Область техники
Изобретение относится к области органической и медицинской химии, а также молекулярной биологии и касается нового класса соединений, которые могут быть использованы в качестве средств для доставки лекарственных или диагностических соединений (агентов) для диагностики или лечения онкологических заболеваний. В частности, диагностики или лечения заболеваний, вызванных клетками, экспрессирующими ПСМА, такими как клетки рака предстательной железы.
Уровень техники
Рак простаты является наиболее распространенным злокачественным заболеванием у мужчин и занимает второе место среди причин смертности в западном мире. По состоянию на 2016 год, общее количество больных раком предстательной железы, зарегистрированных на территории России, составило более 200 тыс. человек. Прирост количества больных по отношению к 2015 году составил 8,0%. В настоящее время не существует радикального лечения нерезектабельных опухолей простаты. Концентрации лекарственных средств, которые могли бы полностью уничтожить опухоль, обычно нельзя достичь из-за ограничивающих дозу побочных действий, таких как токсичность для желудочно-кишечного тракта и костного мозга. Кроме того, у опухолей после продолжительного лечения может развиваться резистентность против противораковых средств. Из-за высокой смертности и болезненности, связанной с прогрессированием заболевания, крайне необходимо развитие новых способов и средств направленной терапии. Поэтому при разработке современных лекарственных средств нацеливание цитотоксических средств на область опухоли может рассматриваться как одна из важнейших задач.
Среди потенциальных маркеров, которые были идентифицированы при раке простаты, наиболее известным, является специфический для простаты мембранный антиген (ПСМА, PSMA). Этот трансмембранный гликопротеид II типа, молекулярной массой примерно 100 кДа, состоит из короткого внутриклеточного участка (аминокислоты 1-18), трансмембранного домена (аминокислоты 19-43) и протяженного внеклеточного домена (аминокислоты 44-750), который экспрессируется главным образом в нормальном эпителии простаты человека, но коррелирует с повышением в раковой простате, включая метастазирование. Поскольку PSMA экспрессируется практически всеми раками простаты и его экспрессия еще больше усиливается в низкодифференцированных, метастатических и гормононезависимых карциномах, он является весьма привлекательной целью для визуализации и терапии простаты.
Наиболее близким к заявляемому соединению является простат-специфический антиген для эндотерапии рака предстательной железы, в одном из частных воплощений изобретения соединения характеризуются наличием в структуре мочевины на основе глутаминовой кислоты и лизина (DCL) ароматического фрагмента при ε-положении лизина и в структуре линкера, в виде дипептида из ароматических аминокислот фенилаланина и тирозина, а также его бром-замещенных аналогов, с различной конфигурацией оптического центра аминокислот, который необходим для соединения с доставляемой молекулой, и хелатирующий агент для включения металла или радиометалла (международная заявка WO 2017165473). Однако, наличие в структуре мочевины на основе глутаминовой кислоты и лизина (DCL), только ароматического фрагмента при омега положении лизина не обеспечивает точное позиционирование соединения в структуре ПСМА, что в свою очередь снижает эффективность связывания препарата с мишенью и эффективность действия соединения в целом. В заявке также раскрыт способ получения соединений, заключающийся в алкилировании на первом этапе три-(трет-бутил) защищенного производного мочевины на основе глутаминовой кислоты и лизина, последующей реакцией ацилирования получившегося продукта Вос-аминопентановой кислотой. Делее проводится удаление Вос-защитной группы в присутствии трифторуксуной кислоты в дихлорметане, с последующим получением конъюгата с хелатирующей группой. Однако в синтезе данного соединения используется в качестве восстанавливающего агента, на стадии алкилирования лизина цианоборгидрид натрия, который является токсичным реагентом. Так же использование ВОС-производного аминопентановой кислоты не позволяет избирательно получить аминопроизводное с защищенными карбоксильными группами, что значительно усложняет дальнейшую модификацию молекулы.
Таким образом, недостатком известных соединений (конъюгатов) является их низкая эффективность связывания с мишенью и, как следствие, необходимость использования доставляемых препаратов в более высоких концентрациях, что может вызывать специфическую цитотоксичность.
Раскрытие изобретения
Задачей настоящего изобретения является разработка новых средств доставки диагностических или терапевтических средств, включающих ПСМА-лиганд с линкером, соединения, являющегося средством для доставки лекарственного или диагностического агента и способ его получения.
Техническим результатом заявляемой группы изобретений является высокая аффинность и селективность действия заявляемых соединений в отношении клеток, экспрессирующих ПСМА. Данные соединения позволяют расшить арсенал перспективных кандитатов для разработки лекарственных и диагностических средств для лечения заболеваний, связанных с высокой экспрессией ПСМА, с меньшей дозировкой при избирательном действии на раковые клетки, не затрагивая здоровые клетки. Новые соединения обладают низкой токсичностью, о чем свидетельствует отсутствие активности на клеточной линии здоровых клеток легочного эпителия человека. Заявляемый способ получения соединений является более экологичным и безопасным за счет применения менее токсичных реагентов. Использование для восстановительного аминирования триацетоксиборогидрида натрия вместо цианоборогидрида натрия позволяет избежать возможности отравления персонала черезвычайно токсичным и летучим циановодородом, который выделяется как побочный продукт при использовании цианоборогидрида натрия. Так же использование азидопроизводного аминопентановой кислоты вместо ВОС-производного при синтезе линкера позволяет избежать использования коррозионной и токсичной трифторуксусной кислоты. Кроме того, использование азидопроизводного аминопентановой кислоты позволяет получить ПСМА вектор с длинным гидрофобным линкером и защищенными карбоксигрупами, что в свою очередь облегчает его модификацию, увеличивает выход и снижает количество используемых в процессе растворителей вследствие значительного увеличения растворимости исходного соединения (ПСМА вектор с длинным гидрофобным линкером и защищенными карбоксигрупами).
Ключевой особенностью заявляемого соединения является наличие в структуре длинного гидрофобного линкера, а также дополнительных ароматических фрагментов, наличие которых, способствует лучшему связыванию заявляемого средства с белковой мишенью, за счет вовлечения дополнительных взаимодействий между соединением и гидрофобными карманами в структуре гидрофобного туннеля белковой мишени.
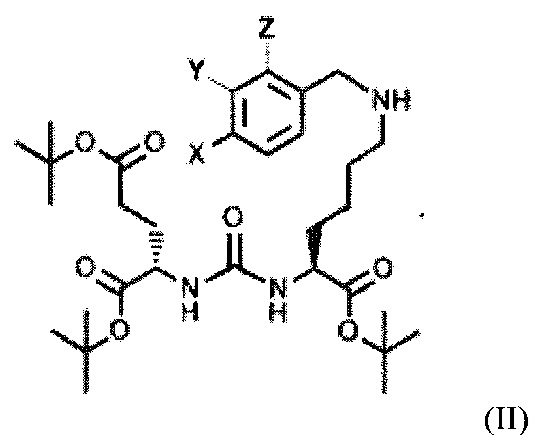
Поставленная задача решается средством для доставки лекарственного или диагностического агента, представляющим собой ковалентно-связанные ПСМА-связывающий лиганд на основе производного мочевины структуры DCL и модифицированный гидрофобный пептидный линкер, включающий фрагменты 6-аминогексановой или 5-аминопентановой или 4-аминобутановой кислот, фрагмент фенилаланина, фрагмент тирозина или его производное, общей формулы:
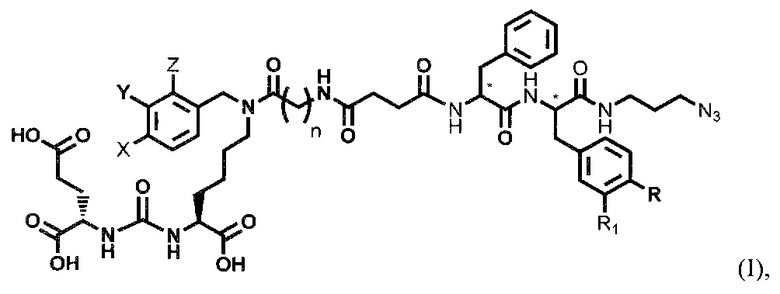
где n=3-5, где X, Y, Z независимо друг от друга представляют собой F, Cl, Br, Н; R=ОН, Н; R1=Н, Br.
Также поставленная задача решается способом получения заявляемого средства, включающим получение тритретбутилового производного ПСМА-связывающего лиганда формулы (2):
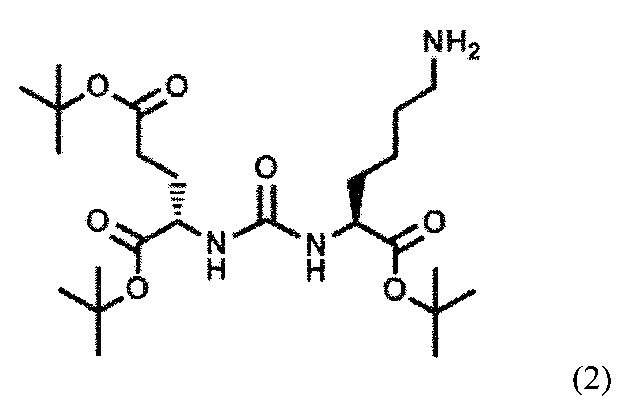
с последующим алкилированием полученного тритретбутилового производного ПСМА-связывающего лиганда с получением соединения общей формулы (II):
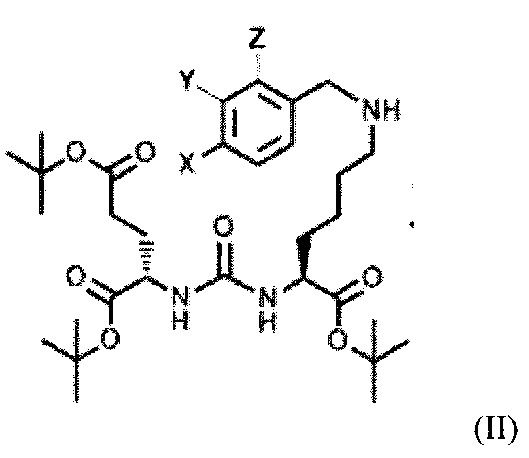 ,
,
где X, Y, Z независимо друг от друга представляют собой F, Cl, Br, Н,
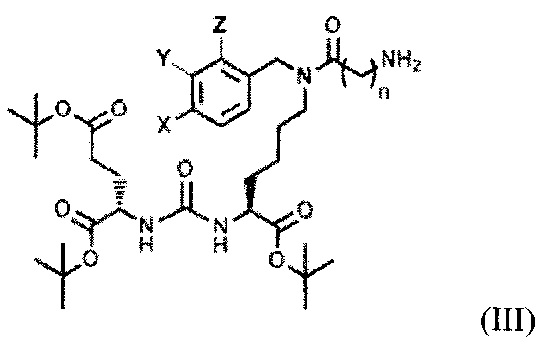
с последующим получением соединения, содержащего алкилированное тритретбутил производное ПСМА-лиганда и фрагмент линкера, представляющего собой алкильный фрагмент, включающий 3-5 атомов углерода общей формулы (III):
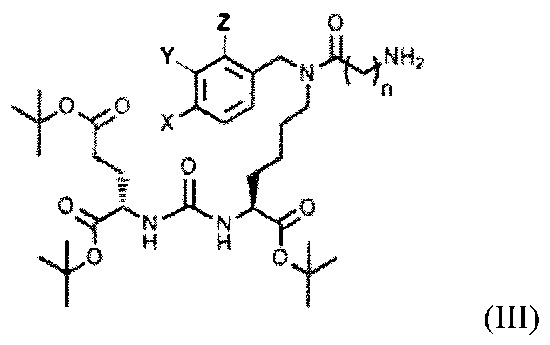 ,
,
где X, Y, Z независимо друг от друга представляют собой F, Cl, Br, Н, n=3-5,
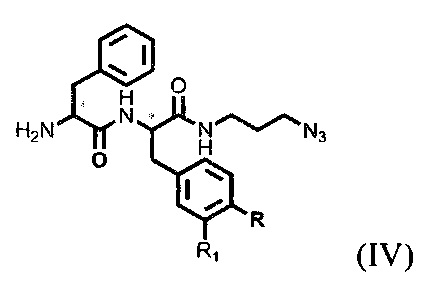
которое модифицируют янтарным ангидридом, с получением производного соединения ацилированного янтарным ангидридом, далее осуществляют получение дипептидов производных ароматических аминокислот, представляющих собой фенилаланил-фенилаланин или фенилаланил-тирозин общей формулы (IV), для связывания с модифицированным фрагментом линкера,
 ,
,
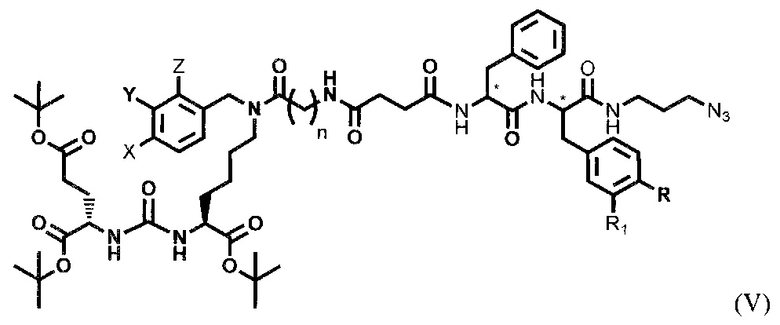
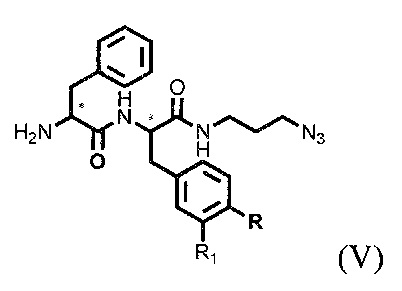
где R=ОН, Н; R1=Н, Br, затем получают тритретбутил производное радикала ПСМА-связывающего лиганда и модифицированного гидрофобного пептидного линкера, включающего фрагменты 6-аминогексановой или 5-аминопентановой или 4-аминобутановой кислот, фрагмент фенилаланина или его производное, фрагмент тирозина или его производное общей формулы (V):
 ,
,
где n=3-5, X, Y, Z независимо друг от друга представляют собой F, Cl, Br, Н; R=ОН, Н; R1=Н, Br
с последующим удалением тритретбутильных защитных групп соединения формулы (V) с получением ковалентно-связанных ПСМА-связывающего лиганда и модифицированного гидрофобного пептидного линкера общей формулы (I).
При этом получение алкилированного тритретбутил производного ПСМА-лиганда осуществляют путем восстановительного аминирования замещенными бензальдегидами, а получение соединения формулы (III) проводят путем ацилирования производными азида бутановой или пентановой или гексановой кислот с получением азидного производного с последующей реакцией восстановления азида до аминогруппы. Реакцию восстановления азида до аминогруппы проводят в присутствии трифенилфосфина и воды в растворе ТГФ или в растворе метанола с использованием водорода в присутствии палладия на углероде в качестве катализатора. Модификацию янтарным ангидридом фрагмента линкера, представляющего собой алкильный фрагмент, включающий 3-5 атомов углерода осуществляют реакцией ацилирования янтарным ангидридом аминогруппы в присутствии ненуклиофильных оснований, в качестве которых используют диизопропилэтиламин или триэтиламин. Получение тритретбутил производного конъюгата формулы (V) осуществляют реакцией ацилирования производным соединения (III), ацилированного янтарным ангидридом, дипептида формулы (IV), а удаление тритретбутильных защитных групп проводят в присутствии 9-11% ТФУ в течение 15-17 часов в дихлорметане.
Поставленная задача также решается применением заявляемого средства для образования конъюгата с лекарственным или диагностическим агентом. При этом лекарственные или диагностические агенты являются средствами для диагностики или лечения заболеваний, вызванных клетками, экспрессирующими ПСМА, таким как рак предстательной железы.
Общая схема синтеза заявляемого средства для доставки лекарственного или диагностического агента представлена на фиг. 1, 2 и 3.
Краткое описание чертежей
На фиг. 1, 2 и 3 представлена общая схема синтеза заявляемого средства.
На фиг. 4 представлены графики интенсивности флуоресцентного сигнала клеток LNCaP, 22Rv1, РС3 при воздействии конъюгатами (80) - №1, (79) - №2 и без воздействия. Данные представлены как среднее со стандартным отклонением, где  - LNCaP,
- LNCaP,  - 22Rv1,
- 22Rv1,  - РС3.
- РС3.
На фиг. 5 представлено изображение клеток 22Rv1 после воздействия коньюгатом формулы (80) в течение одного часа, зеленый цвет маркирует накопление флуоресцентного сигнала FAM (GFP канал), в синий цвет окрашены ядра клеток (DAPI).
На фиг. 6 представлено изображение клеток 22Rv1 после воздействия заявляемым средством (соединение формулы (79)) в течение одного часа, зеленый цвет маркирует накопление флуоресцентного сигнала FAM (GFP канал), в синий цвет окрашены ядра клеток (DAPI).
На фиг. 7 представлено изображение клеток LNCaP после воздействия коньюгатом формулы (80) в течение одного часа, зеленый цвет маркирует накопление флуоресцентного сигнала FAM (GFP канал), в синий цвет окрашены ядра клеток (DAPI).
На фиг. 8 представлено изображение клеток LNCaP после воздействия заявляемым средством (соединение формулы (79)) в течение одного часа, зеленый цвет маркирует накопление флуоресцентного сигнала FAM (GFP канал), в синий цвет окрашены ядра клеток (DAPI).
На фиг. 9 представлено изображение клеток РС3 после воздействия коньюгатом формулы (80) в течение одного часа, зеленый цвет маркирует накопление флуоресцентного сигнала FAM (GFP канал), в синий цвет окрашены ядра клеток (DAPI).
На фиг. 10 представлено изображение клеток РС3 после воздействия коньюгатом формулы (79) в течение одного часа, зеленый цвет маркирует накопление флуоресцентного сигнала FAM (GFP канал), в синий цвет окрашены ядра клеток (DAPI).
На фиг. 11 представлена гистограмма экспрессии сигнала FAM клеточными популяциями в линиях 22RV1, LNCaP, РС3 при воздействии коньюгата формулы (80) - №1 и результаты эксперимента при предварительной инкубации с избытком заявляемого средства - К.
На фиг. 12 показано графическое представление процента клеток, экспрессирующих FAM флуорисцентный сигнал. К - результаты эксперимента при предварительной инкубации с избытком заявляемого средства (соединение формулы (80)) в течение 60 мин.
На фиг. 13 представлена гистограмма экспрессии сигнала FAM клеточными популяциями в линиях 22RV1, LNCaP, РС3 при воздействии коньюгата формулы (79) - №2 и результаты эксперимента при предварительной инкубации с избытком заявляемого средства - К.
На фиг. 14 - графическое представление процента клеток, экспрессирующих FAM флуорисцентный сигнал. К - результаты эксперимента при предварительной инкубации с избытком заявляемого средства (соединение формулы (79)) в течение 60 мин.
На фиг. 15 представлен масс-спектр соединения (82).
На фиг. 16 показан 1Н-ЯМР спектр соединения (83).
На фиг. 17 представлен масс-спектр соединения (83).
На фиг. 18, 19 представлена схема образование коньюгата с лекарственным соединением - доцетакселем.
На фиг. 20 представлена схема синтеза ПСМА-векторного фрагмента на основе дипептида L-фенилаланил-L-тирозина (L-Phe-L-Tyr (33)).
Осуществление изобретения
Ниже приведены определения терминов, которые используются в описании настоящего изобретения.
«ПСМА (PSMA)» - трансмембранный гликопротеид II типа с массой - 100 кДа, состоящий из 750 аминокислот. Данный белок состоит из короткого внутриклеточного участка (1-18 аминокислоты), трансмембранного домна (19-43 аминокислоты) и большого внеклеточного домена (44-750 аминокислоты). Данный белок обладает высокой экспрессией в тканях предстательной железы, в связи с этим является перспективной мишенью для адресной доставки.
В заявляемом техническом решении под средством для доставки лекарственного или диагностического агента, представляющее собой ковалентно-связанные ПСМА-связывающий лиганд и модифицированный гидрофобный пептидный линкер, включающий фрагменты 6-аминогексановой или 5-аминопентановой или 4-аминобутановой кислот, фрагмент фенилаланина, фрагмент тирозина или его производное. Термин «конъюгат» в заявляемом техническом решении означает соединение включающее средство для доставки с лекарственным или диагностическим агентом.
EDC*HCl - 1-Этил-3-(3-диметиламинопропил)карбодиимид гидрохлорид
PFPOH - пентафторфенол
НОВТ - гидроксибензотриазол
HBTU - 3-[Бис(диметиламино)метилиумил]-3Н-бензотриазол-1-оксид гексафторфосфат
EtOAc/MeOH - этилацетат/метанол
DMAP - 4-диметиламинопиридин
DCM - дихлорметан
DIC - диизопропилкарбодиимид
DMF - ДМФА
DIPEA - диизопропилэтиламин
TFA - трифторуксусная кислота
ДХМ - дихлорметан
PBS - натрий-фосфатный буфер
FBS - фетальная телячья сыворотка
PPh3 - трифенилфосфин
РуВОР - бензотриазол-1-ил-окситрипирролидинофосфоний гексафторфосфат
THF - тетрагидрофуран
ВОС2О - ди-третбутил дикарбонат
«IC50» (ПК50) - концентрация соединения, при которой наблюдается 50% ингибирование ферментативной активности
Все используемые реагенты являются коммерчески доступными, выпаривание растворителя осуществляли с использованием роторного испарителя, при пониженном давлении при температуре бани примерно 50°С; контроль за ходом реакции осуществляли при помощи тонкослойной хроматографии (ТСХ), и время реакции указано только для иллюстрации; структуру и чистоту всех выделенных соединений подтверждали, по меньшей мере, одним из следующих методов: ТСХ (пластины для ТСХ с предварительно нанесенным силикагелем 60 F254 Merck), масс-спектрометрия или ядерный магнитный резонанс (ЯМР). Выход продукта приведен только для иллюстрации. Колоночную флэш-хроматографию осуществляли, используя Merck силикагель 60 (230-400 меш ASTM). Масс-спектры высокого разрешения (HRMS) зарегистрированы на спектрометре Bruker microTOF II. Спектры ЯМР регистрировали на приборах Bruker Avance-400 (рабочая частота 400.1 и 100.6 МГц для 1Н и 13С, соответственно) и Agilent 400-MR (рабочая частота 400.0 и 100.6 МГц для 1Н и 13С, соответственно), используя дейтерированный хлороформ (99,8% D) или ДМСО (99,9% D) в качестве растворителя, если не указано иное, относительно тетраметилсилана (TMS) в качестве внутреннего стандарта, миллионных долях (м.д.); обычные используемые сокращения следующие: с - синглет, д - дублет, т - триплет, кв - квартет, м - мультиплет, шир. - широкий и так далее.
В некоторых вариантах воплощения, опухолевые клетки могут экспрессировать PSMA, такие как клетки опухоли предстательной железы или метастазированные клетки опухоли предстательной железы. В других вариантах воплощения, опухоль можно лечить путем прицельного действия на прилегающие или расположенные рядом клетки, которые экспрессируют PSMA. Например, можно прицельно воздействовать на сосудистые клетки, в которых возникает ангиогенез, связанный с опухолью. По существу, все солидные опухоли экспрессируют PSMA в неоваскулатуре. Поэтому способы по настоящему изобретению можно использовать для лечения практически всех солидных опухолей, включая опухоль легкого, почечно-клеточную, глиобластому, поджелудочной железы, мочевого пузыря, саркому, меланому, молочной железы, толстой кишки, зародышевых клеток, феохромоцитому, пищевода и желудка. Также, в соответствии с настоящим изобретением можно лечить некоторые доброкачественные поражения и ткани, включая эндометрий, шванному и хронической пептической язвы пищевода (синдром Баррета).
Лекарственные средства могут вводиться перорально или парентерально (например, внутривенно, подкожно, внутрибрюшинно или местно). Клиническая дозировка средства, содержащего соединение общей формулы (I), у пациентов может корректироваться в зависимости от терапевтической эффективности и биодоступности активных ингредиентов в организме, скорости их обмена и выведения из организма, а также в зависимости от возраста, пола и стадии заболевания пациента.
Заявляемое средство для доставки формулы (I) представляет собой ковалентно связанные лиганд, обеспечивающий селективное связывание с ПСМА, и линкер.
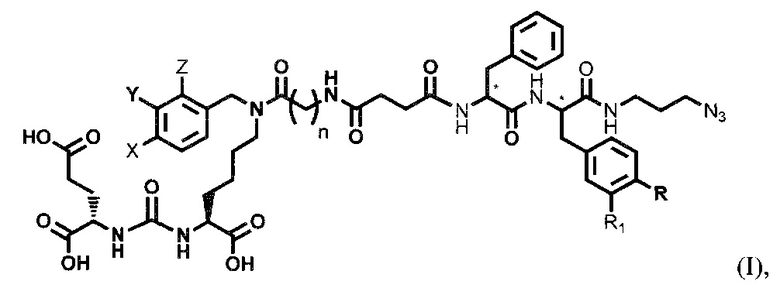
где n=3-5, где X, Y, Z независимо друг от друга представляют собой F, Cl, Br, Н; R=Н, ОН; R1=Н, Br.
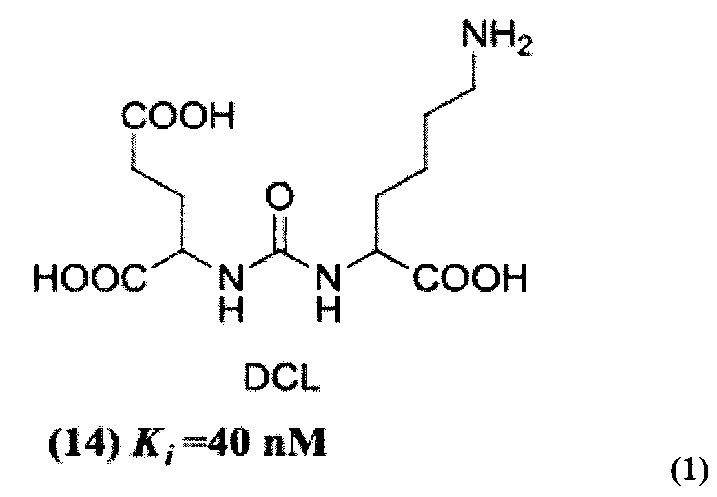
В качестве лиганда выступает соединение, представляющее собой аналог переходного состояния субстрата (NAAG) и являющегося ПСМА-вектором на основе производного мочевины структуры DCL формулы (1), включая модификацию ε-аминогруппы лизина в лиганде ПСМА с использованием различных бензиловых производных, способствующих улучшению связывания с белковой мишенью.
Линкер представляет собой модифицированный гидрофобный пептид, включающий фрагменты 6-аминогексановой или 5-аминонопентановой или 4-аминобутановой кислот, фрагмент янтарной кислоты, фрагмент аминокислоты фенилаланина, производное фенилаланина, с введенными заместителями в ароматический фрагмент, фрагмент 2-азидоэтанола.
Способ синтеза заявляемых соединений заключается в следующем.
Сначала получают тритретбутиловое производное ПСМА-связывающего лиганда формулы (2):
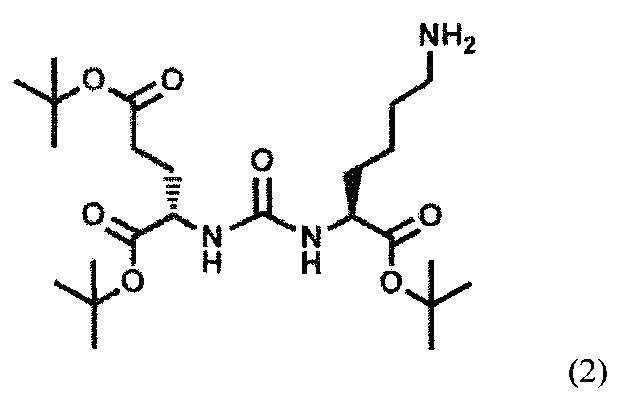
Соединение формулы (2) может быть получено известным из уровня техники способом (Ryan P. Murelli, Andrew X. Zhang, Julien Michel, William L. Jorgensen, David A. Spiege. Chemical Control Over Immune Recognition: A Class of Antibody-Recruiting Molecules (ARMs) that Target Prostate Cancer. J. AM. CHEM. SOC. 2009, 131, 17090-17092). Затем полученное тритретбутиловое производное ПСМА-связывающего лиганда алкилируют с получением соединения формулы (II). Реакцию алкилирования проводят путем восстановительного аминирования замещенными бензальдегидами (Jan Tykvart,  Schimer, Jitka
Schimer, Jitka  , Petr Pachl, Lenka
, Petr Pachl, Lenka  , Pavel Majer, Jan Konvalinka, Pavel
, Pavel Majer, Jan Konvalinka, Pavel  . Rational design of urea-based glutamate carboxypeptidase II (GCPII) inhibitors as versatile tools for specific drug targeting and delivery. Bioorg Med Chem. 2014, 22(15):4099-108.)
. Rational design of urea-based glutamate carboxypeptidase II (GCPII) inhibitors as versatile tools for specific drug targeting and delivery. Bioorg Med Chem. 2014, 22(15):4099-108.)
где X, Y, Z независимо друг от друга представляют собой F, Cl, Br, Н.
Получение соединения формулы (III) проводят путем ацилирования производными азидо- бутановой или пентановой или гексановой кислот с получением азидного производного алкилированного производного ПСМА-лиганда (IV) с последующей реакцией восстановления азида до аминогруппы, при этом реакцию восстановления азида до аминогруппы проводят в присутствии трифенилфосфина и воды в растворе ТГФ или в растворе метанола с использованием водорода в присутствии палладия на углероде в качестве катализатора.
Реакцию ацилирования проводят в среде полярного апротонного растворителя растворяют исходный амин (II), азидо-кислоту и ненуклеофильное основание, взятые из расчета, что на 1 мольный эквивалент амина берут по меньшей мере 1 мольный эквивалент азидокислоты и основания, а также не менее 100 мольных эквивалента полярного апротонного растворителя, к полученной смеси при перемешивании добавляют по меньшей мере 1 мольный эквивалент РуВОР, полученную смесь перемешивают при комнатной температуре до исчезновения исходного амина (II). Далее из полученную реакционной смеси удаляется растворитель при пониженном давлении, и целевой полупродукт выделяется с помощью колоночной хроматографии (Puriflach SILICA-HP 120G, 50 мкм, градиент от 100% петролейного эфира до 100% EtOAc в течение 30 мин, скорость потока = 50 мл/мин). Верхняя граница используемых реагентов не ограничивается, т.к. избыток какого-либо реагента не уменьшает выходов реакций, однако при большом избытке может понадобиться дополнительная очистка продуктов реакций.
Далее реакцию восстановления азидо-группы до амино-группы в среде ТГФ/вода с содержанием воды по меньшей мере 10 об. %, в которой растворяют полученное азидо-производное и трифенилфосфин, взятые из расчета, что на 1 мольный эквивалент азидопроизводного берут по меньшей мере 1,5 мольных эквивалента трифенилфосфина, а также не менее 50 мольных эквивалентов смеси растворителей (ТГФ/вода) из расчета на воду. Реакционную смесь нагревали при температуре не менее 45°С до исчезновения исходного азидо-производного. Растворитель удаляли при пониженном давлении. Очистку проводили методом колоночной хроматографии (триэтиламин: хлористый метилен: метанол; от 1%:98%:1% до 1%:89%:10%).
Предпочтительно в качестве азидо-кислот использовать азидо-бутановую или пентановую или гексановую кислоты.
Предпочтительно в качестве полярного апротонного растворителя использовать ДМФА или ДМСО.
Предпочтительно в качестве ненуклеофильного основания использовать диизопропилэтиламин или триэтиламин.
 ,
,
где X, Y, Z независимо друг от друга представляют собой F, Cl, Br, Н, n=3-5.
Модификация янтарным ангидридом фрагмента линкера, представляющего собой алкильный фрагмент, включающий 3-5 атомов углерода с получением производного соединения (III), ацилированного янтарным ангидридом. При этом модификацию янтарным ангидридом фрагмента линкера, представляющего собой алкильный фрагмент, включающий 3-5 атомов углерода осуществляют реакцией ацилирования янтарным ангидридом аминогруппы в присутствии ненуклиофильных оснований. В качестве ненуклеофильных оснований используют диизопропилэтиламин или триэтиламин.
Реакцию ацилирования янтарным ангидридом проводят в среде неполярного апротонного растворителя путем растворения исходного амина (III), янтарного ангидрида и ненуклеофильного основания, взятых из расчета, что на 1 мольный эквивалент амина берут по меньшей мере 1 мольный эквивалент янтарного ангидрида и ненуклефильного основания, а также не менее 100 мольных эквивалента неполярного апротонного растворителя, полученную смесь перемешивают при комнатной температуре до исчезновения исходного амина (III).
Предпочтительно в качестве неполярного апротонного растворителя использовать дихлорметан или хлороформ.
Предпочтительно в качестве ненуклеофильного основания использовать диизопропилэтиламин или триэтиламин.
Получение дипептидов производных ароматических аминокислот, представляющих собой фенилаланил-фенилаланин или фенилаланил-тирозин, для связывания с модифицированным фрагментом линкера соединения формулы (V).
Синтез дипептидов проиллюстрирован на схемах (а) и (б), где синтез ПСМА-векторного фрагмента на основе дипептида L-Phe-L-Phe - схема (a), a D-Phe-D-Phe - схема (б).
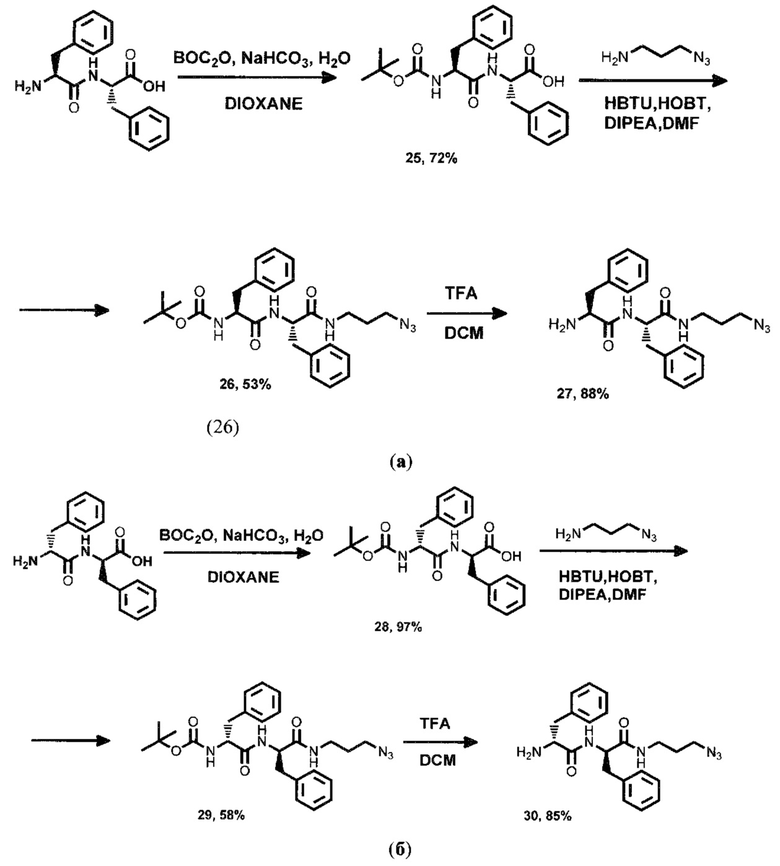
К дипептиду L-Phe-L-Phe в смеси воды с диоксаном, не менее 25 об. % воды в смеси, в присутствии основания в количестве не менее 3-х мольных эквивалентов, добавляли не менее 1 эквивалента ВОС-ангидрида при температуре не выше 5°С. Реакционную смесь оставляют перемешиваться не менее 6 часов при комнатной температуре. Полученную смесь промывали водным раствором соляной кислоты с концентрацией не менее 1 моль/л до не более 4 моль/л, полученный раствор экстрагировали этилацетатом. Органическую фазу осушивали над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный продукт представляет собой белые кристаллы.
Предпочтительно в качестве основания использовать гидрокарбонат натрия, карбонат натрия, гидроксид натрия или гидроксид калия.
Для получения соединения (26), проводили реакцию BOCPhePhe с 3-аминопропилазидом (не менее 1 мольн. экв.) в присутствии не менее 1 эквивалента HBTU, HOBt и DIPEA. После протекания реакции целевой продукт выделяли методом колоночной хроматографии.
Реакцию удаления трет-бутильной защитной группы соединения (26) проводили с помощью раствора трифторуксусной кислоты в дихлорметане, с содержанием кислоты не менее 8 об. %. Реакционную смесь перемешивали, контроль протекания реакции проводили методом ТСХ до исчезновения пятна исходного вещества Rf~0.3. По окончании реакции растворитель удаляли при пониженном давлении и далее полученный продукт промывали диэтиловым эфиром.
Синтез дипептида D-Phe-D-Phe осуществлялся аналогично производному с L-Phe-L-Phe.
Синтез ПСМА-векторного фрагмента на основе дипептида L-фенилаланил-L-тирозина (L-Phe-L-Tyr (33)) осуществлялся по схеме (с) (Фиг. 20). К суспензии L-фенилаланина в смеси растворителей диоксан - вода с содержанием воды не менее 40 об. %, при температуре не более 5°С добавляли основание в количестве не менее одного мольного эквивалента и ди-трет-бутил дикарбонат ВОС2О в количестве не менее одного мольного эквивалента. Полученную смесь перемешивали при комнатной температуре в течение не менее 4 суток. Реакционную смесь концентрировали в вакууме роторного испарителя до удаления органического растворителя. Затем в водный остаток добавляли раствор соляной кислоты с концентрацией не менее 1 моль/л, до рН не более 4 и экстрагировали этилацетатом. Объединенную органическую фазу промывали насыщенным раствором NaHCO3 и NaCl, сушили Na2SO4 и концентрировали в вакууме. Затем повторно переупаривали с дихлорметаном. Продукт реакции получали в виде бесцветного аморфного вещества.
Предпочтительно в качестве основания использовать гидрокарбонат натрия, карбонат натрия, гидроксид натрия или гидроксид калия.
К раствору соединения (31) в дихлорметане добавляли EDC*HCl (не менее 1 экв.), PFPOH (не менее 1 экв.) и перемешивали в течение не менее 12 часов при комнатной температуре. Дальнейшую очистку проводили с помощью колоночной хроматографии на колонке с силикагелем (элюент - дихлорметан). Продукт реакции (желтое маслянистое вещество), растворяли в смеси ТГФ - вода (не менее 30 об. % воды) и добавляли при перемешивании L-тирозин (не менее 1 экв.). К полученному раствору прикапывали раствор ненуклеофильного основания (не менее 1 экв.) и перемешивали в течение не менее 12 часов при комнатной температуре. По окончании реакции реакционную смесь концентрировали в вакууме роторного испарителя до полного удаления органического растворителя. Остаток в колбе подкисляли раствором HCl с концентрацией не менее 1 моль/л до рН не менее 4 и экстрагировали этилацетатом. Объединенную органическую фазу промывали насыщенным раствором NaHCO3 и NaCl, сушили Na2SO4 и концентрировали в вакууме. Полученный бесцветный аморфный остаток растворяли в минимальном количестве дихлорметана и прикапывали при перемешивании гексан, до прекращения выпадения осадка. Выпавший осадок отфильтровывали и ресуспендировали в гексане в УЗИ-бане, затем заново отфильтровывали.
Предпочтительно в качестве ненуклеофильного основания использовать диизопропилэтиламин или триэтиламин
На третьем этапе повторно осуществляли процесс активации карбоксильной группы соединения (32) с последующим взаимодействием с азидопропиламином (не менее 1 экв) в течение не менее 24 часов при комнатной температуре в дихлорметане. По окончании сырую реакционную массу хроматографировали на колонке с силикагелем, в результате получали промежуточный дипептидный амид, который вовлекался в реакцию удаления трет-бутоксикарбонильной защиты действием 10 об.% раствора трифторуксусной кислоты в безводном дихлорметане.
Добавление TFA производили при охлаждении на водяной бане со льдом при температуре не более 10°С с последующим постепенным нагревом реакционной смеси до комнатной температуры. Снятие защиты при комнатной температуре проводили не менее 3 часов.
Таким образом, в результате последовательности реакций по схеме превращения был осуществлен синтез линкера (33), который использован в последующем для получения высокоспецифичных ПСМА-векторов. Разработанные методы синтеза отличаются экологичностью, хорошими выходами целевых продуктов, высокой селективностью процессов и не требуют применения специальной аппаратуры или дорогостоящих реагентов.
Синтез D-Phe-D-Tyr и D-Phe-D-Tyr(Br) проводился аналогично.
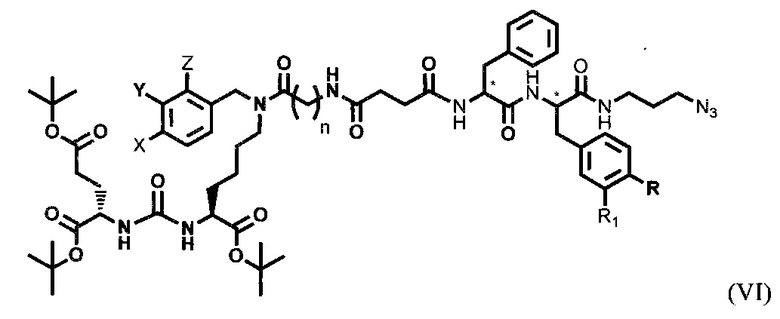
где R=Н, ОН; R1=H, Br.
Получение тритретбутил производного соединения радикала ПСМА-связывающего лиганда и модифицированного гидрофобного пептидного линкера, включающего фрагменты 6-аминогексановой или 5-аминопентановой или 4-аминобутановой кислот, фрагмент фенилаланина или его производное, фрагмент тирозина или его производное общей формулы (VI), проводили/осуществляли реакцией образования амидной связи продукта ацилирования янтарным ангидридом и дипептидом формулы (V).
К раствору продукта ацилирования янтарным ангидридом в ДМФА добавляли не менее 1 эквивалента дипептида, НОВТ, HBTU и ненуклеофильного основания. Смесь перемешивали не менее 24 ч. Далее удаляли растворитель при пониженном давлении. Продукт выделяли с помощью метода колоночной хроматографии. Элюента-EtOAc/МеОН=5:1.
Предпочтительно в качестве ненуклеофильного основания использовали диизопропилэтиламин или триэтиламин
 ,
,
где X, Y, Z независимо друг от друга представляют собой F, Cl, Br, Н, ОН, n=3-5, R=H, ОН; R1=Н, Br.
Получение соединения радикала ПСМА-связывающего лиганда и модифицированного гидрофобного пептидного линкера, общей формулы (I) осуществляли путем удаления тритретбутильных защитных групп соединения формулы (VI). Удаление тритретбутильных защитных групп проводили в присутствии 9-11 об. % ТФУ в течение 15-17 часов в дихлорметане.
Применение соединения общей формулы (I) заключается в получении конъюгата с лекарственным или диагностическим агентом. При этом для получения конечных конъюгатов (с лекарственным или диагностическим препаратом) проводится реакция азид-алкинового циклоприсоединения, между заявляемым соединением (средство доставки, ПСМА-лиганд с ликером), содержащим в своем составе азидо-группу, и доставляемой молекулой (лекарственным или диагностическим средством) (Kolb, Н.С, Finn, М.G. and Sharpless, К.В. (2001), Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angewandte Chemie International Edition, 40: 2004-2021.; Hartmuth С Kolb, K. Barry Sharpless, The growing impact of click chemistry on drug discovery, In Drug Discovery Today, Volume 8, Issue 24, 2003, Pages 1128-1137, ISSN 1359-6446,). В случае отсутствия у доставляемой молекулы фрагмента терминальной тройной связи в структуре, она должна быть модифицирована химически таким образом, чтобы полученное производное содержало терминальную тройную связь. Специалисту в данной области техники известно, каким образом возможно осуществление такой модификации.
Конъюгат может быть представлен общей структурной формулой
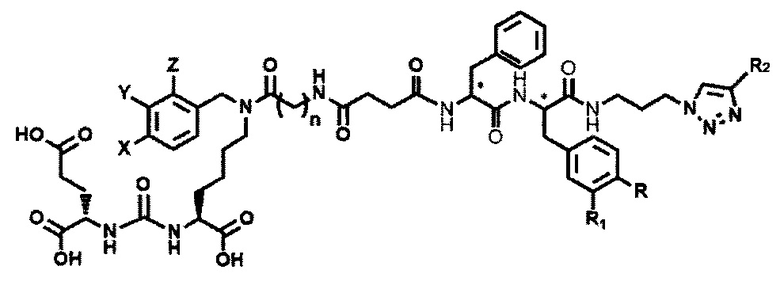 ,
,
где n=3-5;
X, Y, Z независимо друг от друга представляют собой F, Cl, Br, Н;
R=ОН, Н;
R1=Н, Br;
R2 = лекарственный или диагностический агент.
В заявляемом техническом решении раскрыты соединения (средства доставки), способные связываться с ПСМА, нацеливаться на ПСМА для доставки средств диагностики, визуализации и терапевтических (лекарственных средств). Также раскрыты соединения и способы их получения, а также применение соединений для диагностики, визуализации и лечения заболеваний, вызванных патогенными популяциями клеток, которые экспрессируют или сверхэкспрессируют ПСМА. Как показали проведенные эксперименты, заявляемые средства доставки проявляют высокую аффинность в отношении ПСМА, образованные комплексы заявляемого средства доставки с терапевтическим агентом являются эффективными в лечении заболеваний, вызванных патогенными клетками, которые экспрессируют ПСМА, такими как клетки рака предстательной железы.
В качестве лекарственного (терапевтического) средства (агента) могут быть использованы любые молекулы, способные модулировать или модифицировать клеточную функцию. В качестве лекарственных средств могут быть использованы пептиды, олигопептиды, ретроинвертированные олигопептиды, белки, аналоги белка, апопротеины, гликопротеины, ферменты, коферменты, ингибиторы ферментов, аминокислоты и их производные, рецепторы и другие мембранные белки, антигены и антитела против них; гаптены и антитела против них; гормоны, липиды, фосфолипиды, липосомы; токсины; антибиотики; бета-блокаторы; противораковые средства, в том числе химиотерапевтические средства; простагландины и аналоги простогландинов; противовоспалительные вещества; иммуносупрессоры, иммуностимуляторы; минеральные и питательные добавки.
В качестве лекарственных (химиотерапевтических) средств используют соединения, которые являются цитотоксическими, усиливают проницаемость опухоли, ингибируют пролиферацию опухолевых клеток, стимулируют апоптоз, снижают противоапоптическую активность в целевых клетках, применяются для лечения заболеваний, вызванных инфекционными агентами, усиливают эндогенный иммунный ответ, направленный на патогенные клетки, или применимы для лечения болезненного состояния, вызванного патогенными клетками. Такие химиотерапевтические средства могут функционировать при помощи любого из большого разнообразия механизмов действия. Например, цитотоксические соединения могут нарушать любой из целого ряда клеточных механизмов, которые важны для выживания клетки и/или клеточной пролиферации и/или вызывают клеточную смерть или апоптоз. Химиотерапевтические средства включают, но не ограничивают, следующие соединения: адренокортикоиды, кортикостероиды, алкилирующие средства, антиадрогены, антиэстрогены, андрогены, акламицин и производные акламицина, эстрогены, антиметаболиты, такие как цитозинарабинозид, аналоги пурина, аналоги пиримидина, метотрексат, бисульфан, карбоплатина, хлорамбуцил, цисплатин и др. соединения платины, тамоксифен, таксол, паклитаксел, производные паклитаксела, циклофосфамид, дауномицин, ризоксин, растительные алкалоиды, преднизолон, гидроксимочевина, тенипозид, митомицины, дискодермолиды, ингибиторы микротрубочек, эпотилоны, тубулизины, циклопропилбенз [е] индол он, втор-циклопропилбенз [е] индол он, О-Ас-втор-циклопропилбенз[е]индолон, блеомицин, алкалоиды барвинка (винкристин, винбластин, виндезин, винорелбин и их аналоги и производные), азотистые иприты, нитрозомочевины, колхицин и его производные, тритилцистеин, галикондрин В, доластатины, аманитины, камптотецин и его производные, гелданамицин и его производные, эстрамустин, нокодазол, МАР4, колцемид, противовоспалительные средства, ингибиторы пептидной и петидомиметической передачи сигнала, рапамицин, эверолимус, сиролимус, криптофицин, бортезомиб, тиобортезомиб, тубулизин, аминоптерин, доцетаксел, доксорубицин, даунорубицин, верукарин, дидемнин В, гелданомицин, пурваланол А, испинезид, будесонид, дазатиниб, эпотилон, майтанзины, ингибиторы тирозинкиназы, амфотерицинВ, ацикловир, трифлуридин, ганцинкловир, зидовудин, рибавирин и т.п.
В качестве средств визуализации могут быть использованы без ограничения, флуоресцентные средства, такие как Oregon Green (например, Oregon Green 488, Oregon Green 514 и т.п.), AlexaFluor (например, AlexaFluor 488, AlexaFluor 647 и т.п.), флуоресцеин и родственные аналоги, флуоресцентные средства BODIPY (например, BODIPY F1, BODIPY 505 и т.п.), родаминовые флуоресцентные средства (тетраметилродамин и т.п.), флуоресцентные средства DyLight (например, DyLight 680, DyLight 800 и т.п.); циановые красители (например Heptamethine IR-780, Heptamethine IR-808, IRDye® 800CW, DY-675, DY-676, DY-677, DY-678, Cy5, Cy7 и т.п.). Следует принимать во внимание, что конъюгаты терапевтических или диагностических средств с заявляемыми соединениями (средствами доставки) можно применять отдельно или в комбинации с другими соединениями, подходящими для диагностики, визуализации и/или лечения заболеваний, вызванных клетками, экспрессирующими ПСМА, а также в комбинации с другими соединениями, которые вводят для лечения других симптомов заболевания, например, паллиативные средства, действие которых сфокусировано на облегчении симптомов заболевания и/или побочных эффектов терапевтического режима, но не является лечебным. Например, паллиативное лечение включает болеутоляющие средства, средства против тошноты и средства против рвоты.
Ниже представлено более детальное описание заявляемого способа, которое не ограничивает объем притязаний заявляемого изобретения, а демонстрирует возможность осуществления изобретения с достижением заявляемого технического результата.
Пример 1.
(9S,13S)-три-трет-бутил 3,11-диоксо-1-фенил-2-окса-4,10,12-триазапентадекан-9,13,15-трикарбоксилат (1)
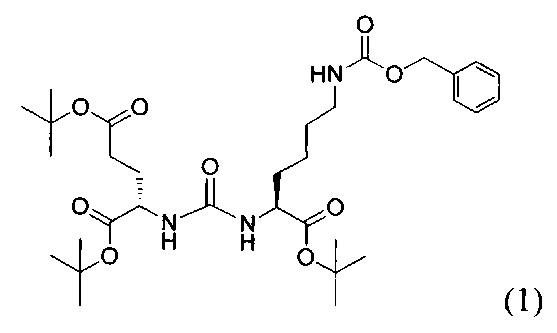
Гидрохлорид ди-трет-бутилового эфира L-глутаминовой кислоты (1.0 г, 3.38 ммоль) и триэтиламин (1,54, 11.09 ммоль) растворили в CH2Cl2 (30 мл), полученный раствор охладили до -78°С. К полученному раствору добавили по каплям раствор трифосгена (341 мг, 1.15 ммоль) в 10 мл CH2Cl2. После добавления раствора трифосгена температура реакции довели до комнатной температуры, после этого раствор перемешивали в течение 30 мин. Далее в реакционную смесь внесли раствор H-Lyc(Z)-Ot-Bu (757 мг, 2.03 ммоль) и триэтиламин (283 мкл, 2.03 ммоль) в 50 мл дихлорметана. Полученную реакционную смесь перемешивали в течение 16 часов при комнатной температуре. После этого реакционную смесь разбавили 50 мл CH2Cl2, и промыли водой (2×100 мл). Объединенные органические фракции высушили над сульфатом натрия. Растворитель удалили при пониженном давлении. Дальнейшую очистку полученной фракции проводили методом колоночной хроматографии (1.5:1 гексан: этилацетат). Таким образом, соединение (1) было выделено в виде бесцветного маслянистого вещества, выход составил 1 г (79%).
2-[3-(5-амино-1-трет-бутоксикарбонилфенил)уреидо]глутаминовой кислоты ди-трет-бутиловый эфир (2)
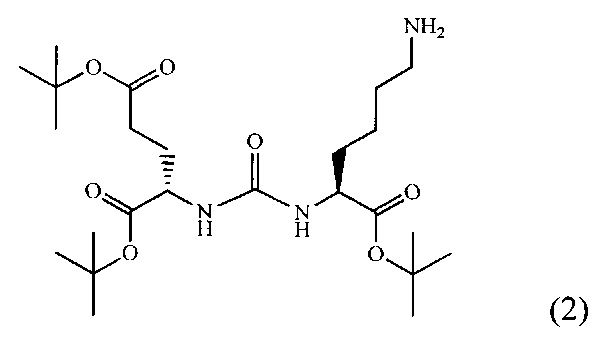
К раствору соединения (1) (1.0 г., 1.61 ммоль) в метаноле (30 мл) было добавлено 100 мг 10% Pd/C. Реакция проводилась в атмосфере водорода (р=1 атм.). Контроль окончания реакции проводился с помощью тонкослойной хроматографии. По окончании реакции полученная реакционная смесь была отфильтрована через диатомитовый порошок Celite. Растворитель был удален при пониженном давлении. Таким образом, соединение (2) было выделено в виде постепенно кристаллизующейся желтой маслянистой жидкости, выход составил 0.762 г (97%).
Ди-трет-бутил-2-(3-(1-(трет-бутокси)-6-((бензил)амино)-1-оксогексан-2-ил)уреидо)пентандиоат (3)
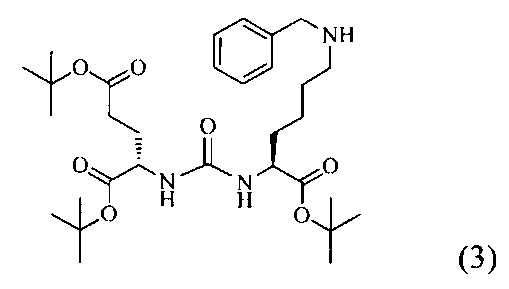
В 15 мл CH2Cl2 растворили соединение (2) 400 мг, 0.820 ммоль и бензальдегид 87 мг; 0.820 ммоль. Реакционную смесь перемешивали в течении 3 часов и добавили триацетоксиборгидрида натрия 261 мг; 1.231 ммоль. Сухой остаток отфильтровали в дихлорметане через вату. Растворитель упарили при пониженном давлении. Очистку с помощью метода колоночной хроматографии (система метанол-дихлорметан, градиент 1:30-1:15). В результате получили 379 мг целевого продукта (3), выход 80%.
Ди-трет-бутил-2-(3-(1-(трет-бутокси)-6-((4-хлоробензил)амино)-1-оксогексан-2-ил)уреидо)пентандиоат (4)
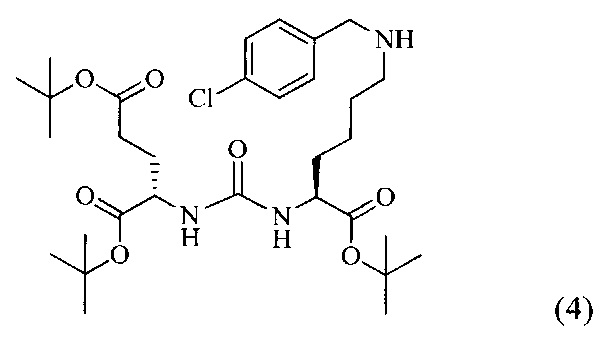
В 5 мл дихлорметана растворили 0,480 г (0,000984 моль, 1 экв) амина (2) и 0,138 г (0,000984 моль, 1 экв) n-хлорбензальдегида. Полученный раствор перемешивали ночь при комнатной температуре, после чего добавили 0,313 г (0,00148 моль, 1,5 экв) триацетоксиборогидрида натрия. Полученный раствор перемешивали в течение 24 ч, после чего растворитель удалили под вакуумом. Сухой остаток ресуспендировали в дихлорметане, осадок отфильтровали через вату, в результате чего получили прозрачный раствор. Растворитель удалили под вакуумом, после чего маслянистый остаток подвергли очистке с помощью метода колоночной хроматографии (система метанол-дихлорметан, градиент 1:30-1:15, нанесение вещества в виде раствора в системе 1:30, 20 г силикагеля).
Таким образом, соединение (4) было выделено в виде желтоватого маслянистого вещества, выход составил 0,41 г (68%).
Ди-трет-бутил-2-(3-(1-(трет-бутокси)-6-((4-бромобензил)амино)-1-оксогексан-2-ил)уреидо)пентандиоат (5)
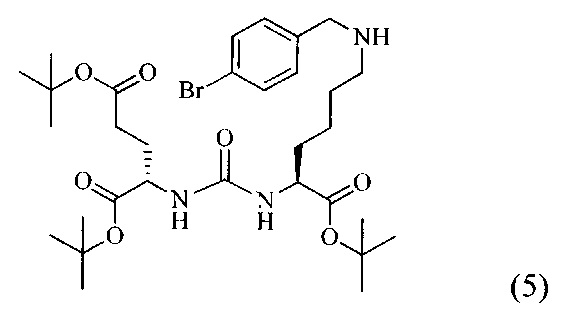
В 5 мл дихлорметана растворили 0,378 г (0,000775 моль, 1 экв) амина (2) и 0,143 г (0,000775 моль, 1 экв) n-бромбензальдегида. Полученный раствор перемешивали ночь при комнатной температуре, после чего было добавили 0,246 г (0,00116 моль, 1,5 экв) триацетоксиборогидрида натрия. Полученный раствор перемешивали в течение 24 ч, после чего растворитель удалили под вакуумом. Сухой остаток ресуспендировали в дихлорметане, осадок отфильтровали через вату, в результате чего получили прозрачный раствор. Растворитель удалили под вакуумом, после чего маслянистый остаток подвергли очистке с помощью метода колоночной хроматографии (система метанол-дихлорметан, градиент 1:30-1:15, нанесение вещества в виде раствора в системе 1:30, 20 г силикагеля).
Таким образом, соединение (5) было выделено в виде желтоватого маслянистого вещества, выход составил 0,313 г (68%).
Ди-трет-бутил-2-(3-(1-(трет-бутокси)-6-((4-гидроксибензил)амино)-1-оксогексан-2-ил)уреидо)пентандиоат (6)
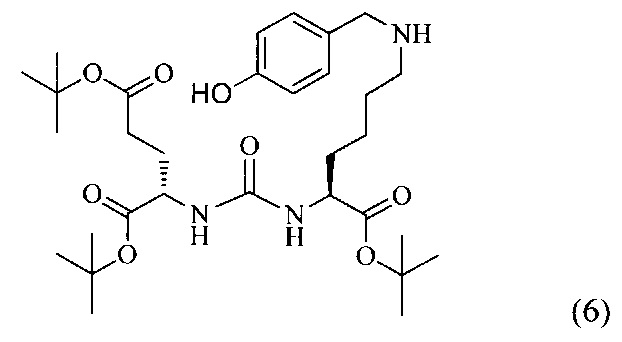
Соединение (2) (443 мг, 0.909 ммоль) растворили в 7 мл CH2Cl2, колбу продули аргоном. Затем добавили эквимолярное количество n-гидроксибензальдегида (111 мг, 0.909 ммоль), перемешивали в течение 2 часов в атмосфере аргона при комнатной температуре. После добавили 1.5 эквивалента триацетоксиборогидрида натрия (288 мг, 1.364 ммоль). Колбу снова продули аргоном и оставили перемешиваться на 20 ч. За ходом реакции следили с помощью тонкослойной хроматографии. По окончании синтеза избыток растворителя упарили, получившуюся смесь разделили с помощью колоночной хроматографии. Выход соединения (6) составил 74% (400 мг, 0.596 ммоль)
Ди-трет-бутил 2-(3-(1-(трет-бутокси)-6-((3-хлорбензил)амино)-1-оксогексан-2-ил)уреидо)пентанедиоат (7)
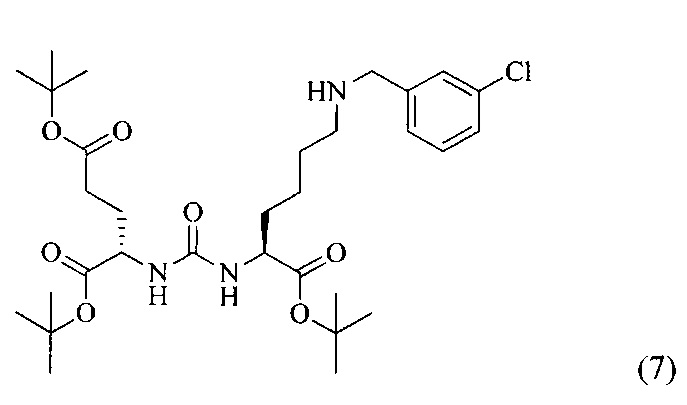
К раствору соединения (2) (400 мг, 0,820 ммоль) в DCM, добавили 3-хлорбензальдегид (115 мг, 0,820 ммоль), полученный раствор перемешивали в течение 3 часов в атмосфере аргона. Далее в реакционную смесь добавили 261 мг (1,230 ммоль) триацетоксиборгидрида, реакцию проводили в течение 16 часов. Полученную реакционную смесь упарили при пониженном давлении, образовавшийся осадок отфильтровали, полученный фильтрат упарили при пониженном давлении. Дальнейшую очистку проводили с помощью колоночной хроматографии с элюентом DCM/MeOH (100/0 до 0/100 в течение 30 минут). Таким образом, соединение (7) было выделено в виде бесцветного маслянистого вещества, выход составил 389 мг (77%).
Ди-трет-бутил 2-(3-(1-(трет-бутокси)-6-((4-хлор-2-фторбензил)амино)-1-оксогексан-2-ил)уреидо)пентанедиоат (8)
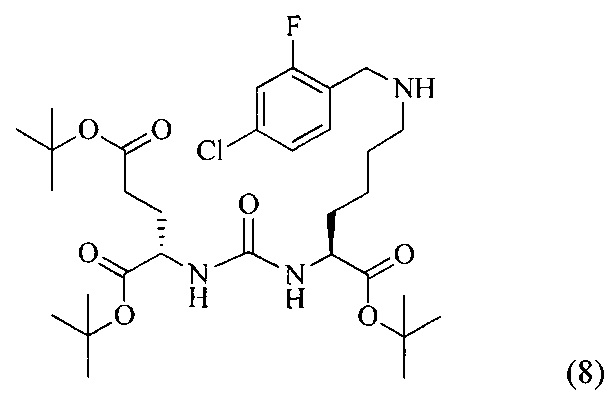
К раствору соединения (2) (400 мг, 0,820 ммоль) в DCM, добавили 2-фтор-4-хлорбензальдегид (130 мг, 0,820 ммоль), полученный раствор перемешивали в течение 3 часов в атмосфере аргона. Далее в реакционную смесь добавили 261 мг (1,230 ммоль) триацетоксиборгидрида, реакцию проводили в течение 16 часов. Полученную реакционную смесь упарили при пониженном давлении, образовавшийся осадок отфильтровали, полученный фильтрат упарили при пониженном давлении. Дальнейшую очистку проводили с помощью колоночной хроматографии с элюентом DCM/MeOH (100/0 до 0/100 в течение 30 минут). В результате был выделено 453 мг (0,719 ммоль) целевого продукта, выход 88% Таким образом, соединение (8) было выделено в виде бесцветного маслянистого вещества, выход составил 453 мг (88%).
Ди-трет-бутил 2-(3-(1-(трет-бутокси)-6-((4-хлор-3-фторбензил)амино)-1-оксогексан-2-ил)уреидо)пентанедиоат (9)
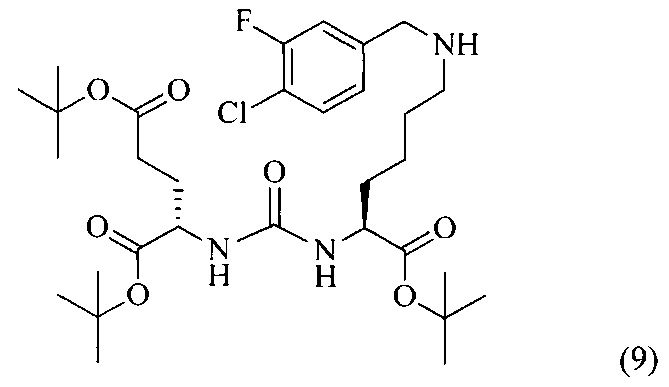
К раствору соединения (2) (400 мг, 0,820 ммоль) в DCM, добавили 3-фтор-4-хлорбензальдегид (130 мг, 0,820 ммоль), полученный раствор перемешивали в течение 3 часов в атмосфере аргона. Далее в реакционную смесь добавили 261 мг (1,230 ммоль) триацетоксиборгидрида, реакцию проводили в течение 16 часов. Полученную реакционную смесь упарили при пониженном давлении, образовавшийся осадок отфильтровали, полученный фильтрат упарили при пониженном давлении. Дальнейшую очистку проводили с помощью колоночной хроматографии с элюентом DCM/MeOH (100/0 до 0/100 в течение 30 минут).
Таким образом, соединение (9) было выделено в виде бесцветного маслянистого вещества, выход составил 467 мг (90%).
(14S,18S)-три-трет-бутил-9-бензил-3,8,16-триоксо-1-фенил-2-окса-4,9,15,17-тетраазаикосан-14,18,10-трикарбоксилат (10)
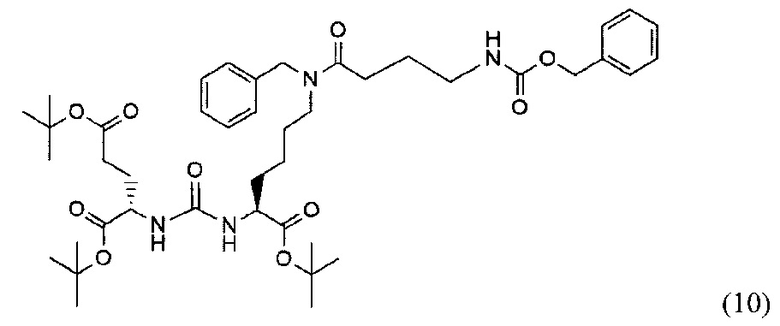
Соединение (3) 366 мг; 0.567 ммоль растворили в 10 мл DMF. При перемешивании добавили DIPEA 307 мкл; 1.792 ммоль, затем 4-(((бензаокси)карбонил)амино)бутановую кислоту. После полного растворения кислоты систему продули аргоном. Добавили РуВОР 463 мг; 0.894 и оставили при перемешивании на сутки. Растворитель упарили при пониженном давлении. Очистку проводили методом колоночной хроматографии (этилацетат : гексан; градиент от 5% до 100% этилацетата). Масса полученного вещества (10) 120 мг; выход 25%.
(S)-ди-трет-бутил-2-(3-((S)-6-(6-азидо-N-бензилгесанамдо)-1-(трет-бутокси)-1-оксогесан-2-ил)уреидо)пентадионат (11)
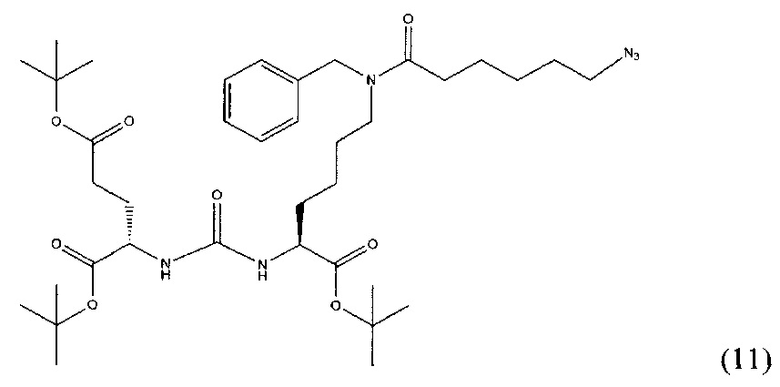
Соединение (3) (379 мг; 0.656 ммоль) растворили в 10 мл DMF и прилили DIPEA 337 мкл; 1.961 ммоль. К полученной смеси добавили 6-азидгексановую кислоту 184 мг; 1.172 ммоль. Систему продули аргоном и добавили РуВОР 512 мг; 0.984 ммоль. Оставили перемешиваться на сутки. Растворитель упарили при пониженном давлении. Очистку проводили методом колоночной хроматографии (этилацетат : гексан; градиент от 5% до 100% этилацетата). В результате выделили 120 мг целевого продукта (11), выход 25%.
Ди-трет-бутил-2-(3-(6-(11-азидо-N-(4-хлорбензил)гексанамидо)-1-(трет-бутокси)-1-оксогексан-2-ил)уреидо)пентандиоат (12)
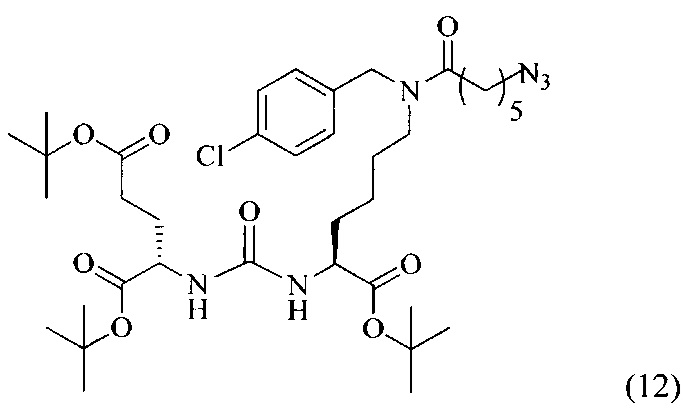
В 5 мл диметилформамида растворили 0,129 г (0,000211 моль, 1 экв.) соединения (4) и 0,066 г (0,000422 моль, 2 экв.) 6-азидогексановой кислоты. Добавили 0,197 г (0,000422 моль, 2 экв.) PyBrOP. Реакционную смесь охлаждали в ледяной бане в течение 5 минут, после чего добавили 220 мкл (0,00127 моль, 6 экв.) диизопропилэтиламина. Реакционную смесь перемешивали при охлаждении в течение 1 минуты, после чего в течение ночи при комнатной температуре. Растворитель удалили под вакуумом, полученный маслянистый остаток подвергли очистке с помощью метода колоночной хроматографии (система этилацетат-петролейный эфир, градиент 1:3 - 1:1, сухое нанесение на 1 г силикагеля, 12 г силикагеля в колонке).
Таким образом, соединение (12) было выделено в виде бесцветного маслянистого вещества, выход составил 0,082 г (52%)
Ди-трет-бутил-2-(3-(6-(11-азидо-N-(4-бромбензил)гексанамидо)-1-(трет-бутокси)-1-оксогексан-2-ил)уреидо)пентандиоат (13)
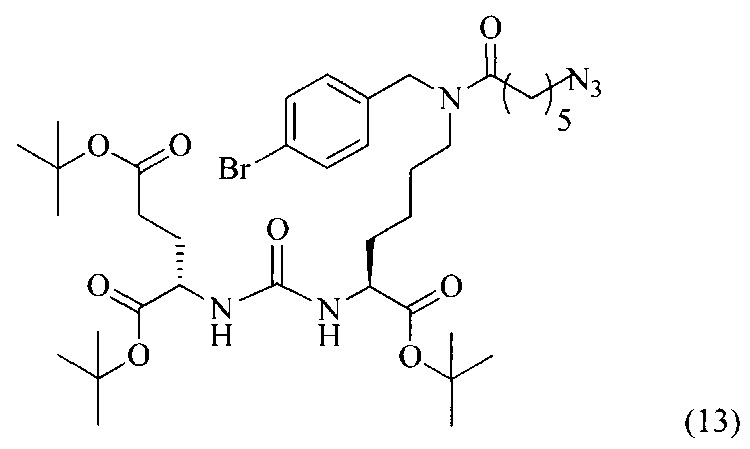
В 5 мл диметилформамида растворили 0,185 г (0,000282 моль, 1 экв.) соединения (5) и 0,133 г (0,000846 моль, 3 экв.) 6-азидогексановой кислоты. Добавили 0,394 г (0,000846 моль, 3 экв.) PyBrOP. Реакционную смесь охлаждали в ледяной бане в течение 5 минут, после чего добавили 442 мкл (0,00254 моль, 9 экв.) диизопропилэтиламина. Реакционную смесь перемешивали при охлаждении в течение 1 минуты, после чего в течение ночи при комнатной температуре. Растворитель удалили под вакуумом, полученный маслянистый остаток подвергли очистке с помощью метода колоночной хроматографии (система этилацетат-петролейный эфир, градиент 1:3 - 1:1, сухое нанесение на 1 г силикагеля, 12 г силикагеля в колонке).
Таким образом, соединение (13) было выделено в виде бесцветного маслянистого вещества, выход составил 0,124 г (55%).
Ди-трет-бутил 2-(3-(6-(6-азидо-N-(4-гидроксибензил)гексанамидо)-1-(трет-бутокси)-1-оксогексан-2-ил)уреидо)пентанедиоат (14)
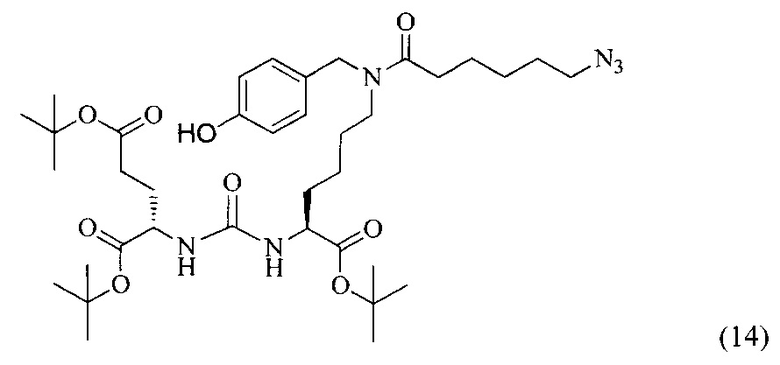
В колбу на 100 мл внесли соединение (6) (354 мг, 0.596 ммоль), добавили 3 эквивалента 6-азидогексановой кислоты (289 мг, 1.839 ммоль), растворенного в 1 мл DMF, затем внесли 9 эквивалентов DIPEA (784.4 мг, 935 мл, 5.368 ммоль) и 20 мл DMF. Реакционную смесь продули аргоном, после добавили 2 эквивалента РуВОР (624 мг, 1.199 ммоль) и оставили перемешивать в атмосфере аргона в течение 20 ч при комнатной температуре. По окончании реакции избыток растворителя упарили, вещество выделили с помощью колоночной хроматографии. Выход целевого продукта (14) составил 43% (190 мг, 0.259 ммоль).
Ди-трет-бутил 2-(3-(6-(6-азидо-N-(3-хлорбензил)гексанамидо)-1-(трет-бутокси)-1-оксогексан-2-ил)уреидо)пентанедиоат (15)
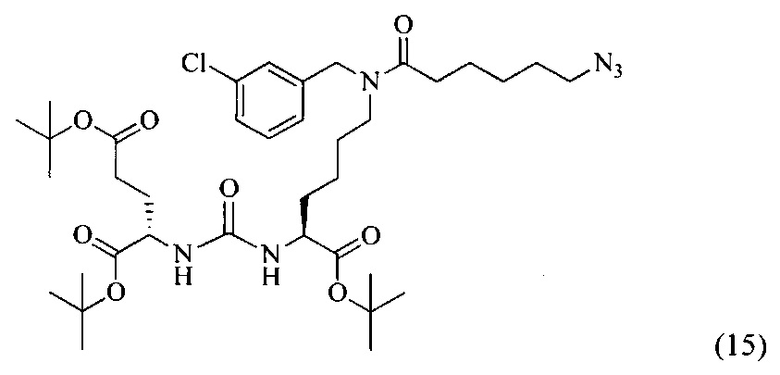
К смеси 6-азидогексановой кислоты (200 мг, 1.27 ммоль) и соединения (7) (389 мг, 0.635 ммоль) в 10 мл DMF добавили РуВОР (496 мг, 0.952 ммоль) и 332 мкл (1.905 ммоль) DIPEA. Полученный раствор перемешивали в течение 16 часов. Реакционную смесь упарили при пониженном давлении. Дальнейшую очистку проводили методом колоночной хроматографии (EtOAc/гексан: 1/2). Таким образом, соединение (15) было выделено в виде бесцветного маслянистого вещества, выход составил 337 г (71%).
Ди-трет-бутил 2-(3-(6-(6-азидо-N-(4-хлор-2-фтор-бензил)гексанамидо)-1-(трет-бутокси)-1-оксогексан-2-ил)уреидо)пентанедиоат (16)
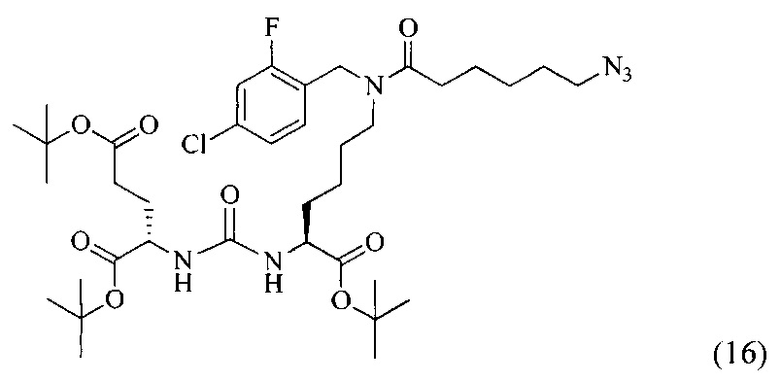
К смеси 6-азидогексановой кислоты (226 мг, 1.438 ммоль) и соединения (8) (453 мг, 0.719 ммоль) в 10 мл DMF добавили РуВОР (561 мг, 1.078 ммоль) и 376 мкл (2.157 ммоль) DIPEA. Полученный раствор перемешивали в течение 16 часов. Реакционную смесь упарили при пониженном давлении. Дальнейшую очистку проводили методом колоночной хроматографии (EtOAc/hexane: 1/2). В результате было выделено 300 мг (0,390 ммоль) целевого продукта, выход составил 54%. Таким образом, соединение (16) было выделено в виде бесцветного маслянистого вещества, выход составил 300 мг (54%).
Ди-трет-бутил 2-(3-(6-(6-азидо-N-(4-хлор-3-фторбензил)гексанамидо)-1-(трет-бутокси)-1-оксогексан)-2-ил)уреидо)пентанедиоат (17)
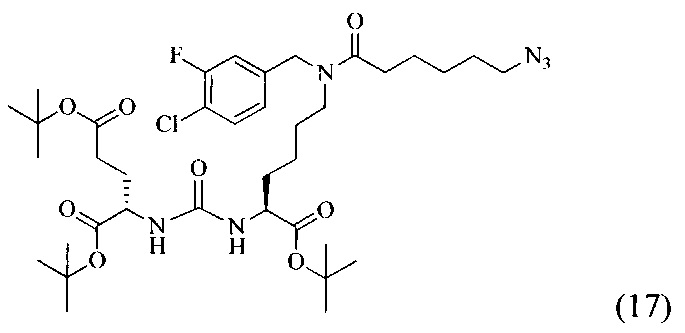
К смеси 6-азидогексановой кислоты (233 мг, 1.482 ммоль) и соединения (9) (467 мг, 0.741 ммоль) в 10 мл DMF добавили РуВОР (578 мг, 1.111 ммоль) и 387 мкл (2.223 ммоль) DIPEA. Полученный раствор перемешивали в течение 16 часов. Реакционную смесь упарили при пониженном давлении. Дальнейшую очистку проводили методом колоночной хроматографии (EtOAc/гексан: 1/2). В результате было выделено 330 мг (0,429 ммоль) целевого продукта, выход составил 58%.
Таким образом, соединение (17) было выделено в виде бесцветного маслянистого вещества, выход составил 330 мг (58%).
(S)-ди-трет-бутил-2-(3-((S)-6-(4-амино-N-бензилбутанамидо)-1-(трет-бутокси)-1-оксогесан-2-ил)уреидо)пентадионат (18)
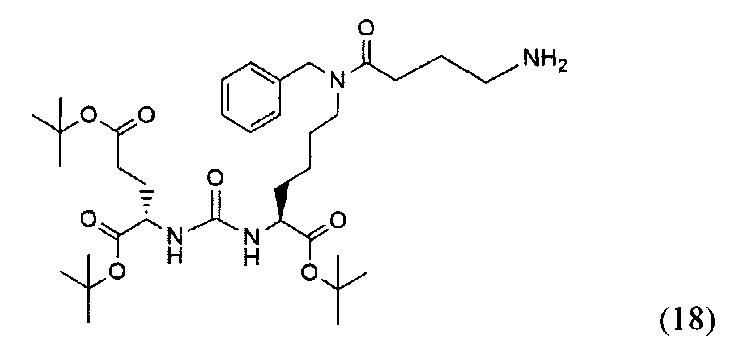
Соединение (10) 120 мг; 0.148 ммоль растворили в 15 мл метанола. После этого добавили 12 мг Pd/C и продули систему водородом. Оставили перемешиваться на сутки. Полученную смесь профильтровали на сорбенте Kiselgur. Растворитель упарили при пониженном давлении. Масса полученного соединения (18) 68 мг, выход 70%.
(S)-ди-трет-бутил 2-(3-((S)-6-(6-амино-N-бензилгексанамидо)-1-(трет-бутокси)-1-оксогексан-2-ил)уреидо)пентанедиоат (19)
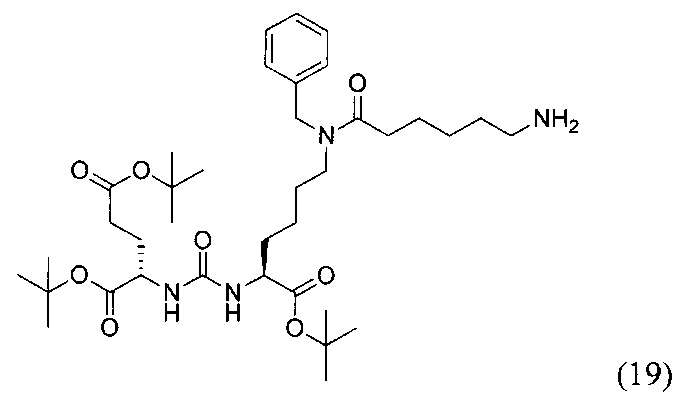
К раствору 161 мг (0,187 ммоль) соединения (11) в 10 мл метанола добавили 21 мг 10% Pd/C в 400 мкл воды. Реакция проводилась в атмосфере водорода (р=1 атм). Контроль окончания реакции проводился с помощью тонкослойной хроматографии. Далее полученная реакционная смесь была отфильтрована через диатомитовый порошок Kiselgur. Растворитель был удален при пониженном давлении. В результате было выделено 96 мг целевого соединения (19), выход 71%.
(S)-Ди-трет-бутил 2-(3-((S)-6-(6-амино-N-(4-хлорбензил)гексанамидо)-1-(трет-бутокси)-1-оксогексан-2-ил)уреидо)пентанедиоат (20)
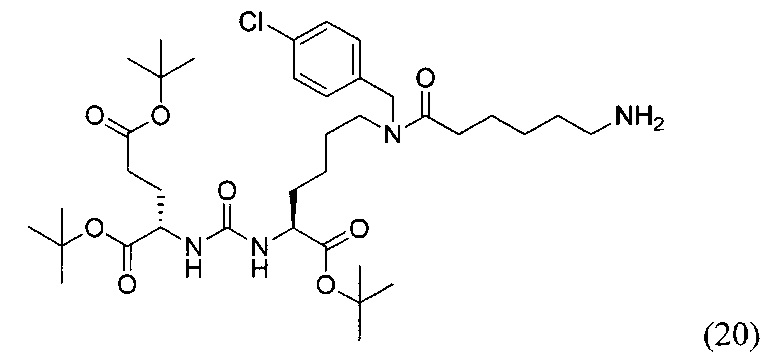
К раствору 135 мг (0,180 ммоль) соединения (12) в 11 мл смеси ТГФ/вода (10/1) было добавлено 95 мг (0,360 ммоль) Ph3P, полученную смесь перемешивали 6 часов при температуре 50°С. Далее удалили растворитель при пониженном давлении и выделяли продукт с помощью метода колоночной хроматографии (Puriflash 50μ 4g, система: 1% раствор TEA в DCM:MeOH, 8% МеОН в течение 10 минут, далее от 8% до 100% МеОН за 1 минуту, промывка МеОН в течение 4 минут). В результате было выделено 45 мг целевого продукта (20), выход 35%.
(S)-Ди-трет-бутокси 2-(3-((S)-6-(6-амино-N-(4-бромбензил)гексанамидо)-1-(трет-бутокси)-1-оксогексан-2-ил)уреидо)пентанедиоат (21)
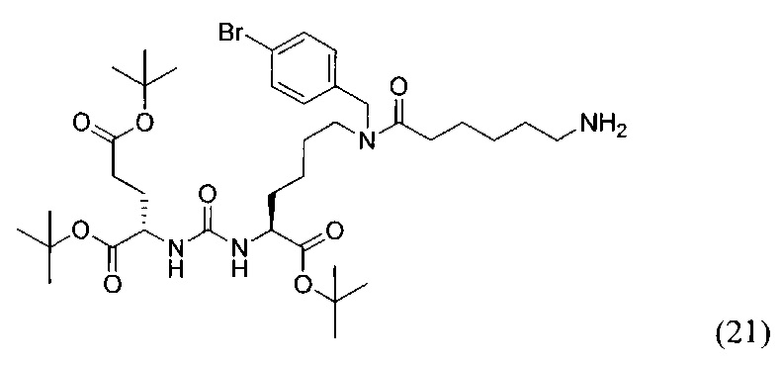
К раствору 128 мг (0,167 ммоль) соединения (13) в 11 мл смеси ТГФ/вода (10/1) было добавлено 88 мг (0,333 ммоль) Ph3P, полученную смесь перемешивали 6 часов при температуре 50°С. Далее удалили растворитель при пониженном давлении и выделяли продукт с помощью метода колоночной хроматографии (Puriflash 50μ 4g, система: 1% раствор TEA в DCM:MeOH, 8% МеОН в течение 10 минут, далее от 8% до 100% МеОН за 1 минуту, промывка МеОН в течение 4 минут). В результате было выделено 55 мг целевого продукта (21), выход 43%.
(S)-ди-трет-бутил 2-(3-((S)-6-(6-амино-N-(3-хлорбензил)гексанамидо)-1-(трет-бутокси)-1-оксогексан-2-ил)уреидо)пентадиоат (22)
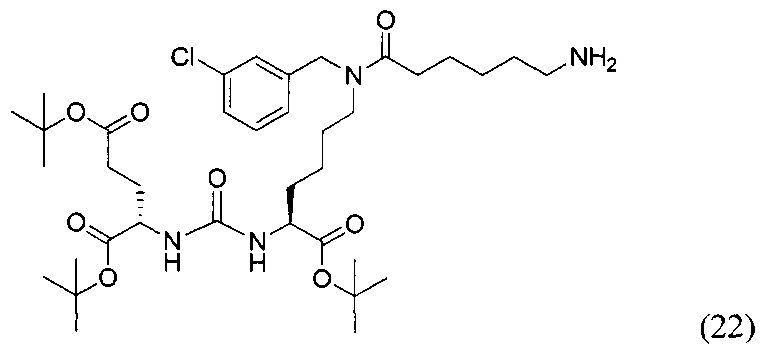
К раствору 159 мг исходного азида (15) в 5 мл THF было добавлено 0.5 мл Н2О и 111 мг PPh3. Реакционную смесь перемешивали в течение 2 ч при 60°С, контроль окончания реакции проводили с помощью ТСХ. После окончания реакции реакционную смесь упарили, и очищали с помощью флеш-хроматографии в системе DCM/MeOH/Et3N (95/4/1). В результате чего было выделено 120 мг целевого продукта (22), выход 78%.
(S)-ди-трет-бутил 2-(3-((S)-6-(6-амино-N-(4-хлор-2-фторбензил)гексанамидо)-1-(трет-бутокси)-1-оксогексан-2-ил)уреидо)пентадиоат (23)
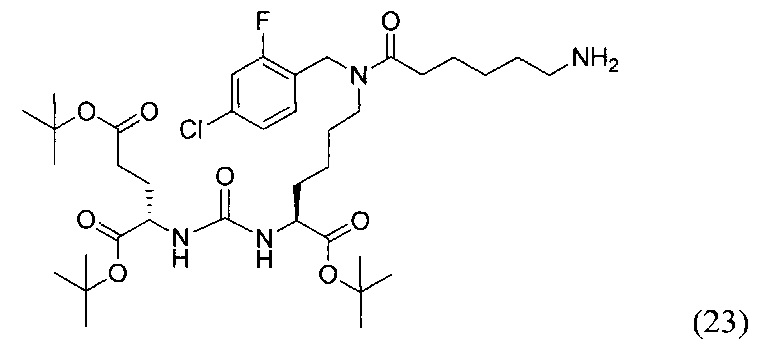
К раствору 139 мг исходного азида (16) в 5 мл THF было добавлено 0.5 мл Н2О и 95 мг PPh3. Реакционную смесь перемешивали в течение 2 ч при 60°С, контроль окончания реакции проводили с помощью ТСХ. После окончания реакции реакционную смесь упарили, и очищали с помощью флеш-хроматографии в системе DCM/MeOH/Et3N (95/4/1). В результате чего было выделено 120 мг целевого продукта (23), выход 78%.
(S)-Ди-трет-бутил 2-(3-((S)-6-(6-амино-N-(4-хлор-3-фторбензил)гексанамидо)-1-(трет-бутокси)-1-оксогексан-2-ил)уреидо)пентанедиоат (24)
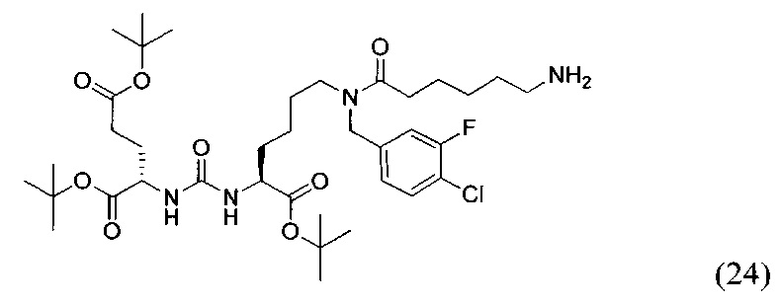
К раствору 227 мг (0,306 ммоль) соединения (17) в 11 мл смеси ТГФ/вода (10/1) было добавлено 161 мг (0,611 ммоль) Ph3P, полученную смесь перемешивали 6 часов при температуре 50°С. Далее удалили растворитель при пониженном давлении и выделяли продукт с помощью метода колоночной хроматографии (Puriflash 50μ 4g, система: 1% раствор TEA в DCM:MeOH, 8% МеОН в течение 10 минут, далее от 8% до 100% МеОН за 1 минуту, промывка МеОН в течение 4 минут). В результате было выделено 138 мг целевого продукта (24), выход 61%.
(S)-2-((S)-2-((трет-бутоксикарбонил)амино)-3-фенилпропанамидо)-3-фенилпропановая кислота (25)
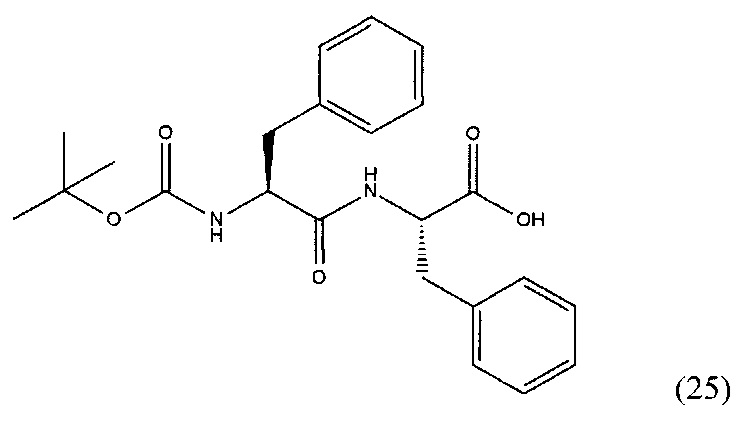
К раствору 1 г 3.2 ммоль дипептида L-Phe-L-Phe в 120 мл смеси диоксан/вода (3/1) было добавлено 958 мг 4.4 ммоль ВОС2О и 920 мг 10.8 NaHCO3, полученную смесь перемешивали в течение 6 часов. Далее реакционную смесь упаривали и растворяли в 50 мл EtOAc и промывали раствором 0.1М HCl (рН=1). Полученную органическую фракцию сушили над Na2SO4 и упаривали при пониженном давлении. В результате чего было выделено 958 мг целевого продукта, выход 72%.
Трет-бутил-((S)-1-(((S)-1-((3-азидопропил)амино)-1-оксо-3-фенилпропан-2-ил)амино)-1-оксо-3-фенилпропан-2-ил)карбамат (26)
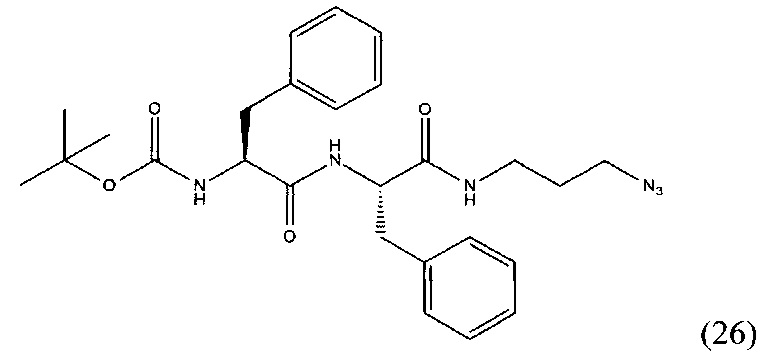
Соединение (25) растворили в 40 мл DMF. Добавили НОВТ, HBTU. К полученной смеси прилили DIPEA. Оставили перемешивать в течении 16 часов, после чего упарили растворитель при пониженном давлении. Очистку проводили методом колоночной хроматографии (этилацетат: гексан; градиент от 5% до 100% этилацетата) В результате выделили 890 мг целевого продукта (26), выход 53%.
(S)-2-амино-N-((S)-1-((3-азидопропил)амино)-1-оксо-3-фенилпропан-2-ил)-3-фенилпропанамид (27)
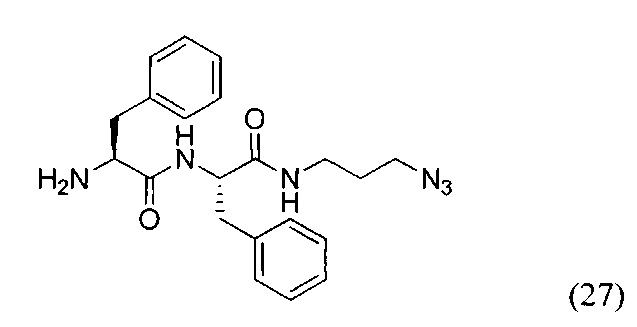
256 мг соединения (26) были растворены в 2700 мкл DCM после чего к реакционной смеси было добавлено 300 мкл TFA. Реакционную смесь перемешивали 2 часа, контроль протекания реакции проводили методом ТСХ. После окончания реакционную смесь промыли диэтиловым эфиром. Полученную маслянистую фракцию растворили в DCM и промыли раствором NaHCO3. Органическую фракцию сушили над Na2SO4. В результате чего выделено ~180 мг целевого продукта (27), выход 88%.
(R)-2-((R)-2-((трет-бутоксикарбонил)амино)-3-фенилпропанамидо)-3-фенилпропановая кислота (28)
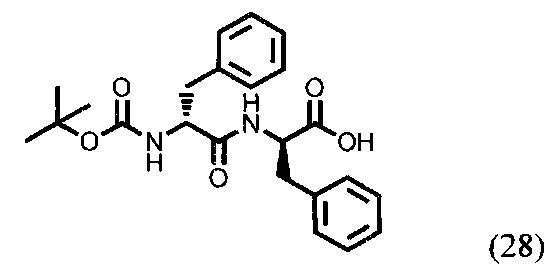
500 мг дипептида D-Phe-D-Phe (1,6 ммоль) растворили в 40 мл смеси диоксан/вода (3/1). К раствору прибавили 479,1 мг ВОС2О (2,2 ммоль) и 460,1 мг NaHCO3 (5,5 ммоль). Данную смесь перемешивали в течении 6 часов, после чего удалили растворитель при пониженном давлении, растворили в 50 мл этилацетата и промывали 0,1 М раствором HCl (рН=1). Органическую фракцию сушили над Na2SO4, после чего удалили растворитель при пониженном давлении. Масса продукта 28 - 638 мг. Выход - 97%.
Трет-бутил ((R)-1-(((R)-1-((3-азидопропил)амино)-1-оксо-3-фенилпропан-2-ил)амино)-1-оксо-3-фенилпропан-2-ил)карбамат (29)
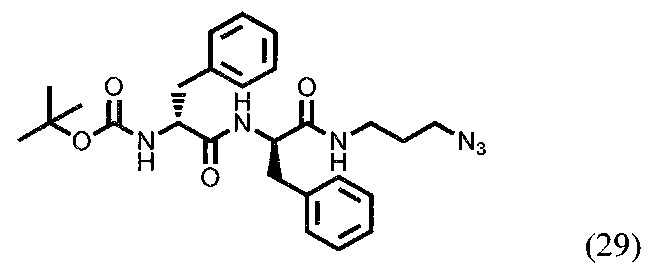
588 мг соединения (28) (1,425 ммоль) растворили в примерно 20 мл ДМФА. К раствору прибавили 649 мг HBTU (1,711 ммоль) и 192 мг НОВТ (1,421 ммоль). К смеси прилили 372 мкл DIPEA (2,136 ммоль). Затем в реакционную смесь ввели 596 мг NH2-(СН2)3-N3 (5,96 ммоль). Реакционная смесь перемешивалась в течении 16 часов, после чего удалили растворитель при пониженном давлении. Очистка вещества производилась с помощью метода колоночной хроматографии (Puriflash 15μ 25g F0025, система этилацетат/петролейный эфир, этилацетат от 5% до 40 в течение 6 минут, от 40% до 60 в течении 15 минут, от 60% до 100% в течении 6 минут, 100% в течении 3 минут). Далее удалили растворитель при пониженном давлении. Конечная масса полученного продукта (29) - 406 мг. Выход - 58%.
(R)-2-амино-N-((R)-1-((3-азидопропил)амино)-1-оксо-3-фенилпропан-2-ил)-3-фенилпроанамид (30)
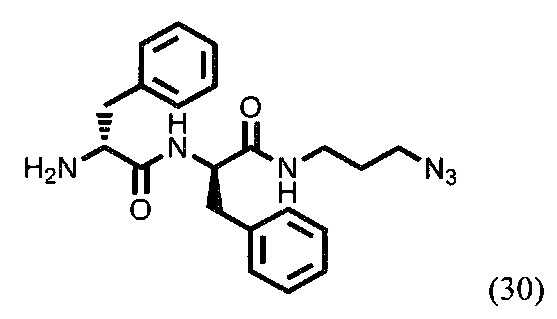
406 мг соединения (29) (0,822 ммоль) растворили в 6585 мкл ДХМ. К раствору прибавили 980 мкл трифторуксусной кислоты. Смесь перемешивали в течении 3 часов. После этого удалили растворитель при пониженном давлении. Затем растворили полученное вещество в ДХМ, добавили насыщенный раствор NaHCO3 в воде, перемешивали в течении 10 минут, экстрагировали и оставили органическую фазу сушиться над Na2SO4 на ночь. Далее удалили растворитель при пониженном давлении. Масса продукта (30) - 274 мг. Выход - 85%.
(S)-2-((трет-бутоксикарбонил)амино)-3-фенилпропановая кислота (31)
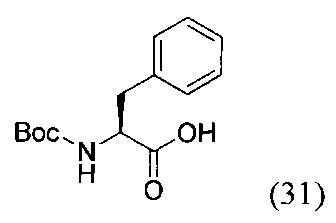
К суспензии аминокислоты L-фенилаланина (1.32 г, 8 ммоль) в 50 мл смеси растворителей ТГФ - вода (3:2) при 0°С добавили NaOH (0.32 г, 8 ммоль, 1 экв.) и ди-трет-бутил дикарбонат ВОС2О (1.74 г, 8 ммоль, 1 экв.). Полученную смесь перемешивали при комнатной температуре в течение 4 суток. Реакционную смесь сконцентрировали в вакууме роторного испарителя до удаления органического растворителя. Затем в водный остаток добавляли 1М раствором соляной кислоты до рН=6 и экстрагировали этилацетатом (3*50 мл). Объединенную органическую фазу промывали насыщенным раствором NaHCO3 и NaCl, сушили Na2SO4 и концентрировали в вакууме. Затем повторно переупаривали с дихлорметаном. Продукт реакции получали в виде бесцветного аморфного вещества (31) с выходом 90% (1.91 г, 7.2 ммоль).
(S)-2-((S)-2-((трет-бутоксикарбонил)амино)-3-фенилпропанамидо)-3-(4-гидроксифенил)пропановая кислота (32)
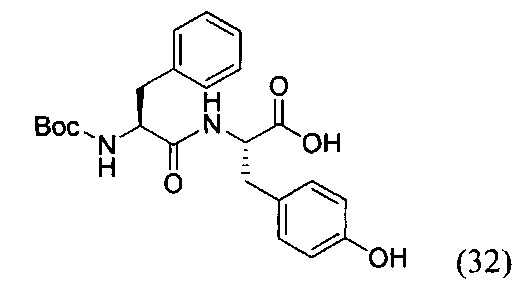
К раствору соединения (31) (1.91 г, 7.2 ммоль) в 50 мл дихлорметана добавили EDC*HCl (1.65 г, 8.6 ммоль, 1.2 экв.), PFPOH (1.31 г, 7.12 ммоль, 1 экв.) и перемешивали в течение 12 часов при комнатной температуре. Затем в реакционную смесь добавляли силикагель (10 г) и полученную суспензию хроматографировали на колонке с силикагелем (элюент - дихлорметан). Продукт реакции (желтое маслянистое вещество) растворяли в 60 мл смеси ТГФ - вода (2:1) и добавляли при перемешивании L-тирозин (1.45 г, 8 ммоль, 1 экв.). К полученному раствору прикапывали DIPEA (1.4 мл, 8 ммоль, 1 экв.) и перемешивали в течение 12 часов при комнатной температуре. По окончании реакции реакционную смесь концентрировали в вакууме роторного испарителя до полного удаления органического растворителя. Остаток в колбе подкисляли 1М раствором HCl до рН=2 и экстрагировали этилацетатом (3*50 мл). Объединенную органическую фазу промывали насыщенным раствором NaHCO3 и NaCl, сушили Na2SO4 и концентрировали в вакууме. Полученный бесцветный аморфный остаток растворяли в минимальном количестве дихлорметана и прикапывали при перемешивании 100 мл гексана. Выпавший осадок отфильтровали и ресуспендировали в 50 мл гексана в УЗИ-бане, затем заново отфильтровали. Получили продукт реакции в виде белого твердого вещества (32) с выходом 52% (1.78 г, 4.16 ммоль).
(S)-1-(((S)-1-((3-азидопропил)амино)-3-(4-гидроксифенил)-1-оксопропан-2-ил)амино)-1-оксо-3-фенилпропан-2-аммоний трифторацетат (33)
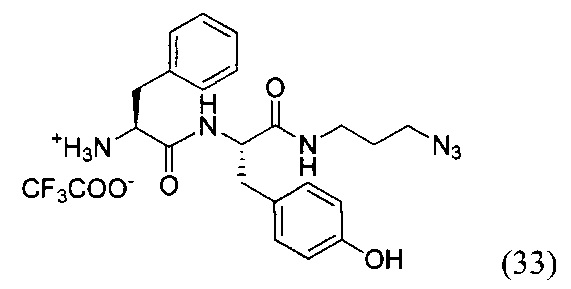
К раствору соединения (32) (1.78 г, 4.16 ммоль) в 50 мл дихлорметана добавили EDC*HCl (0.95 г, 5 ммоль, 1.2 экв.), PFPOH (0.76 г, 4.16 ммоль, 1 экв.) и перемешивали в течение 12 часов при комнатной температуре. Затем в реакционную смесь добавляли силикагель (10 г) и полученную суспензию хроматографировали на колонке с силикагелем (элюент - дихлорметан/этилацетат 40:1). Продукт реакции (желтое маслянистое вещество) растворяли в 60 мл ТГФ и добавляли при перемешивании азидопропиламин (0.42 г, 4 16 ммоль, 1 экв.). К полученному раствору прикапывали DIPEA (0.72 мл, 4.16 ммоль, 1 экв.) и перемешивали в течение 12 часов при комнатной температуре. По окончании реакции реакционную смесь концентрировали в вакууме роторного испарителя и хроматографировали остаток на колонке с силикагелем (элюент - дихлорметан/метанол 40:1). Полученный продукт растворяли в 20 мл безводного дихлорметана и охлаждали на бане со льдом до 0°С. Далее при перемешивании прикапывали 2 мл трифторуксусной кислоты и перемешивали реакционный раствор при комнатной температуре в течение 3-4 часов до полного исчезновения исходного вещества (ТСХ-контроль). По окончании реакции упаривали смесь в вакууме роторного растворителя, переупаривали остаток дважды с дихлорметаном и вакуумировали до полного удаления органического растворителя. Получили продукт реакции в виде желтого аморфного вещества (33) с выходом 57% (1.24 г, 2.37 ммоль).
(R)-2-((трет-бутоксикарбонил)амино)-3-фенилпропановая кислота (34)
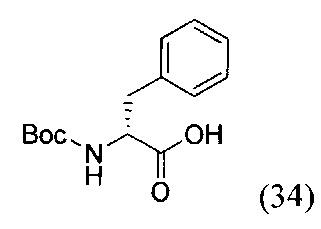
К суспензии аминокислоты D-фенилаланина (4 г, 24 ммоль) в 30 мл смеси растворителей диоксан - вода (1:2) при 0°С добавили NaHCO3 (1.8 г, 30 ммоль, 1.2 экв.) и ди-трет-бутил дикарбонат ВОС2О (5.18 г, 45 ммоль, 1 экв.). Полученную смесь перемешивали при комнатной температуре в течение 2 суток. Реакционную смесь сконцентрировали в вакууме роторного испарителя до удаления органического растворителя. Затем в водный остаток добавляли 1М раствором соляной кислоты до рН=6 и экстрагировали этилацетатом (3*30 мл). Объединенную органическую фазу промывали насыщенным раствором NaHCO3 и NaCl, сушили Na2RO4 и концентрировали в вакууме. Затем повторно переупаривали с дихлорметаном. Продукт реакции получали в виде желтого масла вещества (34) с выходом 97% (6.25 г, 23 ммоль).
(R)-2-((R)-2-((трет-бутоксикарбонил)амино)-3-фенилпропанамидо)-3-(4-гидроксифенил)пропановая кислота (35)
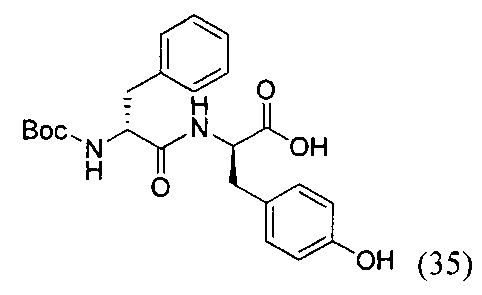
К раствору соединения (34) (6.25 г, 23 ммоль) в 120 мл дихлорметана добавили EDC*HCl (5.28 г, 27 ммоль, 1.2 экв.), PFPOH (4.2 г, 23 ммоль, 1 экв.) и перемешивали в течение 2 часов при комнатной температуре. Затем в реакционную смесь добавляли силикагель (50 г) и полученную суспензию хроматографировали на колонке с силикагелем (элюент - дихлорметан). Продукт реакции (желтое маслянистое вещество) растворяли в 110 мл смеси ТГФ - вода (8:3) и добавляли при перемешивании D-тирозин (8 г, 46 ммоль, 2 экв.). К полученному раствору прикапывали DIPEA (8 мл, 46 ммоль, 2 экв.) и перемешивали в течение 12 часов при комнатной температуре. По окончании реакции реакционную смесь концентрировали в вакууме до полного удаления органического растворителя. Остаток в колбе подкисляли 1М раствором HCl до рН=3 и экстрагировали этилацетатом (3*80 мл). Объединенную органическую фазу промывали насыщенным раствором NaHCO3 и NaCl, сушили Na2RO4 и концентрировали в вакууме. Полученный бесцветный аморфный остаток растворяли в минимальном количестве дихлорметана и прикапывали при перемешивании 100 мл гексана. Выпавший осадок отфильтровали и ресуспендировали в 50 мл диэтилового эфира в УЗИ-бане, затем заново отфильтровали. Получили продукт реакции в виде белого твердого вещества (35) с выходом 84% (8.25 г, 19 ммоль).
(R)-1-(((R)-1-((3-азидопропил)амино)-3-(4-гидроксифенил)-1-оксопропан-2-ил)амино)-1-оксо-3-фенилпропан-2-аммоний трифторацетат (36)
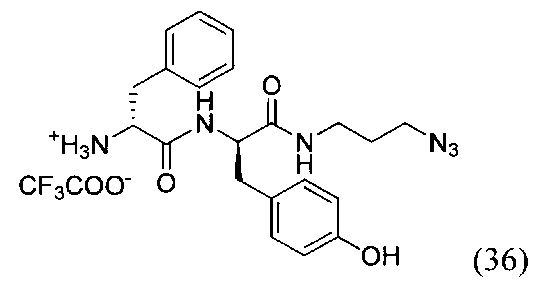
К раствору соединения (35) (8.25 г, 19 ммоль) в 90 мл дихлорметана добавили EDC*HCl (4.4 г, 23 ммоль, 1.2 экв.), PFPOH (3.5 г, 19 ммоль, 1 экв.) и перемешивали в течение 12 часов при комнатной температуре. Затем в реакционную смесь добавляли силикагель (40 г) и полученную суспензию хроматографировали на колонке с силикагелем (элюент - дихлорметан/этилацетат 40:1). Продукт реакции (желтое маслянистое вещество) растворяли в 90 мл ТГФ и добавляли при перемешивании азидопропиламин (1.6 г, 19 ммоль, 1 экв.). К полученному раствору прикапывали DIPEA (4 мл, 23 ммоль, 1 экв.) и перемешивали в течение 12 часов при комнатной температуре. По окончании реакции реакционную смесь концентрировали в вакууме роторного испарителя и хроматографировали остаток на колонке с силикагелем (элюент - дихлорметан/метанол 40:1). Полученный продукт растворяли в 60 мл безводного дихлорметана и охлаждали на бане со льдом до 0°С. Далее при перемешивании прикапывали 4 мл трифторуксусной кислоты и перемешивали реакционный раствор при комнатной температуре в течение 3-4 часов до полного исчезновения исходного вещества (ТСХ-контроль). По окончании реакции упаривали смесь в вакууме роторного растворителя, переупаривали остаток дважды с дихлорметаном и вакуумировали до полного удаления органического растворителя. Полученное масло ресуспендировали в 50 мл диэтилового эфира в УЗИ-бане, затем отфильтровали. Получили продукт реакции в виде белого порошка вещества (36) с выходом 40% (4 г, 7.6 ммоль).
(3S,7S,23S,26S)-три-трет-бутил 31-азидо-12,23,26-трибензил-5,13,18,21,24,27-гексаоксо-4,6,12,17,22,25,28-гептаазагентриаконтан-1,3,7-трикарбоксилат (37)
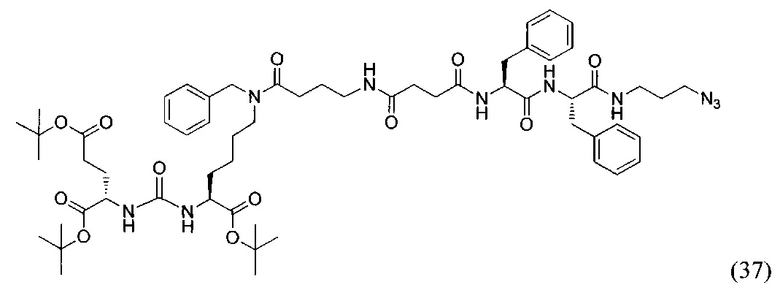
К раствору 60 мг (0,092 ммоль) соединения (18) в 10 мл ДХМ добавили 10 мг (0,096 ммоль) янтарного ангидрида и 23 мкл (0,135) DIPEA. Смесь перемешивали сутки. Далее добавили 5 мл метанола и перемешивали 30 минут. Далее удалили растворитель при пониженном давлении и растворили сухой остаток в 20 мл ДХМ. Раствор промыли трижды 10 мл 0,1 М раствора HCl. Органическую фракцию высушили над Na2SO4 и упарили. Сухой остаток растворили в 10 мл ДМФА и добавили 51 мг (0,160 ммоль соединения (27), 13 мг (0,096 ммоль) HOBt, 55 мг (0,144 ммоль) HBTU, 19 мкл (0,110 ммоль) DIPEA. Смесь перемешивали 24 ч. Далее удалили растворитель при пониженном давлении. Продукт выделяли с помощью метода колоночной хроматографии (Puriflash 50μ 4g, элюент: система петролейный эфир/этилацетат  этилацетат/метанол, от 5% EtOAc до 100% EtOAc в течение 15 минут, 100% EtOAc в течение 1 минуты, от 0% метанола до 100% метанола в течение 5 минут, 100% метанола в течение 5 минут). Элюента для ТСХ - EtOAc/МеОН=5:1. В результате было выделено 51 мг (0,045 ммоль) целевого продукта (37), выход 49%.
этилацетат/метанол, от 5% EtOAc до 100% EtOAc в течение 15 минут, 100% EtOAc в течение 1 минуты, от 0% метанола до 100% метанола в течение 5 минут, 100% метанола в течение 5 минут). Элюента для ТСХ - EtOAc/МеОН=5:1. В результате было выделено 51 мг (0,045 ммоль) целевого продукта (37), выход 49%.
(3S,7S)-три-трет-бутил 33-азидо-12,25,28-трибензил-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатриаконтан-1,3,7-трикарбоксилат (38)
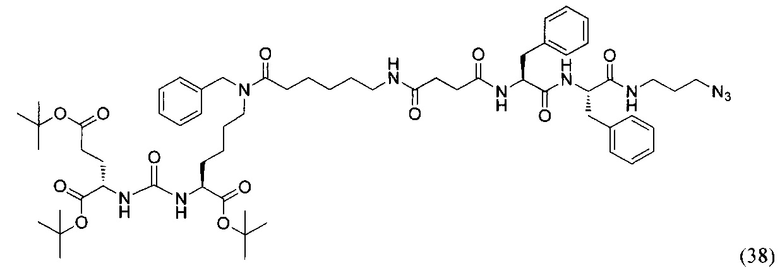
К раствору 88 мг (0,127 ммоль) соединения (19) в 10 мл ДХМ добавили 14 мг (0,134 ммоль) янтарного ангидрида и 34 мкл (0,191) DIPEA. Смесь перемешивали сутки. Далее добавили 5 мл метанола и перемешивали 30 минут. Далее удалили растворитель при пониженном давлении и растворили сухой остаток в 20 мл ДХМ. Раствор промыли трижды 10 мл 0,1 М раствора HCl. Органическую фракцию высушили над Na2SO4 и упарили. Сухой остаток растворили в 10 мл ДМФА и добавили 51 мг (0,160 ммоль) вещества (27), 21 мг (0,152 ммоль) HOBt, 58 мг (0,152 ммоль) HBTU, 26 мкл (0,152 ммоль) DIPEA. Смесь перемешивали 24 ч. Далее удалили растворитель при пониженном давлении. Продукт выделяли с помощью метода колоночной хроматографии (Puriflash 50μ 4g, элюент: система петролейный эфир/этилацетат этилацетат/метанол, от 5% EtOAc до 100% EtOAc в течение 15 минут, 100% EtOAc в течение 1 минуты, от 0% метанола до 100% метанола в течение 5 минут, 100% метанола в течение 5 минут). Элюента для ТСХ - EtOAc/MeOH=5:1. В результате было выделено 44 мг (0,038 ммоль) целевого продукта (38), выход 30%.
(3S,7S,25S,28S)-три-трет-бутил 33-азидо-25,28-дибензил-12-(4-бромбензил)-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатритриаконтан-1,3,7-трикарбоксилат (39)
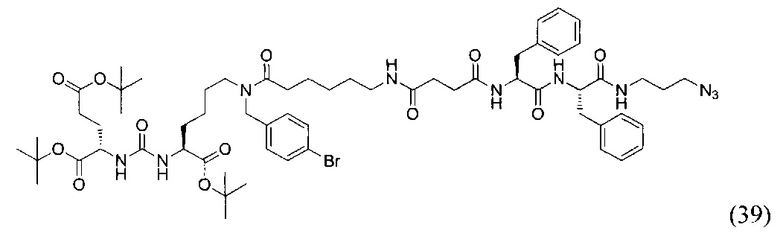
К раствору 50 мг (0,065 ммоль) соединения (21) в 10 мл ДХМ добавили 7 мг (0,068 ммоль) янтарного ангидрида и 23 мкл (0,130 ммоль) DIPEA. Смесь перемешивали сутки. Далее добавили 5 мл метанола и перемешивали 30 минут. Далее удалили растворитель при пониженном давлении и растворили сухой остаток в 20 мл ДХМ. Раствор промыли трижды 10 мл 0,1 М раствора HCl. Органическую фракцию высушили над Na2SO4 и упарили. Сухой остаток растворили в 10 ДМФА и добавили 25 мг (0,049 ммоль) вещества (27), 8 мг (0,060 ммоль) HOBt, 23 мг (0,060 ммоль) HBTU, 20 мкл (0,116 ммоль) DIPEA. Смесь перемешивали 24 ч. Далее удалили растворитель при пониженном давлении. Продукт выделяли с помощью метода колоночной хроматографии (Puriflash 50μ 4g, элюент: система петролейный эфир/этилацетат этилацетат/метанол, от 5% EtOAc до 100% EtOAc в течение 15 минут, 100% EtOAc в течение 1 минуты, от 0% метанола до 100% метанола в течение 5 минут, 100% метанола в течение 5 минут). Элюента для ТСХ - EtOAc/MeOH=5:1. В результате было выделено 35 мг (0,028 ммоль) целевого продукта (39), выход 43%.
(3S,7S,25R,28R)-три-трет-бутил-33-азидо-25,28-дибензил-12-(4-бромбензил)-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатритриаконтан-1,3,7-трикоарбоксилат (40)
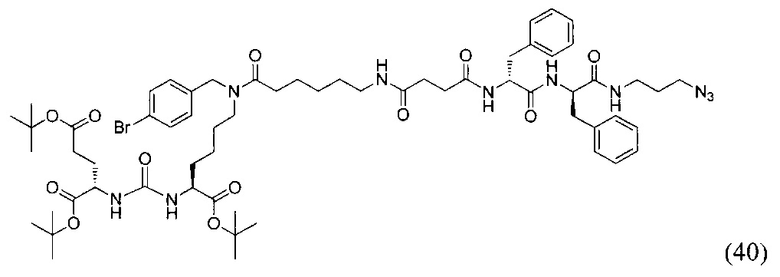
124 мг соединения (21), 0.161 ммоль растворили в 10 мл DCM и систему продули аргоном. Добавили янтарный ангидрид 17 мг, 0,169 ммоль, после этого DIPEA 41 мкл 0.241 ммоль. Оставили при перемешивании на сутки. После добавили 1 мл метанола и оставили при перемешивании на 1,5 часа. Полученную реакционную смесь упарили. Сухой остаток растворили растворили в 2 мл DCM, добавили DIPEA 26 мкл 0.156 ммоль. Систему продули аргоном. Добавили НОВТ 17 мг, 0.117 ммоль, HBTU 44 мг, 0.117 ммоль и оставили перемешиваться на 30 минут. Далее добавили растворенное в 500 мкл DCM вещество (30) 60 мг, 0.077 ммоль. Реакционную смесь оставили перемешиваться на ночь. Растворитель упарили при пониженном давлении. Очистку проводили методом колоночной хроматографии (Puriflash 50μ 4g, элюент: система петролейный эфир/этилацетат этилацетат/метанол, от 5% EtOAc до 100% EtOAc в течение 15 минут, 100% EtOAc в течение 1 минуты, от 0% метанола до 100% метанола в течение 5 минут, 100%) метанола в течение 5 минут). В результате выделили 60 мг целевого продукта (40), выход 31%.
(3S,7S,25R,28R)-три-трет-бутил-33-азидо-25-бензил-12-(-4-бромбензил)-28-(4-гидроксибензил)-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатритриаконтан-1,3,7-трикарбоксилат (41)
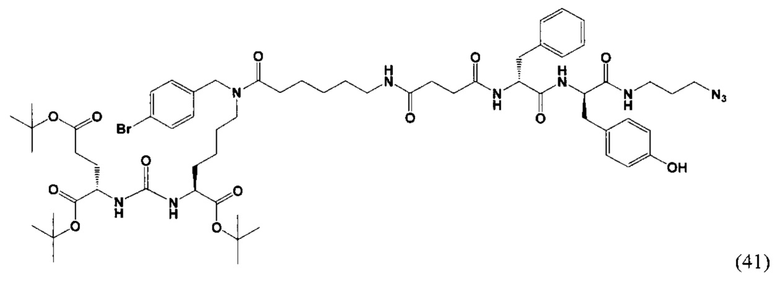
К раствору 69 мг (88,97 мкмоль) вещества (21) в 10 мл ДХМ добавили 9 мг (93,42 мкмоль) янтарного ангидрида и 23 мкл (133,45 мкмоль) DIPEA. Смесь перемешивали сутки. Далее добавили 3 мл метанола и перемешивали 90 минут. Далее удалили растворитель при пониженном давлении. Сухой остаток растворили в 10 мл ДМФА и добавили 31 мкл (177,9 мкмоль) DIPEA, 20 мг (133,4 мкмоль) HOBt, 51 мг (133,4 мкмоль) HBTU, после систему продули аргоном и оставили перемешиваться 60 минут, далее добавили 70 мг (133,4 мкмоль) вещества (36). Смесь перемешивалась 24 ч в инертной атмосфере. Далее удалили растворитель при пониженном давлении. Продукт выделяли с помощью метода колоночной хроматографии (Puriflash 50μ 4g, элюент: система петролейный эфир/этилацетат этилацетат/метанол, от 5% EtOAc до 100% EtOAc в течение 15 минут, 100% EtOAc в течение 1 минуты, от 0% метанола до 100% метанола в течение 5 минут, 100% метанола в течение 5 минут). Элюент для ТСХ - EtOAc/MeOH=5:1.
Таким образом, соединение (41) было выделено в виде желтоватого маслянистого вещества, выход составил 100 мг (89%).
(3S,7S,25R,28R)-три-трет-бутил-33-азидо-25-бензил-28-(-3-бром-4-гидроксибензил)-12-(4-бромбензил)-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатритриаконтан-1,3,7-трикарбоксилат (42)
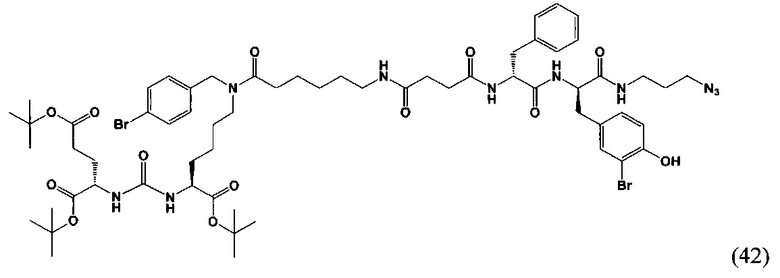
К раствору 41 мг (вещества (21) в 10 мл ДХМ добавили 6 мг (55,78 мкмоль) янтарного ангидрида и 14 мкл (79,68 мкмоль) DIPEA. Смесь перемешивалась сутки. Далее добавили 3 мл метанола и оставили перемешиваться 90 минут. Далее удалили растворитель при пониженном давлении. Сухой остаток растворили в 10 мл ДМФА и добавили 18 мкл (106,25 мкмоль) DIPEA, 12 мг (79,68 мкмоль) HOBt, 30 мг (79,68 мкмоль) HBTU, после систему продули аргоном и оставили перемешиваться 60 минут, далее добавили 48 мг (79,68 мкмоль) (R)-2-амино-N-((R)-1-((3-аазидопропил)амино)-3-(3-бром-4-гидроксифенил)-1-оксопропан-2-ил)-3-фенилпропанамида. Смесь перемешивалась 24 ч в инертной атмосфере. Далее удалили растворитель при пониженном давлении. Продукт выделяли с помощью метода колоночной хроматографии (Puriflash 50μ 4g, элюент: система петролейный эфир/этилацетат этилацетат/метанол, от 5% EtOAc до 100% EtOAc в течение 15 минут, 100% EtOAc в течение 1 минуты, от 0% метанола до 100% метанола в течение 5 минут, 100% метанола в течение 5 минут). Элюент для ТСХ - EtOAc/МеОН=5:1.
Таким образом, соединение (42) было выделено в виде желтоватого маслянистого вещества, выход составил 53 мг (74%).
(3S,7S,25S,28S)-три-трет-бутил-33-азидо-25-бензил-12-(4-бромбензил)-28-(4-гидроксибензил)-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатритриаконтан-1,3,7-трикоарбоксилат (43)
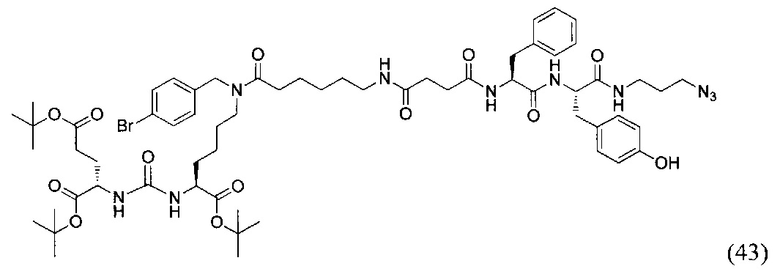
Вещество (21) 124 мг, 0.161 ммоль растворили в 10 мл DCM и систему продули аргоном. Добавили янтарный ангидрид 17 мг, 0,169 ммоль, после этого DIPEA 41 мкл 0.241 ммоль. Оставили при перемешивании на сутки. После добавили 1 мл метанола и оставили при перемешивании на 1,5 часа. Полученную реакционную смесь упарили. Сухой остаток растворили в 2 мл DCM, добавили DIPEA 34 мкл 0.199 ммоль. Систему продули аргоном. Добавили НОВТ 23 мг; 0.148 ммоль, HBTU 56 мг; 0.148 ммоль и оставили перемешиваться на 30 минут. Далее добавили растворенное в 1 мл DMF вещество (33) 60 мг, 0.199 ммоль. Реакционную смесь оставили перемешиваться на ночь. Растворитель упарили при пониженном давлении. Очистку проводили методом колоночной хроматографии (этилацетат/гексан от 5% до 100% этилацетата; метанол : этилацетат; от 0% до 100%). В результате выделили 98 мг целевого продукта (43), выход 39%.
(3S,7S,25S,28S)-три-трет-бутил 33-азидо-25,28-дибензил-12-(-4-хлорбензил)-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатритриаконтан-1,3,7-трикарбоксилат (44)
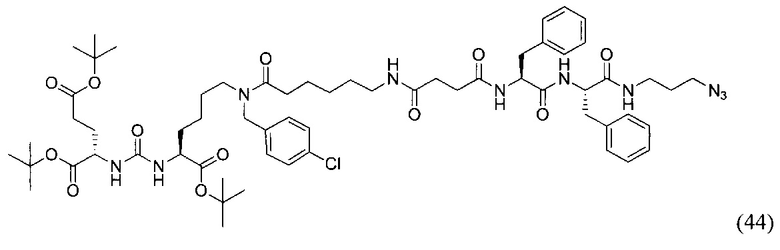
К раствору 70 мг (96,63 мкмоль) вещества (20) в 10 мл ДМФА добавили 72 мг (144,94 мкмоль) вещества (27), 22 мг (144,94 мкмоль) HOBt, 55 мг (144,94 мкмоль) HBTU, 34 мкл (193,26 мкмоль) DIPEA. Смесь перемешивали 24 ч. Далее удалили растворитель при пониженном давлении. Продукт выделяли с помощью метода колоночной хроматографии (Puriflash 50μ 4g, элюент: система петролейный эфир/этилацетат этилацетат/метанол, от 5% EtOAc до 100% EtOAc в течение 15 минут, 100% EtOAc в течение 1 минуты, от 0% метанола до 100% метанола в течение 5 минут, 100% метанола в течение 5 минут). Элюент для ТСХ - EtOAc/MeOH=5:1.
Таким образом, соединение (44) было выделено в виде бесцветного маслянистого вещества, выход составил 59 мг (51%)
(3S,7S,25S,28S)-три-трет-бутил 33-азидо-25-бензил-12-(-4-хлорбензил)-28-(4-гидроксибензил)-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатритриаконтан-1,3,7-трикарбоксилат (45)
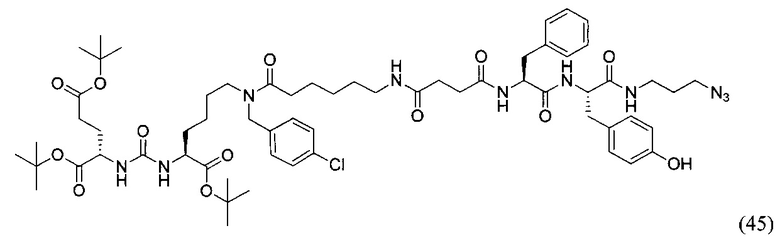
К раствору 56 мг (77,88 мкмоль) соединения (20) в 10 мл ДХМ добавили 8 мг (81,77 мкмоль) янтарного ангидрида и 27 мкл (155,76 мкмоль) DIPEA. Смесь перемешивали сутки. Далее добавили 5 мл метанола и перемешивали 30 минут. Далее удалили растворитель при пониженном давлении и растворили сухой остаток в 20 мл ДХМ. Раствор промыли трижды 10 мл 0,1 М раствора HCl. Органическую фракцию высушили над Na2SO4 и удалили растворитель при пониженном давлении. Сухой остаток растворили в 10 мл ДМФА и добавили 58 мг (110,6 мкмоль) вещества (33), 17 мг (110,6 мкмоль) HOBt, 42 мг (110,6 мкмоль) HBTU, 26 мкл (147,6 мкмоль) DIPEA. Смесь перемешивали 24 ч. Далее удалили растворитель при пониженном давлении. Продукт выделяли с помощью метода колоночной хроматографии (Puriflash 50μ 4g, элюент: система петролейный эфир/этилацетат => этилацетат/метанол, от 5% EtOAc до 100% EtOAc в течение 15 минут, 100% EtOAc в течение 1 минуты, от 0% метанола до 100% метанола в течение 5 минут, 100% метанола в течение 5 минут). Элюента для ТСХ - AtOAc/МеОН=5:1.
Таким образом, соединение (45) было выделено в виде бесцветного маслянистого вещества, выход составил 65 мг (69%).
(3S,7S,25R,28R)-три-трет-бутил 33-азидо-25-бензил-12-(-4-хлорбензил)-28-(4-гидроксибензил)-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатритриаконтан-1,3,7-трикарбоксилат (46)
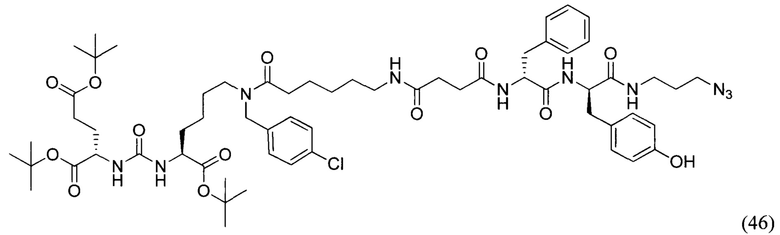
К раствору 62 мг (85,54 мкмоль) соединения (20) в 10 мл ДХМ добавили 9 мг (89,82 мкмоль) янтарного ангидрида и 30 мкл (171,08 мкмоль) DIPEA. Смесь перемешивали сутки. Далее добавили 5 мл метанола и перемешивали 30 минут. Далее удалили растворитель при пониженном давлении и растворили сухой остаток в 20 мл ДХМ. Раствор промыли трижды 10 мл 0,1 М раствора HCl. Органическую фракцию высушили над Na2SO4 и удалили растворитель при пониженном давлении. Сухой остаток растворили в 10 мл ДМФА и добавили 50 мг (122,1 мкмоль) соединения (36), 19 мг (122,1 мкмоль) HOBt, 47 мг (122,1 мкмоль) HBTU, 28 мкл (162,8 мкмоль) DIPEA. Смесь перемешивали 24 ч. Далее удалили растворитель при пониженном давлении. Продукт выделяли с помощью метода колоночной хроматографии (Puriflash 50μ 4g, элюент: система петролейный эфир/этилацетат => этилацетат/метанол, от 5% EtOAc до 100% EtOAc в течение 15 минут, 100% EtOAc в течение 1 минуты, от 0% метанола до 100% метанола в течение 5 минут, 100% метанола в течение 5 минут). Элюент для ТСХ - EtOAc/МеОН=5:1.
Таким образом, соединение (46) было выделено в виде бесцветного маслянистого вещества, выход составил 67 мг (64%).
(3S,7S,25R,28R)-три-трет-бутил 33-азидо-25,28-дибензил-12-(-4-хлорбензил)-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатритриаконтан-1,3,7-трикарбоксилат (47)
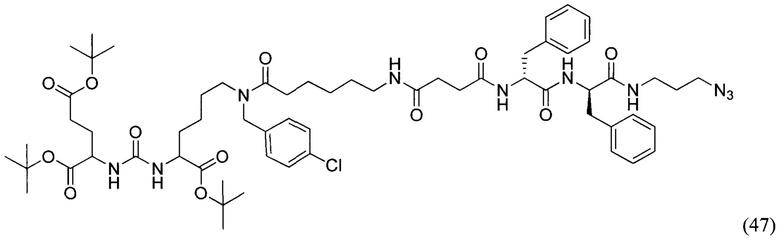
К раствору 47 мг (67,72 мкмоль) соединения (20) в 10 мл ДХМ добавили 7 мг (71,10 мкмоль) янтарного ангидрида и 27 мкл (155,76 мкмоль) DIPEA. Смесь перемешивали сутки. Далее добавили 5 мл метанола и перемешивали 30 минут. Далее удалили растворитель при пониженном давлении и растворили сухой остаток в 20 мл ДХМ. Раствор промыли трижды 10 мл 0,1 М раствора HCl. Органическую фракцию высушили над Na2SO4 и удалили растворитель при пониженном давлении. Сухой остаток растворили в 10 мл ДМФА и добавили 50 мг (96,17 мкмоль) соединение (30), 15 мг (96,17 мкмоль) HOBt, 37 мг (96,17 мкмоль) HBTU, 23 мкл (128,35 мкмоль) DIPEA. Смесь перемешивали 24 ч. Далее удалили растворитель при пониженном давлении. Продукт выделяли с помощью метода колоночной хроматографии (Puriflash 50μ 4g, элюент: система петролейный эфир/этилацетат => этилацетат/метанол, от 5% EtOAc до 100% EtOAc в течение 15 минут, 100% EtOAc в течение 1 минуты, от 0% метанола до 100% метанола в течение 5 минут, 100% метанола в течение 5 минут). Элюент для ТСХ - EtOAc/МеОН=5:1.
Таким образом, соединение (47) было выделено в виде бесцветного маслянистого вещества, выход составил 35 мг (34%)
(3S,7S,25S,28S)-три-трет-бутил 33-азидо-25,28-дибензил-12-(3-хлорбензил)-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатритриаконтан-1,3,7-трикарбоксилат (48)
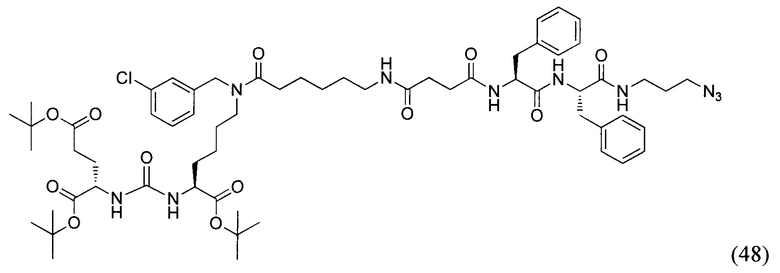
К раствору 88 84 мг (0,069 ммоль) соединения (22) в 10 мл ДХМ добавили 7 мг (0,073 ммоль) янтарного ангидрида и 18 мкл (0,103) DIPEA. Смесь перемешивали сутки. Далее добавили 5 мл метанола и перемешивали 30 минут. Далее удалили растворитель при пониженном давлении и растворили сухой остаток в 20 мл ДХМ. Раствор промыли трижды 10 мл 0,1 М раствора HCl. Органическую фракцию высушили над Na2SO4 и упарили. Сухой остаток растворили в 10 мл ДМФА и добавили 72 мг (0,141 ммоль) соединения (27), 16 мг (0,118 ммоль) HOBt, 72 мг (0,177 ммоль) HBTU, 24 мкл (0,141 ммоль) DIPEA. Смесь перемешивали 24 ч. Далее удалили растворитель при пониженном давлении. Продукт выделяли с помощью метода колоночной хроматографии (Puriflash 50μ 4g, элюент: система петролейный эфир/этилацетат => этилацетат/метанол, от 5% EtOAc до 100% EtOAc в течение 15 минут, 100% EtOAc в течение 1 минуты, от 0% метанола до 100% метанола в течение 5 минут, 100% метанола в течение 5 минут). Элюента для ТСХ - EtOAc/MeOH=5:1. В результате было выделено 76 мг (0,063 ммоль) целевого продукта, выход 92%.
Таким образом, соединение (48) было выделено в виде желтоватого маслянистого вещества, выход составил 76 мг, 0,063 ммоль. (92%)
(3S,2S,28S)-три-трет-бутил-33-азидо-25-бензил-12-(3-хлорбензил)-28-(4-гидроксибензил)-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатритриаконтан-1,3,7-трикоарбоксилат (49)
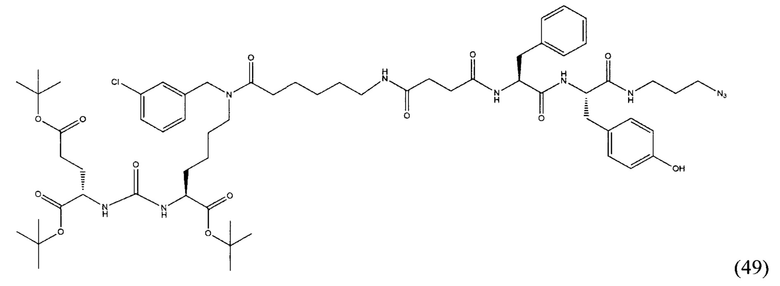
Соединение (22) (160 мг; 0.28 ммоль) растворили в 10 мл DCM и систему продули аргоном. Добавили янтарный ангидрид (37 мг; 0.376), после этого DIPEA (54 мкл, 0.315 ммоль). Оставили при перемешивании на сутки. После добавили 1 мл метанола и оставили при перемешивании на 1,5 часа. Полученную реакционную смесь упарили. Сухой остаток растворили в 2 мл DCM, добавили DIPEA (36 мкл 0.210 ммоль). Систему продули аргоном. Добавили НОВТ (24 мг, 0.158 ммоль), HBTU (59 мг 0.158 ммоль) и оставили перемешиваться на 30 минут. Далее добавили растворенное в 500 мкл DMF соединение (33) (83 мг, 0.158 ммоль). Реакционную смесь оставили перемешиваться на ночь. Растворитель упарили при пониженном давлении. Очистку проводили методом колоночной хроматографии (этилацетат/гексан от 5% до 100% этилацетата; метанол: этилацетат; от 0% до 100%).
Таким образом, соединение (49) было выделено в виде желтоватого маслянистого вещества, выход составил 109 мг.(32%).
(3S,7S,2R,28R)-три-трет-бутил-33-азидо-25-бензил-12-(3-хлорбензил)-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатритриаконтан-1,3,7-трикоарбоксилат (50)
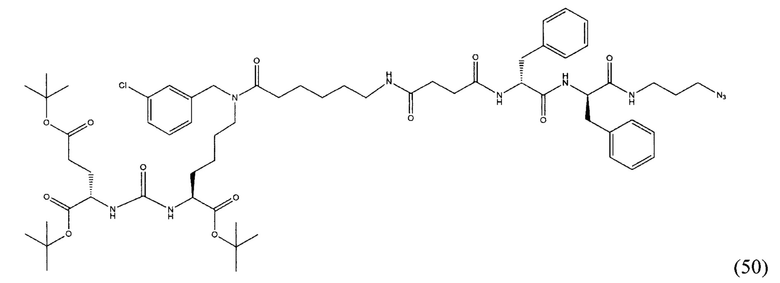
Соединение (22) (62 мг; 0.086 ммоль) растворили в 10 мл DCM и систему продули аргоном. Добавили янтарный ангидрид (90 мг; 0.089 ммоль), после этого DIPEA (22 мкл, 0.129 ммоль). Оставили при перемешивании на сутки. После добавили 1 мл метанола и оставили при перемешивании на 1,5 часа. Полученную реакционную смесь упарили. Сухой остаток растворили в 2 мл DCM, добавили DIPEA (29 мкл 0.145 ммоль). Систему продули аргоном. Добавили НОВТ (20 мг, 0.174 ммоль), HBTU (48 мг 0.174 ммоль) и оставили перемешиваться на 30 минут. Далее добавили растворенное в 500 мкл DMF соединение (30) (64 мг, 0.127 ммоль). Реакционную смесь оставили перемешиваться на ночь. Растворитель упарили при пониженном давлении. Очистку проводили методом колоночной хроматографии (этилацетат/гексан от 5% до 100% этилацетата; метанол: этилацетат; от 0% до 100%).
Таким образом, соединение (50) было выделено в виде желтоватого маслянистого вещества, выход составил 84 мг.(82%).
(3S,7S,2R,28R)-три-трет-бутил-33-азидо-25-бензил-12-(3-хлорбензил)-28-(4-гидроксибензил)-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатритриаконтан-1,3,7-трикоарбоксилат (51)
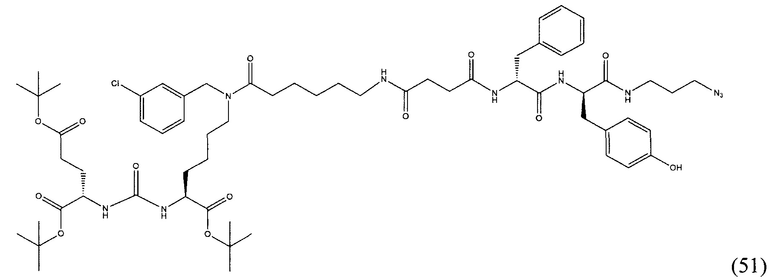
Соединение (22) (62 мг; 0.086 ммоль) растворили в 10 мл DCM и систему продули аргоном. Добавили янтарный ангидрид (90 мг; 0.091), после этого DIPEA (22 мкл, 0.129 ммоль). Оставили при перемешивании на сутки. После добавили 1 мл метанола и оставили при перемешивании на 1,5 часа. Полученную реакционную смесь упарили. Сухой остаток растворили в 2 мл DCM, добавили DIPEA (29 мкл 0.172 ммоль). Систему продули аргоном. Добавили НОВТ (20 мг, 0.158 ммоль), HBTU (48 мг 0.158 ммоль) и оставили перемешиваться на 30 минут. Далее добавили растворенное в 500 мкл DMF соединение (40) (53 мг, 0.158 ммоль). Реакционную смесь оставили перемешиваться на ночь. Растворитель упарили при пониженном давлении. Очистку проводили методом колоночной хроматографии (этилацетат/гексан от 5% до 100% этилацетата; метанол: этилацетат; от 0% до 100%).
Таким образом, соединение (51) было выделено в виде желтоватого маслянистого вещества, выход составил 80 мг (76%).
(3S,7S,2R,28R)-три-трет-бутил-33-азидо-25-бензил-12-(3-хлорбензил)-28-(3-бром-4-гидроксибензил)-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатритриацонат-1,3,7-трикоарбоксилат (52)
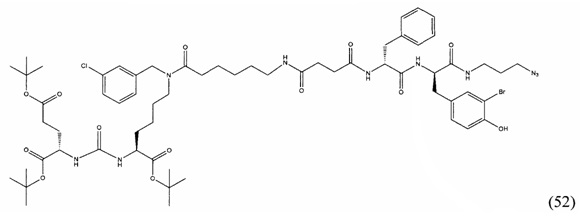
Соединение (22) (52 мг; 0.071 ммоль) растворили в 10 мл DCM и систему продули аргоном. Добавили янтарный ангидрид (75 мг; 0.074 ммоль) после этого DIPEA (18 мкл, 0.106 ммоль). Оставили при перемешивании на сутки. После добавили 1 мл метанола и оставили при перемешивании на 1,5 часа. Полученную реакционную смесь упарили. Сухой остаток растворили в 2 мл DCM, добавили DIPEA (24 мкл 0.145 ммоль). Систему продули аргоном. Добавили НОВТ (17 мг, 0.109 ммоль), HBTU (41 мг 0.109 ммоль) и оставили перемешиваться на 30 минут. Далее добавили растворенный в 500 мкл DMF (R)-2-амино-N-((R)-1-((3-азидопропил)амино-3-(3-бром-4-гидроксифени)-1-оксопропан-2-ил)-3-фенилпропанамид (53 мг, 0.109 ммоль). Реакционную смесь оставили перемешиваться на ночь. Растворитель упарили при пониженном давлении. Очистку проводили методом колоночной хроматографии (этилацетат/гексан от 5% до 100% этилацетата; метанол: этилацетат; от 0% до 100%).
Таким образом, соединение (52) было выделено в виде желтоватого маслянистого вещества, выход составил 78 мг. (84%).
(3S,7S,25S,28S)-три-трет-бутил 33-азидо-25,28-дибензил-12-(4-хлор-2-фторбензил)-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатритриаконтан-1,3,7-трикарбоксилат (53)
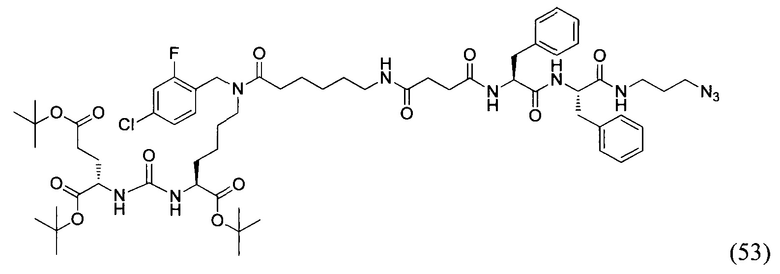
К раствору 86 мг (0,116 ммоль) соединения (23) в 10 мл ДХМ добавили 13 мг (0,127 ммоль) янтарного ангидрида и 22 мкл (0,127) DIPEA. Смесь перемешивали сутки. Далее добавили 5 мл метанола и перемешивали 30 минут. Далее удалили растворитель при пониженном давлении и растворили сухой остаток в 20 мл ДХМ. Раствор промыли трижды 10 мл 0,1 М раствора HCl. Органическую фракцию высушили над Na2SO4 и упарили. Сухой остаток растворили в 10 мл ДМФА и добавили 60 мг (0,152 ммоль) вещества (27), 21 мг (0,152 ммоль) HOBt, 58 мг (0,152 ммоль) HBTU, 27 мкл (0,152 ммоль) DIPEA. Смесь перемешивали 24 ч. Далее удалили растворитель при пониженном давлении. Продукт выделяли с помощью метода колоночной хроматографии (Puriflash 50μ 4g, элюент: система петролейный эфир/этилацетат => этилацетат/метанол, от 5% EtOAc до 100% EtOAc в течение 15 минут, 100% EtOAc в течение 1 минуты, от 0% метанола до 100% метанола в течение 5 минут, 100% метанола в течение 5 минут). Элюента для ТСХ - EtOAc/MeOH=5:1. В результате было выделено 70 мг (0,057 ммоль) целевого продукта (53), выход 49%.
(3S,7S,25S,28S)-три-трет-бутил 33-азидо-25,28-дибензил-12-(4-хлор-3-фторбензил)-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатритриаконтан-1,3,7-трикарбоксилат (54)
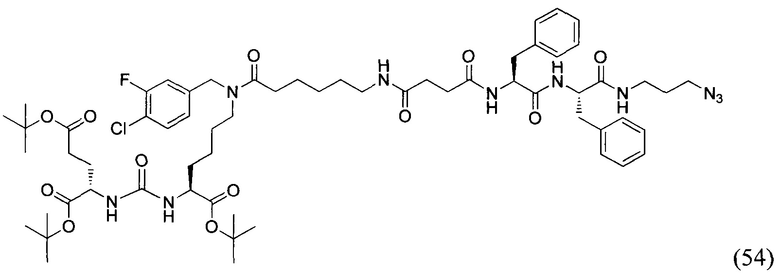
К раствору 130 мг (0,175 ммоль) соединения (24) в 10 мл ДХМ добавили 18 мг (0,184 ммоль) янтарного ангидрида и 46 мкл (0,265 ммоль) DIPEA. Смесь перемешивали сутки. Далее добавили 5 мл метанола и перемешивали 30 минут. Далее удалили растворитель при пониженном давлении и растворили сухой остаток в 20 мл ДХМ. Раствор промыли трижды 10 мл 0,1 М раствора HCl. Органическую фракцию высушили над Na2SO4 и упарили. Сухой остаток растворили в 10 мл ДМФА и добавили 98 мг (0,192 ммоль) вещества (27), 22 мг (0,160 ммоль) HOBt, 91 мг (0,240 ммоль) HBTU, 33 мкл (0,192 ммоль) DIPEA. Смесь перемешивали 24 ч. Далее удалили растворитель при пониженном давлении. Продукт выделяли с помощью метода колоночной хроматографии (Puriflash 50μ 4g, элюент: система петролейный эфир/этилацетат => этилацетат/метанол, от 5% EtOAc до 100% EtOAc в течение 15 минут, 100% EtOAc в течение 1 минуты, от 0% метанола до 100% метанола в течение 5 минут, 100% метанола в течение 5 минут). Элюента для ТСХ - EtOAc/MeOH=5:1. В результате было выделено 65 мг (0,053 ммоль) целевого продукта (54), выход 30%.
(3S,7S,25R,28R)-три-трет-бутил 33-азидо-25,28-дибензил-12-(4-хлорбензил)-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатритриаконтан-1,3,7-трикарбоксилат (55)
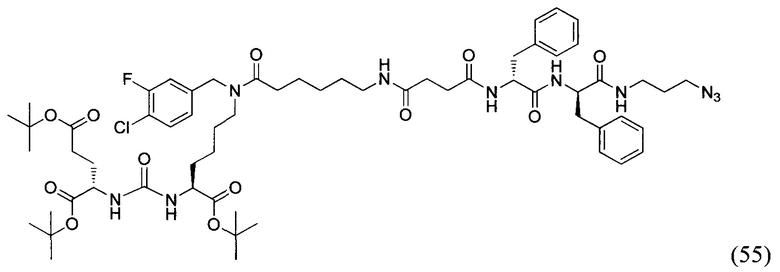
К раствору 156 мг (0,210 ммоль) соединения (24) в 10 мл ДХМ добавили 22 мг (0,221 ммоль) янтарного ангидрида и 55 мкл (0,318 ммоль) DIPEA. Смесь перемешивали сутки. Далее добавили 5 мл метанола и перемешивали 30 минут. Далее удалили растворитель при пониженном давлении и растворили сухой остаток в 20 мл ДХМ. Раствор промыли трижды 10 мл 0,1 М раствора HCl. Органическую фракцию высушили над Na2SO4 и упарили. Сухой остаток растворили в 10 мл ДМФА и добавили 106 мг (0,208 ммоль) соединения (30), 48 мг (0,311 ммоль) HOBt, 118 мг (0,311 ммоль) HBTU, 71 мкл (0,311 ммоль) DIPEA. Смесь перемешивали 24 ч. Далее удалили растворитель при пониженном давлении. Продукт выделяли с помощью метода колоночной хроматографии (Puriflash 50μ 4g, элюент: система петролейный эфир/этилацетат => этилацетат/метанол, от 5% EtOAc до 100% AtOAc в течение 15 минут, 100% AtOAc в течение 1 минуты, от 0% метанола до 100% метанола в течение 5 минут, 100% метанола в течение 5 минут). Элюента для ТСХ - EtOAc/МеОН=5:1. В результате было выделено 217 мг (0,178 ммоль) целевого продукта (55), выход 85%.
(3S,7S,25S,28S)-три-трет-бутил 33-азидо-25-бензил-12-(4-хлор-3-фторбензил)-28-(4-гидроксибензил)-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатриаконтан-1,3,7-трикарбоксилат (56)
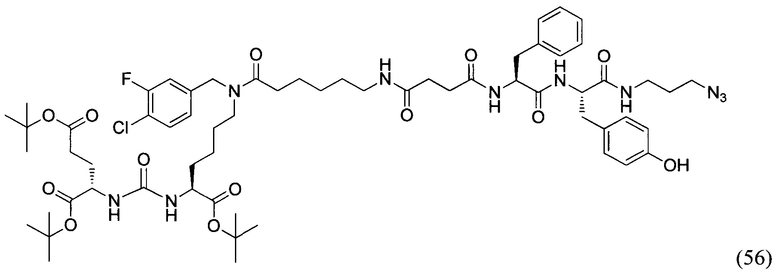
К раствору 156 мг (0,210 ммоль) соединения (24) в 10 мл ДХМ добавили 22 мг (0,221 ммоль) янтарного ангидрида и 55 мкл (0,318 ммоль) DIPEA. Смесь перемешивали сутки. Далее добавили 5 мл метанола и перемешивали 30 минут. Далее удалили растворитель при пониженном давлении и растворили сухой остаток в 20 мл ДХМ. Раствор промыли трижды 10 мл 0,1 М раствора HCl. Органическую фракцию высушили над Na2SO4 и упарили. Сухой остаток растворили в 10 мл ДМФА и добавили 63 мг (0,120 ммоль) соединения (33), 14 мг (0,100 ммоль) HOBt, 57 мг (0,150 ммоль) HBTU, 21 мкл (0,120 ммоль) DIPEA. Смесь перемешивали 24 ч. Далее удалили растворитель при пониженном давлении. Продукт выделяли с помощью метода колоночной хроматографии (Puriflash 50μ 4g, элюент: система петролейный эфир/этилацетат => этилацетат/метанол, от 5% EtOAc до 100% EtOAc в течение 15 минут, 100% EtOAc в течение 1 минуты, от 0% метанола до 100% метанола в течение 5 минут, 100% метанола в течение 5 минут). Элюента для ТСХ - EtOAc/МеОН=5:1. В результате было выделено 217 мг (0,178 ммоль) целевого продукта (56), выход 85%.
(3S,7S,2R,28R)-три-трет-бутил-33-азидо-25-бензил-12-(4-хлор-3-фтор-бензил)-28-(4-гидроксибензил)-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатритриаконтан-1,3,7-трикоарбоксилат (57)
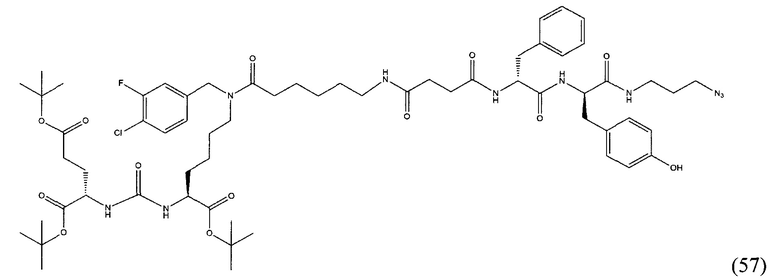
Соединение (24) (159 мг; 0.21 ммоль) растворили в 10 мл DCM и систему продули аргоном. Добавили янтарный ангидрид (22 мг; 0.22 ммоль), после этого DIPEA (54 мкл, 0.315 ммоль). Оставили при перемешивании на сутки. После добавили 1 мл метанола и оставили при перемешивании на 1,5 часа. Полученную реакционную смесь упарили. Сухой остаток растворили в 2 мл DCM, добавили DIPEA (26 мкл, 0.152 ммоль). Систему продули аргоном. Добавили HOBT (17 мг 0.114 ммоль), HBTU (43 мг, 0.114 ммоль) и оставили перемешиваться на 30 минут. Далее добавили растворенное в 500 мкл соединение (35) (60 мг, 0.114 ммоль). Реакционную смесь оставили перемешиваться на ночь. Растворитель упарили при пониженном давлении. Очистку проводили методом колоночной хроматографии (Puriflash 50μ 4g, элюент: система петролейный эфир/этилацетат => этилацетат/метанол, от 5% EtOAc до 100% EtOAc в течение 15 минут, 100% EtOAc в течение 1 минуты, от 0% метанола до 100% метанола в течение 5 минут, 100% метанола в течение 5 минут).
Таким образом, соединение (57) было выделено в виде желтоватого маслянистого вещества, выход составил 93 мг.(36%).
(3S,7S,23S,26S)-31-азидо-12,23,26-трибензил-5,13,18,21,24,27-гексаоксо-4,6,12,17,22,25,28-гептаазагентриаконтан 1,3,7-трикарбоксилат (58)
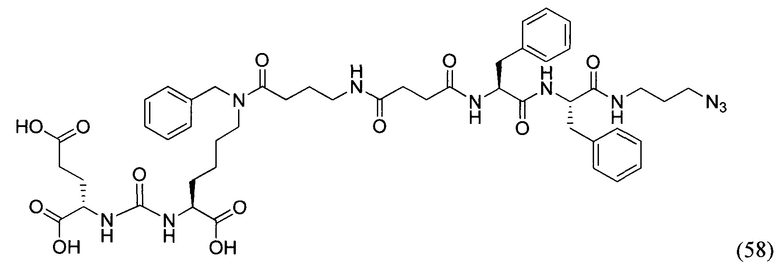
34 мг соединения (37) растворили в 3 мл ДХМ и добавили 335 мкл трифторуксусной кислоты. Смесь перемешивали 13 ч. Далее удалили растворитель при пониженном давлении и трижды переупарили с ДХМ. Продукт осадили эфиром. В результате было выделено 24 мг целевого продукта, выход 83%.
Спектр ЯМР 1Н (400 МГц, ДМСО-d6, δ, м.д.):
8.25-8.27 (м, 1Н, NH), 8.15 (т, J=7.8 Гц, 1Н, NH), 7.96-7.97 (м, 1Н, NH), 7.61-7.62 (м, 1Н, NH), 7.16-7.32 (м, 15Н, Ar), 6.33 (д, J=8.7 Гц, 1Н, CH2Ar), 6.28 (д, J=8.4 Гц, 1Н, CH2Ar), 4.48 (д, J=13.6 Гц, 2Н, СН), 4.37-4.39 (м, 1Н, СН), 4.31 (м, 1Н, CH2), 4.04-4.09 (м, 2Н, СН), 4.00-4.03 (м, 1Н, CH2), 3.16-3.22 (м, 4Н, CH2), 3.02-3.12 (м, 7Н, CH2), 2.18-2.36 (м, 7Н, CH2), 1.89-1.91 (м, 1Н, CH2), 1.56-1.72 (м, 6Н, CH2), 1.44-1.49 (м, 3Н, CH2), 1.16-1.22 (м, 3Н, CH2)
ИК-спектр (ZnSe, см-1): 3305.87 (NH), 3086 (С-Нвал), 2959.79 (Ar), 2098.17 (N3), 1702.84 (C(O)), 1641.61 (С(O) (мочевина)), 1536.51 (С(O)).
ESI-LCMS для C48H62N10O12: m/z рассчитано для [М+Н]+: 971.45, найдено: 971.40
(3S,7S,25S,28S)-33-азидо-12,25,28-трибензил-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатритриаконтан-1,3,7-трикарбоксилат (59)
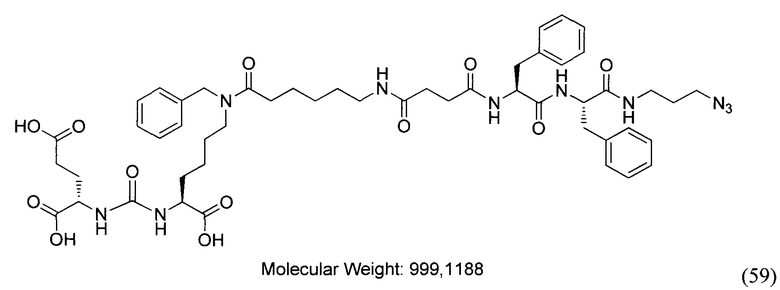
40 мг (0,035 ммоль) соединения (38) растворили в 2.7 мл ДХМ и добавили 300 мкл трифторуксусной кислоты. Смесь перемешивали 16 ч. Далее удалили растворитель при пониженном давлении и трижды переупарили с ДХМ. Продукт осадили эфиром. В результате было выделено 32 мг (0,032 ммоль) целевого продукта, выход 91%.
Спектр ЯМР 1Н (400 МГц, ДМСО-d6, δ, м.д.):
12.43 (уш.с, 3Н, СООН), 8.30 (д, 1Н, J=7.2 Гц, C(O)NH), 8.17 (м, 1Н, C(O)NH), 7.93 (м, 1Н, C(O)NH), 7.58 (м, 1Н, C(O)NH), 7.40-7.10 (м, 16Н, C(O)NH + Ar), 6.30 (м, 2Н, NH(мочевина)), 4.54-4.43 (м, 2Н, ArCH2), 4.45-4.22 (м, 2Н, СН (Phe)), 4.13-3.96 (м, 2Н, СН (Glu, Lys)), 4.08 (м, 2Н, СН, СН), 3.26-2.83 (м, 11Н, CH2), 2.81-2.57 (м, 2Н, CH2), 2.41-2.08 (м, 7Н, CH2), 1.89 (м, 1Н, CH2), 1.70 (м, 1Н, CH2), 1.65-1.33 (м, 8Н, CH2), 1.33-1.11 (м, 5Н, CH2)
ИК спектр 3316 (NH), 3086 (С-Нвал), 2935 (Ar), 2102 (N3), 1729 (C(O)), 1644 (C(O) (мочевина)), 1551 (С(O)), 1293 (Ar)
ESI-LCMS для C50H66N10O12: m/z рассчитано для [M+H]+ 999.49, найдено: 999.40
(3S,7S,25S,28S)-33-азидо-25,28-дибензил-12-(4-бромобензил)-5,13,20,23,26, 29-гексаоксо-4,6,12,19,24,27,30-гептаазатритриаконтан-1,3,7-трикарбоксилат (60)
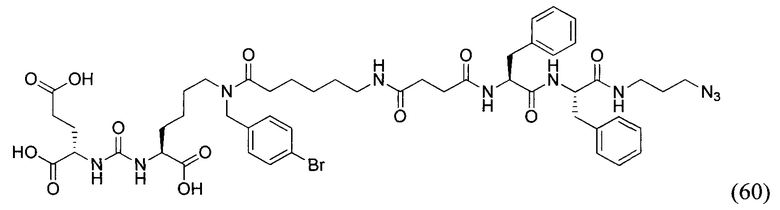
К раствору 23 мг соединения (39) в 3 мл ДХМ добавили 335 мкл трифторуксусной кислоты. Смесь перемешивали 16 ч. Далее удалили растворитель при пониженном давлении и переупарили с ДХМ трижды. Продукт высадили эфиром. В результате было выделено 17 мг целевого соединения, выход 85%.
Спектр ЯМР 1Н (400 МГц, ДМСО-d6, δ, м.д.):
12.43 (уш. с, 2Н, СООН), 8.30-8.38 (м, 1Н, CH(Ph)), 8.08-8.19 (м, 1Н, CH(Ph)), 7.53 (д, J=8.1 Гц, 1Н, CH(Ph)), 7.48 (д, J=7.7 Гц, 1Н, CH(Ph)), 7.19-7.27 (м, 5Н, CH(Ph)), 7.07-7.16 (М, 5Н, CH(Ph)), 6.27-6.34 (м, 2Н, NH), 4.49 (с, 1Н, CH2(Bz)), 4.43 (с, 1Н, CH2(Bz)), 4.37-4.39 (м, 1Н, СН), 4.28-4.39 (м, 2Н, СН), 4.01-4.09 (м, 2Н, СН), 3.34-3.39 (м, 6Н, CH2), 3.22-3.26 (м, 2Н, CH2), 3.13-320 (м, 2Н, CH2), 2.88-3.09 (м, 6Н, CH2), 2.72-2.79 (м, 1Н, CH2), 2.52-2.67 (м, 1Н, CH2), 2.28-2.35 (м, 2Н, CH2), 2.18-2.25 (м, 4Н, CH2), 1.88-1.91 (м, 1Н, CH2), 1.66-1.76 (м, 1Н, CH2), 1.54-1.61 (м, 3Н, CH2), 1.36-1.49 (м, 6Н, CH2), 1.14-1.30 (м, 4Н, CH2)
ИК спектр (ZnSe, см-1): 1639 (NHC(O)NH), 2097 (N3)
ESI-LCMS для C50H65BrN10O12: m/z рассчитано для [M+H]+: 1079.42, найдено: 1079.35.
(3S,7S,25R,28R)-33-азидо-25,28-дибензил-12-(4-бромбензил)-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатритриаконтан-1,3,7-трикарбоксилат (61)
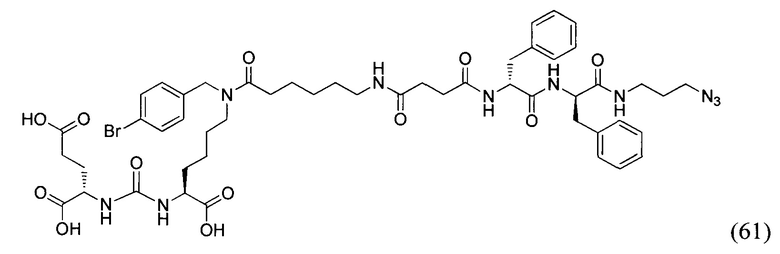
В 4 мл 10% (по объему) раствора трифторуксусной кислоты в DCM растворили 60 мг (0.048 ммоль) соединения (40) и перемешивали 16 часов при комнатной температуре. Растворитель упарили при пониженном давлении. Добавили 4 мл DCM с последующим упариванием для удаления трифторуксусной кислоты, процедуру повторили 5 раз. К полученному маслянистому осадку добавили 5 мл диэтилового эфира, в результате чего выделился белый твердый продукт. Эфир декантировали, процедуру повторили еще 2 раза. Масса полученного вещества 40 мг (0,037 ммоль), выход 77%.
Спектр ЯМР 1H (400 МГц, ДМСО-d6, δ, м.д.):
12.36 (уш.с, 3Н, СООН), 8.30 (дд, 1Н, C(O)NH), 8.17 (дд, 1Н, C(O)NH), 7.79 (м, 1Н, C(O)NH), 7.59-7.47 (м, 3Н, C(O)NH + ArH), 7.21-7.09 (м, 14Н, ArH), 6.29 (м, 2Н, NH(мочевина)), 4.49 (с, 1Н, PhCH2NC(O)), 4.42 (с, 1Н, PhCH2NC(О)), 4.07 (м, 1Н, СН, СН), 4.42 (с, 2Н, CH2Ar), 4.39-4.27 (м, 2Н, CH2Ar), 3.22-2.88 (м, 2Н, CH2), 2.76-2.58 (м, 2Н, CH2), 2.31-2.21 (м, 2Н, CH2), 1.89 (с, 1Н, CH2), 1.22 (м, 18Н, CH2CH)
Спектр ЯМР 13С (100 МГц, ДМСО-d6, δ, м.д.):
207.79 (СООН), 174.55 (C(O)NR2), 174.13 (С(О)NR2), 173.10 (PhCH2NC(O)), 172.48 (PhCH2NC(O)), 171.97 (PhCH2NC(O)), 171.49 (PhCH2NC(O)), 157.66 (NHC(O)NH), 138.37 (Ar), 131.99 (Ar), 131.94 (Ar), 131.60 (Ar), 130.11 (Ar), 129.86 (Ar), 129.57 (Ar), 129.41 (Ar), 129.40 (Ar), 128.57 (Ar), 128.46 (Ar), 126.70 (Ar), 52.06 (CH), 50.95 (СН), 48.63 (CH2NR2), 48.55 (CH2Ar), 47.40 (CH2Ar), 38.10 (CH2), 37.44 (CH2), 36.28 (CH2), 34.37 (CH2), 33.28 (CH2), 33.72 (CH2), 30.90 (CH2), 29.11 (CH2), 28.56 (CH2), 28.14 (CH2), 27.78 (CH2), 25.12 (CH2), 17.61 (CH2), 15.68 (CH2), 14,38 (CH2).
ESI-LCMS для C62H89BrN10O12: m/z рассчитано для [М+Н]+: m/z рассчитано для [М+Н]+ 1078.61, найдено: 1079.30.
(3S,7S,25R,28R)-33-азидо-25-бензил-12-(4-бромбензил)-28-(-4-гидроксибензил-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатритриаконтан-1,3,7-трикарбоксилат (62)
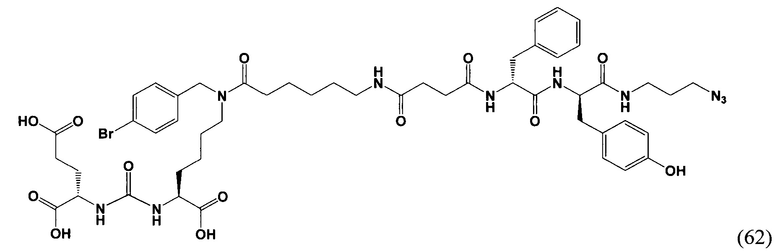
К раствору 90 мг (71,29 мкмоль) соединения (41) в 3,6 мл ДХМ добавили 400 мкл трифторуксусной кислоты. Смесь перемешивали 16 ч. Далее удалили растворитель при пониженном давлении и переупарили с ДХМ трижды. Продукт высадили эфиром и промыли дважды по 10 мл эфира.
Таким образом, соединение (62) было выделено в виде белого аморфного порошка, выход составил 61 мг (77%).
Спектр ЯМР 1H (400 МГц, ДМСО-d6, δ, м.д.):
12.42 (уш.с, 3Н, СООН), 9.17 (уш.с, 1Н, ОН) 8.38-8.20 (м, 1Н, C(O)NH), 8.15-8.00 (м, 1Н, C(O)NH), 7.99-7.85 (м, 1Н, C(O)NH), 7.83-7.67 (м, 1Н, C(O)NH), 7.61-7.43 (м, 2Н, Ar), 7.35-7.04 (м, 9Н, Ar), 7.04-6.92 (м, 2Н, Ar), 6.40-6.20 (м, 2Н, NH(мочевина)), 4.53-4.39 (м, 2Н, ArCH2), 4.37-4.23 (м, 2Н, CH) 4.17-3.95 (м, 2Н, CH), 3.28-2.55 (м, 13Н, CH2), 2.42-2.10 (м, 9Н, CH2), 1.98-1.82 (м, 1Н, CH2), 1.70-1.34 (м, 8Н, CH2), 1.32-1.10 (м, 5Н, CH2).
Спектр ЯМР 13С (100 МГц, ДМСО-d6, δ, м.д.):
174.9 (СООН), 174.6 (C(O)NH), 174.2 (C(O)NH), 172.5 (C(O)NH) 171.5 (C(O)NH), 157.7 (C(O) (мочевина)), 138.5 (Ar), 132.0 (Ar), 131.7 (Ar), 130.5 (Ar), 130.4 (Ar), 130.1 (Ar), 129.6 (Ar), 129.5 (Ar), 129.1 (Ar), 128.5 (Ar), 128.4 (Ar), 115.4 (Ar), 115.3 (Ar), 55.2 (CH), 54.8 (СН), 52.1 (CH), 48.7 (CH), 48.6 (CH), 37.2 (CH2), 36.3 (CH2), 32.2 (CH2), 31.1 (CH2), 30.4 (CH2), 29.5 (CH2), 28.7 (CH2), 28.2 (CH2), 28.0 (CH2), 22.9 (CH2).
ИК спектр (ZnSe, см-1): 3313 (NH), 3088 (С-Н вал), 2937 (Ar), 2100 (N3), 1728 (C(O)), 1643 (C(O)), 1554 (C(O)), 1296 (Ar).
ESI-LCMS для C50H65BrN10O13: m/z рассчитано для [М+Н]+: 1093.39, найдено: 1093.40.
(3S,7S,25R,28R)-33-азидо-25-бензил-28-(-3-бром-4-гидроксибензил)-12-(4-бромбензил)- -5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатритриаконтан-1,3,7-трикарбоксилат (63)
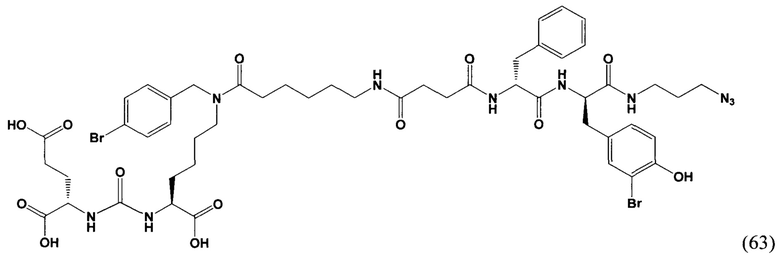
К раствору 49 мг (36,53 мкмоль) соединения (42) в 3,6 мл ДХМ добавили 400 мкл трифторуксусной кислоты. Смесь перемешивали 16 ч. Далее удалили растворитель при пониженном давлении и переупарили с ДХМ трижды. Продукт высадили эфиром и промыли дважды по 10 мл эфира.
Таким образом, соединение (63) было выделено в виде белого аморфного порошка, выход составил 38 мг (88%).
Спектр ЯМР 1Н (400 МГц, ДМСО-d6, δ, м.д.):
12.42 (уш.с, 3Н, СООН), 9.98 (уш.с, 1Н, ОН) 8.40-8.21 (м, 1Н, C(O)NH), 8.21-8.01 (м, 1Н, C(O)NH), 8.00-7.85 (м, 1Н, C(O)NH), 7.84-7.60 (м, 1Н, C(O)NH), 7.58-7.43 (м, 2Н, Ar), 7.41-7.04 (м, 9Н, Ar), 7.04-6.95 (м, 1Н, Ar), 6.88-6.76 (м, 1Н, Ar), 6.43-6.19 (м, 2Н, NH(мочевина)), 4.54-4.22 (м, 4Н, ArCH2 + СН), 4.18-3.90 (м, 2Н, СН), 3.32-2.51 (м, 13Н, CH2), 2.42-2.09 (м, 8Н, CH2), 1.90-1.82 (м, 1Н, CH2), 1.77-1.35 (м, 8Н, CH2), 1.31-1.11 (м, 5Н, CH2).
Спектр ЯМР 13С (100 МГц, ДМСО-d6, δ, м.д.):
174.6 (C(O)NH), 174.2 (C(O)NH), 171.5 (C(O)NH), 157.7 (C(0) (мочевина)), 138.5 (Ar), 138.4 (Ar),133.8 (Ar), 132.0 (Ar), 131.7 (Ar), 130.6 (Ar), 130.2 (Ar), 129.6 (Ar), 129.1 (Ar), 128.4 (Ar), 54.7 (CH), 52.1 (СН), 48.7 (СН), 48.6 (СН), 36.3 (CH2), 32.3 (CH2), 32.2 (CH2), 31.2 (CH2), 31.1 (CH2), 30.3 (CH2), 29.5 (CH2), 28.7 (CH2), 28.0 (CH2), 25.05 (CH2).
ИК спектр (ZnSe, см-1): 3313 (NH), 3086 (С-Н вал), 2937 (Ar), 2100 (N3), 1724 (C(O)), 1643 (C(O)), 1554 (C(O)), 1290 (Ar)
ESI-LCMS для C50H64Br2N10O13: m/z рассчитано для [М+Н]+: 1071.31, найдено: 1071.30.
(3S,7S,25S,28S)-33-азидо-25-бензил-12-(4-бромбензил)-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатритриаконтан-1,317-трикарбоксилат (64)
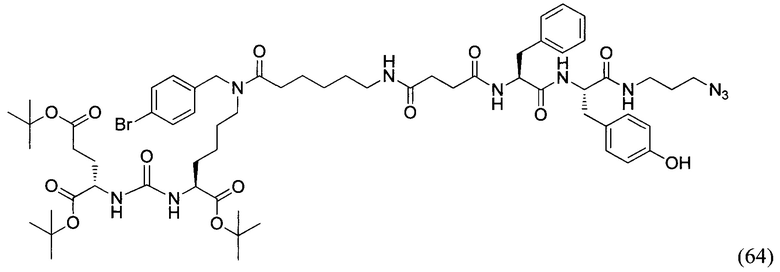
К раствору 86 мг (0,068 ммоль) соединения (43) в 3,6 мл ДХМ добавили 400 мкл трифторуксусной кислоты. Смесь перемешивали 16 ч. Далее удалили растворитель при пониженном давлении и переупарили с ДХМ трижды. Продукт высадили эфиром. В результате было выделено 68 мг (0,062 ммоль) целевого соединения, выход 91%.
Спектр ЯМР 1H (400 МГц, ДМСО-d6, δ, м.д.):
12.52 (уш.с, 3Н, СООН), 8.31 (с, 1Н, C(O)NH), 8.09 (с, 1Н, C(O)NH), 7.91 (с, 1Н, C(O)NH), 7.79 (с, 1Н, C(O)NH), 7.54-7.47 (с, 2Н, C(O)NH + Ar), 7.54-7.47 (с, 2Н, C(O)NH + Ar), 7.15 (м, 7Н, C(O)NH + ArH), 6.30 (м, 1Н, NH(мочевина)), 4.51-4.43 (м, 4Н, PhCH2NC(O)), 4.08 (м, 2Н, СН, СН), 3.26-2.93 (м, 8Н, CH2Ar), 2.71 (м, 2Н, CH2Ar), 2.22 (м, 6Н, CH2), 2.19 (м, 4Н, CH2), 1.90 (с, 1Н, CH2), 1.47 (м, 9Н, CH2), 1.16 (м, 5Н, CH2CH).
Спектр ЯМР 13С (100 МГц, ДМСО-d6, δ, м.д.):
207.18 (СООН), 174.92 (C(O)NR2), 174.63 (C(O)NR2), 174.19 (C(O)NR2), 174.55 (PhCH2NC(O)), 172.48 (PhCH2NC(О)), 171.49 (PhCH2NC(O)), 171.30 (PhCH2NC(O)), 157.71 (NHC(O)NH), 138.47 (Ar), 138.74 (Ar), 131.98 (Ar), 131.66 (Ar), 130.54 (Ar), 130.36 (Ar), 130.15 (Ar), 129.60 (Ar), 129.47 (Ar), 129.08 (Ar), 128.50 (Ar), 128.42 (Ar), 126.68 (Ar), 54.04 (СН), 52.08 (СН), 48.66 (CH2NR2), 36.27 (CH2), 32.34 (CH2), 32.21 (CH2), 31.07 (CH2), 30.33 (CH2), 29.47 (CH2), 28.55 (CH2), 27.97 (CH2), 26.68 (CH2), 25.14 (CH2).
ИК спектр (ZnSe, см-1): 3311 (NH), 2935 (Ar), 2100 (N3), 1643 (C(O)NH), 1292 (Ar), 700 (CH2).
ESI-LCMS для C50H65BrN10O13: m/z рассчитано для [M+H]+ 1094.01, найдено: 1095.25..
(3S,7S,25S,28S)-33-азидо-25,28-бензил-12-(4-хлорбензил)-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатритриаконтан-1,3,7-трикарбоксилат (65).
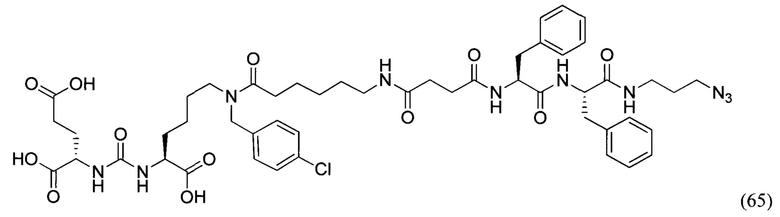
57 мг (28,77 мкмоль) соединения (44) растворили в 3 мл ДХМ и добавили 330 мкл трифторуксусной кислоты. Смесь перемешивали 13 ч. Далее удалили растворитель при пониженном давлении и трижды переупарили с ДХМ. Продукт осадили эфиром и трижды промыли 10 мл диэтилового эфира. В результате было выделено 49 мг целевого продукта, выход 100%.
Таким образом, соединение (65) было выделено в виде белого аморфного порошка, выход составил 49 мг (100%).
Спектр ЯМР 1H (400 МГц, ДМСО-d6, δ, м.д.):
12.36 (уш. с, 2Н, СООН), 8.29-8.37 (м, 1Н, NH), 8.17 (д, J=7.6 Гц, 1Н, NH), 8.02-8.10 (м, 1Н, NH), 7.93 (м, 1Н, NH), 7.40 (д, J=8.2 Гц, 1Н, Ar), 7.34 (д, J=7.9 Гц, 1Н, Ar), 7.23-7.25 (м, 2Н, Ar), 7.19-7.21 (м, 5Н, Ar), 7.17 (м, 3Н, Ar), 6.27-6.33 (м, 1Н, NH), 4.51 (с, 1Н, CH2(Bz)), 4.44 (с, 1Н, CH2(Bz)), 4.29-4.40 (м, 2Н, СН), 4.03-4.08 (м, 2Н, СН), 3.44-3.60 (м, 4Н, CH2), 3.18-3.26 (м, 3Н, CH2), 3.12-3.15 (м, 2Н, CH2), 3.02-3.06 (м, 2Н, CH2), 2.89-3.00 (м, 3Н, CH2), 2.74-2.79 (м, 1Н, CH2), 2.56-2.67 (м, 1Н, CH2), 2.28-2.32 (м, 3Н, CH2), 2.22-2.26 (м, 4Н, CH2), 1.90-1.91 (м, 1Н, CH2), 1.69-1.72 (м, 1Н, CH2), 1.58-1.60 (м, 3Н, CH2), 1.36-1.49 (м, 5Н, CH2), 1.18-1.26 (м, 8Н, CH2)
Спектр ЯМР 13С (100 МГц, ДМСО-d6, δ, м.д.):
174.85 (С(О)ОН), 174.57 (С(О)ОН), 172.41 (С(О)ОН), 172.25 (С(О)ОН), 171.97 (С(О)ОН), 171.66 (С(О)ОН), 171.50 (С(О)ОН), 171.43 (C(O)N), 157.66 (С(O) мочевина), 138.45 (Ar), 138.38 (Ar), 138.45, 130.38 (Ar), 137.51 (Ar), 132.06, 131.81 (Ar), 129.74 (Ar), 129.58 (Ar), 129.52 (Ar), 129.41 (Ar), 128.70, 128.57 (Ar), 128.46 (Ar), 128.37 (Ar), 54.81 (СН), 54.72 (СН), 54.00 (СН), 52.61, 52.49 (СН), 52.05 (СН), 48.64, 48.54 (CH2), 46.74 (CH2), 42.26 (CH2), 38.93 (CH2), 37.99 (CH2), 37.20 (CH2), 36.26 (CH2), 32.20, 32.19 (CH2), 31.09, 31.05 (CH2), 30.31 (CH2), 29.49 (CH2), 28.59 (CH2), 28.55 (CH2), 28.14 (CH2), 27.95 (CH2), 26.65 (CH2), 26.57 (CH2), 25.11 (CH2), 24.98 (CH2), 22.74 (CH2), 19.24 (CH2).
ИК спектр (ZnSe, см-1): 3291 (NH), 3062 (С-Нвал), 2930 (Ar), 2097 (N3), 1722 (C(O)), 1639 (С(O) (мочевина)), 1552 (С(O)), 1252 (Ar)
ESI-HRMS для C50H65ClN10O12: m/z рассчитано для [M+H]+: 1035.4515, найдено: 1035.4551; m/z рассчитано для [M+Na]+: 1056.4398, найдено: 1056.4364.
(3S,7S,25S,28S)-33-азидо-25-бензил-12-(4-хлорбензил)-28-(4-гидроксибензил)-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатритриаконтан-1,3,7-триокарбоксилат (66)
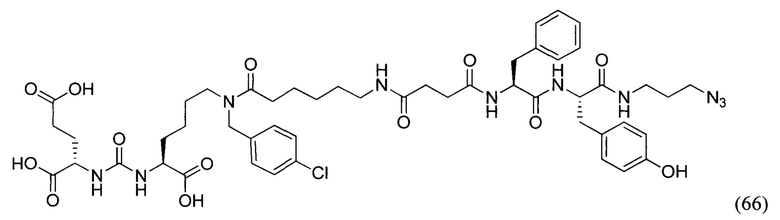
62 мг (50.96 мкмоль) соединения (45) растворили в 3 мл ДХМ и добавили 330 мкл трифторуксусной кислоты. Смесь перемешивали 13 ч. Далее удалили растворитель при пониженном давлении и трижды переупарили с ДХМ. Продукт осадили эфиром и трижды промыли 10 мл диэтилового эфира.
Таким образом, соединение (66) было выделено в виде белого аморфного порошка, выход составил 53 мг (99%).
Спектр ЯМР 1Н (400 МГц, ДМСО-d6, δ, м.д.):
12.40 (уш. с, 3Н, СООН), 8.74-8.81 (м, 1Н, NH), 8.09-8.21 (м, 2Н, NH), 7.79 (м, 1Н, NH), 7.41 (д, J=8.2 Гц, 1Н, Ar), 7.35 (д, J=8.2 Гц, 1Н, Ar), 7.27-7.29 (м, 1Н, Ar), 7.20-7.23 (м, 5Н, Ar), 6.99 (d, J=7.6 Гц, 2Н, Ar), 6.62-6.66 (м, 1Н, Ar), 6.29-6.35 (м, 2Н, Ar), 4.53 (с, 1Н, CH2Ar), 4.46 (с, 1Н, CH2Ar), 4.39-4.41 (м, 1Н, СН), 4.03-4.09 (м, 3Н, СН), 3.14-3.21 (м, 4Н, CH2), 2.95-3.04 (м, 5Н, CH2), 2.54-2.63 (м, 1Н, CH2), 2.49 (м, 6Н, CH2), 2.38-2.40 (м, 2Н, CH2), 2.32-2.35 (м, 1Н, CH2), 2.20-2.29 (м, 4Н, CH2), 1.90-1.92 (м, 1Н, CH2), 1.67-1.72 (м, 1Н, CH2), 1.57-1.60 (м, 2Н, CH2), 1.44-1.55 (м, 4Н, CH2), 1.36-1.40 (м, 2Н, CH2), 1.15-1.30 (м, 5Н, CH2)
Спектр ЯМР 13С (100 МГц, ДМСО-d6, δ, м.д.):
174.68 (С(О)ОН), 174.57 (С(О)ОН), 172.45 (С(О)ОН), 172.22 (С(О)ОН), 171.95 (С(О)ОН), 171.58 (С(О)ОН), 171.37, 171.26 (C(O)N), 157.67 (С(O) мочевина), 156.21 (С(O) мочевина), 138.40 (Ar), 138.00 (Ar), 130.49 (Ar), 130.33 (Ar), 129.74 (Ar), 129.55 (Ar), 129.43 (Ar), 129.02 (Ar), 128.70 (Ar), 128.45 (Ar), 128.37 (Ar), 127.79 (Ar), 126.63 (Ar), 115.36 (Ar), 115.25 (Ar), 65.34 (CH), 55.20 (СН), 54.72 (СН) 52.50 (СН), 52.05 (CH2), 51.69 (CH2), 48.63, 48.56 (CH2), 47.11 (CH2), 37.62, 37.22 (CH2), 36.24 (CH2), 32.72 (CH2), 32.31, 32.19 (CH2), 31.07 (CH2), 30.31 (CH2), 29.43 (CH2), 28.61, 28.57 (CH2), 28.01, 27.95 (CH2), 26.69 (CH2), 25.11, 24.98 (CH2), 22.90 (CH2),15.59 (CH2).
ИК-спектр (ZnSe, см-1): 3238 (NH), 3066 (С-Нвал), 2931 (Ar), 2097 (N3), 1722 (C(O)), 1643 (С(0) (мочевина)), 1548 (С(O)), 1205 (Ar)
ESI-HRMS для C50H65ClN10O13: m/z рассчитано для [М+Н]+: 1052.4498, найдено: 1052.4486.
(3S,7S,25R,28R)-33-азидо-25-бензил-12-(4-хлорбензил)-28-(4-гидроксибензил)-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатритриаконтан-1,3,7-трикарбоксилат (67)
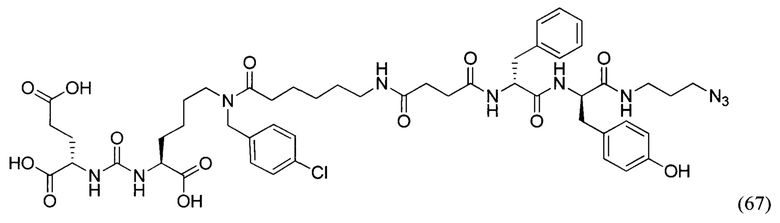
35 мг (28,77 мкмоль) соединения (46) растворили в 3 мл ДХМ и добавили 330 мкл трифторуксусной кислоты. Смесь перемешивали 13 ч. Далее удалили растворитель при пониженном давлении и трижды переупарили с ДХМ. Продукт осадили эфиром и трижды промыли 10 мл диэтилового эфира.
Таким образом, соединение (67) было выделено в виде белого аморфного порошка, выход составил 27 мг (90%)
Спектр ЯМР 1H (400 МГц, ДМСО-d6, δ, м.д.):
12.35 (уш. с, 3Н, СООН), 9.22 (д, J=17 Гц, 1Н, NH), 8.81-8.74 (м, 1Н, NH), 8.21-8.09 (м, 2Н, NH), 7.79 (м, 1Н, NH), 7.41 (д, J=8.2 Гц, 1Н, Ar), 7.35 (д, J=8.2 Гц, 1Н, Ar), 7.29-7.27 (m, 1Н, Ar), 7.23-7.20 (м, 5Н, Ar), 6.99 (д, J=7.6 Гц, 2Н, Ar), 6.66-6.62 (м, 1Н, Ar), 6.35-6.29 (м, 2Н, Ar), 4.53 (с, 1Н, CH2(Bz)), 4.46 (с, 1Н, CH2(Bz)), 4.41-4.39(м, 1Н, СН), 4.09-4.03 (м, 3Н, СН), 3.21-3.14 (м, 4Н, CH2), 3.04-2.95 (м, 5Н, CH2), 2.63 -2.54 (м, 1Н, CH2), 2.49 (м, 6Н, CH2), 2.40-2.38 (м, 2Н, CH2), 2.35-2.32 (м, 1Н, CH2), 2.29-2.20 (м, 4Н, CH2), 1.90-1.92 (м, 1Н, CH2), 1.72-1.67 (м, 1H, CH2), 1.60-1.57 (м, 2Н, CH2), 1.55-1.44 (м, 4Н, CH2), 1.40-1.436 (м, 2Н, CH2), 1.30-1.15 (м, 5Н, CH2)
ИК-спектр (ZnSe, см-1): 3305.97 (NH), 3159.79 (С-Нвал), 2934.16 (Ar), 2098.17 (N3), 1702.84 (C(O)), 1649.20 (С(O) (мочевина)), 1536.51 (С(O)), 1200.47 (Ar)
ESI-HRMS для C50H65ClN10O13: м/z рассчитано для [М+Н]+ 1051.4464, найдено 1051.4419.
(3S,7S,25R,28R)-33-азидо-25-бензил-12-(4-хлорбензил)-28-(4-гидроксибензил)-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатритриаконтан-1,3,7-трикарбоксилат (68)
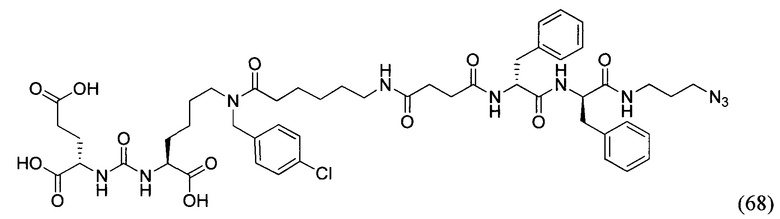
35 мг (29.15 мкмоль) соединения (47) растворили в 3 мл ДХМ и добавили 330 мкл трифторуксусной кислоты. Смесь перемешивали 13 ч. Далее удалили растворитель при пониженном давлении и трижды переупарили с ДХМ. Продукт осадили эфиром и трижды промыли 10 мл диэтилового эфира.
Таким образом, соединение (68) было выделено в виде белого аморфного порошка, выход составил 23 мг (76%).
Спектр ЯМР 1H (400 МГц, ДМСО-д6, δ, м.д.):
12.46 (уш.с., 2Н, СООН), 8.29-8.34 (м, 1H, NH), 8.07-8.14 (м, 2Н, NH), 7.92 (м, 1H, NH), 7.78 (м, 1H, NH), 7.58 (м, 1H, NH), 7.39 (м, 1H, Ar), 7.33 (м, 1H, Ar), 7.21 (м, 10Н, Ar), 7.08 (м, 1H, Ar), 6.28-6.31 (м, 1H, Ar), 4.39-4.51 (м, 4Н, СН + CH2(Ar)), 4.08 (м, 3Н, СН), 3.74 (м, 8Н, CH2), 3.14-3.24 (м, 5Н, CH2), 2.97-3.06 (м, 5Н, CH2), 2.65-2.76 (м, 2Н, CH2), 2.21-2.31 (м, 6Н, CH2), 1.91 (м, 1H, CH2), 1.49-1.70 (м, 8Н, CH2), 1.22 (м, 4Н, CH2) (Рисунок 1.276)
ИК-спектр (ZnSe, см-1): 3311 (NH), 3064 (С-Нвал), 2935 (Ar), 2100 (N3), 1725 (C(O)), 1644 (С(O) (мочевина)), 1554 (С(O)), 1201 (Ar)
ESI-HRMS для C50H65ClN10O12: m/z рассчитано для [М+Н]+ 1035.4515, найдено 1035.4554
(3S,7S,25S,28S)-33-азидо-25,28-дибензил-12-(3-хлорбензил)-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатритриаконтан-1,3,7-трикарбоксилат (69)
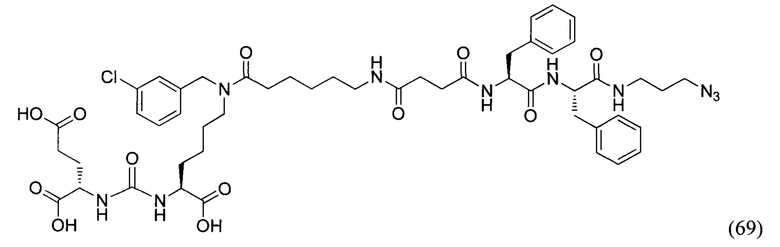
К раствору 77 мг (0,064 ммоль) соединения (48) в 2,7 мл ДХМ добавили 300 мкл трифторуксусной кислоты. Смесь перемешивали 16 ч. Далее удалили растворитель при пониженном давлении и переупарили с ДХМ трижды. Продукт высадили эфиром. В результате было выделено 56 (0,054 ммоль) мг целевого соединения, выход 84%.
Спектр ЯМР 1Н (400 МГц, ДМСО-d6, δ, м.д.):
8.45-8.25 (м, 1Н, C(O)NH), 8.21-8.05 (м, 1Н, C(O)NH), 8,00-7.85 (м, 1Н, C(O)NH), 7.83-7.50 (м, 1Н, C(O)NH), 7.40-7.00 (м, 14Н, Ar), 6.39-6.21 (с, 2Н, NH(мочевина)), 4.58-4.40 (м, 2Н, PhCH2), 4.44-4.22 (м, 2Н, СН), 4.15-3.98 (м, 2Н, СН), 3.29-2.83 (м, 11Н, CH2), 2.82-2.51 (м, 2Н, CH2), 2.41-2.08(м, 7Н, CH2), -1.98-1.84 (м, 1Н, CH2), 1.76-0.93 (м, 17Н, CH2)
Спектр ЯМР 13С (90 МГц, ДМСО-d6, δ, м.д.):
174.91 (C(O)NH), 174.62 (C(O)NH), 174.18 (C(O)NH), 138.42 (Ar), 133.48 (Ar), 130.67 (Ar), 129.56 (Ar), 129.45 (Ar), 128.61 (Ar), 128.50 (Ar), 128.42 (Ar), 127.62 (Ar), 127.62 (Ar), 127.26 (Ar), 126.74 (Ar), 126.51 (Ar), 85.05 (СН), 54.76 (СН), 52.08 (СН), 48.57 (СН), 45.79 (СН), 36.29 (CH2), 32.30 (CH2), 31.13 (CH2), 30.33 (CH2), 28.58 (CH2), 27.98 (CH2), 26.67 (CH2), 13.03 (CH2)
ИК спектр (ZnSe, см-1): 3311 (NH), 3087 (С-Н вал), 2935 (Ar), 2100 (N3), 1727 (С(O)), 1645 (С(O) (мочевина)), 1556 (С(O)), 1292 (Ar)
ESI-HRMS для C50H65ClN10O12: m/z рассчитано для [M+H]+ 1035.4983, найдено: 1035.4515.
52(3S,7S,25S,28S)-33-азидо-25-бензил-12-(3-хлорбензил)-28-(4-гидроксобензил)-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатритриаконтан-1,3,7-трикарбоксилат (70)
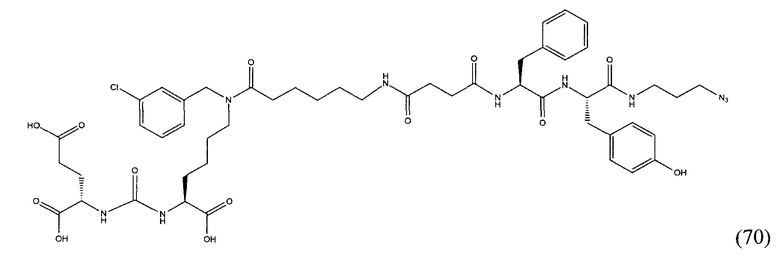
В 4 мл 10% (по объему) раствора трифторуксусной кислоты в DCM растворили соединение (49) (109 мг 0.089 ммоль) и перемешивали 16 часов при комнатной температуре. Растворитель упарили при пониженном давлении. Добавили 4 мл DCM с последующим упариванием для удаления трифторуксусной кислоты, процедуру повторили 5 раз. К полученному маслянистому осадку добавили 5 мл диэтилового эфира, в результате чего выделился белый твердый продукт. Эфир декантировали, процедуру повторили еще 2 раза.
Таким образом, соединение (70) было выделено в виде белого осадка, выход составил 75 мг (80%).
Спектр ЯМР 1H (400 МГц, ДМСО-d6, δ, м.д.):
12.33 (уш.с, 3Н, С(О)ОН), 8.28 (с, 1Н, C(O)NH), 8.07 (с, 1Н, C(O)NH), 7.89 (с, 1Н, C(O)NH), 7.78-7.55 (м, 2Н, C(O)NH + ArH), 7.30-6.99 (м, 13Н, C(O)NH + ArH), 6.67-6.62 (м, 2Н, C(O)NH + ArH), 6.29 (м, 2Н, NH(мочевина)), 4.54 (с, 1Н, PhCH2NC(О)), 4.46 (с, 1Н, PhCH2NC(O)), 4.08 (м, 2Н, СН, СН), 3.62-3.37 (м, 4Н, CH2Ar), 3.51 (м, 9Н, CH2), 3.26-2.93 (м, 2Н, CH2Ar), 3.26-2.99 (м, 16Н, CH2), 2.22 (м, 8Н, CH2), 1.90 (с, 1Н, CH2), 1.24 (м, 18Н, CH2CH)
Спектр ЯМР 13С (101 МГц, ДМСО-d6, м.д.):
175.27 (С(О)ОН), 174.98 (C(O)NR2), 174.56 (C(O)NR2), 171.85 (PhCH2NC(O)), 172.98 (PhCH2NC(O)), 172.37 (PhCH2NC(O)), 171.87 (PhCH2NC(O)), 158.09 (NHC(O)NH), 138.82 (Ar), 131.39 (Ar), 131.04 (Ar), 130.90 (Ar), 130.75 (Ar), 129.96 (Ar), 129.83 (Ar), 128.87 (Ar), 128.79 (Ar), 127.99 (Ar), 127.63 (Ar), 125.08 (Ar), 126.87 (Ar), 115.77 (Ar), 54.41 (СН), 52.46 (СН), 49.04 (CH2NR2), 48.96 (CH2Ar), 47.76 (CH2Ar), 42.67 (CH2), 36.65 (CH2), 32.67 (CH2), 30.72 (CH2), 29.90 (CH2), 29.02 (CH2), 28.59 (CH2), 28.36 (CH2), 27.04 (CH2), 25.53 (CH2), 18.89 (CH2), 17.53 (CH2), 13.30 (CH2)
ИК спектр (ZnSe, см-1): 3314 (NH), 2932 (Ar), 2099 (N3), 1642 (С(O) (мочевина), 1291 (Ar), 699 (CH2)
ESI-HRMS для C50H65ClN10O13: m/z рассчитано для [М+Н]+ 1050.4527, найдено: 1050.4583.
(3S,7S,25R,28R)-33-азидо-25-бензил-12-(3-хлорбензил)-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатритриаконтат-1,3,7-трикарбоксилат (71)
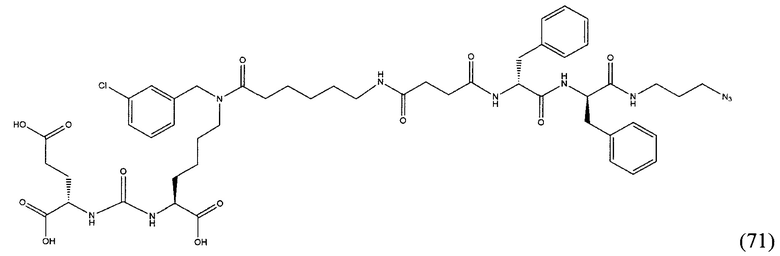
В 4 мл 10% (по объему) раствора трифторуксусной кислоты в DCM растворили соединение (50) (66 мг 0.055 ммоль) и перемешивали 16 часов при комнатной температуре. Растворитель упарили при пониженном давлении. Добавили 4 мл DCM с последующим упариванием для удаления трифторуксусной кислоты, процедуру повторили 5 раз. К полученному маслянистому осадку добавили 5 мл диэтилового эфира, в результате чего выделился белый твердый продукт. Эфир декантировали, процедуру повторили еще 2 раза.
Таким образом, соединение (71) было выделено в виде белого осадка, выход составил 52 мг.(70%).
Спектр ЯМР 1Н (400 МГц, ДМСО-d6, δ, м.д.):
12.44 (уш.с, 3Н, СООН), 8.78 (с, 1Н, C(O)NH), 8.33 (с, 3Н, C(O)NH + Ar), 7.78 (с, 1Н, C(O)NH), 7.18 (м, 14Н, C(O)NH + ArH), 6.27 (м, 1Н, NH(мочевина)), 4.51-4.35 (м, 4Н, PhCH2NC(O)), 4.05 (м, 2Н, СН, СН), 3.19-3.13 (м, 4Н, CH2Ar), 3.10-2.78 (м, 2Н, CH2Ar), 2.30 (м, 4Н, CH2), 2.19 (м, 4Н, CH2), 1.87 (с, 2Н, CH2), 1.47 (м, 9Н, CH2), 1.21 (м, 6Н, CH2CH).
Спектр ЯМР 13С (101 МГц, ДМСО-d6, м.д.):
186.35 (С(О)ОН), 174.97 (C(O)NR2), 174.54 (C(O)NR2), 171.84 (C(O)NR2), 171.84 (PhCH2NC(O)), 169.59 (PhCH2NC(O)), 167.01 (PhCH2NC(O)), 158.07 (NHC(O)NH), 138.81 (Ar), 131.01 (Ar), 129.97 (Ar), 129.92 (Ar), 129.80 (Ar), 128.96 (Ar), 128.87 (Ar), 128.79 (Ar), 127.99 (Ar), 127.99 (Ar), 127.62 (Ar), 127.09 (Ar), 126.87 (Ar), 55.14 (СН), 52.48 (СН), 49.03 (CH2NR2), 49.00 (CH2Ar), 48.94 (CH2Ar), 36.70 (CH2), 36.68 (CH2), 36.32 (CH2), 33.10 (CH2), 32.61 (CH2), 31.51 (CH2), 31.46 (CH2), 31.43 (CH2), 30.72 (CH2), 30.56 (CH2), 29.89 (CH2), 29.56 (CH2), 29.10 (CH2), 29.01 (CH2), 28.95 (CH2), 28.60 (CH2), 28.37 (CH2), 27.02 (CH2), 25.52 (CH2), 23.12 (CH2).
ИК спектр (ZnSe, см-1): 3315.03 (NH), 2933.20 (Ar), 2100.10 (N3), 1643.05 (C(O) (мочевина)), 1292.07 (Ar), 700.03 (CH2)
ESI-LCMS для C50H65ClN10O12: m/z рассчитано для [М+Н]+1033.45, найдено: 1033.35
(3S,7S,25R,28R)-33-азидо-25-бензил-12-(3-хлорбензил)-28-(4-гидроксобензил)-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатритриаконтан-1,3,7-трикарбоксилат (72)
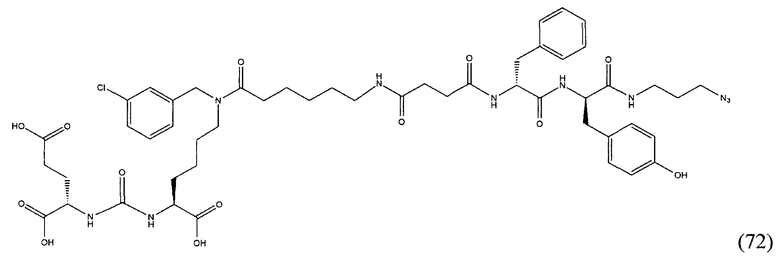
В 4 мл 10% (по объему) раствора трифторуксусной кислоты в DCM растворили соединение (51) (72 мг 0.089 ммоль) и перемешивали 16 часов при комнатной температуре. Растворитель упарили при пониженном давлении. Добавили 4 мл DCM с последующим упариванием для удаления трифторуксусной кислоты, процедуру повторили 5 раз. К полученному маслянистому осадку добавили 5 мл диэтилового эфира, в результате чего выделился белый твердый продукт. Эфир декантировали, процедуру повторили еще 2 раза.
Таким образом, соединение (72) было выделено в виде белого осадка, выход составил 52 мг.(84%).
Спектр ЯМР 1Н (400 МГц, ДМСО-d6, δ, м.д.):
12.44 (уш.с, 3Н, СООН), 8.26 (с, 1Н, C(O)NH), 8.06 (с, 1Н, C(O)NH), 7.87 (с, 1Н, C(O)NH), 7.26-6.97 (м, 16Н, C(O)NH + ArH), 6.60 (м, 1Н, C(O)NH + ArH), 6.27 (м, 1Н, NH(мочевина)), 4.44-4.28 (м, 3Н, PhCH2NC(O)), 4.05 (м, 2Н, СН, СН), 3.50 (м, 4Н, CH2Ar), 3.10-2.78 (м, 2Н, CH2Ar), 2.19 (м, 4Н, CH2), 1.87 (с, 1Н, CH2), 1.47 (м, 5Н, CH2), 1.21 (м, 17Н, CH2CH)
ИК спектр (ZnSe, см-1): 3313 (NH), 3070 (С-Нвал), 2938 (Ar), 2100.10 (N3), 1646 (С(O) (мочевина)), 1261 (Ar), 700 (CH2).
ESI-HRMS для C50H65ClN10O13: m/z рассчитано для [М+Н]+ 1049.4494, найдено: 1049.4498.
(3S,7S,25R,28R)-33-азидо-25-бензил-12-(3-хлорбензил)-28-(3-бром-4-гидроксобензил)-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатритриаконтан-1,3,7-трикарбоксилат (73)
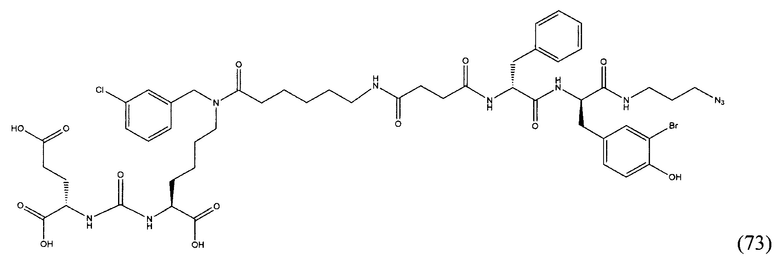
В 4 мл 10% (по объему) раствора трифторуксусной кислоты в DCM растворили вещество (52) (80 мг 0.061 ммоль) и перемешивали 16 часов при комнатной температуре. Растворитель упарили при пониженном давлении. Добавили 4 мл DCM с последующим упариванием для удаления трифторуксусной кислоты, процедуру повторили 5 раз. К полученному маслянистому осадку добавили 5 мл диэтилового эфира, в результате чего выделился белый твердый продукт. Эфир декантировали, процедуру повторили еще 2 раза.
Таким образом, соединение (73) было выделено в виде белого осадка, выход составил 53 мг.(77%).
Спектр ЯМР 1H (400 МГц, ДМСО-d6, δ, м.д.)
12.40 (уш.с, 3Н, С(О)ОН), 8.38 (с, 1Н, C(O)NH), 8.07 (с, 3Н, C(O)NH+Ar), 7.93 (с, 1Н, C(O)NH), 7.34-7.09 (м, 14Н, C(O)NH + ArH), 6.29-6.26 (м, 1Н, NH(мочевина)), 4.54-4.31 (м, 4Н, PhCH2NC(O)), 4.08-4.05 (м, 2Н, СН, СН), 3.59-3.28 (м, 4Н, CH2Ar), 3.17-2.71 (м, 2Н, CH2Ar), 2.21-2.07 (м, 8Н, CH2), 1.62-1.49 (с, 2Н, CH2), 1.29-1.24 (м, 9Н, CH2).
Спектр ЯМР 13С (101 МГц, ДМСО-d6, м.д.):
207.18 (С(О)ОН), 203.53 (С(О)ОН), 198.85 (С(О)ОН), (C(O)NR2), 175.27 (C(O)NR2), 174.98 (С(О)NR2), 174.55 (PhCH2NC(O)), 172.97 (PhCH2NC(O)), 171.67 (PhCH2NC(O)), 158.07 (NHC(O)NH), 139.80 (Ar), 138.74 (Ar), 137.90 (Ar), 131.03 (Ar), 130.91 (Ar), 129.96 (Ar), 129.83 (Ar), 128.87 (Ar), 128.74 (Ar), 127.99 (Ar), 127.63 (Ar), 126.94 (Ar), 126.67 (Ar), 55.05 (СН), 52.46 (СН), 48.96 (CH2NR2), 36.70 (CH2), 30.72 (CH2), 29.91 (CH2), 29.05 (CH2), 28.35 (CH2), 26.96 (CH2), 25.38 (CH2), 24.80 (CH2), 24.01 (CH2), 23.18 (CH2), 23.12 (CH2), 18.78 (CH2), 18.09 (CH2), 17.55 (CH2).
ИК спектр (ZnSe, см-1): 3311.18 (NH), 2935.13 (Ar), 2100.10 (N3), 1643.05 (C(O)NH), 1292.01 (Ar), 700.03 (CH2)
ESI-HRMS для C50H65ClBrN10O13: m/z рассчитано для [М+Н]+ 1131.3690, найдено: 1131.3624; m/z рассчитано для [M+Na]+ 1152.3476, найдено: 1152.3442.
(3S,7S,25S,28S)-33-азидо-25,28-дибензил-12-(4-хлор-2-фторбензил)-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатриаконтан-1,3,7-трикарбоксилат (74)
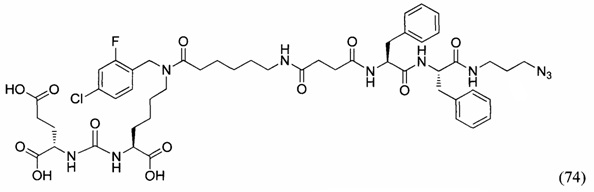
К раствору 70 мг (0,059 ммоль) соединения (53) в 2,7 мл ДХМ добавили 300 мкл трифторуксусной кислоты. Смесь перемешивали 16 ч. Далее удалили растворитель при пониженном давлении и переупарили с ДХМ трижды. Продукт высадили эфиром. В результате было выделено 58 мг (0,055 ммоль) мг целевого соединения, выход 93%.
Спектр ЯМР 1H (400 МГц, ДМСО-d6, δ, м.д.):
8.31 (d, 1Н, J=7.1 Гц, C(O)NH), 8.18 (d, 1H, J=8.3 Гц, C(O)NH), 7,96-7.90 (м, 1Н, C(O)NH), 7,64-7.54 (м, 1Н, C(O)NH), 7,45-7.35 (м, 1Н, Ar), 7.30-7.10 (м, 12Н, Ar), 6,36-6.20 (м, 2H, NH(мочевина)), 4.58-4.43 (м, 2H, ArCH2), 4.42-4.25 (м, 2Н, СН), 4.12-3.95 (м, 2Н, СН), 3.37-2.83 (м, 12Н, CH2), 2.74-2.58 (м, 1Н, CH2), 2.41-2.08 (м, 8Н, CH2), 1.98-1.85 (м, 1Н, CH2), 1.76-1.10 (м, 15Н, CH2)
Спектр ЯМР 13С (90 МГц, ДМСО-d6, δ, м.д.):
176.81 (С(О)ОН), 174.95 (С(О)ОН), 174.61 (C(O)NH), 174.19 (C(O)NH), 173.27 (C(O)NH), 172.02 (C(O)NH), 171.55 (C(O)NH), 171.14 (C(O)NH), 157.70 (C(O) (мочевина)), 138.50 (Ar), 138.42 (Ar), 129.45 (Ar), 128.61 (Ar), 128.50 (Ar), 126.75 (Ar), 126.68 (Ar), 55.36 (СН), 52.59 (СН), 48.57 (СН), 36.29 (CH2), 32.22 (CH2), 31.06 (CH2), 30.34 (CH2), 29.45 (CH2), 28.57 (CH2), 26.65 (CH2), 25.05 (CH2)
ИК спектр (ZnSe, см-1): 3313 (NH), 3084 (С-Нвал), 2932 (Ar), 2098 (N3), 1726 (C(O)), 1641 (С(O) (мочевина)), 1549 (С(O)), 1290 (Ar)
ESI-HRMS для C50H64ClFN10O12: m/z рассчитано для [M+H]+ 1052.4484, найдено: 1052.4484
(3S,7S,25S,28S)-33-азидо-25,28-дибензил-12-(4-хлор-3-фторбензил)-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатриаконтан-1,3,7-трикарбоксилат (75)
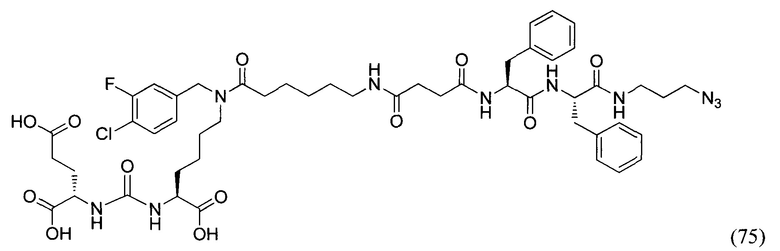
К раствору 80 мг (0,065 ммоль) соединения (54) в 2,7 мл ДХМ добавили 300 мкл трифторуксусной кислоты. Смесь перемешивали 16 ч. Далее удалили растворитель при пониженном давлении и переупарили с ДХМ трижды. Продукт высадили эфиром. В результате было выделено 65 мг (0,062 ммоль) мг целевого соединения, выход 95%.
Спектр ЯМР 1Н (400 МГц, ДМСО-d6, δ, м.д.):
12.43 (уш.с, 3Н, СООН), 8.40-8.27 (m, 1Н, C(O)NH), 8.22-8.07 (m, 1H, C(O)NH), 7.97-7.89 (м, 1H, C(O)NH), 7.84-7.73 (м, 1H, C(O)NH), 7,62-7.52 (м, 1H, Ar), 7.52-7.45 (м, 1H, Ar), 7.30-6.97 (м, 11Н, Ar), 6,36-6.22 (м, 2Н, NH(мочевина)), 4.58-4.43 (м, 2Н, ArCH2), 4.43-4.20 (м, 2Н, СН), 4.12-3.95 (м, 2Н, СН), 3.30-2.82 (м, 10Н, CH2), 2.83-2.70 (м, 1H, CH2), 2.70-2.52 (м, 1H, CH2), 2.41-2.08 (м, 7Н, CH2), 1.98-1.85 (м, 1H, CH2), 1.80-1.11 (м, 16Н, CH2)
Спектр ЯМР 13С (90 МГц, ДМСО-d6, δ, м.д.):
174.92 (C(O)NH), 174.62 (C(O)NH), 174.19 (C(O)NH), 173.28 (C(O)NH), 172.66 (C(O)NH), 172.31 (C(O)NH), 172.03 (C(O)NH), 171.55 (C(O)OH), 171.48 (С(О)ОН), 171.32 (С(О)ОН), 171.14 (С(О)ОН), 157.71 (С(O) (мочевина)), 156.33 (С(O) (мочевина)), 138.50 (Ar), 138.42 (Ar), 131.30 (Ar), 130.91 (Ar), 129.62 (Ar), 129.56 (Ar), 129.45 (Ar), 128.61 (Ar), 128.50 (Ar), 128.42 (Ar), 126.74 (Ar), 126.68 (Ar), 125.05 (Ar), 125.02 (Ar), 55.36 (СН), 54.76 (СН), 52.53 (СН), 52.09 (СН), 48.65 (СН), 48.58 (СН), 47.39 (СН), 36.29 (CH2), 32.29 (CH2), 32.23 (CH2), 31.03 (CH2), 30.34 (CH2), 29.47 (CH2), 28.62 (CH2), 28.22 (CH2), 27.98 (CH2), 26.68 (CH2), 25.10 (CH2), 24.99 (CH2), 22.91 (CH2), 22.74 (CH2), 15.63 (СН2)
ИК спектр (ZnSe, см-1): 3311 (NH), 3086 (С-Нвал), 2934 (Ar), 2099 (N3), 1727 (C(O)), 1642 (С(O) (мочевина)), 1554 (С(O)), 1291 (Ar)
ESI-HRMS для C50H64ClFN10O12: m/z рассчитано для [М+Н]+ 1054.4499, найдено: 1054.4503; m/z рассчитано для [М+K]+ 1091.4121, найдено: 1091.4088
(3S,7S,25R,28R)-33-азидо-25,28-дибензил-12-(4-хлор-3-фторбензил)-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатриаконтан-1,3,7-трикарбоксилат (76)
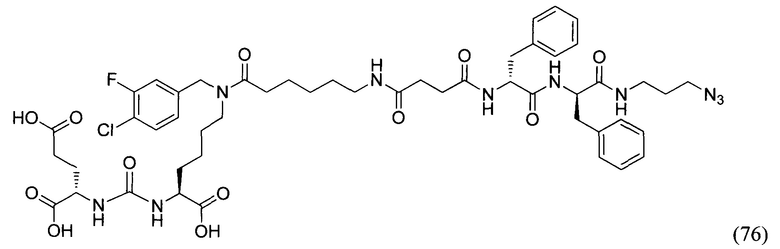
К раствору 217 мг (0,178 ммоль) соединения (55) в 2,7 мл ДХМ добавили 300 мкл трифторуксусной кислоты. Смесь перемешивали 16 ч. Далее удалили растворитель при пониженном давлении и переупарили с ДХМ трижды. Продукт высадили эфиром. В результате было выделено 170 мг (0,161 ммоль) мг целевого соединения, выход 90%.
Спектр ЯМР 1H (400 МГц, ДМСО-d6, δ, м.д.):
12.44 (уш.с, 3Н, СООН), 8.41-8.27 (м, 1Н, C(O)NH), 8.22-8.05 (м, 1Н, C(O)NH), 7.99-7.89 (м, 1Н, C(O)NH), 7.84-7.73 (м, 1Н, C(O)NH), 7.64-7.52 (м, 1Н, Ar), 7.52-7.43 (м, 1Н, Ar), 7.30-6.97 (м, 11Н, Ar), 6.37-6.22 (м, 2Н, NH(мочевина)), 4.58-4.43 (м, 2Н, ArCH2), 4.43-4.21 (м, 2Н, СН), 4.13-3.96 (м, 2Н, СН), 3.30-2.82 (м, 10Н, CH2), 2.80-2.52 (м, 2Н, CH2), 2.41-2.08 (м, 7Н, CH2), 1.98-1.80 (м, 1Н, CH2), 1.79-1.10 (м, 16Н, CH2)
Спектр ЯМР 13С (90 МГц, ДМСО-d6, δ, м.д.):
177.46 (С(О)ОН), 174.92 (С(О)ОН), 174.63 (C(O)NH), 174.19 (C(O)NH), 172.01 (C(O)NH), 171.48 (C(O)NH), 171.13 (C(O)NH), 157.69 (C(O) (мочевина)), 138.42 (Ar), 130.92 (Ar), 129.62 (Ar), 129.57 (Ar), 129.45 (Ar), 128.61 (Ar), 128.50 (Ar), 128.42 (Ar), 126.74 (Ar), 125.05 (Ar), 52.07 (СН), 48.64 (СН), 48.57 (СН), 47.34 (СН), 36.29 (CH2), 31.07 (CH2), 30.34 (CH2), 28.62 (CH2), 27.97 (CH2), 25.09 (CH2), 22.74 (CH2), 20.98 (CH2)
ИК спектр (ZnSe, см-1): 3314 (NH), 3084 (С-Нвал), 2932 (Ar), 2099 (N3), 1727 (C(O)), 1642 (С(O) (мочевина)), 1549 (С(O)), 1291 (Ar)
ESI-HRMS для C50H64ClFN10O12: m/z рассчитано для [M+H]+1051.4450, найдено: 1051.4406
(3S,7S,25S,28S)-33-азидо-25-бензил-12-(4-хлор-3-фторбензил)-28-(4-гидроксибензил)-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатриаконтан-1,3,7-трикарбоксилат (77)
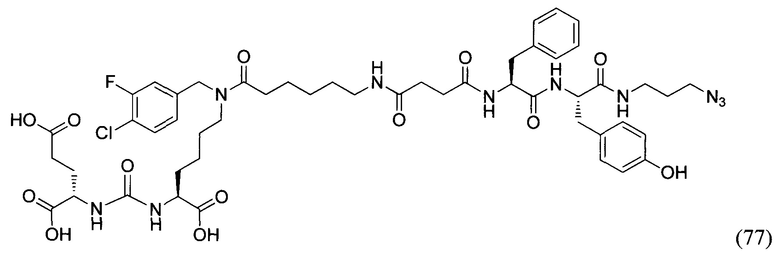
К раствору 63 мг (0,051 ммоль) соединения (56) в 2,7 мл ДХМ добавили 300 мкл трифторуксусной кислоты. Смесь перемешивали 16 ч. Далее удалили растворитель при пониженном давлении и переупарили с ДХМ трижды. Продукт высадили эфиром. В результате было выделено 43 мг (0,040 ммоль) мг целевого соединения, выход 78%.
Спектр ЯМР 1Н (400 МГц, ДМСО-d6, δ, м.д.):
8.41-8.25 (м, 1Н, C(O)NH), 8.21-8.02 (м, 1Н, C(O)NH), 7.99-7.87 (м, 1Н, C(O)NH), 7.84-7.69 (м, 1Н, C(O)NH), 7.65-7.43 (м, 2Н, Ar), 7.30-6.90 (м, 10Н, Ar), 6.69-6.53 (м, 2Н, Ar), 6.42-6.19 (м, 2Н, NH(мочевина)), 4.58-4.40 (м, 2Н, ArCH2), 4.40-4.21 (м, 2Н, СН), 4.13-3.96 (м, 2Н, СН), 3.31-2.52 (м, 14Н, CH2), 2.41-1.98 (м, 8Н, CH2), 1.98-1.80 (м, 1Н, CH2), 1.79-1.10 (м, 17Н, CH2)
Спектр ЯМР 13С (90 МГц, ДМСО-d6, δ, м.д.):
174.91 (C(O)NH), 174.62 (C(O)NH), 174.18 (C(O)NH), 171.48 (C(O)NH), 171.30 (C(O)NH), 157.72 (C(O) (мочевина)), 156.26 (C(O) (мочевина)), 131.29 (Ar), 130.91 (Ar), 130.53 (Ar), 130.37 (Ar), 129.60 (Ar), 129.47 (Ar), 128.50 (Ar), 128.42 (Ar), 126.68 (Ar), 126.60 (Ar), 125.01 (Ar), 119.59 (Ar), 115.40 (Ar), 115.28 (Ar), 52.08 (СН), 48.67 (СН), 48.59 (СН), 47.40 (СН), 36.27 (CH2), 32.30 (CH2), 32.22 (CH2), 30.34 (CH2), 28.64 (CH2), 28.20 (CH2), 27.96 (CH2), 25.09 (CH2), 15.62 CH2)
ИК спектр (ZnSe, см-1): 3310 (NH), 3086 (С-Нвал), 2933 (Ar), 2098 (N3), 1726 (C(O)), 1643 (C(O) (мочевина)), 1555 (C(O)), 1290 (Ar)
ESI-HRMS для C50H64ClFN10O13: m/z рассчитано для [М+Н]+ 1070.4404, найдено: 1070.4406
(3S,7S,25R,28R)-33-азидо-25-бензил-12-(4-хлор-3-фторбензил)-28-(4-гидроксобензил)-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатритриаконтан-1,3,7-трикарбоксилат (78)
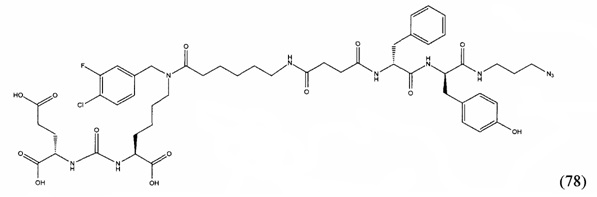
В 4 мл 10% (по объему) раствора трифторуксусной кислоты в DCM растворили соединение (57) (72 мг 0.058 ммоль) и перемешивали 16 часов при комнатной температуре. Растворитель упарили при пониженном давлении. Добавили 4 мл DCM с последующим упариванием для удаления трифторуксусной кислоты, процедуру повторили 5 раз. К полученному маслянистому осадку добавили 5 мл диэтилового эфира, в результате чего выделился белый твердый продукт. Эфир декантировали, процедуру повторили еще 2 раза. Масса полученного вещества 56 мг, выход 89%.
Спектр ЯМР 1H (400 МГц, ДМСО-d6, δ, м.д.):
12.42 (уш.с, 3Н, СООН), 9.15 (с, 1Н, C(O)NH), 8.28 (с, 1Н, C(O)NH), 8.09 (дд, 1Н, C(O)NH), 7.90-7.78 (м, 2Н, C(O)NH + ArH), 7.55-7.79 (м, 1Н, C(O)NH + ArH), 7.20-6.99 (м, 12Н, C(O)NH + ArH), 6.30 (м, 2Н, NH(мочевина)), 4.53 (с, 1Н, PhCH2NC(O)), 4.45 (с, 1Н, PhCH2NC(O)), 4.08 (м, 1Н, СН, СН), 3.51 (м, 9Н, CH2), 4.39-4.27 (м, 2Н, CH2Ar), 4.08 (м, 2Н, СН, СН), 3.51 (м, 9Н, CH2), 3.26-2.99 (м, 16Н, CH2), 2.22 (м, 8Н, CH2), 1.90 (с, 1Н, CH2), 1.24 (м, 18Н, CH2CH).
Спектр ЯМР 13С (101 МГц, ДМСО-d6, м.д.):
208.22 (СООН), 204.88 (СООН), 175.27 (C(O)NR2), 174.98 (C(O)NR2), 174.56 (C(O)NR2), 171.85 (PhCH2NC(O)), 171.68 (PhCH2NC(O)), 158.09 (NHC(O)NH), 138.80 (Ar), 131.66 (Ar), 131.28 (Ar), 130.90 (Ar), 130.74 (Ar), 130.40 (Ar), 129.96 (Ar), 128.83 (Ar), 129.31 (Ar), 128.87 (Ar), 128.79 (Ar), 125.37 (Ar), 116.60 (Ar), 55.72 (СН), 55.46 (СН), 49.04 (CH2NR2), 48.96 (CH2Ar), 47.76 (CH2Ar), 36.55 (CH2), 32.67 (CH2), 32.58 (CH2), 31.51 (CH2), 31.48 (CH2), 30.72 (CH2), 29.88 (CH2), 29.84 (CH2), 29.79 (CH2), 29.02 (CH2), 28.96 (CH2), 31.51 (CH2), 28.34 (CH2), 27.05 (CH2), 25.45 (CH2), 23.11 (CH2), 17.53 (CH2).
ИК спектр (ZnSe, см-1): 3312 (NH), 2938 (Ar), 2099 (N3), 1646 (C(O) (мочевина)), 1293 (Ar), 699 (CH2)
ESI-LCMS для C50H64ClFN10O13: m/z рассчитано для [М+Н]+ 1067.55, найдено: 1067.35
Примеры синтеза конъюгатов.
(3S,7S,25S,28S)-25-бензил-12-(3-хлорбензил)-33-(4-((3',6'-дигидрокси-3-оксо-3H-спиро[изобензофуран-1,9'-ксантен]-5-илкарбоксамидо)метил)-1Н-1,2,3-триазол-1-ил)-28-(4-гидроксибензил)-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатритриаконтан-1,3,7-трикарбоновая кислота (79).
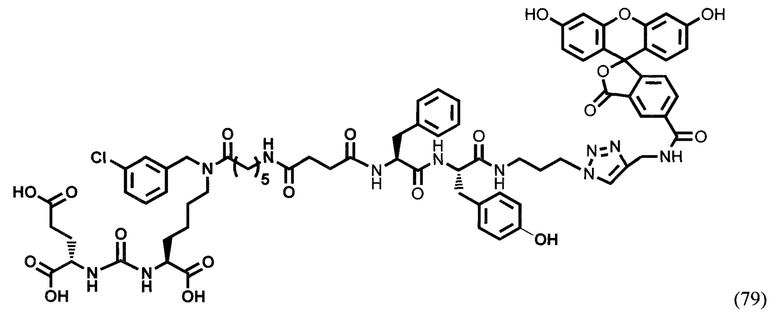
Соединение (70) (32 мг, 0,03 мммоль) растворяли в 5 мл ДМФА и 5 мл воды. Колбу заполняли аргоном. Далее добавляли Fam-5 алкин (12 мг, 0,033 мкмоль), аскорбат натрия (2 мг, 0,012 мкмоль) и пентагидрат сульфата меди (3 мг, 0,012 мкмоль). Смесь перемешивали 24 часа. Далее упаривали растворитель. Дальнейшую очистку проводили с помощью метода обращенной фазовой хроматографии: Puriflash 15C18HP-F0012, система вода/ацетонитрил, от 5% ацетонитрила до 100% ацетонитрила в течение 25 минут, далее 5 минут промывали метанолом, скорость потока - 20 мл/мин. Таким образом, соединение (79) было выделено в виде желтого аморфного порошка, выход составил 35 мг (80%).
Спектр ЯМР 1H (400 МГц, DMSO-d6, δ, м.д.):
12.44 (уш.с, 3Н, СООН), 10.17 (уш. с, 1Н, СОН), 9.38 (с, 1Н, C(O)NH), 8.48 (с, 1Н, C(O)NH), 8.30-8.24 (м, 2Н, C(O)NH) 8.10-8.02 (м, 3Н, C(O)NH), 7.77-7.65 (м, 2Н, C(O)NH + ArH), 7.65-7.64 (м, 2Н, C(O)NH + ArH), 7.35-7.30 (м, 5Н, C(O)NH + ArH), 7.18-7.15 (м, 3Н, C(O)NH + ArH), 4.67-4.54 (м, 5Н, C(O)NH + ArH), 4.31-4.29 (м, 2Н, NH(urea)), 4.55-4.45 (м, 4Н, PhCH2NC(O)), 4.28 (с, 3Н, PhCH2NC(O)), 4.07-4.05 (м, 3Н, СН, СН), 3.17-3.16 (м, 3Н, CH2), 3.00-2.98 (м, 6Н, CH2), 2.70-2.65 (м, 3Н, CH2), 2.23-2.19 (м, 8Н, CH2), 1.91-1.88 (м, 3Н, CH2), 1.49-1.10 (м, 11Н, CH2) (Рисунок 4.8).
ESI-LCMS для C74H80ClN11O19: m/z рассчитано для [М+Н]+ 1462.94; найдено: 1462.55
(3S,7S,25S,28S)-25,28-дибензил-12-(3-хлорбензил)-33-(4-((3',6'-дигидрокси-3-оксо-3Н-спиро[изобензофуран-1,9'-ксантен]-5-илкарбоксамидо)метил)-1Н-1,2,3-триазол-1-ил)-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатритриаконтан-1,3,7-трикарбоновая кислота (80)
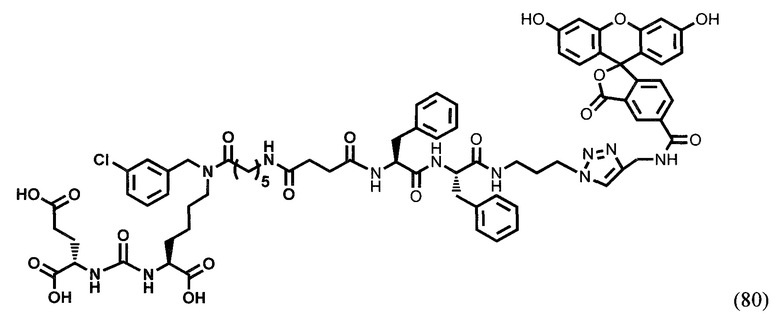
Соединение (74) (33 мг, 0,03 мммоль) растворяли в 5 мл ДМФА и 5 мл воды. Колбу заполняли аргоном. Далее добавляли Fam-5 алкин (12 мг, 0,03 мкмоль), аскорбат натрия (2 мг, 0,012 мкмоль) и пентагидрат сульфата меди (3 мг, 0,012 мкмоль). Смесь перемешивать 24 часа. Далее упарить растворитель. Дальнейшую очистку проводить с помощью метода обращенной фазовой хроматографии: Puriflash 15C18HP-F0012, система вода/ацетонитрил, от 5% ацетонитрила до 100% ацетонитрила в течение 25 минут, далее 5 минут промывка метанолом, скорость потока - 20 мл/мин. Таким образом, соединение (79) было выделено в виде желтого аморфного порошка, выход составил 34 мг (75%).
Образование конъюгата с лекарственным соединением - доцетакселем (DOCETAXEL). Образование конъюгата с заявляемьм средством представлено на схеме на фиг. 18 и 19.
(2aR,4S,4aS,6R,9S,11S,12S,12aR,12bS)-12b-ацетокси-9-(((2R,3S)-3-(трет-бутоксикарбонил)амино)-2-(гекс-5-иноилокси)-3-фенилпропанол)окси)-4,6,11-тригидрокси-4а,8,13,13-тетраметил-5-оксо-2а,3,4,4а,5,6,9,10,11,12,12а,12b-додесагидро-1Н-7,11-метаноциклодека[3,4]бензо[1,2-b]октет-12-ил бензоат (81)
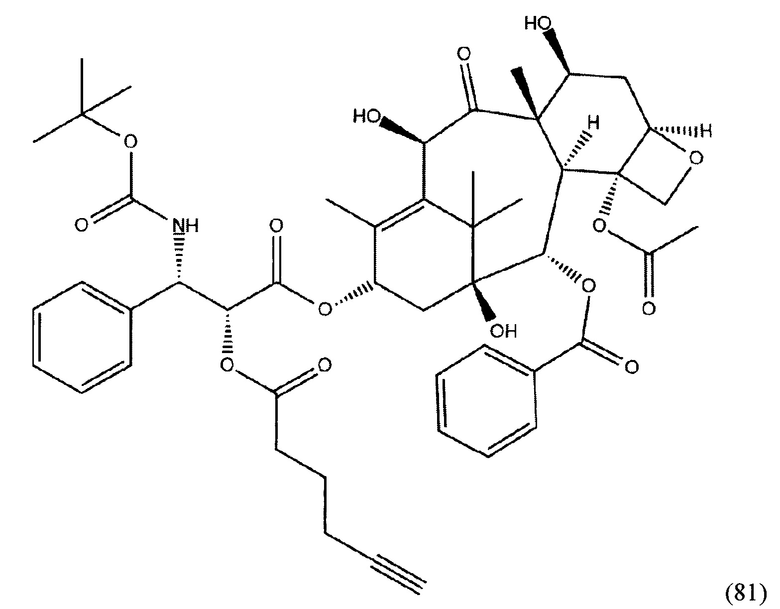
Раствор гексиновой кислоты (114 мг; 1.392 ммоль), доцетаксела (750 мг; 0.932 ммоль) и DMAP (5 мг; 0.044 ммоль) в 50 мл DCM охлаждают до 0°С на ледяной бане. После этого прикалывают DIC 215 мкл; 1.393 ммоль. Реакционную смесь выдерживают 2 часа при 0°С, затем оставляют перемешиваться при комнатной температуре на сутки. После чего упаривают. Очистку проводили методом колоночной хроматографии (система этилацетат : гексан, градиент от 5% до 100% этилацетата). В результате получили 487 мг целевого продукта (81), выход 38%.
(3S,7S,25R,28S)-33-(4-(4-(((2R,3S)-l-(((2aR,4S,4aS,6R,9S,11S,12S,12aR,12bS)-12b-ацетокси-12-(бензилокси)-4,6,11-тригидрокси-4а,8,13,13-тетраметил-5-оксо-2а,3,4,4а,5,6,9,10,11,12,12а,12b-додекагидро-1Н-7,11-метаноциклодека[3,4]бензол[1,2-b]оксет-9-ил)окси)-3-((трет-бутоксикарбонил)амино)-1-оксо-3-фенилпропан-2-ил)окси)-4-оксобутил)-1Н-1,2,3-триазол-1-ил)-12,25,28-трибензил-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатритриаконтан-1,3,7-трикарбоксилат (82)
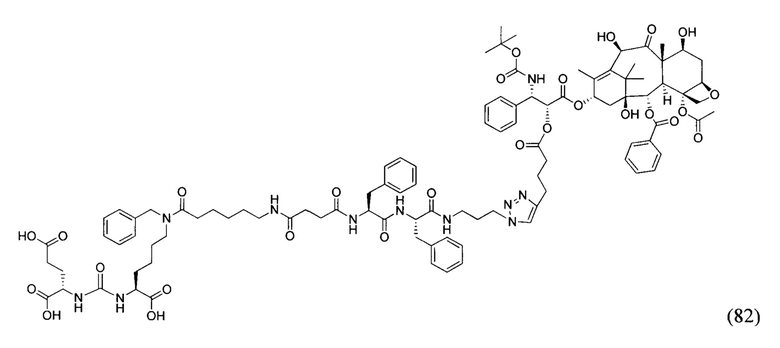
Модифицированный доцетаксел (81) (22 мг; 0.024 ммоль) растворили в 2 мл DMF, добавили соединение (59) (16 мг 0.016 ммоль) и 0,4 мл воды. Систему продули аргоном. Далее к расвору добавили раствор солей аскорбата натрия 2,5 мг 0.0128 ммоль и сульфата меди 1,5 мг; 0.0064. (строго в данной последовательности). Оставили на сутки при перемешивании. Раствор упарили при пониженном давлении. Полученную маслянистую фракцию далее очищали при помощи препаративной обращенно-фазовой хроматографии. Масса полученного вещества 11 мг, выход 90%.
Масс-спектр ESI-MS для C99H125N11O27
m/z рассчитано для [М+Н]+ 1901.11; найдено: 1902.00 (фиг. 15)
(3S,7S,25R,28S)-33-(4-(4-(((2R,3S)-1-(((2aR,4S,4aS,6R,9S,11S,12S,12aR,12bS)-12b-ацетокси-12-(бензилокси)-4,6,11-тригидрокси-4а,8,13,13-тетраметил-5-оксо-2а,3,4,4а,5,6,9,10,11,12,12а,12b-додекагидро-1Н-7,11-метаноциклодека[3,4]бензо[1,2-b]оксе-9-ил)окси)-3-((трет-бутоксикарбонил)амино)-1-оксо-3-фенилпропан-2-ил)оху)-4-оксобутил)-1Н-1,2,3-триазол-1-ил)-25,28-дибензил-12-(4-хлоро-3-фтор6ензил)-5,13,20,23,26,29-гексаоксо-4,6,12,19,24,27,30-гептаазатритриаконтан-1,3,7-трикарбоксилат (83)
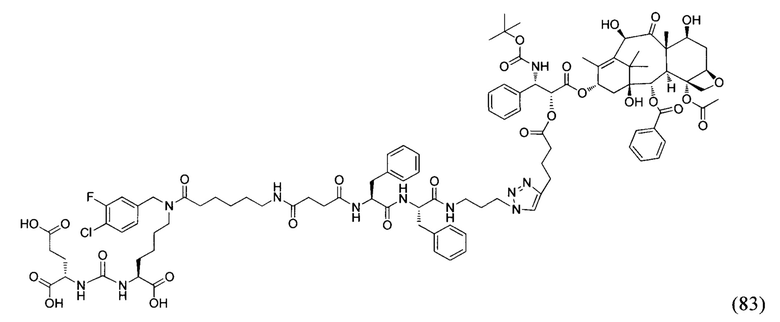
Модифицированный доцетаксел (81) (160 мг; 0.177 ммоль) растворили в 10 мл DMF, добавили соединение (75) (155 мг 0.147 ммоль) и 3 мл воды. Систему продули аргоном. Далее к раствору добавили раствор солей аскорбата натрия 23 мг и сульфата меди 15 мг. Оставили на сутки при перемешивании. Раствор упарили при пониженном давлении. Полученную маслянистую фракцию далее очищали при помощи препаративной обращенно-фазовой хроматографии. Масса полученного вещества 155 мг, выход 90%.
Спектр ЯМР 1Н (400 МГц, ДМСО-d6, δ, м.д.):
12.50 (с, 3Н, СООН), 7.00-8.51 (м, 2Н, NH+Ar), 6.23-6.40 (м, 2Н, NH), 5.70-5.88 (м, 2Н, СН), 5.38 (д, J=7.1 Гц, 1Н, CHOC(O)Ph), 4.95-5,18 (м, 4Н), 4.75-4.95 (м, 2Н), 4.15-4.65 (м, 7Н), 3.92-4.15 (м, 5Н), 3.61 (д, J=6.8 Гц, 1Н, R3CH), 2.85-3.23 (м, 9Н), 2.58-2.85 (м, 4Н, CH2), 2.13-2.41 (м, 12Н, CH2), 1.79-1.99 (м, 6Н, CH2), 1.58-1.75 (м, 6Н, CH2+СН3), 1.43-1.58 (м, 8Н, CH2+СН3), 1.28-1.42 (м, 11Н, CH2+СН3), 1.08-1.27 (м, 6Н, CH2+СН3), 0.96 (с, 6Н, СН3), (фиг. 16-17).
Масс-спектр ESI-MS для C99H123ClFN11O27
m/z рассчитано для [М+Н]+1952.83, найдено: 1952.90
Биологические испытания.
Использованные клеточные линии
LNCaP - андроген-чувствительная клеточная линия аденокарциномы человека, являются адгерентными эпителиальными клетками, растут в виде агрегатов и одиночных клеток. ПСМА-положительные.
Клетки линии LNCaP культивировались в среде RPMI с 10% FBS (Gibco), 1xGlutamax (Gibco) и 1x смесью пенициллин-стрептомицин (Gibco).
Получение ПСМА-содержащего клеточного экстракта.
Для суспендирования клеток с 25 см2 флакона убирали культуральную среду, промывали клетки буфером PBS и инкубировали 5 минут с 0,25% трипсином (1 мл). Трипсин инактивировали полной культуральной средой (2 мл), промывали PBS, переносили 106 клеток в пробирку и добавляли 500 мкл лизирующего буфера (0,5% Triton Х-100, 50 mM Tris-HCl (pH 7,5), 1x Proteinase Inhibition cocktail). Суспензию инкубировали 30 минут во льду и обрабатывали ультразвуком (7 раз по 7 сек с интервалами 20-30 сек) во льду во избежание перегрева. Центрифугировали 10 мин 1000g на 4°С и надосадочный раствор использовали в дальнейшем анализе.
Протокол проведения измерения активности ПСМА.
Анализ ингибирования препаратами ПСМА проводился по нерадиоактивному протоколу с детекцией высвобождаемого в ходе реакции глутамата. Экстракт клеток линии LNCaP (10 μL) смешивался с 2 μL препарата соответствующего разведения. Для первичного тестирования использовалась серия разведении препаратов 2 нМ-100 мкМ с шагом разведения 3-5 раз. К полученной смеси добавлялся 1 μL 100 μМ раствора NAAG. Полученная смесь инкубировалась 2 часа при 37°С. После инкубации смесь разводилась вдвое реакционным буфером (13 μL) из набора Amplex Red Glutamic Acid Kit (Molecular Probes Inc., Eugene) и добавлялась многокомпонентная реакционная смесь для детекции глутамата, приготовленная в соответствии с протоколом производителя (26 μl). Рабочий раствор Amplex Red: 5 мкл Amplex® Red reagent stock solution, 1,25 мкл HRP stock solution, 8 мкл L-glutamate oxidase, 2,5 мкл L-glutamate-pyruvate transaminase, 0,5 мкл L-alanine, 483 мкл 1X Reaction Buffer. Проводилась инкубация 1 час при 37°С. Флуоресценция резоруфина, полученного в результате сопряженных реакций глутамат-детектирующего набора детектировалась на планшетном мультидетекторе VICTORX5 (Perkin Elmer Inc.) при длине возбуждения 555 нм и детекции при 580 нм. В качестве контроля эндогенного уровня глутамата с заменой раствора NAAG на воду.
Оценка ингибирования
Препарат DCL трифторацетат (84) представляет собой известную модификацию вещества DUPA с повышенной растворимостью и является генетическим предшественником большинства протестированных препаратов. Его ингибирующее действие аналогично DUPA и он использовался как контрольный ингибитор в последующих анализах.
Ниже приведено описание ингибирования ПСМА рецептора разработанными соединениями. Чем меньше IC 50 при ингибировании, тем лучше аффинность.
Наибольшее ингибирующее действие показало соединение формулы (70) с линкером, содержащим фрагмент L-Phe-L-Tyr между ядром ингибитора и арильным фрагментом и производным 3-хлорбензальдегида при ε-атоме азота лизина. Также сильным ингибитором является соединение формулы (64) с линкером, содержащим фрагмент L-Phe-L-Tyr между ядром ингибитора и арильным фрагментом и производным 4-бромбензальдегида при ε-атоме азота лизина. В таблице 1 приведены концентрации половинного ингибирования IC50 (нМ) ПСМА протестированными препаратами.
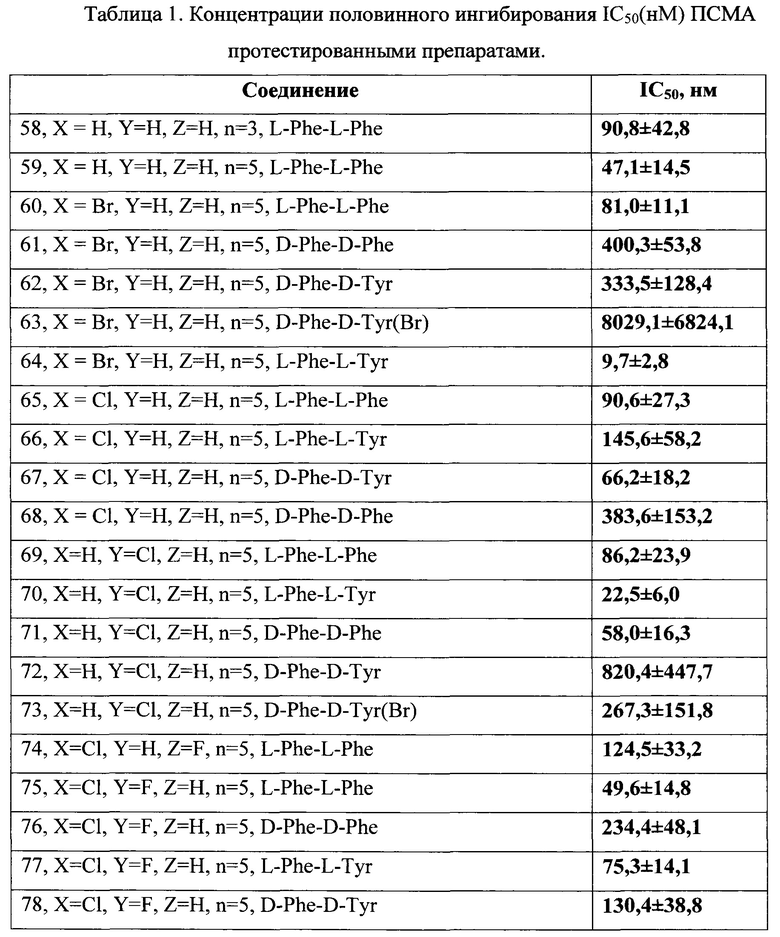
Соединения МА-256, МА-232 - соединения для сравнения, содержащие не модифицированный ПСМА лиганд DCL, и соединение (70) с одним ароматическим фрагментом при ε-NH2 группе, с двумя ароматическими фрагментами в линкере соответственно, формулы которых представлены ниже.
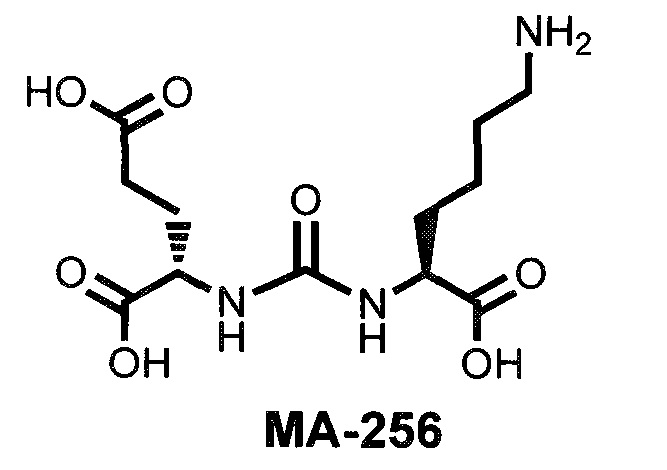
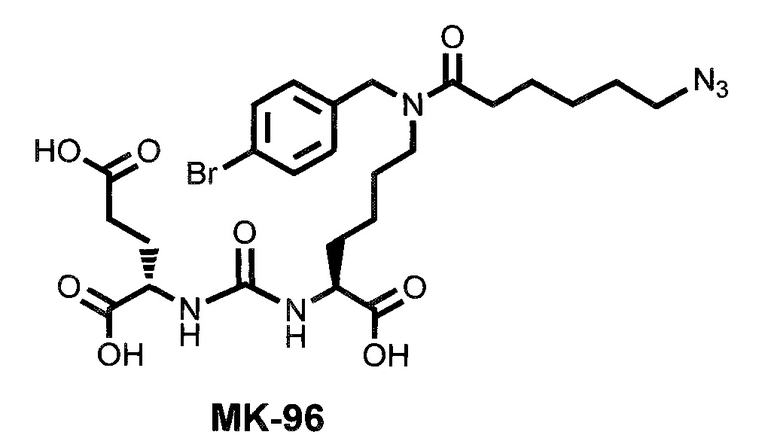
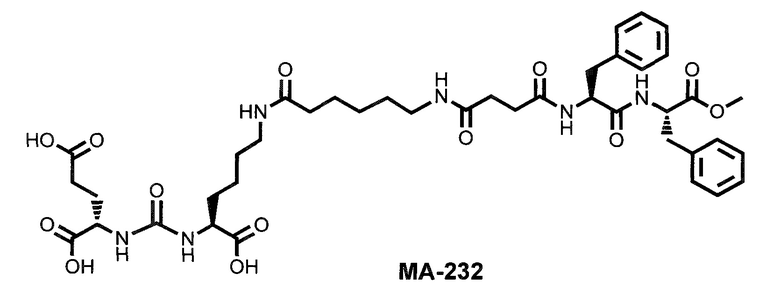
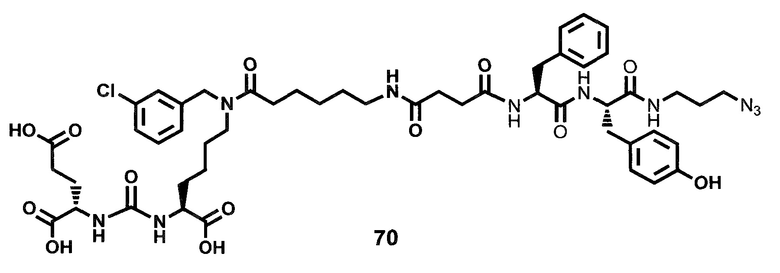
Определение эффективности заявляемого средства в экспериментах in vitro.
Оценка дифференциальной способности конъюгатов на основе заявляемого средства накапливаться в клетках в зависимости от представленности ПСМА на поверхности клеток. Интернализацию флуоресцентного лиганда в клетках детектировали, анализируя интенсивность флуоресцентного сигнала в клетках с использованием изображений, полученных системой визуализации EVOS FL Cell Imaging System с последующей обработкой сигнала в ImageJ. Была проведена оценка эффективности заявялемого средства, включающего флуоресцентную метку FAM и соответствующего структуре коньюгатов (79) и (80). Накопление сигнала флуоресценции, ассоциированного с поступлением меченого FAM конъюгата, проанализировано в трех клеточных линиях
Для проведения анализа использовали следующие реактивы: PBS (фосфатно-солевой буфер) (Gibco), Glutamax (Gibco), FBS (фетальная телячья сыворотка), RPMI 1640 (Gibco), Pen Strep (Gibco), витамины для среды RPMI (100×) (Sigma), Paraformaldehyde, 95% (Sigma Aldrich).
Выбраны три клеточные линии с разным уровнем экспрессии ПСМА на поверхности клеток LNCaP, 22Rv1, PC3. В соответствии с литературными данными наибольший уровень экспрессии ПСМА характерен для LNCaP, низкий уровень экспрессии ПСМА характерен для 22Rv1, PC3 клеточная линия не несет ПСМА, является ПСМА негативной. Использованные клеточные линии: LNCaP - андроген-чувствительная клеточная линия аденокарциномы человека, являются адгерентными эпителиальными клетками, растут в виде агрегатов и одиночных клеток (ПСМА-положительные). 22Rv1 -клеточная линия карциномы человека, являются адгерентными эпителиальными клетками (ПСМА-положительные). PC3 - клеточная линия аденокарциномы человека, являются адгерентными эпителиальными клетками (ПСМА-отрицательные).
При рутинном ведении клетки линии LNCaP, PC3, 22Rv1 культивировали в среде RPMI без фенолового красного с 10% FBS (Gibco), 1xGlutamax (Gibco), 1× смесью пенициллин-стрептомицин (Gibco), 1× смесью витаминов для RPMI среды. Клетки культивировали в матрасиках Т25 в CO2 инкубаторе при 5% CO2 и температуре 37°С до достижения 80% конфлюэнтного монослоя. Для визуализации накопления флуорисцентного сигнала анализируемых коньюгатов с использованием микроскопии были подготовлены препараты стекол с клетками клеточных линий LNCaP, PC3, 22Rv1. Для этого стекла (Corning) покрывали L-полилизином (Sigma) в течении 1 часа, после чего промывали стекла PBS дважды. В лунки 6-луночного планшета помещали покрытое L-полилизином стекло, в каждую лунку вносили клетки (клеточные культуры LNCaP, 22Rv1, PC3) 3×105 клеток в 1,5 мл среды RPMI, инкубировали в стандартных условиях культивирования в течение ночи. Далее клетки отмывали холодной средой RPMI, содержащей 1% фетальной бычьей сыворотки. Для каждого коньюгата в анализ включали клетки LNCaP, клетки 22Rv1, клетки РС3. В лунки, содержащие стекла вносили среду RPMI, содержащей 1% фетальной бычьей сыворотки, с коньюгатами (80 (МА-332), 79 (AY-79)) в концентрации 4 мкМ. Проводили инкубацию в стандартных условиях культивирования в течении 60 минут. В качестве контрольных образцов включали клетки, которые инкубировали со средой RPMI (1% FBS) с эквивалентным содержанием DMSO в течение 60 мин. По истечению 60 мин клетки отмывали холодной средой RPMI, содержащей 1% фетальной бычьей сыворотки и проводили фиксацию клеток при комнатной температуре в течении 8 мин 3,7% парафармальдегидом. Фиксированные клетки отмывали PBS дважды, проводили окраску ядер клеток 4',6-diamidino-2-phenylindole (DAPI) в соответствии с рекомендациями производителя, после заключительной отмывки PBS (дважды) препараты заключали в Mowiol-488.
Анализировали интенсивность флуоресцентного сигнала в клетках с использованием изображений, полученных системой визуализации EVOS FL Cell Imaging System с последующей обработкой сигнала в ImageJ. Результаты исследования представлены на фиг. 4. Как видно на фиг. 4 интенсивность флуоресцентного сигнала при воздействии коньюгатами (80) (МА-332), (79) (AY-79) была наибольшей для клеток LNCaP, наибольшие значения флуоресценции были достигнуты для коньюгата (79). Низкий сигнал флуоресценции, однако отличный от базового уровня автофлуорисценции клеток, был характерен для клеточной линии 22RV1. Для РС3 воздействие коньюгатами не приводило к статистически значимому повышению уровня флуорисценции по сравнению с уровнем автофлуорисценции. Таким образом, для коньюгатов (80) и (79) характерно специфичное проникновение в клетки разных клеточных культур в зависимости от представленности ПСМА на поверхности клеток.
Микрофотографии трех клеточных линий после воздействия исследуемыми коньюгатами на основе заявляемых средств доставки представлены на фиг. 5-10.
В соответствии с проведенными исследованиями по оценке накопления в клетках в зависимости от представленности ПСМА на поверхности клетки, показано, что конъюгаты (80) и (79) являются эффективными ПСМА-векторами, конъюгат (79) обладает выраженной способностью накапливаться в клетках, несущих ПСМА.
Оценка дифференциальной способности анализируемых конъюгатов проникать в клетки.
Проведена оценка дифференциальной способности конъюгатов на основе заявляемого средства проникать в клетки в зависимости от представленности ПСМА на поверхности. Интернализацию флуоресцентного лиганда в клетках детектировали с использованием метода проточной цитофлуориметрии.
Для оценки вклада ПСМА опосредованного захвата лиганда клетками в общую, фиксируемую проточным цитофлуориметром, флуоресценцию, проведено частичное блокирование ПСМА. Частичное блокирование ПСМА было достигнуто путем инкубации анализируемых клеток с избытком лиганда, тождественного по строению, но свободного от флюорисцентной метки.
Выбраны три клеточные линии с разным уровнем экспрессии ПСМА на поверхности клеток LNCaP, 22Rvl, PC3. Для культивирования клеток использовали следующие реактивы: PBS (фосфатно-солевой буфер) (Gibco), Glutamax (Gibco), FBS (фетальная телячья сыворотка), RPMI (Gibco), Pen Strep (Gibco)
При рутинном ведении клетки линии LNCaP, PC3, 22Rvl культивировали в среде RPMI без фенолового красного с 10% FBS (Gibco), IxGlutamax (Gibco), 1× смесью пенициллин-стрептомицин (Gibco), 1× смесью витаминов для RPMI среды. Клетки культивировали в матрасиках Т25 в CO2 инкубаторе при 5% CO2 и температуре 37°С до достижения 80% конфлюэнтного монослоя.
Протокол проведения анализа на проточном цитофлюориметре.
Лунки 12-луночного планшета для дальнейшего культивирования LNCaP клеток покрывали L-полилизином (Sigma) в течении 1 часа, после чего промывали лунки PBS дважды.
Клетки (клеточные культуры Lncap, 22Rv1, PC3) рассаживали в 12-луночный планшет, в лунку помещали 2×105 клеток в 800 мкл среды RPMI, инкубировали в стандартных условиях культивирования в течение ночи. Далее клетки отмывали холодной средой RPMI, содержащей 1% фетальной бычьей сыворотки.
Для каждого коньюгата в анализ включали клетки LNCaP (три повторности), клетки 22Rv1 (три повторности), клетки PC3 (три повторности).
Эксперимент проводили в трех повторностях как при частичном блокировании ПСМА (при внесении избытка анализируемого соединения), так и в эксперименте без дополнительной нагрузки избытком лиганда.
Избыток лиганда достигался внесением к клеткам 720 мкл среды RPMI (1% FBS), содержащей 400 мкМ (100-кратное превышение по концентрации) флуоресцентно немеченого лиганда тождественного по строению анализируемому, инкубацию проводили в стандартных условиях культивирования в течение одного часа В качестве образцов сравнения использовали лунки с клетками, в которые вносили 720 мкл среды RPMI (1% FBS), эти лунки с клетками также инкубировали в стандартных условиях культивирования в течении 1 часа. По истечение времени инкубации в каждую лунку вносили 80 мкл среды RPMI (1% FBS), содержащую анализируемый коньюгат в концентрации 40 мкМ для достижения конечной концентрации в лунке 4 мкМ. Проводили инкубацию в стандартных условиях культивирования в течении 30 минут. В качестве контрольных образцов включали клетки, которые инкубировали со средой RPMI (1% FBS) с эквивалентным содержанием DMSO в течении 30 мин.
Клетки снимали с поверхности пластика трипсинизацией, инактивацию трипсина проводили с использованием PBS, содержащего 10% FBS. Клетки центрифугировали и отмывали дважды в PBS (10% FBS), полученные клетки ресуспендировали в 500 мкл PBS и использовали далее для проточной цитофлуориметрии (Becton Dickinson FACSAria III).
Накопление сигнала флюорисценции, ассоциированного с поступлением меченого FAM коньюгата 80, проанализировано в трех клеточных линиях. Результаты проведения анализа с использованием проточной цитофлюорометрии представлены на фиг. 11 и 12.
Как видно на представленных фигурах процент клеток, накопивших флуоресцентный сигнал, был наибольшим для клеточной линии LNCaP (62,3% клеток), для клеточной линии 22RV1 26,4% клеточной популяции несли флуоресцентный сигнал, наименьшее значение анализируемого параметра было характерно для клеток РС3 (13%). Таким образом, для коньюгата (80) характерно специфичное проникновение в клетки разных клеточных культур в зависимости от представленности ПСМА на поверхности клеток. В эксперименте при частичном блокировании ПСМА путем предварительной инкубации с лигандом ПСМА (69) показано, что в клеточной линии LNCaP, обладающей максимальной представленностью ПСМА на поверхности клеток, наблюдалось выраженное снижение накопления конъюгата (80) клетками (падение накопления сигнала клетками в 10,4 раза). Снижение проникновения коньюгата (80) также было характерно и для клеток 22RV1, однако не столь выраженное. Для клеточной линии РС3 для которой характерно отсутствие ПСМА внесение избытка заявляемого средства - соединения формулы (69) практически не влияло на проникновение в клетки меченого коньюгата (80). Таким образом, в эксперименте с частичным блокированием ПСМА подтверждено избирательное накопление коньюгата (80) клетками в зависимости от представленности на ее поверхности ПСМА.
Накопление сигнала флюорисценции, ассоциированного с поступлением меченого FAM коньюгата (79), проанализировано в трех клеточных линиях. Результаты проведения анализа с использованием проточной цитофлюорометрии представлены на фиг. 13 и 14.
Как видно из фиг. 13 при воздействии коньюгатом (79) процент клеток, накопивших флуоресцентный сигнал, был наибольшим для клеточной линии LNCaP (83,8% клеток), для клеточной линии 22RV1 14,4% клеточной популяции несли флуоресцентный сигнал, наименьшее значение анализируемого параметра было характерно для клеток РС3 (7,6%). Таким образом, для коньюгата (79) характерно специфичное проникновение в клетки разных клеточных культур в зависимости от представленности ПСМА на поверхности клеток. В эксперименте при частичном блокировании ПСМА путем предварительной инкубации с лигандом ПСМА (69) показано, что в клеточной линии LNCaP, обладающей максимальной представленностью ПСМА на поверхности клеток, наблюдалось снижение накопления коньюгата (79) клетками, однако не столь выраженное как это было показано для коньюгата (80). Снижение проникновения коньюгата (79) также было характерно и для клеток 22RV1 и РС3. Таким образом, в эксперименте с частичным блокированием ПСМА подтверждено избирательное накопление коньюгата (79) клетками в зависимости от представленности на ее поверхности ПСМА. Однако в случае коньюгата (79) частичное блокирование ПСМА не было столь эффективным как для коньюгата (80).
В соответствие с проведенными исследованиями по оценке накопления в клетках в зависимости от представленности ПСМА на поверхности клетки, показано, что коньюгаты (80) и (79) являются эффективными ПСМА-векторами.
Оценка токсичности анализируемых конъюгатов.
Клеточные культуры
В данной работе использовались клеточные линии: иммортализованные клетки почки эмбриона человека Hek293T, клетки иммортализованных фибробластов легкого VA-13, рака молочной железы MCF-7 и клетки аденокарциномы легкого человека А549. Клетки культивировались в среде DMEM/F-12 (Gibco, США) с 10% эмбриональной телячьей сыворотки FBS (Gibco, США), стрептомицином (0.05 мг/мл) и пенициллином (50 ед/мл). Культивирование проводилось при 37°С и 5% CO2. Клеточные культуры были проверены на отсутствие микоплазмы.
Проведение тестов на цитотоксичность
МТТ-тест (3-(4,5-диметилтриазол-2-ил)2,5-дифенил тетразолиум бромид - тест) основан на [Ferrari, M., Fomasiero, М.С., and Isetta, А.М. (1990). МТТ colorimetric assay for testing macrophage cytotoxic activity in vitro. J Immunol Methods 131, 165-172.] Клетки Hek293T, А549, MCF7 рассевались в количестве 2500 на лунку 96-луночного планшета, клетки линии VA-13 по 4000 на лунку в среде DMEM/F-12 с 10% эмбриональной телячьей сыворотки FBS (Gibco, США), стрептомицином (0.05 мг/мл) и пенициллином (50 ед/мл). Через 24 часа после посева клеток к ним добавлялись химические препараты, разведенные в соответствующей культуральной среде. Для каждой концентрации препарата использовали три технических реплики. Далее клетки инкубировались 72 часа с препаратами при 37°С и 5% CO2. Затем к клеткам прибавлялся МТТ до 0,5 г/л (использовался 10х кратный раствор в стандартном буфере PBS) и клетки инкубировались еще 2 часа. После инкубации среда отбиралась и осадок растворялся в 140 мкл ДМСО (ООО «Фармамед», Россия) в течение 15 минут при перемешивании на орбитальном шейкере (60 об/мин). Измеряли оптическую плотность каждой лунки при длине волны 565 нм. Каждый эксперимент проводился в трех репликах. Данные обрабатывали с помощью ПО "GraphPad Prism 5" (GraphPad Software, Inc., San Diego, CA), строя зависимости доза-ответ и определяя СС50.
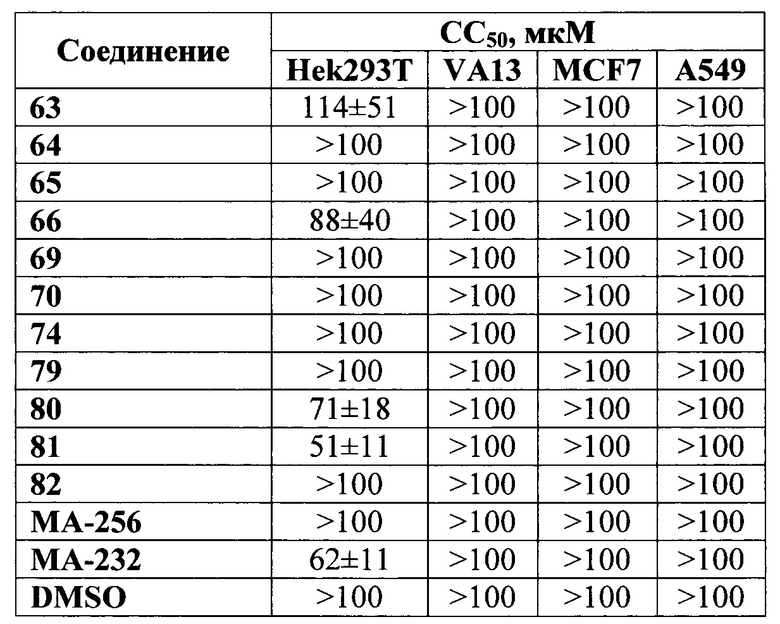
Для ряда лигандов было показано, что данные молекулы не обладают выраженной токсичностью на клеточных линиях Hek293T (эмбриональные клетки почек человека), VA-13 (фибробласты легких), MCF7 (аденокарцинома груди), А549 (карцинома легких), значения поулумаксимальной клеточной токсичности (СС50) на данных клеточных культурах оказывается >50 мкМ.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЕ ПЕПТИДНОЙ ПРИРОДЫ, ОБЛАДАЮЩЕЕ СПОСОБНОСТЬЮ СВЯЗЫВАТЬСЯ С ПСМА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2021 |
|
RU2823164C2 |
| КОНЪЮГАТ ФЛУОРЕСЦЕНТНОГО КРАСИТЕЛЯ С ВЕЩЕСТВОМ ПЕПТИДНОЙ ПРИРОДЫ, ВКЛЮЧАЮЩИМ ПСМА-СВЯЗЫВАЮЩИЙ ЛИГАНД НА ОСНОВЕ ПРОИЗВОДНОГО МОЧЕВИНЫ ДЛЯ ВИЗУАЛИЗАЦИИ КЛЕТОК, ЭКСПРЕССИРУЮЩИХ ПСМА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2019 |
|
RU2713151C1 |
| ЛИГАНДЫ, ТРОПНЫЕ К ПРОСТАТИЧЕСКОМУ СПЕЦИФИЧЕСКОМУ МЕМБРАННОМУ АНТИГЕНУ И ИХ ПРИМЕНЕНИЕ ДЛЯ ПОЛУЧЕНИЯ ДВОЙНЫХ КОНЪЮГАТОВ С ТЕРАПЕВТИЧЕСКИМИ АГЕНТАМИ НА ИХ ОСНОВЕ ДЛЯ КОМБИНИРОВАННОЙ ТЕРАПИИ ПСМА ЭКСПРЕССИРУЮЩИХ ОПУХОЛЕЙ | 2023 |
|
RU2841078C1 |
| СОЕДИНЕНИЕ ДЛЯ ДИАГНОСТИКИ ОПУХОЛЕЙ, ЭКСПРЕССИРУЮЩИХ ПСМА, И КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 2019 |
|
RU2730507C1 |
| КОНЪЮГАТ МОНОМЕТИЛ АУРИСТАТИНА Е ДЛЯ ПОЛУЧЕНИЯ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ РАКА ПРЕДСТАТЕЛЬНОЙ ЖЕЛЕЗЫ | 2019 |
|
RU2729192C1 |
| НОВЫЕ 2',5'-ДИАРИЛСПИРО[ИНДОЛ-3,3'-ПИРРОЛИДИН]-2(1Н)-ОНЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2019 |
|
RU2730287C1 |
| БИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЛЬНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2020 |
|
RU2811612C2 |
| ПРОИЗВОДНЫЕ АРИЛМЕТОКСИ ИЗОИНДОЛИНА И КОМПОЗИЦИИ, ВКЛЮЧАЮЩИЕ ИХ, И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2011 |
|
RU2567753C2 |
| Производное 1'-бромо-2',3',4'-триметоксибензо[5',6':4,5]-(aR, 1S)-1-ацетамидо-6,7-дигидроциклогепта-[3,4-f]-1Н-индола и его применение | 2016 |
|
RU2630303C1 |
| Способ получения производного мочевины с хелатным центром, тропного к простат-специфичному мембранному антигену для связывания технеция-99м/рения для диагностики/лечения рака предстательной железы | 2018 |
|
RU2692126C1 |
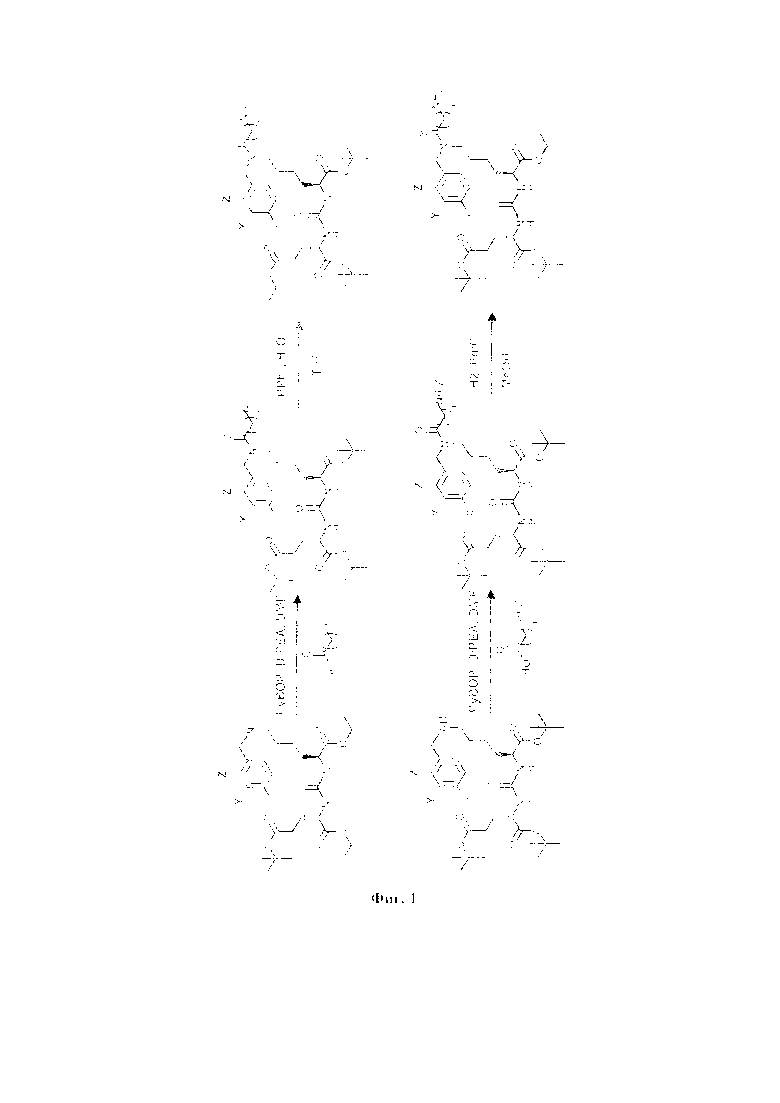

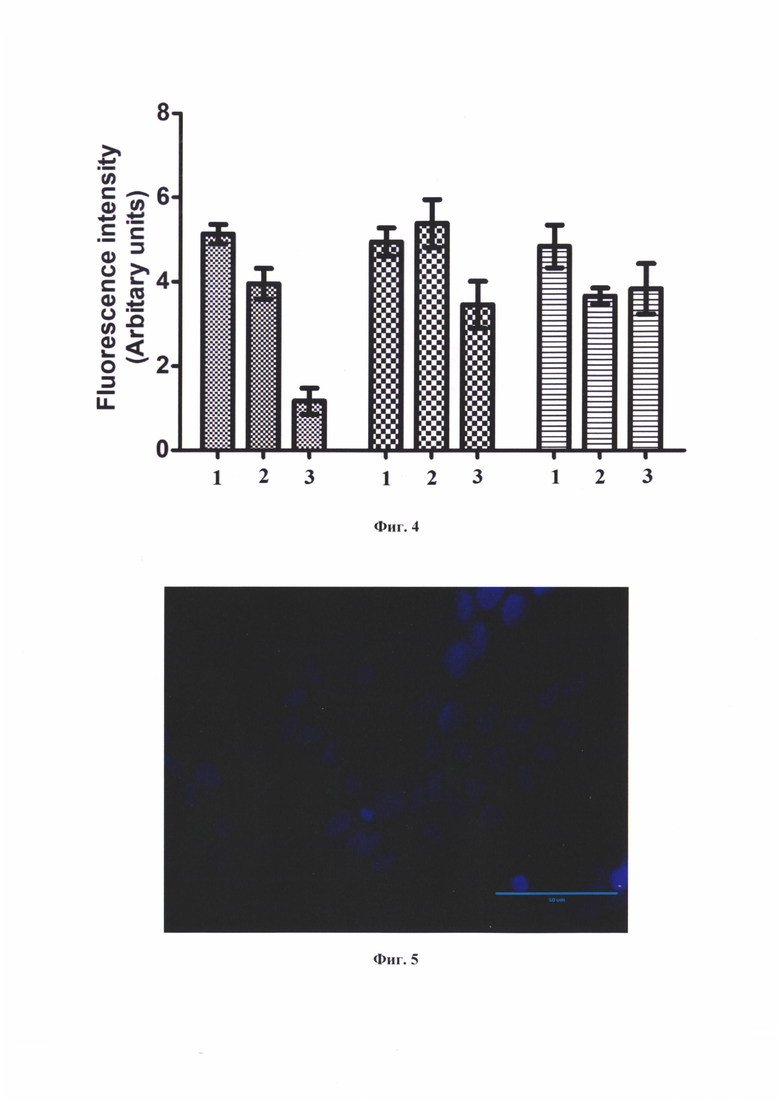



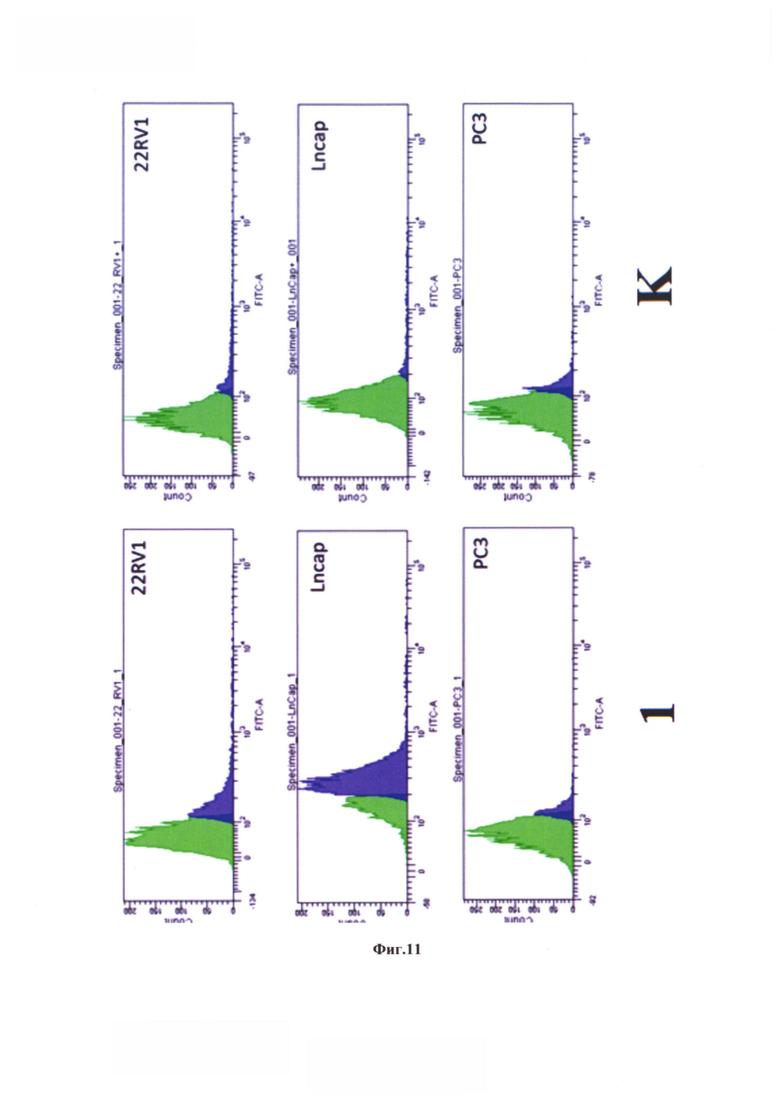
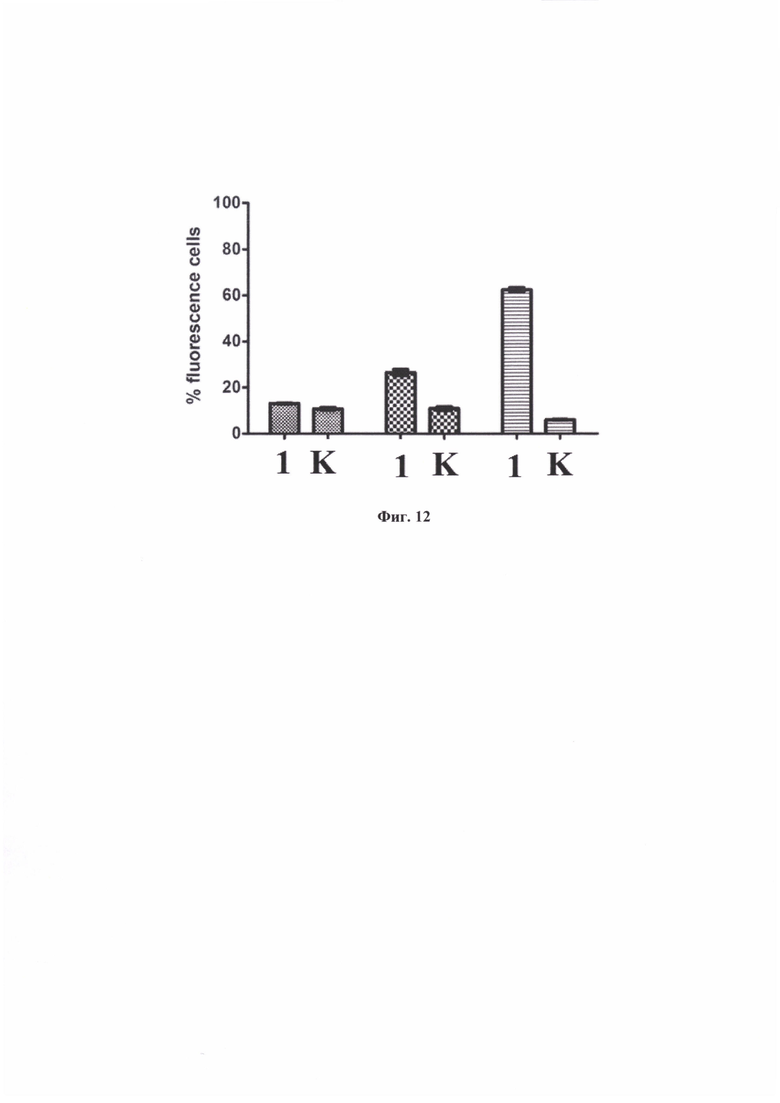
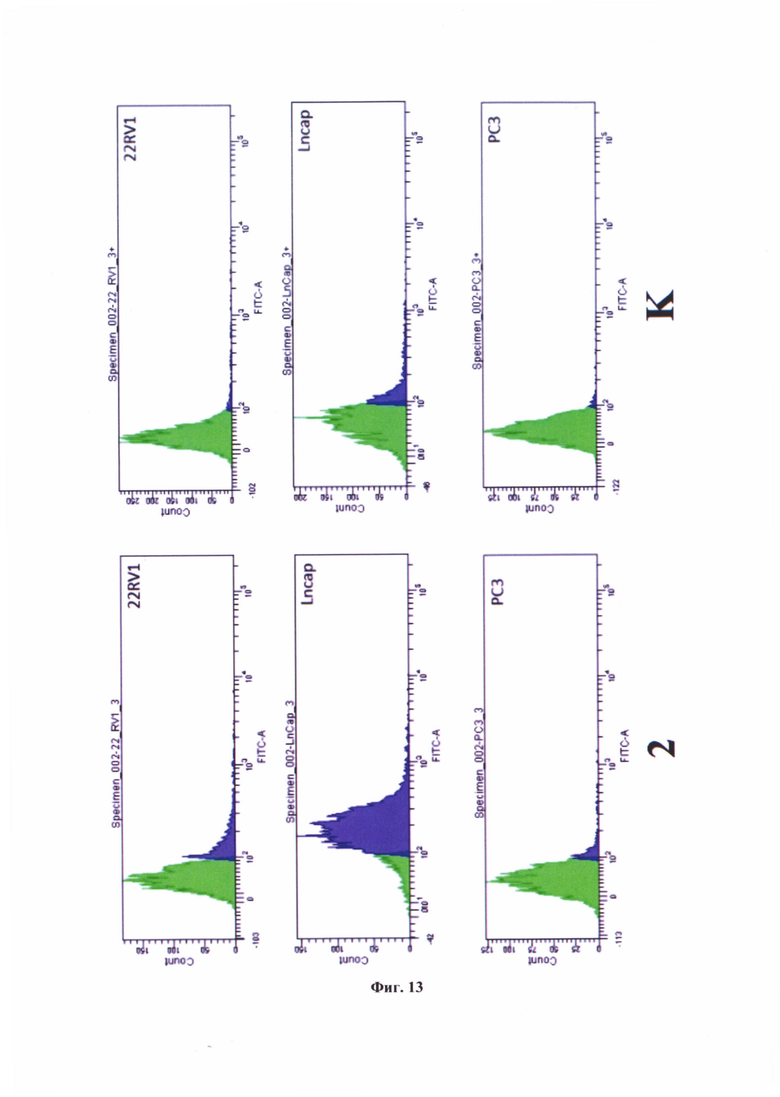
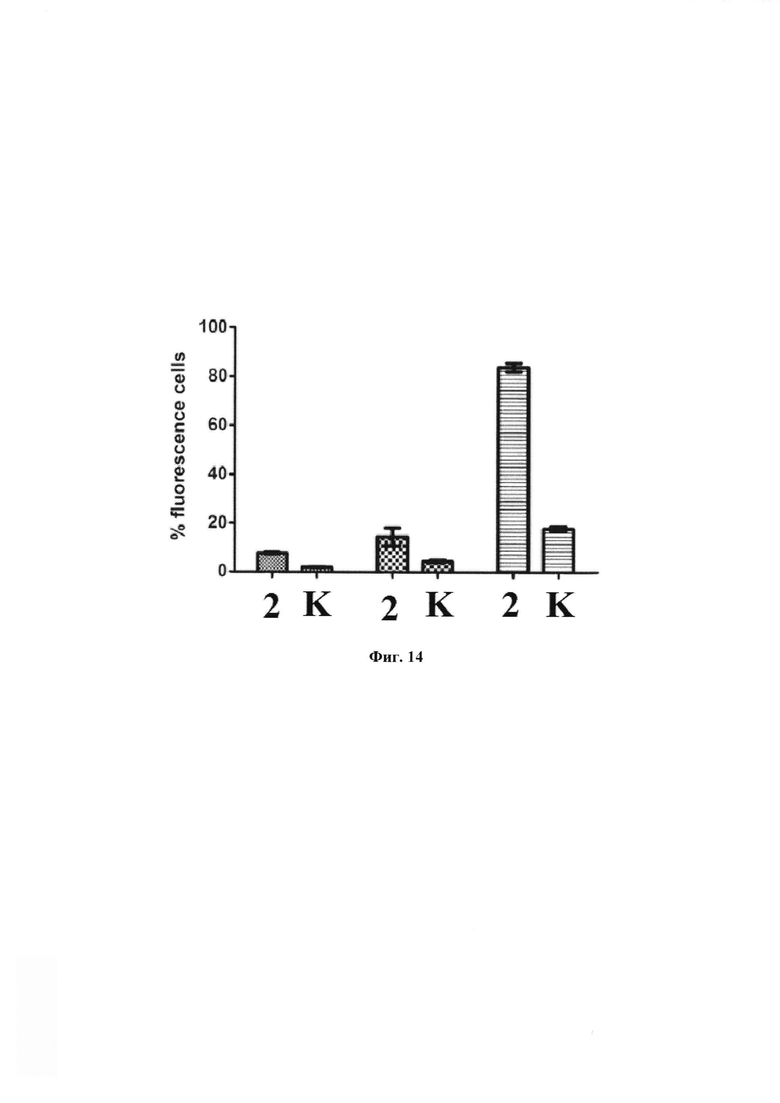

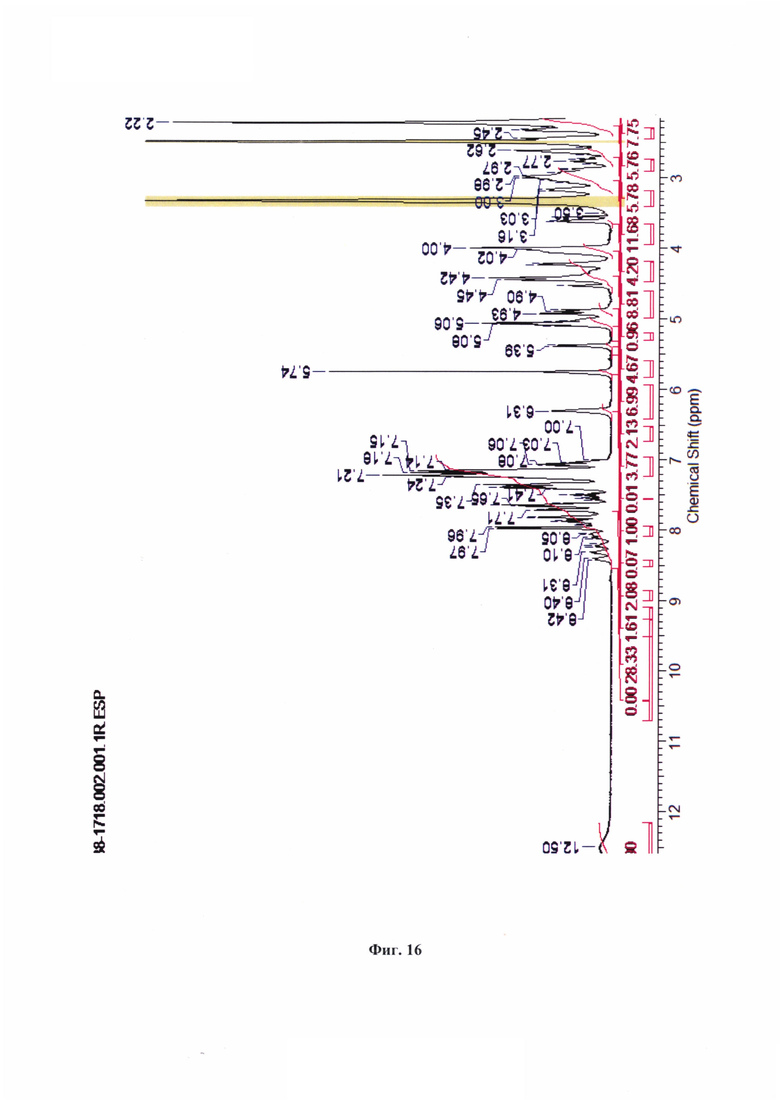
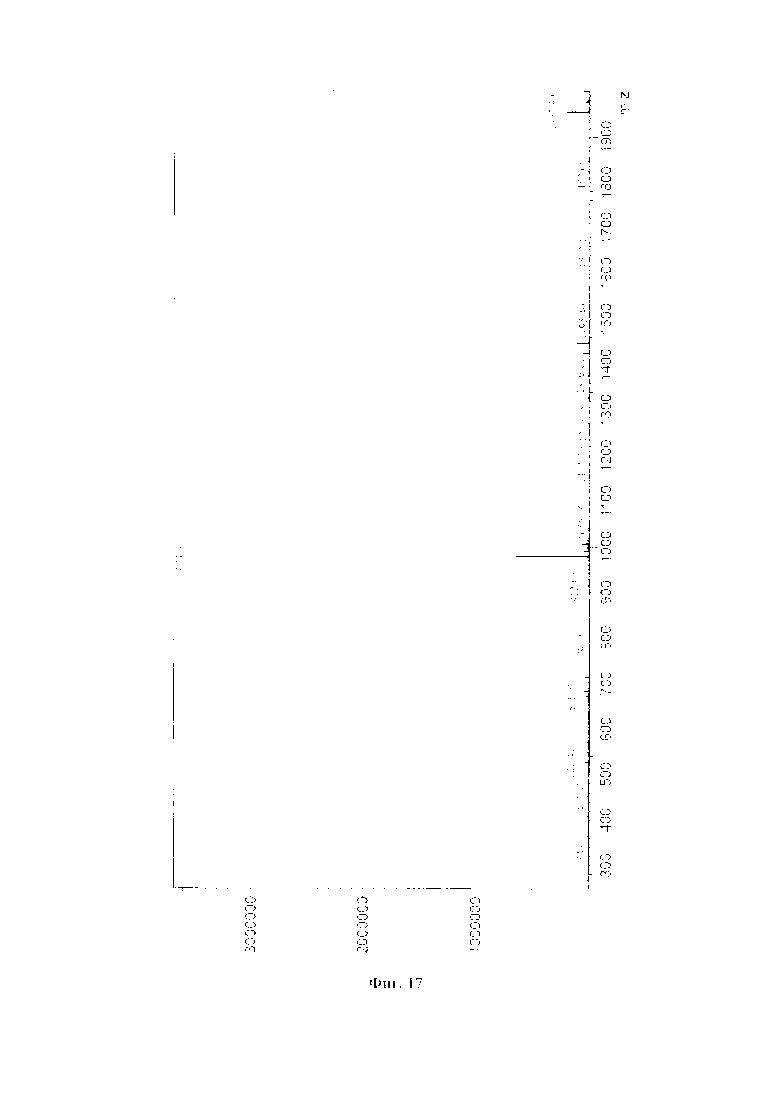


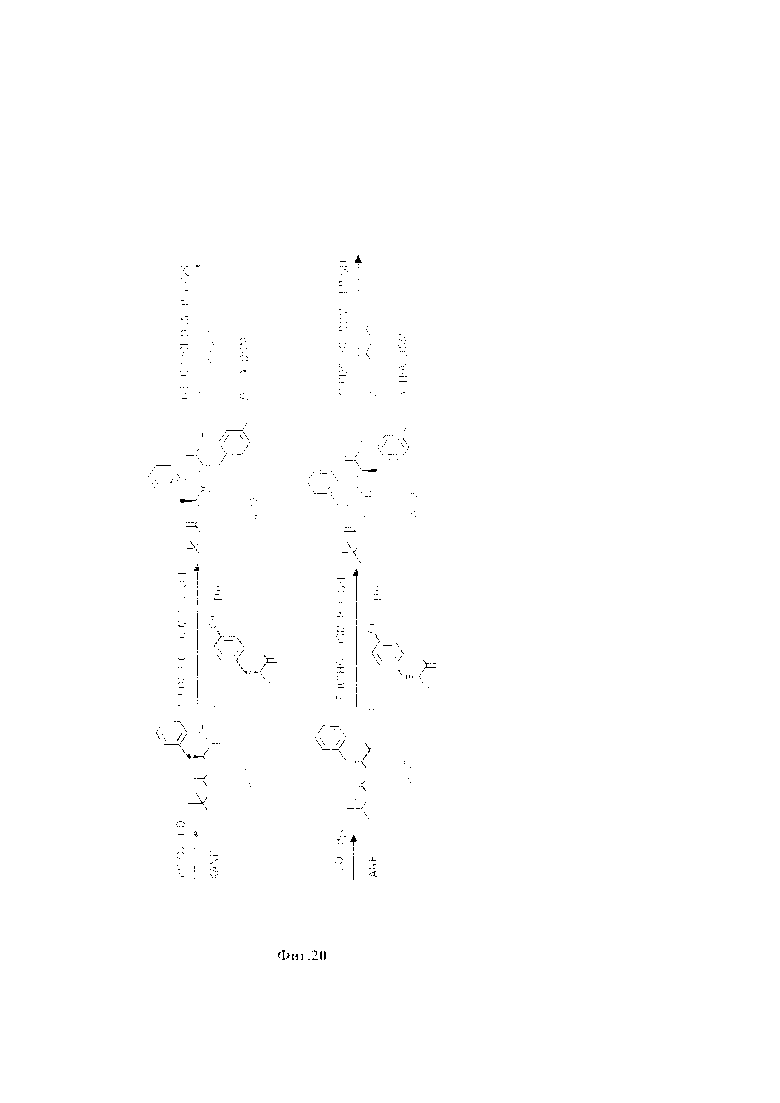
Настоящее изобретение относится к веществу общей формулы
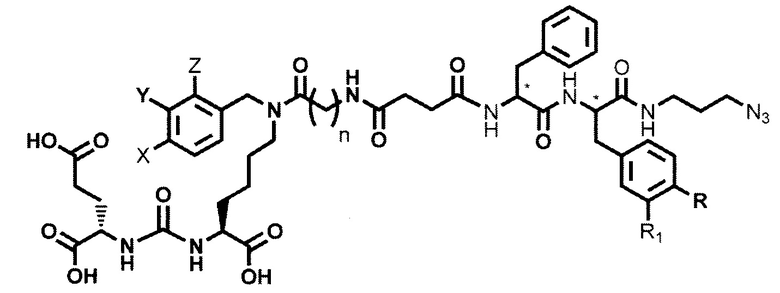 (I),
(I),
где n=3-5; X, Y, Z независимо друг от друга представляют собой F, Cl, Br, Н; R = ОН, Н;
R1 = Н, Br. Также изобретение относится к способу получения такого вещества, к его применению для получения конъюгата с лекарственным или диагностическим агентом для диагностики или лечения заболеваний, вызванных клетками, экспрессирующими простатический специфический мембранный антиген (ПСМА), а также к самому конъюгату. Технический результат – получение новых средств доставки диагностических или терапевтических средств, включающих ПСМА-лиганд с линкером, обладающих низкой токсичностью, высокой аффинностью и селективностью действия в отношении клеток, экспрессирующих ПСМА, что позволяет использовать их для лечения заболеваний, связанных с высокой экспрессией ПСМА, с меньшей дозировкой при избирательном действии на раковые клетки, не затрагивая здоровые клетки. 4 н. и 8 з.п. ф-лы, 20 ил., 1 табл., 1 пр.
1. Вещество общей формулы
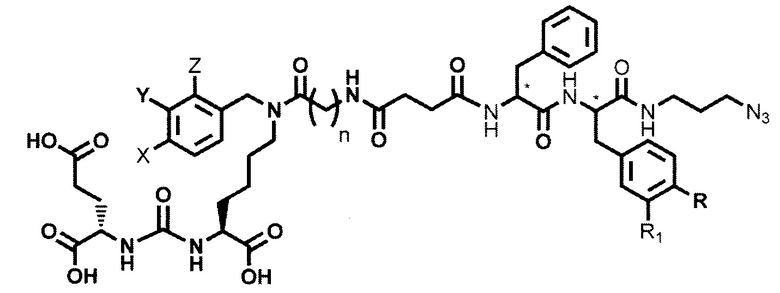 (I),
(I),
где n=3-5;
X, Y, Z независимо друг от друга представляют собой F, Cl, Br, Н;
R = ОН, Н;
R1 = Н, Br.
2. Вещество по п. 1, выбранное из группы:
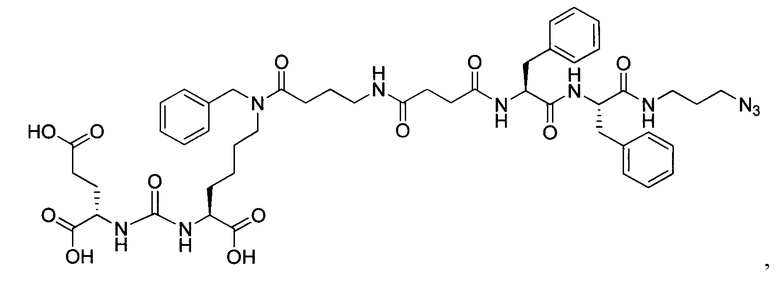
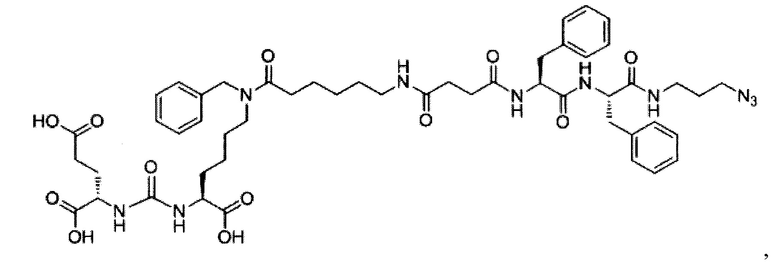
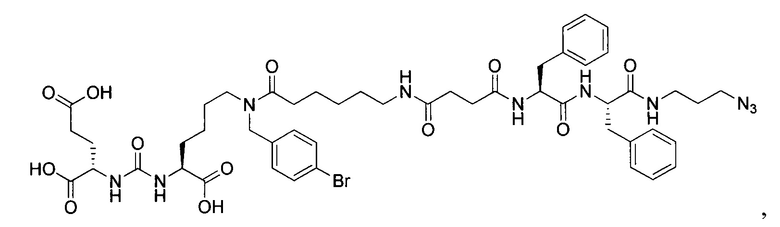
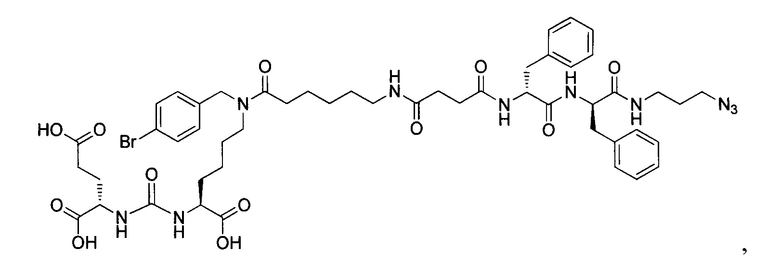
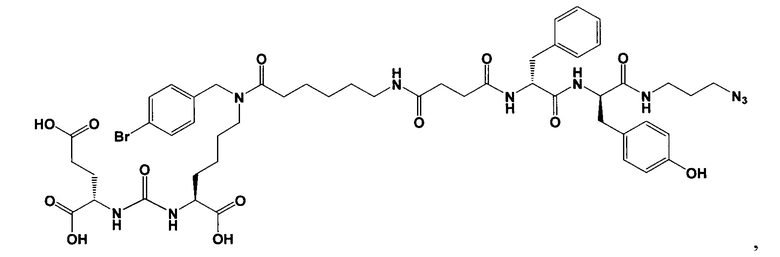
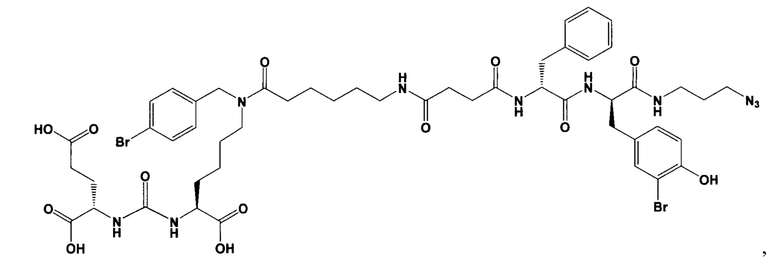
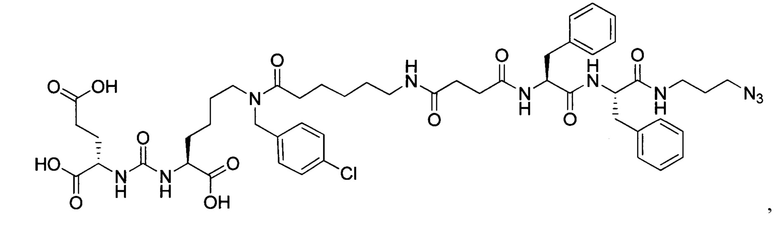
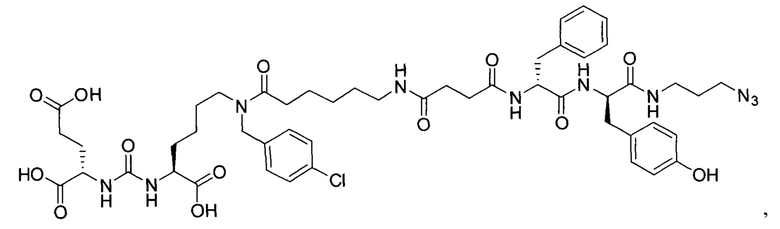
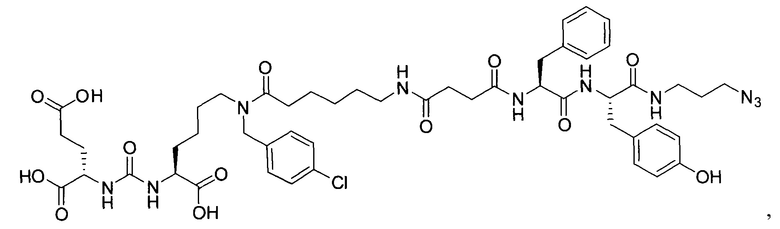
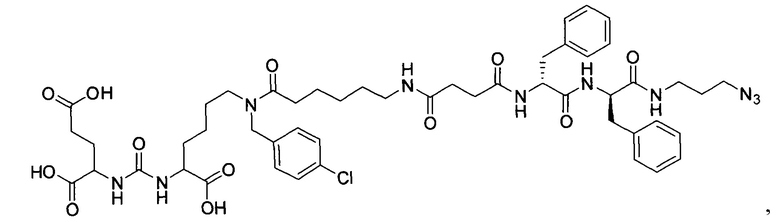
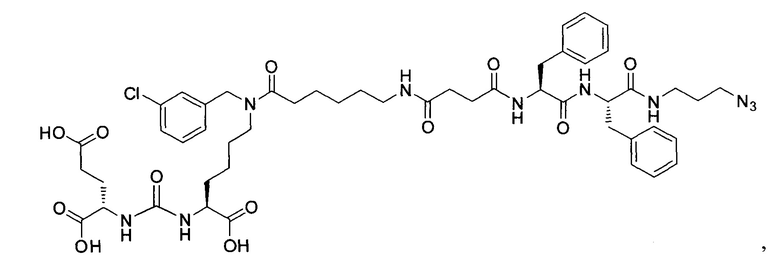
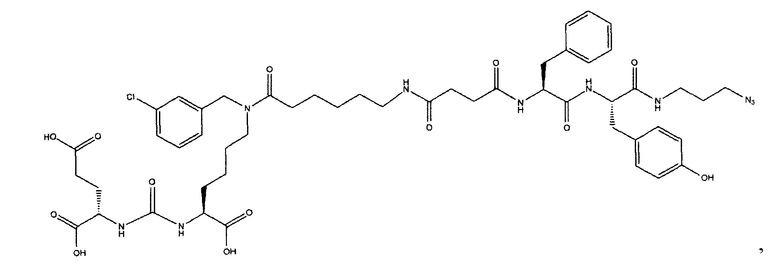
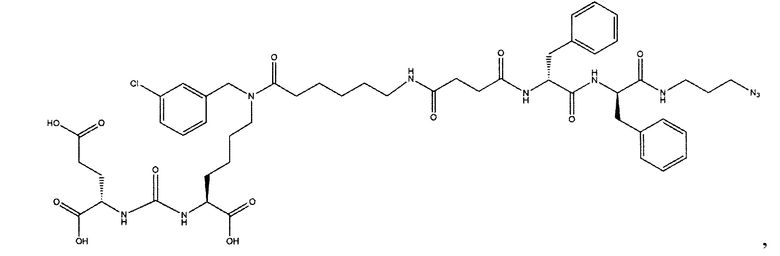
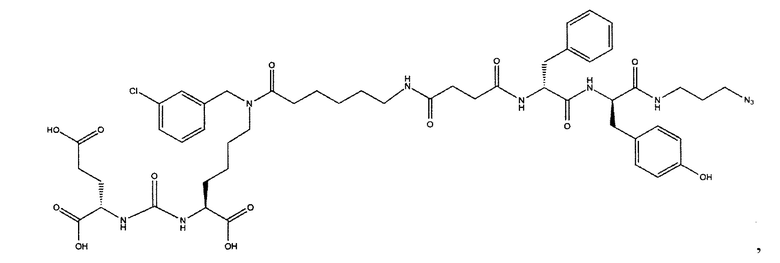
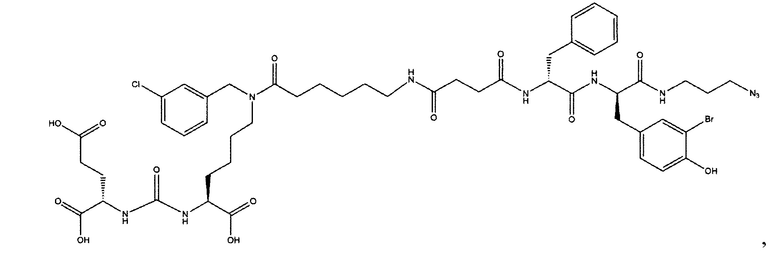
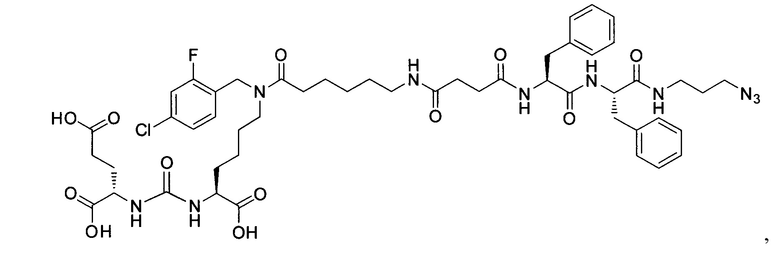
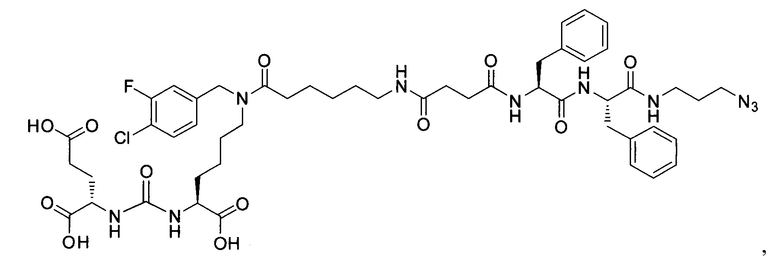
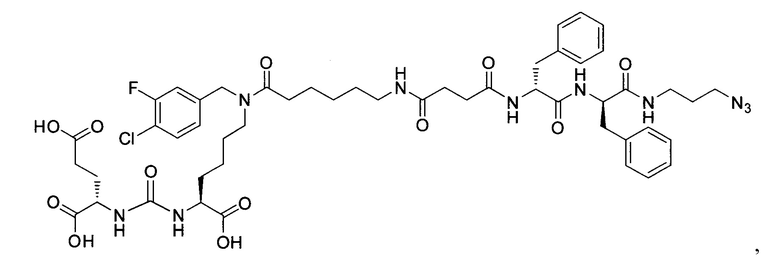
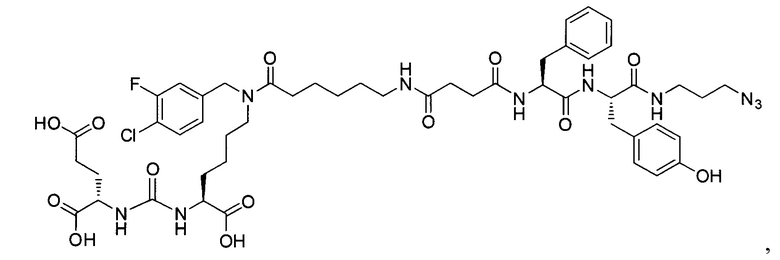
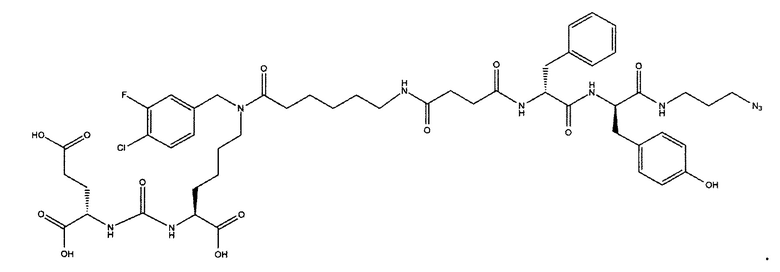
3. Вещество по п. 1 или 2, отличающееся тем, что оно способно связываться с простатическим специфическим мембранным антигеном.
4. Способ получения вещества по п. 1, включающий синтез тритретбутилового производного ПСМА-связывающего лиганда формулы (2):
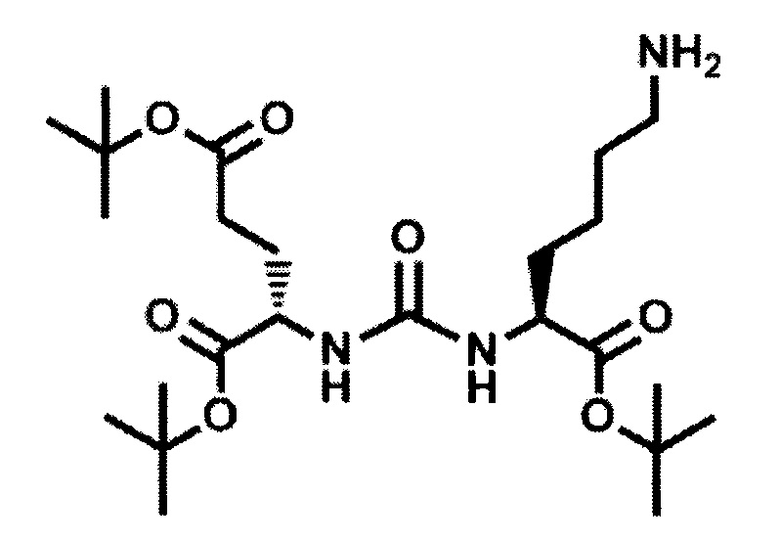
с последующим алкилированием полученного тритретбутилового производного ПСМА-связывающего лиганда с получением соединения формулы (II):
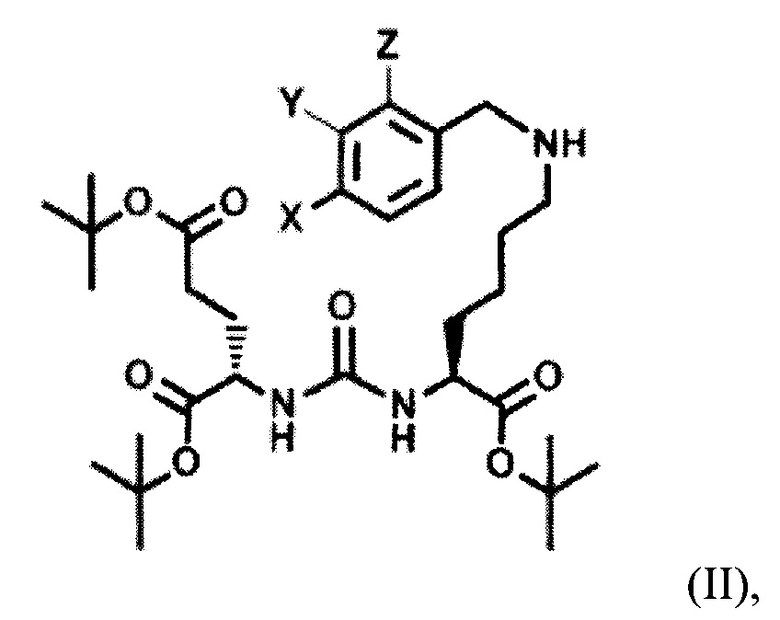
где X, Y, Z независимо друг от друга представляют собой F, Cl, Br, Н
с последующим ацилированием производными азида бутаной или пентановой или гексановой кислот с получением азидного производного алкилированного производного ПСМА-лиганда с дальнейшей реакцией восстановления азида до аминогруппы в присутствии трифенилфосфина и воды в растворе ТГФ или в растворе метанола с использованием водорода в присутствии палладия на углероде в качестве катализатора и получением соединения формулы (III):
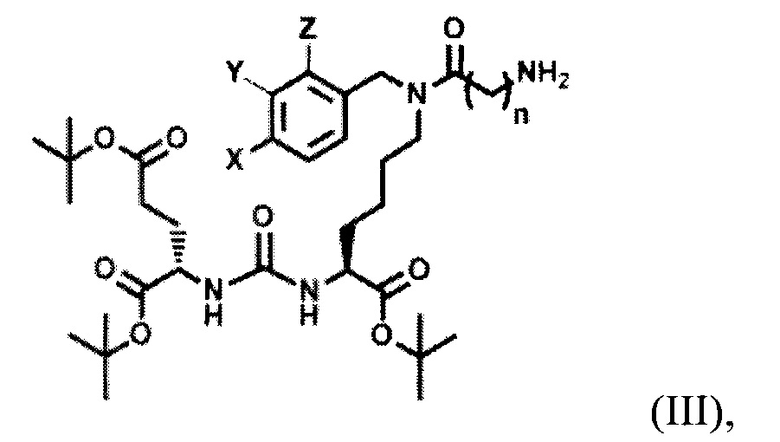
где X, Y, Z представляют собой F, Cl, Br, Н, n=3-5,
которое модифицируют янтарным ангидридом в присутствии диизопропилэтиламина или триэтиламина, с получением производного соединения ацилированного янтарным ангидридом, далее осуществляют получение дипептидов производных ароматических аминокислот, представляющих собой фенилаланил-фенилаланин или фенилаланил-тирозин формулы (IV), для связывания с модифицированным фрагментом линкера,
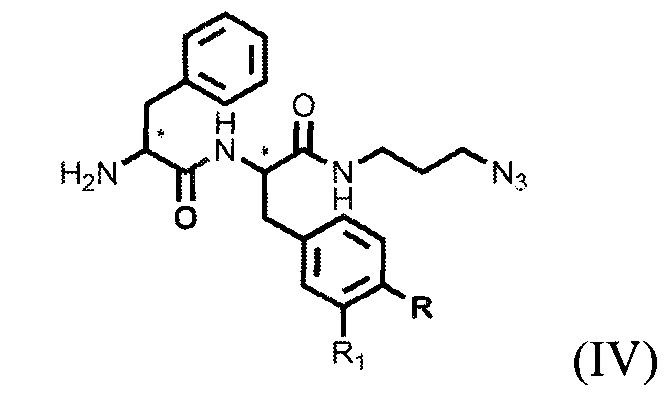
затем получают реакцией ацилирования производным соединения (III), ацилированного янтарным ангидридом, дипептида формулы (IV) тритретбутил производное конъюгата радикала ПСМА-связывающего лиганда и модифицированного гидрофобного пептидного линкера, включающего фрагменты 6-аминогексановой или 5-аминопентановой или 4-аминобутановой кислот, фрагмент фенилаланина или его производное, фрагмент тирозина или его производное общей формулы (V):
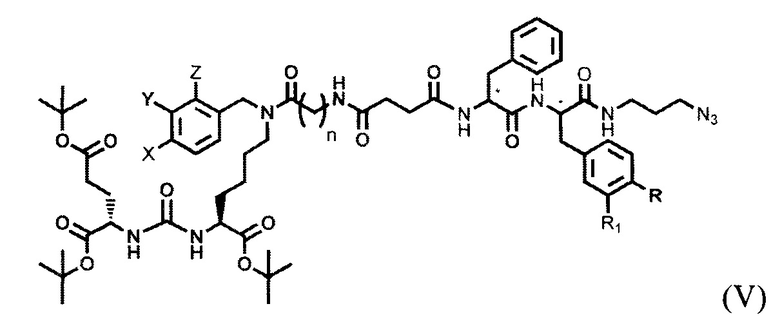 ,
,
где n=3-5;
X, Y, Z независимо друг от друга представляют собой F, Cl, Br, Н;
R = OH, Н;
R1 = Н, Br
с последующим удалением тритретбутильных защитных групп соединения формулы (VI) с получением ковалентно-связанных ПСМА-связывающего лиганда и модифицированного гидрофобного пептидного линкера общей формулы (I).
5. Способ по п. 4, характеризующийся тем, что получение алкилированного тритретбутил производного ПСМА-лиганда осуществляют путем восстановительного аминирования замещенными бензальдегидами.
6. Способ по п. 4, характеризующийся тем, что удаление тритретбутильных защитных групп проводят в присутствии 9-11% ТФУ в течение 15-17 часов в дихлорметане.
7. Применение вещества по п. 1 для получения конъюгата с лекарственным или диагностическим агентом для диагностики или лечения заболеваний, вызванных клетками, экспрессирующими простатический специфический мембранный антиген.
8. Применение по п. 7, характеризующееся тем, что вещество выбрано из группы:
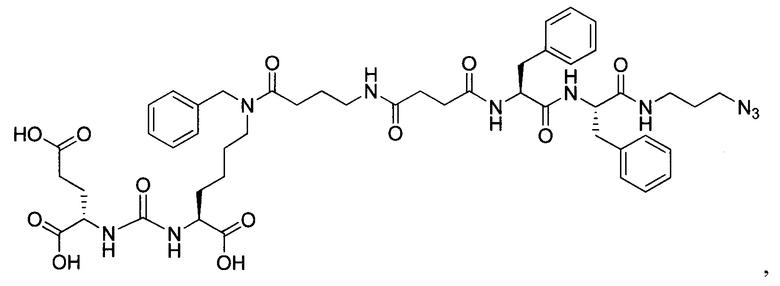
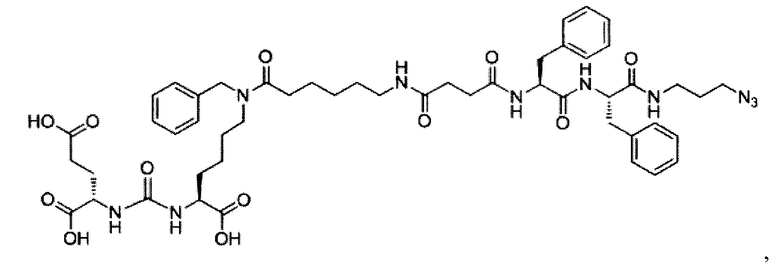
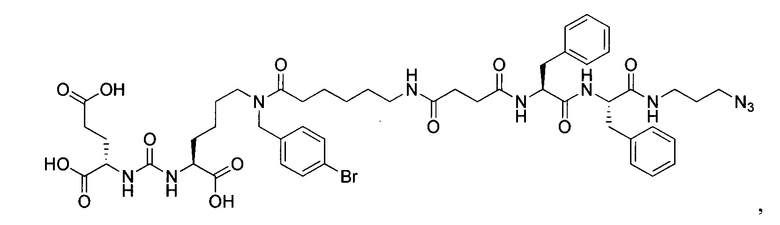
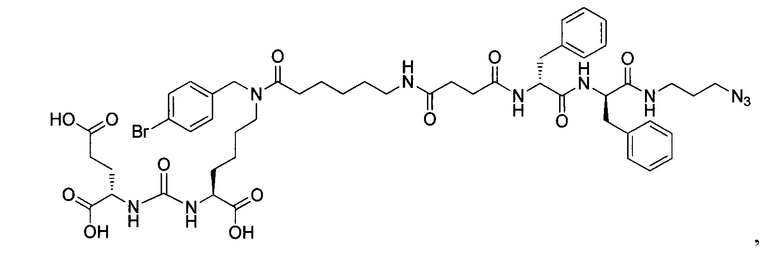
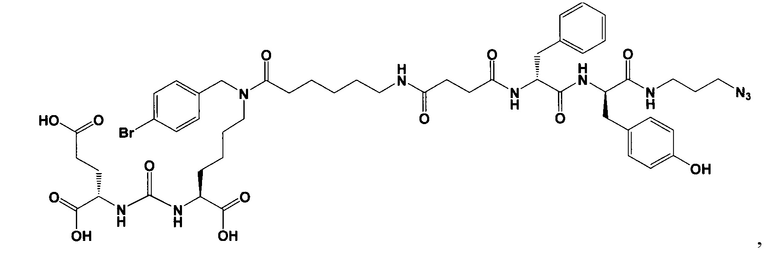
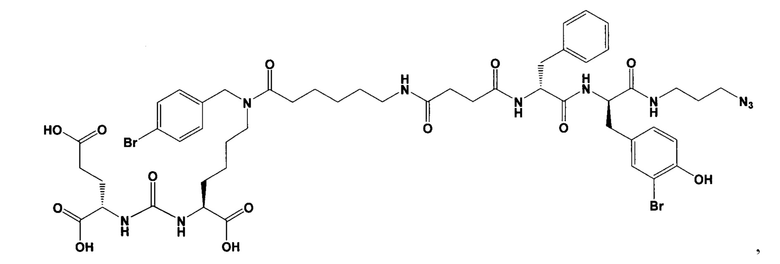
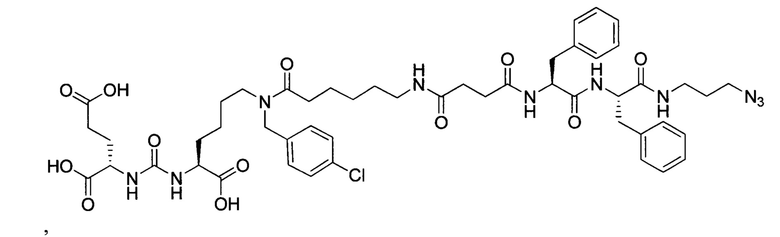
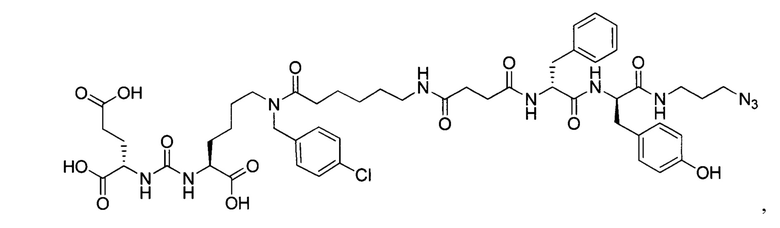
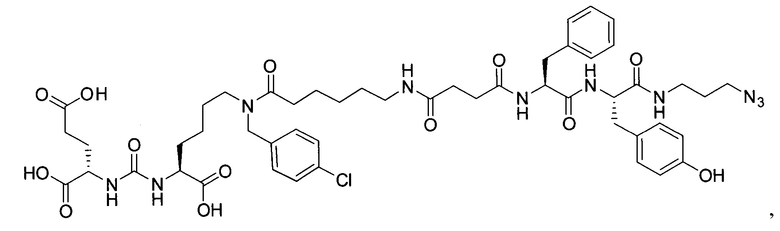
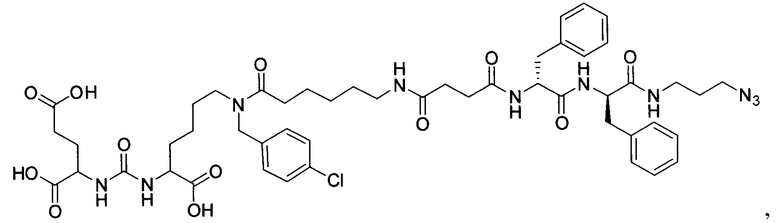
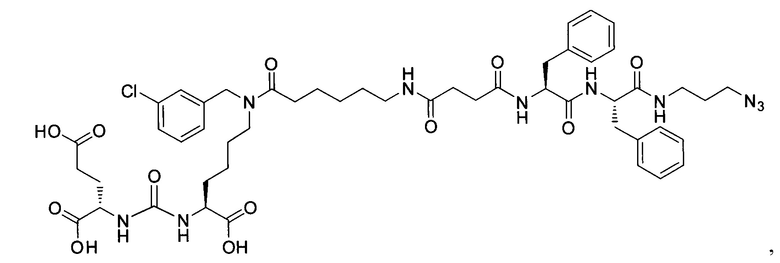
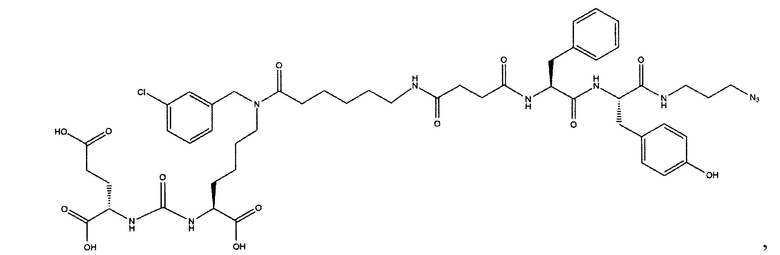
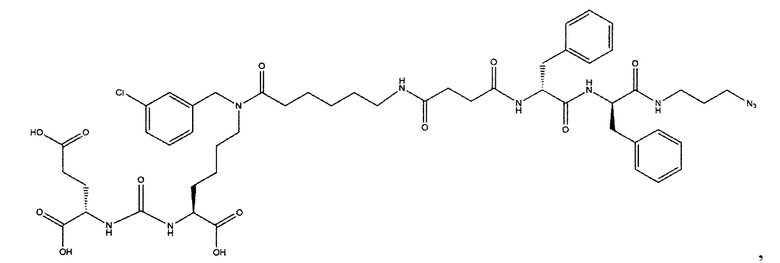
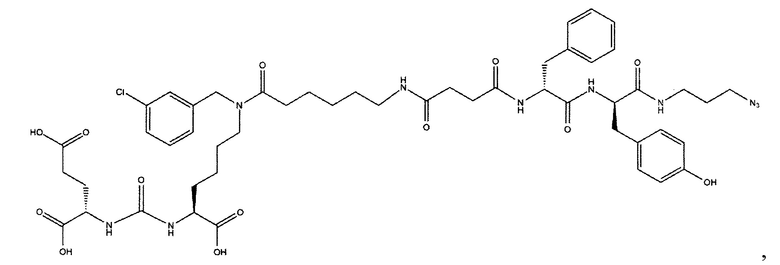
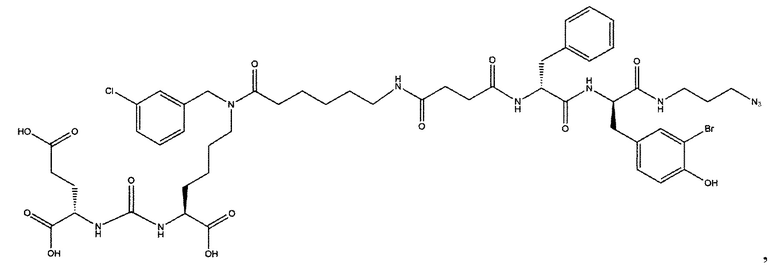
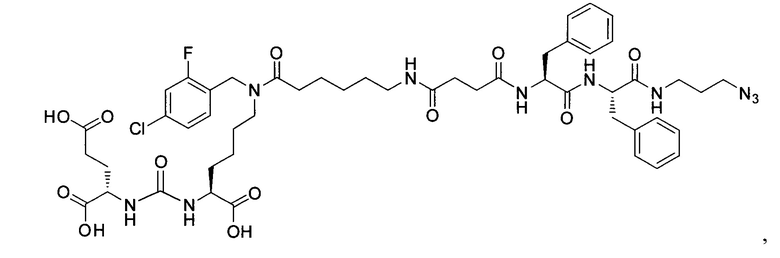
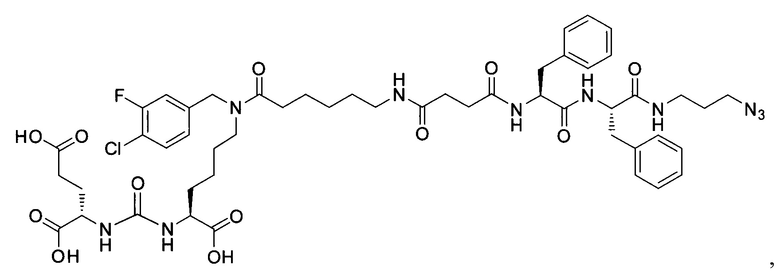
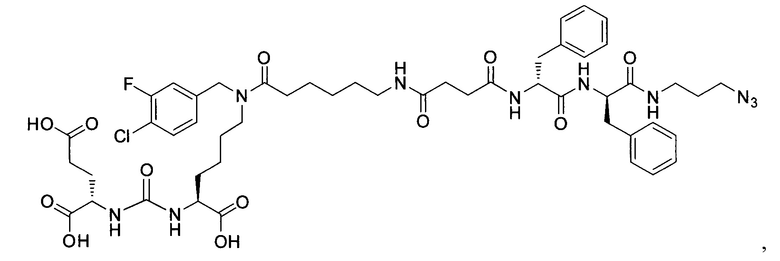
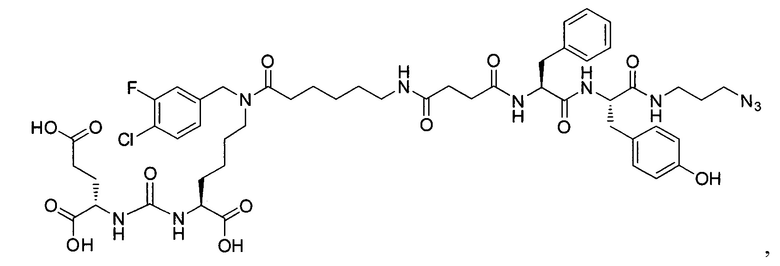
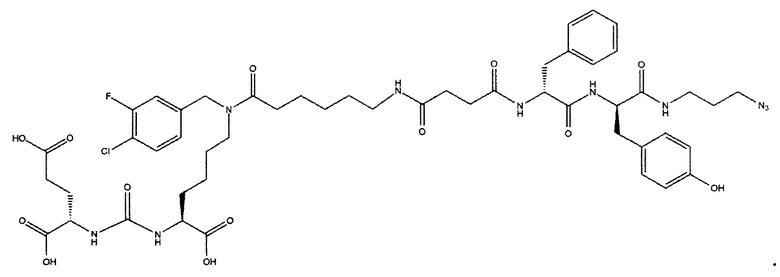
9. Применение по п. 7, характеризующееся тем, что лекарственным агентом является доцетаксел.
10. Применение по п. 7, характеризующееся тем, что таким заболеванием является рак предстательной железы.
11. Конъюгат вещества по п. 1 с лекарственным или диагностическим агентом для диагностики или лечения заболеваний, вызванных клетками, экспрессирующими простатический специфический мембранный антиген
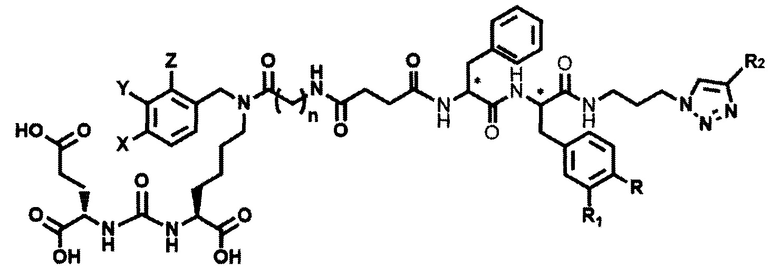 ,
,
где n=3-5;
X, Y, Z независимо друг от друга представляют собой F, Cl, Br, Н;
R = ОН, Н;
R1 = Н, Br;
R2 = лекарственный или диагностический агент.
12. Конъюгат по п. 11, выбранный из группы:
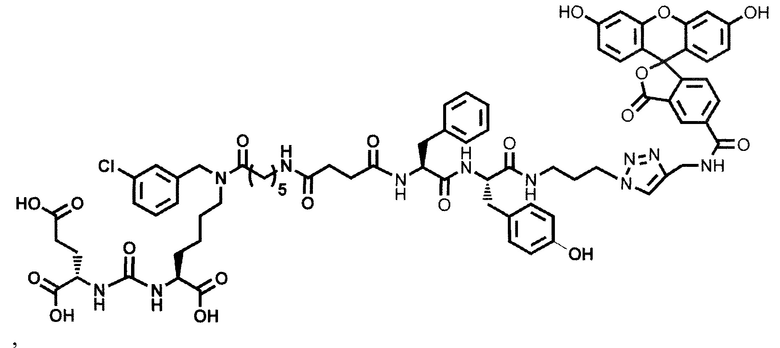
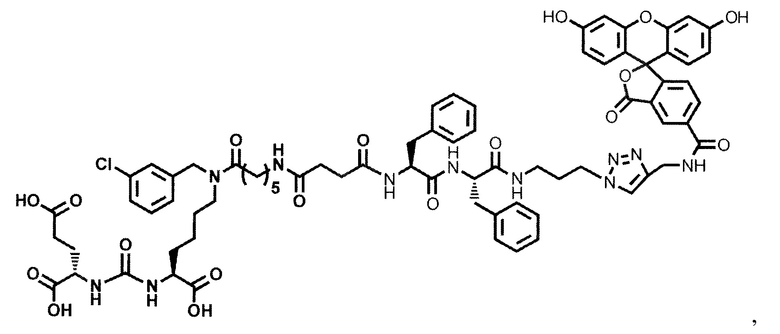
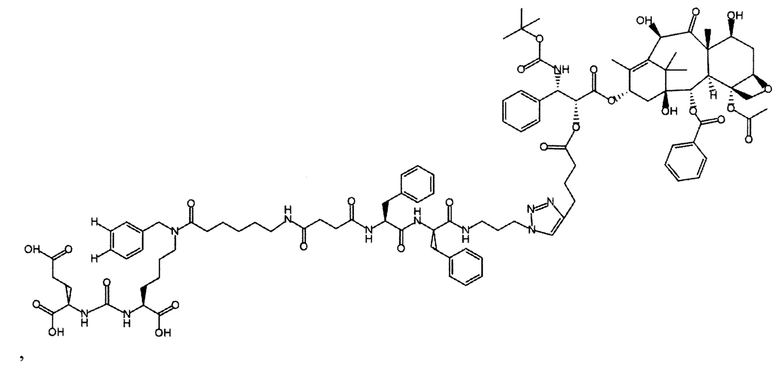
.
| Способ получения цианистых соединений | 1924 |
|
SU2018A1 |
| Автомобиль-сани, движущиеся на полозьях посредством устанавливающихся по высоте колес с шинами | 1924 |
|
SU2017A1 |
| Автомобиль-сани, движущиеся на полозьях посредством устанавливающихся по высоте колес с шинами | 1924 |
|
SU2017A1 |
| Автомобиль-сани, движущиеся на полозьях посредством устанавливающихся по высоте колес с шинами | 1924 |
|
SU2017A1 |
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Автомобиль-сани, движущиеся на полозьях посредством устанавливающихся по высоте колес с шинами | 1924 |
|
SU2017A1 |
| Колосоуборка | 1923 |
|
SU2009A1 |
| АГЕНТЫ, СВЯЗЫВАЮЩИЕСЯ С PSMA, И ИХ ПРИМЕНЕНИЕ | 2009 |
|
RU2494096C2 |

