Область техники, к которой относится изобретение
Настоящее изобретение относится к способу синтеза ненуклеозидного ингибитора обратной транскриптазы. В частности, настоящее изобретение относится к способу синтеза диарилпиримидинового ненуклеозидного ингибитора обратной транскриптазы, такого как этравирин.
Уровень техники
Ненуклеозидные ингибиторы обратной транскриптазы (NNRTI) являются ключевым компонентом высокоактивной антиретровирусной терапии (HAART) из-за их способности нацеливать аллостерический связывающий «карман» на фермент обратную трнаскриптазу, что приводит к развитию широкого спектра активности против мутаций HIV RT. HAART стала стандартом лечения в случае заражения вирусом иимунодефицита человека (ВИЧ) с 1996 и привела в существенному возрастанию выживаемости. Диарилпиримидины представляют собой второе поколение NNRTI и применимы для лечения ВИЧ-инфицированных пациентов с вирусами, устойчивыми к NNRTI. Этравирин (I), ранее ТМС 125 и химически известный как 4-[[6-амино-5-бром-2-[(4-цианофенил)амино]-4-пиримидинил]окси]-3,5-диметилбензонитрил, представляет собой NNRTI, одобренный в 2008 для применения в сочетании с другими антиретровирусными средствами для подвергавшихся лечению взрослых пациентов с ВИЧ-инфекциями с множественной лекарственной устойчивостью. Этравирин продается во всем мире в виде пероральных таблеток и впервые раскрыт De Corte et al. в US 7037917.
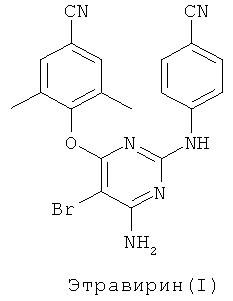
De Corte et al. в US 7037917 предлагают способ получения диарилпиримидиновых производных, согласно которому соединение формулы (II) греют с аммиаком в присутствии инертного растворителя, такого как 1,4-диоксан, в автоклаве при 150°С в течение 4 суток.
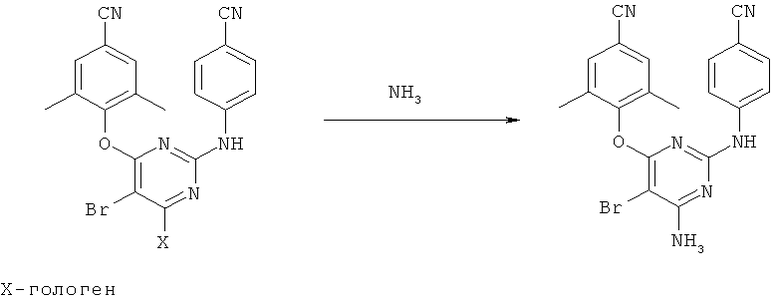
Davis et al. в Drug of the Future, 2005, 30(5): 462-468, раскрывают, что промежуточное соединение (II) можно получить двумя различными способами. Первый способ показывает, что 5-бром-2,4,6-трихлорпиримидин вводят во взаимодействие с 4-аминобензонитрилом с помощью диизопропилэтиламина в кипящем диоксане с образованием диариламина, который затем вводят во взаимодействие с 4-гидрокси-3,5-диметилбензонитрилом, и получают промежуточное соединение формулы (II) (схема 1).
Схема 1
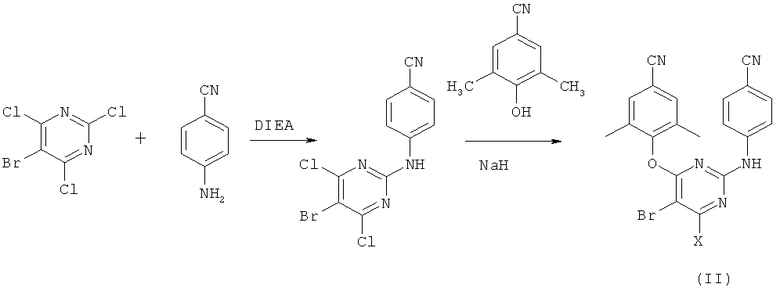
Второй способ синтеза соединения формулы (II) показывает, что 4-гуанидинобензонитрил циклизуют с диэтилмалонатом с помощью этоксида натрия, и получают 4-(4,6-дигидроксипиримидин-2-иламино)бензонитрил, который после обработки РОСl3 дает соответствующее дихлорпроизводное. Дальнейшее бромирование бромом в присутствии бикарбоната натрия в водном метаноле дает 4-(4-бром-4,6-дихлорпиримидин-2-иламино)бензонитрил, который после конденсации с натриевой солью циано-2,6-диметилфенолята в присутствии N-метилпирролидона и диоксана дает промежуточное соединение формулы (II) (схема 2).
Схема 2
Однако вышеуказанная процедура синтеза диарилпиримидиновых NNRTI страдает от недостатка, которым является очень медленная конверсия соединения формулы II в конечное соединение. Взаимодействие соединения формулы (II) с аммиаком даже в кипящем диоксане требует четверо суток для завершения, и полученный выход не является очень удовлетворительным.
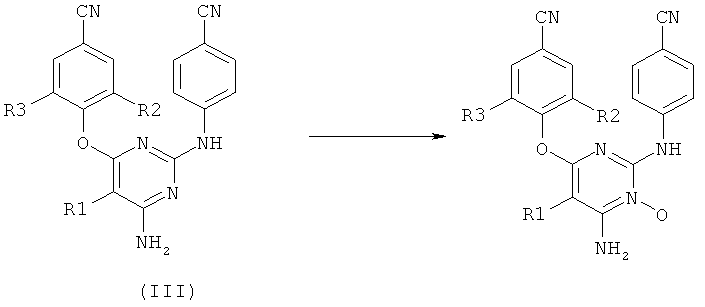
Недавно De Kock et al. в US 2008/0194602 сообщили, что производные диарилпиримидиноксида обладают ингибирующими свойствами на репликацию ВИЧ. Производные диарилпиримидиноксида получают из соответствующих производных диарилпиримидина формулы III с помощью N-окисления третичного азота пиримидинового цикла.
Имеются различные способы, описанные для синтеза производных диарилпиримидина формулы III, которые сведены на схеме 3.
Схема 3
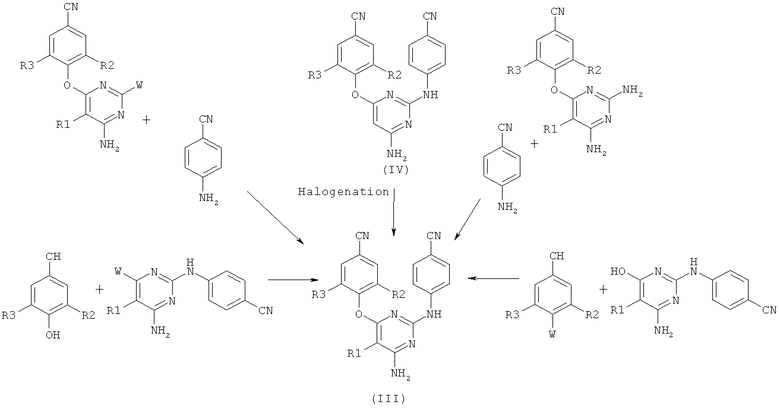
Halogenation - галогенирование
Одним из наиболее предпочтительных способов, описанных для синтеза производных диарилпиримидина формулы III, является галогенирование соединения формулы (IV). Также сообщается, что соединение (IV) получают из 4-аминобензонитрила и цианамида. Такое взаимодействие проводят в воде в присутствии сильной кислоты и получают 4-цианофенилгуанидин, который затем вводят во взаимодействие с алкилмалоновым эфиром в присутствии сильного основания и при повышенной температуре. Затем полученный 4,6-дигидроксипиримидин обрабатывают галогенирующим агентом. Затем производное пиримидина вводят во взаимодействие с 4-замещенным бензонитриллом и затем дополнительно с аммиаком, и получают промежуточные соединения формулы (IV).
Хотя способ, раскрытый в данном случае, относится к синтезу N-оксидных производных, а не конкретно этравирина, этот же способ можно использовать для синтеза этравирина. Однако указанный способ также страдает от такого ограничения, как использование в нем цианамида, который является высокотоксичным соединением.
Таким образом, из описанного выше очевидно, что хотя NNRTI, такие как этравирин, являются главной опорой терапии для лечения ВИЧ-инфекций, однако не сообщается о способах, которые являются безопасными, экономичными и удовлетворительными для коммерческого синтеза таких NNRTI. Следовательно, существует потребность в разработке альтернативного рентабельного и безопасного способа синтеза диарилпиримидиновых NNRTI.
Цели изобретения
Целью настоящего изобретения является простой рентабельный и эффективный способ синтеза диарилпиримидиновых NNRTI.
Другой целью настоящего изобретения является простой рентабельный и эффективный способ синтеза этравирина.
Еще одной целью настоящего изобретения является способ синтеза этравирина с использованием соединения формулы (V), т.е. 4-[(2,6-дихлор)-4-пиримидинилокси]-3,5-диметилбензонитрила.
Еще одной целью настоящего изобретения является способ синтеза соединения формулы (VI) 4-[[6-хлор-2-[(4-цианофенил)амино]-4-пиримидинил]окси]-3,5-диметилбензонитрила из соединения формулы (V), т.е. 4-[(2,6-дихлор)-4-пиримидинилокси]-3,5-диметилбензонитрила.
Раскрытие изобретения
В своих усилиях разработать простой рентабельный способ синтеза этравирина авторы настоящего изобретения обнаружили, что диарилпиримидиновые NNRTI можно получить с использованием недорогого исходного материала, такого как 2,4,6-трихлорпиримидин, и производного гидроксибензонитрила. Затем полученное производное пиримидина обрабатывают подходящими производными аминобензонитрила, и получают промежуточные диарилпиримидины, которые можно легко превратить в диарилпиримидиновые NNRTI, обладающие активностью против ВИЧ.
Авторы настоящего изобретения разработали простой и коммерчески практически осуществимый способ синтеза этравирина. Типичный способ синтеза этравирина включает стадии:
1) конденсации 2,4,6-трихлорпиримидина с 3,5-диметил-4-гидроксибензонитрилом с получением соединения формулы (V);
2) превращения соединения формулы (V) в соединение формулы (VI) конденсацией с 4-аминобензонитрилом;
3) аммонолиза соединения формулы (VI) с образованием соединения формулы (IV); и
4) галогенирования соединения формулы (IV) с образованием этравирина. Настоящее изобретение также относится к новому, простому и эффективному способу конверсии соединения формулы (V) в соединение формулы (VI). Типично, указанный способ включает конденсацию соединения формулы (V) с 4-аминобензонитрилом в подходящем растворителе с образованием соединения формулы (VI).
Таким образом, настоящее изобретение относится к подходящему для крупномасштабного производства простому, экономичному и эффективному способу синтеза диарилпиримидиновых NNRTI.
В другом аспекте настоящее изобретение относится к практически осуществимому коммерчески способу синтеза этравирина.
В еще одном аспекте настоящее изобретение относится к простому способу конверсии соединения формулы (V) в соединение формулы (VI).
Осуществление изобретения
Настоящее изобретение относится к простому, экономичному и практически осуществимому коммерчески способу получения диарилпиримидиновых NNRTI. В частности, настоящее изобретение относится к новому способу синтеза этравирина. Типично, настоящее изобретение относится к способу синтеза этравирина с использованием соединения формулы (V), т.е. 4-[(2,6-дихлор)-4-пиримидинилокси]-3,5-диметилбензонитрила, и 4-аминобензонитрила, как показано на схеме 4.
Схема 4
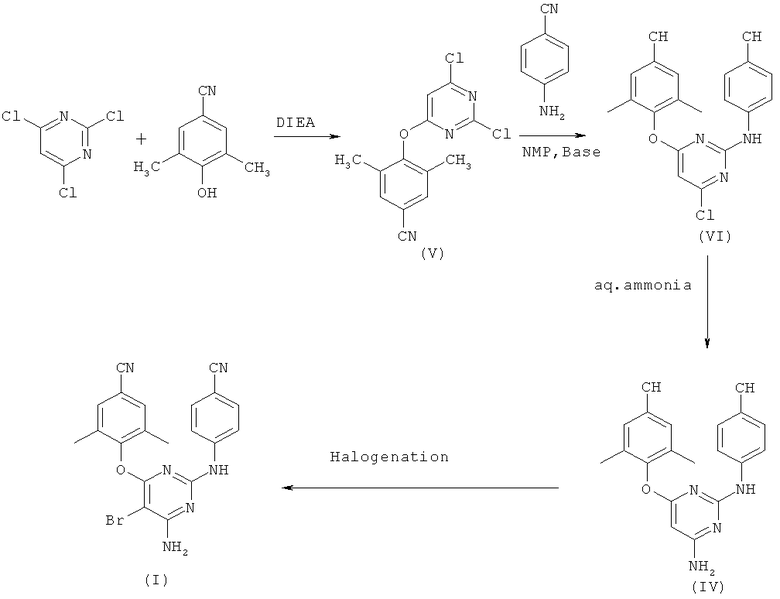
Base - основание, aq.ammonia - водн. аммиак, Halogenation - галогенирование.
В вышеуказанной процедуре 2,4,6-трихлорпиримидин вводят во взаимодействие с 3,5-диметил-4-гидроксибензонитрилом, с образованием соединения формулы (V), в инертном растворителе, таком как этанол, N-метил-2-пирролидон, N,N-диметилформамид, 1,4-диоксан, тетрагидрофуран, диметилсульфоксид, тетралин, сульфолан, ацетонитрил и т.п. Взаимодействие предпочтительно осуществляют при температурах образования флегмы и, необязательно, в присутствии основания. Предпочтительно в качестве растворителя используют 1,4-диоксан и в качестве основания N,N-диизопропилэтиламин.
Затем полученное соединение формулы (V) конденсируют с 4-аминобензонитрилом с образованием соединения формулы (VI). Конденсация соединения формулы (V) с 4-аминобензонитрилом является самой критической стадией по настоящему изобретению. Авторы настоящего изобретения нашли, что кислая среда типично неблагоприятна для такого взаимодействия. Когда указанную реакцию конденсации осуществляют в присутствии кислотного катализатора, такого как 1 N НСl, и в инертном растворителе, таком как диметилформамид или N-метилпирролидон, реакция не протекает так гладко, как желательно для получения нужных продуктов. Даже в присутствии неорганических оснований, таких как гидроксид натрия, гидроксид калия, карбонат натрия, карбонат калия, реакции не удаются. Присутствие органических оснований, таких как диэтиламин, пиридин, дибутилмочевина, также не приводит к инициации реакции конденсации.
Однако авторы настоящего изобретения неожиданно обнаружили, что реакцию конденсации соединения формулы (V) с 4-аминобензонитрилом с образованием соединения формулы (VI) можно осуществить в присутствии алкоксидов, таких как третичный бутоксид калия, третичный бутоксид натрия. Указанную реакцию можно осуществить в присутствии инертного растворителя с использованием алкоксида в качестве основания. Предпочтительно, в качестве растворителя используют N-метилпирролидон, и в качестве основания используют третичный бутоксид калия. Третичный бутоксид калия можно использовать для указанной реакции в количестве до четырех молярных эквивалентов. Предпочтительно, для проведения указанной реакции конденсации используют два молярных эквивалента.
Взаимодействие полученного таким образом соединения формулы (VI) с водным аммиаком в кипящем диоксане дает соединение формулы (IV). Неожиданно обнаружилось, что из-за отсутствия заместителя брома в пиримидиновом цикле в положении 5 указанное взаимодействие проходит до завершения за 10-12 часов вместо 96 часов, как указано в способе известного уровня техники. Предпочтительно для взаимодействия используют 25% водный раствор аммиака. Хотя реакцию осуществляют с использованием диоксана в качестве растворителя и температуры 120-130°С, однако для такого взаимодействия можно использовать другие инертные растворители, указанные в данном описании ранее.
Кроме того, авторы настоящего изобретения обнаружили, что соединение формулы (VI) можно очистить промывкой этилацетатом. Большинство нежелательных примесей и изомеров удаляется промывкой раствора этилацетатом. Типично обработку промывкой этилацетатом проводят при 60-70°С с последующей фильтрацией при комнатной температуре, и получают нужный продукт в чистой форме.
Затем полученное соединение (IV) можно легко превратить в нужный продукт этравирин гелогенированием его свободным галогеном, например, свободным бромом, или используя соединения доноры галогена. Реакцию галогенирования предпочтительно проводят в подходящем инертном растворителе. Предпочтительными растворителями являются метиленхлорид или эфир.
Таким образом, настоящее изобретение относится к эффективному, простому и рентабельному способу синтеза этравирина. Способ типично включает стадии:
1) конденсации 2,4,6-трихлорпиримидина с 3,5-диметил-4-гидроксибензонитрилом в присутствии основания в инертном растворителе с получением соединения формулы (V);
2) превращения соединения формулы (V) в соединение формулы (VI) конденсацией с 4-аминобензонитрилом с использованием алкоксида в качестве основания;
3) необязательно, очистки соединения формулы (VI);
4) аммонолиза соединения формулы (VI) с образованием соединения формулы (IV); и
5) галогенирования соединения формулы (IV) в инертном растворителе с образованием этравирина.
Таким образом, настоящее изобретение также относится к способу получения этравирина с использованием соединения формулы (V) и 4-аминобензонитрила.
Настоящее изобретение также относится к простому способу конденсации соединения формулы (V) с 4-аминобензонитрилом.
Принципы, предпочтительные воплощения и типы операций по настоящему изобретению описаны в описании далее. Однако не следует подразумевать, что изобретение, которое предполагается для охраны, ограничивается определенными раскрытыми формами, так как их следует рассматривать как иллюстративные, а не ограничительные. Вариации и изменения могут быть осуществлены специалистами в данной области техники без отхода от сущности изобретения.
Изобретение поясняется далее с помощью последующих пояснительных примеров, однако указанные примеры никоим образом не должны рассматриваться как ограничивающие объем изобретения.
Примеры
Пример 1
Синтез 4-[(2,6-дихлор)-4-пиримидинилокси]-3,5-диметилбензонитрила (соединение-V)
2,4,6-Трихлорпиримидин (100 г, 0,545 моль) растворяют в 1,4-диоксане (300 мл), и при перемешивании добавляют 3,5-диметил-4-гидроксибензонитрил (80,1 г, 0,545 моль). Добавление к полученному раствору N,N-диизопропилэтиламина (141,00 г, 1,09 моль) осуществляют в течение 30 минут. Реакционную массу греют при 70°С и перемешивают в течение 2,0 часов. Реакционную массу постепенно охлаждают до 15°С, и полученный продукт фильтруют при 12-15°С с последующей промывкой остатка на фильтре 50 мл 1,4-диоксана. Наконец, остаток на фильтре промывают водой (200 мл), и получают нужный продукт. Температура плавления 208-210°С.
Выход 128 г, выход в %=80%.
Пример 2
Синтез 4-[[6-хлор-2-[(4-цианофенил)амино]-4-пиримидинил]окси]-3,5-диметилбензонитрила (соединение - VI)
Соединение - V (100 г, 0,34 моль) растворяют в N-метилпирролидоне (500 мл), и при перемешивании добавляют 4-аминобензонитрил (40,12 г, 0,34 моль). Реакционную массу охлаждают до 0°С. Добавление к полученному раствору трет-бутоксида калия (76,3 г, 0,68 моль) осуществляют порциями в течение 1,0 часа при 0-10°С. Реакционную массу постепенно, в течение 1-2 часов, приводят к комнатной температуре. Затем реакционную массу постепенно добавляют в холодную воду (2,0 л), поддерживая температуру реакционной массы ниже 20°С. Реакционную массу фильтруют, и остаток на фильтре промывают 200 мл воды. Влажный остаток на фильтре снова разводят в 1,0 л воды при температуре ниже 20°С и фильтруют. Полученный продукт очищают с использованием этилацетата (2×300 мл) при 60-70°С с последующей фильтрацией при 10-15°С.
Выход 50 г.
Пример 3
Синтез 4-[[6-амино-2-[(4-цианофенил)амино]-4-пиримидинил]окси]-3,5-диметилбензонитрила (соединение - IV)
К раствору соединения - VI (100 г, 0,266 моль) в 1,4-диоксане (100 мл) добавляют водный аммиак (25%) (600 мл), и реакционную массу нагревают в автоклаве до 120°С и выдерживают при 120-125°С в течение 10-12 часов. Реакционную массу охлаждают до 50°С и снова нагревают до 70-80°С, при которых постепенно добавляют воду (200 мл). Реакционную массу постепенно охлаждают до 10°С и фильтруют, и получают влажный остаток на фильтре, который сушат, и получают нужный продукт.
Выход 75 г, выход в %=80%.
Пример 4
Синтез этравирина
Соединение - IV (100 г, 0,28 моль) растворяют в метиленхлориде (800 мл) и охлаждают до температуры 0-5°С. При 0-5°С постепенно добавляют жидкий бром (47,2 г, 0,294 моль), растворенный в 200 мл метиленхлорида. Реакционную массу перемешивают при 0-5°С в течение 4 час. К реакционной смеси добавляют холодную воду (800 мл), и доводят рН до 9-10 путем постепенного добавления раствора гидроксида натрия при 0-5°С. При 0-5°С добавляют раствор метабисульфита натрия, и реакционную массу перемешивают при 0-10°С в течение 1 часа, поддерживая рН реакционной массы при 8-9. Реакционную массу фильтруют, и остаток на фильтре промывают 200 мл воды. Влажный продукт сушат при 50-60°С и перекристаллизовывают из ацетона.
Выход 100 г.
Температура плавления 252-254°С.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИРУЮЩИЕ ВИЧ 2-(4-ЦИАНОФЕНИЛ)-6-ГИДРОКСИЛАМИНОПИРИМИДИНЫ | 2006 |
|
RU2401261C2 |
| ИНГИБИРУЮЩИЕ ВИЧ ПРОИЗВОДНЫЕ 2-(4-ЦИАНОФЕНИЛАМИНО)-ПИРИМИДИН-ОКСИДА | 2006 |
|
RU2398768C2 |
| Способ получения этравирина | 2019 |
|
RU2728555C1 |
| ПРОТИВОВИРУСНЫЕ СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ВИЧ ИНФЕКЦИИ | 2021 |
|
RU2780103C1 |
| СПОСОБ ПОЛУЧЕНИЯ 4-(1,6-ДИГИДРО-6-ОКСО-2-ПИРИМИДИНИЛ)АМИНО-БЕНЗОНИТРИЛА | 2006 |
|
RU2458056C2 |
| ПРОТИВОВИРУСНЫЕ СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ВИЧ-ИНФЕКЦИИ | 2020 |
|
RU2780101C2 |
| ИНГИБИРУЮЩИЕ ВИЧ ПРОИЗВОДНЫЕ 2-(4-ЦИАНОФЕНИЛАМИНО)ПИРИМИДИНА | 2006 |
|
RU2403245C2 |
| СПОСОБ ПОЛУЧЕНИЯ АРОМАТИЧЕСКОГО ГЕТЕРОЦИКЛИЧЕСКОГО СОЕДИНЕНИЯ, ПРИМЕНЯЕМОГО В КАЧЕСТВЕ СЕЛЕКТИВНОГО ИНГИБИТОРА КИНАЗ JAK3 и/или JAK1, И ПРИМЕНЕНИЕ АРОМАТИЧЕСКОГО ГЕТЕРОЦИКЛИЧЕСКОГО СОЕДИНЕНИЯ | 2015 |
|
RU2671195C2 |
| ПРОИЗВОДНЫЕ 2-(ПИПЕРИДИН-4-ИЛ)-4-ФЕНОКСИ- ИЛИ ФЕНИЛАМИНОПИРИМИДИНА В КАЧЕСТВЕ НЕНУКЛЕОЗИДНЫХ ИНГИБИТОРОВ ОБРАТНОЙ ТРАНСКРИПТАЗЫ | 2007 |
|
RU2469032C2 |
| СПОСОБ ВОССТАНОВЛЕНИЯ КАРБОНИЛСОДЕРЖАЩЕГО ПРОИЗВОДНОГО АКРИДИНА ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМОЙ КИСЛОТНО-АДДИТИВНОЙ СОЛИ | 1991 |
|
RU2069659C1 |
Настоящее изобретение относится к простому и рентабельному способу синтеза диарилпиримидинового ненуклеозидного ингибитора обратной транскриптазы, такого как этравирин. Способ получения этравирина включает стадии a) конденсации 2,4,6-трихлорпиримидина с 3,5-диметил-4-гидроксибензонитрилом в присутствии основания в инертном растворителе с получением 4-[(2,6-дихлор)-4-пиримидинилокси]-3,5-диметилбензонитрила (соединение формулы (V)); b) превращения соединения формулы (V), в 4-[[6-хлор-2-[(4-цианофенил)амино]-4-пиримидинил]окси]-3,5-диметилбензонитрил (соединение формулы (VI)), конденсацией с 4-аминобензонитрилом с использованием алкоксида в качестве основания; c) необязательно, очистки соединения формулы (VI); d) аммонолиза соединения формулы (VI) с образованием 4-[[6-амино-2-[(4-цианофенил)амино]-4-пиримидинил]окси]-3,5-диметилбензонитрила (соединение формулы (IV)) и e) бромирования соединения формулы (IV), в инертном растворителе с образованием этравирина. Предпочтительно стадию (a) осуществляют в присутствии 1,4-диоксана в качестве инертного растворителя, и N,N-диизопропилэтиламина в качестве основания; стадию (b) осуществляют в присутствии третичного бутоксида калия в качестве основания, с использованием инертного растворителя, выбранного из этанола, 1-метил-2-пирролидона, N,N-диметилформамида, 1,4-диоксана, тетрагидрофурана, диметилсульфоксида, тетралина, сульфолана и ацетонитрила. Преимущественно, инертный растворитель на стадии (b) представляет собой 1-метил-2-пирролидон. Стадию (c) обычно осуществляют с использованием для промывки этилацетата. Стадию (d) осуществляют с использованием водного раствора аммиака в 1,4-диоксане при температуре 120-130°C; стадию (e) осуществляют с использованием свободного галогена (брома). Способ позволяет получать целевые и промежуточные продукты с более высоким выходом. При этом повышение общего выхода целевого продукта достигается за более короткое время.
Типично этравирин синтезируют с использованием 2,4,6-трихлорпиримидина и 3,5-диметил-4-гидроксибензонитрила. Кроме того, раскрываются простые способы конденсации 4-аминобензонитрила с соединением формулы (V), т.е. 4-[(2,6-дихлор)-4-пиримидинилокси]-3,5-диметилбензонитрилом. 7 з.п. ф-лы, 4 пр.
1. Способ получения этравирина, включающий стадии:
a) конденсации 2,4,6-трихлорпиримидина с 3,5-диметил-4-гидроксибензонитрилом в присутствии основания в инертном растворителе с получением соединения формулы (V), т.е. 4-[(2,6-дихлор)-4-пиримидинилокси]-3,5-диметилбензонитрила;
b) превращения соединения формулы (V), т.е. 4-[(2,6-дихлор)-4-пиримидинилокси]-3,5-диметилбензонитрила, в соединение формулы (VI), т.е. 4-[[6-хлор-2-[(4-цианофенил)амино]-4-пиримидинил]окси]-3,5-диметилбензонитрил, конденсацией с 4-аминобензонитрилом с использованием алкоксида в качестве основания;
c) необязательно, очистки соединения формулы (VI), т.е. 4-[[6-хлор-2-[(4-цианофенил)амино]-4-пиримидинил]окси]-3,5-диметилбензонитрила;
d) аммонолиза соединения формулы (VI), т.е. 4-[[6-хлор-2-[(4-цианофенил)амино]-4-пиримидинил]окси]-3,5-диметилбензонитрила, с образованием соединения формулы (IV), т.е. 4-[[6-амино-2-[(4-цианофенил)амино]-4-пиримидинил]окси]-3,5-диметилбензонитрила; и
e) бромирования соединения формулы (IV), т.е. 4-[[6-амино-2-[(4-цианофенил)амино]-4-пиримидинил]окси]-3,5-диметилбензонитрила, в инертном растворителе с образованием этравирина.
2. Способ получения этравирина по п. 1, где стадию (a) осуществляют в присутствии 1,4-диоксана в качестве инертного растворителя, и N,N-диизопропилэтиламина в качестве основания.
3. Способ получения этравирина по п. 1, где стадию (b) осуществляют в присутствии третичного бутоксида калия в качестве основания.
4. Способ получения этравирина по п. 1, где стадию (b) осуществляют с использованием инертного растворителя, выбранного из этанола, 1-метил-2-пирролидона, N,N-диметилформамида, 1,4-диоксана, тетрагидрофурана, диметилсульфоксида, тетралина, сульфолана и ацетонитрила.
5. Способ получения этравирина по п. 6, где инертный растворитель представляет собой 1-метил-2-пирролидон.
6. Способ получения этравирина по п. 1, где стадию (c) осуществляют с использованием промывки этилацетатом.
7. Способ получения этравирина по п. 1, где стадию (d) осуществляют с использованием водного раствора аммиака в 1,4-диоксане при температуре 120-130°C.
8. Способ получения этравирина по п. 1, где стадию (e) осуществляют с использованием свободного галогена.
| WO 2006087387 A1, 24.08.2006 | |||
| Колосоуборка | 1923 |
|
SU2009A1 |
| DONALD W.LUDOVICI et al,.Evolution of Anti "HIV Drug Candidates.Part 3: DiarylPyrimidine(DAPY)Analogues, Bioorganic & Medicinal Chemistry Letters, 2001,11,2235-2239 | |||
| BART DESPIEGELEER et al., Synthesis and HPLC-purification of [77 Br]TNCl25-R165335(ETRAVIRINE), | |||

