Изобретение относится к способам получения наноструктурированного олигосахарида хитозана и его производных и может найти применение в медицине, ветеринарии, косметологии, пищевой промышленности и сельском хозяйстве.
УРОВЕНЬ ТЕХНИКИ
Из уровня техники известен способ получения карбоксилсодержащих производных хитозана, включающий взаимодействие хитозана с карбоксилсодержащим реагентом в твердом виде в условиях воздействия сдвиговых напряжений и давления (RU 2100373 C1, ИНСТИТУТ СИНТЕТИЧЕСКИХ ПОЛИМЕРНЫХ МАТЕРИАЛОВ РАН, 27.12.1997). В качестве карбоксилсодержащего реагента используют дикарбоновую кислоту или ангидрид дикарбоновой кислоты (янтарный ангидрид) в количестве 0,25-1,5 моль на 1 моль аминогрупп хитозана.
Известен способ получения водорастворимых форм хитозана, включающий приготовление гомогенного раствора хитозана, его выделение путем обработки щелочным агентом до pH среды 6,8-7,5, аморфизацию хитозана в виде его водной суспензии в кавитационных или механических полях со сдвиговым воздействием, взаимодействие аморфизированного хитозана с неорганическими, органическими кислотами или их ангидридами и выделение целевого продукта из растворов в деминерализованной воде методом распылительной или сублимационной сушки (RU 2215749 С2, КОМАРОВ БОРИС АЛЕКСАНДРОВИЧ, 10.11.2003). Способ позволяет получать хитозан, модифицированный остатками янтарной или L-глутаминовой кислоты.
Известен способ получения прозводных хитозана, в т.ч. сукцината хитозана (SU 508212 A3, Л′ОРЕАЛЬ, 25.03.1976).
Известен способ непрерывного получения микрокристаллического хитозана, включающий осаждение хитозана из его водного раствора в органической или неорганической кислоте с использованием водных растворов гидроксидов щелочных металлов (WO 9100298 A1, FIREXTRA OY; INST WLOKIEN СНЕМ, 10.01.1991).
Известен способ получения хитозановой композиции с размером частиц 0,1-150 мкм, включающий получение твердого хитозана, смачивание его водой или водным раствором и затем смешивание с нежирной кислотой или солью кислоты (US 6323189 B1, NUTRICEUTICALS INC Е, 27.11.2001).
Известен способ получения хитозановых гранул с размером 2-20 пм (WO 0187988 A1, PROCTER & GAMBLE, 22.11.2001), включающий получение водного раствора хитозана, который дополнительно содержит органические или неорганические кислоты и/или их соли, частичную нейтрализацию указанного водного раствора добавлением нейтрализующего агента при сдвиговом перемешивании с дальнейшим образованием гелеподобной суспензии дискретных частиц микрокристаллического хитозана.
Общим недостатком описанных способов является то, что получение хитозановых гранул с размером в нанодиапазоне затруднено, поскольку в отсутствие поверхностно-активных веществ диспергирование кислых растворов приводит к малому процентному выходу гранул.
Из уровня техники известны композиции на основе хитозана для ухода за кожей в виде геля (RU 2085187 C1, ИССЛЕДОВАТЕЛЬСКАЯ КОРПОРАЦИЯ МДТ, 27.07.1997), крема для защиты кожи (RU 2120272 C1, ТОВАРИЩЕСТВО С ОГРАНИЧЕННОЙ ОТВЕТСТВЕННОСТЬЮ ′′БИОКОСМЕТИЧЕСКАЯ ФАБРИКА′′, 20.10.1998), а также средство для лечения инфицированных ран и ожогов (RU 2140264 C1, ТИХООКЕАНСКИЙ ИНСТИТУТ БИООРГАНИЧЕСКОЙ ХИМИИ ДАЛЬНЕВОСТОЧНОГО ОТДЕЛЕНИЯ РАН, 27.10.1999), лекарственное средство, обладающее противовоспалительным и болеутоляющим действием (RU 2154488 C1, МЫНКИНА ГАЛИНА ИВАНОВНА, 20.08.2000).
Основным недостатком перечисленных композиций является то, что все они обладают сравнительно невысокой проникающей способностью из-за большого размера хитозановых гранул и, следовательно, низкой эффективностью.
Наиболее близким техническим решением является способ получения модифицированного хитозанового продукта, имеющего pH-нейтральную реакцию промывочных вод и пластичную структуру осадка, представляющего собой продукты хитозана, находящиеся в виде фрактальных хитозановых частиц с размером нанофракталов не менее 1 нм и не более 5000 нм, и поперечно-сшитых сетчатых полимеров с множеством сферических полостей, имеющих размер не менее 1 нм и не более 50 нм (RU 2313538 C2, МАЙЕР БОРИС ОЛЕГОВИЧ, 04.08.2005).
Известный способ включает приготовление кислого водного раствора хитозана, удаление нерастворившихся частиц фильтрованием и/или осаждением, проведение сшивки хитозана в присутствии гидрофильного органического растворителя и поверхностно-активного вещества в слабокислых, нейтральных или слабощелочных условиях в зависимости от используемого сшивающего агента при непрерывном диспергировании. При этом:
- или кислый водный раствор хитозана первоначально перемешивают в присутствии поверхностно-активного вещества, гидрофильного органического растворителя, хелатокомплексообразующего вещества и сшивающего агента, полученную реакционную смесь выдерживают при температуре не ниже комнатной такое время, чтобы обеспечить проведение хелатокомплексообразования с участием хитозана в качестве лиганда, затем реакционную смесь при диспергировании обрабатывают щелочным агентом, добавляемым по частям или по каплям при таком же диспергировании;
- или кислый водный раствор хитозана первоначально перемешивают в присутствии поверхностно-активного вещества, гидрофильного органического растворителя и хелатокомплексообразующего вещества, полученную реакционную смесь выдерживают при температуре не ниже комнатной такое время, чтобы обеспечить проведение хелатокомплексообразования с участием хитозана в качестве лиганда, затем реакционную смесь при диспергировании обрабатывают щелочным агентом, добавляемым по частям или по каплям, а затем при продолжающемся диспергировании добавляют сшивающий агент.
Поверхностно-активное вещество добавляют в реакционную смесь в концентрации выше критической концентрации мицеллообразования, а хелатокомплексообразующее вещество вводят с пониженной концентрацией, такой, что образующиеся в реакционной смеси агломераты хитозана содержат не более 10 об.% нанофракталов и состоят из поперечно-сшитого сетчатого хитозанового полимера.
В качестве сшивающего агента используют насыщенные или ненасыщенные карбоновые, дикарбоновые или трикарбоновые кислоты, такие как ацетоксиянтарную, гликоуксусную, глутаровую, диацетилвинную, дигликолевую, итаконовую, коричную, кротоновую, лимонную, малеиновую, малоновую, метакриловую, метилянтарную, пропионовую, пропеновую, салициловую, фталевую, фумаровую, цитраконовую, щавелевую, янтарную, или их ангидриды, или их галоидангидриды, или их дигалоидангидриды, или их альдегиды, или их диальдегиды, или их окси- или оксо-производные, или их диглицидные эфиры, или их другие производные, используют также бисимидаты или бисмалеиды.
В качестве гидрофильного органического растворителя используют: спирты, кетоны, альдегиды, ароматические углеводороды, эфиры, галогенпроизводные предельных углеводородов, предпочтительно этанол, метанол, изопропанол, ацетон, ацетонитрил, диоксан или их любую смесь в концентрации не менее 1,0 об.% по отношению к объему реакционной смеси.
В качестве поверхностно-активного вещества используют неионные или цвиттер-ионные поверхностно-активные вещества, или октилглюкозид, или тритон Х-к, где к=45, 114, 100, 102, 165, 305, или ионидет 340, или твин 20, или твин 40, или твин 60, или твин 80, или луброл РХ, или луброл WX, или любую их смесь, или фторсодержащее поверхностно-активное вещество.
В качестве хелатокомплескообразователей используют ионы или соединения щелочных металлов или щелочно-земельных металлов, или переходных металлов, или благородных металлов, кислоты - салициловую, нитрилотриуксусную, этилендиаминтетрауксусную, диэтилентриаминпентауксусную, диамид дитиощавелевой кислоты, транс-1,2-диаминоциклогексантетрауксусную, триэтилентетраамингексауксусную; соли многовалентных кислот; карбонаты, сульфаты, фосфаты или соли щавелевой кислоты кальция или магния; салициловый альдегид, салицилальдоксим.
В зависимости от концентрационных соотношений хелатокомплесксообразователя и поверхностно-активного вещества получают либо модифицированный хитозановый продукт в виде фрактальных хитозановых частиц с размером нанофракталов не менее 1 нм и не более 5000 нм, либо в виде поперечно-сшитого сетчатого полимера с множеством сферических полостей, имеющих размер не менее 1 нм и не более 50 нм.
Основным недостатком указанного способа является то, что полученные хитозановые продукты имеют наименее упорядоченную (аморфизованную) структуру, поскольку сшивка приводит к нарушению регулярности молекулярной структуры и ограничению подвижности структурных элементов хитозана. Также наблюдается и уменьшение сорбционной способности из-за снижения содержания аминогрупп, вероятно, это связано с участием их в образовании азометиновых связей, сшивок, а также уменьшением доступности аминогрупп в сшитом полимере.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В последнее время значительный интерес проявляется к наноструктурированным полимерам. В силу большой удельной поверхности наночастицы проявляют отличные от обычных полимеров свойства и существенное увеличение характерных им. Это открывает большие возможности использования их в различных отраслях науки и техники, в частности при создании лекарственных средств. Такие структуры имеют большое сродство к мембране клетки, а малые размеры позволяют легко проникать в ядро клетки.
Наночастицы, в основном, представляют интерес в качестве вектора для доставки биологически активного материала в клетку. Наночастицы на основе производных хитозана отвечают требованиям, предъявляемым к таким системам. Благодаря высокой плотности заряда на поверхности они активно взаимодействуют с мембраной клеток. Частицы биосовместимы с живыми тканями организма, т.к. не вызывают аллергических реакций и отторжения, постепенно биодеградируют под действием ферментов на аминосахара, которые полностью усваиваются в живом организме. Они нетоксичны и легко выводятся из организма, не вызывая конкурентных побочных реакций. Эффективность действия большинства биологически активных веществ часто ограничивается из-за невозможности подойти к органу-мишени (месту терапевтического действия) с сохранением их изначальных свойств. Применение наночастиц на основе хитозана в качестве подложки для биологически активных веществ должно обеспечить их целевую доставку, снизить применяемую дозу, пролонгировать эффективность действия и обеспечить возможность использования для орального или назального введения как наиболее удобных путей для введения в живой организм. Известно, что введение гидрофобных групп в молекулу хитозана приводит к увеличению стабильности коллоидных систем, полученных на их основе.
Задачей настоящего изобретения является получение нового наноструктурированного производного олигосахарида хитозана с высокой трансдермальной и мембранной проницаемостью, повышенной сорбционной емкостью, содержащего гидрофобный и гидрофильный лиганд, а также растворимого в широком диапазоне pH, в котором хитозановый фрагмент имеет общую формулу (I)
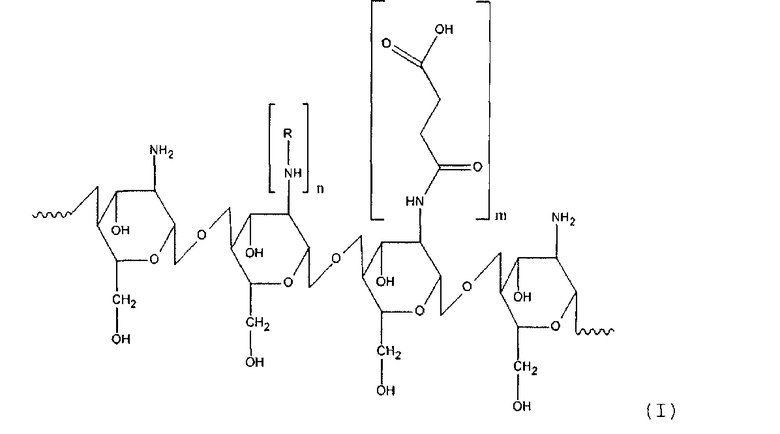
где
R - остаток жирной или аминокислоты,
n составляет от около 12 до около 25% относительно количества моносахаридных остатков хитозана,
m составляет от около 30 до около 60% относительно количества моносахаридных остатков хитозана.
Задача решается получением наноструктурированного олигосахарида хитозана, модифицированного остатками жирной кислоты и ангидридом янтарной кислоты, имеющего рабочую область (растворимого в широком диапазоне pH) pH от около 2 до около 9, предпочтительно от около 4 до около 9, более предпочтительно от около 6 до около 9, наиболее предпочтительно от около 7 до около 8, например около 7.4, где хитозан представлен сферической структурой с размером частиц от около 1 нм до около 2000 нм, предпочтительно от около 1 нм до около 1500 нм, более предпочтительно от около 1 нм до около 900 нм, еще более предпочтительно от около 1 нм до около 400 нм, наиболее предпочтительно от около 1 нм до около 100 нм.
Задача решается также получением наноструктурированного олигосахарида хитозана, модифицированного аминокислотой и ангидридом янтарной кислоты, имеющего рабочую область (растворимого в широком диапазоне pH) pH от около 2 до около 9, предпочтительно от около 4 до около 9, более предпочтительно от около 6 до около 9, наиболее предпочтительно от около 7 до около 8, например около 7.4, где хитозан представлен сферической структурой с размером частиц от около 1 нм до около 2000 нм, предпочтительно от около 1 нм до около 1500 нм, более предпочтительно от около 1 нм до около 900 нм, еще более предпочтительно от около 1 нм до около 400 нм, наиболее предпочтительно от около 1 нм до около 100 нм.
Для получения амфифильного производного хитозана, имеющего содержание гидрофильного лиганда от около 12% до около 25%, предпочтительно от около 15% до около 25%, более предпочтительно от около 18% до около 25%, наиболее предпочтительно от около 20% до около 25%, и содержание гидрофобного лиганда от около 30% до около 60%, предпочтительно от около 35% до около 55%, более предпочтительно от около 40% до около 50%, наиболее предпочтительно от около 4 5% до около 50%, используют исходный хитозан с молекулярной массой (ММ) от около 5 кДа до около 100 кДа, предпочтительно от около 7 кДа до около 80 кДа, более предпочтительно от около 10 кДа до около 60 кДа, наиболее предпочтительно от около 30 кДа до около 50 кДа, например около 23 кДа.
В качестве гидрофильного лиганда могут быть использованы различные химические соединения с определенным набором свойств и их производные.
Примерами жирных кислот являются каприловая, каприновая (декановая), пальмитиновая (насыщенные жирные кислоты) и олеиновая (ненасыщенная жирная кислота).
Для придания заданных свойств к полисахариду последовательно присоединяют два лиганда: остаток жирной или аминокислоты для обеспечения несущих свойств и сродства связывания с мембраной живой клетки, и остаток янтарной кислоты для обеспечения растворимости в более широком диапазоне pH, в том числе при физиологических значениях (pH около 7.4).
Таким образом, настоящее изобретение касается производного хитозана, в котором хитозановый фрагмент имеет общую формулу (I)
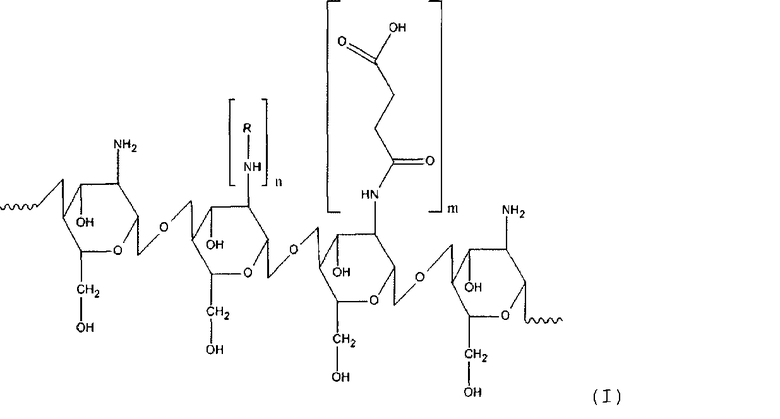
где
R - остаток жирной или аминокислоты,
n для гидрофильного лиганда составляет от около 12 до около 25% относительно количества моносахаридных остатков хитозана,
m для гидрофобного лиганда составляет от около 30 до около 60% относительно количества моносахаридных остатков хитозана.
Указанный остаток жирной кислоты представляет собой остаток каприловой кислоты.
Указанный остаток аминокислоты представляет собой остаток валина.
Указанное производное наноструктурировано.
Указанное производное имеет трансдермальную и/или мембранную проницаемость.
Указанное производное имеет повышенную сорбционную емкость.
Указанное производное растворимо при pH от около 2 до около 9, предпочтительно от около 4 до около 9, более предпочтительно от около 6 до около 9, наиболее предпочтительно от около 7 до около 8.
Указанное производное растворимо при pH около 7.4.
Указанное производное имеет сферическую структуру с размером частиц от около 1 нм до около 2000 нм, предпочтительно от около 1 нм до около 1500 нм, более предпочтительно от около 1 нм до около 900 нм, еще более предпочтительно от около 1 нм до около 400 нм, наиболее предпочтительно от около 1 нм до около 100 нм.
Указанное производное имеет содержание гидрофильного лиганда предпочтительно от около 15% до около 25%, более предпочтительно от около 18% до около 25%, наиболее предпочтительно от около 20% до около 25%.
Указанное производное имеет содержание гидрофобного лиганда предпочтительно от около 35% до около 55%, более предпочтительно от около 40% до около 50%, наиболее предпочтительно от около 45% до около 50%.
Исходный хитозан указанного производного имеет молекулярную массу от около 5 кДа до около 100 кДа, предпочтительно от около 7 кДа до около 80 кДа, более предпочтительно от около 10 кДа до около 60 кДа, наиболее предпочтительно от около 30 кДа до около 50 кДа.
Исходный хитозан указанного производного имеет молекулярную массу около 23 кДа.
Исходный хитозан указанного производного имеет степень деацетилирования 85±2%.
Таким образом, настоящее изобретение касается способа получения производного хитозана, в котором хитозановый фрагмент имеет общую формулу (I)
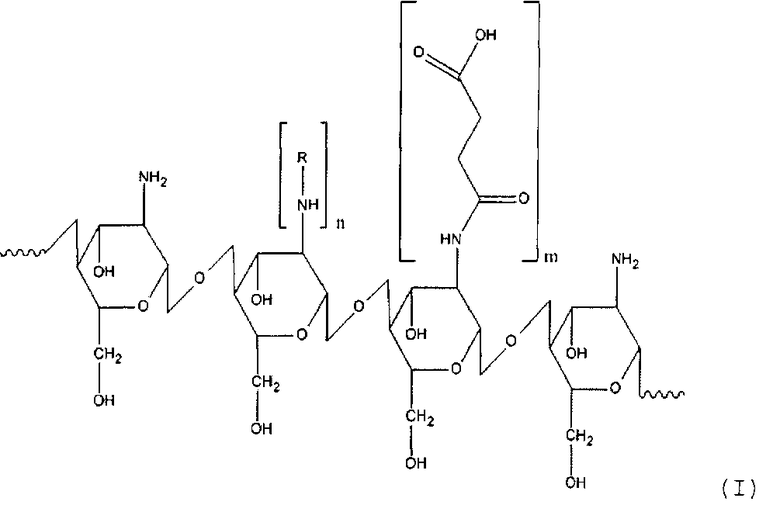
где
R - остаток жирной кислоты,
n для гидрофильного лиганда составляет от около 12 до около 25% относительно количества моносахаридных остатков хитозана,
m для гидрофобного лиганда составляет от около 30 до около 60% относительно количества моносахаридных остатков хитозана, который включает:
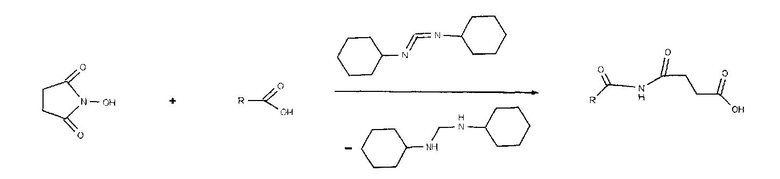
- взаимодействие жирной кислоты с N-гидроксисукцинимидом с получением N-оксисукцинимидного эфира жирной кислоты;
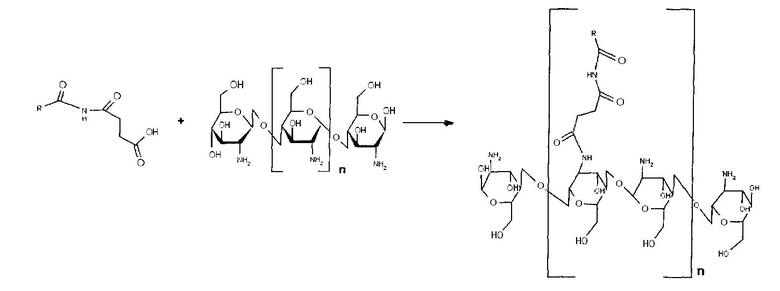
- ацилирование хитозана полученным N-оксисукцинимидным эфиром жирной кислоты;
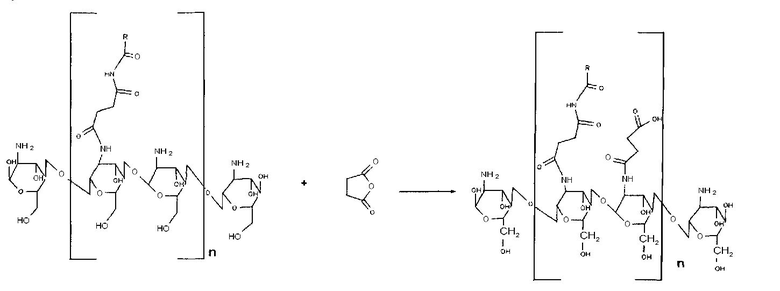
- карбоксиацилирование янтарным ангидридом хитозана, ацилированного N-оксисукцинимидным эфиром жирной кислоты.
Таким образом, настоящее изобретение касается способа получения производного хитозана, в котором хитозановый фрагмент имеет общую формулу (I)
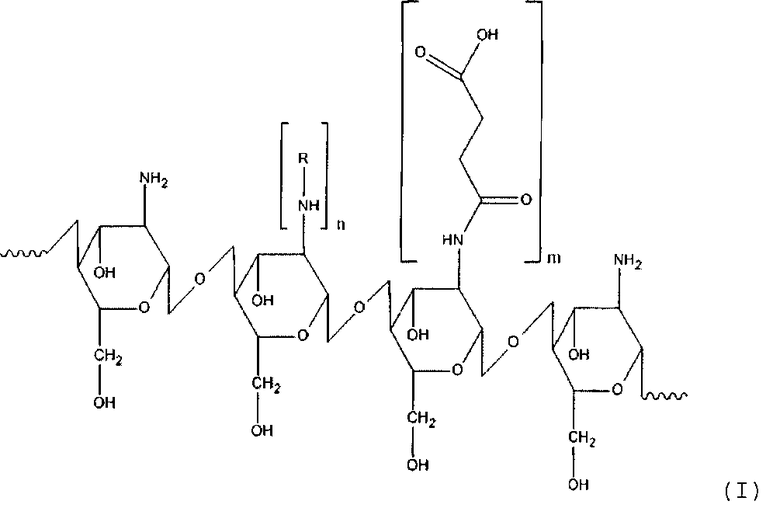
где
R - остаток аминокислоты,
n для гидрофильного лиганда составляет от около 12 до около 25% относительно количества моносахаридных остатков хитозана,
m для гидрофобного лиганда составляет от около 30 до около 60% относительно количества моносахаридных остатков хитозана, который включает:
- взаимодействие аминокислоты с HBTU (N,N,N′,N′-тетраметил-О-(1Н-бензотриазол-1-ил)урониум гексафлюорофосфат) с получением N-оксисукцинимидного эфира аминокислоты;
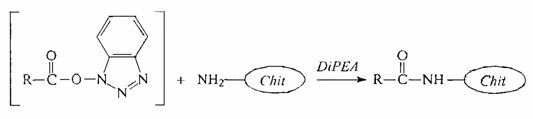
- ацилирование хитозана полученным N-оксисукцинимидным эфиром аминокислоты;
- карбоксиацилирование янтарным ангидридом хитозана, ацилированного N-оксисукцинимидным эфиром аминокислоты.
КРАТКОЕ ОПИСАНИЕ ФИГУР
ФИГ.1 (1-2) Компонентный состав образцов, используемых в экспериментальных испытаниях.
ФИГ.2 (3-4) Компонентный состав образцов, используемых в экспериментальных испытаниях.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В представленных примерах используется фракция олигосахарида хитозана со средней ММ около 23 кДа и степенью деацетилирования 85±2% (произв. Sigma Co.).
Способ получения производного хитозана включает следующие стадии:
1. Взаимодействие жирной кислоты с N-гидроксисукцинимидом в присутствии N,N′-дициклогексилкарбодиимида с получением N-оксисукцинимидного эфира жирной кислоты.
N-Оксисукцинимид растворяли в 10,5 мл сухого этилацетата, добавляли раствор КДИ (N,N′-дициклогексилкарбодиимид) в 6,3 мл этилацетата и жирную кислоту. Раствор перемешивали при КТ (комнатной температуре) в течение 15 ч. Образовавшуюся мочевину отделяли фильтрованием, этилацетат упаривали на роторном испарителе, при этом получали N-оксисукцинимидный эфир жирной кислоты в виде масла, которое хранили в холодильнике.
2. Ацилирование (25/100 МО (молекулярных остатков)) хитозана N-оксисукцинимидным эфиром жирной кислоты.
Хитозан растворяли в 1,05 мл 0,1% уксусной кислоты с добавлением 0,1 мл 0,5% уксусной кислоты и 2 мл метанола. К раствору приливали раствор N-оксисукцинимидного эфира жирной кислоты в 0,53 мл метанола. Раствор перемешивали при КТ в течение 12 часов. Метанол упаривали на роторном испарителе. Продукт высушивали на воздухе.
3. Карбоксиацилирование (30/100) янтарным ангидридом хитозана, ацилированного (25/100 МО) N-оксисукцинимидным эфиром жирной кислоты.
Хитозан, ацилированный (25/100 МО) N-оксисукцинимидным эфиром жирной кислоты, растворяли в 1 мл 0,1% уксусной кислоте с добавлением 4 мл метанола. К раствору добавляли янтарный ангидрид. Раствор перемешивали при КТ в течение 12 часов. При осаждении ацетоном наблюдали почти мгновенное выпадение рыжеватого осадка. Продукт отделяли на центрифуге. Сушили на воздухе.
Альтернативно, при использовании в качестве гидрофобного лиганда аминокислоты способ включает следующие стадии:
1. Получение N-оксисукцинимидного эфира аминокислоты взаимодействием аминокислоты с HBTU (N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate - N,N,N′,N′-тетраметил-О-(1Н-бензотриазол-1-ил)урониум гексафлюорофосфат)

Взаимодействие с HOBt (hydroxybenzotriazole)

N-Оксисукцинимид растворяли в 10,5 мл сухого этилацетата, добавляли раствор КДИ в 6,3 мл этилацетата и аминокислоту. Раствор перемешивали при КТ в течение 15 ч. Образовавшуюся мочевину отделяли фильтрованием, этилацетат упаривали на роторном испарителе, при этом получали N-оксисукцинимидный эфир аминокислоты в виде масла, которое хранили в холодильнике.
2. Ацилирование (25/100 МО) хитозана N-оксисукцинимидным эфиром аминокислоты
Хитозан растворяли в 1,05 мл 0,1% уксусной кислоты с добавлением 0,1 мл 0,5% уксусной кислоты и 2 мл метанола. К раствору приливали раствор N-оксисукцинимидного эфира аминокислоты в 0,53 мл метанола. Раствор перемешивали при КТ в течение 12 часов. Метанол упаривали на роторном испарителе. Продукт высушивали на воздухе.
3. Карбоксиацилирование (30/100) янтарным ангидридом хитозана, ацилированного (25/100 МО) N-оксисукцинимидным эфиром аминокислоты.
Хитозан, ацилированный (25/100 МО) N-оксисукцинимидным эфиром аминокислоты, растворяли в 3 мл 0,1% уксусной кислоте с добавлением 4 мл метанола. К раствору добавляли янтарный ангидрид. Раствор перемешивали при КТ в течение 12 часов. При осаждении ацетоном наблюдали почти мгновенное выпадение белого осадка.

Удаление Fmoc-защитной группы осуществляют стандартными методами в стандартных условиях (Т=40 минут в растворе ДМФА: пиперидин 1:1 на водяной бане при 40°C); продукт промывают ацетоном и лиофильно высушивают.

Для оценки матриц использовали прибор динамического светорассеяния 90Plus S/N, Brookhaven Instruments Corporation, США.
Для определения размера (диаметра) частиц использовали автоматическую функцию 90Plus/BI-MAS, а для определения зета-потенциала (суммарного заряда на поверхности частицы) - ZetaPlus и 90Plus.
Измерения проводили при pH раствора 5.0 и t=25°C
Размер (диаметр) частиц определяли для образцов R2 и R3 (см. выше) в соответствии с Фиг.1(1-2) и 2(3-4) соответственно. Построение модели полученных частиц.
С начала исследований для получения частиц использовали хитозан молекулярной массы около 25 кДа. Подсчитали, что количество моносахаридных звеньев в молекуле такого веса составляет около 156. Используя программы пространственного молекулярного моделирования, измерили, что длина в пространстве хитозанового полимера такой длины составит около 70 нм.
Соответственно, для образца R2 с экспериментальным диаметром частиц в 458 нм по формуле l=2pr, где l - длина окружности, а r - радиус сферы наночастицы, получается l=1438 нм. Таким образом, длина окружности этих сферических наночастиц, в среднем, равна 1438 нм. Сферическая частица используемого здесь полимерного хитозана в 25 кДа с такой окружностью может существовать и представляет собой сферу с окружностью, формируемую, в среднем, 21 полимерной цепью хитозана.
Мы не подвергаем сомнению, что матрицы R2 формируют именно сферу или близко аппроксимируемый сферой многоугольник. Сферическая форма является энергетически низкой формой существования в растворе, а каких-либо структурных особенностей матриц R2, сильно меняющих энергетические предпочтения в формировании ими сферы в растворе, нет.
Для R2, пересчитывая полученные на 100 моносахаридов данные ЯМР (Фиг.1 (1-2)) на исследуемый полимер 156 моносахаридов, получаем, что каждая из такой 21 полимерной цепи окружности наночастицы содержит 18 цепей жирной каприловой кислоты, обращенных внутрь наночастицы, и 45 сукцинильных цепей, обращенных в раствор.
Аналогично провели расчеты для наночастиц образца R3. Частицы из хитозана ММ 25 кДа и экспериментально полученного радиуса в 314 нм также могут существовать. При этом для этого образца, длина окружности наночастицы-сферы составляет 986 нм, и она формируется 14 хитозановыми полимерами указанной ММ. На один хитозановый полимер (в 156 моносахаридов) образца R3 приходится около 38 каприловых остатков и 90 сукцинильных.
Результаты измерений зета-потенциала (на примере образца R3) показали, что сукцинильные цепи, как и предполагалось, выходят на внешнюю поверхность частицы, внося свой вклад отрицательного заряда в суммарный поверхностный заряд и обеспечивая растворимость частиц при физиологических pH 7.4. По проведенным итерациям получили результаты по поверхностному заряду частиц как -7, -5, -6, -1 мВ. Поверхностный заряд наночастиц хитозана, не модифицированного янтарной кислотой, находится в диапазоне от +25 до +54 мВ [11] или от +22 до +28 мВ (в настоящем исследовании приведено ниже). Все приведенные зета-потенциалы были измерены для растворов наночастиц рН 5.0-5.5.
ПРИМЕРЫ
ПРИМЕР 1
Получение N-оксисукцинимидного эфира, каприловой кислоты
N-Оксисукцинимид (367 мг, 3,18 ммоль) растворяли в 10,5 мл сухого этилацетата, добавляли раствор КДИ (639 мг, 3,18 ммоль) в 6,3 мл этилацетата и каприловую кислоту (458 мг, 3,18 ммоль). Раствор перемешивали при КТ в течение 15 ч. Образовавшуюся мочевину отделяли фильтрованием, этилацетат упаривали на роторном испарителе, при этом получали N-оксисукцинимидный эфир каприловой кислоты в виде масла, которое хранили в холодильнике.
Ацилирование (25/100 МО) хитозана N-оксисукцинимидным эфиром каприловой кислоты
Хитозан (23 мг, 1 ммоль) растворяли в 1,05 мл 0,1% уксусной кислоты с добавлением 0,1 мл 0,5% уксусной кислоты и 2 мл метанола. К раствору приливали раствор N-оксисукцинимидного эфира каприловой кислоты (7,3, мг, 30 ммоль) в 0,53 мл метанола. Раствор перемешивали при КТ в течение 12 часов. Метанол упаривали на роторном испарителе. Продукт высушивали на воздухе.
Карбоксиацилирование (30/100) янтарным ангидридом хитозана, ацилированного (25/100 МО) N-оксисукцинимидным эфиром каприловой кислоты
Хитозан, ацилированный (25/100 МО) N-оксисукцинимидным эфиром каприловой кислоты (29,7 мг, 0,7 ммоль), растворяли в 1 мл 0,1% уксусной кислоте с добавлением 4 мл метанола. К раствору добавляли янтарный ангидрид (3,7 мг, 36,9 ммоль). Раствор перемешивали при КТ в течение 12 часов. При осаждении ацетоном наблюдали почти мгновенное выпадение рыжеватого осадка. Продукт отделяли на центрифуге. Сушили на воздухе.
ПРИМЕР 2
Получение N-оксисукцинимидного эфира Nα-трет-бутоксикарбонил-L-валина
N-Оксисукцинимид (367 мг, 3,18 ммоль) растворяли в 10,5 мл сухого этилацетата, добавляли раствор КДИ (639 мг, 3,18 ммоль) в 6,3 мл этилацетата и Nα-трет-бутоксикарбонил-L-валин (690 мг, 3,18 ммоль). Раствор перемешивали при КТ в течение 15 ч. Образовавшуюся мочевину отделяли фильтрованием, этилацетат упаривали на роторном испарителе, при этом получали N-оксисукцинимидный эфир Nα-трет-бутоксикарбонил-L-валина в виде масла, которое хранили в холодильнике.
Ацилирование (25/100 МО) хитозана N-оксисукцинимидным эфиром Nα-трет-бутоксикарбонил-L-валина
Хитозан (23 мг, 1 ммоль) растворяли в 1,05 мл 0,1% уксусной кислоты с добавлением 0,1 мл 0,5% уксусной кислоты и 2 мл метанола. К раствору приливали раствор N-оксисукцинимидного эфира Nα-трет-бутоксикарбонил-L-валина (9,48, мг, 30 ммоль) в 0,53 мл метанола. Раствор перемешивали при КТ в течение 12 часов. Метанол упаривали на роторном испарителе. Продукт высушивали на воздухе.
Карбоксиацилирование (30/100) янтарным ангидридом хитозана, ацилированного (25/100 МО) N-оксисукцинимидным эфиром Nα-трет-бутоксикарбонил-L-валина
Хитозан, ацилированный (25/100 МО) N-оксисукцинимидным Nα-трет-бутоксикарбонил-L-валина (20,7 мг, 0,7 ммоль), растворяли в 3 мл 0,1% уксусной кислоте с добавлением 4 мл метанола. К раствору добавляли янтарный ангидрид (3,7 мг, 36,9 ммоль). Раствор перемешивали при КТ в течение 12 часов. При осаждении ацетоном наблюдали почти мгновенное выпадение белого осадка. Продукт отделяли на центрифуге. Сушили на воздухе.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРИМЕНЕНИЕ ПРОИЗВОДНОГО ХИТОЗАНА ДЛЯ ИЗБИРАТЕЛЬНОЙ ДОСТАВКИ ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ И БИОЛОГИЧЕСКИ АКТИВНЫХ КОМПЛЕКСОВ | 2013 |
|
RU2561062C2 |
| ХИТОЗАНОВЫЙ ПРОДУКТ, СПОСОБ ЕГО ПОЛУЧЕНИЯ (ВАРИАНТЫ) | 2005 |
|
RU2313538C2 |
| ПОЛИМЕРЫ С УГЛЕВОДНЫМИ БОКОВЫМИ ГРУППАМИ И ИХ ПРИМЕНЕНИЕ | 2011 |
|
RU2541534C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПЛЕНОЧНОГО ПОКРЫТИЯ НА ОСНОВЕ ХИТОЗАНА И ПЛЕНОЧНОЕ ПОКРЫТИЕ НА ОСНОВЕ ХИТОЗАНА | 2010 |
|
RU2461575C2 |
| КОНЪЮГАТ МОНОМЕТИЛ АУРИСТАТИНА Е ДЛЯ ПОЛУЧЕНИЯ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ РАКА ПРЕДСТАТЕЛЬНОЙ ЖЕЛЕЗЫ | 2019 |
|
RU2729192C1 |
| СТЕНТ | 2007 |
|
RU2432183C9 |
| КОНЪЮГАТ ФЛУОРЕСЦЕНТНОГО КРАСИТЕЛЯ С ВЕЩЕСТВОМ ПЕПТИДНОЙ ПРИРОДЫ, ВКЛЮЧАЮЩИМ ПСМА-СВЯЗЫВАЮЩИЙ ЛИГАНД НА ОСНОВЕ ПРОИЗВОДНОГО МОЧЕВИНЫ ДЛЯ ВИЗУАЛИЗАЦИИ КЛЕТОК, ЭКСПРЕССИРУЮЩИХ ПСМА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2019 |
|
RU2713151C1 |
| СРЕДСТВО ДЛЯ ЛЕЧЕНИЯ ПОВРЕЖДЕНИЙ НАРУЖНЫХ ТКАНЕЙ ОРГАНИЗМА (ВАРИАНТЫ) И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2014 |
|
RU2578969C1 |
| СЛОЖНЫЙ КАРБОКСИЛАТНЫЙ ЭФИР ПОЛИСАХАРИДА | 2015 |
|
RU2654031C2 |
| СРЕДСТВО ПЕПТИДНОЙ ПРИРОДЫ, ВКЛЮЧАЮЩЕЕ ПСМА-СВЯЗЫВАЮЩИЙ ЛИГАНД НА ОСНОВЕ ПРОИЗВОДНОГО МОЧЕВИНЫ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ ДЛЯ ПОЛУЧЕНИЯ КОНЪЮГАТА С ЛЕКАРСТВЕННЫМ И ДИАГНОСТИЧЕСКИМ АГЕНТОМ | 2018 |
|
RU2697519C1 |


Изобретение относится к производному хитозана, в котором хитозановый фрагмент имеет общую формулу (I), где R - остаток жирной или аминокислоты, n для гидрофильного лиганда составляет от около 12 до около 25% относительно количества моносахаридных остатков хитозана, m для гидрофобного лиганда составляет от около 30 до около 60% относительно количества моносахаридных остатков хитозана. Также изобретение относится к вариантам способа его получения. Изобретение позволяет обеспечить производное хитозана с высокой трансдермальной и мембранной проницаемостью, повышенной сорбционной емкостью, растворимое в широком диапазоне pH. 3 н. и 13 з.п. ф-лы, 2 ил., 2 пр., 1 табл.
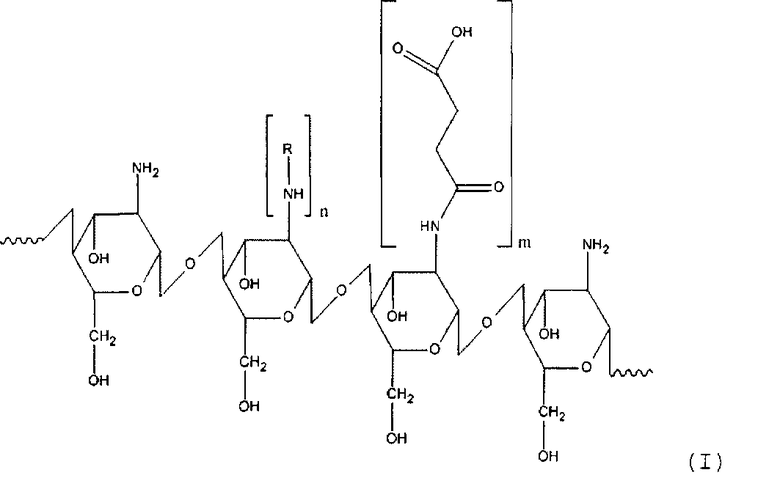
1. Производное хитозана, в котором хитозановый фрагмент имеет общую формулу (I)
где
R - остаток жирной или аминокислоты,
n для гидрофильного лиганда составляет от около 12 до около 25% относительно количества моносахаридных остатков хитозана,
m для гидрофобного лиганда составляет от около 30 до около 60% относительно количества моносахаридных остатков хитозана.
2. Производное хитозана по п.1, отличающееся тем, что указанный остаток жирной кислоты представляет собой остаток каприловой кислоты.
3. Производное хитозана по п.1, отличающееся тем, что указанный остаток аминокислоты представляет собой остаток валина.
4. Производное хитозана по п.1, отличающееся тем, что указанное производное наноструктурировано.
5. Производное хитозана по п.1, отличающееся тем, что указанное производное имеет трансдермальную и/или мембранную проницаемость.
6. Производное хитозана по п.1, отличающееся тем, что указанное производное имеет повышенную сорбционную емкость.
7. Производное хитозана по п.1, отличающееся тем, что указанное производное растворимо при pH от около 2 до около 9, предпочтительно от около 4 до около 9, более предпочтительно от около 6 до около 9, наиболее предпочтительно от около 7 до около 8.
8. Производное хитозана по п.7, отличающееся тем, что указанное производное растворимо при pH около 7.4.
9. Производное хитозана по п.1, отличающееся тем, что указанное производное имеет сферическую структуру с размером частиц от около 1 нм до около 2000 нм, предпочтительно от около 1 нм до около 1500 нм, более предпочтительно от около 1 нм до около 900 нм, еще более предпочтительно от около 1 нм до около 400 нм, наиболее предпочтительно от около 1 нм до около 100 нм.
10. Производное хитозана по п.1, отличающееся тем, что указанное производное имеет содержание гидрофильного лиганда предпочтительно от около 15% до около 25%, более предпочтительно от около 18% до около 25%, наиболее предпочтительно от около 20% до около 25%.
11. Производное хитозана по п.1, отличающееся тем, что указанное производное имеет содержание гидрофобного лиганда предпочтительно от около 35% до около 55%, более предпочтительно от около 4 0% до около 50%, наиболее предпочтительно от около 45% до около 50%.
12. Производное хитозана по п.1, отличающееся тем, что исходный хитозан указанного производного имеет молекулярную массу от около 5 кДа до около 100 кДа, предпочтительно от около 7 кДа до около 80 кДа, более предпочтительно от около 10 кДа до около 60 кДа, наиболее предпочтительно от около 30 кДа до около 50 кДа.
13. Производное хитозана по п.12, отличающееся тем, что исходный хитозан указанного производного имеет молекулярную массу около 23 кДа.
14. Производное хитозана по п.1, отличающееся тем, что исходный хитозан указанного производного имеет степень деацетилирования 85±2%.
15. Способ получения производного хитозана, в котором хитозановый фрагмент имеет общую формулу (I)
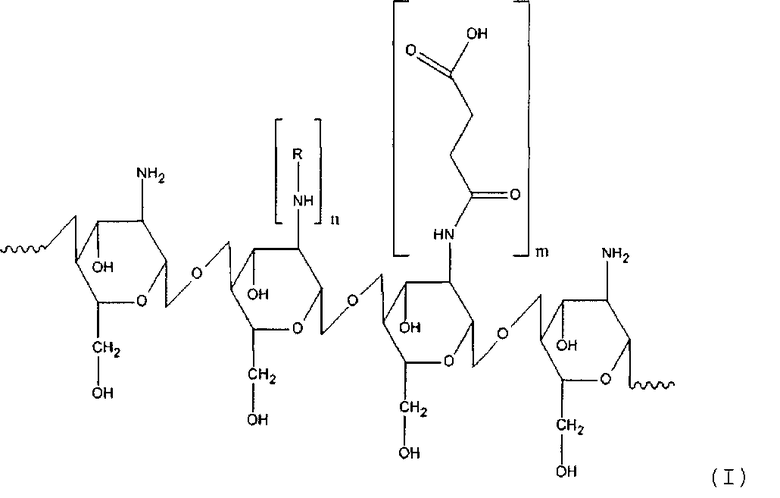
где
R - остаток жирной кислоты,
n для гидрофильного лиганда составляет от около 12 до около 25% относительно количества моносахаридных остатков хитозана,
m для гидрофобного лиганда составляет от около 30 до около 60% относительно количества моносахаридных остатков хитозана, включающий:
- взаимодействие жирной кислоты с N-гидроксисукцинимидом с получением N-оксисукцинимидного эфира жирной кислоты;
- ацилирование хитозана полученным N-оксисукцинимидным эфиром жирной кислоты;
- карбоксиацилирование янтарным ангидридом хитозана, ацилированного N-оксисукцинимидным эфиром жирной кислоты.
16. Способ получения производного хитозана, в котором хитозановый фрагмент имеет общую формулу (I)

где
R - остаток аминокислоты,
n для гидрофильного лиганда составляет от около 12 до около 25% относительно количества моносахаридных остатков хитозана,
m для гидрофобного лиганда составляет от около 30 до около 60% относительно количества моносахаридных остатков хитозана, включающий:
- взаимодействие аминокислоты с HBTU (N,N,N′,N′-тетраметил-О-(1Н-бензотриазол-1-ил)урониум гексафлюорофосфат) с получением N-оксисукцинимидного эфира аминокислоты;
- ацилирование хитозана полученным N-оксисукцинимидным эфиром аминокислоты;
- карбоксиацилирование янтарным ангидридом хитозана, ацилированного N-оксисукцинимидным эфиром аминокислоты.
| ХИТОЗАНОВЫЙ ПРОДУКТ, СПОСОБ ЕГО ПОЛУЧЕНИЯ (ВАРИАНТЫ) | 2005 |
|
RU2313538C2 |
| N-ЦИАНОМЕТИЛИРОВАННЫЕ ХИТОЗАНЫ, ПРОДУКТЫ ИХ ГИДРОЛИЗА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 1995 |
|
RU2158270C2 |
| ВСПСОЮЗКАЯ Т11 ПЛТОЗТКО-б^рПИЧБШЯ E''S ТПОТЕКД11В. К. Ветров | 0 |
|
SU187988A1 |

