Область техники, к которой относится изобретение
Данное изобретение относится к способу получения 4-[(1,6-дигидро-6-оксо-2-пиримидинил)амино]бензонитрила (I) с использованием в качестве исходного реагента эфира 4-оксо-1,6-дигидропиримидинилкарбоновой кислоты (II) или производного гуанидина, которое взаимодействует с эфиром алкоксиметиленмалоновой кислоты с образованием соединения (II), которое превращают в соединение (I), и последовательность этих реакций может быть осуществлена в одном реакторе.
Уровень техники
Вирус, вызывающий синдром приобретенного иммунодефицита (СПИД), обычно известен как вирус иммунодефицита человека (ВИЧ). Распространение ВИЧ вызывало и продолжает вызывать серьезные проблемы со здоровьем во всем мире. Был разработан ряд ингибирующих ВИЧ лекарственных средств, которые в настоящее время применяют для борьбы с вирусом. Было доказано, что эти лекарственные средства являются эффективными в подавлении вируса, в частности, когда их применяют в комбинированной терапии. Однако не существует терапии, которая бы могла полностью удалить вирус из организма.
В настоящее время доступны несколько классов ингибиторов ВИЧ, и исследуются новые классы ингибиторов. Одним таким классом являются ненуклеозидные ингибиторы обратной транскриптазы (NNRTIs). Этот класс включает ряд лекарственных средств, которые применяют в терапии против ВИЧ, в то время как другие NNRTIs находятся на различных стадиях разработки. Одним из таких средств является соединение 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрил, известное также как TMC278. Это соединение, его свойства, а также ряд синтетических подходов к его получению описаны в заявке WO-03/16306. TMC278, которое в настоящее время проходит клинические испытания, не только демонстрирует резко выраженную активность против дикого типа вируса, но также против многих вариантов его мутации.
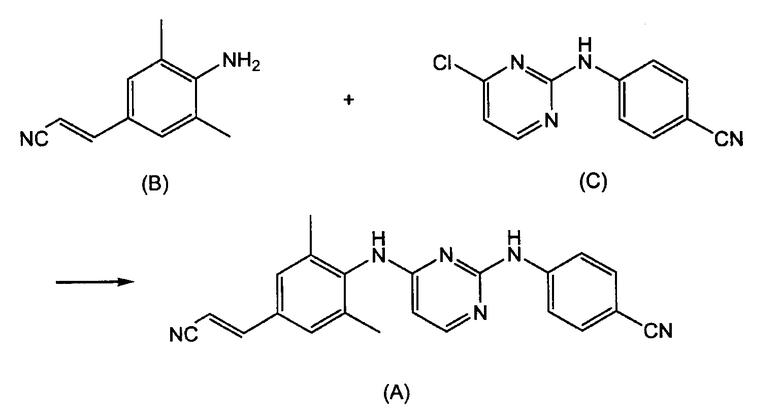
Поэтому существует необходимость в получении больших количеств этого активного ингредиента на основе способов, которые позволяют получать продукт с высоким выходом и с высокой степенью чистоты. Стратегия синтеза, которая была разработана для получения этого соединения, включает взаимодействие (E)-4-амино-3,5-диметилциннамонитрила (B) с анилинопиримидином (C) с получением TMC278, как изображено на следующей схеме реакции, где TMC278 представлено как соединение (A).
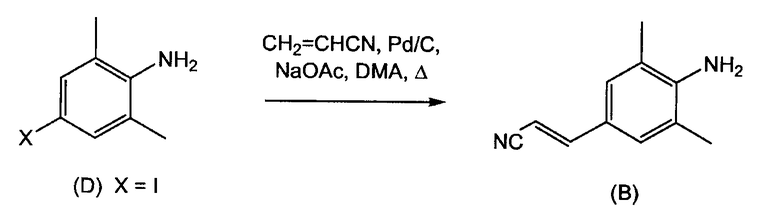
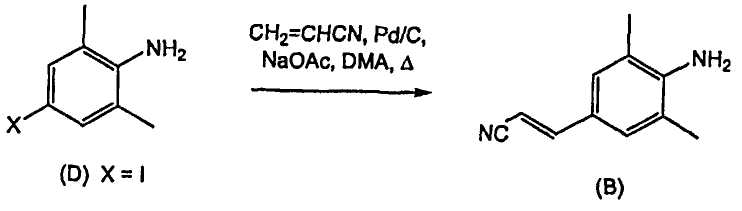
Получение промежуточного соединения (B) было описано в патентной заявке WO-04/016581, которое включает взаимодействие 4-йод-2,6-диметиланилина (D) (X = I) с акрилонитрилом в присутствии палладия на активированном угле, ацетата натрия в качестве основания и диметилацетамида в качестве растворителя.
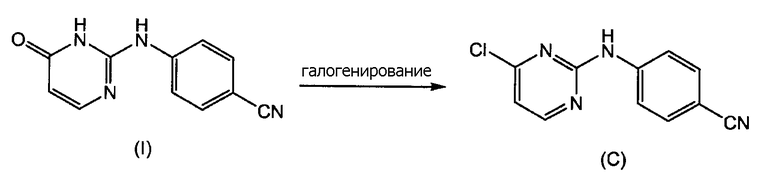
С другой стороны, получение промежуточного соединения (C) было описано в патентной заявке WO-03/16306, которое включает реакцию галогенирования 4-оксо-1,6-дигидропиримидина (I), в частности, POCl3.
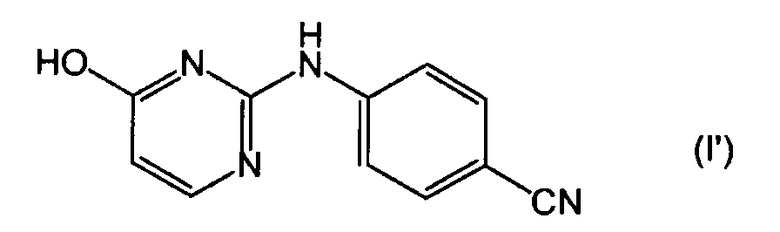
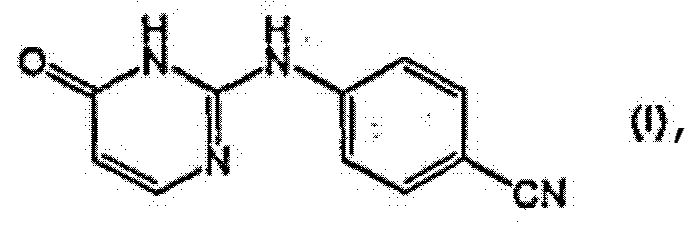
Соединение (I), которое иногда представляют в виде его таутомерной формы (I'):
может быть получено по методике, описанной в публикации Synthetic Communications, 27(11), 1943-1949 (1997):
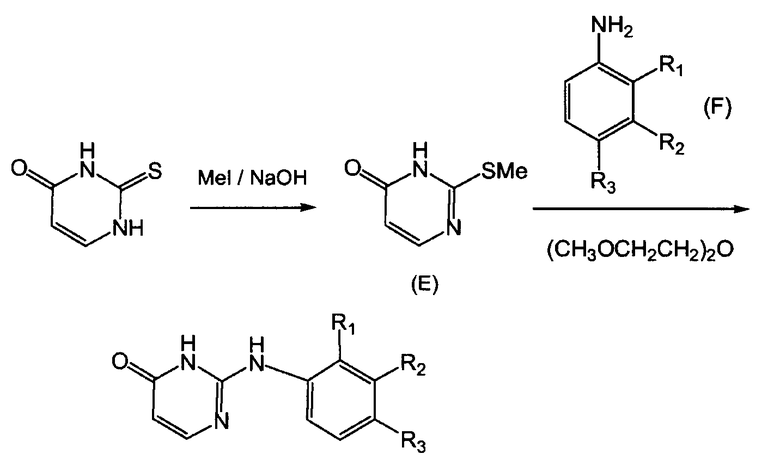
Взаимодействие соединения (E) с соединением (F) приводит к выделению метилмеркаптана, токсичного и исключительно неприятно пахнущего соединения, запах которого обнаруживается уже при концентрациях 2 ppb. Поэтому полное удаление этого меркаптана является требованием, представляющим очень трудную задачу очистки. Это делает такой способ непригодным для промышленного применения.
В патентной заявке WO-00/27825 на стр. 14 раскрыт синтез структурных аналогов соединения (I), содержащих заместитель Y в 5-положении и заместитель Q в 6-положении фрагмента пиримидина. Группа Y в этих структурных аналогах не может быть водородом и, в частности, является галогеном и никогда не является карбоксильной эфирной группой, как указано в способе настоящего изобретения. Более того, раскрываемый синтез в этом литературном источнике не содержит стадию декарбонилирования, которая является важной в способе настоящего изобретения.
В силу того, что вышеупомянутый способ известного уровня техники через промежуточные соединения (E) и (F) может быть применен для получения малых количеств требуемого продукта формулы (I), существует необходимость в способе, который можно было бы масштабировать для производства многих килограмм и больших количеств, который является воспроизводимым, экономически выгодным и с помощью которого получают конечный продукт с высоким выходом и высокой степенью чистоты. Целью настоящего изобретения является разработка такого способа.
Сущность изобретения
Настоящее изобретение относится к способу получения промежуточного соединения в синтезе TMC278, в частности, оно относится к способу получения соединения формулы:
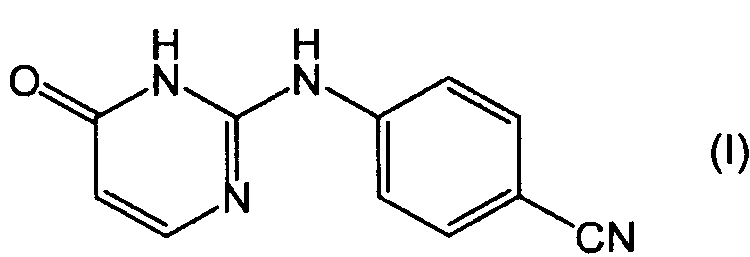
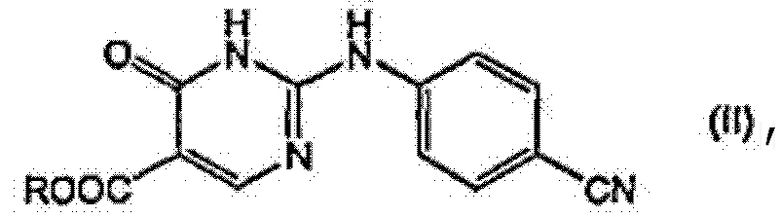
где соединение формулы (I) получают деалкоксикарбонилированием эфира 4-оксо-1,6-дигидропиримидинил-карбоновой кислоты формулы (II):
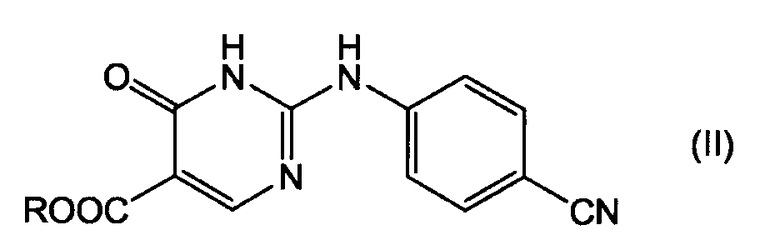 ,
,
где R является C1-4алкилом.
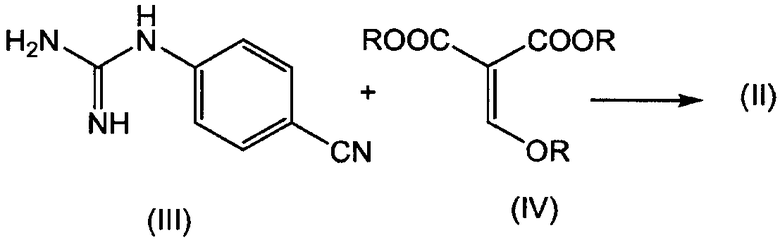
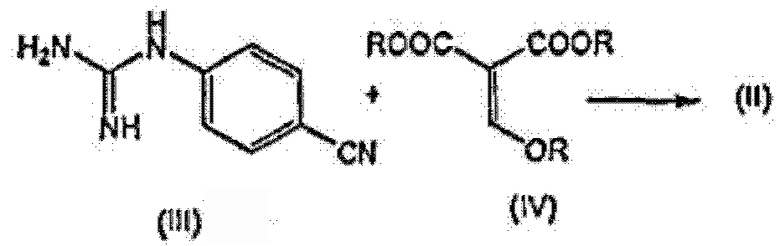
В дополнительном аспекте изобретение относится к способу получения указанного выше соединения формулы (I), в котором соединение формулы (I) получают конденсацией производного гуанидина формулы (III) с эфиром алкоксиметиленмалоновой кислоты формулы (IV), в результате получая указанное выше промежуточное соединение (II), которое деалкоксикарбонилируют с получением требуемого конечного продукта формулы (I), как показано на следующей схеме реакции, где каждый R, независимо от других радикалов R, представляет C1-4алкил:
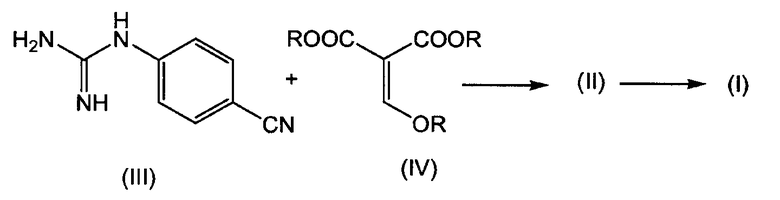
В одном варианте осуществления превращение соединения (III) через соединение (II) с получением соединения (I) проводят в одном реакторе без выделения промежуточного продукта (II).
Подробное описание изобретения
TMC278 существует в стереоизомерных формах, более конкретно в виде E- и Z-изомерных форм. Предпочтительной формой ТМC278 является E-изомер, то есть (E)-4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрил (далее называемый E-TMC278). Другим изомером является Z-изомер TMC278, то есть (Z)-4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрил (который обозначают как Z-TMC278). Всякий раз, когда здесь упоминается 'TMC278', имеется в виду E-форма, так же как и любая смесь обеих форм, которая преимущественно содержит E-форму, например, по меньшей мере, 80%, в частности, по меньшей мере, 90%, более характерно, по меньшей мере, 95% или даже, по меньшей мере, 99% E-формы.
Соединение (I) существует в двух таутомерных формах, то есть формах, имеющих структуру (I) и (I'). Для целей этого описания и формулы изобретения структуры (I) и (I') следует считать как обозначающие одну и ту же химическую структурную единицу, и как (I), так и (I') следует рассматривать в качестве эквивалентных представлений этой химической структурной единицы.
Используемый здесь каждый радикал R, независимо от других, представляет C1-4алкил. Последний определяет линейную или разветвленную цепь насыщенных углеводородных радикалов, имеющих от 1 до 4 углеродных атомов, таких как метил, этил, 1-пропил, 2-пропил, 1-бутил, 2-бутил, 2-метил-1-пропил, 2-метил-2-пропил. Особый интерес представляют C1-4алкильные радикалы, в которых углеродный атом, связанный с атомом кислорода, образует метиленовую группу. Предпочтительно, чтобы C1-4алкил являлся линейным C1-4алкилом (то есть н-C1-4алкилом, таким как н-пропил). Более предпочтительно, чтобы каждый R независимо выбирали из метила и этила. В одном варианте осуществления все радикалы R являются метилом, в другом варианте осуществления все радикалы R являются этилом.
Превращение из соединения (II) в соединение (I) включает реакцию деалкоксикарбонилирования с удалением CO2. Эта реакция может быть осуществлена в условиях, описанных Krapcho (смотрите, например, Krapcho et al., J. Org. Chem., 43, 138-147 (1978); Krapcho, Synthesis, 805-822, 893-914 (1982)).
В предпочтительных вариантах осуществления реакцию проводят в присутствии подходящей соли формулы MX, где M является металлом, аммонием или катионом замещенного аммония и X является анионом, имеющим нуклеофильные свойства. Например, M может быть ион щелочного или щелочноземельного металла, например, ион лития, натрия, калия, магния, кальция. Аммоний или замещенный аммоний включает, например, NH4+, четвертичный аммоний, где аммоний замещен алкилом (предпочтительно C1-4алкилом) и/или бензилом, например, тетра-н-бутиламмоний, триметилбензиламмоний, трибутилбензиламмоний. Подходящими X группами являются галогенидные анионы, в частности, хлорид и бромид; анионы карбоновой кислоты, в частности, C1-4алкилкарбоксилаты, такие как ацетат или пропионат; цианиды. Предпочтительными являются хлорид, бромид, ацетат и цианид, причем ацетат является особенно предпочтительным. Особый интерес представляют соли щелочных металлов с вышеупомянутыми анионами. Конкретными примерами солей MX, которые могут быть использованы, являются цианид натрия, ацетат натрия, хлорид натрия, бромид натрия, цианид калия, ацетат калия, хлорид калия, бромид калия, ацетат тетра-н-бутиламмония, цианид тетра-н-бутиламмония.
Превращение из соединения (II) в соединение (I) может быть проведено в подходящем реакционно инертном растворителе, причем предпочтительными растворителями являются биполярные апротонные растворители, такие как диметилформамид (ДМФ), диметилацетамид (ДМА), триамид гексаметилфосфорной кислоты (ГМФT), N-метилпирролидон (НМП), диметилсульфоксид (ДМСО), ацетонитрил и другие подобные растворители, включая их смеси. Особенно предпочтительным растворителем является НПМ.
В предпочтительном варианте осуществления способа этого изобретения реакцию деалкоксикарбонилирования соединения (II) проводят в НПМ в присутствии ацетатной соли, например, ацетата калия или, в частности, ацетата натрия. Этот конкретный вариант осуществления реакции деалкоксикарбонилирования не был до настоящего времени описан в литературе, и было доказано, что он является очень эффективным.
Реакция деалкоксикарбонилирования может быть проведена при повышенной температуре, например, при температуре в интервале приблизительно от 130°C до температуры кипения реакционной смеси с обратным холодильником, в частности, в интервале приблизительно от 140°C до 170°C, более характерно, в интервале от 140°C до 160°C. Предпочтительно, чтобы реакцию деалкоксикарбонилирования проводили в течение определенного периода времени, например, 24-120 часов, или, в частности, 24-48 часов, или, более характерно, 24-36 часов. В одном варианте осуществления реакцию проводят при повышенном давлении, что позволяет применять еще более высокие температуры, например, вплоть до 200°C, или микроволновый нагрев. Это позволяет уменьшить время реакции до нескольких часов, например, 2-6 часов.
В частности, упомянутые выше предпочтительные реакционные условия, то есть проведение реакции в НМП с ацетатной солью, такой как ацетат натрия или калия, позволяет использовать микроволновый нагрев или использовать вышеупомянутые повышенное давление и более высокие температуры, в результате снижая время реакции и в то же время получая требуемый конечный продукт с высоким выходом и чистотой.
В конце деалкоксикарбонилирования к реакционной смеси может быть добавлена кислота. Предпочтительно, чтобы кислоту добавляли тогда, когда стадия деалкоксикарбонилирования завершена. Предпочтительно, чтобы добавляемой кислотой являлась органическая кислота, такая как алкилугольная кислота, в частности, C1-4алкилугольная кислота, например, уксусная или пропионовая кислота. Не углубляясь в теорию, предполагают, что добавление кислоты разлагает соль пиримидинилоксида формулы
 ,
,
где M определено выше, с получением требуемого конечного продукта формулы (I). Предпочтительно добавлять кислоту к реакционной смеси при повышенной температуре, например, в интервале от 100°C до 150°C, в частности, от 120°C до 140°C.
Выход может быть дополнительно оптимизирован внесением получившегося продукта в низший алканол, такой как C1-4алканол, предпочтительно, этанол, и нагревания, предпочтительно, до кипения с обратным холодильником, алканольной смеси в течение периода, который может изменяться от пары минут до нескольких часов, например, от 10 минут до 3 часов, в частности, от 30 мин до 2 часов.
Промежуточное соединение формулы (II) может быть получено конденсацией производного гуанидина формулы (III) с эфиром алкоксиметиленмалоновой кислоты формулы (IV). Эта реакция может быть проведена в подходящем растворителе, предпочтительно, биполярном апротонном растворителе, таком как любой из растворителей, упомянутых выше в связи с реакцией превращения соединения (II) в соединение (I). Предпочтительным растворителем является НМП. Предпочтительными эфирами формулы (IV) являются диметиловый и диэтиловый эфиры метокси- и этоксиметиленмалоновой кислоты. Метильное производное часто называют как диметил(метоксиметилен)малонат. Взаимодействие соединения (III) с соединением (IV) предпочтительно проводить при повышенной температуре, например, при температуре в интервале от 70°C до 130°C, в частности, от 80°C до 120°C. Смеси позволяют реагировать в течение времени, если время регулируют, достаточного для завершения реакции, которое может изменяться, например, от 30 мин до 3 часов, в частности, от 1 до 2 часов.
Особым аспектом этого изобретения является способ получения соединения формулы (I) из промежуточных соединений (III) и (IV) с получением промежуточного соединения (II) и последующего деалкоксикарбонилирования соединения (II) с получением соединения (I). Каждая из стадий этого способа может быть проведена в одном и том же растворителе или смеси растворителей и с использованием одних и тех же реакционных условий, описанных выше для получения конечного продукта (I) из соединения (II) и реакции соединения (III) с соединением (IV).
В одном варианте осуществления этот способ реализуют в одном реакторе без выделения промежуточного соединения (II). Методика осуществления реакций в одном реакторе может включать использование одного и того же растворителя. Конкретным растворителем, подходящим для осуществления этого способа в одном реакторе, является НПМ. Такой вариант осуществления способа позволяет синтезировать соединение (I) быстро, просто и прямым путем.
В одном варианте осуществления используемую при деалкоксикарбонилировании соль добавляют в реакционную смесь, используемую при конденсации соединения (III) с соединением (IV), то есть в начале синтеза. Это дает практическое преимущество в том, что соль не нужно добавлять в реактор в середине синтеза, то есть после окончания реакции соединения (III) с соединением (IV). Кроме того, было обнаружено, что соль не мешает протеканию реакции конденсации соединения (III) с соединением (IV). Конкретной солью, подходящей для осуществления способа в одном реакторе, является ацетатная соль, такая как ацетат щелочного металла, например, ацетат натрия.
Дополнительной характерной чертой настоящего изобретения является тот факт, что промежуточные соединения формулы (II) представляют собой новые соединения. Поэтому, в дополнительном аспекте, изобретение предлагает соединение формулы (II), имеющее указанную выше химическую структуру, где R определен выше, а также его соли присоединения кислоты. Упоминаемый здесь термин "соли присоединения кислоты" означает, что он включает любые стабильные соли, которые способны образовать промежуточные соединения формулы (II). Предпочтительными являются фармацевтически приемлемые соли присоединения кислоты, которые являются нетоксичными формами солей присоединения кислоты. Соли могут быть легко получены путем обработки основной формы такими соответствующими кислотами, как неорганические кислоты, например, галогенводородные кислоты, например хлористоводородная, бромистоводородная и другие подобные кислоты; серная кислота; азотная кислота; фосфорная кислота и другие подобные кислоты; или органические кислоты, например, уксусная, пропановая, гликолевая, 2-гидроксипропановая, 2-оксопропановая, щавелевая, малоновая, янтарная, малеиновая, фумаровая, яблочная, винная, 2-гидрокси-l,2,3-пропан-трикарбоновая, метансульфоновая, этансульфоновая, бензолсульфоновая, 4-метилбензолсульфоновая, циклогексансульфаминовая, 2-гидроксибензойная, 4-амино-2-гидроксибензойная и другие подобные кислоты. С другой стороны, солевая форма может быть превращена путем обработки щелочью в форму свободного основания. Термин "соли присоединения" также включает гидраты или сольваты, которые способны образовывать соединения формулы (I), включая, например, алкоголяты, такие как метилаты или этилаты.
Способ настоящего изобретения можно масштабировать для производства многих килограмм или больших количеств, и он является воспроизводимым и экономически эффективным. Требуемый конечный продукт получают с высоким выходом и с высокой степенью чистоты. Дополнительными преимуществами, которые следует упомянуть, являются доступность исходных материалов и реагентов, которые могут выпускаться промышленностью или могут быть легко синтезированы.
Промежуточное соединение формулы (I) может также быть использовано в синтезе TMC120, то есть 4-[[4-[(2,4,6-триметилфенил)амино]-2-пиримидинил]амино]бензонитрила, который является дополнительным NNRTI, и в настоящее время разрабатывается в качестве бактерицида для предотвращения передачи ВИЧ инфекции, как раскрыто в патентной заявке WO-03/094920. TMC120, его синтез и свойства описаны в патентной заявке WO-99/50250. Для получения TMC120 соединение (I) превращают в соединение (C), как описано выше, и последнее соединение (C) подвергают взаимодействию с 2,4,6-триметиланилином, получая в результате соединение TMC120. Аналогичные аналоги TMC278 и TMC120 могут быть получены аналогичным способом.
Содержание всей цитируемой литературы приводится в этом описании путем ссылок на нее.
Предполагается, что следующие примеры иллюстрируют настоящее изобретение, но не ограничивают его.
Примеры
Пример 1
Смесь 64 г (0,4 моль) (4-цианофенил)гуанидина, 98,4 г (1,2 моль) ацетата натрия и 76,6 г (0,44 моль) диметил(метоксиметилен)малоната в 600 мл N-метилпирролидона (НМП) нагревали до 100°C и перемешивали в течение 1 часа при этой температуре. Добавляли 64,8 мл деминерализованной воды и реакционную смесь дополнительно нагревали до температуры кипения с обратным холодильником. Около 100 мл растворителя испаряли до тех пор, пока температура реакционной смеси не достигала величины в интервале от 155°C до 160°C. Далее реакционную смесь кипятили с обратным холодильником в течение 30 часов. Давали возможность смеси остыть до 20-25°C и добавляли 25 г средства, облегчающего фильтрацию. После перемешивания смеси в течение 1 часа при 20-25°C отфильтровывали осадок и промывали его 40 мл НМП. Растворитель отгоняли под вакуумом и остаток нагревали до 120°C. Добавляли к нагретому остатку по каплям 300 мл уксусной кислоты (в течение 15 минут) при поддержании температуры 130°C. После добавления уксусной кислоты смесь нагревали до 150°С и перемешивали при этой температуре в течение 15 минут. Затем давали возможность смеси остыть до 20-25°С. Образовавшийся осадок отфильтровывали и промывали этанолом (1×200 мл и 1×80 мл). Добавляли к промытому осадку 400 мл этанола и эту смесь нагревали и кипятили с обратным холодильником в течение 1 часа. После охлаждения до 20-25°С отфильтровывали осадок, промывали его 100 мл этанола и сушили при 50°С под вакуумом в течение 16 часов. Выход: 65,6 г 4-[(1,4-дигидро-4-оксо-2-пиримидинил)-амино]бензонитрила.
Пример 2
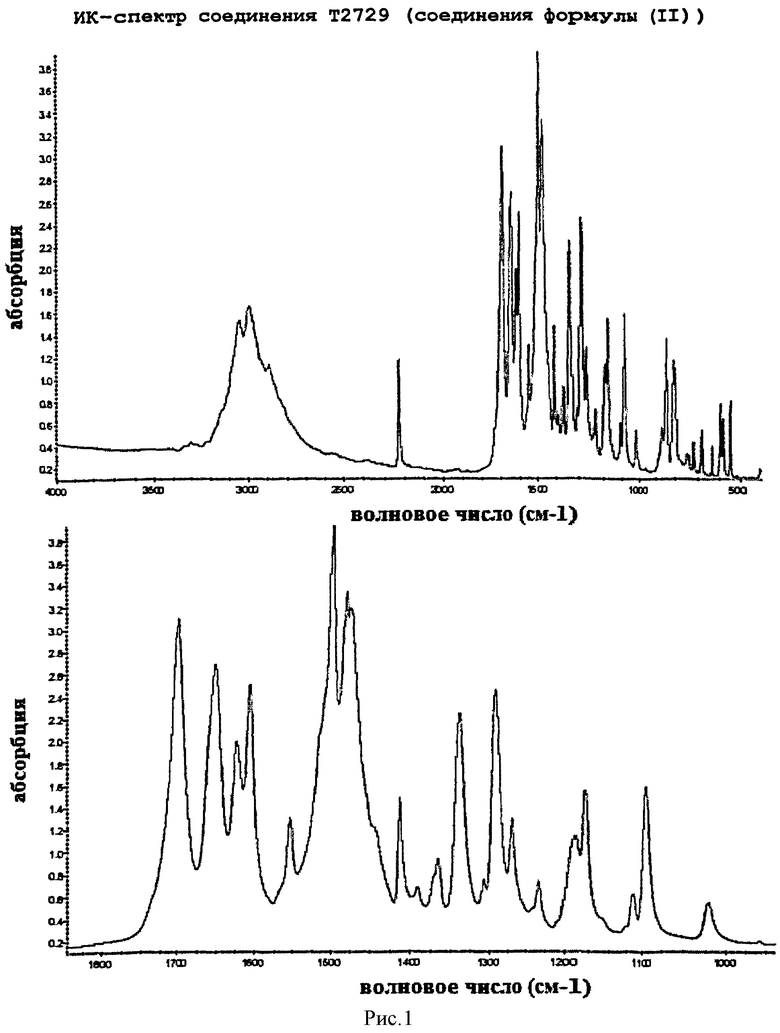
Смесь 64 г (0,4 моль) (4-цианофенил) гуанидина, 98,4 г (1,2 моль) ацетата натрия и 76,6 г (0,44 моль) диэтил(этоксиметилен)малоната в 600 мл N-метилпирролидона (НМП) нагревали до 100°С и перемешивали в течение 1 часа при этой температуре. Добавляли 81 мл деминерализованной воды и реакционную смесь дополнительно нагревали до температуры кипения с обратным холодильником. Около 120 мл растворителя испаряли до тех пор, пока температура реакционной смеси не достигала величины в интервале от 155°С до 160°С. Далее реакционную смесь кипятили с обратным холодильником в течение 72 часов. Давали возможность смеси остыть до 20-25°С и добавляли 25 г средства, облегчающего фильтрацию. После перемешивания смеси в течение 1 часа при 20-25°С отфильтровывали осадок и промывали его 50 мл НМП (полученное промежуточное соединение характеризуется в инфракрасном спектре (рис.1) полосами поглощения карбонильной группы остатка сложного эфира с волновым числом 1702 см-1 и кето-формы указанной структуры с волновым числом 1652 см-1). Растворитель отгоняли под вакуумом и остаток нагревали до 130°С. Добавляли к нагретому остатку по каплям 375 мл уксусной кислоты (в течение 15 минут) при поддержании температуры 130°С. После добавления уксусной кислоты смесь нагревали до 150°С и перемешивали при этой температуре в течение 15 минут. Затем давали возможность смеси остыть до 20-25°С. Образовавшийся осадок отфильтровывали и промывали этанолом (1×250 мл и 1×100 м). Добавляли к промытому осадку 500 мл этанола и эту смесь нагревали и кипятили с обратным холодильником в течение 1 часа. После охлаждения до 20-25°С отфильтровывали осадок, промывали его 100 мл этанола и сушили при 50°С под вакуумом в течение 16 часов. Выход: 80,5 г 4-[(1,4-дигидро-4-оксо-2-пиримидинил)-амино]бензонитрила (75,9%).
| название | год | авторы | номер документа |
|---|---|---|---|
| КРИСТАЛЛИЧЕСКАЯ ФОРМА 4-[[4-[[4-(2-ЦИАНОЭТЕНИЛ)-2,6-ДИМЕТИЛФЕНИЛ]АМИНО]-2-ПИРИМИДИНИЛ]АМИНО]БЕНЗОНИТРИЛА | 2008 |
|
RU2498979C2 |
| 5-ЗАМЕЩЕННЫЕ 1-ФЕНИЛ-1,5-ДИГИДРОПИРИДО[3,2-b]ИНДОЛ-2-ОНЫ И АНАЛОГИ КАК ПРОТИВОВИРУСНЫЕ ПРЕПАРАТЫ | 2005 |
|
RU2362776C2 |
| 6,7,8,9-ЗАМЕЩЕННЫЕ 1-ФЕНИЛ-1,5-ДИГИДРОПИРИДО (3,2-b) ИНДОЛ-2-ОНЫ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ АНТИИНФЕКЦИОННЫХ ФАРМАЦЕВТИЧЕСКИХ СРЕДСТВ | 2005 |
|
RU2377243C2 |
| ЛИОФИЛИЗИРОВАННЫЕ ЛЕКАРСТВЕННЫЕ НАНОСУСПЕНЗИИ | 2012 |
|
RU2643062C2 |
| ПРОИЗВОДНЫЕ 4-АМИНОПИРИМИДИНА | 2008 |
|
RU2489430C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЗОТСОДЕРЖАЩЕГО ГЕТЕРОЦИКЛИЧЕСКОГО СОЕДИНЕНИЯ И ЕГО ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ | 2016 |
|
RU2717830C2 |
| ДИСПЕРГИРУЕМЫЕ КОМПОЗИЦИИ | 2017 |
|
RU2826218C2 |
| Способ получения соединений 7H-пирроло[2,3-d]пиримидина | 2017 |
|
RU2699034C1 |
| КОНДЕНСИРОВАННОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ | 2005 |
|
RU2389731C2 |
| ИНГИБИТОР ГЕКСОН-ГЛЮКОКИНАЗЫ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2797185C2 |
Изобретение относится к улучшенному способу получения 4-[(1,6-дигидро-6-оксо-2-пиримидинил)амино]бензонитрила формулы (I)
где R является С1-4алкилом.
Способ заключается в деалкоксикарбонилировании эфира 4-оксо-1,6-дигидропиримидинилкарбоновой кислоты (II) в присутствии соли MX в подходящем инертном растворителе, где М является металлом или аммонием или замещенным аммонием и X является анионом, имеющим нуклеофильные свойства. Соединение формулы (II) является новым и может быть получено взаимодействием производного гуанидина формулы (III) с эфиром алкоксиметиленмалоновой кислоты эфира (IV) по следующей схеме:
Полученное указанным способом соединение (I) подвергают галогенированию для получения соединения (С), которое используют в реакции с соединением (В) для получения Е-4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила (ТМС278), обладающего противовирусным действием в случае ВИЧ-инфекции. Способ позволяет получить конечный продукт с высоким выходом и высокой степенью чистоты. 3 н. и 11 з.п. ф-лы, 2 пр., 1 ил.
1. Способ получения соединения формулы
в котором соединение формулы (I) получают деалкоксикарбонилированием эфира 4-оксо-1,6-дигидропиримидинил-карбоновой кислоты формулы (II):
где R является С1-4алкилом,
в присутствии соли в подходящем инертном растворителе,
где соль представлена формулой MX, где М является металлом или аммонием или замещенным аммонием, и X является анионом, имеющим нуклеофильные свойства.
2. Способ получения соединения формулы (I) по п.1, в котором соединение формулы (II) получают конденсацией гуанидина формулы (III) с эфиром алкоксиметиленмалоновой кислоты формулы (IV) в присутствии подходящего инертного растворителя, как изображено на следующей схеме реакции, где каждый R независимо выбран из С1-4алкила:
3. Способ по п.2, в котором превращение из соединения (III) через соединение (II) с получением соединения (I) проводят в одном реакторе без выделения промежуточного соединения (II).
4. Способ по пп.1-3, в котором R является метилом или этилом.
5. Способ по пп.1 и 2, в котором М является щелочным металлом, аммонием или замещенным аммонием, и X является галогенидом, цианидом или С1-4алкилкарбоксилатом.
6. Способ по п.5, в котором М является щелочным металлом, и X является С1-4алкилкарбоксилатом.
7. Способ по пп.1-3, в котором процесс осуществляют в биполярном апротонном растворителе.
8. Способ по п.7, в котором растворитель выбирают из диметилформамида (ДМФ), диметилацетамида (ДМА), триамида гексаметилфосфорной кислоты (ГМФТ), N-метилпирролидона (НМП), диметилсульфоксида (ДМСО) и ацетонитрила.
9. Способ по п.1, в котором в конце стадии деалкоксикарбонилирования к реакционной смеси добавляют кислоту.
10. Способ по п.9, в котором в конце стадии деалкоксикарбонилирования к реакционной смеси добавляют алкилугольную кислоту.
11. Соединение формулы
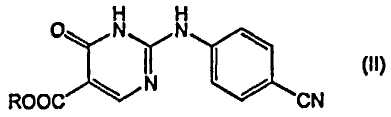
или его фармацевтически приемлемая соль, где R является С1-4алкилом.
12. Соединение по п.11, где R является метилом.
13. Соединение по п.11, где R является этилом.
14. Способ получения Е-4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила (ТМС278), включающий взаимодействие (Е)-4-амино-3,5-диметилциннамонитрила (В) с анилинопиримидином (С) с получением ТМС278, как изображено на следующей схеме реакции, где ТМС278 представлено как соединение (А):
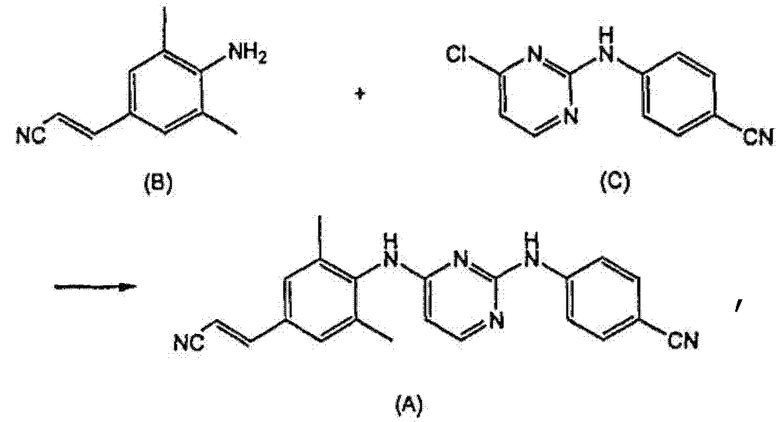
где используют соединение (В), полученное взаимодействием 4-йод-2,6-диметиланилина (D) (Х=1) с акрилонитрилом в присутствии палладия на активированном угле, ацетата натрия в качестве основания и диметилацетамида в качестве растворителя
и где используют соединение (С), полученное реакцией галогенирования 4-оксо-1,6-дигидропиримидина (I) с помощью РОСl3
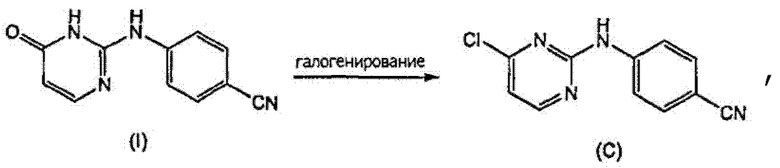
где соединение (I) получено способом по п.1.
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| WO 00/27825 A1, 18.05.2000 | |||
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| ПРОИЗВОДНЫЕ ЧЕТВЕРТИЧНОГО АМИНОПИРИМИДИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1990 |
|
RU2108329C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ АРОМАТИЧЕСКИХ НИТРИЛОВ | 1991 |
|
RU2037483C1 |

