ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
[0001] Настоящее изобретение относится к магнитной композиции, в которой частицы магнитного комплексного соединения металл-сален, покрытые диспергирующим агентом, диспергированы в растворителе, и способу получения такой магнитной композиция.
УРОВЕНЬ ТЕХНИКИ
[0002] Обычно комплексное соединение металл-сален известно как магнитное органическое соединение (международная публикация WO 2010/058520). Комплексное соединение металл-сален можно направить к являющемуся мишенью пораженному участку ткани, и оно может агрегироваться в этом участке ткани введением комплексного соединения металл-сален человеку или животному и затем созданием внешним образом магнитного поля для данного комплексного соединения металл-сален. В результате этого фармакологические действия комплексного соединения металл-сален могут суммироваться и проявляться у являющегося мишенью пораженного участка ткани. Например, противораковое действие известно как фармакологические воздействия комплексного соединения металл-сален. Кроме того, в указанной выше международной публикации описано также, что лекарственные молекулы можно направить к являющемуся мишенью участку при помощи внешнего магнитного поля сочетанием лекарственных молекул с комплексным соединением металл-сален. Другими словами комплексное соединение металл-сален может служить в качестве носителя лекарственных молекул.
[0003] Кроме того, вносится на рассмотрение обзорная статья об органических магнитных веществах и описывается, что магнетизм продуцируется полимерными материалами посредством синтеза “молекул с высоким спином”, имеющих более параллельные спины, чем спины у обычных металлических магнитных веществ (см., например, непатентный документ 1).
[0004] Кроме того, включен также способ, в котором платину, содержащуюся в цисплатине, заменяют другим элементом (см., например, непатентный документ 2).
ПЕРЕЧЕНЬ ССЫЛОК
Патентный документ
[0005] [Патентный документ 1] Международная патентная публикация WO 2010/058520.
[Непатентный документ 1] Hizu Iwamura “Molecular Design Aimed at Organic Ferromagnetic Substances", Feb. 1989 issue, p.p. 76-88.
[Непатентный документ 2] Kristy Cochran et al., Structural Chemistry, 13 (2002), p.p. 133-140.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
ПРОБЛЕМЫ, КОТОРЫЕ РАЗРЕШАЮТСЯ ИЗОБРЕТЕНИЕМ
[0006] После изучения предшествующего уровня техники автор настоящей заявки на патент изобретения решил проблему невозможности гарантированного направления соединения металл-сален к участку, в котором создают магнитное поле, даже если животному вводят инъекцию комплексного соединения металл-сален и затем создают для животного магнитное поле.
[0007] Кроме того, ни в непатентном документы 1, ни в непатентном документе 2 не указывается намагничивание самого лекарственного средства.
[0008] Таким образом, задачей настоящего изобретения является предоставление магнитной композиции, которую можно гарантированно направить к являющемуся целью участку, который предпочтительно обрабатывают при помощи магнитного поля, и способа получения такой магнитной композиции.
РАЗРЕШЕНИЕ ПРОБЛЕМЫ
[0009] Для достижения данной цели настоящее изобретение отличается тем, что оно представляет собой магнитную композицию, полученную диспергированием магнитных частиц, которые являются комплексным соединением металл-сален и которые покрыты диспергирующим агентом, в полярном растворителе при помощи диспергирующего агента.
[0010] В результате интенсивного исследования автор данной заявки на патент обнаружил, что диспергируемость комплексного соединения металл-сален в растворителе для инъекций и внутривенных вливаний является недостаточной, и, если в растворителе для инъекций или внутривенных вливаний, в котором диспергировано комплексное соединение металл-сален, создают магнитное поле, будет иметь место феномен, заключающийся в том, что комплексное соединение металл-селен агломерирует. Даже если магнитное поле создают по направлению к являющемуся целью участку, магнитное поле будет легко оказывать влияние на периферическую часть являющегося целью участка. Поэтому комплексное соединение металл-сален будет агломерироваться в периферической части этого участка вследствие влияния магнитного поля до достижения целевого участка, и комплексное соединение металл-сален не может обладать способностью двигаться в капиллярах (5-10 мкм) из периферической части к целевому участку.
[0011] Поскольку магнитную композицию согласно настоящему изобретению получают диспергированием магнитных частиц, которые покрыты диспергирующим агентом, в полярном растворителе при помощи диспергирующего агента, комплексное соединение металл-сален может быть достаточно диспергированным в полярном растворителе. Поэтому, если даже комплексное соединение металл-сален подвергают воздействию магнитного поля в периферической части целевого участка (поврежденного целевого участка), оно может надежно достичь целевого участка без агломерации.
[0012] Кроме того, комплексное соединение металл-сален, содержащееся в магнитной композиций настоящего изобретения, может проявлять неагломерирующее свойство и не будет агломерировать в капиллярах в окружающей среде магнитного поля. Соответственно этому, например, если даже магнитное поле создают после введения, например, растворителя для инъекций или внутривенных вливаний, в котором манитная композиция согласно настоящему изобретению диспергируется, в организм, оно возможно гарантированно предотвращает агломерацию комплексного соединения металл-сален. Поэтому комплексное соединение металл-сален может двигаться в микрокапиллярах (5-10 мкм), так что становится более гарантированным возможное достижение комплексным соединением металл-сален целевого участка.
[0013] Кроме того, настоящее изобретение предлагает способ получения магнитной композиции, включающий в себя первую стадию смешивания комплексного соединения металл-сален с диспергирующим агентом в органическом растворителе и покрытия комплексного соединения металл-сален диспергирующим агентом и вторую стадию диспергирования комплексного соединения металл-сален в полярном растворителе.
[0014] Указанным способом получения можно получить магнитную композицию диспергированием магнитных частиц, которые получают покрытием комплексного соединения металл-сален диспергирующим агентом, в полярном растворителе при помощи диспергирующего агента.
БЛАГОПРИЯТНЫЕ ДЕЙСТВИЯ ИЗОБРЕТЕНИЯ
[0015] Согласно настоящему изобретению, можно предоставить магнитную композицию, содержащую комплексное соединение металл-сален, которую гарантированно можно направить магнитным полем к являющемуся мишенью участку, который нужно предпочтительно лечить, и способ получения магнитной композиции.
ОПИСАНИЕ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
[0016] Комплексное соединение металл-сален, которое применяют для данного изобретения, является структурой, в которой лиганд сален (N,N'-бис(салицилиден)этилендиамин) координирует с металлом или его производными. Конкретный пример комплексного соединения металл-сален описан в указанной выше международной публикации WO 2010/058520. Соединения, полученные смешиванием функциональных молекул, таких как лекарственные молекулы, с комплексной структурой металл-сален включены в комплекс металл-сален настоящего изобретения.
[0017] Стадию покрытия комплексного соединения металл-сален диспергирующим агентом выполняют в органическом растворителе с применением диспергирующего агента, обладающего сродством к органическому растворителю. Затем, когда комплексное соединение металл-сален покрыто диспергирующим агентом, его отделяют и вводят в полярный растворитель. Продукт связывания между комплексным соединением металл-сален образуется на основе взаимодействия Ван-дер-Ваальса и электростатического взаимодействия.
[0018] Согласно предпочтительному варианту, полярная группа диспергирующего агента предпочтительно должна быть защищена защитной группой. Когда защитная группа, которая защищает полярную группу диспергирующего агента, удаляется полярным растворителем, полярная группа диспергирующего агента входит в ионизированном состоянии и диспергирует комплексное соединение металл-сален в полярном растворителе, таком как физиологический солевой раствор.
[0019] Когда комплексное соединение металл-сален диспергируется в полярном растворителе, комплексное соединение металл-сален приобретает форму наночастиц, средний диаметр которых должен быть предпочтительно 10 нм или больше и 500 нм или меньше, и, когда растворитель для инъекций или внутривенных введений или подобных введений, к которому добавляют эти наночастицы, вводят в организм, агломерация комплексного соединения металл-сален можно предотвратить, даже если магнитное поле применяют в капиллярах внутри тела (другими словами не проявляются агломерирующие свойства).
[0020] Примеры диспергирующего агента, применяемого для покрытия комплексного соединения металл-сален, конкретно не ограничивают, пока диспергирующий агент может диспергировать комплексное соединение металл-сален в полярном растворителе, таком как физиологический солевой раствор, однако, предпочтительным является диспергирующий агент для наночастиц металла. Примеры этого типа диспергирующего агента описаны, например, в публикации выложенной заявки на патент Японии (Kokai) № 2011-68988 и в публикации выложенной заявки на патент Японии (Kokai) № 2008-127241.
[0021] Если магнитное поле, создаваемое для комплексного соединения металл-сален, является сильным, имеется возможность того, что комплексное соединение металл-сален может агломерировать. С другой стороны, если интенсивность магнитного поля является низкой, имеется возможность того, что комплексное соединение металл-сален не может быть направлено к целевому участку. С этой точки зрения предпочтительным диапазоном интенсивности магнитного поля является от 0,3 Тл до 1 Тл. Кроме того, предпочтительный диапазон процентного содержания комплексного соединения металл-сален в магнитной композиции составляет 10% или больше и 60% или меньше.
ПРИМЕРЫ
[0022] [Пример получения комплексного соединения железо-сален]
Далее комплексное соединение железо-сален, имеющее аминогруппу в качестве заместителя, получали, как описано ниже.
[0023] Стадия 1:
[Химическая реакция 1]
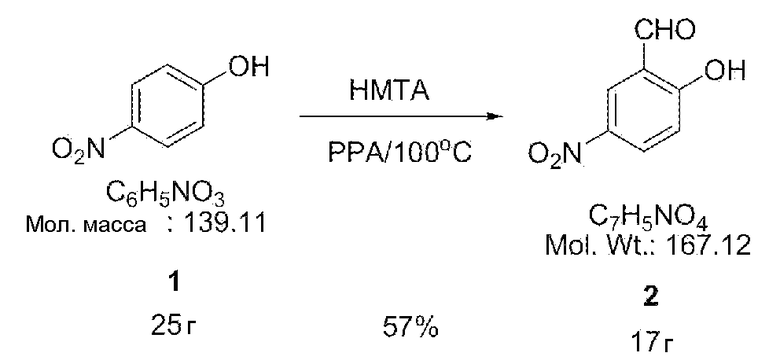
[0024] Смесь 4-нитрофенола (25 г, 0,18 моль), гексаметилентетрамина (25 г, 0,18 моль) и полифосфорной кислоты (200 мл) перемешивали в течение 1 часа при 100°С. Затем смесь вводили в 500 мл этилацетата и 1 л воды и перемешивали до тех пор, пока смесь не становилась полностью растворенной. Кроме того, в результате добавления к указанному выше раствору дополнительных 400 мл этилацетата раствор разделялся, например, на две фазы. Водную фазу удаляли из двухфазной системы и оставшееся соединение промывали дважды растворителем солевым раствором и в результате сушки MgSO4 получали 17 г синтезированного соединения 2 (выход 57%).
[0025] Стадия 2:
[Химическая реакция 2]
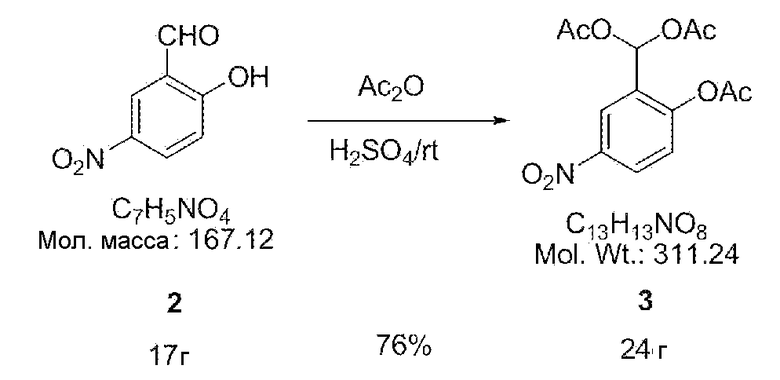
[0026] Затем соединение 2 (17 г, 0,10 моль), уксусный ангидрид (200 мл) и серную кислоту (15 мл) перемешивали в течение 1 часа при комнатной температуре. Полученный раствор перемешивали в ледяной воде (2 л) в течение 0,5 часа и проводили гидролиз. После фильтрования полученного раствора и сушки продукта получали белое порошкообразное вещество. В результате перекристаллизации порошка в растворе, содержащем этилацетат, получали 24 г соединения 3 (выход 76%) в виде белых кристаллов.
[0027] Стадия 3:
[Химическая реакция 3]
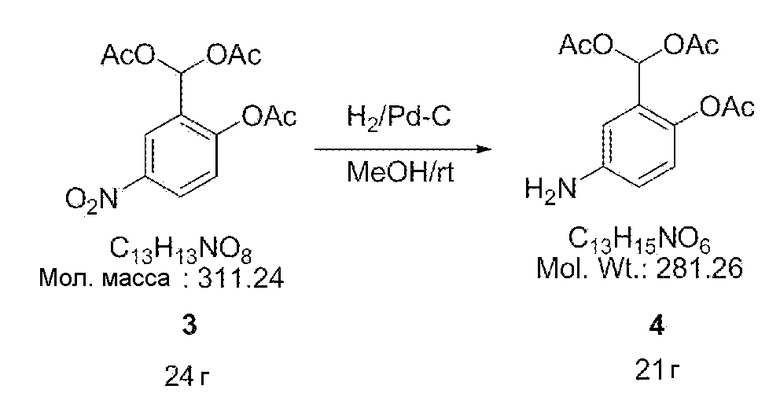
[0028] Далее смесь угля (2,4 г), содержащего 10% палладия, и метанола (500 мл) применяли для восстановления соединения 3 (24 г, 77 ммоль) в атмосфере водорода на протяжении ночи при давлении 1,5 атм. После завершения реакции продукт фильтровали фильтром и получали синтезированное соединение 4 (21 г) в виде коричневого масла.
[0029] Стадии 4,5:
[Химическая реакция 4]
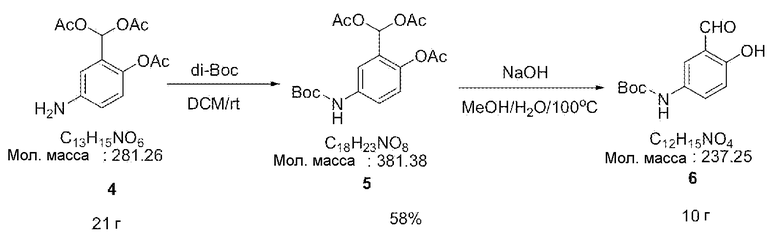
[0030] Затем соединение 4 (21 г, 75 ммоль) и ди(трет-бутил)дикарбонат (18 г, 82 ммоль) вводили к дихлорметан (DCM) (200 мл) и полученную смесь перемешивали на протяжении ночи в атмосфере азота. Полученный раствор упаривали в вакууме и затем растворяли в метаноле (100 мл). Затем добавляли гидроксид натрия (15 г, 374 ммоль) и воду (50 мл) и смесь кипятили с обратным холодильником в течение 5 часов. Затем продукт охлаждали, фильтровали фильтром, промывали водой и сушили в вакууме, тем самым получая коричневое соединение. Полученное соединение дважды подвергали флэш-хроматографии с применением силикагеля, получая при этом 10 г соединения 6 (выход 58%).
[0031] Стадия 6:
[Химическая реакция 5]
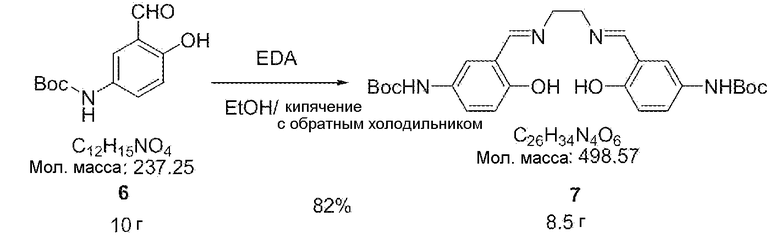
[0032] Соединение 6 (10 г, 42 ммоль) вводили в 400 мл обезвоженного этанола, кипятили с обратным холодильником при нагревании и добавляли несколько капель этилендиамина (1,3 г, 21 ммоль) в 20 мл обезвоженного этанола и смесь перемешивали в течение 0,5 часа. Смешанный раствор охлаждали в сосуде (соединение льдом) и перемешивали в течение 15 минут. Затем продукт промывали 200 мл этанола и фильтровали и сушили в вакууме, тем самым получая 8,5 г синтезированного соединения 7 (выход 82%).
[0033] Стадия 7:
[Химическая реакция 6]
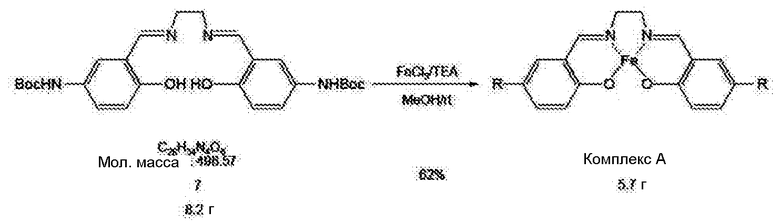
[0034] Соединение 7 (8,2 г, 16 ммоль)и триэтиламин (22 мл, 160 ммоль) вводили в безводный метанол (50 мл) и смешивали с раствором, полученным добавлением FeCl3 (2,7 г, 16 ммоль) к 10 мл метанола в атмосфере азота. В результате перемешивания продукта при комнатной температуре в атмосфере азота получали коричневое соединение. Продукт затем сушили в вакууме. Полученное соединение разбавляли 400 мл дихлорметана, промывали дважды солевым раствором, сушили в вакууме и получали комплексное соединение железо-сален (комплекс А; однако, R=H).
[0035] [Пример получения комплексного соединения кобальт-сален]
После получения соединения 7 указанным выше способом соединение 7 (8,2 г, 16 ммоль) и триэтиламин (22 мл, 160 ммоль) вводили в безводный метанол (50 мл) и смешивали в атмосфере азота с раствором, полученным добавлением CoCl2 (хлорид кобальта (II) от Alfa Aesar) (2,7 г, 16 ммоль) к метанолу (10 мл). После этого получали комплексное соединения кобальт-сален таким же методом, как метод получения комплексного соединения железо-сален.
[0036] [Пример получения комплексного соединения никель-сален]
После получения соединения 7 указанным выше способом соединение 7 (8,2 г, 16 ммоль) и триэтиламин (22 мл, 160 ммоль) вводили в безводный метанол и смешивали в атмосфере азота с раствором, полученным добавлением (50 мл), NiCl2 (хлорид никеля(II) от Alfa Aesar) (2,7 г, 16 ммоль) к метанолу (10 мл). Затем таким же способом, как способ получения комплексного соединения железо-сален, получали комплексное соединение никель-сален.
[0037] [Пример получения комплексного соединения молибден-сален]
После получения соединения 7 описанным выше способом соединение 7 (8,2 г, 16 ммоль) и триэтиламин (22 мл, 160 ммоль) вводили в безводный метанол (50 мл) и смешивали в атмосфере азота с раствором, полученным добавлением MoCl3 (хлорид молибдена(III) от Alfa Aesar) (2,7 г, 16 ммоль) к метанолу (10 мл). Затем таким же способом, как способ получения комплексного соединения железо-сален, получали комплексное соединение молибден-сален.
[0038] [Пример получения комплексного соединения рутений-сален]
После получения соединения 7 указанным выше способом соединение 7 (8,2 г, 16 ммоль) и триэтиламин (22 мл, 160 ммоль) вводили в безводный метанол (50 мл) и смешивали в атмосфере азота с раствором, полученным добавлением RuCl3 (хлорид рутения(III) от Alfa Aesar) (2,7 г, 16 ммоль) к метанолу (10 мл). Затем таким же способом, как способ получения комплексного соединения железо-сален, получали комплексное соединение рутений-сален.
[0039] [Пример получения комплексного соединения родий-сален]
После получения соединения 7 описанным выше способом соединение 7 (8,2 г, 16 ммоль) и триэтиламин (22 мл, 160 ммоль) вводили в безводный метанол (50 мл) и смешивали в атмосфере азота с раствором, полученным добавлением RhCl3 (хлорид родия(III) от Alfa Aesar) (2,7 г, 16 ммоль) к метанолу (10 мл). Затем таким же способом, как способ получения комплексного соединения железо-сален, получали комплексное соединение родий-сален.
[0040] [Пример получения комплексного соединения палладий-сален]
После получения соединения 7 описанным выше способом соединение 7 (8,2 г, 16 ммоль) и триэтиламин (22 мл, 160 ммоль) вводили в безводный метанол (50 мл) и смешивали в атмосфере азота с раствором, полученным добавлением PdCl2 (хлорид палладия(II) от Alfa Aesar) (2,7 г, 16 ммоль) к метанолу (10 мл). Затем таким же способом, как способ получения комплексного соединения железо-сален, получали комплексное соединение палладий-сален.
[0041] [Пример получения комплексного соединения вольфрам-сален]
После получения соединения 7 указанным выше способом соединение 7 (8,2 г, 16 ммоль) и триэтиламин (22 мл, 160 ммоль) вводили в безводный метанол (50 мл) и смешивали в атмосфере азота с раствором, полученным добавлением WCl6 (хлорид вольфрама(III) от Alfa Aesar) (2,7 г, 16 ммоль) к метанолу (10 мл). Затем таким же способом, как способ получения комплексного соединения железо-сален, получали комплексное соединение вольфрам-сален.
[0042] [Пример получения комплексного соединения рений-сален]
После получения соединения 7 описанным выше способом соединение 7 (8,2 г, 16 ммоль) и триэтиламин (22 мл, 160 ммоль) вводили в безводный метанол (50 мл) и смешивали в атмосфере азота с раствором, полученным добавлением ReCl5 (хлорид рения(V) от Alfa Aesar) (2,7 г, 16 ммоль) к метанолу (10 мл). Затем таким же способом, как способ получения комплексного соединения железо-сален, получали комплексное соединение рений-сален.
[0043] [Пример получения комплексного соединения осмий-сален]
После получения соединения 7 описанным выше способом, соединение 7 (8,2 г, 16 ммоль) и триэтиламин (22 мл, 160 ммоль) вводили в безводный метанол (50 мл) и смешивали в атмосфере азота с раствором, полученным добавлением тригидрата соединения осмий-сален (тригидрат хлорида осмия(III) от Alfa Aesar) (2,7 г, 16 ммоль) к метанолу (10 мл). Затем таким же способом, как способ получения комплексного соединения железо-сален, получали комплексное соединение осмий-сален.
[0044] [Пример получения комплексного соединения индий-сален]
После получения соединения 7 описанным выше способом соединение 7 (8,2 г, 16 ммоль) и триэтиламин (22 мл, 160 ммоль) вводили в безводный метанол (50 мл) и смешивали в атмосфере азота с раствором, полученным добавлением IrCl3 (хлорид иридия(III) от Alfa Aesar) (2,7 г, 16 ммоль) к метанолу (10 мл). Затем таким же способом, как способ получения комплексного соединения железо-сален, получали комплексное соединение иридий-сален.
[0045] [Пример получения комплексного соединения платина-сален]
После получения соединения 7 описанным выше способом соединение 7 (8,2 г, 16 ммоль) и триэтиламин (22 мл, 160 ммоль) вводили в безводный метанол (50 мл) и смешивали в атмосфере азота с раствором, полученным добавлением PtCl2 (хлорид платины(II) от Alfa Aesar) (2,7 г, 16 ммоль) к метанолу (10 мл). Затем таким же способом, как способ получения комплексного соединения железо-сален, получали комплексное соединение платина-сален.
[0046] [Пример получения комплексного соединения неодим-сален]
После получения соединения 7 описанным выше способом соединение 7 (8,2 г, 16 ммоль) и триэтиламин (22 мл, 160 ммоль) вводили в безводный метанол (50 мл) и смешивали в атмосфере азота с раствором, полученным добавлением NdCl3 (хлорид неодима(III) от Alfa Aesar) (2,7 г, 16 ммоль) к метанолу (10 мл). Затем таким же способом, как способ получения комплексного соединения железо-сален, получали комплексное соединение неодим-сален.
[0047] [Пример получения комплексного соединения самарий-сален]
После получения соединения 7 описанным выше способом соединение 7 (8,2 г, 16 ммоль) и триэтиламин (22 мл, 160 ммоль) вводили в безводный метанол (50 мл) и смешивали в атмосфере азота с раствором, полученным добавлением ReCl5 (хлорид рения(V) от Alfa Aesar) (2,7 г, 16 ммоль) к метанолу (10 мл). Затем таким же способом, как способ получения комплексного соединения железо-сален, получали комплексное соединение самарий-сален.
[0048] [Пример получения комплексного соединения европий-сален]
После получения соединения 7 описанным выше способом соединение 7 (8,2 г, 16 ммоль) и триэтиламин (22 мл, 160 ммоль) вводили в безводный метанол (50 мл) и смешивали в атмосфере азота с раствором, полученным добавлением ReCl5 (хлорид рения(V) от Alfa Aesar) (2,7 г, 16 ммоль) к метанолу (10 мл). Затем таким же способом, как способ получения комплексного соединения железо-сален, получали комплексное соединение европий-сален.
[0049] [Пример получения комплексного соединения гадолиний-сален]
После получения соединения 7 описанным выше способом соединение 7 (8,2 г, 16 ммоль) и триэтиламин (22 мл, 160 ммоль) вводили в безводный метанол (50 мл) и смешивали в атмосфере азота с раствором, полученным добавлением GdCl3 (хлорид гадолиния(III) от Alfa Aesar) (2,7 г, 16 ммоль) к метанолу (10 мл). Затем таким же способом, как способ получения комплексного соединения железо-сален, получали комплексное соединение гадолиний-сален.
[0050]
Затем будут объясняться примеры получения (примеры) жидкой дисперсии магнитных наночастиц, в которой диспергировано комплексное соединение железо-сален. Сначала будет объясняться случай, в котором применяют диспергирующий агент, полярная группа которого защищена защитной группой.
[Пример случая, когда защитную группу Вос-защитную группу заменяют на систему амина]
Раствор в ДМСО (1 М, объем 1 мл) (Вос-аминоокси)уксусной кислоты (Fluka Corp.), которая является диспергирующим агентом, добавляли к комплексному соединению железо-сален (приблизительно 10 мг/мл), которое суспендировали в 10 мл хлороформа; и полученный раствор энергично перемешивали в течение 3-5 часов при комнатной температуре, получая при этом магнитные наночастицы, которые обладают диспергируемостью в хлороформе. Затем добавляли 10 мл 1 н. раствора хлорида водорода и полученную смесь энергично перемешивали в двухфазной системе. В это время магнитные наночастицы передвигались к водной фазе, в результате чего образуется водный раствор магнитных наночастиц, обладающий водной диспергируемостью. При этих условиях рН водной фазы регулировали до 2 или менее. (Вос-аминоокси)уксусная кислота ([трет-бутоксикарбонил)аминоокси]уксусная кислота) является защитной группой, описанной ранее, и ДМСО является диспергирующим агентом.
[0051] Даже когда магнит (интенсивность магнитного поля приблизительно составляет 0,5 Т) помещали ближе к таким образом полученной жидкой дисперсии магнитных наночастиц, агломерацию частиц не наблюдали. Кроме того, распределение частиц проверяли трансмиссионным электронным микроскопом, средний диаметр частиц был приблизительно 80 нм.
[0052] [Пример случая, когда защитную группу Fmoc заменяют на систему амина]
Раствор в ДМСО (концентрация 1 М; объем 1 мл) Fmoc-Asp-OH (Watanabe Chemical), который был диспергирующим агентом, добавляли к комплексу железо-сален (приблизительно 10 мг/мл), который был диспергирован в 10 мл хлороформа, и полученный раствор энергично перемешивали в течение 3-5 часов при комнатной температуре, тем самым получая магнитные наночастицы, которые обладают диспергируемостью в хлороформе. Затем добавляли 10 мл 1 н. водного гидроксида натрия и полученную смесь энергично перемешивали в двухфазной системе. В это время магнитные наночастицы передвигались к водной фазе, в результате чего образуется водный раствор магнитных наночастиц, обладающий водной диспергируемостью. При этих условиях рН водной фазы регулировали до 8 или более. Даже когда магнит (интенсивность магнитного поля приблизительно 0,5 Тл) помещали ближе к таким образом полученной жидкой дисперсии магнитных наночастиц, агломерацию частиц не наблюдали. Кроме того, распределение частиц проверяли трансмиссионным электронным микроскопом, средний диаметр частиц был приблизительно 70 нм. Fmoc-Asp-OH (N-[(9H-флуорен-9-илметокси)карбонил]-(L)-аспарагиновая кислота) представляет собой защитную группу, описанную ранее.
[0053] [Пример случая, когда защитную этиловую сложноэфирную группу заменяют на карбоксигидразиновую группу]
Раствор в ДМСО (концентрация 1 М; объем 1 мл) моноэтиладипата (Tokyo Chemical Industry Co., Ltd.), который был диспергирующим агентом, добавляли к комплексу железо-сален (приблизительно 10 мг/мл), который был суспендирован в 10 мл хлороформа; и полученный раствор энергично перемешивали в течение 3-5 часов при комнатной температуре, в результате чего получая магнитные наночастицы, которые обладают диспергируемостью в хлороформе. Затем добавляли 10 мл моногидрата гидразина и полученную смесь энергично перемешивали. В это время в растворе хлороформа образовывались осадки. Супернатантную жидкость отбрасывали один раз и добавляли дистиллированную воду, в результате чего получали водный раствор магнитных наночастиц, обладающий водной диспергируемостью. В это время рН водной фазы регулировали до 3 или менее. Даже когда магнит (интенсивность магнитного поля приблизительно 0,5 Тл) помещали ближе к таким образом полученной жидкой дисперсии магнитных наночастиц, агломерацию частиц не наблюдали. Кроме того, распределение частиц проверяли трансмиссионным электронным микроскопом, средний диаметр частиц был приблизительно 90 нм. Моноэтиладипат является описанной ранее защитной группой.
[0054] [Пример случая, когда защитную этиловую сложноэфирную группу заменяют, например, группу карбоновой кислоты]
Раствор в ДМСО (концентрация 1 М; объем 1 мл) моноэтиладипата (Tokyo Chemical Industry Co., Ltd.), который был диспергирующим агентом, добавляли к комплексу железо-сален (приблизительно 10 мг/мл), который был суспендирован в 10 мл хлороформа; и полученный раствор энергично перемешивали в течение 3-5 часов при комнатной температуре, в результате чего получали магнитные наночастицы, которые обладают диспергируемостью в хлороформе. Затем добавляли 10 мл 1 н. водного гидроксида натрия и полученную смесь энергично перемешивали. В это время магнитные наночастицы передвигались к водной фазе, в результате чего образуется водный раствор магнитных наночастиц, обладающий водной диспергируемостью. В это время рН водной фазы регулировали до 8 или более. Даже когда магнит (интенсивность магнитного поля приблизительно 0,5 Тл) помещали ближе к таким образом полученной жидкой дисперсии магнитных наночастиц, агломерацию частиц не наблюдали. Кроме того, распределение частиц проверяли трансмиссионным электронным микроскопом, средний диаметр частиц был приблизительно 80 нм.
[0055] Далее, комплексное соединение металл-сален, другое, чем комплексное соединение железо-сален (соответствующие комплексные соединения металл-сален, полученные выше в примерах), применяли в виде комплексного соединения металл-сален, и жидкие дисперсии магнитных наночастиц получали соответственно согласно примерам. Затем, когда магнит (интенсивность магнитного поля приблизительно 0,5 Тл) помещали ближе к каждой из жидких дисперсий магнитных наночастиц, агломерацию частиц не наблюдали. Кроме того, распределение частиц проверяли трансмиссионным электронным микроскопом, средний диаметр частиц был приблизительно 100-600 нм.
Изобретение относится к медицине, в частности к жидкому лекарственному средству, в котором после введения в организм магнитные частицы движутся по капиллярам к являющемуся мишенью участку под действием магнитного поля, приложенного к поврежденному участку. Жидкое лекарственное средство получено диспергированием магнитных частиц, которые получают покрытием комплексного соединения металл-сален диспергирующим агентом в полярном растворителе при помощи диспергирующего агента. Осуществление изобретения позволяет предотвратить агломерацию частиц и обеспечить терапевтический эффект при их движении по капиллярам. 4 з.п. ф-лы.
1. Жидкое лекарственное средство, полученное диспергированием магнитных частиц, которые получают покрытием комплексного соединения металл-сален диспергирующим агентом в полярном растворителе при помощи диспергирующего агента,
где после введения в организм жидкого лекарственного средства магнитные частицы движутся по капиллярам к являющемуся мишенью участку, который является поврежденным участком, магнитное поле прикладывают к являющемуся мишенью участку снаружи организма, при этом магнитные частицы диспергированы в полярном растворителе таким образом, что они не подвергаются агломерации, даже находясь под действием магнитного поля в капиллярах в периферийной области до того, как достигнут являющегося мишенью участка.
2. Жидкое лекарственное средство по п. 1, где диспергирующий агент выбран из группы состоящей из (Boc-аминоокси)уксусной кислоты в ДМСО, Fmoc-Asp-OH в ДМСО или моноэтиладипата в ДМСО
3. Жидкое лекарственное средство по п. 1, где интенсивность магнитного поля составляет от 0,3 Тл до 1 Тл.
4. Жидкое лекарственное средство по п. 1 или 3, где диаметр каждого из капилляров составляет от 5 мкм до 10 мкм.
5. Жидкое лекарственное средство по любому из пп. 1-3, где диаметр частиц комплексного соединения металл-сален находится между 10 нм или более и 500 нм или менее.
| US 20090169484 А1, 02.07.2009 | |||
| WO2006133354 A2, 14.12.2006 | |||
| 0 |
|
SU158406A1 | |

