ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННУЮ ЗАЯВКУ
Настоящая заявка является частичным продолжением заявки на патент США № 12/029283 (U.S. Patent Application Serial No. 12/029283), озаглавленной "Способ непрерывного получения органических карбонатов или органических карбаматов и твердые катализаторы для него", зарегистрированной 11 февраля 2008 года автором изобретения J. Yong Ryu, содержание которой приводится здесь путем ссылки на нее.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Раскрываемые в описании изобретения варианты осуществления в целом относятся к способам и твердым катализаторам для реакций, включающих алкоголиз, переэтерификацию и диспропорционирование. Более конкретно, раскрываемые в описании изобретения варианты осуществления относятся к способам непрерывного получения органических карбонатов, органических карбаматов и других продуктов путем алкоголиза, переэтерификации и/или диспропорционирования на твердом катализаторе. В частности, раскрываемые в описании изобретения варианты осуществления относятся к способам получения диарилкарбонатов.
УРОВЕНЬ ТЕХНИКИ
Переэтерификация, или реакция обмена эфиров со спиртами (реакция алкоголиза), является важным классом реакций, которые могут быть ускорены с помощью как кислотных, так и щелочных катализаторов. Примеры переэтерификации, обычно, включают химические реакции с участием органических карбонатов и эфиров карбоновых кислот в качестве реагентов, продуктов, или и тех, и других. Другие реакции переэтерификации включают получение биодизельного топлива путем переэтерификации триглицеридов с этанолом или метанолом. Обычно, алкоголиз представляет собой реакцию, при которой одна или более функциональных групп соединения замещаются с помощью алкоксильной или арилоксильной группы спирта (алкил- или арилгидроксильного соединения). Примеры алкоголиза включают химические реакции с участием мочевины, в которой аминогруппы замещаются с помощью алкоксильных групп с получением органических карбаматов и карбонатов.
Эфиры карбоновых кислот получают путем переэтерификации эфира карбоновой кислоты со спиртом в присутствии кислотного и щелочного катализатора. Серная кислота (гомогенный катализатор) и кислотные ионообменные смолы (твердые катализаторы) являются предпочтительными катализаторами. Растворимые основания, такие как NaOH и KOH, различные Na/K алкоксиды или амины (гомогенные катализаторы), и различные основные ионообменные смолы (твердые катализаторы) являются предпочтительными щелочными катализаторами. Катализаторами для переэтерификации карбоновых эфиров могут являться как гомогенные катализаторы, так и гетерогенные катализаторы, но щелочные катализаторы обычно являются более эффективными, чем кислотные катализаторы. Например, длинноцепочечные алкилметакриловые эфиры получают путем реакции обмена метилметакрилата с длинноцепочечным спиртом в присутствии щелочного катализатора.
Биодизельное топливо может быть получено путем переэтерификации растительных масел (триглицеридов) с метанолом или этанолом с использованием гомогенного щелочного катализатора, такого как метоксид натрия или ацетат кальция, раскрытого в Патентах США №№ 6712867 и 5525126, и щелочного твердого катализатора, такого как смесь оксидов цинка и алюминия или алюминат цинка (оксид цинка, нанесенный на оксид алюминия и прокаленный при высокой температуре). Например, твердые катализаторы из алюмината цинка раскрыты в Патенте США № 5908946 и заявке на Патент США № 2004/0034244.
В Патенте США № 5908946 описан двухстадийный способ получения эфиров путем реакции растительных масел или животных масел со спиртом в присутствии твердых катализаторов, таких как оксид цинка или алюминаты цинка типа шпинели. На первой стадии, конверсию триглицерида проводят при высокой степени превращения, обычно выше чем 90%. На второй стадии подвергают превращению оставшиеся триглицериды, диглицериды и моноглицериды. Переэтерификации проводят при температуре от 230 до 245°C при давлении около 5,2 бар (около 72,5 фунт/дюйм2). Высокая степень конверсии требует относительно низких объемных скоростей вводимой смеси (0,5 час-1 или более низкая объемная скорость).
В Патенте США № 6147196 раскрыт способ получения эфиров жирных кислот высокой чистоты из растительного или животного масла в присутствии гетерогенного катализатора (алюмината цинка). Заявка на Патент США № 2004/0034244 относится к технологической схеме получения алкиловых эфиров из растительного или животного масла и спирта в присутствии гетерогенного катализатора (алюмината цинка). Эфиры получают путем переэтерификации в двух реакторах с неподвижным слоем катализатора. Высокая конверсия триглицирида достигалась в первом реакторе. После разделения глицерина от реакционного потока первой переэтерификации, оставшиеся непрореагировавший триглицерид, диглицерид и моноглицерид превращают в эфиры во втором реакторе. Переэтерификацию проводят при 200°C, давлении около 62 бар (900 фунт/дюйм2) и объемной скорости 0,5 час-1.
В публикации W. Xie et al., J. Mol. Cat. A: Chem. 246, 2006, pp. 24-32 обсуждается метанолиз соевого масла в присутствии Mg-Al катализатора из прокаленного гидроталькита. Прокаленные гидрокалькиты с Mg/Al отношением 3,0, получаемые прокаливанием при 500°C, являются катализатором, который может обеспечивать высокую щелочность и отличную каталитическую активность для этой реакции. В этой публикации приводятся данные по создаваемой прокаленными при различных температурах гидроталькитами щелочности в растворе.
Дизельные двигатели выбрасывают больше твердых частиц и NOx, чем бензиновые двигатели. Сообщается, что диалкилкарбонаты эффективно снижают содержания твердых частиц в выхлопе дизельного двигателя. Согласно Патенту США № 5954280 мочевина и аммиак являются эффективными средствами для снижения содержания NOx. Но использование мочевины и аммиака для дизельного двигателя создает проблемы или неудобства их применения на практике. В Патенте США № 6017368 раскрыт этилкарбамат в качестве эффективного средства для снижения NOx в выхлопе дизельных двигателей. В Патенте США № 4731231 (1988) сообщается, что сублимируемая циануровая кислота может быть эффективным средством для предотвращения или снижения выброса NOx. Высокотемпературная сублимация циануровой кислоты приводит к образованию изоциановой кислоты (HNCO), которая, по-видимому, содействует предотвращению выброса NOx. В патентных документах EP 0363681 и EP 0636681 раскрыт карбонатный эфир алифатического триола или тетраола в качестве компонента малодымящих смазок.
N-арилметилкарбамат получают реакцией ароматического амина с диметилкарбонатом, обычно в присутствии щелочного катализатора ввиду низких скоростей реакции в отсутствие катализатора. N-арилметилкарбамат может быть подвергнут разложению при повышенных температурах с получением ароматического изоцианата. Например, толуолдикарбамат получают путем реакции толуолдиамина с диметилкарбонатом в присутствии катализатора. Разложение толуолдикарбамата при повышенных температурах дает толуолдиизоцианат.
Органические карбонаты (диэфиры угольной кислоты) являются ценными соединениями, которые могут применяться в качестве растворителей, алкилирующих реагентов, карбонилирующих реагентов, реагентов сополимеризации, присадок к топливу, и так далее. Диметилкарбонат (ДМК) является важным диалкилкарбонатом, широко используемым в качестве сырья для получения дифенилкарбоната (ДФК, диарилкарбоната). Существуют различные способы промышленного получения ДМК. В одном таком промышленном способе, ДМК получают путем переэтерификации циклического карбоната с метанолом в присутствии гомогенного катализатора. Несмотря на то, что в патентах может раскрываться применение гомогенных катализаторов или гетерогенных катализаторов для переэтерификации циклического карбоната с метанолом, в настоящее время в промышленности не применяют гетерогенный или твердый катализатор для получения ДМК, по-видимому, вследствие короткого срока службы гетерогенных катализаторов в таких способах. ДФК обычно сополимеризуют с диолом, таким как бисфенол A, с получением поликарбонатов. Поликарбонаты используют в различных специальных областях применения, таких как диски памяти, ветровые стекла, конструкционные пластики, оптические материалы, и так далее.
Современные методы получения диарилкарбонатов с использованием нефосгенового способа позволяют получать ароматические карбонаты, такие как ДФК, путем переэтерификации ДМК с фенолом с получением метилфенилкарбоната и метанола и затем диспропорционирования метилфенилкарбоната с получением ДФК и ДМК в присутствии гомогенных металлоорганических катализаторов при использовании нескольких соединенных последовательно реакторов реакционной дистилляции. Предпочтительным гомогенным катализатором является алкоксид титана. Например, такие способы раскрыты в Патентах США № 4045464, 4554110, 5210268 и 6093842. Гомогенные катализаторы извлекают из части продуктовых потоков с самой высокой молекулярной массой в виде твердого вещества, которое может затем быть превращено в растворимый гомогенный катализатор, который рециркулируют.
Использование гомогенного катализатора при получении ДФК часто требует разделение гомогенного катализатора от продукта, особенно когда катализаторы используют при относительно высоких объемных скоростях введения сырья. Для устранения этого и других недостатков, связанных с использованием гомогенных катализаторов для получения диарилкарбонатов, в Патентах США № 5354923 и 5565605, и в патентном документе PCT Application Publication WO 03/066569 раскрыты альтернативные способы, в которых используют гетерогенные катализаторы. Например, в Патенте США № 5354923 раскрыты катализаторы из оксида титана в порошковой форме при иллюстрации получения ЭФК, МФК и ДФК из ДЭК или ДМК и фенола. В Патенте США № 5565605 раскрыты микропористые материалы, содержащие элементы Группы 4, в качестве катализаторов для переэтерификации и диспропорционирования. Однако твердые катализаторы в порошкообразной форме являются обычно неприемлемыми или менее предпочтительными для крупнотоннажного промышленного производства ДФК или метилфенилкарбоната. В патентном документе WO03/066569 раскрыт способ непрерывного получения ДФК в присутствии гетерогенного катализатора, приготовленного путем нанесения оксида титана на оксид кремния, в двустадийном способе с неподвижным слоем путем реакции ДМК с фенолом.
В публикации Z-H Fu and Y. Ono, J. Mol. Catal. A. Chemical, 118 (1997), pp. 293-299 и в патентном документе JP Application No. HEI 07-6682 раскрыты гетерогенные катализаторы для получения дифенилкарбоната путем переэтерификации ДМК с фенолом в МФК и диспропорционирования МФК в ДФК в присутствии MoO3 или V2O5, нанесенных на неорганический носитель, такой как оксид кремния, оксид циркония или оксид титана. Переэтерификацию и диспропорционирование проводят в системе реактор-дистилляционная колонна, состоящей из реактора и дистилляционной колонны, служащей для удаления побочных продуктов путем дистилляции.
В заявке на Патент США № 2007/0093672 ('672) (в настоящий момент Патент США № 7378540) и № 2007/0112214 ('214) (в настоящий момент Патент США № 7288668) раскрыты способы получения различных органических карбонатов, таких как диарилкарбонаты, включая ДФК, в присутствии гетерогенных катализаторов. В публикации '214, необходимые реакции (переэтерификацию и диспропорционирование) проводят в жидкой фазе в присутствии гетерогенного катализатора. Несколько реакторов с неподвижным слоем для реакций переэтерификации и диспропорционирования подсоединены к единственной дистилляционной колонне, в которой низкокипящие соединения, такие как этанол и ДЭК, удаляются в виде фракции в верхней части колонны, и более высококипящие соединения, включая ДФК, удаляются в виде смешанной кубовой фракции. ДФК затем извлекают из кубовой фракции.
В публикации '672 раскрывается способ получения диарилкарбонатов и диалкилкарбонатов путем осуществления необходимых реакций в двухфазной (пар и жидкость) системе на различных твердых катализаторах для переэтерификации и диспропорционирования. Химические реакции, позволяющие получать органические карбонаты, проводят в ряду последовательно соединенных реакторов с неподвижным слоем, при этом осуществляя выделение легкокипящего продукта из жидкой фазы в паровую фазу, для того чтобы сдвинуть неблагоприятное равновесие реакции в сторону образования требуемого продукта. Способ, в частности, применяют для получения алкиларилкарбонатов, таких как ЭФК (этилфенилкарбонат), и диарилкарбонатов, таких как ДФК (дифенилкарбонат). Способ также применяют для получения диалкилкарбонатов, таких как ДЭК. Расположенные в ряд реакторы с неподвижным слоем подсоединяют в различных местах к единственной дистилляционной колонне через потоки боковой фракции и возвратные потоки. Дистилляционная колонна также содержит ступени сепарации выше подсоединения последнего реактора в ряду и ниже подсоединения первого реактора в ряду. Гетерогенные катализаторы могут быть приготовлены путем нанесения одного или двух оксидов Ti, Zr, Nb, Hf, Ta, Mo, V, Sb, и других металлов на пористые носители, такие как силикагель. Гетерогенные катализаторы могут быть также приготовлены путем прививки одного или более металлоорганических соединений Ti, Zr, Nb, Hf, Ta, Mo, V, Sb и других металлов на пористый носитель, который имеет поверхностные гидроксильные группы или смесь гидроксильных и алкоксильных групп.
Другие различные способы получения органических карбонатов на гетерогенных катализаторах раскрыты в Патентах США № 5231212, 5498743 и 6930195.
В публикации P. Ball et al., C1 Mol. Chem. Vol. 1, 1984, pp. 95-108 приведены данные по исследованию химии получения диалкилкарбоната в присутствии различных гомогенных или гетерогенных катализаторов. Например, диметилкарбонат получают путем алкоголиза мочевины. Сообщается о диметоксиде дибутилолова, как об особенно эффективном катализаторе. Сообщается, что гетерогенные катализаторы являются также эффективными в отношении указанных химических реакций в присутствии сокатализаторов, таких как 4-диметиламинопиридин и PPh3. Рассмотренными гетерогенными катализаторами являются Al2O3, Sb2O3 и оксид кремния. Плавленый SiO2 не является катализатором, но становится каталитически активным в присутствии PPh3.
В Патенте США № 7074951 диалкилкарбонат получают путем алкоголиза мочевины с помощью спирта в присутствии гомогенного катализатора из комплекса олова в присутствии электродонорного высококипящего растворителя, такого как триглим. В этом патенте также продемонстрирована возможность непрерывного получения ДМК в течение приблизительно 1500 часов.
В патентном документе EP 1629888 и публикации D. Wang et al., Fuel Processing Tech. 88, 8, 2007, pp. 807-812 показано, что ДМК и ДЭК могут быть получены в присутствии оксида цинка и оксида цинка, нанесенного на оксид кремния. Но в этих публикациях полностью отсутствует информация о стабильности катализатора или продолжительности работы катализатора.
Дезактивация катализатора в процессе протекания реакций переэтерификации и диспропорционирования может быть вызвана отложением полимеров с высокой молекулярной массой на поверхности и порах катализатора. Скорость дезактивации катализатора в результате отложения полимера повышается с увеличением концентрации алкиларилкарбоната и диарилкарбоната или их обоих в реакционной смеси. Деполимеризация полимеров на поверхности гетерогенных катализаторов раскрыта в публикации '672. Однако деполимеризация может приводить только к частичному восстановлению активности твердого катализатора.
В Патентах США № 6768020 и 6835858 раскрыты способы получения диалкилкарбонатов и побочного продукта пропиленгликоля путем реакции пропиленкарбоната с ДМК, водой, или с обоими, в присутствии твердого катализатора, такого как оксид лантана и оксид цинка, нанесенный на оксид алюминия, оксид кремния и другие носители. Проблема нестабильности катализатора частично решается в Патенте США № 6768020 путем нанесения большого количества оксида лантана на носитель, такой как оксид алюминия и оксид кремния.
Методом, позволяющим компенсировать дезактивацию катализатора, является повышение температуры реакции по мере дезактивации катализатора. Этот метод, к сожалению, часто ускоряет дезактивацию гетерогенных катализаторов.
Для промышленного производства, в котором используется гетерогенный катализатор, обычно требуется стабильная на протяжении длительного времени рабочая характеристика твердого катализатора. Затраты на катализатор, простои производства в связи с заменой катализатора и другие известные в технике факторы выдвигает для гетерогенных катализаторов в качестве необходимого требование минимального срока службы обычно в течение более чем 3 месяцев, 6 месяцев, или года, в зависимости от процесса.
Несмотря на то, что гетерогенный катализ возможен при проведении различных реакций переэтерификации, как это описано выше в различных патентах и публикациях, в этих источниках не приводятся данные о продолжительности срока службы или продолжительности рабочего цикла катализатора. Автору настоящего изобретения из личного опыта известно, что такие гетерогенные катализаторы имеют неприемлемо короткие продолжительности цикла работы.
Следовательно, существует необходимость в способах переэтерификации и/или диспропорционирования с использованием гетерогенных катализаторов с улучшенной характеристикой их работы.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В одном аспекте, раскрываемые в изобретении варианты осуществления относятся к способу алкоголиза, где способ включает: введение реагентов и следового количества растворимого металлоорганического катализатора в реактор, включающий твердый катализатор алкоголиза; где растворимый металлоорганический катализатор и твердый катализатор алкоголиза каждый независимо включает элемент Группы II - Группы VI. В некоторых вариантах осуществления, твердый катализатор и металлоорганическое соединение могут включать один и тот же элемент Группы II - Группы VI.
В другом аспекте, раскрываемые в изобретении варианты осуществления относятся к способу получения диалкилкарбонатов, где способ включает: введение спирта и реагента, подвергаемого алкоголизу, включающего, по меньшей мере, либо мочевину, либо органический карбамат, или циклический карбонат, в первую реакционную зону, включающую твердый катализатор алкоголиза; введение растворимого металлоорганического катализатора в первую реакционную зону, где твердый катализатор алкоголиза и растворимый металлоорганический катализатор каждый независимо включает элемент Группы II - Группы VI.
В другом аспекте, раскрываемые в изобретении варианты осуществления относятся к способу получения диарилкарбоната, где способ включает: введение ароматического гидроксисоединения и диалкилкарбоната в первую реакционную зону, включающую твердый катализатор переэтерификации; и введение растворимого металлоорганического катализатора в первую реакционную зону, где твердый катализатор переэтерификации и растворимый металлоорганический катализатор каждый независимо включает элемент Группы II - Группы VI.
В другом аспекте, раскрываемые в изобретении варианты осуществления относятся к способу получения алкиларилкарбоната, где способ включает: введение ароматического гидроксисоединения и диалкилкарбоната в первую реакционную зону, включающую твердый катализатор переэтерификации; и введение растворимого металлоорганического катализатора в первую реакционную зону, где твердый катализатор переэтерификации и растворимый металлоорганический катализатор каждый независимо включает элемент Группы II - Группы VI.
В другом аспекте, раскрываемые в изобретении варианты осуществления относятся к способу получения биодизельного топлива, где способ включает: введение спирта и глицерида в первую реакционную зону, включающую твердый катализатор переэтерификации; и введение растворимого металлоорганического катализатора в первую реакционную зону, где твердый катализатор переэтерификации и растворимый металлоорганический катализатор каждый независимо включает элемент Группы II - Группы VI.
В другом аспекте, раскрываемые в изобретении варианты осуществления относятся к способу получения алкиларилкарбоната, где способ включает: введение ароматического гидроксисоединения и диалкилкарбоната в первую реакционную зону, включающую твердый катализатор переэтерификации; и введение растворимого металлоорганического катализатора в первую реакционную зону, где твердый катализатор переэтерификации и растворимый металлоорганический катализатор каждый независимо включает элемент Группы II - Группы VI.
В другом аспекте, раскрываемые в изобретении варианты осуществления относятся к способу получения биодизельного топлива, где способ включает: введение спирта и глицерида в первую реакционную зону, включающую твердый катализатор переэтерификации; и введение растворимого металлоорганического катализатора в первую реакционную зону, где твердый катализатор переэтерификации и растворимый металлоорганический катализатор каждый независимо включает элемент Группы II - Группы VI
В другом аспекте, раскрываемые в изобретении варианты осуществления относятся к способу реактивации отработанного твердого катализатора алкоголиза, где способ включает: удаление полимерных материалов, отложившихся на катализаторе; и повторное нанесение каталитически активных металлов на твердый катализатор.
В другом аспекте, раскрываемые в изобретении варианты осуществления относятся к способам получения диарилкарбоната, включающим: реакцию эпоксида и диоксида углерода в первой реакционной зоне с образованием первого реакционного продукта, включающего циклический карбонат; переэтерификацию циклического карбоната с этанолом в присутствии первого катализатора переэтерификации во второй реакционной зоне с образованием второго реакционного продукта, включающего диэтилкарбонат и гликоль; разделение второго реакционного продукта с извлечением первой фракции диэтилкарбоната и первой фракции гликоля; переэтерификацию, по меньшей мере, части первой фракции диэтилкарбоната с арилгидроксисоединением в присутствии катализатора второй переэтерификации в третьей реакционной зоне с образованием третьего реакционного продукта, включающего этиларилкарбонат и этанол; разделение третьего реакционного продукта с извлечением фракции этиларилкарбоната и первой фракции этанола; диспропорционирование, по меньшей мере, части фракции этиларилкарбоната в присутствии катализатора диспропорционирования в четвертой реакционной зоне с образованием четвертого реакционного продукта, включающего диарилкарбонат и диэтилкарбонат; разделение четвертого реакционного продукта с извлечением фракции диарилкарбоната и второй фракции диэтилкарбоната; рециркуляцию, по меньшей мере, части первой фракции этанола во вторую реакционную зону; и рециркуляцию, по меньшей мере, части второй фракции диэтилкарбоната в третью реакционную зону.
В другом аспекте, раскрываемые в изобретении варианты осуществления относятся к способу получения диарилкарбоната, включающему: реакцию аммиака и диоксида углерода в первой реакционной зоне с образованием первого реакционного продукта, включающего мочевину; переэтерификацию мочевины с этанолом в присутствии катализатора первой переэтерификации во второй реакционной зоне с образованием второго реакционного продукта, включающего диэтилкарбонат и аммиак; разделение второго реакционного продукта с извлечением первой фракции диэтилкарбоната и первой фракции аммиака; переэтерификацию, по меньшей мере, части первой фракции диэтилкарбоната с арилгидроксисоединением в присутствии катализатора второй переэтерификации в третьей реакционной зоне с образованием третьего реакционного продукта, включающего этиларилкарбонат и этанол; разделение третьего реакционного продукта с извлечением фракции этиларилкарбоната и фракции этанола; диспропорционирование, по меньшей мере, части фракции этиларилкарбоната в присутствии катализатора диспропорционирования в четвертой реакционной зоне с образованием четвертого реакционного продукта, включающего диарилкарбонат и диэтилкарбонат; разделение четвертого реакционного продукта с извлечением фракции диарилкарбоната и второй фракции диэтилкарбоната; рециркуляцию, по меньшей мере, части фракции этанола во вторую реакционную зону; и рециркуляцию, по меньшей мере, части второй фракции диэтилкарбоната в третью реакционную зону.
Другие аспекты и преимущества будут очевидны из следующего далее описания и прилагаемых пунктов формулы изобретения.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На фигуре 1 приведена упрощенная принципиальная технологическая схема, иллюстрирующая способ получения диарилкарбонатов согласно раскрываемым в изобретении вариантам осуществления.
На фигуре 2 приведена упрощенная принципиальная технологическая схема, иллюстрирующая способ получения диарилкарбонатов согласно раскрываемым в изобретении вариантам осуществления.
На фигуре 3 приведена упрощенная принципиальная технологическая схема, иллюстрирующая способ получения диарилкарбонатов согласно раскрываемым в изобретении вариантам осуществления.
На фигуре 4 графически представлена переэтерификация с использованием гомогенного катализатора.
На фигуре 5 графически представлена активность катализатора после повторной активации катализатора согласно раскрываемым в изобретении вариантам осуществления.
На фигуре 6 графически представлена активность твердого катализатора при введении в реактор следового количества растворимого металлоорганического катализатора согласно раскрываемым в изобретении вариантам осуществления.
На фигуре 7 приводится графически представленное сравнение активности гетерогенного катализатора с активностью твердого катализатора при введении в реактор следового количества растворимого металлоорганического катализатора согласно раскрываемым в изобретении вариантам осуществления.
На фигуре 8 графически представлена активность твердого катализатора при введении в реактор следового количества растворимого металлоорганического катализатора согласно раскрываемым в изобретении вариантам осуществления.
На фигурах 9A и 9B графически представлена активность твердого катализатора в процессе получения ЭФК и ДФК, соответственно, при введении в реактор следового количества растворимого металлоорганического катализатора согласно раскрываемым в изобретении вариантам осуществления.
На фигуре 10 графически представлена активность гетерогенного катализа в процессе получения ДФК при прививке катализатора одновременно с проведением реакции переэтерификации.
На фигуре 11 графически представлена конверсия ЭФК в ДФК и ДЭК в отсутствии твердых катализаторов согласно раскрываемым в изобретении вариантам осуществления.
На фигуре 12 графически представлены результаты алкоголиза пропиленкарбоната с этанолом с получением ДЭК и пропиленгликоля в присутствии твердого катализатора согласно раскрываемым в изобретении вариантам осуществления.
На фигуре 13 представлены результаты получения ДЭК с использованием гомогенного катализатора.
На фигуре 14 представлены результаты получения ДЭК с использованием твердого катализатора согласно раскрываемым в изобретении вариантам осуществления.
На фигуре 15 приведена упрощенная принципиальная технологическая схема получения диалкилкарбонатов с использованием твердого катализатора согласно раскрываемым в изобретении вариантам осуществления.
На фигуре 16 представлены результаты получения ДЭК из этилкарбамата с использованием твердого катализатора согласно раскрываемым в изобретении вариантам осуществления.
На фигуре 17 представлены результаты алкоголиза масла канолы с метанолом с использованием твердого катализатора согласно раскрываемым в изобретении вариантам осуществления.
На фигуре 18 приведена упрощенная принципиальная технологическая схема непрерывного получения ДЭК и пропиленгликоля в качестве побочного продукта путем осуществления алкоголиза пропиленкаробоната с этанолом в присутствии твердого катализатора согласно раскрываемым в изобретении вариантам осуществления.
На фигуре 19 приведена упрощенная принципиальная технологическая схема, иллюстрирующая способ получения дифенилкарбоната (ДФК) согласно раскрываемым в изобретении вариантам осуществления.
На фигуре 20 приведена упрощенная принципиальная технологическая схема, иллюстрирующая совмещенный способ получения дифенилкарбоната (ДФК) согласно раскрываемым в изобретении вариантам осуществления.
На фигуре 21 графически представлены результаты алкоголиза пропиленкарбоната с этанолом с получением ДЭК и пропиленгликоля в присутствии твердого катализатора согласно раскрываемым в изобретении вариантам осуществления.
На фигуре 22 представлены результаты получения ДЭК из этилкарбамата с использованием твердого катализатора согласно раскрываемым в изобретении вариантам осуществления.
На фигуре 23 графически представлены результаты каталитической переэтерификации ДЭК с фенолом с получением ЭФК, промежуточного продукта в способе получения ДФК.
На фигуре 24 графически представлены результаты каталитического диспропорционирования ЭФК с получением ДФК и ДЭК.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В одном аспекте, раскрываемые в изобретении варианты осуществления относятся к процессам алкоголиза, переэтерификации и/или диспропорционирования с использованием твердых катализаторов. Используемый в описании изобретения термин "алкоголиз" обозначает различные химические реакции, в которых органическое гидроксильное соединение (спирт) принимает участие в качестве одного или двух реагентов с образованием продукта и побочного продукта. Алкоголиз может быть охарактеризован как разрыв связей (C - Y) между углеродным атомом и гетероатомом Y молекул с помощью молекулы спирта (ROH). Реакциями алкоголиза являются реакции,Alcoholyses в которых участвуют карбонильные группы молекулы, и карбонильная группа сама по себе сохраняется в молекуле продукта. Поэтому, атом C в связи C - Y является углеродным атомом карбонильной группы молекулы. Обычно алкоголиз является обратимой реакцией и может быть представлен следующим образом:
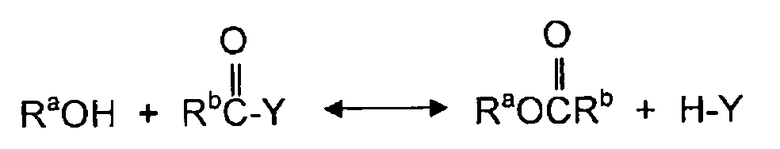 ,
,
где Y является гетероатомом или гетероатомом функциональной группы, и Rb является алкилом, арилом, или функциональной группой, имеющей один или более гетероатомов.
Примерами реакций алкоголиза являются реакция спирта с диэфирами угольной кислоты, эфирами карбоновой кислоты, мочевиной и карбаматами. Алкоголиз диалкилкарбоната (часто называемой в литературе переэтерификацией) с фенолом дает алкиларилкарбонат и спирт. Алкоголиз эфира карбоновой кислоты со спиртом производит обмен алкильной группы эфира с алкильной группой молекулы спирта и дает в результате молекулу нового спирта. Алкоголиз мочевины со спиртом спирт дает органический карбамат и аммиак. Алкоголиз органического карбамата со спиртом приводит к образованию диалкилкарбоната и аммиака. Конкретными примерами реакций алкоголиза являются переэтерификация ДЭК с фенолом с получением ЭФК и этанола, алкоголиз мочевины или органического карбамата со спиртом с получением органического карбамата или диалкилкарбоната и аммиака, переэтерификация триглицерида с метанолом с получением метиловых эфиров (биодизельного топлива) и глицерина.
Несмотря на то, что при диспропорционировании асимметрических диэфиров угольной кислоты и при реакции диалкилкарбоната с органическим амином спирт не участвует в качестве реагента, тем не менее, для удобства эти типы реакций в описании изобретения также относят к реакциям алкоголиза, так как RA группы (R является алкилом или арилом, и A является атомом кислорода или атомом азота) принимают участие в механизмах реакций на молекулярном уровне. Поэтому, в случае необходимости, при описании различных вариантов осуществления термины переэтерификация и диспропорционирование используются в качестве синонимов термина алкоголиз. Ряд упомянутых выше реакций алкоголиза могут быть представлены следующими уравнениями реакций:
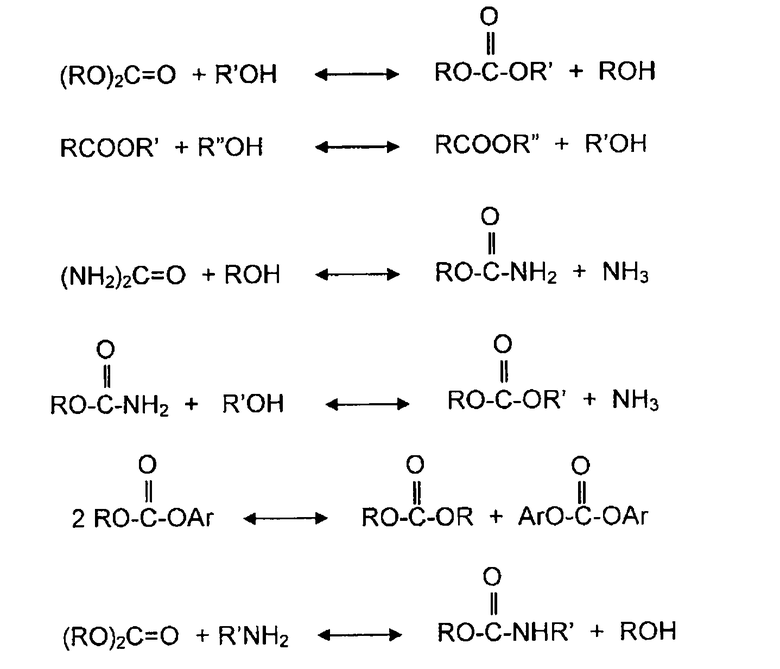
В другом аспекте, раскрываемые в изобретении варианты осуществления относится к новому способу сохранения каталитической активности твердого катализатора в течение длительного производственного цикла. Время цикла или продолжительность цикла работы твердого катализатора определяется в описании изобретения как период времени, в течение которого можно непрерывно без каких-либо остановок использовать твердый катализатор для конкретной химической реакции. Например, если для катализатора требуется регенерация или замена после непрерывного использования в течение 6 месяцев, продолжительность или время цикла работы катализатора составляет 6 месяцев. Согласно раскрываемому изобретением способу, в различных вариантах осуществления, твердые катализаторы для процессов алкоголиза могут сохранять каталитическую активность в течение продолжительного времени цикла работы, например, в течение более чем 3 месяцев, 6 месяцев, 1 года, 1,5 лет и 2 лет или более.
В процессе переэтерификации ДЭК с фенолом, автором настоящего изобретения наблюдалась дезактивация гетерогенных катализаторов (оксида титана и смеси оксидов ниобия и титана, нанесенных на оксид кремния), которую он описал в Испытании 4 в публикации '672. Деполимеризация отложения полимера на катализаторе для повышения активности катализатора была также проиллюстрирована в Испытании 6B в публикации '672. Однако регенерация катализатора путем деполимеризации приводила только к частичному восстановлению исходной активности катализатора. На тот момент, природа дезактивации катализатора была еще не полностью выяснена.
Неожиданно было установлено, что гетерогенные катализаторы переэтерификации, такие как гетерогенные катализаторы для получения ДФК, теряют свою активность вследствие двух основных причин: отложения полимера и вымывания компонента каталитически активного металла. Гетерогенные катализаторы для получения диалкилкарбоната путем переэтерификации циклического карбоната со спиртом теряют свою активность вследствие вымывания компонентов каталитически активных металлов.
В процессе реакций алкоголиза или переэтерификации на гетерогенных катализаторах, компоненты каталитически активных металлов на твердых катализаторах могут вымываться из гетерогенных металлоксидных катализаторов и металлоорганических катализаторов, иммобилизируемых на различных пористых носителях, в реакционной среде при реакционных условиях, что приводит к постоянной дезактивации катализатора. Это влечет за собой неприемлемо короткий срок службы промышленных гетерогенных катализаторов, которые могут быть использованы для непрерывного получения различных органических карбонатов. Кроме того, как уже упоминалось выше, отложение полимера может также отрицательно воздействовать на рабочую характеристику катализатора переэтерификации. Еще одним видом дезактивации катализатора является отравление катализатора.
Гетерогенный катализатор, который предполагается использовать в промышленном реакторе с неподвижным слоем, должен иметь приемлемую долговечность, как с точки времени рабочего цикла, так и суммарного срока службы. При отсутствии отравления, и в случае, если на гетерогенном катализаторе не происходит отложение полимеров или оно является небольшим, долговечность катализатора может определяться скоростью вымывания компонента активного металла из гетерогенного катализатора.
Раскрываемые в изобретении варианты осуществления относятся к способам поддержания постоянной или почти постоянной активности твердого катализатора в течение длительного периода времени, приемлемого для непрерывного получения различных органических соединений в промышленном масштабе. Такие способы могут, в частности, применяться для непрерывного получения различных органических карбонатов, таких как диарилкарбонаты, диалкилкарбонаты и алкиларилкарбонаты, а также в других реакциях переэтерификации, таких как получение биодизельного топлива. Выбранные раскрываемые в изобретении варианты осуществления относятся к способам поддержания стабильной активности катализатора в течение продолжительного периода времени в крупных промышленных реакторах для непрерывного получения органических карбонатов, эфиров карбоновых кислот, или органических карбаматов.
Новый способ поддержания каталитической активности твердого катализатора в течение длительного времени рабочего цикла заключается в добавлении следового количества растворимых компонентов активного металла в жидкий поток сырья, вводимого в реактор, содержащий твердый катализатор, что дает в результате постоянную или почти постоянную активность катализатора в течение продолжительных периодов времени рабочего цикла. Неожиданно было установлено, что добавление следового количества растворимых компонентов активного металла в поток жидкого сырья, вводимого в реактор, содержащий твердый катализатор, может эффективно восполнять потери металла вследствие вымывания металла из твердых катализаторов, например, за счет повторного осаждения компонента активного металла на катализаторе, что дает в результате постоянную или почти постоянную активность катализатора в течение продолжительных периодов времени рабочего цикла. Например, проводимые автором настоящего изобретения исследования свидетельствуют о том, что активность катализатора может поддерживаться в течение более чем одного года путем добавления следового количества растворимых соединений активного металла в жидкий поток сырья, вводимого в реакторы с неподвижным или подвижным слоем, содержащие твердый катализатор.
Количество соединения активного металла, требуемое для поддержания активности твердого катализатора, может составлять от менее чем 1 ppm до приблизительно 3000 ppm, в зависимости от конкретного компонента активного металла, реагентов и других вводимых компонентов. Количество активного металла во вводимом сырье, например, при получении органических карбонатов путем переэтерификации, может быть на один, два или более порядков ниже, чем концентрация гомогенного катализатора, вводимого в сравнимом способе, использующем только гомогенный катализатор. В некоторых вариантах осуществления, соединение активного металла может быть введено в количестве от 1 до 400 частей на миллион (ppm) по массе; в других вариантах осуществления, от 10 до 300 частей на миллион (ppm) по массе; в других вариантах осуществления, от 15 до 200 частей на миллион (ppm) по массе; в других вариантах осуществления, от 20 до 150 частей на миллион (ppm) по массе; и в еще одних вариантах осуществления, от 30 до 100 частей на миллион (ppm) по массе, от суммарной массы жидкости, входящей в каталитическую реакционную зону.
Например, когда твердый катализатор включает активный металл, такой как металл Группы II-VI, для поддержания активности твердого катализатора в реактор может быть введено следовое количество растворимого металлоорганического катализатора, имеющего такой же активный металл Группы II-VI. В качестве конкретного примера, когда твердый катализатор включает титан в качестве активного металла, может быть использован растворимый металлоорганический катализатор, содержащий титан.
При использовании нескольких соединенных последовательно реакторов, например, реактора переэтерификации, соединенного последовательно с реактором диспропорционирования, следовое количество растворимого металлоорганического катализатора может быть введено в один или оба реактора для поддержания активности катализатора в соответствующих реакторах. В некоторых вариантах осуществления, путем введения следового количества растворимого металлоорганического катализатора только в первый реактор из нескольких соединенных последовательно реакторов, активность твердого катализатора может быть поддержана в каждом из этих реакторов. Например, когда реактор переэтерификации включает твердый катализатор из смеси оксидов титана и ниобия, а реактор диспропорционирования содержит твердый алкоксид титана, привитый на оксид кремния, добавление следового количества растворимого титанорганического соединения или растворимого соединения титана, такого как оксиалкоксид титана, в первый реактор может привести к удлинению времени рабочего цикла обоих типов твердых катализаторов.
В случае необходимости, растворимый металлоорганический катализатор может быть извлечен и рециркулирован. В некоторых вариантах осуществления, извлечение активного металла из выходящих из реактора потоков с целью его рециркуляции может быть экономически нецелесообразным. В случае извлечения, компонент активного металла в выходящем из реактора потоке может быть извлечен из потока тяжелого кубового остатка в виде твердого материала и превращен, например, путем реакции извлеченного твердого материала с органическим карбонатом или смесью органического карбоната и спирта при повышенной температуре, в растворимый металлоорганический катализатор, который может быть рециркулирован в реактор. Извлекаемое металлоорганическое соединение может являться, например, алкоксидом металла, алкоксиалкилкарбонатом металла (солью металла с моноэфиром угольной кислоты), оксиалкоксидом металла или их смесью.
Достигаемый таким образом продолжительный срок службы твердого катализатора алкоголиза может в результате позволять рентабельно осуществлять процессы получения органических карбонатов на твердом катализаторе, наряду с алкоголизом и/или другими процессами переэтерификации. Значительная экономия может быть достигнута вследствие удлиненного времени рабочего цикла катализаторов и более низких требований к процессу разделения (меньшее количество технологических операций, что приводит к экономии, связанной с возможными капиталовложениями и эксплуатационными расходами).
Отложение полимера на твердых катализаторах также может вызывать потерю каталитической активности. В таких случаях, дезактивированный катализатор может быть регенерирован с помощью способов деполимеризации, раскрытых в описании настоящего изобретения и в патентном документе U.S. Patent Application Publication No. 2007/0093672. Деполимеризация также может приводить к потере металла. В случае если после деполимеризации гетерогенный катализатор не восстанавливает каталитическую активность до приемлемого уровня от исходной активности, для гетерогенного катализатора может потребоваться повторное нанесение металла, например, с помощью раскрываемых в изобретении способов реактивации катализатора.
Во всех случаях дезактивации катализатора, вызванной либо отложением полимера, либо вымыванием металла, активность катализатора может быть восстановлена путем использования раскрываемых в изобретении способов регенерации и реактивации катализаторов. Реактивация катализатора состоит из двух стадий: деполимеризации и кондиционирования поверхности на первой стадии, и повторного нанесения металла на второй стадии. На первой стадии, дезактивированный твердый катализатор подвергают деполимеризации для удаления полимеров с твердого катализатора и кондиционированию поверхности путем сушки. На второй стадии, осуществляют повторное нанесение компонента активного металла для компенсации потери металла. Реактивация дезактивированного катализатора будет обсуждена более подробно позже.
С точки зрения реактивации и/или регенерации катализатора, целесообразно иметь несколько реакторов, соединенных параллельно, для того чтобы осуществлять непрерывное производство продукта во время проведения процессов реактивации и восстановления каталитической активности катализаторов.
Как описано выше, раскрываемые изобретением процессы алкоголиза, переэтерификации, и диспропорционирования на твердом катализаторе могут включать введение реагентов и следового количества растворимых соединений активного металла в реактор, содержащий твердый катализатор, и контактирование реагентов в присутствии твердого катализатора для алкоголиза, переэтерификации, или диспропорционирования, по меньшей мере, части реагентов. Такие процессы алкоголиза, переэтерификации, или диспропорционирования могут включать, наряду с другими реакциями, например, реакции получения диалкилкарбонатов, диарилкарбонатов, алкиларилкарбонатов, биодизельных топлив, органических эфиров и N-арилалкилкарбаматов.
Выше были в общих чертах описаны реакции алкоголиза, переэтерификации и диспропорционирования, а ниже подробно описывается применение этих процессов в производстве органических карбонатов. Как уже было отмечено выше, в патентных документах U.S. Patent Application Publications 2007/0093672 ('672) и 2007/0112214 ('214) раскрыты способы получения органических карбонатов с использованием гетерогенных катализаторов. Содержание каждого из них приводится в описании изобретения путем ссылки на эти патентные документы.
Получение органического карбоната и органического карбамата
Органические карбонаты или органические карбаматы можно непрерывно получать путем использования систем с одним или несколькими реакторами в присутствии твердого катализатора или двух различных твердых катализаторов. Твердый катализатор или твердые катализаторы требуют добавления следового количества растворимого соединения активного металла в водимый в реактор поток сырья для увеличения времени рабочего цикла катализатора. Твердые катализаторы могут находиться в любой физической форме и могут включать различные металлоорганические соединения, иммобилизируемые на пористых носителях, и/или оксиды, содержащие элемент или несколько элементов Группы II, III, IV, V и VI, нанесенные на подходящий пористый носитель. Катализаторами могут являться либо кислотные катализаторы, либо щелочные катализаторы. Суммарное количество каталитически активного металла или компонентов металла в нанесенном катализаторе, в некоторых вариантах осуществления, может составлять приблизительно от 0,02 масс.% до приблизительно 20 масс.%; в других вариантах осуществления, приблизительно от 0,05 масс.% до приблизительно 10 масс.%. Другим типом твердых каталитических материалов, применяемых в раскрываемых в изобретении вариантах осуществления, являются материалы типа металл - органические каркасы (MOFs), которые включают один или более элементов из Групп II-VI и органические каркасы. Согласно различным вариантам осуществления, MOFs могут служить как в качестве твердых катализаторов, так и в качестве носителей для катализаторов.
Реакторы, используемые в раскрываемых в изобретении вариантах осуществления, могут включать любые физические устройства или комбинацию двух или более устройств. Реакторы могут иметь различные внутренние устройства для разделения в системе пар-жидкость и перемещения пара/жидкости.
В результате добавления следового количества растворимого соединения активного металла в вводимый поток сырья, активность катализатора может поддерживаться в течение чрезвычайно длительного времени рабочего цикла. Например, добавление следовых количеств растворимого соединения активного металла в потоки, вводимые в реактор с неподвижным слоем для получения смесей этилфенилкарбоната и дифенилкарбоната, может позволять достигать продолжительности рабочего цикла более чем 14 месяцев. Такая стабильная работа катализатора может в результате приводить к более высокой производительности по требуемому продукту. В вариантах осуществления, в которых применяют несколько последовательно соединенных реакторов, следовое количество компонента активного металла может быть добавлено только в поток, вводимый в первый реактор. В случае системы с несколькими соединенными параллельно реакторами, следовое количество компонента активного металла может быть добавлено во все реакторы.
Компоненты активных металлов могут включать соединение или смесь соединений, содержащих один или более металлов Группы II, III, IV, V и VI Периодической таблицы элементов. Примеры активных металлов включают Mg, Ca, Zn, La, Ac, Ti, Zr, Hf, V, Nb, Ta, Cr, Nb, W, Sn, Pb, Sb и другие металлы. Соединение активного металла должно быть растворимо в реакционной смеси или, по меньшей мере, должно образовывать эмульсию/коллоидный раствор. Количество следов металла во вводимом потоке может быть достаточно низким, для того чтобы не было необходимости с экономической точки зрения в извлечении металла из технологического потока для его рециркуляции, хотя может быть выбран и вариант извлечения металла.
В случае необходимости, дезактивированный в реакторе катализатор может быть реактивирован прямо в реакторе в течение относительно короткого промежутка времени, для того чтобы быть готовым для замены другого работающего реактора или начать работу. Поэтому, в зависимости от продолжительности рабочего цикла катализатора и других факторов, варианты осуществления раскрываемых в изобретении способов могут требовать наличие запасного реактора.
Раскрываемые в изобретении способы могут, в частности, применяться для непрерывного получения диарилкарбонатов, таких как дифенилкарбонат (ДФК), алкиларилкарбонатов, таких как этилфенилкарбонат (ЭФК), или диалкилкарбонатов, таких как диэтилкарбонат (ДЭК) или диметилкарбонат (ДМК). Реакция получения диарилкарбоната может быть проведена в нескольких реакционных зонах, например, в первой и второй реакционных зонах. Первая реакционная зона служит в основном для проведения переэтерификации диалкилкарбоната с ароматическим спиртом с получением алкиларилкарбоната, хотя может также образовываться небольшое количество диарилкарбоната. Вторая реакционная зона служит для проведения диспропорционирования алкиларилкарбоната с получением диарилкарбоната и диалкилкарбоната. Присутствие твердого катализатора во второй реакционной зоне не является обязательным, хотя может быть выбрано и использование твердого катализатора.
Диалкилкарбонаты, такие как ДМК или ДЭК, могут быть получены путем проведения переэтерификации циклического карбоната, такого как пропиленкарбонат или этиленкарбонат, с метанолом или этанолом подобным образом. Реакции получения диарилкарбоната и диалкилкарбоната проводят в системе с несколькими реакторами и с оборудованием для разделения и извлечения продуктов из реакционных смесей. Непрореагировавшие реагенты и промежуточные соединения могут быть извлечены для рециркуляции или подвергнуты конечной обработке путем проведения второго диспропорционирования или второй переэтерификации. Непрореагировавший фенол в жидкой реакционной смеси из зоны переэтерификации может быть отделен либо до проведения диспропорционирования алкилфенилкарбоната, либо после проведения диспропорционирования. Кроме того, существуют различные варианты удаления побочного продукта алкилфенилового эфира из реакционной системы путем продувки. Соответствующая компоновка реакторов с установками для разделения материалов хорошо известна обычным специалистам в этой области.
Для сдвига равновесия в требуемом направлении, реакции предпочтительно проводить в системе со смешанными фазами, в которой реагенты и продукты находится в жидком и парообразном состоянии. В качестве варианта, реакция может быть осуществлена, например, в жидкой фазе, когда сдвиг равновесия не дает никакого положительного эффекта или он очень небольшой вследствие того, что температуры кипения реакционных продуктов являются более высокими, чем предпочтительный интервал температур для проведения реакции.
Раскрываемые в изобретении варианты осуществления могут также применяться при получении органических карбонатов, таких как этилфенилкарбонат, метилфенилкарбонат, и дифенилкарбонат, путем проведения переэтерификации диалкилкарбонатов, таких как диэтилкарбонат или диметилкарбонат, с фенолом, и диспропорционирования алкиларилкарбоната, такого как этилфенилкарбонат или метилфенилкарбонат, с получением дифенилкарбоната.
Раскрываемые в изобретении варианты осуществления могут также применяться при получении диалкилкарбоната, такого как диметилкарбонат или диэтилкарбонат, путем переэтерификации циклического карбоната со спиртом. В других вариантах осуществления получения диалкилкарбонатов, диалкилкарбонат может быть получен путем алкоголиза мочевины со спиртом в присутствии твердого катализатора. Например, в Патенте США № 7074951, диалкилкарбонат получают путем использования гомогенного катализатора из оловоорганического комплекса в присутствии высококипящего электродонорного растворителя; такой способ может быть осуществлен на твердых катализаторах согласно раскрываемым в изобретении вариантам осуществления. Различные органические карбаматы, такие как N-арилалкилкарбамат, могут быть также эффективно получены путем реакции диалкилкарбоната с ароматическим амином в присутствии твердого катализатора согласно раскрываемым в изобретении вариантам осуществления.
Для проведения описанных в изобретении реакций может быть использован любой тип реакторов. Примеры реакторов, подходящих для проведения реакций, относящихся к реакциям органического карбоната или органических карбаматов, могут включать реакторы с дистилляционной колонной, реакторы с дистилляционной колонной с разделительной стенкой, традиционные трубчатые реакторы с неподвижным слоем, барботажные реакторные колонны, суспензионные реакторы, оборудованные дистилляционной колонной или без дистилляционной колонны, реакторы с пульсирующим потоком, каталитические дистилляционные колонны, в которых суспензия твердых катализаторов стекает в колонне сверху вниз, или любую комбинацию этих реакторов.
Многореакторные системы, применяемые в раскрываемых в изобретении вариантах осуществления, для первой реакционной зоны могут включать несколько реакторов, соединенных последовательно, или несколько реакторов, соединенных параллельно. Если продукт получают из реагентов через образование промежуточного соединения, такого как алкиларилкарбонат, первая реакционная зона может служить, главным образом, для получения промежуточного соединения, хотя одновременно в первой реакционной зоне может образовываться и незначительное количество конечного продукта реакции.
Технологический поток из первой реакционной зоны, после отгонки спирта и диалкилкарбоната, поступает во вторую реакционную зону, в которой получают диарилкарбонат вместе с побочным продуктом диалкилкарбонатом. При отгонке реакционного продукта с более низкой температурой кипения из каталитической реакционной зоны, одновременно может быть проведена переэтерификация, что приводит к сдвигу равновесия в сторону прямой реакции.
Реакции, с помощью которых получают органические карбонаты или органические карбаматы, обычно проводят, в некоторых вариантах осуществления, при температуре в интервале приблизительно от 104°C до приблизительно 260°C (приблизительно от 220°F до приблизительно 500°F); в других вариантах осуществления, приблизительно от 121°C до приблизительно 232°C (приблизительно от 250°F до приблизительно 450°F). Давление, при котором проводят реакцию, зависит от температур кипения реагентов и продуктов, типа используемого реактора и от того, присутствует ли в реакционной зоне жидкая фаза или двойная фаза (пар/жидкость). Обычно, давление в реакторе может находиться, в некоторых вариантах осуществления, в интервале от давления ниже атмосферного до приблизительно 22 бар (приблизительно 319 фунт/дюйм2); и в других вариантах осуществления, приблизительно от 0,005 бар до приблизительно 17 бар (приблизительно от 0,1 фунт/дюйм2 до приблизительно 250 фунт/дюйм2). В классе вариантов осуществления, реакции могут проводиться с использованием подходящего растворителя, который отрицательно не влияет на процесс разделения продуктов реакции.
В выбранных вариантах осуществления, раскрываемые в изобретении варианты осуществления применяются, в частности, для непрерывного получения диарилкарбонатов из диалкилкарбоната и ароматического гидроксисоединения, например, получения дифенилкарбоната (ДФК) из диалкилкарбоната и фенола. Одним направлением получения ДФК является реакция диэтилкарбоната (ДЭК) с фенолом в присутствии одного или более твердых катализаторов. Преимущества получения ДФК путем использования ДЭК могут включать экономию энергии и экономию конструкционного материала при сооружении установки, так как нет необходимости в выделении веществ из азеотропной смеси. Для получения всех материалов необходимы затраты энергии. Поэтому, считается, что экономия конструкционных материалов и энергии является характеристиками "экологически чистого производства". В отличие от этого, в современном промышленном нефосгеновом способе получения ДФК используют ДМК в качестве одного из исходных веществ. ДМК и метанол необходимо выделять из технологического потока, образующего азеотропную смесь, путем экстрактивной дистилляции с растворителем. Эксплуатация установок экстрактивной дистилляции является энергоемкой. Несмотря на то, что получение ДФК через ДМК является возможным, тем мне менее, использование ДЭК может быть предпочтительным вследствие экономии энергии и материалов.
Раскрываемые в изобретении варианты осуществления могут также применяться для получения диалкилкарбонатов путем переэтерификации циклического карбоната со спиртом, таким как этанол или метанол.
Получение ДФК из диалкилкарбоната и фенола включает две реакционные стадии; переэтерификацию в первой реакционной зоне и затем диспропорционирование во второй реакционной зоне. Реакции могут быть описаны следующими уравнениями:
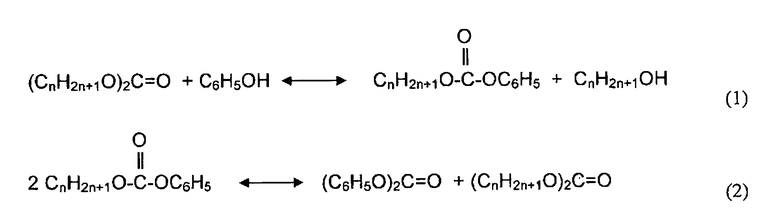 ,
,
где суммарная реакция может быть записана как:

Реакцией (1) является переэтерификация диалкилкарбоната с фенолом с образованием алкилфенилкарбоната и спирта. Реакция (2) включает диспропорционирование алкилфенилкарбоната с образованием дифенилкарбоната и диалкилкарбоната. Обе стадии реакции являются обратимыми. Однако диспропорционирование является значительно более термодинамически выгодной реакцией, чем переэтерификация. Переэтерификацию проводят главным образом в первой реакционной зоне, которая может включать один единственный реактор или многореакторную систему. Реакция диспропорционирования может быть в основном проведена во второй реакционной зоне.
Получение диалкилкарбоната путем переэтерификации циклического карбоната со спиртом также является двухстадийной обратимой реакцией. Для переэтерификации циклического карбоната со спиртом могут быть использованы как кислотные, так и щелочные катализаторы.
Согласно раскрываемым в изобретении вариантам осуществления, для достижения увеличенной продолжительности рабочего цикла катализатора, во вводимый в реактор сырьевой поток добавляют следовое количество растворимого соединения металла. Для переэтерификации циклического карбоната со спиртом с получением диалкилкарбоната и диола, могут быть использованы либо твердый щелочной, либо кислотный катализатор. Переэтерификация может быть также проведена путем замены части спирта водой. В качестве варианта, переэтерификация может быть проведена на первой стадии, а затем на второй стадии может быть проведена реакция непрореагировавшего циклического карбоната и промежуточного продукта со смесью вода-спирт с получением гликоля в качестве основного продукта реакции. Добавление воды существенно повышает конверсию циклического карбоната или производительность по диолу. Однако преимущество добавления воды выражается только в пониженном выходе диалкилкарбоната.
Катализаторы, применяемые для получения органического карбоната и органического карбамата
Описанные выше катализаторы, применяемые для получения органического карбоната и органического карбамата, могут включать нанесенные на носитель твердые катализаторы, имеющие один или более активных металлов из Групп II, III, IV, V и VI Периодической таблицы элементов. Один тип катализатора, применяемого в раскрываемых в изобретении вариантах осуществления, включает металлоорганическое соединение или несколько металлоорганических соединений упомянутых выше элементов, иммобилизируемых на пористом носителе. Пористые носители, применяемые в раскрываемых в изобретении вариантах осуществления, могут включать поверхностные функциональные группы, такие как гидроксильные группы, алкоксильные группы, смесь гидроксильных и алкоксильных групп, хлор, и другие группы. Примеры носителей могут включать оксид кремния, оксид кремния-оксид алюминия, оксид титана, оксид циркония, или цеолитные материалы, такие как MCM-41, MCM-48, SBA-15, и другие, и композитные материалы, включающие связующее вещество и цеолит.
Другие возможные носители могут включать углерод и/или углеродистые материалы. Углеродные и углеродистые носители могут иметь поверхностные функциональные группы, такие как гидроксильные, карбонильные, или и те, и другие, для удержания металлоорганических соединений на поверхности, как это уже было обсуждено ранее. Для приготовления нанесенных на носитель металлоксидных, гидроксидных, или оксигидроксидных катализаторов, может не быть необходимости в присутствии поверхностных функциональных групп, хотя в некоторых вариантах осуществления они могут быть использованы. Углеродистые носители могут быть приготовлены путем контролируемой термической дегидратации углеводов, таких как древесина, скорлупа кокосового ореха, крахмалы, целлюлоза, смеси крахмала и целлюлозы, сахара, метилцеллюлоза и другие подобные материалы, при повышенных температурах. Углеродистые носители могут быть нанесенными или нанесенными. Для приготовления нанесенного углеродистого материала, углеводы могут быть нанесены на подходящий пористый носитель и затем подвергнуты контролируемой термической гидратации при повышенной температуре, например, температуре в интервале приблизительно от 250°C до 1000°C, в инертной атмосфере или в атмосфере, состоящей из инертного газа и небольшого количества кислорода, водяного пара или их смеси. Носители для углеродистых материалов могут включать любые неорганические материалы, такие как оксид алюминия, оксид титана, оксид кремния, оксид циркония, синтетические и природные глины, включая оксиды кремния-оксиды алюминия, и другие известные в технике носители.
Носители, в некоторых вариантах осуществления, могут требовать удаления конденсированной воды в порах перед контактированием металлоорганических соединений с носителями с целью их иммобилизации на поверхности носителей. Под конденсированной водой на носителе в описании изобретения подразумевают то количество воды, которое может быть удалено путем сушки носителя при температуре в интервале приблизительно от 50°C до приблизительно 400°C в токе сухого газа или под вакуумом, в зависимости от химического состава носителя. Используемые в изобретении твердые катализаторы могут быть приготовлены путем иммобилизации одного или двух металлоорганических соединений или путем реакции одного или нескольких растворимых соединений металла с поверхностными функциональными группами носителя, что приводит к созданию активных каталитических мест на пористом твердом носителе. Иммобилизация может быть осуществлена, например, используя такие методы, как прививка, связывание, адсорбция, и так далее. Например, методы приготовления катализатора с использованием металлоорганических соединений, таких как алкоксиды титана, на пористых носителях были раскрыты в публикации '672.
Второй тип катализатора, применяемого в раскрываемых в изобретении вариантах осуществления, включает оксид металла, смешанные оксиды металлов, или оксигидроксиды, нанесенные на пористый носитель. Примеры этого типа катализатора также раскрываются в публикации '672.
Носители могут быть в форме таблеток, экструдатов, сфер, гранул, сот и в других подобных формах, с размерами приблизительно от 1 мм до приблизительно 5 мм для различных реакторов с неподвижным слоем. В качестве варианта, для использования в качестве носителя может быть выбрана ткань или сетка, изготовленная из стекловолокна или углеродного волокна или из того, и другого, вместе со структурированными насадочными материалами, которым соответствующим образом придают требуемые формы и размеры в зависимости от типа реакторов. Носители в порошковой форме или в форме микросфер могут быть также использованы для приготовления катализаторов, применяемых в суспензионном реакторе или в реакторе с мешалкой.
Приготовление описанных выше катализаторов второго типа может не требовать носителя, имеющего поверхностную гидроксильную группу. Однако носители, содержащие поверхностные функциональные группы, такие как оксид кремния, углеродистый материал, оксид алюминия, и другие носители, могут быть также использованы для приготовления катализатора гидроксид металла/оксид путем прививки алкоксидов металла, таких как алкоксид титана, на оксид кремния, и затем путем обработки паром или гидролиза и/или сушки при температуре приблизительно от 90°C до приблизительно 500°C.
Другой метод приготовления металлоксидных или оксигидроксидных катализаторов включает нанесение соли требуемого элемента или смеси солей двух различных элементов на носитель с последующим прокаливанием при температуре от 300°C до 1000°C для разложения солей до оксидов металлов.
При условиях проведения процесса, переэтерификация и диспропорционирование в каталитической реакционной зоне могут происходить одновременно, по мере того как возрастает концентрация алкиларилкарбоната в реакционной среде. Вымывание и отложение полимера, две обсуждаемые выше причины дезактивации катализатора, также происходят одновременно в условиях осуществления реакции. В то время как отложение полимера не приводит к постоянному повреждению катализатора, вымывание компонента активного металла из гетерогенных катализаторов в условиях осуществления реакции реально приводит к постоянному повреждению катализатора. При низких степенях конверсии при переэтерификации или при низких концентрациях алкиларила и диарилкарбонатов, дезактивация катализатора вызывается главным образом в результате вымывания из твердого катализатора компонентов активного металла катализатора в реакционную среду. Другими словами, причиной постоянной дезактивации катализатора при всех условиях осуществления реакции является вымывание металла.
По мере того как степень конверсии в процессе переэтерификации возрастает, отложение полимера на катализаторе приводит к еще более быстрой дезактивации катализатора. Отложение полимера является, в основном, результатом протекания нежелательных побочных реакций алкиларила и диарилкарбонатов (и вероятно следовых количеств примесей полигидроксилароматических соединений во вводимом феноле и образующихся в незначительных количествах в результате побочных реакций). Поэтому, для непрерывного получения дифенилкарбоната из фенола и диалкилкарбоната, например, диэтилкарбоната или диметилкарбоната, в присутствии гетерогенных катализаторов, может возникать необходимость решения проблемы дезактивации катализаторов, вызываемой как (1) отложением полимера, так и (2) выщелачиванием/вымыванием компонентов активного металла катализатора. Проблема отложения полимера может решаться путем упоминаемого выше регулирования конверсии, концентрации ароматических карбонатов, или и того и другого, в каталитической реакционной зоне, и путем реактивации катализатора, например, раскрытой в публикации '672. Проблема вымывания решается путем добавления следового количества описанного выше растворимого металлоорганического катализатора.
Иммобилизация (например, прививка, связывание, адсорбция и так далее) металлоорганических соединений или растворимых соединений металлов на носителе, таком как оксид кремния или углеродистый материал, для алкоголиза и/или переэтерификации диалкилкарбоната с фенолом может быть осуществлена на одной стадии в реакционной зоне, или на нескольких стадиях в реакционной зоне. Примеры раскрываемых металлоорганических соединений включают алкоксиды металлов, алкоксихлориды, оксиалкоксиды, карбоксилаты, карбонаты и другие соединения элементов Группы II, III, IV, V и VI. Примеры активных металлов включают Mg, Ca, Zn, La, Ac, Ti, Zr, Hf, V, Nb, Ta, Cr, Mo, W, Sn, Pb, Sb и другие металлы. Различные варианты осуществления включают алкоксиды олова, алкоксиды алкилолова, оксиды алкилолова, гидроксиды алкилолова, дихлорид диалкилолова, трихлорид алкилолова и смеси этих соединений, а также оксиалкоксиды металлов [(RO)nMO] и алкоксигидроксиды металлов [(RO)nM(OH)x] или олигомеры этих оксиалкоксидов и алкоксигидроксидов, где M является элементом Группы IV, V или VI, n=2, 3 или 4, x=0, 1, 2 или 3, и n+x=4, 5 или 6. В выбранных вариантах осуществления, металлоорганическим соединением может являться один или более из алкоксидов или феноксидов титана, алкиларилтитанатов, или титановых солей моноэфиров угольных кислот. Следует иметь в виду, что алкоксиды металлов включают мономеры, различные олигомеры, или смесь различных мономерных и олигомерных соединений, зависящих от длины углеродной цепи и структуры алкильной группы алкоксида или арилоксида [смотрите, например, публикацию Coordin. Chem. Rev., 2 (1967) 299-318; J. Chem. Soc, 3977 (1955)].
Описываемые в изобретении алкоксиды переходных металлов включают все соединения мономера и различные олигомеры. Например, в то время как этоксид титана [Ti(OEt)4] существует в основном в виде тримера в кипящем этаноле или бензоле, стерически затрудненные алкоксиды титана, такие как изопропоксид титана, являются мономерными в кипящих углеводородных растворах. Например, считается, что изопропоксид титана существует в основном в виде мономера в кипящем толуольном растворе.
Пористые носители, применяемые в различных раскрываемых в изобретении вариантах осуществления, могут иметь поверхностные гидроксильные группы, алкоксильные группы, или и те, и другие. Для приготовления пористого носителя, пористые металлоксидные носители, такие как оксид титана, оксид циркония, оксид молибдена и оксид ванадия, могут быть подвергнуты обработке с помощью потока, содержащего один или более спирт, органический карбонат, такой как диметилкарбонат, диэтилкарбонат, и так далее, в некоторых вариантах осуществления при температуре, в интервале приблизительно от 130°C до приблизительно 400°C, в других вариантах осуществления, от 150°C до 350°C, в паровой фазе, жидкой фазе, или системе пар-жидкость. Поток может содержать воду, в некоторых вариантах осуществления, от 0 масс.% до приблизительно 20 масс.%; в других вариантах осуществления, от 0 масс.% до приблизительно 10 масс.%; и в еще одних вариантах осуществления, приблизительно от 0,005 масс.% до приблизительно 5 масс.%. Так как вода имеет низкую растворимость в ДМК и ДЭК, поток может содержать соответствующие количества метанола и/или этанола в качестве растворителя для воды. В некоторых вариантах осуществления, могут быть использованы выпускаемый промышленностью силикагель или оксид кремния, имеющий поверхностные гидроксильные группы. Может быть проведена необязательная обработка оксида кремния с помощью воды, водяного пара или их смеси при температуре приблизительно от 80°C до приблизительно 500°C с последующей сушкой при температуре, в некоторых вариантах осуществления, приблизительно от 70° до приблизительно 800°C, и в других вариантах осуществления, приблизительно от 80°C до приблизительно 500°C.
Оксисилоксановые и силоксановые соединения переходных металлов, иммобилизированные на пористых носителях или металлоксидных катализаторах, описанных выше, могут также быть использованы для приготовления твердых катализаторов. Примерами оксисилоксановых и силоксановых соединений являются (RO)n-xM[-O-Si(O-R)3]x, M(O-SiR3)n, (R3SiO)n-2MO, и так далее, где каждый R является независимо алкильной или арильной группой, n=3, 4, или 5, x=1 или 2, n+x=4, 5 или 6, и M является описанным выше переходным металлом Группы IV, V и VI. Другие соединения кремний-металл входят в объем раскрываемых в изобретении вариантов осуществления, при условии, что их иммобилизация обеспечивает в результате каталитическую активность твердых катализаторов. Оксисилоксановые и силоксановые соединения переходных металлов могут быть также использованы в качестве растворимых металлоорганических катализаторов при аппаратурном оформлении способа, раскрытого в публикациях '672 и '214, а также в реакторах с реакционной дистилляционной колонной. Для приготовления иммобилизируемых твердых катализаторов могут быть также использованы различные олигомерные и полимерные гетерооксисилоксаны или гетеросилоксаны переходных металлов, или они могут быть использованы в различных вариантах осуществления в качестве растворимых металлоорганических катализаторов. Как было описано выше, диспропорционирование ЭФК или МФК в ДФК и ДЭК или ДМК может быть осуществлено в отсутствие твердого катализатора во второй реакционной зоне, и применяемые активные каталитические соединения включают оксисилоксановые или силоксановые соединения переходных металлов, таких как Ti.
Металлоксиды и алкоксиды силоксанов могут включать различные олигомеры. Сведения о различных олигомерах можно найти в публикациях D. C. Bradley, Coordin. Chem. Rev., 2 (1967) p.p. 299-318; J. Chem. Soc, (1955) 3977. Для достижения стабильной активности катализатора, для добавления во вводимый в реакционную зону поток можно выбрать следовое количество одного из этих соединений.
При проведении диспропорционирования алкиларилкарбонатов с получением диарилкарбоната и диалкилкарбоната во второй реакционной зоне в присутствии гомогенного катализатора, гомогенный катализатор может представлять собой смесь алкиларилтитанатов, титановых солей моноэфиров угольных кислот, и силоксановых соединений титана, рассмотренных выше. Следует иметь в виду, что гомогенные катализаторы могут образовываться из твердых катализаторов и растворимых катализаторов, используемых в реакционной зоне переэтерификации.
Так как различные раскрываемые в изобретении металлоорганические соединения чувствительны к воздействию влаги, присутствующей во вводимом потоке сырья, то важно контролировать содержание воды во вводимом в реакционную зону потоке сырья. В некоторых вариантах осуществления, содержание воды во вводимом потоке сырья составляет менее чем приблизительно 700 частей на миллион (ppm); в других вариантах осуществления, менее чем приблизительно 600 ppm. Твердый иммобилизированный на носителе металлалкоксидный катализатор может быть приготовлен непосредственно внутри реактора, или может быть приготовлен вне реактора. Для приготовления непосредственно внутри реактора, заданное количество подходящего носителя помещают в реактор, затем сушат при соответствующей температуре для удаления, по меньшей мере, части любой конденсированной воды. Затем носитель контактируют с раствором, содержащим растворимый алкоксид металла или смешанные алкоксиды переходных металлов или металлов, при температуре в интервале, в некоторых вариантах осуществления, приблизительно от температуры окружающей среды до приблизительно 260°C (500°F), и в других вариантах осуществления, приблизительно от 37°C до приблизительно 204°C (приблизительно от 100°F до приблизительно 400°F). Контактирование может быть осуществлено в течение периода времени, в некоторых вариантах осуществления, приблизительно от 5 минут до приблизительно 24 часов, и в других вариантах осуществления, приблизительно от 15 минут до приблизительно 15 часов, и этот период времени может зависеть от температуры и концентрации компонента активного металла в растворе. После удаления избытка раствора алкоксида металла из реактора, катализатор в реакторе может быть промыт с помощью растворителя (обычно такого же растворителя, который использовали для приготовления раствора алкоксида металла), перед его использованием в реакциях диспропорционирования или переэтерификации. Растворителем может являться спирт, эфир, углеводород, смесь углеводородов и спирта, или смесь диалкилкарбоната и фенола или спирта, или смеси всех этих веществ.
В качестве варианта, металлоксидные, смешанные металлоксидные, или металлгидроксидные катализаторы, в которых металлом является один или более металл из Групп II, III, IV, V и VI Периодической таблицы элементов, могут быть также использованы согласно раскрываемым в изобретении вариантам осуществления. В технике известен ряд металлоксидных катализаторов. Например, согласно публикации P. Iengo et al., Appl Catal. A: General 178 (1999) 97-109, катализаторы из оксида титана, нанесенные на оксид кремния, могут быть приготовлены путем прививки изопропоксида титана и затем путем обработки водяным паром/прокаливания, при этом нанесенный катализатор имеет сильно модифицированную поверхность исходного оксида кремния, что дает в результате катализатор, который отличается от катализаторов, получаемых путем импрегнирования и соосаждения.
Для приготовления нанесенных металлических или смешанных металлгидроксидных или металлоксигидроксидных катализаторов, можно подвергать гидролизу описанные выше привитые металлалкоксидные катализаторы и затем сушить при температуре в интервале приблизительно от 50°C до приблизительно 110°C. В некоторых вариантах осуществления, в сушке может не быть необходимости.
Перед осуществлением реакций получения органических карбонатов может быть проведено предварительное кондиционирование нанесенных металлоксидных катализаторов. Предварительное кондиционирование проводят путем контактирования пористых металлоксидных катализаторов, таких как оксид титана, оксид циркония, оксид молибдена, или оксид ванадия, с потоком, содержащим органический карбонат, такой как диметилкарбонат, диэтилкарбонат, и так далее, в некоторых вариантах осуществления, при температуре в интервале приблизительно от 125°C до приблизительно 450°C, и в других вариантах осуществления, приблизительно от 150°C до приблизительно 350°C, где органический карбонат может находиться в паровой фазе, жидкой фазе, или смешанной фазе. Предварительное кондиционирование может быть проведено в течение периода времени, в некоторых вариантах осуществления, приблизительно от 2 минут до приблизительно 50 часов, и в других вариантах осуществления, приблизительно от 4 минут до приблизительно 24 часов. Поток, содержащий органический карбонат, может включать воду и спирт, где вода может присутствовать, в некоторых вариантах осуществления, приблизительно больше чем от 0 масс.% до приблизительно 10 масс.%, и в других вариантах осуществления, приблизительно от 0,005 масс.% до приблизительно 4 масс.%. В результате предварительного кондиционирования может быть повышена селективность катализатора. После предварительного кондиционирования, металлоксидные катализаторы могут быть высушены при температуре приблизительно от 80°C до приблизительно 300°C в потоке инертного газа в течение периода времени приблизительно от 2 минут до приблизительно 6 часов.
Два типа смешанных металлоксидных катализаторов могут быть использованы для переэтерификации циклического карбоната со спиртом. Первый тип смешанных металлоксидных катализаторов может включать один или более элементов из Групп III, IV, V, и VI Периодической таблицы элементов, нанесенных на носитель. Второй тип смешанных металлоксидных катализаторов включает твердый щелочной катализатор, который содержит один или два элемента из Группы II Периодической таблицы элементов и лантаниды или актиниды на носителе. Необязательно, но можно использовать гидроксид четвертичного аммония, привитый на носитель из оксида кремния или связанный с носителем из оксида кремния. Оксидные катализаторы обычно наносят на оксид алюминия или оксид кремния, или приготавливают в форме смешанного оксида или твердого раствора. Элементы, применяемые для второго типа твердых катализаторов, могут включать Mg, Ca, Zn, La, и так далее.
Компоненты активных металлов второго типа катализаторов могут также вымываться в условиях реакции переэтерификации, что приводит к дезактивации катализатора. К тому же было обнаружено, что носители из оксида кремния могут также подвергаться вымыванию, только значительно более медленно, чем компоненты активных металлов Группы II. Так как примеси щелочных металлов в носителе из оксида кремния могут увеличивать вымывание оксида кремния в реакционную среду, весьма желательно, чтобы содержание примесей щелочных металлов в носителе из оксида кремния было минимальным. Путем добавления следового количества растворимых металлоорганических катализаторов в вводимый поток сырья, может быть увеличена продолжительность рабочего цикла твердого катализатора в реакторах с неподвижным слоем. Примеры таких растворимых соединений включают 2-метоксиэтоксид цинка, 2-метоксиэтоксид кальция, 2-метоксипропоксид цинка, этоксид цинка, алкоксиалкилкарбонат цинка, 2-метоксипропоксид кальция, этоксид кальция, алкоксиалкилкарбонат кальция, 2-метоксиэтоксид магния, 2-метоксипропоксид магния, этоксид магния, бутоксид магния, алкоксиалкилкарбонат магния, алкоксид лантана, алкоксиалкилкарбонат лантана, и наряду с другими, пропиленглицериды или гликоляты Mg, Ca и Zn. Может быть также использована смесь этих соединений.
Растворимые соединения Ca, Mg, Zn и La могут быть получены путем реакции оксидов или гидроксидов этих металлов со спиртом, органическим карбонатом, или смесью органического карбоната и спирта при температуре, в некоторых вариантах осуществления, приблизительно от 105°C (221°F) до приблизительно 260°C (500°F), в других вариантах осуществления, приблизительно от 149°C (300°F) до приблизительно 227°C (440°F), в системе с жидкой фазой или со смешанной фазой (жидкость и пар). Приготовленные таким способом растворы могут быть использованы для добавления следовых количеств этих металлов во вводимый в реактор поток сырья с целью увеличения времени рабочего цикла катализатора. Суммарное количество активного металла или компонентов металла на твердом металлалкоксидном, металлгидроксидном, или металлоксидном катализаторе может составлять приблизительно от 0,02 масс.% до приблизительно 20 масс.%, предпочтительно, приблизительно от 0,05 масс.% до приблизительно 12 масс.%.
Увеличенная продолжительность рабочего цикла катализатора и срока его службы
Раскрываемые в изобретении твердые катализаторы могут иметь продолжительные рабочие циклы и могут быть подвергнуты многократной регенерации и реактивации, которые в результате приводят к длительным срокам службы катализатора. Раскрываемые в изобретении методы увеличения продолжительности рабочего цикла и реактивации катализатора позволяют применять катализаторы, которые обычно представляют интерес только для лабораторных синтезов, для промышленного получения различных органических карбонатов. Предполагается, что, вне зависимости от того, являются ли катализаторы в исходном состоянии нанесенными металлоксидными катализаторами, или металлалкоксидными катализаторами, иммобилизированными на оксиде кремния, при стационарном режиме активными катализаторами являются металлоорганические соединения, иммобилизированные на оксиде кремния. Для иллюстрации преимущества добавления во вводимое сырье следового количества активных металлов, были проведены различные эксперименты, которые будут более подробно описаны ниже. Вкратце, в одном эксперименте, катализатор из оксида титана (6 масс.% Ti), нанесенного на силикагель, подвергали регенерации путем деполимеризации после эксплуатации в течение приблизительно 350 часов, и восстанавливали меньше чем наполовину его исходную активность при переэтерификации диэтилкарбоната с фенолом. Было обнаружено, что больше половины количества Ti было вымыто из катализатора в реакционную среду в процессе его эксплуатации. В другом эксперименте, катализатор из бутоксида титана (4 масс.% Ti), привитый на силикагеле, потерял больше 90% Ti после эксплуатации в течение только 171 часа при диспропорционировании этилфенилкарбоната. После эксплуатации в течение 173 часов, другой катализатор из оксида титана (5,7 масс.%), нанесенного на силикагель, использованный для переэтерификации пропиленкарбоната с этанолом для получения диэтилкарбоната и пропиленгликоля, потерял 35% количества Ti на катализаторе. Из этих результатов совершенно очевидно, что как нанесенный катализатор из оксида титана, так и привитый катализатор из алкоксида титана, не пригодны для применения в промышленных реакторах для непрерывного получения органических карбонатов, таких как диалкилкарбонат, алкилфенилкарбонат и диарилкарбонат, вследствие постоянной дезактивации при коротком сроке службы. Судя по всему, органические карбонаты и/или реакционные смеси являются достаточно реакционно-способными для того, чтобы вызывать медленное образование растворимых металлоорганических катализаторов в реакционной среде в результате реакции с твердыми катализаторами.
Было также установлено, что потоки паров ДМК и ДЭК могут реагировать с оксидом кремния или оксидом титана с образованием тетраалкилортооксида кремния и тетраалкоксида титана при температуре приблизительно выше чем 350°C. Реакции ДМК и ДЭК с оксидом кремния протекают легче в присутствии каталитического количества щелочного металла в оксиде кремния. Поэтому, было необходимо разработать простой метод реактивации катализатора и метод поддержания постоянных поверхностных концентраций компонентов активного металла на катализаторе в течение достаточно длительного времени рабочего цикла, приемлемого для промышленных реакторов.
Регенерация путем деполимеризации и повторного нанесения металла решает проблемы, связанные с отложением полимера. Однако регенерация путем деполимеризации не может решить проблемы, связанные с непрерывным вымыванием активных металлов из гетерогенных катализаторов в условиях проведения реакции. Для достижения длительного рабочего цикла катализатора, приемлемого для крупномасштабных промышленных реакторов, должна быть решена проблема непрерывной потери активного металла гетерогенными катализаторами. Было обнаружено, что эффект вымывания металла из гетерогенных катализаторов может быть компенсирован в случае многореакторной системы с последовательным соединением реакторов путем добавления следовых количеств соединений активного металла в поступающий поток (потоки) в первый реактор. В результате добавления следового количества растворимого соединения активного металла, вымывание металла и его повторное нанесение взаимно уравновешиваются или почти уравновешиваются, что эффективно способствует поддержанию постоянного числа активных центров на твердых катализаторах, приводя к стабильной активности катализатора в течение длительного времени рабочего цикла катализатора. Следует иметь в виду, что растворимый компонент металла, вымываемый из твердого катализатора в первом реакторе, является смесью различных видов соединений металла. Виды соединений металла в смеси не являются обязательно одинаковыми для видов соединений металла, поступающих в первый реактор. Вымывание металла и повторное его нанесение взаимно уравновешиваются точно так же во втором реакторе. Для многореакторных систем с параллельным соединением реакторов, может потребоваться добавление следового количества соединения активного металла во все входящие потоки в первую основную реакционную зону. Поэтому, реактивация катализатора (деполимеризация/кондиционирование поверхности и повторное нанесение металла непосредственно в реакторе) и добавление следового количества соединения активного металла и затем повторное нанесение металла могут решить проблему, как вымывания металла, так и отложения полимера. Реактивация катализатора может быть проведена в две стадии: (1) деполимеризация/кондиционирование поверхности катализатора и (2) повторное нанесение компонентов активных металлов. Кондиционирование поверхности катализатора является необходимым для иммобилизации алкоксида титана на поверхности носителя из оксида кремния. Свежий повергнутый иммобилизации или реактивированный катализатор непрерывно теряет каталитическую активность, что приводит к неприемлемому короткому времени рабочего цикла, которое не удовлетворяет требованиям для крупнотоннажных промышленных реакторов. Падение исходной активности катализатора почти в два раза при переэтерификации диалкилкарбоната с фенолом происходит в течение 80-150 часов рабочего цикла, что, несомненно, не отвечает требованиям для промышленного реактора, работающего в непрерывном режиме. Путем добавления следовых количеств растворимого соединения активного металла и проведения двухстадийной реактивации, можно увеличить продолжительность рабочего цикла катализатора и проводить реактивацию катализатора большое количество раз при непрерывном получении различных органических карбонатов.
Деполимеризация дезактивированного катализатора может быть осуществлена путем контактирования катализатора с потоком, содержащим гидроксисоединение или смесь гидроксисоединений, непосредственно в реакторе при температуре, в некоторых вариантах осуществления, от 102°C (215°F) до 316°C (600°F), в других вариантах осуществления, от 104°C (220°F) до 232°C (450°F), в течение периода времени, в некоторых вариантах осуществления, приблизительно от 10 минут до приблизительно 50 часов, и в других вариантах осуществления, от 30 минут до 15 часов. Деполимеризация может быть проведена в паровой фазе, в жидкой фазе, смешанной фазе, или сначала в жидкой фазе, а затем в паровой фазе, или в обратном порядке. Продукты деполимеризации могут включать фенол, спирт, диоксид углерода, полигидроксибензол, диалкилкарбонат, алкилфенилкарбонат, и соединения с большой молекулярной массой.
Примерами гидроксисоединений, используемых для деполимеризации на катализаторе, являются спирты (предпочтительно, метанол или этанол), вода, или их смесь. Если используют диметилкарбонат в качестве одного из исходных реагентов при получении метилфенилкарбоната и дифенилкарбоната, для деполимеризации могут быть использованы метанол или смесь воды и метанола. Если в качестве одного из исходных реагентов используют диэтилкарбонат, для деполимеризации могут быть использованы этанол или смесь воды и этанола. Может быть также использована смесь метанола и этанола. При использовании для деполимеризации воды, содержание воды в смеси, в некоторых вариантах осуществления, может находиться в интервале от больше чем ноль масс.% до меньше чем 100 масс.%; в других вариантах осуществления, от 10 ppm по массе до 15 масс.%; и в еще одних вариантах осуществления, от 15 ppm по массе до 5 масс.%. Азеотропная смесь воды (4 масс.%) и этанола является очень эффективной для деполимеризации, в случае когда в качестве одного из исходных реагентов используют диэтилкарбонат. В некоторых вариантах осуществления, смесь воды и спирта может быть предпочтительней, чем только одна вода или спирт, в силу того, что эта смесь позволяет осуществлять и кондиционирование поверхности катализатора для повторного нанесения компонента активного металла. Кроме того, смесь воды и спирта может быть более эффективной для деполимеризации и кондиционирования поверхности, чем только один спирт или вода.
При деполимеризации может использоваться растворитель. Подходящие растворители могут включать бензол, толуол, ксилолы, пентан, гексан, октан, декан, тетрагидрофуран, эфир, и другие растворители, или любые смеси этих растворителей. Концентрация растворителя в смеси при деполимеризации может составлять от 0 масс.% до приблизительно 90 масс.%.
После деполимеризации, катализатор может быть подвергнут сушке для удаления избытка воды на катализаторе и регулирования количества поверхностных гидроксильных групп перед повторным нанесением компонента активного металла на катализатор. Сушка непосредственно в реакторе может быть проведена, в некоторых вариантах осуществления, при температуре приблизительно от 49°C (120°F) до приблизительно 427°C (800°F), в других вариантах осуществления, от 65°C (150°F) до 316°C (600°F), в потоке инертного газа, в течение периода времени приблизительно от 15 минут до 40 часов при давлении окружающей среды или при давлении ниже атмосферного, перед проведением повторного нанесения активного компонента катализатора. Неудовлетворительное предварительное кондиционирование поверхности катализатора может приводить только к частичному восстановлению активности катализатора. Раскрываемый в изобретении метод деполимеризации может быть использован в любом способе получения ароматических карбонатов или для любой реакции, в которой органические карбонаты принимают участие в качестве реагентов, продуктов, или и тех, и других.
Раскрываемый в изобретении метод деполимеризации может быть также использован для реакций получения органических карбонатов в присутствии гомогенного катализатора. Для регенерации гомогенной каталитической системы, раствор спирта должен быть практически сухим, так чтобы содержание воды не могло превышать приблизительно 0,01 масс.%. Поэтому, раскрываемый в изобретении метод регенерации катализатора может быть использован для любого способа получения органических карбонатов.
Выходящий из реактора поток в процессе деполимеризации может содержать следовое количество компонента активного металла, в зависимости от того, как осуществляют деполимеризации. Этот поток может также содержать в качестве основных продуктов деполимеризации фенол, ДЭК, небольшие количества фенетола, ЭФК и соединения с более высокими молекулярными массами. В случае необходимости, можно попытаться извлечь из этого потока полезные компоненты, такие как фенол, этанол, алкилфенилкарбонат и ДЭК.
Повторное нанесение компонента активного металла на подвергнутую деполимеризации и кондиционированию поверхность носителя может быть проведено таким же образом, как описанная выше иммобилизация алкоксида металла на носителе. Иммобилизация алкоксида металла на носителе может быть проведена на одной стадии или на нескольких стадиях. Реактор, содержащий реактивированный катализатор, готов далее к очередному запуску в эксплуатацию.
Описанное выше добавление следовых количеств растворимых компонентов активных металлов катализаторов во вводимый поток позволяет в результате достигать стабильной работы катализатора в течение продолжительного времени рабочего цикла. Например, проводили переэтерификацию ДЭК с фенолом в прямоточном реакторе с неподвижным слоем с восходящим потоком второй фазы при добавлении во вводимый сырьевой поток приблизительно от 45 частей на миллион (ppm) до приблизительно 60 частей на миллион (ppm) по массе Ti. Заметных признаков дезактивации катализатора не было отмечено на протяжении более чем 14 месяцев непрерывной работы.
Описываемое в изобретении добавление следового количества соединений активных металлов может быть использовано для непрерывного промышленного получения различных органических карбонатов или карбаматов. Реакции получения органических карбонатов могут быть осуществлены только в одном реакторе, в нескольких реакторах, соединенных последовательно, или в многореакторной системе с соединенными параллельно реакторами, в зависимости от требования конкретной реакционной системы. Например, реакции могут быть проведены только в одном каталитическом реакторе с дистилляционной колонной, или в нескольких соединенных последовательно каталитических реакторах с дистилляционными колоннами, в которых размещены твердый катализатор или два различных твердых катализатора. Необязательно, но для производства органического карбоната могут быть также использованы несколько суспензионных реакторов, соединенных последовательно. Добавление следового количества растворимого компонента активного металла во вводимый поток сырья может осуществляться только в первый реактор из соединенных последовательно нескольких реакторов. Требуемое следовое количество компонента активного металла во вводимом потоке сырья зависит от конкретного активного элемента металла для конкретных вводимых компонентов. Для переэтерификации диалкилкарбоната с фенолом, это количество может составлять, в некоторых вариантах осуществления, приблизительно от 15 ppm до приблизительно 400 частей на миллион (ppm) по массе; в других вариантах осуществления, приблизительно от 20 ppm до приблизительно 300 частей на миллион (ppm); и в еще одних вариантах осуществления, приблизительно от 25 частей на миллион (ppm) до приблизительно 200 частей на миллион (ppm), в зависимости от металла. Например, для вводимого потока сырья, состоящего из диэтилкарбоната и фенола, требуемое количество Ti может составлять, в некоторых вариантах осуществления, приблизительно от 20 частей на миллион (ppm) до приблизительно 150 частей на миллион (ppm); и в еще одних вариантах осуществления, от 30 частей на миллион (ppm) до 100 частей на миллион (ppm) по массе. Количество компонента активного металла в вводимом потоке сырья приблизительно на один или два порядка меньше, чем концентрация гомогенного катализатора известного уровня техники в реакционной среде.
Концентрация Ti в выходящем из реактора потоке находится обычно в интервале приблизительно от 20 частей на миллион (ppm) до приблизительно 100 частей на миллион (ppm), в зависимости от величины концентрации активного металла во вводимом в реактор потоке сырья. При этой концентрации, обычно экономически нецелесообразно извлекать Ti из выходящих из реактора потоков для его рециркуляции, хотя может быть выбран и такой подход. Компонент активного металла в выходящем из реактора потоке может быть извлечен из потоков тяжелого кубового остатка колонны извлечения неочищенного ДФК в виде твердого материала и превращен в растворимый металлоорганический катализатор для повторного использования путем реакции с органическим карбонатом или смесью органического карбоната и спирта при повышенной температуре. Извлекаемым металлоорганическим соединением может быть алкоксид металла, алкоксиалкилкарбонат металла (соль металла и моноэфира угольной кислоты) или их смесь.
Для извлечения растворимого компонента активного металла из потока кубового остатка колонны извлечения ДФК в виде твердого материала, может быть подвергнут обработке сбрасываемый поток тяжелого кубового остатка из колонны извлечения ДФК с помощью горячей воды или смеси водяного пара и воды для осаждения компонента металла в виде твердого вещества. В случае твердых титансодержащих катализаторов, твердый титансодержащий осадок в водной фазе отделяют от жидкости с помощью традиционных методов, таких как фильтрация или центрифугирование. Отделенное твердое вещество превращают в растворимый материал путем обработки с помощью жидкого потока, содержащего диалкилкарбонат или смесь диалкилкарбоната и спирта, при температуре в некоторых вариантах осуществления, от 121 до 343°C (от 250 до 650°F) при повышенном давлении в течение периода времени от 10 минут до 80 часов, и в других вариантах осуществления, от 20 минут до 45 часов. Давление поддерживается достаточно высоким, для того чтобы диалкилкарбонат или смесь спирта и диалкилкарбоната могла, по меньшей мере, частично, находиться в виде жидкости в реакторе, таком как, автоклав, трубчатые реакторы, реакторная система с дистилляционной колонной, и так далее. Необязательно, но жидкий поток может содержать инертный растворитель, такой как бензол, толуол, гексан, гептан, эфир, и другие растворители. Примерами жидкого потока являются смеси этанола и ДЭК, или метанола и ДМК. Содержание диалкилкарбоната в смеси спирта и диалкилкарбоната может изменяться от 0,1 масс.% до меньше чем 100 масс.%.
Реакции получения органических карбонатов или карбаматов могут быть проведены только в одном реакторе, или в нескольких соединенных последовательно реакторах, при различных компоновках реакторов с соответственно расположенными дистилляционными колоннами для экономически эффективного разделения продуктов реакции и для рециркуляции непрореагировавших реагентов. В качестве варианта, реакции могут быть проведены только в одном реакторе или в нескольких соединенных параллельно реакторах. Специалистами в этой области могут быть предложены различные другие компоновки реакторов и дистилляционной колонны.
Реакция может быть проведена только в одной каталитической дистилляционной колонне, в нескольких соединенных последовательно каталитических дистилляционных колоннах, в нескольких соединенных последовательно трубчатых или корпусных реакторах с неподвижным слоем или любой комбинации различных типов реакторов. Когда для получения ДФК используют три каталитические дистилляционные колонны, твердый катализатор размещают в первых двух соединенных последовательно реакторах для переэтерификации. Третий реактор с дистилляционной колонной может содержать твердый катализатор или, в качестве варианта, может не содержать твердый катализатор. Диспропорционирование в третьем реакторе может быть проведено путем использования только растворимого гомогенного катализатора, присутствующего в реакционной среде.
Реакции, осуществляемые в традиционных трубчатых реакторах с неподвижным слоем, могут быть проведены в режиме восходящего потока или в режиме нисходящего потока. Например, реакции получения алкиларилкарбоната, такого как ЭФК, и диарилкарбоната, такого как ДФК, могут быть проведены в жидкой фазе, но могут быть также проведены и в системе со смешанной фазой в присутствии одного или более твердых катализаторов. Два соединенных последовательно реактора для переэтерификации могут поочередно становиться то первым, то вторым реактором, для увеличения продолжительности рабочего цикла. Третий реактор для диспропорционирования ЭФК с получением ДФК и ДЭК может функционировать в нижней половине колонны извлечения фенола, которая может эксплуатироваться при давлениях ниже атмосферного давления. В выбранных вариантах осуществления, раскрываемые в изобретении способы могут применяться для получения дифенилкарбоната путем проведения переэтерификации диэтилкарбоната с фенолом, и затем путем диспропорционирования этилфенилкарбоната.
В жидкую реакционную среду, вводимую в первую реакционную зону, может быть добавлено следовое количество растворимого соединения активного металла, например, такого как этилфенилтитанат или этилкарбонат этоксититана, или смесь алкоксида титана и алкилкарбоната алкоксититана. В качестве варианта, диспропорционирование может быть проведено в каталитической дистилляционной колонне, в которую вводят в соответствующей точке выше средней части колонны поток кубового остатка из колонны извлечения фенола. Диспропорционирование может быть также проведено в каталитической дистилляционной колонне, в которую непосредственно вводят поток кубового остатка из реактора переэтерификации без удаления фенола (фенол может быть удален из потока кубового остатка из колонны диспропорционирования ЭФК). Первая реакционная зона может включать две соединенные последовательно каталитические дистилляционные колонны или две соединенные параллельно каталитические дистилляционные колонны для переэтерификации ДЭК с фенолом. Вторая реакционная зона может включать каталитический реактор с дистилляционной колонной для диспропорционирования ЭФК в ДФК и ДЭК. Может быть выбран ДМК вместо ДЭК, и МФК вместо ЭФК. В каталитические реакторы с дистилляционной колонной для первой реакционной зоны может быть загружен один или более твердых катализаторов, таких как алкоксид титана, иммобилизированный на носителе из оксида кремния, или оксид титана, нанесенный на носитель из оксида кремния. Обычно, существуют два альтернативных способа непрерывного получения дифенилкарбоната, которые могут быть использованы в случае, когда используют больше чем два реактора.
В первом способе непрерывного получения дифенилкарбоната, могут использоваться, в различных вариантах осуществления, от трех до семи каталитических реакторов с дистилляционной колонной, в выбранных вариантах осуществления, от трех до четырех дистилляционных колонн, одна или более из которых могут служить в качестве запасных реакторов для замены наименее активного реактора из нескольких работающих реакторов. Из нескольких реакторов с дистилляционной колонной, для получения в основном ЭФК могут быть использованы от двух до шести реакторов. Оставшиеся каталитические реакторы с дистилляционной колонной могут служить в качестве второй реакционной зоны, в которой, главным образом, происходит диспропорционирование ЭФК в ДФК и ДЭК. ДЭК и, по меньшей мере, часть фенола в потоке из первой реакционной зоны удаляют из выходящего потока высококипящей фракции первой реакционной зоны перед поступлением во вторую реакционную зону. В качестве варианта, удаление фенола из выходящего потока высококипящей фракции первой реакционной зоны может быть отложено до момента после проведения диспропорционирования, в зависимости от концентрации фенола в этом потоке. По мере того как катализатор стареет в процессе работы, активность катализатора медленно снижается. Для нескольких последовательно соединенных реакторов существует три различных варианта поочередного использования режима для получения ароматических карбонатов и режима для реактивации катализатора:
(1) поочередное периодическое использование всех реакторов в последовательном порядке после истечения заданного срока эксплуатации путем вывода из эксплуатации реактора с самым продолжительным периодом работы для реактивации катализатора и, одновременно, путем ввода в эксплуатацию реактора, имеющего свежий или реактивированный катализатор, в качестве первого реактора в ряду соединенных последовательно нескольких реакторов (то есть, новый реактор → в качестве первого реактора, первый реактор → в качестве второго реактора; второй реактор → в качестве третьего реактора и третий реактор → на реактивацию или замену катализатора), или, необязательно, путем ввода в эксплуатацию нового реактора в качестве последнего реактора в ряду, при этом второй реактор становится первым реактором (прямая последовательность, представленная в обратном направлении);
(2) реакторы разделяют на две группы: реакторов первой реакционной зоны и реакторов второй реакционной зоны, при этом каждая группа имеет запасной реактор для поочередного использования для эксплуатации и для замены катализатора/реактивации катализатора;
(3) выведение из эксплуатации реактора с самой низкой активностью катализатора в ряду нескольких последовательно соединенных реакторов для реактивации катализатора, по мере необходимости, и введение в эксплуатацию запасного реактора (в котором катализатор уже был реактивирован) для замены реактора, выведенного из эксплуатации.
В альтернативном способе, два соединенных последовательно реактора используют в качестве первой реакционной зоны. Эту последовательность между первым реактором и вторым реактором периодически меняют в ряду двух реакторов после их эксплуатации в течение заданного периода времени, например, каждые 6000 часов; это чередование повторяют столько раз, сколько это необходимо. Запасной реактор для второй реакционной зоны отсутствует. Этот режим эксплуатации возможен вследствие добавления следовых количеств соединений активных металлов в первый реактор в ряду. ДЭК и фенол в потоке из первой реакционной зоны удаляют путем дистилляции, и затем во второй реакционной зоне оставшийся поток подвергают диспропорционированию ЭФК с получением ДФК. Существуют два способа осуществления диспропорционирования.
(1) В первом способе, диспропорционирование проводят в присутствии твердого катализатора в реакторе с неподвижным слоем, таком как каталитический дистилляционный реактор. Предусмотрен запасной реактор для замены реактора, находящегося в эксплуатации. Дезактивированный катализатор подвергают описанной выше реактивации.
(2) Во втором способе, диспропорционирование проводят в каталитическом дистилляционном реакторе в отсутствие твердого катализатора, и запасной реактор не предусмотрен. Растворимые соединения активного металла в потоке, выходящем из первой реакционной зоны, служат в качестве гомогенного катализатора для реакции диспропорционирования.
Следует иметь в виду, что каталитическая дистилляционная колонна, в которой либо присутствует, либо отсутствует твердый катализатор, сконструирована для второй реакционной зоны таким образом, чтобы секция верхней половины колонны (секция извлечения фенола) служила в основном для отгонки фенола из входящей реакционной смеси из первой реакционной зоны, а секция нижней половины колонны служила в основном для осуществления диспропорционирования ЭФК или МФК. В альтернативном технологическом решении способа, колонна извлечения фенола и каталитическая дистилляционная колонна разделены на две отдельные колонны, хотя при этом может возникать некоторый дисбаланс в нижней секции колонны извлечения фенола. Как уже было указано выше, в зависимости от концентрации фенола во входящем потоке сырья, извлечение фенола может быть отложено до момента после проведения диспропорционирования, хотя некоторое количество фенола может быть удалено в каталитической дистилляционной колонне вместе с ДЭК в потоке отводимого из верхней части колонны пара. Каталитическая дистилляционная колонна для диспропорционирования может эксплуатироваться при давлении ниже атмосферного давления.
На фигуре 1 приведена упрощенная принципиальная схема, иллюстрирующая способ непрерывного получения ДФК с использованием трех каталитических дистилляционных колонн согласно раскрываемым в изобретении вариантам осуществления. Две последовательно соединенные каталитические дистилляционные колонны служат в качестве первой реакционной зоны для переэтерификации ДЭК с фенолом с получением ЭФК и этанола в присутствии твердого катализатора, и одна каталитическая дистилляционная колонна служит в качестве второй реакционной зоны для диспропорционирования ЭФК с получением ДФК и ДЭК.
Далее иллюстрируется способ получения ДФК из ДЭК и фенола согласно раскрываемым в изобретении вариантам осуществления, показанный на фигуре 1. C1 и C2 обозначают каталитические дистилляционные колонны для проведения переэтерификации; C3 обозначает колонну извлечения этанола; C4 обозначает колонну извлечения ДЭК (колонну отдувки фенетола); C5 обозначает каталитическую дистилляционную колонну для диспропорционирования и извлечения фенола; C6 обозначает колонну извлечения ЭФК; и C7 обозначает колонну извлечения ДФК.
Колонны C1 и C2 являются последовательно соединенными каталитическими дистилляционными колоннами, в которых расположены структурированные насадки в реакционной зоне R1 и R2, соответственно. Специально структурированные насадки содержат твердый катализатор. Потоки сырья 1 и 4, содержащие фенол и ДЭК, соответственно, подают на тарелку в верхней секции каталитических дистилляционных колонн C1 и C2, выше каталитических реакционных зон R1 и R2. Мольное отношение ДЭК к фенолу в сырьевых потоках свежего ДЭК и свежего фенола может составлять приблизительно 1:2. Однако мольные отношения ДЭК к фенолу в каталитических реакционных зонах R1 и R2 регулируются таким образом, чтобы они составляли, в некоторых вариантах осуществления, от приблизительно 12:1 до приблизительно 1:2,5; в других вариантах осуществления, приблизительно от 10:1 до приблизительно 1:2; и в еще одних вариантах осуществления, приблизительно от 7:1 до приблизительно 1:1.
Растворимый металлоорганический катализатор также вводят на тарелку в верхней части колонны C1 через линию подачи 3. Например, в случае титансодержащего твердого катализатора в реакционных зонах R1 и R2, в верхнюю часть первого каталитического реактора с дистилляционной колонной C1 может быть введен раствор, содержащий растворимое соединение титана, такое как Ti(OEt)4-x(OPh)x (где x является 0, 1, 2, 3 или 4), или соли титана и моноэфиров угольной кислоты, такие как этилкарбонаты этоксититана, или их смесь. Растворителем для получения раствора катализатора может служить, например, ДЭК, смешанный раствор ДЭК и фенола, смешанный раствор ДЭК и этанола, или смешанный раствор ДЭК, этанола и фенола.
Скорость подачи раствора катализатора может регулироваться так, чтобы концентрация титана в жидком потоке над катализатором в первом колонном реакторе составляла, в некоторых вариантах осуществления, приблизительно от 20 частей на миллион (ppm) до приблизительно 100 частей на миллион (ppm) активного металла (титана, для примеров растворов катализатора, приведенных в предыдущем параграфе) по массе; в других вариантах осуществления, приблизительно от 25 частей на миллион (ppm) до приблизительно 80 частей на миллион (ppm) по массе; и в еще одних вариантах осуществления, приблизительно от 30 частей на миллион (ppm) до приблизительно 70 частей на миллион (ppm) по массе.
Отводимые сверху колонны потоки пара 6 и 14 из каталитических дистилляционных колонн C1 и C2 направляют в колонну извлечения этанола C3 по линии 8. Этот отводимый сверху колонны поток может также содержать незначительные количества побочных продуктов, таких как диэтиловый эфир и диоксид углерода, и следовое количество фенола. Диэтиловый эфир и диоксид углерода могут быть удалены в виде отводимого сверху колонны потока пара 9. Этанол может быть извлечен сбоку из колонны C3 по линии 10. В потоке кубового остатка 11 может быть рециркулирован ДЭК из колонны C3 в каталитические реакторы с дистилляционной колонной C1 и C2 по линиям 12 и 13, соответственно.
Колонна C1 может эксплуатироваться так, чтобы температура каталитической реакционной зоны R1 находилась в интервале приблизительно от 160°C до приблизительно 210°C (приблизительно от 320°F до приблизительно 410°F). Абсолютное давление в верхней части колонны C1 может находиться в интервале приблизительно от 2 бар до приблизительно 4,8 бар (приблизительно от 14,7 фунт/дюйм2 до приблизительно 55 фунт/дюйм2). Поток кубового остатка 7 из первой каталитической дистилляционной колонны C1 может быть введен в верхнюю часть каталитической дистилляционной колонны C2, которая может эксплуатироваться так, чтобы температура в каталитической реакционной зоне могла находиться в интервале приблизительно от 162°C до приблизительно 216°C (приблизительно от 325°F до приблизительно 420°F), и колонна может эксплуатироваться при давлении в верхней части колонны в интервале от ниже атмосферного давления, приблизительно от 1 бар (14,5 фунт/дюйм2), до приблизительно 4,5 бар (51 фунт/дюйм2). Необязательно, но небольшие порции потока рециркулируемого или свежего ДЭК могут быть введены в колонну C1 и C2 по линиям 4a и 4b, соответственно.
Концентрация ЭФК повышается по мере того как поток проходит сверху вниз через тарелки в каталитических дистилляционных колоннах C1 и C2. По мере того как в колонных реакторах C1 и C2 происходит до некоторой степени диспропорционирование ЭФК в ДФК и ДЭК, концентрация ДФК также повышается. Поток кубового остатка 15 из реактора с дистилляционной колонной C2 направляют в колонну C4 извлечения ДЭК, в которой ДЭК извлекают из отводимого сверху колонны потока 16. Колонна C4 может эксплуатироваться в интервале температур приблизительно 127°C до приблизительно 204°C (приблизительно от 260°F до приблизительно 400°F) при абсолютном давлении в верхней части колонны приблизительно от 0,3 бар (приблизительно 4 фунт/дюйм2) до приблизительно 1,5 бар (приблизительно 22 фунт/дюйм2). Поток 16 может быть введен в колонну извлечения этанола C3 для разделения ДЭК и фенола, который может находиться в потоке 16, из которой ДЭК и фенол могут быть направлены на рециркуляцию по линиям 11, 12, 13 в колонны C1 и C2.
Отводимый сверху колонны C4 поток 16 может также содержать ДЭК и небольшие количества фенетола, фенола и этанола. Боковой поток 18 из колонны C4 может быть использован в качестве потока продувки фенетола, для минимизации накопления фенетола в системе.
Поток кубового остатка 17 из колонны C4 содержит соединения гомогенного катализатора, попадающие из колонн C1 и C2. Поток кубового остатка 17 может быть введен в соответствующем месте в верхнюю часть дистилляционной колонны C5. Колонна C5 может быть использована для проведения диспропорционирования ЭФК и может необязательно содержать гетерогенный катализатор в реакционной зоне R3.
Колонна C5 может быть предназначена и может эксплуатироваться для достижения двух целей: удаления фенола в потоке 17 и побочного продукта ДЭК из диспропорционирования ЭФК в качестве отводимого сверху потока 19; и диспропорционирования ЭФК с образованием ДФК. Колонну C5 эксплуатируют так, чтобы температура зоны гомогенной каталитической реакции R3 находилась в интервале приблизительно от 165°C до приблизительно 210°C (приблизительно от 330°F до приблизительно 410°F), и абсолютное давление в верхней части колонны составляло приблизительно от 0,07 бар (приблизительно 1 фунт/дюйм2) до приблизительно 0,6 бар (9 фунт/дюйм2).
Отводимый сверху колонны C5 поток 19, содержащий ДЭК и фенол, может быть рециркулирован в колонны C1 и C2 с помощью потоков 20 и 21, соответственно. Поток кубового остатка 22 (который содержит ДФК, непрореагировавший ЭФК, фенол, фенетол, продукты с более высокой молекулярной массой и растворимый Ti катализатор) из колонны C5 вводят в колонну C6 извлечения ЭФК, которая может эксплуатироваться при температуре приблизительно от 168°C до приблизительно 213°C (приблизительно от 335°F до приблизительно 415°F) и при давлениях ниже атмосферного давления в интервале приблизительно от 0,03 бар (приблизительно 0,4 фунт/дюйм2) до приблизительно 0,55 бар (8 фунт/дюйм2).
Поток кубового остатка 25, содержащий ЭФК, из колонны C6 вводят в колонну C7 извлечения ДФК для извлечения ДФК в качестве бокового потока 27. Колонну C7 извлечения ДФК эксплуатируют под высоким вакуумом (например, <0,03 бар (<0,4 фунт/дюйм2). Отводимый сверху колонны поток 26 может быть объединен с отводимым сверху колонны C6 извлечения ЭФК потоком 23 и рециркулирован в колонну C5 по линии 24.
Поток кубового остатка 28 колонны C7 извлечения ДФК, содержащий соединения с более высокими температурами кипения и растворимый катализатор, может быть подвергнут процессу разделения или может быть сброшен в качестве отходов. Если требуется, то в случае, когда, например, используют и вводят в реактор титановые катализаторы, титан в виде растворимого Ti катализатора (Ti(OEt)4 или смесь Ti(OEt)4 и этилкарбонатов этоксида титана) может быть извлечен с целью рециркулирования, как это было обсуждено ранее. В качестве одного метода удаления отходов, поток 28 может быть направлен в установку очистки титана для извлечения титана Ti. ДФК может быть извлечен из потока 22 в дополнительных линиях технологического оборудования для извлечения и очистки, которые известны обычным специалистам в этой области.
В качестве уже упомянутого выше варианта, диспропорционирование ЭФК в ДФК и ДЭК может быть осуществлено в присутствии твердого катализатора в каталитической дистилляционной колонне C5. Однако твердые катализаторы в колонне C5 могут дезактивироваться быстрее, чем твердые катализаторы во второй каталитической дистилляционной колонне C2, показанной на фигуре 1.
Как уже обсуждалось выше, когда колонна C5 содержит твердый катализатор, для поддержания достаточной каталитической активности в процессе производства могут быть использованы различные варианты циклической работы реакторов с дистилляционной колонной. Для циклической работы реакторов могут быть предусмотрены не показанные на фигуре соответствующие запорно-регулирующие клапаны и сеть трубопроводов, известные специалистам в этой области.
На фигуре 2 приведена альтернативная схема технологического процесса, в которой одинаковые численные обозначения представляют такие же части схемы, как на фигуре 1. Для проведения переэтерификации и диспропорционирования в ней предусмотрено аналогичное количество каталитических дистилляционных колонн и колонн для разделения веществ, так же как на фигуре 1. Однако часть отводимого сверху каталитической дистилляционной колонны C5 потока 19 может быть рециркулирована назад в колонну C4 извлечения ДЭК по линии 30. Рециркуляция по линии 30 позволяет в результате использовать альтернативный способ продувки фенетола.
На фигуре 3 приведена еще одна альтернативная схема технологического процесса согласно раскрываемым в изобретении вариантам осуществления, аналогичная представленным схемам на фигурах 1 и 2, в которой одинаковые численные обозначения представляют одинаковые такие же части схемы, как на фигурах 1 и 2. Первая каталитическая дистилляционная колонна C1 эксплуатируется более или менее аналогичным образом, как в предыдущих случаях (фигуры 1 и 2). Однако эксплуатация второй каталитической дистилляционной колонны C2 и колонны C4 извлечения ДЭК осуществляется отличным от предыдущих случаев способом. Колонна C2 может эксплуатироваться при более высокой температуре и более низком давлении, чем в предыдущих случаях. Поток 13 рециркуляции ДЭК вводят в нижнюю часть колонны C2. Необязательно, но часть потока 4 свежего ДЭК может также быть введена в нижнюю часть колонны C2. Вследствие эксплуатации при более высокой температуре и более низком давлении, поток кубового остатка 15 колонны C2 содержит меньше ДЭК. Отводимый сверху колонны C2 поток 14 содержит, наряду с другими компонентами, фенетол. Поток 14 может быть введен в колонну C4, где поток кубового остатка 17 колонны C4 является потоком продувки фенетола.
Согласно раскрываемым в изобретении вариантам осуществления, из образующегося сырого ДФК может быть получен чрезвычайно чистый ДФК. Высокочистый ДФК может быть получен путем фракционной кристаллизации с использованием смесей углеводород-эфир, таких как смеси гексан-диэтиловый эфир. В некоторых вариантах осуществления, в очищенном ДФК единственной обнаруживаемой примесью, помимо фенола, является ксантон в количестве приблизительно до 0,5 частей на миллион (ppm) по массе. Фенол в очищенном ДФК может присутствовать в следовом количестве, таком как приблизительно от 5 до приблизительно 17 частей на миллион (ppm) по массе. Анализ микропримесей в ДФК, полученном согласно раскрываемым в изобретении вариантам осуществления, указывает на то, что чистота получаемого ДФК является значительно более высокой, чем чистота для ДФК, который может быть поставлен известными производителями химических реактивов для лабораторных исследований.
ПРИМЕРЫ
За исключением эксперимента 1, все реакции переэтерификации ДЭК с фенолом проводили в реакторе с восходящим потоком при температуре кипения реагентов. Поэтому, в каталитической реакционной зоне существовали одновременно паровая и жидкая фазы. Реактор с неподвижным слоем имел диаметр 1,3 см (1/2 дюйма) и длину 6,5 см (25 дюймов). Реактор имел раздельно регулируемый нагрев верхней и нижней зоны. Реактор с неподвижным слоем устанавливали вертикально. Объем твердых катализаторов составлял 25 мл.
Сравнительный эксперимент 1
Эксперименты по переэтерификации ДЭК с фенолом проводили в присутствии гомогенного катализатора из алкоксида титана с использованием автоклава с мешалкой объемом 50 мл. Автоклав заполняли приблизительно 35 мл смеси ДЭК/фенол, приведенной в таблице 1. Автоклав погружали в масляную баню для регулирования температуры реакции. После проведения реакции, автоклав удаляли из масляной бани и резко охлаждали холодной водой. Присутствие дифенилового эфира в реакционной смеси не обнаруживалось. Результаты проведения реакций приведены в таблице 1.
Когда концентрации Ti в водимых растворах сырья составляли 4767 частей на миллион (ppm) по массе, максимальная конверсия фенола после приблизительно 3 часов проведения реакции составляла приблизительно 15% при очень низкой селективности (<20,5 моль. %) по ЭФК и ДФК. Когда время реакции составляло 2 часа, конверсия фенола составляла менее чем 5%; селективность была выше, но все еще неудовлетворительной (63 моль.%). Когда концентрацию катализатора понижали до 42 частей на миллион (ppm) Ti по массе, селективность значительно повышалась, но была неудовлетворительной конверсия.
Сравнительный эксперимент 2
Цель проведения этого эксперимента заключалась в получении экспериментальных данных для гомогенного катализатора для сравнения с результатами примеров, согласно раскрываемым в изобретении вариантам осуществления. Твердый катализатор в реакторе отсутствовал. Пространство объемом 25 мл для твердого катализатора в реакторе было незаполненным. Реакционную смесь 73,3 масс.% ДЭК и 26,7 масс.% фенола (мольное отношение ДЭК/PhOH равно 2,19), содержащую различные количества гомогенного катализатора Ti(OEt)4-x(OPh)x (x= ~2), пропускали через реактор в режиме восходящего потока при различных реакционных условиях в течение от 0 до 768 часов работы и затем в течение от 1266 до 1362 часов работы. Концентрации Ti во вводимой смеси изменялось от 59 ppm до 709 ppm Ti по массе, как показано на фигуре 4. Объемная скорость составляла 0,5 мл/мин для большей части времени работы. Динамика изменения скорости подачи смеси приведена в таблице 2.
Переэтерификация
Из продуктов переэтерификации отгоняли этанол и затем добавляли ДЭК для корректировки мольного отношения ДЭК/PhOH до величины 2,19 путем добавления ДЭК для получения второго сырья для переэтерификации. Второе сырье для переэтерификации содержало в среднем приблизительно 3,4 масс.% ЭФК, приблизительно 250 ppm фенетола и приблизительно 300 ppm ДФК по массе. Динамика изменения концентраций гомогенного катализатора во втором сырье для переэтерификации приведена в таблице 3.

Используя эти смеси сырья, проводили вторую переэтерификацию в течение от 768 до 1266 часов работы. Конверсия фенола на фигуре 4 в течение этого периода времени работы представляет собой суммарную конверсию в результате проведения 1-ой и 2-ой переэтерификации.
Интервал реакционных температур составлял от 174°C до 210°C (от 345°F до 410°F), как показано на фигуре 4. Интервал давлений в реакторе составлял приблизительно от 2,9 до приблизительно 5,5 бар (приблизительно от 27 фунт/дюйм2 до приблизительно 65 фунт/дюйм2). Все реакции проводили в условиях кипения реагентов. Поэтому, реакцию осуществляли в системе со смешанной фазой (пар и жидкость). Температуры на фигуре 4 являются температурами в нижней части реактора.
Когда концентрация катализатора в реакционной смеси выше чем 118 ppm Ti, отмечались отрицательные воздействия катализатора на конверсию фенола. Причина этого отрицательного воздействия до конца не понятна, но оно может быть следствием воздействия двух этоксильных групп Ti катализатора. Кроме того, когда концентрация Ti во вводимом сырье составляла более чем 300 ppm, возникали проблемы, связанные с закупоркой трубопровода в выходящей из реактора линии, вследствие осаждения Ti катализатора. Поэтому, для решения проблем закупорки трубопровода устанавливали пару проходных фильтров. Когда концентрация Ti составляла 59 ppm, влияние температуры на конверсию фенола было умеренным, что указывает на низкое значение энергии активации реакции переэтерификации ДЭК с фенолом. Самое высокое значение конверсии фенола для первой переэтерификации составляло приблизительно 11,3 моль.% для концентрации 337 ppm Ti при 204°C (400°F) и 4,5 бар (50 фунт/дюйм2). Самое высокое значение конверсии фенола для второй переэтерификации составляло приблизительно 14,5 моль.% при 193°C (380°F) и 2,9 бар (27 фунт/дюйм2) для концентрации 188 ppm Ti. Эксперимент также подтвердил наличие низкой конверсии при реакции в жидкой фазе (410°C и 7,9 бар (100 фунт/дюйм2)), что и ожидалось.
Эксперимент 3
Целями этого эксперимента являлась демонстрация (1) метода приготовления н-бутоксида титана, иммобилизированного на носителе из силикагеля, непосредственно в реакторе, (2) метода реактивации катализатора и (3) использования для переэтерификации двухфазного реактора с неподвижным слоем. Двойную фазу из пара и жидкости в каталитической реакционной зоне создавали в результате кипения реакционной смеси.
45,74 г гранулированного силикагеля (+8 меш) подвергали обработке с помощью раствора гидроксида натрия (7,5 г NaOH в 550 мл воды) при температуре приблизительно 42°C в течение 7 минут при перемешивании при температуре окружающей среды. Силикагель промывали, сначала холодной водой, а затем горячей водой (приблизительно 80°C), для удаления следовых количеств натрия на оксиде кремния. Полученный обработанный силикагель сушили при 125°C в течение 2 часов и затем при 300°C в течение 2 часов в токе азота. Высушенный носитель из силикагеля содержал 23 частей на миллион (ppm) Na по массе. Обработанный носитель из силикагеля имел следующие характеристики: удельная поверхность 291 м2/г по БЭТ, объем пор 1,052 см3/г и средний диаметр пор 16,3 нм.
Загружали в реактор 25 мл носителя из высушенного гранулированного силикагеля (приблизительно 9,3 г). Раствор н-бутоксида титана готовили путем растворения 27 г н-бутоксида титана в 500 мл осушенного толуола. Раствор н-бутоксида титана помещали в резервуар. После циркуляции раствора н-бутоксида титана через реактор в режиме восходящего потока при 15 мл/мин и температуре окружающей среды в течение 15 минут, реактор нагревали до 168°C (335°F) при давлении приблизительно 5,5 бар (65 фунт/дюйм2). Циркуляцию продолжали при 168°C (335°F) в течение 4,5 часов, и затем реактор охлаждали. После удаления избытка раствора из реактора, нанесенный катализатор промывали с помощью восходящего потока осушенного толуола при 4 мл/мин в течение 1,5 часов. Промытый катализатор сушили при 168°C (335°F) в восходящем потоке 350 см3/мин газообразного азота в течение 2 часов. Приготовленный непосредственно в реакторе катализатор из н-буктоксида титана, привитого на носитель из гранулированного силикагеля, подвергали испытаниям при переэтерификации ДЭК с фенолом.
1-ый цикл переэтерификации: Переэтерификацию ДЭК с фенолом проводили в присутствии твердого катализатора, приготовленного непосредственно в реакторе с неподвижном слоем, при кипении реагентов. Смесь ДЭК и фенола (25,57 масс.% фенола и 74,43 масс.% ДЭК; мольное отношение ДЭК/PhOH 2,32) пропускали через слой твердого катализатора в режиме восходящего потока при 168°C (335°F), 2,4 бар (20 фунт/дюйм2) и при скорости подачи сырья 0,2 мл/мин. Это испытание представляло собой 1-ый цикл переэтерификации, и результаты приведены на фигуре 5. Катализатор достигал своей максимальной активности (12 моль.% конверсии фенола) приблизительно через 40 часов работы. Приблизительно через 80 часов работы, катализатор терял большую часть своей активности. Этот дезактивированный катализатор подвергали 1-ой реактивации следующим образом.
1-ая реактивация катализатора: Реактивация катализатора включает две стадии; деполимеризацию/кондиционирование поверхности катализатора и повторное нанесение активного металлического титана на катализатор. После опорожнения реактора, катализатор промывали восходящим потоком осушенного толуола (300 мл) при температуре окружающей среды, и затем толуол выводили из реактора. 0,19 г н-бутоксида титана растворяли в 2 литрах раствора этанола, приготовленного путем смешения 400 мл этанола и 1700 мл толуола. Раствор титана пропускали через реактор в режиме восходящего потока с объемной скоростью 2,2 мл/мин при 168°C (335°F) и 12 бар (160 фунт/дюйм2) в течение 13,5 часов. После удаления избытка раствора титана из реактора, катализатор сушили при 168°C (335°F) при давлении окружающей среды в восходящем потоке 200 см3/мин азота в течение 45 минут. Осуществляли циркуляцию раствора н-бутоксида титана (135 г н-бутоксида титана в 2 литрах толуола) через реактор при объемной скорости 15 мл/мин в режиме восходящего потока при комнатной температуре в течение 20 минут и затем при 168°C (335°F) и 10,7 бар (140 фунт/дюйм2) в течение 4 часов. После охлаждения, из реактора удаляли избыток раствора. Катализатор промывали с помощью 4 мл/мин толуола в режиме восходящего потока в течение 1,5 часов. Промытый катализатор сушили при 168°C (335°F) в восходящем потоке 300 см3/мин газообразного азота в течение 2 часов. Реактивированный катализатор использовали во втором цикле переэтерификации следующим образом.
2-ой цикл переэтерификации: Переэтерификацию проводили так же, как в 1-ом цикле. Результат приведен на фигуре 5. Реактивированный катализатор функционировал не так хорошо как в 1-ом цикле, и катализатор потерял активность приблизительно через 40 часов работы. Затем была проведена 2-ая реактивация катализатора следующим образом.
2-ая реактивация катализатора: После удаления материала из реактора, катализатор в реакторе промывали в восходящем потоке осушенного толуола при 10 мл/мин при температуре окружающей среды в течение 30 минут, и затем толуол удаляли из реактора. Катализатор в реакторе сушили при 168°C (335°F) в восходящем потоке газообразного азота при 250 см3/мин в течение 1 часа. Раствор, приготовленный путем смешения 8 мл воды, 500 мл этанола и 1100 мл толуола, пропускали через реактор в режиме восходящего потока при 2,2 мл/мин при 168°C (335°F) и 12 бар (160 фунт/дюйм2) в течение 12,1 часа. После удаления избытка раствора из реактора, катализатор сушили при 168°C (335°F) при давлении окружающей среды в восходящем потоке 200 см3/мин азота в течение 1 часа. Осуществляли циркуляцию раствора н-бутоксида титана (135 г н-бутоксида титана в 2 литрах толуола) через реактор в режиме восходящего потока 15 мл/мин при комнатной температуре в течение 20 минут и затем при 168°C (335°F) и 10,7 бар (140 фунт/дюйм2) в течение 6 часов. После охлаждения, избыток раствора титана удаляли из реактора. Катализатор промывали восходящим потоком 4 мл/мин толуола в течение 1,5 часов. Промытый катализатор сушили при 168°C (335°F) в восходящем потоке 300 см3/мин газообразного азота в течение 2 часов. Реактивированный катализатор использовали в третьем цикле реакции переэтерификации следующим образом.
3-ий цикл переэтерификации: Переэтерификацию проводили так же, как в 1-ом цикле. Результат приведен на фигуре 5. Реактивированный катализатор функционировал так же, как в 1-ом цикле. Тем не менее, катализатор потерял активность приблизительно через 90 часов работы.
Дезактивированный катализатор в реакторе был подвергнут еще двум реактивациям при аналогичных условиях и затем еще двум переэтерификациям, с аналогичными результатами. После 5-го цикла реакций переэтерификации, катализатор был подвергнут 5-ой реактивации следующим образом. Данные по реактивации катализатора с 3-ей реактивации по 5-ую реактивацию описаны ниже.
3-ая реактивация катализатора. После удаления из реактора всего материала, оставшегося после 3-его цикла переэтерификации, катализатор в реакторе промывали в восходящем потоке осушенного толуола при 10 мл/мин при температуре окружающей среды в течение 1 часа, и затем избыток толуола удаляли из реактора. Катализатор в реакторе сушили при 157°C (315°F) в восходящем потоке газообразного азота при 250 см3/мин в течение 1 часа. Раствор, приготовленный путем смешения 8 мл воды, 500 мл этанола и 1100 мл толуола, пропускали через реактор в режиме восходящего потока при 2,2 мл/мин при 157°C (315°F) и 2,7 бар (25 фунт/дюйм2) в течение 12,1 часа. После удаления избытка раствора из реактора, катализатор сушили при 149°C (300°F) при давлении окружающей среды в восходящем потоке 200 см3/мин азота в течение 1 часа. Осуществляли циркуляцию раствора н-бутоксида титана (135 г н-бутоксида титана в 2 литрах толуола) через реактор в режиме восходящего потока со скоростью 15 мл/мин при комнатной температуре в течение 20 минут и затем при 157°C (315°F) и 7,2 бар (90 фунт/дюйм2) в течение 6 часов. После охлаждения, избыток раствора удаляли из реактора. Катализатор промывали восходящим потоком толуола со скоростью 4 мл/мин в течение 1,5 часов. Промытый катализатор сушили при 163°C (325°F) в восходящем потоке 300 см3/мин газообразного азота в течение 2 часов.
Реактивированный катализатор использовали в 4-ом цикле переэтерификации. Рабочая характеристика реактивированного катализатора была аналогична рабочей характеристике во 2-ом цикле переэтерификации. Результат на фигуре 5 не показан.
4-ая реактивация катализатора: После удаления из реактора материала, оставшегося после 4-го цикла переэтерификации, катализатор в реакторе промывали в восходящем потоке осушенного толуола при скорости 10 мл/мин при температуре окружающей среды в течение 1 часа, и затем избыток толуола удаляли из реактора. Катализатор в реакторе сушили при 157°C (315°F) в восходящем потоке газообразного азота при скорости 250 см3/мин в течение 1 часа. Раствор, приготовленный путем смешения 8 мл воды, 500 мл этанола и 1100 мл толуола, пропускали через реактор в режиме восходящего потока при 2,2 мл/мин при 157°C (315°F) и 2,7 бар (25 фунт/дюйм2) в течение 12,1 часа. После удаления избытка раствора из реактора, катализатор сушили при 149°C (300°F) при давлении окружающей среды в восходящем потоке 200 см3/мин азота в течение 1 часа. Осуществляли циркуляцию раствора н-бутоксида титана (135 г н-бутоксида титана в 2 литрах толуола) через реактор в режиме восходящего потока при скорости 15 мл/мин при комнатной температуре в течение 20 минут и затем при 157°C (315°F) и 7,2 бар (90 фунт/дюйм2) в течение 6 часов. Катализатор промывали восходящим потоком 4 мл/мин толуола в течение 1,5 часов. Промытый катализатор сушили при 163°C (325°F) в восходящем потоке 300 см3/мин газообразного азота в течение 2 часов. Реактивированный катализатор затем использовали в 5-ом цикле переэтерификации. Рабочая характеристика реактивированного катализатора была аналогична рабочей характеристике во 3-ом цикле переэтерификации. Результат на фигуре 5 не показан.
5-ая реактивация катализатора: После удаления из реактора материала, катализатор в реакторе промывали в восходящем потоке осушенного толуола при 10 мл/мин при температуре окружающей среды в течение 1 часа, и затем избыток толуола удаляли из реактора. Катализатор в реакторе сушили при 124°C (255°F) в восходящем потоке газообразного азота при 250 см3/мин в течение 1 часа. Пропускали через реактор восходящий поток воды со скоростью 0,3 мл/мин при 152-154°C (305-310°F) и давлении окружающей среды в течение 6 часов. Катализатор, обработанный в реакторе водяным паром, сушили в восходящем потоке газообразного азота при 100 см3/мин в течение 1 часа 20 минут при 146-149°C (295-300°F). Осуществляли циркуляцию раствора н-бутоксида титана (135 г н-бутоксида титана в 1600 мл толуола) через реактор в режиме восходящего потока при скорости 15 мл/мин при комнатной температуре в течение 20 минут и затем при 127°C (260°F) и 3,1 бар (30 фунт/дюйм2) в течение 6 часов. После охлаждения, избыток раствора удаляли из реактора. Катализатор промывали восходящим потоком 4 мл/мин толуола в течение 1,5 часов. Промытый катализатор сушили при 138°C (280°F) в восходящем потоке 300 см3/мин газообразного азота в течение 2 часов. Реактивированный катализатор затем использовали в 6-ом цикле переэтерификации следующим образом.
6-ой цикл переэтерификации: Переэтерификацию проводили так же, как в 1-ом цикле. Результат приведен на фигуре 5. Реактивированный катализатор функционировал так же, как в 1-ом цикле. Примечательно то, что катализатор терял активность более медленно.
Приведенные выше эксперименты доказывают возможность реактивации непосредственно в реакторе дезактивированного катализатора из алкоксида титана, иммобилизированного на носителе из силикагеля. Однако продолжительность рабочего цикла катализатора может быть слишком короткой с точки зрения применения на практике этой технологии в крупнотоннажных промышленных реакторах для непрерывного получения ароматических карбонатов.
Эксперимент 4
Целью этого эксперимента являлась демонстрация увеличенной продолжительности рабочего цикла катализатора, которая достигалась за счет добавления следового количества (42 частей на миллион (ppm) Ti по массе) растворимого соединения Ti (н-бутоксида титана) во вводимый поток сырья. Дезактивированный катализатор после 6-го цикла переэтерификации в эксперименте 3 снова подвергали 7-ой реактивации катализатора следующим образом. После удаления материала из реактора, катализатор в реакторе промывали в восходящем потоке осушенного толуола при 10 мл/мин при температуре окружающей среды в течение 1 часа, и затем избыток толуола удаляли из реактора. Катализатор в реакторе сушили при 124°C (255°F) в восходящем потоке газообразного азота при 250 см3/мин в течение 1 часа. Смешанный раствор воды (4 масс.%) в этаноле пропускали через реактор в режиме восходящего потока при 1,4 мл/мин при 154°C (310°F) и давлении окружающей среды в течение 6 часов. Катализатор в реакторе сушили в восходящем потоке 150 см3/мин газообразного азота в течение 1 часа 25 минут при 154°C (310°F). Раствор н-бутоксида титана (67,5 г н-бутоксида титана в 800 мл толуола) пропускали через реактор в режиме восходящего потока со скоростью 15 мл/мин при комнатной температуре в течение 20 минут, и затем при 127°C (260°F) при 3,4 бар (35 фунт/дюйм2) в течение 6 часов. После охлаждения, избыток раствора удаляли из реактора. Катализатор промывали восходящим потоком 4 мл/мин толуола в течение 1,5 часов. Промытый катализатор сушили при 138°C (280°F) в восходящем потоке 300 см3/мин газообразного азота в течение 2 часов. Реактивированный катализатор использовали в 7-ом цикле переэтерификации следующим образом.
7-ой цикл переэтерификации проводили путем добавления следового количества (42 ppm Ti по массе) н-бутоксида титана в вводимые потоки сырья при различных реакционных условиях. В этом эксперименте использовали две различные смеси сырья. Смешанный раствор сырья 19,73 масс.% фенола и 80,27 масс.% ДЭК (мольное отношение ДЭК/PhOH 3,24) использовали в течение первых 593 часов работы, а затем использовали 25,83 масс.% фенола и 74,17 масс.% ДЭК (мольное отношение ДЭК/PhOH 2,29) в течение оставшегося времени до момента прекращения эксперимента на 749 часу работы. Н-бутоксид титана смешивали с предварительно смешанными растворами сырья ДЭК/PhOH. Скорости подачи сырья составляли 0,2 мл/мин в течение первых 353 часов работы, 0,3 мл/мин в течение от 353 до 401 часов работы, и затем 0,2 мл/мин до конца эксперимента. Анализ микропримесей в образцах продукта, взятых в различное время в процессе работы, показал содержание 21 частей на миллион (ppm) Ti после 48 часов работы, 44 частей на миллион (ppm) Ti через 305 часов, 44 частей на миллион (ppm) Ti через 449 часов, 31 частей на миллион (ppm) Ti через 491 час, 51 частей на миллион (ppm) Ti через 593 часов, 51 частей на миллион (ppm) Ti через 713 часов и 31 частей на миллион (ppm) Ti через 749 часов. Результат этого эксперимента приведен на фигуре 6. Температуры, приведенные на фигуре 6, являлись температурами, измеряемыми в нижней части слоя катализатора. Температура, измеряемая в верхней части слоя катализатора, составляла обычно на 1,5-3°C (3-5°F) ниже температуры в нижней части реактора, в зависимости от концентрации этанола в потоке продукта, что указывает на испарение этанола в верхней части слоя катализатора. Более низкие температуры выходящего из реактора потока в верхней части слоя катализатора отмечались в случае, когда концентрации этанола в продукте были выше, чем приблизительно 1,2 масс.%. Единственным обнаруживаемым побочным продуктом являлся фенетол. Селективность образования фенетола в расчете на фенол составляла менее 0,3 моль.%.
Как показано на фигуре 6, дезактивации катализатора не происходило на протяжении всего периода времени работы (749 часов), что убедительно доказывает возможность увеличения продолжительности рабочего цикла катализатора с величины менее чем 80 часов до 749 часов и более путем добавления 42 частей на миллион (ppm) Ti по массе в вводимый поток сырья.
После завершения работы катализатор в реакторе подвергали анализу, и он представлял собой в основном желтые гранулы, но некоторая часть гранул была темно-коричневого цвета. Для сравнения, феноксид титана имеет интенсивный оранжевый цвет или цвет янтаря. Анализ отработанного катализатора дал содержание 0,55 масс.% Ti в катализаторе. Этот факт был весьма неожиданным.
Эксперимент 5
Целями этого эксперимента являлась демонстрация (1) необходимости предварительной подготовки катализатора перед проведением реакций, (2) реактивации катализатора, (3) увеличения продолжительности рабочего цикла катализатора и (4) необходимости регулирования содержания воды (меньше чем приблизительно 650 частей на миллион (ppm) по массе) в сырье. В этом эксперименте, для приготовления носителя использовали гранулы оксида кремния, на которые прививали н-бутоксид титана.
Носитель из гранул оксида кремния (0,3 см (1/8 дюйма), 555 частей на миллион (ppm) Na и 2500 частей на миллион (ppm) Al по массе, удельная поверхность 280 м2/г по БЭТ и объем пор 1 см3/г) использовали для приготовления катализатора с иммобилизированным на поверхности н-бутоксидом титана. 100 г гранул оксида кремния обрабатывали при приблизительно 52°C в течение 5 минут при перемешивании с помощью раствора гидроксида натрия (10 г NaOH в 570 мл воды). Оксид кремния тщательно промывали холодной водой и затем горячей водой (приблизительно 80°C) для удаления следовых количеств натрия на оксиде кремния. Обработанный оксид кремния сначала сушили при комнатной температуре, затем сушили при 130°C в течение 1,5 часов, и затем при 150°C в течение 1 часа в вакуумном сушильном шкафу. Высушенный носитель из оксида кремния имел 150 частей на миллион (ppm) Na по массе. Приготовленный носитель из оксида кремния имел следующие характеристики: удельная поверхность 252 м2/г по БЭТ, объем пор 1,035 см3/г и средний диаметр пор 15,7 нм.
Загружали в реактор 25 мл высушенных гранул оксида кремния (9,72 г). Резервуар для раствора катализатора заполняли раствором н-бутоксида титана, приготовленного путем растворения 135 г н-бутоксида титана в 1600 мл толуола. Осуществляли циркуляцию этого раствора катализатора через реактор в режиме восходящего потока при скорости 15 мл/мин при комнатной температуре в течение 20 минут, и затем при 135°C (275°F) и 3,4 бар (35 дюйм2) в течение 6 часов. После охлаждения, избыток раствора катализатора удаляли из реактора и затем приготовленный непосредственно в реакторе катализатор промывали восходящим потоком толуола при объемной скорости 4 мл/мин в течение 1,5 часов при температуре окружающей среды. После удаления избытка толуола из реактора, катализатор сушили при 138°C (280°F) в течение 2 часов в восходящем потоке 350 см3/мин газообразного азота. Полученный катализатор использовали в 1-ом цикле переэтерификации следующим образом.
1-ый цикл переэтерификации: 1-ый цикл переэтерификации проводили без введения растворимых соединений титана в поток сырья при кипении реагентов при 168°C (335°F) и 2,4 бар (20 фунт/дюйм2) при введении сырья в режиме восходящего потока со скоростью 0,2 мл/мин. Состав сырья представлял собой 26,07 масс.% фенола и 73,93 масс.% ДЭК (мольное отношение ДЭК/фенол 2,56). Результат приведен на фигуре 7. Во время работы происходила дезактивация катализатора. Через 100 часов работы катализатор имел низкую активность.
1-ая реактивация катализатора: После удаления материала из реактора, катализатор в реакторе промывали восходящим потоком осушенного толуола 10 мл/мин при температуре окружающей среды в течение 1 часа, и затем избыток толуола удаляли из реактора. Катализатор в реакторе сушили при 124°C (255°F) в восходящем потоке 250 см3/мин азота в течение 1 часа. Смешанный раствор воды (4 масс.%) и этанола пропускали через реактор в режиме восходящего потока при 2,2 мл/мин при 154°C (310°F) и давлении окружающей среды в течение 6 часов. Катализатор сушили при 154°C (310°F) в восходящем потоке 150 см3/мин газообразного азота в течение 1 часа 25 минут. Осуществляли циркуляцию раствора н-бутоксида титана (67,5 г н-бутоксида титана в 800 мл толуола) через реактор при объемной скорости 15 мл/мин в режиме восходящего потока при комнатной температуре в течение 20 минут, и затем при 134°C (275°F) и 3,4 бар (35 фунт/дюйм2) в течение 6 часов. После охлаждения, из реактора удаляли избыток раствора. Катализатор промывали восходящим потоком толуола при 4 мл/мин в течение 1,5 часов. Промытый катализатор сушили при 138°C (280°F) в восходящем потоке 300 см3/мин газообразного азота в течение 2 часов и использовали во втором цикле переэтерификации следующим образом.
2-ой цикл переэтерификации: Реактивированный катализатор использовали во 2-ом цикле переэтерификации с таким же вводимым раствором сырья и при таких же условиях, как и при 1-ом цикле переэтерификации. Результат приведен на фигуре 7. Были получены такие же результаты, как при 1-ом цикле переэтерификации.
2-ая реактивация катализатора. 2-ою реактивацию катализатора проводили так же, как 1-ую реактивацию катализатора.
3-ий цикл переэтерификации. Реактивированный катализатор, полученный при 2-ой реактивации катализатора, использовали в 3-ем цикле переэтерификации с растворимыми соединениями титана, добавляемыми к такому же вводимому раствору сырья и при таких же условиях, как при 1-ом цикле переэтерификации. Результаты 3-его цикла переэтерификации приведены на фигуре 7. Был получен такой же результат, как в случае 1-го цикла переэтерификации, но катализатор сохранял стабильную каталитическую активность в течение длительного периода времени. Анализ микропримесей в образце, взятом через 270 часов работы, показал содержание 47 ppm Ti по массе. После того как был взят образец через 270 часов работы, резервуар для сырья был повторно заполнен новым сырьем. К сожалению, сырье становилось мутным при смешении с н-бутоксидом титана. По-видимому, помутнение было связано с непредвиденным более высоким содержанием воды в растворе сырья по сравнению с предыдущими растворами сырья. Активность катализатора быстро снижалась в случае использования этого нового раствора сырья. Анализ микропримесей в смешанном продукте в случае использования этого нового сырья показал содержание 9 ppm Ti по массе. Был сделан вывод о том, что содержание воды во вводимом потоке сырья должно поддерживаться на уровне ниже чем 650 частей на миллион (ppm) по массе.
Холостой эксперимент (без осуществления иммобилизации алкоксида Ti на катализаторе): В аналогичный реактор загружали 25 мл (9,54 г) носителя из гранулированного оксида кремния, приготовленного путем обработки гранул оксида кремния с помощью раствора гидроксида натрия (10 г NaOH в 570 мл воды) при приблизительно 52°C в течение 5 минут при перемешивании. Оксид кремния тщательно промывали холодной водой и затем горячей водой (приблизительно 80°C) для удаления следовых количеств натрия на оксиде кремния. Обработанный оксид кремния сначала сушили при комнатной температуре, затем сушили при 130°C в течение 1,5 часов и затем при 150°C в течение 1 часа в вакуумном сушильном шкафу. Катализатор не подвергали процедуре прививки на носитель. Реакцию переэтерификации проводили с содержанием 42 частей на миллион (ppm) Ti по массе при таком же, как выше, составе сырья и при аналогичных условиях (при кипении реагентов при 168°C (335°F) и 2,4 бар (20 фунт/дюйм2) при введении сырья в режиме восходящего потока при объемной скорости 0,2 мл/мин, где состав сырья представлял собой 26,07 масс.% фенола и 73,93 масс.% ДЭК (мольное отношение ДЭК/фенол 2,56). Результаты приведены на фигуре 7. За время работы конверсия фенола составляла менее чем 2%.
Эта последовательность проведения эксперимента (эксперимента 5) убедительно продемонстрировала возможность реактивации дезактивированного катализатора и увеличения продолжительности рабочего цикла катализатора до более чем 250 часов. Холостой эксперимент ясно показал необходимость предварительного приготовления катализатора перед проведением переэтерификации. В качестве варианта, возможно инициирование переэтерификации с помощью предварительно приготовленного привитого катализатора из алкоксида титана вне реактора. Этот эксперимент также указывает на необходимость регулирования содержания воды в сырье до значения ниже, чем приблизительно 650 частей на миллион (ppm) по массе для сохранения стабильной активности катализатора.
Эксперимент 6
Цель этого эксперимента заключалась в демонстрации непрерывного получения ароматических карбонатов в нескольких соединенных последовательно реакторах в присутствии катализатора из оксида титана, нанесенного на носитель из оксида кремния. Аналогичный, как в эксперименте 3, гранулированный силикагель (40,7 г) обрабатывали с помощью раствора гидроксида натрия (6,86 г NaOH в 500 мл воды) при температуре окружающей среды в течение 7 минут при перемешивании. Силикагель сначала тщательно промывали холодной водой и затем горячей водой (приблизительно 80°C) для удаления следовых количеств натрия на оксиде кремния. Обработанный силикагель сушили при 140°C в течение 2 часов, при 345°C в течение 3 часов и затем при 375°C в течение 2 часов. 30 мл (10,99 г) силикагеля импрегнировали с помощью раствора н-бутоксида титана, приготовленного путем растворения 4,71 г н-бутоксида титана в 80 мл осушенного толуола. Пропитанный носитель из силикагеля прокаливали при 500°C в течение 3 часов. Содержание титана в катализаторе из оксида титана, нанесенного на оксид кремния, составляло 5,48% Ti по массе, если исходить из используемого количества н-бутоксида титана. Загружали в реактор 25 мл (9,8 г) катализатора из оксида титана, нанесенного на оксид кремния. Переэтерификацию ДЭК с фенолом проводили при различных условиях. Сырьем в период работы от 0 часов до 308 часов являются отличающиеся друг от друга смеси ДЭК и фенола. Эти смеси сырья использовали для проведения 1-ой переэтерификации ДЭК с фенолом. Содержание титана во вводимых растворах ДЭК/PhOH (в период работы от 0 часов до 308 часов), приготавливаемых путем добавленимя в них исходного раствора Ti(OEt)4-x(OPh)x (где x= ~2), составляло 59 ppm Ti по массе. Исходный раствор Ti(OEt)4-x(OPh)x готовили путем отгонки этанола из раствора, приготовленного путем смешения соответствующего количества тетраэтоксида титана со смешанным раствором ДЭК и фенола (PhOH) (25 масс.%) при температуре от 120°C до 125°C в течение приблизительно 3 часов. Составы сырья для периода работы от 308 часов до 986 часов приготавливали путем отгонки этанола из смешанных продуктов 1-ой переэтерификации. Эти составы сырья использовали для проведения 2-ой переэтерификации, которая эквивалентна реакциям во втором реакторе в ряду, или эквивалентна реакциям на нескольких ступенях ниже точки ввода сырья многоступенчатой каталитической дистилляционной колонны. Составы сырья для периода работы от 986 часов до 1136 часов приготавливали путем отгонки этанола из смешанных продуктов 2-ой переэтерификации. Эти составы сырья использовали для проведения 3-ей переэтерификации. Растворимый компонент титанового катализатора не добавляли в составы сырья для 2-ой или 3-ей переэтерификации. Составы сырья приведены в таблице 4. Переэтерификацию проводили при 185°C (365°F), 2,9 бар (27 фунт/дюйм2) и скорости подачи сырья 0,24 мл/мин. Результат этого эксперимента приведен на фигуре 8. Конверсия фенола на фигуре 8 представляет собой суммарную конверсию фенола в результате 1-ой переэтерификации, 2-ой переэтерификации и 3-ей переэтерификации. В процессе работы (в течение 1362 часов непрерывной работы) признаки дезактивации катализатора отсутствовали. Исследование катализатора, извлеченного из реактора после окончания работы, показало наличие небольшого отложения полимеров с более высокими молекулярными массами. Анализ катализатора дал содержание 2,3% Ti по массе, что указывало на то, что потери Ti вследствие его вымывания в поток продукта составляли приблизительно 58%. Анализ следовых количеств Ti в потоках продукта, проведенный на момент 686, 887 и 1293 часов работы, показал 75, 57 и 78 частей на миллион (ppm) Ti по массе, соответственно.
Результат этого эксперимента однозначно продемонстрировал возможность получения ароматических карбонатов в нескольких последовательно соединенных реакторах с продолжительным рабочим циклом катализатора путем добавления в вводимый поток сырья следового количества растворимого соединения Ti. Кроме того, этот эксперимент позволяет сделать вывод об отсутствии необходимости в наличии больших количеств оксида титана на катализаторе, так как любое избыточное количество оксида титана может вымываться в результате образования растворимых титанорганических соединений. Продолжительность рабочего цикла катализатора до момента времени, когда требуется проведение реактивации катализатора, является более чем достаточной. Суммарная селективность для ЭФК и ДФК составляла приблизительно от 98 моль.% до приблизительно 93%, в зависимости от степени превращения фенола и от условий работы.
Эксперимент 7
Целью этого эксперимента являлась демонстрация возможности непрерывного получения ароматических карбонатов в нескольких соединенных последовательно реакторах путем проведения переэтерификации ДЭК с фенолом в присутствии катализатора из этоксида титана, иммобилизированного на носителе из силикагеля.
Этот эксперимент состоит из двух частей: эксперимента 7A и эксперимента 7B. В эксперименте 7A, иммобилизацию этоксида Ti на носителе из силикагеля осуществляли до проведения переэтерификации. Вводимое сырье содержало различные количества растворимого соединения Ti(OEt)4-x(OPh)x (где x= ~2). В эксперименте 7B, в пространство в реакторе объемом 25 мл загружали 25 мл носителя из силикагеля, и переэтерификацию проводили без прививки тетраэтоксида Ti на носитель из оксида кремния.
Эксперимент 7A
Носителем, используемым для приготовления катализатора непосредственно в реакторе, являлись гранулы силикагеля сферической формы (диаметром 1,7-4 мм). Этот носитель из силикагеля имел приблизительно 6 гидроксильных групп на нм2, удельную поверхность 392 м2/г по БЭТ, объем пор 0,633 см3/г, средний диаметр пор 6,48 нм и кажущуюся объемную плотность приблизительно 0,58 г/мл (ABD). Этот носитель из силикагеля (25 мл; 14,46 г) загружали в реактор. Осуществляли циркуляцию раствора этоксида титана (45,25 г этоксида титана в 800 мл толуола) через реактор при объемной скорости 15 мл/мин в режиме восходящего потока при температуре окружающей среды в течение 20 минут и затем при 135°C (275°F) и 3,4 бар (35 фунт/дюйм2) в течение 6 часов, для прививки этоксида титана на носитель из силикагеля. После охлаждения, из системы удаляли избыток раствора, и затем катализатор промывали толуолом при 4 мл/мин в течение 1,5 часов. Промытый катализатор сушили при 138°C (280°F) в течение 2 часов в потоке 300 см3/мин газообразного азота.
Реакции получения ЭФК и ДФК проводили при различных условиях. Результат представлен на фигурах 9A и 9B. Все реакции первой переэтерификации проводили при содержании 59 ppm Ti в виде Ti(OEt)4-x(OPh)x (где x= ~2), добавленного в вводимый поток сырья, за исключением периода работы от 709 часов до 799 часов, когда добавляли 151 ppm Ti. Эксперимент начинали при 185°C (365°F), 2,9 бар (27 фунт/дюйм2) и 0,24 мл/мин для первой переэтерификации. После первых 50 часов работы, температуру медленно понижали до 174°C (345°F), а скорость подачи сырья медленно повышали до 0,5 мл/мин в течение 96 часов работы. В дальнейшем и 1-ую, и 2-ую переэтерификации проводили при условии 174°C (345°F), 2,9 бар (27 фунт/дюйм2) и 0,5 мл/мин. Путем отгонки этанола из смешанных продуктов 1-ой, 2-ой и 3-ей переэтерификаций, приготавливали смеси сырья для 2-ой, 3-ей и 4-ой переэтерификаций. В интервале времени работы от 973 часов до 1064 часов, проводили смешанную переэтерификацию и диспропорционирование при 174°C (345°F) и 2,4 бар (20 фунт/дюйм2), 0,5 мл/мин скорости подачи сырья. Состав сырья для реакции представлял собой 18,553% ДЭК, 0,108% этилбутилкарбоната, 0,283% фенетола, 0,182% неиндентифицированных соединений, 57,508% фенола, 22,03% ЭФК, 0,054% п-феноксифенилметилкарбоната и 1,282% ДФК по массе. Результат показывает, что основной реакцией является диспропорционирование. Но анализ данных позволяет также сделать вывод о необходимости удаления ДЭК из сырья для диспропорционирования. На фигуре 9A, 2-ые переэтерификации проводили при 174°C (345°F), 2,4 бар (20 фунт/дюйм2) и 0,5 мл/мин при концентрации Ti в сырье от 44 частей на миллион (ppm) Ti до 69 частей на миллион (ppm) Ti по массе. На фигуре 9B, 2-ые переэтерификации проводили при 174°C (345°F), 2,9 бар (27 фунт/дюйм2) и 0,5 мл/мин при концентрации Ti в сырье от 45 частей на миллион (ppm) Ti до 75 частей на миллион (ppm) Ti по массе. 3-ью переэтерификацию проводили при 174°C (345°F), 2,5 бар (22 фунт/дюйм2) и 0,5 мл/мин при концентрации Ti в сырье от 52 частей на миллион (ppm) Ti до 74 частей на миллион (ppm) Ti по массе. 4-ую переэтерификацию проводили при 174°C (345°F), 2,4 бар (20 фунт/дюйм2) и 0,5 мл/мин при концентрации Ti в сырье от 51 частей на миллион (ppm) Ti до 73 частей на миллион (ppm) Ti по массе.
Селективность по ароматическим карбонатам понижалась с увеличением конверсии фенола. Суммарная селективность по ЭФК и ДФК при 1-ой переэтерификации составляет приблизительно 99 моль.% в расчете на фенол. Суммарная селективность по ЭФК и ДФК при 4-ой переэтерификации составляла от 94 моль.% до 95 моль.% в расчете на фенол.
Твердый катализатор проработал в течение более 14 месяцев до момента прекращения эксперимента, причина которого не была связана с падением активности катализатора. Данные на фигурах 9A и 9B позволяют однозначно сделать вывод о том, что в течение 14 месяцев или наблюдалась незначительная дезактивация катализатора, или дезактивация катализатора полностью отсутствовала. Анализы двух образцов катализатора, осторожно взятых сверху и снизу слоя катализатора, дают для обоих образцов катализатора (после прокаливания при 550°C) одинаковое количество 0,28 масс.% Ti. Этот эксперимент убедительно доказывает возможность достижения продолжительного рабочего цикла катализатора (в течение более чем 14 месяцев) в результате добавления во вводимый поток сырья следового количества растворимого соединения титана.
Эксперимент 7B (холостой эксперимент)
Целью этого эксперимента являлась попытка иммобилизации алкоксида Ti на носителе из оксида кремния при проведении переэтерификации. В поток сырья вводили различные количества соединения Ti(OEt)x(OPh)4-x. Переэтерификацию ДЭК с фенолом проводили при 174°C (345°F) и 2,9 бар (27 фунт/дюйм2). Результат приведен на фигуре 10.
При сравнении холостых экспериментов для эксперимента 5 (фигура 7) и эксперимента 7B (фигура 10) с результатом на фигурах 9A и 9B, становится очевидной необходимость иммобилизации алкоксида титана на носителе из силикагеля перед проведением переэтерификаций. При сопоставлении данных сравнительных экспериментов 1 и 2 (фигура 4) с данными на фигурах 9A и 9B, также становится очевидным преимущество новой технологии использования твердого катализатора с добавлением следового количества растворимого активного металлоорганического соединения в сырье перед технологиями известного уровня техники.
Эксперимент 8
Цель этого эксперимента заключалась в демонстрации диспропорционирования ЭФК в ДФК и ДЭК в отсутствие твердого катализатора, но в присутствии растворимых каталитических компонентов Ti. Сырье для диспропорционирования приготавливали путем отгонки этанола, ДЭК и части фенола из смешанных продуктов с 4-ой переэтерификации в эксперименте 7 в атмосфере азота. Гомогенный Ti катализатор в смесях сырья образовывался из смешанных продуктов 4-ой переэтерификации. С смеси сырья никакого дополнительного растворимого Ti катализатора не добавляли. В две смеси сырья добавляли толуол для создания паровой фазы при температуре кипения в реакторе. Первый состав сырья представлял собой 16,26% толуола, 1,61% ДЭК, 49,33% фенола, 30,91% ЭФК и 0,78% ДФК по массе и остальную часть составляли побочные продукты, включающие следовые количества МФК. Второй состав сырья представлял собой 16,15% толуола, 1,61% ДЭК, 49,28% фенола, 31,08% ЭФК и 0,80% ДФК по массе и остальную часть составляли побочные продукты, включающие следовые количества МФК. Концентрация гомогенного катализатора в первом и во втором составах сырья составляла 180 частей на миллион (ppm) и 200 частей на миллион (ppm) Ti по массе, соответственно.
Диспропорционирование проводили в реакторе с незаполненной зоной катализатора объемом 25 мл (в отсутствие твердого катализатора) при 179°C (355°F) и 2,9 бар (27 фунт/дюйм2). Скорость подачи сырья в режиме восходящего потока составляла 0,5 мл/мин в течение первых 72 часов работы и затем 0,60 мл/мин в режиме восходящего потока в дальнейшем. Результаты проведения реакций диспропорционирования приведены на фигуре 11. Результаты эксперимента указывают, что помимо ДФК также образуются небольшие количества ЭФК. Ксантон являлся единственным новым побочным продуктом, образующимся в процессе диспропорционирования в количестве приблизительно 35 частей на миллион (ppm) по массе. Дифениловый эфир не обнаруживался ни в одном из анализируемых образцов. Селективность по всем побочным продуктам составляла от 3,0 моль.% до 3,3 моль.%. Этот эксперимент убедительно продемонстрировал возможность диспропорционирования ЭФК с получением ДФК и ДЭК согласно раскрываемым в изобретении вариантам осуществления.
Эксперимент 9
Этот эксперимент демонстрирует проведение очистки ДФК. Смешанный продукт диспропорционирования из эксперимента 8 подвергали дистилляции для удаления этанола, ДЭК и значительной части фенола с помощью лабораторного дистилляционного оборудования. Остающийся в дистилляционной колбе материал имел следующий состав: 0,024% EtOH, 0,204% ДЭК, 0,017% фенетола, 1,619% неидентифицированных продуктов, 12,563% фенола, 25,377% ЭФК, 59,474% ДФК и 0,723% продуктов с более высокими молекулярными массами. В результате проведения вакуумной дистилляции получали сырой ДФК (отбираемый при температуре пара от 235 до 245°C). Состав этого сырого ДФК представлял собой 0,535% неидентифицированных продуктов, 2,112% фенола, 0,013% фенилового эфира, 0,030% ЭФК, 94,555% ДФК, 0,026% ксантона и 2,73% продуктов с более высокими молекулярными массами. Этот сырой ДФК перекристаллизовывали в смеси 5 масс.% диэтилового эфира в гексане пять раз. Конечный продукт ДФК содержал примеси 0,4 частей на миллион (ppm) ксантона и 11,6 частей на миллион (ppm) фенола по массе. Никаких других примесей с помощью следового анализа обнаружено не было. Этот продукт ДФК имел более высокую чистоту, чем ДФК высокой чистоты, продаваемый на рынке (28,7 частей на миллион (ppm) неидентифицированных продуктов и 67,2 частей на миллион (ppm) фенола по массе).
Получение диалкилкарбонатов путем переэтерификации циклического карбоната со спиртом
Диалкилкарбонаты получают в непрерывном режиме путем проведения переэтерификации циклического карбоната со спиртами в присутствии твердых катализаторов. Как было обсуждено выше, раскрываемые в изобретении варианты осуществления могут, в частности, применяться для непрерывного получения диалкилкарбонатов, таких как ДМК, ДЭК и так далее. Известен ряд гомогенных катализаторов для переэтерификации. При получении диалкилкарбонатов путем проведения переэтерификации циклического карбоната со спиртом в присутствии нанесенных металлоксидных или смешанных металлоксидных катализаторов или твердого катализатора, приготовленного путем иммобилизации гомогенного катализатора на пористом носителе, катализаторы имеют неприемлемо короткий рабочий цикл для использования в крупнотоннажных промышленных реакторах. Постоянная дезактивация катализатора, происходящая при контакте с органическими карбонатами, является следствием вымывания активных каталитических компонентов из гетерогенных катализаторов в реакционную среду. Поэтому, диалкилкарбонат, такой как ДМК, обычно получают путем проведения переэтерификации в присутствии гомогенного катализатора.
Однако раскрываемые в изобретении варианты осуществления предлагают способы получения диалкилкарбоната в присутствии твердого катализатора. Твердые катализаторы могут включать один или более элементов из Групп II, III, IV, V и VI Периодической таблицы элементов. Первый тип твердого катализатора включает одно или более металлоорганических соединений упомянутых выше элементов, иммобилизированных на пористом носителе, который может иметь поверхностные функциональные группы, такие как гидроксильные, карбонильные, алкоксильные, смесь гидроксильных и алкоксильных групп, хлор и так далее. Носители могут включать оксид кремния, оксид титана, цеолитные материалы, такие как MCM-41, MCM-48, SBA-15, углерод и/или углеродистые материалы и так далее. Второй тип твердого катализатора включает оксиды металлов, гидроксиды или оксигидроксиды одного или более из упомянутых выше элементов, нанесенных на пористый носитель. Для поддержания стабильной каталитической активности, во вводимый поток сырья добавляют следовое количество растворимого каталитического компонента. В результате этого, продолжительность рабочего цикла катализатора может быть увеличена до значений, при которых он может применяться в промышленном реакторе.
Переэтерификация может быть проведена в любых физических устройствах, таких как традиционные реакторы с неподвижным слоем, каталитические дистилляционные колонны, реакторы для проведения реакции при кипении реагентов, дистилляционные колонны с разделительной стенкой, реактор с пульсирующими потоками, или любая комбинация из этих устройств. Примеры комбинации могут включать реактор с неподвижным слоем для проведения реакции при кипении реагентов, за которым следует каталитический реактор с дистилляционной колонной. Переэтерификация циклических карбонатов с первичным спиртом, таким как этанол или метанол, может быть проведена в две стадии, на которых присутствуют два промежуточных соединения этил пропиленгликолькарбоната. Кроме того, реакционный продукт содержит небольшие количества побочных продуктов, таких как изомеры этилового эфира пропиленгликоля, образующиеся в результате O-алкилирования пропиленгликоля с помощью ДЭК. Переэтерификация может быть проведена только в одной реакционной зоне или в нескольких реакционных зонах.
На фигуре 18 приведена упрощенная принципиальная технологическая схема непрерывного получения ДЭК и пропиленгликоля в качестве побочного продукта путем проведения переэтерификации пропиленкарбоната с этанолом в присутствии твердого катализатора согласно раскрываемым в изобретении вариантам осуществления. Переэтерификация, как показано, может быть проведена в каталитическом дистилляционном реакторе 101 при температуре приблизительно от 149°C до приблизительно 177°C (приблизительно от 300°F до 350°F) при давлении приблизительно от давлений ниже атмосферного давления до приблизительно 11,4 бар (то есть, приблизительно от 7 фунт/дюйм2 до 165 фунт/дюйм2), в зависимости от состава реакционной смеси. Помимо каталитического дистилляционного реактора 101, способ включает две дистилляционные колонны, 102 и 114. Каталитическая дистилляционная колонна 101 включает реакционную зону RZ, в которой может быть расположен твердый катализатор. Вводимый свежий пропиленкарбонат 105 объединяют с рециркулируемым потоком 117, и объединенный поток 106 вводят в каталитическую дистилляционную колонну 101 в подходящем месте над слоем твердого катализатора реакционной зоны RZ.
Отбираемый сверху колонны 101 поток 107, смесь этанола, ДЭК и легкокипящих веществ, таких как диоксид углерода, вводят в колонну 102 извлечения ДЭК, для отделения ДЭК от более легкокипящих компонентов. Отбираемый сверху колонны 102 поток 108 может быть введен в цилиндрический газожидкостной сепаратор 110 для отделения жидкого этанола от газов, удаляемых по линии 111. Жидкий этанол, извлеченный из сепаратора 110, в потоке 112 объединяют с вводимым потоком 103 свежего этанола, и объединенный поток 104 нагревают для получения паров этанола, вводимых в каталитическую дистилляционную колонну 101 в соответствующем месте ниже реакционной зоны RZ. Поток кубового остатка 109 дистилляционной колонны 102 содержит продукт, ДЭК, который может быть направлен в резервуар для хранения (не показан) или в другие следующие далее процессы переработки.
Поток кубового остатка 113 из каталитической дистилляционной колонны 101, который содержит пропиленгликоль, пропиленкарбонат, промежуточные продукты реакции и побочные продукты, такие как 1-этокси-2-пропанол, соединения с более высокими температурами кипения, и так далее, и следовое количество катализатора, вводят во вторую дистилляционную колонну 114 для извлечения отбираемого сверху потока 115, содержащего пропиленгликоль, 1-этокси-2-пропанол и так далее. Пропиленгликоль может быть извлечен из смеси в потоке 115 путем дистилляции (не показано). Поток кубового остатка 116 колонны 114 рециркулируют в каталитическую дистилляционную колонну 101 по линиям 117 и 106. Часть потока кубового остатка 116 может быть отдута из системы с помощью потока 118 для предотвращения накопления более высококипящих соединений в системе.
Следовое количество растворимого металлоорганического катализатора вводят в каталитическую дистилляционную колонну 101 выше каталитической реакционной зоны по линии 119. В некоторых вариантах осуществления, раствор катализатора вводят при такой скорости, чтобы жидкая реакционная смесь, стекающая в каталитическую реакционную зону RZ, содержала следовое количество, обычно от 5 частей на миллион (ppm) до приблизительно 100 частей на миллион (ppm) металла по массе, растворимого компонента металла, такого как соединения Mg, Ca, Zn, La, Ac, или Ti.
Получение диалкилкарбонатов иллюстрируются с помощью следующих экспериментов.
Эксперимент 10
Цель этого эксперимента заключалась в демонстрации переэтерификации пропиленкарбоната с этанолом с получением ДЭК и пропиленгликоля в присутствии твердого катализатора. Твердый катализатор приготавливают непосредственно в реакторе путем иммобилизации этоксида титана на носителе из силикагеля.
В реактор загружали 25 мл носителя из сферического силикагеля (диаметром 1,7-4 мм). Масса носителя составляла 10,097 г. Этот носитель из силикагеля имел около 6 гидроксильных групп на нм2, удельную площадь поверхности 314 м2/г по БЭТ, объем пор 1,055 см3/г и средний диаметр пор 13,46 нм. Осуществляли циркуляцию раствора этоксида титана (40 г этоксида титана в 800 мл толуола) в режиме восходящего потока через реактор при скорости 15 мл/мин при температуре окружающей среды в течение 30 минут и затем при 135°C (275°F) и 3,4 бар (35 фунт/дюйм2) в течение 6 часов, для прививки этоксида титана на носитель из силикагеля. После охлаждения, удаляли из реактора избыток раствора, и затем катализатор промывали с помощью толуола при скорости подачи 4 мл/мин в течение 1,5 часов. Промытый катализатор сушили при 138°C (280°F) в течение 2 часов в восходящем потоке газообразного азота при скорости подачи 300 см3/мин.
Готовили смешанные растворы пропиленкарбоната и этанола, и добавляли 45 частей на миллион (ppm)Ti в виде этоксида титана в смешанные растворы вводимого сырья. Переэтерификации проводили с использованием различных смесей сырья в восходящем потоке жидкой фазы при 174°C (345°F) и 17,9 бар (245 фунт/дюйм2). Условия работы реактора приведены в таблице 5. Результаты этого эксперимента приведены на фигуре 12.
в сырье
Результаты этого эксперимента однозначно доказывают, что ДЭК (диалкилкарбонат) может быть получен путем проведения переэтерификации циклического карбоната, такого как пропиленкарбонат, с этанолом в присутствии твердого катализатора из алкоксида Ti, иммобилизированного на носителе из силикагеля путем добавления следового количества растворимого соединения Ti в поток сырья. Без добавления следового количества Ti в поток сырья, активность катализатора быстро падает, как показано на фигуре 12, в течение часов работы 1397-1469.
Эксперимент 11
Целью этого эксперимента является демонстрация переэтерификации пропиленкарбоната с этанолом с получением ДЭК и пропиленгликоля в присутствии твердого катализатора. Эксперимент состоит из двух частей: сравнительных экспериментов 11A (не в соответствии с изобретением) и 11B.
Сравнительный эксперимент 11A
Переэтерификацию проводили в присутствии гомогенного катализатора третбутоксида магния. Мольное отношение этанол/пропиленкарбонат в сырьевой смеси составляло 6,85. Концентрация гомогенного катализатора составляла 57 ppm Mg по массе. Переэтерификацию проводили при 168°C (335°F), 17,9 бар (245 фунт/дюйм2) и 0,5 мл/мин. Результат приведен на фигуре 13. Средняя конверсия пропиленкарбоната составляет около 24,3 моль.%. Средняя селективность по ДЭК и пропиленгликолю составляет 95,7 и 94,3 моль.%, соответственно.
Эксперимент 11B
Переэтерификацию проводили в присутствии твердого катализатора. Исходным твердым катализатором являлся MgO, нанесенный на носитель из силикагеля. Раствор нитрата магния готовили путем растворения 10,098 г Mg(NO3)2∙6H2O в 22,73 г деионизированной воды. Нитрат магния наносили на 30 мл (11,777 г) такого же носителя из силикагеля, как в эксперименте 10, путем постепенного импрегнирования. Импрегнированный продукт сушили при 100°C в вакуумном сушильном шкафу в течение одного часа и затем прокаливали при 510°C в течение 2 часов с получением MgO, нанесенного на силикагель. Загружали в реактор 25 мл (10,77 г) этого поверхностного смешанного оксидного катализатора из MgO и силикагеля. Переэтерификацию пропиленкарбоната с этанолом проводили при различных условиях, приведенных в таблице 6.
(час)
в сырье
(масс. ppm)
Продукты реакции содержали 1-этокси-2-пропанол и дипропиленгликоль в качестве побочного продукта. Диэтиловый эфир не был обнаружен ни в одном образце. Результат также приведен на фигуре 14. Средняя селективность по ДЭК и пропиленгликолю для 1-ой переэтерификации составляет 95,3 моль.% и 94,8 моль.%, соответственно. Обычно, селективность медленно уменьшается с ростом конверсии пропиленкарбоната. Кроме того, селективность возрастает с увеличением мольного отношения EtOH/пропиленкарбонат. Средняя селективность по ДЭК и пропиленгликолю для 2-ой переэтерификации составляет 94,0 моль.% и 92,8 моль.%, соответственно.
Получение диалкилкарбонатов из мочевины и спирта согласно раскрываемым в изобретении вариантам осуществления
Согласно упомянутым выше публикациям, например, P. Ball et al. и D. Wang et al, гетерогенные катализаторы, применяемые для получения диалкилкарбонатов из мочевины и спирта, могут включать Al2O3, Sb2O3, и оксид кремния. Плавленый SiO2 не является катализатором, но может становиться каталитически активным в присутствии PPh3, ZnO и MgO, нанесенных на оксид кремния, могут также быть использованы для получения диалкилкарбонатов из мочевины и спирта.
Однако металлоксидные катализаторы, такие как ZnO или MgO, вымываются из твердых катализаторов в условиях проведения реакции, что приводит к постоянной дезактивации катализатора. Продолжительность рабочего цикла катализатора является очень важной характеристикой при промышленном получении диалкилкарбонатов с использованием каталитического реактора с дистилляционной колонной, так как каталитическая дистилляция обеспечивает быстрое удаление ДМК или ДЭК и аммиака из жидкой каталитической реакционной среды, что повышает производительность и селективность по диалкилкарбонату. Кроме того, описанные выше гетерогенные катализаторы не являются такими же эффективными, как гомогенный катализатор из диметоксида дибутилолова.
Согласно раскрываемым в изобретении вариантам осуществления, диалкилкарбонаты можно непрерывно получать путем алкоголиза мочевины со спиртом в две стадии в присутствии твердого катализатора. Обе стадии реакции являются обратимыми. Примеры спиртов, применяемых для получения диалкилкарбонатов, включают метанол, этанол, пропанол и другие спирты. На первой стадии реакции, мочевина реагирует со спиртом в реакционной дистилляционной колонне (предварительном реакторе), служащей в качестве первой реакционной зоны, либо в отсутствие, либо в присутствии катализатора, с образованием алкилкарбамата и аммиака. Присутствие катализатора не является необходимым на первой стадии реакции. Кроме того, в первой реакционной зоне из потоков сырья удаляют примеси, такие как вода и карбамат аммония, в виде CO2 и аммиака, тем самым защищая катализаторы для проводимых последующих реакций. На второй стадии реакции, алкилкарбамат, образовавшийся в первой реакционной зоне, реагирует со спиртом с образованием диалкилкарбоната и аммиака в присутствии твердого катализатора в одной или более каталитических дистилляционных колоннах (в основных реакторах), которые служат в качестве второй реакционной зоны. Эта двухступенчатая реакция может быть проиллюстрирована следующим образом:
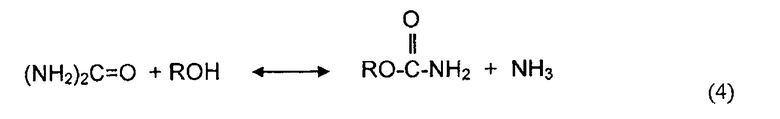
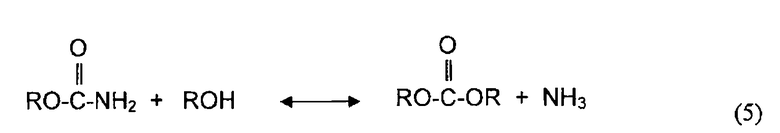
Твердый катализатор может быть приготовлен путем, например, иммобилизации титанорганического соединения на носителе, таком как оксид кремния или углеродистый материал. В начале реакции присутствуют два типа катализаторов. Первым типом катализатора является алкоксид металла, соль металла и моноэфира угольной кислоты, или их смесь, иммобилизированные на носителе, таком как оксид кремния или углеродистый материал. Вторым типом катализатора является оксид металла, нанесенный на носитель, такой как оксид кремния, оксид алюминия, оксид титана, оксид циркония, углеродистый материал и другие носители. Компонентами активных металлов могут быть элементы, такие как Sn, Sb, Ti, Zr, Zn, Mg, Ca, и другие элементы.
И в этом случае, небольшое количество растворимого соединения металла добавляют в реакционный поток, входящий в реактор, для поддержания каталитической активности в течение увеличенной продолжительности рабочего цикла. При этом продолжительность рабочего цикла катализатора может быть увеличена до значения, которое приемлемо при применении в промышленных процессах. Очевидно, что при стационарном режиме, действующим катализатором является алкоксид металла, алкоксиалкилкарбонат металла (соль моноэфира угольной кислоты), или их олигомеры и смеси, иммобилизированные на носителях. Концентрация растворимого металлоорганического катализатора, такого как диалкоксид дибутилолова, в реакционной смеси составляет значительно более низкую величину, чем для гомогенного катализатора, используемого в Патенте США № 7074951.
Высококипящий растворитель, такой как триглим, может быть использован в качестве растворителя на второй стадии, и он служит в качестве сокатализатора для повышения скорости реакции и селективности. Важно отметить, что вследствие высокой температуры кипения растворителя, реакция может быть проведена при низком давлении, что способствует быстрому удалению ДЭК и аммиака из жидкой реакционной среды в паровую фазу вместе с избытком паров этанола в качестве отдувочного газа, что в результате приводит к высокой производительности и селективности по ДЭК. Раскрываемые в изобретении варианты осуществления предлагают альтернативный усовершенствованный способ получения диалкилкарбоната в присутствии твердого катализатора. Этот способ может являться "более экологически чистым" способом, так как концентрация катализатора из олова в технологическом потоке значительно ниже, чем в способах на основе гомогенного катализатора, как это показано в примере 14 ниже. Следовое количество растворимого каталитического соединения в технологическом потоке остается в системе. Не требуется никакого извлечения или разделения катализатора из растворимого соединения в технологическом потоке.
На фигуре 15 показана технологическая схема способа непрерывного получения ДЭК согласно раскрываемым в изобретении вариантам осуществления. Реактор с двумя различными диаметрами с дистилляционной колонной 36 используют в качестве предварительного реактора для удаления примесей в потоках сырья и для превращения мочевины в этилкарбамат (ЭК). Раствор мочевины приготавливают в цилиндрической емкости 34 путем смешения потока мочевина 31 и потока этанола 33. Поток этанола 33 может включать поток свежего этанола 32 и поток рециркулируемого этанола 74.
Раствор мочевины 35 из цилиндрической емкости 34 вводят посередине верхней более узкой части колонного реактора с двумя различными диаметрами 36. Реактор 36 служит в качестве предварительного реактора для очистки от примесей (воды и карбамата аммония) сырьевых потоков этанола и мочевины, и для превращения мочевины в ЭК. Поток пара 37 из предварительного реактора 36 состоит из аммиака, диоксида углерода и этанола. Очищенный смешанный раствор выводят из предварительного реактора 36 в виде потока кубового остатка 38. Поток 38 вводят в основной реактор 39 (каталитическую дистилляционную колонну) в месте над каталитической реакционной зоной 39R, содержащей твердый катализатор.
Поток рециркуляции этанола 40 вводят в реактор 39 в виде перегретых паров этанола в месте ниже каталитической реакционной зоны 39R. Поток кубового остатка из каталитической дистилляционной колонны 39 рециркулируют через линии 42, 44 и 78 в верхнюю часть колонны 39 в месте выше точки подачи линии 38. Небольшую часть потока 43 из петли рециркуляции объединяют с потоком кубового остатка 65 из колонны извлечения ДЭК 63 в поток 66, который вводят в реактор очистки 67 в положении выше каталитической реакционной зоны, которая является еще одной небольшой каталитической дистилляционной колонной, содержащей твердый катализатор. Небольшая часть потока 43 может включать этанол, аммиак, этиламин, диэтиловый эфир, ДЭК, этилкарбамат, этил-N-этилкарбамат, триглим (ТГ), соединения с более высокими молекулярными массами и следовое количество растворимого компонента катализатора. Поток кубового остатка 65 из колонны извлечения ДЭК 63 может включать этилкарбамат, этил-N-этилкарбамат, ТГ, и следовое количество катализатора. Отводимый сверху поток 68 из реактора очистки 67 может включать аммиак, этиламин, CO2, диэтиловый эфир, этанол и ДЭК. Поток кубового остатка 69 из реактора очистки 67 может включать аммиак, этиламин, CO2, диэтиловый эфир, этанол, этил-N-этилкарбамат, этилкарбамат, гетероциклические соединения и следовое количество растворимого компонента катализатора.
Поток кубового остатка 69 из реактора 67 охлаждают для осаждения гетероциклических соединений в системе охлаждения/фильтрации 70. Осажденный твердый побочный продукт выводят из системы 70 по линии 71. Жидкий поток 72 из системы 70 разделяют на два потока 77 и 78 для рециркуляции в реактор очистки 67 и основной реактор 39, соответственно.
Отводимый сверху поток 41 из основного реактора 39 может быть объединен с отводимым сверху потоком 68 из реактора очистки 67 в поток 42. Отводимый сверху поток 41 из основного реактора 39 может включать аммиак, CO2, этиламин, диэтиловый эфир, этанол, этилкарбамат, этил-N-этилкарбамат, ДЭК, ТГ и следовое количество катализатора. Объединенный поток 42 вводят в дистилляционную колонну 43, в которой разделяют низкокипящие и высококипящие соединения. Отводимый сверху поток 44 из дистилляционной колонны 43, который может включать аммиак, CO2, этиламин, диэтиловый эфир и этанол, объединяют с отводимым сверху потоком 37 из предварительного реактора 36 в поток 45 для введения в дистилляционную колонну 46.
Отводимый сверху поток 47 из дистилляционной колонны 46 охлаждают с целью инициирования протекания реакции CO2 с аммиаком с образованием карбамата аммония. Карбамат аммония осаждают в жидком аммиаке и выводят в виде твердого вещества по линии 49 из системы охлаждения/фильтрации 48. Поток жидкого аммиака 50 из системы охлаждения/фильтрации 48 направляют в емкость для хранения аммиака.
Поток кубового остатка 51 из колонны 46 может включать этиламин, диэтиловый эфир, этанол и следы ДЭК. Поток 51 вводят в колонну извлечения этиламина 52. Отводимый сверху поток этиламина 53 направляют в емкость для хранения. Поток кубового остатка 54 из колонны 52 объединяют с потоком кубового остатка 55 из дистилляционной колонны 43 в поток 56. Поток 56 вводят в колонну извлечения эфира 57. Эфир выводят из дистилляционной колонны 57 в виде отводимого сверху потока 58, который направляют в емкость для хранения эфира. Поток кубового остатка 59 из дистилляционной колонны 57 вводят в дистилляционную колонну 60 (колонну извлечения этанола).
Извлекаемый этанол в виде отводимого сверху потока 61 рециркулируют в основной реактор 39, реактор очистки 67 и предварительный реактор 36 (или цилиндрическую емкость 34). Поток рециркулируемого этанола 74 является незначительной частью отводимого сверху потока 61 дистилляционной колонны 60 (колонны извлечения этанола). Поток 61 разделяют на три потока: 40, 73 и 74. Поток 73 рециркулируют в реактор очистки 67. Поток 74 рециркулируют в цилиндрическую емкость 34 для приготовления раствора мочевины. Поток 40, который может являться основной частью потока 61, рециркулируют в основной реактор 39. Поток кубового остатка 62 из колонны извлечения этанола 60 вводят в колонну извлечения ДЭК 63. Продукт ДЭК извлекают в виде отводимого сверху потока 64 из дистилляционной колонны 63 и направляют в емкость для хранения ДЭК. Поток кубового остатка 65 из колонны 63 может включать этилкарбамат, этил-N-этилкарбамат, ТГ и следовое количество растворимого компонента катализатора. Этот поток 65 направляют в реактор очистки 67 по линии 66.
ДМК может быть получен из метанола и мочевины способом, который аналогичен способу получения ДЭК, приведенному на фигуре 15. Однако следует иметь в виду, что конечный продукт ДМК извлекают из технологического потока, содержащего азеотропную смесь метанол-ДМК. Извлечение ДМК путем разрушения азеотропной смеси метанол-ДМК с помощью метода экстрактивной дистилляции с растворителем подробно описано в литературе, например, в Патенте США № 7074951.
Эксперимент 12
Целью этого эксперимента является демонстрация реакции этилкарбамата с этанолом с получением ДЭК и аммиака в присутствии твердого катализатора. Твердый катализатор готовили путем иммобилизации диметоксида дибутилолова на силикагеле непосредственно в реакторе.
Загружали в реактор 25 мл (14,79 г) носителя из силикагеля сферической формы, используемого в эксперименте 7A. Раствор диметоксида дибутилолова приготавливали путем смешения 87 г диметоксида дибутилолова в 2 л осушенного толуола. Реактор заполняли этим раствором в режиме восходящего потока при температуре и давлении окружающей среды. Реактор медленно нагревали до 110°C (230°F) при пропускании этого раствора в режиме восходящего потока при 2 мл/мин. При 110°C (230°F) в реакторе создавали давление 3,4 бар (35 фунт/дюйм2) и затем продолжали нагревать до 135°C (275°F). При 135°C (275°F) и 3,4 бар (35 фунт/дюйм2) пропускали через реактор раствор диметоксида дибутилолова в режиме восходящего потока при 0,5 мл/мин в течение 6 часов. После охлаждения, удаляли из реактора избыток раствора и затем промывали катализатор осушенным толуолом в режиме восходящего потока при 4 мл/мин в течение 1,5 часов. Промытый катализатор сушили при 104°C (220°F) при давлении окружающей среды в нисходящем потоке N2 при скорости подачи 300 см3/мин в течение 2 часов.
Реакцию проводили путем пропускания раствора 13,2% этилкарбамата, 31,36% триглима и 55,44% этанола по массе в режиме восходящего потока с азотом через слой твердого катализатора в реакторе с кипящими реагентами. Можно также проводить реакцию в реакторе с нисходящим потоком. В этот раствор добавляли следовые количества диметоксида дибутилолова. Условия реакции приведены в таблице 7, и результаты этого эксперимента показаны на фигуре 16. Анализ продуктов реакции показал присутствие следовых количеств этил-N-этилкарбамата и диэтилового эфира. Селективность по ДЭК в расчете на этилкарбамат составляла от 98,5 моль.% до 99,9 моль.% при общей тенденции понижения селективности по мере увеличения конверсии этилкарбамата.
(фунт/
дюйм2)
(ppm Sn по массе)
Этот эксперимент убедительно показал возможность получения ДЭК из мочевины и этанола. На первой стадии, этилкарбамат получали путем реакции мочевины с этанолом в отсутствие катализатора (смотрите Патент США 7074951). На второй стадии, ДЭК получали путем проведения реакции этилкарбамата с этанолом в присутствии твердого катализатора и с добавлением следового количества растворимого металлоорганического катализатора в потоки сырья для компенсации потери металла вследствие его вымывания. При промышленном производстве ДЭК вторую стадию предпочтительно проводить в одной или более каталитических дистилляционных колоннах.
Совмещение способов получения диалкилкарбоната и диарилкарбоната
Раскрывается совмещенный способ получения диарилкарбонатов, который не требует использования установки экстрактивной дистилляции с растворителем для разделения азеотропных технологических потоков, что приводит к экономии энергии и затрат на установку оборудования, и который снижает выброс парникового газа CO2 в атмосферу. Поэтому, совмещенный способ является более экологически безопасным по сравнению с традиционными способами получения диарилкарбонатов.
Более высокая экологическая безопасность способа может достигаться за счет совмещения способа получения диалкилкарбонатов и способа получения диарилкарбонатов согласно раскрываемым в изобретении вариантам осуществления. Например, диалкилкарбонаты могут быть получены в начале совмещенного способа, по меньшей мере, часть которых может затем быть использована в конце совмещенного способа для получения диарилкарбонатов. Как описано выше, диалкилкарбонаты могут быть получены с помощью одного из двух способов: (1) переэтерификацией циклического карбоната, который может быть получен из эпоксида и диоксида углерода, и (2) реакцией между мочевиной и спиртом, где мочевина может быть синтезирована из аммиака и диоксида углерода. Диалкилкарбонаты, полученные в начале совмещенного процесса с помощью, по меньшей мере, одного из двух упомянутых выше способов, могут затем быть подвергнуты реакции с арилгидроксисоединением в конце совмещенного способа для получения диарилкарбонатов.
Следовательно, совмещенные способы получения диарилкарбонатов согласно раскрываемым в изобретении вариантам осуществления могут быть созданы на основе диалкилкарбонатов, получаемых исключительно из этанола и CO2. Побочный продукт этанол со стадии процесса переэтерификации ДЭК в ЭФК (этилфенилкарбонат) рециркулируют на получение ДЭК. ЭФК подвергают диспропорционированию с получением ДФК и побочного продукта ДЭК. ДЭК получают из диоксида углерода и этанола через промежуточные соединения. Для получения ДЭК не требуется использование установки экстрактивной дистилляции, так как этанол и ДЭК не образуют азеотроп (в отличие от метанола и ДМК, которые образуют азеотроп). Способы получения ДФК из диоксида углерода и фенола могут быть осуществлены с использованием различных многостадийных схем реакций, которые подробно описаны ниже.
Преимущества совмещенных способов получения ДФК из ДЭК согласно раскрываемым в изобретении вариантам осуществления могут включать экономию энергии и экономию конструкционного материала при создании установки. Способы согласно вариантам осуществления не требуют выделения веществ из составов, которые образуют азеотроп, и, следовательно, потребность в оборудовании и потреблении энергии в раскрываемых в изобретении способах может быть снижена по сравнению с получением ДФК из ДМК, которое приводит к образованию азеотропа (ДМК и метанола).
Применение совмещенного способа получения ДЭК и ДФК согласно раскрываемым в изобретении вариантам осуществления может дополнительно обеспечивать несколько преимуществ по сравнению с отдельно используемыми способами. Одним таким преимуществом совмещения является возможность рециркуляции этанола, побочного продукта, получаемого в конце процесса получения ДФК, в начало процесса получения ДЭК в качестве сырья. В результате чего, могут быть снижены суммарные затраты на сырье. В некоторых вариантах осуществления, весь или часть рециркулируемого побочного продукта этанола может быть объединена с подпитывающим потоком свежего этанола для введения в процесс получения ДЭК. В других вариантах осуществления, только рециркулируемого побочного продукта этанола может быть достаточно для введения в качестве сырья в процесс получения ДЭК. В качестве варианта, может быть выбрано применение биоэтанола вместо синтетического этанола для получения ДЭК при пуске совмещенной установки получения ДФК и/или в качестве подпитывающего этанола.
Другим преимуществом использования совмещенного способа получения ДЭК и ДФК является экономия затрат на энергию и конструкционные материалы, связанная с сушкой этанола, вводимого в качестве сырья в процесс получения ДЭК. Например, этанол является гигроскопическим соединением, которое имеет тенденцию поглощать воду из окружающей среды, такую как атмосферная влага. Примеси воды, содержащиеся в этаноле, вводимом в качестве сырья, могут оказывать отрицательное влияние на процесс получения ДЭК, например, путем отравления или дезактивации катализатора и путем забивки технологических трубопроводов. Поэтому, свежий вводимый в качестве сырья этанол в установку получения ДЭК обычно требует осушки для получения высокочистого этанола путем отделения от него примесей воды. В отличие от этого, этанол, извлекаемый в качестве побочного продукта в конце процесса получения ДФК, может содержать немного или совсем не содержать примесей воды. Поэтому, в случае рециркуляции этанола в процесс получения ДЭК, затраты на осушение, включающие затраты на конструкционные материалы и энергетические затраты, могут быть значительно снижены или даже полностью исключены.
Еще одним преимуществом совмещенного способа получения ДЭК и ДФК может являться экономия затрат на строительство и экономия эксплуатационных затрат, возникающая вследствие исключения излишнего технологического оборудования. Например, процесс получения ДЭК и ДФК может требовать сепаратор для отделения ДЭК от этанола. В процессе получения ДЭК, выходящий из реактора поток может содержать как ДЭК, так и непрореагировавший этанол, который может быть необходимо отделять. В процессе получения ДФК, выходящий из реактора поток низкокипящих соединений, извлекаемых при переэтерификации, может содержать как ДЭК, так и этанол, который может быть также необходимо отделять. Однако, совмещение способов получения ДЭК и ДФК согласно раскрываемым в изобретении вариантам осуществления, может позволять использовать только одну систему для разделения ДЭК и этанола, что в результате приводит к экономии затрат на строительство и экономии эксплуатационных затрат.
Один способ получения диарилкарбонатов согласно раскрываемым в изобретении вариантам осуществления включает: 1) реакцию диоксида углерода с эпоксидом с получением циклического карбоната, 2) переэтерификацию циклического карбоната с этанолом с получением диэтилкарбоната и 3) переэтерификацию диэтилкарбоната с образованием этиларилкарбоната, и 4) диспропорционирование этиларилкарбоната с образованием диарилкарбоната. Каждая из этих реакций может быть проведена в одной или более реакционных зонах, между которыми или в которых может быть реализована одна или более стадий разделения для разделения продуктов реакции, реагентов и/или побочных продуктов реакции.
Например, диоксид углерода, производимый на установке синтеза аммиака, установке получения синтез-газа, или в электрогенераторе, может быть подвергнут контактированию с эпоксидом, таким как, по меньшей мере, один из оксида этилена и оксида пропилена, в реакторе синтеза циклического карбоната с получением циклического карбоната. Выходящий из реактора синтеза циклического карбоната поток, содержащий циклический карбонат, может быть подвергнут контактированию с этанолом в присутствии катализатора переэтерификации согласно раскрываемым в изобретении вариантам осуществления с получением диэтилкарбоната и гликоля. Продукт диэтилкарбонат и непрореагировавший этанол могут быть извлечены и разделены. Диэтилкарбонат может затем быть использован в реакции переэтерификации для получения этилфенилкарбоната, и для дальнейшего получения дифенилкарбоната из этилфенилкарбоната с помощью реакции диспропорционирования согласно раскрываемым в изобретении вариантам осуществления. Этанол может быть возвращен в систему для переэтерификации циклического карбоната с получением диэтилкарбоната.
Другой способ получения диарилкарбонатов согласно раскрываемым в изобретении вариантам осуществления включает использование мочевины, которая может быть получена путем реакции диоксида углерода с аммиаком. Алкоголиз мочевины с этанолом может затем давать диэтилкарбонат, который может быть подвергнут описанным выше переэтерификации и диспропорционированию с образованием диарилкарбоната. Каждая из этих реакций может быть проведена в одной или более реакционных зонах, между которыми или в которых может быть реализована одна или более стадий разделения для разделения продуктов реакции, реагентов и/или побочных продуктов реакции.
Далее с помощью фигуры 19 иллюстрируется упрощенная технологическая блок-схема совмещенного способа получения диарилкарбонатов согласно раскрываемым в изобретении вариантам осуществления. Как показано на фигуре 19, диарилкарбонат, такой как ДФК, получают из CO2, эпоксида и фенола.
Диоксид углерода и эпоксид, такой как оксид этилена или оксид пропилена, вводят в реакционную зону 203 по линиям 201 и 202 для получения циклического карбоната, такого как этиленкарбонат или пропиленкарбонат. Поток циклического карбоната 204 из зоны синтеза 203 и этанол посредством потока 207 вводят в реакторную систему каталитической дистилляции 205 для проведения переэтерификации с этанолом в присутствии катализатора переэтерификации. Подпитывающий поток свежего этанола 206, в случае необходимости, может быть объединен с потоками рецикла этанола 220 и/или 210 для введения в реакционную зону 205 посредством потока 207. Отводимый сверху поток 208 из реакционной зоны 205 направляют в зону первого разделения 209, которая может представлять собой, например, дистилляционную колонну, для отделения продукта ДЭК от этанола. Отводимый сверху поток этанола 210 из зоны разделения 209 рециркулируют назад в зону переэтерификации 205. Поток ДЭК 211 (жидкий поток кубового остатка) из первой зоны разделения 209 подают в реакционную зону второй переэтерификации 217.
Поток кубового остатка 212 из зоны переэтерификации 205 направляют в зону второго разделения 213, которая может включать одну или более дистилляционных колонн, для разделения продукта гликоля от непрореагировавшего циклического карбоната и промежуточных продуктов реакции. Продукт гликоль из зоны разделения 213 выводят через линию 214. Оставшийся жидкий поток соединений с более высокими температурами кипения 215, включающий циклический карбонат, рециркулируют назад в зону первой переэтерификации 205.
Поток свежего фенола 216 вводят в зону второй переэтерификации 217, которая включает, по меньшей мере, одну реакторную систему каталитической дистилляции, для одновременного проведения переэтерификации ДЭК с фенолом в присутствии катализатора с получением ЭФК и побочного продукта этанола и отделения ЭФК от этанола и непрореагировавшего ДЭК. Отводимый сверху поток 218, который содержит этанол, ДЭК, легкокипящие соединения и небольшое количество фенола, из зоны переэтерификации 217 направляют в зону третьего разделения 219. Разделение этанола и ДЭК, содержащихся в потоке 218, не требует применения экстрактивной дистилляции, как это уже упоминалось выше.
Поток этанола 220 рециркулируют в зону первой переэтерификации 205 по линии 207. Легкокипящие соединения удаляются через линию 221. Поток кубового остатка 222, который содержит ДЭК и фенол, рециркулируют назад в зону второй переэтерификации 217 по линии 222. Поток кубового остатка 223 из зоны второй переэтерификации 217, который содержит продукт ЭФК, фенол и небольшие количества ДФК, направляют в зону диспропорционирования ЭФК 224, которая включает, по меньшей мере, одну реакторную систему каталитической дистилляции, работающую под низким вакуумом. Отводимый сверху поток 225 из реакционной зоны 224, который содержит ДЭК, фенол и небольшое количество этанола, рециркулируют назад в верхнюю часть каталитической дистилляционной колонны зоны переэтерификации 217. Поток кубового остатка 226 из реакционной зоны 224 направляют в зону четвертого разделения 227, которая включает две или более дистилляционных колон для разделения материалов в потоке 226. Поток 228 из зоны разделения 227, который содержит непрореагировавший ЭФК и следовое количество фенола, рециркулируют назад в верхнюю часть каталитической дистилляционной колонны реакционной зоны диспропорционирования 224.
Поток конечного продукта ДФК 229 из зоны разделения 227 направляют в емкость для хранения ДФК. Высококипящие соединения в потоке 226 выводят из зоны 227 по линии 230 в качестве отходов или для дальнейшей переработки, если в этом есть необходимость. В качестве дополнительного варианта, гликоль, извлекаемый из зоны разделения 213, может быть подвергнут дегидратации с образованием эпоксида и воды в реакторе дегидратации 232, откуда эпоксид может быть рециркулирован для получения циклического карбоната, как это было описано выше. В результате рециркуляции гликоля в способе получения ДФК практически отсутствуют побочные продукты. При определенных обстоятельствах, могут быть востребованными способы без получения побочных продуктов, например, в случае отсутствия спроса на гликоль на рынках сбыта.
Далее с помощью фигуры 20 иллюстрируется упрощенная технологическая блок-схема совмещенного способа получения диарилкарбонатов согласно раскрываемым в изобретении вариантам осуществления, на которой одинаковые численные обозначения представляют одинаковые части схем на другой фигуре. Диарилкарбонат, такой как ДФК, получают из CO2 и фенола альтернативным способом. Аммиак используют в этом альтернативном способе для получения промежуточного газа-носителя для транспорта ДЭК, что является отличием от схемы процесса, показанного на фигуре 19.
Поток диоксида углерода 240 и поток аммиака 260, который является объединенным потоком подпитывающего потока аммиака 241 и потока рециркулируемого аммиака 250, вводят в установку синтеза мочевины 242. Побочный продукт H2O в синтезе мочевины удаляют по линии 243 из установки синтеза 242. Поток продукта мочевины 244 из зоны синтеза 242 и вводимый поток этанола 247, который является объединенным потоком рециркулируемого этанола 253 и подпитывающего потока этанола 246, вводят в реакционную зону алкоголиза мочевины 245.
Реакция алкоголиза мочевины в зоне 245 является двухстадийной реакцией. На первой стадии, проводят первый алкоголиз мочевины с этанолом с получением этилкарбамата (C2H5O-CONH2) и с удалением примесей, таких как карбамат аммония с побочным продуктом аммиаком, обычно в отсутствие катализатора. Мочевина может быть растворена в этаноле, и полученный раствор мочевины перекачивают в реакционную дистилляционную колонну. Одновременно, перегретые пары этанола вводят в нижнюю часть колонны для извлечения побочного продукта аммиака из жидкой фазы в паровую фазу для удаления аммиака в качестве части отводимого сверху потока. Фракция кубового остатка, извлекаемая из колонны, включает этилкарбамат, незначительное количество мочевины и этанол. На второй стадии проводят второй алкоголиз этилкарбамата и оставшейся мочевины с этанолом в присутствии катализатора с получением ДЭК и побочного продукта аммиака в другой реакционной дистилляционной колонне.
Поток продуктов 248 из зоны алкоголиза мочевины 245 направляют в зону разделения 249, которая включает одну или несколько дистилляционных колонн. Аммиак из зоны разделения 249 рециркулируют по линиям 250 и 260 в зону синтеза мочевины 242. Извлекаемый поток этанола 251 рециркулируют назад в зону алкоголиза мочевины 245 по линии 251. ДЭК из зоны разделения 249 становится сырьевым потоком ДЭК 252 для проведения переэтерификации с фенолом в реакционной зоне 217. Побочный продукт этанол реакции переэтерификации ДЭК с фенолом в реакционной зоне 217 рециркулируют назад в зону алкоголиза мочевины 245 для получения ДЭК по линиям 253 и 247.
Оставшиеся стадии способа получения ДФК с этого момента являются идентичными по описанию приведенным выше стадиям на фигуре 19. В качестве варианта, мочевина может приобретаться у производителя мочевины, и этому производителю может осуществляться возврат побочного продукта аммиака в качестве частичной оплаты. Это может быть дешевле с точки зрения стоимости мочевины и более эффективно с точки зрения энергозатрат, чем эксплуатация небольшой установки синтеза мочевины непосредственно на месте производства ДФК.
Как уже упоминалось выше, способы получения ДФК, описанные с помощью фигур 19 и 20, могут быть осуществлены практически без введения свежего этанола. Этанол сначала расходуется в процессе переэтерификации мочевины или циклических карбонатов для получения ДЭК, а затем этанол образуется в процессе переэтерификации ДЭК с арилгидроксильным соединением, таким как фенол, для получения диарилкарбоната, такого как ДФК. Так как этанол расходуется и образуется при приблизительно одинаковом мольном отношении, исключая расход этанола в реакции образования побочных продуктов, раскрываемые в изобретении способы могут быть осуществлены с практически замкнутым циклом по этанолу. В силу этого, затраты на этанол в качестве сырья и затраты на его предварительную подготовку (осушение) могут быть в значительной степени снижены по сравнению со способом отдельного получения ДЭК.
Реакционные зоны для проведения упомянутых выше реакций переэтерификации и диспропорционирования могут содержать один или несколько твердых катализаторов и могут состоять из одного или более реакторов. Реактор может иметь любую физическую форму для различных режимов работы для проведения переэтерификации в жидкой фазе или в присутствии двойной фазы жидкости и пара. Для проведения описываемых в изобретении реакций может быть использован любой тип реакторов. Примеры реакторов, подходящих для проведения реакций, в частности, реакций органического карбоната или органических карбаматов, могут включать реакторы с дистилляционной колонной, реакторы с дистилляционной колонной с разделительной стенкой, традиционные трубчатые реакторы с неподвижным слоем, барботажные реакторные колонны, суспензионные реакторы, оборудованные дистилляционной колонной, или без дистилляционной колонны, реакторы с пульсирующим потоком, каталитические дистилляционные колонны, в которых суспензия твердого катализатора стекает в колонне сверху вниз, или любую комбинацию этих реакторов.
Катализатор, применяемый для реакций переэтерификации и диспропорционирования для получения ДЭК, ЭФК и ДФК в этих совмещенных способах, может являться описанным выше катализатором, включающим один или более гомогенных катализаторов, гетерогенный катализатор, и твердый катализатор согласно раскрываемым в изобретении и подробно описанным выше вариантам осуществления. Гетерогенный катализатор может иметь любую физическую форму, такую как, например, порошок, для эксплуатации в реакторе с мешалкой или суспензионном каталитическом реакторе с дистилляционной колонной, или такую форму, как сферы, гранулы, таблетки, экструдаты, ткань, сетка и другие формы.
ПРИМЕРЫ
Все реакции в эксперименте проводили в каталитической дистилляционной реакционной системе, содержащей одноступенчатую вертикально установленную дистилляционную колонну, имеющую диаметр 1,3 см (1/2 дюйма) и высоту 6,5 см (25 дюймов), с неподвижным слоем катализатора и работающую как реактор с восходящим потоком при кипении реагентов, в котором сосуществуют паровая и жидкая фазы. Реактор имел отдельно регулируемые верхнюю и нижнюю зоны нагрева. Объем твердых катализаторов составлял 25 мл.
Следующие примеры экспериментов иллюстрируют варианты осуществления согласно настоящему изобретению получения дифенилкарбоната (ДФК) либо из диэтилкарбоната (ДЭК) и фенола, либо из диоксида углерода и фенола, без необходимости использования экстрактивной дистилляции с растворителем для разделения азеотропа.
Пример 13
Этот пример иллюстрирует вариант осуществления согласно настоящему изобретению получения диэтилкарбоната (ДЭК), представленный на фигуре 19. Переэтерификацию пропиленкарбоната (циклического карбоната) с этанолом в присутствии твердого катализатора с получением ДЭК проводили при различных условиях, приведенных в таблице 8 ниже.
в сырье
Исходным твердым катализатором переэтерификации являлся MgO, нанесенный на силикагель. Раствор нитрата магния приготавливали путем растворения 10,1 г Mg(NO3)2∙6H2O в 22,7 г деионизированной воды. Нитрат магния наносили на 30 мл (11,8 г) носителя из силикагеля путем импрегнирования. Носитель из силикагеля (сферы с диаметром ~3 мм) имели удельную поверхность 314 м2/г по БЭТ, объем пор 1,06 см3/г и средний диаметр пор 13,46 нм. Продукт импрегнирования сушили при 100°C в вакуумном сушильном шкафу в течение одного часа, и затем прокаливали при 510°C в течение 2 часов с получением MgO, нанесенного на силикагеле. В реактор загружали 25 мл (10,77 г) катализатора MgO, нанесенного на силикагель.
Во вводимые реакционные растворы также добавляли третбутоксид магния в различных следовых количествах для достижения стабильной активности катализатора, как показано в таблице 8 выше. Обычно, добавление следового количества растворимого металлоорганического соединения щелочноземельного металла, такого как алкоксид, гликольоксид, или их смесь, может использоваться для достижения увеличенной продолжительности и стабильности рабочего цикла катализатора, как это описано выше для раскрываемых в изобретении вариантов осуществления.
Продукты реакции переэтерификации включали 1-этокси-2-пропанол и дипропиленгликоль в качестве побочного продукта. Диэтиловый эфир ни в одном образце продукта не обнаруживали. Результаты эксперимента 13 также приведены на фигуре 21. Средние значения селективности образования ДЭК и пропиленгликоля при первой переэтерификации составляли 95,3 мольных процента и 94,8 мольных процента, соответственно. Значения селективности медленно снижались с ростом конверсии пропиленкарбоната и повышались с ростом мольного отношения этанола к пропиленкарбонату. Например, средние значения селективности образования ДЭК и пропиленгликоля при второй переэтерификации составляли 94,0 мольных процента и 92,8 мольных процента, соответственно.
Пример 14
Этот пример иллюстрирует показанный на фигуре 20 вариант осуществления согласно настоящему изобретению получения диэтилкарбоната (ДЭК). Алкоголиз этилкарбамата с этанолом с получением ДЭК и побочного продукта аммиака в присутствии твердого катализатора проводили при различных условиях, приведенных в таблице 9 ниже:
(бар (фунт/дюйм2))
(масс.ppm Sn)
Твердый катализатор приготавливали путем иммобилизации диметоксида дибутилолова на силикагеле непосредственно в реакторе. Для приготовления катализатора, загружали в реактор 25 мл (14,79 г) носителя сферической формы из силикагеля, имеющего приблизительный диаметр 3 мм. Носитель из силикагеля имел приблизительно 6 гидроксильных групп на нм2, удельную поверхность 392 м2/г по методу БЭТ, объем пор 0,633 см3/г, средний диаметр пор 6,48 нм и кажущуюся объемную плотность приблизительно 0,58 г/мл.
Раствор диметоксида дибутилолова приготавливали путем смешения 87 г диметоксида дибутилолова с 2 литрами осушенного толуола. Наполняли этим раствором реактор в режиме восходящего потока при температуре и давлении окружающей среды. Реактор медленно нагревали до температуры 110°C (230°F) при добавлении раствора диметоксида дибутилолова в режиме восходящего потока при скорости 2 мл/мин. При 110°C (230°F), в реакторе повышали давление до 3,4 бар (35 фунт/дюйм2) и затем повышали температуру до 135°C (275°F).
При поддержании в реакторе температуры 135°C (275°F) и давления 3,4 бар (35 фунт/дюйм2), через реактор пропускали раствор диметоксида дибутилолова в режиме восходящего потока при 0,5 мл/мин в течение 6 часов. После охлаждения, из реактора удаляли избыток раствора и затем промывали катализатор осушенным толуолом в режиме восходящего потока при 4 мл/мин в течение 1,5 часов. Промытый катализатор сушили при температуре 104°C (220°F) и давлении окружающей среды путем продувки азотом в режиме нисходящего потока при 300 см3/мин в течение 2 часов.
Реакцию алкоголиза этилкарбамата с этанолом для получения ДЭК проводили путем пропускания раствора 13,2 массовых процентов этилкарбамата, 31,36 массовых процентов триглима и 55,44 массовых процентов этанола в режиме восходящего потока с газообразным азотом через слой описанного выше твердого катализатора в условиях кипения реагентов. Реакция может быть также проведена в реакторе с нисходящим потоком. Следовые количества диметоксида дибутилолова добавляли в этот раствор в процессе реакции.
Условия реакции приведены в таблице 9 выше, и результаты этого эксперимента показаны на фигуре 22. Селективность образования ДЭК в расчете на этилкарбамат составляла от 98,5 мольных процентов до 99,9 мольных процентов при общей тенденции снижения селективности с ростом конверсии этилкарбамата.
Пример 15
Этот пример иллюстрирует показанный на фигуре 19 вариант осуществления согласно настоящему изобретению получения этилфенилкарбоната (ЭФК). Переэтерификацию ДЭК с фенолом для получения этилфенилкарбоната (ЭФК) проводили в присутствии катализатора из этоксида титана, иммобилизированного на носителе из силикагеля.
В реактор загружали такой же носитель из силикагеля, как в примере 14 выше, в количестве 25 мл (14,47 г), и затем проводили иммобилизацию этоксида титана. Раствор этоксида титана приготавливали путем растворения 45,25 г этоксида титана в 800 мл толуола. Затем осуществляли циркуляцию раствора этоксида титана в режиме восходящего потока через реактор при скорости 15 мл/мин и при температуре и давлении окружающей среды в течение 30 минут. Этоксид титана затем прививали на носитель из силикагеля при 135°C (275°F) и 3,4 бар (35 фунт/дюйм2) в течение 17 часов. После охлаждения, из реактора удаляли избыток раствора этоксида титана, и катализатор промывали толуолом при скорости 4 мл/мин в течение 1,5 часов. Промытый катализатор сушили при 138°C (280°F) в течение 4 часов в потоке газообразного азота при скорости 300 см3/мин.
Проводили переэтерификацию ДЭК с фенолом с получением этилфенилкарбоната (ЭФК) в условиях кипения реагентов в реакторе при температуре 345°C и давлении 27 фунт/дюйм2 путем подачи в реактор раствора 23 массовых процентов ДЭК со скоростью 0,5 мл/мин.
Для стабилизации работы катализатора в течение всех 860 часов непрерывной работы, в поток сырья вводили титан в форме Ti(OEt)4-x(OPh)x (где x=2) при концентрации около 55 масс.ppm. Суммарная селективность реакции переэтерификации по ЭФК и ДФК составляла около 99 мольных процентов. Результат этого эксперимента показан на фигуре 23.
Пример 16
Этот пример иллюстрирует показанный на фигуре 19 вариант осуществления согласно настоящему изобретению получения дифенилкарбоната (ДФК). Диспропорционирование ЭФК с получением ДФК и ДЭК проводили в присутствии растворимого титанового катализатора.
Сырье для реакции диспропорционирования готовили путем отгонки в атмосфере азота этанола, ДЭК и части фенола из смешанного продукта переэтерификации, который получали с помощью экспериментов, аналогичных эксперименту, описанному в примере 15 выше. Гомогенный титановый катализатор в смеси сырья для реакции диспропорционирования изначально вводили в реактор переэтерификации, описанный в примере 15 выше. Никакого дополнительного титанового катализатора в смеси сырья не добавляли. Толуол добавляли в две смеси сырья для создания паровой фазы в реакторе, работающем при кипении реагентов.
Первый состав сырья представлял собой 16,26 массовых процентов толуола, 1,61 массовых процентов ДЭК, 49,33 массовых процентов фенола, 30,91 массовых процентов ЭФК, 0,78 массовых процентов ДФК, и оставшаяся часть приходилась на побочные продукты реакции переалкилирования, такие как МФК. Первый состав сырья представлял собой 16,15 массовых процентов толуола, 1,61 массовых процентов ДЭК, 49,28 массовых процентов фенола, 31,08 массовых процентов ЭФК, 0,80 массовых процентов ДФК, и оставшаяся часть приходилась на побочные продукты реакции переалкилирования, такие как МФК. Концентрация гомогенного катализатора в первом и втором сырье составляла 180 массовых ppm и 200 массовых ppm титана, соответственно.
Реакцию диспропорционирования проводили в реакторе с незаполненным катализаторным пространством объемом 25 мл (в отсутствие твердого катализатора) при 179°C (355°F) и 2,9 бар (27 фунт/дюйм2). Скорости подачи сырья составляли 0,5 мл/мин в режиме восходящего потока в течение первых 72 часов работы и затем 0,60 мл/мин в режиме восходящего потока в дальнейшем. Результаты проведения реакций диспропорционирования приведены на фигуре 24. Ксантон являлся единственным новым побочным продуктом, образующимся в процессе диспропорционирования в количестве около 35 масс. ppm. Дифениловый эфир не был обнаружен ни в одном из анализов образцов продукта. Селективность по всем побочным продуктом составляла от 3,0 мольных процентов до 3,3 мольных процентов.
Получение биодизельного топлива согласно раскрываемым в изобретении вариантам осуществления
Биодизельное топливо получают путем проведения переэтерификации растительных масел или животных жиров с метанолом в присутствии гомогенных катализаторов и твердых катализаторов. Сырьем для получения биодизельного топлива являются растительные масла и животные жиры, которые являются эфирами высших жирных кислот. Термин жир (растительное или животное масло в случае жидкости) обычно включает в себя эфиры (глицериды) жирных кислот с глицерином, а термин воск относится к эфирам других спиртов. Основной химической реакцией, происходящей при получении биодизельного топлива, является каталитическая реакция природных эфиров (в основном, глицеридов) с первичным спиртом (обычно, с метанолом или этанолом). В качестве катализатора может быть использован спиртовой раствор основания (обычно NaOH, KOH, метоксида калия, или метоксида натрия). Поэтому, биодизельное топливо является смесью метиловых или этиловых эфиров различных насыщенных и ненасыщенных жирных кислот. Побочным продуктом является глицерин, который составляет от 6 до 25 масс.%. Биодизельное топливо может также содержать незначительные количества некоторых жирных кислот (продуктов гидролиза эфиров), в зависимости от количества воды в сырье или используемого катализатора.
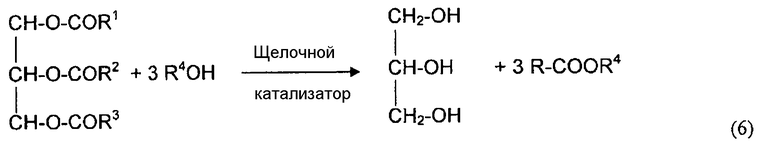
где R4OH = метанол или этанол; R=R1, R2 или R3.
Алкильные группы R1, R2 и R3 продукта из природного глицерида обычно отличаются по длине цепи и степени ненасыщенности. Алкильные группы обычно являются линейными и имеют четное число углеродных атомов от 4 до 26. Исключением является разветвленная изовалерьяновая кислота (CH3)2CHCH2COOH, которая встречается в относительно больших количествах у дельфинов. Некоторые ненасыщенные жирные кислоты имеют две или три двойных связи в алкильных цепях. Ненасыщенные жирные кислоты имеют более низкие температуры плавления, чем их насыщенные аналоги. Длина цепи ненасыщенных жирных кислот обычно находится в интервале C10-C24. Масло канолы имеет более высокую степень ненасыщенности в цепи длиной C16-C20, чем кукурузное масло.
Обычно, щелочные катализаторы являются более эффективными для переэтерификации карбоновых эфиров со спиртом, чем кислотные катализаторы. Гетерогенные катализаторы, раскрываемые в известном уровне техники (смотрите раздел описания заявки "УРОВЕНЬ ТЕХНИКИ"), также являются щелочными катализаторами. К сожалению, активные компоненты катализатора вымываются из твердого катализатора в условиях проведения реакции, что приводит к дезактивации катализатора. Катализаторы из алюмината цинка являются не очень активным катализатором и требуют более высоких температур реакции и более низких скоростей подачи сырья, чем более щелочные катализаторы, такие как MgO или CaO. Но последние вымываются из твердого катализатора даже быстрее, чем алюминаты цинка.
Переэтерификация растительных масел или животных жиров может быть проведена с метанолом или этанолом в присутствии твердого катализатора в реакторе с кипящими реагентами, в реакторе с пульсирующим потоком, или каталитической дистилляционной колонне со следовым количеством растворимого каталитического компонента в смеси сырья в результате одностадийной или двухстадийной реакции. Исходные катализаторы могут включать оксиды металлов, такие как оксид магния, оксид кальция, оксид цинка, оксид натрия, оксид калия, оксид лантана, и другие оксиды, нанесенные на носитель, такой как оксид кремния, оксид алюминия, углерод и/или углеродистый материал. Предпочтительно, чтобы углерод и углеродистые носители имели поверхностные функциональные группы, такие как гидроксильные или карбонильные, или и те, и другие, для иммобилизации металлоорганических соединений на поверхности носителя.
Для приготовления нанесенных оксидов металлов, гидроксидов или оксигидроксидов, наличие поверхностных функциональных групп необязательно. Углеродистые носители могут быть приготовлены путем регулируемого термического разложения углеводов, таких как древесина, скорлупа кокосового ореха, крахмал, целлюлоза, смесь крахмала и целлюлозы, сахар, метилцеллюлоза и другие углеводы, при повышенных температурах. Углеродистые носители могут быть либо ненанесенными, либо нанесенными. Для приготовления нанесенного углеродистого материала, углеводы осаждают на подходящий пористый носитель и затем подвергают регулируемому термическому разложению при повышенной температуре от 300°C до 1000°C в инертной атмосфере или атмосфере, состоящей из инертного газа, небольшого количества кислорода или водяного пара, или того, и другого. Носителем для углеродистых материалов могут быть любые неорганические материалы, такие как оксид алюминия, оксид титана, оксид кремния, оксид циркония, глины, оксид кремния-оксид алюминия, и другие.
В двухстадийном способе, конверсия триглицерида на выходе из первого реактора может составлять более чем 90%. Оставшийся непрореагировавший триглицерид, диглицерид и моноглицерид в потоке продукта реакции из реактора первой переэтерификации могут быть подвергнуты полной конверсии в реакторе второй переэтерификации. Так как переэтерификация является двухфазной реакцией, проведение переэтерификации в реакторе с кипящими реагентами или в реакторе с пульсирующим потоком будет способствовать возвратно-поступательному движению крупных молекул триглицерида, метиловых эфиров и клейкого глицерина через поры катализатора из среды жидких продуктов внутрь гранул катализатора, где происходит большинство каталитических реакций, что в итоге будет приводить к высокой производительности. Так как раскрываемые в изобретении катализаторы имеют высокую активность, переэтерификация может быть проведена при более низкой температуре и давлении, что означает более низкие затраты на строительство и энергозатраты.
Добавление растворимого каталитического компонента во вводимый в реакторы поток сырья составляет, в некоторых вариантах осуществления приблизительно от 0,5 ppm до приблизительно 500 ppm по массе; в других вариантах осуществления, приблизительно от 5 ppm до приблизительно 250 ppm по массе и в других вариантах осуществления, от 10 ppm до 50 ppm по массе. Примеры растворимых каталитических соединений включают, наряду с другими, 2-метоксиэтоксид цинка, 2-метоксиэтоксид кальция, 2-метоксипропоксид цинка, этоксид цинка, алкоксиалкилкарбонат цинка, 2-метоксипропоксид кальция, этоксид кальция, метоксид кальция, алкоксиалкилкарбонат кальция, 2-метоксиэтоксид магния, 2-метоксипропоксид магния, этоксид магния, метоксид магния, бутоксид магния, алкоксиалкилкарбонат магния, алкоксид лантана, алкоксиалкилкарбонат лантана, соли цинка и карбоновых кислот, соли магния и карбоновых кислот, соли кальция и карбоновых кислот, и глицериды Mg, Ca, и Zn. Могут также быть использованы их смеси. Растворимые соединения Ca, Mg, Zn и La могут быть получены путем реакции оксида или гидроксида этих металлов с органическим карбонатом, или смесью органического карбоната и спирта, или с карбоновыми кислотами, или смесью органической карбоновой кислоты и спирта, такого как метанол, 2-метоксиэтанол, и другие, при температуре от 93°C до 260°C (200°F до 500°F), предпочтительно, от 121°C до 232°C (250°F до 450°F) в жидкой фазе или в присутствии жидкости и пара. Необязательно, но может быть выбран вариант извлечения компонентов металла с целью их рециркулирования. Такие приготовленные растворы применяют для добавления следового количества этих металлов во вводимый в реактор поток сырья с целью достижения длительного рабочего цикла катализатора. Суммарное количество активного металла или компонентов металла на твердом алкоксиде металла, гидроксиде металла или металлоксидном катализаторе составляет, в некоторых вариантах осуществления, приблизительно от 0,05 масс.% до приблизительно 20 масс.%, и в других вариантах осуществления, приблизительно от 0,07 масс.% до приблизительно 12 масс.%.
Необязательно, но помимо реакции переэтерификации с метанолом, все или часть ди- или моноглициридов могут быть превращены в органические карбонаты или органические карбаматы, или и в те, и другие, путем реакции с ДМК, метил 2-этил-1-гексилкарбонатом, метилкарбамат, 2-этил-1-гексилкарбаматом, мочевиной, или их смесью, во втором реакторе или, необязательно, в третьем реакторе. Полученные органические карбонаты и карбаматы могут служить в качестве присадки к биодизельному топливу для снижения содержания в выхлопных газах твердых частиц, выброса NOx, или для повышения цетанового числа дизельного топлива.
Так как природные растительные масла могут содержать различные незначительные количества свободных жирных кислот, свободные жирные кислоты необходимо удалять путем предварительной обработки перед проведением переэтерификации со спиртом в присутствии твердого щелочного катализатора. Примером таких методов предварительной обработки является этерификация свободных жирных кислот с метанолом в присутствии кислотного катализатора. Одним таким кислотным катализатором является сульфоновая кислота, иммобилизированная на углеродистом носителе. Носитель может включать носители, приготавливаемые путем регулируемого термического разложения скорлупы кокосового ореха или углеводов, нанесенных или осажденных на пористом носителе. Проведение этерификации свободных жирных кислот со спиртом в присутствии твердого кислотного катализатора в каталитической дистилляционной колонне имеет преимущества, которыми являются непрерывное удаление воды из реакционной зоны в виде отводимого сверху колонны потока, сдвиг равновесия реакции этерификации в сторону образования конечных продуктов и исключение применения отдельной стадии сушки продукта этерификации перед проведением переэтерификации триглицерида со спиртом. Еще одним важным преимуществом является меньшее время проведения этерификации.
Все реакции переэтерификации в следующих примерах проводили в реакторах с нисходящим потоком. Размер реактора с неподвижным слоем составлял 1,3 см (1/2 дюйма) в диаметре и 53,3 см (21 дюйм) высотой. Реактор имел раздельно регулируемый нагрев верхней и нижней зоны. Поток вводимого метанола и поток растительного масла (6 масс.% метанола в растительном масле) раздельно подавали в верхнюю часть реактора, где эти два потока опускались вниз в каталитическую реакционную зону. Следовые количества растворимого каталитического компонента вводили в поток метанола или в поток, содержащий частично конвертированный продукт. Объем твердых катализаторов составлял 15 мл.
Эксперимент 17
Целью этого эксперимента являлась демонстрация переэтерификации масла канолы с метанолом в присутствии твердого катализатора в реакторе с кипящими реагентами с нисходящим потоком или каталитическом дистилляционном реакторе. Твердым катализатором являлся MgO, ненанесенный на силикагель.
Раствор нитрата магния готовили путем растворения 10,96 г Mg(NO3)2∙6H2O в 24 г деионизированной воды. 30 мл (11,91 г) носителя из силикагеля сферической формы (диаметр 1,7-4 мм; около 6 гидроксильных групп на нм2, удельная поверхность 314 м2/г по методу БЭТ, объем пор 1,055 см3/г и средний диаметр пор 13,46 нм) импрегнировали упомянутым выше раствором нитрата магния с помощью метода пропитки по влагоемкости. Сферический носитель из силикагеля получали с помощью метода падения масляной капли. После сушки импрегнированного продукта при 100°C в течение 1 часа, его затем прокаливали при 510°C в течение 2 часов.
Загружали в реактор 15 мл (6,30 г) катализатора MgO/SiO2. Вводимое в качестве сырья масло канолы (приобретенное в местном продовольственном магазине) приготавливали путем смешения метанола (5,39 масс.%) с маслом канолы (94,61 масс.%). Кислотность свободной жирной кислоты в этом сырье составляло 0,48 мг KOH/г. Переэтерификацию масла канолы с метанолом проводили при 165°C (330°F) и 19,4 бар (267 фунт/дюйм2) путем введения в реактор масла конолы и метанола при скорости каждого потока 0,2 мл/мин. Этоксид магния растворяли в вводимом в качестве сырья метаноле, для того чтобы иметь 28 ppm Mg по массе в каталитической реакционной зоне.
Выходящие потоки состояли из двух прозрачных слоев. Верхние слои содержали продукт метиловые эфиры и небольшие количества непрореагировавших триглицеридов. Среднее содержание непрореагировавших триглицеридов в реакционных продуктах, исключая метанол из верхних слоев, составляло около 1,2 масс.%. Нижний слой содержит большую часть непрореагировавших триглицеридов. Результат приведен на фигуре 17, и он указывает на стабильную работу катализатора.
Эксперимент 18
Целью этого эксперимента была демонстрация превращения оставшихся непрореагировавших или частично прореагировавших материалов в выходящем из первого реактора потоке (состоящем из двух слоев при отстаивании) во втором реакторе с нисходящим потоком при кипении реагентов или каталитическом дистилляционном реакторе, или необязательной рециркуляции в единственный реактор переэтерификации.
Раствор нитрата магния готовили путем растворения 9,04 г Mg(NO3)2∙6H2O в 19,1 г деионизированной воды. 22 мл (9,14 г) носителя из силикагеля сферической формы (размер 9-14 меш, удельная поверхность 309 м2/г по методу БЭТ и объем пор 1,03 см3/г) импрегнировали упомянутым выше раствором нитрата магния с помощью метода пропитки по влагоемкости. После сушки импрегнированного продукта при 150°C в течение 1 часа, его затем прокаливали при 510°C в течение 2 часов. Готовый катализатор содержал 4,5% Mg по массе.
15 мл (7,2 г) катализатора MgO/SiO2 загружали в такой же реактор, как тот, который использовали в эксперименте 13. Два слоя смешанного продукта из реакции первой переэтерификации масла канолы с метанолом отделяли от смешанного продукта с помощью делительной воронки для использования в качестве сырья в реакции второй переэтерифакации. Состав сырья из нижнего слоя смешанного продукта представлял собой 25,4 масс.% триглицеридов, 8,5 масс.% диглицеридов, 3,1 масс.% моноглицеридов, 0,1 масс.% глицерина, 47,1 масс.% метиловых эфиров и 15,8 масс.% метанола. Сырье содержало около 8,5 ppm растворимых соединений Mg по массе и имело кислотность свободных жирных кислот 0,32 мг KOH/г. Переэтерификации проводили при 160°C (320°F) и 19,5 бар (268 фунт/дюйм2) путем подачи сырья со скоростью 0,12 мл/мин и метанола со скоростью 0,10 мл/мин в реактор с нисходящим потоком, работающий при температурах кипения реагентов. Ни в один из двух потоков сырья не вводили дополнительного количества алкоксида Mg. Выходной поток из реактора представлял собой прозрачный светло-желтый раствор (один слой).
Состав сырья из верхнего слоя смешанного продукта представлял собой 1,12 масс.% триглицеридов, 0,57 масс.% диглицеридов, 3,78 масс.% моноглицеридов, 7,47 масс.% метиловых эфиров, 0,03 масс.% глицерина и 87,03 масс.% метанола. Кислотность свободных жирных кислот в этом сырье составляла 0,51 мг KOH/г. Переэтерификацию проводили над тем же катализатором при той же температуре и давлении при скорости подачи сырья 0,2 мл/мин. Дополнительного количества метанола в реактор не подавали. Эти два продукта конечной переэтерификации сырья в виде нижнего и верхнего слоя смешанного продукта объединяли для отгонки избытка метанола и для извлечения неочищенного биодизельного топлива. Извлекаемое неочищенное биодизельное топливо содержало 0,36 масс.% непрореагировавших триглицеридов и имело кислотность свободных жирных кислот 0,74 мг KOH/г.
Приведенные выше результаты убедительно продемонстрировали возможность получения биодизельного топлива путем проведения переэтерификации растительного масла со спиртом, таким как метанол, в присутствии твердого катализатора.
Катализаторные системы
Как описано для различных вариантов осуществления в описании изобретения, реакционные зоны или слои катализатора в реакторах могут включать один или более катализаторов алкоголиза согласно раскрываемым в изобретении вариантам осуществления, включая реакционные зоны или слои катализатора, имеющие два или более гетерогенных катализаторов, твердых катализаторов, или их комбинации. Например, катализатор может включать два или более оксидов металлов, то есть, смешанный металлоксидный катализатор, имеющий два или более различных оксидов металлов, нанесенных или иммобилизированных на носителе, который может также быть оксидом металла или смешанным оксидом металла, который является пористым.
Используемые в изобретении термин твердые или гетерогенные катализаторы относится не только к компонентам активных металлов катализатора, но также к свойствам носителя, который может сам по себе быть отличительным признаком катализатора. Например, два различных носителя катализатора, которые имеют различные физические свойства, такие как кристалличность, пористость, плотность, размер и так далее, но имеют одинаковый химический состав, определяются в изобретении как два различных носителя.
Аналогично, твердые или гетерогенные катализаторы могут отличаться по концентрации активного компонента на носителе, например, 1 масс.% оксида металла против 3 масс.% оксида металла на носителе. Несмотря на то, что каждый катализатор может иметь аналогичный оксид металла и аналогичный состав и структуру носителя, содержание металла может давать в результате значительно отличающуюся характеристику катализатора.
Таким образом, "два или более" катализаторов, используемые в раскрываемых в изобретении вариантах осуществления, могут включать катализаторы, имеющие несколько активных компонентов, различные структуры/составы носителя, различные содержания активных компонентов, или любую комбинацию или изменение элементов металлов, содержаний металлов и носителей.
В реакционных зонах, имеющих два или более катализаторов согласно раскрываемым в изобретении вариантам осуществления, могут эффективно использоваться отличающиеся по характеристикам катализаторы. Например, реакционная зона каталитической дистилляции может быть специально спроектирована для использования в ней нескольких катализаторов, обеспечивающих требуемую конверсию при минимальной полимеризации или отложении. Например, катализаторы, имеющие более низкую активность и/или высокую селективность, могут быть использованы в тех местах слоя, где может быть самой высокой концентрация предшественников полимера. В качестве варианта, катализаторы, имеющие большой средний диаметр пор или другие свойства, позволяющие эффективно осуществлять движение жидкости в реакторе с дистилляционной колонной с целью отмывки катализатора от примесей, могут быть использованы ближе к нижней части реакционной зоны, где концентрация предшественников полимеров может быть самой высокой. Катализаторы, имеющие высокую активность или высокую удельную поверхность, могут быть избирательно расположены в тех местах слоя, где концентрация предшественника полимера является низкой. Таким образом, слой катализатора может быть специально приспособлен для обеспечения требуемой конверсии (активности и селективности) и небольшого отложения полимера или полного отсутствия такого отложения полимера на катализаторах внутри слоя, что увеличивает продолжительность рабочего цикла катализатора и улучшает характеристику работы реактора.
Вода
Как уже было упомянуто выше, содержание влаги в потоке сырья может регулироваться, в некоторых вариантах осуществления, до менее чем приблизительно 700 ppm и в других вариантах осуществления, до менее чем 600 ppm. Неожиданно было обнаружено, что использование следовых количеств воды в потоке сырья или в реакционной зоне может увеличивать продолжительность рабочего цикла катализатора и/или снижать накопление высокомолекулярных полимерных материалов на твердом катализаторе, что может в результате приводить к замедлению скоростей дезактивации катализатора.
Не ссылаясь на какую-либо одну теорию, тем не менее, следует отметить, что механизм улучшения характеристики работы катализатора при использовании гетерогенных и гомогенных катализаторов в присутствии воды еще полностью не ясен. Однако следует отметить, что (a) отложение полимеров на катализаторе дезактивирует катализатор, и (b) вымывание активных металлов из твердого катализатора в условиях реакции приводит к постоянной дезактивации катализатора. Это позволяет сделать вывод, что использование следовых количеств воды может (a) приводить к деполимеризации полимеров, которые могут образовываться во время реакций переэтерификации и/или реакций диспропорционирования в раскрываемых в изобретении вариантах осуществления, и/или (b) принимать участие в реакции иммобилизации, нанесения, или связывания одного или более вымываемых катализаторов или добавляемых гомогенных катализаторов на носителе в условиях реакции. Следовое количество воды в количестве менее чем приблизительно 600 ppm по массе в сырье или добавленное путем непосредственного или косвенного введения воды в реакционную зону, может вызывать эффект деполимеризации, эффект реактивации катализатора непосредственно в реакторе, или комбинацию этих эффектов.
Однако следует избегать слишком большого количества воды в потоке сырья или реакционной зоне, так как это может вызывать осаждение растворимого катализатора или образование геля на твердом катализаторе, или и то, и другое, что может приводить к возникновению нежелательных проблем, связанных с работой реактора и оптимальной работой катализатора. Следовое количество воды в потоке сырья или реакционной зоне в раскрываемых в изобретении вариантах осуществления могут находиться в интервале приблизительно от 1 части на миллион (ppm) до приблизительно 600 частей на миллион (ppm; в других вариантах осуществления, приблизительно от 2 частей на миллион (ppm) до 500 частей на миллион (ppm); в других вариантах осуществления, приблизительно от 5 частей на миллион (ppm) до приблизительно 400 частей на миллион (ppm) и в еще одних вариантах осуществления приблизительно от 50 частей на миллион (ppm) до приблизительно 250 частей на миллион (ppm), по массе.
Как показано на фигурах 19 и 20, вода может образовываться в качестве побочного продукта реакции в каждой технологической схеме получения диарилкарбонатов согласно раскрываемым в изобретении вариантам осуществления. Что касается способа на фигуре 19, то вода может образовываться в процессе дегидратации гликоля для получения эпоксида. Что касается способа на фигуре 20, то вода может образовываться в процессе синтеза мочевины из аммиака и диоксида углерода. Вода, извлеченная с одной из этих реакционных стадий, может быть использована в качестве добавляемой воды в одну или более реакционных зон переэтерификации и/или диспропорционирования для поддержания содержания воды внутри соответствующих реакционных зон в описанных выше интервалах.
Внутренние устройства, используемые в реакторах
Как описано выше, реакторы, используемые в раскрываемых в изобретении вариантах осуществления, для проведения переэтерификации в жидкой фазе или в присутствии двойной фазы жидкости и пара могут иметь любую физическую форму для различных рабочих режимов. Для проведения описанных в изобретении реакций может быть использован любой тип реакторов. Примеры реакторов, подходящих для проведения реакций с участием органического карбоната или органических карбаматов, могут включать реакторы с дистилляционной колонной, реакторы с дистилляционной колонной с разделительной стенкой, традиционные трубчатые реакторы с неподвижным слоем, барботажные реакторные колонны, суспензионные реакторы, оборудованные дистилляционной колонной или без дистилляционной колонны, реакторы с пульсирующим потоком, каталитические дистилляционные колонны, в которых суспензия твердых катализаторов стекает вниз по колонне, или любую комбинацию этих реакторов.
Реакторы, используемые в раскрываемых в изобретении вариантах осуществления, могут дополнительно включать любое внутреннее физическое устройство или комбинацию двух или более внутренних устройств для разделения пара и жидкости и направления движения пара и жидкости внутри реактора. Различные внутренние устройства могут быть любой формы или являться любым физическим устройством, и они могут выполнять несколько функций при условии, что такое устройство или такие устройства обеспечивают как эффективное разделение пара и жидкости, так и движение пара и жидкости.
В случае обратимой эндотермической реакции, одновременное решение обеих задач эффективного разделения пара и жидкости и движения пара и жидкости является необходимым для высокой конверсии, но при этом оно усложняется в результате охлаждающего действия реакционной среды вследствие воздействия двух объединенных эффектов: поглощения тепла при парообразовании и поглощения тепла при протекании реакции. Для достижения высокой конверсии, один из продуктов реакции должен быть удален из жидкой реакционной среды в паровую фазу, и затем пар должен быть быстро удален из реакционной зоны, что требует затрат тепла на испарение и на эффективное перемещение пара. Примером такого случая является переэтерификация диалкилкарбоната, такого как ДЭК, с фенолом. Результатом является охлаждение жидкой реакционной среды, что в свою очередь приводит к снижению конверсии. Теплота парообразования и эндотермический тепловой эффект реакции вступают в конфликт друг с другом, что в результате приводит к более низкой конверсии. В традиционном трубчатом реакторе идеального вытеснения может быть использована внутренняя система подогрева или могут быть использованы несколько небольших реакторов с периодическим подогревом между реакторами.
Может быть использовано внутреннее вспомогательное устройство или устройства в реакционной зоне, которые эффективно разделяют газ и жидкость и, кроме того, обеспечивают теплом эндотермическую реакцию. Таким устройством может быть тарелка для перераспределения жидкости вместе с устройством или устройствами для каталитической дистилляционной колонны, которое внутри подает тепло в середину реакционной зоны. Такое устройство или устройства должны способствовать более эффективному разделению пара от жидкости, а также обеспечивать теплом эндотермическую реакцию, что приводит в результате к высокой конверсии. Использование такого устройства или устройств в реакционной зоне каталитической дистилляционной колонны должно обеспечивать существенный положительный эффект независимо от того, используется ли твердый катализатор, гомогенный катализатор, или и тот, и другой, при переэтерификации диалкилкарбоната, такого как ДЭК, с фенолом, или циклического карбоната с этанолом.
В случае экзотермических реакций, можно использовать внутренне охлаждающее устройство для предотвращения протекания нежелательной реакции или осушения реакционной зоны (низкой скорости движения жидкости), которые обычно приводят к низкой селективности и/или неудовлетворительной работе катализатора. Внутреннее устройство должно быть эффективным для разделения парожидкостной фазы и способствовать движению пара и жидкости.
Внутренние устройства подогрева или охлаждения могут быть размещены на любой высоте внутри колонны, например, вверху ректификационной секции колонны, такой как реактор переэтерификации, с помощью которых частично конденсируют ДЭК и главным образом конденсируют фенол внутри колонны.
Как описано выше, раскрываемые в изобретении варианты осуществления позволяют достигать продолжительных рабочих циклов катализаторов для различных твердых катализаторов путем введения следового количества растворимых металлоорганических катализаторов в сырье. Другие раскрываемые в изобретении варианты осуществления могут включать способ непрерывного получения органического карбоната или органического карбамата при постоянной скорости процесса; способы приготовления непосредственно в реакторе иммобилизированных твердых катализаторов, способы поддержания постоянной активности катализатора в течение длительных рабочих циклов и сроков службы, для того чтобы их можно было использовать в промышленных реакторах с неподвижным слоем; и способ реактивации дезактивированных твердых катализаторов непосредственно в реакторе.
Положительным эффектом раскрываемых в изобретении вариантов осуществления является то, что они предлагают катализаторы переэтерификации, имеющие увеличенную продолжительность рабочего цикла, в силу чего снижаются эксплуатационные расходы, связанные с частыми остановками реакторов и заменами катализатора. Кроме того, вследствие использования следового количества растворимого металлоорганического катализатора, может быть значительно снижена необходимость в удалении гомогенного катализатора из различных потоков продуктов.
Несмотря на то что изобретение включает ограниченное число вариантов осуществления, для специалистов в этой области, которым понятна сущностью этого изобретения, является очевидным, что могут быть предложены другие варианты осуществления, которые не выходят за рамки объема настоящего изобретения. Соответственно, объем изобретения может быть ограничен только прилагаемыми пунктами формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ НЕПРЕРЫВНОГО ПОЛУЧЕНИЯ ОРГАНИЧЕСКИХ КАРБОНАТОВ ИЛИ ОРГАНИЧЕСКИХ КАРБАМАТОВ И ТВЕРДЫЕ КАТАЛИЗАТОРЫ ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2009 |
|
RU2447056C1 |
| СПОСОБ УДАЛЕНИЯ ПРИМЕСИ АЛКАНОЛА ИЗ ПОТОКА ОРГАНИЧЕСКОГО КАРБОНАТА | 2009 |
|
RU2507192C2 |
| СПОСОБ ПОЛУЧЕНИЯ ДИАЛКИЛКАРБОНАТОВ | 2005 |
|
RU2367648C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКАНДИОЛА И ДИАЛКИЛКАРБОНАТА | 2010 |
|
RU2531620C2 |
| СПОСОБ ПОЛУЧЕНИЯ ДИАРИЛКАРБОНАТОВ ИЛИ АЛКИЛАРИЛКАРБОНАТОВ ИЗ ДИАЛКИЛКАРБОНАТОВ | 2008 |
|
RU2490251C9 |
| СПОСОБ ПОЛУЧЕНИЯ ДИАРИЛКАРБОНАТА | 2008 |
|
RU2474571C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПО МЕНЬШЕЙ МЕРЕ ОДНОГО ДИАРИЛКАРБОНАТА ПО МЕНЬШЕЙ МЕРЕ ИЗ ОДНОГО ДИАЛКИЛКАРБОНАТА И ПО МЕНЬШЕЙ МЕРЕ ИЗ ОДНОГО АРОМАТИЧЕСКОГО ГИДРОКСИСОЕДИНЕНИЯ | 2009 |
|
RU2515993C2 |
| СПОСОБ ПОЛУЧЕНИЯ АРОМАТИЧЕСКОГО КАРБОНАТА | 1991 |
|
RU2041869C1 |
| СПОСОБ И УСТРОЙСТВО ДЛЯ ОЧИСТКИ ДИАЛКИЛКАРБОНАТОВ | 2010 |
|
RU2564035C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКАНДИОЛА И ДИАЛКИЛКАРБОНАТА | 2008 |
|
RU2461541C2 |
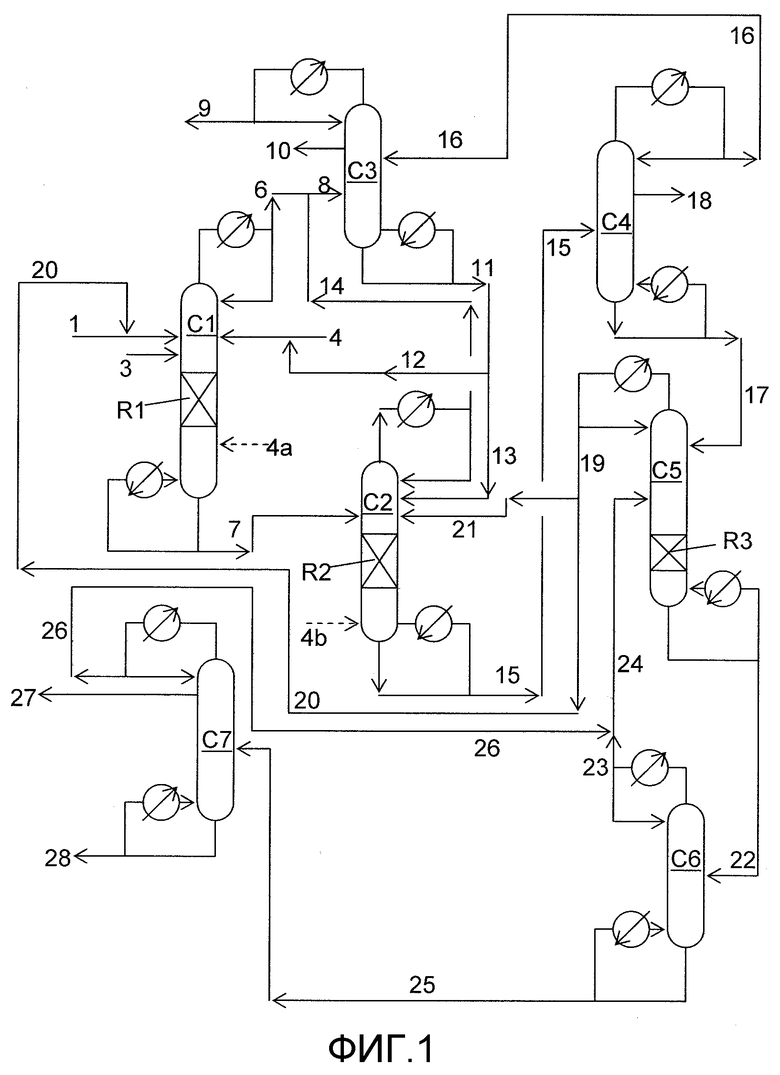

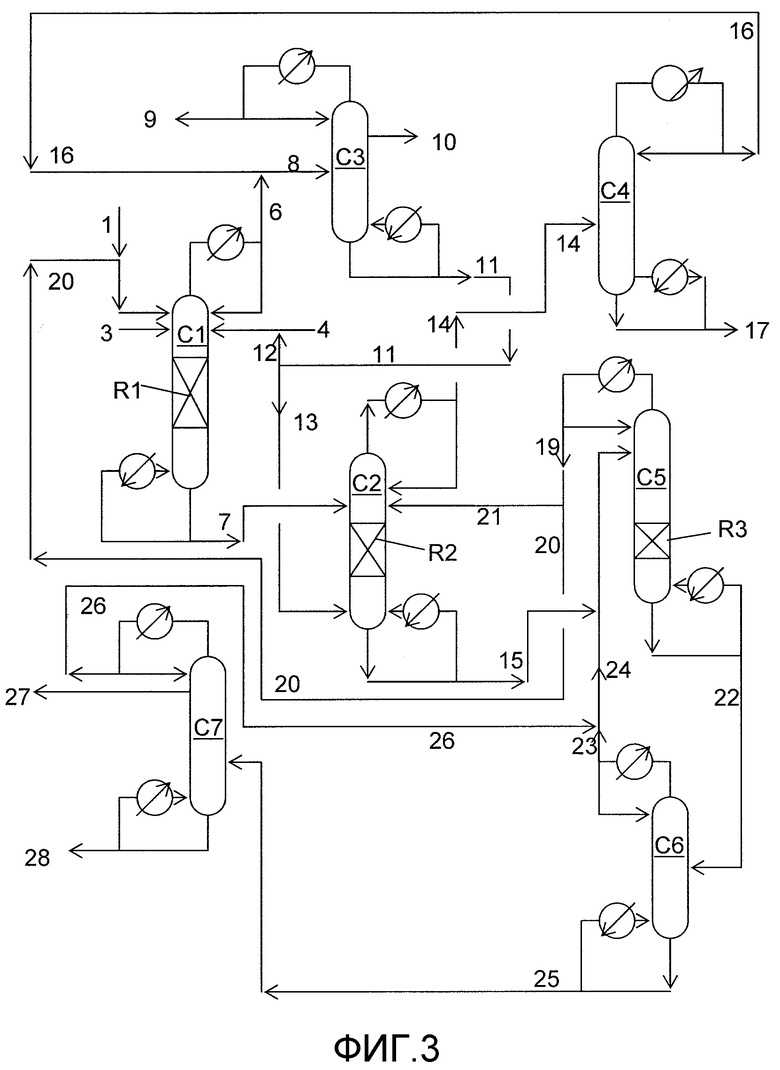
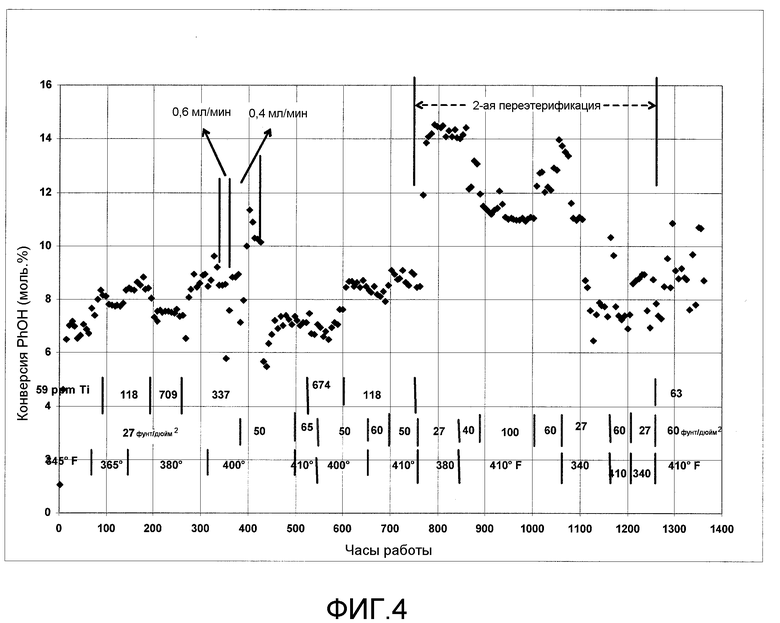
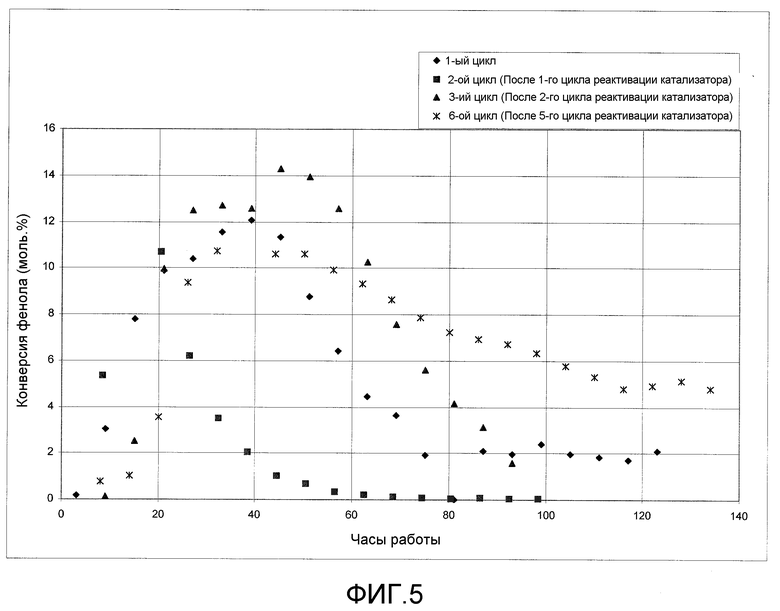
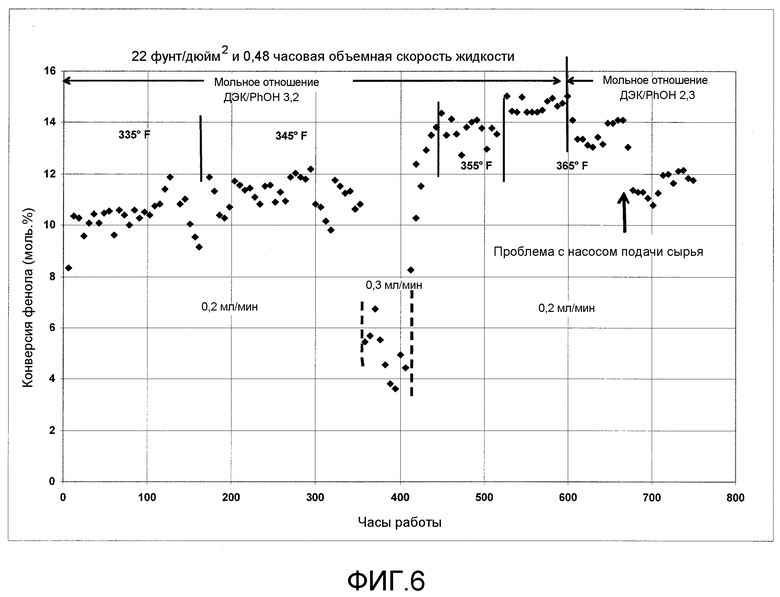
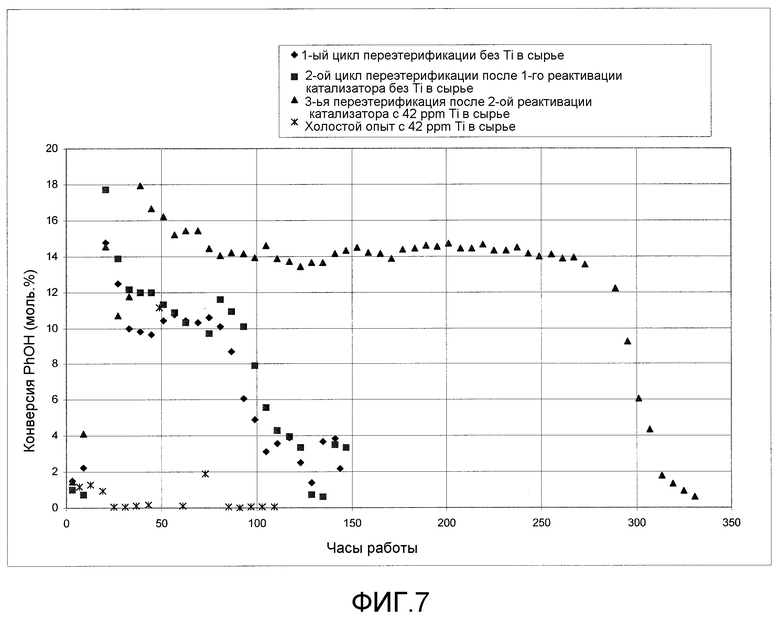
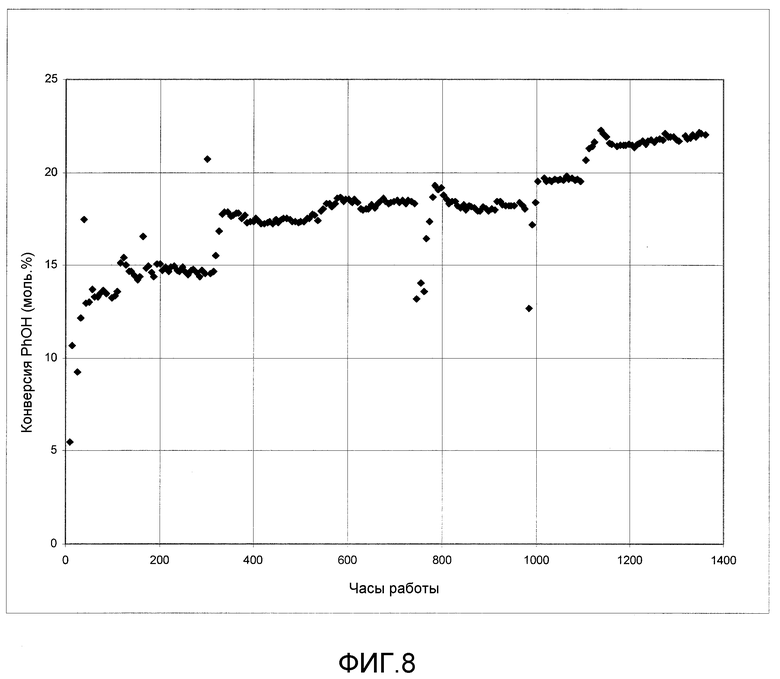
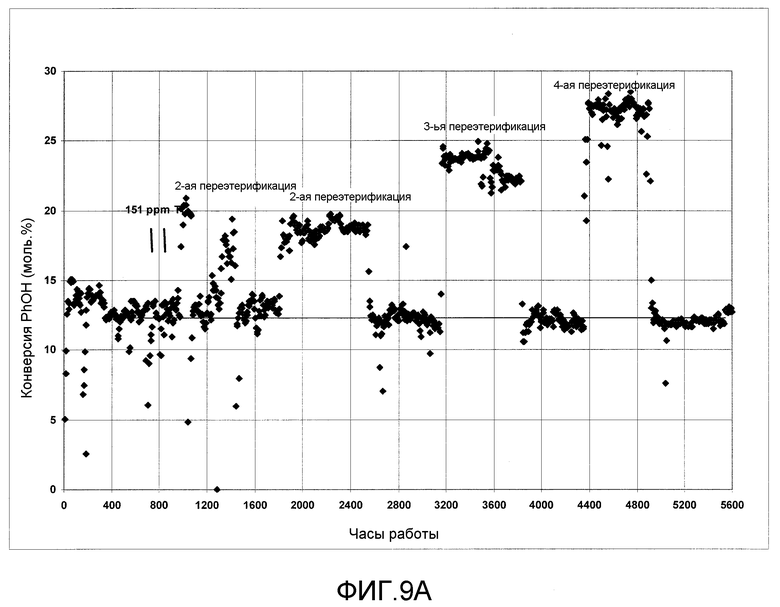
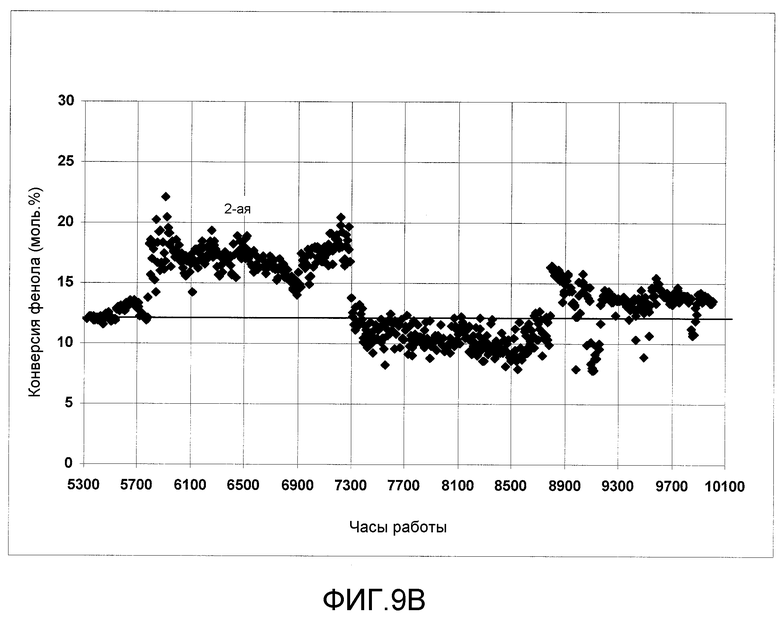
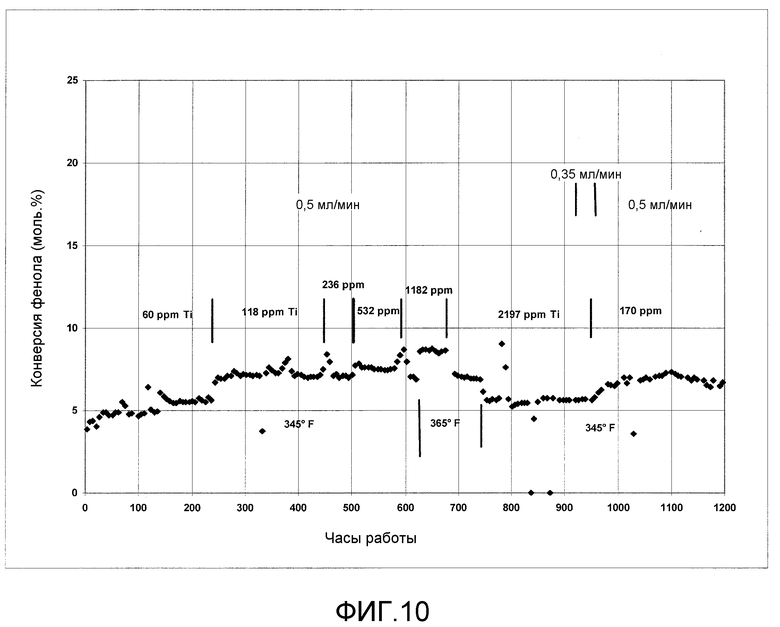
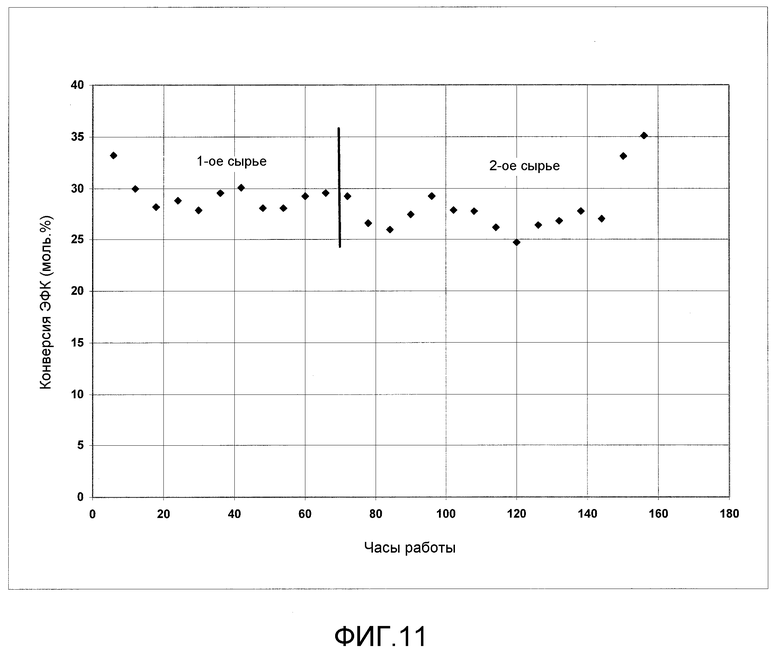
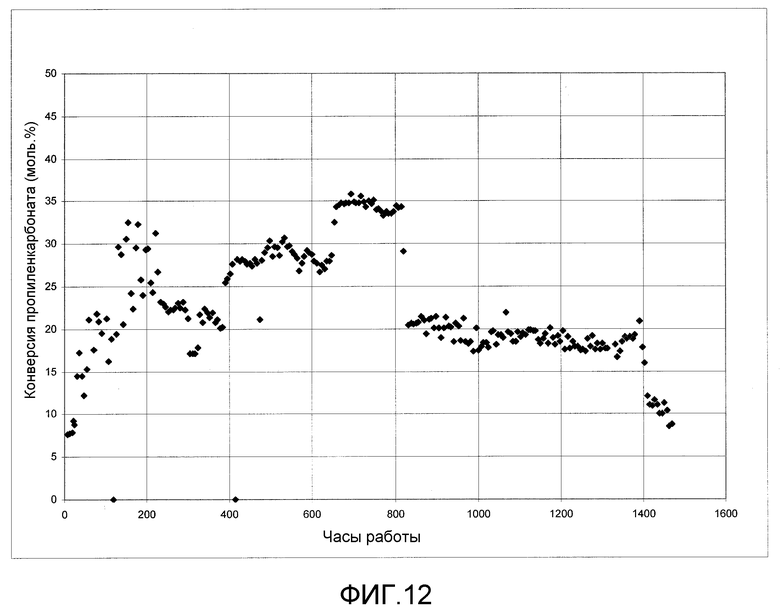
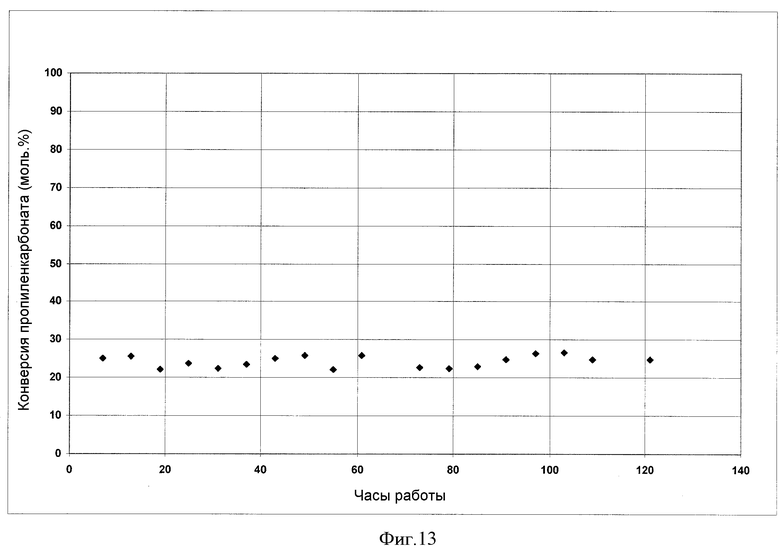
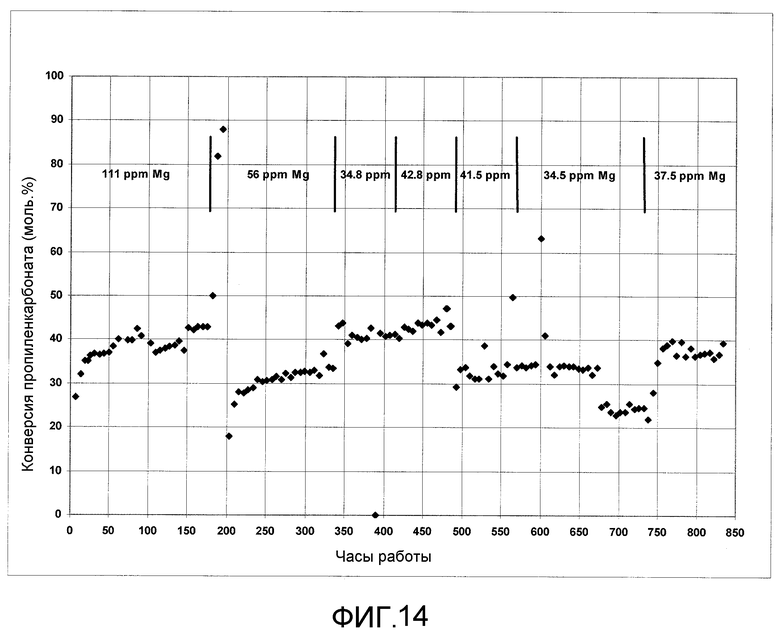
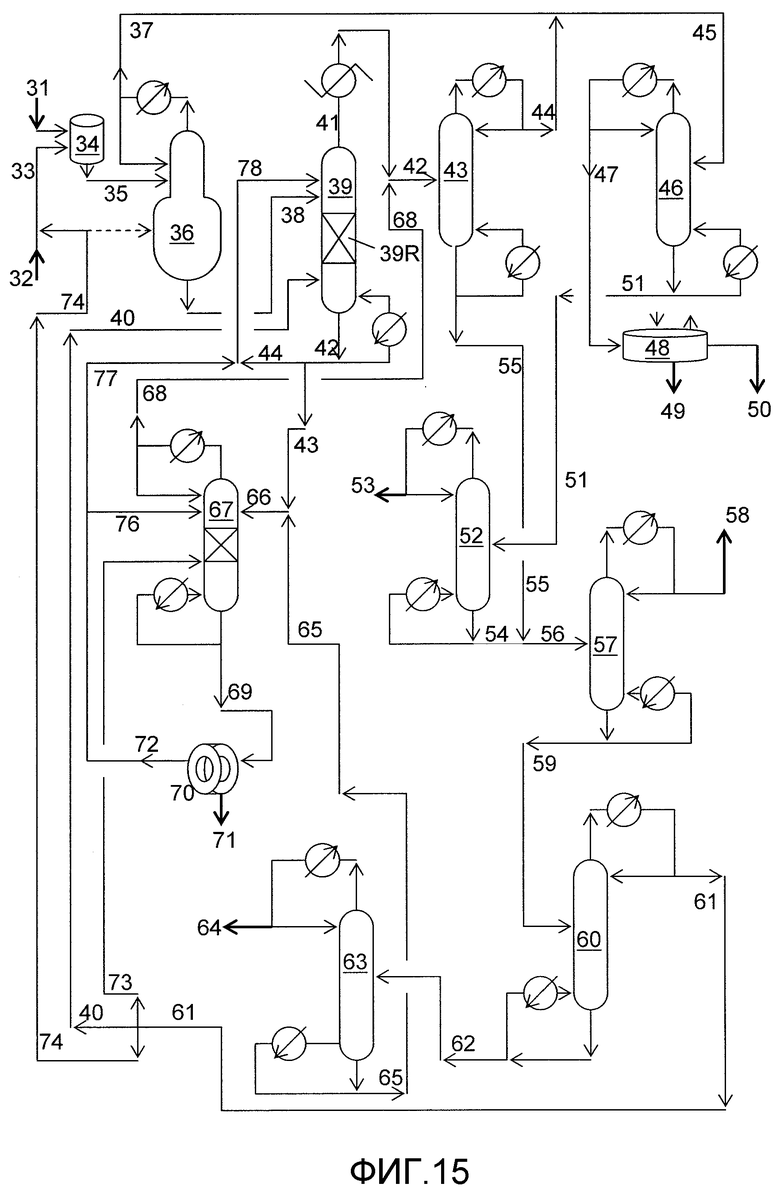
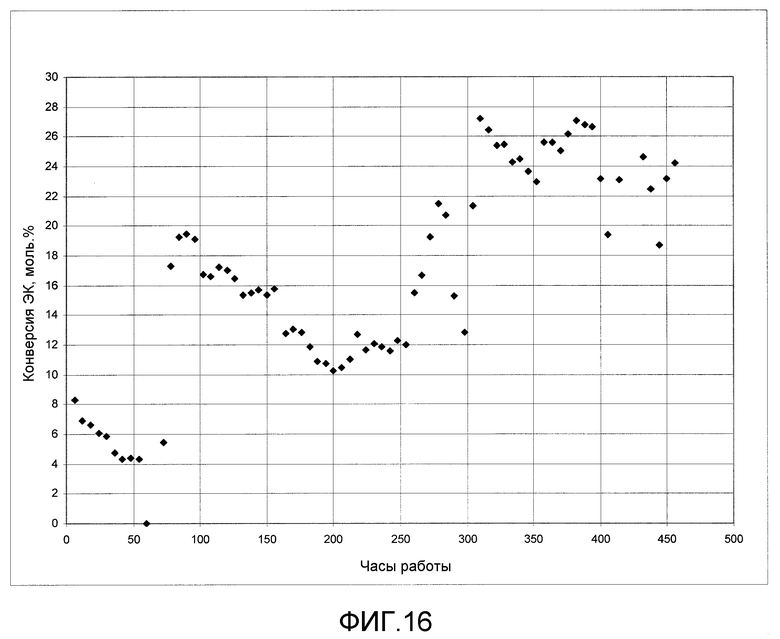
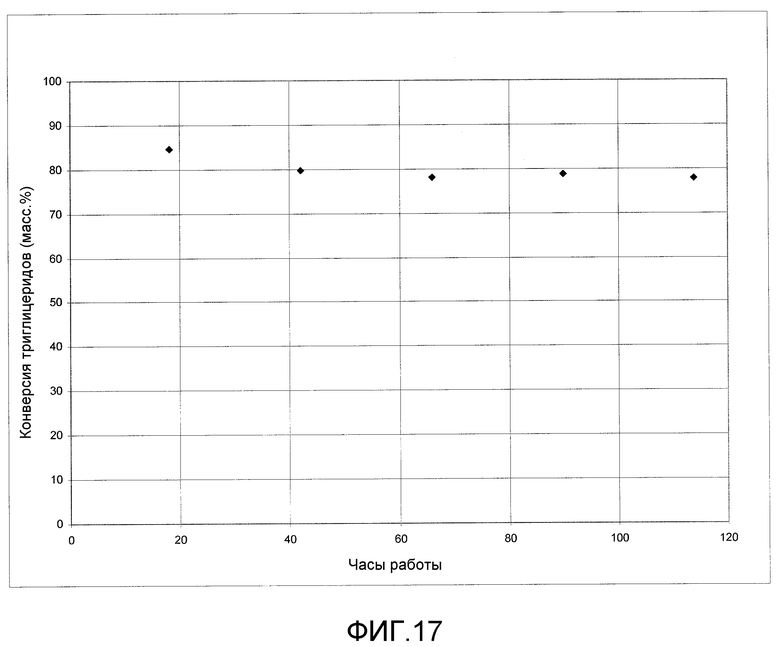
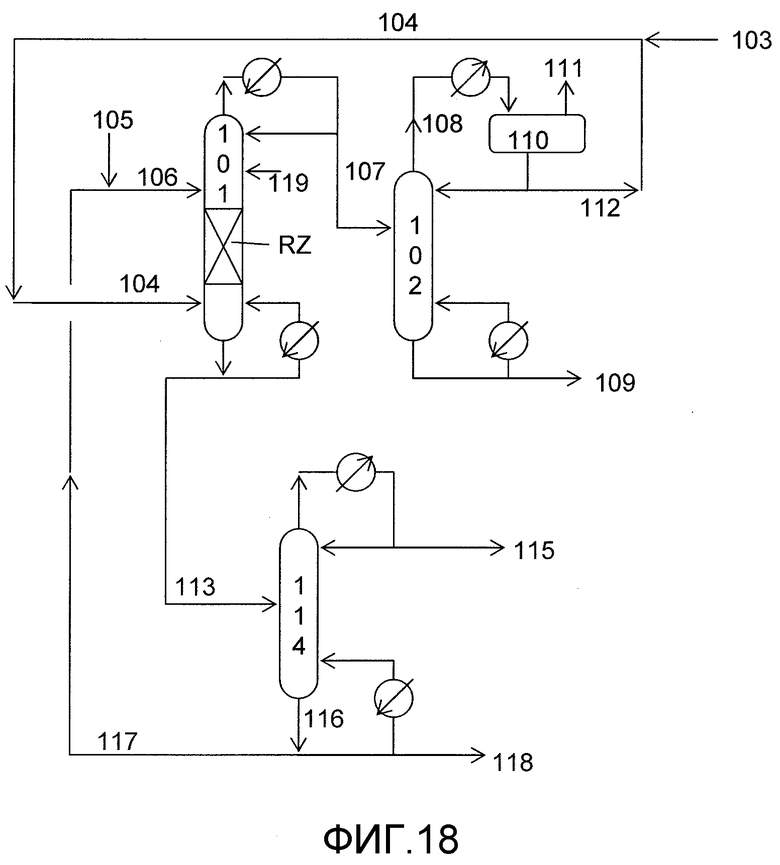
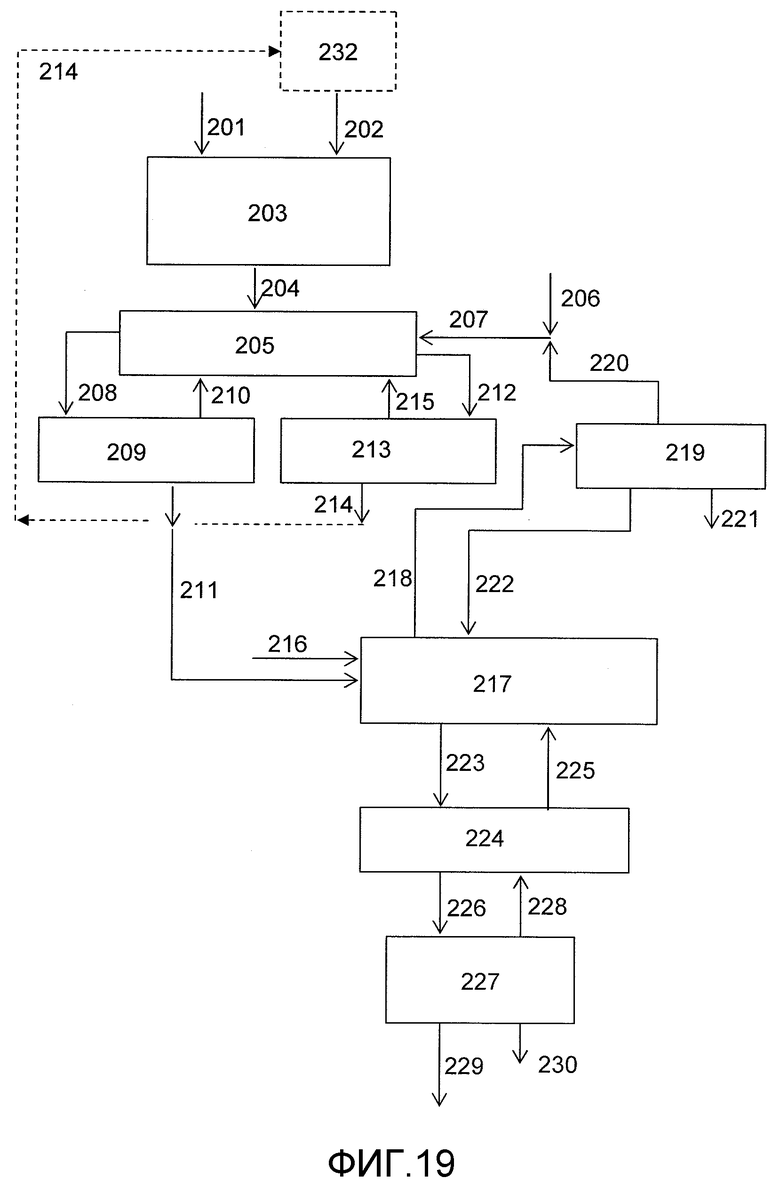

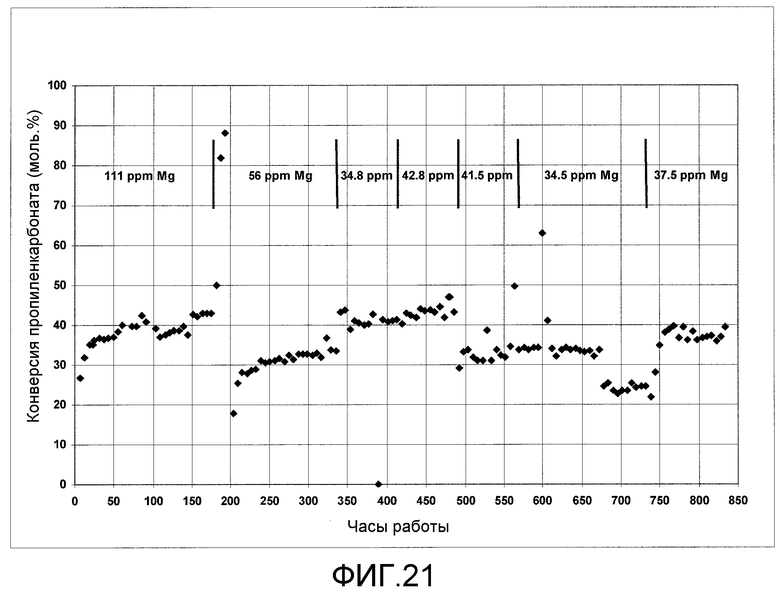
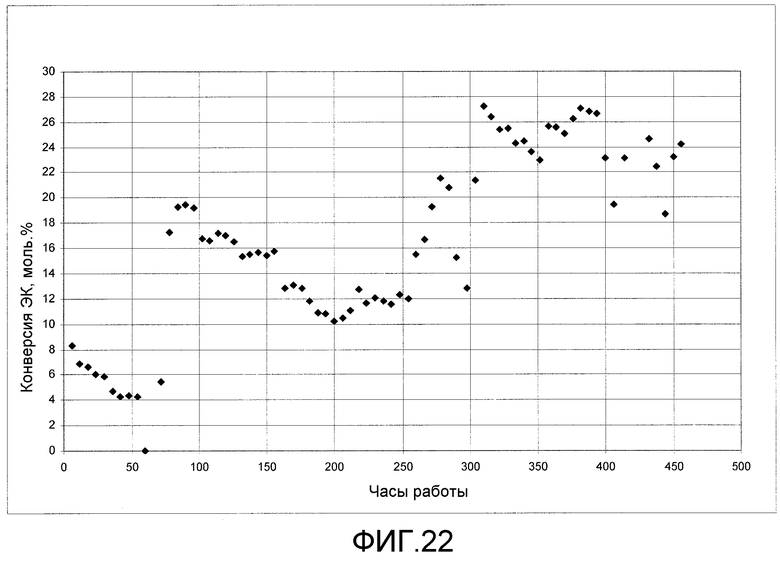
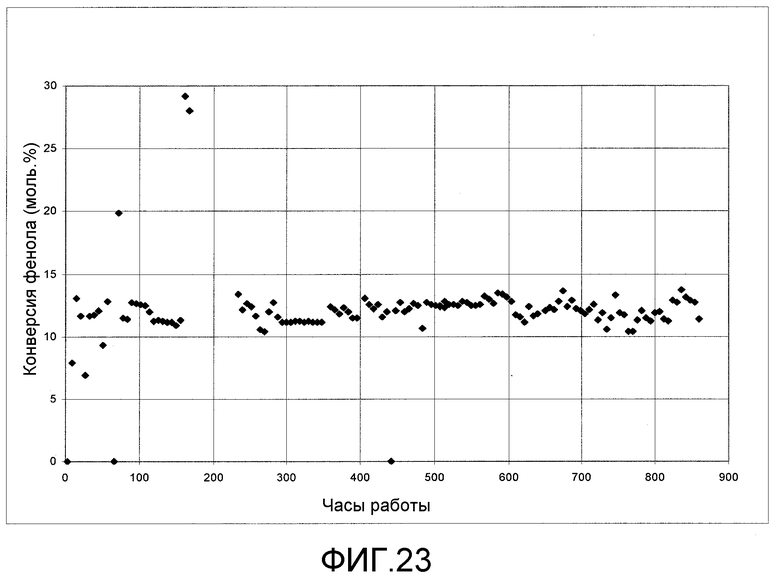
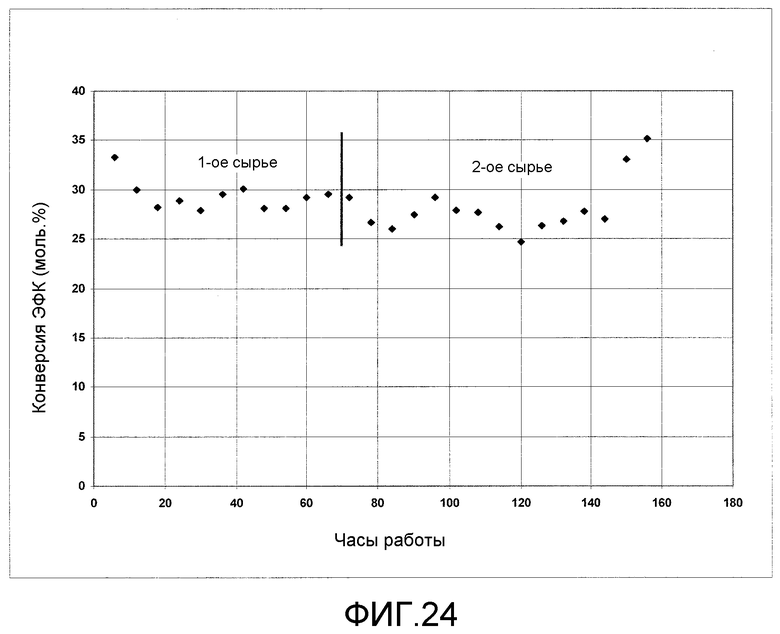
Изобретение относится к способам получения диарилкарбонатов, которые позволяют получать диарилкарбонаты из газов, вызывающих парниковый эффект, таких как диоксид углерода. Способ получения диарилкарбоната включает реакцию эпоксида и диоксида углерода в первой реакционной зоне с образованием первого реакционного продукта, включающего циклический карбонат; переэтерификацию циклического карбоната с этанолом в присутствии катализатора первой переэтерификации во второй реакционной зоне с образованием второго реакционного продукта, включающего диэтилкарбонат и гликоль; разделение второго реакционного продукта с извлечением первой фракции диэтилкарбоната и первой фракции гликоля; переэтерификацию, по меньшей мере, части первой фракции диэтилкарбоната с арилгидроксисоединением в присутствии катализатора второй переэтерификации в третьей реакционной зоне с образованием третьего реакционного продукта, включающего этиларилкарбонат и этанол; разделение третьего реакционного продукта с извлечением фракции этиларилкарбоната и первой фракции этанола; диспропорционирование, по меньшей мере, части фракции этиларилкарбоната в присутствии катализатора диспропорционирования в четвертой реакционной зоне с образованием четвертого реакционного продукта, включающего диарилкарбонат и диэтилкарбонат; разделение четвертого реакционного продукта с извлечением фракции диарилкарбоната и второй фракции диэтилкарбоната; рециркуляцию, по меньшей мере, части первой фракции этанола во вторую реакционную зону и рециркуляцию, по меньшей мере, части второй фракции диэтилкарбоната в третью реакционную зону. Либо способ получения диарилкарбоната включает реакцию аммиака и диоксида углерода в первой реакционной зоне с образованием первого реакционного продукта, включающего мочевину; переэтерификацию мочевины с этанолом в присутствии катализатора первой переэтерификации во второй реакционной зоне с образованием второго реакционного продукта, включающего диэтилкарбонат и аммиак; разделение второго реакционного продукта с извлечением первой фракции диэтилкарбоната и первой фракции аммиака; переэтерификацию, по меньшей мере, части первой фракции диэтилкарбоната с арилгидроксисоединением в присутствии катализатора второй переэтерификации в третьей реакционной зоне с образованием третьего реакционного продукта, включающего этиларилкарбонат и этанол; разделение третьего реакционного продукта с извлечением фракции этиларилкарбоната и фракции этанола; диспропорционирование, по меньшей мере, части фракции этиларилкарбоната в присутствии катализатора диспропорционирования в четвертой реакционной зоне с образованием четвертого реакционного продукта, включающего диарилкарбонат и диэтилкарбонат; разделение четвертого реакционного продукта с извлечением фракции диарилкарбоната и второй фракции диэтилкарбоната; рециркуляцию, по меньшей мере, части фракции этанола во вторую реакционную зону и рециркуляцию, по меньшей мере, части второй фракции диэтилкарбоната в третью реакционную зону. Раскрываемые способы эффективно объединяют в одно целое производство диэтилкарбоната и диарилкарбоната, исключая при этом необходимость экстракционной дистилляции с растворителем, которую обычно применяют при получении диарилкарбонатов из диметилкарбоната, обеспечивая совмещение реакционного и разделительного оборудования и наилучшее использование сырья и снижая издержки производства и капитальные вложения для таких способов. В некоторых вариантах осуществления раскрываемые в изобретении способы могут быть осуществлены, например, с практически замкнутым циклом по этанолу. 2 н. и 27 з.п. ф-лы, 24 ил., 9 табл., 18 пр.
1. Способ получения диарилкарбоната, включающий:
реакцию эпоксида и диоксида углерода в первой реакционной зоне с образованием первого реакционного продукта, включающего циклический карбонат;
переэтерификацию циклического карбоната с этанолом в присутствии катализатора первой переэтерификации во второй реакционной зоне с образованием второго реакционного продукта, включающего диэтилкарбонат и гликоль;
разделение второго реакционного продукта с извлечением первой фракции диэтилкарбоната и первой фракции гликоля;
переэтерификацию, по меньшей мере, части первой фракции диэтилкарбоната с арилгидроксисоединением в присутствии катализатора второй переэтерификации в третьей реакционной зоне с образованием третьего реакционного продукта, включающего этиларилкарбонат и этанол;
разделение третьего реакционного продукта с извлечением фракции этиларилкарбоната и первой фракции этанола;
диспропорционирование, по меньшей мере, части фракции этиларилкарбоната в присутствии катализатора диспропорционирования в четвертой реакционной зоне с образованием четвертого реакционного продукта, включающего диарилкарбонат и диэтилкарбонат;
разделение четвертого реакционного продукта с извлечением фракции диарилкарбоната и второй фракции диэтилкарбоната;
рециркуляцию, по меньшей мере, части первой фракции этанола во вторую реакционную зону; и
рециркуляцию, по меньшей мере, части второй фракции диэтилкарбоната в третью реакционную зону.
2. Способ по п.1, дополнительно включающий
дегидратацию, по меньшей мере, части первой фракции гликоля с образованием эпоксида и воды;
разделение воды от эпоксида;
рециркуляцию, по меньшей мере, части эпоксида в первую реакционную зону.
3. Способ по п.1, в котором катализатор первой переэтерификации и катализатор второй переэтерификации каждый независимо включает, по меньшей мере, либо один твердый катализатор переэтерификации, либо один растворимый металлоорганический катализатор, или их комбинации.
4. Способ по п.3, в котором катализатор первой переэтерификации включает твердый катализатор переэтерификации, где способ дополнительно включает введение следового количества растворимого металлоорганического катализатора во вторую реакционную зону.
5. Способ по п.4, в котором растворимый металлоорганический катализатор вводят в количестве от 1 ppm до 200 ppm от суммарной массы реагентов.
6. Способ по п.3, в котором катализатор второй переэтерификации включает твердый катализатор переэтерификации, где способ дополнительно включает введение следового количества растворимого металлоорганического катализатора в третью реакционную зону.
7. Способ по п.6, в котором растворимый металлоорганический катализатор вводят в количестве от 1 части на миллион (ppm) до 200 частей на миллион (ppm) от суммарной массы реагентов.
8. Способ по п.7, дополнительно включающий поддержание концентрации воды в третьей реакционной зоне в интервале от 1 части на миллион (ppm) до 600 частей на миллион (ppm) по массе.
9. Способ по п.3, в котором твердый катализатор переэтерификации и растворимый металлоорганический катализатор каждый независимо включает, по меньшей мере, один элемент Группы II - Группы VI.
10. Способ по п.1,
в котором вторая реакционная зона включает реакторную систему каталитической дистилляции для одновременной переэтерификации циклического карбоната с этанолом и разделения второго реакционного продукта;
в котором третья реакционная зона включает реакторную систему каталитической дистилляции для одновременной переэтерификации диэтилкарбоната с арилгидроксильным соединением и разделения третьего реакционного продукта;
в котором четвертая реакционная зона включает реакторную систему каталитической дистилляции для одновременного диспропорционирования этиларилкарбоната и разделения четвертого реакционного продукта;
в котором первая фракция диэтилкарбоната включает диэтилкарбонат и этанол, где способ дополнительно включает:
разделение первой фракции диэтилкарбоната с извлечением третьей фракции диэтилкарбоната и второй фракции этанола и
рециркуляцию второй фракции этанола во вторую реакционную зону;
в котором первая фракция гликоля включает гликоль и циклический карбонат, где способ дополнительно включает:
разделение первой фракции гликоля с извлечением второй фракции гликоля и фракции циклического карбоната и
рециркуляцию фракции циклического карбоната во вторую реакционную зону;
в котором фракция диарилкарбоната включает диарилкарбонат, этиларилкарбонат, арилгидроксильное соединение и побочные продукты реакции, где способ дополнительно включает:
разделение фракции диарилкарбоната с извлечением фракции продукта диарилкарбоната, фракции побочных продуктов реакции, включающей соединения с большей молекулярной массой, чем диарилкарбонат, и, по меньшей мере, одной рециркулируемой фракции, включающей, по меньшей мере, либо один этиларилкарбонат, либо одно арилгидроксильное соединение; и
рециркуляцию, по меньшей мере, одного рецикла в четвертую реакционную зону; и
в котором первая фракция этанола включает этанол и диэтилкарбонат, где способ дополнительно включает:
разделение первой фракции этанола с извлечением третьей фракции этанола и четвертой фракции диэтилкарбоната;
рециркуляцию четвертой фракции диэтилкарбоната в третью реакционную зону; и
введение, по меньшей мере, части третьей фракции этанола в качестве, по меньшей мере, части первой фракции этанола, рециркулируемой во вторую реакционную зону.
11. Способ по п.10, дополнительно включающий
дегидратацию, по меньшей мере, части второй фракции гликоля с образованием эпоксида и воды;
разделение воды от эпоксида;
рециркуляцию, по меньшей мере, части эпоксида в первую реакционную зону.
12. Способ по п.1, в котором, по меньшей мере, одна из второй, третьей и четвертой реакционных зон включает реакторную систему каталитической дистилляции, и в котором, по меньшей мере, одна реакторная система каталитической дистилляции включает внутреннее теплообменное устройство, обеспечивающее разделение пара и жидкости и направление потока пара и жидкости внутри реакторной системы каталитической дистилляции.
13. Способ по п.1, в котором, по меньшей мере, одна из второй реакционной зоны, третьей реакционной зоны и четвертой реакционной зоны включает два или более катализаторов переэтерификации для увеличения срока службы катализатора и уменьшения загрязнения катализатора.
14. Способ по п.13, в котором два или более катализаторов переэтерификации включают, по меньшей мере, либо отличающиеся компоненты активного металла, либо отличающиеся композиции носителя, любо отличающиеся свойства носителя.
15. Способ по п.1, где способ осуществляют с практически замкнутым циклом по этанолу.
16. Способ получения диарилкарбоната, включающий
реакцию аммиака и диоксида углерода в первой реакционной зоне с образованием первого реакционного продукта, включающего мочевину;
переэтерификацию мочевины с этанолом в присутствии катализатора первой переэтерификации во второй реакционной зоне с образованием второго реакционного продукта, включающего диэтилкарбонат и аммиак;
разделение второго реакционного продукта с извлечением первой фракции диэтилкарбоната и первой фракции аммиака;
переэтерификацию, по меньшей мере, части первой фракции диэтилкарбоната с арилгидроксисоединением в присутствии катализатора второй переэтерификации в третьей реакционной зоне с образованием третьего реакционного продукта, включающего этиларилкарбонат и этанол;
разделение третьего реакционного продукта с извлечением фракции этиларилкарбоната и фракции этанола;
диспропорционирование, по меньшей мере, части фракции этиларилкарбоната в присутствии катализатора диспропорционирования в четвертой реакционной зоне с образованием четвертого реакционного продукта, включающего диарилкарбонат и диэтилкарбонат;
разделение четвертого реакционного продукта с извлечением фракции диарилкарбоната и второй фракции диэтилкарбоната;
рециркуляцию, по меньшей мере, части фракции этанола во вторую реакционную зону и
рециркуляцию, по меньшей мере, части второй фракции диэтилкарбоната в третью реакционную зону.
17. Способ по п.16, дополнительно включающий рециркуляцию, по меньшей мере, части фракции аммиака в первую реакционную зону.
18. Способ по п.16, в котором катализатор первой переэтерификации и катализатор второй переэтерификации каждый независимо включает, по меньшей мере, либо один твердый катализатор переэтерификации, либо один растворимый металлоорганический катализатор, или их комбинации.
19. Способ по п.18, в котором катализатор первой переэтерификации включает твердый катализатор переэтерификации, где способ дополнительно включает введение следового количества растворимого металлоорганического катализатора во вторую реакционную зону.
20. Способ по п.19, в котором растворимый металлоорганический катализатор вводят в количество от 1 части на миллион (ppm) до 200 частей на миллион (ppm) от суммарной массы реагентов.
21. Способ по п.18, в котором катализатор второй переэтерификации включает твердый катализатор переэтерификации, где способ дополнительно включает введение следового количества растворимого металлоорганического катализатора в третью реакционную зону.
22. Способ по п.21, в котором растворимый металлоорганический катализатор вводят в количество от 1 части на миллион (ppm) до 200 частей на миллион (ppm) от суммарной массы реагентов.
23. Способ по п.22, дополнительно включающий поддержание концентрации воды в третьей реакционной зоне в интервале от 1 части на миллион (ppm) до 600 частей на миллион (ppm) по массе.
24. Способ по п.18, в котором твердый катализатор переэтерификации и растворимый металлоорганический катализатор каждый независимо включает, по меньшей мере, один элемент Группы II - Группы VI.
25. Способ по п.16:
в котором вторая реакционная зона включает:
первую зону реакции и дистилляции мочевины с катализатором или без катализатора для переэтерификации, по меньшей мере, части мочевины с этанолом с образованием пятого реакционного продукта, включающего этилкарбамат и аммиак;
вторую зону реакции мочевины, включающую реакторную систему каталитической дистилляции, для переэтерификации этилкарбамата в пятом реакционном продукте с этанолом с образованием шестого реакционного продукта, включающего диэтилкарбонат и аммиак; и
одну или более ректификационных колонн для разделения пятого и шестого реакционных продуктов с извлечением первой фракции диэтилкарбоната и фракции аммиака;
в котором третья реакционная зона включает реакторную систему каталитической дистилляции для одновременной переэтерификации диэтилкарбоната арилгидроксильным соединением и разделения третьего реакционного продукта;
в котором четвертая реакционная зона включает реакторную систему каталитической дистилляции для одновременного диспропорционирования этиларилкарбоната и разделения четвертого реакционного продукта;
в котором первая фракция диэтилкарбоната включает диэтилкарбонат и этанол, где способ дополнительно включает:
разделение первой фракции диэтилкарбоната с извлечением третьей фракции диэтилкарбоната и второй фракции этанола и
рециркуляцию второй фракции этанола во вторую реакционную зону;
и
в котором фракция диарилкарбоната включает диарилкарбонат, этиларилкарбонат, арилгидроксильное соединение и побочные продукты реакции, где способ дополнительно включает:
разделение фракции диарилкарбоната с извлечением фракции продукта диарилкарбоната, фракции побочных продуктов реакции, включающей соединения с большей молекулярной массой, чем диарилкарбонат, и, по меньшей мере, одной рециркулируемой фракции, включающей, по меньшей мере, либо один этиларилкарбонат, либо одно арилгидроксильное соединение; и
рециркуляцию, по меньшей мере, одного рецикла в четвертую реакционную зону.
26. Способ по п.16, в котором, по меньшей мере, одна из второй, третьей и четвертой реакционных зон включает реакторную систему каталитической дистилляции, и в котором, по меньшей мере, одна реакторная система каталитической дистилляции включает внутреннее теплообменное устройство, обеспечивающее разделение пара и жидкости и направление потока пара и жидкости внутри реакторной системы каталитической дистилляции.
27. Способ по п.16, в котором, по меньшей мере, одна из второй реакционной зоны, третьей реакционной зоны и четвертой реакционной зоны включает два или более катализаторов переэтерификации для увеличения срока службы катализатора и уменьшения загрязнения катализатора.
28. Способ по п.27, в котором два или более катализаторов переэтерификации включают, по меньшей мере, либо отличающиеся компоненты активного металла, либо отличающиеся композиции носителя, любо отличающиеся свойства носителя.
29. Способ по п.16, где способ осуществляют с практически замкнутым циклом по этанолу.
| US 2007093672 A1, 26.04.2007 | |||
| US 2004059083 A1, 25.03.2004 | |||
| US 2005203307 A1, 15.09.2005 | |||
| US 7141641 B2, 28.11.2006 | |||
| US 20060094893 A1, 04.05.2006 | |||
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| US 5498743 A1, 12.03.1996 | |||
| СПОСОБ ПОЛУЧЕНИЯ АРОМАТИЧЕСКОГО КАРБОНАТА | 2004 |
|
RU2329250C2 |

