ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
Данная заявка испрашивает преимущество предварительной заявки на патент США № 62/584589, поданной 10 ноября 2017 г., и предварительной заявки на патент США № 62/743864, поданной 10 октября 2018 г.; каждая из которых включена в данный документ ссылкой во всей своей полноте.
ОБЛАСТЬ ТЕХНИКИ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к препаратам с пролонгированным высвобождением для лечения или предотвращения повреждения суставов, вызванного артритом, травмой сустава или травмой хряща.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Артрит - это воспаление одного или нескольких суставов, которым в мире страдает около 350 миллионов человек. Остеоартрит (ОА) - это наиболее распространенная форма артрита и характеризуется медленным дегенеративным разрушением сустава, затрагивающим как суставный хрящ, так и субхондральную кость, лежащую под суставным хрящом. Повреждение сустава (например, острая травма сустава, такая как разрыв мениска или связки, или внутрисуставный перелом) также может привести к артриту, например, к посттравматическому артриту. Поскольку суставный хрящ имеет ограниченную способность к восстановлению, даже небольшое невыявляемое повреждение зачастую может со временем ухудшиться и привести к возникновению ОА.
В PCT/US15/30303, которая полностью включена в настоящее описание ссылкой, описаны N-(3,4-дихлорфенил)-3-(пиридин-4-ил)-7-оксабицикло[2.2.1]гептан-2-карбоксамид (соединение А) и другие соединения, которые полезны для предотвращения, улучшения или лечения артрита и/или травмы суставов.
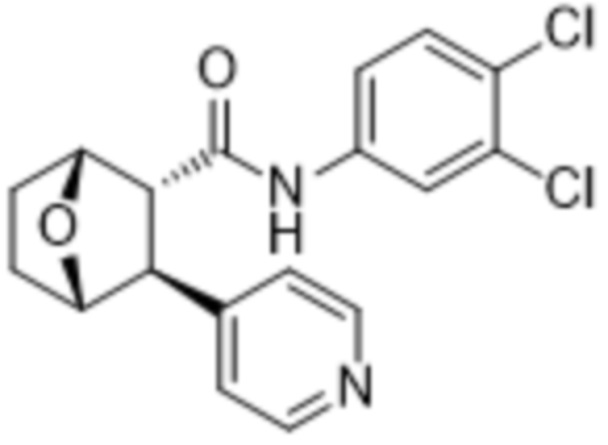 (Соединение A)
(Соединение A)
Соединение A является слабым основанием с pKa 5,2 и плохо растворимо в водных растворах с нейтральным pH и щелочным pH, но его растворимость сильно зависит от pH. Растворимость увеличивается с понижением рН. Соединение А может быть составлено в виде препаратов с быстрым высвобождением для внутрисуставной инъекции. Однако из-за высокого обмена жидкости между синовиальной жидкостью и кровотоком препараты с быстрым высвобождением имеют короткий период полувыведения в синовиальной среде и требуют частых инъекций.
Таким образом, по-прежнему существует потребность в препаратах, которые как можно дольше поддерживают уровни содержания лекарственной субстанции в синовиальной полости, достаточные для лечения хронических симптомов.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В настоящем изобретении предложены препараты с пролонгированным высвобождением, содержащие N-(3,4-дихлорфенил)-3-(пиридин-4-ил)-7-оксабицикло[2.2.1]гептан-2-карбоксамид или его фармацевтически приемлемую соль.
В одном аспекте настоящее изобретение относится к фармацевтическому составу, содержащему водную суспензию: (i) кристаллического N-(3,4-дихлорфенил)-3-(пиридин-4-ил)-7-оксабицикло[2.2.1]гептан-2-карбоксамида или его фармацевтически приемлемой соли; и (ii) поверхностно-активного вещества, содержащего водорастворимый сополимер, характеризующийся растворимостью в воде > 5% при 25oC.
В данном документе описаны различные пронумерованные варианты осуществления настоящего изобретения. Признаки, описанные в каждом варианте осуществления, можно объединять с другими описанными признаками с получением дополнительных вариантов осуществления настоящего изобретения. В данном документе процентное содержание ингредиентов в составах приведено в % (мас./об.), если не указано иное.
Вариант осуществления 1. Фармацевтический состав, как описано выше. В одном варианте осуществления состав представляет собой препарат для инъекций с пролонгированным высвобождением.
Вариант осуществления 2. Фармацевтический состав согласно варианту осуществления 1, где указанное поверхностно-активное вещество (ПАВ) представляет собой водорастворимый сополимер, гидрофильно-липофильный баланс (ГЛБ) которого имеет значение не менее 18.
Вариант осуществления 3. Фармацевтический состав согласно варианту осуществления 1 или 2, где указанное ПАВ представляет собой водорастворимый сополимер, средняя молекулярная масса которого находится в пределах 7500-15000 дальтон.
Вариант осуществления 4. Фармацевтический состав согласно варианту осуществления 1 или 2, где указанное ПАВ представляет собой водорастворимый сополимер, средняя молекулярная масса которого находится в пределах 8000-13000 дальтон.
Вариант осуществления 5. Фармацевтический состав согласно любому из вариантов осуществления 1-4, где указанное ПАВ представляет собой водорастворимый сополимер, характеризующийся растворимостью в воде >5% при 25oC при 1 атм.
Вариант осуществления 6. Фармацевтический состав согласно любому из вариантов осуществления 1-4, где указанное ПАВ представляет собой водорастворимый сополимер, характеризующийся растворимостью в воде >10% при 25oC при 1 атм.
Вариант осуществления 7. Фармацевтический состав согласно любому из вариантов осуществления 1-6, где указанное ПАВ представляет собой водорастворимый блок-сополимер и, необязательно, водорастворимый тройной блок-сополимер.
Вариант осуществления 8. Фармацевтический состав согласно любому из вариантов осуществления 1-7, где указанное ПАВ представляет собой водорастворимый блок-сополимер формулы
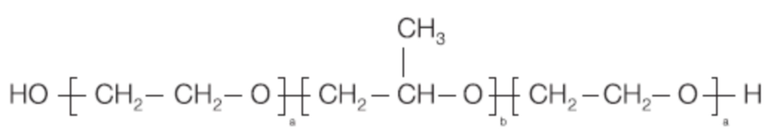
где
а равно 75-101; и
b равно 25-60.
Вариант осуществления 9. Фармацевтический состав согласно варианту осуществления 8, где указанный водорастворимый блок-сополимер представляет собой полоксамер 188.
Вариант осуществления 10. Фармацевтический состав согласно варианту осуществления 8, где указанный водорастворимый блок-сополимер представляет собой полоксамер 407.
Вариант осуществления 11. Фармацевтический состав согласно любому из вариантов осуществления 8-10, где концентрация указанного водорастворимого блок-сополимера составляет не менее 0,025% (мас./об.), например, в интервале 0,025-2% (мас./об.), в интервале 0,025-1% (мас./об.), или предпочтительно - в интервале 0,05-1% (мас./об.).
Вариант осуществления 12. Фармацевтический состав согласно варианту осуществления 1, где указанное ПАВ дополнительно содержит лаурилсульфат натрия. В одном варианте осуществления концентрация указанного лаурилсульфата натрия составляет не менее 0,05% (мас./об.); более конкретно она находится в интервале 0,05-0,5% (мас./об.).
Вариант осуществления 13. Фармацевтический состав согласно любому из вариантов осуществления 1-12, дополнительно содержащий стабилизатор суспензии. В одном варианте осуществления концентрация указанного стабилизатора суспензии составляет 0,1-10% (мас./об.).
Вариант осуществления 14. Фармацевтический состав согласно варианту осуществления 13, в котором указанный стабилизатор суспензии выбран из: (i) поливинилпирролидона со средней молекулярной массой от 1 до 10 кДа и, необязательно, от 2 до 5 кДа; (ii) карбоксиметилцеллюлозы со средней молекулярной массой от 25 до 2500 кДа и, необязательно, от 75 до 125 кДа; и (iii) их комбинации.
Вариант осуществления 15. Фармацевтический состав согласно варианту 14, где концентрация указанного поливинилпирролидона составляет 1-5% (мас./об.) и, необязательно, примерно 2-4% (мас./об.); и концентрация указанной карбоксиметилцеллюлозы составляет 0,5-2% (мас./об.) и, необязательно, составляет 0,75-1,5% (мас./об.).
Вариант осуществления 16. Фармацевтический состав согласно варианту осуществления 15, где указанным поливинилпирролидоном является PVP-12.
Вариант осуществления 17. Фармацевтический состав согласно варианту осуществления 1, где указанная водная суспензия содержит: (i) 1-400 мг кристаллического N-(3,4-дихлорфенил)-3-(пиридин-4-ил)-7-оксабицикло[2.2.1]гептан-2-карбоксамида в 1 мл указанной водной суспензии; (ii) 0,05-1% (мас./об.) водорастворимого блок-сополимера, необязательно, где указанным водорастворимым блок-сополимером является полоксамер 188 или полоксамер 407; и (iii) 1-5% (мас./об.) поливинилпирролидона, необязательно, где указанным поливинилпирролидоном является PVP-K12.
Вариант осуществления 18. Фармацевтический состав согласно варианту осуществления 17, содержащий 10-30 мг или примерно 25 мг кристаллического N-(3,4-дихлорфенил)-3-(пиридин-4-ил)-7-оксабицикло[2.2.1]гептан-2-карбоксамида в 1 мл указанной водной суспензии.
Вариант осуществления 19. Фармацевтический состав согласно варианту осуществления 18, содержащий 0,05-0,15% (мас./об.), 0,08-0,12% (мас./об.) или примерно 0,1% (мас./об.) полоксамера 407.
Вариант осуществления 20. Фармацевтический состав согласно варианту осуществления 17, содержащий вплоть до 75 мг, необязательно, 40-60 мг или примерно 50 мг кристаллического N-(3,4-дихлорфенил)-3-(пиридин-4-ил)-7-оксабицикло[2.2.1]гептан-2-карбоксамида в 1 мл указанной водной суспензии.
Вариант осуществления 21. Фармацевтический состав согласно варианту осуществления 17, содержащий 80-120 мг или примерно 100 мг кристаллического N-(3,4-дихлорфенил)-3-(пиридин-4-ил)-7-оксабицикло[2.2.1]гептан-2-карбоксамида в 1 мл указанной водной суспензии.
Вариант осуществления 22. Фармацевтический состав согласно любому из вариантов осуществления 20-21, содержащий 0,1-0,3% (мас./об.) или примерно 0,2% (мас./об.) полоксамера 407.
Вариант осуществления 23. Фармацевтический состав согласно варианту осуществления 17, содержащий 150-250 мг или примерно 200 мг кристаллического N-(3,4-дихлорфенил)-3-(пиридин-4-ил)-7-оксабицикло[2.2.1]гептан-2-карбоксамида в 1 мл указанной водной суспензии.
Вариант осуществления 24. Фармацевтический состав согласно варианту осуществления 23, содержащий 0,1-0,5% (мас./об.) или примерно 0,2-0,4% (мас./об.) полоксамера 407.
Вариант осуществления 25. Фармацевтический состав согласно варианту осуществления 17, содержащий 350-450 мг или примерно 400 мг кристаллического N-(3,4-дихлорфенил)-3-(пиридин-4-ил)-7-оксабицикло[2.2.1]гептан-2-карбоксамида в 1 мл указанной водной суспензии.
Вариант осуществления 26. Фармацевтический состав согласно варианту осуществления 25, содержащий 0,6-1% (мас./об.), 0,7-0,9% (мас./об.) или примерно 0,8% (мас./об.) полоксамера 407.
Вариант осуществления 27. Фармацевтический состав согласно варианту осуществления 18 или 19, содержащий 1,5-2,5% (мас./об.) или примерно 2% (мас./об.) PVP-K12.
Вариант осуществления 28. Фармацевтический состав согласно любому из вариантов осуществления 20-26, содержащий 1-5% (мас./об.) или примерно 2-4% (мас./об.) PVP-K12.
Вариант осуществления 29. Фармацевтический состав согласно варианту осуществления 1, где указанная водная суспензия содержит: (i) 1-100 мг кристаллического N-(3,4-дихлорфенил)-3-(пиридин-4-ил)-7-оксабицикло[2.2.1]гептан-2-карбоксамида в 1 мл указанной водной суспензии; (ii) 0,5-1,5 мг или примерно 1 мг полоксамера 407 в 1 мл указанной водной суспензии; и (iii) 15-25 мг или примерно 20 мг PVP-K12 в 1 мл указанной водной суспензии.
Вариант осуществления 30. Фармацевтический состав согласно варианту осуществления 29, содержащий 20-30 мг или примерно 25 мг кристаллического N-(3,4-дихлорфенил)-3-(пиридин-4-ил)-7-оксабицикло[2.2.1]гептан-2-карбоксамида в 1 мл указанной водной суспензии.
Вариант осуществления 31. Фармацевтический состав согласно любому из вариантов осуществления 1-30, где указанный кристаллический N-(3,4-дихлорфенил)-3-(пиридин-4-ил)-7-оксабицикло[2.2.1]гептан-2-карбоксамид имеет микрокристаллическую или микронизированную форму.
Вариант осуществления 32. Фармацевтический состав согласно любому из вариантов осуществления 1-30, где указанный кристаллический N-(3,4-дихлорфенил)-3-(пиридин-4-ил)-7-оксабицикло[2.2.1]гептан-2-карбоксамид имеет распределение частиц по размерам D90, равное 50 микрон или меньше, измеренное по дифракции лазерного излучения в суспензии при длине волны 610-650 нм и, в частности, при примерно 630-635 нм.
Вариант осуществления 33. Фармацевтический состав согласно любому из вариантов осуществления 1-30, где указанный кристаллический N-(3,4-дихлорфенил)-3-(пиридин-4-ил)-7-оксабицикло[2.2.1]гептан-2-карбоксамид имеет распределение частиц по размерам D90, равное 25 микрон или меньше, измеренное по дифракции лазерного излучения в суспензии при длине волны 610-650 нм и, в частности, при примерно 630-635 нм.
Вариант осуществления 34. Фармацевтический состав согласно любому из вариантов осуществления 1-33, где указанный N-(3,4-дихлорфенил)-3-(пиридин-4-ил)-7-оксабицикло[2.2.1]гептан-2-карбоксамид дополнительно определен как (1R,2R,3S,4S)-N-(3,4-дихлорфенил)-3-(пиридин-4-ил)-7-оксабицикло[2.2.1]гептан-2-карбоксамид.
Вариант осуществления 35. Фармацевтический состав согласно любому из вариантов осуществления 1-34, дополнительно содержащий буфер, способный поддерживать рН суспензии при значении 6-8.
Вариант осуществления 36. Фармацевтический состав согласно любому из вариантов осуществления 1-35, где указанный состав подходит для внутрисуставной инъекции.
Вариант осуществления 37. Фармацевтический состав согласно варианту осуществления 36, где указанный состав подходит для внутрисуставной инъекции в синовиальную полость, особенно в коленный сустав пациента, страдающего от артрита, травмы сустава или травмы хряща. В одном варианте осуществления состав подходит для внутрисуставной инъекции в синовиальную полость пациента, страдающего остеоартритом. В другом варианте осуществления состав подходит для внутрисуставной инъекции в синовиальную полость пациента, страдающего травматическим артритом. В другом варианте осуществления состав подходит для внутрисуставной инъекции в синовиальную полость пациента, страдающего аутоиммунным артритом.
Вариант осуществления 38. Фармацевтический состав согласно варианту осуществления 37, где указанный состав обеспечивает пролонгированное высвобождение N-(3,4-дихлорфенил)-3-(пиридин-4-ил)-7-оксабицикло[2.2.1]гептан-2-карбоксамида в синовиальной полости в течение более одного часа, 24 часов, 7 дней, 14 дней или 30 дней.
Вариант осуществления 39. Фармацевтический состав согласно любому из вариантов осуществления 1-35, где указанная композиция подходит для введения иглой калибра 22-31, иглой калибра 29-31 или предпочтительно иглой 30-го калибра.
Вариант осуществления 40. Комбинация, включающая фармацевтический состав согласно любому из вариантов осуществления 1-35 и второе терапевтическое средство.
Вариант осуществления 41. Набор, содержащий фармацевтический состав согласно любому из вариантов осуществления 1-35 и, по меньшей мере, инструкцию по применению или иглу.
Вариант осуществления 42. Фармацевтический состав согласно любому из вариантов осуществления 1-35 и, необязательно, в комбинации со вторым терапевтическим средством, для лечения, улучшения или предотвращения повреждения или травмы сустава, такого как артрит (остеоартрит, травматический артрит или аутоиммунный артрит, такой как системный ревматоидный артрит); дегенеративное заболевание диска; острая травма сустава или хряща.
Вариант осуществления 43. Фармацевтический состав согласно любому из вариантов осуществления 1-35 и, необязательно, в комбинации со вторым терапевтическим средством для индуцирования образования гиалинового хряща или для индуцирования дифференцировки хондроцитов.
Вариант осуществления 44. Применение фармацевтического состава согласно любому из вариантов осуществления 1-35 и, необязательно, в комбинации со вторым терапевтическим средством, для изготовления лекарственного средства для лечения повреждения или травмы сустава, такого как артрит (остеоартрит, травматический артрит или аутоиммунный артрит, такой как системный ревматоидный артрит); дегенеративное заболевание диска; острая травма сустава или хряща.
Вариант осуществления 45. Применение фармацевтического состава согласно любому из вариантов осуществления 1-35 и, необязательно, в комбинации со вторым терапевтическим средством для изготовления лекарственного средства для индуцирования образования гиалинового хряща или для индуцирования дифференцировки хондроцитов.
Вариант осуществления 46. Способ лечения, улучшения или предотвращения острого повреждения или травмы сустава у субъекта, нуждающегося в этом, включающий введение фармацевтического состава согласно любому из вариантов осуществления 1-35 и, необязательно, в комбинации со вторым терапевтическим средством нуждающемуся в этом субъекту; тем самым осуществляя лечение, улучшение или предотвращение острого повреждения или травмы сустава у указанного субъекта.
Вариант осуществления 47. Способ согласно варианту осуществления 46, где указанный фармацевтический состав вводят указанному субъекту, причем доза кристаллического N-(3,4-дихлорфенил)-3-(пиридин-4-ил)-7-оксабицикло[2.2.1]гептан-2-карбоксамида составляет не более 25 мг, не более 75 мг, не более 100 мг, не более 200 мг или не более 400 мг.
Вариант осуществления 48. Способ согласно варианту осуществления 46 или варианту осуществления 47, включающий инъекцию фармацевтического состава в синовиальную полость индивидуума, страдающего остеоартритом.
Вариант осуществления 49. Способ индуцирования образования гиалинового хряща или дифференцировки хондроцитов, включающий осуществление контакта хондрогенных клеток-предшественников с терапевтически эффективным количеством фармацевтического состава согласно любому из вариантов осуществления 1-35 и, необязательно, в комбинации со вторым терапевтическим средством; тем самым индуцируя образование внеклеточного матрикса гиалинового хряща.
Вариант осуществления 50. Способ согласно варианту осуществления 49, где указанную стадию контакта у млекопитающего осуществляют in vitro или in vivo; и при проведении стадии in vivo у млекопитающего должны быть в наличии стволовые клетки.
Вариант осуществления 51. Способ согласно варианту осуществления 49 или 50, где указанная стадия осуществления контакта происходит в матриксе или биосовместимом каркасе.
Вариант осуществления 52. Фармацевтический состав согласно варианту осуществления 42 или 43, применение согласно варианту осуществления 44 или 45 или способы согласно любому из вариантов осуществления 46-51, где указанное второе терапевтическое средство выбирают из ангиопоэтин-подобного белка 3 (ANGPTL3), инсулинового фактора роста (IGF1), SM04690, ингибитора Янус-киназы, перорального кальцитонина лососевых рыб, SD-6010, витамина D3, гидролизованного коллагена, костного морфогенетического белка 7 (BMP7), амидацетата ТР508, неомыляемых веществ авокадо и сои (ASU), стероида, нестероидного противовоспалительного средства (НПВС), гиалуроновой кислоты, картогенина и TPX-100.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фигура 1 (ФИГ.1) представляет собой электронно-микроскопическое изображение cоединения А через три месяца при 40oC.
На фигуре 2 (ФИГ. 2) сравнивается концентрация соединения А в плазме крови крысы после внутрисуставного введения трех препаратов соединения А с пролонгированным высвобождением: суспензии микрокристаллов, суспензии микрочастиц сополимера лактида с гилколидом (PLGA) и липосомальной суспензии многослойных везикул (MLV).
На фигуре 3 (ФИГ. 3) сравнивается профиль in vivo суспензии микрокристаллов с пролонгированным высвобождением соединения А (250 мкг) с препаратом раствора с быстрым высвобождением (91 нг).
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В настоящем изобретении предложены препараты с пролонгированным высвобождением, содержащие суспензию микрокристаллов N-(3,4-дихлорфенил)-3-(пиридин-4-ил)-7-оксабицикло[2.2.1]гептан-2-карбоксамида или его фармацевтически приемлемой соли и поверхностно-активное вещество (ПАВ), содержащее водорастворимый сополимер.
Настоящее изобретение решает множество проблем, связанных с приготовлением суспензии микрокристаллов соединения A, включая указание на формообразующие, которые стабилизируют лекарственную суспензию и обеспечивают приемлемые характеристики при изготовлении (смешивание/наполнение) и введении (точность дозирования, проходимость через иглу) высококонцентрированной суспензии кристаллов (до 100 мг/мл), включая, но не ограничиваясь этим, следующее:
- Аспекты смешивания:
o Быстрая суспендируемость лекарственной субстанции (ЛС) (хорошая смачиваемость ЛС)
o Медленное осаждение исходной суспензии (предотвращение агрегации микрокристаллов ЛC) для обеспечения точности дозирования в ходе производства в соответствии с Правилами производства и контроля качества лекарственных средств (GMP)
- Аспекты после обработки в автоклаве или хранения:
o Химическая стабильность лекарственной субстанции;
o Способность к быстрому повторному суспендированию микрокристаллов
o Проходимость через иглу 30-го калибра для безболезненной инъекции в колено
o Медленное осаждение для обеспечения точности дозирования
o Отсутствие крупных кристаллов (созревание Освальда) или агрегатов
Определения
Используемый здесь термин «пролонгированное высвобождение» относится к лекарственной форме, которая специально модифицирована для замедления скорости высвобождения лекарственной субстанции по сравнению с той, которая наблюдается для лекарственной формы с быстрым высвобождением. Схема высвобождения при дозировании с пролонгированным высвобождением может начинаться со взрывного эффекта, который имитирует быстрое высвобождение, с последующим более медленным высвобождением оставшейся в лекарственной форме лекарственной субстанции.
Используемый здесь термин «примерно» означает, что величина находится в пределах статистически значимого интервала значений, обычно в пределах 10%. Такой интервал может находиться в пределах ошибки эксперимента, типичной для стандартных методов, используемых для измерения и/или определения заданного значения или интервала. В одном варианте осуществления интервал находится в пределах 5% от указанного значения. В другом варианте осуществления интервал находится в пределах 1% от указанного значения. В еще одном варианте осуществления интервал находится в пределах 0,5% от указанного значения.
Используемый здесь термин «субъект» относится к приматам (например, людям - мужчинам или женщинам), собакам, кроликам, морским свинкам, свиньям, крысам, мышам и лошадям. В определенных вариантах осуществления субъектом является примат. В других вариантах осуществления субъектом является человек.
Применяемый в данном документе термин «лечить», «осуществление лечения» или «лечение» любого заболевания или нарушения относится к облегчению или снижению тяжести заболевания или нарушения (т. е. замедлению или остановке развития заболевания или по меньшей мере одного из его клинических симптомов); или облегчению или уменьшению по меньшей мере одного физического параметра или биомаркера, ассоциированного с заболеванием или нарушением, включая таковые, которые могут быть не очевидными для пациента.
В данном контексте термин «предотвращать», «предотвращающий» или «предотвращение» любого заболевания или нарушения относится к профилактическому лечению заболевания или нарушения или замедлению начала или прогрессирования заболевания или нарушения.
В данном контексте субъект «нуждается в» лечении, если в результате такого лечения такой субъект получит пользу с биологической, медицинской точки зрения, или улучшится качество его жизни.
В данном контексте термин «терапевтически эффективное количество» фармацевтического состава относится к количеству состава, которое будет вызывать биологический или медицинский ответ у субъекта, например, снижение или подавление активности фермента или белка, или уменьшать тяжесть симптомов, облегчать состояние, замедлять или сдерживать прогрессирование заболевания, или предотвращать заболевание и т.д. В одном неограничивающем варианте осуществления термин «терапевтически эффективное количество» относится к количеству cоединения А препарата с пролонгированным высвобождением, которое при введении субъекту является эффективным в отношении (1) по меньшей мере частичного облегчения, подавления, предотвращения и/или снижения тяжести повреждения сустава в результате травмы сустава и артрита. В другом неограничивающем варианте осуществления выражение «терапевтически эффективное количество» относится к количеству cоединения А препарата с пролонгированным высвобождением, которое при введении в клетку, или ткань, или неклеточный биологический материал, или среду эффективно для стимулирования хондрогенеза.
Используемые здесь термины «лечить», «проведение лечения», «лечение», а также «улучшать» и «улучшение» относятся к любым признакам успеха в лечении или улучшении состояния травмы, патологии, состояния или симптома (например, боли), включая любые объективные или субъективные параметры, такие как ослабление боли; ремиссия; уменьшение выраженности симптомов или облегчение переносимости симптома, травмы, патологии или состояния для пациента; уменьшение частоты или продолжительности симптома или состояния; или, в некоторых ситуациях, предотвращение появления симптома или состояния. Лечение или улучшение симптомов может быть основано на любом объективном или субъективном параметре; включая, например, на основании результата медицинского осмотра.
Используемый здесь термин «введение» относится к введению в определенный сустав.
Применяемый в данном документе термин «фармацевтическая композиция» относится к соединению или его фармацевтически приемлемой соли вместе с по меньшей мере одним фармацевтически приемлемым носителем в подходящей для перорального или парентерального введения форме.
Применяемый в данном документе термин «фармацевтически приемлемый носитель» относится к веществу, пригодному для составления или применения фармацевтического состава, и включает, например, подходящие разбавители, растворители, дисперсионные среды, ПАВ, антиоксиданты, консерванты, изотонические средства, буферные средства, эмульгаторы, средства, замедляющие абсорбцию, соли, стабилизаторы лекарственных средств, связующие средства, формообразующие, разрыхляющие средства, смазочные средства, смачивающие средства, подсластители, ароматизирующие средства, красители и их комбинации, которые должны быть известны специалистам в данной области техники (см., например, Remington The Science and Practice of Pharmacy, 22nd Ed. Pharmaceutical Press, 2013, pp. 1049-1070).
Используемые в данном документе термины в форме единственного числа, «упомянутый» и подобные термины, используемые в контексте настоящего изобретения (в частности, в контексте формулы изобретения), следует истолковывать как охватывающие и формы единственного числа, и формы множественного числа, если в данном документе не указано иное или нет явного противоречия контексту.
Настоящее изобретение относится к препаратам с пролонгированным высвобождением, содержащим N-(3,4-дихлорфенил)-3-(пиридин-4-ил)-7-оксабицикло[2.2.1]гептан-2-карбоксамид или его фармацевтически приемлемую соль, в частности, препарату с пролонгированным высвобождением для инъекции, подходящему для внутрисуставной инъекции в сустав пациента, страдающего от артрита, травмы сустава или травмы хряща.
В одном аспекте настоящее изобретение относится к фармацевтическому составу, содержащему водную суспензию: (i) кристаллического N-(3,4-дихлорфенил)-3-(пиридин-4-ил)-7-оксабицикло[2.2.1]гептан-2-карбоксамида или его фармацевтически приемлемой соли; и (ii) поверхностно-активного вещества, содержащего водорастворимый сополимер, характеризующийся растворимостью в воде > 5% при 25oC.
В одном варианте осуществления фармацевтический состав включает ПАВ, которое представляет собой водорастворимый блок-сополимер, гидрофильно-липофильный баланс (ГЛБ) которого имеет значение не менее 18. Гидрофильно-липофильный баланс (ГЛБ) поверхностно-активного вещества является мерой степени, в которой он является гидрофильным или липофильным, его определяют путем вычисления значений для различных областей молекулы с использованием методов, известных специалистам в данной области (например, Техническая информация о полоксамерах типа BASF® PLURACARE® L/F, 04_070801e-01, июль 2009 г.; Техническая информация о BASF® Kolliphor® P, 03_111136e-03).
Примеры водорастворимых блок-сополимеров, которые могут подходить для использования с фармацевтическими композициями, раскрытыми в данном документе, включают, но не ограничиваются ими, полоксамер 188 (LUTROL® F-68), полоксамер 237 (PLURONIC® F-87), полоксамер 338 (PLURONIC® F-108) и полоксамер 407 (PLURONIC® F-127 или LUTROL® F-127) или их смесь. Концентрация полоксамера в общем случае составляет примерно > 0,025%; например, 0,025-2% (мас./об.).
В другом варианте осуществления фармацевтический состав может дополнительно содержать стабилизатор суспензии, такой как поливинилпирролидин (PVP), карбоксиметилцеллюлоза или их комбинация. В конкретных вариантах осуществления дополнительный стабилизатор суспензии представляет собой PVP-K12, отдельно или в комбинации с карбоксиметилцеллюлозой.
В еще одном варианте осуществления фармацевтический состав дополнительно содержит подходящий буфер, способный поддерживать рН водной суспензии при физиологически приемлемом значении рН, равном 6-8 или примерно 7,2-7,4. В одном варианте осуществления фармацевтический состав содержит фосфатный буфер. Для использования в фармацевтическиих составах, раскрываемых в данном документе, можно рассматривать другие известные буферные агенты, включая, но не ограничиваясь ими, соли органических кислот, TRIS или гидрохлорид трометамина. В другом варианте осуществления фармацевтический состав содержит NaCl для инъекций.
Фармацевтические составы настоящего изобретения можно вводить одновременно с одним или несколькими другими терапевтическими средствами, или до или после них. Фармацевтические составы настоящего изобретения можно вводить отдельно или вместе с одним или несколькими терапевтическими средствами одним и тем же или разными путями введения. Терапевтическое средство представляет собой, например, химическое соединение, пептид, антитело, фрагмент антитела или нуклеиновую кислоту, которые проявляют терапевтическое действие или усиливают терапевтическое действие при введении пациенту в комбинации с препаратом пролонгированного высвобождения соединения А или его фармацевтической соли.
В одном варианте осуществления изобретение относится к продукту, содержащему препарат с пролонгированным высвобождением соединения А или его фармацевтически приемлемой соли и по меньшей мере одно другое терапевтическое средство в качестве комбинированной лекарственной формы для одновременного, раздельного или последовательного применения в терапии. В одном варианте осуществления терапия представляет собой лечение повреждения сустава в результате травмы сустава или артрита. Продукты, предоставляемые в виде комбинированной лекарственной формы, включают состав, содержащий препарат с пролонгированным высвобождением соединения А или его фармацевтически приемлемой соли и другое(ие) терапевтическое(ие) средство(а) вместе в одном и том же фармацевтическом составе; или препарат с пролонгированным высвобождением соединения А или его фармацевтически приемлемой соли и другое(ие) терапевтическое(ие) средство(а) в отдельной форме, например, в форме набора.
В одном варианте осуществления изобретение относится к препарату с пролонгированным высвобождением соединения А или его фармацевтически приемлемой соли в комбинации со вторым терапевтическим средством. Вторым средством может быть одно или несколько дополнительных средств для дифференцировки хондроцитов. Примеры средства для дифференцировки хондроцитов включают, но не ограничиваются ими, ангиопоэтин-подобный белок 3 (ANGPTL3), инсулиновыйого фактор роста (IGF1), SM04690 (ингибитор Wnt), ингибиторы Янус-киназы (такие как руксолитиниб, тофацитиниб, барицитиниб), пероральный кальцитонин лососевых рыб, SD-6010 (ингибитор iNOS), витамин D3 (холекальциферол), гидролизованный коллаген, костный морфогенетический белок 7 (BMP7), ацетат русалатида, неомыляемые вещества авокадо и сои (ASU), стероид, нестероидное противовоспалительное средство (НПВС), гиалуроновую кислоту, картогенин и TPX-100.
Фармацевтический состав или комбинация настоящего изобретения может быть в стандартной лекарственной форме с 0,5-1000 мг активного(ых) ингредиента(ов) для субъекта весом примерно 50-70 кг; например, в стандартной лекарственной форме с содержанием активного(ых) ингредиента(ов) в интервале 0,5-500 мг, 0,5-250 мг, 0,5-150 мг, 0,5-100 мг или 0,5-50 мг. Терапевтически эффективная доза соединения, фармацевтического состава или их комбинаций зависит от вида субъекта, веса тела, возраста и индивидуального состояния, нарушения или заболевания, лечение которых осуществляют, или их тяжести. Квалифицированный лечащий врач, клиницист или ветеринар может легко определить эффективное количество каждого из активных ингредиентов, необходимое для предотвращения, лечения или подавления прогрессирования нарушения или заболевания.
Свойства вышеприведенных дозировок являются очевидными в тестах in vitro и in vivo с использованием млекопитающих для наилучших результатов, например, мышей, крыс, собак, обезьян, или выделенных органов, тканей и их препаратов. Составы настоящего изобретения можно применять in vitro в виде растворов, например водных растворов, и in vivo либо энтерально, либо парентерально, для наилучших результатов - внутривенно, например, в виде суспензии или водного раствора. Дозировка in vitro может быть в пределах значений молярной концентрации от примерно 10-3 моль/л до 10-9 моль/л.
В одном варианте осуществления фармацевтический состав настоящего изобретения вводят внутрисуставно, причем доза активного ингредиента составляет не более 25 мг (например, cоединения A); например, примерно 0,5 мг, 2,5 мг, 7,5 мг, 15 мг или 25 мг активного ингредиента. В другом варианте осуществления фармацевтический состав настоящего изобретения вводят внутрисуставно, причем доза активного ингредиента составляет не более 75 мг; например, примерно 40 мг, 50 мг, 60 мг или 75 мг активного ингредиента. В еще одном варианте осуществления фармацевтический состав настоящего изобретения вводят внутрисуставно, причем доза активного ингредиента составляет не более 100 мг; например, от 50 до 100 мг активного ингредиента. В еще одном варианте осуществления фармацевтический состав настоящего изобретения вводят внутрисуставно, причем доза активного ингредиента составляет не более 200 мг или 400 мг активного ингредиента.
В другом аспекте изобретение относится к набору, включающему два или несколько отдельных фармацевтических состава, по меньшей мере один из которых содержит препарат с пролонгированным высвобождением cоединения А или его фармацевтически приемлемой соли. В одном варианте осуществления набор содержит средства для раздельного вмещения указанных композиций, такие как контейнер, разделенная бутылка или разделенный пакет из фольги. Примером такого набора является блистерная упаковка, как правило, применяемая для упаковки таблеток, капсул и т.п.
Набор согласно настоящему изобретению можно применять для введения различных лекарственных форм, например, для перорального и парентерального применения, для введения отдельных составов с различными интервалами между введениями доз или для титрования отдельных составов один относительно другого. В целях содействия соблюдению режима лечения набор согласно настоящему изобретению, как правило, содержит инструкции по введению.
В видах комбинированной терапии согласно настоящему изобретению составы данного изобретения и другое терапевтическое средство могут быть изготовлены и/или составлены одним и тем же или разными производителями. Кроме того, соединение настоящего изобретения и другое терапевтическое средство можно объединять в комбинированной терапии: (i) до того, как комбинированный продукт попадает к врачам (например, в случае набора, содержащего соединение настоящего изобретения и другое терапевтическое средство); (ii) самими врачами (или под наблюдением врача) незадолго до введения; (iii) в самих пациентах, например во время последовательного введения соединения настоящего изобретения и другого терапевтического средства.
ПРИМЕРЫ
Следующие примеры иллюстрируют изобретение и не ограничивают изобретение. Специалист в данной области поймет, что процентное содержание (% (мас./об.)) в фармацевтических составах данного изобретения дополнительно включает приемлемые вариации в пределах статистически значимого диапазона и в пределах приемлемой ошибки эксперимента, например, в пределах 10% от указанного значения и, более предпочтительно, в пределах 5%, 1% или 0,5% от указанного значения.
Сокращения
Используются следующие сокращения.
(CAS 9281-44-2; полярные липиды Avanti)
АНАЛИЗЫ
Было разработано несколько анализов, чтобы проверить, соответствуют ли препараты требованиям для внутрисуставного введения.
Суспендируемость. Этот анализ измеряет способность препарата смачивать микрокристаллы cоединения А и поддерживать частицы в суспендированном состоянии. Суспензии микрокристаллов соединения А непрерывно перемешивали в течение 5, 15, 30 и 60 минут. В конце периода перемешивания каждый раствор оценивали по следующим критериям: (i) была ли получена гомогенная суспензия; (ii) прилипают ли частицы лекарственного средства к стеклянной поверхности флакона; (iii) достигнута ли гомогенная суспензия, но осадок прилип к дну; или (iv) образовала ли лекарственная субстанция большие агломераты.
Седиментация (осаждение). Этот анализ измеряет скорость осаждения исходной суспензии. Медленное осаждение указывает на меньшую агрегацию частиц лекарственного средства, обеспечивая лучшую точность дозирования. После вышеуказанной стадии перемешивания образцам препаратов давали отстояться в течение 5, 15, 30 и 60 минут. В конце каждого периода каждый образец оценивали по тем же критериям суспендируемости.
Проходимость через иглу. Этот анализ определяет, можно ли быстро доставить препарат в сустав, не причиняя боли пациенту. Гомогенную суспензию извлекали с помощью шприца с иглой 30-го калибра и снова вводили обратно в тот же флакон. Проходимость через иглу определяли субъективно, исходя из легкости выталкивания суспензии из иглы.
Пример 1. Приготовление микрокристаллов соединения А
За производственной партией cоединения А наблюдали под электронным микроскопом (ФИГ. 1). Частицы были нерегулярными и столбчатыми, и их длина варьировалась от примерно 100 нм до 140 мкм, но большинство частиц были размером менее 50 мкм. Эти частицы имеют тенденцию к агломерации.
Соединение А можно микронизировать с использованием любых способов, известных специалистам в данной области. Неизмельченное соединение А подвергают процессу уменьшения размера для получения требуемого размера частиц. Уменьшение размера соединения А можно выполнять, используя любые известные способы, такие как технология размола на струйной мельнице и, в частности, технология размола на спиральной струйной мельнице, технология размола на противоточной струйной мельнице с кипящим слоем или технология размола на струйной мельнице с кольцевой камерой.
В одном варианте осуществления соединение A имеет распределение частиц по размерам D90<50 мкм. В другом варианте осуществления соединение A имеет распределение частиц по размерам D90<30 мкм, < 25 мкм, < 20 мкм или < 15 мкм.
В других вариантах осуществления соединение А микронизировали с образованием микрокристаллов с максимальными диаметрами частиц, охарактеризованными в Таблице 1.
Таблица 1
Пример 2. Скрининг препаратов с PVP-K12 или КМЦ
Суспензии микрокристаллов соединения А, полученные в примере 1, готовили с растворами, содержащими PVP-K12 или натриевую соль карбоксиметилцеллюлозы (КМЦ). Способность к повторному суспендированию препаратов исследовали путем повторения центрифугирования и суспендирования с помощью встряхивания. Препараты, содержащие КМЦ, после центрифугирования повторно суспензии не образовывали. Препараты, содержащие PVP-K12, исследовали дополнительно.
Пример 3. Препараты P1-P8 (PVP-K12 с F68, F127, SDS или яичным ФХ)
Растворы плацебо P1-P8 готовили в соответствии с составом (мас./об.%), приведенным в Таблице 2.
Таблица 2
Препараты готовили во флаконах R2 с примерно 5 мг микрокристаллов соединения A и заполняли растворами плацебо P1-P8 до достижения концентрации лекарственной субстанции 5 мг/мл. Препараты исследовали на способность к повторному суспендированию и скорость осаждения в соответствии с последовательными стадиями, перечисленными в столбце 1 Таблицы 3. После каждой стадии наблюдали за мутностью образцов, которую регистрировали с помощью фотографий и сводили в Таблицу 3.
Таблица 3
в течение ночи
1 ч
4 ч
1 ч (2000 г)
в течение ночи
1 ч
4 ч
1 ч (2000 г)
Испытание на проходимость через иглу дает примерное представление о том, можно ли плавно вводить препарат в сустав. Для каждого флакона суспензию отбирали в шприц через иглу 30 калибра и снова вводили обратно в тот же флакон. Легкость или сложность извлечения содержимого классифицировали как «приемлемо» или «с трудом».
Тепловую стерилизацию проводили при 122,5°C в течение 20 минут. Наблюдаемые суспендируемость и седиментация были одинаковыми у образцов до тепловой стерилизации и после тепловой стерилизации. Таким образом, тепловая стерилизация, по-видимому, не влияла на разрушение и агломерацию микрокристаллов. Оптические микрофотографии получали для каждого повторно суспендированного препарата, которые подвергали скринингу на предмет частиц со средним размером > 50 мкм; ни в одном из препаратов не было обнаружено частиц со средним размером > 50 мкм. Распределение микронизированных кристаллов по размерам было однородным во всех случаях (микрофотографии не показаны).
Препараты, содержащие SDS и яичный ФХ (P5-P8), демонстрировали плохую способность к повторному суспендированию после 4 часов центрифугирования. С другой стороны, препараты, содержащие F68 и F127 (P1-P4), показали улучшенную способность к повторному суспендированию, и их исследовали дополнительно.
Пример 4. Препараты P9-P16 (PVP-K12 с F68 или F127)
Растворы плацебо P9-P16 готовили в соответствии с составом (%(мас./об.)), приведенным в Таблице 4.
Таблица 4
Препараты готовили во флаконах R2 с добавлением примерно 5 мг микрокристаллов соединения A и заполняли растворами плацебо P9-P16 до достижения концентрации лекарственной субстанции 5 мг/мл. Препараты исследовали на способность к повторному суспендированию и скорость осаждения в соответствии с последовательными стадиями, перечисленными в столбце 1 Таблицы 5. После каждой стадии наблюдали за мутностью образцов и результаты сводили в Таблицу 5.
Таблица 5
Из Таблицы 5 видно, что все препараты, содержащие F68 и F127, проявляли хорошую способность к повторному суспендированию после четырехчасового центрифугирования. Однако препараты, содержащие F127 (P13-P16), были способны повторно суспендировать микрокристаллы в течение более короткого времени и дольше сохранять микрокристаллы в суспендированном состоянии по сравнению с препаратами, содержащими F68.
Пример 5. Препараты P17-P36 (PVP-K12 и F127)
Препараты с P17 по P36, с или без F127, были разработаны, как описано в Таблице 6, с ингредиентами, приведенными в % (мас./об.). Используемый здесь термин «соед. A» относится к соединению A.
Таблица 6
2%
0,05%
0,01%
0,003%
0,001%
0,875%
5,45%
рН 7,0
5 мг/мл
Вышеуказанные препараты P17-P36 приготовили из их отдельных растворов плацебо путем добавления целевых количеств микрокристаллов. Растворы плацебо (средства доставки) готовили из стандартных растворов каждого ингредиента. Ингредиенты (в % (мас./об.) и объем (в мл) растворов ингредиентов, необходимых для приготовления 50 мл раствора плацебо для каждого из препаратов, и pH растворов плацебо, перечислены в Таблице 7.
Таблица 7
8,0%
1,0%
0,1%
17,5 мг/мл
109 мг/мл
рН 7,0
(мл)
основной массы плацебо
В Таблице 8 описано приготовление исходных растворов для каждого формообразующего.
Таблица 8
50 мМ pH 7,0
Для каждого раствора плацебо, в объем 50 мл вносили лекарственную субстанцию (5 мг/мл) и добавляли соответствующее плацебо согласно объему. Для последующих анализов готовили два флакона с не менее чем 1 мл каждого препарата. Первый ряд флаконов проанализировали для определения их характеристик суспендируемости и седиментации. Препараты непрерывно перемешивали в течение 5, 15, 30 и 60 минут. После 60 минут непрерывного перемешивания содержимое флаконов 17, 18, 18А, 19, 22, 23А, 27, 28 было гомогенным. Значения pH, измеренные в конце цикла перемешивания, хорошо соотносились с данными, взятыми для исходных растворов плацебо.
Препараты испытывали в различных анализах для определения проходимости через иглу, скорости осаждения и суспендируемости. Для определения проходимости через иглу каждый образец препарата отбирали в шприц через иглу 30-го калибра и снова вводили обратно в тот же флакон. Все препараты имели приемлемую проходимость через иглу, даже несмотря на то, что отбор образцов происходил медленно. Затем образцам препаратов давали возможность осаждаться в течение 60 минут. Через 60 минут содержимое флаконов 17, 18, 18А, 22, 23, 23А, 24, 27, 28А, 32 и 33А все еще находились в суспендированном состоянии; содержимое флаконов 19, 20, 21, 25, 26 и 28 начинало осаждаться; а в остальных флаконах произошло полное осаждение. Флаконы укупоривали закатыванием и обрабатывали в автоклаве при 122,5oC в течение 20 минут. Все образцы были прозрачными, что указывало на осаждение микрокристаллов. После встряхивания флакона вручную несколько раз только содержимое флаконов P17, P18, 23A, P27 и P28 повторно суспендировалось до гомогенной суспензии. Второй ряд флаконов подвергали пяти циклам термоциклирования, каждый примерно по 12 часов при 2-8oC и 50oC, и оценивали, как описано выше. Образцы встряхивали вручную для повторного суспендирования частиц, повторное образование суспензии наблюдали только во флаконах P17, 18, 19, 27 и 28. Однако большинство препаратов (но не все) повторно суспендировались после более энергичного встряхивания. Проходимость через иглу была приемлемой для содержимого всех флаконов, за исключением P20 и P22.
Был проведен еще один анализ на седиментацию. Через 60 минут содержимое флаконов P17, P18A, P22, P23, P23A, P24, P27, P28A, P32 и P33A все еще находилось в суспендированном состоянии. Образцы переносили в пробирки Эппендорфа и центрифугировали при 4000 об/мин в течение 4 часов. Впоследствии образцы встряхивали для повторного суспендирования. Более половины образцов суспендировались повторно после некоторого энергичного встряхивания (интенсивного перемешивания), но только содержимое флаконов P17, P22, P27, P32 и P33A стабильно суспендировались повторно через 60 минут и после центрифугирования.
Пример 6. Иллюстративный препарат с пролонгированным высвобождением
В примере 6 предложен иллюстративный состав в качестве примера, а не для ограничения изобретения. Фармацевтические составы данного изобретения дополнительно включают приемлемые варианты, известные специалисту в данной области.
Неожиданно выбранная суспензия микрокристаллов оказалась пригодной для окончательной стерилизации влажным теплом и не продемонстрировала каких-либо признаков роста кристаллов (созревания Освальда) или агрегации во время стерилизации и в продолжение хранения, что позволило точно дозировать ее через тонкую иглу (30-го калибра (30G)). Местного раздражения тканей не наблюдалось. В одном варианте осуществления состав содержит 25 мг/мл микрокристаллов соединения A в виде однородной суспензии в забуференном носителе, указанном выше.
Пример 7. Сравнительные данные препаратов пролонгированного высвобождения
Были приготовлены и испытаны несколько препаратов со свойствами пролонгированного высвобождения. В каждой рецептуре препарата использовалась максимально возможная нагрузка по лекарственной субстанции для получения максимально возможного терапевтического эффекта.
Суспензии микрокристаллов
Соединение А (250 мкг) суспендировали в 25 мкл забуференного носителя из примера 6.
Суспензия микрочастиц PLGA
Соединение A (2% (мас./об.)) и PLGA (12 кДа; соотношение L:G 1:1) в дихлорметане эмульгировали в водном растворе, содержащем 1% (мас./об.) поливинилового спирта (PVA), для стабилизации исходной эмульсии. Выпаривание дихлорметана при нагревании привело к получению соединения A, молекулярно диспергированного в микрочастицах PLGA, которые затем промыли в воде и высушили. Были получены микрочастицы PLGA, имеющие распределение частиц по размерам D50=40 мкм и D90=57 мкм.
Липосомальная суспензия
Липосомный препарат использовали для солюбилизации соединения А в двойном липидном слое и для получения возможного замедленного высвобождения. Кроме того, липосома должна способствовать смазке в месте инъекции. Липосомную рецептуру получали методиками, описанными ниже:
Первая методика
1. Липид DMPC (100 мг) и немикронизированное соединение A (3 мг) растворяли в 10 мл EtOH:DCM 1:1
2. EtOH и DCM удаляли на роторном испарителе в течение 30 минут при 40°C и 250 мбар (140 об/мин)
3. Следы EtOH и DCM удаляли за 10 минут при 40°C и 40 мбар (140 об/мин)
4. Пленку липид/соединение А повторно гидратировали в 1 мл фосфатно-солевого буфера (PBS) или сахаров в воде
5. Образцы выдавливали через 1 мкм фильтр для получения однородных размеров
6. Образцы замораживали при -20°С
7. Образцы лиофилизировали в течение ночи
8. Образцы повторно суспендировали в стерильной воде
Вторая методика
1. Соединение A и липид DMPC растворяли в трет-бутаноле
2. Образцы замораживали при -20°С
3. Образцы лиофилизировали в течение ночи
4. Образцы повторно суспендировали в PBS
Исходные материалы можно стерилизовать фильтрацией солюбилизированных в трет-бутаноле DMPC и соединения A. После лиофилизации порошок можно восстанавливать для производства многослойных везикул (MLV). Используемые липиды (DMPC) в продуктовой форме (MLV) имеют преимущества с точки зрения потенциально лучшего удержания в месте инъекции благодаря их большому размеру и противовоспалительным свойствам. Утечка соединения А была протестирована на мышиной модели, где наблюдалось быстрое высвобождение соединения А.
Сравнительные данные
Соединение A вводили внутрисуставно (IA) в различных препаратах с пролонгированным высвобождением самцам крыс Lewis, по 3 животных на препарат. Суспензию микрочастиц PLGA и липосомальную суспензию MLV вводили в количестве, близком к максимально возможной дозе, которая может быть ограничена емкостью нагрузки лекарственного средства. Образцы плазмы крови брали серийно после инъекций IA вплоть до 480 часов (20 дней, n=3 в каждый момент времени).
Концентрации соединения А в плазме крови определяли количественно с применением анализа жидкостной хроматографии/масс-спектрометрии (ЖХ/МС/МС). Двести пикограмм на миллилитр (пг/мл) верапамила гидрохлорида (Sigma-Aldrich, V4629, CAS 152-11-4) в ацетонитриле/метаноле, 3/1 (об./об.), использовали в качестве внутреннего стандарта и растворителя для осаждения плазмы крови. К 20 мкл каждого образца плазмы добавляли 100 мкл раствора внутреннего стандарта для осаждения белков матрикса. Образец интенсивно перемешивали в течение 5 минут, затем центрифугировали на Eppendorf Centrifuge 5810R (Eppendorf, Гамбург, Германия) при 4000 об/мин в течение 5 минут при 10oC. Аликвоту надосадочной жидкости (80 мкл) переносили в чистый 96-луночный планшет и смешивали с 75 мкл воды Milli-Q. Перемешанные образцы (5 мкл) впрыскивали в аналитическую колонку ZORBAX® SB-C8 (2,1 х 30 мм, 3,5 мкм, Agilent Technologies Inc., Пало-Альто, Калифорния, США), используя градиентный режим с расходом 700 мкл/мин (см. таблицу ниже). Использовали подвижные фазы, состоящие из 0,05% муравьиной кислоты в воде (растворитель A) и 0,05% муравьиной кислоты в ацетонитриле (растворитель B). Соединение А и внутренний стандарт элюировали при времени удерживания 1,40 и 1,45 минуты, соответственно.
ВЭЖХ градиент
Система ВЭЖХ, состоящая из бинарного насоса серии Agilent 1260 (Agilent Technologies Inc.), микро-вакуумного дегазатора серии Agilent 1260 (Agilent Technologies Inc.), аналитического автоматического пробоотборника CTC PAL-HTC-xt (LEAP Technologies, Карборро, Северная Каролина, США), была подключена к тройному квадрупольному масс-спектрометру Applied Biosystems SCIEX 5500 (AB Sciex LLC., Фостер-Сити, Калифорния, США). Масс-спектральные анализы осуществляли с применением ионизации электрораспылением (ESI) в режиме регистрации положительных ионов. Интегрирование пиков соединения А (363,03 > 156,00) и внутреннего стандарта (455,40 > 165,10) проводили, используя программное обеспечение AnalystTM 1.5. Нижний предел количественного определения - НПКО (LLOQ) в плазме крови составлял 5 пг/мл. Известные количества соединения А прибавляли в плазму крови для создания образцов контроля качества с известными концентрациями 20, 80, 800, 4000 и 20000 пг/мл.
В моменты времени до 480 часов образцы плазмы крови крыс отбирали и анализировали на концентрацию лекарственного средства. В Таблице 9 приведены параметры pK в плазме крови после внутрисуставного введения. Как показано на ФИГ. 2 и в Таблице 9 суспензия микрокристаллов превзошла по своим показателям другие препараты, позволив получить самую высокую дозу лекарства, самые высокие Cmax и воздействие лекарственного средства от времени (AUC) до конца периода наблюдения. Профиль микросуспензии ( ) достигает максимальной Cmax, поскольку нагрузка по лекарственному средству была самой высокой среди протестированных систем. Из-за более длительного времени пребывания суспензии микрокристалов соединения А профиль суспензии микрокристаллов соединения А дополнительно исследовали in vivo.
) достигает максимальной Cmax, поскольку нагрузка по лекарственному средству была самой высокой среди протестированных систем. Из-за более длительного времени пребывания суспензии микрокристалов соединения А профиль суспензии микрокристаллов соединения А дополнительно исследовали in vivo.
Таблица 9
(мкг)
(ч)
(нМ)
(ч)
(ч*нМ/
мкг)
AUC 0-беск. (ч*нМ
/мкг)
Сmax (нМ/мкг)
Пример 8. Сравнительные данные препаратов быстрого и пролонгированного высвобождения
В исследовании in vivo сравнивали препараты быстрого высвобождения (IR) и пролонгированного высвобождения (ER) в доставляемых дозировках, которые можно осуществить с соответствующими типами препаратов, в модели разрыва мениска крысы (RMT) у самцов Lewis в возрасте 36 недель после однократной внутрисуставной инъекции через 4 недели и по окончании испытания через 12 недель (т.е. через 8 недель после инъекции). На фигуре 3 сравниваются профили in vivo препарата ER соединения A (250 мкг) и препарата IR (91 нг). Как показано на фигуре3, суспензия микрокристаллов с пролонгированным высвобождением была эффективна при восстановлении хряща и, следовательно, обеспечивает преимущество благодаря сниженной частоте дозирования (т.е. меньшего количества инъекций) без снижения эффективности лечения.
Пример 9. Препараты с пролонгированным высвобождением, содержащие соединение А (25 мг/мл)
Соединение А (25 мг/мл) диспергировали в водном забуференном носителе, содержащем следующие наполнители, в присутствии безводного фосфата динатрия (5 мМ), NaOH (1 н.), HCl 25% (1 н.) в качестве регуляторов рН, чтобы довести рН до физиологического рН 7, и воды для инъекций.
Чтобы увеличить концентрацию лекарственной субстанции, 50 мг порошка соединения А добавляли к 2 мл раствора носителя, как описано выше. Раствор перемешивали в течение ночи при 500 об/мин магнитной мешалкой и получали гомогенную суспензию индивидуально диспергированных микрокристаллов без видимых агрегатов или агломератов.
Пример 10. Препарат с пролонгированным высвобождением, содержащий соединение А (50-400 мг/мл)
Составы, содержащие носитель с практически такой же композицией, что и в примере 10, с различными концентрациями PVP K12 и полоксамера 407, подвергали скринингу для оценки влияния концентрации формообразующего на осаждение и повторное суспендирование составов, содержащих соединение A, при концентрациях от примерно 50 мг/мл до примерно 400 мг/мл. Следующие составы (1)-(4) были получены в виде неагломерированных суспензий, которые легко подвергались повторному суспендированию после осаждения (например, во время обработки в автоклаве или при хранении в течение нескольких дней).
Пример 11. Карбоксиметилцеллюлоза как дополнительный стабилизатор суспензии
Карбоксиметилцеллюлозу добавляли к суспензии, содержащей микрокристаллы соединения А (25 мг/мл), в забуференном носителе примера 6. Удивительно, что повторное суспендирование не подвергавшихся и подвергавшихся обработке в автоклаве образцов улучшалось в присутствии КМЦ (1% и 1,5% (мас./об.).
Понятно, что примеры и варианты осуществления, описанные здесь, предназначены только для иллюстративных целей. Также понятно, что специалисты в данной области техники могут предпринять различные модификации или изменения описанных примеров и вариантов осуществления, не выходя за рамки сущности и охвата настоящей заявки и формулы описанного здесь изобретения. Все публикации, патенты и заявки на патенты, цитируемые в данном документе, включены в данный документ с помощью ссылки для всех целей.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЛЕКАРСТВЕННАЯ ФОРМА ДЛЯ КОМБИНАЦИИ hGH И rhIGF-1 | 2010 |
|
RU2558821C2 |
| ЛИОФИЛИЗИРОВАННЫЕ ЛЕКАРСТВЕННЫЕ НАНОСУСПЕНЗИИ | 2012 |
|
RU2643062C2 |
| КОМПОЗИЦИИ АНТАГОНИСТОВ РЕЦЕПТОРОВ CGRP | 2014 |
|
RU2690006C2 |
| ПРОИЗВОДНЫЕ БОРОНОВОЙ КИСЛОТЫ | 2018 |
|
RU2793315C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА, СПОСОБ ИХ ПОЛУЧЕНИЯ, СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ СИНТЕЗА | 1993 |
|
RU2119917C1 |
| СПОСОБ ПОЛУЧЕНИЯ МИКРОСФЕР ДЛЯ ПРИГОТОВЛЕНИЯ ИНЪЕЦИРУЕМОЙ ЛЕКАРСТВЕННОЙ ФОРМЫ ДИКЛОФЕНАКА, КОМПОЗИЦИЯ И ЛЕКАРСТВЕННАЯ ФОРМА | 2013 |
|
RU2524649C1 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛИНА, ОБЛАДАЮЩИЕ CB-АНТАГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ | 2005 |
|
RU2357958C2 |
| ТЕТРАЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ РЕЦЕПТОРОВ CB КАННАБИНОИДОВ С ВЫСОКОЙ СЕЛЕКТИВНОСТЬЮ К ПОДТИПАМ РЕЦЕПТОРОВ CB/CB | 2005 |
|
RU2354650C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ИНЪЕКЦИЙ И ИСПОЛЬЗОВАНИЕ ТАКОВЫХ | 2020 |
|
RU2829447C2 |
| ЛЕКАРСТВЕННАЯ ДОЗИРОВАННАЯ ФОРМА, КОТОРАЯ СОДЕРЖИТ 6'-ФТОР-(N-МЕТИЛ- ИЛИ N, N-ДИМЕТИЛ-)-4-ФЕНИЛ-4', 9'-ДИГИДРО-3'Н-СПИРО[ЦИКЛОГЕКСАН-1, 1'-ПИРАНО[3, 4, b]ИНДОЛ]-4-АМИН | 2011 |
|
RU2589830C2 |



Настоящее изобретение относится к фармацевтическому составу, содержащему водную суспензию: (i) кристаллического N-(3,4-дихлорфенил)-3-(пиридин-4-ил)-7-оксабицикло[2.2.1]гептан-2-карбоксамида или его фармацевтически приемлемой соли; и (ii) поверхностно-активного вещества, содержащего водорастворимый сополимер, характеризующийся растворимостью в воде > 5% при 25oC. Составы обеспечивают пролонгированное высвобождение N-(3,4-дихлорфенил)-3-(пиридин-4-ил)-7-оксабицикло[2.2.1]гептан-2-карбоксамида и подходят для внутрисуставной инъекции в сустав пациента, страдающего от артрита, травмы сустава или травмы хряща. 3 н. и 14 з.п. ф-лы, 3 ил., 9 табл., 11 пр.
1. Фармацевтический состав, подходящий для внутрисуставной инъекции, содержащий водную суспензию (i) 1-400 мг кристаллического N-(3,4-дихлорфенил)-3-(пиридин-4-ил)-7-оксабицикло[2.2.1]гептан-2-карбоксамида; (ii) 0,05-1% (мас./об.) водорастворимого блок-сополимера, где водорастворимый блок-сополимер представляет собой полоксамер 407, и (iii) 1-5% (мас./об.) поливинилпирролидона, имеющего среднюю молекулярную массу 1-10 кДа.
2. Фармацевтический состав по п. 1, содержащий 2,5 мг, 7,5 мг, 15 мг или 25 мг кристаллического N-(3,4-дихлорфенил)-3-(пиридин-4-ил)-7-оксабицикло[2.2.1]гептан-2-карбоксамида.
3. Фармацевтический состав по п. 1, содержащий 40 мг, 50 мг, 60 мг или 75 мг кристаллического N-(3,4-дихлорфенил)-3-(пиридин-4-ил)-7-оксабицикло[2.2.1]гептан-2-карбоксамида.
4. Фармацевтический состав по п. 1, содержащий от 50 до 100 мг кристаллического N-(3,4-дихлорфенил)-3-(пиридин-4-ил)-7-оксабицикло[2.2.1]гептан-2-карбоксамида.
5. Фармацевтический состав по п. 1, содержащий 25 мг кристаллического N-(3,4-дихлорфенил)-3-(пиридин-4-ил)-7-оксабицикло[2.2.1]гептан-2-карбоксамида.
6. Фармацевтический состав по п. 1, содержащий (i) 10-30 мг кристаллического N-(3,4-дихлорфенил)-3-(пиридин-4-ил)-7-оксабицикло[2.2.1]гептан-2-карбоксамида в 1 мл указанной водной суспензии и (ii) 0,05-0,15% (мас./об.), предпочтительно 0,1 (мас./об.) полоксамера 407.
7. Фармацевтический состав по п. 1, где указанный поливинилпирролидон представляет собой PVP-K12.
8. Фармацевтический состав по п. 7, содержащий 1,5-2,5% (мас./об.) PVP-K12.
9. Фармацевтический состав по п. 1, содержащий (i) 150-250 мг, предпочтительно, 200 мг кристаллического N-(3,4-дихлорфенил)-3-(пиридин-4-ил)-7-оксабицикло[2.2.1]гептан-2-карбоксамида в 1 мл указанной водной суспензии; (ii) 0,1-0,5% (мас./об.), предпочтительно, 0,2-0,4% (мас./об.) полоксамера 407; и (iii) 1-5% (мас./об.), предпочтительно 2-4% (мас./об.) PVP-K12.
10. Фармацевтический состав по п. 1, содержащий (i) 350-400 мг, предпочтительно, 400 мг кристаллического N-(3,4-дихлорфенил)-3-(пиридин-4-ил)-7-оксабицикло[2.2.1]гептан-2-карбоксамида в 1 мл указанной водной суспензии; (ii) 0,6-1% (мас./об.), предпочтительно, 0,7-0,9% (мас./об.), предпочтительно 0,8% (мас./об.) полоксамера 407; и (iii) 1-5% (мас./об.), предпочтительно, 2-4% (мас./об.) PVP-K12.
11. Фармацевтический состав по п. 1, дополнительно содержащий лаурилсульфат натрия, где концентрация указанного лаурилсульфата натрия составляет не менее 0,05% (мас./об.), предпочтительно, 0,05-0,5% (мас./об.).
12. Фармацевтический состав по любому из пп. 1-11, где указанный кристаллический N-(3,4-дихлорфенил)-3-(пиридин-4-ил)-7-оксабицикло[2.2.1]гептан-2-карбоксамид имеет микрокристаллическую или микронизированную форму.
13. Фармацевтический состав по любому из пп. 1-12, где указанный N-(3,4-дихлорфенил)-3-(пиридин-4-ил)-7-оксабицикло[2.2.1]гептан-2-карбоксамид дополнительно определен как (1R,2R,3S,4S)-N-(3,4-дихлорфенил)-3-(пиридин-4-ил)-7-оксабицикло[2.2.1]гептан-2-карбоксамид.
14. Фармацевтический состав по любому из пп. 1-13, дополнительно содержащий буфер, способный поддерживать рН суспензии при значении 6-8.
15. Способ лечения, улучшения или предотвращения острого повреждения или травмы сустава у субъекта, нуждающегося в этом, включающий введение фармацевтического состава согласно любому из пп. 1-14 нуждающемуся в этом субъекту; тем самым осуществляя лечение, улучшение или предотвращение острого повреждения или травмы сустава у указанного субъекта.
16. Способ по п. 15, где указанный фармацевтический состав вводят указанному субъекту, причем доза кристаллического N-(3,4-дихлорфенил)-3-(пиридин-4-ил)-7-оксабицикло[2.2.1]гептан-2-карбоксамида составляет не более 25 мг, не более 75 мг, не более 100 мг, не более 200 мг или не более 400 мг.
17. Набор для внутрисуставной инъекции, содержащий фармацевтический состав по любому из пп. 1-14 и, по меньшей мере, инструкцию по применению или иглу.
| WO 2015175487 A1, 2015.11.19 | |||
| СПОСОБЫ И КОМПОЗИЦИИ, СОДЕРЖАЩИЕ КЛОНИДИН, ПРЕДНАЗНАЧЕННЫЕ ДЛЯ ЛЕЧЕНИЯ ПОСЛЕОПЕРАЦИОННОЙ БОЛИ | 2009 |
|
RU2510263C2 |
| WO 2012172072 A1, 2012.12.20. | |||

