Настоящее изобретение относится к фармацевтическому депо, содержащему N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамид или его фармацевтически приемлемую соль, и к применению этого фармацевтического депо.
В WO-A-2005/061465 раскрыты амидные производные, включая N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамид и его фармацевтически приемлемые соли, и сообщается, что амидные производные являются ингибиторами синтеза цитокинов, таких как фактор некроза опухолей (в дальнейшем TNF), например TNFα, и различных членов семейства интерлейкинов (в дальнейшем IL), например IL-1, IL-6 и IL-8 (особенно IL-1). В частности, и без стремления предполагать, что амидные производные, раскрытые в WO-A-2005/061465, обладают фармакологической активностью только вследствие их воздействия на единственный биологический процесс, считают, что амидные производные ингибируют эффекты цитокинов вследствие подавления фермента р38 киназы. р38 киназа (известная также как цитокинсупрессивный связывающий белок, в дальнейшем CSBP) и реактивирующая киназа (в дальнейшем RK) представляют собой членов семейства ферментов митогенактивируемых протеинкиназ (в дальнейшем MAP), которые, как известно, активируются физиологическим стрессом, таким как стресс, индуцированный ионизирующим излучением, цитотоксическими агентами и токсинами, например эндотоксинами, такими как бактериальный липополисахарид, и рядом агентов, таких как цитокины, например TNFα и IL-1. Известно, что киназа р38 фосфорилирует некоторые внутриклеточные белки, которые вовлечены в каскад ферментных стадий, что влечетза собой биосинтез и выделение цитокинов, таких как TNFa и IL-1.
Таким образом, полагают, что амидные производные, раскрытые в WO-A-2005/061465, являются полезными в лечении заболеваний или медицинских состояний, при которых имеет место избыточное продуцирование цитокинов, например избыточное продуцирование TNFα или IL-1. Такие заболевания и медицинские состояния включают воспалительные и аллергические заболевания, такие как воспаление суставов (особенно ревматоидный артрит, остеоартрит и подагра). Для лечения заболеваний и медицинских состояний, таких как воспаление суставов, было бы желательно вводить амидное производное непосредственно в область (такую как сустав), требующую лечения, предпочтительно таким образом, чтобы обеспечить контролируемое и/или непрерывное высвобождение амидного производного в этом месте. Таким образом, существует потребность в препарате или композиции, содержащих Л/-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамид или его фармацевтически приемлемую соль в форме, подходящей для такого введения, например для фармацевтического депо.
Хотя в WO-A-2005/061465 высказано предположение, что раскрытые амидные производные можно включать в фармацевтическую композицию, например, в форме, подходящей для перорального или местного применения, для введения посредством ингаляции или инсуффляции, или для парентерального введения, в WO-A-2005/061465 отсутствует раскрытие фармацевтического депо, содержащего амидное производное, как оно раскрыто, не говоря уже о фармацевтическом депо, содержащем N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамид или его фармацевтически приемлемую соль.
Согласно настоящему изобретению предлагается фармацевтическое депо, содержащее (1) N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамид или его фармацевтически приемлемую соль в качестве фармацевтического агента (РА) и (2) полимер, который разлагается с созданием кислого микроклимата, где РА высвобождается из полимера при разложении полимера.
В фармацевтическое депо по настоящему изобретению фармацевтический агент (в дальнейшем РА) представляет собой N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамид или его фармацевтически приемлемую соль. Таким образом, ссылки в данном описании на РА включают соединение N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамид само по себе, а также его фармацевтически приемлемые соли.
Специалисту будет очевидно, что фармацевтическое депо представляет собой композицию, которая высвобождает РА, главным образом фармацевтически эффективное количество РА (в данном описании N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамида или его фармацевтически приемлемой соли) постепенно, таким образом, чтобы обеспечивать введение с контролируемым и/или непрерывным высвобождением РА, содержащегося в нем.
N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамид имеет структуру:
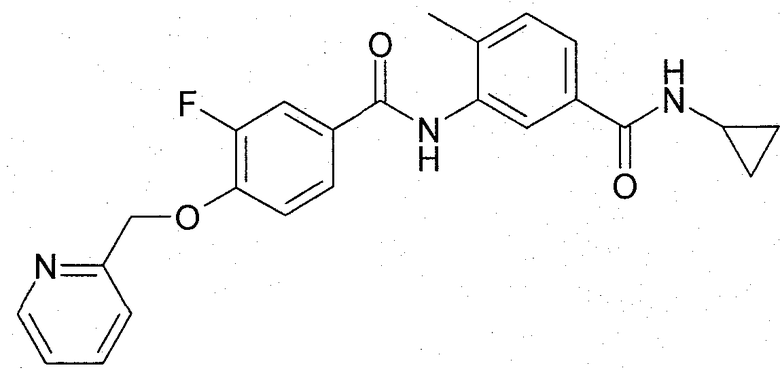
и раскрыт как пример (5-у) в WO-A-2005/061465.
Подходящие фармацевтически приемлемые соли N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамида для включения в фармацевтическое депо по настоящему изобретению, руководствуясь обоснованной медицинской оценкой, являются подходящими для введения субъекту, например теплокровному животному, такому как человек, без нежелательных фармакологических действий и без чрезмерной токсичности. Подходящие фармацевтически приемлемые соли включают соли присоединения кислоты, например соли присоединения с неорганической или органический кислотой, такой как соляная, бромистоводородная, серная, фосфорная, трифторуксусная, лимонная, малеиновая, винная, фумаровая, гемифумаровая, янтарная, гемиянтарная, миндальная, метансульфоновая, диметансульфоновая, этан-1,2-сульфоновая, бензолсульфоновая, салициловая или 4-толуолсульфоновая кислота. Предпочтительная соль присоединения кислоты представляет собой соль присоединения с соляной кислотой, то есть с получением гидрохлорида N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамида.
N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамид и его фармацевтически приемлемые соли можно синтезировать из подходящих исходных веществ, используя стандартные методики органический химии, например раскрытые в WO-A-2005/061465. Например, N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамид может быть получен взаимодействием N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-гидроксибензамида с гидрохлоридом 2-хлорметил-пиридина в присутствии подходящего основания (такого как карбонат калия). Взаимодействие N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамида с соляной кислотой дает соль гидрохлорид.
Фармацевтическое депо по настоящему изобретению делает возможным введение РА, используя контролируемое и/или непрерывное высвобождение композиции таким образом, чтобы поддерживать терапевтический уровень РА в течение продолжительного периода времени. Это является выгодным, так как снижает частоту дозирования и обеспечивает удобный способ введения РА, который является особенно желательным для введения РА непосредственно в сустав, то есть посредством внутрисуставного введения. Композиции с контролируемым и/или непрерывным высвобождением также могут снижать тяжесть и частоту любых нежелательных побочных эффектов, ассоциированных с конкретным РА. Улучшения в удобстве введения и пониженная частота и тяжесть побочных эффектов в свою очередь улучшают соблюдение пациентом предписанного режима лечения.
Обнаружено, что многие соединения, которые представляют собой РА, являются неподходящими для включения в фармацевтические депо, в первую очередь из-за таких факторов, как нестабильность соединений в композициях, требуемых для контролируемого и/или непрерывного высвобождения и/или для внутрисуставного введения. Авторы настоящего изобретения неожиданно обнаружили, что РА, включенный в фармацевтическое депо по настоящему изобретению, является гидролитически стабильным в кислом микроклимате, созданном при разложении полимера, включенного в него, и из этого следует, что РА является подходящим для включения в фармацевтическое депо. Кроме того, авторы настоящего изобретения неожиданно обнаружили, что РА, включенный в фармацевтическое депо по настоящему изобретению, может быть обеспечен с непрерывной высокой местной концентрацией РА в месте введения, например в суставе, чтобы обеспечивать эффективное контролируемое и/или непрерывное высвобождение РА. Другими словами, фармацевтическое депо является эффективным для медленного высвобождения РА таким образом, чтобы обеспечивать длительно действующий эффект.
Предпочтительно РА может быть включен в фармацевтическое депо по настоящему изобретению без какой-либо химической модификации, необходимой до его включения в депо.
Специалисту понятно, что "фармацевтический агент" (или РА) представляет собой агент, который вызывает фармакологический эффект у субъекта, которому его вводят, например у теплокровного животного, такого как человек, например таким образом, чтобы лечить или предупреждать заболевание или медицинское состояние. Как рассматривалось выше, РА в фармацевтическом депо по настоящему изобретению представляет собой N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамид или его фармацевтически приемлемую соль, которые, как полагают, вызывают фармакологический эффект путем ингибирования эффектов цитокинов (таких как TNF, например TNFα, и различных членов семейства IL, например IL-1, IL-6 и IL-8) посредством ингибирования фермента р38 киназы.
РА, который включают в фармацевтическое депо по настоящему изобретению, является эффективным в лечении воспалительного заболевания или состояния, например состояния, вызванного воспалением сустава, такого как остеоартрит, при котором может иметь место как острое, так и хроническое синовиальное воспаление. Остеоартрит (также известный как дегенеративный артрит или дегенеративное суставное заболевание) представляет собой наиболее распространенную форму артрита со множеством страдающих во всем мире, и улучшенные композиции для доставки РА для лечения остеоартрита являются в высокой степени желаемыми.
Специалисту понятно, что РА присутствует в фармацевтическом депо по настоящему изобретению в терапевтически эффективном количестве.
"Терапевтически эффективное количество" представляет собой любое количество РА (например, которое содержится в фармацевтическом депо), которое при введении субъекту, страдающему от заболевания или медицинского состояния, против которого РА эффективен, вызывает ослабление, ремиссию или регрессию заболевания или медицинского состояния.
Терапевтически эффективное количество РА, которое включено в фармацевтическое депо, обязательно варьирует в зависимости от природы и тяжести расстройства, подлежащего лечению, и конкретного находящегося на лечении пациента, согласно хорошо известным принципам медицины. Кроме того, терапевтически эффективное количество РА, которое включено в фармацевтическое депо, обязательно варьирует согласно желаемому профилю контролируемого и/или непрерывного высвобождения, например, в зависимости от периода времени, в течение которого требуется высвобождение РА, и от концентрации РА, желаемой в течение этого времени.
В дополнение к РА, фармацевтическое депо по настоящему изобретению содержит полимер, который разлагается с созданием кислого микроклимата, например полимер, который разлагается в присутствии воды с созданием кислого микроклимата. Под ним авторы изобретения подразумевают полимер, который разлагается или разрушается химически с обеспечением кислого рН в небольшой локализованной области (такой как сустав), в которую вводят фармацевтическое депо. Предпочтительно кислый рН является по существу одинаковым в локализованной области и отличается от окружающей области, которая может находиться при физиологическом рН (обычно примерно рН 7,4). Кислый рН обычно равен рН меньше чем примерно 7,4, например рН в интервале от примерно 1 до примерно 7, таком как от примерно 3 до примерно 7; удобным образом от примерно 1 до менее чем 7 или от примерно 3 до менее чем 7.
Обычно РА диспергирован или инкапсулирован в полимере так, что РА непрерывно высвобождается из полимера по мере того, как полимер разлагается с течением времени с созданием кислого микроклимата. Было обнаружено, что РА, который включен в фармацевтическое депо по настоящему изобретению, является гидролитически стабильным в кислом микроклимате, который создается при разложении полимера. Высвобождение PA из полимера обеспечивает контролируемое и/или непрерывное высвобождение РА из фармацевтического депо в организме субъекта, например теплокровного животного, такого как человек, которому вводят фармацевтическое депо. Предпочтительно при разложении полимера и высвобождении РА достигается высокая локальная концентрация (то есть концентрация в области, в которую вводят фармацевтическое депо, такой как сустав) для обеспечения желаемого терапевтического эффекта и низкая системная концентрация для ослабления любой нежелательной системной токсичности РА. Таким образом, фармацевтическое депо доставляет РА субъекту в концентрациях, эффективных для лечения конкретного заболевания или медицинского состояния в течение длительного периода времени.
В фармацевтическом депо по настоящему изобретению может быть использован любой подходящий полимер при условии, что этот полимер разлагается с созданием кислого микроклимата, то есть при введении субъекту, например теплокровному животному, такому как человек, и является биоразлагаемым и биосовместимым.
Специалисту понятно, что под термином "биосовместимый" авторы изобретения подразумевают вещество, которое является совместимым с живой тканью или с живой системой, не являясь токсичным, повреждающим или физиологически реактивным, и не вызывает иммунологического отторжения.
Под термином "биоразлагаемый" авторы изобретения подразумевают вещество, которое разлагается в биологической окружающей среде.
Например, полимер может являться "биоразлагаемым" так, что весь полимер подвергается биоразложению и не нуждается в удалении после использования, то есть после того как весь РА будет высвобожден. Такие полимеры могут содержать гидролизуемые и ферментативно расщепляемые сложно-эфирные связи, которые разлагаются в биологических условиях (например в присутствии воды и биологических ферментов, находящихся в тканях теплокровных животных, таких как люди) с образованием нетоксичных, биосовместимых и/или биоразлагаемых продуктов. Альтернативно полимер может являться "биоразлагаемым" в силу присущего ему конечного периода полураспада в биологической окружающей среде. Например, полимер может иметь период полураспада от 1 до 12 месяцев, такой как от 1 до 6 месяцев.
Обычно полимер включает по меньшей мере одну кислотную функциональную группу или по меньшей мере одну функциональную группу, которая может взаимодействовать с образованием кислотной функциональной группы, где такая кислотная функциональная группа представляет собой группу, которая способна отдавать протон основной функциональной группе, такой как амин. Примеры подходящих кислотных функциональных групп включают группы карбоновой кислоты (то есть -СO2Н) и группы сульфоновой кислоты (то есть -S(O)2OH). Примеры подходящих функциональных групп, которые могут взаимодействовать с образованием кислотной функциональной группы, включают сложные эфиры (то есть RC(O)OR, где R может представлять собой алкил или арил), которые могут взаимодействовать с водой с образованием группы соответствующей карбоновой кислоты и спирта. Предпочтительно полимер выбирают таким образом, чтобы он разлагался и высвобождал РА в течение периода от примерно 30 до 90 суток. Например, полимер может разлагаться и высвобождать РА в течение периода примерно 30, примерно 60 или примерно 90 суток. Например, полимер может разлагаться и высвобождать РА в течение периода примерно 120, примерно 150 или примерно 180 суток.
Подходящие полимеры включают полиэфир жирной гидроксикислоты и его производные (например полимолочной кислоты, полигликолевой кислоты, полилимонной кислоты, полияблочной кислоты, поли-β-гидроксимасляной кислоты, полимер α-капролактона с раскрытием кольца, сополимер молочной кислоты и гликолевой кислоты, сополимер 2-гидроксимасляной кислоты и гликолевой кислоты, сополимер полимолочной кислоты и полиэтиленгликоля или сополимер полигликолевой кислоты и полиэтиленгликоля), полимер алкил-α-цианоакрилата (например поли(бутил-2-цианоакрилат)), полиалкиленоксалат (например политриметиленоксалат или политетраметиленоксалат), поли-орто-эфир, поликарбонат (например полиэтиленкарбонат или полиэтиленпропиленкарбонат), поли-орто-карбонат, полиаминокислоту (например, поли-γ-L-аланин, поли-γ-бензил-L-глутаминовую кислоту или поли-γ-метил-L-глутаминовую кислоту), сложный эфир гиалуроновой кислоты и тому подобное, и можно использовать один или более из таких полимеров.
Если полимеры представляют собой сополимеры, они могут представлять собой случайные, блок- и привитые сополимеры. Если молекулы указанных выше α-гидроксикарбоновых кислот, гидроксидикарбоновых кислот и гидрокситрикарбоновых кислот обладают оптической активностью, может быть использован любой из D-изомеров, L-изомеров и DL-изомеров. В числе прочих предпочтительными являются полимер α-гидроксикарбоновой кислоты (предпочтительно полимер молочной кислоты и гликолевой кислоты), ее сложного эфира, сложных эфиров поли-α-цианоакриловой кислоты и так далее, и наиболее предпочтительны сополимер молочной кислоты и гликолевой кислоты (также называемый поли(лактид-со-гликолид) или поли(молочной-со-гликолевой кислоты), и в дальнейшем называемый PLGA). Таким образом, в одном аспекте полимер представляет собой PLGA. При использовании в данном описании термин PLGA включает полимеры молочной кислоты (также называемые полилактид, поли(молочная кислота) или PLA).
Подходящие полимеры PLGA могут иметь молярное соотношение молочная кислотатликолевая кислота в интервале от 100:0 до 50:50, целесообразно в интервале от 95:5 до 50:50. Например, полимер PLGA может иметь молярное соотношение молочная кислотатликолевая кислота составляющее 95:5 или 50:50.
Подходящие полимеры PLGA могут иметь длину блока в интервале от 1 до 5, предпочтительно от 2 до 4.
Подходящие полимеры PLGA могут иметь средневзвешенную молекулярную массу от примерно 3000 до примерно 50000, предпочтительно от примерно 4000 до примерно 40000, и более предпочтительно от примерно 5000 до примерно 30000 Дальтон. Степень дисперсности (среднемассовая молекулярная масса/среднечисловая молекулярная масса, в дальнейшем называемая полидисперсностью) может меняться от примерно 1,2 до примерно 4,0, предпочтительно от примерно 1,3 до примерно 3,5.
Специалисту понятно, что среднемассовую молекулярную массу, среднечисловую молекулярную массу и полидисперсность можно определять любым подходящим способом или средствами, например посредством гель-хроматографии (SEC) полистиреновых эталонных веществ с узкой полидисперсностью и с пиковыми молекулярными массами 1000000, 130000 50000, 20000, 10000, 5000, 2000 и 580 соответственно. Определение можно выполнять, используя SEC-колонку Mixed Bed D 5 мкм (произведенную Polymer Laboratories Ltd., UK), и используя 5%-ныи метанол в тетрагидрофуране в качестве подвижной фазы.
PLGA может быть получен любым общепринятым способом или может иметься в продаже. Например, PLGA может быть образован полимеризацией с раскрытием кольца при помощи подходящего катализатора на основе циклического лактида, гликолида и так далее (смотри Encyclopedic Handbook of Biomaterials and Bioengineering Part A: Materials, Volume 2, Marcel Dekker, Inc. (1995); EP-0058481B2; Effects of polymerisation variables on PLGA properties: molecular weight, composition and chain structure and Dorta et al, Int. J. Pharm., 100, pp 9-14 (1993)).
Считают, что PLGA является биоразлагаемым благодаря разложению всей твердой полимерной композиции в результате разрыва гидролизуемых и ферментативно расщепляемых сложноэфирных связей в биологических условиях (например в присутствии воды и биологических ферментов, находящихся в тканях теплокровных животных, таких как люди) с образованием молочной кислоты и гликолевой кислоты. И молочная кислота, и гликолевая кислота являются водорастворимыми, нетоксичными продуктами нормального метаболизма, которые могут далее подвергаться биоразложению с образованием диоксида углерода и воды. Другими словами, считается, что PLGA разлагается посредством гидролиза его сложноэфирных групп в присутствии воды, например, в организме теплокровного животного, такого как человек, с образованием молочной кислоты и гликолевой кислоты и созданием кислого микроклимата. Молочная и гликолевая кислоты представляют собой побочные продукты различных метаболических путей в организме теплокровного животного, такого как человек, в нормальных физиологических условиях и поэтому являются хорошо переносимыми при минимальной системной токсичности.
Полимер используют в любой подходящей форме, в которой РА может быть диспергирован или инкапсулирован до разложения полимера. Например, фармацевтическое депо может содержать полимер в форме микрочастиц или наночастиц, либо в жидкой форме, с диспергированным или инкапсулированным в нем РА.
Подходящие микрочастицы обычно имеют средний размер частиц в интервале от 0,1 до 1000 мкм, предпочтительно от 1 до 750 мкм и более предпочтительно от 10 до 500 мкм.
Подходящие наночастицы обычно имеют средний размер частиц в интервале от 1 до 2000 нм, предпочтительно от 10 до 1000 нм, и более предпочтительно от 50 до 500 нм.
В частности, микрочастицы являются по существу сферическими по форме (то есть являются микросферами).
Если полимер представлен в форме микрочастиц, то микрочастицы можно получать, используя любой подходящий способ, такой как выпаривание растворителя или способ экстракции растворителем. Например, в способе выпаривания растворителя РА и полимер могут быть растворены в подходящем летучем органическом растворителе (например кетоне, таком как ацетон, галогенированном углеводороде, таком как хлороформ или метиленхлорид, галогенированном ароматическом углеводороде, циклическом простом эфире, таком как диоксан, сложном эфире, таком как этилацетат, нитриле, таком как ацетонитрил, или спирте, таком как этанол) и диспергированы в водной фазе, содержащей подходящий стабилизатор эмульсии (например поливиниловый спирт, PVA). Органический растворитель затем выпаривают с образованием микрочастиц с РА, инкапсулированным в них. В способе экстракции растворителем РА и полимер могут быть растворены в полярном растворителе (таком как ацетонитрил, дихлорметан, метанол, этилацетат или метилформиат) и затем диспергированы в водной фазе (такой как раствор вода/PVA). Получают эмульсию с образованием микрочастиц с РА, инкапсулированным в них. Распылительная сушка является альтернативным способом изготовления для получения микрочастиц.
В одном аспекте фармацевтическое депо может содержать полимер (такой как PLGA, как описано выше) в форме микрочастиц с РА, инкапсулированным в них. Например, фармацевтическое депо может содержать полимер PLGA, имеющий молярное соотношение лактид тликолид 50:50, в форме микрочастиц с РА, инкапсулированным в них. Такое фармацевтическое депо может являться подходящим для контролируемого и/или непрерывного высвобождения РА в течение периода примерно 30 суток. Кроме того, в качестве примера, фармацевтическое депо может содержать полимер PLGA, имеющий молярное соотношение лактидтликолид равное 95:5, в форме микрочастиц с РА, инкапсулированным в них. Такое фармацевтическое депо может быть подходящим для контролируемого и/или непрерывного высвобождения РА в течение периода от примерно 60 до 90 суток. Такое фармацевтическое депо может также являться подходящим для контролируемого и/или непрерывного высвобождения РА в течение периода вплоть до 120, вплоть до 150, или вплоть до 180 суток.
Фармацевтическое депо может содержать РА и полимер в любых подходящих количествах. Например, фармацевтическое депо может содержать от 1 до 30% по массе РА и от 70 до 99% по массе полимера.
Например, если фармацевтическое депо по настоящему изобретению содержит микрочастицы PLGA, то PLGA может присутствовать в количестве, варьирующем от примерно 70% до примерно 99% по массе от массы микрочастиц. Такое количество PLGA можно использовать, когда в микрочастицы загружено от примерно 1% до примерно 30% по массе РА. Кроме того, данное количество полимера рассчитано для микрочастиц, содержащих РА и PLGA, но не другие фармацевтическое эксципиенты, например, используемые для суспендирования микрочастиц перед лиофилизацией. PLGA можно использовать в количестве от примерно 88% до примерно 90% по массе от массы микрочастиц, когда от примерно 10% до примерно 12% по массе РА загружено в микрочастицы. Состав полимера обычно зависит от значительности фармакологической активности используемого РА и скорости и продолжительности высвобождения РА.
Фармацевтическое депо может дополнительно содержать подходящий фармацевтически приемлемый разбавитель или носитель, которые должны смешиваться с водой. Подходящие разбавители или носители включают, например, подходящие модифицирующие пористость агенты (такие как хлорид натрия), которые быстро растворяют уходящие поры, и/или подходящие пластификаторы для модификации скорости диффузии и/или снижения пористости (смотри, например, Burgess, D.J., Hickey, A.J, Drugs and Pharmaceutical Sciences (149) pp 305-353).
Разбавитель или носитель можно включать в фармацевтическое депо в любом подходящем количестве. Например, разбавитель или носитель может быть включен в количестве от 0 до 50% по массе от массы общей композиции.
Предпочтительно фармацевтическое депо не содержит дополнительный разбавитель или носитель.
Фармацевтическое депо обычно предлагается для местной доставки в желаемое место лечения, такое как в сустав.
Фармацевтическое депо можно приготовить в виде препарата для введения посредством инъекции, такой как внутрисуставная инъекция. Таким образом, в частности, фармацевтическое депо может быть представлено в инъецируемой форме (то есть в виде инъецируемого фармацевтического депо). Под "инъецируемым" авторы изобретения подразумевают, что фармацевтическое депо можно набирать в шприц и инъецировать субъекту, например теплокровному животному, такому как человек, не вызывая вредных эффектов из-за присутствия твердого вещества в депо. Например, фармацевтическое депо можно инъецировать в сустав, такой как воспаленный сустав. Другими словами, предлагается фармацевтическое депо для внутрисуставной инъекции. Подходящие суставы включают коленные, тазобедренные, плечевые, голеностопные, локтевые, лучезапястные, суставы пальцев стопы, суставы пальцев рук и дугоотросточные суставы позвоночника. Фармацевтическое депо остается в суставе после инъецирования туда и выполняет местную доставку РА контролируемым и непрерывным образом, предпочтительно в течение периода времени, варьирующего от 30 до 90 суток. Фармацевтические депо, которые выполняют местную доставку РА контролируемым и непрерывным образом в течение периода вплоть до 90 суток, является полезным, так как это минимизирует количество местных инъекций, которые требуется сделать в сустав, что делает такое депо удовлетворяющим существующим рекомендациям для внутрисуставной терапии, согласно которым рекомендовано не превышать трех - четырех небольших (примерно 2 мл) местных инъекций в сустав в год в связи с возможными вредными эффектами.
Фармацевтическое депо можно приготовить в виде препарата для инъекции во внутрисуставное пространство пораженного сустава, например в содержащую синовиальную жидкость область пораженного сустава, такую как очаг остеоартрита. Специалисту понятно, что синовиальная жидкость содержится в центральном пространстве сустава, ограниченном противолежащими костями суставов. Авторы настоящего изобретения обнаружили, что при инъецировании фармацевтического депо в синовиальную жидкость РА высвобождается и по существу проникает в окружающую ткань при исключительно незначительных количествах, проникающих в кровоток, то есть достигается высокая местная концентрация РА в области, в которую вводят фармацевтическое депо (такой как сустав), и низкая системная концентрация. Кроме того, фармацевтическое депо обеспечивает приемлемый "всплеск" (то есть высвобождение РА) в первые сутки после введения, который является выгодным в использовании и неожиданным в свете известного уровня техники, например US 6217911, где сообщается, что предпочтительным является высвобождение с незначительным «всплеском» или его отсутствием. Эффективный профиль высвобождения, обеспечиваемый фармацевтическим депо по настоящему изобретению, не мог быть предсказан на основании известного уровня техники и способствует эффективности фармацевтического депо.
Предпочтительно фармацевтическое депо обеспечивает непрерывную высокую местную концентрацию РА в суставном сочленении после введения его туда посредством инъекции, например выше 100 наномолей.
Инъецируемые фармацевтические депо могут содержать суспензию или дисперсию РА и полимерную комбинацию в фармацевтически приемлемом разбавителе или носителе, который должен смешиваться с водой. Подходящие разбавители или носители включают водные разбавители или носители, такие как изотонический водный раствор усилителя вязкости (такого как натриевая карбоксиметилцеллюлоза), поверхностно-активное вещество (такое как полисорбат 80) и/или регулятор тоничности (такой как хлорид натрия). Инъецируемые фармацевтические депо могут содержать дополнительные активные агенты, такие как местные анестетики.
Фармацевтическое депо по настоящему изобретению можно приготовить в виде препарата для лечения человека или использования в ветеринарии. Например, может предлагаться фармацевтическое депо, приготовленное в виде препарата для внутрисуставной инъекции для лечения человека или использования в ветеринарии.
В настоящем изобретении дополнительно предлагается фармацевтическое депо, как определено в данном описании, для применения в ингибировании эффектов цитокинов, например посредством ингибирования фермента р38 киназы, у субъекта.
Согласно другому аспекту настоящего изобретения предложено применение фармацевтического депо, как определено в данном описании, для ингибирования эффектов цитокинов, например посредством ингибирования фермента р38 киназы, у субъекта.
Согласно другому аспекту настоящего изобретения предложено применение фармацевтического депо, как определено в данном описании, в изготовлении лекарственного средства для использования в ингибировании эффектов цитокинов, например посредством ингибирования фермента р38 киназы, у субъекта.
Согласно другому аспекту настоящего изобретения предложен способ ингибирования эффектов цитокинов, например посредством ингибирования фермента р38 киназы, у субъекта, нуждающегося в этом, включающий введение указанному субъекту фармацевтического депо, как определено в данном описании.
В настоящем изобретении дополнительно предложено фармацевтическое депо, как определено в данном описании, для применения в предупреждении или лечении воспалительного заболевания, такого как остеоартрит, у субъекта.
Согласно другому аспекту настоящего изобретения предложено применение фармацевтического депо, как определено в данном описании, для предупреждения или лечения воспалительного заболевания, такого как остеоартрит, у субъекта.
Согласно другому аспекту настоящего изобретения предложено применение фармацевтического депо, как определено в данном описании, в изготовлении лекарственного средства для использования в предупреждении или лечении воспалительного заболевания, такого как остеоартрит, у субъекта.
Согласно другому аспекту настоящего изобретения предложен способ предупреждения или лечения воспалительного заболевания, такого как остеоартрит, у субъекта, нуждающегося в этом, включающий введение указанному субъекту фармацевтического депо, как определено в данном описании.
"Субъект", которому вводят фармацевтическое депо по изобретению, представляет собой животное, особенно теплокровное животное, такое как домашнее животное или человек, особенно человек.
Далее изобретение иллюстрируют следующими не ограничивающими примерами.
Пример 1
Готовили фармацевтическое депо, которое содержало микрочастицы PLGA с инкапсулированным в них N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамидом в качестве РА.
(1) Приготовление микрочастиц
60 мг N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамида и 340 мг PLGA (молярное соотношение лактидтликолид составляло 50:50, а молекулярная масса - 19,5) растворяли в смеси дихлорметан/метанол с соотношением 3:1 (2 мл). Затем этот раствор диспергировали в водной фазе из 0,5%-ного (масс./об.) PVA с высоким усилием сдвига с образованием эмульсии. Высокое усилие сдвига создавали с помощью статического смесителя с высокой скоростью потока водной фазы, например 1000 мл/мин. Полученную эмульсию добавляли к воде (1250 мл) при 30°С и перемешивали при 500 об/мин (используя мешалку Heidolph RZR1) в течение 1 часа. Полученную суспензию охлаждали на ледяной бане и микрочастицы оставляли оседать в течение 45 минут. Приблизительно 90% (по объему) надосадочной жидкости удаляли, стараясь не потревожить осевшие микрочастицы. Добавляли воду (1 л) и процесс повторяли. Приблизительно 95% (по объему) надосадочной жидкости удаляли и микрочастицы переносили в стеклянную пробирку. Цикл промывания/осаждения повторяли дополнительно 2 раза и микрочастицы переносили во флакон для сублимационной сушки с минимальным объемом воды. Флакон быстро замораживали в жидком азоте и микрочастицы подвергали сублимационной сушке в течение 48 часов.
(2) Протокол высвобождения in vitro
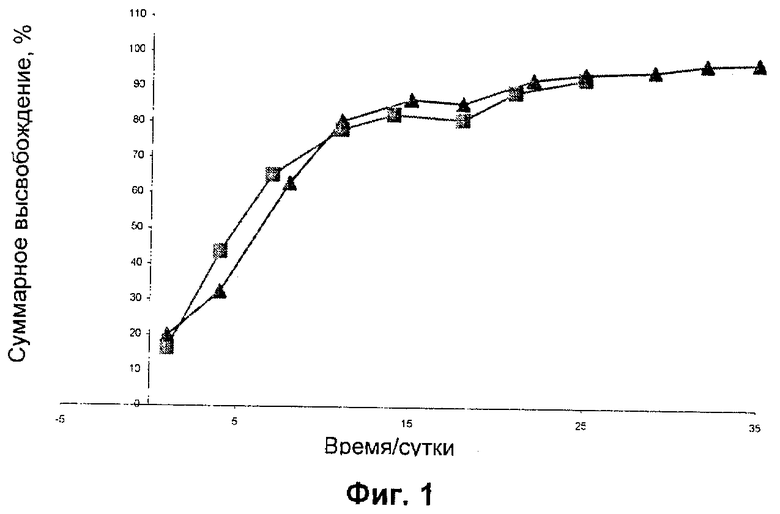
0,8 мг микрочастиц, содержащих N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамид в 50:50 PLGA, суспендировали в PBS (забуференный фосфатом физиологический раствор), содержащем 0,1% масс./об. Твин 80 (20 мл). Полученную суспензию хранили статически при 37°С, и образцы отбирали через 24 часа путем отбора среды (1 мл) с последующим добавлением на среду (1 мл) для гарантии того, что объем среды сохраняется постоянным в ходе эксперимента. Образцы отбирали с регулярными интервалами (смотри Фиг.1) вплоть до момента, когда депо больше не высвобождало N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамид, и анализировали при помощи ВЭЖХ. Результаты показаны ниже в Таблице 1.
Микрочастицы с PLGA 50:50 обеспечивали высокие эффективности инкапсулирования, давая нагрузки N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамида примерно 13%. Данные профиля высвобождения in vitro показаны в Фиг.1. Исследования высвобождения in vitro показали, что N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамид в микрочастицах PLGA 50:50 имел приемлемый «всплеск» в первые сутки и высвобождался в течение 1 месяца т vitro. Две партии, изготовленные с использованием PLGA 50:50 (Таблица 1), показали хорошую воспроизводимость.
Пример 2
Готовили фармацевтическое депо, которое содержало микрочастицы PLGA с инкапсулированным в них N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамидом в качестве РА. (1) Приготовление микрочастиц 60 мг N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамида и 340 мг PLGA (с молярным соотношением лактидтликолид 95:5 и молекулярной массой 23 кДа) растворяли в смеси дихлорметан/метанол с соотношением 3:1 (2 мл). Затем данный раствор диспергировали в водной фазе из 0,5%-ного (масс./об.) PVA с высоким усилием сдвига с образованием эмульсии. Высокое усилие сдвига создавали посредством использования статического смесителя с высокой скоростью потока водной фазы, например 1000 мл/мин. Полученную эмульсию добавляли к воде (1250 мл) при 30°С и перемешивали при 500 об/мин (используя мешалку Heidolph RZR1) в течение 1 часа. Полученную суспензию охлаждали на ледяной бане и микрочастицы оставляли оседать в течение 45 минут. Приблизительно 90% по объему надосадочной жидкости удаляли, стараясь не потревожить осевшие микрочастицы. Добавляли воду (1 л) и повторяли процесс. Приблизительно 95% (по объему) надосадочной жидкости удаляли и микрочастицы переносили в стеклянную пробирку. Цикл промывания/осаждения дополнительно повторяли 2 раза и микрочастицы переносили во флакон для сушки сублимацией с минимальным объемом воды. Флакон быстро замораживали в жидком азоте и микрочастицы сушили сублимацией в течение 48 часов.
(2) Протокол высвобождения in vitro
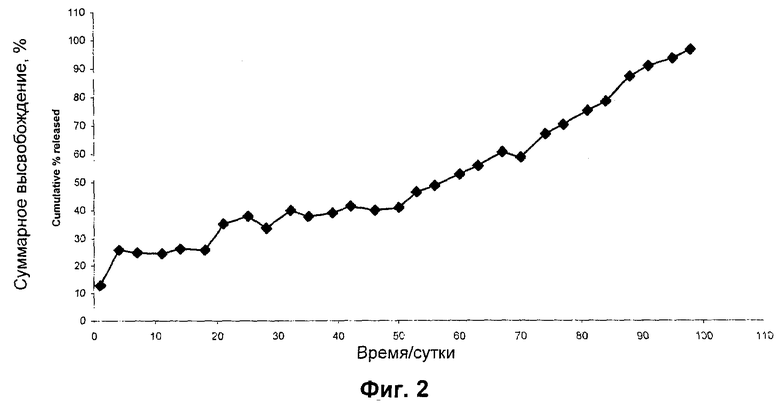
0,8 мг микрочастиц, содержащих N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамид в PLGA 95:5, суспендировали в PBS, содержащем 0,1% (масс./об.) Твин 80 (20 мл). Полученную суспензию хранили статически при 37°С и образцы отбирали через 24 часа путем отбора среды (1 мл) с последующим добавлением на среду (1 мл) для гарантии того, что объем среды сохраняется постоянным в ходе эксперимента. Образцы отбирали с регулярными интервалами (смотри Фиг.2) вплоть до момента, когда депо больше не высвобождало N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамид, и анализировали при помощи ВЭЖХ. Результаты показаны ниже в Таблице 2.
Микрочастицы с N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамидом обеспечивали высокие эффективности инкапсулирования, давая нагрузки РА примерно 13%. Данные о профиле полного высвобождения in vitro показаны на Фиг.2. Исследования высвобождения in vitro показали, что N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамид в микрочастицах PLGA 95:5 имел приемлемый «всплеск» в первые сутки и высвобождался в течение 3 месяцев in vitro.
Пример 3
Изучали характеристики высвобождения in vivo N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамида в микрочастицах PLGA 50:50 у крыс.
N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамид не в виде препарата инъецировали внутрисуставно (15 нг в 5 мкл инъекции в PBS) крысам и определяли концентрации в синовиальной жидкости через 15, 30 и 60 минут после дозирования. Отбирали образцы синовиальной жидкости из коленного сустава крыс, используя способ «промывания колена» (knee wash). Колено обнажали и делали поперечный разрез к сухожилию коленной чашечки проксимально к большеберцовой кости. Полость колена вскрывали рассечением и проводили лаваж колена между мыщелками большеберцовой кости и бедренной кости при помощи 3×25 мкл PBS, используя пипетку эппендорф. Фармакокинетические параметры РА в синовиальной жидкости (SF), рассчитанные из данного эксперимента, показаны ниже в Таблице 3.
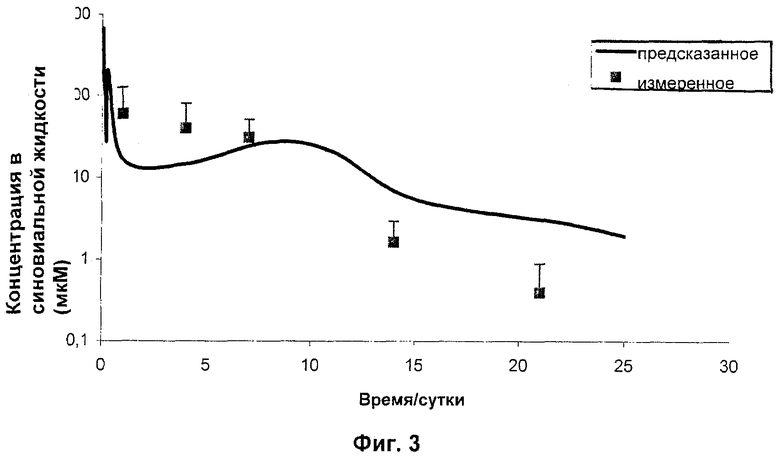
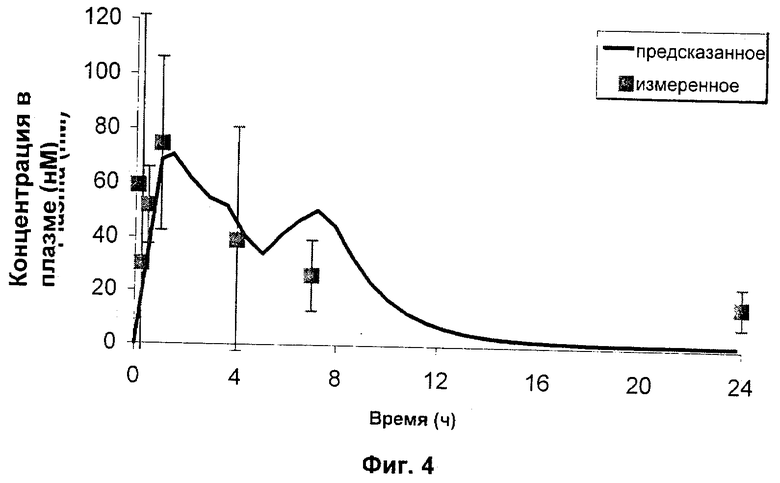
Затем N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамид в качестве РА в микрочастицах PLGA 50:50 (как получено в Примере 1) дозировали внутрисуставно (200 мкг в 30 мкл) крысам и определяли концентрации в синовиальной жидкости на сутки 1, 4, 7, 14 и 21 после дозирования. Данные, полученные в этом исследовании, представлены графически на Фиг.4 совместно с моделью ожидаемых концентраций в синовиальной жидкости на основе характеристик высвобождения in vitro для данной композиции (смотри Пример 1) и рассчитанным клиренсом высвобожденного лекарственного средства из синовиальной жидкости (Сl=61 мкл/час).
Эти данные ясно демонстрируют, что N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамид в качестве РА в микрочастицах PLGA 50:50 при инъецировании внутрисуставно крысам может поддерживать высвобождение в синовиальную жидкость в течение 21 суток. Кроме того, хорошее совпадение между предсказанными концентрациями и измеренными концентрациями показывает, что анализ высвобождения in vitro является хорошим средством прогноза поведения in vivo для данной композиции.
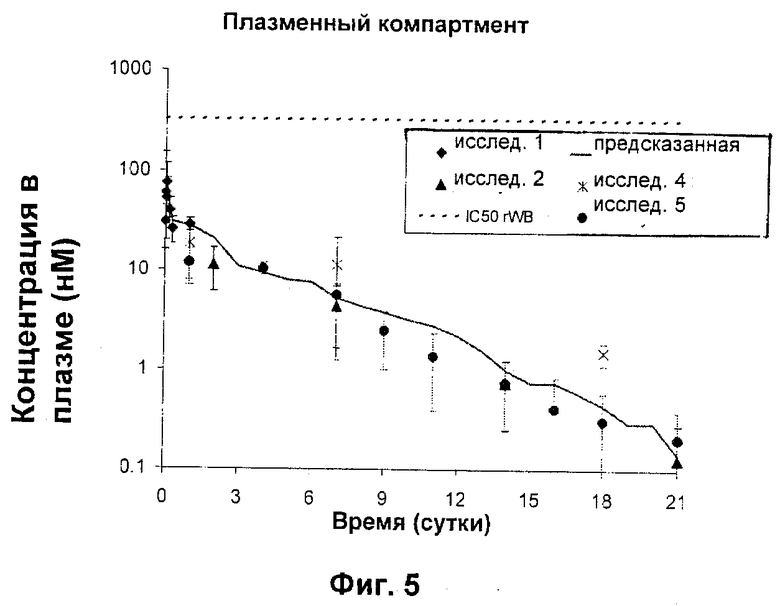
Затем N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамид в качестве РА в микрочастицах PLGA 50:50 (как в Примере 1) дозировали внутрисуставно (200 мкг) крысам и определяли концентрации в плазме вплоть до 24 часов и на 21-е сутки после дозирования. Полученные данные представлены графически на Фиг.4 и Фиг.5, соответственно, совместно с моделью ожидаемых концентраций в плазме на основе характеристик высвобождения в виде «всплеска» in vitro, рассчитанного клиренса высвобожденного лекарственного средства из синовиальной жидкости (Сl=61 мкл/час) и системных фармакокинетических параметров данного соединения у крыс (Сl=14 мл/мин/кг, Vdss=1,7 л/кг).
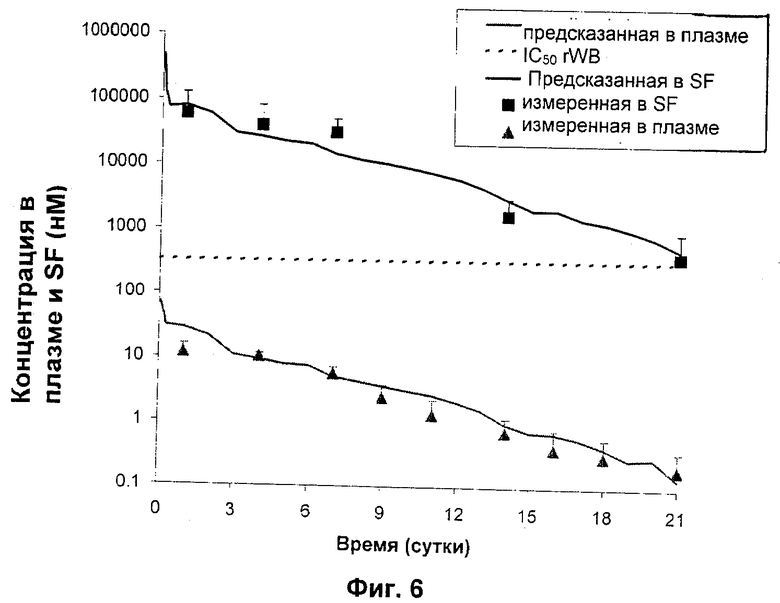
Как показано на Фиг.4 и 5, концентрации РА в плазме находились в наномолярном диапазоне (по сравнению с микромолярным диапазоном для синовиальной жидкости, как показано на Фиг.3), подтверждая представление о том, что внутрисуставная доставка посредством фармацевтического депо по настоящему изобретению может эффективно ослаблять системное воздействие даже во время пикового выброса РА из депо-композиций. Обобщенные результаты этих исследований представлены в одном и том же масштабе на Фиг.6 (где "предсказанная в SF" представляет собой верхнюю линию, а "ожидаемая в плазме" представляет собой нижнюю линию). Микрочастицы, инъецированные внутрисуставно, показали только небольшое количество потери РА вследствие эффектов «всплеска», приводящее к низким концентрациям в плазме и таким образом минимизирующее риск токсичности.
Таким образом, фармацевтическое депо, содержащее РА в микрочастицах PLGA, при инъецировании внутрисуставно (200 мкг) крысам может поддерживать высвобождение в синовиальную жидкость в течение вплоть до 21 суток и давать очень низкие концентрации в плазме вскоре после дозирования вследствие пониженного эффекта «всплеска». Кроме того, анализ высвобождения in vitro является хорошим средством прогноза поведения in vivo для микрочастиц PLGA 50:50.
Пример 4
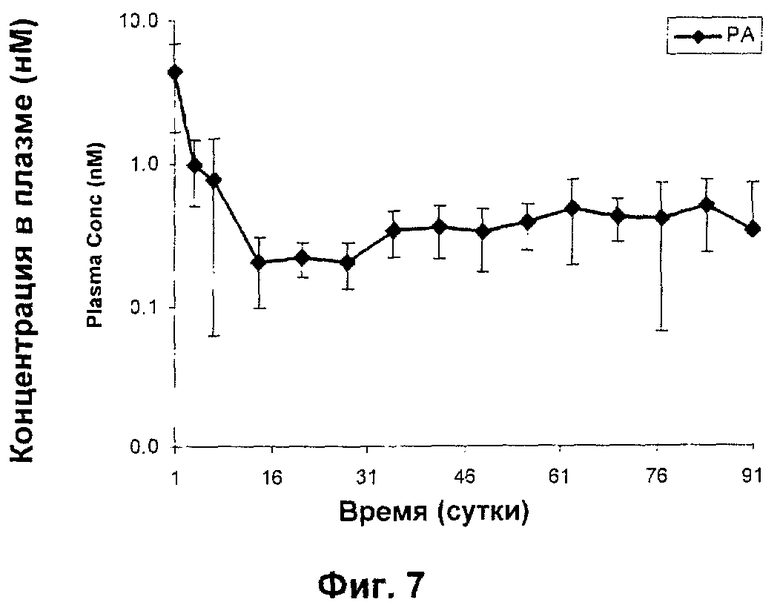
Изучали характеристики высвобождения N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамида в микрочастицах PLGA 50:50 in vivo у крыс.
N-{5-[(Циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамид в качестве РА в микрочастицах PLGA 50:50 (как в Примере 2) дозировали внутрисуставно (200 мкг) крысам и определяли концентрации в плазме вплоть до 91 суток после дозирования. Полученные данные графически представлены на Фиг.7 (который показывает профиль высвобождения in vivo РА в микрочастицах PLGA 95:5 у крыс).
Таким образом, фармацевтическое депо, содержащее РА в микрочастицах PLGA, при инъецировании внутрисуставно (200 мкг) крысам демонстрирует профиль высвобождение в плазме в течение 91 суток, дающий очень низкие концентрации в плазме вскоре после дозирования вследствие пониженного эффекта «всплеска».
Пример 5
Изучали длительную эффективность N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамида в качестве РА в фармацевтическом депо.
Все исследования выполняли на модели суставной боли у крыс под действием монойодоацетата (МIА) в качестве средства скрининга на анальгетическое действие в отношении боли, являющейся результатом суставного воспаления и разрушения (смотри Ivanavicius et al., 2007 Pain 128 р272). Модель MIA индуцирует начальный синовит (3-е сутки) с последующей прогрессивной потерей суставного хряща и патологию субхондральной кости к 14-м сутками.
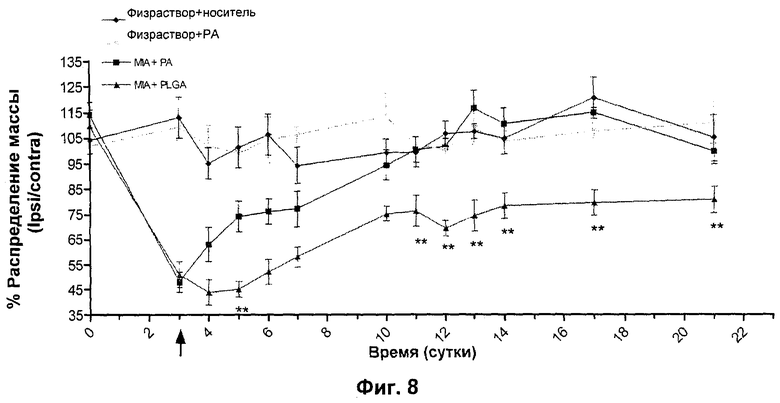
N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамид готовили в виде препарата в микросферах PLGA (50:50 PLGA как в Примере 1) и испытывали в модели MIA. Крысам внутрисуставно инъецировали MIA в сутки 0. Через трое суток после введения MIA (для того, чтобы заболевание развилось) животным внутрисуставно инъецировали препарат N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамида в PLGA 50:50 (200 мкг/30 мкл) или композицию в микросферах (30 мкл). Данные представлены на Фиг.3, и эти данные ясно показывают, что имеет место безотлагательная и длительная эффективность после инъекции приготовленного в виде препарата N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамида. Такая нормализация асимметрии нагрузки на лапу является статистически значимой через 48 часов после дозирования и от 6-х суток после дозирования вплоть до окончания исследования (18-е сутки после дозирования) и показана графически на Фиг.8.
Это демонстрирует длительную эффективность, достигаемую с использованием приготовленного в виде препарата N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамида в микросферах PLGA. Имела место полная инверсия асимметрии нагрузки на лапу.
Пример 6
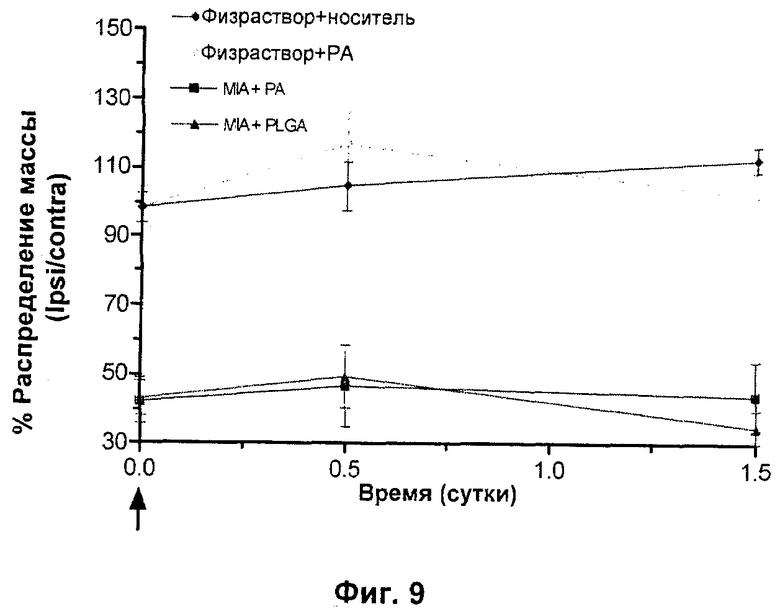
Исследование, аналогичное Примеру 5, выполняли для анализа эффектов N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамида не в виде препарата. Дозу 29 мкг/мл (69 мкМ) М-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамида доставляли в инъекции объемом 5 мкл с получением концентрации Смин 1 мкМ через 1,5 часа. Дозирование выполняли через 3 суток после MIA в тот же самый момент времени, в который дозировали приготовленный в виде препарата N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамид в качестве РА. Данные представлены на Фиг.9, и они ясно показывают отсутствие эффективности N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамида не в виде препарата в модели MIA на 3 сутки, свидетельствуя об абсолютной необходимости в N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамиде, представленном в виде препарата - депо, в суставе в течение длительных периодов времени для достижения фармакодинамического эффекта.
Изобретение относится к области фармацевтики и представляет собой фармацевтическое депо, изготовленное для введения посредством внутрисуставной инъекции в сустав субъекта, страдающего остеоартритом, содержащее микрочастицы или наночастицы, составленные из N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамида или его фармацевтически приемлемой соли и биоразлагаемого сополимера молочная кислота-гликолевая кислота. Изобретение обеспечивает контролируемое высвобождение активного агента и длительное действие без токсических побочных эффектов. 2 н. и 11 з.п. ф-лы, 5 пр., 3 табл., 9 ил.
1. Фармацевтическое депо, изготовленное для введения посредством внутрисуставной инъекции в сустав субъекта, страдающего остеоартритом, содержащее микрочастицы или наночастицы, составленные из N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамида или его фармацевтически приемлемой соли и биоразлагаемого сополимера молочная кислота-гликолевая кислота.
2. Фармацевтическое депо по п.1, где сополимер молочная кислота-гликолевая кислота имеет молярное соотношение молочная кислота:гликолевая кислота в интервале от 100:0 до 50:50.
3. Фармацевтическое депо по п.1, где сополимер молочная кислота-гликолевая кислота имеет молярное соотношение молочная кислота:гликолевая кислота, равное 95:5.
4. Фармацевтическое депо по п.1, где сополимер молочная кислота-гликолевая кислота имеет молярное соотношение молочная кислота:гликолевая кислота, равное 50:50.
5. Фармацевтическое депо по п.1, дополнительно содержащее усилитель вязкости.
6. Фармацевтическое депо по п.5, где усилитель вязкости представляет собой натриевую карбоксиметилцеллюлозу.
7. Фармацевтическое депо по п.1, дополнительно содержащее поверхностно-активное вещество.
8. Фармацевтическое депо по п.7, где поверхностно-активное вещество представляет собой полисорбат 80.
9. Фармацевтическое депо по п.1, где указанное соединение инкапсулировано в сополимере молочная кислота-гликолевая кислота.
10. Фармацевтическое депо по п.9, где указанное соединение диспергировано в сополимере молочная кислота-гликолевая кислота.
11. Фармацевтическое депо по любому из пп.1-10, которое изготовлено для контролируемого и/или непрерывного высвобождения N-{5-[(циклопропиламино)карбонил]-2-метилфенил}-3-фтор-4-(пиридин-2-илметокси)бензамида в течение периода от примерно 30 до 90 суток.
12. Применение фармацевтического депо по любому из пп.1-11 для лечения остеоартрита.
13. Применение по п.12 для лечения остеоартрита коленного сустава.
| US 20070135440 A1, 14.06.2007 | |||
| US 2004151753 A1, 05.08.2004 | |||
| US 20060173050 A1, 03.08.2006 | |||
| US 20040082585 A1, 29.04.2004 | |||
| US 20050049288 A1, 03.03.2005 | |||
| ЩЕТОЧНОЕ УПЛОТНЕНИЕ | 1987 |
|
SU1484033A1 |
| S | |||
| BOZDAG, S | |||
| CALIS, H.S | |||
| KAS, M.T | |||
| ERCAN, I | |||
| PEKSOY, A.A | |||
| HINCAL / In vitro evaluation and intra-articular administration of biodegrable microspheres containing | |||

