Область техники, к которой относится изобретение
Варианты осуществления, описанные в настоящем документе, в целом, относятся к способам и твердым катализаторам для реакций, включающих алкоголиз, переэтерификацию и диспропорционирование. Более конкретно, варианты осуществления, описанные в настоящем документе, относятся к способам непрерывного получения органических карбонатов, органических карбаматов и других продуктов посредством алкоголиза, переэтерификации и/или диспропорционирования над твердым катализатором в виде неподвижного слоя. Растворимое металлорганическое соединение может непрерывно вводиться в реактор при очень низких уровнях для поддержания активности твердого катализатора в течение большой продолжительности цикла.
Уровень техники
Переэтерификация или реакция обмена сложных эфиров с помощью спиртов (реакция алкоголиза) представляет собой важный класс реакций, которые могут катализироваться с помощью как кислотных, так и основных катализаторов. Примеры переэтерификации, как правило, включают химические реакции, в которых участвуют органические карбонаты и сложные эфиры карбоновых кислот в качестве реагентов, продуктов или как тех, так и других. Другие реакции переэтерификации включают получение биодизельного топлива посредством переэтерификации триглицеридов с помощью этанола или метанола. Алкоголиз, как правило, представляет собой реакцию, где одна или несколько функциональных групп соединения заменяются алкокси или арилокси группами алкоголя (алкил- или арилгидроксильного соединения). Примеры алкоголиза включают химические реакции, в которых участвует мочевина, где аминовые группы заменяются алкокси группами с получением органических карбаматов и карбонатов.
Сложные эфиры карбоновых кислот получают посредством переэтерификации сложного эфира карбоновой кислоты с помощью спирта в присутствии кислотного и основного катализатора. Серная кислота (гомогенная) и кислотные смолы (твердые) являются предпочтительными кислотными катализаторами. Растворимые основания, такие как NaOH и KOH, различные алкоксиды или амины Na/K (гомогенные) и различные основные смолы (твердые) являются предпочтительными основными катализаторами. Хотя катализаторы могут представлять собой либо гомогенный катализатор, либо гетерогенный катализатор для переэтерификации сложных эфиров карбоновых кислот, основные катализаторы являются, как правило, более эффективными, чем кислотные катализаторы. Например, длинноцепные сложные алкилметакриловые эфиры получают посредством реакции обмена метилметакрилата с помощью длинноцепного спирта в присутствии основного катализатора.
Биодизельное топливо может быть получено посредством переэтерификации растительных масел (триглицеридов) с помощью метанола или этанола посредством использования гомогенного основного катализатора, такого как метоксид натрия или ацетат кальция, как описано в патентах США № 6712867 и 5525126, и основного твердого катализатора, такого как смешанный оксид из оксида цинка и оксида алюминия или алюмината цинка (оксид цинка на носителе из оксида алюминия, кальцинированные при высокой температуре). Твердые катализаторы на основе алюмината цинка описаны, например, в патенте США № 5908946 и в публикации заявки на патент США № 2004/0034244.
Патент США № 5908946 описывает двухстадийный способ получения сложных эфиров посредством взаимодействия растительных масел или животных масел со спиртом в присутствии твердых катализаторов, таких как оксид цинка или алюминаты цинка типа шпинели. На первой стадии, вызывается преобразование триглицерида до высокой степени преобразования, обычно выше чем 90%. На второй стадии, преобразуются остальные триглицериды, диглицериды и моноглицериды. Реакции переэтерификации осуществляют при температуре от 230 до 245°C, примерно при 5,2 бар (примерно 725 фунт/кв.дюйм абс.). Высокая степень преобразования требует относительно низких скоростей потока смеси исходных материалов (часовая объемная скорость 0,5 час-1 или ниже).
Патент США № 6147196 описывает способ получения сложных эфиров жирных кислот с высокой чистотой из растительного или животного масла в присутствии гетерогенного катализатора (алюмината цинка). Публикация заявки на патент США №2004/0034244 относится к технологической схеме получения сложных алкиловых эфиров из растительного или животного масла и спирта в присутствии гетерогенного катализатора (алюмината цинка). Сложные эфиры получают посредством переэтерификации в двух реакторах с неподвижным слоем. Высокое преобразование триглицерида получают в первом реакторе. После выделения глицерина из первого потока реакции переэтерификации, оставшиеся непреобразованные триглицериды, диглицериды и моноглицериды преобразуются в сложные эфиры во втором реакторе. Переэтерификацию осуществляют при 200°C, примерно при 62 бар (900 фунт/кв.дюйм абс.) и при часовой объемной скорости 0,5 час-1.
W. Xie et al. (J. Mol. Cat. A: Chem. 246, 2006, p. 24-32) обсуждают метанолиз соевого масла в присутствии кальцинированного гидротальцитного катализатора Mg-Al. Кальцинированные гидротальциты с отношением Mg/Al 3,0, полученные в результате кальцинирования при 500°C представляют собой катализатор, который может дать высокую основность и превосходную каталитическую активность для этой реакции. Они сообщают о растворимой основности гидротальцитов кальцинированных при различных температурах.
Дизельные двигатели выбрасывают больше частиц и NOx, чем бензиновые двигатели. Сообщается, что диалкилкарбонаты являются эффективными при уменьшении уровня частиц в выхлопе дизельного двигателя. В соответствии с патентом США № 5954280, мочевина и аммиак являются эффективными агентами, понижающими уровень NOx. Но использование мочевины и аммиака для дизельного двигателя имеет практические проблемы или неудобства. Патент США № 6017368 описывает этилкарбамат как эффективный при понижении уровней NOx от дизельных двигателей. Патент США № 4731231 (1988) сообщает, что сублимированная циануровая кислота может представлять собой эффективный агент для устранения или понижения уровня NOx. Высокотемпературное сублимирование циануровой кислоты производит изоциановую кислоту (HNCO), которая, как считается, является ответственной за устранение NOx. Европейские патенты EP 0363681 и EP 0636681 описывают сложный карбонатный эфир алифатического триола или тетраола в качестве компонента смазывающих агентов с низким уровнем дыма.
N-арилметилкарбамат получают по реакции ароматического амина с диметилкарбонатом, как правило, в присутствии основного катализатора, из-за низкой скорости реакции в отсутствие катализатора. N-арилметилкарбамат может разлагаться с получением ароматического изоцианата при повышенной температуре. Например, толуолдикарбамат получают посредством взаимодействия толуолдиамина с диметилкарбонатом в присутствии катализатора. Разложение толуолдикарбамата при повышенной температуре дает толуолдиизоцианат.
Органические карбонаты (сложные диэфиры угольной кислоты) являются пригодными для использования соединениями, которые могут использоваться в качестве растворителей, алкилирующих агентов, агентов для карбонилирования, агентов сополимеризации, присадок к топливу и тому подобное. Диметилкарбонат (DMC) представляет собой важный диалкилкарбонат, обычно используемый в качестве исходного материала для получения дифенилкарбоната (DPC, диарилкарбонат). Имеются различные способы промышленного получения DMC. В одном из таких промышленных способов, DMC получают посредством переэтерификации циклического карбоната с помощью метанола в присутствии гомогенизированного катализатора. Хотя патенты могут описывать использование гомогенных катализаторов или гетерогенных катализаторов для переэтерификации циклического карбоната с помощью метанола, в настоящее время нет промышленного опыта, где гетерогенный или твердый катализатор используют для получения DMC, вероятно, из-за малой продолжительности цикла гетерогенных катализаторов для таких способов. DPC обычно сополимеризуется с диолом, таким как бисфенол A, для получения поликарбонатов. Поликарбонаты используются в различных специальных применениях, таких как диски памяти, ветровые стекла, технологические пластики, оптические материалы и тому подобное.
Современные технологии для получения диарилкарбонатов с использованием нефосгенового способа производят ароматические карбонаты, такие как DPC, посредством переэтерификации DMC с помощью фенола с получением метилфенилкарбоната и метанола, с последующим диспропорционированием метилфенилкарбоната с получением DPC и DMC в присутствии гомогенных металлорганических катализаторов посредством использования ряда из множества реакторов реакционной дистилляции. Предпочтительный гомогенный катализатор представляет собой алкоксид титана. Такие способы описаны, например, в патентах США № 4045464, 4554110, 5210268 и 6093842. Гомогенные катализаторы извлекают из самой тяжелой части потоков продуктов как твердый продукт, который может затем преобразовываться в растворимый гомогенный катализатор для рециклирования.
Использование гомогенного катализатора при получении DPC часто требует отделения гомогенного катализатора от продукта, в особенности, когда катализаторы используются при относительно высоких скоростях введения исходных материалов. Для уменьшения этого и других недостатков, связанных с использованием гомогенных катализаторов для получения диарилкарбонатов, патенты США № 5354923 и 5565605 и публикация заявки PCT WO03/066569 описывают альтернативные способы, где используются гетерогенные катализаторы. Например, патент США № 5354923 описывает катализаторы на основе оксида титана в форме порошка для демонстрации получения EPC, MPC и DPC из DEC или DMC и фенола. Патент США № 5565605 описывает микропористые материалы, содержащие элементы Группы 4, в качестве катализаторов для переэтерификации и диспропорционирования. Однако твердые катализаторы в форме порошка, как правило, являются непригодными или менее предпочтительными для промышленного получения DPC или метилфенилкарбоната в больших объемах. WO03/066569 описывает способ непрерывного получения DPC в присутствии гетерогенного катализатора, полученного посредством нанесения оксида титана на носителе из диоксида кремния в двухстадийном способе с неподвижным слоем, посредством взаимодействия DMC с фенолом.
Z-H Fu and Y. Ono (J. MoI. Catal. A.Chemical, 118 (1997), p. 293-299) и заявка JP № HEI 07-6682 описывают гетерогенные катализаторы для получения дифенилкарбоната посредством переэтерификации DMC с помощью фенола до MPC и диспропорционирования MPC до DPC в присутствии MoO3 или V2O5, нанесенных на неорганический носитель, такой как диоксид кремния, диоксид циркония или диоксид титана. Переэтерификация и диспропорционирование осуществляют в башенном реакторе реакционной дистилляции, состоящем из реактора и дистилляционной башни, с удалением побочных продуктов посредством дистилляции.
Публикации заявок на патент США № 2007/0093672 ('672) и 2007/0112214 ('214) (теперь патент США № 7288668) описывают способы получения различных органических карбонатов, таких как диарилкарбонаты, включая DPC, в присутствии гетерогенных катализаторов. В публикации '214, необходимые реакции (переэтерификации и диспропорционирования) осуществляют в жидкой фазе в присутствии гетерогенного катализатора. Множество реакторов с неподвижным слоем для реакций переэтерификации и диспропорционирования соединены с одной дистилляционной колонной, где легкие соединения, такие как этанол и DEC, удаляют как фракции из верхней части колонны, а более высококипящие соединения, включая DPC, удаляют как смешанную кубовую фракцию. Затем DPC извлекают из кубовой фракции.
Публикация '672 описывает способ получения диарилкарбонатов и диалкилкарбонатов посредством осуществления необходимых реакций в духфазной (пары и жидкость) реакции над различными твердыми катализаторами для переэтерификации и диспропорционирования. Химические реакции, дающие органические карбонаты, осуществляются в ряду реакторов с неподвижным слоем, в то же время, осуществляя выделение легкого побочного продукта в жидкой фазе в паровую фазу, чтобы сдвинуть неблагоприятное равновесие реакции в направлении желаемого продукта. Способ является в особенности пригодным для получения алкиларилкарбонатов, таких как EPC (этилфенилкарбонат), и диарилкарбонатов, таких как DPC (дифенилкарбонат). Способ является также пригодным для получения диалкилкарбонатов, таких как DEC. Ряд реакторов с неподвижным слоем соединяются в различных положениях на одной дистилляционной колонне посредством боковых выходных потоков и обратных потоков. Дистилляционная колонна также содержит разделительные ступени выше последнего реактора в ряду и ниже первого реактора в ряду. Гетерогенные катализаторы могут быть получены посредством осаждения одного или двух оксидов металлов из Ti, Zr, Nb, Hf, Ta, Mo, V, Sb и тому подобное на пористых носителях, таких как силикагель. Гетерогенные катализаторы могут также быть получены посредством прививки одного или нескольких металлорганических соединений из элементов Ti, Zr, Nb, Hf, Ta, Mo, V, Sb и тому подобное на пористом носителе, который имеет поверхностные гидроксильные группы или смесь гидроксильных и алкоксигрупп.
Различные другие способы получения органических карбонатов с помощью гетерогенных катализаторов описываются в патентах США № 5231212, 5498743 и 6930195.
P. Ball et al. (C1 Mol. Chem. Vol. 1, 1984, p. 95-108) изучали химию получения диалкилкарбоната в присутствии различных гомогенных или гетерогенных катализаторов. Например, диметилкарбонат получают посредством алкоголиза мочевины. Диметоксид дибутилолова, как сообщается, является особенно эффективным катализатором. Сообщается, что гетерогенные катализаторы также являются эффективными для химии в присутствии сокатализаторов, таких, как 4-диметиламинопиридин и PPh3. Гетерогенные катализаторы, о которых сообщалось, представляют собой Al2O3, Sb2O3 и диоксид кремния. Плавленный SiO2 не является катализатором, но становится каталитическим в присутствии PPh3.
В патенте США № 7074951, диалкилкарбонат получают посредством алкоголиза мочевины с помощью спирта в присутствии гомогенного катализатора на основе комплекса олова в присутствии высококипящего растворителя, содержащего атом - донор электронов, такого как триглим. Этот патент также демонстрирует способность непрерывного получения DMC в течение примерно 1500 часов.
Европейский патент EP 1629888 и D. Wang et al. (Fuel Processing Tech. 88, 8, 2007, p. 807-812) описывают, что DMC и DEC могут быть получены в присутствии оксида цинка и оксида цинка на носителе из диоксида кремния. Эти публикации ничего не говорят о стабильности катализатора или продолжительности цикла катализатора.
Дезактивирование катализатора во время реакций переэтерификации и диспропорционирования может вызываться осаждением тяжелых полимеров на поверхности катализатора и в порах. Скорость дезактивирования катализатора под действием осаждения полимера увеличивается с увеличением концентрации алкиларилкарбоната и диарилкарбоната или как того, так и другого в реакционной смеси. Деполимеризация полимеров на гетерогенных катализаторах описывается в публикации '672. Однако деполимеризация может приводить только к частичному восстановлению активности твердого катализатора.
Патенты США № 6768020 и 6835858 описывают способы получения диалкилкарбонатов и побочного продукта пропиленгликоля посредством реакции пропиленкарбоната с DMC, водой или как с тем, так и с другим, в присутствии твердого катализатора, такого как оксид лантана и оксид цинка на носителе из оксида алюминия, диоксида кремния и тому подобное. Проблема нестабильности катализатора частично решается в патенте США № 6768020 посредством осаждения большого количества оксида лантана на носитель, такой как оксид алюминия и диоксид кремния.
Приемлемая методика для компенсации дезактивирования катализатора представляет собой постепенный подъем температуры реакции, когда дезактивируется катализатор. Это методика, к сожалению, часто ускоряет дезактивирование гетерогенных катализаторов.
Для промышленного производства, использующего гетерогенный катализатор, как правило, требуются долговременные стабильные рабочие характеристики твердого катализатора. Стоимость катализатора, перерывы в работе, связанные с заменой катализатора, и другие факторы, как известно в данной области, диктуют, чтобы гетерогенные катализаторы имели минимальный срок службы, как правило, больше чем 3 месяц, 6 месяцев или год, в зависимости от способа.
Хотя гетерогенный катализ различных реакций переэтерификации является возможным, как описывается в различных патентах и публикациях, выше, они не сообщают ничего о сроке службы или продолжительности цикла катализатора. Согласно опыту авторов настоящего изобретения такие гетерогенные катализаторы имеют нежелательно короткие продолжительности цикла.
Соответственно, имеется необходимость в способах переэтерификации и/или диспропорционирования, использующих гетерогенные катализаторы с улучшенными рабочими характеристиками катализатора.
Сущность изобретения
В одном из аспектов, варианты осуществления, описанные в настоящем документе, относятся к способу алкоголиза, способ включает: введение реагентов и микроскопического количества растворимого металлорганического соединения в реактор, содержащий твердый катализатор алкоголиза; где растворимое металлорганическое соединение и твердый катализатор алкоголиза, каждый, независимо содержит элемент Группы II - Группы VI. Твердый катализатор и металлорганическое соединение могут содержать один и тот же элемент Группы II - Группы VI в некоторых вариантах осуществления.
В другом аспекте, варианты осуществления, описанные в настоящем документе, относятся к способу получения диалкилкарбонатов, способ включает: введение спирта и реагента алкоголиза, содержащего, по меньшей мере, одно соединение из мочевины, органического карбамата и циклического карбоната, в первую реакционную зону, содержащую твердый катализатор алкоголиза; введение растворимого металлорганического соединения в первую реакционную зону, где твердый катализатор алкоголиза и растворимое металлорганическое соединение, каждый, независимо содержат элемент Группы II - Группы VI.
В другом аспекте, варианты осуществления, описанные в настоящем документе, относятся к способу получения диарилкарбоната, способ включает: введение ароматического гидроксисоединения и диалкилкарбоната в первую реакционную зону, содержащую твердый катализатор переэтерификации; и введение растворимого металлорганического соединения в первую реакционную зону, где твердый катализатор переэтерификации и растворимое металлорганическое соединение, каждый, независимо содержат элемент Группы II - Группы VI.
В другом аспекте, варианты осуществления, описанные в настоящем документе, относятся к способу получения алкиларилкарбоната, способ включает: введение ароматического гидроксисоединения и диалкилкарбоната в первую реакционную зону, содержащую твердый катализатор переэтерификации; и введение растворимого металлорганического соединения в первую реакционную зону, где твердый катализатор переэтерификации и растворимое металлорганическое соединение, каждый, независимо содержат элемент Группы II - Группы VI.
В другом аспекте, варианты осуществления, описанные в настоящем документе, относятся к способу получения биодизельного топлива, способ включает: введение спирта и глицерида в первую реакционную зону, содержащую твердый катализатор переэтерификации; и введение растворимого металлорганического соединения в первую реакционную зону, где твердый катализатор переэтерификации и растворимое металлорганическое соединение, каждый, независимо содержат элемент Группы II - Группы VI.
В другом аспекте, варианты осуществления, описанные в настоящем документе, относятся к способу получения алкиларилкарбоната, способ включает: введение ароматического гидроксисоединения и диалкилкарбоната в первую реакционную зону, содержащую твердый катализатор переэтерификации; и введение растворимого металлорганического соединения в первую реакционную зону, где твердый катализатор переэтерификации и растворимое металлорганическое соединение, каждый, независимо содержат элемент Группы II - Группы VI.
В другом аспекте, варианты осуществления, описанные в настоящем документе, относятся к способу получения биодизельного топлива, способ включает: введение спирта и глицерида в первую реакционную зону, содержащую твердый катализатор переэтерификации; и введение растворимого металлорганического соединения в первую реакционную зону, где твердый катализатор переэтерификации и растворимое металлорганическое соединение, каждый, независимо содержат элемент Группы II - Группы VI.
В другом аспекте, варианты осуществления, описанные в настоящем документе, относятся к способу реактивирования отработанного твердого катализатора алкоголиза, способ включает: удаление полимерных материалов, осажденных на катализаторе; и повторное осаждение каталитически активных металлов на твердый катализатор.
Другие аспекты и преимущества будут понятны из следующего далее описания и прилагаемой формулы изобретения.
Краткое описание чертежей
Фигура 1 представляет собой упрощенную блок-схему способа, иллюстрирующую способ получения диарилкарбонатов в соответствии с вариантами осуществления, описанными в настоящем документе.
Фигура 2 представляет собой упрощенную блок-схему способа, иллюстрирующую способ получения диарилкарбонатов в соответствии с вариантами осуществления, описанными в настоящем документе.
Фигура 3 представляет собой упрощенную блок-схему способа, иллюстрирующую способ получения диарилкарбонатов в соответствии с вариантами осуществления, описанными в настоящем документе.
Фигура 4 представляет собой графическое представление переэтерификации с использованием гомогенного катализатора.
Фигура 5 представляет собой графическое представление активности катализатора после реактивирования катализатора в соответствии с вариантами осуществления, описанными в настоящем документе.
Фигура 6 представляет собой графическое представление активности твердого катализатора, когда микроскопическое количество растворимого металлорганического соединения добавляют в реактор в соответствии с вариантами осуществления, описанными в настоящем документе.
Фигура 7 графически сравнивает активность гетерогенного катализатора с активностью твердого катализатора, когда микроскопическое количество растворимого металлорганического соединения добавляют в реактор в соответствии с вариантами осуществления, описанными в настоящем документе.
Фигура 8 представляет собой графическое представление активности твердого катализатора, когда микроскопическое количество растворимого металлорганического соединения добавляют в реактор в соответствии с вариантами осуществления, описанными в настоящем документе.
Фигуры 9A и 9B представляют собой графические представления активности твердого катализатора во время получения EPC и DPC, соответственно, когда микроскопическое количество растворимого металлорганического соединения добавляют в реактор в соответствии с вариантами осуществления, описанными в настоящем документе.
Фигура 10 представляет собой графическое представление активности гетерогенного катализатора во время получения DPC, когда прививают катализатор, осуществляя одновременно реакцию переэтерификации.
Фигура 11 графически иллюстрирует преобразование EPC в DPC и DEC в отсутствие твердых катализаторов в соответствии с вариантами осуществления, описанными в настоящем документе.
Фигура 12 графически представляет результаты алкоголиза пропиленкарбоната с помощью этанола с получением DEC и пропиленгликоля в присутствии твердого катализатора в соответствии с вариантами осуществления, описанными в настоящем документе.
Фигура 13 представляет результаты получения DEC с использованием гомогенного катализатора.
Фигура 14 представляет результаты получения DEC с использованием твердого катализатора в соответствии с вариантами осуществления, описанными в настоящем документе.
Фигура 15 представляет собой упрощенную блок-схему способа получения диалкилкарбонатов с использованием твердого катализатора в соответствии с вариантами осуществления, описанными в настоящем документе.
Фигура 16 представляет результаты получения DEC из этилкарбамата с использованием твердого катализатора в соответствии с вариантами осуществления, описанными в настоящем документе.
Фигура 17 представляет результаты алкоголиза масла канолы с помощью метанола с использованием твердого катализатора в соответствии с вариантами осуществления, описанными в настоящем документе.
Фигура 18 представляет собой упрощенную блок-схему способа непрерывного получения DEC и побочного продукта пропиленгликоля посредством осуществления алкоголиза пропиленкарбоната с помощью этанола в присутствии твердого катализатора в соответствии с вариантами осуществления, описанными в настоящем документе.
Подробное описание
В одном из аспектов, варианты осуществления, описанные в настоящем документе, относятся к способам алкоголиза, переэтерификации и/или диспропорционирования с использованием твердых катализаторов. Как используется в настоящем документе, алкоголиз определяется как процесс, представляющий собой различные химические реакции, где органическое соединение гидроксила (спирт) участвует как один из двух реагентов с получением продукта и побочного продукта. Алкоголиз может определяться как разрыв связей (C - Y) между атомом углерода и гетероатомом Y молекул с помощью молекулы спирта (ROH). Алкоголиз представляет собой реакции, в которых участвуют карбонильные группы молекулы и сама карбонильная группа удерживается в молекуле продукта. По этой причине атом C в связи C - Y представляет собой атом углерода карбонильной группы молекулы. Как правило, алкоголиз представляет собой обратимую реакцию и может быть представлен следующим образом:
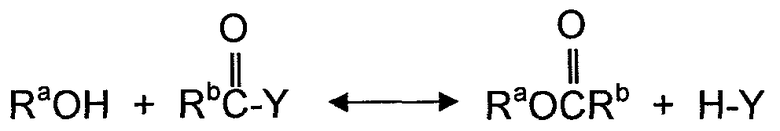
где Y представляет собой гетероатом или гетероатом функциональной группы и Rb представляет собой алкил, арил или функциональную группу, имеющую один или несколько гетероатомов.
Примеры реакций алкоголиза представляют собой реакцию спирта со сложными диэфирами угольной кислоты, сложными эфирами карбоновых кислот, мочевиной и карбаматами. Алкоголиз диалкилкарбоната (часто упоминается в литературе как переэтерификация) с помощью фенола дает алкиларилкарбонат и спирт. Алкоголиз сложного эфира карбоновой кислоты с помощью спирта обменивает алкильную группу сложного эфира с алкильной группой молекулы спирта и дает новую молекулу спирта. Алкоголиз мочевины с помощью спирта дает органический карбамат и аммиак. Алкоголиз органического карбамата с помощью спирта дает диалкилкарбонат и аммиак. Конкретные примеры реакций алкоголиза представляют собой переэтерификацию DEC с помощью фенола с получением EPC и этанола, алкоголиз мочевины или органического карбамата с помощью спирта, с получением органического карбамата или диалкилкарбоната и аммиака, переэтерификацию триглицерида с помощью метанол, с получением сложных метиловых эфиров (биодизельного топлива) и глицерина.
Хотя диспропорционирование несимметричных сложных диэфиров угольной кислоты и реакция диалкилкарбоната с органическим амином не включают спирта в качестве реагента, здесь считается, что эти типы реакций также определяются как реакции алкоголиза, также для удобства, поскольку Группы RA (R представляет собой алкил или арил, и A представляет собой атом кислорода или атом азота) участвуют в механизмах реакции на молекулярном уровне. По этой причине, переэтерификацию и диспропорционирование используют как синонимы алкоголиза, когда это необходимо для описания различных вариантов осуществления. Несколько реакций алкоголиза, рассмотренных выше, могут быть представлены следующими уравнениями реакций:
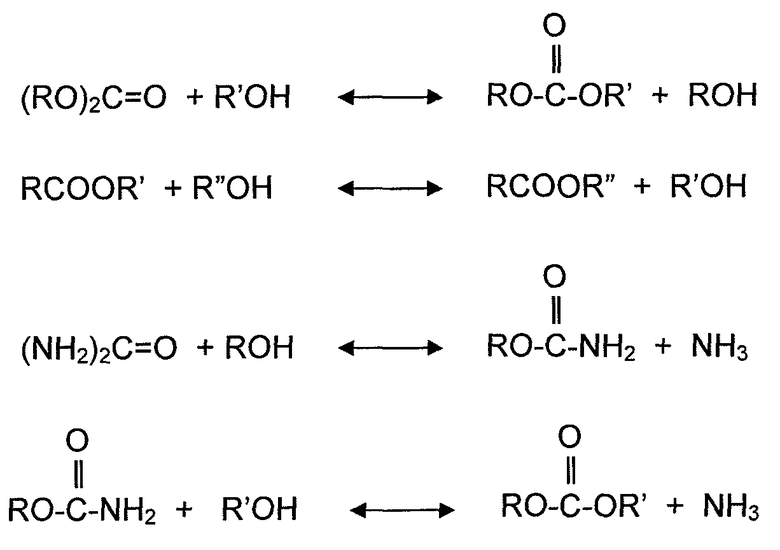
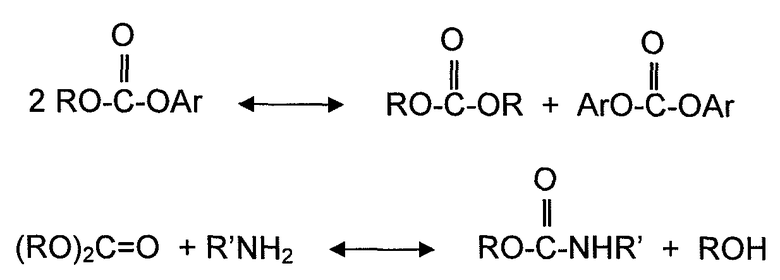
В другом аспекте, варианты осуществления, описанные в настоящем документе, относятся к новой методике поддержания активности катализатора для твердого катализатора в течение увеличенного времени цикла. Время цикла или продолжительность цикла твердого катализатора в настоящем документе определяется как период времени, в течение которого твердый катализатор может непрерывно использоваться без перерыва для желаемой химической реакции. Например, если катализатор требует регенерации или замены катализатора после непрерывного использования в течение 6 месяцев, продолжительность или время цикла катализатора составляет 6 месяцев. В соответствии с методикой, описанной в настоящем документе, твердые катализаторы для способов алкоголиза, могут сохранять активность катализатора в течение увеличенного времени цикла, такого как большее чем 3 месяца, 6 месяцев, 1 год, 1,5 года и 2 года или более, в различных вариантах осуществления.
Во время переэтерификации DEC с помощью фенола, дезактивирование гетерогенных катализаторов (оксида титана и смешанного оксида ниобия и оксида титан, иммобилизованного на диоксиде кремния) наблюдалось автором настоящего изобретения, и об этом сообщается в Исследовании 4 публикации '672. Деполимеризация осажденного полимера на катализаторе для улучшения активности катализатора также демонстрируется в Исследовании 6B публикации '672. Однако регенерация катализатора посредством деполимеризации приводит только к частичному восстановлению исходной активности катализатора. Природа дезактивирования катализатора не является полностью понятной в настоящее время.
Неожиданно обнаружено, что гетерогенные катализаторы переэтерификации, такие как гетерогенные катализаторы для получения DPC, дезактивируются по двум основным причинам: из-за осаждения полимера и выщелачивания компонента каталитически активного металла. Гетерогенные катализаторы для получения диалкилкарбоната посредством переэтерификации циклического карбоната с помощью спирта дезактивируется, прежде всего, из-за выщелачивания компонентов каталитически активного металла.
Во время реакций алкоголиза или переэтерификации над гетерогенными катализаторами, компоненты каталитически активного металла на твердых катализаторах могут выщелачиваться из гетерогенных катализаторов на основе оксида металла и металлорганических катализаторов, иммобилизованных на различных пористых носителях, в реакционную среду при условиях реакции, приводя к непрерывному дезактивированию катализатора. Это приводит к появлению неприемлемо короткой жизни катализатора для промышленных гетерогенных катализаторов, которые могут использоваться для непрерывного получения различных органических карбонатов. В дополнение к этому, как рассматривается выше, осаждение полимера может также влиять на рабочие характеристики катализатора переэтерификации. Еще один режим дезактивирования катализатора представляет собой отравление.
Гетерогенный катализатор, который должен использоваться в промышленном реакторе с неподвижным слоем должен иметь разумный срок службы как относительно времени цикла, так и общего времени службы. В отсутствие отравления, и если имеется слабое осаждение полимеров на гетерогенном катализаторе, или оно отсутствует, скорость растворения компонента активного металла из гетерогенного катализатора может определять срок службы катализатора.
Варианты осуществления, описанные в настоящем документе, относятся к способам поддержания постоянной или почти постоянной активности твердого катализатора в течение продолжительного периода времени, приемлемого для непрерывного получения различных органических соединений в промышленном масштабе. Такие способы могут быть особенно пригодными для непрерывного получения различных органических карбонатов, таких как диарилкарбонаты, диалкилкарбонаты и алкиларилкарбонаты, а также при других реакциях переэтерификации, таких как получение биодизельного топлива. Выбранные варианты осуществления, описанные в настоящем документе, относятся к способам поддержания стабильной активности катализатора в течение продолжительного периода времени для больших промышленных реакторов для непрерывного получения органических карбонатов, сложных эфиров карбоновых кислот или органических карбаматов.
Новая технология поддержания активности катализатора для твердого катализатора в течение увеличенного времени цикла заключается в том, что добавление микроскопического количества растворимого компонента активных металлов в поток жидких исходных материалов в реактор, содержащий твердый катализатор может давать в результате постоянную или почти постоянную активность катализатора в течение продолжительных периодов времен цикла. Неожиданно обнаружено, что добавление микроскопического количества растворимых компонентов активных металлов в поток жидких исходных материалов в реактор, содержащий твердый катализатор, может эффективно компенсировать потерю металла из-за выщелачивания металла из твердых катализаторов, например, посредством повторного осаждения компонента активного металла на катализаторе, что приводит к получению постоянной или почти постоянной активности катализатора в течение продолжительных периодов времени цикла. Например, настоящие результаты автора настоящего изобретения показывают, что активность катализатора может поддерживаться в течение более чем одного года посредством добавления микроскопического количества растворимых соединений активных металлов в поток жидких исходных материалов, вводимый в реакторы с неподвижным слоем, содержащие твердый катализатор.
Количество соединения активного металла, необходимое для поддержания активности твердого катализатора, может находиться в пределах от менее чем 1 м.д. до примерно 3000 м.д., в зависимости от конкретного компонента активного металла, реагентов и других компонентов исходных материалов. Количество активного металла в исходных материалах, например, для получения органических карбонатов посредством переэтерификации, может быть на один, два или более порядков величины меньше, чем концентрация гомогенного катализатора, вводимого для сравнимого способа с использованием только гомогенного катализатора. В некоторых вариантах осуществления, соединение активного металла может вводиться при отношении от 1 до 400 м.д. масс; от 10 до 300 м.д. масс, в других вариантах осуществления; от 15 до 200 м.д. масс, в других вариантах осуществления; от 20 до 150 м.д. масс, в других вариантах осуществления; и от 30 до 100 м.д. масс, в других вариантах осуществления, по отношению к общей массе жидкости, поступающей в зону каталитической реакции.
Например, когда твердый катализатор содержит активный металл, такой как металл Группы II-VI, микроскопическое количество растворимого металлорганического соединения, имеющего такой же активный металл Группы II-VI, может вводиться в реактор для поддержания активности твердого катализатора. В качестве конкретного примера, когда твердый катализатор содержит в качестве активного металла титан, может использоваться растворимое металлорганическое соединение, содержащее титан.
Когда используют ряд реакторов, например, реактор переэтерификации, соединенный последовательно с реактором диспропорционирования, микроскопическое количество растворимого металлорганического соединения может вводиться в один или оба реактора для поддержания активности катализатора в соответствующих реакторах. В некоторых вариантах осуществления, посредством введения микроскопического количества растворимого металлорганического соединения только в первый реактор в ряду реакторов, активность твердого катализатора может поддерживаться в каждом из реакторов. Например, когда реактор переэтерификации содержит твердый катализатор на основе смешанного оксида титана и ниобия, и реактор диспропорционирования содержит твердый алкоксид титана, привитой на диоксид кремния, добавление микроскопического количества растворимого органического соединения титана, такого как оксид титана, в первый реактор, может увеличить время цикла обоих типов твердых катализаторов.
Растворимое металлорганическое соединение может извлекаться и рециклироваться, если это желательно. В некоторых вариантах осуществления, может быть экономически невыгодным извлечение активного металла из потоков эффлюента из реактора для рециклирования. Когда он извлекается, компонент активного металла из потока эффлюента из реактора может извлекаться из тяжелого кубового потока как твердый материал и преобразовываться в растворимое металлорганическое соединение, которое может рециклироваться в реактор, например, посредством взаимодействия извлеченного твердого материала с органическим карбонатом или смесью органического карбоната и спирта при повышенной температуре. Извлеченное металлорганическое соединение, например, может представлять собой алкоксид металла, алкоксиалкилкарбонат металла (соль металла и сложного моноэфира угольной кислоты) или их смесь.
Получаемое таким образом увеличенное время жизни твердого катализатора алкоголиза может приводить к получению промышленно жизнеспособных способов с твердыми катализаторами для получения органических карбонатов, среди алкоголиза и/или других способов переэтерификации. Значительная экономия может быть реализована благодаря увеличению времени цикла катализаторов и уменьшению требований к разделению (меньше рабочих узлов, что приводит к потенциальной экономии капитальных затрат и затрат на работу).
Осаждение полимера на твердых катализаторах может также вызывать потерю активности катализатора. В таком случае, дезактивированный катализатор может регенерироваться посредством методик деполимеризации, описанных в настоящем документе и в публикации заявки на патент США № 2007/0093672. Деполимеризация также может вызывать потерю металла. В случае следующей деполимеризации, когда гетерогенный катализатор не восстанавливает активности катализатора до приемлемого уровня исходной активности, гетерогенный катализатор может потребовать повторного осаждения металла, например, с помощью технологий реактивирования катализатора, описанных в настоящем документе.
Вызывается ли дезактивирование катализатора осаждением полимера и выщелачиванием металла, активность катализатора может восстанавливаться с помощью технологий регенерации и реактивирования катализатора, описанных в настоящем документе. Реактивирование катализатора состоит из двух стадий: деполимеризации и кондиционирования поверхности на первой стадии и повторного осаждения металла на второй стадии. На первой стадии, дезактивированный твердый катализатор подвергается деполимеризации для удаления полимеров на твердом катализаторе, а затем кондиционированию поверхности посредством сушки. На второй стадии, осуществляют повторное осаждение компонента активного металла для компенсации потерь металла. Реактивирование дезактивированного катализатора будет обсуждаться более подробно позже.
Когда рассматривается реактивирование и/или регенерация катализатора, может быть выгодным иметь множество параллельных реакторов, с тем чтобы сделать возможным непрерывное производство во время способов реактивирования и восстановления катализатора.
Как описано выше, твердофазные способы алкоголиза, переэтерификации и диспропорционирования, описанные в настоящем документе, могут включать введение реагентов и микроскопического количества растворимого соединения активного металла в реактор, содержащий твердый катализатор, и осуществление контакта реагентов в присутствии твердого катализатора для алкоголиза, переэтерификации или диспропорционирования, по меньшей мере, части реагентов. Такие способы алкоголиза, переэтерификации или диспропорционирования могут включать, например, реакции для получения диалкилкарбонатов, диарилкарбонатов, алкиларилкарбонатов, биодизельных топлив, сложных органических эфиров и N-арилалкилкарбаматов, среди прочих реакций.
Хотя они описываются выше по отношению к реакциям алкоголиза, переэтерификации и диспропорционирования, в целом, расширение таких способов на получение органических карбонатов подробно объясняется ниже. Публикации заявок на патент США 2007/0093672 ('672) и 2007/0112214 ('214), как отмечено выше, описывают способы получения органических карбонатов с использованием гетерогенных катализаторов. Каждая из них тем самым включается в качестве ссылки.
Получение органического карбоната и органического карбамата
Органические карбонаты или органические карбаматы могут непрерывно получаться посредством использования систем из одного или множества реакторов в присутствии твердого катализатора или двух различных твердых катализаторов. Твердый катализатор или катализаторы требуют добавления микроскопического количества растворимого соединения активного металла в поток исходных материалов для реактора для получения увеличенного времени цикла катализатора. Твердые катализаторы могут иметь любую физическую форму и могут содержать различные металлорганические соединения, иммобилизованные на пористых носителях, и/или оксиды, содержащие элемент или множество элементов Групп II, III, IV, V и VI, нанесенные на соответствующий пористый носитель. Катализаторы могут представлять собой либо кислотные катализаторы, либо основные катализаторы. Общее количество каталитически активного металла или компонентов металла на катализаторе на носителе может находиться в пределах от примерно 0,02 мас.% до примерно 20 мас.%, в некоторых вариантах осуществления; от примерно 0,05 мас.% до примерно 10 мас.%, в других вариантах осуществления.
Реакторы, используемые в вариантах осуществления, описанных в настоящем документе, могут включать любые физические устройства или сочетание двух или более устройств. Реакторы могут иметь различные внутренние устройства для разделения пар-жидкость и прохождения паров/жидкости.
Посредством добавления микроскопического количества растворимого соединения активного металла в поток исходных материалов, стабильная активность катализатора может поддерживаться в течение неожиданно больших времен циклов. Например, добавление микроскопического количества растворимого соединение активного металла в потоки, вводимые в реактор с неподвижным слоем, для получения смесей этилфенилкарбоната и дифенилкарбоната, могут приводить к получению времени цикла более чем 14 месяцев на один рабочий цикл. Такие стабильные рабочие характеристики катализатора могут приводить к повышению производительности относительно желаемого продукта. В вариантах осуществления, имеющих ряд реакторов, микроскопическое количество компонента активного металла может добавляться только в поток исходных материалов в первый реактор. Для системы из множества параллельных реакторов, микроскопическое количество компонента активного металла может добавляться во все реакторы.
Компоненты активного металла могут содержать соединение или смесь соединений, содержащих один или несколько металлов Группы II, III, IV, V и VI Периодической таблицы. Примеры активных металлов включают Mg, Ca, Zn, La, Ac, Ti, Zr, Hf, V, Nb, Ta, Cr, Nb, W, Sn, Pb, Sb и тому подобное. Соединение активного металла должно быть растворимым в реакционной смеси или, по меньшей мере, образовывать эмульсию. Количества микроэлемента металла в потоке исходных материалов могут быть достаточно низкими с тем, чтобы не было экономической необходимости в извлечении металла из технологического потока для рециклирования, хотя можно выбрать и осуществление этого процесса.
Если это необходимо, дезактивированный катализатор в реакторе может реактивироваться in situ через относительно короткое время, с тем, чтобы он был готов заменить другой реактор на обслуживании, или для начала нового обслуживания. По этой причине, варианты осуществления способов, описанных в настоящем документе, могут потребовать запасного реактора, в зависимости от продолжительности цикла катализатора и других факторов.
Способы, описанные в настоящем документе, могут быть особенно пригодными для непрерывного получения диарилкарбонатов, таких как дифенилкарбонат, алкиларилкарбонатов, таких как этилфенилкарбонат или диалкилкарбонатов, таких как диэтилкарбонат или диметилкарбонат. Реакция для получения диарилкарбоната может осуществляться во множестве реакционных зон, таких как первая и вторая реакционная зона. Первая реакционная зона служит для осуществления, прежде всего, переэтерификации диалкилкарбоната с помощью ароматического спирта с получением алкиларилкарбоната, хотя может также быть получено малое количество диарилкарбоната. Вторая реакционная зона служит для осуществления диспропорционирования алкиларилкарбоната с получением диарилкарбоната и диалкилкарбоната. Присутствие твердого катализатора во второй реакционной зоне не является необходимым, хотя можно выбрать и использование твердого катализатора.
Диалкилкарбонаты, такие как DMC или DEC, могут быть получены посредством осуществления переэтерификации циклического карбоната, такого как пропиленкарбонат или этиленкарбонат, с помощью метанола или этанола подобным же образом. Реакции, производящие диарилкарбонат и диалкилкарбонат, осуществляют в системе с множеством реакторов с узлами для разделения материалов для извлечения продуктов из реакционных смесей. Непрореагировавшие реагенты и промежуточные соединения могут извлекаться для рециклирования или подвергаться конечной обработке посредством осуществления второго диспропорционирования или второй переэтерификации. Непрореагировавший фенол в жидкой реакционной смеси из зоны переэтерификации может отделяться либо до осуществления диспропорционирования алкилфенилкарбоната, либо после осуществления диспропорционирования. В дополнение к этому, имеются различные возможности для удаления продувкой побочного продукта простого алкилфенилового эфира из реакционной системы. Соответствующее расположение реакторов с узлами разделения материала известно специалистам в данной области.
Реакции предпочтительно осуществляют в многофазной системе, где реагенты и продукты являются жидкими и парообразными, чтобы сдвинуть равновесие в желаемом направлении. Альтернативно, можно осуществлять реакцию в жидкой фазе, например, когда имеется малая выгода от сдвига равновесия реакции из-за температур кипения продуктов реакции, более высоких, чем предпочтительный диапазон температур осуществления реакции, или ее вообще нет.
Варианты осуществления, описанные в настоящем документе, могут быть также пригодными для получения органических карбонатов, таких как этилфенилкарбонат, метилфенилкарбонат и дифенилкарбонат, посредством осуществления переэтерификации диалкилкарбонатов, таких как диэтилкарбонат или диметилкарбонат, с помощью фенола и диспропорционирования алкиларилкарбоната, такого как этилфенилкарбонат или метилфенилкарбонат, с получением дифенилкарбоната.
Варианты осуществления, описанные в настоящем документе, могут быть также пригодными для получения диалкилкарбоната, такого как диметилкарбонат или диэтилкарбонат, посредством переэтерификации циклического карбоната с помощью спирта. В других вариантах осуществления для получения диалкилкарбонатов, диалкилкарбонат может быть получен посредством алкоголиза мочевины с помощью спирта в присутствии твердого катализатора. Например, в патенте США № 7074951, диалкилкарбонат получают посредством использования гомогенного катализатора на основе оловоорганического комплекса в присутствии высококипящего растворителя, содержащего атом - донор электрона; такой способ может осуществляться над твердыми катализаторами в соответствии с вариантами осуществления, описанными в настоящем документе. Различные органические карбаматы, такие как N-арилалкилкарбамат, могут быть также преимущественно получены посредством взаимодействия диалкилкарбоната с ароматическим амином в присутствии твердого катализатора в соответствии с вариантами осуществления, описанными в настоящем документе.
Любой тип реакторов может использоваться для осуществления реакций, описанных в настоящем документе. Примеры реакторов, пригодных для осуществления реакций, включающих в себя реакции органического карбоната или органических карбаматов, могут включать реакторы в виде дистилляционных колонн, реакторы в виде дистилляционных колонн с разделительной стенкой, традиционные трубчатые реакторы с неподвижным слоем, реакторы в виде барботажных колонн, суспензионные реакторы, снабженные или не снабженные дистилляционной колонной, импульсные проточные реакторы, каталитические дистилляционные колонны, где суспендированные твердые катализаторы стекают вниз по колонне, или любое сочетание этих реакторов.
Системы с множеством реакторов, пригодные для использования в вариантах осуществления, описанных в настоящем документе, могут содержать ряд из множества последовательных реакторов или множество параллельных реакторов для первой реакционной зоны. Если продукт получают из реагентов посредством промежуточного продукта, такого как алкиларилкарбонат, первая реакционная зона может служить, прежде всего, для получения промежуточного продукта, хотя малое количество конечного продукта реакции может одновременно получаться в первой реакционной зоне.
Технологический поток из первой реакционной зоны, после отделения любого спирта и диалкилкарбоната, поступает во вторую реакционную зону, где получают диарилкарбонат вместе с побочным продуктом диалкилкарбоната. Во время отделения более легкого продукта реакции из зоны каталитической реакции, одновременно может осуществляться переэтерификация, при сдвиге равновесия реакции в направлении прямой реакции.
Реакции для получения органических карбонатов или органических карбаматов, как правило, осуществляют при температуре в пределах от примерно 104°C до примерно 260°C (примерно 220°F до примерно 500°F), в некоторых вариантах осуществления; от 121°C до примерно 232°C (примерно 250°F до примерно 450°F), в других вариантах осуществления. Давление для реакции зависит от температур кипения реагентов и продуктов, от типа реактора, который должен использоваться, и от того существует ли жидкость или смешанная фаза (пар/жидкость) в реакционной зоне. В целом, давление реактора может находиться в пределах от давления ниже атмосферного до примерно 22 бар (примерно 319 фунт/кв.дюйм абс.), в некоторых вариантах осуществления; и от примерно 0,005 бар до примерно 17 бар (0.1 фунт/кв.дюйм абс. до примерно 250 фунт/кв.дюйм абс.), в других вариантах осуществления. В некотором классе вариантов осуществления, реакции могут осуществляться с использованием соответствующего растворителя, который не влияет на разделение продуктов реакции.
Среди выбранных вариантов осуществления, варианты осуществления, описанные в настоящем документе, являются особенно пригодными для непрерывного получения диарилкарбонатов из диалкилкарбоната и ароматического гидроксисоединения, такого как получение дифенилкарбоната (DPC) из диалкилкарбоната и фенола. Один из путей получения DPC представляет собой реакцию диэтилкарбоната (DEC) с фенолом в присутствии одного или нескольких твердых катализаторов. Преимущества получения DPC посредством использования DEC могут включать экономию энергии и экономию материалов для конструирования установки, поскольку выделение материалов от азеотропной смеси не является необходимым. Все материалы требуют энергии для их получения. Таким образом, экономия конструкционных материалов и энергии считается "зеленой". В противоположность этому, современный промышленный способ получения DPC без фосгена использует DMC в качестве одного из исходных материалов. DMC и метанол должны отделяться от технологического потока, образующего азеотропную смесь, посредством экстракционной дистилляции растворителя. Работающие узлы экстракционной дистилляции потребляют много энергии. Хотя является возможным получение DPC через DMC, использование DEC может быть предпочтительным из-за экономии энергии и материалов.
Варианты осуществления, описанные в настоящем документе, могут быть также пригодными для получения диалкилкарбонатов посредством переэтерификации циклического карбоната с помощью спирта, такого как этанол или метанол.
Получение DPC из диалкилкарбоната и фенола включает в себя две стадии реакции; переэтерификацию в первой реакционной зоне и последующее диспропорционирование во второй реакционной зоне. Реакции могут иллюстрироваться следующим образом:
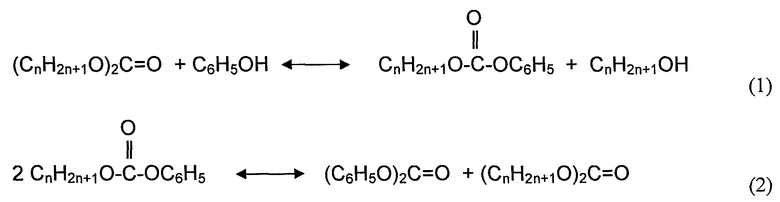
где общая реакция может иллюстрироваться как:

Реакция (1) представляет собой переэтерификацию диалкилкарбоната с помощью фенола с получением алкилфенилкарбоната и спирта. Реакция (2) включает в себя диспропорционирование алкилфенилкарбоната с получением дифенилкарбоната и диалкилкарбоната. Обе стадии реакции представляют собой равновесные реакции. Однако диспропорционирование является термодинамически гораздо более благоприятным, чем переэтерификация. Переэтерификацию в основном осуществляют в первой реакционной зоне, которая может содержать один реактор или систему с множеством реакторов. Реакция диспропорционирования может затем осуществляться в основном во второй реакционной зоне.
Получение диалкилкарбоната посредством переэтерификации циклического карбоната с помощью спирта также представляет собой двухстадийную равновесную реакцию. Как кислотные, так и основные катализаторы могут использоваться для переэтерификации циклического карбоната с помощью спирта.
В соответствии с вариантами осуществления, описанными в настоящем документе, для получения увеличенной продолжительности цикла катализатора, в поток исходных материалов для реактора добавляют микроскопическое количество растворимого соединения металла. Для переэтерификации циклического карбоната с помощью спирта с получением диалкилкарбоната и диола может использоваться твердый либо основный, либо кислотный катализатор. Можно также осуществлять переэтерификацию с помощью замены части спирта водой. Альтернативно, переэтерификация может осуществляться на первой стадии, с последующей реакцией непреобразованного циклического карбоната и промежуточного продукта со смесью вода-спирт с получением гликоля в качестве главного продукта реакции на второй стадии. Добавление воды существенно увеличивает преобразование циклического карбоната или производительность по отношению к диолу. Однако преимущество воды реализуется только в уменьшении выхода диалкилкарбоната.
Катализаторы, пригодные для получения органического карбоната и органического карбамата
Как описано выше, катализаторы пригодные для получения органического карбоната и органического карбамата, могут включать твердые катализаторы на носителе, имеющие один или несколько активных металлов из Групп II, III, IV, V и VI Периодической таблицы. Один из типов катализатора, пригодных для использования в вариантах осуществления, описанных в настоящем документе, включает металлорганическое соединение или множество металлорганических соединений указанных выше элементов, иммобилизованных на пористом носителе. Пористые носители, пригодные для использования в вариантах осуществления, описанных в настоящем документе, могут содержать поверхностные функциональные группы, такие как гидроксильные группы, алкоксигруппы, смеси гидроксильных и алкоксигрупп, хлор и тому подобное. Примеры носителей могут включать диоксид кремния, диоксид кремния - оксид алюминия, оксид титана, оксид циркония или цеолитные материалы, такие как MCM-41, MCM-48, SBA-15 и тому подобное, и композитные материалы, содержащие связующее и цеолит.
Альтернативные носители могут включать углерод и/или углеродистые материалы. Углеродные и углеродистые носители могут иметь поверхностные функциональные группы, такие как гидроксил, карбонил, или как то, так и другое, для иммобилизации металлорганических соединений на поверхности, как обсуждалось ранее. Для получения катализаторов на основе оксида, гидроксида или оксигидроксида металла на носителе, поверхностные функциональные группы могут быть не нужны, хотя могут быть пригодными для использования в некоторых вариантах осуществления. Углеродистые носители могут быть получены посредством контролируемого термического разложения таких углеводов, как древесина, скорлупа кокосового ореха, крахмалы, целлюлоза, смесь крахмала и целлюлозы, сахар, метилцеллюлоза и тому подобное, при повышенных температурах. Углеродистые носители могут либо наноситься на носитель, либо нет. Для получения углеродистого материала на носителе, углеводы могут осаждаться на соответствующий пористый носитель с последующим контролируемым термическим дегидратированием при повышенной температуре, такой как температура, находящаяся в пределах от примерно 250°C до 1000°C, в инертной атмосфере или в атмосфере, состоящей из инертного газа и малого количества кислорода, водяного пара или их смеси. Носители для углеродистых материалов могут включать любые неорганические материалы, такие как оксид алюминия, диоксид титана, диоксид кремния, диоксид циркония, синтетические и природные глины, включая оксиды кремния-алюминия, и другие носители, как известно в данной области.
Носители, в некоторых вариантах осуществления, могут потребовать удаления конденсированной воды в порах перед приведением в контакт металлорганических соединений с носителями для осуществления иммобилизации. Конденсированная вода на носителе определяется в настоящем документе как содержание воды, которое может удаляться посредством сушки носителя при температуре в пределах от примерно 50°C до примерно 400°C в потоке сухого газа или в вакууме, в зависимости от химической композиции носителя. Твердые катализаторы, используемые в настоящем документе, могут быть получены посредством иммобилизации одного или двух металлорганических соединений, имеющих активные центры катализатора, на пористом твердом носителе. Иммобилизация может осуществляться, например, посредством использования таких технологий, как прививка, прикрепление, адсорбция и тому подобное. Например, технологии получения катализатора для металлорганических соединений, таких как алкоксиды титана на пористых носителях, описаны в публикации '672.
Второй тип катализатора, пригодного для использования в вариантах осуществления, описанных в настоящем документе, включает оксид металла, смешанные оксиды металлов, или оксигидроксиды, осажденные на пористом носителе. Примеры этого типа катализатора также описаны в публикации '672.
Носители могут находиться в форме гранул, экструдатов, сфер, гранул, сот и тому подобное, размерами в пределах от примерно 1 мм до примерно 5 мм для различных реакторов с неподвижным слоем. Носители в форме порошков или микросфер также могут использоваться для получения катализаторов, которые должны использоваться для суспензионного или перемешиваемого реактора.
Получение второго типа катализаторов, описанных выше, может и не потребовать носителя, имеющего поверхностную гидроксильную группу. Однако носители, содержащие поверхностные функциональные группы, такие как диоксид кремния, углеродистый материал, оксид алюминия и тому подобное также могут использоваться для получения катализатора на основе гидроксида/оксида металла посредством прививки алкоксидов металлов, таких как алкоксид титана, на диоксиде кремния, с последующей обработкой водяным паром или гидролизом, и/или сушкой при температуре от примерно 90°C до примерно 500°C.
Другой способ получения катализаторов на основе оксида или оксигидроксида металла включает осаждение соли желаемого элемента или смеси солей двух различных элементов на носителе, с последующим кальцинированием при температуре от 300°C до 1000°C для разложения солей до оксидов металлов.
При условиях способа, переэтерификация и диспропорционирование в зоне каталитической реакции могут осуществляться одновременно, когда концентрация алкиларилкарбоната в реакционной среде увеличивается. Две причины дезактивирования катализатора, обсуждаемые выше, выщелачивание и осаждение полимера, также возникают одновременно при условиях реакции. В то время как осаждение полимера не вызывает необратимого повреждения катализатора, выщелачивание компонента активного металла из гетерогенных катализаторов при условиях реакции приводит к необратимому повреждению катализатора. При низких уровнях преобразования для переэтерификации или при низких концентрациях алкиларил- и диарилкарбонатов, дезактивирование катализатора вызывается по большей части растворением компонентов катализатора на основе активного металла из твердого катализатора в реакционной среде. Другими словами, причиной необратимого дезактивирования катализатора при всех условиях реакции является выщелачивание металла.
Когда вклад переэтерификации усиливается, осаждение полимера на катализаторе вызывает еще более быстрое дезактивирование катализатора. Осаждение полимера является, прежде всего, результатом нежелательных побочных реакций алкиларил- и диарилкарбонатов (и потенциально, микроскопических количеств примесей полигидроксилароматических соединений, находящихся в фенольных исходных материалах и производимых нежелательными побочными реакциями в очень малых количествах). По этой причине, для непрерывного получения дифенилкарбоната из фенола и диалкилкарбоната, такого как диэтилкарбонат или диметилкарбонат, в присутствии гетерогенных катализаторов, может быть необходимым компенсировать дезактивирование катализатора, вызываемого как (1) осаждением полимера, так и (2) растворением/выщелачиванием компонентов катализаторов на основе активных металлов. Осаждение полимера может быть компенсировано посредством контроля преобразования, концентрации ароматических карбонатов или как того, так и другого, в зоне каталитической реакции, как рассматривается выше, и с помощью реактивирования катализатора, такой как описано в публикации '672. Выщелачивание компенсируется посредством добавления микроскопического количества растворимого металлорганического соединения, как описано выше.
Иммобилизация (например, прививка, прикрепление, адсорбция и тому подобное) металлорганических соединений на носителе, таком как диоксид кремния или углеродистый материал для алкоголиза и/или переэтерификации диалкилкарбоната с помощью фенола, может осуществляться в одной ступени реакционной зоны или во множестве ступеней реакционных зон. Примеры описанных металлорганических соединений включают алкоксиды, алкоксихлориды, карбоксилаты, карбонаты и тому подобное металлов, элементов Группы II, III, IV, V и VI. Примеры активных металлов включают Mg, Ca, Zn, La, Ac, Ti, Zr, Hf, V, Nb, Ta, Cr, Mo, W, Sn, Pb, Sb и тому подобное. В различных вариантах осуществления включаются, алкоксиды олова, алкоксиды алкилолова, оксиды алкилолова, гидроксиды алкилолова, дихлорид диалкилолова, трихлорид алкилолова и смеси этих частиц, а также оксиалкоксиды металлов [(RO)nMO] и алкоксигидроксиды металла [(RO)nM(OH)x] или олигомеры этих оксиалкоксидов и алкоксигидроксида, где M представляет собой элемент Группы IV, V или VI, n = 2, 3 или 4, x = 0, 1, 2 или 3, и n + x = 4, 5 или 6. В выбранных вариантах осуществления, металлорганическое соединение может представлять собой одно или несколько соединений из алкоксида или феноксида титана, алкиларилтитаната или соли титана и сложного моноэфира угольной кислоты. Необходимо понять, что алкоксиды металлов включают мономеры, различные олигомеры или смесь различных мономерных и олигомерных частиц, в зависимости от длины углеродной цепи и структуры алкильной группы на алкоксиде или арилоксиде [смотри, например, Coordin. Chem. Rev., 2 (1967) 299-318; J. Chem. Soc, 3977 (1955)].
Как описано в настоящем документе, алкоксиды переходных металлов включают все частицы мономера и различных олигомеров. Например, в то время как этоксид титана [Ti(OEt)4] существует в кипящем этаноле или бензоле по большей части как тример, стерически затрудненные алкоксиды титана, такие как изопропоксид титана, являются мономерными в кипящих растворах в углеводородах. Например, изопропоксид титана, как предполагается, существует в кипящем толуоловом растворе по большей части как мономер.
Пористые носители, используемые в различных вариантах осуществления, описанных в настоящем документе, могут иметь поверхностные гидроксильные группы, алкоксигруппы или как те, так и другие. Для получения пористого носителя, пористые носители на основе оксида металлов, таких как оксид титана, оксид циркония, оксид молибдена и оксид ванадия, могут обрабатываться потоком, содержащим одно или несколько соединений из спирта, органического карбоната, такого как диметилкарбонат, диэтилкарбонат и тому подобное, при температуре в пределах от примерно 130°C до примерно 400°C, в некоторых вариантах осуществления, от 150°C до 350°C, в других вариантах осуществления, в паровой фазе, жидкой фазе или в системе пар-жидкость. Поток может содержать воду, от 0 мас.% до примерно 20 мас.%, в некоторых вариантах осуществления; от 0 мас.% до примерно 10 мас.%, в других вариантах осуществления; и от примерно 0,005 мас.% до примерно 5 мас.%, в других вариантах осуществления. Поскольку вода имеет низкую растворимость в DMC и DEC, поток может содержать соответствующие количества метанола и/или этанола в качестве растворителя для воды. Коммерчески доступный силикагель или диоксид кремния, имеющий поверхностные гидроксильные группы, может использоваться в некоторых вариантах осуществления. Необязательно, можно осуществлять обработку диоксида кремния жидкой водой, паром или их смесью при температуре от примерно 80°C до примерно 500°C с последующей сушкой при температуре от примерно 70° до примерно 800°C, в некоторых вариантах осуществления, и от примерно 80°C до примерно 500°C, в других вариантах осуществления.
Силооксановые и силоксановые соединения переходных металлов также могут использоваться для получения твердых катализаторов, иммобилизованных на пористых носителях, или катализаторов на основе оксидов металлов, как описано выше. Примеры силооксановых и силоксановых соединений представляют собой (RO)n-xM[-O-Si(O-R)3]x, M(O-SiR3)n, (R3SiO)n-2MO и тому подобное, где каждый из R независимо представляет собой алкильную или арильную группу, n = 3, 4, или 5, x = 1 или 2, n + x = 4, 5 или 6, и M представляет собой переходной металл Группы IV, V или VI, как описано выше. Другие соединения кремний - металл находятся в рамках вариантов осуществления, описанных в настоящем документе, поскольку иммобилизация приводит к появлению каталитической активности твердых катализаторов. Силооксановые и силоксановые соединения переходных металлов также могут использоваться в качестве растворимых металлорганических соединений в технологических системах, описанных в публикациях '672 и '214, а также в реакторах в виде колонн для реакционной дистилляции. Различные олигомерные и полимерные гетеросилооксаны или гетеросилоксаны переходных металлов также могут использоваться для получения иммобилизованных твердых катализаторов или могут использоваться в качестве растворимых металлорганических соединений в различных вариантах осуществления. Как описано выше, диспропорционирование EPC или MPC до DPC и DEC или DMC может осуществляться в отсутствие твердого катализатора во второй реакционной зоне, и пригодные для использования каталитически активные частицы включают силооксановые или силоксановые соединения переходного металла, такого как Ti.
Оксиды металлов и алкоксиды силоксанов могут включать различные олигомеры.
Различные олигомеры можно найти в публикации Bradely [D. C. Bradley, Coordin. Chem. Rev., 2 (1967) p. 299-318); J. Chem. Soc, (1955) 3977]. Можно выбрать микроскопическое количество одного из этих соединений для добавления в исходные материалы для реакционной зоны, для получения стабильной активности катализатора.
При осуществлении диспропорционирования алкиларилкарбонатов с получением диарилкарбоната и диалкилкарбоната во второй реакционной зоне в присутствии гомогенного катализатора, гомогенный катализатор может представлять собой смесь алкиларилтитанатов, солей титана и сложных моноэфиров угольной кислоты и силоксановых соединений титана, обсуждаемых выше. Понятно, что гомогенные катализаторы могут происходить из твердых катализаторов и растворимых катализаторов, используемых в реакционной зоне переэтерификации.
Поскольку различные металлоорганические соединения, описанные в настоящем документе, являются чувствительными к влажности в потоке исходных материалов, важно контролировать содержание воды в потоке исходных материалов в реакционную зону. В некоторых вариантах осуществления, содержание влажности в потоке исходных материалов меньше примерно чем 700 м.д.; в других вариантах осуществления, меньше примерно чем 600 м.д.
Твердый катализатор на основе алкоксида металла, иммобилизованный на носителе, может быть получен посредством технологий in situ внутри реактора или может быть получен вне реактора. Для получения in situ, заданное количество соответствующего носителя помещают в реактор, с последующей сушкой при соответствующей температуре для удаления, по меньшей мере, части любой конденсированной воды. Затем носитель приводится в контакт с раствором, содержащим растворимый алкоксид металла или смешанные алкоксиды металла, для переходного металла или металлов, при температуре в пределах от примерно температуры окружающей среды до примерно 260°C (500°F), в некоторых вариантах осуществления, и от примерно 37°C до примерно 204°C (от примерно 100°F до примерно 400°F), в других вариантах осуществления. Приведение в контакт может осуществляться в течение периода времени от примерно 5 минут до примерно 24 часов, в некоторых вариантах осуществления, и от примерно 15 минут до примерно 15 часов, в других вариантах осуществления, и может зависеть от температуры и концентрации компонента активного металла в растворе. После удаления из реактора избытка раствора алкоксида металла, катализатор в реакторе может промываться растворителем (обычно таким же растворителем, как тот, который используют для получения раствора алкоксида металла) перед использованием в реакциях диспропорционирования или переэтерификации. Растворитель может представлять собой спирт, простой эфир, углеводород, смесь углеводородов и спирта или смесь диалкилкарбоната и фенола или спирта, или смеси их всех.
Альтернативно, катализаторы на основе оксида металла, смешанного оксида металла или гидроксида металла, где металл представляет собой один или несколько металлов из Групп II, III, IV, V и VI Периодической таблицы, также может использоваться в соответствии с вариантами осуществления, описанными в настоящем документе. Некоторые катализаторы на основе оксида металла известны в данной области. Например, в соответствии с P. Iengo et al., Appl Catal. A: General 178 (1999) 97-109, катализаторы на основе оксида титана на носителе из диоксида кремния могут быть получены посредством прививки изопероксида титана, а затем обработки водяным паром/кальцинирования катализатора на носителе, имеющем сильно модифицированную исходную поверхность диоксида кремния, что приводит к получению катализатора, отличного от тех, которые получают посредством пропитки и совместной преципитации.
Для получения катализаторов на основе гидроксида или оксигидроксида металла или смеси металлов на носителе, можно гидролизовать катализаторы на основе привитого алкоксида металла, как описано выше, с последующей сушкой при температуре в пределах от примерно 50°C до примерно 110°C, в некоторых вариантах осуществления, сушка может быть не нужна.
Предварительное кондиционирование катализаторов на основе оксидов металла без носителя может осуществляться перед осуществлением реакций для получения органических карбонатов. Предварительное кондиционирование осуществляют посредством приведения в контакт катализаторов на основе пористых оксидов металлов, таких как оксид титана, оксид циркония, оксид молибдена или оксид ванадия, с потоком, содержащим органический карбонат, такой как диметилкарбонат, диэтилкарбонат и тому подобное, при температуре в пределах от примерно 125°C до примерно 450°C, в некоторых вариантах осуществления, и от примерно 150°C до примерно 350°C, в других вариантах осуществления, где органический карбонат может находиться в паровой фазе, в жидкой фазе или смешанной фазе. Предварительное кондиционирование может осуществляться в течение периода от примерно 2 минут до примерно 50 часов, в некоторых вариантах осуществления, и от примерно 4 минут до примерно 24 часов, в других вариантах осуществления. Поток, содержащий органический карбонат, может содержать воду и спирт, где вода может присутствовать от более чем 0 мас.% до примерно 10 мас.%, в некоторых вариантах осуществления, и от примерно 0,005 мас.% до примерно 4 мас.%, в других вариантах осуществления. Селективность катализатора может быть улучшена посредством предварительного кондиционирования. После предварительного кондиционирования, катализаторы на основе оксидов металлов могут сушиться при температуре от примерно 80°C до примерно 300°C в потоке инертного газа в течение периода времени от примерно 2 минут до примерно 6 часов.
Два типа катализаторов на основе смешанных оксидов металлов могут использоваться для переэтерификации циклического карбоната с помощью спирта. Первый тип катализаторов на основе смешанных оксидов металлов может содержать один или несколько элементов из Групп III, IV, V, и VI Периодической таблицы, нанесенных на носитель. Второй тип смешанных оксидов содержит твердый основный катализатор, который содержит один или два элемента из Группы II Периодической таблицы и лантаноиды или актиноиды на носителе. Необязательно, можно использовать гидроксид четвертичного аммония, привитой или закрепленный на носителе из диоксида кремния. Катализаторы на основе оксида обычно наносятся на носитель из оксида алюминия или диоксида кремния или получаются в форме смешанного оксида или твердого раствора. Элементы, пригодные для второго типа твердых катализаторов, могут включать Mg, Ca, Zn, La и тому подобное.
Компоненты активных металлов для второго типа катализаторов могут также выщелачиваться при условиях реакции переэтерификации, что приводит к дезактивированию катализатора. На самом деле, обнаружено, что носители из диоксида кремния также могут выщелачиваться, только с гораздо меньшей скоростью, чем компоненты активных металлов Группы II. Поскольку примеси щелочных металлов на носителе из диоксида кремния могут увеличить растворение диоксида кремния в реакционной среде, сведение к минимуму примесей щелочных металлов в носителе из диоксида кремния является в высшей степени желательным. Посредством добавления микроскопического количества растворимого металлорганического соединения в поток исходных материалов, продолжительность цикла твердого катализатора для реакторов с неподвижным слоем может быть увеличена. Примеры таких растворимых соединений включают цинк 2-метоксиэтоксид, кальций 2-метоксиэтоксид, цинк 2-метоксипропоксид, цинк этоксид, цинк алкоксиалкилкарбонат, кальций 2-метоксипроксид, кальций этоксид, кальций алкоксиалкилкарбонат, магний 2-метоксиэтоксид, магний 2-метоксипроксид, магний этоксид, магний бутоксид, магний алкоксиалкилкарбонат, лантан алкоксид, лантан алкоксиалкилкарбонат и пропиленглицериды Mg, Ca и Zn, среди прочего. Может также использоваться их смесь.
Растворимые соединения Ca, Mg, Zn и La могут быть получены посредством взаимодействия оксидов или гидроксидов этих металлов со спиртом, органическим карбонатом или смесью органического карбоната и спирта при температуре от примерно 105°C (221°F) до примерно 260°C (500°F), в некоторых вариантах осуществления, от примерно 149°C (300°F) до примерно 227°C (440°F), в других вариантах осуществления, в жидкой фазе или в двухфазной системе (жидкость и пары). Растворы, приготовленные таким способом, могут быть пригодными для добавления микроскопических количеств этих металлов в поток исходных материалов в реактор, с тем, чтобы получить большое время цикла. Общее количество активного металла или компонентов металла на твердом катализаторе на основе алкоксида металла, гидроксида металла или оксида металла, может находиться в пределах от примерно 0,02 мас.% до примерно 20 мас.%, предпочтительно, от примерно 0,05 мас.% до примерно 12 мас.%.
Улучшенная продолжительность цикла и время службы катализатора
Твердые катализаторы, описанные в настоящем документе, могут иметь большие продолжительности цикла и могут быть способны подвергаться регенерации и реактивированию катализатора много раз, что приводит к получению больших времен службы катализатора. Технологии продления продолжительности цикла катализатора и реактивирования катализаторов, описанные в настоящем документе, делают катализаторы, как правило, интересные для лабораторных целей, пригодными для промышленного получения различных органических карбонатов. Предполагается, что начиная либо с катализаторов на основе оксидов металлов на носителе, либо с катализаторов на основе алкоксидов металлов, иммобилизованных на диоксиде кремния, в стационарном состоянии, активные катализаторы представляют собой частицы металлорганического соединения, иммобилизованного на диоксиде кремния. Для иллюстрации выгоды добавления микроскопического количества активных металлов в исходные материалы, осуществляют различные эксперименты, и ниже, они будут описаны более подробно. Вкратце, в одном из экспериментов, катализатор на основе оксида титана (6 мас.% Ti) на носителе из силикагеля регенерируют посредством деполимеризации после службы примерно 350 часов, и он восстанавливает менее половины от своей исходной активности переэтерификации диэтилкарбоната с помощью фенола. Обнаружено, что в течение времени службы более половины Ti выщелачивается из катализатора в реакционную среду. В другом эксперименте, катализатор на основе бутоксида титана (4 мас.% Ti), привитого на силикагеле, теряет более 90% Ti после использования в течение всего лишь 171 часа для диспропорционирования этилфенилкарбоната. После использования в течение 173 часов, другой катализатор на основе оксида титана (5,7 мас.%) на носителе из силикагеля, используемый для переэтерификации пропиленкарбоната с помощью этанола с получением диэтилкарбоната и пропиленгликоля, теряет 35% Ti на катализаторе. Из этих данных вполне понятно, что как катализатор на основе оксида титана на носителе, так и катализатор на основе привитого алкоксида титана являются непригодными для промышленных реакторов для непрерывного получения органических карбонатов, таких как диалкилкарбонат, алкилфенилкарбонат и диарилкарбонат, из-за необратимого дезактивирования катализатора за короткое время службы. Видно, что органические карбонаты и/или реакционные смеси являются достаточно химически активными, чтобы вызвать медленное образование растворимого металлорганического соединения в реакционной среде посредством взаимодействия с твердыми катализаторами.
Известно также, что потоки паров DMC и DEC могут взаимодействовать с диоксидом кремния или оксидом титана с образованием тетраалкилортооксида кремния и тетраалкоксида титана при температуре выше примерно чем 350°C. Реакции DMC и DEC с диоксидом кремния осуществляются легче в присутствии каталитического количества щелочного металла на диоксиде кремния. По этой причине, необходимо найти технологию для простого реактивирования катализатора и способ для поддержания постоянных поверхностных концентраций компонентов активного металла на катализаторе в течение достаточно большого времени цикла, которое является приемлемым для промышленных реакторов.
Регенерация катализатора посредством деполимеризации и повторного осаждения металла компенсирует проблемы, связанные с осаждением полимера. Однако регенерация посредством деполимеризации не может компенсировать проблемы, связанные с непрерывным выщелачиванием активных металлов из гетерогенных катализаторов при условиях реакции. Непрерывные потери активного металла из гетерогенных катализаторов должны быть компенсированы для получения большой продолжительности цикла катализатора, пригодной для реакторов промышленного масштаба. Обнаружено, что воздействие выщелачивания металла из гетерогенных катализаторов может быть нейтрализовано посредством добавления микроскопических количеств соединений активного металла в поток (потоки) исходных материалов в первый реактор в случае системы множества последовательных реакторов. Посредством добавления микроскопического количества растворимого соединения активного металла, выщелачивание и повторное осаждение металла компенсируют или почти компенсируют друг друга, эффективно поддерживая постоянное количество активных центров на твердых катализаторах, приводя к получению постоянной активности катализатора в течение большого времени цикла катализатора. Понятно, что растворимый компонент металла, выщелачивающийся из твердого катализатора в первом реакторе, представляет собой смесь различных частиц соединений металлов. Частицы соединений металлов в смеси необязательно являются идентичными частицам соединений металлов, поступающим в первый реактор. Выщелачивание и повторное осаждение металла во втором реакторе также компенсируют друг друга подобным же образом. Для параллельных систем из множества реакторов, может потребоваться добавление микроскопического количества соединения активного металла во все потоки исходных материалов и в первую главную реакционную зону. По этой причине, реактивирование катализатора (деполимеризация/кондиционирование поверхности in situ и повторное осаждение металла) и добавление микроскопического количества соединения активного металла с последующим повторным осаждением металла может компенсировать как выщелачивание металла, так и осаждение полимера. Реактивирование катализатора может осуществляться в две стадии: (1) деполимеризация/кондиционирование поверхности катализатора и (2) повторное осаждение компонентов активного металла. Кондиционирование поверхности катализатора необходимо для иммобилизации алкоксида титана на поверхности носителя из диоксида кремния. Свежий иммобилизованный катализатор или реактивированный катализатор постоянно теряет каталитическую активность, что приводит к получению неприемлемо короткого времени цикла, которое непригодно для больших промышленных реакторов. Потеря исходной активности катализатора примерно до половины активности для переэтерификации диалкилкарбоната с помощью фенола занимают примерно 80-150 часов времени рабочего цикла, что явно неадекватно для непрерывной работы промышленного реактора. Посредством добавления микроскопического количества растворимого соединения активного металла и осуществления двухстадийного реактивирования, теперь возможно увеличение продолжительности цикла катализатора и осуществление многократного реактивирования катализатора для непрерывного получения различных органических карбонатов.
Деполимеризация дезактивированного катализатора может осуществляться посредством приведения в контакт катализатора с потоком, содержащим гидроксисоединение или смесь гидроксисоединений in situ при температуре от 102°C (215°F) до 316°C (600°F), в некоторых вариантах осуществления, от 104°C (220°F) до 232°C (450°F), в других вариантах осуществления, в течение периода времени от примерно 10 минут до примерно 50 часов, в некоторых вариантах осуществления, и от 30 минут до 15 часов, в других вариантах осуществления. Деполимеризация может осуществляться в паровой фазе, в жидкой фазе, в смешанной фазе или сначала в жидкой фазе, а затем в паровой фазе, или в обратном порядке. Продукты деполимеризации могут включать фенол, спирт, диоксид углерода, мультигидроксибензол, диалкилкарбонат, алкилфенилкарбонат и более тяжелые соединения.
Примеры гидроксисоединений, которые должны использоваться для деполимеризации на катализаторе, представляют собой спирты (предпочтительно метанол или этанол), воду или их смесь. Если в качестве одного из исходных материалов для получения метилфенилкарбоната и дифенилкарбоната используют диметилкарбонат, для деполимеризации может использоваться метанол или смесь воды и метанола. Если в качестве одного из исходных материалов используют диэтилкарбонат, для деполимеризации может использоваться этанол или смесь воды и этанола. Можно также использовать смесь метанола и этанола. Когда для деполимеризации используют воду, содержание воды в смеси может находиться в пределах от более чем ноль мас.% до менее чем 100 мас.%, в некоторых вариантах осуществления; от 10 м.д. масс до 15 мас.%, в других вариантах осуществления; и от 15 м.д. масс до 5 мас.%, в других вариантах осуществления. Азеотропная смесь воды (4 мас.%) и этанола является очень эффективной при деполимеризации, когда в качестве одного из исходных материалов используют диэтилкарбонат. В некоторых вариантах осуществления, смесь воды и спирта может быть предпочтительной по сравнению с водой или спиртом по отдельности, делая возможным кондиционирование поверхности катализатора для повторного осаждения компонента активного металла. В дополнение к этому, смесь воды и спирта может быть более эффективной при деполимеризации и кондиционировании поверхности, чем спирт или вода по отдельности.
При деполимеризации можно также использовать растворитель. Пригодные для использования растворители могут включать бензол, толуол, ксилолы, пентан, гексан, октан, декан, тетрагидрофуран, простой эфир и тому подобное, или любые смеси таких растворителей. Концентрация растворителя в смеси для деполимеризации может находиться в пределах от 0 мас.% до примерно 90 мас.%.
Деполимеризованный катализатор может сушиться для удаления избытка воды на катализаторе и для контроля заселенности поверхностных гидроксильных групп перед повторным осаждением компонента активного металла на катализатор. Сушка in situ может осуществляться при температуре от примерно 49°C (120°F) до примерно 427°C (800°F), в некоторых вариантах осуществления, от 65°C (150°F) до 316°C (600°F), в других вариантах осуществления, в потоке инертного газа, в течение периода от примерно 15 минут до 40 часов при давлении окружающей среды или при давлении ниже атмосферного, перед осуществлением повторного осаждения компонента активного катализатора. Неправильное предварительное кондиционирование поверхности катализатора может приводить к частичному только восстановлению активности катализатора. Технологии деполимеризации, описанные в настоящем документе, могут использоваться в любом способе получения ароматических карбонатов или в любой реакции, где органические карбонаты участвуют как реагенты, продукты или как то, так и другое.
Описанная технология деполимеризации может быть также пригодной для реакций, производящих органические карбонаты в присутствии гомогенного катализатора. Для регенерации системы гомогенных катализаторов, спиртовой раствор должен быть достаточно сухим так, чтобы содержание воды не могло превышать примерно 0,01 мас.%. По этой причине, технология регенерации катализатора, описанная в настоящем документе, может быть пригодной для любого способа получения органических карбонатов.
Поток эффлюента из реактора во время деполимеризации может содержать микроскопическое количество компонента активного металла, в зависимости от того, как осуществляют деполимеризацию. Этот поток может также содержать фенол, DEC, малые количества фенетола, EPC и более тяжелые соединения в качестве главных продуктов деполимеризации. Если это желательно, можно пытаться извлечь из этого потока пригодные для использования компоненты, такие как фенол, этанол, алкилфенилкарбонат и DEC.
Повторное осаждение компонента активного металла на деполимеризованный и имеющий кондиционированную поверхность носитель может осуществляться способом, сходным с иммобилизацией алкоксида металла на носитель, как описано выше. Иммобилизация алкоксида металла на носитель может осуществляться на одной стадии или на множестве стадий. После этого реактор, содержащий реактивированный катализатор, опять готов к использованию.
Добавление микроскопических количеств растворимых компонентов активных металлов катализаторов в поток исходных материалов, как описано выше, может давать стабильные рабочие характеристики катализатора в течение пролонгированного времени цикла. В качестве примера, переэтерифакацию DEC с помощью фенола осуществляют в однопроходном реакторе с неподвижным слоем с восходящим потоком с добавлением от примерно 45 до примерно 60 м.д. масс Ti в поток исходных материалов. Имеются небольшие признаки дезактивирования катализатора в течение 14 месяцев непрерывного рабочего цикла.
Добавление микроскопического количества соединений активного металла, как описано в настоящем документе, может быть пригодным для непрерывного промышленного получения различных органических карбонатов или карбаматов. Реакции, производящие органические карбонаты, могут осуществляться в одном реакторе, в ряду из множества последовательных реакторов или в системе множества параллельных реакторов, как диктует конкретная система реакций. Например, реакции могут осуществляться в отдельном реакторе в виде каталитической дистилляционной колонны или в ряду из множества реакторов в виде каталитических дистилляционных колонн, где размещается твердый катализатор или два различных твердых катализатора. Необязательно, ряд из множества последовательных суспензионных реакторов также может использоваться для получения органического карбоната. Добавление микроскопического количества растворимого компонента активного металла в поток исходных материалов может происходить только в первом реакторе в ряду из множества последовательных реакторов. Желаемая величина микроскопического количества компонента активного металла в потоке исходных материалов зависит от конкретного элемента активного металла для конкретных компонентов исходных материалов. Для переэтерификации диалкилкарбоната с помощью фенола, она может находиться в пределах от примерно 15 м.д. до примерно 400 м.д. масс, в некоторых вариантах осуществления; от примерно 20 м.д. до примерно 300 м.д., в других вариантах осуществления; и от примерно 25 м.д. до примерно 200 м.д., в других вариантах осуществления, в зависимости от металла. Для потока исходных материалов, состоящего из диэтилкарбоната и фенола, например, желаемое количество Ti может составлять от примерно 20 м.д. масс до примерно 150 м.д. масс, в некоторых вариантах осуществления; и от 30 м.д. до 100 м.д. масс, в других вариантах осуществления. Количество компонента активного металла в потоке исходных материалов приблизительно на один или два порядка величины ниже, чем концентрация гомогенного катализатора в реакционной среде, известной из литературы.
Концентрация Ti в потоке эффлюента из реактора находится обычно в пределах от примерно 20 м.д. до примерно 100 м.д., в зависимости от величины концентрации активного металла в потоке исходных материалов в реактор. На этом уровне, как правило, не является экономически выгодным извлечение Ti из потоков эффлюента из реактора для рециклирования, хотя можно выбрать и осуществление этого процесса. Компонент активного металла в потоке эффлюента из реактора может извлекаться из тяжелых кубовых потоков колонны для извлечения сырого DPC в виде твердого материала и преобразовываться в растворимое металлорганическое соединение, которое должно повторно использоваться посредством взаимодействия с органическим карбонатом или смесью органического карбоната и спирта при повышенной температуре. Извлеченное металлорганическое соединение может представлять собой алкоксид металла, алкоксиалкилкарбонат металла (соль металла и сложного моноэфира угольной кислоты) или их смесь.
Для извлечения растворимого компонента активного металла в кубовом потоке колонны для извлечения DPC в виде твердого материала, кубовый поток тяжелых отходов из колонны для извлечения DPC может обрабатываться горячей водой или смесью водяного пара и воды для осаждения компонента металла в виде твердого продукта. В случае катализаторов, содержащих твердый продукт титана, преципитат твердого титана в водной фазе выделяется из жидкости посредством использования обычных способов, таких как фильтрование или центрифуга. Выделенный твердый продукт преобразуется в растворимый материал посредством обработки с помощью жидкого потока, содержащего диалкилкарбонат или смесь диалкилкарбоната и спирта при температуре от 121 до 343°C (250 - 650°F) под давлением, в течение периода от 10 минут до 80 часов, в некоторых вариантах осуществления, и от 20 минут до 45 часов, в других вариантах осуществления. Давление является достаточно высоким, так что диалкилкарбонат или смесь спирта и диалкилкарбоната должна, по меньшей мере, частично, существовать как жидкость в реакционной емкости, такой как автоклав, трубчатый реактор и тому подобное. Необязательно, жидкий поток может содержать инертный растворитель, такой как бензол, толуол, гексан, гептан, простой эфир и тому подобное. Примеры жидкого потока представляют собой смеси этанола и DEC или метанола и DMC. Содержание диалкилкарбоната в смеси спирта и диалкилкарбоната может находиться в пределах от 0,1 мас.% до менее чем 100 мас.%.
Реакции, дающие органические карбонаты или карбаматы, могут осуществляться в одном реакторе или в ряду из множества последовательных реакторов в различных системах реакторов с расположенными соответствующим образом дистилляционными колоннами для экономически эффективного разделения продуктов реакции и для рециклирования непрореагировавших реагентов. Альтернативно, реакции могут осуществляться в отдельных реакторах или во множестве параллельных реакторов. Различные другие системы реакторов и дистилляционные колонны могут быть разработаны специалистами в данной области.
Реакция может осуществляться в отдельной каталитической дистилляционной колонне, в ряду из множества каталитических дистилляционных колонн, в ряду из множества трубчатых или имеющих форму танка реакторов с неподвижным слоем или в любом сочетании различных типов реакторов. Когда для получения DPC используют три каталитических дистилляционных колонны, твердый катализатор помещают в первые два реактора в ряду для переэтерификации. Третий реактор в виде дистилляционной колонны может содержать твердый катализатор или, альтернативно, может не содержать твердого катализатора. Диспропорционирование в третьем реакторе может осуществляться посредством использования только растворимого гомогенного катализатора, присутствующего в реакционной среде.
Реакции, осуществляемые в традиционных трубчатых реакторах с неподвижным слоем, могут осуществляться в режиме восходящего потока или нисходящего потока. Реакции, дающие алкиларилкарбонат, такой как EPC, и диарилкарбоната, такой как DPC, например, могут осуществляться в жидкой фазе, но также могут осуществляться в двухфазной системе в присутствии одного или нескольких твердых катализаторов. Два реактора в ряду для переэтерификации могут периодически меняться в качестве первого и второго реактора для продления времени цикла. Третий реактор для диспропорционирования EPC с получением DPC и DEC может работать в нижней половине колонны для извлечения фенола, которая может работать при давлениях ниже атмосферного. В выбранных вариантах осуществления, способы, описанные в настоящем документе, могут быть пригодными для получения дифенилкарбоната посредством осуществления переэтерификации диэтилкарбоната с помощью фенола с последующим диспропорционированием этилфенилкарбоната.
Микроскопическое количество растворимого соединения активного металла, такого, например, как этилфенилтитанат или этоксититан этилкарбонат, или смесь алкоксида титана и алкоксититана алкилкарбоната может добавляться в жидкую реакционную среду, вводимую в первую реакционную зону. Альтернативно, диспропорционирование может осуществляться в каталитической дистилляционной колонне, в которую кубовый поток из колонны для извлечения фенола вводят в соответствующей точке, в секции, находящейся в верхней половине колонны. Диспропорционирование может также осуществляться в каталитической дистилляционной колонне, в которую непосредственно вводится кубовый поток из реактора переэтерификации без удаления фенола (фенол может извлекаться из кубового потока из колонны диспропорционирования EPC). Первая реакционная зона может содержать две последовательные каталитические дистилляционные колонны или две параллельные каталитические дистилляционные колонны для переэтерификации DEC с помощью фенола. Вторая реакционная зона может содержать реактор в виде каталитической дистилляционной колонны для диспропорционирования EPC до DPC и DEC. Можно выбрать DMC вместо DEC и MPC вместо EPC. Реакторы в виде каталитических дистилляционных колонн для первой реакционной зоны могут загружаться одним или несколькими твердыми катализаторами, такими как алкоксид титана, иммобилизованный на носителе из диоксида кремния или оксида титана, нанесенный на носитель из диоксида кремния. В целом, имеются два альтернативных способа непрерывного получения дифенилкарбоната, которые могут использоваться, в случае, когда используется более двух реакторов.
В первом способе непрерывного получения дифенилкарбоната, может иметься от трех до семи реакторов в виде каталитических дистилляционных колонн в различных вариантах осуществления, в выбранных вариантах осуществления - от трех до четырех каталитических дистилляционных колонн. Из этих реакторов в виде каталитических дистилляционных колонн, один или несколько могут служить в виде запасных реакторов для замены последнего активного реактора из множества реакторов, которые работают. Из множества реакторов в виде дистилляционных колонн, от двух до шести реакторов могут использоваться в основном для получения EPC. Оставшиеся реакторы в виде каталитических дистилляционных колонн могут служить в качестве второй реакционной зоны, где происходит в основном диспропорционирование EPC до DPC и DEC. DEC и, по меньшей мере, часть фенола в потоке из первой реакционной зоны удаляются из потока тяжелого эффлюента из первой реакционной зоны перед поступлением во вторую реакционную зону. Альтернативно, удаление фенола в потоке тяжелого эффлюента из первой реакционной зоны может откладываться до окончания диспропорционирования, в зависимости от концентрации фенола в этом потоке. Когда используемый катализатор состаривается, активность катализатора медленно дезактивируется. Имеются три различные возможности для ротации множества реакторов в ряду между работой для получения ароматических карбонатов и реактивированием катализатора:
(1) циклическая ротация всех реакторов в последовательном порядке после данного срока службы, при этом самый старый реактор выходит из работы для реактивирования катализатора, при этом в работу вводится реактор, имеющий свежий или реактивированный катализатор, как первый реактор в ряду из множества реакторов (то есть новый → первый реактор, первый реактор → второй; второй → третий; и третий → реактивирование или замена катализатора) или необязательно вводится в работу новый реактор как последний реактор в ряду, когда второй реактор становится первым реактором (представлена обратная или прямая последовательность);
(2) реакторы разделяют на две группы из реакторов первой реакционной зоны и второй реакционной зоны, при этом каждая группа имеет запасной реактор для ротации между работой и заменой/реактивированием катализатора;
(3) выведение последнего активного реактора в ряду из множества реакторов из работы для реактивирования катализатора, когда это необходимо, и введение в работу запасного реактора (когда катализатор уже реактивирован) для замены реактора, выведенного из работы.
В альтернативном способе, два последовательных реактора используют в качестве первой реакционной зоны. Последовательность двух реакторов периодически изменяется, между первым реактором и вторым реактором, после работы в течение заданного периода времени, скажем, каждые 6000 часов; эта ротация повторяется столько раз, сколько нужно. Запасного реактора для второй реакционной зоны нет. Этот тип работы возможен благодаря добавлению микроскопических количеств соединений активных металлов в первый реактор в ряду. DEC и фенол в потоке из первой реакционной зоны удаляют посредством дистилляции, а затем оставшийся поток подвергают диспропорционированию EPC с получением DPC во второй реакционной зоне. Имеются два пути осуществления диспропорционирования.
(1) В первом способе, диспропорционирование осуществляют в присутствии твердого катализатора в реакторе с неподвижным слоем, таком как реактор каталитический дистилляции. Имеется запасной реактор для замены работающего реактора. Дезактивированный катализатор подвергают реактивированию катализатора, описанному ранее.
(2) Во втором способе, диспропорционирование осуществляют в реакторе каталитической дистилляции в отсутствие твердого катализатора, и запасного реактора нет. Частицы активного растворимого металла в потоке, поступающие из первой реакционной зоны, служат в качестве гомогенного катализатора для реакции диспропорционирования.
Понятно, что каталитическая дистилляционная колонна, где твердый катализатор либо присутствует, либо отсутствует, для второй реакционной зоны, конструируется так, что секция в верхней половине колонны (секция извлечения фенола) служит, прежде всего, для отгонки фенола в поступающей реакционной смеси из первой реакционной зоны, а секция нижней половины служит, прежде всего, для осуществления диспропорционирования EPC или MPC. В альтернативной схеме способа, колонна для извлечения фенола и каталитическая дистилляционная колонна разделены на две колонны, хотя некоторое диспропорционирование может осуществляться в нижней секции колонны для извлечения фенола. Как утверждается выше, в зависимости от концентрации фенола в поступающем потоке исходных материалов, извлечение фенола может быть отложено до окончания диспропорционирования, хотя некоторая часть фенола может отделяться в каталитической дистилляционной колонне как поток паров из верхней части колонны вместе с DEC. Каталитическая дистилляционная колонна для диспропорционирования может работать при давлении ниже атмосферного.
Фигура 1 представляет собой упрощенную блок-схему, иллюстрирующую способ непрерывного получения DPC с помощью трех каталитических дистилляционных колонн в соответствии с вариантами осуществления, описанными в настоящем документе. Две каталитические дистилляционные колонны в ряду служат в качестве первой реакционной зоны для переэтерификации DEC с помощью фенола с получением EPC и этанола в присутствии твердого катализатора, и одна каталитическая дистилляционная колонна служит в качестве второй реакционной зоны для диспропорционирования EPC с получением DPC и DEC.
Обращаясь теперь к фигуре 1, здесь иллюстрируется способ получения DPC из DEC и фенола в соответствии с вариантами осуществления, описанными в настоящем документе. C1 и C2 представляют собой каталитические дистилляционные колонны для осуществления переэтерификации; C3 представляет собой колонну для извлечения этанола; C4 представляет собой колонну для извлечения DEC (барботажная колонна для фенетола); C5 представляет собой каталитическую дистилляционную колонну для диспропорционирования и извлечения фенола; C6 представляет собой колонну для извлечения EPC и C7 представляет собой колонну для извлечения DPC.
Колонны C1 и C2 представляют собой ряд каталитических дистилляционных колонн, где структурированные насадочные устройства размещаются в реакционной зоне R1 и R2 соответственно. Специально структурированные насадочные устройства содержат твердый катализатор. Фенол и DEC, содержащие потоки исходных материалов 1 и 4 соответственно вводятся в поддон в верхней секции каталитических дистилляционных колонн C1 и C2, над зонами каталитических реакций R1 и R2. Молярное отношение DEC к фенолу в потоках исходных материалов свежего DEC и свежего фенола может составлять приблизительно 1:2. Молярные отношения DEC к фенолу в зонах каталитических реакций R1 и R2, однако, контролируется так, чтобы оно составляло от примерно 12:1 до примерно 1:2,5, в некоторых вариантах осуществления; от примерно 10:1 до примерно 1:2, в других вариантах осуществления; и от примерно 7:1 до примерно 1:1, в других вариантах осуществления.
Растворимое металлорганическое соединение также вводится в поддон в верхней секции C1 через проточную линию 3. Например, для твердого катализатора, содержащего титан, в реакционных зонах R1 и R2, раствор, содержащий растворимое соединение титана, такое как Ti(OEt)4-x(OPh)x (где x равен 0, 1, 2, 3 или 4), или соли титана и сложных моноэфиров угольной кислоты, такие как этоксититан этилкарбонаты, или их смеси могут вводиться в верхнюю часть первого реактора в виде каталитической дистилляционной колонны C1. Растворитель для раствора катализатора может представлять собой, например, DEC, смешанный раствор DEC и фенола, смешанный раствор DEC и этанола или смешанный раствора DEC, этанола и фенола.
Скорость потока раствора катализатора может контролироваться так, что концентрация титана в жидком потоке над катализатором в первом реакторе в виде колонны составляет от примерно 20 м.д. до примерно 100 м.д. масс активного металла (титана, для иллюстративных растворов катализаторов, перечисленных в предыдущем абзаце), в некоторых вариантах осуществления; от примерно 25 м.д. до примерно 80 м.д. масс, в других вариантах осуществления; и от примерно 30 м.д. до примерно 70 м.д. масс, в других вариантах осуществления.
Потоки паров из верхней части колонны 6 и 14 из каталитических дистилляционных колонн C1 и C2 направляются в колонну для извлечения этанола C3 через проточную линию 8. Этот поток из верхней части колонны также может содержать микроскопические количества побочных продуктов, таких как простой диэтиловый эфир и диоксид углерода, и микроскопическое количество фенола. Простой диэтиловый эфир и диоксид углерода могут удаляться как поток паров из верхней части колонны 9. Этанол может извлекаться посредством боковой откачки из колонны C3 через проточную линию 10. Кубовый поток 11 может рециклировать DEC из колонны C3 в реакторы в виде каталитической дистилляционной колонны C1 и C2 через проточные линии 12 и 13 соответственно.
Колонна C1 может работать так, что температура зоны каталитической реакционной R1 находится в пределах от примерно 160°C до примерно 210°C (примерно 320°F до примерно 410°F). Давление к верхней части колонны в колонне C1 может находиться в пределах от примерно 2 бар, абсолютное, до примерно 4,8 бар, абсолютное, (от примерно 14,7 фунт/кв.дюйм в датчике до примерно 55 фунт/кв.дюйм в датчике). Кубовый поток 7 из первой каталитической дистилляционной колонны C1 может вводиться в верхнюю часть каталитической дистилляционной колонны C2, которая может работать так, что температура в зоне каталитической реакции может находиться в пределах от примерно 162°C до примерно 216°C (от примерно 325°F до примерно 420°F), и колонна может работать при давлении в верхней части колонны в пределах от более низкого, чем атмосферное, примерно 1 бар (0 фунт/кв.дюйм в датчике), до примерно 4,5 бар (51 фунт/кв.дюйм в датчике). Необязательно малые доли потока рециклированного или свежего DEC могут вводиться в колонны C1 и C2 через проточные линии 4a и 4b соответственно.
Концентрация EPC увеличивает при движении вниз по ступеням в каталитических дистилляционных колоннах C1 и C2. Поскольку в реакторах в виде колонн C1 и C2 происходит некоторое диспропорционирование EPC до DPC и DEC, концентрация DPC также увеличивается. Кубовый поток 15 из реактора в виде дистилляционной колонны C2 направляется в колонну для извлечения DEC C4, где DEC извлекается в потоке паров из верхней части колонны 16. Колонна C4 может работать при температуре от примерно 127°C до примерно 204°C (от примерно 260°F до примерно 400°F) при давлении верхней части колонны от примерно 0,3 бар (примерно 4 фунт/кв.дюйм, абс.) до примерно 1,5 бар (примерно 22 фунт/кв.дюйм, абс.). Поток 16 может вводиться в колонну для извлечения этанола C3 для разделения DEC и фенола, которые могут находиться в потоке 16, где DEC и фенол могут рециклироваться через линии 11, 12, 13 в C1 и C2.
Поток из верхней части колонны 16, из колонны C4 может также содержать DEC и малые количества фенетола, фенола и этанола. Боковой поток 18 из колонны C4 может использоваться в качестве потока для барботирования фенетола, сводя к минимуму накопление фенетола в системе.
Кубовый поток 17 из колонны C4 содержит частицы гомогенного катализатора, поступающие из колонн C1 и C2. Кубовый поток 17 может вводиться в соответствующие положения в верхней секции дистилляционной колонны C5. Колонна C5 может использоваться для осуществления диспропорционирования EPC и может необязательно содержать гетерогенный катализатор в реакционной зоне R3.
Колонна C5 может конструироваться и работать, чтобы она служила для двух целей: для удаления фенола в потоке 17 и побочного продукта DEC от диспропорционирования EPC в виде потока из верхней части колонны 19 и для диспропорционирования EPC с образованием DPC. Колонна C5 работает так, что температура зоны гомогенной каталитической реакции R3 находится в пределах от примерно 165°C до примерно 210°C (от примерно 330°F до примерно 410°F) и давление в верхней части колонны составляет от примерно 0,07 бар (примерно 1 фунт/кв.дюйм абс.) до примерно 0,6 бар (9 фунт/кв.дюйм абс.).
Поток паров из верхней части колонны 19 из C5, содержащий DEC и фенол, может рециклироваться в колонны C1 и C2 с помощью потоков 20 и 21 соответственно. Кубовый поток из C5 22 (который содержит DPC, непреобразованный EPC, фенол, фенетол, тяжелые фракции и растворимый катализатор на основе Ti) из C5 вводят в колонну для извлечения EPC C6, которая может работать при температуре от примерно 168°C до примерно 213°C (от примерно 335°F до примерно 415°F) и при давлении ниже атмосферного в пределах от примерно 0,03 бар (примерно 0,4 фунт/кв.дюйм абс.) до примерно 0,55 бар (8 фунт/кв.дюйм абс.).
Кубовый поток 25 из колонны для EPC C6 вводят в колонну для извлечения DPC C7, для извлечения DPC в виде бокового потока 27. Колонна для извлечения DPC C7 работает при высоком вакууме (например, < 0,03 бар (< 0,4 фунт/кв.дюйм абс.). Поток из верхней части колонны 26 может объединяться с потоком из верхней части колонны 23 для EPC C6 и рециклироваться в колонну C5 через линию 24.
Кубовый поток 28 из колонны для извлечения DPC C7, содержащий тяжелые фракции и растворимый катализатор, может извлекаться или утилизироваться. Если это желательно, когда катализаторы на основе титана используются и вводятся в реактор, например, можно извлекать титан как растворимый катализатор на основе Ti (Ti(OEt)4 или смесь Ti(OEt)4 и этоксититан этилкарбонатов) для рециклирования, как обсуждалось ранее. В качестве одного из способов, поток 28 может направляться в установку для рафинирования титана, для извлечения Ti. Можно извлекать DPC из потока 22 в чередующихся операциях извлечения и очистки, которые известны специалистам в данной области.
Альтернативно, как рассматривается выше, диспропорционирование EPC до DPC и DEC может осуществляться в присутствии твердого катализатора в каталитической дистилляционной колонне C5. Твердые катализаторы в C5 могут, однако, дезактивироваться быстрее, чем твердые катализаторы во второй каталитической дистилляционной колонне C2, как иллюстрируется на фигуре 1.
Как обсуждалось выше, когда C5 содержит твердый катализатор, различные возможности могут использоваться для циклирования реакторов в виде дистилляционных колонн для того, чтобы поддержать достаточную каталитическую активность для способа. Достаточное количество клапанов и труб, которые не иллюстрируются, могут быть предусмотрены, чтобы сделать возможным циклирование реакторов, и это известно специалистам в данной области.
Фигура 2 представляет собой блок-схему альтернативного способа, где сходные номера представляют сходные детали. Имеется сходное количество каталитических дистилляционных колонн для осуществления переэтерификации и диспропорционирования и колонн для разделения материалов, как на фигуре 1. Однако некоторая доля потока из верхней части колонны 19 из каталитической дистилляционной колонны C5 может рециклироваться обратно в колонну для извлечения DEC C4 через линию 30. Рециклирование через линию 30 может таким образом сделать возможным альтернативный способ барботирования фенетола.
Фигура 3 иллюстрирует другую блок-схему альтернативного способа в соответствии с вариантами осуществления, описанными в настоящем документе, сходную с фигурами 1 и 2, где сходные номера представляют сходные детали. Первая каталитическая дистилляционная колонна C1 работает способом более или менее сходным с предыдущими случаями (фигуры 1 и 2). Однако работа второй каталитической дистилляционной колонны C2 и колонны для извлечения DEC C4 осуществляется способами, отличными от предыдущих случаев. Колонна C2 может работать при более высокой температуре и при более низком давлении, чем в предыдущих случаях. Поток рециклированного DEC 13 вводят в нижнюю часть колонны C2. Необязательно, часть потока свежего DEC 4 также может вводиться в нижнюю часть колонны C2. Благодаря работе при более высокой температуре и при более низком давлении, кубовый поток 15 из C2 содержит меньше DEC. Поток 14 из верхней части колонны C2 содержит, среди других компонентов, фенетол. Поток 14 может вводиться в колонну C4, где кубовый поток 17 из C4 представляет собой поток барботирования фенетола.
Исключительно чистый DPC может быть получен из сырых продуктов DPC, полученных в соответствии с вариантами осуществления, описанными в настоящем документе. DPC высокой чистоты может быть получен посредством фракционной кристаллизации с использованием смесей углеводород - простой эфир, таких как смеси гексан - диэтиловый простой эфир. В некоторых вариантах осуществления, единственная детектируемая примесь, кроме фенола, в очищенном продукте DPC представляет собой ксантон в количестве до примерно 0,5 м.д. масс. Фенол в очищенном DPC может присутствовать в микроскопическом количестве, например, от примерно 5 до примерно 17 м.д. масс. Анализы микроскопических примесей в DPC, полученном в соответствии с вариантами осуществления, описанными в настоящем документе, показывают, что чистота полученного DPC гораздо выше, чем для DPC, который может быть получен от обычных поставщиков лабораторных химикалиев.
ПРИМЕРЫ
За исключением Эксперимента 1, все реакции переэтерификации DEC с помощью фенола осуществляют в реакторе с кипящим слоем с восходящим потоком. По этой причине, паровая и жидкая фазы сосуществуют в зоне каталитической реакции. Размер реактора с неподвижным слоем составляет диаметр 1,3 см (1/2 дюйма) и длина 6,5 см (25 дюймов). Реактор имеет контролируемые по-отдельности верхнюю и нижнюю зоны нагрева. Реактор с неподвижным слоем устанавливают вертикально. Объем твердых катализаторов составляет 25 мл.
Сравнительный эксперимент 1
Эксперименты для переэтерификации DEC с помощью фенола осуществляют в присутствии гомогенного катализатора на основе алкоксида титана посредством использования перемешиваемого 50-мл автоклавного реактора. Автоклав заполняют примерно 35 мл смеси DEC/фенол, как приведено в таблице 1. Автоклав погружают в масляную баню для контроля температуры реакции. После осуществления реакции, автоклав удаляют из масляной бани и гасят холодной водой. Простой дифениловый эфир не наблюдается в какой-либо реакционной смеси. Результаты реакций приведены в таблице 1.
°С (°F)
(% моль)
ность для EPC и DPC
(% моль)
Когда концентрации Ti в растворах исходных материалов составляют 4767 м.д. масс, максимальное преобразование фенола при времени реакции примерно 3 часов составляет примерно 15% при очень плохой селективности (<20,5% моль) относительно EPC и DPC. Когда время реакции составляет 2 часа, преобразование фенола составляет меньше чем 5%; селективность лучше, но по-прежнему плохая (63% моль). Когда концентрацию катализатора уменьшают до 42 м.д. Ti масс, селективность значительно улучшается, но преобразование плохое.
Сравнительный эксперимент 2
Целью настоящего эксперимента является получение экспериментальных данных по гомогенному катализатору в качестве эталона для сравнения с результатами примеров в соответствии с вариантами осуществления, описанными в настоящем документе. Твердого катализатора в реакторе нет. 25-мл пространство для твердого катализатора в реакторе пустое. Реакционные смеси из 73,3 мас.% DEC и 26,7 мас.% фенола (молярное отношение DEC/PhOH 2,19), имеющие различные количества гомогенного катализатора Ti(OEt)4-x(OPh)x (x = ~2) пропускают через восходящий поток реактора при различных условиях реакции, в течение 0-768 часов непрерывной работы, а затем от 1266 до 1362 часов непрерывной работы. Концентрации Ti в смеси исходных материалов находятся в пределах от 59 м.д. до 709 м.д. масс Ti, как показано на фигуре 4. Скорость потока составляет 0,5 мл/мин для большей части времени опыта. История скорости потока исходных материалов приведена в таблице 2.
Этанол в смеси продуктов переэтерификации отгоняют, а затем добавляют DEC для доведения молярного отношения DEC/PhOH до 2,19 посредством добавления DEC с получением исходных материалов для второй переэтерификации. Исходные материалы для второй переэтерификации имеют примерно 3,4 мас.% EPC, примерно 250 м.д. фенетола и примерно 300 м.д. масс DPC, в среднем. История концентрации гомогенных катализаторов в исходных материалах для второй переэтерификации приведена в таблице 3.
Используя эти смеси исходных материалов, вторую переэтерификацию осуществляют в течение времени непрерывной работы от 768 до 1266 часов. Преобразование фенола на фигуре 4 в течение этого периода времени представляет собой общее преобразование в течение как 1-й, так и 2-й переэтерификации.
Диапазон температур реакции составляет от 174°C до 210°C (345°F - 410°F), как показано на фигуре 4. Диапазон давлений реактора составляет от примерно 2,9 до примерно 5,5 бар (от примерно 27 фунт/кв.дюйм в датчике до примерно 65 фунт/кв.дюйм в датчике). Все реакции осуществляют в условиях кипения. Таким образом, реакцию осуществляют как реакцию в двухфазной системе (пары и жидкость). Температуры на фигуре 4 представляют собой температуры в нижней части реактора.
Когда концентрация катализатора в реакционной смеси выше чем 118 м.д. Ti, имеются отрицательные воздействия катализатора на преобразование фенола. Причина этого отрицательного воздействия не является полностью понятной, но может представлять собой воздействие двух этоксигрупп катализатора на основе Ti. Также, когда концентрация Ti в исходных материалах выше примерно чем 300 м.д., возникают проблемы забивания трубопроводов в линиях для эффлюента из реактора из-за осаждения катализатора на основе Ti. По этой причине, устанавливают in-line пару фильтров для решения проблем с забиванием трубопроводов. Когда концентрация Ti составляет 59 м.д., воздействие температуры на преобразование фенола является умеренным, указывая на низкую энергию активации переэтерификации DEC с помощью фенола. Самое высокое преобразование фенола в течение первой переэтерификации составляет примерно 11,3% моль при 337 м.д. Ti при 204°C (400°F) и 4,5 бар (50 фунт/кв.дюйм в датчике). Самое высокое преобразование фенола в течение второй переэтерификации составляет примерно 14,5% моль при 193°C (380°F) и 2,9 бар (27 фунт/кв.дюйм в датчике) при концентрации Ti 188 м.д. Эксперимент также говорит о более низком преобразовании для жидкофазной реакции (410°C и 7,9 бар (100 фунт/кв.дюйм в датчике)), как и ожидалось.
Эксперимент 3
Целями настоящего эксперимента являются демонстрация (1) технологии получения in situ н-бутоксида титана, иммобилизованного на носителе из силикагеля, (2) технологии реактивирования катализатора и (3) рабочих характеристик двухфазного реактора переэтерификации с неподвижным слоем. Двойную фазу из паров и жидкости в зоне каталитической реакции создают посредством кипения реакционной смеси.
45,74 г гранулированного силикагеля (+8 меш) обрабатывают раствором гидроксида натрия (7,5 г NaOH в 550 мл воды) примерно при температуре 42°C в течение 7 минут при перемешивании при температуре окружающей среды. Силикагель промывают, сначала холодной водой, а затем горячей водой (примерно 80°C), для удаления микроскопических количеств натрия на диоксиде кремния. Полученный обработанный силикагель сушат при 125°C в течение 2 часов, а затем при 300°C в течение 2 часов с продувкой азотом. Носитель из высушенного силикагеля имеет 23 м.д. масс Na. Носитель из обработанного силикагеля имеет следующие свойства: удельная площадь поверхности согласно БЭТ 291 м2/г, объем пор 1,052 см3/г и средний диаметр пор 16,3 нм.
25 мл высушенного носителя из гранулированного силикагеля (примерно 9,3 г) загружают в реактор. Раствор н-бутоксида титана приготавливают посредством растворения 27 г н-бутоксида титана в 500 мл сухого толуола. Раствор н-бутоксида титана помещают в резервуар. После циркулирования раствора н-бутоксида титана в восходящем потоке реактора при скорости 15 мл/мин и при температуре окружающей среды в течение 15 минут, реактор нагревают до 168°C (335°F) при давлении примерно 5,5 бар (65 фунт/кв.дюйм в датчике). Циркуляцию продолжают при 168°C (335°F) в течение 4,5 часов, а затем реактор охлаждают. После удаления избытка раствора из реактора, катализатор на носителе промывают с помощью восходящего потока сухого толуола при 4 мл/мин в течение 1,5 часа. Промытый катализатор сушат при 168°C (335°F) в потоке газообразного азота (восходящий поток), 350 см3/мин, в течение 2 часов. Полученный в результате приготовленный in situ катализатор на основе н-бутоксида титана, привитого на носитель из гранулированного силикагеля, исследуют относительно переэтерификации DEC с помощью фенола.
Переэтерификация 1-го цикла: Переэтерификацию DEC с помощью фенола осуществляют в присутствии приготовленного in situ твердого катализатора в реакторе с кипящим слоем, в реакторе с неподвижным слоем. Смесь DEC и фенола (25,57 мас.% фенола и 74,43 мас.% DEC; молярное отношение DEC/PhOH 2,32) пропускают через восходящий поток слоя твердого катализатора при 168°C (335°F), 2,4 бар (20 фунт/кв.дюйм в датчике) и при скорости потока исходных материалов 0,2 мл/мин. Это исследование составляет 1-й цикл переэтерификации и результаты иллюстрируются на фигуре 5. Катализатор достигает своей максимальной активности (преобразование фенола 12% моль) примерно через 40 часов непрерывной работы. После времени непрерывной работы примерно 80 часов, катализатор теряет большую часть своей активности. Этот дезактивированный катализатор подвергают 1-му реактивированию следующим образом.
1-е реактивирование катализатора: Реактивирование катализатора включает две стадии; деполимеризацию катализатора/ кондиционирование поверхности и повторное осаждение активного металлического титана на катализатор. После дренирования реактора, катализатор промывают с помощью восходящего потока сухого толуола (300 мл) при температуре окружающей среды, а затем из реактора удаляют толуол. 0,19 г н-бутоксида титана растворяют в 2 литрах раствора этанола, приготовленного посредством смешивания 400 мл этанола и 1700 мл толуола. Раствор титана проходит через реактор при скорости восходящего потока 2,2 мл/мин при 168°C (335°F) и 12 бар (160 фунт/кв.дюйм в датчике) в течение 13,5 часов. После удаления избытка раствора титана из реактора, катализатор сушат при 168°C (335°F) при давлении окружающей среды в восходящем потоке азота, 200 см3/мин, в течение 45 минут. Раствор н-бутоксида титана (135 г н-бутоксида титана в 2 литрах толуола) циркулирует через реактор в восходящем потоке, 15 мл/мин, при комнатной температуре в течение 20 минут, а затем при 168°C (335°F) и 10,7 бар (140 фунт/кв.дюйм в датчике) в течение 4 часов. После охлаждения, избыток раствора удаляют из реактора. Катализатор промывают с помощью восходящего потока толуола, 4 мл/мин, в течение 1,5 часов. Промытый катализатор сушат при 168°C (335°F) в восходящем потоке газообразного азота, 300 см3/мин, в течение 2 часов. Реактивированный катализатор используют во втором цикле переэтерификации следующим образом.
Переэтерификация 2-го цикла: Переэтерификацию осуществляют способом, идентичным способу 1-го цикла. Результат иллюстрируется на фигуре 5. Реактивированный катализатор работает не так хорошо, как в 1-м цикле, и катализатор отказывает после всего лишь примерно 40 часов непрерывной работы. Реактивирование 2-го катализатора осуществляют затем следующим образом.
2-е реактивирование катализатора: После удаления материала из реактора, катализатор в реакторе промывают сухим толуолом в восходящем потоке, 10 мл/мин, при температуре окружающей среды в течение 30 мин, а затем толуол удаляют из реактора. Катализатор в реакторе сушат при 168°C (335°F) в восходящем потоке газообразного азота, 250 см3/мин, в течение 1 часа. Раствор, приготовленный посредством смешивания 8 мл воды, 500 мл этанола и 1100 мл толуола пропускают через реактор в восходящем потоке, 2,2 мл/мин, при 168°C (335°F) и 12 бар (160 фунт/кв.дюйм в датчике) в течение 12,1 часа. После удаления избытка раствора из реактора, катализатор сушат при 168°C (335°F) при давлении окружающей среды в восходящем потоке азота, 200 см3/мин, в течение 1 часа. Раствор н-бутоксида титана (135 г н-бутоксида титана в 2 литрах толуола) циркулирует через реактор в восходящем потоке, 15 мл/мин, при комнатной температуре в течение 20 минут, а затем при 168°C (335°F) и 10,7 бар (140 фунт/кв.дюйм в датчике) в течение 6 часов. После охлаждения, избыток раствора титана удаляют из реактора. Катализатор промывают в восходящем потоке толуола, 4 мл/мин, в течение 1,5 часов. Промытый катализатор сушат при 168°C (335°F) в восходящем потоке газообразного азота, 300 см3/мин, в проточном режиме в течение 2 часов. Реактивированный катализатор используют в третьем цикле реакции переэтерификации следующим образом.
Переэтерификация 3-го цикла: Переэтерификацию осуществляют способом, идентичным способу 1-го цикла. Результат иллюстрируется на фигуре 5. Реактивированный катализатор работает не так хорошо, как катализатор в 1-м цикле. Тем не менее, катализатор отказывает после примерно 90 часов непрерывной работы.
Дезактивированный катализатор в реакторе подвергают воздействию еще двух реактивирований катализатора при сходных условиях, а затем еще двух переэтерификаций, со сходными результатами. После 5-го цикла реакций переэтерификации, катализатор подвергают 5-му реактивированию катализатора, следующим образом. История реактивирований катализатора, от 3-го до 5-го, описана ниже.
3-е реактивирование катализатора. После удаления всего материала в реакторе, оставшегося после переэтерификаци 3-го цикла, катализатор в реакторе промывают в восходящем потоке сухого толуола, 10 мл/мин, при температуре окружающей среды в течение 1 часа, а затем избыток толуола удаляют из реактора. Катализатор в реакторе сушат при 157°C (315°F) в восходящем потоке газообразного азота, 250 см3/мин, в течение 1 часа. Раствор, приготовленный посредством смешивания 8 мл воды, 500 мл этанола и 1100 мл толуола, пропускают через реактор в восходящем потоке, 2,2 мл/мин, при 157°C (315°F) и 2,7 бар (25 фунт/кв.дюйм в датчике) в течение 12,1 часа. После удаления избытка раствора из реактора, катализатор сушат при 149°C (300°F) при давлении окружающей среды в восходящем потоке азота, 200 см3/мин, в течение 1 часа. Раствор н-бутоксида титана (135 г н-бутоксида титана в 2 литрах толуола) циркулирует через реактор в восходящем потоке, 15 мл/мин, при комнатной температуре в течение 20 минут, а затем при 157°C (315°F) и 7,2 бар (90 фунт/кв.дюйм в датчике) в течение 6 часов. После охлаждения, избыток раствор удаляют из реактора. Катализатор промывают в восходящем потоке толуола, 4 мл/мин, в течение 1,5 часов. Промытый катализатор сушат при 163°C (325°F) в восходящем потоке газообразного азота, 300 см3/мин, в течение 2 часов.
Реактивированный катализатор подвергают 4-й переэтерификации. Рабочие характеристики реактивированного катализатора сходны с переэтерификацией 2-го цикла. Результат не показан на фигуре 5.
4-е реактивирование катализатора: После удаления материала, оставшегося в реакторе от переэтерификации 4-го цикла, катализатор в реакторе промывают в восходящем потоке сухого толуола, 10 мл/мин, при температуре окружающей среды в течение 1 часа, а затем избыток толуола удаляют из реактора. Катализатор в реакторе сушат при 157°C (315°F) в восходящем потоке газообразного азота, 250 см3/мин, в течение 1 часа. Раствор, приготовленный посредством смешивания 8 мл воды, 500 мл этанола и 1100 мл толуола, пропускают через реактор в восходящем потоке, 2,2 мл/мин, при 157°C (315°F) и 2,7 бар (25 фунт/кв.дюйм в датчике) в течение 12,1 часа. После удаления избытка раствора из реактора, катализатор сушат при 149°C (300°F) при давлении окружающей среды в восходящем потоке азота, 200 см3/мин, в течение 1 часа. Раствор н-бутоксида титана (135 г н-бутоксид титана в 2 литрах толуола) циркулирует через реактор в восходящем потоке, 15 мл/мин, при комнатной температуре в течение 20 минут, а затем при 157°C (315°F) и 7,2 бар (90 фунт/кв.дюйм в датчике) в течение 6 часов. После охлаждения, избыток раствора удаляют из реактора. Катализатор промывают в восходящем потоке сухого толуола, 4 мл/мин, в течение 1,5 часов. Промытый катализатор сушат при 163°C (325°F) в восходящем потоке газообразного азота, 300 см3/мин, в течение 2 часов. Реактивированный катализатор используют затем в 5-м цикле переэтерификации. Рабочие характеристики катализатора сходны с характеристиками переэтерификации 3-го цикла. Результат не показан на фигуре 5.
5-е реактивирование катализатора: После удаления материала из реактора, катализатор в реакторе промывают в восходящем потоке сухого толуола, 10 мл/мин, при температуре окружающей среды в течение 1 часа, а затем избыток толуола удаляют из реактора. Катализатор в реакторе сушат при 124°C (255°F) в восходящем потоке газообразного азота, 250 см3/мин, в течение 1 часа. Воду пропускают через реактор в нисходящем потоке, 0,3 мл/мин, при 152-154°C (305-310°F) и при давлении окружающей среды в течение 6 часов. Обработанный водяным паром катализатор в реакторе сушат в нисходящем потоке газообразного азота, 100 см3/мин, в течение 1 часа 20 минут при 146-149°C (295-300°F). Раствор н-бутоксида титана (135 г н-бутоксида титана в 1600 мл толуола) циркулирует через реактор в восходящем потоке, 15 мл/мин, при комнатной температуре в течение 20 минут, а затем при 127°C (260°F) и 3,1 бар (30 фунт/кв.дюйм в датчике) в течение 6 часов. После охлаждения, избыток раствора удаляют из реактора. Катализатор промывают в восходящем потоке толуола, 4 мл/мин, в течение 1,5 часов. Промытый катализатор сушат при 138°C (280°F) в 300 см3/мин в потоке восходящего газообразного азота в течение 2 часов. Реактивированный катализатор затем подвергают 6-й переэтерификации следующим образом.
Переэтерификация 6-го цикла: Переэтерификацию осуществляют способом, идентичным способу 1-го цикла. Результат иллюстрируется на фигуре 5. Реактивированный катализатор работает так же хорошо, как и в 1-м цикле. Интересно, катализатор дезактивируется с меньшей скоростью.
Указанные выше эксперименты демонстрируют, что возможно реактиврировать дезактивированный катализатор на основе алкоксида титана, иммобилизованный на носителе из силикагеля in situ. Однако продолжительность цикла катализатора может быть слишком короткой для осуществления этой технологии в больших промышленных реакторах для непрерывного получения ароматических карбонатов.
Эксперимент 4
Целью этого эксперимента является демонстрация увеличения продолжительности цикла катализатора, получаемого посредством добавления микроскопического количества (42 м.д. масс Ti) растворимого соединения Ti (н-бутоксида титана) в поток исходных материалов. Дезактивированный катализатор от переэтерификации 6-го цикла в Эксперименте 3 опять подвергают 7-му реактивированию катализатора следующим образом. После удаления материала в реакторе, катализатор в реакторе промывают в восходящем потоке сухого толуола, 10 мл/мин, при температуре окружающей среды в течение 1 часа, а затем избыток толуола удаляют из реактора. Катализатор в реакторе сушат при 124°C (255°F) в потоке восходящем газообразного азота, 250 см3/мин, в течение 1 часа. Смешанный раствор воды (4 мас.%) в этаноле пропускают через реактор в нисходящем потоке, 1,4 мл/мин, при 154°C (310°F) и при давлении окружающей среды в течение 6 часов. Катализатор в реакторе сушат в нисходящем потоке газообразного азота, 150 см3/мин, в течение 1 часа 25 минут при 154°C (310°F). Раствор н-бутоксида титана (67,5 г н-бутоксида титана в 800 мл толуола) пропускают через реактор в восходящем потоке, 15 мл/мин, при комнатной температуре в течение 20 минут, а затем при 127°C (260°F) при 3,4 бар (35 фунт/кв.дюйм в датчике) в течение 6 часов. После охлаждения, избыток раствора удаляют из реактора. Катализатор промывают в восходящем потоке толуола, 4 мл/мин, в течение 1,5 часа. Промытый катализатор сушат при 138°C (280°F) в восходящем потоке газообразного азота, 300 см3/мин, в течение 2 часов. Реактивированный катализатор подвергают 7-му циклу переэтерификации следующим образом.
7-й цикл переэтерификации осуществляют посредством добавления микроскопического количества (42 м.д. масс Ti) н-бутоксида титана в потоки исходных материалов при различных условиях реакции. В этом эксперименте используют две различные смеси исходных материалов. Смешанный раствор исходных материалов из 19,73 мас.% фенола и 80,27 мас.% DEC (молярное отношение DEC/PhOH 3,24) используют в течение первых 593 часов непрерывной работы, а затем 25,83 мас.% фенола и 74,17 мас.% DEC (молярное отношение DEC/PhOH 2,29) - в течение остального опыта до выключения, при 749 часах непрерывной работы. н-Бутоксид титана подмешивают в предварительно смешанные растворы исходных материалов DEC/PhOH. Скорости введения составляют 0,2 мл/мин в течение первых 353 часов непрерывной работы, 0,3 мл/мин, от 353 до 401 часов непрерывной работы, а затем 0,2 мл/мин, до конца опыта. Анализ микроскопических примесей в образцах продукта, отобранных в различные моменты непрерывной работы, показывает 21 м.д. Ti при 48 часов, 44 м.д. Ti при 305 часов, 44 м.д. Ti при 449 часов, 31 м.д. Ti при 491 часов, 51 м.д. Ti при 593 часов, 51 м.д. Ti при 713 часов и 31 м.д. Ti при 749 часов. Результат настоящего эксперимента иллюстрируется на фигуре 6. Температуры, приведенные на фигуре 6, представляют собой данные для температуры в нижней части катализатора. Данные для температуры в верхней части слоя катализатора обычно на 1,5-3°C (3-5°F), ниже, чем температура в нижней части реактора, в зависимости от концентрации этанола в потоке продукта, указывая на испарение этанола в верхней секции слоя катализатора. Более низкие температуры эффлюента из реактора в верхней части слоя катализатора становятся заметными, если концентрации этанола в продукте выше примерно, чем 1,2 мас.%. Фенетол представляет собой единственный детектируемый побочный продукт. Селективность для фенетола по отношению к фенолу меньше чем 0,3% моль.
Как показано на фигуре 6, нет дезактивирования катализатора во время всего времени опыта (749 часов), что успешно демонстрирует, что продолжительность цикла катализатор может быть увеличена от менее чем 80 часов до 749 часов или более посредством добавления 42 м.д. масс растворимого Ti в поток исходных материалов.
Катализатор в реакторе анализируют в конце опыта, и он представляет собой в основном желтые гранулы, при этом некоторые гранулы катализатора являются темно-коричневыми по цвету. Феноксид титана имеет интенсивный оранжевый или янтарный цвет, для сравнения. Анализ отработанного катализатора показывает 0,55 мас.% Ti на катализаторе. Это является неожиданным открытием.
Эксперимент 5
Целями настоящего эксперимента является демонстрация (1) необходимости предварительного формирования катализатора перед осуществлением реакций, (2) реактивирования катализатора, (3) увеличения времени цикла катализатора и (4) необходимости контроля содержания воды (меньше примерно, чем 650 м.д. масс) в исходных материалах. В этом эксперименте, гранулы оксида кремния используют для получения носителя, на который прививают н-бутоксид титана.
Носитель в виде гранул оксида кремния (0,3 см (1/8 дюйм), 555 м.д. Na и 2500 м.д. Al масс, удельная площадь поверхности согласно БЭТ 280 м2/г и объем пор 1 см3/г) используют для получения катализатора на основе иммобилизованного н-бутоксида титана. 100 г гранул оксида кремния обрабатывают примерно при 52°C в течение 5 минут при перемешивании раствором гидроксида натрия (10 г NaOH в 570 мл воды). Диоксид кремния тщательно промывают холодной водой, а затем горячей водой (примерно 80°C) для удаления микроскопических количеств натрия на диоксиде кремния. Обработанный оксид кремния сначала сушат при комнатной температуре, затем сушат при 130°C в течение 1,5 часов, а затем при 150°C в течение 1 часа в вакуумной печи. Высушенный носитель из диоксида кремния имеет 150 м.д. масс Na. Приготовленный носитель из диоксида кремния имеет следующие свойства: удельная площадь поверхности согласно БЭТ 252 м2/г, объем пор 1,035 см3/г и средний диаметр пор 15,7 нм.
25 мл гранул сухого оксида кремния (9,72 г) загружают в реактор. Резервуар для раствора катализатора заполняют раствором н-бутоксида титана, приготовленного посредством растворения 135 г н-бутоксида титана в 1600 мл толуола. Этот раствор катализатора циркулирует в восходящем потоке через реактор при скорости потока 15 мл/мин при температуре окружающей среды в течение 20 минут, а затем при 135°C (275°F) и 3,4 бар (35 фунт/кв.дюйм в датчике) в течение 6 часов. После охлаждения, избыток раствора катализатора удаляют из реактора, а затем приготовленный in situ катализатор промывают в восходящем потоке толуола при температуре окружающей среды при скорости потока 4 мл/мин в течение 1,5 часов. После удаления избытка толуола из реактора, катализатор сушат при 138°C (280°F) в течение 2 часов в восходящем потоке газообразного азота, 350 см3/мин. Полученный катализатор используют в переэтерификации 1-го цикла следующим образом.
Переэтерификация 1-го цикла: Переэтерификацию 1-го цикла осуществляют без инжекции частиц растворимого титана в поток исходных материалов, при условиях температуры кипения 168°C (335°F) и 2,4 бар (20 фунт/кв.дюйм в датчике) при скорости введения восходящего потока 0,2 мл/мин. Композиция исходного материала составляет 26,07 мас.% фенола и 73,93 мас.% DEC (молярное отношение DEC/фенол 2,56). Результат иллюстрируется на фигуре 7. Катализатор дезактивируется во время непрерывной работы. После примерно 100 часов непрерывной работы, катализатор имеет низкую активность.
1-е реактивирование катализатора: После удаления материала из реактора, катализатор в реакторе промывают в восходящем потоке сухого толуола, 10 мл/мин, при температуре окружающей среды в течение 1 часа, а затем избыток толуола удаляют из реактора. Катализатор в реакторе сушат при 124°C (255°F) в газообразном азоте, протекающем при 250 см3/мин в восходящем потоке в течение 1 часа. Смешанный раствор воды (4 мас.%) и этанола пропускают через реактор в восходящем потоке, 2,2 мл/мин, при 154°C (310°F) и давлении окружающей среды в течение 6 часов. Катализатор сушат при 154°C (310°F) в восходящем потоке азота, 150 см3/мин, в течение 1 часа 25 минут. Раствор н-бутоксида титана (67,5 г н-бутоксида титана в 800 мл толуола) циркулирует через реактор в восходящем потоке, 15 мл/мин, при комнатной температуре в течение 20 минут, а затем при 134°C (275°F) и 3,4 бар (35 фунт/кв.дюйм в датчике) в течение 6 часов. После охлаждения, избыток раствора удаляют из реактора. Катализатор промывают в восходящем потоке толуола, 4 мл/мин, в течение 1,5 часов. Промытый катализатор сушат при 138°C (280°F) в восходящем потоке газообразного азота, 300 см3/мин, в течение 2 часов и используют во втором цикле реакции переэтерификации.
Переэтерификация 2-го цикла: Реактивированный катализатор подвергают переэтерификации 2-го цикла с таким же раствором исходных материалов при условиях, идентичных условиям переэтерификации 1-го цикла. Результат иллюстрируется на фигуре 7. Получают результаты, сходные с результатами переэтерификации 1-го цикла.
2-е реактивирование катализатора. 2-е реактивирование катализатора осуществляют способом, идентичным способу 1-го реактивирования катализатора.
Переэтерификация 3-го цикла. Реактивированный катализатор, полученный от 2-го реактивирования катализатора, подвергают 3-му циклу переэтерификации с добавлением частиц растворимого титана в такой же раствор исходных материалов при условиях, идентичных условиям переэтерификации 1-го цикла. Результаты переэтерификации 3-го цикла иллюстрируются на фигуре 7. Получают результат, сходный с результатом переэтерификации 1-го цикла, но катализатор поддерживает постоянную активность катализатора в течение продолжительного периода времени. Анализ микроскопических примесей образца, отбираемого через 270 часов непрерывной работы, показывает 47 м.д. масс Ti. После отбора образцов через 270 часов непрерывной работы, резервуар для исходных материалов повторно заполняют новыми исходными материалами. К сожалению, исходные материалы становятся мутными, когда их смешивают с н-бутоксидом титана. Считается, что мутность вызывается неожиданно более высоким содержанием воды в растворе исходных материалов, чем в предыдущих растворах исходных материалов. Активность катализатора быстро уменьшается при использовании этого нового раствора исходных материалов. Анализ микроскопических примесей композитного продукта для этих новых исходных материалов показывает 9 м.д. масс Ti. Обнаружено, что содержание воды в потоке исходных материалов должно поддерживаться меньшим, примерно чем 650 м.д. масс.
Холостой опыт (без осуществления иммобилизации катализатора на основе алкоксида Ti): В этот же реактор загружают 25 мл (9,54 г) носителя в виде гранул оксида кремния, приготовленного посредством обработки гранул оксида кремния раствором гидроксида натрия (10 г NaOH в 570 мл воды) примерно при 52°C в течение 5 минут при перемешивании. Диоксид кремния тщательно промывают холодной водой, а затем горячей водой (примерно 80°C) для удаления микроскопических количеств натрия на диоксиде кремния. Обработанный диоксид кремния сначала сушат при комнатной температуре, затем сушат при 130°C в течение 1,5 часов, а затем при 150°C в течение 1 часа в вакуумной печи. Катализатор на носитель не прививают. Реакцию переэтерификации осуществляют с помощью 42 м.д. масс Ti при той же композиции исходных материалов, как рассмотрено выше, при идентичных условиях (при условиях температуры кипения 168°C (335°F) и 2,4 бар (20 фунт/кв.дюйм в датчике) при скорости восходящего потока исходных материалов 0,2 мл/мин, где композиция исходных материалов составляет 26,07 мас.% фенола и 73,93 мас.% DEC (молярное отношение DEC/фенол 2,56). Результаты иллюстрируются на фигуре 7. Преобразование фенола в течение опыта составляет меньше чем 2%.
Эта серия экспериментов (Эксперимент 5) успешно демонстрирует, что является возможным реактивирование дезактивированного катализатора и продление времени цикла катализатора до более чем 250 часов. Холостой опыт ясно демонстрирует, что необходимо приготовить катализатор перед осуществлением переэтерификации. Альтернативно, можно выбрать инициирование переэтерификации с помощью предварительно приготовленного катализатора на основе привитого алкоксида титана вне реактора. Этот эксперимент также показывает, что может быть необходимым контроль содержания воды в исходных материалах до менее примерно чем 650 м.д. масс для поддержания постоянной активности катализатора.
Эксперимент 6
Целью настоящего эксперимента является демонстрация непрерывного получения ароматических карбонатов в ряду из множества последовательных реакторов в присутствии катализатора на основе оксида титана, нанесенного на носитель из диоксида кремния. Такой же гранулированный силикагель (40,7 г) как в Эксперименте 3 обрабатывают раствором гидроксида натрия (6,86 г NaOH в 500 мл воды) при температуре окружающей среды в течение 7 минут, при перемешивании. Силикагель сначала тщательно промывают холодной водой, а затем горячей водой (примерно 80°C), для удаления микроскопических количеств натрия на диоксиде кремния. Обработанный силикагель сушат при 140°C в течение 2 часов, при 345°C в течение 3 часов, а затем при 375°C в течение 2 часов. 30 мл (10,99 г) пропитывают раствором н-бутоксида титана, приготовленного посредством растворения 4,71 г н-бутоксида титана в 80 мл сухого толуола. Пропитанный носитель из силикагеля кальцинируют при 500°C в течение 3 часов. Содержание титана на катализаторе на основе оксида титана на носителе из диоксида кремния составляет 5,48 мас.% Ti по отношению к количеству используемого н-бутоксида титана. 25 мл (9,8 г) катализатора на основе оксида титана на носителе из диоксида кремния загружают в реактор. Переэтерификацию DEC с помощью фенола осуществляют при различных условиях. Исходные материалы в течение 0 часов - 308 часов непрерывной работы представляют собой две различные смеси DEC и фенола. Эти исходные материалы используют для осуществления 1-й переэтерификаци DEC с помощью фенола. Содержание титана в растворах исходных материалов DEC/PhOH (от 0 до 308 часов непрерывной работы) составляет 59 м.д. масс Ti, его получают посредством смешивания исходного раствора Ti(OEt)4-x(OPh)x (где x=~2). Исходный раствор Ti(OEt)4-x(OPh)x приготавливают посредством отгонки этанола из раствора, приготовленного посредством смешивания соответствующего количества тетраэтоксида титана в смешанном растворе DEC и фенола (PhOH) (25 мас.%) при 120°C - 125°C в течение примерно 3 часов. Исходные материалы для 308 часов - 986 часов непрерывной работы получают посредством отгонки этанола из смешанных продуктов от 1-й переэтерификации. Эти исходные материалы используют для осуществления 2-х переэтерификаций, которые эквивалентны реакциям во втором реакторе в ряду или на некоторых ступенях ниже точки введения многоступенчатой каталитической дистилляционной колонны. Исходные материалы для 986 часов - 1136 часов непрерывной работы получают посредством отгонки этанола из смешанных продуктов из 2-й переэтерификации. Эти исходные материалы используют для осуществления 3-х переэтерификаций. Компонент растворимого катализатора на основе титана не подмешивают в исходные материалы для 2-й или 3-й переэтерификации. Композиции исходных материалов приведены в таблице 4. Переэтерификацию осуществляют при 185°C (365°F), 2,9 бар (27 фунт/кв.дюйм в датчике) и при скорости введения 0,24 мл/мин. Результат настоящего эксперимента иллюстрируется на фигуре 8. Преобразование фенола на фигуре 8 представляет собой общее преобразование фенола для 1-й переэтерификации - 3-й переэтерификации. Указаний на дезактивирование катализатора в течение опыта (1362 часов непрерывной работы) нет. Исследование катализатора, извлеченного из реактора по окончании опыта, показывает небольшое осаждение тяжелых полимеров. Анализ катализатора показывает 2,3 мас.% Ti, показывая примерно 58% потерь Ti из-за выщелачивания в поток продукта. Анализ микроскопических примесей Ti в потоках продукта, отобранных после 686, 887 и 1293 часов непрерывной работы, показывают 75, 57 и 78 м.д. масс Ti соответственно.
Результат этого эксперимента ясно демонстрирует непрерывное получение ароматических карбонатов посредством использования ряда из множества реакторов с большим временем цикла катализатора посредством добавления микроскопического количества растворимого соединения Ti в поток исходных материалов. Также, этот эксперимент может говорить о том, что большие количества оксида титана на катализаторе могут быть не нужны, поскольку любое избыточное количество оксида титана может смываться посредством образования растворимых органических соединений титана. Время цикла катализатора является более чем достаточным для времени, необходимого для реактивирования катализатора. Объединенная селективность относительно EPC и DPC составляет от примерно 98% моль до примерно 93% по отношению к преобразованному фенолу и в зависимости от условий опыта.
Эксперимент 7
Целью настоящего эксперимента является демонстрация непрерывного получения ароматических карбонатов в ряду из множества реакторов посредством осуществления переэтерификации DEC с помощью фенола в присутствии катализатора на основе этоксида титана, иммобилизованного на носителе из силикагеля.
Этот эксперимент состоит из двух частей; Эксперимент 7A и 7B. В Эксперименте 7A, осуществляют иммобилизацию этоксида Ti на носителе из силикагеля перед осуществлением переэтерификации. Исходные материалы содержит различные количества растворимого соединения Ti(OEt)4-x(OPh)x (где x=~2). В Эксперименте 7B, 25-мл пространство в реакторе заполняют 25 мл носителя из силикагеля и переэтерификацию осуществляют без прививки тетраэтоксида Ti на носителе из диоксида кремния.
Эксперимент 7A
Носитель, используемый для приготовления катализатора in situ, представляет собой сферы силикагеля сферической формы (диаметр 1,7-4 мм). Этот носитель из силикагеля имеет примерно 6 гидроксильных групп на нм2, удельную площадь поверхности согласно БЭТ 392 м2/г, объем пор 0,633 см3/г, средний диаметр пор 6,48 нм и ABD (видимую насыпную плотность) примерно 0,58 г/мл. Этот носитель из силикагеля (25 мл; 14 46 г) загружают в реактор. Раствор этоксида титана (45,25 г этоксида титана в 800 мл толуола) циркулирует в восходящем потоке, 15 мл/мин, через реактор при температуре окружающей среды в течение 20 минут, а затем при 135°C (275°F) при 3,4 бар (35 фунт/кв.дюйм в датчике) в течение 6 часов для прививки этоксида титана на носитель из силикагеля. После охлаждения, избыток раствора в системе удаляют, а затем катализатор промывают толуолом, 4 мл/мин, в течение 1,5 часов. Промытый катализатор сушат при 138°C (280°F) в течение 2 часов при скорости потока 300 см3/мин в потоке газообразного азота.
Реакции, дающие EPC и DPC, осуществляют при различных условиях. Результат иллюстрируется на фигуре 9A и 9B. Все реакции первой переэтерификации осуществляют при 59 м.д. Ti в виде этоксида Ti, добавляемого в поток исходных материалов, за исключением периода от 709 часов до 799 часов непрерывной работы, когда добавляют 151 м.д. Ti. Опыт начинают при 185°C (365°F), 2,9 бар (27 фунт/кв.дюйм в датчике) и 0,24 мл/мин, для первой переэтерификации. После первых 50 часов непрерывной работы, температуру медленно понижают до 174°C (345°F) и скорость потока исходных материалов медленно повышают до 0,5 мл/мин, в течение следующих 96 часов непрерывной работы. После этого, вся 1-я и 2-я переэтерификация осуществляются при условиях 174°C (345°F), 2,9 бар (27 фунт/кв.дюйм в датчике) и 0,5 мл/мин. Посредством отгонки этанола от смешанных продуктов из 1-й, 2-й и 3-й переэтерификации, получают смеси исходных материалов для 2-й, 3-й и 4-й переэтерификации. В течение от 973 часов до 1064 часов непрерывной работы, смешанную реакцию переэтерификации и диспропорционирования осуществляют при 174°C (345°F) и 2,4 бар (20 фунт/кв.дюйм в датчике), при скорости потока исходных материалов 0,5 мл/мин. Композиция исходных материалов для реакции представляет собой 18,553 мас.% DEC, 0,108 мас.% этилбутилкарбоната, 0,283 мас.% фенетола, 0182 мас.% неизвестных веществ, 57,508 мас.% фенола, 22,03 мас.% EPC, 0,054% п-феноксифенилметилкарбоната и 1,282 мас.% DPC. Результат показывает, что главная реакция представляет собой диспропорционирование. Но анализ данных говорит также о необходимости удаления DEC из исходных материалов диспропорционирования. На фигуре 9A, 2-ю переэтерификацию осуществляют при 174°C (345°F), 2,4 бар (20 фунт/кв.дюйм в датчике) и 0,5 мл/мин, при этом концентрация Ti в исходных материалах находится в пределах от 44 м.д. масс Ti до 69 м.д. масс Ti. На фигуре 9B, 2-ю переэтерификацию осуществляют при 174°C (345°F), 2,9 бар (27 фунт/кв.дюйм в датчике) и 0,5 мл/мин, при этом концентрация Ti в исходных материалах находится в пределах от 45 м.д. масс Ti до 75 м.д. масс Ti. 3-ю переэтерификацию осуществляют при 174°C (345°F), 2,5 бар (22 фунт/кв.дюйм в датчике) и 0,5 мл/мин, при этом концентрация Ti в исходных материалах находится в пределах от 52 м.д. масс Ti до 74 м.д. масс Ti. 4-ю переэтерификацию осуществляют при 174°C (345°F), 2,4 бар (20 фунт/кв.дюйм в датчике) и 0,5 мл/мин, при этом концентрация Ti в исходных материалах находится в пределах от 51 м.д. масс Ti до 73 м.д. масс Ti.
Селективность ароматических карбонатов уменьшается вместе с преобразованием фенола. Объединенная селективность по отношению к EPC и DPC во время 1-й переэтерификации составляет примерно 99% моль по отношению к фенолу. Объединенная селективность по отношению к EPC и DPC во время 4-й переэтерификации составляет от 94 моль до 95% моль по отношению к фенолу.
Твердый катализатор находится в работе в течение 14 месяцев до окончания эксперимента, не связанного с активностью катализатора. Фигуры 9A и 9B четко говорят о том, что имеется малое дезактивирование катализатора в течение 14 месяцев или оно вообще отсутствует. Анализы двух образцов катализатора, осторожно отобранных из верхней и нижней части слоя катализатора, показывают одинаковое количество 0,28 мас.% Ti (по отношению к кальцинированному при 550°C) на обоих образцах катализатора. Этот эксперимент успешно демонстрирует, что продолжительный цикл катализатора (более чем 14 месяц) может быть получен посредством добавления микроскопического количества растворимого соединения титана в поток исходных материалов.
Эксперимент 7B (холостой опыт)
Целью настоящего эксперимента является попытка иммобилизации алкоксида Ti на носителе из диоксида кремния, осуществляя при этом переэтерификацию. Различные количества растворимого соединения Ti(OEt)x(OPh)4-x добавляют в поток исходных материалов. Переэтерификацию DEC с помощью фенола осуществляют при 174°C (345°F) и 2,9 бар (27 фунт/кв.дюйм в датчике). Результат иллюстрируется на фигуре 10.
Если сравнивать холостые опыты в Эксперименте 5 (фигура 7) и в Эксперименте 7B (фигура 10) с результатом (фигуры 9A и 9B), имеется четкая необходимость в иммобилизации алкоксида титана на носителе из силикагеля перед осуществлением переэтерификации. Если сравнивать Сравнительные эксперименты 1 и 2 (фигура 4) с фигурами 9A и 9B, также ясно демонстрируется превосходство новой технологии твердого катализатора с добавлением микроскопического количества растворимого активного металлорганического соединения к исходным материалам по сравнению с данными, известными из литературы.
Эксперимент 8
Целью настоящего эксперимента является демонстрация диспропорционирования EPC в DPC и DEC в отсутствие твердого катализатора, но в присутствии растворимых каталитических компонентов на основе Ti. Исходные материалы для диспропорционирования получают посредством отгонки этанола, DEC и части фенола из смешанных продуктов от 4-й переэтерификации в Эксперименте 7 в атмосфере азота. Гомогенный катализатор на основе Ti в смесях исходных материалов происходит из смешанных продуктов 4-й переэтерификации. Дополнительного растворимого катализатора на основе Ti к смесям исходных материалов не добавляют. Толуол добавляют к двум смесям исходных материалов для создания паровой фазы для реактора с кипящим слоем.
Первая композиция исходных материалов представляет собой 16,26 мас.% толуола, 1,61 мас.% DEC, 49,33 мас.% фенола, 30,91 мас.% EPC и 0,78 мас.% DPC, и остаток представляет собой побочные продукты, включая микроскопические количества MPC. Вторая композиция исходных материалов представляет собой 16,15 мас.% толуола, 1,61 мас.% DEC, 49,28 мас.% фенола, 31,08 мас.% EPC и 0,80 мас.% DPC, и остаток представляет собой побочные продукты, включая микроскопические количества MPC. Концентрация гомогенного катализатора в первых и вторых исходных материалах составляет 180 м.д. и 200 м.д. масс Ti соответственно.
Диспропорционирование осуществляют в реакторе с 25-мл пустым пространством для катализатора (в отсутствие твердого катализатора) при 179°C (355°F) и 2,9 бар (27 фунт/кв.дюйм в датчике). Скорость восходящего потока исходных материалов составляют 0,5 мл/мин для первых 72 часов непрерывной работы, а затем, после этого, скорость восходящего потока составляет 0,60 мл/ми. Результаты реакций диспропорционирования иллюстрируются на фигуре 11. Экспериментальные результаты показывают, что получаются также малые количества EPC, в дополнение к DPC. Остаточный ксантон представляет собой единственный новый побочный продукт, получаемый во время диспропорционирования в количестве примерно 35 м.д. масс. Простой дифениловый эфир при анализе каких-либо образцов не детектируется. Селективность по отношению ко всем побочным продуктам составляет от 3,0% моль до 3,3% моль. Этот эксперимент демонстрирует успешное диспропорционирование EPC с получением DPC и DEC в соответствии с вариантами осуществления, описанными в настоящем документе.
Эксперимент 9
Этот эксперимент демонстрирует очистку DPC. Смешанный продукт от диспропорционирования из Эксперимента 8 дистиллируют для удаления этанола, DEC и значительной части фенола посредством использования лабораторного оборудования для дистилляции. Оставшийся материал в дистилляционной колбе имеет следующую композицию: 0,024% EtOH, 0,204% DEC, 0,017% фенетола, 1,619% неизвестных веществ 12,563% фенола, 25,377% EPC, 59,474% DPC и 0,723% тяжелых фракций. Посредством осуществления вакуумной дистилляции, получают сырой DPC (выходящий при температуре паров от 235 до 245°C). Композиция этого сырого DPC составляет 0,535% неизвестных веществ 2,112% фенола, 0,013% простого фенилового эфира, 0,030% EPC, 94,555% DPC, 0,026% ксантона и 2,73% тяжелых фракций. Этот сырой DPC перекристаллизуют пять раз в смеси из 5 мас.% простого диэтилового эфира в гексане. Конечный продукт DPC имеет примеси из 0,4 м.д. ксантона и 11,6 м.д. масс фенола. Другие примеси не детектируются с помощью анализа микроскопических примесей. Этот продукт DPC имеет более высокую чистоту, чем DPC высокой чистоты, доступный на рынке (28,7 м.д. масс неизвестных веществ и 67,2 м.д. масс фенола).
Получение диалкилкарбонатов посредством переэтерификации циклического карбоната с помощью спирта
Диалкилкарбонаты непрерывно получают посредством осуществления переэтерификации циклического карбоната с помощью спиртов в присутствии твердых катализаторов. Как описано выше, варианты осуществления, описанные в настоящем документе, могут быть особенно пригодными для непрерывного получения диалкилкарбонатов, таких как DMC, DEC и тому подобное. Имеется ряд гомогенных катализаторов для переэтерификации. Когда диалкилкарбонаты получают посредством осуществления переэтерификации циклического карбоната с помощью спирта в присутствии катализаторов на основе оксида металла или смешанного оксида металла на носителе или твердого катализатора, полученного посредством иммобилизации гомогенного катализатора на пористом носителе, катализаторы имеют неприемлемо короткую продолжительность цикла для работы больших промышленных реакторов. Необратимое дезактивирование катализатора, участвующего в работе с органическими карбонатами, вызывается выщелачиванием активных каталитических компонентов из гетерогенных катализаторов в реакционную среду. По этой причине, диалкилкарбонат, такой как DMC, обычно получают посредством осуществления переэтерификации в присутствии гомогенного катализатора.
Однако варианты осуществления, описанные в настоящем документе, предлагают способы получения диалкилкарбоната в присутствии твердого катализатора. Твердые катализаторы могут содержать один или несколько элементов из Групп II, III, IV, V и VI Периодической таблицы. Первый тип твердого катализатора содержит одно или несколько металлорганических соединений указанных выше элементов, иммобилизованных на пористом носителе, который может иметь поверхностные функциональные группы, такие как гидроксил, карбонил, алкокси, смесь гидроксила и алкокси, хлор и тому подобное. Носители могут содержать диоксид кремния, оксид титана, цеолитные материалы, такие как MCM-41, MCM-48, SBA-15, углерод и/или углеродистые материалы и тому подобное. Второй тип твердого катализатора содержит оксиды, гидроксиды или оксигидроксиды металлов, одного или нескольких из указанных выше элементов, осажденные на пористом носителе. Для поддержания стабильной активности катализатора, микроскопическое количество растворимого каталитического компонента добавляют в поток исходных материалов. Если делать это, продолжительность цикла катализатора может быть увеличена, так что она станет пригодной для промышленного реактора.
Переэтерификация может осуществляться в любом физическом устройстве, таком как традиционные реакторы с неподвижным слоем, каталитические дистилляционные колонны, реакторы с кипящим слоем, дистилляционные колонны с разделительной перегородкой, импульсные проточные реакторы, или любое сочетание этих устройств. Примеры сочетания могут включать реактор с кипящим слоем, реактор с неподвижным слоем, за которым следует реактор в виде каталитической дистилляционной колонны. Переэтерификация циклических карбонатов с помощью первичного спирта, такого как этанол или метанол, может осуществляться как двухстадийная реакция, где имеются два промежуточных соединения карбонатов этилпропиленгликоля. Также продукт реакции содержит малые количества побочных продуктов, таких как изомеры простого этилового эфира пропиленгликоля, получаемые посредством O-алкилирования пропиленгликоля с помощью DEC. Переэтерификация может осуществляться в одной реакционной зоне или во множестве реакционных зон.
Фигура 18 иллюстрирует блок-схему упрощенного способа непрерывного получения DEC и побочного продукта пропиленгликоля посредством осуществления переэтерификации пропиленкарбоната с помощью этанола в присутствии твердого катализатора в соответствии с вариантами осуществления, описанными в настоящем документе. Переэтерификация, как иллюстрируется, может осуществляться в реакторе каталитической дистилляции 101 при температуре от примерно 149°C до примерно 177°C (примерно 300°F - 350°F) при давлении от примерно 2 бар до примерно 11,4 бар (от примерно 45 фунт/кв.дюйм в датчике до 150 фунт/кв.дюйм в датчике) в зависимости от композиции реакционной смеси. В дополнение к реактору каталитической дистилляции 101, способ включает две дистилляционных колонны, 102 и 114. Каталитическая дистилляционная колонна 101 содержит реакционную зону RZ, в которой может находиться твердый катализатор. Исходные материалы свежего пропиленкарбоната 105 объединяются с рециклированным потоком 117, и объединенный поток 106 вводится в каталитическую дистилляционную колонну 101 в соответствующем положении выше реакционной зоны со слоем твердого катализатора RZ.
Поток 107 из верхней части колонны 101, смесь этанола, DEC и легких фракций, таких как диоксид углерода, вводится в колонну для извлечения DEC 102, для отделения DEC от более легких компонентов. Поток 108 из верхней части колонны 102 может вводиться в барабан для разделения газ-жидкость 110 для отделения жидкого этанола от газов, откачиваемых через линию 111. Жидкий этанол, извлекаемый из барабана 110 в поток 112, объединяется с потоком исходных материалов свежего этанола 103, и объединенный поток 104 нагревают с получением паров этанола, которые вводят в каталитическую дистилляционную колонну 101 в соответствующем положении ниже реакционной зоны RZ. Кубовый поток 109 дистилляционной колонны 102 содержит продукт, DEC, который может направляться в танк-хранилище (не показан) или в другие последующие процессы.
Кубовый поток 113 из каталитической дистилляционной колонны 101, который содержит пропиленгликоль, пропиленкарбонат, промежуточные продукты реакции и побочные продукты, такие как 1-этокси-2-пропанол, тяжелые фракции и тому подобное, и микроскопическое количество катализатора, вводятся во вторую дистилляционную колонну 114 для извлечения потока из верхней части колонны 115, содержащего пропиленгликоль, 1-этокси-2-пропанол и тому подобное. Пропиленгликоль может извлекаться из смеси в потоке 115 посредством отгонки (не показано). Кубовый поток 116 из колонны 114 рециклируется в каталитическую дистилляционную колонну 101 через линии 117 и 106. Часть кубового потока 116 может продуваться из системы посредством потока 118 для предотвращения накопления тяжелых фракций в системе.
Микроскопическое количество растворимого металлорганического соединения вводится в каталитическую дистилляционную колонну 101 выше зоны каталитической реакции через линию 119. В некоторых вариантах осуществления, раствор катализатора вводится при такой скорости, что жидкая реакционная смесь, стекающая вниз из зоны каталитической реакции RZ, содержит микроскопическое количество, как правило, от 5 м.д. до примерно 100 м.д. масс металла, растворимого металлического компонента, такого как соединения Mg, Ca, Zn, La, Ac или Ti.
Получение диалкилкарбонатов иллюстрируется с помощью следующих экспериментов.
Эксперимент 10
Целью настоящего эксперимента является демонстрация переэтерификации пропиленкарбоната с помощью этанола с получением DEC и пропиленгликоля в присутствии твердого катализатора. Твердый катализатор приготавливают in situ посредством иммобилизации этоксида титана на носителе из силикагеля.
Реактор заполняют 25 мл сферического носителя из силикагеля (диаметром 1,7-4 мм). Масса носителя составляет 10,097 г. Этот носитель из силикагеля имеет примерно 6 гидроксильных групп на нм2, удельную площадь поверхности согласно БЭТ 314 м2/г, объем пор 1,055 см3/г и средний диаметр пор 13,46 нм. Раствор этоксида титана (40 г этоксида титана в 800 мл толуола) циркулирует в восходящем потоке через реактор, 15 мл/мин, при температуре окружающей среды в течение 30 минут, а затем при 135°C (275°F) и 3,4 бар (35 фунт/кв.дюйм в датчике) в течение 6 часов для прививки этоксида титана на носителе из силикагеля. После охлаждения, избыток раствора удаляют из реактора, а затем катализатор промывают толуолом, 4 мл/мин, в течение 1,5 часов. Промытый катализатор сушат при 138°C (280°F) в течение 2 часов в потоке газообразного азота, 300 см3/мин.
Приготавливают смешанные растворы пропиленкарбоната и этанола, и 45 м.д. Ti в виде этоксида титана подмешивают в смешанные растворы исходных материалов. Переэтерификацию осуществляют для различных смесей исходных материалов в жидкой фазе, в восходящем потоке при 174°C (345°F) и 17,9 бар (245 фунт/кв.дюйм в датчике). Условия опыта приведены в таблице 5. Результаты настоящего эксперимента иллюстрируются на фигуре 12.
Результаты настоящего эксперимента ясно демонстрируют, что DEC (диалкилкарбонат) может быть получен посредством осуществления переэтерификации циклического карбоната, такого как пропиленкарбонат, с помощью этанола в присутствии твердого катализатора на основе алкоксида Ti, иммобилизованного на носителе из силикагеля, посредством добавления микроскопического количества растворимого соединения Ti в поток исходных материалов. Без добавления микроскопического количества Ti в поток исходных материалов, активность катализатора быстро уменьшается, как показано на фигуре 12, в течение 1397-1469 часов опыта.
Эксперимент 11
Целью настоящего эксперимента является демонстрация переэтерификации пропиленкарбоната с помощью этанола с получением DEC и пропиленгликоля в присутствии твердого катализатора. Эксперимент состоит из двух частей; Сравнительных экспериментов 11A (не по настоящему изобретению) и 11B.
Сравнительный эксперимент 11A
Переэтерификацию осуществляют в присутствии гомогенного трет-бутоксида магния. Молярное отношение этанола/пропиленкарбоната в смеси исходных материалов составляет 6,85. Концентрация гомогенного катализатора составляет 57 м.д. масс Mg. Переэтерификацию осуществляют при 168°C (335°F), 17,9 бар (245 фунт/кв.дюйм в датчике) и 0,5 мл/мин. Результат иллюстрируется на фигуре 13. Среднее преобразование пропиленкарбоната составляет примерно 24,3% моль. Средняя селективность по отношению к DEC и пропиленгликолю составляет 95,7 и 94,3% моль соответственно.
Эксперимент 11B
Переэтерификацию осуществляют в присутствии твердого катализатора. Исходный твердый катализатор представляет собой MgO, нанесенный на носитель из силикагеля. Раствор нитрата магния приготавливают посредством растворения 10,098 г Mg(NO3)2·6H2O в 22,73 г деионизованной воды для начальной пропитки. Нитрат магния осаждают на 30 мл (11,777 г) такого же носителя из силикагеля, как используется в Эксперименте 10, посредством начальной пропитки. Продукт пропитки сушат при 100°C в вакуумной печи в течение одного часа, с последующим кальцинированием при 510°C в течение 2 часов с получением MgO на носителе из силикагеля. 25 мл (10,77 г) этого катализатора со смешанным оксидом на поверхности из MgO и силикагеля загружают в реактор. Переэтерификацию пропиленкарбоната осуществляют с помощью этанола при различных условиях, приведенных в таблице 6.
Продукты реакции содержат 1-этокси-2-пропанол и дипропиленгликоль в качестве побочных продуктов. Простой диэтиловый эфир в каком-либо образце продукта не детектируется. Результат также иллюстрируются на фигуре 14. Средняя селективность по отношению к DEC и пропиленгликолю для 1-й переэтерификации составляет 95,3% моль и 94,8% моль соответственно. В целом, селективность медленно уменьшается вместе с преобразованием пропиленкарбоната. Также, селективность увеличивается вместе с молярным отношением EtOH/пропиленкарбонат. Средняя селективность по отношению к DEC и пропиленгликолю для 2-й переэтерификации составляет 94,0% моль и 92,8% моль соответственно.
Получение диалкилкарбонатов из мочевины и спирта в соответствии с вариантами осуществления, описанными в настоящем документе
В соответствии с публикациями, такими как P. Ball et al. и D. Wang et al, как упоминается выше, гетерогенные катализаторы, пригодные для получения диалкилкарбонатов из мочевины и спирта, могут содержать Al2O3, Sb2O3 и диоксид кремния. Плавленный SiO2 не является катализатором, но может стать каталитическим в присутствии PPh3. ZnO и MgO на носителе из диоксида кремния могут также использоваться для получения диалкилкарбонатов из мочевины и спирта.
Однако катализаторы на основе оксидов металла, такие как ZnO или MgO, выщелачиваются из твердых катализаторов при условиях реакции, что приводит к необратимому дезактивированию катализатора. Продолжительность цикла катализатора является очень важной при промышленном получении диалкилкарбонатов с использованием реактора в виде каталитической дистилляционной колонны, поскольку каталитическая дистилляция предусматривает быстрое удаление DMC или DEC и аммиака из жидкой среды каталитической реакции, улучшая производительность и селективность по отношению к диалкилкарбонату. В дополнение к этому, описанные выше гетерогенные катализаторы не являются такими же эффективными как гомогенный катализатор на основе диметоксида дибутилолова.
В соответствии с вариантами осуществления, описанными в настоящем документе, диалкилкарбонаты могут быть непрерывно получены, если вызывать алкоголиз мочевины с помощью спирта в двух стадиях в присутствии твердого катализатора. Обе стадии реакции представляют собой равновесные реакции. Примеры спиртов, используемых для получения диалкилкарбонатов, включают метанол, этанол, пропанол и тому подобное. На первой стадии реакции, мочевина взаимодействует со спиртом в реакционной дистилляционной колонне (предварительный реактор), служащей в качестве первой реакционной зоны, либо в отсутствие, либо в присутствии катализатора, с получением алкилкарбамата и аммиака. Катализатор не является необходимым для реакции первой стадии. Примеси в потоках исходных материалов, такие как вода и карбамат аммония, так же удаляются, как CO2 и аммиак, в первой реакционной зоне, защищая размещенные далее катализаторы. В реакции второй стадии, алкилкарбамат, полученный в первой реакционной зоне, взаимодействует со спиртом с получением диалкилкарбоната и аммиака в присутствии твердого катализатора в одной или нескольких каталитических дистилляционных колоннах (первичные реакторы), которые служат в качестве второй реакционной зоны. Двухстадийная реакция может иллюстрироваться следующим образом:
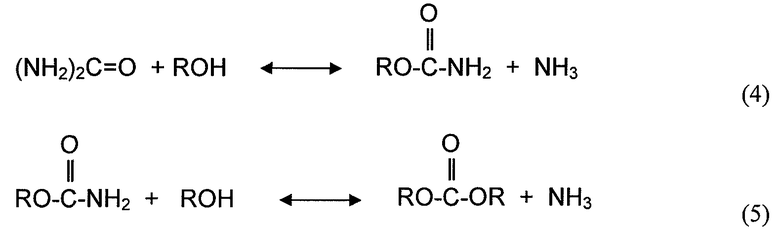
Твердый катализатор может быть получен посредством иммобилизации органического соединения олова, органического соединения сурьмы и тому подобное, на носителе, таком как диоксид кремния или углеродистый материал. Примеры соединений олова и сурьмы представляют собой алкоксиды алкилолова, алкоксиды олова, алкоксиды сурьмы и тому подобное. Другие типы твердого катализатора представляют собой оксид олова или оксид сурьмы, нанесенные на носитель. В начале реакции имеются два типа катализаторов. Первый тип катализатора представляет собой алкоксид металла, соль металла и сложного моноэфира угольной кислоты или их смесь, иммобилизованную на носителе, таком как диоксид кремния или углеродистый материал. Второй тип катализатора представляет собой оксид металла, нанесенный на носитель, такой как диоксид кремния, оксид алюминия, диоксид титана, диоксид циркония, углеродистый материал и тому подобное. Компоненты активных металлов могут представлять собой элементы, такие как Sn, Sb, Ti, Zr, Zn, Mg, Ca и тому подобное.
Опять же, малое количество растворимого соединение металла добавляют в реакционный поток, поступающий в реактор, для поддержания активности катализатора в течение большой продолжительности цикла. Делая это, продолжительность цикла катализатора может увеличиться, чтобы он был пригодным для использования в промышленных способах. Предполагается, что рабочий катализатор в стационарных условиях представляет собой алкоксид металла, алкоксиалкилкарбонат металла (соль со сложным эфиром угольной кислоты) или их олигомеры или смеси, иммобилизованные на носителе. Концентрация растворимого металлорганического соединения, такого как диалкоксид дибутилолова, в реакционной смеси значительно ниже, чем для гомогенного катализатора, используемого в патенте США № 7074951.
Высококипящий растворитель, такой как триглим, может использоваться в качестве растворителя во второй стадии, и служит в качестве сокатализатора для улучшения скорости и селективности реакции. Важно, что благодаря высокой температуре кипения растворителя реакция может осуществляться при низком давлении, которое помогает при быстром удалении DEC и аммиака из жидкой реакционной среды в паровую фазу вместе с избытком паров этанола в качестве газа носителя при разделении, что приводит к получению высокой производительности и селективности по отношению к DEC. Варианты осуществления, описанные в настоящем документе, предлагают альтернативный улучшенный способ получения диалкилкарбоната в присутствии твердого катализатора. Этот способ может представлять собой "более зеленый" способ, поскольку концентрация катализатора на основе олова в технологическом потоке значительно уменьшается. Микроскопическое количество растворимого каталитического соединения в технологическом потоке остается в системе. Какое-либо извлечение или отделение растворимого соединения катализатора в технологическом потоке не является необходимым.
Фигура 15 иллюстрирует блок-схему способа непрерывного получения DEC в соответствии с вариантами осуществления, описанными в настоящем документе. Реактор 36 в виде дистилляционной колонны с двумя разными диаметрами используют в качестве предварительного реактора для удаления примесей в потоках исходных материалов и для преобразования мочевины в этилкарбамат (EC). Раствор мочевины приготавливают в барабане 34 посредством смешивания исходных материалов мочевины 31 и потока этанола 33. Поток этанола 33 может содержать свежие исходные материалы этанола 32 и этанол из рециклированного потока 74.
Раствор мочевины 35 из барабана 34 вводится в среднюю часть верхней, более узкой секции колонны реактора в виде башни с двумя разными диаметрами 36. Реактор 36 служит в качестве предварительного реактора для удаления примесей (воды и карбамата аммония) в исходных материалах, этаноле и мочевине и для преобразования мочевины в EC. Поток паров 37 из предварительного реактора 36 состоит из аммиака, диоксида углерод и этанола. Очищенный смешанный раствор удаляют из предварительного реактора 36 как кубовый поток 38. Поток 38 вводится в первичный реактор 39 (каталитическую дистилляционную колонну) в положении выше зоны каталитической реакционной 39R, содержащей твердый катализатор.
Поток рециклированного этанола 40 вводится в реактор 39 как перегретые пары этанола в положении ниже зоны каталитической реакции 39R. Кубовый поток из каталитической дистилляционной колонны 39 рециклируется в положении выше точки введения исходных материалов линии 38 в верхнюю часть колонны 39 через линии 42, 44 и 78. Небольшой перекрестный поток 43 из контура рециклирования объединяется с кубовым потоком 65 из колонны для извлечения DEC 63 в поток 66, который вводится в реактор очистки 67 в положении выше зоны каталитической реакции, он представляет собой другую малую каталитическую дистилляционную колонну, содержащую твердый катализатор. Перекрестный поток 43 может включать этанол, аммиак, этиламин, простой диэтиловый эфир, DEC, этилкарбамат, N-этил этилкарбамат, триглим (TG), тяжелые фракции и микроскопическое количество компонента растворимого катализатора. Кубовый поток 65 из колонны для извлечения DEC 63 может содержать этилкарбамат, N-этилэтилкарбамат, TG и микроскопическое количество катализатора. Поток из верхней части колонны 68 из реактора очистки 67 может содержать аммиак, этиламин, CO2, простой диэтиловый эфир, этанол и DEC. Кубовый поток 69 из реактора очистки 67 может содержать аммиак, этиламин, CO2, простой диэтиловый эфир, этанол, N-этилэтилкарбамат, этилкарбамат, гетероциклические соединения и микроскопическое количество компонента растворимого катализатора.
Кубовый поток 69 из реактора 67 охлаждают для преципитации гетероциклических соединений в системе охлаждения/фильтра 70. Преципитированный твердый побочный продукт удаляют из системы 70 через линию 71. Жидкий поток 72 из системы 70 разделяют на два потока 77 и 78 для рециклирования в реактор очистки 67 и первичный реактор 39 соответственно.
Поток из верхней части колонны 41 из первичного реактора 39 может объединяться с потоком из верхней части колонны 68 из реактора очистки 67 в виде потока 42. Поток из верхней части колонны 41 из первичного реактора 39 может содержать аммиак, CO2, этиламин, простой диэтиловый эфир, этанол, этилкарбамат, N-этил этилкарбамат, DEC, TG и микроскопическое количество катализатора. Объединенный поток 42 вводится в дистилляционную колонну 43, где разделяются легкие фракции и более тяжелые соединения. Поток из верхней части колонны 44 из дистилляционной колонны 43, который может содержать аммиак, CO2, этиламин, простой диэтиловый эфир и этанол, объединяют с потоком из верхней части колонны 37 из предварительного реактора 36 в виде потока 45 для введения в дистилляционную колонну 46.
Поток из верхней части колонны 47 из дистилляционной колонны 46 охлаждают, чтобы вызывать реакцию CO2 с аммиаком с образованием карбамата аммония. Карбамат аммония преципитирует в жидком аммиаке и удаляется как твердые продукты через линию 49 из системы охлаждения/фильтра 48. Поток жидкого аммиака 50 из системы охлаждения/фильтра 48 направляется в танк-хранилище для аммиака.
Кубовый поток 51 из колонны 46 может содержать этиламин, простой диэтиловый эфир, этанол и микроскопическое количество DEC. Поток 51 вводится в колонну для извлечения этиламина 52. Поток этиламина из верхней части колонны 53 направляется в танк-хранилище. Кубовый поток 54 из колонны 52 объединяется с кубовым потоком 55 из дистилляционной колонны 43 в виде потока 56. Поток 56 вводится в колонну для извлечения простого эфира 57. Простой эфир удаляют из дистилляционной колонны 57 как поток из верхней части колонны 58, который направляется в танк-хранилище для простого эфира. Кубовый поток 59 из дистилляционной колонны 57 вводится в дистилляционную колонну 60 (колонну для извлечения этанола).
Извлеченный этанол как поток из верхней части колонны 61 рециклируется в первичный реактор 39, реактор для очистки 67 и предварительный реактор 36 (или барабан 34). Рециклированный поток этанола 74 представляет собой малую часть потока из верхней части колонны 61 из дистилляционной колонны 60 (колонны для извлечения этанола). Поток 61 разделяют на три потока, 40, 73 и 74. Поток 73 рециклируют в реактор очистки 67. Поток 74 рециклируют в барабан 34 для получения раствора мочевины. Поток 40, который может представлять собой главную часть потока 61, рециклируется в первичный реактор 39. Кубовый поток 62 из колонны для извлечения этанола 60 вводится в колонну для извлечения DEC 63. Продукт DEC извлекают как поток из верхней части колонны 64 из дистилляционной колонны 63 и направляют в танк-хранилище для DEC. Кубовый поток 65 из колонны 63 может содержать этилкарбамат, N-этилэтилкарбамат, TG и микроскопическое количество компонента растворимого катализатора. Этот поток 65 направляется в реактор очистки 67 через линию 66.
DMC может быть получен из метанола и мочевины способом, сходным со способом получения DEC, как описано по отношению к фигуре 15. Однако понятно, что конечный продукт DMC извлекают из технологического потока, имеющего азеотропную смесь метанол - DMC. Извлечение DMC посредством разрушения азеотропности смеси метанол - DMC с помощью технологии экстракционной дистилляции в растворителе хорошо документировано, как описано в патенте США № 7074951.
Эксперимент 12
Целью настоящего эксперимента является демонстрация реакции этилкарбамата с этанолом с получением DEC и аммиака в присутствии твердого катализатора. Твердый катализатор приготавливают посредством иммобилизации диметоксида дибутилолова на силикагеле с помощью технологии in situ.
25 мл (14,79 г) носителя из силикагеля сферической формы, используемого в Эксперименте 7A, загружают в реактор. Раствор диметоксида дибутилолова приготавливают посредством смешивания 87 г диметоксида дибутилолова в 2 литрах сухого толуола. Реактор заполняют восходящим потоком этого раствора при температуре и давлении окружающей среды. Реактор медленно нагревают до 110°C (230°F), в то же время протекает восходящий поток этого раствора, 2 мл/мин. При 110°C (230°F), реактор помещают под давление 3,4 бар (35 фунт/кв.дюйм в датчике), а затем продолжают нагревать до 135°C (275°F). При 135°C (275°F) и 3,4 бар (35 фунт/кв.дюйм в датчике), раствор диметоксида дибутилолова пропускают через реактор в восходящем потоке, 0,5 мл/мин, в течение 6 часов. После охлаждения, избыток раствора в реакторе удаляют, а затем катализатор промывают восходящим потоком сухого толуола, 4 мл/мин, в течение 1,5 часов. Промытый катализатор сушат при 104°C (220°F) при давлении окружающей среды в нисходящем потоке N2, 300 см3/мин, в течение 2 часов.
Реакцию осуществляют посредством прохождения раствора 13,2 мас.% этилкарбамата, 31,36 мас.% триглима и 55,44 мас.% этанола с помощью восходящего потока газообразного азота через слой твердого катализатора в реакторе с кипящим слоем. Можно также осуществлять реакцию в реакторе с нисходящим потоком. Микроскопические количества диметоксида дибутилолова подмешивают к этому раствору. Условия реакции приведены в таблице 7, и результаты этого исследования иллюстрируются на фигуре 16. Анализы продуктов реакции указывают на микроскопические количества N-этил этилкарбамата и простого диэтилового эфира. Селективность по отношению к DEC по отношению к этилкарбамату находится в пределах от 98,5% моль до 99,9% моль с общей тенденцией уменьшения селективности, когда увеличивается преобразование этилкарбамата.
°C (°F)
Этот эксперимент успешно демонстрирует, что DEC может быть получен из мочевины и этанола. На первой стадии, этилкарбамат получают посредством взаимодействия мочевины с этанолом в отсутствие катализатора (смотри патент США № 7074951). На второй стадии, DEC получают посредством осуществления реакции этилкарбамата с этанолом в присутствии твердого катализатора и с добавлением микроскопического количества растворимого металлорганического соединения в потоки исходных материалов для компенсации потерь металла из-за выщелачивания. Промышленное получение DEC во второй стадии предпочтительно осуществляют в одной или нескольких каталитических дистилляционных колоннах.
Получение биодизельного топлива в соответствии с вариантами осуществления, описанными в настоящем документе
Биодизельное топлив получают посредством осуществления переэтерификации растительных масел и животных жиров с помощью метанола в присутствии гомогенного катализатора и твердого катализатора. Исходные материалы для получения биодизельного топлива представляют собой растительные масла и животные жиры, которые представляют собой сложные эфиры высших жирных кислот. Термин жир (растительное или животное масло, если он жидкий) обычно ограничен сложными эфирами (глицеридами) жирных кислот и глицерина, а термин воск - сложными эфирами других спиртов. Основной химический механизм, вовлеченный в получение биодизельного топлива, представляет собой каталитическую реакцию обмена природных сложных эфиров (главным образом, глицеридов) и первичного спирта (как правило, метанола или этанола). Спиртовой раствор основания (обычно NaOH, KOH, метоксида калия или метоксида натрия) может использоваться в качестве катализатора. Следовательно, биодизельное топливо представляет собой смесь сложных метиловых или этиловых эфиров различных насыщенных и ненасыщенных жирных кислот. Побочный продукт представляет собой глицерин, который составляет от 16 до 25 мас.%. Биодизельное топливо может также содержать некоторые жирные кислоты (продукты гидролиза сложных эфиров) в малых количествах, в зависимости от количества воды в используемых исходных материалах или катализаторе.
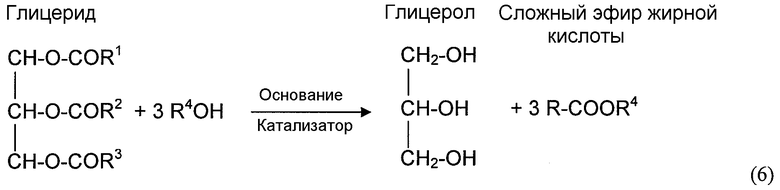
где R4OH = метанол или этанол; R = R1, R2 или R3,
Алкильные группы R1, R2 и R3 природного продукта глицерида, в целом, различаются по длине цепи и степени ненасыщенности. Алкильные группы обычно имеют прямую цепь и имеют четное количество атомов углерода от 4 до 26. Исключение представляет собой разветвленная изовалериановая кислота (CH3)2CHCH2COOH, которая встречается в относительно больших количествах у дельфинов. Некоторые ненасыщенные жирные кислоты имеют две или три двойных связи в алкильных цепях. Ненасыщенные жирные кислоты имеют более низкие температуры плавления, чем их насыщенные аналоги. Длина цепи ненасыщенной жирной кислоты, как правило, находится в пределах C10-C24. Масло канолы имеет более высокую степень ненасыщености при длине цепи C16-C20, чем кукурузное масло.
В целом, основные катализаторы являются более эффективными при переэтерификации сложных эфиров карбоновых кислот с помощью спирта, чем кислотные катализаторы. Гетерогенные катализаторы, описанные в литературе (смотри Уровень техники) также представляют собой основные катализаторы. К сожалению, активные каталитические компоненты выщелачиваются из твердого катализатора при условиях реакции, приводя к дезактивированию катализатора. Катализаторы на основе алюмината цинка представляют собой не очень активный катализатор и требуют более высоких температур реакций и более низких скоростей введения исходных материалов, чем более основные катализаторы, такие как MgO или CaO. Но последние выщелачиваются из твердого катализатора еще быстрее, чем алюминаты цинка.
Переэтерификация растительных масел или животных жиров может осуществляться с помощью метанола или этанола в присутствии твердого катализатора в реакторе с кипящим слоем, импульсном проточном реакторе, или в каталитической дистилляционной колонне с помощью введения микроскопического количества растворимого каталитического компонента в смеси исходных материалов при одностадийной или двухстадийной реакции. Исходные катализаторы могут включать оксиды металлов, такие как оксид магния, оксид кальция, оксид цинка, оксид натрия, оксид калия, оксид лантана и тому подобное, нанесенные на носитель, такой как диоксид кремния, оксид алюминия, углерод и/или углеродистый материал. Углерод и углеродистые носители предпочтительно будут иметь поверхностные функциональные группы, такие как гидроксил или карбонил, или как то, так и другое, для иммобилизации металлорганических соединений на поверхности носителя.
Для получения оксидов, гидроксидов или оксигидроксидов металлов на носителях, поверхностные функциональные группы могут быть не нужны. Углеродистые носители могут быть получены посредством контролируемого термического дегидратирования углеводов, таких как древесина, скорлупа коксового ореха, крахмал, целлюлоза, смесь крахмала и целлюлозы, сахар, метилцеллюлоза и тому подобное, при повышенных температурах. Углеродистые носители могут либо наноситься на носители, либо не наноситься. Для получения углеродистого материала на носителе, углеводы наносят на соответствующий пористый носитель с последующим контролируемым термическим дегидратированием при повышенной температуре от 300°C до 1000°C в инертной атмосфере или в атмосфере, состоящей из инертного газа, малого количества кислорода или водяного пара, или как того, так и другого. Носитель для углеродистых материалов может представлять собой любые неорганические материалы, такие как оксид алюминия, диоксид титана, диоксид кремния, диоксид циркония, глины, диоксид кремния - оксид алюминия и тому подобное.
В двухстадийном способе, преобразование триглицерида в первом реакторе может быть выше примерно чем 90%. Оставшийся непреобразованный триглицерид, диглицерид и моноглицерид в потоке продукта реакции из первого реактора переэтерификации может преобразовываться до завершения во втором реакторе переэтерификации. Поскольку переэтерификация представляет собой двухфазную реакцию, осуществление переэтерификации в реакторе с кипящим слоем или в импульсном проточном реакторе будет облегчать перенос больших молекул триглицеридов, сложных метиловых эфиров и липкого глицерина через поры катализатора туда и обратно между объемной жидкой средой и внутренним пространством гранул катализатора, где происходит большая часть каталитических реакций, что приводит к высокой производительности. Поскольку катализаторы, описанные в настоящем документе, имеют высокую активность, переэтерификация может осуществляться при более низкой температуре и давлении, что означает более низкую стоимость строительства и стоимость использования.
Добавление растворимого каталитического компонента в поток исходных материалов в реакторы составляет от примерно 0,5 м.д. масс до примерно 500 м.д. масс, в некоторых вариантах осуществления; от примерно 5 м.д. масс до примерно 250 м.д. масс, в других вариантах осуществления; и от 10 м.д. масс до 50 м.д. масс, в других вариантах осуществления. Примеры растворимых каталитических соединений включают 2-метоксиэтоксид цинка, 2-метоксиэтоксид кальция, 2-метоксипропоксид цинка, этоксид цинка, алкоксиалкилкарбонат цинка, 2-метоксипроксид кальция, этоксид кальция, метоксид кальция, алкоксиалкилкарбонат кальция, 2-метоксиэтоксид магния, 2-метоксипроксид магния, этоксид магния, метоксид магния, бутоксид магния, алкоксиалкилкарбонат магния, алкоксид лантана, алкоксиалкилкарбонат лантана, соли цинка и карбоновой кислоты, соли магния и карбоновой кислоты, соли кальция и карбоновой кислоты и глицериды Mg, Ca, и Zn, среди прочего. Их смеси также могут использоваться. Растворимые соединения Ca, Mg, Zn и La могут быть получены посредством взаимодействия оксида или гидроксида этих металлов с органическим карбонатом или смесью органического карбоната и спирта или карбоновых кислот или смеси органической карбоновой кислоты и спирта, такого как метанол, 2-метоксиэтанол и тому подобное, при температуре от 93°C до 260°C (200°F - 500°F), предпочтительно, от 121°C до 232°C (250°F - 450°F) в жидкой фазе или в присутствии жидкости и паров. Необязательно, можно выбрать извлечение компонентов металлов для рециклирования. Такие приготовленные растворы являются пригодными для добавления микроскопического количества этого металла в поток исходных материалов в реактор для получения большого времени цикла катализатора. Общее количество активного металла или компонентов металла на твердом катализаторе на основе алкоксида металла, гидроксида металла или оксида металла составляет от примерно 0,05 мас.% до примерно 20 мас.%, в некоторых вариантах осуществления, и от примерно 0,07 мас.% до примерно 12 мас.%, в других вариантах осуществления.
Необязательно, все ди- или моноглицериды, или их часть, могут преобразовываться в органические карбонаты или органические карбаматы, или как те, так и другие, посредством реакции с DMC, метил 2-этил-1-гексилкарбонатом, метилкарбаматом, 2-этил-1-гексилкарбаматов, мочевиной или их смесью в дополнение к переэтерификации с помощью метанола во втором реакторе или, необязательно, в третьем реакторе. Полученные органические карбонаты и карбаматы могут служить в качестве агента присадки к биодизельному топливу для уменьшения количества уходящих частиц, выбросов NOx или для улучшения цетанового числа дизельного топлива.
Поскольку природные растительные масла могут содержать различные малые количества свободных жирных кислот, свободные жирные кислоты должны удаляться посредством предварительной обработки перед осуществлением переэтерификации с помощью спирта в присутствии твердого основного катализатора. Пример таких способов предварительной обработки представляет собой этерификацию свободных жирных кислот с помощью метанола в присутствии кислотного катализатора. Один из таких кислотных катализаторов представляет собой сульфоновую кислоту, иммобилизованную на углеродистом носителе. Носитель может включать те носители, которые получают посредством контролируемого термического дегидратирования скорлупы кокосового ореха или углеводов, нанесенных или осажденных на пористый носитель. Осуществление этерификации свободных жирных кислот с помощью спирта в присутствии твердого кислотного катализатора в реакторе каталитической дистилляции имеет преимущества, которые представляют собой непрерывное удаление воды из реакционной зоны как потока из верхней части колонны, доведение этерификации до завершения и устранение отдельной стадии сушки продукта этерификации перед осуществлением переэтерификации триглицерида с помощью спирта. Другое важное преимущество представляет собой уменьшение времен этерификации.
Все реакции переэтерификаци в следующих далее примерах осуществляют в реакторах с нисходящим потоком. Размеры реактора с неподвижным слоем составляют диаметр 1,3 см (1/2 дюйма) и длину 53,3 см (21 дюймов). Реактор имеет контролируемые по-отдельности верхнюю и нижнюю зоны нагрева. Поток исходных материалов метанола и поток растительного масла (6 мас.% метанола в растительном масле) по-отдельности закачиваются в верхнюю секцию реактора, откуда два потока стекают вниз в зону каталитической реакции. Микроскопические количества растворимого каталитического компонента подмешиваются в поток метанола или поток, уже содержащий частично преобразованный продукт. Объем твердого катализатора составляет 15 мл.
Эксперимент 13
Целью настоящего эксперимента является демонстрация переэтерификации масла канолы с помощью метанола в присутствии твердого катализатора в реакторе с кипящим слоем с нисходящим потоком или в реакторе каталитической дистилляции. Твердый катализатор представляет собой MgO на носителе из силикагеля.
Раствор нитрата магния приготавливают посредством растворения 10,96 г Mg(NO3)2·6H2O в 24 г деионизованной воды. 30 мл (11,91 г) носителя в виде сфер силикагеля (диаметр 1,7-4 мм; примерно 6 гидроксильных групп на нм2, удельная площадь поверхности согласно БЭТ 314 м2/г, объем пор 1,055 см3/г и средний диаметр пор 13,46 нм), их пропитывают указанным выше раствором нитрата магния с помощью технологии начального смачивания. Носитель из сфер силикагеля получают с помощью технологии капания в масло. После сушки продукта пропитки при 100°C в течение 1 часа, его затем кальцинируют при 510°C в течение 2 часов.
15 мл (6,30 г) катализатора на основе MgO/SiO2 загружают в реактор. Исходные материалы масла канолы (купленное в местном магазине розничной торговли) получают посредством смешивания метанола (5,39 мас.%) с маслом канолы (94,61 мас.%). Кислотное число свободных жирных кислот этих исходных материалов составляет 0,48 мг KOH/г. Переэтерификацию масла канолы с помощью метанола осуществляют при 165°C (330°F) и 19,4 бар (267 фунт/кв.дюйм в датчике) посредством введения исходных материалов масла канолы и метанола при скорости потока 0,2 мл/мин, каждого. Этоксид магния растворяют в исходных материалах метанола для получения 28 м.д. масс Mg в зоне каталитической реакции.
Потоки эффлюента состоят из двух прозрачных слоев. Верхние слои содержат продукты сложных метиловых эфиров и малые количества непреобразованных триглицеридов. Среднее содержание непреобразованных триглицеридов в продуктах реакции, исключая метанол из верхних слоев, составляет примерно 1,2 мас.%. Нижний слой содержит большую часть непреобразованных триглицеридов. Результат иллюстрируется на фигуре 17, который показывает стабильные рабочие характеристики катализатора.
Эксперимент 14
Целью настоящего эксперимента является демонстрация преобразования оставшихся непреобразованных или частично преобразованных материалов в потоке эффлюента (два слоя при стоянии) из первого реактора, во втором реакторе с кипящим слоем с нисходящим потоком или в реакторе каталитической дистилляции, или необязательное рециклирование в переднюю часть отдельного реактора переэтерификации.
Раствор нитрата магния приготавливают посредством растворения 9,04 г Mg(NO3)2·6H2O в 19,1 г деионизованной воды. 22 мл (9,14 г) носителя в виде сфер из силикагеля (9-14 меш, удельная площадь поверхности согласно БЭТ 309 м2/г и 1,03 объем пор см3/г), их пропитывают указанным выше раствором нитрата магния посредством технологии начального смачивания. После сушки продукта пропитки при 150°C в течение 1 часа, его кальцинируют при 510°C в течение 2 часов. Готовый катализатор содержит 4,5 мас.% Mg.
15 мл (7,2 г) катализатора на основе MgO/SiO2 загружают в такой же реактор, как используется в Эксперименте 13. Два слоя смешанного продукта от первой реакции переэтерификации масла канолы с метанолом отделяются от смешанного продукта посредством использования разделительной воронки для использования в качестве исходных материалов для второй реакции переэтерификации. Композиция исходных материалов на основе кубового смешанного продукта составляет 25,4 мас.% триглицеридов, 8,5 мас.% диглицеридов, 3,1 мас.% моноглицеридов, 0,1 мас.% глицерин, 47,1 мас.% сложных метиловых эфиров и 15,8 мас.% метанола. Исходные материалы содержат примерно 8,5 м.д. масс растворимых частиц Mg и имеют кислотное число 0,32 мг KOH/г свободных жирных кислот. Переэтерификацию осуществляют при 160°C (320°F) и 19,5 бар (268 фунт/кв.дюйм в датчике) посредством прокачки исходных материалов 0,12 мл/мин и метанола 0,10 мл/мин в реакторе с кипящим слоем с нисходящим потоком. Дополнительного алкоксида Mg не добавляют ни к одному из двух потоков исходных материалов. Поток эффлюента из реактора представляет собой прозрачный ярко-желтый раствор (один слой).
Композиция исходных материалов на основе смешанного продукта из верхней части представляет собой 1,12 мас.% триглицеридов, 0,57 мас.% диглицеридов, 3,78 мас.% моноглицеридов, 7,47 мас.% сложных метиловых эфиров, 0,03 мас.% глицерина, 87,03 мас.% метанола. Кислотное число этих исходных материалов составляет 0,51 мг KOH/г свободных жирных кислот. Переэтерификацию осуществляют над таким же катализатором и при такой же температуре и давлении при скорости потока исходных материалов 0,2 мл/мин. Дополнительного метанола в реактор не закачивают. Эти два конечных продукта переэтерификации из исходных материалов на основе смешанного кубового продукта и смешанного продукта из верхней части объединяют для отгонки избытка метанола и для извлечения сырого биодизельного топлива. Извлеченное сырое биодизельное топливо содержит 0,36 мас.% непреобразованных триглицеридов и имеет кислотное число 0,74 мг KOH/г свободных жирных кислот.
Рассмотренный выше экспериментальный результат успешно демонстрирует, что биодизельное топливо может быть получено посредством осуществления переэтерификации растительного масла с помощью спирта, такого как метанол, в присутствии твердого катализатора.
Как описано выше, варианты осуществления, описанные в настоящем документе, обеспечивают увеличение времен циклов катализаторов для различных твердых катализаторов посредством введения микроскопического количества растворимого металлорганического соединения вместе с исходными материалами. Другие варианты осуществления, описанные в настоящем документе, могут включать способ непрерывного получения органического карбоната или органического карбамата при стабильной скорости; технологии получения катализатора in situ для иммобилизованных твердых катализаторов, технологии для поддержания стабильной активности катализатора в течение длительных времен цикла катализатора и времен службы, с тем, чтобы они были пригодны для промышленных реакторов с неподвижным слоем; и способ реактивирования дезактивированных твердых катализаторов in situ.
Преимущественно, варианты осуществления, описанные в настоящем документе, могут обеспечить катализаторы переэтерификации, имеющие увеличенную продолжительность цикла, понижая тем самым рабочие затраты, связанные с частыми выключениями и заменами катализатора. В дополнение к этому, благодаря микроскопическому количеству используемого растворимого металлорганического соединения, удаление гомогенного катализатора из различных потоков продуктов может быть существенно уменьшено.
Хотя описание включает ограниченное количество вариантов осуществления, специалисты в данной области, имея преимущество настоящего описания, заметят, что могут быть разработаны и другие варианты осуществления, которые не выходят за рамки настоящего описания. Соответственно, рамки должны ограничиваться только прилагаемой формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ДИФЕНИЛКАРБОНАТА | 2010 |
|
RU2528048C2 |
| СПОСОБ ПОЛУЧЕНИЯ ДИАРИЛКАРБОНАТА | 2008 |
|
RU2474571C2 |
| КАТАЛИТИЧЕСКАЯ КОНВЕРСИЯ ОРГАНИЧЕСКОГО КАРБОНАТА | 2003 |
|
RU2320633C2 |
| СПОСОБ УДАЛЕНИЯ ПРИМЕСИ АЛКАНОЛА ИЗ ПОТОКА ОРГАНИЧЕСКОГО КАРБОНАТА | 2009 |
|
RU2507192C2 |
| СПОСОБ ПОЛУЧЕНИЯ АРОМАТИЧЕСКОГО КАРБОНАТА | 1991 |
|
RU2041869C1 |
| СПОСОБ ПОЛУЧЕНИЯ ДИАРИЛКАРБОНАТОВ ИЛИ АЛКИЛАРИЛКАРБОНАТОВ ИЗ ДИАЛКИЛКАРБОНАТОВ | 2008 |
|
RU2490251C9 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОЦИАНАТОВ С ИСПОЛЬЗОВАНИЕМ ДИАРИЛКАРБОНАТА | 2008 |
|
RU2523201C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПО МЕНЬШЕЙ МЕРЕ ОДНОГО ДИАРИЛКАРБОНАТА ПО МЕНЬШЕЙ МЕРЕ ИЗ ОДНОГО ДИАЛКИЛКАРБОНАТА И ПО МЕНЬШЕЙ МЕРЕ ИЗ ОДНОГО АРОМАТИЧЕСКОГО ГИДРОКСИСОЕДИНЕНИЯ | 2009 |
|
RU2515993C2 |
| СПОСОБ И УСТРОЙСТВО ДЛЯ ОЧИСТКИ ДИАЛКИЛКАРБОНАТОВ | 2010 |
|
RU2564035C2 |
| СПОСОБ ПОЛУЧЕНИЯ АРОМАТИЧЕСКОГО КАРБОНАТА | 2005 |
|
RU2372322C2 |
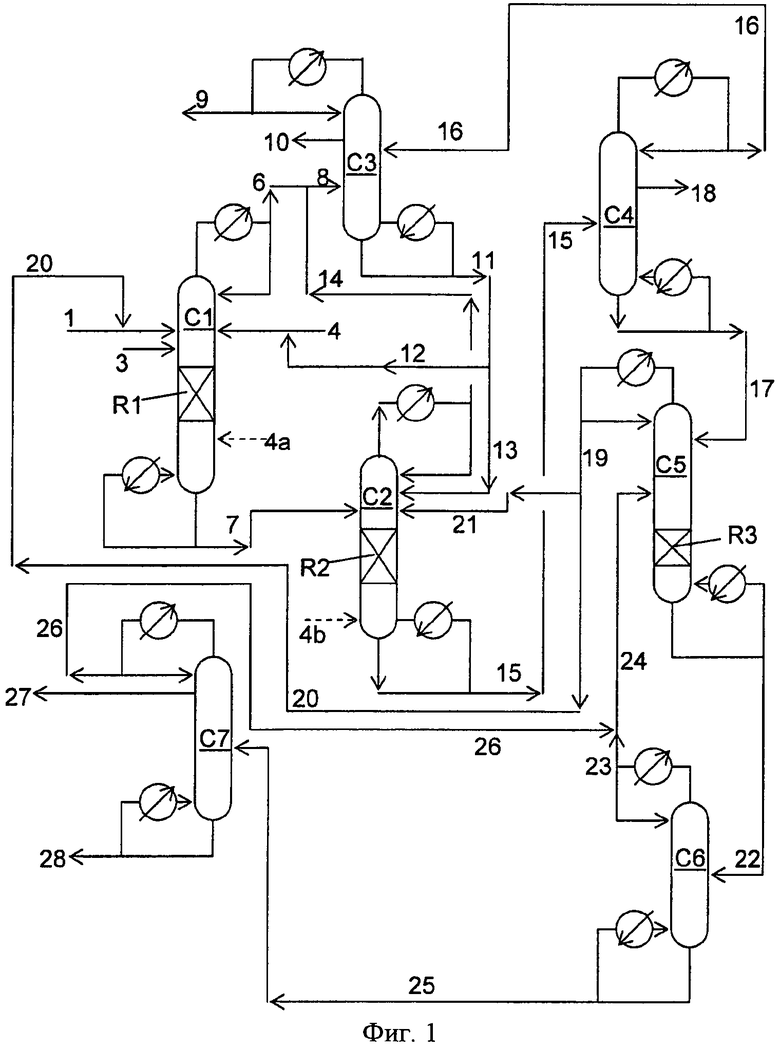
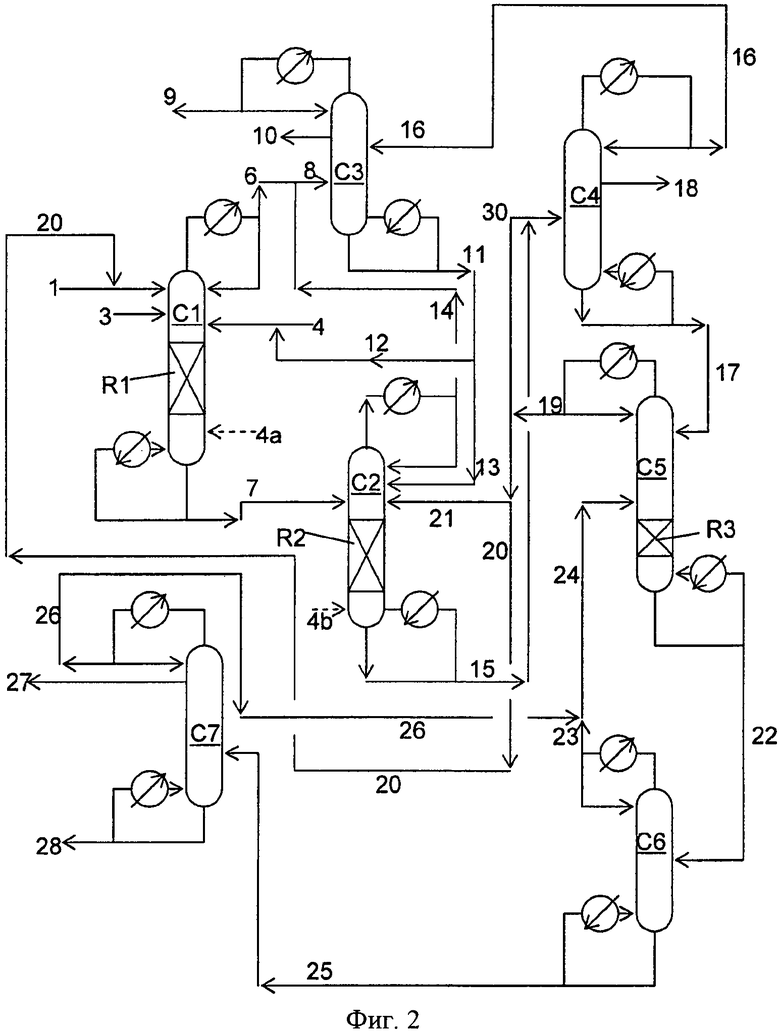
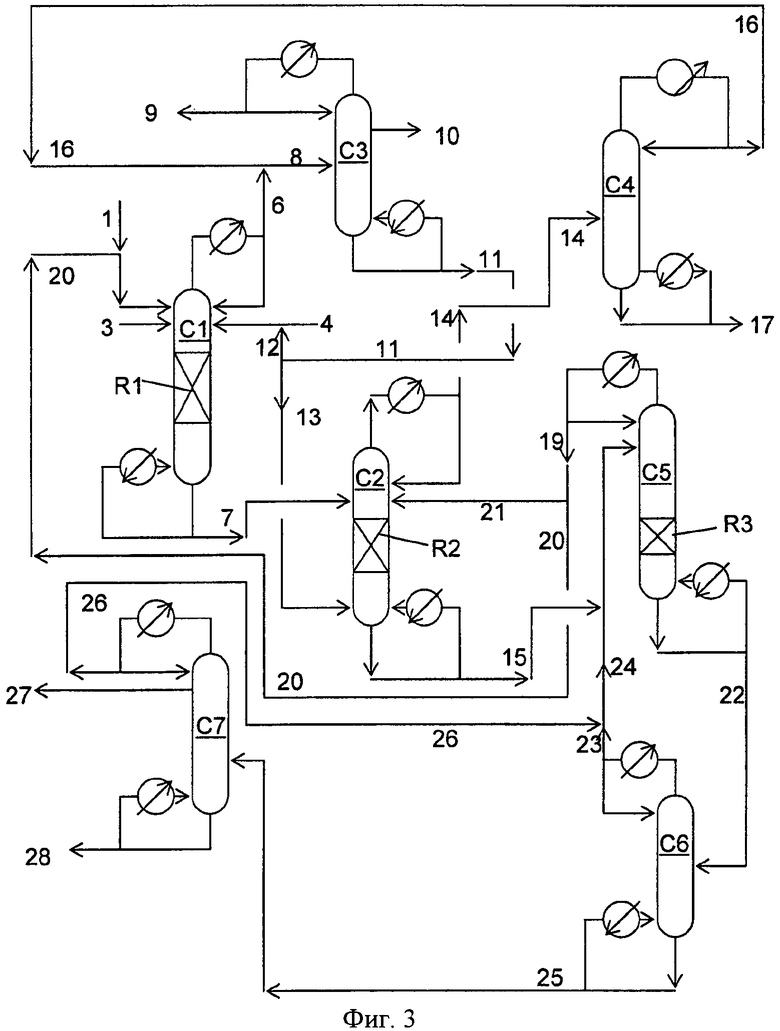
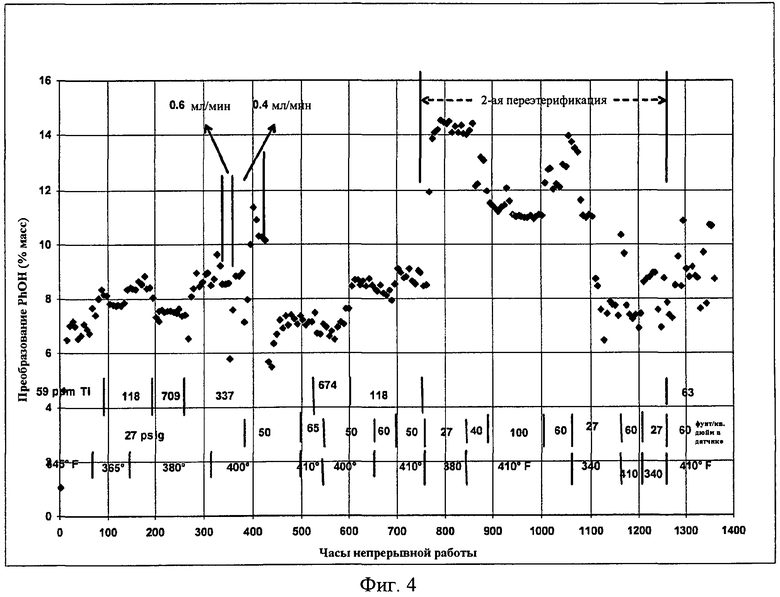
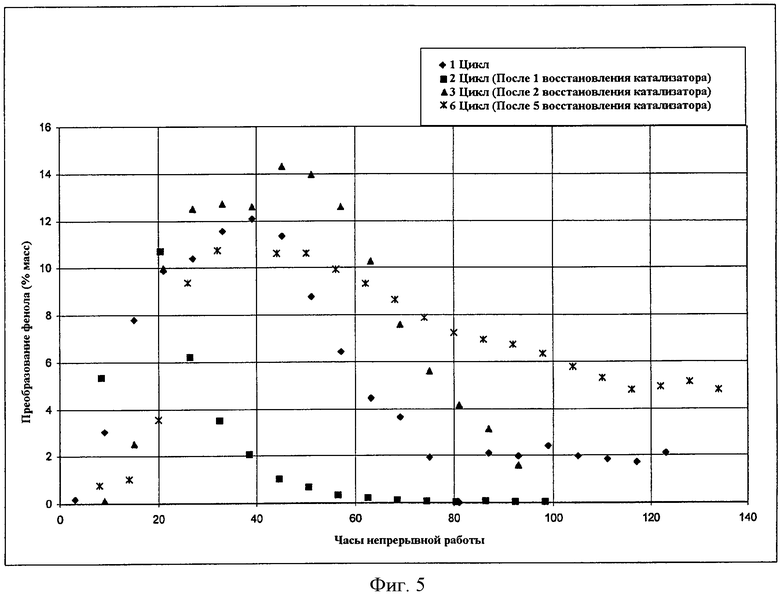
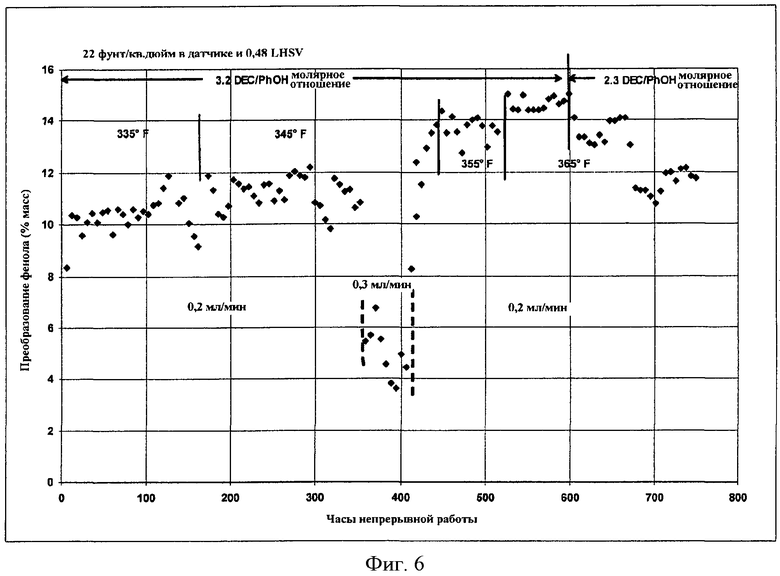
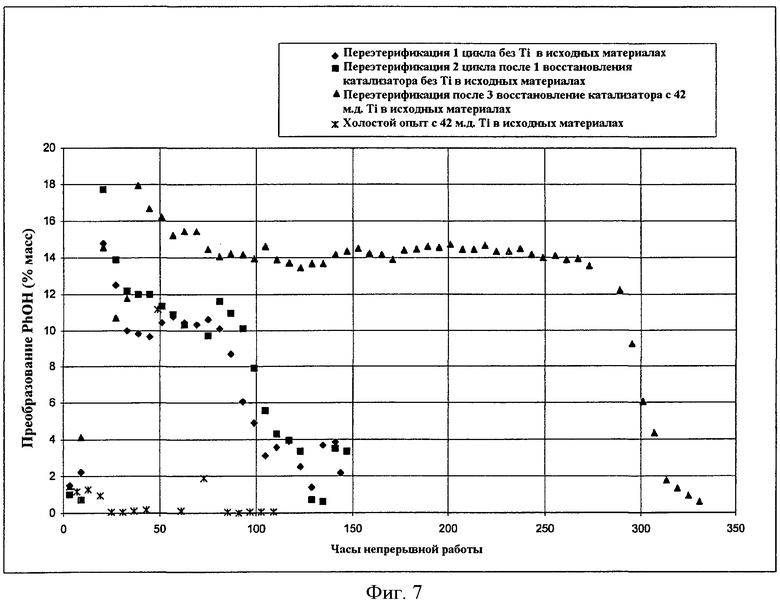
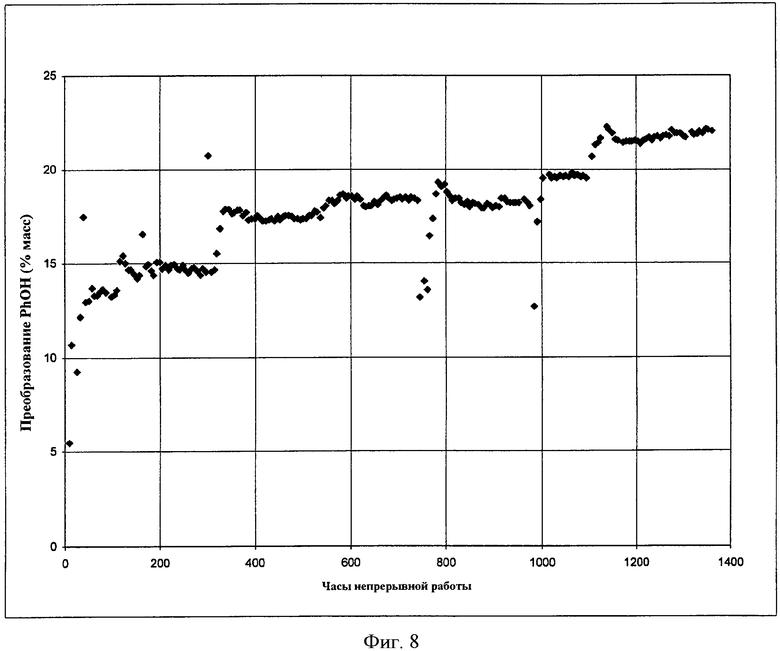
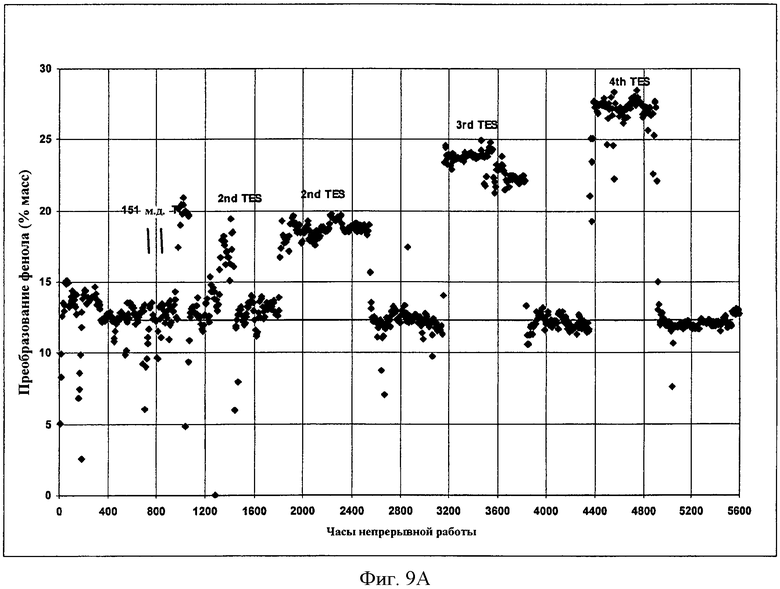
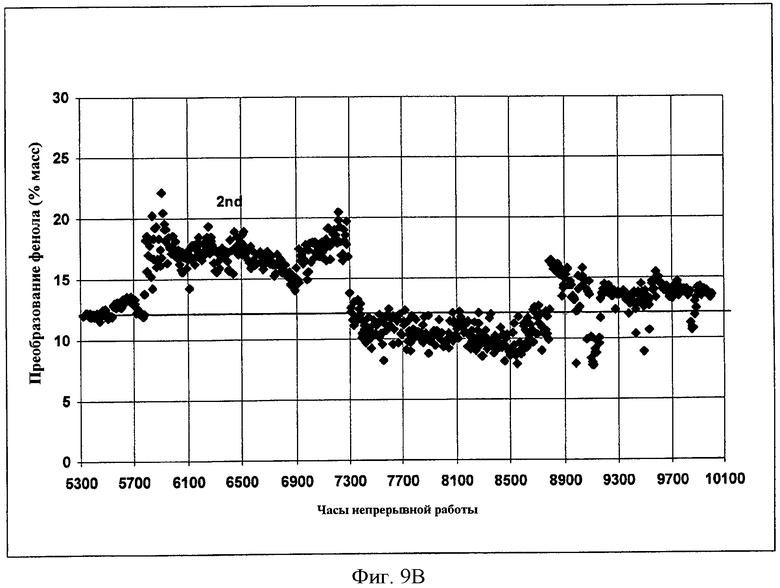
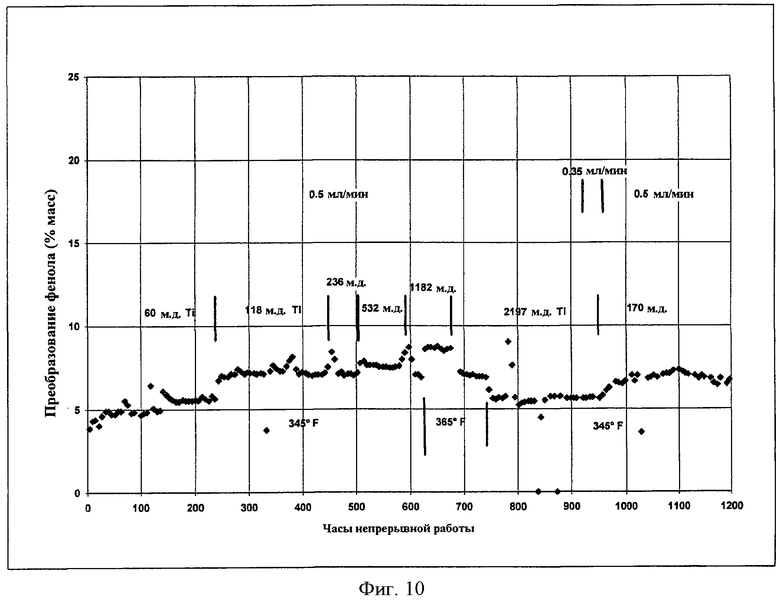
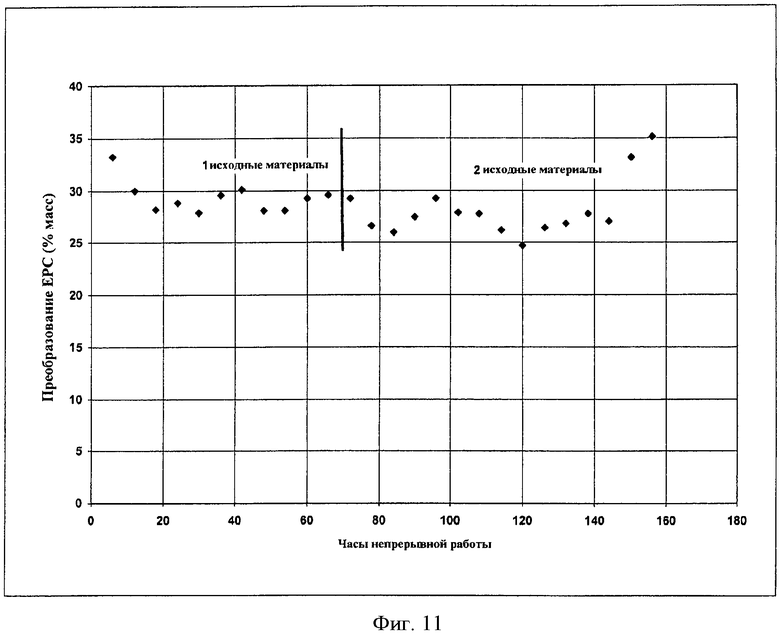
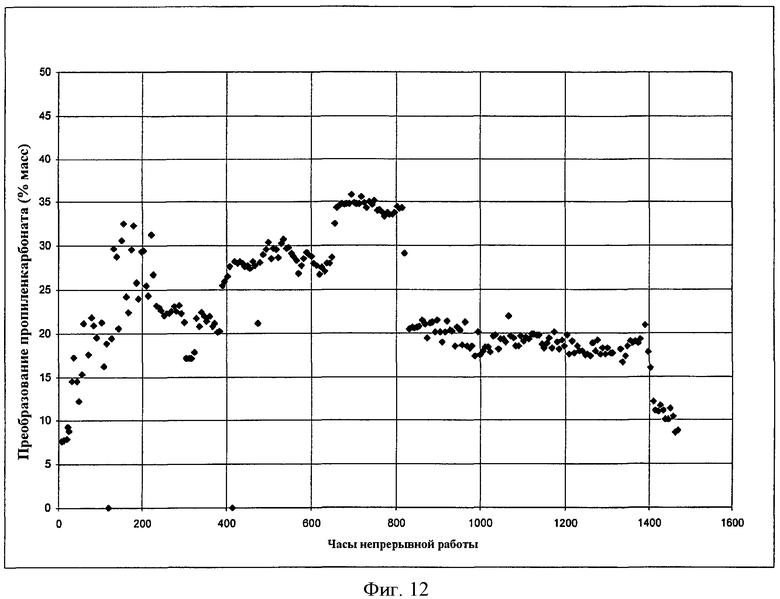
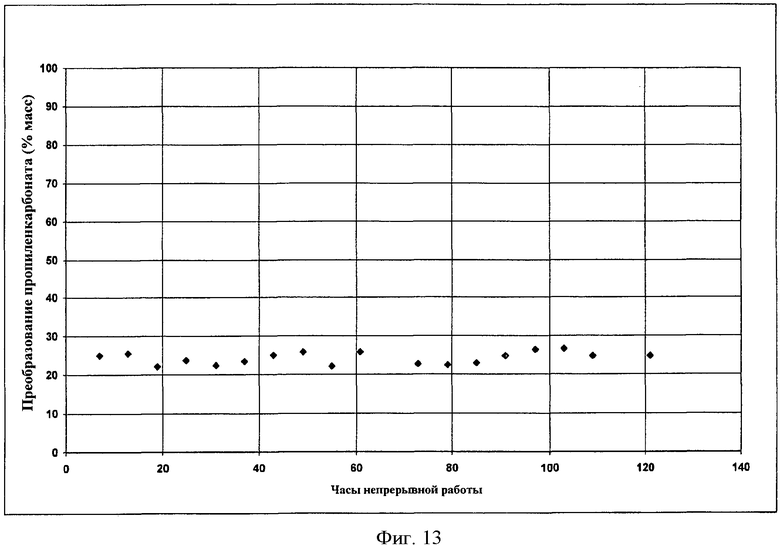
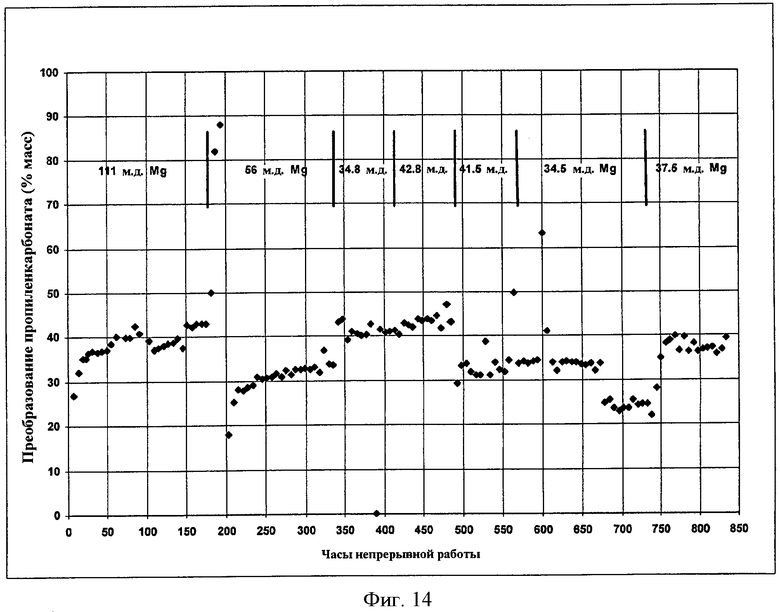
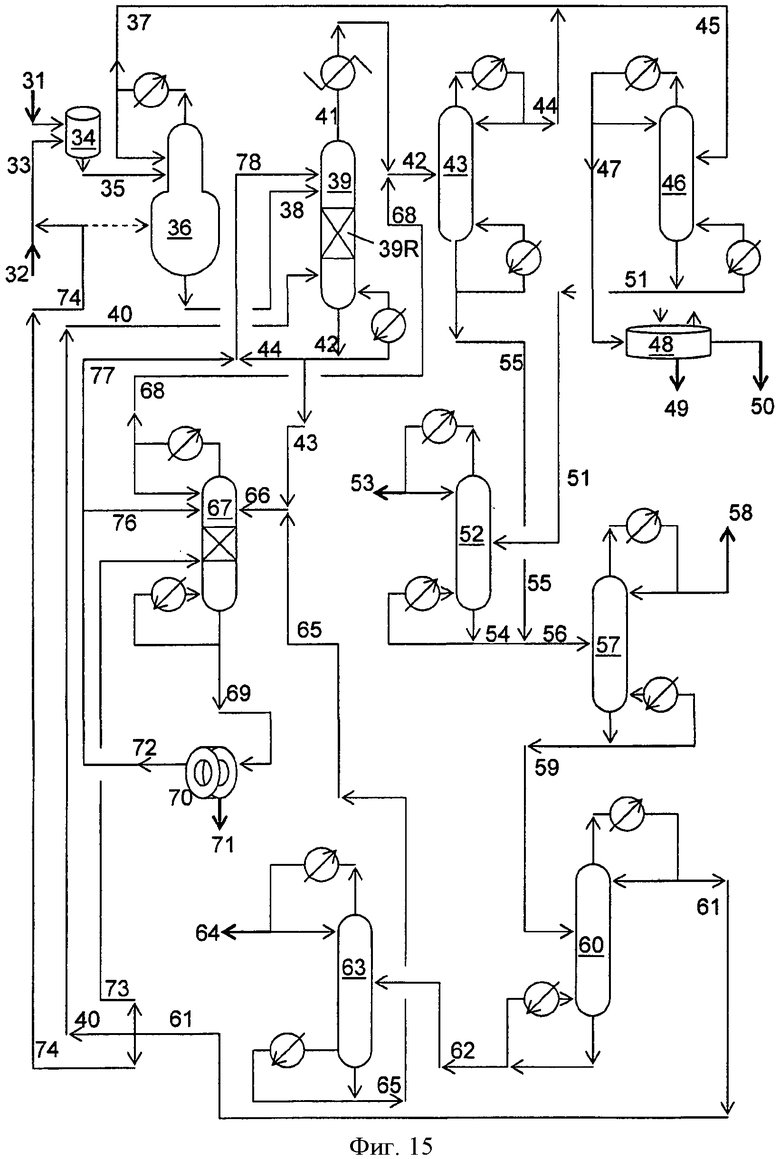
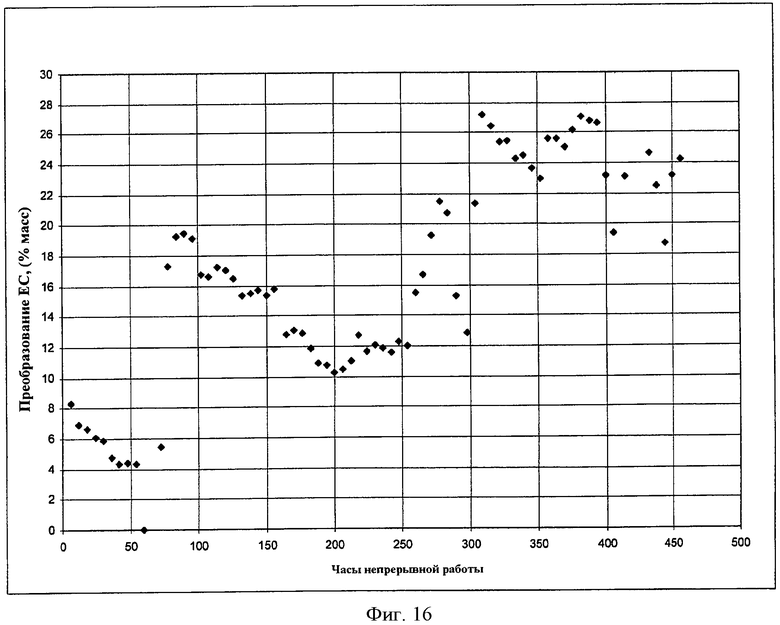
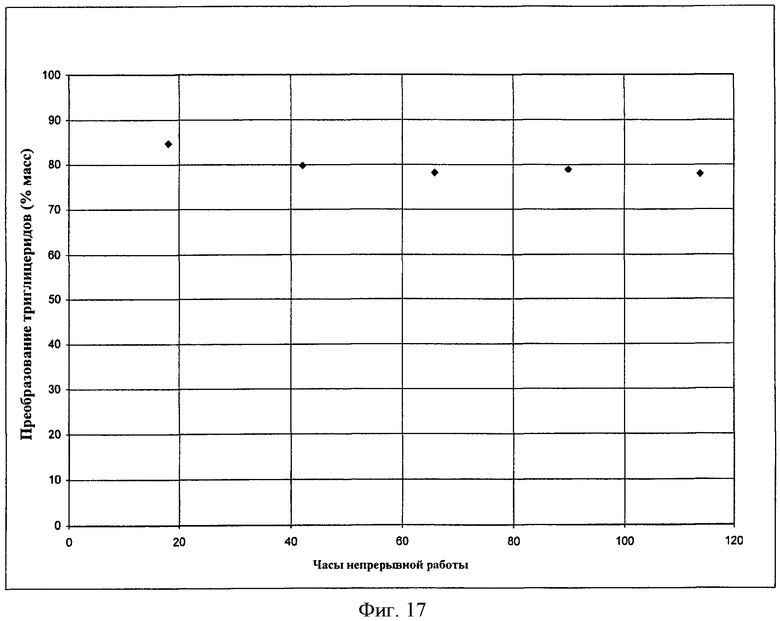
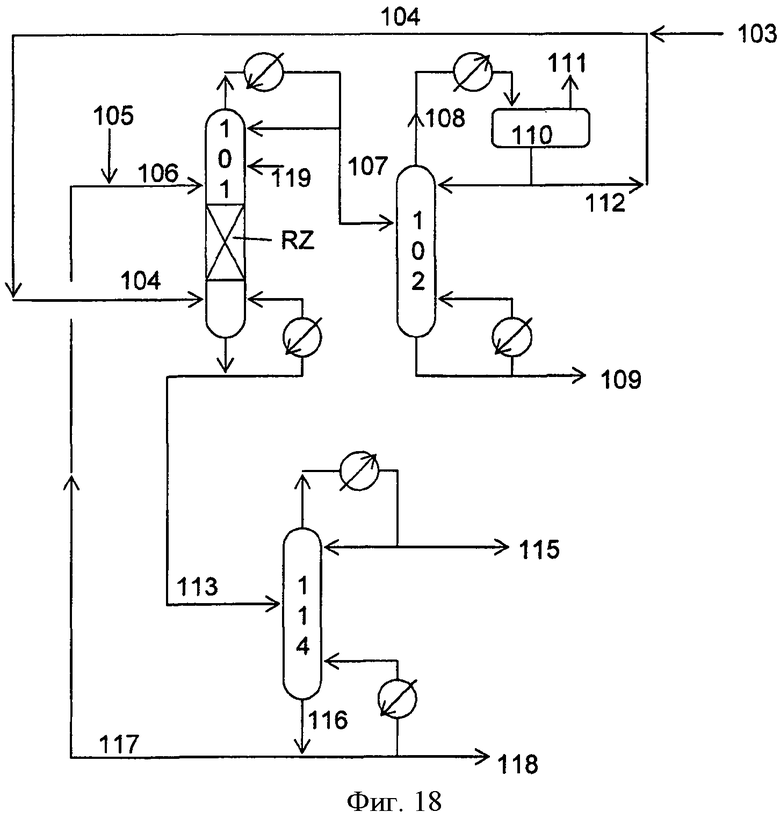
Изобретение относится к способам получения органических карбонатов и карбаматов. Описан способ алкоголиза, включающий: введение реагентов и микроскопического количества растворимого металлорганического соединения, растворимого в реагентах, в реактор, содержащий твердый катализатор алкоголиза, где микроскопическое количество находится в пределах от примерно 1 м.д. до примерно 3000 м.д. масс по отношению к общей массе водимых реагентов; где растворимое металлорганическое соединение и твердый катализатор алкоголиза, каждый, независимо содержат элемент Группы II - Группы VI. Описан способ получения диалкилкарбонатов, включающий: введение спирта и реагента алкоголиза, содержащего, по меньшей мере, одно соединение из мочевины, органического карбамата и циклического карбоната, в присутствии описанной выше каталитической системы. Описан способ получения диарилкарбоната, включающий: введение ароматического гидрокси соединения и диалкилкарбоната в присутствии описанной выше каталитической системы. Описан способ получения алкиларилкарбоната, включающий: введение ароматического гидрокси соединения и диалкилкарбоната в присутствии описанной выше каталитической системы. Описан способ получения биодизельного топлива, включающий: введение спирта и глицерида в присутствии описанной выше каталитической системы. Описанные выше способы предусматривают реактивирование отработанного твердого катализатора алкоголиза, включающего: удаление полимерных материалов, осажденных на катализаторе; и повторное осаждение каталитически активных металлов на твердый катализатор. Технический результат - увеличение продолжительности цикла способа алкоголиза. 6 н. и 30 з.п. ф-лы, 7 табл., 18 ил., 14 пр.
1. Способ алкоголиза, включающий: введение реагентов и микроскопического количества растворимого металлорганического соединения, растворимого в реагентах, в реактор, содержащий твердый катализатор алкоголиза, где микроскопическое количество находится в пределах от примерно 1 м.д. до примерно 3000 м.д. масс по отношению к общей массе вводимых реагентов; где растворимое металлорганическое соединение и твердый катализатор алкоголиза каждый независимо содержит элемент Группы II - Группы VI.
2. Способ по п.1, в котором алкоголиз представляет собой переэтерификацию.
3. Способ по п.1, в котором алкоголиз представляет собой диспропорционирование.
4. Способ по п.1, в котором растворимое металлорганическое соединение и твердый катализатор алкоголиза каждый содержит один и тот же элемент Группы II - Группы VI.
5. Способ по п.1, включающий: приведение в контакт реагентов в присутствии твердого катализатора алкоголиза для алкоголиза, по меньшей мере, части реагентов; и извлечение эффлюента из реактора, содержащего растворимое металлорганическое соединение, продукт алкоголиза и непрореагировавшие реагенты.
6. Способ по п.1, где способ алкоголиза включает, по меньшей мере, одну из реакций алкоголиза, переэтерификации и диспропорционирования с получением, по меньшей мере, одного продукта из диалкилкарбоната, диарилкарбоната, алкиларилкарбоната, биодизельного топлива, органического сложного эфира и органического карбамата.
7. Способ по п.1, в котором растворимое металлорганическое соединение вводится в количестве в пределах от 1 м.д. до 2000 м.д. масс по отношению к общей массе реагентов исходных материалов.
8. Способ по п.1, в котором твердый катализатор алкоголиза содержит, по меньшей мере, одно соединение из иммобилизованного органического соединения титана и соединения титана на носителе, и растворимое металлорганическое соединение содержит соединение титана, растворимое в реагентах из исходных материалов.
9. Способ по п.1, в котором твердый катализатор алкоголиза содержит, по меньшей мере, одно соединение из иммобилизованного органического соединения кальция и соединения кальция на носителе, и растворимое металлорганическое соединение содержит соединение кальция, растворимое в реагентах из исходных материалов.
10. Способ по п.1, в котором твердый катализатор алкоголиза содержит, по меньшей мере, одно соединение из иммобилизованного органического соединения магния и соединения магния на носителе, и растворимое металлорганическое соединение содержит соединение магния, растворимое в реагентах из исходных материалов.
11. Способ по п.1, в котором твердый катализатор алкоголиза содержит, по меньшей мере, одно соединение из иммобилизованного органического соединения цинка и соединения цинка на носителе, и растворимое металлорганическое соединение содержит соединение цинка, растворимое в реагентах из исходных материалов.
12. Способ по п.1, в котором твердый катализатор алкоголиза содержит, по меньшей мере, одно соединение из иммобилизованного органического соединения олова и соединения олова на носителе, и растворимое металлорганическое соединение содержит соединение олова, растворимое в реагентах из исходных материалов.
13. Способ по п.1, в котором твердый катализатор алкоголиза содержит, по меньшей мере, одно соединение из иммобилизованного органического соединения сурьмы и соединения сурьмы на носителе, и растворимое металлорганическое соединение содержит соединение сурьмы, растворимое в реагентах из исходных материалов.
14. Способ по п.3, дополнительно включающий извлечение, по меньшей мере, части растворимого металлорганического соединения из эффлюента из реактора.
15. Способ по п.14, дополнительно включающий рециклирование, по меньшей мере, части извлекаемого растворимого металлорганического соединения в реактор алкоголиза.
16. Способ получения диалкилкарбонатов, включающий: введение спирта и реагента алкоголиза, содержащего, по меньшей мере, одно соединение из мочевины, органического карбамата и циклического карбоната, в первую реакционную зону, содержащую твердый катализатор алкоголиза; введение микроскопического количества растворимого металлорганического соединения, растворимого в реагентах, в первую реакционную зону, где твердый катализатор алкоголиза и растворимое металлорганическое соединение каждый независимо содержит элемент Группы II - Группы VI, где микроскопическое количество находится в пределах от примерно 1 м.д. до примерно 3000 м.д. масс по отношению к общей массе вводимых реагентов.
17. Способ по п.16, в котором спирт представляет собой алкиловый спирт.
18. Способ по п.16, дополнительно включающий: приведение в контакт спирта и реагента алкоголиза в присутствии твердого катализатора алкоголиза при условиях взаимодействия, по меньшей мере, части реагента алкоголиза со спиртом с образованием, по меньшей мере, одного соединения из алкиларилкарбоната и диалкилкарбоната; извлечение из первой реакционной зоны эффлюента, содержащего растворимое металлорганическое соединение.
19. Способ по п.18, в котором эффлюент из реактора дополнительно содержит алкиларилкарбонат, дополнительно включающий: введение, по меньшей мере, части эффлюента из первой реакционной зоны во вторую реакционную зону; диспропорционирование, по меньшей мере, части алкиларилкарбоната с образованием диалкилкарбоната.
20. Способ по п.19, в котором диалкилкарбонат включает, по меньшей мере, одно соединение из диэтилкарбоната и диметилкарбоната.
21. Способ получения диарилкарбоната, включающий: введение ароматического гидрокси соединения и диалкилкарбоната в первую реакционную зону, содержащую твердый катализатор переэтерификации; введение микроскопического количества растворимого металлорганического соединения, растворимого в реагентах, в первую реакционную зону, где твердый катализатор переэтерификации и растворимое металлорганическое соединение каждый независимо содержит элемент Группы II - Группы VI, где микроскопическое количество находится в пределах от примерно 1 м.д. до примерно 3000 м.д. масс по отношению к общей массе вводимых реагентов.
22. Способ по п.21, дополнительно включающий: приведение в контакт ароматического гидроксильного соединения и диалкилкарбоната в присутствии твердого катализатора переэтерификации при условиях для взаимодействия, по меньшей мере, части диалкилкарбоната с ароматическим гидроксильным соединением с образованием, по меньшей мере, одного соединения из алкиларилкарбоната и диарилкарбоната; извлечение из первой реакционной зоны эффлюента, содержащего гомогенный катализатор.
23. Способ по п.22, в котором эффлюент из реактора дополнительно содержит алкиларилкарбонат, дополнительно включающий: введение, по меньшей мере, части эффлюента из первой реакционной зоны во вторую реакционную зону; диспропорционирование, по меньшей мере, части алкиларилкарбоната с образованием диарилкарбоната.
24. Способ по п.21, в котором ароматическое гидроксильное соединение включает фенол.
25. Способ по п.21, в котором диарилкарбонат включает дифенилкарбонат.
26. Способ получения алкиларилкарбоната, включающий: введение ароматического гидрокси соединения и диалкилкарбоната в первую реакционную зону, содержащую твердый катализатор переэтерификации; введение микроскопического количества растворимого металлорганического соединения, растворимого в реагентах, в первую реакционную зону, где твердый катализатор переэтерификации и растворимое металлорганическое соединение каждый независимо содержит элемент Группы II - Группы VI, где микроскопическое количество находится в пределах от примерно 1 м.д. до примерно 3000 м.д. масс по отношению к общей массе вводимых реагентов.
27. Способ по п.26, дополнительно включающий: приведение в контакт ароматического гидроксильного соединения и диалкилкарбоната в присутствии твердого катализатора переэтерификации при условиях для взаимодействия, по меньшей мере, части диалкилкарбоната с ароматическим гидроксильным соединением с образованием алкиларилкарбоната; извлечение из первой реакционной зоны эффлюента, содержащего гомогенный катализатор.
28. Способ получения биодизельного топлива, включающий: введение спирта и глицерида в первую реакционную зону, содержащую твердый катализатор переэтерификации; введение микроскопического количества растворимого металлорганического соединения, растворимого в реагентах, в первую реакционную зону, где твердый катализатор переэтерификации и растворимое металлорганическое соединение каждый независимо содержит элемент Группы II - Группы VI, где микроскопическое количество находится в пределах от примерно 1 м.д. до примерно 3000 м.д. масс по отношению к общей массе вводимых реагентов.
29. Способ по п.28, дополнительно включающий: приведение в контакт спирта и глицерида в присутствии твердого катализатора переэтерификации при условиях для взаимодействия, по меньшей мере, части спирта с глицеридом с образованием, по меньшей мере, одного соединения из глицерина и сложного эфира жирной кислоты; извлечение из первой реакционной зоны эффлюента, содержащего растворимое металлорганическое соединение.
30. Способ по одному из пп.1, 16, 21, 26 или 28, включающий реактивирование отработанного твердого катализатора алкоголиза, содержащий: удаление полимерных материалов, осажденных на катализаторе; и повторное осаждение каталитически активных металлов на твердый катализатор.
31. Способ по п.30, в котором удаление и повторное осаждение осуществляют в реакторе переэтерификации in situ.
32. Способ по п.30, в котором удаление включает приведение в контакт твердого катализатора с раствором, содержащим, по меньшей мере, одно соединение из спирта и воды.
33. Способ по п.32, в котором раствор дополнительно содержит растворитель.
34. Способ по п.33, в котором растворитель содержит, по меньшей мере, одно соединение из бензола, толуола, ксилола, гексана, октана, декана, тетрагидрофурана и простого эфира.
35. Способ по п.30, дополнительно включающий кондиционирование твердого катализатора перед повторным осаждением.
36. Способ по п.35, в котором кондиционирование включает сушку твердого катализатора при температуре от примерно 49°C до примерно 427°C (от примерно 120°F до примерно 800°F) в потоке инертного газа.
| СПОСОБ ПОЛУЧЕНИЯ АРОМАТИЧЕСКОГО КАРБОНАТА | 1991 |
|
RU2041869C1 |
| US 7074951 B2, 11.07.2006 | |||
| Способ контроля магнитных свойств аморфных металлических сплавов | 1988 |
|
SU1629888A1 |
| US 6712867 B1, 30.03.2004 | |||
| US 20070093672, 26.04.2007 | |||
| СПОСОБ РЕГЕНЕРАЦИИ СЕРЕБРЯНОГО КАТАЛИЗАТОРА ПОЛУЧЕНИЯ ФОРМАЛЬДЕГИДА | 2003 |
|
RU2242281C1 |
| WO 2006068053, 29.06.2006 | |||
| КАТАЛИЗАТОРЫ ЭТЕРИФИКАЦИИ | 1998 |
|
RU2181307C2 |
| СПОСОБ ПЕРЕЭТЕРИФИКАЦИИ ЖИРА И/ИЛИ МАСЛА БИОЛОГИЧЕСКОГО ПРОИСХОЖДЕНИЯ ПУТЕМ АЛКОГОЛИЗА | 2000 |
|
RU2263660C2 |

