ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к новым циклопентил- и циклогептилпиразолам, которые полезны при лечении или профилактике заболеваний, на которые действуют FXR модуляторы, и в частности полезны при лечении дислипидемии.
Более конкретно, настоящее изобретение относится к новым производным циклопентил- и циклогептилпиразолов формулы

где А представляет собой -СН2- или -(СН2)3-, и R1-R4 являются такими, как определено ниже, или их фармацевтически активным солям, их получению, фармацевтическим композициям их содержащим, способам их применения и их применению в качестве лекарственных средств для лечения заболеваний, на которые действуют FXR модуляторы.
Настоящие соединения являются селективными модуляторами фарнезоидного Х-рецептора, в частности агонистами фарнезоидного Х-рецептора.
УРОВЕНЬ ТЕХНИКИ
Фарнезоидный Х-рецептор (FXR) является членом суперсемейства ядерных гормональных рецепторов транскрипционных факторов. FXR впервые был идентифицирован как рецептор, активируемый фарнезолом, а дальнейшие исследования выявили главную роль FXR в качестве рецептора желчных кислот [Makishima, М., Okamoto, A.Y., Repa, J. J., Tu, H., Learned, R.M., Luk, A., Hull, M.V., Lustig, K.D., Mangelsdorf, D.J. and Shan, B. (1999) Identification of a nuclear receptor for bile acids. Science 284, 1362-5]. FXR экспрессируется в печени, кишечнике, почках и надпочечниках. У человека были клонированы четыре сплайсированных изоформы.
Среди основных желчных кислот хенодезоксихолевая кислота является наиболее сильным агонистом FXR. Связывание желчных кислот или синтетических лигандов с FXR индуцирует транскрипцию малого гетеродимерного партнера (small heterodimer partner, SHP), нетипичного представителя семейства ядерных рецепторов, которые связывают несколько других гормональных ядерных рецепторов, включая LRH-1 и LXR альфа, и блокирует их транскрипционные функции [Lu, Т.Т., Makishima, М., Repa, J.J., Schoonjans, К., Kerr, Т.A., Auwerx, J.and Mangelsdorf, D.J. (2000) Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol Cell 6, 507-15]. CYP7A1 и CYP8B являются ферментами, вовлеченными в синтез желчных кислот в печени. FXR подавляет их экспрессию через активацию SHP пути. FXR непосредственно индуцирует экспрессию транспортеров семейства ABC, экспортирующих желчные кислоты в гепатоцитах, включая экспортирующую соли желчных кислот помпу (АВСВ11) и ассоциированный с мультилекарственной резистентностью белок 2 (АВСС2) [Kast, Н.R., Goodwin, В., Таrr, Р.Т., Jones, S.A., Anisfeld, А. М., Stoltz, С.М., Tontonoz, Р., Kliewer, S., Willson, Т. М. and Edwards, P. А. (2002) Regulation of multidrug resistance-associated protein 2 (ABCC2) by the nuclear receptors pregnane X receptor, farnesoid X-activated receptor, and constitutive androstane receptor. J Biol Chem 277, 2908-15; Ananthanarayanan, M., Balasubramanian, N., Makishima, M., Mangelsdorf, D.J. and Suchy, F. J. (2001) Human bile salt export pump promoter is transactivated by the farnesoid X receptor/bile acid receptor. J Biol Chem 276, 28857-65]. Мыши с нокаутированным геном FXR обладают сниженной резистентностью к гепатотоксичности, индуцированной желчными кислотами, и на моделях холестаза у животных было показано, что синтетические FXR агонисты являются гепатопротекторами [Liu, Y., Binz, J., Numerick, M.J., Dennis, S., Luo, G., Desai, В., MacKenzie, К. I., Mansfield, T.A., Kliewer, S.A., Goodwin, B. and Jones, S.A. (2003) Hepatoprotection by the farnesoid X receptor agonist GW4064 in rat models of intra- and extrahepatic cholestasis. J Clin Invest 112, 1678-87; Sinai, C.J., Tohkin, M., Miyata, M., Ward, J.M., Lambert, G. and Gonzalez, F.J. (2000) Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell 102, 731-44]. Приведенные данные показывают, что FXR защищает гепатоциты от токсичности желчных кислот посредством супрессирования как клеточного синтеза и импорта желчных кислот, так и стимулирования их билиарной экскрекции.
Процесс энтерогепатической циркуляции желчных кислот является также и основным регулятором гомеостаза сывороточного холестерина. После биосинтеза из холестерина в печени желчные кислоты секретируются с желчью в просвет тонкой кишки для участия в расщеплении и абсорбции жиров и жирорастворимых витаминов. Соотношение различных желчных кислот определяет гидрофильность пула желчных кислот и его способность растворять холестерин. Активация FXR увеличивает гидрофильность пула, уменьшает кишечную растворимость холестерина, эффективно блокирует его абсорбцию. Как предполагают, уменьшение абсорбции приводит к пониженному уровню холестерина в плазме. Более того, прямые ингибиторы абсорбции холестерина, такие как эзитимиб, уменьшают уровень холестерина в плазме, обеспечивая некоторое подтверждение данной гипотезе. Однако эзитимиб обладает ограниченной эффективностью, что по-видимому связано с активацией в результате обратной связи синтеза холестерина в клетках, пытающихся таким образом компенсировать истощение холестерина. Последние исследования показали, что FXR противодействует этому эффекту частично прямым ингибированием экспрессии HMGCoA редуктазы посредством сигнального пути, в котором участвуют SHP и LRH1 [Datta, S., Wang, L, Moore, D. D. and Osborne, T. F. (2006) Regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase promoter by nuclear receptors liver receptor homologue-1 and small heterodimer partner: a mechanism for differential regulation of cholesterol synthesis and uptake. J Biol Chem 281, 807-12]. FXR также уменьшает печеночный синтез триглицеридов ингибированием экспрессии SREBP1-C посредством альтернативного сигнального пути, в котором участвуют SHP и LXR-альфа. Таким образом, соединения, которые модулируют активность FXR, могут высокоэффективно терапевтически действовать на снижение холестерина и триглицеридов плазмы крови, чем текущие лекарства.
Большинство пациентов с ишемической болезнью сердца обладают высоким плазматическим уровнем атерогенных ЛПНП (липопротеинов низкой плотности). Ингибиторы HMGCoA редуктазы (статины) являются эффективными нормализаторами уровней холестерина ЛПНП, при этом уменьшая риск сердечно-сосудистых патологий, таких как инсульт и инфаркт миокарда, только на 30%. Необходима дополнительная терапия, направленная на дальнейшее снижение атерогенных ЛПНП, также как и других липидных факторов риска, таких как высокий уровень триглицеридов в плазме и низкий уровень Х-ЛПВП.
Большая часть пациентов с сахарным диабетом 2-го типа в США обладают ненормальной концентрацией липопротеинов в плазме крови. Превышение общего холестерина >240 мг/дл наблюдается у 37% мужчин с диабетом и 44% у женщин с диабетом и превышение холестерина ЛПНП>160 мг/дл составляет 31% и 44%, соответственно в данной популяции. Диабет представляет собой заболевание, при котором уменьшена способность пациентов контролировать уровень глюкозы в крови в результате частичного ухудшения ответа на инсулин. Диабет 2-го типа (СД2), также называемый инсулиннезависимый сахарный диабет (NIDDM), составляет 80-90% от всех случаев диабета в развитых странах. При СД2 островки Лангерганса в поджелудочной железе производят инсулин, но в первичных целевых тканях для инсулина (мышцы, печень и жировая ткань) сильная резистентность к его эффектам. Организм компенсирует это продуцированием большего количества инсулина в конечном счете приводя к снижению способности продуцировать инсулин поджелудочной железой. Таким образом, СД2 является сердечно-сосудистым метаболическим синдромом, связанным с сопутствующей патологией, включая дислипидемию и инсулиновую резистентность, а также гипертонию, эндотелиальную дисфункцию и воспалительный атеросклероз.
Терапией первой линии при дислипидемии и диабете является диета с низким содержанием глюкозы и жиров, физические упражнения и снижение веса. Вовлеченность пациентов может уменьшаться и тогда становится необходимым лечение различных метаболических недостатков, например, с помощью липид-модулирующих агентов, таких как статины и фибраты, гипогликемических средств, таких как сульфонилмочевины и метформин, или сенситайзеров инсулина тиазолидиндионового класса PPAR-гамма агонистов. Последние исследования подтвердили, что модуляторы FXR могут обладать расширенным терапевтическим действием, обеспечивая улучшенную нормализацию уровня как холестерина ЛПНП, так и триглицеридов, которая в настоящее время возможна только при использовании комбинации существующих лекарственных средств и, вдобавок, может устранять эффекты обратной связи на гомеостаз клеточного холестерина.
Новые соединения согласно настоящему изобретению превосходят соединения, известные в уровне техники, поскольку они связываются с и избирательно модулируют FXR с высокой эффективностью. Следовательно, уменьшается абсорбция холестерина, понижается уровень холестерина ЛПНП и триглицеридов, и воспалительный атеросклероз ослабевает. Поскольку на FXR модуляцию реагируют множественные аспекты комбинированных дислипидемии и гомеостаза холестерина, ожидается, что они обладают расширенным терапевтическим действием по сравнению с известными в уровне техники соединениями.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Если не указано иного, следующие определения даны для иллюстрирования и определения значений и охвата различных терминов, использованных здесь для описания изобретения.
В настоящем изобретении термин "низший" используется для обозначения группы, состоящей из одного-семи, предпочтительно от одного до четырех атомов углерода.
Термин "галоген" относится к фтору, хлору, брому и йоду, предпочтительно к фтору, хлору и брому.
Термин "алкил", один или в комбинации с другими группами, относится к разветвленно- или линейноцепочечному моновалентному насыщенному алифатическому углеводородному радикалу из от одного до 20 атомов углерода, предпочтительно от одного до 16 атомов углерода, наиболее предпочтительно от одного до 10 атомов углерода. Термин "С1-10-алкил" относится к разветвленно- или линейноцепочечному моновалентному насыщенному алифатическому углеводородному радикалу из от одного до 10 атомов углерода, такому как, например, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил, пентил, 1,1,3,3-тетраметил-бутил и аналогичным. В частности, термин "алкил" включает низшие алкильные группы, как описано далее.
Термин "низший алкил" или "С1-7-алкил", один или в комбинации, обозначает разветвленно- или линейноцепочечную алкильную группу с от 1 до 7 атомами углерода, предпочтительно разветвленно- или линейноцепочечную алкильную группу с от 1 до 6 атомами углерода и особенно предпочтительно разветвленно- или линейноцепочечную алкильную группу с от 1 до 4 атомами углерода. Примерами разветвленно- или линейноцепочечных С1-7алкильных групп являются метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, изомерные пентилы, изомерные гексилы и изомерные гептилы, особенно метил и этил, и наиболее предпочтительно метил.
Термин "циклоалкил" или "С3-7-циклоалкил" означает насыщенную карбоциклическую группу, содержащую от 3 до 7 атомов углерода, такую как циклопропил, циклобутил, циклопентил, циклогексил или циклогептил. В частности, термин "циклоалкил" означает циклогексил.
Термин "низший циклоалкилалкил" или "С3-7-циклоалкил-С1-7-алкил" относится к низшей алкильной группе, как определено выше, в которой по меньшей мере один атом водорода низшей алкильной группы замещен циклоалкилом. Примером является циклопропилметил.
Термин "низший алкокси" или "С1-7-алкокси" относится к группе R'-O-, в которой R' представляет собой низший алкил, и термин "низший алкил" был определен ранее. Примерами низших алкокси групп являются метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, втор-бутокси и трет-бутокси, особенно метокси и этокси.
Термин "циклоалкилокси" или "С3-7-циклоалкилокси" относится к группе R"-O-, в которой R" представляет собой циклоалкил. Примерами циклоалкилокси групп являеются циклопропокси, циклобутилокси, циклопентилокси, циклогексилокси и циклогептилокси.
Термин "алкоксициклоалкил" означает насыщенную С3-7-циклоалкильную группу, как определено выше, при этом один из 3-7 углеродных атомов замещен атомом кислорода. Примерами "алкоксициклоалкильных" групп являются оксиранил, оксетан, тетрагидрофуран и тетрагидропиран, особенно оксиран.
Термин "низший галогеналкил " или "галоген-С1-7-алкил" относится к низшей алкильной группе, как определено выше, в которой по меньшей мере один атом водорода в составе низшей алкильной группы замещен атомом галогена, в частности фтором или хлором, в особенности фтором. Среди галогенированных низших алкильных групп находятся трифторметил, дифторметил, трифторэтил, 2,2-дифторэтил, фторметил и хлорметил.
Термин "низший галогеналкокси" или "галоген-С1-7-алкокси" относится к низшей алкокси группе, как определено выше, в которой по меньшей мере один атом водорода низшей алкокси группы замещен атомом галогена, в частности фтором или хлором, особенно фтором. Среди предпочтительных галогенированных низших алкокси-групп находятся трифторметокси, дифторметокси, фторметокси и хлорметокси.
Термин "карбоксил" обозначает группы-СООН.
Термин "низший алкоксикарбонил" или "С1-7-алкоксикарбонил" относится к группе -CO-OR', в которой R' представляет собой низший алкил, и термин "низший алкил" был определен ранее. Примерами низших алкоксикарбонильных групп являются метоксикарбонил или этоксикарбонил.
Термин "низший алкоксикарбонилалкил " или "С1-7-алкоксикарбонил-С1-7-алкил" обозначает низшую алкильную группу, как определено выше, в которой один атом водорода низшей алкильной группы замещен С1-7-алкоксикарбонилом. Примером низшей алкоксикарбонилалкильной группы является -СН2-СООСН3.
Термин "низший алкоксикарбонилалкокси" или "С1-7-алкоксикарбонил-С1-7-алкокси" относится к низшей алкокси группе, как определено выше, в которой один атом водорода низшей алкокси группы замещен С1-7-алкоксикарбонилом. Примером низшей алкоксикарбонилалкокси группы является трет-бутоксикарбонилметокси (-O-СН2-СОО-С(СН3)3).
Термин "низший карбоксиалкил " или "карбокси-С1-7-алкил" относится к низшей алкильной группе, как определено выше, в которой по меньшей мере один атом водорода низшей алкильной группы замещен карбоксильной группой. Среди низших карбоксиалкильных групп находятся карбоксиметил (-СН2-СООН) и карбоксиэтил (-СН2-СН2-СООН), в частности карбоксиметил.
Термин "низший карбоксиалкокси" или "карбокси-С1-7-алкокси" относится к низшей алкокси группе, как определено выше, в которой по меньшей мере один атом водорода низшей алкокси группы замещен карбоксильной группой. Примером низшей карбоксиалкокси группы является карбоксиметокси (-O-СН2-СООН).
Термин "гетероциклил" относится к 5-6-членному моноциклическому кольцу или 8-10-членному би- или трицикличному кольцу, которое может содержать 1, 2 или 3 атома, выбранных из азота, кислорода и/или серы, такому как морфолинил, тиоморфолинил, 1,1-диоксо-тиоморфолинил, пиперидинил, 2-оксо-пиперидинил, пирролидинил, 2-оксо-пирролидинил, пиперазин-2-он, 8-окса-3-аза-бицикло [3.2.1]октил, пиперазинил, тетрагидрофуранил и тетрагидропиранил. В частности, термин "гетероциклил" относится к тетрагидрофуранилу и тетрагидропиранилу.
Термин "защитная группа" относится к группам, которые используются для временной защиты функциональных групп, в частности гидроксигрупп.Типичными примерами защитных групп являются бензил, пара-метоксибенщил, трет-бутилдиметилсилил, трет-бутилдифенилсилил и (для защиты аминогрупп) Воc и бензилоксикарбонил.
Соединения формулы I могут образовывать фармацевтически приемлемые соли. В качестве примера таких фармацевтически приемлемых солей могут выступать кислотно-аддитивные соли соединений формулы I и физиологически совместимыми минеральными солями, такими как соляная кислота, серная кислота, сернистая или фосфорная кислота; или органических кислот, таких как метансульфоновая кислота, р-толуолсульфоновая кислота, уксусная кислота, молочная кислота, трифторуксусная кислота, лимонная кислота, фумаровая кислота, малеиновая кислота, винная кислота, янтарная кислота или салициловая кислота. Термин "фармацевтически приемлемые соли " относится к таким солям соединений формулы I, в которых присутствующая группа СООН может образовывать соли с основаниями. Примером таких солей являются такие соли как щелочноземельные и аммонийные соли, такие как, например, соли Na, К, Са и соли триметиламмония. Термин " фармацевтически приемлемые соли " также относится к таким солям.
Более подробно, настоящее изобретение раскрывает соединения формулы
где
А представляет собой -СН2- или -(СН2)3-,
R1 представляет собой кольцо, выбранное из фенила, нафтила или гетероарила, указанное кольцо необязательно замещено 1-3 заместителями, независимо выбранными из группы, состоящей из низшего алкила, галогена, низшего галогеналкила, гидрокси, низшего алкокси, низшего галогеналкокси и циано;
R2 выбран из группы, состоящей из водорода, метила, этила, гидрокси, метокси, фтора, фторметила, дифторметила и трифторметила;
R3 выбран из группы, состоящей из
незамещенного циклоалкила или циклоалкила, замещенного 1-4 группами, независимо выбранными из метила или фтора, низшего циклоалкилалкила,
незамещенного фенила или фенила, замещенного 1-3 заместителями, независимо выбранными из группы, состоящей из низшего алкила, галогена, низшего галогеналкила, гидрокси, низшего алкокси, низшего галогеналкокси и циано и гетероциклила;
R4 выбран из группы, состоящей из -C(O)-NH-R5,
-CR7R8-OR6, -O-(CR7R8)n-R6;
-CR7R8-SR6, -CR7R8-SO-R6, -CR7R8-SO2-R6,
-CR7R8-NH-R6; -CH=CH-R6 и -(CH2)2-R6,
где
n представляет собой 0 или 1,
R5 выбран из группы, состоящей из низшего алкила, циклоалкила, низшего циклоалкилалкила,
циклоалкила, замещенного 1-3 заместителями, независимо выбранными из группы, состоящей из гидрокси, карбоксила, тетразолила, низшего карбоксиалкила, низшего алкоксикарбонила, низшего алкоксикарбонилалкила, низшего карбоксиалкокси и низшего алкоксикарбонилалкокси,
незамещенного фенила и фенила, замещенного 1-3 заместителями, независимо выбранными из группы, состоящей из низшего алкила, галогена, низшего галогеналкила, гидрокси, низшего алкокси, низшего галогеналкокси, карбоксила, тетразолила, низшего алкокси карбонила, низшего алкоксикарбонилалкила, низшего карбоксиалкила, низшего карбоксиалкокси, низшего алкоксикарбонилалкокси, циано и циклоалкилокси, где циклоалкильная группа замещена карбоксилом, низшим алкоксикарбонилом или тетразолилом;
незамещенного пиридила или пиридила, замещенного радикалом, выбранным из группы, состоящей из карбоксила, низшего алкоксикарбонила или тетразолила;
R6 выбран из группы, состоящей из
низшего алкила, циклоалкила, низшего циклоалкилалкила,
циклоалкила, замещенного 1-3 заместителями, независимо выбранными из группы, состоящей из гидрокси, карбоксила, тетразолила, низшего карбоксиалкила, низшего алкоксикарбонила, низшего алкоксикарбонилалкила, низшего карбоксиалкокси и низшего алкоксикарбонилалкокси,
гетероциклила,
незамещенного пиридила или пиридила, замещенного карбоксилом, низшим алкоксикарбонилом или тетразолилом,
незамещенного фенила и фенила, замещенного 1-3 заместителями, независимо выбранными из группы, состоящей из низшего алкила, галогена, низшего галогеналкила, гидрокси, низшего алкокси, низшего галогеналкокси, карбоксила, тетразолила, низшего алкоксикарбонила, низшего алкоксикарбонилалкила, низшего карбоксиалкила, низшего карбоксиалкокси, низшего алкоксикарбонилалкокси, циано и циклоалкилокси, где циклоалкильная группа замещена карбоксилом, низшим алкоксикарбонилом или тетразолилом; и
R7 и R8 независимо выбраны из группы, состоящей из водорода, низшего алкила и низшего галогеналкила, или
R7 и R8 вместе с атомом углерода, к которому они присоединены, образуют циклоалкильное или алкоксициклоалкильное кольцо;
или их фармацевтически приемлемые соли.
Настоящее изобретение обеспечивает соединения формулы I, в которых R1 представляет собой кольцо, выбранное из фенила, нафтила или гетероарила, указанное кольцо необязательно замещено 1-3 заместителями, независимо выбранными из группы, состоящей из низшего алкила, галогена, низшего галогеналкила, гидрокси, низшего алкокси, низшего галогеналкокси и циано. Настоящее изобретение также обеспечивает соединения формулы I, в которых R1 представляет собой фенольное кольцо, указанное кольцо может быть незамещенным или замещенным 1-3 заместителями, независимо выбранными из группы, состоящей из низшего алкила, галогена, низшего галогеналкила, гидрокси, низшего алкокси, низшего галогеналкокси и циано. Особенно, настоящее изобретение обеспечивает соединения формулы I, в которых R1 представляет собой фенил или фенил, замещенный галогеном.
Соединениями формулы I в соответствии с настоящим изобретением" являются также те, у которых R2 выбран из группы, состоящей из водорода, метила, этила, гидрокси, метокси, фтора, фторметила, дифторметила и трифторметила. Настоящее изобретение также обеспечивает соединения формулы I, в которых R2 является водородом.
Более того, соединениями формулы I в соответствии с настоящим изобретением являются те, у которых R3 выбран из группы, состоящей из
незамещенного циклоалкила или циклоалкила, замещенного 1-4 группами, независимо выбранными из метила или фтора, низшего циклоалкилалкила,
незамещенного фенила или фенила, замещенного 1-3 заместителями, независимо выбранными из группы, состоящей из низшего алкила, галогена, низшего галогеналкила, гидрокси, низшего алкокси, низшего алогеналкокси и циано и гетероциклила.
Настоящее изобретение относится также к соединениям формулы I, в которых R3 представляет собой циклоалкил.
Частной группой соединений настоящего изобретения являются те соединения, у которых R4 выбран из группы, состоящей из -C(O)-NH-R5, -CR7R8-OR6 и -CR7R8-SR6, где
R5 выбран из группы, состоящей из
низшего алкила, циклоалкила, низшего циклоалкилалкила,
циклоалкила, замещенного 1-3 заместителями, независимо выбранными из группы, состоящей из гидрокси, карбоксила, тетразолила, низшего карбоксиалкила, низшего алкоксикарбонила, низшего алкоксикарбонилалкила, низшего карбоксиалкокси и низшего алкоксикарбонилалкокси,
незамещенного фенила и фенила, замещенного 1-3 заместителями, независимо выбранными из группы, состоящей из низшего алкила, галогена, низшего галогеналкила, гидрокси, низшего алкокси, низшего галогеналкокси, карбоксила, тетразолила, низшего алкокси карбонила, низшего алкоксикарбонилалкила, низшего карбоксиалкила, низшего карбоксиалкокси, низшего алкоксикарбонилалкокси, циано и циклоалкилокси, где циклоалкильная группа замещена карбоксилом, низшим алкоксикарбонилом или тетразолилом; и
незамещенного пиридила или пиридила, замещенного радикалом, выбранным из группы, состоящей из карбоксила, низшего алкоксикарбонила или тетразолила;
R6 выбран из группы, состоящей из
низшего алкила, циклоалкила, низшего циклоалкилалкила,
циклоалкила, замещенного 1-3 заместителями, независимо выбранными из группы, состоящей из гидрокси, карбоксила, тетразолила, низшего карбоксиалкила, низшего алкоксикарбонила, низшего алкоксикарбонилалкила, низшего карбоксиалкокси и низшего алкоксикарбонилалкокси, гетероциклила,
незамещенного пиридила или пиридила, замещенного карбоксилом, низшим алкоксикарбонилом или тетразолилом,
незамещенного фенила и фенила, замещенного 1-3 заместителями, независимо выбранными из группы, состоящей из низшего алкила, галогена, низшего галогеналкила, гидрокси, низшего алкокси, низшего галогеналкокси, карбоксила, тетразолила, низшего алкокси карбонила, низшего алкоксикарбонилалкила, низшего карбоксиалкила, низшего карбоксиалкокси, низшего алкоксикарбонилалкокси, циано и циклоалкилокси, где циклоалкильная группа замещена карбоксилом, низшим алкоксикарбонилом или тетразолилом; и
R7 и R8 независимо выбраны из группы, состоящей из водорода, низшего алкила и низшего галогеналкила, или
R7 и R8 вместе с атомом углерода, к которому они присоединены, образуют циклоалкильное или алкоксициклоалкильное кольцо.
Таким образом, настоящее изобретение обеспечивает соединения формулы I в соответствии с любым из пунктов 1-6, где R4 представляет собой -C(O)-NH-R5 и R5 выбран из группы, состоящей из
низшего алкила, циклоалкила, низшего циклоалкилалкила,
циклоалкила, замещенного 1-3 заместителями, независимо выбранными из группы, состоящей из гидрокси, карбоксила, тетразолила, низшего карбоксиалкила, низшего алкоксикарбонила, низшего алкоксикарбонилалкила, низшего карбоксиалкокси и низшего алкоксикарбонилалкокси,
незамещенного фенила и фенила, замещенного 1-3 заместителями, независимо выбранными из группы, состоящей из низшего алкила, галогена, низшего галогеналкила, гидрокси, низшего алкокси, низшего галогеналкокси, карбоксила, тетразолила, низшего алкоксикарбонила, низшего алкоксикарбонилалкила, низшего карбоксиалкила, низшего карбоксиалкокси, низшего алкоксикарбонилалкокси, циано и циклоалкилокси, где циклоалкильная группа замещена карбоксилом, низшим алкоксикарбонилом или тетразолилом; и
незамещенного пиридила или пиридила, замещенного радикалом, выбранным из группы, состоящей из карбоксила, низшего алкоксикарбонила или тетразолила.
Более подробно, настоящее изобретение обеспечивает соединения, в которых R4 представляет собой -C(O)-NH-R5, и R5 выбран из группы, состоящей из циклоалкила, незамещенного фенила и фенила, замещенного 1-3 заместителями, независимо выбранными из группы, состоящей из низшего алкила, галогена, низшего галогеналкила, гидрокси, низшего алкокси, низшего галогеналкокси, карбоксила, тетразолила, низшего алкоксикарбонила, низшего алкоксикарбонилалкила, низшего карбоксиалкила, низшего карбоксиалкокси, низшего алкоксикарбонилалкокси, циано и циклоалкилокси, где циклоалкильная группа замещена карбоксилом, низшим алкоксикарбонилом или тетразолилом.
Настоящее изобретение также обеспечивает соединения формулы I, в которых R4 представляет собой -C(O)-NH-R5, и R5 выбран из группы, состоящей из циклоалкила и фенила, замещенного 1-3 заместителями, независимо выбранными из группы, состоящей из низшего алкила, галогена, карбоксила, тетразолила, низшего алкоксикарбонила, низшего карбоксиалкокси, низшего алкоксикарбонилалкокси и циано.
Настоящее изобретение дополнительно обеспечивает соединения формулы I, в которых R4 представляет собой -C(O)-NH-R5 и R5 представляет собой циклоалкил или фенил, замещенный 1-3 заместителями, независимо выбранными из группы, состоящей из галогена, карбоксила, низшего алкоксикарбонила, низшего карбоксиалкокси и низшего алкоксикарбонилалкокси.
Также раскрыты соединения формулы I по любому из пунктов 1-6, в которых R4 представляет собой -CR7R8-OR6 и в которых
R6 выбран из группы, состоящей из
низшего алкила, циклоалкила, низшего циклоалкилалкила,
циклоалкила, замещенного 1-3 заместителями, независимо выбранными из группы, состоящей из гидрокси, карбоксила, тетразолила, низшего карбоксиалкила, низшего алкоксикарбонила, низшего алкоксикарбонилалкила, низшего карбоксиалкокси и низшего алкоксикарбонилалкокси,
гетероциклила,
незамещенного пиридила или пиридила, замещенного карбоксилом, низшим алкоксикарбонилом или тетразолилом,
незамещенного фенила и фенила, замещенного 1-3 заместителями, независимо выбранными из группы, состоящей из низшего алкила, галогена, низшего галогеналкила, гидрокси, низшего алкокси, низшего галогеналкокси, карбоксила, тетразолила, низшего алкокси карбонила, низшего алкоксикарбонилалкила, низшего карбоксиалкила, низшего карбоксиалкокси, низшего алкоксикарбонилалкокси, циано и циклоалкилокси, где циклоалкильная группа замещена карбоксилом, низшим алкоксикарбонилом или тетразолилом; и
R7 и R8 представляют собой водород.
В частности, настоящее изобретение обеспечивает соединения формулы I, в которых R4 представляет собой -CR7R8-OR6, R6 выбран из группы, состоящей из циклоалкила и фенила, замещенного 1-3 заместителями, независимо выбранными из группы, состоящей из низшего алкила, галогена, карбоксила, тетразолила, низшего алкоксикарбонила, низшего карбоксиалкокси, низшего алкоксикарбонилалкокси и циано, a R7 и R8 представляют собой водород.
Настоящее изобретение дополнительно обеспечивает соединения формулы I, в которых А представляет собой -СН2-. Этими соединениями являются те, которые имеют формулу I-I
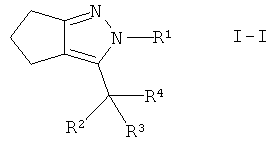 .
.
Настоящее изобретение также обеспечивает соединения формулы I, в которых А представляет собой -(СН2)3-. Этими соединениями являются те, которые имеют формулу I-II
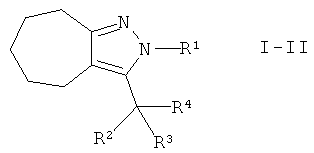 .
.
Дополнительно настоящее изобретение относится к соединениям формулы I, которые выбраны из группы, состоящей из
4-{2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2-циклогексил-этокси}-3,5-диметил-бензойной кислоты метилового эфира,
4-{2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2-циклогексил-этокси}-3,5-диметил-бензойной кислоты,
4-{2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2-циклогексил-этокси}-3-фторбензонитрила,
2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2,N-дициклогексил-ацетамида,
2-(4-хлорфенил)-3-{1-циклогексил-2-[2-фторо-4-(2Н-тетразол-5ил)-фенокси]-этил}-2,4,5,6-тетрагидро-циклопентапиразола,
4-{2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2-циклогексил-ацетиламино}-3-фторбензойной кислоты,
6-{2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2-циклогексил-этокси}-никотиновой кислоты,
4-{2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2-циклогексил-этилсульфанил}-бензойной кислоты,
2-(4-{2-[2-(4-хлорфенил)-2,4,5,6,7,8-гексагидро-циклогептапиразол-3-ил]-2-циклогексил-ацетиламино}-3-фторфенокси)-2-метил-пропионовой кислоты этилового эфира,
2-(4-{2-[2-(4-хлорфенил)-2,4,5,6,7,8-гексагидро-циклогептапиразол-3-ил]-2-циклогексил-ацетиламино}-3-фторфенокси)-2-метилпропионовой кислоты,
2-[2-(4-хлорфенил)-2,4,5,6,7,8-гексагидро-циклогептапиразол-3-ил]-2,N-дициклогексил-ацетамида,
4-{2-[2-(4-хлорфенил)-2,4,5,6,7,8-гексагидро-циклогептапиразол-3-ил]-2-циклогексил-ацетиламино}-3-фторбензойной кислоты метилового эфира,
4-{2-[2-(4-хлорфенил)-2,4,5,6,7,8-гексагидро-циклогептапиразол-3-ил]-2-циклогексил-ацетиламино}-3-фторбензойной кислоты,
4-{2-[2-(4-хлорфенил)-2,4,5,6,7,8-гексагидро-циклогептапиразол-3-ил]-2-циклогексил-этокси}-3-фторбензонитрила,
4-{2-[2-(4-хлорфенил)-2,4,5,6,7,8-гексагидро-циклогептапиразол-3-ил]-2-циклогексил-этокси}-3,5-диметил-бензойной кислоты,
2-(4-хлорфенил)-3-{1-циклогексил-2-[2-фторо-4-(1Н-тетразол-5ил)-фенокси]-этил}-2,4,5,6,7,8-гексагидро-циклогептапиразола,
или их фармацевтически приемлемым солям.
В частности, настоящее изобретение относится к соединениям формулы I, выбранным из группы, состоящей из
4-{2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2-циклогексил-этокси}-3,5-диметил-бензойной кислоты,
4-{2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2-циклогексил-этокси}-3-фторбензонитрила,
2-(4-хлорфенил)-3-{1-циклогексил-2-[2-фторо-4-(2Н-тетразол-5ил)-фенокси]-этил}-2,4,5,6-тетрагидро-циклопентапиразола,
4-{2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2-циклогексил-ацетиламино}-3-фторбензойной кислоты,
6-{2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2-циклогексил-этокси}-никотиновой кислоты,
4-{2-[2-(4-хлорфенил)-2,4,5,6,7,8-гексагидро-циклогептапиразол-3-ил]-2-циклогексил-ацетиламино}-3-фторбензойной кислоты,
2-(4-хлорфенил)-3-{1-циклогексил-2-[2-фторо-4-(1Н-тетразол-5ил)-фенокси]-этил}-2,4,5,6,7,8-гексагидро-циклогептапиразола,
или их фармацевтически приемлемым солям.
Особенно, настоящее изобретение относится к соединению формулы I, которое представляет собой
4-{2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2-циклогексил-этокси}-3,5-диметил-бензойную кислоту.
Настоящее изобретение также относится к соединению формулы I, которое представляет собой
2-(4-хлорфенил)-3-{1-циклогексил-2-[2-фторо-4-(2Н-тетразол-5ил)-фенокси]-этил}-2,4,5,6-тетрагидро-циклопентапиразол.
Кроме того, настоящее изобретение относится к способу получения соединений формулы I, которые определены выше, при этом способ содержит взаимодействие карбоновой кислоты формулы II
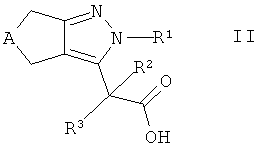 ,
,
где А и R1-R3 такие, как определено здесь ранее, с амином формулы III
 ,
,
где R5 такой, как определено здесь ранее, в присутствии связывающего агента в щелочной среде с получением соединения формулы Iа
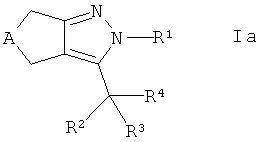 ,
,
где R4 представляет собой -C(O)-NH-R5, и, если требуется, конвертирование полученного соединения в его фармацевтически приемлемую соль.
Подходящими связующими агентами являются, например, N,N'-карбонилдиимидазол (CDI), N,N'-дициклогексилкарбодиимид (DCC), N-(3-диметиламинопролил)-N'-этил-карбодиимид-гидрохлорид (EDCI), O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилуроний тетрафторборат - (TBTU), 1-[бис(диметиламино)метилен]-1Н-1,2,3-триазоло[4,5-b]пиридиний-3-оксида гексафторфосфат (HATU) или бензотриазол-1-илокситрис(диметиламино)фосфония гексафторфосфат (ВОР), с предпочтительным использованием EDCI, TBTU или ВОР. В щелочной среде означает присутствие основания, такого как диизопропилэтиламин, триэтиламин, N-метилморфолин, необязательно в присутствии 4-диметиламинопиридина или HOBt (1-гидроксибензотриазол). Реакция проводится в подходящем растворителе, таком как, например, дихлорметан, ДМФ (диметилформамид), ДМА (диметилацетамид) или диоксан при температуре между 0°С и температурой окружающей среды.
Альтернативно, в настоящем изобретении предложен способ получения соединений формулы I, которые определены выше, который содержит
взаимодействие спирта формулы IV
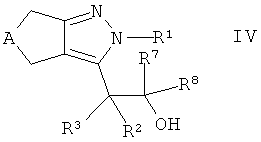 ,
,
где А, R1-R3 и R7, R8 такие, как определено здесь ранее, с соединением формулы V
 ,
,
где R6 является таким, как определено здесь ранее, и X обозначает галидный, мезилатный или тозилатный остаток, или в случае, когда R6 относится к фенилу или фенилу, замещенному как определено в настоящем изобретении ранее, X обозначает гидроксигруппу,
с получением соединения формулы Ib
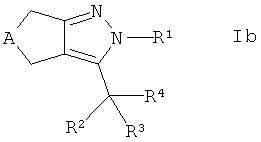 ,
,
где R4 представляет собой -CR7R8-OR6, и, если требуется, конвертирование полученного соединения в его фармацевтически приемлемую соль.
Соединения формулы V, где X обозначает галидный, мезилатный или тозилатный остаток, могут взаимодействовать с соединениями формулы IV в присутствии слабого основания, такого как карбонат цезия или калия, в растворителе, таком как N,N-диметилформамид, ацетонитрил, ацетон или метилэтилкетон, при температуре в диапазоне от комнатной до 140°С, предпочтительно около 50°С, поскольку соединения формулы V, где X обозначает гидроксигруппу могут взаимодействовать с соединениями формулы IV в присутствии трифенилфосфина и ди-трет-бутил-, диизопропил- или диэтил-азодикарбоксилата или в присутствии трибутилфосфина и N,N,N',N'-тетраметил азодикарбоксамида, предпочтительно в растворителе аналогичном толуолу, дихлорметану или тетрагидрофурану при температуре окружающей среды.
Альтернативно, настоящее изобретение относится к способу получения соединений формулы I, которые определены выше, который содержит взаимодействие спирта формулы IV
,
где А, R1-R3 и R7, R8 такие, как определено здесь ранее, с низшим алкил-, низшим фторалкил- или фенилсульфоновой кислоты хлоридом или ангидридом в присутствии основания с получением интермедиата
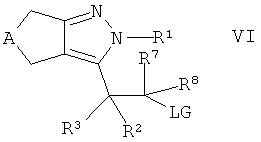
где LG обозначает -ОSО2-низший алкил, -ОSО2-низший фторалкил или -ОSО2-фенил, и взаимодействие интермедиата в присутствии основания с тиолом
 ,
,
где R6 является таким, как определено ранее, с получением соединения формулы Ic
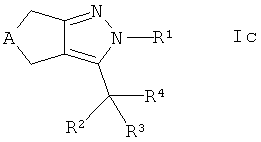 ,
,
где R4 представляет собой -CR7R8-SR6, и, если требуется, конвертирование полученного соединения в его фармацевтически приемлемую соль.
Альтернативно, в настоящем изобретении предложен способ получения соединений формулы I, которые определены выше, который содержит взаимодействие спирта формулы VIII
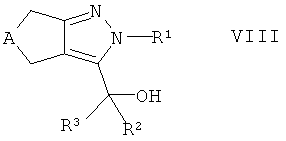 ,
,
где А и R1-R3 такие, как определено здесь ранее, с соединением формулы IX
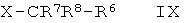 ,
,
где R6-R8 такие, как определено в п.1, а X обозначает галидный, мезилатный или тозилатный остаток,
с получением соединения формулы Id
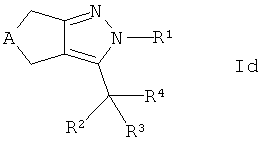
где R4 представляет собой -CR7R8-R6, и, если требуется, конвертирование полученного соединения в его фармацевтически приемлемую соль.
Более детально, соединения формулы I, которые являются объектом настоящего изобретения, могут быть получены так, как показано на схемах А, В, С, D, Е, F, G, Н, I и J, с помощью способов, данных в примерах или аналогичными способами. Если не указано иного, A, R1, R2, R2', R3, R3', R4, R4', R5, R5', R6, R7, R8 и n являются такими, как описано выше. Исходные соединения являются как коммерчески доступными или описанными в литературе, так и могут быть получены способами, хорошо известными из уровня техники.
Схема А
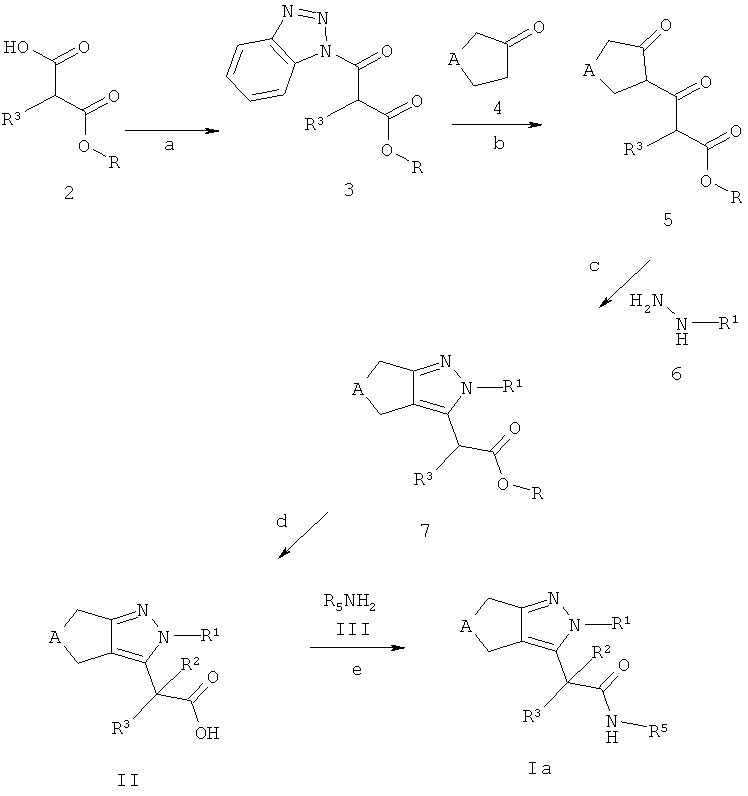
Альтернативно, циклопентил- и циклопиразолы формулы Iа могут быть получены из 2-замещенных моноэфиров малоновой кислоты 2 (R, например, относится к C1-7-алкилу, Схема А). Производные малоновой кислоты 2 являются коммерчески доступными, описаны в литературе или могут быть синтезированы способами, хорошо известными квалифицированным специалистам. Для облегчения конверсии производных малоновой кислоты 2 в бис-кето эфиры 5 кислотная группа соединений 2 может, например, быть трансформирована в бензотриазол-1-ил амиды 3 (стадия а). Эта трансформация может быть достигнута, например, посредством i) обработки кислот 2 тионилхлоридом, предпочтительно при кипячении с обратным холодильником с получением соответствующего хлорангидрида (альтернативный способ: карбоновая кислота 2, СН2Сl2, (СlСО)2, ДМФ, комнатная температура); и ii) последующего взаимодействия с 1,2,3-бензотриазол в присутствии основания, такого как триэтиламин или аналогично, предпочтительно в растворителе, аналогичном дихлорметану, при температуре между -20°С и температурой окружающей среды. Бензотриазолы 3 могут затем быть превращены в бис-кето эфиры 5 посредством взаимодействия с депротонированным кетоном (полученном из кетона 4), предпочтительно в растворителе, таком как тетрагидрофуран или аналогичном (стадия b). Депротонирование может быть выполнено с использованием основания, такого как диизопропиламид лития в растворителе, таком как тетрагидрофуран или аналогичном, при температуре между -78°С и температурой окружающей среды. Кетоны 4 являются коммерчески доступными, описаны в литературе или могут быть синтезированы способами, хорошо известными квалифицированным специалистам. Конденсация бис-кетонов 5 с (гетеро)ароматическими гидразинами 6 или солью, например, хлористоводородная соль (гетеро)ароматических гидразинов 6 дает циклопентил- или циклогептилпиразоловые эфиры 7 (стадия с). Предпочтительно, такая конденсация проводится в растворителе, таком как этанол и аналогичном, при температуре кипения используемого растворителя. (Гетеро)ароматические гидразины 6 или соответствующие соли (гетеро)ароматических гидразинов являются коммерчески доступными, описаны в литературе или могут быть синтезированы способами, хорошо известными квалифицированным специалистам. Эфиры 7 могут быть омылены с формированием кислот формулы II, с использованием, например, водных LiOH, NaOH или КОН в тетрагидрофуране/этаноле или другом подходящем растворителе при температуре между 0°С и температурой кипения используемого растворителя (стадия d). Кислоты формулы II - после необходимого активирования - могут быть сопряжены с аминами формулы III с получением амидов формулы Ia с использованием обычных процедур пептидного связывания, описанных в литературе (стадия е). Активация карбоновых кислот формулы II может осуществляться с использованием способов, хорошо известных квалифицированным специалистам (например, хлорангидриды карбоновых кислот: 1. Карбоновая кислота, CH2Cl2, (ClCO)2, ДМФ, комнатная температура; или 2. Карбоновая кислота, тионил хлорид, кипячение с обратным холодильником). Альтернативно, карбоновые кислоты формулы II могут быть активированы in situ и трансформированы в конечные продукты формулы Iа с использованием связующих реагентов, таких как, например, N,N'-карбонилдиимидазол (CDI), N,N'-дициклогексилкарбодиимид (DCC), N-(3-диметиламинопролил)-N'-этил-карбодиимид-гидрохлорид (EDCI), O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилуроний тетрафторборат (TBTU), 1-[бис(диметиламино)метилен]-1Н-1,2,3-триазоло[4,5-b]пиридиний-3-оксида гексафторфосфат (HATU) или бензотриазол-1-илокситрис(диметиламино)фосфония гексафторфосфат (ВОР). Предпочтительно использовать EDCI, TBTU или ВОР. Реакция проводится в присутствии основания, такого как диизопропилэтиламин, триэтиламин, N-метилморфолин, необязательно в присутствии 4-диметиламинопиридина или HOBt (1-гидроксибензотриазол), в растворителе, таком как дихлорметан, ДМФ, ДМА или диоксан, при температуре между 0°С и температурой окружающей среды. Амины формулы III являются коммерчески доступными, описаны в литературе или могут быть получены способами, хорошо известными квалифицированным специалистам.
Амиды формулы Ia могут включать карбоновые эфиры, которые могут быть гидролизованы до соответствующих кислот с использованием стандартных способов, например, обработкой гидроокисью щелочного металла, аналогичной LiOH или NaOH в смеси полярных растворителей, такой как тетрагидрофуран/этанол/вода или обработкой соляной кислотой в диоксане в случае, например, трет-бутиловых эфиров. Необязательно, 4,5,6,7-тетрагидроиндазолы формулы Ia могут содержать циано-группы, которые могут быть конвертированы в соответствующие тетразолы с использованием стандартных процедур, например, обработкой азидом натрия в присутствии кислоты Льюиса в воде или органическом растворителе, аналогичном дихлорметану, при температуре между 0°С и и температурой кипения растворителя.
Если одно из исходных соединений, соединение формулы 2, 4, 6 или III, содержит одну или больше функциональных групп, которые являются нестабильными или реактивны в условиях проведения реакции на одной или больше реакционных стадий, перед проведением критической стадии могут быть введены подходящие защитные группы (PG) (как описано, например, в "Protective Groups in Organic Chemistry" под авторством T.W.Greene и P.G.M. Wutts, 2nd Ed., 1991, Wiley N.Y.) с использованием способов, хорошо известных из уровня техники. Такие защитные группы могут быть удалены на последующих стадиях синтеза с использованием стандартных методов, описанных в литературе.
Если одно или более соединений формул 2-7, II или III содержат хиральные центры, циклопентил- или циклогептилпиразолы формулы Iа могут быть получены в виде смеси диастереомеров или энантиомеров, которые могут быть разделены способами, хорошо известными в уровне техники, например (хиральной) ВЭЖХ или кристаллизацией. Рацемические соединения могут быть разделены на антиподы через диастереомерные соли посредством кристаллизации, например, с оптически чистыми аминами (такими как, например, (R) или (S)-1-фенил-этиламин, (R) или (S)-1-нафтален-1-ил-этиламин, бруцин, хинин или хинидин) или разделением антиподов посредством специальных хроматографических методов с использованием как хиральных адсорбентов, так и хиральных элюентов.
Схема В
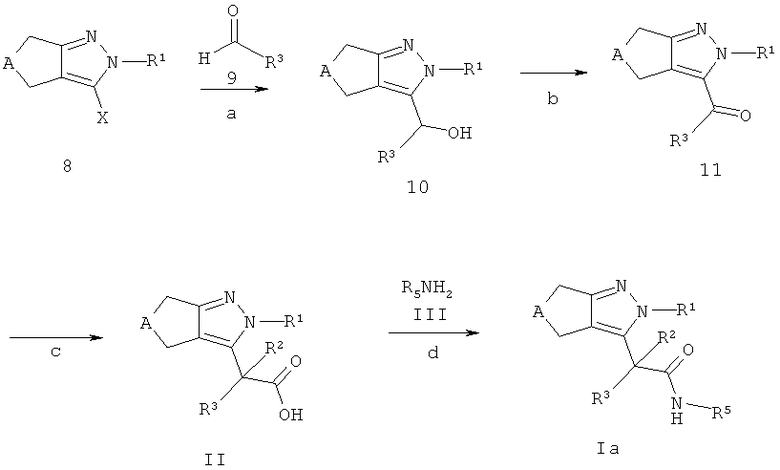
Циклопентил- и циклопиразолы 8, где X соответствует Н, Cl, Вr или I, описаны в литературе, могут быть получены способами, хорошо известными квалифицированным специалистам, или методами, описанными на схеме Е. Циклопентил- и циклопиразолы 8 могут быть конвертированы в спирты 10, например, обработкой сильным основанием, таким как н-бутиллитий в растворителе, аналогичном тетрагидрофурану, предпочтительно при температуре между -78°С и 0°С и последующим добавлением альдегида формулы 9 (стадия а). Альдегиды 9 являются коммерчески доступными, описаны в литературе или могут быть получены способами, хорошо известными квалифицированным специалистам.
Спирты 10 могут быть окислены в кетоны 11, применяя стандартные, описанные в литературе процедуры, например, 2-йодоксибензойной кислотой в смеси тетрагидрофурана и диметилсульфоксида, предпочтительно при температуре между 0°С и температурой окружающей среды (стадия b).
Кетоны 11 могут быть превращены в кислоты формулы II с использованием, например, следующей последовательности реакций: i) взаимодействие кетонов 11 с триметилсилилцианидом с использованием каталитического количества йодида цинка (II) с получением соответствующих триметилсиланилокси-ацетонитрилов, предпочтительно при температуре между температурой окружающей среды и 50°С; ii) последующее однореакционное восстановление с хлоридом олова (II) и гидролиза в кислоты формулы II в смеси растворителей, состоящей из концентрированной водной соляной кислоты и уксусной кислоты, предпочтительно при температуре кипения используемой смеси растворителей (стадия с).
Кислоты формулы II - после соответствующей активации - могут быть сопряжены с аминами формулы III с получением амидов формулы Ia (соединения формулы I, где R4 соответствует -C(O)-NH-R5), используя стандартные процедуры пептидного связывания, описанные в литературе (стадия d). Активация карбоновых кислот формулы II может быть осуществлена способами, хорошо известными квалифицированным специалистам. Например, карбоновые кислоты формулы II могут быть превращены в хлорангидриды карбоновой кислоты посредством растворения кислоты в дихлорметане и ее взаимодействия с (ClCO)2 в ДМФ при комнатной температуре или ее взаимодействия с чистым тионилхлоридом при температуре кипения растворителя. Альтернативно, карбоновые кислоты формулы II могут быть активированы in situ и трансформированы в конечные продукты формулы Ia с использованием связующих реагентов, таких как, например, N,N'-карбонилдиимидазол (CDI), N,N'-дициклогексилкарбодиимид (DCC), N-(3-диметиламинопролил)-N-этил-карбодиимид-гидрохлорид (EDCI), 0-(бензотриазол-1-ил)-N,N,N',N'-тетраметилуроний тетрафторборат (TBTU), 1-[бис(диметиламино)метилен]-1Н-1,2,3-триазоло[4,5-Ь]пиридиний-3-оксида гексафторфосфат (HATU) или бензотриазол-1-илокситрис(диметиламино)фосфония гексафторфосфат (ВОР). Предпочтительно используют EDCI, TBTU или ВОР. Реакция проводится в присутствии основания, такого как диизопропилэтиламин, триэтиламин, N-метилморфолин, необязательно в присутствии 4-диметиламинопиридина или HOBt (1-гидроксибензотриазол), в растворителе, таком как дихлорметан, ДМФ, ДМА или диоксан, при температуре между 0°С и температурой окружающей среды.
Амины формулы III являются коммерчески доступными, описаны в литературе или могут быть получены способами, хорошо известными квалифицированным специалистам. Для введения остатка R2 ≠ водороду карбоновые кислоты формулы II могут, например, i) быть конвертированы в соответствующие эфиры карбоновой кислоты с использованием стандартных литературных способов (например, нагревание кислоты формулы II с первичным или вторичным спиртом в присутствии катализатора, такого как серная кислота, предпочтительно при кипячении с обратным холодильником); ii) обработка полученного эфира основанием и алкилирующим реагентом, используя способы, известные квалифицированному в уровне технике специалисту (например, диизопропиламид лития в качестве основания и алкилгалида в качестве алкилирующего реагента в растворителе, таком как тетрагидрофуран при температуре между -78°С и температуре кипения используемого растворителя). Необязательно, такое алкилирование может быть проведено энантиоселективно или диастереоселективно с использованием спиртов, которые содержат хиральный центр на стадии этерификации и/или хирального катализатора на стадии алкилирования; iii) омыление эфира с формированием замещенных карбоновых кислот формулы II (например, используя водные LiOH, NaOH или КОН в тетрагидрофуране/этаноле или другой подходящий растворитель). Кислоты формулы II с R2=F могут, например, быть синтезирвоаны через прямое фторирование соответствующего силилкетенацеталя с помощью 1-хлорметил-4-фтор-1,4-диазонийбицикло [2.2.2]октан бис-(тетрафторбората) следуя способу, описанному у F. Zhang, J.Z. Song, Tetrahedron Lett. 2006, 47, 7641-7644.
Амиды формулы Ia могут содержать карбоновые эфиры, которые могут быть гидролизованы в соответствующие кислоты посредством стандартных процедур, например, обработкой гидроксидами щелочных металлов, таких как LiOH или NaOH в смеси полярных растворителей аналогичных тетрагидрофуран/этанол/вода или обработкой соляной кислотой в диоксане, в случае, например, трет-бутиловых эфиров. Опционально, циклопентил- или циклогептилпиразолы формулы Iа могут содержать циано-группы, которые могут быть конвертированы в соответствующие тетразолы, используя стандартные процедуры, например, обработкой азидом натрия в присутствии кислоты Льюиса в воде или органическом растворителе аналогичном дихлорметану при температуре между 0°С и температурой кипения растворителя.
Если одно из исходных соединений, соединения формул 8, 9 или III, содержит одну или больше функциональных групп, которые являются нестабильными или реактивны в условиях проведения реакции на одной или больше реакционных стадий, перед проведением критической стадии могут быть введены подходящие защитные группы (PG) (как описано, например, в "Protective Groups in Organic Chemistry" под авторством T.W. Greene и P.G.M. Wutts, 2nd Ed., 1991, Wiley N.Y.) с использованием способов, хорошо известных из уровня техники. Такие защитные группы могут быть удалены на последующих стадиях синтеза с использованием стандартных методов, описанных в литературе.
Если одно или более соединений формул 8-11, II или III содержат хиральные центры, циклопентил- или циклогептилпиразолы формулы Ia могут быть получены в виде смеси диастереомеров или энантиомеров, которые могут быть разделены способами, хорошо известными в уровне техники, например (хиральной) ВЭЖХ или кристаллизацией. Рацематы могут быть разделены на антиподы через диастереомерные соли посредством кристаллизации, например, с оптически чистыми аминами (такими как, например, (R) или (S)-1-фенил-этиламин, (R) или (S)-1-нафтален-1-ил-этиламин, бруцин, хинин или хинидин) и разделением антиподов посредством специальных хроматографических методов с использованием как хиральных адсорбентов, так и хиральных элюентов.
Схема С
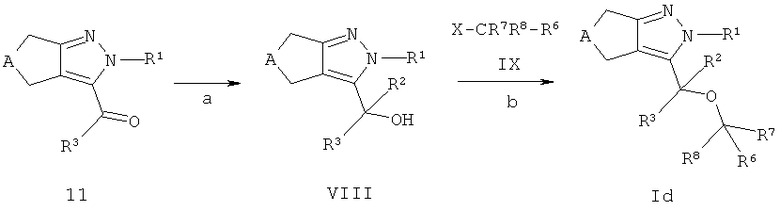
Циклопентил- и циклогептилпиразоловые эфиры формулы Ib (соединения формулы I где R4 представляет собой -O-CR7R8-R6) могут быть получены из кетонов 11 (Схема В). Кетоны 11 могут быть конвертированы в спирты формулы IV (для R2=Н эквивалентно соединению 10 на схеме В), применяя стандартные способы, описанные в литературе (стадия а). Обработка кетонов 11 алкиллитиевым реагентом R2Li в растворителе аналогичном эфиру или тетрагидрофурану дает третичные спирты формулы VIII (стадия а); обработка кетонов 11 алюмогидридом лития в растворителе аналогичном тетрагидрофурану или диэтиловому эфиру или борогидридом натрия в растворителях аналогичных этанолу или метанолу, предпочтительно при температуре между -15°С и 40°С, дает спирты формулы VIII с R2=Н (стадия а). Спиртовые соединения формулы VIII которые содержат хиральный центр, необязательно могут быть разделены на оптически чистые антиподы способами, хорошо известными в уровне техники, например, хроматографией на хиральных ВЭЖХ колонках, или дериватизацией с оптически чистыми кислотами с образованием эфиров, которые могут быть разделены обычной ВЭЖХ хроматографией и могут быть затем конвертированы обратно в энантиомерно чистые спирты формулы VIII. Альтернативно, восстановление кетонов 11 в соответствующие вторичные спирты формулы IV также может проводиться энантиоселективно с получением (R)- или (S)-спиртов формулы IV, например, обработкой боран-диметилсульфоксидным комплексом и (S)- или (R)-2-метил-СВS-оксазоборолидином ((S)- или (R)-1-метил,3,3-дифенил-тетрагидро-пирроло (1,2-с)(1,3,2)оксазоборолом) в качестве хирального катализатора в тетрагидрофуране, предпочтительно при температуре между -78°С и температурой окружающей среды, в соответствии с Corey и соавт. (Е.J.Corey, R.К.Bakshi, S.Shibata, J. Am. Chem. Soc. 1987, 109, 5551-5553), или обработкой (+)-или (-)-В-хлордиизопинокамфенил-бораном (DIP-CI), в соответствии с Brown и соавт. (P.V.Ramachandran, В.Gong, А.V.Teodorovic, Н.С.Brown, Tetrahedron: A symmetry 1994, 5, 1061-1074).
Спирты формулы VIII конденсируют с соединениями формулы IX в соответствии с широко известными способами. Если X является галидным, мезилатным или тозилатным остатком, спирты формулы VIII могут взаимодействовать с соединениями формулы IX в растворителях, аналогичных N.N-диметилформамиду, ацетонитрилу, ацетону или метилэтилкетону в присутствии слабого основания, такого как карбонат цезия или калия, при температуре в диапазоне от комнатной до 140°С, предпочтительно около 50°С, с получением эфиров соединений формулы Id (стадия b).
Эфиры формулы Id могут включать эфиры карбоновых кислот, которые могут быть гидролизованы в соответствующие кислоты посредством стандартных процедур, например, обработкой гидроксидами щелочных металлов, таких как LiOH или NaOH, в смеси полярных растворителей, аналогичной тетрагидрофуран/этанол/вода или обработкой соляной кислотой в диоксане, например, в случае трет-бутиловых эфиров. Необязательно, циклопентил- или циклогептилпиразолы формулы Id также могут содержать циано-группы, которые могут быть конвертированы в соответствующие тетразолы, используя стандартные процедуры, например, обработкой азидом натрия в присутствии кислоты Льюиса в воде или органическом растворителе аналогичном дихлорметану при температуре между 0°С и температурой кипения растворителя.
Если одно из исходных соединений, соединения формул 11 или IX, содержит одну или больше функциональных групп, которые являются нестабильными или реактивны в условиях проведения реакции на одной или больше реакционных стадий, перед проведением критической стадии могут быть введены подходящие защитные группы (PG) (как описано, например, в "Protective Groups in Organic Chemistry" под авторством T.W.Greene и P.G.M.Wutts, 2nd Ed., 1991, Wiley N.Y.) с использованием способов, хорошо известных из уровня техники. Такие защитные группы могут быть удалены на последующих стадиях синтеза с использованием стандартных методов, описанных в литературе.
Если соединения формулы 11, VIII или IX содержат хиральные центры, циклопентил- или циклогептилпиразолы формулы Ib могут быть получены в виде смеси диастереомеров или энантиомеров, которые могут быть разделены способами, хорошо известными в уровне техники, например (хиральной) ВЭЖХ или кристаллизацией. Рацематы могут быть разделены на антиподы через диастереомерные соли посредством кристаллизации, например, с оптически чистыми аминами (такими как, например, (R) или (S)-1-фенил-этиламин, (R) или (S)-1-нафтален-1-ил-этиламин, бруцин, хинин или хинидин) или разделением антиподов посредством специальных хроматографических методов с использованием как хиральных адсорбентов, так и хиральных элюентов.
Схема D
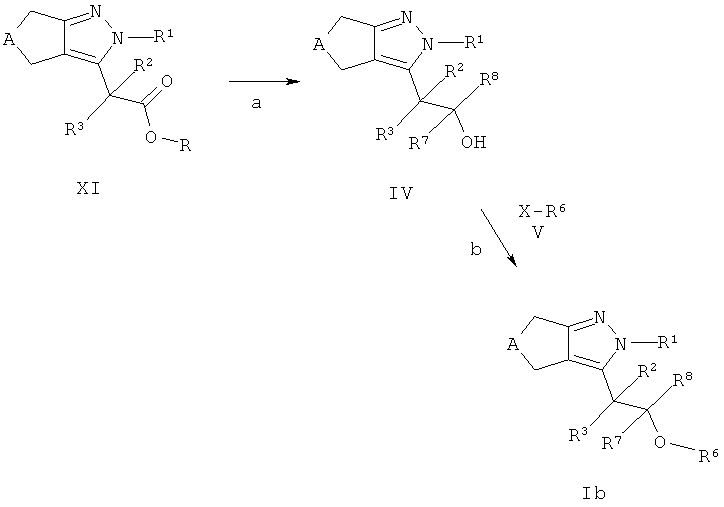
Циклопентил- и циклогептилпиразоловые эфиры формулы Ib (соединения формулы I где R4 представляет собой -CR7R8-OR6) могут быть получены из кислот формулы XI (R=Н, соединения формулы II на схемах А и В) или эфиров формулы XI (R, например, соответствует С1-7-алкилу, соединение 7 на схеме А). Кислоты формулы XI (R=Н) могут быть конвертированы в эфиры (R, например, эквивалентно С1-7-алкилу) с использованием стандартных описанных в литературе способов, например нагреванием кислоты формулы XI (R=Н) с первичным или вторичным спиртом в присутствии катализатора, такого как серная кислота, предпочтительно при кипячении с обратным холодильником. Кислоты формулы XI (R=Н) затем могут быть превращены в первичные спирты формулы IV (R7=Н, R8=Н), например, с использованием диборана в тетрагидрофуране (стадия а). Эфиры формулы XI (R, например, эквивалентно С1-7-алкилу) могут быть восстановлены, например, гидридом алюмолития в растворителях, аналогичных эфиру или тетрагидрофурану, до спиртов формулы IV с R7=R8=Н (стадия а). Альтернативно, заместители R7 и/или R8, отличные от водорода, могут вводться в кислоты формулы XI (R=Н) посредством i) обработки R7Li, необязательно в присутствии соли Си (I), в эфире или тетрагидрофуране с получением алкилкетонов-COR7; ii) последующей реакции с R8Li или гидридом алюмолития в эфире или тетрагидрофуране (стадия а). Спиртовые соединения формулы IV которые содержат хиральный центр, необязательно могут быть разделены на оптически чистые антиподы способами, хорошо известными в уровне техники, например, хроматографией на хиральных ВЭЖХ колонках, или дериватизацией с оптически чистыми кислотами с образованием эфиров, которые могут быть разделены обычной хроматографией ВЭЖХ и затем конвертированы обратно в энантиомерно чистые спирты формулы IV. Восстановление алкилкетонов-COR7 в соответствующие вторичные спирты формулы IV на Схеме D также может быть проведено энантиоселективно с получением (R)- или (S)-спиртов формулы IV, например, обработкой боран-диметилсульфоксидным комплексом и (S)- или (R)-2-метил-СВЭ-оксазоборолидином в качестве хирального катализатора в тетрагидрофуране, предпочтительно при температуре между -78°С и температурой окружающей среды, в соответствии с Corey и соавт. (Е. J.Corey, R.К.Bakshi, S.Shibata, J. Am. Chem. Soc. 1987, 109, 5551-5553), или обработкой (+)- или (-)-В-хлордиизопинокамфенил-бораном (DIP-CI), в соответствии с Brown и соавт.(Р.V.Ramachandran, В. Gong, А. V. Teodorovic, Н.С.Brown, Tetrahedron: Asymmetry 1994, 5, 1061-1074).
Спирты формулы IV конденсируют с соединениями формулы V в соответствии с широко известными способами: если X представляет собой гидрокси-группу и R6 представляет собой арильную систему, то, например, посредством реакции Мицунобу, с использованием трифенилфосфина и ди-трет-бутил-, диизопропил- или диэтил-азодикарбоксилата в качестве реагента, или используя трибутилфосфин и N,N,N',N'-тетраметил азодикарбоксамид; это превращение предпочтительно осуществляется в растворителе, аналогичном толуолу, дихлорметану или тетрагидрофурану при температуре окружающей среды (стадия b). Альтернативно, если X является галидным, мезилатным или тозилатным остатком, спирты формулы IV могут взаимодействовать с соединениями V (R6 не является арильной системой) в растворителях, аналогичных N,N-диметилформамиду, ацетонитрилу, ацетону или метилэтилкетону в присутствии слабого основания, такого как карбонат цезия или калия, при температуре в диапазоне от комнатной до 140°С, предпочтительно около 50°С, с получением эфиров соединений Ic (стадия b).
Эфиры формулы Ib могут включать эфиры карбоновых кислот, которые могут быть гидролизованы в соответствующие кислоты посредством стандартных процедур, например, обработкой гидроксидами щелочных металлов, таких как LiOH или NaOH, в смеси полярных растворителей, аналогичной тетрагидрофуран/этанол/вода, или обработкой соляной кислотой в диоксане, например, в случае трет-бутиловых эфиров. Необязательно, циклопентил- или циклогептилпиразолы формулы Ic могут содержать циано-группы, которые могут быть конвертированы в соответствующие тетразолы используя стандартные процедуры, например, обработкой азидом натрия в присутствии кислоты Льюиса в воде или органическом растворителе, аналогичном дихлорметану, при температуре между 0°С и температурой кипения растворителя.
Если одно из исходных соединений, соединения формул V или XI, содержит одну или больше функциональных групп, которые являются нестабильными или реактивны в условиях проведения реакции на одной или больше реакционных стадий, перед проведением критической стадии могут быть введены подходящие защитные группы (PG) (как описано, например, в "Protective Groups in Organic Chemistry" под авторством T.W.Greene и P.G.M. Wutts, 2nd Ed., 1991, Wiley N.Y.) с использованием способов, хорошо известных из уровня техники. Такие защитные группы могут быть удалены на последующих стадиях синтеза с использованием стандартных методов, описанных в литературе.
Если соединения формулы V, IV и XI содержат хиральные центры, циклопентил- или циклогептилпиразолы формулы Ic могут быть получены в виде смеси диастереомеров или энантиомеров, которые могут быть разделены способами, хорошо известными в уровне техники, например (хиральной) ВЭЖХ или кристаллизацией. Рацематы могут быть разделены на антиподы через диастереомерные соли посредством кристаллизации, например, с оптически чистыми аминами (такими как, например, (R) или (S)-1-фенил-этиламин, (R) или (S)-1-нафтален-1-ил-этиламин, бруцин, хинин или хинидин) или разделением антиподов посредством специальных хроматографических методов с использованием как хиральных адсорбентов, так и хиральных элюентов.
Схема Е
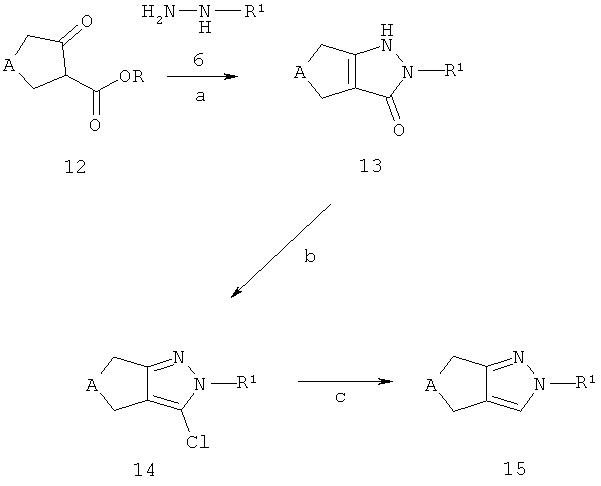
2-замещенные циклопентил- и циклопиразолы 14 и 15 (соответствующие соединениям 8 на схеме В) могут быть получены из эфиров циклопентанон- или циклогептанон-2-карбоновой кислоты 12 (R представляет собой, например, C1-7-алкил), как описано в схеме Е. Эфиры циклопентанон- или циклогептанон-2-карбоновой кислоты 12 являются коммерчески доступными, описаны в литературе или могут быть синтезированы способами, хорошо известными квалифицированным специалистам. Конденсация кето-эфиров 12 с (гетеро)ароматическими гидразинами 6 или солями, например, хлористоводородной солью, (гетеро)ароматических гидразинов 6 приводит к получению 2-замещенных пиразол-3-онов 13 (стадия а). Предпочтительно, такая конденсация проводится в растворителе, таком как толуол и аналогичных, при температуре кипения используемого растворителя. (Гетеро)ароматические гидразины 6 или соответствующие соли (гетеро)ароматических гидразинов являются коммерчески доступными, описаны в литературе или могут быть синтезированы способами, хорошо известными квалифицированным специалистам. Пиразол-3-оны 13 могут быть конвертированы в 2-замещенные 3-хлорпиразолы 14, например, обработкой оксихлоридом фосфора в присутствии каталитического количества N,N-диметиланилина, предпочтительно при кипячении с обратным холодильником (стадия b). Превращение 3-хлорпиразолов 14 в 2-замещенные пиразолы 15 может, например, быть достигнута использованием газообразного водорода в присутствии катализатора на основе переходного металла, такого как палладий на угле (стадия с).
Если одно из исходных соединений, соединения формул 12 или 6, содержит одну или больше функциональных групп, которые являются нестабильными или реактивны в условиях проведения реакции на одной или больше реакционных стадий, перед проведением критической стадии могут быть введены подходящие защитные группы (PG) (как описано, например, в "Protective Groups in Organic Chemistry" под авторством T.W. Greene и P.G.M. Wutts, 2nd Ed., 1991, Wiley N.Y.) с использованием способов, хорошо известных из уровня техники. Такие защитные группы могут быть удалены на последующих стадиях синтеза с использованием стандартных методов, описанных в литературе.
Если соединения 12, 6 или 13 содержат хиральные центры, 2-замещенные пиразолы 14 и 15 могут быть получены в виде смеси диастереомеров или энантиомеров, которые могут быть разделены способами, хорошо известными в уровне техники, например (хиральной) ВЭЖХ или кристаллизацией. Рацематы могут быть разделены на их антиподы посредством специальных хроматографических методов с использованием как хиральных адсорбентов, так и хиральных элюентов.
Соединения общей структуры Ic-Ig могут быть получены в соответствии со схемой F из промежуточных соединений типа 16. Интермедиа™ 16 могут быть получены в случае, если LG обозначает - ОSО2алкил, -ОSО2фторалкил или -ОSО2арил, обработкой спирта IV (Схема D), например, хлоридом или хлорангидридом алкил-, фторалкил- или арилсульфоновой кислоты в подходящем растворителе, таком как, например, дихлорметан и использовании подходящего основания, такого как, например, основание Хунига или пиридина (стадия а). Взаимодействие промежуточных соединений 16 с, например, необязательно замещенными алкил- или арил-тиолами 17 (VII) с подходящим основанием, таким как, например, гидрид натрия, в подходящем растворителе, таком как, например, N,N-диметилформамид, приводит к образованию соединений Ic (стадия b). Соединения Iс могут быть конвертированы в соединения Iе через окисление атома серы окислительным агентом, таким как, например, 3-хлорпероксибензойная кислота, в подходящем растворителе, таком как, например, дихлорметан (стадия с). В случае, когда соединения Iс и Iе несут эфирную группу карбоновой кислоты, она может быть отщеплена способами, известными квалифицированному специалисту (и как, например, описано в "Protective Groups in Organic Chemistry" под авторством T.W. Greene и P.G.M. Wutts, 2nd Ed., 1991, Wiley N.Y.), с получением соответствующих карбоновых кислот. Наприимер, бензиловый эфир можно расщепить каталитическим гидрированием с использованием подходящего катализатора, такого как, например, палладий на угле, в подходящем растворителе, таком как, например, метанол, этанол, этилацетат, тетрагидрофуран или смеси указанных растворителей. Алкиловые эфиры, такие как, например, метиловый или этиловый эфир, могут быть расщеплены в щелочной среде (например, гидроксидом лития или натрия в полярном растворителе, таком как, например, метанол, вода или тетрагидрофуран или смеси указанных растворителей). Трет-бутиловый эфир можно расщепить, например, в кислотной среде (например, используя трифторуксусную кислоту, необязательно в подходящем растворителе, таком как, например дихлорметан и необязательно с использованием нуклеофильного акцептора, такого как, например, 1,3-диметоксибензола или тиоанизола, или используя концентрированную уксусную кислоту в тетрагидрофурану или муравьиную кислоту в подходящем растворителе, аналогичном спирту, например изопропаноле). Аллиловый эфир можно расщепить, например, в реакции с катализатором на основе переходного металла с использованием, например, тетракис(трифенилфенил)палладий в качестве катализатора вместе с пирролидином или морфолином в тетрагидрофуране в качестве растворителя.
Схема F
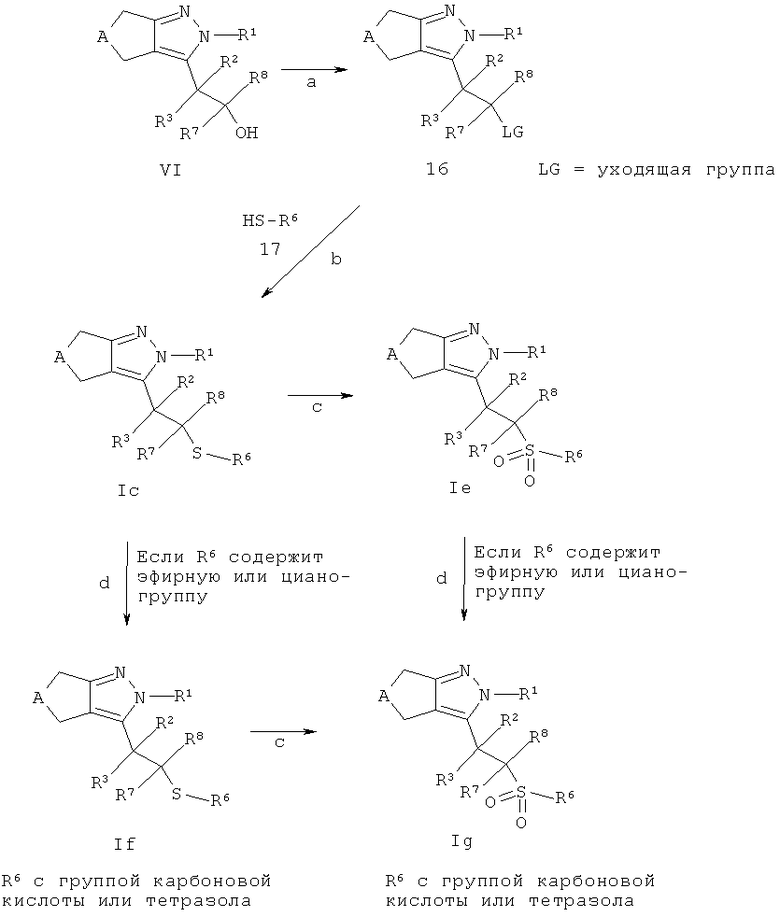
Необязательно, соединения Ic и Iе также могут содержать циано-группы, которые могут быть гидролизованы в карбоновые кислоты в щелочной среде (например, с водным раствором гидроксида натрия или лития) или в кислотной среде (например, соляной или серной кислоте) или могут быть конвертированы в соответствующие тетразолы используя стандартные процедуры, такие как, например, обработку азидом натрия в присутствии кислоты Льюиса или хлорида аммония в воде или органическом растворителе, аналогичном дихлорметану или N,N-диметилформамиду, при температуре между 0°С и температурой кипения растворителя с получением соединений If и Ig (стадия d).
Альтернативно, соединения формулы Ig могут быть синтезированы посредством окисления соединений If (стадия с) с использованием способов, описанных выше.
Если одно из исходных соединений, соединения формул IV или 17 (VII), содержит одну или больше функциональных групп, которые являются нестабильными или реактивны в условиях проведения реакции на одной или больше реакционных стадий, перед проведением критической стадии могут быть введены подходящие защитные группы (PG) (как описано, например, в "Protective Groups in Organic Chemistry" под авторством T.W. Greene и P.G.M. Wutts, 2nd Ed., 1991, Wiley N.Y.) с использованием способов, хорошо известных из уровня техники. Такие защитные группы могут быть удалены на последующих стадиях синтеза с использованием стандартных методов, описанных в литературе.
Если соединения формулы IV и 17 (VII) содержат хиральные центры, циклопентил- или циклогептилпиразолы формулы Ic, Ie, If и Ig могут быть получены в виде смеси диастереомеров или энантиомеров, которые могут быть разделены способами, хорошо известными в уровне техники, например (хиральной) ВЭЖХ или кристаллизацией. Рацематы могут быть разделены на антиподы через диастереомерные соли посредством кристаллизации, например, с оптически чистыми аминами (такими как, например, (R) или (S)-1-фенил-этиламин, (R) или (S)-1-нафтален-1-ил-этиламин, бруцин, хинин или хинидин) или разделением антиподов посредством специальных хроматографических методов с использованием как хиральных адсорбентов, так и хиральных элюентов.
Схема G
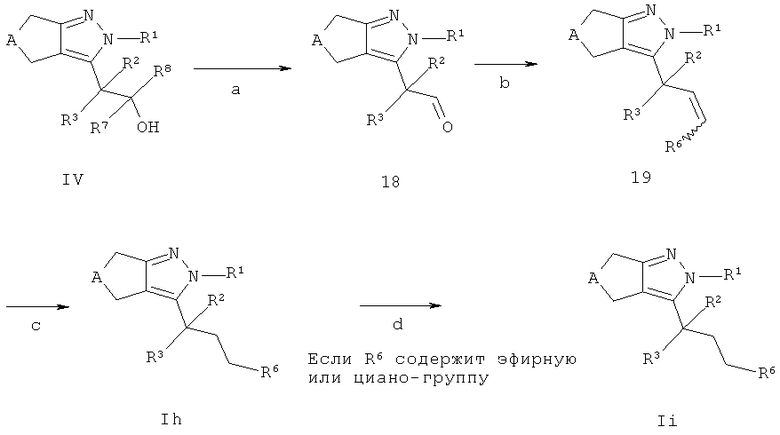
Соединения общей структуры In и Ii, в которых R7=R8=Н могут быть получены в соответствии со схемой G. Альдегиды 18 могут быть получены окислением промежуточных соединений IV (стадия а). Реакции данного типа являются известными квалифицированным специалистам и широко используются и описаны в литературе (например, "March's Advanced Organic Chemistry" под авторством М. В. Smith и J. March, 7th ed., 2007, Wiley & Sons N.Y.). Например, интермедиат IV может быть окислен, например, 1,1,1-триацетокси-1,1-дищидро-1,2-бензидиоксол-3(1Н)-оном в подходящем растворителе, таком как, например, дихлорметан или хлороформ. Промежуточные соединения 19 доступны, например, в результате реакции Виттинга, которая хорошо известна квалифицированным специалистам. Например, промежуточное соединение 18 взаимодействует с необязательно замещенным бензил-трифенилфосфония хлоридом или бромидом (как доступных коммерчески, так и синтезируемых способами, известными в уровне техники) в присутствии подходящего основания и растворителя, такого как, например, трет-бутилат калия, бутиллитий или гидрид натрия в, например, тетрагидрофуране (стадия b). В зависимости от условий реакции интермедиаты 19 могут находиться в форме цис, транс или смеси цис/транс изомеров. Промежуточные соединения 19 могут быть превращены в соединения Ih посредством, например, каталитического гидрирования с использованием катализатора на основе переходного металла, такого как, например, палладий или палладий на в подходящем растворителе, таком как, например, этилацетат, метанол или этанол или смеси указанных растворителей (стадия с).
Необязательно соединения Ih могут содержать эфирную или циано-группу, которые могут быть конвертированы в соответствующие группы карбоновой кислоты или тетразола, соответственно, с использованием условий, описанных выше, с получением соединений Н (стадия d).
Если одно из исходных соединений, соединения формул IV или замещенный бензил-трифенилфосфония хлорид или бромид, содержит одну или больше функциональных групп, которые являются нестабильными или реактивны в условиях проведения реакции на одной или больше реакционных стадий, перед проведением критической стадии могут быть введены подходящие защитные группы (PG) (как описано, например, в "Protective Groups in Organic Chemistry" под авторством T.W.Greene и P.G.M. Wutts, 2nd Ed., 1991, Wiley N.Y.) с использованием способов, хорошо известных из уровня техники. Такие защитные группы могут быть удалены на последующих стадиях синтеза с использованием стандартных методов, описанных в литературе.
Если соединения формулы IV и замещенный бензил-трифенилфосфония хлорид или бромид содержат хиральные центры, циклопентил- или циклогептилпиразолы формулы Ih и Ii могут быть получены в виде смеси диастереомеров или энантиомеров, которые могут быть разделены способами, хорошо известными в уровне техники, например (хиральной) ВЭЖХ или кристаллизацией. Рацематы могут быть разделены на антиподы через диастереомерные соли посредством кристаллизации, например, с оптически чистыми аминами (такими как, например, (R) или (S)-1-фенил-этиламин, (R) или (S)-1-нафтален-1-ил-этиламин, бруцин, хинин или хинидин) или разделением антиподов посредством специальных хроматографических методов с использованием как хиральных адсорбентов, так и хиральных элюентов.
Схема Н
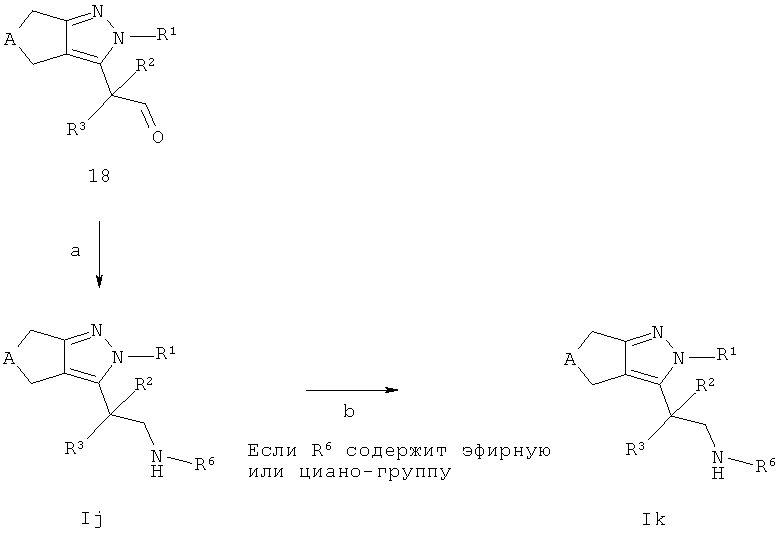
Соединения общей формулы Ij и Ik, в которых R7=R8=Н могут быть получены, как описано на схеме Н. Промежуточные соединения 18 (полученные как описано на схеме G) могут взаимодействовать с алкил- или необязательно замещенным ариламином в присутствии восстанавливающего агента, такого как, например, цианоборогидрида, триацетоксиборогидрида натрия или ди-n-бутилолова дихлорида с трифенилсиланом в подходящем растворителе, таком как, например, тетрагидрофуран, с получение соединений Ij (стадия а). В тех случаях, когда соединения Ij содержат эфирные или циано-группы, они могут быть конвертированы в соответствующие группы карбоновой кислоты или тетразола (стадия b), соответственно, с использованием условий, описанных выше.
Если одно из исходных соединений, соединения формул 18 или алкил- или необязательно замещенный ариламин, содержит одну или больше функциональных групп, которые являются нестабильными или реактивны в условиях проведения реакции на одной или больше реакционных стадий, перед проведением критической стадии могут быть введены подходящие защитные группы (PG) (как описано, например, в "Protective Groups in Organic Chemistry" под авторством T.W. Greene и P.G.M. Wutts, 2nd Ed., 1991, Wiley N.Y.) с использованием способов, хорошо известных из уровня техники. Такие защитные группы могут быть удалены на последующих стадиях синтеза с использованием стандартных методов, описанных в литературе.
Если соединения формулы 18 и алкил- или необязательно замещенный ариламин содержат хиральные центры, циклопентил- или циклогептилпиразолы формулы Ij и Ik могут быть получены в виде смеси диастереомеров или энантиомеров, которые могут быть разделены способами, хорошо известными в уровне техники, например (хиральной) ВЭЖХ или кристаллизацией. Рацематы могут быть разделены на антиподы через диастереомерные соли посредством кристаллизации, например, с оптически чистыми аминами (такими как, например, (R) или (S)-1-фенил-этиламин, (R) или (S)-1-нафтален-1-ил-этиламин, бруцин, хинин или хинидин) или разделением антиподов посредством специальных хроматографических методов с использованием как хиральных адсорбентов, так и хиральных элюентов.
Альтернативно, соединения II и Im могут быть получены в соответствии со схемой I, карбоновые кислоты VIII (R=Н, см. Схема D) могут быть превращены в промежуточные соединения 20 посредством, например, обработки кислотной группы VIII активирующим агентом, таким как, например, N-гидроксибензотриазол моногидрат, необязательно вместе с 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорид, в присутствии основания, такого как, например, этил диизопропиламина, в подходящем растворителе, таком как, например, N,N-диметилформамид и источнике аммония, таком как, например, хлорид аммония (стадия а). Амидная группа в промежуточных соединениях 20 может быть конвертирована в соответствующие амины посредством, например, обработки восстанавливающим агентом, таким как, например, алюмогидрид лития, в подходящем растворителе, таком как, например, тетрагидрофуран, с получением промежуточного соединения 21 с R7=R8=Н (стадия b). Промежуточные соединения 21, с R7 и R8, как определено выше, альтернативно могут быть получены из промежуточного соединения 16 (полученного как описано на схеме F) посредством конвертирования его в азид (интермедиат 22, стадия е) посредством, например, реакции с азидом натрия в подходящем растворителе, таком как, например, N,N-диметилформамид, и восстановления азида в амин (стадия f) посредством, например, каталитического гидрирования способами, аналогичными описанным выше. Промежуточные соединения 21 могут быть превращены в соединения формулы II через алкилирование или восстановительное аминирование в соответствии со способами, описанными выше (стадия с). В случае, когда соединения II содержат эфирные или циано-группы, они могут быть конвертированы в соответствующие группы карбоновой кислоты или тетразола, соответственно, используя условия, описанные выше, с получением соединений Im (стадия d), где R6 содержит группу карбоновой кислоты или тетразола.
Если одно из исходных соединений, соединения формул VIII, 16 или алкилирующий реагент, содержит одну или больше функциональных групп, которые являются нестабильными или реактивны в условиях проведения реакции на одной или больше реакционных стадий, перед проведением критической стадии могут быть введены подходящие защитные группы (PG) (как описано, например, в "Protective Groups in Organic Chemistry" под авторством T.W.Greene и P.G.M. Wutts, 2nd Ed., 1991, Wiley N.Y.) с использованием способов, хорошо известных из уровня техники. Такие защитные группы могут быть удалены на последующих стадиях синтеза с использованием стандартных методов, описанных в литературе.
Если соединения формулы VIII, 16 или алкилирующие реагенты содержат хиральные центры, циклопентил- или циклогептилпиразолы формулы II и Im могут быть получены в виде смеси диастереомеров или энантиомеров, которые могут быть разделены способами, хорошо известными в уровне техники, например (хиральной) ВЭЖХ или кристаллизацией. Рацематы могут быть разделены на антиподы через диастереомерные соли посредством кристаллизации, например, с оптически чистыми аминами (такими как, например, (R) или (S)-1-фенил-этиламин, (R) или (S)-1-нафтален-1-ил-этиламин, бруцин, хинин или хинидин) или разделением антиподов посредством специальных хроматографических методов с использованием как хиральных адсорбентов, так и хиральных элюентов.
Схема I
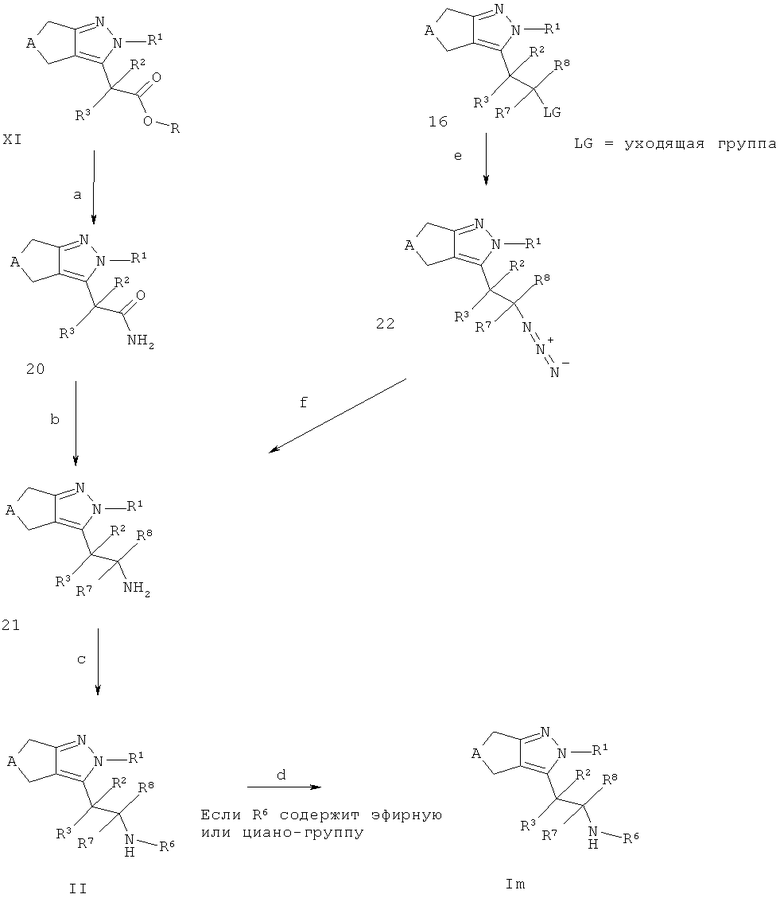
Схема J
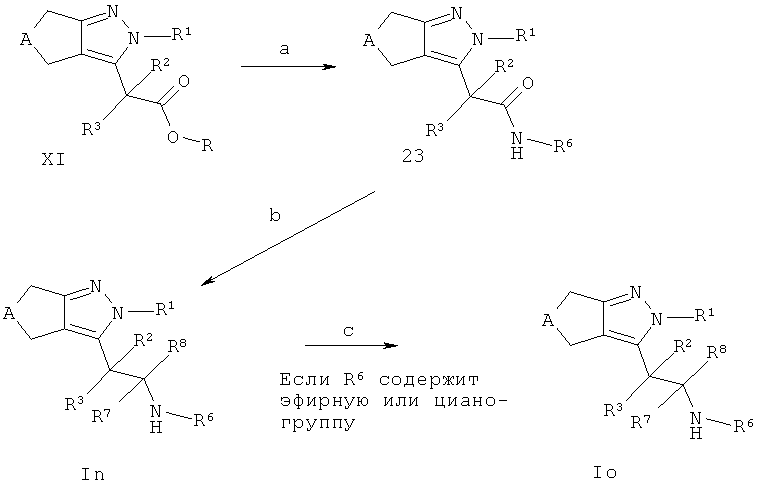
Соединения In и Ir также могут быть получены в соответствии со Схемой J, если заместители R1-R8 являются стабильными при восстановительных условиях, применяемых на стадии b. Амидное связывание промежуточных соединений XI (R=Н) с необязательно незамещенными аминами R6NH2 (как коммерчески доступными, так и получаемые способами, описанными в ссылках или способами, известными из уровня техники) дает соединения 23 (стадия а). Амидное связывание такого типа широко описано в литературе (например, Comprehensive Organic Transformations: A Guide to Functional Group Preparations, 2nd Edition, Richard C. Larock, John Wiley & Sons, New York, NY. 1999) и может быть выполнено использованием связующих реагентов, таких как, например, N,N-карбонилдиимидазол (CDI), 1-гидрокси-1,2,3-бензотриазол (НОВТ) или О-(бензотриазол-1-ил)-N,N,N',N'-тетраметилуроний тетрафторборат (TBTU), в подходящем растворителе, таком как, например, N,N-диметилформамид (ДМФ) или диоксан, необязательно в присутствии основания (например, триэтиламина, диизопропилэтиламина или 4-(диметиламино)пиридина). Альтернативно, промежуточные соединения 28 могут быть получены конвертирорванием промежуточных соединений XI (R=Н) в соответствующие хлорангидриды посредством обработки, например, тионилхлоридом, необязательно в растворителе, таком как, например, дихлорметан, и взаимодействием хлорангидрида с необязательно замещенными циклоалкил/(гетеро)арил аминами в подходящем растворителе, таком как, например, дихлорметан и основания, такого как, например, триэтиламин, диизопропилэтиламин пиридина или 4-(диметиламино)пиридин. Конверсия промежуточных соединений 23 в соединения In с R7=R8=Н (стадия b) может быть осуществлена, например, посредством обработки промежуточных соединений 23 подходящим восстанавливающим агентом, таким как, например, алюмогидрид лития, ди-изобутилалюмогидрид или диметилсульфид боргидрид или тетрагидрофурановым комплексом, в подходящем растворителе, таком как, например, диэтиловый эфир, трет-бутилметиловый эфир или тетрагидрофуран, при температуре между 0°С и температурой кипения растворителя. Конверсия соединений In в Iо, где R6 обозначает группу карбоновой кислоты или тетразола (стадия d), может быть выполнена в соответствии со способами, описанными выше.
Если одно из исходных соединений, соединения формул XI или амины R6NH2, содержит одну или больше функциональных групп, которые являются нестабильными или реактивны в условиях проведения реакции на одной или больше реакционных стадий, перед проведением критической стадии могут быть введены подходящие защитные группы (PG) (как описано, например, в "Protective Groups in Organic Chemistry" под авторством T.W. Greene и P.G.M. Wutts, 2nd Ed., 1991, Wiley N.Y.) с использованием способов, хорошо известных из уровня техники. Такие защитные группы могут быть удалены на последующих стадиях синтеза с использованием стандартных методов, описанных в литературе.
Если соединения формулы XI или амины R6NH2 содержат хиральные центры, циклопентил- или циклогептилпиразолы формулы In и Io могут быть получены в виде смеси диастереомеров или энантиомеров, которые могут быть разделены способами, хорошо известными в уровне техники, например (хиральной) ВЭЖХ или кристаллизацией. Рацематы могут быть разделены на антиподы через диастереомерные соли посредством кристаллизации, например, с оптически чистыми аминами (такими как, например, (R) или (S)-1-фенил-этиламин, (R) или (S)-1-нафтален-1-ил-этиламин, бруцин, хинин или хинидин) или разделением антиподов посредством специальных хроматографических методов с использованием как хиральных адсорбентов, так и хиральных элюентов.
Если желательно или необходимо, функциональные группы, присутствующие в составе соединений формулы I (такие как -СO2алкил, аминогруппы, циано-группы и другие), могут быть превращены в другие функциональные группы, используя обычные стандартные способы, известные квалифицированному специалисту (например, восстановление -СO2алкила до -СН2ОН с LiAlH4, гидролиз -СO2алкила до -СO2Н и последующая необязательная конверсия в амид, ацилирование аминогрупп).
Как описано выше, было найдено, что новые соединения согласно настоящему изобретению связывают и избирательно активируют рецепторы FXR. Они, таким образом, могут использоваться при лечении или профилактике заболеваний и состояний, на которые действуют FXR модуляторы. В частности, FXR модуляторы являются агонистами FXR.
"Заболевания, на которые действуют FXR модуляторы", включают увеличенный уровень жиров и холестерина, в частности, высокий уровень холестерина-ЛПНП, высокий уровень триглицеридов, низкий уровень холестерина-ЛПВП, дислипидемию, заболевания, связанные с всасыванием холестерина, атеросклеротические заболевания, облитерирующий артериит, ишемический инсульт, диабет, в частности, инсулиннезависимый сахарный диабет, метаболический синдром, диабетическая нефропатия, ожирение, холестериновый желчный камень, холестаз/фиброз печени, неалкогольный стеатогепатит (NASH), неалкогольная жировая дистрофия печени (NAFLD), псориаз, опухоли, в частности, опухоли желудочно-кишечного тракта, остеопороз, болезнь Паркинсона и болезнь Альцгеймера.
В частности, заболеваниями (и состояниями), на которые действуют модуляторы FXR, являются высокий уровень холестерина-ЛПНП, высокий уровень триглицеридов, дислипидемия, холестериновый желчный камень, опухоль, инсулиннезависимый сахарный диабет и метаболический синдром. В частности, заболевания, на которые действуют модуляторы FXR, это высокий уровень холестерина-ЛПНП, высокий уровень триглицеридов и дислипидемия.
Настоящее изобретение также относится к фармацевтическим композициям, содержащим соединения, указанные выше, и фармацевтически приемлемый носитель и/или адъювант.
Настоящее изобретение также охватывает соединения, которые описаны выше, для применения в качестве терапевтически активных веществ, особенно для применения в лечении или профилактике увеличенного уровня жиров и холестерина, в частности, высокого уровня холестерина-ЛПНП, высокого уровня триглицеридов, низкого уровня холестерина-ЛПВП, дислипидемии, заболеваний, связанных с всасыванием холестерина, атеросклеротических заболеваний, облитерирующего артериита, ишемического инсульта, диабета, в частности, инсулиннезависимого сахарного диабета, метаболического синдрома, диабетической нефропатии, ожирения, холестеринового желчного камня, холестаза/фиброза печени, неалкогольного стеатогепатита (NASH), неалкогольной жировой дистрофии печени (NAFLD), псориаза, опухоли, в частности, опухоли желудочно-кишечного тракта, остеопороза, болезни Паркинсона и болезни Альцгеймера.
В другом воплощении, настоящее изобретение относится к способу терапевтического или профилактического лечения заболеваний, на которые действуют модуляторы FXR, в частности для терапевтического или профилактического лечения увеличенного уровня жиров и холестерина, в частности, высокого уровня холестерина-ЛПНП, высокого уровня триглицеридов, низкого уровня холестерина-ЛПВП, дислипидемии, заболеваний, связанных с всасыванием холестерина, атеросклеротических заболеваний, облитерирующего артериита, ишемического инсульта, диабета, в частности, инсулиннезависимого сахарного диабета, метаболического синдрома, диабетической нефропатии, ожирения, холестеринового желчного камня, холестаза/фиброза печени, неалкогольного стеатогепатита (NASH), неалкогольной жировой дистрофии печени (NAFLD), псориаза, опухолей, в частности, опухолей желудочно-кишечного тракта, остеопороза, болезни Паркинсона и болезни Альцгеймера, при этом способ содержит введения указанного выше соединения человеку или животному.
Настоящее изобретение также охватывает применение соединений, описанных выше, для терапевтического или профилактического лечения заболеваний, на которые действуют модуляторы FXR, в частности для терапевтического или профилактического лечения увеличенного уровня жиров и холестерина, в частности, высокого уровня холестерина-ЛПНП, высокого уровня триглицеридов, низкого уровня холестерина-ЛПВП, дислипидемии, заболеваний, связанных с всасыванием холестерина, атеросклеротических заболеваний, облитерирующего артериита, ишемического инсульта, диабета, в частности, инсулиннезависимого сахарного диабета, метаболического синдрома, диабетической нефропатии, ожирения, холестеринового желчного камня, холестаза/фиброза печени, неалкогольного стеатогепатита (NASH), неалкогольной жировой дистрофии печени (NAFLD), псориаза, опухолей, в частности, опухолей желудочно-кишечного тракта, остеопороза, болезни Паркинсона и болезни Альцгеймера.
Настоящее изобретение также относится к применению соединений, описанных выше, для изготовления лекарственного средства для терапевтического или профилактического лечения заболеваний, на которые действуют модуляторы FXR, в частности для терапевтического или профилактического лечения увеличенного уровня жиров и холестерина, в частности, высокого уровня холестерина-ЛПНП, высокого уровня триглицеридов, низкого уровня холестерина-ЛПВП, дислипидемии, заболеваний, связанных с всасыванием холестерина, атеросклеротических заболеваний, облитерирующего артериита, ишемического инсульта, диабета, в частности, инсулиннезависимого сахарного диабета, метаболического синдрома, диабетической нефропатии, ожирения, холестеринового желчного камня, холестаза/фиброза печени, неалкогольного стеатогепатита (NASH), неалкогольной жировой дистрофии печени (NAFLD), псориаза, опухолей, в частности, опухолей желудочно-кишечного тракта, остеопороза, болезни Паркинсона и болезни Альцгеймера. В частности, в настоящем изобретении предложено применение соединений формулы I для изготовления лекарственного средства для терапевтического или профилактического лечения высокого уровня холестерина-ЛПНП, высокого уровня триглицеридов и дислипидемии, в особенности дислипидемии. Такие лекарственные средства содержат соединения, как описано выше.
Также настоящим изобретением предусмотрена комбинационная терапия с использованием одного или более соединений формулы I или композиций на их основе или их фармацевтически активных производных, в комбинации с одним или боле соединением, выбранным из группы, состоящей из следующих соединений: ингибиторы биосинтеза холестерина (ГМГ КоА-редуктазы, например, ловастатин, симвастатин, правастатин, флувастатин, аторвастатин, церивастатин, нисвастатин и ривастатин); ингибиторы сквален-эпоксидазы (например, тербинафин); агенты, повышающие уровень ЛПВП в плазме (например, ингибиторы БПЭХ (белок переноса эфира холестерина), например, анацетрапиб, R1658); агонисты гамма-рецепторов человека, активирующих пролиферацию пероксисом (PPAR) (например, тиазолидиндионы, например, розиглитазон, троглитазон, и пиоглитазон); PPAR альфа-агонистов (например, клофибрат, фенофибрат и гемфибронзил); двойные PPAR альфа/гамма агонисты (например, мураглитазар, алеглитазар, пелиглитазар); вещества, усиливающие экскрецию желчи (например, анионообменные смолы, или четвертичные амины (например, холестирамин или колестипол)); ингибиторы транспорта желчных кислот (BATi), никотиновая кислота, ниацинамид, ингибиторы всасывания холестерина (например, эзетимиб); ацил-коэнзим А; ингибиторы холестерин-О-ацилтрансферазы (АСАТ) (например, авазимиб); селективные модуляторы рецепторов эстрогена (например, ралоксифен или тамоксифен); LXR альфа- или бета-агонисты, антагонисты или частичные агонисты (например, 22 (Р)-гидроксихолестерин, 24 (S)-гидроксихолестерин, Т0901317 или GW3965); ингибиторы микросомального белка-транспортера триглицеридов (МТР), противодиабетические агенты, такие как, например, инсулин и аналоги инсулина (например, инсулин Лизпро, ингаляционные препараты, содержащие инсулин, препараты сульфонилмочевины и аналоги (например, толазамид, хлорпропамида, глипизид, глимепирид, глибурид, глибенкламид, толбутамид, ацетогексамид, глипизид), бигуаниды (например, метформин или метформина гидрохлорид, фенформин, буформин) альфа-2-антагонисты и имидазолины (например, мидаглизол, изаглидол, дериглидол, идазоксан, эфароксан, флупароксан), тиазолидиндионы (например, гидрохлорид пиоглитазона, розиглитазона малеат, циглитазон, троглитазон или балаглитазон), ингибиторы альфа-глюкозидазы (например, миглитол, акарбоза, эпалрестат или воглибоз), меглитиниды (например, репаглинид или натеглинид), ингибиторы DPP-4 (например, ситаглиптина фосфат, саксаглиптин, вилдаглиптин, алоглиптин или денаглиптин), инкретины (например, агонисты рецепторов глюкагон-подобного пептида-1 (GLP-1) (например, Эксенатид (Byetta™), NN2211 (Лираглютид), GLP-1 (7-36) амид и его аналоги, GLP-1 (7-37) и его аналоги, AVE-0010 (ZP-10), R1583 (Таспоглютид), GSK-716155 (fk.buk.nbl, GSK / Human Genome Sciences), BRX-0585 (Pfizer/Biorexis) и CJC-1134-PC (экзендин-4: PC-DAC™ и глюкозо-зависимый инсулинотропный пептид (GIP)); агонисты амилина (например, Прамлинтид, АС-137); стимуляторы секреции инсулина (например, линоглирид, натеглинид, репаглинид, митиглинида кальция гидрат или меглитинид; SGLT-2 ингибиторы (например, дапаглифлозин (BMS), серглифлозин (Kissei), AVE 2268 (Sanofi-Aventis); активаторы глюкокиназы, такие как соединения, описанные, например, в WO 00/58293 А1, средства для похудения, такие как агонист фактора роста нервов (например, аксокин), агонисты гормона роста (например, AOD-9604), ингибиторы поглощения адреналина (например, GW-320659), ингибиторы 5-НТ (серотонин) обратного захвата/транспорта (например, прозак), ингибитора 5-HT/NA (серотонина/норадреналина) обратного захвата (например, сибутрамин), ингибиторы обратного захвата DA (дофамина) (например, бупроприон), блокаторы обратного захвата 5-НТ, NA и DA, стероидные растительные экстракты (например, Р57), антагонисты NPY1 или 5 (нейропептида Y Y1 или Y5), агонисты NPY2 (нейропептида Y Y2), агонисты МС4 (меланокортин 4), агонисты ССК-А (холецистокинина-А), антагонисты/обратные агонисты GHSRIa (рецептор стимуляции секреции гормона роста), грелиновые антитела, антагонисты MCH1R (рецептор 1 меланин-концентрирующего гормона) (например, SNAP 7941), агонисты/антагонисты MCH2R (рецептор 2 меланин-концентрирующего гормона), обратные агонисты или антагонисты НЗ (рецептор гистамина 3), агонисты Н1 (рецептор гистамина 1), ингибиторы FAS (синтаза жирных кислот), ингибиторы АСС-2 (ацетил-КоА карбоксилаза-1), агонисты □ 3 (бета-адренергических рецепторов, 3), ингибиторы DGAT-2 (диацилглицерол ацилтрансферазы 2), ингибиторы DGAT-1 (диацилглицерол ацилтрансферазы 1), агонисты CRF (кортикотропин рилизинг-фактор), антагонисты Галанина, активаторы UCP-1 (разобщающий белок 1), 2 или 3, лептин или производные лептина, опиоидные антагонисты, антагонисты орексина, агонисты BRS3 агонисты GLP-1 (глюкагон-подобный пептид-1), агонисты IL-6, агонисты a-MSH, антагонисты AGRP, агонисты BRS3 (рецептор бомбезина, подтип 3), агонисты 5-НТ1 В, антагонисты РОМС, CNTF (цилиарный нейротрофический фактор или производные CNTF), NN2211, топирамат, глюкокортикоидный антагонист, агонисты экзендина-4, агонисты 5-НТ 2С (рецептор серотонина 2С) (например, лоркасерин), ингибиторы PDE (фосфодиэстеразы), ингибиторы транспортера жирных кислот, ингибиторы транспортера дикарбоксилата, ингибиторы транспортера глюкозы, обратные агонисты или антагонисты СВ-1 (рецептор каннабиноида-1) (например, SR141716), ингибиторы липазы (например, орлистат); ингибиторы циклооксигеназы-2 (СОХ-2) (например, рофекоксиб и целекоксиб); ингибиторы тромбина (например, гепарин, аргатробан, мелагатран, дабигатран), ингибиторы агрегации тромбоцитов (например, антагонисты рецептора фибриногена гликопротеина IIb/IIIa или аспирин), витамин В6 и их фармацевтически приемлемые соли, витамин В 12; фолиевая кислота или ее фармацевтически приемлемая соль или эфир, витамины-антиоксиданты, такие как витамин С и Е и бета-каротин, бета-блокаторы (например, антагонисты ангиотензин II рецепторов, такие как лозартан, ирбесартан или валсартан; ингибиторы ингиотензинпревращающего фермента, такие как эналаприл и каптоприл; блокаторы кальциевых каналов, такие как нифедипин и дилтиазам; антагонисты рецепторов эндотелия; аспирин; агенты, отличные от LXR лигандов, которые повышают экспрессию гена АТФ-связывающего кассетного транспортера-AI; и бисфосфонатные соединения (например, алендронат натрия).
Следующие тесты проводились для определения активности соединений формулы I. Сведения справочного характера относительно анализа связывания можно найти у Nichols JS и соавт. "Development of a scintillation proximity assay for peroxisome proliferator-activated receptor gamma Iigand binding domain", (1998) Anal. Biochem. 257:112-119.
Были сконструированы экспрессионные векторы для бактерий и млекопитающих с целью продукции белков глутатион-s-трансцеразы (GST) и Gal4 домена, связывающего ДНК (GAL) слитого с лигандсвязывающим доменом (LBD) человеческого FXR (аа 193-473). Для этого часть последовательностей, кодирующих FXR LBD, амплифицировали с помощью ПЦР из полноразмерного клона и затем субклонировали в плазмидный вектор. Конечный клон был проверен с помощью анализа секвенирования ДНК.
Индукция, экспрессия и последующая очистка слитого белка GST-LBD проводились в штамме клеток Е.coli BL21(pLysS) стандартными способами (Current Protocols in Molecular Biology, Wiley Press, под ред. Ausubel и соавт).
Анализ радиолигандного связывания
Связывание тестируемых веществ с FXR лигандсвязывающим доменом определяли в анализе замещения радиолиганда. Анализ проводился в буфере, состоящем из 50 мМ Hepes, pH 7.4, 10 мМ NaCl, 5 мМ MgCl2. Для каждой реакционной лунки в 96-луночном планшете 40 нМ слитого GST-FXR LBD белка связывали с 10 мкг шариками SPA из силиката иттрия, покрытыми глутатионом (Pharmacia Amersham) в конечном объеме 50 мкл посредством встряхивания. Добавили радиолиганд (например, 40 нМ) 2,N-дициклогексил-2-[2-(2,4 диметоксифенил)-бензоимидазол-1-ил]-ацетамид и реакцию проводили при комнатной температуре в течение 30 минут в присутствии тестируемых соединений с последующим подсчетом сцинтиляционного сближения. Все анализы связывания проводили в 96-луночных планшетах и количество связавшегося лиганда измеряли на счетчике Packard TopCount с использованием планшет OptiPlates (Packard). Кривая зависимости доза-эффект строилась в диапазоне концентраций от 6×10-9 М до 2.5×10-5 М.
Анализ гена-репортера трансрипции люциферазы
Клетки почки новорожденного хомяка (ВНК21 АТСС CCL10) выращивали в DMEM среде, содержащей 10% FBS, при 37°С в атмосфере 95%O2:5%СO2. Клетки засевали в 6-луночные планшеты с плотностью 105 клеток/планшет и затем трансфицировали pFA-FXR-LBD или экспрессионной плазмидой плюс репортерной плазмидой. Трансфекцию выполняли с помощью реагента Fugene 6 (Roche Molecular Biochemicals) в соответствии с предлагаемым протоколом. После шести часов трансфекции клетки собрали трипсинизацией и засеяли в 96-луночные планшеты с плотностью 104 клеток/планшет. После 24 часов для прикрепления клеток среду удалили и заменили ее 100 мкл среды, свободной от фенолового красного и содержащей тестовые соединения или тестовые лиганды (конечная концентрация DMSO: 0.1%). После 24-часового инкубирования клеток с тестируемыми соединениями отобрали 50 мкл супернатанта и затем добавили 50 мкл реагента Luciferase Constant-Light Reagent (Roche Molecular Biochemicals) для лизирования клеток и запуска люциферазной реакции. Люминисценция, как мера люциферазной активности, определялась на счетчике Packard TopCount. Активация транскрипции в присутствии тестируемых соединений выражалась как кратные изменения люминесценции в сравнении с люминесценцией клеток, инкубированных без тестируемых соединений. Значения ЕС50 считали с использованием программы XLfit (ID Business Solutions Ltd. UK).
Соединения, соответствующие формуле I, обладают активностью по меньшей мере в одном из вышеуказанных анализах (ЕС50 или IC50), предпочтительно в диапазоне от 0.5 нМ до 10 мкМ, наиболее предпочтительно от 0.5 нМ до 100 нМ.
Например, соединения формулы I согласно настоящему изобретению показали следующие значения IC50 в анализе связывания, описанном выше:
Соединения формулы I и их фармацевтически активные соли могут применяться в качестве лекарственных средств, например, в форме фармацевтических препаратов для энтерального, парентерального или местного применения. Они могут вводиться, например, перорально, например, в форме таблеток, покрытых оболочкой таблеток, драже, твердых и мягких желатиновых капсул, растворов, эмульсий или суспензий, ректально, например, в форме суппозиториев, парентерально, например, в форме растворов или суспензий для инъекций или инфузионных растворов, или местно, например, в форме мази, крема или масла. В частности, соединения формулы I могут применяться для перорального введения.
Производство фармацевтических препаратов может быть выполнено таким способом, которые знакомы любому квалифицированному специалисту путем создания, на основе описанных соединений формулы I и их фармацевтически приемлемых солей, необязательно в комбинации с другими терапевтически ценными веществами, галеновых форм для введения вместе с подходящими, нетоксичными, инертными, терапевтически совместимыми твердыми или жидкими носителями и, при желании, обычных фармацевтических вспомогательных веществ.
Подходящими носителями являются не только неорганические носители, но также и носители из органических веществ. Так, например, лактоза, кукурузный крахмал или его производные, тальк, стеариновая кислота или ее соли могут быть использованы в качестве материала-носителя для таблеток, покрытых оболочкой таблеток, драже и твердых желатиновых капсул. Подходящими носителями для мягких желатиновых капсул являются, например, растительные масла, воски, жиры и полутвердые и жидкие полиолы (в зависимости от природы активного ингредиента, однако, носитель может быть не нужен в случае мягких желатиновых капсул). Подходящими материалами носителей для производства растворов и сиропов являются, например, вода, полиолы, сахароза, инвертный сахар и аналогичные. Подходящими материалами носителей для инъекционных растворов являются, например, вода, спирты, полиолы, глицерин и растительные масла. Подходящими носителями для суппозиториев являются, например, природные или гидрогенизированные масла, воска, жиры и полужидкие или жидкие полиолы. Подходящими материалами носителей для местных препаратов являются глицериды, полусинтетические и синтетические глицериды, гидрогенизированные масла, жидкие воска, жидкие парафины, жидкие жирные спирты, стеролы, полиэтиленгликоли и производные целлюлозы.
В качестве вспомогательных фармацевтических веществ используют обычные стабилизаторы, консерванты, увлажняющие и эмульгирующие агенты, улучшающие однородность средства, ароматизаторы, соли для изменения осмотического давления, буферные вещества, растворители, красители и маскирующие агенты, а также антиоксиданты.
Дозировка соединений формулы I может изменяться в широких пределах в зависимости от заболевания, которое нужно контролировать, возраста и индивидуальных особенностей пациентов, а также от способа введения, и будет, конечно, подбираться к индивидуальным требованиям в каждом конкретном случае. Для взрослых пациентов подходит доза в диапазоне от 1 до 1000 мг, особенно от 1 до 300 мг. В зависимости от степени тяжести заболевания и точного фармакокинетического профиля соединение может вводиться единоразово или несколькими дозами в день, например от 1 до 3 единиц дозирования.
Фармацевтические препараты предпочтительно содержат около 1-500 мг, более предпочтительно 1-100 мг соединения формулы I.
Следующие примеры помогают пояснить настоящее изобретение более детально. Они, однако, ни в коей мере не ограничивают каким-либо образом объем настоящего изобретения.
Примеры
Сокращения:
CH2Cl2 = дихлорметан, d = день, DCM = дихлорметан, DIPEA = N,N-диизопропилэтиламин, ДМАР = 4-(диметиламино)-пиридин, ДМФ = N,N-диметилформамид, DMSO = диметилсульфоксид, ее = энантиомерный избыток, Et3N = триэтиламин, ЕtOАс = этилацетат, h = час, HATU = 2-(1Н-7-азабензотриазол-1-ил)-1,1,3,3-тетраметилуроний гексафторфосфат, HCl = соляная кислота, ВЭЖХ=высокоэффективная жидкостная хроматография, iPrOAc = изопропилацетат, LDA = диизопропиламид лития, LiHMDS = гексаметилдисилазид лития, МеОН = метанол, мин = минуты, NaHCO3 = бикарбонат натрия, NaOH = гидроксид натрия, Na2SO4 = сульфат натрия, кол. = количественно, rt = комнатная температура, ТВМЕ = трет-бутилметиловый эфир, ТГФ = тетрагидрофуран, ТСХ = тонкослойная хроматография.
Пример 1
4-{2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2-циклогексил-этокси)-3,5-диметил-бензойной кислоты метиловый эфир
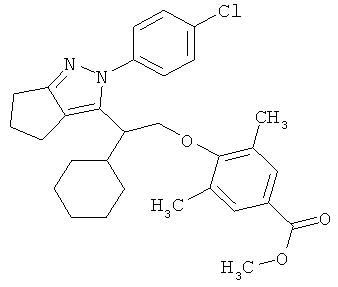
1.1 3-бензотриазол-1-ил-2-циклогексил-3-оксо-пропионовой кислоты этиловый эфир
Раствор 2-циклогексил-малоновой кислоты моноэтилового эфира (2.9 г, 14 мМ; CAS Reg. No. 147596-63-2) в тионилхлориде (29 мл) нагревали при кипячении с обратным холодильником в течение 2 ч. Растворитель удалили при пониженном давлении с получением хлоркарбонил-циклогексил-уксусной кислоты этилового эфира. 1,2,3-бензотриазол (1.47 г, 12 мМ) растворили при температуре окружающей среды в атмосфере аргона в CH2Cl2 (45 мл). Добавили Et3N (1.86 мл, 13 мМ) и раствор хлоркарбонил-циклогексил-уксусной кислоты этилового эфира в CH2Cl2 (4 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 14 ч, погасили реакцию ледяной водной 2 н HCl и экстрагировали дважды iPrOAc. Объединенные экстракты промыли ледяным раствором вода/1 н водная HCl, ледяной смесью вода/солевой раствор 1/1 и высушили над Na2SO4. После фильтрации растворитель удалили при пониженном давлении с получением желтого масла, которое очистили с помощью хроматографии на колонках (силикагель, iPrOAc / гептан) с получением соединения, указанного в заголовке (1.05 г, 3.3 мМ; 25%) в виде желтого масла. MS: m/e=316.2 [М+Н+].
1.2 2-циклогексил-3-оксо-3-(2-оксо-циклопентил)-пропионовой кислоты этиловый эфир
К -78°С холодному растовру LDA (2 М раствор в гептан/этилбензенЛТФ, 15.7 мл, 31 мМ) в ТГФ (96 мл) в атмосфере аргона добавили раствор циклопентанона (2.78 мл, 31 мМ; CAS Reg. No. 120-92-3) в ТГФ (72 мл) в течение 25 мин. Смесь перемешивали в течение 2 ч при -78°С. Добавили раствор 3-бензотриазол-1-ил-2-циклогексил-3-оксо-пропионовой кислоты этилового эфира (9 г, 29 мМ) в ТГФ (63 мл) и раствор перемешивали при температуре окружающей среды в течение 14 ч. Добавили ледяную воду, смесь влили в ледяную смесь вода/солевой раствор 1/1 и экстрагировали дважды ТВМЕ. Объединенные экстракты промыли ледяной смесью вода/солевой раствор 1/1 и высушили над Na2SO4. После фильтрации растворитель удалили при пониженном давлении с получением соединения, указанного в заголовке (8.8 г; кол.), которое использовалось на следующей стадии без дополнительной очистки.
1.3 [2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-циклогексил-уксусной кислоты этиловый эфир
(4-хлорфенил)-гидразин (5.63 г, 31 мМ; CAS Reg. No. 1073-69-4) добавили к раствору 2-циклогексил-3-оксо-3-(2-оксо-циклопентил)-пропионовой кислоты этилового эфира (8.8 г, 31 мМ) в этаноле (200 мл). Реакционную смесь нагревали при кипячении с обратным холодильником в течение 6 ч. Растворитель удалили при пониженном давлении. Остаток суспендировали в дихлорметане и отфильтровали. Фильтрат высушили досуха при пониженном давлении с получением коричневого масла, которое очистили с помощью хроматографии на колонках (силикагель, iPrOAc/гептан) с получением соединения, указанного в заголовке (6.26 г, 16 мМ; 51%) в виде оранжевого масла. MS: m/e=388.4 [М+Н+].
1.4 2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2-циклогексил-этанол
Раствор [2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-циклогексил-уксусной кислоты этилового эфира (500 мг, 1.2 мМ) в ТГФ (25 мл) добавили в течение 20 мин к ледяной суспензии алюмогидрида лития (66 мг, 1.7 мМ) в ТГФ (25 мл). Раствор перемешивали при 0°С в течение 45 мин, отфильтровали через Speedex и фильтрат высушили досуха при пониженном давлении. Остаток поместили в ледяную смесь вода/солевой раствор 1/1 и iPrOAc. Слои разделили и водный слой экстрагировали еще раз iPrOAc. Объединенные экстракты высушили над Na2S04. После фильтрации растворитель удалили при пониженном давлении с получением соединения, указанного в заголовке (336 мг, 0.97 мМ; 79%) в виде желтого масла, которое использовалось на следующей стадии без дополнительной очистки. MS: m/e=345.2 [М+Н+].
1.5 4-{2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2-циклогексил-этокси}-3,5-диметил-бензойной кислоты метиловый эфир
К раствору 2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2-циклогексил-этанола (100 мг, 290 мкМ) в ТГФ (2 мл) добавили 4-гидрокси-3,5-диметил-бензойной кислоты метиловый эфир (57 мг, 319 мкМ; CAS Reg. No. 34137-14-9) и трифенилфосфин (91 мг, 348 мкМ) при температуре окружающей среды в атмосфере аргона. Смесь охладили до 0°С, добавили ди-трет-бутил азодикарбоксилат (80 мг, 348 мкМ) и суспензию перемешивали в течение 48 ч при температуре окружающей среды. Растворитель удалили при пониженном давлении с получением осадка, который очистили с помощью препаративной обратнофазовой ВЭЖХ с использованием градиента ацетонитрил/вода в качестве элюента с получением соединения, указанного в заголовке (19 мг, 37 мкМ; 13%) в виде желтого осадка. MS: m/e=507.2 [М+Н+].
Пример 2
4-{2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил1-2-циклогексил-этокси)-3,5-диметил-бензойная кислота
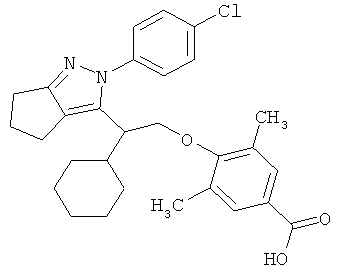
К раствору 4-{2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2-циклогексил-этокси}-3,5-диметил-бензойной кислоты метилового эфира (19 мг, 37 мкМ; пример 1.5) в ТГФ (0.7 мл) и МеОН (0.3 мл) добавили 1 н водный раствор гидроксида лития (450 мкл, 450 мкМ) при температуре окружающей среды в атмосфере аргона. Реакционную смесь перемешивали в течение 14 ч при температуре окружающей среды и влили в ледяной раствор вода/1 н водная HCl 1 / 1. Смесь экстрагировали дважды iPrOAc. Объединенные экстракты промыли ледяной смесью вода/солевой раствор 1/1 и высушили над Na2S04. После фильтрации растворитель удалили при пониженном давлении с получением соединения, указанного в заголовке (25 мг; кол.) в виде бесцветного масла.
Пример 3
4-{2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2-ииклогексил-этокси)-3-фторбензонитрил
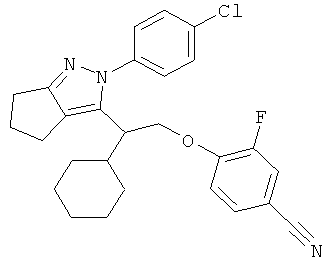
По аналогии со способом, описанным в примере 1.5, 2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2-циклогексил-этанол (пример 1.4) конденсировали с 3-фторо-4-гидрокси-бензонитрилом (CAS Reg. No. 405-04-9) в присутствии трифенилфосфина и ди-трет-бутил азодикарбоксилата в ТГФ с получением соединения, указанного в заголовке, в виде белого осадка. MS: m/e=464.2 [М+Н+].
Пример 4
2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2,N-дициклогексил-ацетамид
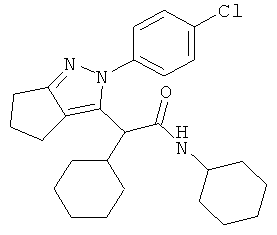
4.1 [2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-циклогексил-уксусная кислота
Раствор [2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-циклогексил-уксусной кислоты этилового эфира (1 г, 2.6 мМ; пример 1.3) в МеОН (56 мл) и 4 н водный NaOH (9.7 мл, 39 мМ) перемешивали при температуре окружающей среды в течение 14 ч. Растворитель удалили при пониженном давлении, добавили ледяную воду/ ТВМЕ 1/1 и слои разделили. Водный слой экстрагировали еще раз ТВМЕ. Водный слой подкислили с помощью 1 н водного раствора HCl и экстрагировали дважды iPrOAc. Объединенные экстракты промыли ледяной смесью вода/солевой раствор 1/1 и высушили над Na2SO4. После фильтрации растворитель удалили при пониженном давлении с получением соединения, указанного в заголовке (570 мг, 1.59 мМ; 61%) в виде желтого масла, которое было достаточно чистым для использования на следующей стадии. MS: m/e=359.2 [М+Н+].
4.2 [2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-циклогексил-уксусной кислоты пентафторфениловый эфир
К раствору [2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-циклогексил-уксусной кислоты (300 мг, 836 мкМ) в ДМФ (3 мл) добавили пиридин (70 мкл, 920 мкМ) и пентафторфенил трифторацетат (290 мкл, 1.7 мМ) при температуре окружающей среды в атмосфере аргона. Реакционную смесь перемешивали при температуре окружающей среды в течение 12 ч, влили в ледяную воду / 0.1 н HCl 1/1 и экстрагировали дважды iPrOAc. Объединенные экстракты промыли ледяной смесью вода/ насыщенные водный раствор NНСO3 1/1, ледяной смесью вода/солевой раствор 1/1 и высушили над Na2SO4. После фильтрации растворитель удалили при пониженном давлении с получением [2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-циклогексил-уксусной кислоты пентафторфенилового эфира в виде оранжевого масла (762 мг; кол.), которое непосредственно использовалось на следующей стадии без дополнительной очистки.
4.3 2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2,N-дициклогексил-ацетамид
К суспензии [2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-циклогексил-уксусной кислоты пентафторфенилового эфира (95 мг, 181 мкМ) в ДМФ (0.9 мл) добавили циклогексиламин (30 мкл, 271 мкМ; CAS Reg. No. 108-91-8) и ДМАР (66 мг, 543 мкМ) при температуре окружающей среды в атмосфере аргона. Реакционную смесь перемешивали при температуре окружающей среды в течение 30 ч. Растворитель удалили при пониженном давлении с получением коричневого масла, которое очистили с помощью препаративной тонкослойной хроматографии (силикагель, iPrOAc/гептан) с получением соединения, указанного в заголовке (32 мг, 75 мкМ; 40%) в виде коричневого масла. MS: m/e=440.3 [М+Н+].
Пример 5
2-(4-хлорфенил)-3-{1-циклогексил-2-[2-фторо-4-(2Н-тетразол-5ил)-фенокси]-этил)-2,4,5.6-тетрагидро-циклопентапиразол
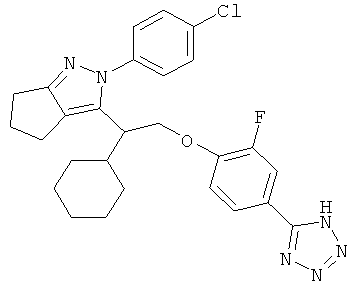
Азид натрия (14 мг, 215 мкМ) и гидрохлорид триэтиламина (29 мг, 215 мкМ) добавили к раствору 4-{2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2-циклогексил-этокси}-3-фторбензонитрила (20 мг, 43 мкМ; пример 3) в ДМФ (0.5 мл). Раствор перемешивали при 120°С в течение 14 ч, влили в ледяной раствор вода/1 н водная HCl 1/1 и экстрагировали дважды iPrOAc. Объединенные экстракты промыли ледяной смесью вода/солевой раствор 1/1 и высушили над Na2S04. Растворитель удалили при пониженном давлении с получением осадка, который кристаллизовался из смеси гептан/дихлорметан с получением соединения, указанного в заголовке (13 мг, 26 мкМ; 59%) в виде бледно-белого осадка. MS: m/e=507.2 [М+Н+].
Пример 6
4-{2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2-ииклогексил-ацетиламино)-3-фторбензойная кислота
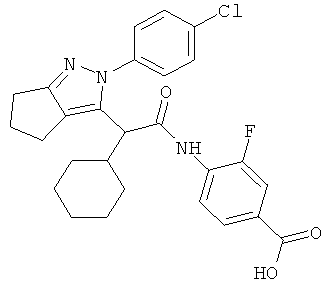
6.1 4-{2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2-циклогексил-ацетиламино}-3-фторбензойной кислоты метиловый эфир
По аналогии со способом, описанным в примере 4.3, [2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-циклогексил-уксусной кислоты пентафторфениловый эфир (пример 4.2) взаимодействовал с 4-амино-3-фторбензойной кислоты метиловым эфиром (CAS Reg. No. 185629-32-7) в присутствии ДМАР в ДМФ с получением соединения, указанного в заголовке в виде бесцветного масла. MS: m/e=510.2 [М+Н+].
6.2 4-{2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2-циклогексил-ацетиламино}-3-фторбензойная кислота
По аналогии со способом, описанным в примере 2, 4-{2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2-циклогексил-ацетиламино}-3-фторбензойной кислоты метиловый эфир гидролизовали с использованием водного раствора гидроксида лития в ТГФ и МеОН с получением соединения, указанного в заголовке в виде коричневого осадка. MS: m/e=494.2 [М-Н-].
Пример 7
6-{2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2-циклогексил-этокси}-никотиновая кислота
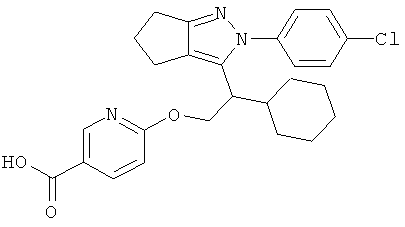
7.1 Метансульфоновой кислоты 2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2-циклогексил-этиловый эфир
К раствору 2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2-циклогексил-этанола (250 мг, 0.72 мМ; пример 1.4) в безводном дихлорметане (5 мл) добавили триэтиламин (0.32 мл, 1.19 мМ) при 25°С. По каплям добавили метилсульфонил хлорид (0.16 мл, 0.725 мМ) при 0°С. Реакционную смесь оставили перемешиваться при 25°С в течение 2 ч, разбавили водой (5 мл) и водный слой экстрагировали дихлорметаном (3×10 мл). Объединенные органические слои промыли последовательно ледяной водой (10 мл), 10% водным раствором NaHCO3 (10 мл), солевым раствором (10 мл) и в заключение высушили над Na2SO4. Растворитель удалили при пониженном давлении с получением соединения, указанного в заголовке в виде светло-желтого масла (250 мг, 0,58 мМ; 81%), которое было достаточно чистым для использования на следующей реакционной стадии.
7.2 6-{2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2-циклогексил-этокси}-никотиновой кислоты метиловый эфир
К раствору метил-6-гидроксиникотината 4 (80 мг, 0.19 мМ; CAS Reg. No. 10128-91-3) в безводном ДМФ (3 мл) добавили сухой К2СO3 (29 мг, 0.226 мМ) при 0°С. Реакционную смесь перемешивали в течение 15 минут при 0°С. Метансульфоновой кислоты 2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2-циклогексил-этиловый эфир (35 мг, 0.227 мМ) растворили в безводном ДМФ (1 мл) и добавили при 0°С. Реакционную смесь нагревали до 100°С в закрытой колбе в течение 12 ч. Добавили 10% водный раствор лимонной кислоты (10 мл) и EtOAc (5 мл) в реакционную смесь. Слои разделили и водный слой экстрагировали еще раз EtOAc (5 мл). Объединенные органические слои промыли солевым раствором (5 мл) и высушили над Na2SO4. Растворитель удалили при пониженном давлении с получением остатка, который очистили с помощью хроматографии на колонках через силикагель (20% ЕtOАс/гексан) с получением соединения, указанного в заголовке (60.2 мг, 0.12 мМ; 67%) в виде бледно-белого осадка.
7.3 6-{2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2-циклогексил-этокси}-никотиновая кислота
По аналогии со способом, описанным в примере 2, 6-{2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2-циклогексил-этокси}-никотиновой кислоты метиловый эфир гидролизовали, используя водный раствор гидроксида натрия в МеОН с получением соединения, указанного в заголовке в виде бледно-белого осадка. MS: m/e=466.2 [М+Н+].
Пример 8
4-(2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил1-2-циклогексил-этилсульфанилУбензойная кислота
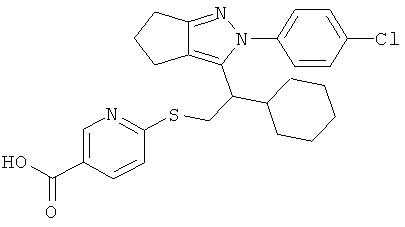
8.1 4-{2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2-циклогексил-этилсульфанил}-бензойной кислоты метиловый эфир
По аналогии со способом, описанным в примере 7.2, метансульфоновой кислоты 2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2-циклогексил-этиловый эфир (пример 7.1) взаимодействовал с 4-меркапто-бензойной кислоты метиловым эфиром (CAS Reg. No. 6302-65-4) в присутствии К2СO3 в ДМФ с получением соединения, указанного в заголовке в виде бледно-белого осадка.
8.2 4-{2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2-циклогексил-этилсульфанил}-бензойная кислота
По аналогии со способом, описанным в примере 2, 4-{2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2-циклогексил-этилсульфанил}-бензойной кислоты метиловый эфир гидролизовали, используя водный раствор гидроксида натрия в МеОН с получением соединения, указанного в заголовке в виде бледно-белого осадка. MS: m/e=481.4 [М+Н+].
Пример 9
2-(4-{2-[2-(4-хлорфенил)-2,4,5,6,7,8-гексагидро-циклогептапиразол-3-ил]-2-циклогексил-ацетиламино)-3-фторфенокси)-2-метил-пропионовой кислоты этиловый эфир
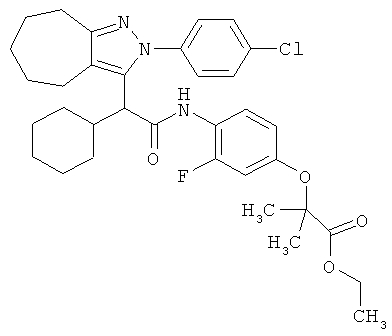
9.1 2-циклогексил-3-оксо-3-(2-оксо-циклогептил)-пропионовой кислоты этиловый эфир
По аналогии со способом, описанным в примере 1.2, 3-бензотриазол-1-ил-2-циклогексил-3-оксо-пропионовой кислоты этиловый эфир (пример 1.1) обработали LDA и затем проводили взаимодействие с циклогептаноном (CAS Reg. No. 502-42-1) в ТГФ с получением соединения, указанного в заголовке в виде желтого масла.
9.2 [2-(4-хлорфенил)-2,4,5,6,7,8-гексагидро-циклогептапиразол-3-ил]-циклогексил-уксусной кислоты этиловый эфир
По аналогии со способом, описанным в примере 1.3, 2-циклогексил-3-оксо-3-(2-оксо-циклогептил)-пропионовой кислоты этиловый эфир конденсировали с (4-хлорфенил)-гидразином (CAS Reg. No. 1073-69-4) в этаноле с получением соединения, указанного в заголовке в виде оранжевого масла. MS: m/e=415.3 [М+Н+].
9.3 [2-(4-хлорфенил)-2,4,5,6,7,8-гексагидро-циклогептапиразол-3-ил]-циклогексил-уксусная кислота
Раствор [2-(4-хлорфенил)-2,4,5,6,7,8-гексагидро-циклогептапиразол-3-ил]-циклогексил-уксусной кислоты этилового эфира (337 мг, 812 мкМ) в МеОН (19 мл) и 4 н водный NaOH (3.05 мл, 12.2 мкМ) нагревали в течение 14 ч при кипячении с обратным холодильником. Растворитель удалили при пониженном давлении, добавили ледяной раствор вода / 2 н водный раствор HCl/iPrOAc 1/1/2 и слои разделили. Водный слой экстрагировали еще раз iPrOAc. Объединенные экстракты промыли ледяной смесью вода/солевой раствор 1/1 и высушили над Na2S04. После фильтрации растворитель удалили при пониженном давлении с получением соединения, указанного в заголовке (284 мг, 734 мкМ; 90%) в виде бледно-белого осадка, который был достаточно чистым для использования на следующей стадии. MS: m/e=387.2 [М+Н
9.4 2-(3-фторо-4-нитрофенокси)-2-метил-пропионовой кислоты этиловый эфир
Карбонат калия (3.96 г, 29 мМ) и 2-бромо-2-метилпропионовой кислоты этиловый эфир (4.47 г, 23 мМ; CAS Reg. No. 600-00-0) добавили к раствору 3-фторо-4-нитрофенола (3 г, 19 мМ; CAS Reg. No. 394-41-2) в DMSO (50 мл). Смесь перемешивали в течение 18 ч при 100°С. Затем добавили 10% водную лимонную кислоту и EtOAc и слои разделили. Органический слой промыли солевым раствором и высушили над MgSО4. Осадок отфильтровали и фильтрат сконцентрировали при пониженном давлении. Остаток очистили хроматографией на колонках (силикагель, EtOAc/гептан) с получением соединения, указанного в заголовке (1.19 г, 4.4 мМ; 23%) в виде желтого масла.
9.5 2-(4-амино-3-фторфенокси)-2-метил-пропионовой кислоты этиловый эфир
10% палладий на угле (200 мг) добавили к раствору 2-(3-фторо-4-нитрофенокси)-2-метил-пропионовой кислоты этилового эфира (1.15 г, 4 мМ) в этаноле (20 мл). Суспензию гидрогенезировали при давлении водорода 1.7 бар в течение 8 ч при температуре окружающей среды. Добавили этилацетат (100 мл), осадок отфильтровали и фильтрат высушили досуха при пониженном давлении с получением соединения, указанного в заголовке (1.23 г, кол.), которое использовалось на следующей стадии без дополнительной очистки.
9.6 2-(4-{2-[2-(4-хлорфенил)-2,4,5,6,7,8-гексагидро-циклогептапиразол-3-ил]-2-циклогексил-ацетиламино}-3-фторфенокси)-2-метил-пропионовой кислоты этиловый эфир
Раствор [2-(4-хлорфенил)-2,4,5,6,7,8-гексагидро-циклогептапиразол-3-ил]-циклогексил-уксусной кислоты (40 мг, 103 мкМ) в тионилхлориде (2 мл) нагревали при кипячении с обратным холодильником в течение 45 мин. Растворитель удалили при пониженном давлении и полученный сырьевой [2-(4-хлорфенил)-2,4,5,6,7,8-гексагидро-циклогептапиразол-3-ил]-циклогексил-ацетил хлорид растворили в CH2Cl2 (1 мл) и добавили к раствору 2-(4-амино-3-фторфенокси)-2-метил-пропионовой кислоты этиловый эфир (37 мг, 155 мкМ) и ДМАР (38 мг, 310 мкМ) in CH2Cl2 (1 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 14 ч. Ледяной смесью вода/солевой раствор 1/1 добавили и смесь экстрагировали дважды iPrOAc. Объединенные экстракты промыли ледяной смесью вода/солевой раствор 1/1 и высушили над Na2SО4. После фильтрации растворитель удалили при пониженном давлении с получением коричневого масла, которое очистили препаративной обратнофазовой ВЭЖХ с использованием градиента ацетонитрил/вода в качестве элюента с получением соединения, указанного в заголовке (15 мг, 25 мкМ; 24%) в виде коричневого осадка. MS: m/e=610.3 [М+Н+].
Пример 10
2-(4-{2-[2-(4-хлорфенил)-2,4,5,6.7.8-гексагидро-циклогептапиразол-3-ил]-2-циклогексил-ацетиламино)-3-фторфенокси)-2-метил-пропионовая кислота
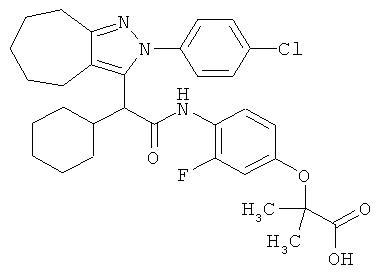
По аналогии со способом, описанным в примере 2, 2-(4-{2-[2-(4-хлорфенил)-2,4,5,6,7,8-гексагидро-циклогептапиразол-3-ил]-2-циклогексил-ацетиламино}-3-фторфенокси)-2-метил-пропионовой кислоты этиловый эфир (пример 9.6) гидролизовали, используя водный раствор гидроксида лития в ТГФ и МеОН с получением соединения, указанного в заголовке в виде красного осадка. MS: m/e=582.4 [М+Н+].
Пример 11
2-[2-(4-хлорфенил)-2.4.5,6.7.8-гексагидро-циклогептапиразол-3-ил]-2,N-дициклогексил-ацетамид
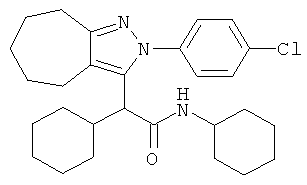
По аналогии со способом, описанным в примере 9.6, [2-(4-хлорфенил)-2,4,5,6,7,8-гексагидро-циклогептапиразол-3-ил]-циклогексил-уксусную кислоту (пример 9.3) конвертировали в соответствующий ангидрид с помощью тионилхлорида, который затем взаимодействовал с циклогексиламином (CAS Reg. No. 108-91-8) в присутствии ДМАР с получением соединения, указанного в заголовке в виде бледно-белого осадка. MS: m/e=468.3 [М+Н+].
Пример 12
4-{2-[2-(4-хлорфенил)-2,4,5,6,7,8-гексагидро-циклогептапиразол-3-ил]-2-ииклогексил-аиетиламино)-3-Фторбензойной кислоты метиловый эфир
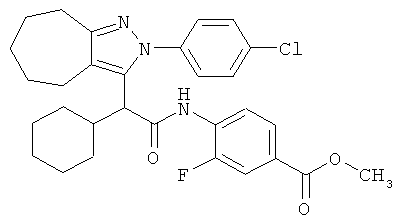
По аналогии со способом, описанным в примере 9.6, [2-(4-хлорфенил)-2,4,5,6,7,8-гексагидро-циклогептапиразол-3-ил]-циклогексил-уксусная кислота (пример 9.3) конвертировали в соответствующий ангидрид с помощью тионилхлорида, который затем взаимодействовал с 4-амино-3-фторбензойной кислоты метиловым эфиром (CAS Reg. No. 185629-32-7) в присутствии ДМАР с получением соединения, указанного в заголовке, в виде бледно-белого осадка. MS: m/e=538.3 [М+Н+].
Пример 13
4-{2-[2-(4-хлорфенил)-2,4,5,6,7,8-гексагидро-циклогептапиразол-3-ил]-2-циклогексил-ацетиламино)-3-фторбензойная кислота
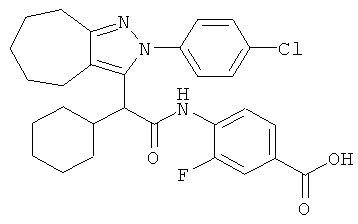
По аналогии со способом, описанным в примере 2, 4-{2-[2-(4-хлорфенил)-2,4,5,6,7,8-гексагидро-циклогептапиразол-3-ил]-2-циклогексил-ацетиламино}-3-фторбензойной кислоты метиловый эфир (пример 12) гидролизовали, используя водный раствор гидроксида лития в ТГФ и МеОН с получением соединения, указанного в заголовке в виде белого осадка. MS: m/e=524.2 [М+Н+].
Пример 14
4-(2-[2-(4-хлорфенил)-2,4,5,6,7,8-гексагидро-циклогептапиразол-3-ил]-2-циклогексил-этокси}-3-Фторбензонитрил
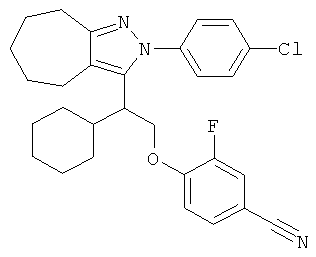
14.12-[2-(4-хлорфенил)-2,4,5,6,7,8-гексагидро-циклогептапиразол-3-ил]-2-циклогексил-этанол
Боран-тетрагидрофурановый комплекс (920 мкл, 920 мкМ; 1 М раствор в ТГФ) добавили к ледяному раствору [2-(4-хлорфенил)-2,4,5,6,7,8-гексагидро-циклогептапиразол-3-ил]-циклогексил-уксусной кислоты (143 мг, 370 мкМ; пример 9.3) в ТГФ (1.5 мл). Раствор перемешивали при температуре окружающей среды в течение 14 ч и охладили до 0***С. Добавили метанол (1.5 мл) и воду (1.5 мл) и смесь экстрагировали дважды iPrOAc. Объединенные экстракты промыли солевым раствором и высушили над Na2SО4. Растворитель удалили при пониженном давлении, остаток растворили в метаноле (4.5 мл) и нагревали при кипячении с обратным холодильником в течение 30 мин. Растворитель удалили при пониженном давлении и очистили остаток с помощью препаративной тонкослойной хроматографии (силикагель, iPrOAc / гептан) с получением соединения, указанного в заголовке (70 мг, 188 мкМ; 51%) в виде бесцветного масла. MS: m/e=373.2 [М+Н+].
14.24-{2-[2-(4-хлорфенил)-2,4,5,6,7,8-гексагидро-циклогептапиразол-3-ил]-2-циклогексил-этокси}-3-фторбензонитрил
По аналогии со способом, описанным в примере 1.5, 2-[2-(4-хлорфенил)-2,4,5,6,7,8-гексагидро-циклогептапиразол-3-ил]-2-циклогексил-этанол конденсировали с 3-фторо-4-гидрокси-бензонитрилом (CAS Reg. No. 405-04-9) в присутствии трифенилфосфина и ди-трет-бутил азодикарбоксилата в ТГФ с получением соединения, указанного в заголовке в виде бледно-белого осадка. MS: m/e=492.2 [М+Н+].
Пример 15
4-(2-[2-(4-хлорфенил)-2,4,5,6,7,8-гексагидро-циклогептапиразол-3-ил]-2-циклогексил-этокси)-3,5-диметил-бензойная кислота
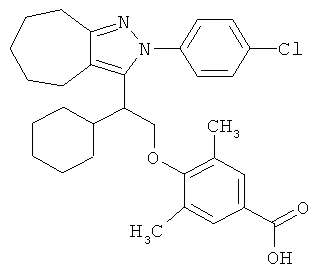
15.14-{2-[2-(4-хлорфенил)-2,4,5,6,7,8-гексагидро-циклогептапиразол-3-ил]-2-циклогексил-этокси}-3,5-диметил-бензойной кислоты метиловый эфир
По аналогии со способом, описанным в примере 1.5, 2-[2-(4-хлорфенил)-2,4,5,6,7,8-гексагидро-циклогептапиразол-3-ил]-2-циклогексил-этанол конденсировали с 4-гидрокси-3,5-диметил-бензойной кислоты метиловым эфиром (CAS Reg. No. 34137-14-9) в присутствии трифенилфосфина и ди-трет-бутил азодикарбоксилата в ТГФ с получением соединения, указанного в заголовке в виде белого осадка. MS: m/e=535.2 [М+Н+].
15.24-{2-[2-(4-хлорфенил)-2,4,5,6,7,8-гексагидро-циклогептапиразол-3-ил]-2-циклогексил-этокси}-3,5-диметил-бензойная кислота
По аналогии со способом, описанным в примере 2, 4-{2-[2-(4-хлорфенил)-2,4,5,6,7,8-гексагидро-циклогептапиразол-3-ил]-2-циклогексил-этокси}-3,5-диметил-бензойной кислоты метиловый эфир гидролизовали, используя водный раствор гидроксида лития в ТГФ и МеОН с получением соединения, указанного в заголовке, в виде бледно-белого осадка. MS: m/e=507.2 [М+Н+].
Пример 16
2-(4-хлорфенил)-3-(1-циклогексил-2-Г2-фторо-4-(1Н-тетразол-5ил)-фенокси1-этил)-2.4.5,6.7,8-гексагидро-циклогептапиразоле
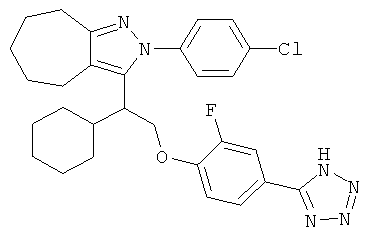
По аналогии со способом, описанным в примере 5, 4-{2-[2-(4-хлорфенил)-2,4,5,6,7,8-гексагидро-циклогептапиразол-3-ил]-2-циклогексил-этокси}-3-фторбензонитрил (пример 14.2) обработали азидом натрия и гидрохлоридом триэтиламина в ДМФ с получением соединения, указанного в заголовке в виде белого осадка. MS: m/e=535.7 [М+Н+].
Пример А
Покрытые пленкой таблетки, содержащие следующие ингредиенты, могут производиться стандартными способами:
Активный ингредиент просеивают и смешивают с микрокристаллической целлюлозой и смесь гранулируют с раствором поливинилпирролидона в воде. Гранулят смешивают с карбоксиметилкрахмалом натрия и стератом магния и прессуют с получением ядра массой 120 или 350 мг соответственно. Ядра покрывают водным раствором/суспензией вышеприведенного пленочного покрытия.
Пример В
Капсулы, содержащие следующие ингредиенты, могут производиться стандартными способами:
Компоненты просеивают, смешивают и наполняют ими капсулы 2-го размера.
Пример С
Растворы для инъекций могут обладать следующим составом:
Активный ингредиент растворяют в смеси полиэтиленгликоля 400 и воды для инъекций (часть). pH доводят до 5.0 добавлением уксусной кислоты. Объем доводят до 1.0 мл добавлением оставшегося количества воды. Раствор фильтруют, разливают в ампулы используя подходящее количество и стерилизуют.
Пример D
Мягкие желатиновые капсулы, содержащие следующие ингредиенты, могут производиться стандартными способами:
Активный ингредиент растворяют в теплой расплавленной массе остальных ингредиентов и этой смесью наполняют мягкие желатиновые капсулы подходящего размера. Заполненные мягкие желатиновые капсулы обрабатывают в соответствии с обычными процедурами.
Пример Е
Саше, содержащие следующие ингредиенты, могут производиться стандартными способами:
Активный ингредиент смешивают с лактозой, микрокристаллической целлюлозой и карбоксиметилцеллюлозой натрия и гранулируют со смесью поливинилпирролидона в воде. Гранулят смешивают со стеаратом магния и вкусовой добавкой и наполняют саше.
Настоящее изобретение относится к новым циклопентил- и циклогептилпиразоловым производным формулы I, где А и R1-R4 определены в формуле изобретения, или их фармацевтически приемлемым солям. Соединения формулы (I) являются модуляторами активности FXR. Изобретение также относится к фармацевтической композиции их содержащей и применению указанных соединений. 4 н. и 10 з.п. ф-лы, 16 пр. получения соединений и 4 пр. фармацевтической композиции.
1. Соединения формулы
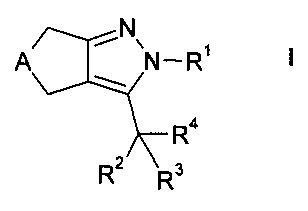
где
А представляет собой -СН2- или -(СН2)3-;
R1 представляет собой фенил, замещенный 1-3 атомами галогена;
R2 представляет собой водород;
R3 представляет собой С3-7циклоалкил;
R4 выбран из группы, состоящей из -C(O)-NH-R5, -CR7R8-OR6, -CR7R8-SR6;
R5 выбран из группы, состоящей из С3-7циклоалкила и фенила, замещенного 1-3 заместителями, независимо выбранными из группы, состоящей из галогена, карбоксила, C1-7алкоксикарбонила, карбоксиС1-7алкокси, С1-7алкоксикарбонилС1-7алкокси;
R6 выбран из группы, состоящей из пиридила, замещенного карбокси или C1-7алкоксикарбонилом, и фенила, замещенного 1-3 заместителями, независимо выбранными из группы, состоящей из С1-7алкила, галогена, C1-7галогеналкила, карбоксила, тетразолила, С1-7алкоксикарбонила и циано; и
R7 и R8 представляют собой водород;
или их фармацевтически приемлемые соли.
2. Соединения формулы I по п. 1, где R4 представляет собой -C(O)-NH-R5 и R5 выбран из группы, состоящей из С3-7циклоалкила и фенила, замещенного 1-3 заместителями, независимо выбранными из группы, состоящей из галогена, карбоксила, С1-7алкоксикарбонила, карбоксиС1-7алкокси, C1-7алкоксикарбонилС1-7алкокси.
3. Соединения формулы I по п. 2, где R5 представляет собой С3-7циклоалкил или фенил, замещенный 1-3 заместителями, независимо выбранными из группы, состоящей из галогена, карбокси, С1-7алкоксикарбонила, карбоксиС1-7алкокси и алкоксикарбонилС1-7алкокси.
4. Соединения формулы I по п. 1, где R4 представляет собой -CR7R8-OR6 и где
R6 выбран из группы, состоящей из пиридила, замещенного карбокси или C1-7алкоксикарбонилом, и фенила, замещенного 1-3 заместителями, независимо выбранными из группы, состоящей из С1-7алкила, галогена, C1-7галогеналкила, карбоксила, тетразолила, С1-7алкоксикарбонила и циано; и
R7 и R8 представляют собой водород.
5. Соединения формулы I по п. 4, где R6 выбран из группы, состоящей из фенила, замещенного 1-3 заместителями, независимо выбранными из группы, состоящей из С1-7алкила, галогена, карбокси, тетразолила, С1-7алкоксикарбонила и циано.
6. Соединения формулы I по п. 1, где А представляет собой -СН2-.
7. Соединения формулы I по п. 1, где А представляет собой -(СН2)3-.
8. Соединения формулы I по п. 1, выбранные из группы, состоящей из
4-{2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2-циклогексил-этокси}-3,5-диметил-бензойной кислоты метилового эфира,
4-{2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2-циклогексил-этокси}-3,5-диметил-бензойной кислоты,
4-{2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2-циклогексил-этокси}-3-фторбензонитрила,
2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2,N-дициклогексил-ацетамида,
2-(4-хлорфенил)-3-{1-циклогексил-2-[2-фторо-4-(2Н-тетразол-5-ил)-фенокси]-этил}-2,4,5,6-тетрагидро-циклопентапиразола,
4-{2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2-циклогексил-ацетиламино}-3-фторбензойной кислоты,
6-{2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2-циклогексил-этокси}-никотиновой кислоты,
4-{2-[2-(4-хлорфенил)-2,4,5,6-тетрагидро-циклопентапиразол-3-ил]-2-циклогексил-этилсульфанил}-бензойной кислоты,
2-(4-{2-[2-(4-хлорфенил)-2,4,5,6,7,8-гексагидро-циклогептапиразол-3-ил]-2-циклогексил-ацетиламино}-3-фторфенокси)-2-метил-пропионовой кислоты этилового эфира,
2-(4-{2-[2-(4-хлорфенил)-2,4,5,6,7,8-гексагидро-циклогептапиразол-3-ил]-2-циклогексил-ацетиламино}-3-фторфенокси)-2-метил-пропионовой кислоты,
2-[2-(4-хлорфенил)-2,4,5,6,7,8-гексагидро-циклогептапиразол-3-ил]-2,N-дициклогексил-ацетамида,
4-{2-[2-(4-хлорфенил)-2,4,5,6,7,8-гексагидро-циклогептапиразол-3-ил]-2-циклогексил-ацетиламино}-3-фторбензойной кислоты метилового эфира,
4-{2-[2-(4-хлорфенил)-2,4,5,6,7,8-гексагидро-циклогептапиразол-3-ил]-2-циклогексил-ацетиламино}-3-фторбензойной кислоты,
4-{2-[2-(4-хлорфенил)-2,4,5,6,7,8-гексагидро-циклогептапиразол-3-ил]-2-циклогексил-этокси}-3-фторбензонитрила,
4-{2-[2-(4-хлорфенил)-2,4,5,6,7,8-гексагидро-циклогептапиразол-3-ил]-2-циклогексил-этокси}-3,5-диметил-бензойной кислоты,
2-(4-хлорфенил)-3-{1-циклогексил-2-[2-фторо-4-(1Н-тетразол-5-ил)-фенокси]-этил}-2,4,5,6,7,8-гексагидро-циклогептапиразола,
или их фармацевтически приемлемые соли.
9. Соединения формулы I по любому из пп. 1-8 для применения в качестве модулятора FXR.
10. Соединения формулы I по любому из пп. 1-8 для применения при лечении или профилактике заболеваний, на которые действуют модуляторы FXR.
11. Фармацевтическая композиция для модуляции активности FXR, содержащая соединения формулы I по любому из пп. 1-8 и фармацевтически приемлемый носитель или адъювант.
12. Способ лечения или профилактики заболеваний, на которые действуют модуляторы FXR, при этом способ содержит введение соединений формулы I по пп. 1-8 человеку или животному.
13. Применение соединений формулы I по любому из пп. 1-8 при изготовлении лекарственного средства для лечения или профилактики заболеваний, на которые действуют модуляторы FXR.
14. Применение по п. 13 при изготовлении лекарственного средства для лечения или профилактики заболеваний, на которые действуют модуляторы FXR.
| EA 200870570 А1, 28.04.2009, WO 2007/052843 A1, 10.05.2007, JP 2001-064176 A, 13.03.2001 |

