Настоящее изобретение относится к 2-(4-цианофенил)-6-гидроксиламинопиримидинам, обладающим ингибирующими репликацию ВИЧ (вирус иммунодефицита человека) свойствами. Данное изобретение, далее, относится к способам получения этих пиримидинов и фармацевтическим композициям, содержащим эти соединения, и их применению для предотвращения или лечения инфекции ВИЧ.
Резистентность вируса ВИЧ к доступным в настоящее время лекарственным средствам против ВИЧ продолжает оставаться основной причиной неблагоприятного исхода терапии. Это привело к введению комбинированной терапии из двух или более анти-ВИЧ-агентов, как правило, имеющих различный профиль активности. Значительный прогресс достигнут путем введения HAART-терапии (высокоактивная антиретровирусная терапия), которая приводит к существенному снижению заболеваемости и смертности в случае подвергаемых с ее помощью лечению ВИЧ-пациентов. HAART-терапия включает различные комбинации ингибиторов нуклеозидной обратной транскриптазы (NRTI), ингибиторов ненуклеозидной обратной транскриптазы (NNRTI) и ингибиторов протеазы (РI). Современные директивы в отношении антиретровирусной терапии рекомендуют такую схему тройной комбинированной терапии даже для первоначального лечения. Однако эти мультилекарственные терапии не всегда эффективны и никогда полностью не уничтожают ВИЧ. Описано, что половина пациентов, подвергаемых комбинированной анти-ВИЧ-терапии, совершенно не поддается лечению, главным образом, вследствие резистентности вируса к одному или более используемым лекарственным средствам. Переход к альтернативным комбинациям, как правило, обусловливает временное облегчение, но любая форма продолжительного лечения по окончании не будет иметь успеха вследствие развития резистентности ко многим лекарственным средствам. Кроме того, показано, что резистентный вирус переносится на вновь инфицируемых индивидуумов, что приводит к вариантам строго ограниченной терапии для этих, не поддающихся воздействию лекарственных средств, пациентов.
Ферменты-мишени в случае вируса ВИЧ способны мутировать в таком направлении, что известные лекарственные средства становятся менее эффективными или даже неэффективными против этих мутантных вирусов ВИЧ. Или, иначе говоря, вирус ВИЧ вызывает даже возрастающую резистентность против доступных лекарственных средств. Все больше штаммов ВИЧ, резистентных к NNRTI, обнаруживаемых у пациентов, не поддающихся анти-ВИЧ-терапии, являются дважды или даже многократно мутировавшими штаммами. Такие штаммы-мутанты ВИЧ имеют две или более мутации в гене обратной транскриптазы и, следовательно, проявляют сильную резистентность по отношению к базирующейся на NNRTI терапии.
Вследствие их способности к быстрой мутации и провоцированию резистентности по отношению к существующим лекарственным терапиям имеется непрерывная потребность в новых комбинациях активных ингредиентов, которые эффективны против ВИЧ. В особенности, существует постоянная необходимость в новых типах эффективных против ВИЧ активных ингредиентов, различающихся по химической структуре и профилю активности, для применения в новых видах комбинированной терапии. Существует особая необходимость в новых типах эффективных против ВИЧ активных ингредиентов, которые активны против дважды или многократно мутировавших штаммов ВИЧ. Обнаружение таких активных ингредиентов, следовательно, является очень желательной для достижения целью.
Настоящее изобретение относится к отдельной новой серии бисарилзамещенных производных пиримидина, которые могут найти применение в ВИЧ-терапии, в особенности в качестве нового компонента лекарственных комбинаций. Бисарилзамещенные пиримидины, обладающие ингибирующими репликацию ВИЧ свойствами, известны из WO 00/27825.
Новая серия производных пиримидина согласно настоящему изобретению проявляет превосходные в отношении ингибирования репликации ВИЧ свойства, в особенности против штаммов ВИЧ, имеющих двойные или многократные мутации в гене обратной транскриптазы.
Настоящее изобретение относится к соединению формулы (I):
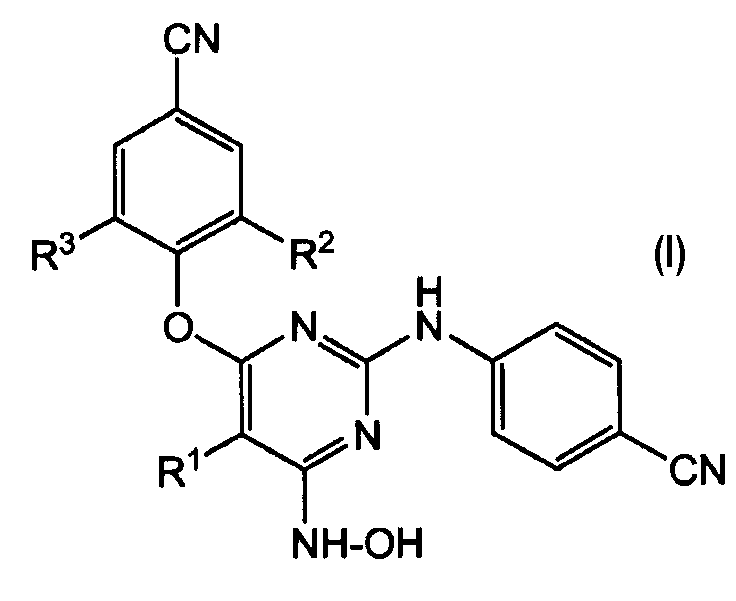
его фармацевтически приемлемым аддитивным солям или его стереохимически изомерной форме, где
R1 означает галоген;
R2 и R3, каждый независимо, означает С1-6-алкил.
Как используется в данном контексте, термин «С1-4-алкил» означает насыщенные углеводородные радикалы с линейной или разветвленной цепью, имеющие 1-4 атома углерода, такие как метил, этил, 1-пропил, 2-пропил, 1-бутил, 2-бутил, 2-метил-1-пропил и т.п.; термин «С1-6-алкил» включает С1-4-алкильные радикалы и их высшие гомологи, имеющие 5 или 6 атомов углерода, такие как 1-пентил, 2-пентил, 3-пентил, 1-гексил, 2-гексил, 2-метил-1-бутил, 2-метил-1-пентил и т.п. Представляющими интерес среди С1-6-алкильных радикалов являются С1-4-алкильные радикалы.
Термин «галоген» включают фтор, хлор, бром и йод.
Для терапевтического применения соли соединений формулы (I) представляют собой такие, где противоион является фармацевтически приемлемым. Однако соли кислот и оснований, которые являются фармацевтически неприемлемыми, также могут находить применение, например при получении или очистке фармацевтически приемлемого соединения. Все соли, являются ли они фармацевтически приемлемыми или нет, входят в рамки настоящего изобретения.
Термин «фармацевтически приемлемые аддитивные соли», как используется в данном контексте, включает терапевтически активные, нетоксичные, кислотно-аддитивные солевые формы, которые способны образовывать соединения формулы (I). Последние пригодным образом могут быть получены путем обработки основной формы такими подходящими кислотами, как неорганические кислоты, например галогенводородные кислоты, например соляная кислота, бромистоводородная кислота и т.п.; серная кислота; азотная кислота; фосфорная кислота и т.п.; или органические кислоты, например уксусная кислота, пропановая кислота, гидроксиуксусная кислота, 2-гидроксипропановая кислота, 2-оксопропановая кислота, щавелевая кислота, малоновая кислота, янтарная кислота, малеиновая кислота, фумаровая кислота, яблочная кислота, винная кислота, 2-гидрокси-1,2,3-пропантрикарбоновая кислота, метансульфоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота, 4-метилбензолсульфоновая кислота, циклогексансульфаминовая кислота, 2-гидроксибензойная кислота, 4-амино-2-гидроксибензойная кислота и т.п. кислоты. Наоборот, солевая форма может быть превращена в форму свободного основания путем обработки щелочью. Соединения формулы (I), содержащие кислотные протоны, можно превращать в их терапевтически активные, нетоксичные, аддитивные солевые формы с металлом или амином путем обработки соответствующими органическими или неорганическими основаниями.
Термин «аддитивная соль» также включает гидраты и аддитивные формы с растворителями, которые способны образовывать соединения формулы (I). Примерами таких форм являются, например, гидраты, алкоголяты и т.п.
Некоторые из соединений формулы (I) и их аддитивных солей могут иметь один или более хиральных центров и существовать в виде стереохимически изомерных форм. Стереоизомеры могут существовать, когда R2 и R3 означают С4-6-алкил. Термин «стереохимически изомерные формы», как используется в данном контексте, включает все возможные стереоизомерные формы, которые могут иметь соединения формулы (I) и их аддитивные соли. Подразумевают, что стереохимически изомерные формы соединений формулы (I) входят в рамки данного изобретения.
Предпочтительные подгруппы этих соединений являются такими соединениями формулы (I), как описано выше, или любой подгруппой соединений формулы (I), описанной в данном контексте, где R1 означает хлор или бром, более предпочтительно, где R1 означает бром.
Другие предпочтительные подгруппы соединений являются такими соединениями формулы (I), как описано выше, или любой подгруппой соединений формулы (I), описанной в данном контексте, где R2 и R3 означают С1-4-алкил, более предпочтительно, где R2 и R3 означают метил. Представляющими особый интерес являются такие соединения, где R1 означает бром, а R2 и R3 означают метил; или где R1 означает хлор, а R2 и R3 означают метил.
Вообще, соединения формулы (I) можно получать путем взаимодействия производного пиримидина формулы (III) с защищенным гидроксиламином формулы NH2OP, таким образом получая промежуточный продукт (II), из которого впоследствии удаляют защитную группу. Группа W в производных пиримидина формулы (III) представляет собой подходящую удаляемую группу, такую как галоген, например хлор или бром, предпочтительно, она представляет собой хлор.

В альтернативном воплощении промежуточный продукт (III) может быть введен во взаимодействие с гидроксиламином при непосредственном получении соединений формулы (I), как указано на следующей реакционной схеме:

Подходящие защитные группы (обозначаемые в виде Р на вышеуказанной схеме) включают любую из гидроксилзащитных групп, используемых в уровне техники, включая таковые, которые могут быть удалены путем кислотного расщепления, такие как метоксиэтоксиметил (МЕМ), тетрагидропиранил (ТНР), трет-бутил (t-Bu) и т.п., или путем гидрирования, такие как бензил (Bz) и т.п., триалкилсилильные группы, такие как триметилсилил (TMS), трет-бутилдиметилсилил (TBDMS), триизопропилсилил (TIPS), трет-бутилдифенилсилил и т.п., которые можно отщеплять в кислых или щелочных условиях. Предпочтительной является ТНР-группа.
Взаимодействие исходного вещества (III) с защищенным гидроксиламином NH2OP можно осуществлять в подходящем растворителе, предпочтительно в присутствии основания, которое можно добавлять к улавливаемой кислоте, которая высвобождается во время протекания реакции, как, например, карбонат или гидрокарбонат щелочного металла, такой как карбонат калия, или органические основания, такие как триалкиламины, например триэтиламин. Подходящие растворители включают, например, ацетонитрил, спирты, например, этанол, 2-пропанол; полярные апротонные растворители, такие как N,N-диметилформамид, N,N-диметилацетамид, 1-метил-2-пирролидинон, ацетонитрил; простые эфиры, такие как 1,4-диоксан, монометиловый эфир пропиленгликоля, тетрагидрофуран. Предпочтительными эфирами являются простые эфиры, в частности тетрагидрофуран.
Группу Р в таким образом полученном промежуточном продукте (II) можно затем удалять с помощью известных в уровне техники способов. В случае, когда группа Р представляет собой ТНР, ее можно без труда удалять в кислых условиях, таких как при использовании галогенводородных кислот, таких как соляная кислота, при использовании сульфоновых кислот, однако, также при использовании смол с кислотными группами, таких как ионообменные смолы, содержащие сульфогруппу.
Соединения формулы (I) также можно получать непосредственно из соединения (III), используя гидроксиламин. Эту реакцию можно проводить, используя подобные условия как таковые, в случае соединения (III) с использованием защищенного гидроксиламина.
В вышеуказанных способах синтеза для получения соединений формулы (I) и также в следующих способах получения промежуточных продуктов R1 означает галоген, но также может представлять собой предшественник галоген-группы, такой как гидроксильная группа или защищенная гидроксильная группа (например, бензилокси), которую можно превращать в галоген-группу с помощью галогенирующего агента, такого как POCl3 или POBr3. Это может способствовать избежанию нежелательных побочных реакций.
Промежуточные продукты формулы (II) также можно получать путем взаимодействия промежуточного продукта формулы (IV) или формулы (VI) с промежуточным продуктом формулы (V) или формулы (VII), как показано на следующей реакционной схеме, где R1, R2 и R3 имеют значения, как определено для соединений формулы (I) или их любой подгруппы, и W представляет собой подходящую удаляемую группу, такую как, например, галоген, например хлор, бром и т.п. Промежуточные продукты формулы (II) можно превращать в конечные продукты формулы (I) путем снятия защиты. Альтернативно, можно использовать промежуточные продукты (IV) или (VI), в которых гидроксиламиногруппа является незащищенной, так что непосредственно получают соединения формулы (I).
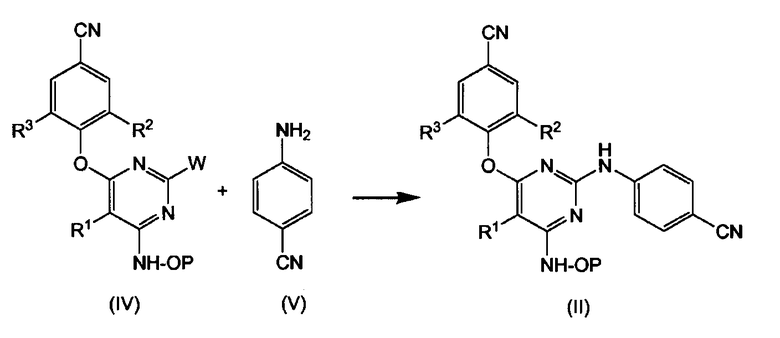

Реакцию производного пиримидина (IV), соответственно (VI), с цианоанилином (V), соответственно с цианофенильным производным (VII), предпочтительно проводят в подходящем растворителе, таком как, например, ацетонитрил, спирт, такой как, например, этанол, 2-пропанол, N,N-диметилформамид, N,N-диметилацетамид, 1-метил-2-пирролидинон; простой эфир, такой как 1,4-диоксан, монометиловый эфир пропиленгликоля. Взаимодействия можно проводить в кислых условиях, которые можно получать за счет добавляемых количеств подходящей кислоты, например камфорсульфокислоты, и подходящего растворителя, такого как, например, тетрагидрофуран или спирт, например этанол, 1- или 2-пропанол, или при использовании подкисленных растворителей, как, например, соляная кислота, растворенная в алканоле, таком как этанол, 1- или 2-пропанол.
В альтернативном воплощении незащищенные производные пиримидина (IV), т.е. промежуточные продукты (IV), где Р означает водород, могут быть введены во взаимодействие с соединением (V), таким образом непосредственно получая конечные продукты формулы (I). Для избежания побочных реакций предпочтительным является использование защищенных промежуточных продуктов (IV) и затем удаление группы Р.
Промежуточные продукты формулы (II) также можно получать путем взаимодействия цианофенильного производного (VIII) c производным пиримидина (IX) или путем взаимодействия цианофенильного производного (Х) с производным пиримидина (XI), как показано на следующих схемах.


На этих реакционных схемах R1, R2 и R3 имеют значения, как определено для соединений формулы (I) или любой их подгруппы, Р представляет собой защитную группу, как описано выше, и W представляет собой подходящую удаляемую группу, как описано выше. Эти реакции предпочтительно проводят в подходящем растворителе, в частности любом из растворителей, указанных выше в связи с реакцией соединения (IV) с соединением (V).
В альтернативном воплощении незащищенные производные пиримидина (IX) или (XI), т.е. промежуточные продукты (IX) или (XI), где Р означает водород, могут быть введены во взаимодействие с соединениями (VIII) или (X), таким образом непосредственно получая конечные продукты формулы (I). Для избежания побочных реакций предпочтительным является использование защищенных промежуточных продуктов (IX) или (XI) и затем удаление группы Р.
Соединения формулы (I), кроме того, можно получать путем превращения соединений формулы (I) друг в друга согласно известным в уровне техники реакциям превращения групп. Например, хлор-аналоги можно превращать в соответствующие бром-аналоги или, наоборот, путем реакций обмена галогена.
Некоторые из соединений формулы (I) и их некоторые промежуточные продукты-предшественники могут содержать асимметрический атом. Стереохимически чистые изомерные формы вышеуказанных соединений и вышеуказанных промежуточных продуктов можно получать путем использования известных из уровня техники способов.
Синтез некоторых промежуточных продуктов, используемых согласно предыдущим реакционным схемам, описан в дальнейшем, где на реакционных схемах R1, R2 и R3 имеют значения, как определено для соединений формулы (I) или любой их подгруппы, а W представляет собой подходящую удаляемую группу, в частности хлор или бром.
Исходные вещества формулы (III) можно получать, как описано в WO-00/27825. В особенности, их можно получать, как показано на следующей схеме:

Защищенный 4-цианофенол (VIII) вводят во взаимодействие с производным пиримидина (XII), где каждый W независимо представляет собой удаляемую группу, такую, как указанная выше.
Промежуточные продукты формулы (IV) можно получать, как показано на следующей реакционной схеме:

Подобным образом можно получать промежуточные продукты (VI), исходя из пиримидина (XIV), как показано на следующей схеме:
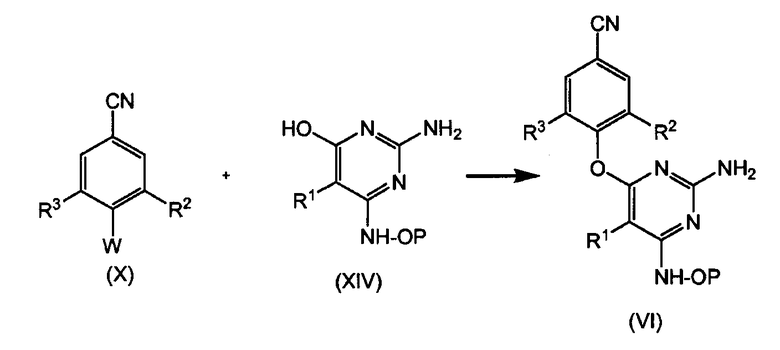
В случае вышеуказанного взаимодействия аминогруппу можно защищать или не защищать с помощью подходящей защитной группы. В альтернативном воплощении можно осуществлять взаимодействие незащищенных производных пиримидина (XIII) или (XIV), т.е. промежуточных продуктов (XIII) или (XIV), где Р означает водород, с соединениями (VIII) или (X). Для избежания побочных реакций предпочтительным является использование защищенных промежуточных продуктов (XIII) или (XIV) и затем удаление группы Р.
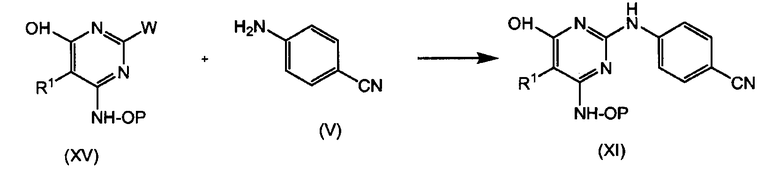
Промежуточные продукты (IX) можно получать путем конденсации производного пиримидина (XIII) с цианоанилином (V), как показано на следующей схеме. Если желательно избежать побочных реакций, W-группа, которая является нереагирующей, и/или R1 могут быть предшественником галогена, как указано выше.

Промежуточные продукты (XI) можно получать путем конденсации производного пиримидина (XV) с цианоанилином (V), как показано на следующей схеме. Если желательно избежать побочных реакций, гидроксигруппа в соединении (XV) может быть защищена и/или R1 может быть предшественником галогена, как указано выше.
В дальнейшем аспекте данное изобретение относится к химическому соединению формулы (II):

или его кислотно-аддитивной соли или его стереохимически изомерной форме, где R1, R2 и R3 имеют значения, как определено в данном описании и формуле изобретения, а Р означает гидроксизащитную группу. Предпочтительными кислотно-аддитивными солями являются фармацевтически приемлемые кислотно-аддитивные соли, в частности таковые, как указано выше. Защитная группа Р может представлять собой такую, как описано выше.
Еще в одном дальнейшем аспекте данное изобретение относится к химическому соединению формулы (III), где W означает хлор, R1 означает бром, а R2 и R3 означают метил, соединению, которое может быть представлено формулой (III-a):

или его кислотно-аддитивной соли. Предпочтительными кислотно-аддитивными солями являются фармацевтически приемлемые кислотно-аддитивные соли, в частности таковые, указанные выше.
Соединения формулы (I) проявляют антиретровирусные свойства (ингибирующие обратную транскриптазу свойства), в частности против вируса иммунодефицита человека (ВИЧ), который является этиологическим агентом синдрома приобретенного иммунодефицита (СПИД) у людей. Вирус ВИЧ предпочтительно инфицирует человеческие Т-4-клетки и разрушает их или изменяет их нормальную функцию, в особенности координацию иммунной системы. В результате инфицированный пациент всегда имеет уменьшающееся количество Т-4-клеток, которые, кроме того, ведут себя анормально. Следовательно, иммунологическая защитная система неспособна бороться с инфекциями и новообразованиями, и ВИЧ-инфицированный субъект обычно умирает от инфекций, вызываемых условно-патогенными микроорганизмами, таких как пневмония, или от раковых заболеваний. Другие состояния, связанные с ВИЧ-инфекцией, включают тромбоцитопению, саркому Капоши и инфекцию центральной нервной системы, характеризующуюся прогрессирующей демиелинизацией, которая приводит, в результате, к деменции и симптомам, таким как прогрессирующая дизартрия, атаксия и дезориентация. ВИЧ-инфекция, далее, также ассоциируется с периферической невропатией, прогрессирующей генерализованной лимфаденопатией (PGL) и СПИД-связанным комплексом (ARC).
Соединения согласно настоящему изобретению также проявляют активность против резистентных к (многим) лекарственным средствам штаммов ВИЧ, в частности резистентных к (многим) лекарственным средствам штаммов ВИЧ-1, более того, в особенности, соединения согласно настоящему изобретению проявляют активность против штаммов ВИЧ, главным образом, штаммов ВИЧ-1, которые обладают приобретенной резистентностью к одному или более известным из уровня техники ингибиторам ненуклеозидной обратной транскриптазы. Известные в уровне техники ингибиторы ненуклеозидной обратной транскриптазы представляют собой ингибиторы ненуклеозидной обратной транскриптазы, иные, чем соединения согласно настоящему изобретению, и известны квалифицированному специалисту в данной области, в частности, как коммерчески доступные ингибиторы ненуклеозидной обратной транскриптазы. Соединения согласно настоящему изобретению также обладают незначительной или «не связующей» аффинностью к человеческому α-1-кислому гликопротеину; человеческий α-1-кислый гликопротеин не подвергается или только слабо подвергается воздействию анти-ВИЧ-активности соединений согласно настоящему изобретению.
Вследствие их антиретровирусных свойств, особенно их анти-ВИЧ-свойств, главным образом, их анти-ВИЧ-1-активности, соединения формулы (I), их фармацевтически приемлемые аддитивные соли и их стереохимически изомерные формы пригодны для лечения индивидуумов, инфицированных ВИЧ, и для профилактики этих инфекций. Вообще соединения согласно настоящему изобретению могут быть пригодны для лечения теплокровных животных, инфицированных вирусами, существование которых опосредуется или зависит от фермента обратной транскриптазы. Состояния, которые можно предотвращать или лечить с помощью соединений согласно настоящему изобретению, главным образом, являются состояниями, связанными с ВИЧ и другими патогенными ретровирусами, включая СПИД, СПИД-связанный комплекс (ARC), прогрессирующую генерализованную лимфаденопатию (PGL), а также хронические заболевания центральной нервной системы, вызываемые ретровирусами, такие как, например, опосредуемая ВИЧ деменция и рассеянный склероз.
Соединения согласно настоящему изобретению или любую их подгруппу, следовательно, можно использовать в качестве лекарственных средств против вышеуказанных состояний. Вышеуказанное использование в качестве лекарственного средства или способ лечения включает введение ВИЧ-инфицированным субъектам эффективного количества соединения для борьбы с состояниями, связанными с ВИЧ и другими патогенными ретровирусами, главным образом, с ВИЧ-1. В частности, соединения формулы (I) можно использовать для получения лекарственного средства в целях лечения или предотвращения ВИЧ-инфекций.
В дальнейшем аспекте данного изобретения предусматривается способ лечения теплокровных животных, включая людей, страдающих от вирусных инфекций, или к способу предохранения теплокровных животных, включая людей, от страдания от вирусных инфекций, главным образом, от ВИЧ-инфекций. Вышеуказанный способ включает введение, предпочтительно пероральное введение, эффективного количества соединения формулы (I), его фармацевтически приемлемой аддитивной соли или его возможной стереоизомерной формы теплокровным животным, включая людей.
В другом аспекте соединения формулы (I) или их любая подгруппа пригодны в случае способа предупреждения, лечения или борьбы с инфекцией или заболеванием, ассоциированным с инфицированием млекопитающего мутантным вирусом ВИЧ, включающего введение вышеуказанному млекопитающему эффективного количества соединения формулы (I) или его любой подгруппы.
В другом аспекте соединения формулы (I) или их любая подгруппа пригодны в случае способа предупреждения, лечения или борьбы с инфекцией или заболеванием, ассоциированным с инфицированием млекопитающего резистентным ко многим лекарственным средствам вирусом ВИЧ, включающего введение вышеуказанному млекопитающему эффективного количества соединения формулы (I) или его любой подгруппы.
Еще в одном другом аспекте соединения формулы (I) или их любая подгруппа пригодны в случае способа ингибирования репликации вируса ВИЧ, в частности вируса ВИЧ, обладающего обратной транскриптазой мутантного ВИЧ, еще, в частности, обратной транскриптазой мутантного ВИЧ, резистентного ко многим лекарственным средствам, включающего введение млекопитающему, нуждающемуся в этом, эффективного количества соединения формулы (I) или его любой подгруппы.
Предпочтительно, млекопитающим, как указано в способах согласно данному изобретению, является человек.
Настоящее изобретение также относится к композициям для лечения вирусных инфекций, содержащим терапевтически эффективное количество соединения формулы (I) и фармацевтически приемлемый носитель или разбавитель.
Соединения согласно настоящему изобретению или их любая подгруппа могут быть использованы для получения различных фармацевтических форм для целей введения. В качестве подходящих композиций можно назвать все композиции, обычно применяемые для систематически вводимых лекарственных средств. Для получения фармацевтических композиций согласно данному изобретению эффективное количество конкретного соединения, необязательно в форме аддитивной соли, в качестве активного ингредиента, комбинируют, при тщательном смешении, с фармацевтически приемлемым носителем, причем носитель может иметь большое разнообразие форм в зависимости от готовой лекарственной формы, желательной для введения. Эти фармацевтические композиции желательны в стандартной лекарственной форме, пригодной, в особенности, для перорального введения, ректального введения, чрескожного введения или парентеральной инъекции. Например, при получении композиций в виде пероральной лекарственной формы можно использовать любую из обычных фармацевтических сред, такую как, например, вода, гликоли, масла, спирты и т.п., в случае пероральных жидких готовых лекарственных форм, таких как суспензии, сиропы, эликсиры, эмульсии и растворы; или твердые носители, такие как крахмалы, сахара, каолин, разбавители, смазочные вещества, связующие вещества, дезинтегрирующие агенты и т.п., в случае порошков, пилюль, капсул и таблеток.
Вследствие легкости их введения таблетки и капсулы представляют собой наиболее выгодные пероральные стандартные лекарственные формы, в случае которых обычно используют твердые фармацевтические носители. В случае парентеральных композиций носитель обычно включает стерильную воду, по меньшей мере, большей частью, несмотря на то, что могут быть включены другие ингредиенты, например, для способствования растворимости. Могут быть получены инъецируемые растворы, например в которых носитель включает физиологический раствор, раствор глюкозы или смесь физиологического раствора и раствора глюкозы. Также могут быть получены инъецируемые суспензии, в случае которых могут быть использованы подходящие жидкие носители, суспендирующие агенты и т.п. Также сюда входят препараты в твердой форме, которые предназначены для превращения незадолго до использования в жидкие готовые лекарственные формы. В композициях, пригодных для чрескожного введения, носитель необязательно включает улучшающий пенетрацию агент и/или пригодный смачиватель, необязательно комбинированный с пригодными добавками любой природы в небольших количествах, причем добавки не оказывают значительного пагубного воздействия на кожу. Вышеуказанные добавки могут облегчать введение в кожу и/или могут быть полезны для получения желательных композиций. Эти композиции можно вводить различными путями, например в виде трансдермального пластыря, в виде притирания, в виде мази. Соединения согласно настоящему изобретению также можно вводить путем ингаляции или инсуффляции посредством способов и при использовании готовых лекарственных форм, применяемых в уровне техники для введения этим путем. Таким образом, вообще, соединения согласно настоящему изобретению можно вводить в легкие в форме раствора, суспензии или сухого порошка. Любая система, разработанная для введения растворов, суспензий или сухих порошков посредством пероральной или назальной ингаляции или инсуффляции, пригодна для введения соединений согласно настоящему изобретению.
Для способствования растворимости соединений формулы (I) в композиции можно включать подходящие ингредиенты, например циклодекстрины. Соответствующими циклодекстринами являются α-, β-, γ-циклодекстрины или их простые эфиры и смешанные простые эфиры, где одна или более гидроксильных групп ангидроглюкозных звеньев циклодекстрина замещены С1-6-алкилом, особенно метилом, этилом или изопропилом, как, например, статистически метилированный β-CD; гидрокси-С1-6-алкилом, особенно гидроксиэтилом, гидроксипропилом или гидроксибутилом; карбокси-С1-6-алкилом, особенно карбоксиметилом или карбоксиэтилом; С1-6-алкилкарбонилом, особенно ацетилом. В особенности заслуживающими внимания в качестве комплексообразователей и/или солюбилизаторов являются β-CD, статистически метилированный β-CD, 2,6-диметил-β-CD, 2-гидроксиэтил-β-CD, 2-гидроксипропил-β-CD и (2-карбоксиметокси)пропил-β-CD, и, в частности, 2-гидроксипропил-β-CD (2-НР-β-CD). Другим типом замещенных циклодекстринов являются сульфобутилциклодекстрины.
Термин «смешанный простой эфир» означает производные циклодекстрина, где, по меньшей мере, две гидроксильные группы циклодекстрина превращены в простые эфиры с разными группами, такими, как, например, гидроксипропил и гидроксиэтил.
Среднее молярное замещение (M.S.) используют в качестве меры среднего числа молей алкоксильных единиц на моль ангидроглюкозы. Средняя степень замещения (D.S.) относится к среднему числу замещенных гидроксильных групп на звено ангидроглюкозы. Значения М.S. и D.S. могут быть определены различными аналитическими методами, такими как ядерный магнитный резонанс (ЯМР), масс-спектрометрия (МС) и инфракрасная спектроскопия (ИК). В зависимости от используемого метода могут быть получены незначительно различающиеся значения для одного данного производного циклодекстрина. Предпочтительно, при измерении с помощью масс-спектрометрии, значение М.S. колеблется в пределах от 0,125 до 10, а значение D.S. колеблется в пределах от 0,125 до 3.
Другие пригодные композиции для перорального или ректального введения включают частицы, состоящие из твердой дисперсии, содержащей соединение формулы (I) и один или более подходящих фармацевтически приемлемых, растворимых в воде полимеров.
Термин «твердая дисперсия», используемый выше, определяет систему в твердом состоянии (в отличие от жидкого или газообразного состояния), включающую, по меньшей мере, два компонента, в случае соединения формулы (I) и растворимого в воде полимера, где один компонент более или менее равномерно диспергирован в другом компоненте или компонентах (в случае включения дополнительных, фармацевтически приемлемых для получения готовых лекарственных форм агентов, обычно известных в уровне техники, таких как пластификаторы, консерванты и т.п.). Когда вышеуказанная дисперсия компонентов является такой, что система химически и физически совершенно однородна или гомогенна или состоит из одной фазы, как определено в термодинамике, такую твердую дисперсию можно назвать «твердым раствором». Твердые растворы являются предпочтительными физическими системами, так как компоненты в них обычно без труда биодоступны организмам, в которые они введены.
Термин «твердая дисперсия» также включает дисперсии, которые являются во всех отношениях менее гомогенными, чем твердые растворы. Такие дисперсии не являются химически и физически совершенно однородными или включают более чем одну фазу, например, системы, имеющие области или небольшие зоны, где аморфное, микрокристаллическое или кристаллическое соединение формулы (I), или аморфный, микрокристаллический или кристаллический водорастворимый полимер, или они оба, более или менее равномерно диспергированы в другой фазе, включающей растворимый в воде полимер или соединение формулы (I), или твердый раствор, содержащий соединение формулы (I) и растворимый в воде полимер. Вышеуказанные области являются зонами твердой дисперсии, отчетливо заметными за счет физической особенности: малые по размеру и по случаю и статистически распределенные в твердой дисперсии.
Существуют различные способы получения твердых дисперсий, включающие экструзию из расплава, сушку распылением и растворение-выпаривание. После образования твердых дисперсий полученные продукты необязательно можно измельчать и просеивать. Продукт в виде твердой дисперсии можно измельчать или размалывать до частиц, имеющих размер менее чем 600 мкм, предпочтительно менее чем 400 мкм и наиболее предпочтительно менее чем 125 мкм. Частицы, полученные, как описано выше, можно затем использовать для получения обычными способами фармацевтических лекарственных форм, таких как таблетки и капсулы.
Водорастворимые полимеры в частицах представляют собой полимеры, которые имеют кажущуюся вязкость, когда растворены при температуре 20°С в водном растворе с концентрацией 2% (масс./об.), составляющую 1-5000 мПа·с, более предпочтительно 1-700 мПа·с и наиболее предпочтительно 1-100 мПа·с. Например, пригодные водорастворимые полимеры включают алкилцеллюлозы, гидроксиалкилцеллюлозы, гидроксиалкилалкилцеллюлозы, карбоксиалкилцеллюлозы, соли щелочных металлов карбоксиалкилцеллюлоз, карбоксиалкилалкилцеллюлозы, сложные эфиры карбоксиалкилцеллюлозы, крахмалы, пектины, производные хитина, ди-, олиго- и полисахариды, такие как трегалоза, альгиновая кислота или их соли щелочных металлов или аммониевые соли, каррагенаны, галактоманнаны, трагакант, агар-агар, гуммиарабик, гуаровую смолу и гуммиксантан, полиакриловые кислоты и их соли, полиметакриловые кислоты и их соли, сополимеры на основе метакрилатов, поливиниловый спирт, поливинилпирролидон, сополимеры поливинилпирролидона с винилацетатом, комбинации поливинилового спирта и поливинилпирролидона, полиалкиленоксиды и сополимеры этиленоксида и пропиленоксида. Предпочтительными растворимыми в воде полимерами являются гидроксипропилметилцеллюлозы.
Также один или более циклодекстринов можно использовать в качестве растворимого в воде полимера при получении вышеуказанных частиц, как описано в WO 97/18839. Эти циклодекстрины включают фармацевтически приемлемые незамещенные и замещенные циклодекстрины, известные в уровне техники, более предпочтительно, α-, β- или γ-циклодекстрины, или их фармацевтически приемлемые производные.
Замещенные циклодекстрины, которые можно использовать для получения вышеописанных частиц, включают простые полиэфиры, описанные в патенте США 3459731. Далее, замещенные циклодекстрины являются таковыми, описанными выше, в качестве агентов для способствования растворимости соединений формулы (I).
Соотношение соединения формулы (I) к растворимому в воде полимеру можно широко варьировать. Например, можно использовать соотношения от 1/100 до 100/1. Представляющие интерес соотношения соединения формулы (I) к циклодекстрину составляют от примерно 1/10 до 10/1. Представляющие больший интерес соотношения составляют от примерно 1/5 до 5/1.
Далее, может быть пригодно использование соединений формулы (I) для получения готовой лекарственной формы в виде наночастиц, которые обладают модификатором поверхности, адсорбированным на их поверхности, в количестве, достаточном для поддержания эффективного среднего размера частиц, менее чем 1000 нм. Подразумевают, что пригодные модификаторы поверхности включают таковые, которые физически сцепляются с поверхностью соединения формулы (I), но не за счет химической связи с вышеуказанным соединением, и которые могут быть выбраны из известных органических и неорганических фармацевтических эксципиентов. Такие эксципиенты включают различные полимеры, олигомеры с низкой молекулярной массой, натуральные продукты и поверхностно-активные вещества. Предпочтительные модификаторы поверхности включают неионогенные и анионогенные поверхностно-активные вещества.
Еще другой, представляющий интерес путь использования соединений формулы (I) для получения готовых лекарственных форм, включает фармацевтическую композицию, где соединения формулы (I) вводят в гидрофильные полимеры, и эту смесь наносят в качестве покровной пленки на множество маленьких гранул, таким образом, получая композицию, которую можно пригодным образом перерабатывать и которая пригодна для получения фармацевтических лекарственных форм для перорального введения. Такие гранулы включают центральное, округленное или сферическое ядро, покровную пленку из гидрофильного полимера и соединение формулы (I), и, необязательно, герметизирующий покровный слой. Вещества, пригодные для использования в качестве ядер в гранулах, разнообразны, при условии, что вышеуказанные вещества являются фармацевтически приемлемыми и имеют подходящие размеры и плотность. Примерами таких веществ являются полимеры, неорганические вещества, органические вещества и сахариды и их производные.
Особенно преимущественным является приготовление вышеуказанных фармацевтических композиций в стандартной лекарственной форме для легкости введения и единообразия дозировки. Стандартная лекарственная форма, как используется в данном контексте, относится к физически дискретным единицам, пригодным в качестве стандартных доз, причем каждая стандартная доза содержит предопределенное количество активного ингредиента, рассчитанное для достижения желательного терапевтического эффекта, в сочетании с требующимся фармацевтическим носителем. Примерами таких стандартных лекарственных форм являются таблетки (включая таблетки с насечкой или с нанесенным покрытием), капсулы, пилюли, пакетированные порошки, облатки, суппозитории, инъецируемые растворы или суспензии и т.п., и их сегрегированные множества.
Квалифицированный специалист при лечении ВИЧ-инфекции может определить эффективное суточное количество из результатов теста, представленного в данном контексте. Вообще, предполагают, что эффективное суточное количество составляет от 0,01 мг/кг до 50 мг/кг массы тела, более предпочтительно, от 0,1 мг/кг до 10 мг/кг массы тела. Оно может быть подходящим для введения требуемой дозы в виде двух, трех, четырех или более субдоз через соответствующие интервалы в течение суток. Вышеуказанные субдозы можно получать в виде стандартных лекарственных форм, например, содержащих 1-1000 мг, и, в особенности, 5-200 мг активного ингредиента на стандартную лекарственную форму.
Точная дозировка и частота введения зависят от используемого конкретного соединения формулы (I), конкретного излечиваемого состояния, тяжести излечиваемого состояния, возраста, массы тела и общего физического состояния конкретного пациента, а также использования другой лекарственной терапии для индивидуума, как хорошо известно квалифицированному специалисту в данной области. Кроме того, очевидно, что вышеуказанное эффективное суточное количество может быть уменьшено или увеличено, в зависимости от ответной реакции подвергаемого лечению субъекта и/или в зависимости от оценки врача, назначающего прием соединений согласно настоящему изобретению. Указанные выше диапазоны эффективного суточного количества, следовательно, являются только рекомендуемыми и не предназначены для ограничения рамок настоящего изобретения или использования данного изобретения в любом объеме.
Соединения формулы (I) согласно настоящему изобретению можно использовать индивидуально или в комбинации с другими терапевтическими агентами, такими как противовирусные средства, антибиотики, иммуномодуляторы или вакцины для лечения вирусных инфекций. Их можно также использовать отдельно или в комбинации с другими профилактическими агентами для предотвращения вирусных инфекций. Соединения согласно настоящему изобретению можно использовать в вакцинах и способах защиты индивидуумов от вирусных инфекций на длительный период времени. Соединения можно использовать в таких вакцинах или индивидуально или совместно с другими соединениями согласно данному изобретению или совместно с другими противовирусными агентами, до известной степени совместимыми со стандартным применением ингибиторов обратной транскриптазы в вакцинах. Таким образом, соединения согласно настоящему изобретению можно комбинировать с фармацевтически приемлемыми вспомогательными средствами, обычно применяемыми в вакцинах, и вводить в профилактически эффективных количествах для защиты индивидуумов на длительный период времени против ВИЧ-инфекций.
Также комбинации одного или более дополнительных антиретровирусных соединений и соединения формулы (I) можно использовать в качестве лекарственного средства. Таким образом, настоящее изобретение также относится к продукту, содержащему (а) соединение формулы (I) и (b) одно или более дополнительных антиретровирусных соединений, в качестве комбинированного препарата для одновременного, раздельного или последовательного использования при анти-ВИЧ-лечении. Различные лекарственные средства можно комбинировать в одном препарате вместе с фармацевтически приемлемыми носителями. Вышеуказанные другие антиретровирусные соединения могут быть известными антиретровирусными соединениями, такими как сурамин, пентамидин, тимопентин, кастаноспермин, декстран (декстрансульфат), фоскарнет-натрий (тринатрийфосфоноформиат); ингибиторы нуклеозидной обратной транскриптазы (NRTI), например зидовудин (AZT), диданозин (ddI), залцитабин (ddC), ламивудин (3ТС), ставудин (d4T), эмтрицитабин (FTC), абакавир (АВС), D-D4FC (Reverset™), аловудин (MIV-310), амдоксовир (DAPD), элвуцитабин (АСН-126443) и т.п.; ингибиторы ненуклеозидной обратной транскриптазы (NNRTI), такие как деларвидин (DLV), эфавиренц (EFV), невирапин (NVP), каправирин (СРV), каланолид А, ТМС 120, этравирин (ТМС 125), ТМС278, BMS-561390, DPC-083 и т.п.; ингибиторы нуклеотидной обратной транскриптазы (NtRTI), например тенофовир (TDF) и тенофовирдизопроксилфумарат и т.п.; соединения типа TIBO (тетрагидроимидазо[4,5,1-jk][1,4]бензодиазепин-2(1Н)-он и -тион), например (S)-8-хлор-4,5,6,7-тетрагидро-5-метил-6-(3-метил-2-бутенил)имидазо[4,5,1-jk][1,4]бензодиазепин-2(1Н)-тион; соединения типа α-АРА (α-анилинофенилацетамид), например α-[(2-нитрофенил)амино]-2,6-дихлорбензолацетамид и т.п.; ингибиторы трансактивирующих белков, такие как ТАТ-ингибиторы, например RO-5-3335; REV-ингибиторы; ингибиторы протеазы, например ритонавир (RTV), саквинавир (SQV), лопинавир (АВТ-378 или LPV), индинавир (IDV), ампренавир (VX-478), ТМС-126, ВМS-232632, VX-175, DMP-323, DMP-450 (мозенавир), нелфинавир (AG-1343), атазанавир (BMS 232632), палинавир, ТМС-114, RO033-4649, фозампренавир (GW433908 или VX-175), Р-1946, BMS 186318, SC-55389a, L-756423, типранавир (PNU-140690), BILA 1096 BS, U-140690 и т.п.; ингибиторы «проникновения», которые включают ингибиторы гибридизации (например, Т-20, Т-1249); ингибиторы присоединения и ингибиторы корецептора; последние включают антагонисты ССR5 и антагонисты CXR4 (например, AMD-3100); примерами ингибиторов «проникновения» являются энфувиртид (ENF), GSK-873140, PRO-542, SCH-417690, TNX-355, маравирок (UK-427857); ингибитор матурации, например РА-457 (Panacos Pharmaceuticals); ингибиторы вирусной интегразы; ингибиторы рибонуклеотидредуктазы (клеточные ингибиторы), например гидроксимочевина, и т.п.
При введении соединений согласно настоящему изобретению с другими противовирусными агентами, которые нацелены на различные события в вирусном жизненном цикле, может быть усилен терапевтический эффект этих соединений. Комбинированные терапии, как описано выше, оказывают синергическое действие на ингибирование ВИЧ-репликации, так как каждый компонент комбинации действует на другой участок ВИЧ-репликации. Использование таких комбинаций может снижать дозировку используемого стандартного антиретровирусного агента, которая требуется для желательного терапевтического или профилактического эффекта, по сравнению с той, когда агент вводят в качестве монотерапии. Эти комбинации могут снижать или исключать побочные эффекты одной стандартной антиретровирусной терапии, когда нет взаимного влияния противовирусной активности агентов. Эти комбинации снижают потенциал резистентности к терапиям с использованием одного агента, в то время как происходит минимизация любой связанной с этим токсичности. Эти комбинации также могут повышать эффективность стандартного агента без увеличения ассоциированной токсичности.
Соединения согласно настоящему изобретению также можно вводить в комбинации с иммуномодулирующими агентами, такими как, например, левамизол, бропиримин, антитело против человеческого альфа-интерферона, альфа-интерферон, интерлейкин-2, метионинэнкефалин, диэтилдитиокарбамат, фактор некроза опухоли, налтрексон и т.п.; антибиотиками, такими как, например, пентамидинизетиорат и т.п.; холинергическими агентами, такими как, например, такрин, ривастигмин, донепезил, галантамин и т.п.; блокаторами NMDA-канала, такими как, например, мемантин; для предотвращения или борьбы с инфекцией и заболеваниями или симптомами заболеваний, связанными с ВИЧ-инфекциями, такими как СПИД и ARC, например деменция. Соединение формулы (I) также можно комбинировать с другим соединением формулы (I).
Несмотря на то что настоящее изобретение направлено в основном на использование соединений согласно настоящему изобретению для предотвращения или лечения ВИЧ-инфекций, соединения согласно настоящему изобретению также можно использовать в качестве ингибирующих агентов для других вирусов, которые зависят от подобных обратных транскриптаз в отношении обязательных событий в их жизненном цикле.
Производные пиримидина согласно данному изобретению не только благоприятно действуют в том, что касается их способности к ингибированию репликации вируса иммунодефицита человека (ВИЧ), но также проявляют улучшенную способность к ингибированию репликации мутантных штаммов, в частности штаммов, которые имеют двойные или многократные мутации в вирусном геноме, кодирующем обратную транскриптазу. Соединения согласно данному изобретению, следовательно, могут найти применение при лечении пациентов, инфицированных ВИЧ, который стал резистентным к одному или более известным лекарственным средствам NNRTI (лекарственные средства как ингибиторы ненуклеозидной обратной транскриптазы), причем штаммы относятся к штаммам ВИЧ, резистентным к лекарственному средству или ко многим лекарственным средствам.
Следующие примеры предназначены для пояснения настоящего изобретения и не ограничивают его объема охраны.
ПРИМЕРЫ
Пример 1
К 2,11 г 4-[(4,6-дихлор-2-пиримидинил)амино]бензонитрила (0,00796 моль) добавляют 500 мл СНСl3 и смесь перемешивают в течение 30 минут до растворения почти всего исходного соединения. В виде одной порции добавляют 1-бром-2,5-пирролидиндион (0,0397 моль) и реакционную смесь перемешивают при комнатной температуре. Спустя примерно 30 минут смесь становится раствором светло-оранжевого цвета, и со временем реакционная смесь становится все более и более красноватой. Спустя 40 часов TСХ (тонкослойная хроматография) и ВЭЖХ/МС показывают, что реакция завершена. Реакционную смесь очищают с помощью колоночной флэш-хроматографии на силикагеле, используя СН2Сl2 в качестве элюента. Фракции, содержащие желательный продукт, отделяют, и растворитель выпаривают. Остаток перекристаллизовывают из ацетонитрила, получая 1,51 г 4-[(5-бром-4,6-дихлор-2-пиримидинил)амино]бензонитрила (выход 55%) (промежуточный продукт 1).
Пример 2
Смесь 0,47 г 4-гидрокси-3,5-диметилбензонитрила (0,00320 моль) и 1,4-диоксана (3 мл) вносят в пробирку под давлением, в атмосфере аргона. Добавляют 0,13 г 60%-ного NaH (0,00320 моль) и смесь перемешивают в течение 2 минут. Добавляют 1-метил-2-пирролидинон (3 мл) и смесь перемешивают в течение 10 минут. Добавляют промежуточный продукт 1 (0,00291 моль) и смесь нагревают в герметично закрытой пробирке при температуре 155°С в течение 16 часов. Смесь выливают в воду (15 мл). Пробирку промывают водой, 1,4-диоксаном (11 мл) и снова водой, и промывные жидкости объединяют с водной фазой, в которую была вылита реакционная смесь. Таким образом, полученный раствор перемешивают в течение 15 минут и помещают в холодильник. Полученное вещество отфильтровывают, получая 1,43 г 4-[[5-бром-6-хлор-2-[(4-цианофенил)амино]-4-пиримидинил]окси]-3,5-диметилбензонитрила (выход 45%) (промежуточный продукт 2).
Пример 3
Синтез 4-[[6-тетрагидропиранилоксиламино-5-бром-2-[(4-цианофенил)амино]-4-пиримидинил]окси]-3,5-диметилбензонитрила
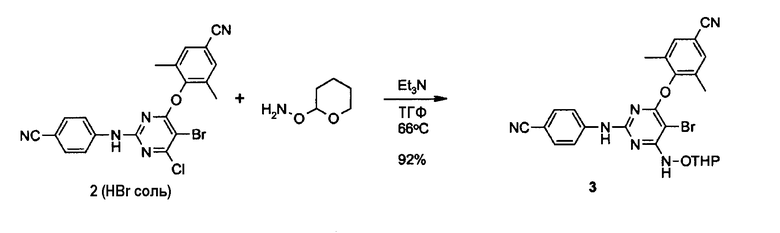
Смесь гидробромида промежуточного продукта 2 (1,55 г, 2,89 ммоль), полученного, как описано в примере 2, триэтиламина (1,43 мл, 1,04 г, 10,2 ммоль, 3,5 экв.) и О-(тетрагидро-2Н-пиран-2-ил)гидроксиламина (2,00 г, 17,1 ммоль, 5,9 экв.) кипятят с обратным холодильником в ТГФ (30 мл) в течение ночи. Анализ LCMS (жидкостная хроматография/масс-спектрометрия) показывает 50%-ную конверсию. Добавляют другую порцию О-(тетрагидро-2Н-пиран-2-ил)гидроксиламина (1,00 г, 8,54 ммоль, 3 экв.), и реакционную смесь кипятят с обратным холодильником в течение двух суток. Анализ LCMS показывает полную конверсию. Реакционную смесь охлаждают, добавляют силикагель и выпаривают ТГФ. С помощью колоночной хроматографии, используя смесь гептан/EtOAc = 2/1, содержащую 0,3% триэтиламина, получают 2,7 г масла желтого цвета, которое обрабатывают EtOAc, дважды промывают насыщенным раствором NH4Cl, водой и рассолом. После сушки над Na2SO4 получают 1,43 г (выход 92%) пены желто-белого цвета (промежуточный продукт 3).
Анализ LCMS (4 мл/мин, линейный градиент, t0 100% 10 мМ водная НСООН/ацетонитрил до t10 100% 10 мМ водная НСООН/ацетонитрил, UV-DAD): чистота 98%, t=7,2 мин, масс-спектр, m/z=533, 535 [М-Н]-.
Пример 4
Синтез 4-[[6-гидроксиламино-5-бром-2-[(4-цианофенил)амино]-4-пиримидинил]окси]-3,5-диметилбензонитрила

Смесь промежуточного продукта 3 (1,40 г, 2,61 ммоль) и Amberlyst-15 (1,10 г, 80 мас.%) в метаноле перемешивают в течение ночи при комнатной температуре в атмосфере аргона. Amberlyst-15 отфильтровывают и фильтрат концентрируют, обрабатывают ТГФ и «связывают» с силикагелем. С помощью колоночной хроматографии, используя смесь гептан/EtOAc = 1/1, достигают только частичного разделения. Чистые фракции выпаривают и оставшееся твердое вещество перемешивают в дихлорметане и отфильтровывают, получая 250 мг (выход 21%) соединения 1 в виде твердого вещества белого цвета. Смешанные фракции также содержат желательный продукт, но их далее не очищают и не анализируют; температура плавления - 211оС (разложение).
1Н-ЯМР (300 МГц, ДМСО): δ 2,14 (с, 6Н), 7,41 (д, 2Н), 7,55 (д, 2Н), 7,75 (с, 2Н), 9,08 (с, 1Н), 9,88 (с, 1Н), 9,94 (с, 1Н).
Анализ LCMS (1 мл/мин, линейный градиент, t0 95% 10 мМ водная НСООН/ацетонитрил до t15 5% 10 мМ водная НСООН/ацетонитрил, UV-DAD): чистота 98%, t=9,90 мин, масс-спектр, m/z=449, 451 [М-Н]-.
Пример 5
Готовые лекарственные формы
Капсулы
Соединение 1, которое является соединением, описанным в примере 4, растворяют в органическом растворителе, таком как этанол, метанол или метиленхлорид, предпочтительно в смеси этанола и метиленхлорида. Полимеры, такие как сополимер поливинилпирролидона и винилацетата (PVP-VA) или гидроксипропилметилцеллюлоза (НРМС), обычно с кажущейся вязкостью 5 мПа·с, растворяют в органических растворителях, таких как этанол, метанол и метиленхлорид. Преимущественно полимер растворяют в этаноле. Растворы полимера и соединения смешивают и затем подвергают распылительной сушке. Соотношение соединение/полимер выбирают от 1/1 до 1/6. Промежуточными диапазонами могут быть 1/1,5 и 1/3. Подходящее соотношение может составлять 1/6. Полученным путем распылительной сушки порошком, твердой дисперсией, затем наполняют капсулы для введения. Количество лекарственного средства, загружаемого в одну капсулу, колеблется в пределах от 50 мг до 100 мг, в зависимости от используемого размера капсулы.
Таблетки с пленочным покрытием
Получение ядра таблетки
Смесь 100 г соединения 1, 570 г лактозы и 200 г крахмала тщательно перемешивают и после этого увлажняют раствором 5 г додецилсульфата натрия и 10 г поливинилпирролидона примерно в 200 мл воды. Влажную порошковую смесь просеивают, сушат и снова просеивают. Затем добавляют 100 г микрокристаллической целлюлозы и 15 г гидрогенизированного растительного масла. Всю смесь тщательно перемешивают и прессуют в таблетки, получая 10000 таблеток, каждая из которых содержит 10 мг активного ингредиента.
Нанесение покрытия
К раствору 10 г метилцеллюлозы в 75 мл денатурированного этанола добавляют раствор из 5 г этилцеллюлозы в 150 мл дихлорметана. Затем добавляют 75 мл дихлорметана и 2,5 мл 1,2,3-пропантриола. 10 г полиэтиленгликоля расплавляют и растворяют в 75 мл дихлорметана. Последний раствор добавляют к предшествующему раствору и затем добавляют 2,5 г октадеканоата магния, 5 г поливинилпирролидона и 30 мл концентрированной суспензии красителя и всю смесь гомогенизируют. На ядра таблеток наносят покрытие с помощью таким образом полученной смеси в глазировочном аппарате.
Пример 6
Антивирусный спектр
Вследствие увеличивающегося появления резистентных к лекарственным средствам штаммов ВИЧ соединения согласно настоящему изобретению тестировали в отношении их эффективности против клинически выделенных штаммов ВИЧ с некоторыми скрытыми мутациями. Эти мутации связаны с резистентностью к ингибиторам обратной транскриптазы и приводят к вирусам, которые проявляют различные степени фенотипической перекрестной резистентности к коммерчески доступным в настоящее время лекарственным средствам, таким как, например, AZT и делавирдин.
Противовирусную активность соединения согласно настоящему изобретению оценивали в присутствии ВИЧ дикого типа и мутантов ВИЧ, образующихся за счет мутаций в гене обратной транскриптазы. Активность соединений оценивали, используя клеточный анализ, и остаточную активность выражали в значениях рЕС50. В нижеприводимой таблице, в колонках IIIB и A-G, перечислены значения рЕС50 против различных штаммов IIIB, A-G.
Штамм IIIB представляет собой штамм ВИЧ-LAI дикого типа;
штамм А содержит мутацию Y181С в обратной транскриптазе ВИЧ;
штамм В содержит мутацию К103N в обратной транскриптазе ВИЧ;
штамм С содержит мутацию L100I в обратной транскриптазе ВИЧ;
штамм D содержит мутации Y188L и S162K в обратной транскриптазе ВИЧ;
штамм Е содержит мутации L100I и К103N в обратной транскриптазе ВИЧ;
штамм F содержит мутации К101Е и К103N в обратной транскриптазе ВИЧ.
Штамм G содержит мутации L100I, К103N, E138G, V179I, Y181С, L214F, V276V/I и A327A/V в обратной транскриптазе ВИЧ.
Соединение А является соединением, которое описано в WO00/27825 и имеет следующую структуру:

По сравнению со справочным соединением А соединение 1 показывает улучшенную активность против двойных мутантных штаммов, таких как штаммы Е и F. Соединение 1, в особенности, показывает улучшенную активность против многократно мутированного штамма G.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИРУЮЩИЕ ВИЧ ПРОИЗВОДНЫЕ 2-(4-ЦИАНОФЕНИЛАМИНО)ПИРИМИДИНА | 2006 |
|
RU2403245C2 |
| 5-ЗАМЕЩЕННЫЕ ПИРИМИДИНЫ, ИНГИБИРУЮЩИЕ ВИЧ | 2005 |
|
RU2410379C2 |
| 5-(ГИДРОКСИМЕТИЛЕН- И АМИНОМЕТИЛЕН)ЗАМЕЩЕННЫЕ ПИРИМИДИНЫ, ИНГИБИРУЮЩИЕ ВИЧ | 2007 |
|
RU2452737C2 |
| ИНГИБИРУЮЩИЕ ВИЧ ПРОИЗВОДНЫЕ 2-(4-ЦИАНОФЕНИЛАМИНО)-ПИРИМИДИН-ОКСИДА | 2006 |
|
RU2398768C2 |
| ВИЧ-ИНГИБИРУЮЩИЕ 5-КАРБО- ИЛИ ГЕТЕРОЦИКЛИЧЕСКИЕ ЗАМЕЩЕННЫЕ ПИРИМИДИНЫ | 2005 |
|
RU2403244C2 |
| 5-АМИДО-ЗАМЕЩЕННЫЕ ПИРИМИДИНЫ, ИНГИБИРУЮЩИЕ ВИЧ | 2007 |
|
RU2480464C2 |
| 5-ГЕТЕРОЦИКЛИЛПИРИМИДИНЫ, ИНГИБИРУЮЩИЕ ВИЧ | 2005 |
|
RU2405778C2 |
| 1,5,6-ЗАМЕЩЕННЫЕ 2-ОКСО-3-ЦИАНО-1,6А-ДИАЗАТЕТРАГИДРОФЛУОРАНТЕНЫ | 2006 |
|
RU2389730C2 |
| ПРОИЗВОДНЫЕ (1,10b-ДИГИДРО-2-(АМИНОАЛКИЛФЕНИЛ)-5Н-ПИРАЗОЛО[1,5-c][1,3]БЕНЗОКСАЗИН-5-ИЛ)ФЕНИЛМЕТАНОНА В КАЧЕСТВЕ ИНГИБИТОРОВ ВИРУСНОЙ РЕПЛИКАЦИИ ВИЧ | 2006 |
|
RU2416615C2 |
| 6,7,8,9-ЗАМЕЩЕННЫЕ 1-ФЕНИЛ-1,5-ДИГИДРОПИРИДО (3,2-b) ИНДОЛ-2-ОНЫ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ АНТИИНФЕКЦИОННЫХ ФАРМАЦЕВТИЧЕСКИХ СРЕДСТВ | 2005 |
|
RU2377243C2 |
Изобретение относится к новым соединениям формулы (I) и их фармацевтически приемлемым аддитивным солям, обладающим свойствами ингибитора репликации ВИЧ.
В формуле 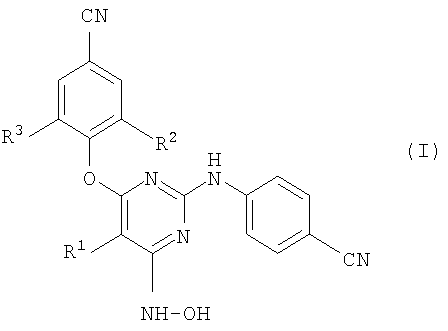 R1 означает галоген; R2 и R3 каждый независимо означают C1-6-алкил. Изобретение относится также к способу получения этих соединений и фармацевтической композиции. 3 н. и 4 з.п. ф-лы.
R1 означает галоген; R2 и R3 каждый независимо означают C1-6-алкил. Изобретение относится также к способу получения этих соединений и фармацевтической композиции. 3 н. и 4 з.п. ф-лы.
1. Соединение формулы (I)

или его фармацевтически приемлемая аддитивная соль, где
R1 означает галоген;
R2 и R3, каждый независимо, означают C1-6-алкил.
2. Соединение по п.1, где R1 означает хлор или бром.
3. Соединение по п.1, где R1 означает бром.
4. Соединение по любому из пп.1-3, где R2 и R3 означают метил.
5. Фармацевтическая композиция, ингибирующая репликацию ВИЧ, содержащая фармацевтически приемлемый носитель и, в качестве активного ингредиента, терапевтически эффективное количество соединения по любому из пп.1-4.
6. Способ получения соединения по любому из пп.1-4, отличающийся тем, что осуществляют взаимодействие промежуточного продукта (III) с защищенным гидроксиламином с получением промежуточного продукта (II) и последующее удаление защиты в промежуточном продукте (II) с получением конечных продуктов (I):

где R1, R2 и R3 имеют значения, как описано в любом из пп.1-4, Р представляет собой гидроксизащитную группу, выбранную из метоксиэтоксиметила (MEM), тетрагидропиранила (ТНР), трет-бутила (t-Bu), бензила (Bz), триметилсилила (TMS), трет-бутилдиметилсилила (TBDMS), триизопропилсилила (TIPS), трет-бутилдифенилсилила; и
W представляет собой уходящую группу;
и, в случае необходимости, получение фармацевтически приемлемой кислотно-аддитивной солевой формы соединений формулы (I) путем их обработки фармацевтически приемлемой кислотой.
7. Способ по п.6, где Р означает гидроксизащитную группу, в частности Р означает тетрагидро-2Н-пиран-2-ил.
| WO 00/27825 A1, 18.05.2000 | |||
| D.W.LUDOVICI et al, Evolution of Anti-HIV Drug Candidates | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Diarylpyrimidine (DAPY) Analogues, BIOORG | |||
| & MED | |||
| CHEM | |||
| LETT., 2001, 11, 2235-2239 | |||
| Прибор для измерения и масштабного вычерчивания линий и углов и для вычерчивания дуг круга | 1925 |
|
SU2973A1 |
| ЗАМЕЩЕННЫЕ ПИРИМИДИНТИОАЛКИЛЬНЫЕ ИЛИ АЛКИЛЭФИРНЫЕ СОЕДИНЕНИЯ И СПОСОБ ИНГИБИРОВАНИЯ ОБРАТНОЙ ТРАНСКРИПТАЗЫ ВИРУСОВ | 1996 |
|
RU2167155C2 |

