Изобретение относится к фармацевтической промышленности и касается новых производных на основе антибиотика азитромицина и гликопептидных антибиотиков, и способа их получения.
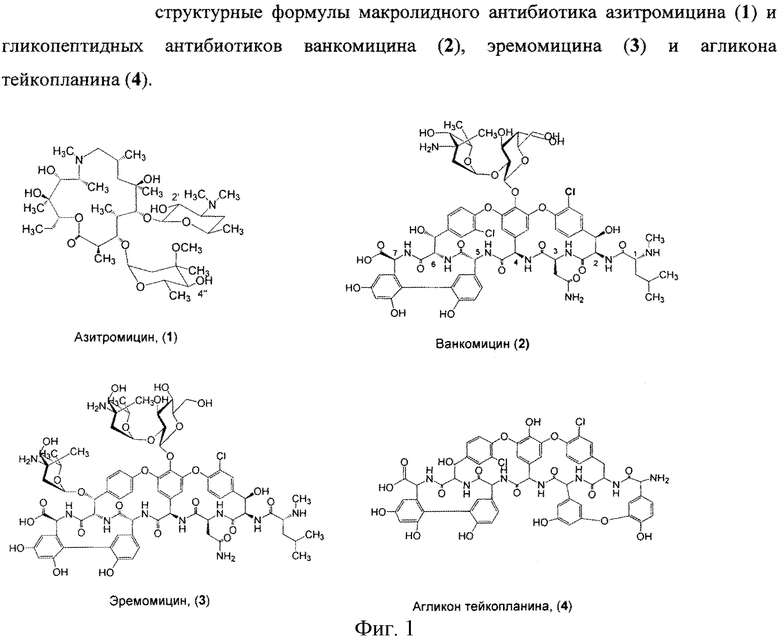
Азитромицин (1) (Фигура 1) является полусинтетическим антибиотиком, первым представителем подкласса азалидов, несколько отличающихся по структуре от классических макролидов, основу химической структуры которых составляет макроциклическое лактонное кольцо. Получен модификацией 14-членных макролидов путем включения атома азота в лактонное кольцо между 9 и 10 атомами углерода, кольцо при этом превращается в 15-членное. Данная структурная перестройка обусловливает значительное повышение кислотоустойчивости препарата - в 300 раз по сравнению с эритромицином. Антибиотик широкого спектра действия действует бактериостатически. Связываясь с 50S-субъединицей рибосом, угнетает пептидтранслоказу на стадии трансляции, подавляет синтез белка, замедляет рост и размножение бактерий, в высоких концентрациях оказывает бактерицидный эффект. Действует на вне- и внутриклеточных возбудителей. Неактивен в отношении грамположительных бактерий, устойчивых к эритромицину.
Обладает уникальной по сравнению с другими макролидами способностью накапливаться в органах и тканях. Азитромицин активно поглощается разными клетками, включая лейкоциты, фибробласты, макрофаги и фагоциты, и вместе с ними транспортируется к месту инфекции (воспаления) [Lalak N.J., Morris D.L., Azithromycin Clinical Pharmacokinetics, Clinical Pharmacokinetics, 1993, V. 25, P. 370-374; Gladue R.P, Bright G.M., Isaacson R.E., Newborg M.F. In vitro and in vivo uptake of azithromycin (CP-62,993) by phagocytic cells: possible mechanism of delivery and release at sites of infection. Antimicrobial Agents Chemotherapy, 1990, V. 34, P. 1056-1060]. Азитромицин относится к тканевым антибиотикам, так как его концентрации в сыворотке крови значительно ниже тканевых. Он хорошо распределяется в организме, создавая высокие концентрации в различных тканях и органах (в том числе в предстательной железе), особенно при воспалении. При этом азитромицин проникает внутрь клеток и создает высокие внутриклеточные концентрации. Метаболизируется в печени при участии микросомальной системы цитохрома Р-450, метаболиты выводятся преимущественно с желчью. Период полувыведения азитромицина 55 ч. При почечной недостаточности этот параметр не изменяется [Практическое руководство по антиинфекционной химиотерапии. НИИАХ СГМА. 2007. Под редакцией Л.С. Страчунского, Ю.Б. Белоусова, C.H. Козлова].
Ванкомицин (2) представляет собой трициклический гептапептид, к которому присоединен дисахарид, состоящий из аминодезоксисахара (ванкозамина) и D-глюкозы (формула 1) [Sztaricskai F., Pelyva′s-Ferenczik I.. Chemistry of carbohydrate components. In Glycopeptide Antibiotics, 1st edn (Ed. Nagarajan R.), 1994, Marcel Dekker, New York, NY, USA.].
Эремомицин (3) - оригинальный отечественный антибиотик, агликон которого отличается от агликона ванкомицина отсутствием атома хлора в боковом ароматическом радикале аминокислоты 6, структурой дисахаридной цепи (2-(O-(α-L-эремозаминил)-β-D-глюкопиранозил) и наличием третьего углеводного остатка (L-эремозаминил) в боковом радикале аминокислоты 6 (формула 1) [Gause G.F., Brazhnikova M.G., Lomakina N.N., Berdnikova T.F., Fedorova G.B., Tokareva N.L., Borisova V.N., Batta G. Eremomycin - new glycopeptide antibiotics. Chemical properties and structure. J. Antibiotics, 1989, V. 42, P. 1790-1799].
Агликон тейкопланина (4) представляет собой гептапептид, в котором в отличие от агликонов ваномицина и эремомицина, боковые радикалы аминокислот 1 и 3 являются ароматическими и соединены между собой эфирной связью (формула 1).
Ванкомицин применяется в клинической практике с 1958 г., тейкопланин - с середины 80-х годов. В последнее время интерес к гликопептидам возрос в связи с увеличением частоты нозокомиальных инфекций грамположительными микроорганизмами. Гликопептиды активны в отношении грамположительных аэробных и анаэробных микроорганизмов, включая метициллин-устойчивые Staphylococcus aureus (MSRA), метициллин-устойчивые Staphylococcus epidermidis (MRSE), стрептококков, пневмококков (включая АРП), энтерококков, включая резистентных к ампицилину и аминогликозидам, пептострептококков, листерий, коринебактерий, клостридий (включая С. difficile). Грамотрицательные микроорганизмы устойчивы к гликопептидам.
Гликопептиды нарушают синтез клеточной стенки бактерий. Оказывают бактерицидное действие, однако в отношении энтерококков, некоторых стрептококков и коагулазонегативных стафилококков действуют бактериостатически. Гликопептиды практически не всасываются при приеме внутрь. Биодоступность тейкопланина при внутримышечном введении составляет около 90%. Гликопептиды не метаболизируются, выводятся почками в неизмененном виде, поэтому при почечной недостаточности требуется коррекция доз. Препараты не удаляются при гемодиализе. Период полувыведения ванкомицина при нормальной функции почек составляет 6-8 ч, тейкопланина - от 40 ч до 70 ч. Длительный период полувыведения тейкопланина дает возможность назначать его один раз в сутки [Практическое руководство по антиинфекционной химиотерапии. НИИАХ СГМА. 2007. Под редакцией Л.С. Страчунского, Ю.Б. Белоусова, C.H. Козлова].
По спектру антимикробной активности ванкомицин и тейкопланин сходны, однако имеются некоторые различия в уровне природной активности и приобретенной резистентности. Их успешное применение в клинике поставлено под угрозу распространением устойчивых к ванкомицину грамположительных бактерий: энтерококков (VRE) и ванкомицин-резистентных Staphilococcus aureus (VRSA) [Mendez-Alvarez S., Perez-Hernandez X., Claverie-Martin F. Glycopeptide resistance in enterococci // Internat. Microbiol. - 2000. - V. 3. - P. 71-80; Cetinkaya Y., Falk P., Mayhall C.G. Vancomycin-resistant Enterococci // Clin. Microboil. Rev. - 2000. - V. 13. - P. 686-707]. Штаммы VRE, и в особенности VRSA, устойчивы к подавляющему большинству применяемых антибиотиков, и поэтому распространение таких микроорганизмов представляет очень серьезную проблему.
Широкое распространение резистентности к антибиотикам среди возбудителей заболеваний привело к утрате клинической значимости ряда лекарственных препаратов и послужило стимулом для поиска новых эффективных антимикробных агентов. Одной из перспективных стратегий, направленной на создание препаратов, активных в отношении резистентных микроорганизмов, является создание антибиотиков двойного действия - «химерных» антибиотиков. Они состоят из молекул разных антибиотиков, связанных между собой различными способами через так называемые «спейсеры». Они обладают расширенным спектром действия по сравнению с исходными антибиотиками и замедляют развитие антибиотикорезистентности.
Настоящее изобретение призвано получить антибиотики двойного действия на основе макролидного антибиотика азитромицина и гликопептидных антибиотиков, обладающие антибактериальной активностью.
Объединение в одной молекуле азитромицина с другим антибактериальным агентом потенциально может расширить спектр действия полученного химерного антибиотика по сравнению со спектрами действия исходных антибиотиков. Фармакокинетические свойства такого химерного антибиотика могут превосходить фармакокинетические свойства второго антибактериального агента, использованного для его синтеза.
Гликопептидные антибиотики выбраны в качестве второго антибактериального агента при синтезе химерных антибиотиков на основе азитромицина, поскольку они активны в отношении грамположительных аэробных и анаэробных микроорганизмов и являются препаратами выбора при инфекциях, вызванных широко распространенными в клиниках резистентными грамположительными бактериями.
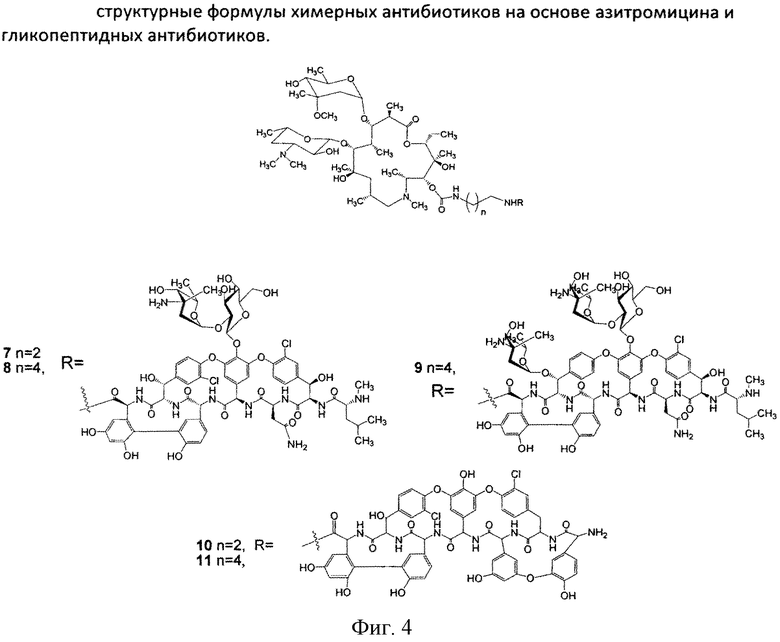
Изобретение включает соединения, соответствующие структурной формуле, изображенной на фигуре 2, где n=1-10, a R представляет собой остаток гликопептидного антибиотика, такого как ванкомицин, или эремомицин, или агликон тейкопланина.
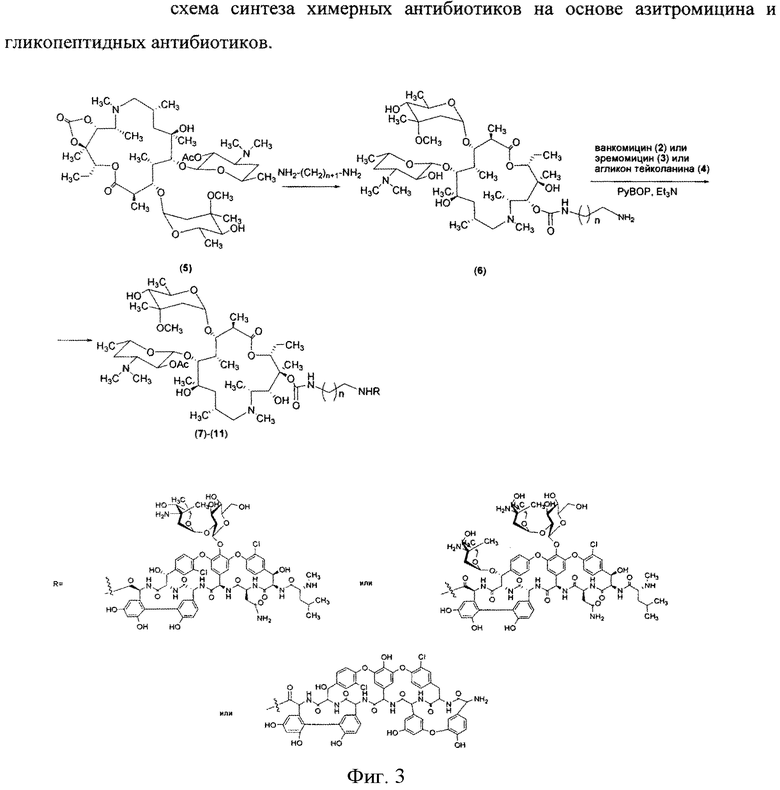
Изобретение также включает в себя способ получения антибиотиков на основе азитромицина и гликопептидных антибиотиков (Фигура 1), заключающийся в проведении реакции ацилирования 11-O-(ω-аминоалкилкарбамоил)азитромицина (6) гликопептидным антибиотиком (эремомицином (2), или ванкомицином (3), или агликоном тейкопланина (4)) в присутствии конденсирующего агента (Фигура 3).
11-O-(ω-Аминоалкилкарбамоил)азитромицин (6) получен из 2′-O-ацетил-11,12-циклического карбоната азитромицина (5) методом, аналогичным описанному в литературе [X. Li, S. Ma, M. Yan, Y. Wang, S Ma. Synthesis and antibacterial evaluation of novel 11,4′′-disubstituted azithromycin analogs with greatly improved activity against erythromycin-resistant bacteria. European Journal of Medicinal Chemistry, 2013, V. 59, pp 209-217].
В этой работе описано получение 11,4′′-дизамещенных производных азитромицина - производных 11-O-(аминофенилкарбамоил)-4′′-O-(амино)карбамоилазитромицина, которые показали высокую активность в отношении эритромицин-чувствительных штаммов S. pneumoniae и значительно улучшенную активность в отношении эритромицин-резистентных штаммов S. pneumonia (МПК 2÷16 мкг/мл против 256 мкг/мл).
Реакцию ацилирования 11-O-(ω-аминоалкилкарбамоил)азитромицина (6) (Фиг. 3, Схема 1) гликопептидным антибиотиком с получением соединения формулы 2 проводят в присутствии конденсирующих агентов, известных из уровня техники и применяемых для образования амидной связи, например бензотриазол-1-ил-окси-триспирролидинофосфоний гексафторфосфата (РуВОР) или O-(бензотриазол-1-ил)-N,N,N′,N′-бис(тетраметилен)) гексафторфосфат мочевины (HBPyU). Реакцию ацилирования 11-O-(ω-аминоалкилкарбамоил)азитромицина (6) (Фигура 3) гликопептидным антибиотиком с получением соединения структурной формулы, изображенной на фигуре 2, и показанного на фигуре 4, проводят в растворителе, выбираемом из метанола, этанола, Ν,Ν-диметилформамида или диметилсульфоксида.
Соединения структурной формулы, изображенной на фигуре 2, обладают выраженной антибактериальной активностью, в том числе в отношении штаммов, устойчивых к ванкомицину (см. Пример 2) и могут быть использованы для лечения инфекционных заболеваний.
Вспомогательные средства
Эремомицин сульфат получен на опытной установке НИИ по изысканию новых антибиотиков им. Г.Ф. Гаузе РАМН. Ванкомицин гидрохлорид был коммерческим продуктом фирмы Aldrich (США). Агликон тейкопланина был получен от фирмы Lepetit Research Center (Gerenzano (Varese), Италия). Бензотриазол-1-ил-окси-триспирролидинофосфоний гексафторфосфат (РуВОР), O-(бензотриазол-1-ил)-N,N,N′,N′-бис(тетраметилен)) гексафторфосфат мочевины (HBPyU) были коммерческими продуктами фирмы Acros. Все растворы высушивали над сульфатом натрия и упаривали при температуре не выше 40°C.
Тонкослойную хроматографию осуществляли на пластинках с силикагелем G60 (Merck) в смеси растворителей: система (A) AcOEt-nPrOH-NH4OH, 1:1:2. Для препаративной очистки использовали колоночную хроматографию на силанизированном силикагеле Merck с размером частиц 0.040-0.063 µм.
Аналитическую ВЭЖХ осуществляли на хроматографе LC-10 (Shimadzu, Япония) с использованием УФ-детектора и колонки Kromasil 100-С18 4×250 мм, размер частиц 6 мкм (АО БиоХимМак СТ, РФ). Подвижной фазой служили системы, состоящие из двух компонентов А и Б:
Система (В): А (0.2% HCOONH4 рН 4.5) и Б (MeCN), изократический режим 8% ацетонитрила от 0 до 5 минут, затем линейный градиент концентрации ацетонитрила 8→70% от 5 до 40 мин, скорость потока 1.0 мл/мин.
ИК-спектры снимали в таблетке KBr на спектрофотометре DTGS. Температуры плавления получены на приборе Buchi SMP-20.
Масс-спектры при ионизации электрораспылением (ESI) получали на приборе Finnigan MAT 900S (Германия).
Примеры получения производных на основе азитромицина и гликопептидных антибиотиков по настоящему изобретению и изучение их антибактериальной активности:
Пример 1. Общая методика получения химерных антибиотиков на основе остатков азитромицина и гликопептидного антибиотика.
2′-O-ацетил-11,12-циклического карбоната азитромицина (5).
К раствору азитромицина (5 г, 3.27 ммоль) в 24 мл этилацетата добавляли K2CO3 (0.64 г, 4.63 ммоль), нагревали смесь до кипения и затем медленно, в течение 20 мин при кипячении добавляли 1.6 г (18.2 ммоль) этиленкарбоната. Далее смесь кипятили 24 ч, затем этилацетат упаривали. Остаток растворяли в дихлорметане (30 мл) при комнатной температуре, затем добавляли уксусный ангидрид (0.61 мл, 4.37 ммоль) и триэтиламин (1.8 мл, 13 ммоль), реакционную массу премешивали 24 ч при комнатной температуре. К реакционной смеси добавляли 5% водный раствор NaHCO3 (30 мл), и водный раствор экстагировали дихлорметаном (3×10 мл). Объединенные слои дихлорметана высушивали безводным Na2SO4 и высушивали в вакууме. Далее остаток растворяли в хлороформе и наносили на хроматографическую колонку (45×1.5 см) с силикагелем Merck, уравновешенную хлороформом, затем элюировали хлороформом (40 мл), затем смесью хлороформ-метанол (10:1). Фракции, содержащие целевое вещество, упаривали и высушивали в вакууме.
11-O-(ω-Аминоалкилкарбамоил)азитромицин (6).
2′-O-ацетил-11,12-циклического карбоната азитромицина (5) растворяли в минимальном объеме Να,Νω-диаминоалкана и перемешивали при комнатной температуре 48 ч. Далее в реакционную смесь добавляли CHCl3 (20 мл) и воду (20 мл), смесь встряхивали. Органический слой отделяли и промывали водой 6 раз. Органические слои объединяли и далее добавляли воду и 0.5 n HCl, встряхивая слои так, чтобы рН водного слоя составил 8. Органический слой отделяли, добавляли Na2SO4, выдерживали 1 ч, осадок Na2SO4 отфильтровывали, промывая хлороформом. Органический слой упаривали и высушивали в вакууме.
Общая методика проведения реакции ацилирования 11-O-(ω-аминоалкилкарбамоил)азитромицина (6) гликопептидными антибиотиками (ванкомицином или эремомицином или аглконом тейкопланина), отщепления ацетильной группы и очистки.
К раствору гликопептидного антибиотика (0.47 ммоль) в ДМСО (7 мл) добавляли 11-O-(ω-аминоалкилкарбамоил)азитромицина (6) (0.5 экв., 0.235 ммоль), значение рН реакционной смеси доводили до ~7.5 добавлением Et3N. Порциями в течение 1 ч добавляли РуВОР (1.1 экв., 0.26 ммоль), поддерживая рН реакционной смеси ~7.5 добавлением Et3N. Реакционную смесь перемешивали в течение 20 часов при комнатной температуре, затем добавляли пятикратный объем диэтилового эфира. Полученную смесь интенсивно перемешивали, затем эфирный слой удаляли. Процедуру повторяли дважды, до получения вязкого масла, затем добавляли метанол (0.5 мл), ацетон (2 мл) и избыток диэтилового эфира, выпавший осадок отфильтровывали, промывали диэтиловым эфиром и высушивали. Продукт далее очищали методом колоночной хроматографии на силинизированном силикагеле. Вещество растворяли в 30% водном растворе МеОН, добавляли 1 см3 силанизированного силикагеля и высушивали эту смесь в вакууме, далее наносили ее на колонку с силанизированным силикагелем, уравновешенную водой. Элюцию осуществляли сначала водой (100 мл), затем 0.05 M раствором CH3COOH, затем системой МеОН-0.05 M CH3COOH (30:70) (100 мл) для соединений 7-9 или системой МеОН-0.05 M CH3COOH (30:70) (100 мл) для соединений 10-11. Фракции, содержащее целевое вещество, объединяли, упаривали в роторном испарителе с добавлением n-BuOH, к остатку добавляли ацетон и диэтиловый эфир. Выпавший осадок отфильтровывали, промывали ацетоном и высушивали в вакууме. Физико-химические данные для производных 7-11 (Фигура 4) представлены в Таблице 1.

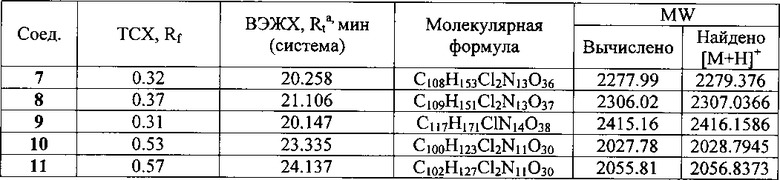
Пример 2. Изучение антибактериальной активности антибиотиков на основе азитромицина и гликопептидных антибиотиков
Антибактериальная активность производных 7-11 в сравнении с азитромицином (1) и ванкомицином (2) изучена на панели штаммов грамположительных и грамотрицательных бактерий (Таблица 2).
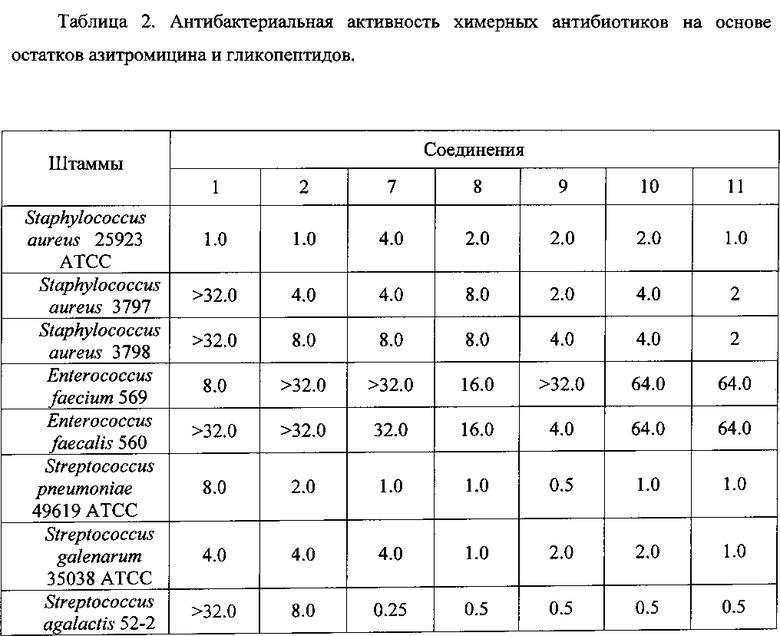
Полученные данные свидетельствуют, что все синтезированные химерные антибиотики на основе азитромицина и гликопептидов 7-11 не уступали или превосходили азитромицин и ванкомицин по антибактериальной активности в отношении всех изученных штаммов грамположительных бактерий. Ценной является высокая активность производных (выше, чем у азитромицина и ванкомицина) в отношении пневмокков, являющихся причиной большинства случаев менингитов, внебольничных пневмоний и ряда гнойно-септических инфекций. Производные на основе эремомицина и азитромицина оказались активны в отношении штаммов Enterococcus faecium и Enterococcus faecalis, устойчивых к ванкомицину.
Таким образом, предложенный способ получения антибиотиков на основе азитромицина и гликопептидных антибиотиков позволяет получать новые соединения, обладающие высокой антибактериальной активностью, в том числе, в отношении ряда штаммов Enterococcus, устойчивых к ванкомицину.
| название | год | авторы | номер документа |
|---|---|---|---|
| ХИМЕРНЫЕ АНТИБИОТИКИ НА ОСНОВЕ ГЛИКОПЕПТИДОВ И 11,12-ЦИКЛИЧЕСКОГО КАРБОНАТА АЗИТРОМИЦИНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2014 |
|
RU2570425C1 |
| НОВЫЕ ПРОИЗВОДНЫЕ ЭРЕМОМИЦИНА И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2019 |
|
RU2708628C1 |
| НОВОЕ ПОЛУСИНТЕТИЧЕСКОЕ ПРОИЗВОДНОЕ ЭРЕМОМИЦИНА И ЕГО ПРИМЕНЕНИЕ | 2016 |
|
RU2641912C1 |
| N-(ГУАНИДИНОАЛКИЛ)АМИДЫ ЭРЕМОМИЦИНА И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2022 |
|
RU2815029C1 |
| НОВЫЕ ПРОИЗВОДНЫЕ АМИДА ЭРЕМОМИЦИНА И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2020 |
|
RU2751334C1 |
| СОРБЕНТ ДЛЯ РАЗДЕЛЕНИЯ ОПТИЧЕСКИХ ИЗОМЕРОВ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2004 |
|
RU2255802C1 |
| Штамм Amycolatopsis orientalis - продуцент антибиотика диметилванкомицина и способ получения антибиотика | 2016 |
|
RU2633511C1 |
| ШТАММ AMYCOLATOPSIS ORIENTALIS SUBSP. EREMOMYCINI ВКПМ-S892 - ПРОДУЦЕНТ АНТИБИОТИКА ЭРЕМОМИЦИНА И СПОСОБ ПОЛУЧЕНИЯ АНТИБИОТИКА ЭРЕМОМИЦИНА | 1997 |
|
RU2110578C1 |
| АНТИБИОТИК "ЭРЕМОМИЦИН" И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1981 |
|
SU1475150A1 |
| Соль гликопептидного антибиотика и фармацевтические композиции на ее основе | 2023 |
|
RU2827992C1 |
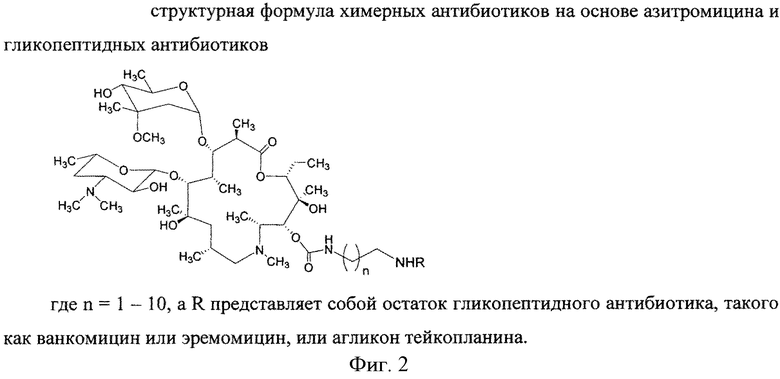
Изобретение относится к новым производным азитромицина и способу их получения, которые могут найти применение в фармацевтической промышленности и медицине. Предложены соединения, соответствующие формуле 2
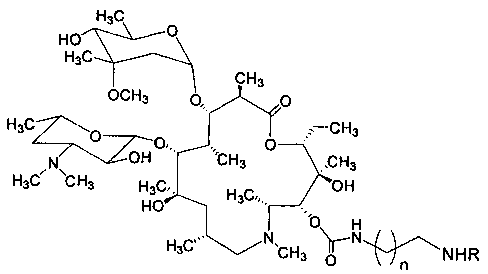 ,
,
где n=1-10, a R - остаток ванкомицина, эремомицина или агликона тейкопланина. Способ получения соединений формулы 2 включает проведение реакции ацилирования 11-O-(ω-аминоалкилкарбамоил)азитромицина (6) гликопептидным антибиотиком (эремомицином, или ванкомицином, или агликоном тейкопланина) в присутствии конденсирующего агента. 2 н.п. ф-лы, 4 ил., 2 пр., 2 табл.
1. Соединения, соответствующие формуле 2
,
где n=1-10, a R представляет собой остаток гликопептидного антибиотика, такого как ванкомицин, или эремомицин, или агликон тейкопланина.
2. Способ получения антибиотиков на основе азитромицина и гликопептидных антибиотиков по п. 1, заключающийся в проведении реакции ацилирования 11-O-(ω-аминоалкилкарбамоил)азитромицина (6) гликопептидным антибиотиком (эремомицином, или ванкомицином, или агликоном тейкопланина) в присутствии конденсирующего агента.
| WO 2014080251 A1, 30.05.2014 | |||
| WO 2004112713 A2, 29.12.2004 | |||
| WO 2011132171 A1, 27.10.2011 | |||
| US 6437119 B1, 20.08.2002 | |||
| АНТИМИКРОБНОЕ СРЕДСТВО И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ЭФФЕКТИВНОЕ КОЛИЧЕСТВО АНТИМИКРОБНОГО СРЕДСТВА | 2005 |
|
RU2278675C1 |

