собой ди-трет-бутил(4R)-4-(3-{[(4-метилфенил)сульфонил]окси}пропил)-N-тритил-L-глутамат.

Предпочтительное соединение формулы I представляет собой ди-трет-бутил(4R)-4-{3-[(2-нафтилсульфонил)окси]пропил}-N-тритил-L-глутамат
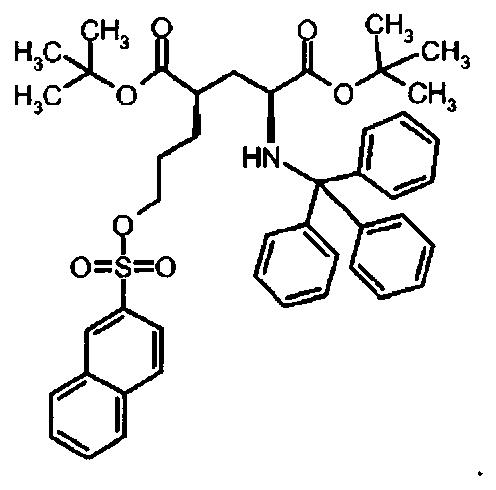
Второй аспект настоящего изобретения относится к соединениям формулы I, Ia или Ib в твердом виде. Предпочтительно, настоящее изобретение относится к твердым соединениям формулы I, Ia или Ib, как перечислено выше.
Кроме того, изобретение относится к способам получения кристаллической формы соединений формулы I, Ia или Ib. Способы кристаллизации хорошо известны специалисту в данной области техники.
В предпочтительном варианте осуществления, настоящее изобретение относится к кристаллическим соединениям формулы I, Ia или Ib.
Предпочтительно, следующее соединение находится в кристаллической форме ди-трет-бутил(4S)-4-{3-[(2-нафтилсульфонил)окси]пропил}-N-тритил-L-глутамата.
Предпочтительно, следующее соединение находится в кристаллической форме ди-трет-бутил(4R)-4-{3-[(2-нафтилсульфонил)окси]пропил}-N-тритил-L-глутамата.
В третьем аспекте, изобретение относится к способам получения соединений формулы I.
Способ получения соединений формулы I
Способ получения соединений формулы I осуществляют сульфонилированием гидроксигруппы в формуле II с пригодным сульфонилгалогенидом (предпочтительно, сульфонилхлоридом) или ангидридом с приемлемым заместителем А, чтобы образовать соединение формулы I, как определено выше.
Способ получения соединений формулы I включает стадию:
- сульфонилирования соединения формулы II с сульфонилгалогенидом (предпочтительно, сульфонилхлоридом) или сульфонилангидридом, которые имеют приемлемый заместитель А.
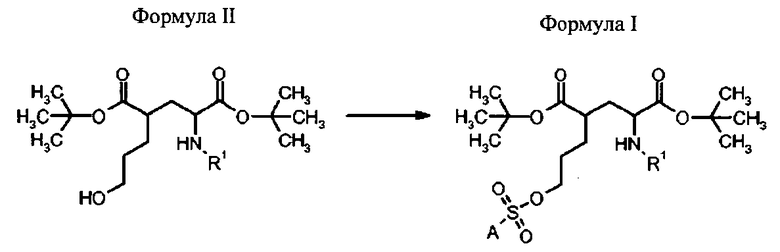
в которой R1 представляет собой трифенилметил (тритил), А выбран из группы:
a) моноциклический арил,
b) бициклический арил,
c) биарил,
d) моноциклический гетероарил, и
e) бициклический гетероарил
по выбору, А несет один или несколько заместителей, выбранных из группы, включающей:
a) галоген,
b) нитро,
c) алкил,
d) трифторметил, и
e) Z,
Z представляет собой

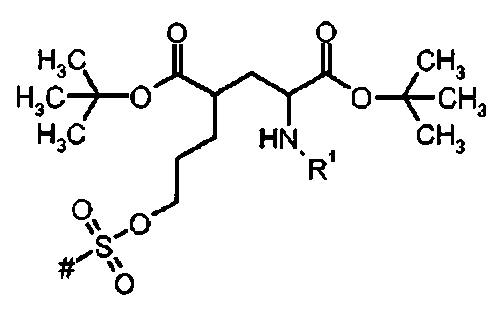
R1 представляет собой трифенилметил (тритил),
# указывает положение связи с А.
В другом варианте осуществления, бис-сульфонилгалогенид X-SO2-A-SO2-X вступает в реакцию с двумя молекулами соединения формулы II, чтобы получить соединение формулы I, в которой А представляет собой замещенный посредством Z, как описано выше. X представляет собой галоген, предпочтительно X представляет собой хлор.
Способ получения соединений формулы Ia
Предпочтительно, способ осуществляют путем реакции соединений формулы IIa для получения соединений формулы Ia с (2S′,4S)-конфигурацией
- сульфонилирование соединения формулы IIa с сульфонилгалогенидом (предпочтительно, сульфонилхлоридом) или сульфонилангидридом, которые имеют приемлемый заместитель А.

в которых А и R1 определены выше.
Способ получения соединений формулы Ib
Предпочтительно, способ осуществляют путем реакции соединений формулы IIb для получения соединений формулы Ib с (2S,4R)-конфигурацией
- сульфонилирование соединения формулы IIb с сульфонилгалогенидом (предпочтительно, сульфонилхлоридом) или сульфонилангидридом, которые имеют приемлемый заместитель А.

в которых А и R1 определены выше.
В другом предпочтительном варианте осуществления, способ осуществляют путем реакции смеси соединений формулы IIa и IIb для получения смеси соединений формулы Ia с (2S,4S)-конфигурацией и соединений формулы Ib с (2S,4R)-конфигурацией, которые могут быть разделены способами, известными специалисту в данной области техники (например, хроматографией, кристаллизацией), чтобы получить выделенные соединения формулы Ia и выделенные соединения формулы Ib
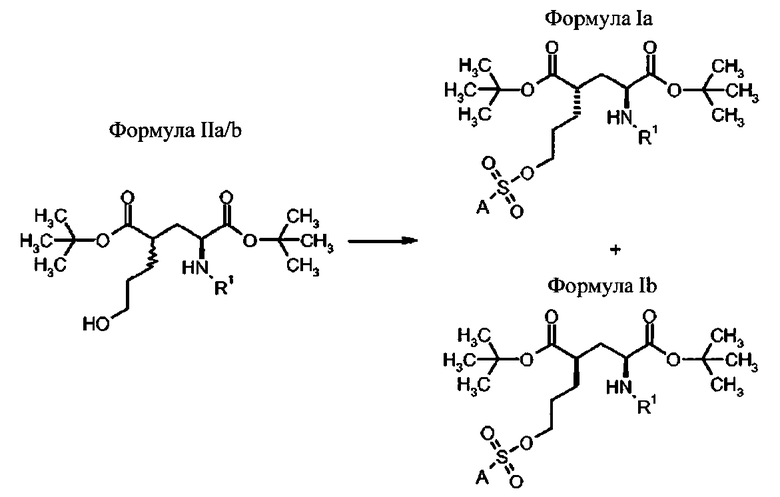
в которых А и R1 определены выше.
Реагенты, растворители и условия, которые могут быть использованы для данного сульфонилирования являются общепринятыми и хорошо известны специалисту в данной области техники. (J. March, Advanced Organic Chemistry, 4 th ed. 1992, John Wiley & Sons, cc. 352 и на др. ее).
Сульфонилирование соединений формулы II до получения соединений формулы I осуществляют в приемлемом инертном растворителе, в присутствии приемлемого основания, по выбору в микроволновом реакторе, в случае если реакцию проводят при повышенной температуре, температуре между - 10°C и 150°C и под давлением до 5 бар.
Пригодными инертными растворителями являются амиды, такие как N,N-диметилформамид, N,N-диметилацетамид, или N-метилпирролидинон, простые эфиры, такие как тетрагидрофуран, 1,2-диметоксиэтан, или диоксан, галогенированные углеводороды, такие как дихлорметан или хлороформ, или другие, такие как ацетонитрил.
Пригодные основания представляют собой щелочные карбонаты, такие как карбонат натрия или карбонат калия, щелочные бикарбонаты, такие как бикарбонат калия, или органические основания, такие как триэтиламин, N,N-диизопропилэтиламин, пиридин, N-метилморфолин, N-метилпиперидин, или DBU (1,8-диазабицикло(5.4.0)-ундец-7-ен).
Предпочтительными инертными растворителями являются дихлорметан или тетрагидрофуран.
Предпочтительными основаниями являются триэтиламин, N,N-диизопропилэтиламин или пиридин.
Предпочтительные признаки и варианты осуществления, описанные для соединений общей формулы I, Ia, Ib, II, IIa и IIb, включены в данное описание.
В четвертом аспекте изобретение относится к способам получения соединений формулы IV-F18.
Способ получения IV-F18: посредством прямого мечения соединений формулы I
Прямой способ получения соединений формулы IV-F18 содержит стадии:
- взаимодействия соединения формулы I с 18F-фторирующим агентом, с целью получения соединения формулы III-F18, и
- снятия защиты с полученного соединения формулы III-F18 для получения соединения формулы IV-F18,
в которой
соединение формулы III-F18 представляет собой

в которой R1 представляет собой трифенилметил (тритил),
и соединение формулы IV-F18 представляет собой

По выбору способ осуществляют посредством очистки соединения формулы IV-F18 путем твердофазной экстракции. Предпочтительно используют картриджи или колонки для твердофазной экстракции.
Предпочтительно, прямой способ получения соединений формулы IVa-F18 содержит стадии:
- взаимодействия соединения формулы Ia с 18F-фторирующим агентом, с целью получения соединения формулы IIIa-F18, и
- снятия защиты с полученного соединения формулы IIIa-F18 для получения соединения формулы IVa-F18,
в которой
соединение формулы IIIa-F18 представляет собой
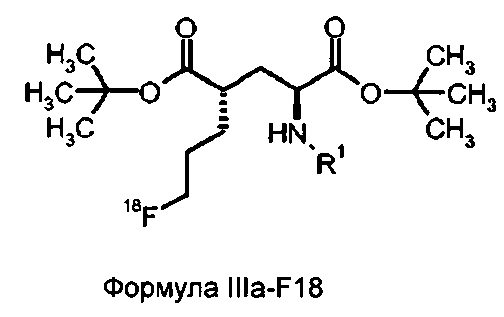
R1 представляет собой трифенилметил (тритил), и
соединение формулы IVa-F18 представляет собой
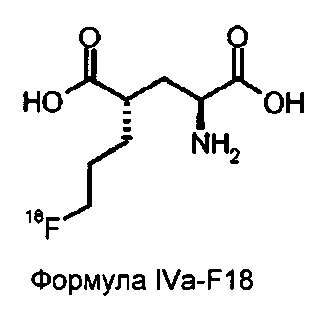
По выбору способ осуществляют посредством очистки соединения формулы IVa-F18 путем твердофазной экстракции. Предпочтительно используют картриджи или колонки для твердофазной экстракции.
Примерами 18F-фторирующего агента являются, но не ограничиваются ними, K18F, H18F, Rb18F, Cs18F, Na18F.
По выбору, 18F-фторирующий агент содержит хелатообразующий агент, такой как криптанд (например: 4,7,13,16,21,24-гексаокса-1,10-диазабицикло[8.8.8]-гексакозан - Kryptofix®) или краун-эфир (например: 18-краун-6).
Также 18F-фторирующий агент может представлять собой тетраалкиламмониевую соль 18F- или тетраалкилфосфониевую соль 18F-, которые известны специалисту в данной области техники, например: [18F]фторид тетрабутиламмония, [18F]фторид тетрабутилфосфония.
Предпочтительно, 18F-фторирующий агент представляет собой Cs18F, K18F, [18F]фторид тетрабутиламмония.
Реагенты, растворители и условия, которые могут быть использованы для подобного фторирования, являются распространенными и хорошо известны специалисту в данной области техники. См., например, J. Fluorine Chem., 27 (1985): 177-191; Coenen, Fluorine-18 Labeling Methods: Features and Possibilities of Basic Reactions, (2006), in: Schubiger P.A., Friebe M., Lehmann L, (eds), PET-Chemistry - The Driving Force in Molecular Imaging. Springer, Berlin, Heidelberg, cc.15-50). Предпочтительно, используемыми в данном способе растворителями являются ДМФ, ДМСО, ацетонитрил, ДМА, ТГФ, или их смеси, предпочтительно растворитель представляет собой ацетонитрил.
Нагревание можно осуществлять путем обычного нагревания или микроволнового нагревания.
В другом предпочтительном варианте осуществления, прямой способ получения соединений формулы IVb-F18 содержит стадии:
- взаимодействия соединения формулы Ib с 18F-фторирующим агентом с целью получения соединения формулы IIIb-F18, и
- снятия защиты с полученного соединения формулы IIIb-F18 для получения соединения формулы IVb-F18,
в которой
соединение формулы IIIb-F18 представляет собой

R1 представляет собой трифенилметил(тритил), и
соединение формулы IVb-F18 представляет собой

По выбору способ осуществляют посредством очистки соединения формулы IVb-F18 путем твердофазной экстракции. Предпочтительно используют картриджи или колонки для твердофазной экстракции.
Примерами 18F-фторирующего агента являются, но не ограничиваются ними, K18F, H18F, Rb18F, Cs18F, Na18F.
По выбору, 18F-фторирующий агент содержит хелатообразующий агент, такой как криптанд (например: 4,7,13,16,21,24-гексаокса-1,10-диазабицикло[8.8.8]-гексакозан - Kryptofix®) или краун-эфир (например: 18-краун-6).
Также 18F-фторирующий агент может представлять собой тетраалкиламмониевую соль 18F- или тетраалкилфосфониевую соль 18F-, которые известны специалисту в данной области техники, например: [18F]фторид тетрабутиламмония, [18F]фторид тетрабутилфосфония.
Предпочтительно, 18F-фторирующий агент представляют собой Cs18F, K18F, [18F]фторид тетрабутиламмония.
Реагенты, растворители и условия, которые могут быть использованы для подобного фторирования, являются распространенными и хорошо известны специалисту в данной области техники. См., например, J. Fluorine Chem., 27 (1985): 177-191; Coenen, Fluorine-18 Labeling Methods: Features and Possibilities of Basic Reactions, (2006), in: Schubiger P.A., Friebe M., Lehmann L, (eds), PET-Chemistry - The Driving Force in Molecular Imaging. Springer, Berlin Heidelberg, cc.15-50). Предпочтительно, используемыми в данном способе растворителями являются ДМФ, ДМСО, ацетонитрил, ДМА, ТГФ, или их смеси, предпочтительно растворитель представляет собой ацетонитрил.
Нагревание можно осуществлять путем обычного нагревания или микроволнового нагревания.
В предпочтительном варианте осуществления, соединение формулы IV получают путем взаимодействия соединения формулы I с [18F]фторирующим реагентом. Затем защитные группы отщепляют кислотным гидролизом, и соединение формулы очищают твердофазной экстракцией.
Более предпочтительно, [18F]фторирующий реагент представляет собой комплекс калий/[18F]фторид/криптофикс.
Более предпочтительно, взаимодействие соединения формулы I с реагентом [18F]фторид осуществляют в ацетонитриле в качестве растворителя.
Более предпочтительно используют 1-25 мкмоль, еще более предпочтительно 1-20 мкмоль, и еще более предпочтительно 5-10 мкмоль соединения формулы I.
Более предпочтительно, соединение формулы I взаимодействует с реагентом [18F]фторид при 60-160°C, более предпочтительно при 80-140°C, еще более предпочтительно при 100-140°C.
Более предпочтительно для кислотного гидролиза применяют HCl, H2SO4 или H3PO4. Еще более предпочтительно для кислотного гидролиза применяют 1М-4М HCl.
Более предпочтительно для очистки соединения формулы IV применяют катионообменный материал. Еще более предпочтительно для очистки соединения формулы IV применяют МСХ картридж(и).
Более предпочтительно для очистки соединения формулы IV применяют пористый углеродный материал. Еще более предпочтительно для очистки соединения формулы IV применяют картридж(и) Hypercarb.
В одном предпочтительном варианте осуществления соединение формулы I представляет собой соединение формулы Ia и соединение формулы IV представляет собой соединение формулы IVa.
В одном предпочтительном варианте осуществления соединение формулы I представляет собой соединение формулы Ib и соединение формулы IV представляет собой соединение формулы IVb.
В одном предпочтительном варианте осуществления, основание добавляют после кислотного гидролиза. Более предпочтительно после кислотного гидролиза добавляют NaOH. Еще более предпочтительно добавляют 1М-6М NaOH и смесь нагревают при 60°C-100°C.
Предпочтительные признаки и варианты осуществления, описанные для соединений общей формулы I, Ia, Ib, III-F18, IIIa-F18, IIIb-F18, IV-F18, IVa-F18 и IVb-F18, включены в данное описание.
В пятом аспекте изобретение относится к соединениям формулы II

в которой R1 представляет собой трифенилметил (тритил), и
к отдельным изомерам, таутомерам, диастереомерам, энантиомерам, стереоизомерам, стереоизомерным смесям или их смесям и их приемлемым солям.
Предпочтительно, соединения формулы II относятся к соединениям с (2S′,4S)-конфигурацией (соединение формулы IIa)

в которой R1 представляет собой трифенилметил (тритил), соответствующий ди-трет-бутил(4S)-4-(3-гидроксипропил)-]N-тритил-L-глутамату.
В другом предпочтительном варианте осуществления, соединения формулы II относятся к соединениям с (2S,4R)-конфигурацией (соединение формулы IIb)
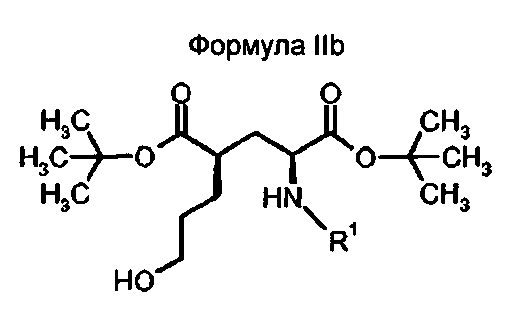
в которой R1 представляет собой трифенилметил (тритил), соответствующий ди-трет-бутил(4R)-4-(3-гидроксипропил)-N-тритил-L-глутамату.
В другом предпочтительном варианте осуществления, соединения формулы II относятся к соединениям с (2S)-конфигурацией (соединение формулы IIa/b)

в которой R1 представляет собой трифенилметил (тритил), соответствующий ди-трети-бутил 4-(3-гидроксипропил)-N-тритил-L-глутамау.
В шестом аспекте, изобретение относится к защищенным соединениям формулы III-F

в которой R1 представляет собой трифенилметил (тритил),
F означает атом фтора, и
к отдельным изомерам, таутомерам, диастереомерам, энантиомерам, стереоизомерам, стереоизомерным смесям или их смесям и их приемлемым солям.
Предпочтительно, F представляет собой 18F или 19F.
Более предпочтительно, F представляет собой 18F (соединение формулы III-F18).
Предпочтительно, соединения формулы III относятся к соединениям с (2S,4S)-конфигурацией (соединение формулы IIIa-F)

в которой
R1 представляет собой трифенилметил (тритил), и
F означает атом фтора.
Предпочтительно, F представляет собой 18F в соединении формулы IIIa-F. Предпочтительное соединение формулы IIIa-F18 представляет собой ди-трет-бутил(4S)-4-(3-[18F]фторпропил)-N-тритил-L-глутамат.
В другом предпочтительном варианте осуществления соединения формулы III относятся к соединениям с (2S,4R)-конфигурацией (соединение формулы IIIb-F)

в которой
R1 представляет собой трифенилметил (тритил), и
F означает атом фтора.
Предпочтительно, F представляет собой 18F в соединении формулы IIIb-F.
Предпочтительное соединение формулы IIIb-F18 представляет собой ди-трет-бутил(4R)-4-(3-[18F]фторпропил)-N-тритил-L-глутамат.
В седьмом аспекте, изобретение относится к композиции, содержащей соединение формулы I, Ia, II, IIa, III-F, IIIa-F, IIIa-F18, IVa-F или IVa-F18, как определено в указанных выше аспектах и охватывает варианты осуществления. Предпочтительно, композиция содержит соединение формулы I, Ia, Ib, II, IIa, IIb, III-F, IIIa-F, IIIb-F, IIIa-F18, IIIb-F18, IVa-F18 или IVb-F18, как определено в указанных выше аспектах и охватывает варианты осуществления. Более предпочтительно, композиция содержит соединение формулы I, Ia, Ib, II, IIa, IIb, III-F, IIIa-F, IIIb-F, IIIa-F18, IIIb-F18, как определено в указанных выше аспектах и охватывает варианты осуществления.
В первом варианте осуществления, изобретение относится к композиции, содержащей соединение формулы I или Ia или IIb и приемлемые реагенты для реакции введения радиоактивной метки фтора и/или вспомогательные вещества, в числе других, носители, растворители или стабилизаторы.
Специалисту в данной области техники хорошо известны вспомогательные вещества, которые пригодны для целевых фармацевтических составов, препаратов или композиций, исходя из его/ее экспертных знаний.
Предпочтительно, композиция содержит приведенные в качестве примеров соединения, стереоизомеры и их смеси, и их приемлемые соли, и приемлемые носители или разбавители, как описано выше.
Во втором варианте осуществления, изобретение относится к композиции, содержащей соединение формулы II или IIa или IIb, как описано выше и по выбору пригодные вспомогательные вещества. Такие вспомогательные вещества включают, в числе других, носители, растворители или стабилизаторы.
Специалисту в данной области техники хорошо известны вспомогательные вещества, которые пригодны для целевых фармацевтических составов, препаратов или композиций, исходя из его/ее экспертных знаний.
В третьем варианте осуществления, изобретение относится к композиции, содержащей соединение формулы IV-F18 или IVa-F18 или IVb-F18, и фармацевтически пригодные вспомогательные вещества. Введение соединений, фармацевтических композиций или комбинаций в соответствии с изобретением осуществляют любым из общепринятых способов введения, доступных из уровня техники. Предпочтение отдается внутривенному введению.
В восьмом аспекте, изобретение относится к набору, включающему одну ампулу или более чем одну ампулу, которая содержит предопределенное количество соединений формулы I, предпочтительно соединений формулы Ia или Ib. Более предпочтительно, набор содержит соединения формулы Ia.
По выбору набор содержит приемлемый носитель, разбавитель, наполнитель или вспомогательное вещество.
Предпочтительно, набор содержит предопределенное количество соединения формулы I и один или несколько картриджей/колонок для твердофазной экстракции для очистки соединения формулы IV-F18.
Предпочтительно, набор содержит физиологически приемлемую основу или носитель и необязательно вспомогательные вещества и консерванты, реагенты, пригодные для осуществления описанных в данном тексте реакций и/или чтобы создать реагенты для 18F введения метки. Кроме того, набор может содержать инструкции по его применению.
Общий синтез соединений согласно изобретению
Определения
Термины, используемые в настоящем изобретении, определены ниже, но не ограничивают объем притязаний изобретения.
Используемое в данном описании понятие "предшественник" относится к соединению, которое может быть применено в качестве исходного материала для реакции введения радиоактивной метки, где соответствующую уходящую группу предшественника замещают радиоактивным изотопом [18F].
Используемое в данном описании понятие "аминозащитная группа" относится к химической структурной единице (такой как, например, трифенилметил), химически связанной с аминогруппой, которая ингибирует участие этой аминогруппы в химических реакциях (см. Greene′s Protective Groups in Organic Synthesis, P. Wuts, T. Greene (Wiley)).
Используемое в данном описании понятие "гидроксильная защитная группа" относится к химической структурной единице (такой как, например, трет-бутил), химически связанной с гидроксильной группой, которая ингибирует участие этой гидроксильной группы в химических реакциях (см. Greene′s Protective Groups in Organic Synthesis, P. Wuts, T. Greene (Wiley)).
Используемое в данном описании понятие "алкил" относится к C1-С6 алкильной группе с прямой цепью или разветвленной цепью, такой как, например, метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, пентил, изопентил, неопентил. Предпочтительно, алкил представляет собой C1-С3 алкил с прямой цепью или разветвленной цепью.
"Арил" представляет собой моно- или бициклический ароматический, карбоциклический двухвалентный радикал, имеющий, как правило, от 6 до 10 атомов углерода, по выбору замещенных от одного до четырьмя "заместителями"; в качестве примера и предпочтительно фенил или нафтил.
"Биарил" представляет собой ароматический радикал, замещенный одинаковым ароматическим радикалом. Предпочтительно, биарил представляет собой бифенил.
"Гетероарил" представляет собой ароматический, моно- или бициклический двухвалентный радикал, имеющий, как правило, от 5 до 10, предпочтительно от 5 до 6, кольцевых атомов и до 3, предпочтительно 1, гетероатомов из рядов, состоящих из S, О и N; в качестве примера и включая, но не ограничиваясь ними, тиенил, фурил, пирролил, тиазолил, оксазолил, имидазолил, пиридил, пиримидил, пиридазинил, индолил, индазолил, бензофуранил, бензотиофенил, хинолинил, изохинолинил, триазолил, причем указанный "гетероарил" по выбору замещен от одного до четырьмя "заместителями". Предпочтительно, "гетероарил" представляет собой пиридил или хинолинил.
Используемое в данном описании понятие "арилсульфонил" относится к арильным группам, соответствующим образом связанным с соответствующим скелетом сульфонильной группой, т.е. -S(=O)2-O, с арильной частью, которая была определена выше, такой как, например, n-толуолсульфонил.
Термин "галоген" относится к фтору, хлору, брому, и йоду.
В случаях, когда используют понятие "замещенный", то оно предназначено для указания, что один или несколько атомов водородов на атоме, указанном в выражении с использованием "замещенный", замещен/замещены одной или множеством частей из группы, содержащей галоген, гидроксил, нитро, C1-С6-алкилкарбонил, циано, трифторметил, C1-С6-алкилсульфонил, C1-С6-алкил, С1-С6-алкокси и C1-С6-алкилсулфьанил, при условии, что не превышается обычная валентность соответствующего атома, и что замещение приводит к химически стабильному соединению, т.е. к соединению, которое является достаточно прочным, чтобы перенести выделение из реакционной смеси до пригодной степени чистоты, и приготовление в виде фармацевтической композиции.
Используемый в данном описании Cn-Cm указывает диапазон числа атомов углерода, соответствующая часть может представлять, в качестве пояснения, но не ограничиваясь, например С1-С6-алкил или С1-С6-алкокси, которые могут иметь 1, 2, 3, 4, 5, или 6 атомов углерода, не охватывая необязательное дополнительное замещение.
Если хиральные центры или другие формы изомерных центров по-другому не определены в соединении, предлагаемом в настоящем изобретении, то все формы таких стереоизомеров, включая энантиомеры и диастереоизомеры, включены в данное описание. Соединения, содержащие хиральные центры, могут применяться в виде рацемической смеси или в виде энантиомерно обогащенной смеси или в виде диастереомерной смеси или в виде диастереомерно обогащенной смеси, или эти изомерные смеси могут быть разделены с применением хорошо известных методик, и отдельный стереоизомер может быть использован единственным. В случаях, когда соединения могут находиться в таутомерных формах, то каждую таутомерную форму предполагается включить в объем настоящего изобретения, находясь ли в равновесии или преимущественно в одной форме.
Используемое в данном описании понятие "растворители" относится к неорганическим, таким как вода, а также к органическим соединениям, таким как ацетонитрил и к их смесям, применяемым для растворения других твердых, жидких или газообразных соединений.
Набор
Используемое в данном описании понятие "набор" относится к множеству материалов (таких как фильтры) и химикаты (такие как предшественник или растворители), необходимых для осуществления единого процесса введения радиоактивной метки.
Введение радиоактивной метки
Используемое в данном описании понятие "введение радиоактивной метки" относится к химическому процессу, при котором радиоактивный изотоп (такой как 18F) присоединяется к выбранной молекуле (такой как предшественник).
Снятие защиты
Используемое в данном описании понятие "снятие защиты" относится к одной или нескольким химической(им) реакции(ям), при которых удаляют защитную химическую группу, такую как тритил из молекулы и восстанавливают функциональную группу молекулы, такую как аминогруппу.
Десилилирование
Используемое в данном описании понятие "десилилирование" относится к одной или нескольким химической(им) реакции(ям), при которых силильную группу R3 - Si, такую как трет-бутилдиметилсилил удаляют из молекулы и замещают протоном.
Кристаллизация
Используемое в данном описании понятие "кристаллизация" относится к физико-химическому процессу, при котором твердые кристаллы осаждаются из раствора, расплава или газа.
Используемое в данном описании понятие "несет" означает, какой из эквивалентов подлежит замещению.
Экспериментальная часть
Сокращения
Общая часть
Все растворители и химикаты были получены из коммерчески доступных источников и применяли без дополнительной очистки. Использовали безводные растворители и инертную атмосферу (азот или аргон), если не указано иное. В предшествующей таблице перечислен список сокращений, использованных в данном разделе и в разделе Промежуточные продукты и Примеры, поскольку не объясняются в рамках основной части описания. Формы пиков ЯМР отражены так, как они появляются в спектрах, возможные эффекты более высокого порядка не принимали во внимание.
Реакции контролировали способами, известными специалисту в данной области техники, такими как тонкослойная хроматография на подходящих стационарных фазах, таких как алюминиевые или стеклянные пластины, покрытые силикагелем, или ВЭЖХ/УФ анализы.
Соединения и промежуточные продукты, полученные в соответствии со способами, предлагаемыми в изобретении, могут потребовать очистки. Очистка органических соединений хорошо известна специалисту в данной области техники и может быть несколько способов очистки одного того же соединения. В некоторых случаях очистка не нужна. В определенных случаях соединения могут быть очищены кристаллизацией. В некоторых случаях примеси можно удалить измельчением в порошок с применением пригодного растворителя. В некоторых случаях, соединения можно очистить колоночной хроматографией. Колоночная хроматография, которая упоминается в дальнейшем в данном описании, обычно относится к препаративной жидкостной хроматографии на подходящей стационарной фазе, такой как коммерчески доступный силикагель или предварительно упакованные картриджи с силикагелем, например силикагель Merck 60 (230-400 ячеек) и элюенты, такие как градиенты этилацетата/н-гексана.
Введение радиоактивной метки:
Все химикаты были приобретены в коммерчески доступных источниках, Aldrich и Merck, и использовали без дополнительной очистки.
Радиохимический синтез осуществляли с применением модуля GE MX tracerlab. Аналитическую ВЭЖХ осуществляли на системе Agilent 1200. ВЭЖХ растворители были приобретены у Aldrich.
Общий синтез
А. Алкилирование скелета глутамата
Соединения согласно изобретению могут быть получены путем алкилирования производных глутамата А-1, как показано на схеме 2.

Схема 2. Алкилирование скелета глутамата (RA1 представляет собой гидроксильную защитную группу, RA2 представляет собой уходящую группу, RA3 представляет собой аминозащитную группу).
RA2 ведет себя как уходящая группа (например, Br, I, сульфонат) и RA1 представляет собой защитную группу. Алкилирование производных глутамата описано в литературных источниках, например: М.A. Brimble et al., Bioorg. Med. Chem. 2005, 13, 519-523; S. Hanessian et al., J. Org. Chem. 2005, 70, 5070-5085; S. Hanessian et al., Org. Lett. 2004, 6, 4683-4686; J. Zhang et al., Tetrahedron Lett. 2003, 44, 1413-1415. Хорошо известно, что алкилирование селективно обеспечивает соединения А-3, если R1 представляет собой защитную группу карбаматного типа (например Boc, CBz). Смеси А-3 и А-4 можно получить и выделить хроматографическими способами, если применяют другие защитные группы (например, RA3 = тритил).
Специалисту в данной области техники хорошо известны способы превращения соединений формул А-3 в соединения формулы Па, включая например:
- отщепление аминозащитной группы RA3 и введение аминозащитной группы R1 (например, введение группы тритила через трифенилметилхлорид);
- отщепление гидроксильной защитной группы RA1 (например, дисилилирование с использованием TBAF).
Дополнительные способы синтеза IIa хорошо известны специалисту в данной области техники, например аллирование А-1 с применением аллилбромида и последующим гидроборированием.
Б. Синтез сульфонатов
Предшественники для 18F-алкильных соединений общей формулы I и Ia могут быть синтезированы из соответствующих гидроксильных соединений общей формулы II и IIa в соответствии со способами, известными из уровня техники (J. March, Advanced Organic Chemistry, 4-е изд-е 1992, John Wiley & Sons, cc.352 и на др. ее).
В. 18F фторирование
Радиохимический синтез 18F-меченого соединения согласно изобретению можно осуществить с помощью множества способов, с применением известных методов, описанных в литературных источниках и базах данных, доступных специалисту в данной области техники.
Более конкретно, соединения согласно общим формулам III-F18 и IV-F18 можно синтезировать, исходя из I, как показано на схеме 4. Подобные нуклеофильные фторирования известны специалисту в данной области техники и также описаны в литературных источниках, для обзора и приведенных там ссылок см., например Cai et ai, Eur. J. Org. Chem., 2008, 2853; Ametamey et al., Chem. Rev., 2008, 108, 1501, Miller et ai, Angew. Chem. Int. Ed. 2008, 47, 8998.

Схема 4. Синтез 18F-меченых соединений формулы III-F18 и IV-F18.
Способы ВЭЖХ
Способ А1 (аналитика ди-трет-бутил(4S)-4-(3-гидроксипропил)-N-тритил-L-глутамата.
Колонка: ChiralPak IA, 4,6×250 мм
Подвижная фаза: 5% IPA/n-гептан
Скорость потока: 1 мл/мин
Длина волны: 214/254 нм
Способ А2 (для Id)
Колонка: X-Bridge
Подвижная фаза: ацетонитрил/вода 20:80 до 100% воды
Скорость потока: 1 мл/мин
Длина волны: 214 нм
Способ A3 (для от Ia до Ic и от Ie до Ij)
Колонка: X-Bridge
Подвижная фаза: ацетонитрил/вода 15:80 до 100% воды
Скорость потока: 1 мл/мин
Длина волны: 214 нм
Способ А4 (19F-фторирование)
Колонка: Phenomenex Lux 5U Amylose-2
Подвижная фаза: 10% IP A/Hex
Скорость потока: 1 мл/мин
Длина волны: 214 нм
Способ А5 (введение радиоактивной метки 18F)
Колонка: Phenomenex Luna 5µ С18(2); 250*4,6 мм
Подвижная фаза: А: Na2HPO4 10 мМ pH 7,4, В: ацетонитрил
Градиент: 0 мин 12% В, 15 мин 12% В, 16 мин 100% В, 18 мин 100% В, 20 мин 12% В, 23 мин 12% В
Скорость потока: 1,2 мл/мин
Длина волны: 340 нм
Получение производных: 10 мл раствора продукта смешивают с 30 мл реагента ОРА (Thermo Scientific, №.:26015). Через 1 мин реакции при комнатной температуре раствор подвергают ВЭЖХ.
Получение промежуточных продуктов I
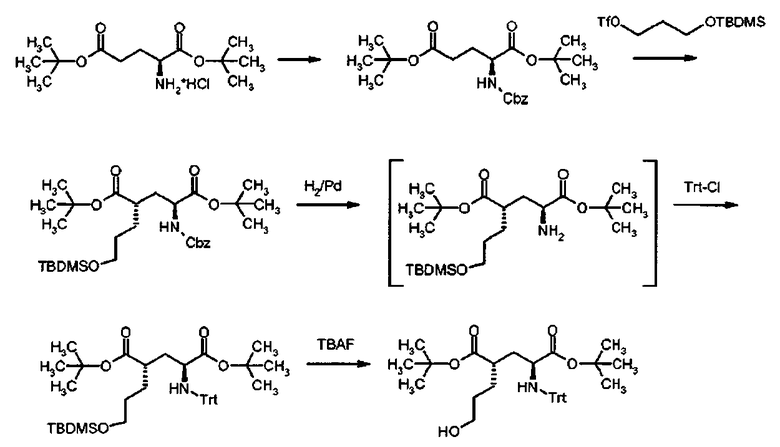
1. Cbz защита
К раствору ди-трет-бутил L-глутамата гидрохлорида (3 г, 10,14 ммоль) и ДИПЭА (5.3 мл, 30,4 ммоль) в дихлорметане (60 мл) добавляли раствор бензилхлороформиата (1,74 мл, 12,2 ммоль) в дихлорметане (30 мл). Раствор перемешивали в течение 30 мин при комнатной температуре. После выпаривания растворителей, осадок вносили в смесь этилацетата и воды. Органическую фазу отделяли, промывали водой и рассолом, и высушивали над сульфатом натрия. После фильтрования, раствор выпаривали, и сырой продукт очищали флеш-хроматографией (этилацетат/н-гексан: от 10/90 до 20/80), чтобы получить целевой продукт (3.65 г, 91%) в виде бесцветного масла.
1Н ЯМР (400 МГц, CDCl3) δ част. на млн. 1.43 (s, 9Н), 1.46 (s, 9Н), 1.84-1.96 (m, 1Н), 2.06-2.18 (m, 1H), 2.20-2.40 (m, 2Н), 4.20-4.30 (q, J=8.0 Гц, 1Н), 5.10 (s, 2Н), 5.34 (d, J=8.0 Гц, 1Н), 7.27-7.40 (m, 5Н).
2. Алкилирование
Раствор ди-трет-бутил N-[(бензилокси)карбонил]-L-глутамата (4.77 г, 12.12 ммоль) в ТГФ (76 мл) охлаждали до -78°C и 1.0 М раствор бис(триметилсилил)амида лития (2S.45 мл, 25.45 ммоль) добавляли медленно в ТГФ. Раствор перемешивали в течение 45 мин при -78°C, и раствор 3-(трет-бутилдиметилсилилокси)пропила трифторметансульфоната (5.08 г, 15.76 ммоль) в ТГФ (2S мл) добавляли по каплям при -78°C. После перемешивания в течение 2 ч, реакционную смесь резко охлаждали 2.0 N водным раствором NH4Cl, и нагревали до комнатной температуры, и концентрировали под вакуумом. Полученный водный раствор экстрагировали этилацетатом, объединенную органическую фазу промывали водой и рассолом, и высушивали над сульфатом натрия. После фильтрования, раствор выпаривали, и сырой продукт очищали флеш-хроматографией (этилацетат/н-гексан 10/90), чтобы получить целевой продукт (4.62 г, 67%) в виде бесцветного масла.
1Н ЯМР (400 МГц, CDCl3) 6 част. на млн. 0.04 (s, 6Н), 0.88 (s, 9Н), 1.42 (s, 9Н), 1.45 (s, 9Н), 1.48-1.62 (m, 4Н), 1.75-1.86 (m, 1Н), 1.90-2.00 (m, 1H), 2.30-2.40 (m, 1Н), 3.50-3.62 (m, 2Н), 4.16-4.25 (q, J=8.8 Гц, 1H), 5.10 (s, 2Н), 5.14 (d, J=8.8 Гц, 1Н), 7.28-7.38 (m, 5Н); 13С ЯМР (100 МГц, CDCl3) δ-5.30, 18.31, 25.93, 27.95, 28.03, 29.12, 30.01, 34.32, 43.14, 53.75, 62.71, 66.89, 80.68, 82.12, 110.00, 128.09, 128.12, 128.46, 136.27, 156.02, 171.53, 174.93; МС (ESI, режим определения положительных ионов) C30H51NO7Si: m/z 588.5 [(M+Na]+].
3. Снятие защиты Cbz и защита тритила
К раствору ди-трет-бутил(4S)-N-[(бензилокси)карбонил]-4-(3-{[трет-бутил(диметил)силил]окси}пропил)-L-глутамата (4.158 г, 7.349 ммоль) в МеОН (140 мл) добавляли 10% Pd/C (2.346 г, 2.2046 ммоль) под атмосферой аргона. После промывки газообразным водородом, раствор суспендировали в течение 18 ч при комнатной температуре. После фильтрования с целитом, раствор выпаривали. Осадок растворяли в дихлорметане (130 мл). Добавляли ДИПЭА (3.5 мл, 20.337 ммоль) и трифенилметилхлорид (2.268 г, 8.135 ммоль). Реакционную смесь перемешивали в течение 2 ч при комнатной температуре, и затем добавляли воду. Реакционную смесь экстрагировали дихлорметаном. Объединенный органический раствор промывали водой, и высушивали над сульфатом натрия. После фильтрования, раствор выпаривали и сырой продукт очищали флеш-хроматографией (этилацетат/н-гексан: 5/95), чтобы получить целевой продукт (3.64 г, 79% общий выход) в виде бесцветного масла.
1Н ЯМР (400 МГц, CDCl3) δ 0.05 (s, 6Н), 0.90 (s, 9Н), 1.16 (s, 9Н), 1.33 (s, 9Н), 1.46-1.72 (m, 5Н), 2.12-2.22 (m, 1H), 2.28-2.40 (m, 1H), 2.70-2.82 (m, 1Н), 3.20-3.30 (m, 1Н), 3.59 (t, J=5.6 Гц, 2Н), 7.15-7.20 (m, 3Н), 7.20-7.28 (m, 6Н), 7.42-7.52 (m, 6Н); 13С ЯМР (100 МГц, CDCl3) δ -5.26, 18.35, 25.98, 27.87, 28.06, 29.93, 30.41, 39.04, 42.67, 55.27, 62.84, 71.14, 80.04, 80.84, 126.31, 127.79, 128.89, 146.35, 174.58, 174.67; МС (ESI) C41H59NO5Si: m/z 696.9 [(M+Na)+]
4. Десилилирование
К раствору ди-трет-бутил(4S)-4-(3-{[трет-бутил(диметил)силил]окси}пропил)-N-тритил-L-глутамата (3.64 г, 5.40 ммоль) в ТГФ (40 мл)добавляли TBAF (1.0 М в ТГФ, 10.8 мл, 10.8 ммоль). Раствор перемешивали в течение 1.5 ч при комнатной температуре. После выпаривания растворителя, сырой продукт очищали флеш-хроматографией (этилацетат/н-гексан 40/60), чтобы получить целевой продукт (2.55 г, 84%) в виде твердого вещества белого цвета.
1Н ЯМР (400 МГц, CDCl3) δ част. на млн. 1.15 (s, 9Н), 1.32 (s, 9Н), 1.50-1.76 (m, 5Н), 2.10-2.20 (m, 1Н), 2.30-2.40 (m, 1Н), 2.70-2.82 (m, 1H), 3.20-3.30 (m, 1Н), 3.61 (t, J=5.6 Гц, 2Н), 7.12-7.18 (m, 3Н), 7.20-7.28 (m, 6Н), 7.42-7.50 (m, 6Н); 13С ЯМР (100 МГц, CDCl3) δ 27.86, 28.04, 29.59, 30.26, 39.10, 42.63, 55.27, 62.49, 71.16, 80.33, 80.96, 126.34, 127.80, 128.87, 146.29, 174.63, 174.68; МС (ESI) C35H45NO5: m/z 582.6 [(M+Na)+]
Анализ хиральной ВЭЖХ ди-трет-бутил(4S)-4-(3-гидроксипропил)-N-тритил-L-глутамата проводили в соответствии со способом А1 (время удержания: 7-8 мин).
Общие методики
19F-фторирование:
Предшественник (0.01 ммоль) растворяли в ацетонитриле (0.5 мл), и добавляли раствор 1.0 М TBAF/ацетонитрил (20 мкл, 0.02 ммоль). Реакционную смесь перемешивали при 80°C в течение 2 ч 40 мкл раствора отбирали в 5, 10, 20, 40, 60, 90, и 120 мин для анализа ВЭЖХ (способ А4).
18F-фторирование:
[18F]фторид (380-1400 МБк) улавливали на QMA картридже (Waters, SepPak light). Активность вымывали с помощью 0.6 мл раствора kryptofix 2.2.2/карбонат калия (3 мг/0.6 мг) в ацетонитрил/вода в реакционном сосуде. Смесь высушивали (95°C, поток азота, вакуум). 6 мг предшественника в 1.5 мл ацетонитрила добавляли к сухому остатку, и полученный раствор перемешивали при 120°C (выводимая на экран температура реактора) в течение 5 мин. Затем добавляли прибл. 1.5 мл 2 М HCl. Смесь нагревали при 120°C в течение 4,2 мин
Реакционную смесь разбавляли с 10 мл воды и переносили на 2 МСХ картриджи (Waters, картридж для экстракции Oasis МСХ plus). Промывали с помощью 10 мл воды и впоследствии вымывали с помощью 15 мл фосфатного буфера (содержащего 10,5 мг Na2HPO4×2Н2О, 9 мг NaCl). Раствор продукта пропускали через картридж Hypercarb (Thermo Scientific, Hypersep Hypercarb 500 мг/6 мл) в сосуд с конечным продуктом.
Аналитику ВЭЖХ полученного продукта осуществляют с использованием способа А5.
Идентичность IV-F18 подтверждали посредством совместного элюирования с образцовым соединением IV-F19 и УФ обнаружением при 340 нм (время удержания: 12-13 мин).
Примерные соединения согласно изобретению (соединения-предшественники) I
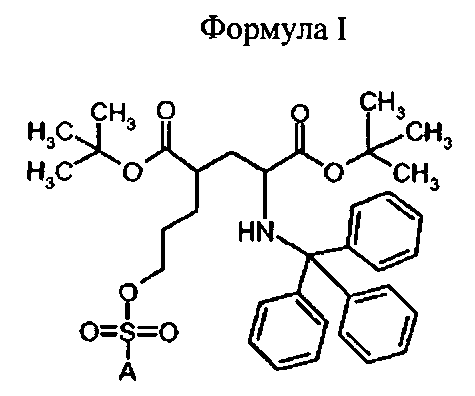



Ia Ди-трет-бутил(4S)-4-(3-{[(4-нитрофенил)сульфонил]окси}пропил)-N-тритил-L-глутамат
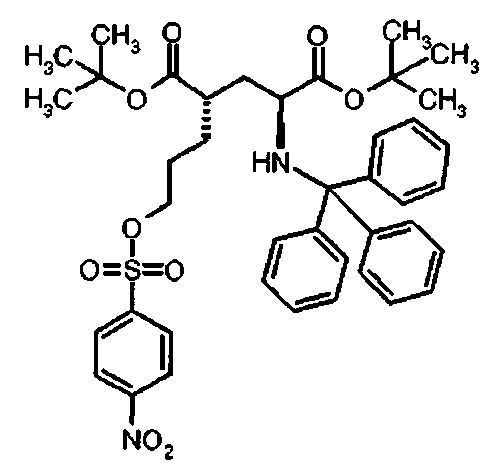
К раствору ди-трет-бутил(4S)-4-(3-гидроксипропил)-N-тритил-L-глутамата (212,6 мг, 0,38 ммоль) и триэтиламина (159 мкл, 1.14 ммоль) в дихлорметане (5 мл) добавляли 4-нитробензолсульфонилхлорид (126 мг, 0,57 ммоль) при 0°C. Реакционную смесь перемешивали при 0°C в течение 2 ч и затем добавляли воду. Органический слой отделяли, и водный слой экстрагировали дихлорметаном. Объединенный органический раствор высушивали над сульфатом натрия, и концентрировали в вакууме. Остаток очищали колоночной флеш-хроматографией (этилацетат/н-гексан=15/85), чтобы получить целевой продукт (231 мг, 82%) в виде твердого вещества белого цвета.
1Н ЯМР (400 МГц, CDCl3) δ част. на млн. 1.14 (s, 9Н), 1.30 (s, 9Н), 1.50-1.73 (m, 5Н), 2.00-2.12 (m, 1H), 2.22-2.32 (m, 1Н), 2.75 (d, J=9.2 Гц, 1Н), 3.20-3.27 (m, 1H), 4.12 (t, J=6.4 Гц, 2Н), 7.14-7.19 (m, 3Н), 7.20-7.27 (m, 6Н), 7.42-7.47 (m, 6Н), 8.09 (d, J=8.8 Гц, 2Н), 8.38 (d, J=8.8 Гц, 2Н); 13С ЯМР (100 МГц, CDCl3) 5 26.63, 27.83, 28.00, 29.03, 38.57, 42.20, 55.16, 71.18, 71.34, 80.64, 81.05, 124.48, 126.41, 127.83, 128.81, 129.20, 141.86, 146.17, 173.87, 174.33; МС (ESI, режим определения положительных ионов) C41H48N2O9S: m/z 767.6 [M+Na]+.
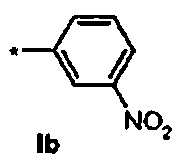
Ib Ди-трет-бутил(4S)-4-(3-{[(3-нитрофенил)сульфонил]окси}пропил)-N-тритил-L-глутамат

К раствору ди-трет-бутил(4S)-4-(3-гидроксипропил)-N-тритил-L-глутамата (206.2 мг, 0.37 ммоль) и триэтиламина (154 мкл, 1.10 ммоль) в дихлорметане (5 мл) добавляли 3-нитробензолсульфонилхлорид (122 мг, 0.55 ммоль) при 0°C. Реакционную смесь перемешивали при 0°C в течение 2 ч и затем добавляли воду. Органический слой отделяли, и водный слой экстрагировали дихлорметаном. Объединенный органический раствор высушивали над сульфатом натрия, и концентрировали в вакууме. Остаток очищали колоночной флеш-хроматографией (этилацетат/н-гексан = 20/80), чтобы получить целевой продукт (215 мг, 78%) в виде твердого вещества белого цвета.
1Н ЯМР (400 МГц, CDCl3) δ част. на млн. 1.14 (s, 9Н), 1.30 (s, 9Н), 1.50-1.73 (m, 5Н), 2.03-2.12 (m, 1Н), 2.23-2.32 (m, 1Н), 2.75 (d, J=8.4 Гц, 1Н), 3.20-3.27 (m, 1H), 4.13 (t, J=6.4 Гц, 2Н), 7.14-7.19 (m, 3Н), 7.20-7.27 (m, 6Н), 7.42-7.47 (m, 6Н), 7.77 (t, J=8.2 Гц, 1Н), 8.22 (dq, J=0.8, 8.0 Гц, 1Н), 8.50 (dq, J=0.8, 8.0 Гц, 1Н), 8.75 (t, J=1.8 Гц, 1Н); 13С ЯМР (100 МГц, CDCl3) δ 26.63, 27.82, 27.97, 29.03, 38.65, 42.24, 55.12, 71.14, 71.32, 80.62, 81.03, 123.13, 126.38, 127.80, 128.18, 128.81, 130.74, 133.24, 138.31, 146.17, 173.84, 174.34; МС (ESI, режим определения положительных ионов) C41H48N2O9S: m/z 767.8 [M+Na]+.
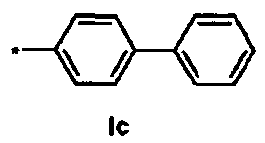
Ic Ди-трет-бутил(4S)-4-{3-[(бифенил-4-илсульфонил)окси]пропил}-N-тритил-L-глутамат
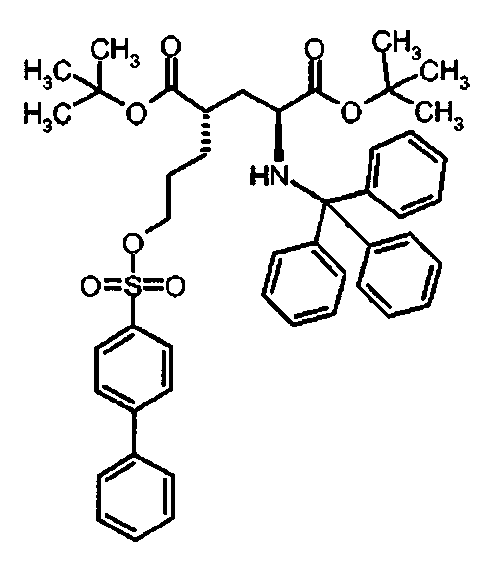
К раствору ди-трет-бутил(4S)-4-(3-гидроксипропил)-N-тритил-L-глутамата (202.8 мг, 0.36 ммоль) и триэтиламина (151 мкл, 1.09 ммоль) в дихлорметане (5 мл) добавляли бифенил-4-сульфонилхлорид (137 мг, 0.54 ммоль) при 0°C. Реакционную смесь перемешивали при 0°C в течение 5 ч и затем добавляли воду. Органический слой отделяли, и водный слой экстрагировали дихлорметаном. Объединенный органический раствор высушивали над сульфатом натрия, и концентрировали в вакууме. Остаток очищали колоночной флеш-хроматографией (этилацетат/н-гексан = 10/90), чтобы получить целевой продукт (236 мг, 84%) в виде твердого вещества белого цвета.
1Н ЯМР (400 МГц, CDCl3) δ част. на млн. 1.14 (s, 9Н), 1.30 (s, 9Н), 1.50-1.73 (m, 5Н), 2.03-2.12 (m, 1Н), 2.23-2.32 (m, 1Н), 2.70-2.80 (m, 1Н), 3.18-3.27 (m, 1H), 4.06 (t, J=6.4 Гц, 2Н), 7.12-7.17 (m, 3Н), 7.20-7.27 (m, 6Н), 7.40-7.52 (m, 9Н), 7.58-7.62 (m, 2Н), 1.12-1.16 (m, 2Н), 7.94-7.98 (m, 2Н); 13С ЯМР (100 МГц, CDCl3) δ 26.66, 27.85, 28.00, 29.26, 38.74, 42.35, 55.14, 70.38, 71.16, 80.51, 80.99, 126.37, 127.38, 127.81, 127.86, 128.39, 128.70, 128.84, 129.10, 134.50, 139.04, 146.21, 146.72, 173.98, 174.41; МС (ESI, режим определения положительных ионов) C47H53NO7S: m/z 798.5 [M+Na]+.
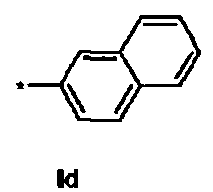
Id Ди-трет-бутил(4S)-4-{3-[(2-нафтилсульфонил)окси]пропил}-N-тритил-L-глутамат

К раствору ди-трет-бутил(4S)-4-(3-гидроксипропил)-N-тритил-L-глутамата (217.5 мг, 0.39 ммоль) и триэтиламина (160 мкл, 1.17 ммоль) в дихлорметане (5.0 мл) добавляли нафталин-2-сульфонилхлорид (155.4 мг, 0.58 ммоль) при 0°C. Реакционную смесь перемешивали при 0°C в течение 3 ч и затем добавляли воду. Органический слой отделяли, и водный слой экстрагировали дихлорметаном. Объединенный органический раствор высушивали над сульфатом натрия, и концентрировали в вакууме. Остаток очищали колоночной флеш-хроматографией (этилацетат/н-гексан = 12/88), чтобы получить целевой продукт (289 мг, 82%) в виде твердого вещества белого цвета (т.п.=119.3°C).
1Н ЯМР (400 МГц, CDCl3) δ 1.12 (s, 9Н), 1.27 (s, 9Н), 1.50-1.70 (m, 5Н), 2.00-2.10 (m, 1H), 2.22-2.32 (m, 1Н), 2.74 (d, J=8.8 Гц, 1Н), 3.14-3.24 (m, 1H), 4.04 (t, J=6.4 Гц, 2Н), 7.10-7.16 (m, 3Н), 7.18-7.24 (m, 6Н), 7.40-7.46 (m, 6Н), 7.60-7.72 (m, 2Н), 7.85 (dd, J=1.6, 8.0 Гц, 1Н), 7.93 (d, J=8.0 Гц, 1Н), 7.96-8.02 (m, 2Н), 8.48 (d, J=1.2 Гц, 1Н); 13С ЯМР (100 МГц, CDCl3) δ 26.6, 27.8, 27.9, 29.2, 38.7, 42.3, 55.1, 70.4, 71.1, 80.5, 80.9, 122.5, 126.3, 127.8, 128.0, 128.8, 129.3, 129.7, 131.9, 132.8, 135.2, 146.2, 173.9, 174.4; МС (ESI, режим определения положительных ионов) C45H51NO7S: m/z 772.9 [M+Na]+.
Ie Ди-трет-бутил(4S)-4-{3-[(1-нафтилсульфонил)окси]пропил}-N-тритил-L-глутамат
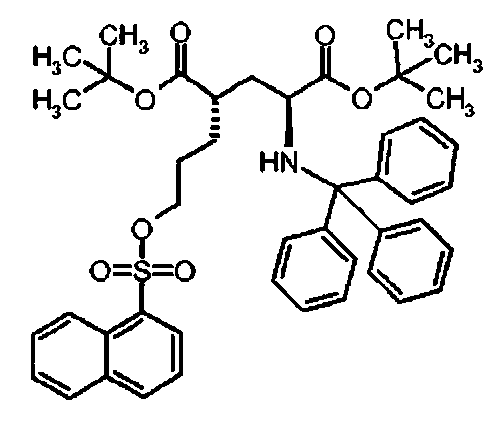
К раствору ди-трет-бутил(4S)-4-(3-гидроксипропил)-N-тритил-L-глутамата (216.8 мг, 0.39 ммоль) и триэтиламина (160 мкл, 1.16 ммоль) в дихлорметане (5.0 мл) добавляли нафталин-1-сульфонилхлорид (131.7 мг, 0.58 ммоль) при 0°C. Реакционную смесь перемешивали при 0°C в течение 3 ч и затем добавляли воду. Органический слой отделяли, и водный слой экстрагировали дихлорметаном. Объединенный органический раствор высушивали над сульфатом натрия, и концентрировали в вакууме. Остаток очищали колоночной флеш-хроматографией (этилацетат/н-гексан = 12/88), чтобы получить целевой продукт (248 мг, 85%) в виде твердого вещества белого цвета.
1Н ЯМР (400 МГц, CDCl3) δ 1.12 (s, 9Н), 1.25 (s, 9Н), 1.48-1.64 (m, 5Н), 1.96-2.18 (m, 1Н), 2.16-2.26 (m, 1H), 2.73 (d, J=9.2 Гц, 1Н), 3.10-3.20 (m, 1H), 3.90-4.00 (m, 2Н), 7.10-7.16 (m, 3Н), 7.18-7.24 (m, 6Н), 7.40-7.46 (m, 6Н), 7.56 (t, J=7.6 Гц, 1Н), 7.62 (t, J=8.0 Гц, 1Н), 7.69 (t, J=7.6 Гц, 1H), 7.96 (d, J=8.0 Гц, 1Н), 8.13 (d, J=8.4 Гц, 1H), 8.28 (d, J=7.2 Гц, 1H), 8.60 (d, J=8.4 Гц, 1H); 13C ЯМР (100 МГц, CDCl3) δ 26.5, 27.8, 27.9, 29.2, 38.7, 42.3, 55.0, 70.5, 71.1, 80.4, 80.9, 124.0, 124.9, 126.3, 127.2, 127.8, 128.4, 128.7, 128.80, 128.83, 130.4, 131.2, 134.1, 135.2, 146.2, 173.9, 174.4; MC (ESI, режим определения положительных ионов) C45H51NO7S: m/z 772.8 [M+Na]+.
If Ди-трет-бутил(4S)-4-{3-[(хинолин-8-илсульфонил)окси]пропил}-N-тритил-L-глутамат

К раствору ди-трет-бутил(4S)-4-(3-гидроксипропил)-N-тритил-L-глутамата (203.4 мг, 0.36 ммоль) и триэтиламина (150 мкл, 1.09 ммоль) в дихлорметане (5.0 мл) добавляли хинолин-8-сульфонилхлорид (124.1 мг, 0.55 ммоль) при 0°C. Реакционную смесь перемешивали при 0°C в течение 3 ч и при комнатной температуре в течение ночи и затем добавляли воду. Органический слой отделяли, и водный слой экстрагировали дихлорметаном. Объединенный органический раствор высушивали над сульфатом натрия, и концентрировали в вакууме. Остаток очищали колоночной флеш-хроматографией (МеОН/CH2Cl2=1/99), чтобы получить целевой продукт (140 мг, 51%) в виде твердого вещества белого цвета.
1Н ЯМР (400 МГц, CDCl3) δ 1.11 (s, 9Н), 1.26 (s, 9Н), 1.46-1.74 (m, 5Н), 2.00-2.30 (m, 1Н), 2.20-2.28 (m, 1Н), 2.72 (d, J=9.2 Гц, 1Н), 3.12-3.22 (m, 1Н), 4.31 (t, J=6.4 Гц, 2Н), 7.12-7.16 (m, 3Н), 7.20-7.26 (m, 6Н), 7.40-7.46 (m, 6Н), 7.56 (dd, J=8.4, 4.2 Гц, 1Н), 7.53-7.68 (m, 1H), 8.12 (dd, J=8.2, 1.6 Гц, 1H), 8.26 (dd, J=2.0, 8.2 Гц, 1H), 8.50 (dd, J=7.2, 1.6 Гц, 1H), 9.16 (dd, J=1.6,4.2 Гц, 1Н); 13С ЯМР (100 МГц, CDCl3) 5 26.9, 27.8, 27.9, 29.4, 38.8, 42.4, 55.1, 71.1, 71.5, 80.4, 80.9, 122.4, 125.2, 126.3, 127.8, 128.8, 129.0, 133.1, 134.6, 136.4, 146.2, 151.9, 173.9, 174.4; МС (ESI, режим определения положительных ионов) C44H50N2O7S: m/z 773.9 [M+Na]+.
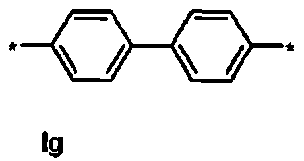
Ig тетра-трет-бутил(2S,4S,2′S,4′S)-2,2′-[бифенил-4,4′-диилбис(сульфонилоксипропан-3,1-диил)]бис[4-тритиламино)пентандиоат]

К раствору ди-трет-бутил(4S)-4-(3-гидроксипропил)-N-тритил-L-глутамата (209.6 мг, 0.37 ммоль, 2.2 екв.) и триэтиламина (140 мкл, 1.02 ммоль) в дихлорметане (5.0 мл) добавляли бифенил-4-4′-дисульфонилхлорид (60 мг, 0.17 ммоль) при 0°C. Реакционную смесь перемешивали при 0°C в течение 3 ч и при комнатной температуре в течение ночи и затем добавляли воду. Органический слой отделяли, и водный слой экстрагировали дихлорметаном. Объединенный органический раствор высушивали над сульфатом натрия, и концентрировали в вакууме. Остаток очищали колоночной флеш-хроматографией (этилацетат/н-гексан = 25/75), чтобы получить целевой продукт (98.7 мг, 41%) в виде твердого вещества белого цвета.
1Н ЯМР (400 МГц, CDCl3) δ 1.16 (s, 9Н), 1.30 (s, 9Н), 1.50-1.76 (m, 5Н), 2.04-2.14 (m, 1Н), 2.24-2.34 (m, 1Н), 2.75 (d, J=9.2 Гц, 1Н), 3.18-3.28 (m, 1Н), 4.08 (t, J=6.4 Гц, 2Н), 7.12-7.18 (m, 3Н), 7.20-7.26 (m, 6Н), 7.40-7.46 (m, 6Н), 7.72 (d, J=8.4 Гц, 2Н), 8.00 (d, J=8.4 Гц, 1Н); 13С ЯМР (100 МГц, CDCl3) 5 26.6, 27.8, 28.0, 29.2, 38.6, 42.3, 55.1, 70.6, 71.2, 80:5, 81.0, 126.4, 127.8, 128.2, 128.6, 128.8, 136.1, 144.4, 146.2, 174.0, 174.4; МС (ESI, режим определения положительных ионов) C82H96N2O14S2: m/z 1420.6 [M+Na]+.
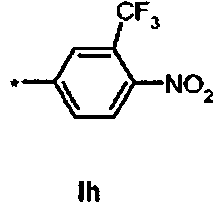
Ih Ди-трет-бутил(4S)-4-[3-({[4-нитро-3-(трифторметил)фенил]сульфонил}окси)пропил]-N-тритил-L-глутамат

К раствору ди-трет-бутил(4S)-4-(3-гидроксипропил)-N-тритил-L-глутамата (439 мг, 0.78 ммоль) и триэтиламина (330 мкл, 2.35 ммоль) в дихлорметане (7.0 мл) добавляли 4-нитро 3-(трифторметил)бензолсульфонилхлорид (340.7 мг, 1.18 ммоль) при 0°C. Реакционную смесь перемешивали при 0°C в течение 45 мин и затем добавляли воду. Органический слой отделяли, и водный слой экстрагировали дихлорметаном. Объединенный органический раствор высушивали над сульфатом натрия, и концентрировали в вакууме. Остаток очищали колоночной флеш-хроматографией (этилацетат/н-гексан=15/85), чтобы получить целевой продукт (4 ч, 470 мг, 74%) в виде твердого вещества светло-желтого цвета.
1Н ЯМР (400 МГц, CDCl3) δ 1.14 (s, 9Н), 1.30 (s, 9Н), 1.52-1.80 (m, 5Н), 2.04-2.14 (m, 1H), 2.24-2.34 (m, 1H), 2.76 (d, J=8.8 Гц, 1Н), 3.20-3.28 (m, 1Н), 4.17 (t, J=6.0 Гц, 2Н), 7.16-7.20 (m, 3Н), 7.20-7.28 (m, 6Н), 7.42-7.48 (m, 6Н), 7.97 (d, J=8.4 Гц, 1H), 8.23 (dd, J=2.0, 8.4 Гц, 1H), 8.34 (d, J=1.6 Гц, 1H); MC (ESI, режим определения положительных ионов) C42H47F3N2O9S: m/z 835.4 [M+Na]+.
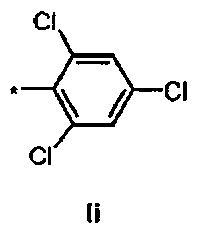
Ii Ди-трет-бутил(4S)-4-(3-{[(2,4,6-трихлорфенил)сульфонил]окси}пропил)-N-тритил-L-глутамат
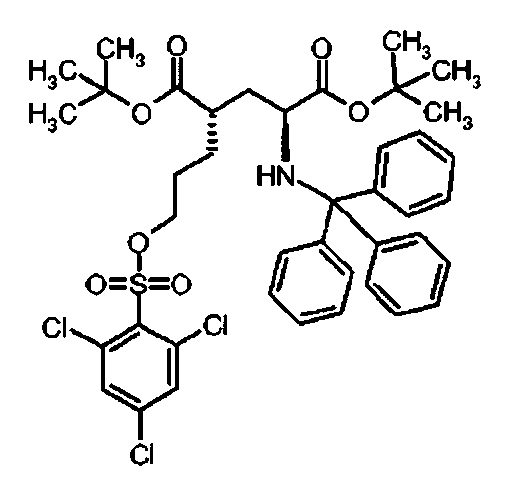
К раствору ди-треш-бутил(4S)-4-(3-гидроксипропил)-N-тритил-L-глутамата (438 мг, 0.78 ммоль) и триэтиламина (327 мкл, 2.35 ммоль) в дихлорметане (7.0 мл) добавляли 2,4,6-трихлорбензилсульфонилхлорид (328.6 мг, 1.17 ммоль) при 0°C. Реакционную смесь перемешивали при 0°C в течение 1 ч и затем добавляли воду. Органический слой отделяли, и водный слой экстрагировали дихлорметаном. Объединенный органический раствор высушивали над сульфатом натрия, и концентрировали в вакууме. Остаток очищали колоночной флеш-хроматографией (этилацетат/н-гексан = 10/90), чтобы получить целевой продукт (415 мг, 66%) в виде твердого вещества белого цвета.
1Н ЯМР (400 МГц, CDCl3) δ 1.15 (s, 9Н), 1.31 (s, 9Н), 1.52-1.80 (m, 5Н), 2.04-2.16 (m, 1Н), 2.26-2.36 (m, 1Н), 2.77 (d, J=9.6 Гц, 1Н), 3.18-3.24 (m, 1Н), 4.15 (t, J=6.4 Гц, 2Н), 7.12-7.18 (m, 3Н), 7.20-7.28 (m, 6Н), 7.42-7.48 (m, 6Н), 7.50 (s, 2Н); 13С ЯМР (100 МГц, CDCl3) δ 26.5, 27.8, 28.0, 29.2, 38.7, 42.2, 55.1, 71.1, 71.4, 80.5, 81.0, 126.3, 127.8, 128.8, 130.9, 131.2, 136.7, 139.3, 146.1, 173.8, 174.3; МС (ESI, режим определения положительных ионов) C41H46C13NO7S: m/z 826.3 [M+Na]+.
Ij Ди-трет-бутил(4S)-4-(3-{[(7-нитро-2-нафтил)сульфонил]окси}пропил)-N-тритил-L-глутамат

К раствору ди-трет-бутил(4S)-4-(3-гидроксипропил)-N-тритил-L-глутамата (486 мг, 0.84 ммоль) и триэтиламина (350 мкл, 2.58 ммоль) в дихлорметане (7.0 мл) добавляли 5-нитро-нафталин-2-сульфонил хлорид (340.8 мг, 1.25 ммоль) при 0°C. Реакционную смесь перемешивали при 0°C в течение 2 ч и добавляли воду. Органический слой отделяли, и водный слой экстрагировали дихлорметаном. Объединенный органический раствор высушивали над сульфатом натрия, и концентрировали в вакууме. Остаток очищали колоночной флеш-хроматографией (этилацетат/н-гексан = 20/80), чтобы получить целевой продукт (616 мг, 93%) в виде твердого вещества белого цвета.
1Н ЯМР (400 МГц, CDCl3) δ 1.12 (s, 9Н), 1.28 (s, 9Н), 1.48-1.74 (m, 5Н), 2.00-2.12 (m, 1Н), 2.20-2.30 (m, 1Н), 2.74 (d, J=8.0 Гц, 1Н), 3.12-3.24 (m, 1H), 4.11 (t, J=6.4 Гц, 2Н), 7.10-7.18 (m, 3Н), 7.18-7.26 (m, 6Н), 7.38-7.46 (m, 6Н), 7.73 (t, J=7.6 Гц, 1Н), 8.04-8.10 (m, 1H), 8.25 (d, J=8.4 Гц, 1Н), 8.43 (d, J=7.6 Гц, 1Н), 8.58 (s, 1H), 8.77 (d, J=9.2 Гц, 1Н); 13С ЯМР (100 МГц, CDCl3) δ 26.6, 27.8, 28.0, 29.1, 38.6, 42.2, 55.1, 70.9, 71.1, 80.5, 81.0, 125.4, 125.9, 126.3, 127.01, 127.04, 127.8, 128.8, 129.8, 133.0, 134.8, 135.8, 146.2, 146.4, 173.9, 174.3; МС (ESI, режим определения положительных ионов) C45H50N2O9S: m/z 817.5 [M+Na]+.
Кристаллизация
Кристаллизацию осуществляли для соединения Id. Для данной кристаллизации применяли 2% простой эфир/гексан. Кристаллизация была получена для соединения Id.
19F-фторирование примерных соединений
19F-фторирование осуществляли, как описано в "Общих методиках". Развитие реакции проверяли через 5, 10, 20, 40, 60, 90, и 120 мин. Составляя процентное отношение превращения в зависимости от времени, подсчитывали скорость реакции. Для подсчитывания относительных скоростей реакции, самую медленную реакцию (19F-фторирование If) определяли как 1. Самое быстрое превращение было обнаружено для соединений Ia, Ib и в особенности для Ig. Соединения Ic, Id, Ie, Ii и Ij показывали похожие скорости реакции по сравнению с If.
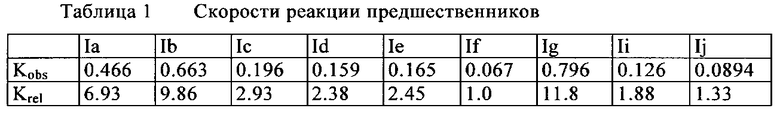
Данные соответствуют скоростям реакции, измеренным для 19F-фторирования предшественников.
18F-фторирование примерных соединений I
18F-фторирование осуществляли, как описано в "Общих методиках". Радиохимические выходы и чистоту определяли, как показано в таблице 2.
Радиохимический выход подсчитывали с помощью соотношения радиоактивности продукта и начальной радиоактивности. Обе измеряют с применением дозкалибратора (MED Nuklearmedizin Technik Dresden). Радиохимическую чистоту определяют с помощью аналитической ВЭЖХ (способ А5).
Таблица 2 показывает, что для всех соединений были получены высокие радиохимические выходы (38-56% n.d.c).
Кроме того, таблица 2 показывает, что введение радиоактивной метки получено с высокой стереохимической чистотой для соединений Ia-Ij (93/7-99/1).
Устойчивость примерных соединений
Устойчивость соединений формулы I проверяли в твердом виде при двух различных температурах: 0°C и - 20°C. Предшественники подвергали испытанию еженедельно в течение 4 недель. До начала исследования чистоту предшественников определяли индивидуально посредством анализа ВЭЖХ.
Отбор соединений
1. Твердое состояние: 3-5 мг соответствующего предшественника от Ia до Ij помещали в 8 янтарных пробирок, которые заполняли газом Ar и закупоривали. Каждые четыре пробирки, содержащие предшественник, хранили при 0°C и - 20°C. Каждую неделю в течение 4 недель, 1 мг предшественника растворяли в ацетонитриле (1.0 мл). 10 мкл раствора впрыскивали в ВЭЖХ (способ А2 или A3, соответственно).
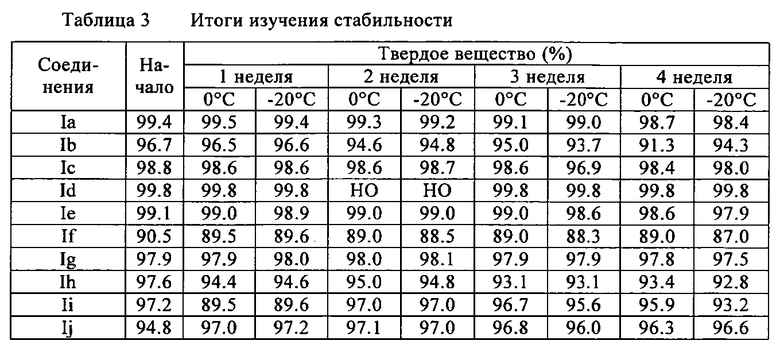
НО: не определено
Получение промежуточных продуктов II

1. Защита тритила
Тритилхлорид (2.05 г.7.36 ммоль) добавляли к раствору ди-трет-бутил L-глутамата гидрохлорида (2.15 г, 7.27 ммоль) и триэтиламина (5 мл, 36 ммоль) в дихлорметане (20 мл). Раствор перемешивали в течение ночи (16 ч) при комнатной температуре. Раствор промывали раствором бикарбоната натрия (3×10 мл) и вода (2×5 мл). После высушивания над сульфатом натрия растворитель выпаривали. Сырой продукт очищали флеш-хроматографией (этилацетат/н-гексан: от 2/98 до 3/97), чтобы получить ди-трет-бутил N-тритил-L-глутамат (3.2 г, 88%) в виде пены белого цвета.

ди-трет-бутил N-тритил-L-глутамат
1Н ЯМР (400 МГц, CDCl3) δ част. на млн. 1.17 (s, 9Н), 1.47 (s, 9Н), 1.90-1.20 (m, 3Н), 2.51 (ddd, 1H), 2.76 (br. d, 1H), 3.37 (br. s, 1Н), 7.16-7.21 (m, 3Н), 7.24-7.29 (m, 6Н), 7.51 (br. d, 6Н).
2. Алкилирование
Раствор ди-трет-бутил N-тритил-L-глутамата (1.99 г, 1.85 ммоль) в ТГФ (50 мл) охлаждали до -70°C и 1.0 М раствор бис(триметилсилил)амида лития (47 мл, 47 ммоль) в ТГФ добавляли медленно (в течение периода в 20 мин). Раствор перемешивали в течение 2 ч при -70°C, и по каплям добавляли аллилбромид (1.44 г, 11.9 ммоль) при -70°C. После перемешивания в течение 1.5 ч, реакционную смесь резко охлаждали насыщенным водным раствором NH4Cl, и нагревали до комнатной температуры, и концентрировали под вакуумом. Полученный водный раствор экстрагировали дихлорметаном, объединенную органическую фазу промывали водой и высушивали над сульфатом натрия. После фильтрования, раствор выпаривали и сырой продукт очищали флеш-хроматографией (силикагель, этилацетат/н-гексан), чтобы получить ди-трет-бутил 4-аллил-N-тритил-L-глутамат (1.01 г, 46%) в виде смеси (4S,4S)/(2S,4R) диастереоизомеров.

ди-трет-бутил 4-аллил-А/-тритил-L-глутамат
1Н ЯМР (400 МГц, CDCl3) δ част. на млн. 1.16 (s, 9Н), 1.45 (s, 9Н), 1.69-1.77 (m, 1H), 2.10-2.37 (m, 3Н), 2.43-2.51 (m, 1Н), 2.74 (br. d, 1Н), 3.26-3.33 (m, 1Н), 4.96-5.06 (m, 2H), 5.63-5.76 (m, 1H), 7.14-7.18 (m, 3H), 7.21-7.27 (m, 6H), 7.45-7.51 (m, 6H).
MC (ES+) C35H43NO4: m/z 541 [M]+.
Специалисту в данной области техники известны методы разделения диастереоизомеров (например, хроматографические методы), обеспечивающие получение чистых изомеров (2S/2R) и (2S/4S), которые в дальнейшем могут быть преобразованы в изомерно чистые соединения, подобно тому, как описано в последующих стадиях ниже.
3. Гидроборирование
Комплекс боргдрид-тетрагидрофуран (1 М, 2,8 мл, 2.8 ммоль) добавляли по каплям к раствору ди-трет-бутил4-аллил-N-тритил-L-глутамат (1.00 г, 1.85 ммоль) в ТГФ (10 мл) при 0°C. Полученную смесь перемешивали в течение 2 ч при 0°C и в течение 16 ч при комнатной температуре. Раствор охлаждали до 0°C. Добавляли по каплям NaOH (1 М, 3 мл) и Н2О2 (30% в воде, 3 мл). Смесь перемешивали при 0°C в течение 1 ч. Добавляли воду (5 мл) и смесь концентрировали под сниженным давлением. Водный остаток экстрагировали этилацетатом. Объединенную органическую фракцию промывали рассолом, высушивали над сульфатом натрия, отфильтровывали и концентрировали. Сырой продукт очищали флеш-хроматографией (силикагель, этилацетат/гексан), чтобы получить ди-трет-бутил4-(3-гидроксипропил)-N-тритил-L-глутамат (0.46 г, 44%) в виде смеси (4S,4S)/(2S,4R) диастереоизомеров.

ди-трет-бутил 4-(3-гидроксипропил)-N-тритил-L-глутамат
1Н ЯМР (400 МГц, CDCl3) δ част. на млн. 1.16 (s, 9Н), 1.47 (s, 9Н), 1.48-1.78 (m, 5Н), 2.06-2.20 (m, 1Н), 2.35-2.45 (m, 1Н), 2.70-2.82 (m, 1Н), 3.23-3.34 (m, 1H), 3.55-3.67 (m, 2Н), 7.12-7.20 (m, 3Н), 7.21-7.30 (m, 6Н), 7.45-7.53 (m, 6Н). МС (ES+) C35H45NO5: m/z 560 [М]+.
Специалисту в данной области техники известны методы разделения диастереоизомеров (например, хроматографические методы), обеспечивающие получение чистых изомеров (2S/2R) и (2S/4S), которые в дальнейшем могут быть преобразованы в изомерно чистые соединения, подобно тому, как описано в последующих стадиях ниже.
Примерные соединения согласно изобретению (соединения-предшественники) II
Ди-трет-бутил(4S)-4-{3-[(2-нафтилсульфонил)окси]пропил}-N-тритил-L-глутамат (Id) и ди-трет-бутил(4R)-4-{3-[(2-нафтилсульфонил) окси]пропил}-N-тритил-L-глутамат (Ik)
При 0°C триэтиламин (0.68 мл, 4.90 ммоль) и нафталин-2-сульфонилхлорид (0.370 г, 1.63 ммоль) добавляли к раствору ди-трет-бутил4-(3-гидроксипропил)-N-тритил-L-глутамата (0.457 г, 0.816 ммоль) в дихлорметане (10 мл). Полученную смесь перемешивали при 0°C в течение 2 ч и в течение 16 ч при комнатной температуре. Раствор концентрировали и сырой продукт очищали флеш-хроматографией (силикагель, этилацетат/гексан), чтобы получить ди-трет-бутил4-{3-[(2-нафтилсульфонил)окси]пропил}-N-тритил-L-глутамат (0.479 мг, 78%) в виде смеси (4S,4S)/(2S,4R) диастереоизомеров. Изомеры разделяли хиральной ВЭЖХ (Chiralpak 1С 5 мкм 250×30 мм, этанол/метанол 1:1, 30 мл/мин):
Ди-трет-бутил(4S)-4-{3-[(2-нафтилсульфонил)окси]пропил}-N-тритил-L-глутамат (Id): 80 мг, 13%,
Ди-трет-бутил(4R)-4-{3-[(2-нафтилсульфонил)окси]пропил}-N-тритил-L-глутамат (Ik): 323 мг, 53%.

Ди-трет-бутил(4S)-4-{3-[(2-нафтилсульфонил)окси]пропил}-N-тритил-L-глутамат (Id)

Ди-трет-бутил(4R)-4-{3-[(2-нафтилсульфонил)окси]пропил}-N-тритил-L-глутамат (Ik)
Id:
1Н ЯМР (400 МГц, CDCl3) δ 1.12 (s, 9Н), 1.27 (s, 9Н), 1.50-1.70 (m, 5Н), 2.00-2.10 (m, 1H), 2.22-2.32 (m, 1H), 2.74 (d, J=8.8 Гц, 1H), 3.14-3.24 (m, 1H), 4.04 (t, J=6.4 Гц, 2H), 7.10-7.16 (m, 3H), 7.18-7.24 (m, 6H), 7.40-7.46 (m, 6H), 7.60-7.72 (m, 2H), 7.85 (dd, J=1.6, 8.0 Гц, 1H), 7.93 (d, J=8.0 Гц, 1H), 7.96-8.02 (m, 2H), 8.48 (d, J=1.2 Гц, 1H).
MC (ES+) C45H51NO7S: m/z 750 [M]+.
Ik:
1H ЯМР (400 МГц, CDCl3) δ 1.14 (s, 9H), 1.41 (s, 9H), 1.43-1.52 (m, 3H), 1.55-1.64 (m, 2H), 2.10 (ddd, 1H), 2.31-2.37 (m, 1H), 2.71 (br. d, 1H), 3.22 (td, 1H), 4.03 (t, 2H), 7.16 (d, 3H), 7.20-7.25 (m, 6H), 7.45-7.49 (m, 6H), 7.65 (ddd, 1H), 7.69 (ddd, 1H), 7.84 (dd, 1H), 7.93 (d, 1H), 7.76 (d, 2H), 7.99 (dd, 1H), 8.49 (d, 1H).
MC (ES+) C45H51NO7S: m/z 750 [M]+.
Ди-трет-бутил (4S)-4-{3-([(4-метилфенил)сульфонил]пропил}-N-тритил-L-глутамат (Im) и ди-трет-бутил (4R)-4-{3-{[(4-метилфенил)сульфонил)окси]пропил}-N-тритил-L-глутамат (In)
При 0°C триэтиламин (0.31 мл, 2.2 ммоль) и 4-метилбензолсульфонилхлорид (0.141 г, 0.74 ммоль) добавляли к раствору ди-трет-бутил4-(3-гидроксипропил)-N-тритил-L-глутамат (0.239 г, 0.427 ммоль) в дихлорметане (10 мл). Полученную смесь перемешивали при 0°C в течение 2 ч и в течение 16 ч при комнатной температуре. Раствор концентрировали и сырой продукт очищали флеш-хроматографией (силикагель, этилацетат/гексан), чтобы получить ди-трет-бутил 4-{3-{[(4-метилфенил)сульфонил)окси]пропил}-N-тритил-L-глутамат (0.255 мг, 67%) в виде смеси (4S,4S)/(2S,4R) диастереоизомеров. Изомеры разделяли с помощью хиральной ВЭЖХ (Chiralpak AD-H 5 мкм 250×20 мм, гексан/2-пропанол 9:1, 25 мл/мин):
Ди-трет-бутил (4S)-4-{3-{[(4-метилфенил)сульфонил]пропил}-N-тритил-L-глутамат (Im): 34 мг (11%)
Ди-трет-бутил(4R)-4-{3-{[(4-метилфенил)сульфонил)окси]пропил}-N-тритил-L-глутамат (In): 127 мг (42%).

Ди-трет-бутил (4S)-4-{3-{[(4-метилфенил)сульфонил]пропил}-N-тритил-L-глутамат (Im)
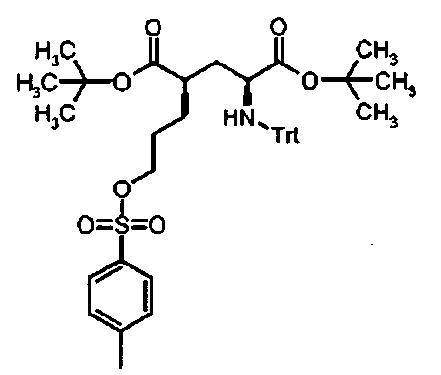
Ди-трет-бутил (4R)-4-{3-{[(4-метилфенил)сульфонил)окси]пропил}-N-тритил-L-глутамат (In)
Im
1НЯМР (400 МГц, CDCl3) δ 1.14 (s, 9H), 1.30 (s, 9H), 1.45-1.68 (m, 5H), 2.03-2.15 (m, 1H), 2.22-2.31 (m, 1H), 2.44 (s, 3H), 2.75 (mc, 1H), 3.21 (dd, 1H), 4.00 (t, 2H), 7.12-7.18 (m, 3H), 7.21-7.28 (m, 6H), 7.33 (d, 2H), 7.41-7.47 (m, 6H), 7.78 (d, 2H).
In
1H ЯМР (400 МГц, CDCl3) δ 1.15 (s, 9H), 1.42 (s, 9H), 1.48-1.65 (m, 5H), 2.10 (ddd, 1H), 2.34 (dt, 1H), 2.44 (s, 3H), 2.71 (br. s, 1H), 3.23 (br. s, 1H), 3.95 (t, 2H), 7.13-7.18 (m, 3H), 7.21-7.29 (m, 6H), 7.32 (d, 2H), 7.43-7.48 (m, 6H), 7.76 (d, 2H).
18F-фторирование примерных соединений II
Введение радиоактивной метки в ди-трет-бутил (4R)-4-{3-{[(4-метилфенил)сульфонил)окси]пропил}-N-тритил-L-глутамат (In)
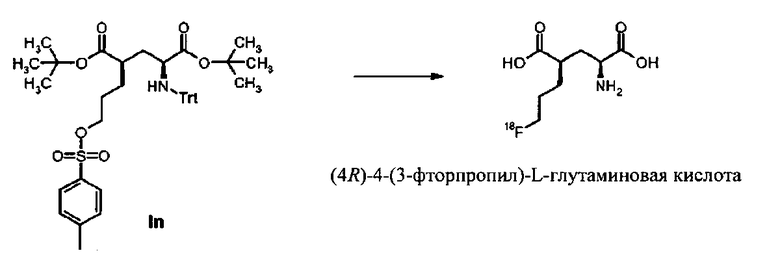
Введение радиоактивной метки осуществляли на синтезаторе GE Tracerlab MX. [18F]фторид (968 МБк) улавливали на картридже для анионного обмена (QMA light, Waters). Активность элюировали раствором 5 мг криптофикса и 1 мг карбоната калия в 600 мкл ацетонитрил/вода (1:1). Смесь высушивали нагреванием под легким потоком азота и вакуумом. Сушку повторяли после добавления ацетонитрила. 5.9 мг ди-трет-бутил (4R)-4-{3-{[(4-метилфенил)сульфонил)окси]пропил}-]N-тритил-L-глутамат (In) добавляли в 1.5 мл ацетонитрил и смесь нагревали при 120°C в течение 5 мин После добавления 2 мл HCl (2 М), смесь нагревали в течение 5 мин при 130°C. Добавляли 1.5 мл NaOH (4 М) и смесь нагревали в течение 5 мин при 70°C. Сырой продукт разбавляли с 2 мл HCl (2 М) и водой (до 30 мл) и пропускали через два картриджа МСХ (МСХ plus, Waters). Картриджи промывали водой (30 мл), и меченный радиоактивным изотопом продукт элюировали из картриджей МСХ через картридж Hypercarb (Hypercarb 500 мг, Thermo Scientific) с 15 мл фосфатным буфером (7 г Na2HPO4 2Н2О; 6 г NaCl in 1 л Н2О) в пробирку с продуктом, чтобы получить 381 МБк (34% d.c.) (4R)-4-(3-фторпропил)-L-глутаминовой кислоты. Было определено, что радиохимическая чистота составляет >96% посредством радио-ВЭЖХ (Luna 5 µ С18(2); 250*4,6 мм; 5 µ; Phenomenex; 12-100% ацетонитрил в 0.01 М Na2HPO4; предколоночная дериватизация с фторальдегидом, реактивный раствор о-фталальдегида; Thermo Scientific).
Введение радиоактивной метки в ди-трет-бутил (4S)-4-{3-[(2-нафтилсульфонил)окси]пропил}-N-тритил-L-глутамат Id

Введение радиоактивной метки осуществляли на синтезаторе GE Tracerlab MX. [18F]фторид (2915 МБк) улавливали на картридже для анионного обмена (QMA light, Waters). Активность элюировали раствором 3 мг криптофикса и 0.6 мг карбоната калия в 800 мкл ацетонитрил/вода (1:1). Смесь высушивали нагреванием под легким потоком азота и вакуумом. Сушку повторяли после добавления ацетонитрила. 6 мг ди-трет-бутил(4S)-4-{3-[(2-нафтилсульфонил)окси]пропил}-N-тритил-L-глутамата (Id) в 1,5 мл ацетонитрила добавляли и смесь нагревали при 130°C в течение 5 мин. После добавления 2 мл HCl (2 М), смесь нагревали в течение 10 мин при 120°C. Сырой продукт разбавляли водой (до 30 мл) и пропускали через два картриджа МСХ (МСХ plus, Waters). Картриджи промывали водой (30 мл), и меченный радиоактивным изотопом продукт элюировали из картриджей МСХ через картридж Hypercarb (Hypercarb 500 мг, Thermo Scientific) с 10 мл фосфатного буфера (7 г Na2HPO4 2Н2О; 6 г NaCl в 1 л Н2О) в пробирку с продуктом, чтобы получить 1168 МБк (40% n.d.c.) (4S)-4-(3-фторпропил)-L-глутаминовой кислоты. Было определено, что радиохимическая чистота составляет >96% посредством радио-ВЭЖХ >95% посредством радио-ВЭЖХ (Advanced Chromatography Technologies АСЕ 5 С18 250×4.6 мм; 2-100% В в 0.04М Na2HPO4; В: 45% ацетонитрил, 45% метанол, 10% вода; предколоночная дериватизация с реактивным раствором о-фталальдегида; Agilent).
Введение радиоактивной метки в ди-трет-бутил(4R)-4-{3-[2-нафтилсульфонил)окси]пропил}-N-тритил-L-глутамат Ik

Введение радиоактивной метки осуществляли на синтезаторе GE Tracerlab MX. [18F]фторид (9400 МБк) улавливали на картридже для анионного обмена (QMA light, Waters). Активность элюировали раствором 3 мг криптофикса и 0.6 мг карбоната калия в 800 мкл ацетонитрил/вода (1:1). Смесь высушивали нагреванием под легким потоком азота и вакуумом. Сушку повторяли после добавления ацетонитрила. 6 мг ди-трет-бутил (4R)-4-{3-[(2-нафтилсульфонил)окси]пропил}-N-тритил-L-глутамата (Ik) в 1.5 мл ацетонитрила добавляли и смесь нагревали при 130°C в течение 5 мин. После добавления 2 мл HCl (2 М), смесь нагревали в течение 10 мин при 120°C. Сырой продукт разбавляли с водой (до 30 мл) и пропускали через два картриджа МСХ (МСХ plus, Waters). Картриджи промывали водой (30 мл) и меченный радиоактивным изотопом продукт элюировали из картриджей МСХ через картридж Hypercarb (Hypercarb 500 мг, Thermo Scientific) с 10 мл фосфатного буфера (7 г Na2HPO4 2Н2О; 6 г NaCl в 1 л Н2О) в пробирку с продуктом, чтобы получить 5100 МБк (54% n.d.c.) (4R)-4-(3-фторпропил)-L-глутаминовой кислоты. Было определено, что радиохимическая чистота составляет >96% посредством радио-ВЭЖХ, >95% посредством радио-ВЭЖХ (Advanced Chromatography Technologies АСЕ 5 С18 250×4.6 мм; 2-100% В в 0.04 М Na2HPO4; В: 45% ацетонитрил, 45% метанол, 10% вода; предколоночная дериватизация с реактивным раствором о-фталальдегида; Agilent).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПАРА-АМИНОБЕНЗИЛЬНЫЕ ЛИНКЕРЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ В КОНЪЮГАТАХ | 2021 |
|
RU2839675C1 |
| СОЕДИНЕНИЯ, ПРИГОДНЫЕ ДЛЯ ТЕРАПИИ ПРОТИВ ВИЧ | 2019 |
|
RU2806030C2 |
| 3-АЗАБИЦИКЛО[3.1.0]ГЕКСИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ МЕТАБОТРОПНЫХ ГЛУТАМАТНЫХ РЕЦЕПТОРОВ | 2009 |
|
RU2510396C2 |
| СПОСОБ ПОЛУЧЕНИЯ F-БФА И ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ | 2019 |
|
RU2776178C1 |
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ БЕНЗОПИРАНА В КАЧЕСТВЕ ПРОТИВОАРИТМИЧЕСКИХ АГЕНТОВ | 2005 |
|
RU2380370C2 |
| Аналоги сплицеостатина | 2013 |
|
RU2618523C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ КАК ИНГИБИТОРЫ TRK | 2019 |
|
RU2803817C2 |
| АЗААДАМАНТАНОВЫЕ ПРОИЗВОДНЫЕ И СПОСОБЫ ПРИМЕНЕНИЯ | 2007 |
|
RU2450002C2 |
| АНТИБАКТЕРИАЛЬНЫЕ АГЕНТЫ | 1999 |
|
RU2246941C2 |
| КОНДЕНСИРОВАННОЕ ХИНОЛИНОВОЕ ПРОИЗВОДНОЕ И ЕГО ПРИМЕНЕНИЕ | 2005 |
|
RU2384571C2 |
Настоящее изобретение относится к соединению формулы I
Формула I

в которой R1 представляет собой трифенилметил (тритил), А выбран из группы: a) моноциклический С6-10-арил, b) бициклический С6-10-арил, c) С12-20-биарил, d) моноциклический гетероарил и e) бициклический гетероарил, где гетероарил представляет собой ароматический, моно- или бициклический двухвалентный радикал, имеющий от 5 до 10 кольцевых атомов и гетероатом N, по выбору, А несет один или несколько заместителей, выбранных из группы, включающей: a) галоген, b) нитро, c) алкил, d) трифторметил и e) Z, где Z представляет собой

R1 представляет собой трифенилметил (тритил), # указывает положение связи с А, и отдельным изомерам, таутомерам, диастереомерам, энантиомерам, стереоизомерам, их смесям и их приемлемым солям. Также предложены способ получения соединений формулы I, меченные фтором производные глутамата, композиции. Изобретение позволяет предоставить устойчивые, оптически чистые, твердые и достаточно реакционноспособные предшественники для изготовления меченных фтором производных глутамата. 9 н. и 8 з.п. ф-лы, 3 табл., 14 пр.
1. Соединение формулы I
Формула I
в которой
R1 представляет собой трифенилметил (тритил),
А выбран из группы:
a) моноциклический С6-10-арил,
b) бициклический С6-10-арил,
c) С12-20-биарил,
d) моноциклический гетероарил и
e) бициклический гетероарил,
где гетероарил представляет собой ароматический, моно- или бициклический двухвалентный радикал, имеющий от 5 до 10 кольцевых атомов и гетероатом N,
по выбору, А несет один или несколько заместителей, выбранных из группы, включающей:
a) галоген,
b) нитро,
c) алкил,
d) трифторметил и
e) Z,
где Z представляет собой
R1 представляет собой трифенилметил (тритил),
# указывает положение связи с А, и
отдельные изомеры, таутомеры, диастереомеры, энантиомеры, стереоизомеры, их смеси и их приемлемые соли.
2. Соединение по п.1, в котором
А выбран из группы:
a) фенил,
b) бифенил,
c) нафтил и
d) хинолинил,
по выбору, А несет один или несколько заместителей, выбранных из группы, включающей:
a) галоген,
b) нитро,
c) С1-С3 алкил,
d) трифторметил и
e) Z.
3. Соединение по п. 1 или 2 с (2S,4S)-конфигурацией (соединение формулы Ia)

R1 и А являются такими, как определены в п.1 или 2.
4. Соединение по п.3, выбранное из приведенного ниже перечня ди-трет-бутил (4S)-4-(3-{[(4-нитрофенил)сульфонил]окси}пропил)-N-тритил-L-глутамат
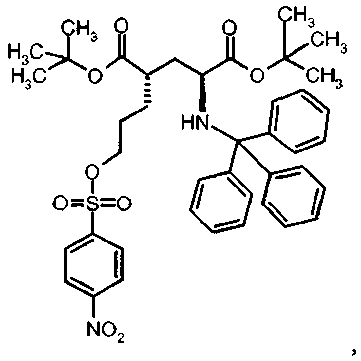
ди-трет-бутил (4S)-4-(3-{[(3-нитрофенил)сульфонил]окси}пропил)-N-тритил-L-глутамат
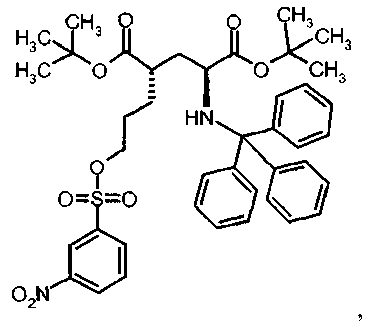
ди-трет-бутил (4S)-4-{3-[(бифенил-4-илсульфонил)окси]пропил}-N-тритил-L-глутамат
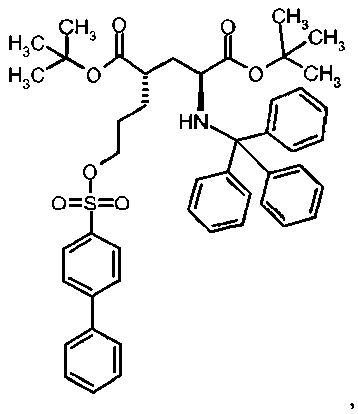
ди-трет-бутил (4S)-4-{3-[(2-нафтилсульфонил)окси]пропил}-N-тритил-L-глутамат

ди-трет-бутил (4S)-4-{3-[(1-нафтилсульфонил)окси]пропил}-N-тритил-L-глутамат

ди-трет-бутил (4S)-4-{3-[(хинолин-8-илсульфонил)окси]пропил}-N-тритил-L-глутамат

ди-трет-бутил (4S)-4-(3-{[(2,4,6-трихлорфенил)сульфонил]окси}пропил)-N-тритил-L-глутамат

тетра-трет-бутил (2S,4S,2′S,4′S)-2,2′-[бифенил-4,4′-диилбис(сульфонилоксипропан-3,1-диил)]бис[4-(тритиламино)пентандиоат]

ди-трет-бутил (4S)-4-(3-{[(7-нитро-1-нафтил)сульфонил]окси}пропил)-N-тритил-L-глутамат

ди-трет-бутил (4S)-4-[3-({[4-нитро-3-(трифторметил)фенил]сульфонил}окси)пропил]-N-тритил-L-глутамат

ди-трет-бутил (4S)-4-(3-{[(4-метилфенил)сульфонил]окси}пропил)-N-тритил-L-глутамат
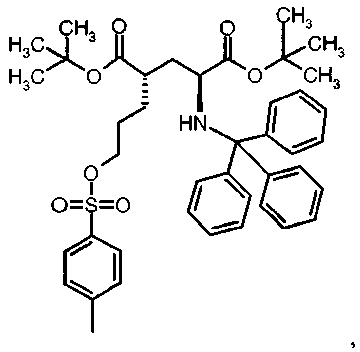
ди-трет-бутил (4S)-4-{3-[(2-нафтилсульфонил)окси]пропил}-N-тритил-L-глутамат

5. Соединение формулы I или Ia по п.3 в твердом виде.
6. Способ получения соединений формулы I, включающий стадию сульфонилирования соединения формулы II с сульфонилгалогенидом (предпочтительно сульфонилхлоридом) или сульфонилангидридом, которые имеют приемлемый заместитель А,
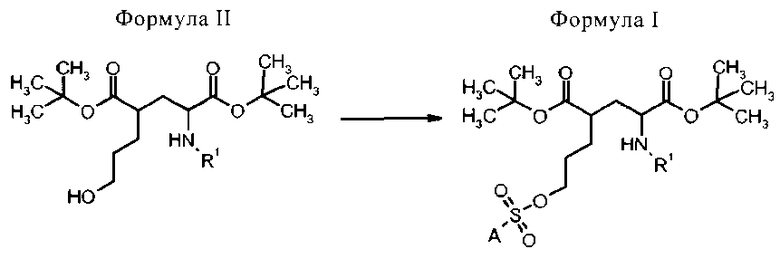
в которой R1 представляет собой трифенилметил (тритил),
А выбран из группы:
a) моноциклический С6-10-арил
b) бициклический С6-10-арил,
c) С12-20-биарил,
d) моноциклический гетероарил и
e) бициклический гетероарил,
где гетероарил представляет собой ароматический, моно- или бициклический двухвалентный радикал, имеющий от 5 до 10 кольцевых атомов и гетероатом N,
по выбору, А несет один или несколько заместителей, выбранных из группы, включающей:
a) галоген,
b) нитро,
c) алкил,
d) трифторметил и
e) Z,
где Z представляет собой

R1 представляет собой трифенилметил (тритил) и
# указывает положение связи с А.
7. Способ по п.6 получения соединения с (2S,4S)-конфигурацией (соединение формулы Ia)

R1 и А являются такими, как определены в п.1 или 2.
8. Способ получения соединений формулы IV-F18, включающий стадии
- взаимодействия соединения формулы I по п.1 с 18F-фторирующим агентом, чтобы получить соединение формулы III-F18, и
- снятия защиты с полученного соединения формулы III-F18 для получения соединения формулы IV-F18,
в котором
соединение формулы III-F18 представляет собой
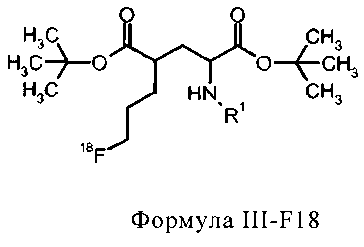
в которой R1 представляет собой трифенилметил (тритил),
и соединение формулы IV-F18 представляет собой

9. Способ по п.8, в котором полученное соединение представляет собой соединение с (2S,4S)-конфигурацией (формула IVa-F18), включающий стадии
- взаимодействия соединения формулы Ia по п.3 с 18F-фторирующим агентом, чтобы получить соединение формулы IIIa-F18, и
- снятия защиты с полученного соединения формулы IIIa-F18 для получения соединения формулы IVa-F18,
в котором
соединение формулы IIIa-F18 представляет собой

R1 представляет собой трифенилметил (тритил), и
соединение формулы IVa-F18 представляет собой
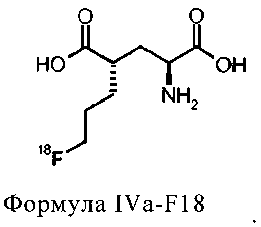
10. Соединение формулы II
Формула II

в которой R1 представляет собой трифенилметил (тритил), и отдельные изомеры, таутомеры, диастереомеры, энантиомеры, стереоизомеры, их смеси и их приемлемые соли.
11. Соединение по п.10 с (2S,4S)-конфигурацией (соединение формулы IIa)
Формула IIа

в которой R1 представляет собой трифенилметил (тритил), соответствующий ди-трет-бутил (4S)-4-(3-гидроксипропил)-N-тритил-L-глутамату.
12. Соединение формулы III-F
Формула III-F
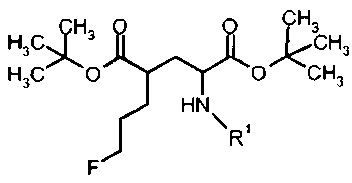
в которой R1 представляет собой трифенилметил (тритил),
F представляет собой 18F, и отдельные изомеры, таутомеры, диастереомеры, энантиомеры, стереоизомеры, их смеси и их приемлемые соли.
13. Соединение по п.12 с (2S,4S)-конфигурацией (соединение формулы IIIa-F)
Формула IIIa-F

14. Композиция, содержащая соединение формулы I или Ia и приемлемые реагенты для реакции введения радиоактивной метки фтора и вспомогательные вещества, предназначенная для получения соединений III-F, IIIa-F или IIIa-F18.
15. Композиция, содержащая соединение формулы II или IIa и вспомогательные вещества, предназначенная для получения соединений формулы I или Ia.
16. Композиция, содержащая соединение формулы III-F, IIIa-F или IIIa-F18 и вспомогательные вещества, предназначенная для получения соединений формулы IV-F18 или IVa-F18.
17. Набор для получения реагентов для введения метки 18F, включающий одну ампулу или более чем одну ампулу, содержащую предопределенное количество соединений формулы I.
| WO 2009141090 A1, 26.11.2009;WO 2010000409 A2, 07.01.2010 | |||
| [F-18] МЕЧЕННАЯ L-ГЛЮТАМИНОВАЯ КИСЛОТА, [F-18] МЕЧЕННЫЙ L-ГЛЮТАМИН, ИХ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ, А ТАКЖЕ СПОСОБ ИХ ПОЛУЧЕНИЯ | 2006 |
|
RU2395489C2 |

