Область техники, к которой относится изобретение
Настоящее изобретение относится к фармацевтической композиции для орального введения, которая включает часть с модифицированным высвобождением, способную регулировать высвобождение тамсулозина, и часть с немедленным высвобождением, способную быстро высвобождать солифенацин.
В частности, настоящее изобретение относится к фармацевтической композиции для орального введения, которая в одной препаративной форме включает часть с модифицированным высвобождением, содержащую тамсулозин, полимер, образующий гидрогель, и гидрофильное основание и часть с немедленным высвобождением, содержащую солифенацин и гидрофильное вещество.
Предпосылки к созданию изобретения
Тамсулозин представляет собой (R)-5-(2-{[2-(2-этоксифенокси)этил]амино}пропил)-2-метоксибензол-1-сульфонамид, имеющий следующую структурную формулу. Указанное соединение, а также его фармацевтически приемлемые соли ранее раскрыты в патентной литературе 1.
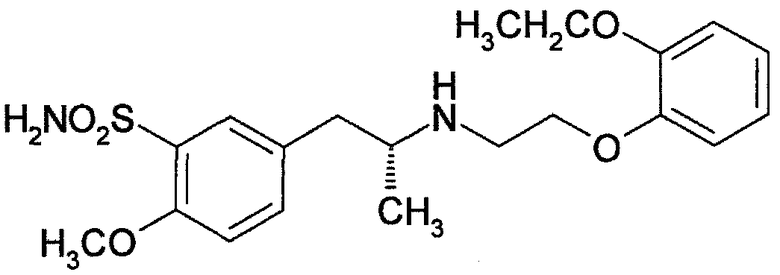
Известно, что тамсулозин или его соли обладают способностью блокировать адренергический рецептор α1A. В частности, гидрохлорид тамсулозина активен при блокировании рецепторов α1 в мочеиспускательном канале и предстательной железе и широко используется в качестве средства для лечения дизурии, связанной с доброкачественной гиперплазией предстательной железы, которое снижает давление в предстательной железе в профиле давления в мочеиспускательном канале. Клинические исследования подтверждают, что гидрохлорид тамсулозина эффективен при лечении симптомов со стороны нижних мочевыводящих путей и, таким образом, гидрохлорид тамсулозина является чрезвычайно полезным лекарственным средством для использования в клинических условиях. Тамсулозин поставляется на рынок как Harnal (зарегистрированный товарный знак) в Японии, Flomax (зарегистрированный товарный знак) в Соединенных Штатах и Omnic (зарегистрированный товарный знак) в Европе.
Солифенацин представлен следующей химической формулой и имеет химическое название (R)-хинуклидин-3-ил (S)-1-фенил-1,2,3,4-тетрагидроизохинолин-2-карбоксилат.
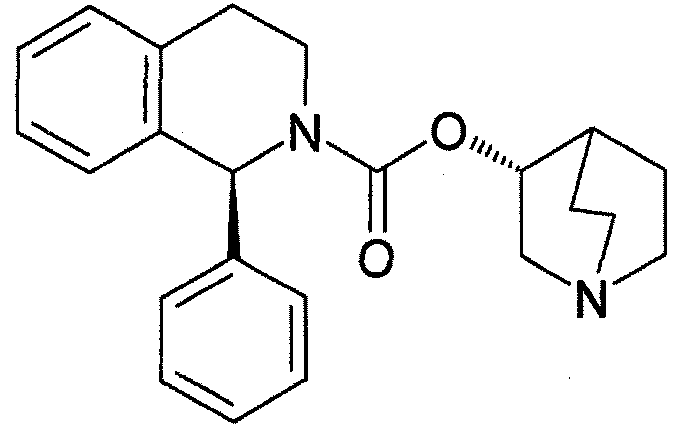
Известно, что солифенацин или его соли проявляют великолепную селективную антагонистическую активность против мускариновых рецепторов M3 и пригодны в качестве средства для предотвращения или лечения заболеваний мочеиспускательной системы, таких как недержание мочи и поллакиурия при неврогенной поллакиурии, нейрогенном мочевом пузыре, ночном недержании мочи, слабом мочевом пузыре, цитоспазме и хроническом цистите; заболеваний дыхательных путей, таких как хронические обструктивные заболевания легких, хронический бронхит, астма и ринит; и заболеваний пищеварительной системы, таких как синдром раздраженного кишечника, спастический колит и дивертикулит (см. патентный документ 2).
В частности, солифенацин обладает высокой селективностью к рецепторам M3, расположенным в гладких мышцах, тканях желез и т.д., по сравнению с рецепторами M2, расположенными в сердце и т.д., и пригоден в качестве антагониста рецептора M3, оказывающего меньшее побочное воздействие на сердце и т.д., в частности, пригоден в качестве средства для предотвращения или лечения недержания мочи и поллакиурии, хронических обструктивных заболеваний легких, хронического бронхита, астмы, ринита и т.п. В качестве средства для лечения неотложных позывов к мочеиспусканию, частоты мочеиспускания и недержания мочи при гиперактивности мочевого пузыря солифенацин поставляется на рынок как Vesicare (зарегистрированный товарный знак) в Японии, VESIcare (зарегистрированный товарный знак) в Соединенных Штатах и Vesicare (зарегистрированный товарный знак) в Европе.
Препаративная форма с модифицированным высвобождением, содержащая тамсулозин или его фармацевтически приемлемую соль, известна (например, из патентных документов 3 и 4) и поставляется на рынок как Omnic OCAS (зарегистрированный товарный знак).
В настоящем изобретении предлагается препаративная форма с модифицированным высвобождением, с профилем концентрации в крови показывающим меньшее отношение пикового значения к минимальному значению, чем у обычной препаративной формы с модифицированным высвобождением. Препаративная форма с модифицированным высвобождением не только уменьшает вероятность возникновения побочных эффектов, таких как ортостатическая анемия, но и, как ожидается, будет увеличивать дозу или поддерживать эффективность в течение длительного периода времени. Кроме того, можно избежать влияния пищи на концентрацию лекарственного средства в крови и, как ожидается, будет достигнут высокий профиль безопасности с точки зрения соблюдения пациентом режимов дозирования лекарственного средства (патентный документ 5).
Для лечения симптомов со стороны нижних мочевыводящих путей, связанных с доброкачественной гиперплазией предстательной железы, раскрывается фармацевтическая композиция, содержащая тамсулозин или его фармацевтически приемлемую соль и солифенацин или его фармацевтически приемлемую соль, в частности, фармацевтическая композиция для лечения симптомов со стороны нижних мочевыводящих путей, связанных с доброкачественной гиперплазией предстательной железы, и настоящее изобретение относится к комбинированному использованию обоих лекарственных средств (патентный документ 6).
Тамсулозин или его фармацевтически приемлемая соль эффективны при лечении симптомов опорожнения, напротив, солифенацин или его фармацевтически приемлемая соль эффективны при лечении симптомов наполнения и, таким образом, оба соединения оказывают противоположное действие. Однако комбинированное использование обоих лекарственных средств неожиданно привело к дальнейшему снижению интенсивности симптомов наполнения и не повлияло на снижение интенсивности симптомов опорожнения, по сравнению с введением каждого лекарственного средства по отдельности.
Поскольку было подтверждено, что комбинированная терапия с использованием тамсулозина или его фармацевтически приемлемой соли или солифенацина или его фармацевтически приемлемой соли клинически эффективна при лечении симптомов со стороны нижних мочевыводящих путей, связанных с доброкачественной гиперплазией предстательной железы, то желательно разработать для данной области медицины комбинированную препаративную форму (т.е. одну препаративную форму), содержащую оба лекарственных средства, с целью улучшения соблюдения пациентом режимов дозирования лекарственного средства. В качестве варианта комбинированной препаративной формы по настоящему изобретению, с целью обеспечить эффективное воздействие, которое проявляется при комбинированном использовании, удерживая при этом на низком уровне возможные побочные эффекты, и в течение длительного времени поддерживать эффективность воздействия, может быть предложена комбинированная препаративная форма с модифицированным высвобождением, содержащая тамсулозин, вместе с обычной препаративной формой (препаративной формой с немедленным высвобожденимем), содержащей солифенацин. Однако поскольку скорости растворения лекарственного средства в обеих препаративных формах отличаются от друг друга, то даже в том случае, когда из обеих препаративных форм готовят единую препаративную форму (т.е. комбинированную препаративную форму), желательно, чтобы скорость высвобождения лекарственного средства в каждой препаративной форме, содержащейся в единой препаративной форме, не сильно изменилась.
Список литературы
Патентная литература:
[патентный документ 1] Не прошедшая экспертизу заявка на патент Японии (Kokai) № 56-110665
[патентный документ 2] Патент США № 6017927 (соответствует международной публикации WO 96/20194)
[патентный документ 3] Международная публикация WO 94/06414
[патентный документ 4] Международная публикация WO 2004/078212
[патентный документ 5] Опубликованная заявка на патент США 2005-0100603
[патентный документ 6] Международная публикация WO 2009/013846
Сущность изобретения
Техническая задача
Авторы настоящего изобретения используют те же самые компоненты рассматриваемых лекарственных форм, т.е. Vesicare (название лекарственной формы) с большой скоростью высвобождения лекарственного средства (85% в течение 30 мин) и препаративную форму с модифицированным высвобождением лекарственного средства Omnic OCAS (название лекарственной формы), для приготовления единой препаративной формы (т.е. комбинированной препаративной формы) (приведенный ниже сравнительный пример 1).
В частности, изготавливают двухслойные таблетки, состоящие из части с модифицированным высвобождением, которая содержит тамсулозин, полиэтиленоксид, полиэтиленгликоль и стеарат магния, и части с немедленным высвобождением, которая содержит солифенацин, лактозу, кукурузный крахмал, гидроксипропилметилцеллюлозу и стеарат магния. Было проведено исследование растворимости с использованием полученных двухслойных таблеток, и неожиданно было обнаружено:
[1] скорость растворения солифенацина снижается и составляет менее чем 85% в течение 30 мин, и
[2] максимальный процент растворения солифенацина составляет менее чем 90%.
Когда скорость растворения или максимальный процент растворения солифенацина снижается, это вызывает озабоченность, что это приводит к уменьшению его доступности в организме, т.е. биодоступности, а в результате фармакологические эффекты, эквивалентные тем, которые проявляются при комбинированном использовании известных препаративных форм (варианты препаративных форм с одним лекарственным средством), не могут быть получены.
Целью настоящего изобретения является разработка для данной области медицины единой препаративной формы (комбинированной препаративной формы), которая включает часть с модифицированным высвобождением, содержащую тамсулозин, и часть с немедленным высвобождением, содержащую солифенацин, в частности, (1) разработка единой препаративной формы (комбинированной препаративной формы), в которой скорости растворения обоих лекарственных средств (в частности, скорость растворения солифенацина в части с немедленным высвобождением) аналогичны скоростям растворения известных препаративных форм с одним лекарственным средством, и (2) разработка единой препаративной формы (комбинированной препаративной формы), имеющей максимальный процент растворения обоих лекарственных средств (в частности, максимальный процент растворения солифенацина в части с немедленным высвобождением), равный 90% или более, биодоступность которой эквивалентна биодоступности известных препаративных форм с одним лекарственным средством.
Решение задачи
Вышеуказанные результаты, т.е. замедление скорости растворения солифенацина из двухслойной таблетки и уменьшение максимального процента растворения солифенацина были в высшей степени неожиданными для авторов настоящего изобретения, поскольку растворимость солифенацина в воде составляет 610 мг/мл, и таким образом, солифенацин считается водорастворимым соединением в соответствии с уравнением растворимости, приведенным в Фармакопее Японии, и, кроме того, известная препаративная форма [Vesicare (зарегистрированный товарный знак)] разработана как препаративная форма с немедленным высвобождением.
Вследствие различного поведения в среде живого организма введенной двухслойной таблетки и каждой введенной отдельно препаративной формы (препаративной формы с одним лекарственным средством) следует указать, что часть с модифицированным высвобождением располагается рядом с частью с немедленным высвобождением и, таким образом, солифенацин, растворившийся из части с немедленным высвобождением, находится вблизи части с модифицированным высвобождением, содержащей другое лекарственное средство.
В частности, неожиданным является то, что появляется возможность для “растворимого” в воде солифенацина растворяться и включаться в препаративную форму части с модифицированным высвобождением, содержащей полимер, образующий гидрогель.
Времена распада немедленно высвобождаемых препаративных форм с одним лекарственным средством, которые приготовлены из тех же самых компонентов, что и препаративные формы из смеси порошков для части с немедленным высвобождением, которые готовят, соответственно, в рассмотренных ниже примерах 1-3 и сравнительном примере 1, приведены на Фиг.1, а времена высвобождения лекарственного средства из немедленно высвобождаемых препаративных форм с одним лекарственным средством, приведены на Фиг.2. Когда используют те же самые компоненты (солифенацин, лактозу, кукурузный крахмал, гидроксипропилметилцеллюлозу и стеарат магния), что и в известной препаративной форме, которые приведены на указанных фигурах в виде смеси порошков для части с немедленным высвобождением, описанной в сравнительном примере 1, то указанная препаративная форма показывает быстрый распад и быстрое высвобождение лекарственного средства. Этот результат отличен от результата для единой препаративной формы (комбинированной препаративной формы) и, таким образом, результат для двухслойной таблетки в качестве единой препаративной формы (комбинированной препаративной формы) был в высшей степени неожиданным.
В данных обстоятельствах авторы настоящего изобретения сосредоточили внимание на увеличении скорости высвобождения солифенацина и повышении максимального процента растворения солифенацина и провели соответствующие интенсивные исследования; таким образом было осуществлено настоящее изобретение.
В настоящем изобретении предлагается:
[1] фармацевтическая композиция для орального введения, включающая (1) часть с модифицированным высвобождением, которая включает тамсулозин или его фармацевтически приемлемую соль, и (2) часть с немедленным высвобождением, которая включает солифенацин или его фармацевтически приемлемую соль, и гидрофильное вещество;
[2] фармацевтическая композиция для орального введения по п.[1], где часть с немедленным высвобождением распадается и/или растворяется прежде, чем часть с модифицированным высвобождением образует гель;
[3] фармацевтическая композиция для орального введения по пп.[1] или [2], где 70% или более солифенацина растворяется в течение 15 мин;
[4] фармацевтическая композиция для орального введения по п.[3], где 90% или более солифенацина растворяется в течение 60 мин;
[5] фармацевтическая композиция для орального введения по п.[4], где 70% или более солифенацина растворяется в течение 15 мин и 90% или более солифенацина растворяется в течение 60 мин;
[6] фармацевтическая композиция для орального введения по п.[5], где 85% или более солифенацина растворяется в течение 30 мин и 90% или более солифенацина растворяется в течение 60 мин;
[7] фармацевтическая композиция для орального введения по любому из пп.[1]-[6], где гидрофильное вещество представляет собой одно соединение или два или несколько соединений, выбранных из группы, состоящей из полиэтиленгликоля, мальтозы, поливинилпирролидона и маннита;
[8] фармацевтическая композиция для орального введения по любому из пп.[1]-[7], где гидрофильное вещество составляет от 5% масс. до 99% масс.
[9] фармацевтическая композиция для орального введения по любому из пп.[1]-[8], где гидрофильное вещество или два или несколько гидрофильных веществ, выбранных из группы, состоящей из полиэтиленгликоля, мальтозы, поливинилпирролидона и маннита, используют в качестве связующего вещества;
[10] фармацевтическая композиция для орального введения по любому из пп.[1]-[9], где маннит в качестве гидрофильного вещества используют как наполнитель;
[11] фармацевтическая композиция для орального введения по любому из пп.[1]-[10], где часть с модифицированным высвобождением содержит полимер, образующий гидрогель;
[12] фармацевтическая композиция для орального введения по п.[11], где полимер, образующий гидрогель, имеет вязкость 4000 мПа·с или более в 1%-ном водном растворе (25°С);
[13] фармацевтическая композиция для орального введения по п.[12], где полимер, образующий гидрогель, представляет собой один полимер или два или несколько полимеров, выбранных из группы, состоящей из полиэтиленоксида, гидроксипропилметилцеллюлозы, натрий карбоксиметилцеллюлозы и карбоксивинилового полимера;
[14] фармацевтическая композиция для орального введения по п.[13], где полимером, образующим гидрогель, является полиэтиленоксид;
[15] фармацевтическая композиция для орального введения по п.[14], где средневязкостная молекулярная масса полиэтиленоксида составляет 5000000 или более;
[16] фармацевтическая композиция для орального введения по любому из пп.[11]-[15], где полимер, образующий гидрогель, составляет от 5% масс. до 95% масс.
[17] фармацевтическая композиция для орального введения по любому из пп.[1]-[16], где часть с модифицированным высвобождением дополнительно включает добавку, которая позволяет воде проникать в препаративную форму;
[18] фармацевтическая композиция для орального введения по п.[17], где добавка, которая позволяет воде проникать в препаративную форму, имеет такую растворимость, что количество воды, необходимое для растворения 1 г указанной добавки, составляет 5 мл или менее;
[19] фармацевтическая композиция для орального введения по п.[18], где добавка, которая позволяет воде проникать в препаративную форму, составляет от 3% масс. до 80% масс.
[20] фармацевтическая композиция для орального введения по любому из пп.[1]-[19], которая представляет собой фармацевтическую композицию для лечения симптомов со стороны нижних мочевыводящих путей, связанных с доброкачественной гиперплазией предстательной железы; и
[21] фармацевтическая композиция для орального введения по любому из пп.[1]-[20], где фармацевтическая композиция является таблеткой.
Преимущества изобретения
В настоящем изобретении предлагается фармацевтическая композиция для орального введения, включающая часть с модифицированным высвобождением, которая включает тамсулозин или его фармацевтически приемлемую соль, и часть с немедленным высвобождением, которая включает солифенацин или его фармацевтически приемлемую соль, и гидрофильное вещество. Фармацевтическая композиция по настоящему изобретению способна высвобождать лекарственное средство аналогично каждой из препаративных форм с одним лекарственным средством и, таким образом, единая препаративная форма (комбинированная препаративная форма) способна оказывать фармакологическое действие, эквивалентное фармакологическому действию, которое могут оказать препаративные формы с одним лекарственным средством. Кроме того, ожидается, что она улучшит соблюдения пациентом режимов дозирования лекарственного средства, поскольку количество препаративных форм, которые необходимо принимать, уменьшается.
Краткое описание чертежей
[Фиг.1] Фигура 1 представляет собой диаграмму, которая показывает времена распада препаративных форм с немедленным высвобождением, содержащих одно лекарственное средство, которые приготовлены из тех же компонентов, что и смешанные порошки для части с немедленным высвобождением, полученной в примерах 1-3 по настоящему изобретению и сравнительном примере 1, соответственно.
[Фиг.2] Фигура 2 представляет собой график, который показывает профили высвобождения для препаративных форм с немедленным высвобождением, содержащих одно лекарственное средство, которые приготовлены из тех же компонентов, что и смешанные порошки для части с немедленным высвобождением, полученной в примерах 1-3 по настоящему изобретению и сравнительном примере 1, соответственно.
[Фиг.3] Фигура 3 представляет собой график, который показывает профили растворения фармацевтических композиций, полученных в примерах 1-3 по настоящему изобретению и сравнительном примере 1, соответственно.
Что касается времени распада каждой препаративной формы с немедленным высвобождением, содержащей одно лекарственное средство, которое указано на фигуре 1, и высвобождения лекарственного средства из каждой препаративной формы с немедленным высвобождением, содержащей одно лекарственное средство, которое указано на фигуре 2, то препаративная форма с немедленным высвобождением, содержащая одно лекарственное средство, которая приготовлена из тех же компонентов, что и смешанные порошки для части с немедленным высвобождением, полученной в сравнительном примере 1, демонстрирует быстрый распад и быстрое высвобождение лекарственного средства, где растворение завершается в течение 15 мин, так же как и препаративные формы с немедленным высвобождением, содержащие одно лекарственное средство, которые приготовлены из тех же компонентов, что и смешанные порошки для части с немедленным высвобождением, приготовленной в примерах 1-3, соответственно.
Что касается единых препаративных форм (комбинированных препаративных форм), как показано на фигуре 3, то хотя препаративные формы с немедленным высвобождением, содержащие одно лекарственное средство, демонстрируют такой же быстрый распад и такое же быстрое растворение, в фармацевтической препаративной форме, полученной в сравнительном примере 1, водорастворимый солифенацин растворяется и включается в часть с модифицированным высвобождением, содержащую полимер, образующий гидрогель, и неожиданно степень высвобождения лекарственного средства не достигает 90%, и полное высвобождение лекарственного средства не наблюдается.
Описание предпочтительных вариантов осуществления настоящего изобретения
Ниже разъясняются предпочтительные варианты осуществления настоящего изобретения.
Термин “препаративная форма, содержащая одно лекарственное средство” в данном описании означает вариант осуществления препаративной формы, содержащей лекарственное средство.
Термин “комбинированная препаративная форма” в данном описании также обозначают как “единая препаративная форма”, и он означает вариант осуществления препаративной формы, содержащей два или более лекарственных средств. Термин “комбинированная препаративная форма” включает препаративную форму, которая содержит функционально различные препаративные формы, такие как часть с модифицированным высвобождением и часть с немедленным высвобождением по настоящему изобретению.
Термин “часть с модифицированным высвобождением” в данном описании означает вариант воплощения настоящего изобретения, который содержится в единой препаративной форме, и часть, которая контролирует высвобождение лекарственного средства.
Термин “часть с немедленным высвобождением” в данном описании означает вариант воплощения настоящего изобретения, который содержится в единой препаративной форме, и часть, которая быстро высвобождает лекарственное средство из фармацевтической композиции (в случае “растворимого” лекарственного средства термин “высвобождение” имеет практически то же самое значение, что и “растворение”).
Термин “максимальный процент растворения” в данном описании означает такой процент растворения, когда степень растворения лекарственного средства из фармацевтической композиции достигает плато в испытании на растворимость в заданных условиях.
Далее поясняется фармацевтическая композиция для орального введения по настоящему изобретению.
Тамсулозин или его фармацевтически приемлемую соль, которые могут применяться по настоящему изобретению, легко получают в соответствии со способами, приведенными в JP 56-110665 и JP 62-114952, или в соответствии с модифицированными вариантами указанных способов.
Тамсулозин может образовывать фармацевтически приемлемые соли с различными неорганическими и органическими кислотами. Указанные фармацевтически приемлемые соли могут использоваться в настоящем изобретении. Примеры солей включают соли с неорганическими кислотами, такими как хлористоводородная кислота, серная кислота и фосфорная кислота; соли с органическими кислотами, такими как фумаровая кислота, яблочная кислота, лимонная кислота и янтарная кислота; соли с щелочными металлами, такими как натрий и калий; и соли с щелочноземельными металлами, такими как кальций и магний. Гидрохлорид тамсулозина может использоваться в другом варианте осуществления настоящего изобретения. Указанные соли могут быть получены обычными способами.
Дозу тамсулозина или его фармацевтически приемлемой соли можно соответствующим образом определить для каждого пациента в зависимости, например, от пути введения, симптомов заболевания, возраста и пола пациента, лечение которого проводят, и т.п. Когда гидрохлорид тамсулозина назначают взрослому орально, то дневная доза активного ингредиента составляет от приблизительно 0,1 мг до 1,6 мг, и его вводят орально раз в день.
Солифенацин или его фармацевтически приемлемую соль, которые могут применяться по настоящему изобретению, легко получают в соответствии со способом, приведенным в WO 96/20194, или в соответствии с модифицированными вариантами указанных способов.
Солифенацин может образовывать фармацевтически приемлемые соли с различными неорганическими и органическими кислотами. Указанные фармацевтически приемлемые соли могут использоваться в настоящем изобретении. Примеры солей включают кислотно-аддитивные соли с неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, иодистоводородная кислота, серная кислота, азотная кислота и фосфорная кислота; и кислотно-аддитивные соли с органическими кислотами, такими как муравьиная кислота, уксусная кислота, пропионовая кислота, щавелевая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, молочная кислота, яблочная кислота, лимонная кислота, винная кислота, угольная кислота, пикриновая кислота, метансульфоновая кислота, этансульфоновая кислота и глутаминовая кислота. Сукцинат солифенацина может использоваться в другом варианте осуществления настоящего изобретения. Указанные соли могут быть получены обычным способом.
Дозу солифенацина или его фармацевтически приемлемой соли можно соответствующим образом определить для каждого пациента в зависимости, например, от пути введения, симптомов заболевания, возраста и пола пациента, лечение которого проводят, и т.п. Когда сукцинат солифенацина назначают взрослому орально, то дневная доза активного ингредиента составляет от приблизительно от 0,01 мг/кг до 100 мг/кг, и его вводят один раз в день или делят на отдельные дозы от двух до четырех доз в день. В качестве альтернативы, когда дозу вводят взрослому внутривенно, количество лекарственного средства составляет от 0,01 мг/кг на дозу до 10 мг/кг на дозу, и указанную дозу вводят один раз в день или несколько раз в день.
Содержание каждого лекарственного средства специально не ограничивается, при условии, что оно представляет собой количество, эффективное для лечения или предотвращения заболевания. Содержание лекарственного средства в препаративной форме, например, составляет 85% масс. или менее, 80% масс. или менее в другом варианте осуществления настоящего изобретения, 50% масс. или менее в другом варианте осуществления настоящего изобретения, и 10% масс. или менее еще в одном варианте осуществления настоящего изобретения.
“Часть с модифицированным высвобождением” по настоящему изобретению включает тамсулозин или его фармацевтически приемлемую соль, полимер, образующий гидрогель, (в данном описании иногда называют полимером, который образует гидрогель) и гидрофильное основание (в данном описании иногда называют добавкой, которая позволяет воде проникать в препаративную форму). “Часть с модифицированным высвобождением ” определяют в данном описании как часть, для которой точка, соответствующая растворению 50% лекарственного средства из фармацевтической композиции, составляет от 3 час до 15 час с момента начала проведения испытаний растворимости, когда испытания растворимости проводят в соответствии с тестом на растворимость, способ 2 (способ с использованием лопастной мешалки, скорость вращения от 50 об/мин до 200 об/мин), который описан в Фармакопее Японии, или когда испытания растворимости проводят в соответствии с тестом на растворимость, способ 1 (способ в барабане, скорость вращения от 50 об/мин до 200 об/мин), который описан в Фармакопее Японии.
Полимер, образующий гидрогель, который может использоваться по настоящему изобретению, специально не ограничивается, при условии, что он обладает такими свойствами, как вязкость и т.п. во время гелеобразования, сохраняющими в определенной степени форму почти полностью превратившейся в гель препаративной формы от воздействия сокращений в пищеварительном тракте, которые сопровождают переваривание пищи и перемещают превратившуюся в гель препаративную форму к толстому кишечнику в нижней части пищеварительного тракта. В качестве полимера, образующего гидрогель, может использоваться полимер, имеющий высокую вязкость во время гелеобразования, такой как полимер с вязкостью 4000 мПа·с или более в 1%-ном водном растворе при 25°C.
Свойства полимера зависят от его молекулярной массы, и в качестве полимера, образующего гидрогель, который может использоваться по настоящему изобретению, предпочтителен полимер с более высокой молекулярной массой. Примеры полимера с более высокой молекулярной массой включают полиэтиленоксид со средней молекулярной массой 5000000 или более, со средней молекулярной массой 7000000 или более в другом варианте осуществления настоящего изобретения, со средней молекулярной массой от 5000000 до 8000000 в другом варианте осуществления настоящего изобретения, и со средней молекулярной массой от 7000000 до 8000000 в еще одном варианте осуществления настоящего изобретения.
Примеры полимеров включают:
полиэтиленоксид (PEO) [например, продукты с названиями Polyox WSR-308 (средняя молекулярная масса: 8000000, вязкость: 10000-15000 мПа·с (1%-ный водный раствор при 25°C)), Polyox WSR-303 (средняя молекулярная масса: 7000000, вязкость: 7500-10000 мПа·с (1%-ный водный раствор при 25°C)), Polyox WSR Coagulant (средняя молекулярная масса: 5000000, вязкость: 5500-7500 мПа·с (так же, как указано выше)), которые все изготавливает компания Dow];
гидроксипропилметилцеллюлозу (HPMC) [например, продукты с названиями Metolose 90SH100000 (вязкость: 4100-5600 мПа·с (1%-ный водный раствор при 20°C)) и Metolose 90SH30000 (вязкость: 25000-35000 мПа·с (2%-ный водный раствор при 20°C)), которые все изготавливает компания Shin-Etsu Chemical Co., Ltd.];
натрий карбоксиметилцеллюлозы (CMC-Na) [например, продукты с названиями Sunrose F-1000MC (средняя молекулярная масса: 420,000, вязкость: 8000-12000 мПа·с (так же, как указано выше)) и HEC DAICEL SE900 (средняя молекулярная масса: 1560000, вязкость: 4000-5000 мПа·с (так же, как указано выше)), которые изготавливает компания Daicel chemical Industries, Ltd.]; и
карбоксивиниловые полимеры [например, Carbopol 940 (средняя молекулярная масса: приблизительно 2500000), изготавливает компания B.F. GoodRich Chemical)]. В другом варианте осуществления настоящего изобретения может применяться полиэтиленоксид.
Указанные полимеры, образующие гидрогель, могут использоваться индивидуально или в виде подходящей комбинации из двух или нескольких полимеров, образующих гидрогель. Когда смешивают два или большее количество полимеров, вязкость или средняя молекулярная масса которых выходит за границы вышеуказанных конкретных диапазонов, а полученная смесь имеет свойства, соответствующие вышеуказанным конкретным диапазонам, то смесь полимеров включают в полимеры, образующие гидрогель, которые могут применяться по настоящему изобретению.
Для того чтобы высвободить лекарственное средство в толстом кишечнике человека, предпочтительно, часть образовавшей гель препаративной формы сохраняется в толстом кишечнике по прошествии, по крайней мере, от 6 до 8 часов, более предпочтительно, по прошествии 12 часов или более после введения препаративной формы. Приготовление образующей гидрогель препаративной формы, обладающей указанными свойствами, меняется в зависимости от размера препаративной формы, типа полимеров, свойств лекарственного средства и добавки, которая позволяет воде проникать в препаративную форму, содержания полимера и т.п. Для препаративной формы, составляющей 600 мг или менее на таблетку, содержание полимера, образующего гидрогель, по отношению к массе препаративной формы составляет, например, от 5% масс. до 95% масс. и, предпочтительно, составляет от 10% масс. до 90% масс. в другом варианте осуществления настоящего изобретения, а содержание полимера, образующего гидрогель, по отношению к массе части с модифицированным высвобождением, составляет, например, от 10% масс. до 95% масс. и, предпочтительно, составляет от 15% масс. до 90% масс. в другом варианте осуществления настоящего изобретения. Количество на одну препаративную форму, предпочтительно, составляет 40 мг или более, и 60 мг или более в другом варианте осуществления настоящего изобретения. Когда содержание ниже чем указанные значения, то возникает вероятность того, что достаточное пролонгированное высвобождение в течение длительного времени не достигается вследствие эрозии в пищеварительном тракте.
Добавка, позволяющая воде проникать в препаративную форму (ее также обозначают как “гидрофильное основание”), которая может использоваться по настоящему изобретению, специально не ограничивается, при условии, что она представляет собой вещество, способное придавать препаративной форме свойства, обеспечивающие проникновение воды в препаративную форму. Примеры гидрофильного основания включают вещество, обладающее такой растворимостью, что количество воды, необходимое для растворения 1 г гидрофильного основания при 20±5°С составляет 5 мл или менее и 4 мл или менее в другом варианте осуществления настоящего изобретения. Когда гидрофильное основание имеет большую растворимость в воде, то эффект, который позволяет воде проникать в препаративную форму, усиливается.
Примеры гидрофильного основания включают:
водорастворимый полимер, такой как полиэтиленгликоль [PEG: например, продукты с названиями PEG 400, PEG 1500, PEG 4000, PEG 6000 и PEG 20000 (изготавливаются компанией NOF Corporation)] и поливинилпирролидон [PVP: например, продукт с названием PVP K30 (изготавливается компанией BASF)];
сахарные спирты, такие как D-сорбит и ксилит;
сахариды, такие как сахароза, безводная мальтоза, D-фруктоза, декстран (например, Dextran 40) и глюкоза;
поверхностно-активные вещества, такие как полиоксиэтилен-полиокиспропиленгликоль [например, Pluronic F68 (изготавливается компанией Asahi Denka и т.д.)];
соли, такие как хлорид натрия и хлорид магния;
органические кислоты, такие как лимонная кислота и винная кислота;
аминокислоты, такие как глицин, β-аланин и гидрохлорид лизина; и
аминосахариды, такие как меглумин.
В другом варианте осуществления настоящего изобретения могут применяться PEG6000, поливинилпирролидон, D-сорбит и т.п. Указанные гидрофильные основания могут использоваться отдельно или в виде комбинации из двух или большего количества гидрофильных оснований.
Содержание гидрофильного основания может меняться в зависимости от различных факторов, таких как свойства (растворимость, терапевтический эффект и т.п.) и содержание лекарственного средства, растворимость гидрофильного основания, свойства полимера, образующего гидрогель, состояние субъекта, которому будет вводиться композиция, и т.п., однако предпочтительным является количество, при котором гелеобразование почти полностью достигается во время удерживания препаративной формы в верхней части пищеварительного тракта. Время удерживания лекарственного средства в верхней части пищеварительного тракта меняется в зависимости от химических веществ и индивидуумов, однако время удерживания после введения собакам и людям составляет приблизительно 2 часа и приблизительно от 4 до 5 часов, соответственно [Br. J. Clin. Pharmac., (1988) 26, 435-443]. У людей, содержание гидрофильного основания, предпочтительно, представляет собой количество, при котором гелеобразование препаративной формы почти полностью достигается через 4-5 час после ее введения. Содержание составляет, например, от приблизительно 3% масс. до 80% масс. (от приблизительно 3% масс. до 60% масс. в другом варианте осуществления настоящего изобретения) от массы препаративной формы, и например, от приблизительно 5% масс. до 80% масс. (от приблизительно 5% масс. до 60% масс. в другом варианте осуществления настоящего изобретения) от массы части с модифицированным высвобождением.
Гидрофильное вещество, образующее "часть с немедленным высвобождением", которое используют в настоящем изобретении, специально не ограничивается, при условии, что часть с немедленным высвобождением может распадаться и/или растворяться. В другом варианте осуществления настоящего изобретения гидрофильное вещество специально не ограничивается, при условии, что часть с немедленным высвобождением может практически полностью распасться прежде, чем часть с модифицированным высвобождением образует гель. Состояние "почти полностью" можно определить или оценить по моменту, когда гидрофильное вещество, которое образует матрицу части с немедленным высвобождением, визуально почти полностью распадается в условиях проведения теста на растворимость. Состояние "почти полностью" можно определить или оценить на основе результатов испытания на растворимость, когда 70% или более лекарственного средства (85% или более в другом варианте осуществления настоящего изобретения и 90% или более в еще одном варианте осуществления настоящего изобретения) растворяется в течение 15 мин, или когда 85% или более лекарственного средства (90% или более в другом варианте осуществления настоящего изобретения) растворяется в течение 30 мин, или когда 90% или более лекарственного средства растворяется в течение 60 мин. Максимальный процент растворения части с немедленным высвобождением определяют, установив процент растворения лекарственного средства из фармацевтической композиции через 60 мин в условиях, когда испытания растворимости проводят в соответствии с тестом на растворимость, способ 2 (способ с использованием лопастной мешалки, скорость вращения от 50 об/мин до 200 об/мин), который описан в Фармакопее Японии, или когда испытания растворимости проводят в соответствии с тестом на растворимость, способ 1 (способ в барабане, скорость вращения от 50 об/мин до 200 об/мин), который описан в Фармакопее Японии.
Примеры гидрофильного вещества включают D-маннит, мальтозу, полиэтиленгликоль и поливинилпирролидон.
Часть с немедленным высвобождением содержит солифенацин или его фармацевтически приемлемую соль и гидрофильное вещество. Гидрофильное вещество может выполнять функции наполнителя и/или связующего. В качестве наполнителя могут использоваться D-маннит, мальтоза, полиэтиленгликоль или поливинилпирролидон. В качестве связующего могут использоваться мальтоза, полиэтиленгликоль или поливинилпирролидон. Примеры полиэтиленгликоля (PEG) включают PEG 400, PEG 1500, PEG 4000, PEG 6000 и PEG 20000 (торговое название, каждое их них изготавливается компанией NOF Corporation). Примеры поливинилпирролидона (PVP) включают Kollidon K25 и Kollidon K90 (торговые названия, каждое их них изготавливается компанией BASF).
Что касается состояния гидрофильного вещества, которое находится в части с немедленным высвобождением, то настоящее изобретение включает вариант воплощения, в котором гидрофильное вещество равномерно распределено в части с немедленным высвобождением, и вариант воплощения, в котором гидрофильное вещество равномерно располагается на границе части с немедленным высвобождением.
Гидрофильные вещества могут использоваться отдельно или в виде соответствующей комбинации двух или большего количества гидрофильных веществ.
Содержание гидрофильного вещества составляет, например, от 2% масс. до 40% масс. (от 4% масс. до 35% масс. в другом варианте осуществления настоящего изобретения) по отношению к препаративной форме, и от 5% масс. до 95% масс. (от 10% масс. до 90% масс. в другом варианте осуществления настоящего изобретения и от 20% масс. до 80% масс. в еще одном варианте осуществления настоящего изобретения) по отношению к части с немедленным высвобождением.
Различные фармацевтические добавки, если необходимо, могут соответствующим образом использоваться при получении фармацевтической композиции по настоящему изобретению, и они специально не ограничиваются при условии, что они фармацевтически и фармакологически приемлемы. Примеры фармацевтической добавки включают связующее, стабилизатор, разрыхлитель, подкислитель, шипучее средство, искусственный подсластитель, отдушку, лубрикант, краситель, буферную добавку, антиоксидант и поверхностно-активное вещество.
Примеры связующего включают гуммиарабик, гидроксипропилметилцеллюлозу, гидроксипропилцеллюлозу и гидроксиэтилцеллюлозу.
Примеры стабилизатора включают желтый оксид железа(III), красный оксид железа(III) и черный оксид железа. В качестве стабилизатора для полиэтиленоксида фармацевтическая композиция для орального введения по настоящему изобретению, предпочтительно, содержит желтый оксид железа(III) и/или красный оксид железа(III). Когда стабилизатор добавляют в матрицу путем физического смешивания, содержание стабилизатора составляет, например, от 1% масс. до 20% масс. (от 3% масс. до 15% масс. в другом варианте осуществления настоящего изобретения) по отношению к массе препаративной формы. Например, содержание красного оксида железа(III) составляет, например, от 5% масс. до 20% масс. (от 10% масс. до 15% масс. в другом варианте осуществления настоящего изобретения) по отношению к массе препаративной формы. Содержание желтого оксида железа(III) составляет, например, от 1% масс. до 20% масс. (от 3% масс. до 10% масс. в другом варианте осуществления настоящего изобретения). Когда на стабилизатор наносят пленочное покрытие, то его содержание составляет, например, от 0,3% масс. до 2% масс. (от 0,5% масс. до 1,5% масс. в другом варианте осуществления настоящего изобретения) по отношению к массе таблетки. В этом случае концентрация желтого оксида железа(III) или красного оксида железа(III), содержавшегося в пленке, составляет, например, от 5% масс. до 50% масс. (от 10% масс. до 20% масс. в другом варианте осуществления настоящего изобретения). Термин "путем физического смешивания с матрицей" в данном описании означает, например, способ, при котором лекарственное средство, полиэтиленоксид и оксид(ы) железа(III) диспергированы равномерно и в итоге лекарственное средство и оксид железа(III), равномерно диспергированы в полиэтиленоксиде, как главной основе препаративной формы. Термин "пленочное покрытие" в данном описании означает, например, способ, в котором оксид(ы) железа(III) растворены или суспендированы в растворе водорастворимого полимера, таком как гидроксипропилметилцеллюлоза, а предварительно приготовленные таблетки покрывают указанным раствором или суспензией, чтобы сформировать тонкий слой. Желтый оксид железа(III) и/или красный оксид железа(III), которые могут использоваться по настоящему изобретению, могут находиться в любом месте препаративной формы. Оксид(ы) железа(III) могут, например, попасть в пленку при нанесении пленочного покрытия и т.п., в гранулы при гранулировании и т.п., в матрицу (например, располагаться вокруг полиэтиленоксида) и т.п.
Примеры разрыхлителя включают кукурузный крахмал, картофельный крахмал, кальциевое производное кармеллозы, натриевое производное кармеллозы и гидроксипропилцеллюлозу с низкой степенью замещения.
Примеры подкислителя включают лимонную кислоту, винную кислоту и яблочную кислоту.
Примеры шипучего вещества включают бикарбонат натрия.
Примеры искусственного подсластителя включают натриевое производное сахарина, динатрий глицирринат, аспартам, стевию и тауматин.
Примеры отдушки включают лимон, лимон-лайм, апельсин и ментол.
Примеры лубриканта включают стеарат магния, стеарат кальция, сложные эфиры сахарозы с жирными кислотами, полиэтиленгликоль, тальк и стеариновую кислоту.
Примеры красителя включают пищевой краситель желтый №4, пищевой краситель желтый №5, пищевой краситель красный №3, пищевой краситель красный №102 и пищевой краситель синий №3.
Примеры буферной добавки включают лимонную кислоту, янтарную кислоту, фумаровую кислоту, винную кислоту, аскорбиновую кислоту и их соли; глутаминовую кислоту, глутамин, глицин, аспарагиновую кислоту, аланин, аргинин и их соли; и оксид магния, оксид цинка, гидроксид магния, фосфорную кислоту, борную кислоту и их соли.
Примеры антиоксиданта включают аскорбиновую кислоту, дибутилгидрокситолуол и пропилгаллат.
Примеры поверхностно-активного вещества включают полисорбат 80, лаурилсульфат натрия и полиоксиэтилированное гидрированное касторовое масло.
Указанные фармацевтические добавки могут соответствующим образом добавляться индивидуально или же в виде комбинации двух или большего количества фармацевтических добавок в соответствующем количестве.
Каждая фармацевтическая добавка может содержаться в таком количестве, чтобы достигалось требуемое действие композиции по настоящему изобретению.
Примеры фармацевтической композиции (препаративной формы) по настоящему изобретению включают таблетки, которые готовят известным способом, например, многослойные таблетки, такие как двухслойная таблетка, в которой часть с модифицированным высвобождением и часть с немедленным высвобождением представляют собой тонкие слои, многослойную таблетку, в которой ламинированию подвергают множество частей с модифицированным высвобождением и частей с немедленным высвобождением, и трехслойную таблетку, в которой не содержащий лекарственного средства слой гидрофильного вещества и/или нерастворимого в воде вещества располагается в сэндвич-структуре между частью с модифицированным высвобождением и частью с немедленным высвобождением; таблетка, покрытая оболочкой сухим способом, в которой сердцевину таблетки образует часть с модифицированным высвобождением, а часть с немедленным высвобождением выполняет роль внешнего покрытия; и таблетка с пленочным покрытием, в которой часть с модифицированным высвобождением в качестве сердцевины покрыта частью с немедленным высвобождением путем нанесения пленочного покрытия.
Ниже подробно поясняется способ приготовления фармацевтической композиции по настоящему изобретению.
(1) Стадии измельчения и смешивания
Устройство и способ, которые используют на стадии измельчения, специально не ограничиваются, при условии, что лекарственные средства и соответствующие добавки можно фармацевтически измельчить. Примеры устройства включают молотковую мельницу, шаровую мельницу, вихревую мельницу и коллоидную мельницу. Условия измельчения можно выбрать соответствующим образом, и они специально не ограничиваются.
Устройство и способ, которые используют на стадии смешивания после проведения стадии измельчения, специально не ограничиваются, при условии, что компоненты могут быть однородно смешаны фармацевтически.
(2) Часть с модифицированным высвобождением: стадия гранулирования
Устройство и способ, которые используют на данной стадии, специально не ограничиваются, при условии, что полимер, образующий гидрогель, может быть гранулирован с использованием распыляемой жидкости.
Примеры гранулирования включают способ гранулирования при быстром перемешивании, способ гранулирования измельчением, способ гранулирования в псевдоожиженном слое, экструзионный способ гранулирования, способ гранулирования в барабане и способ гранулирования распылением; и устройства, которые используют в указанных способах. В другом варианте осуществления настоящего изобретения может применяться способ гранулирования в псевдоожиженном слое и соответствующее устройство, а в еще одном варианте осуществления настоящего изобретения может применяться способ гранулирования в псевдоожиженном слое в барабане и соответствующее устройство. Полученный гранулированный продукт можно высушить. Способ сушки специально не ограничивается, при условии, что гранулированный продукт можно высушить фармацевтически.
(3) Часть с немедленным высвобождением: стадия гранулирования
Устройство и способ, которые используют на данной стадии, специально не ограничиваются, при условии, что лекарственное средство может быть гранулировано с использованием распыляемой жидкости.
Примеры гранулирования включают способ гранулирования в псевдоожиженном слое, способ гранулирования в расплаве, способ гранулирования при быстром перемешивании, способ гранулирования измельчением, экструзионный способ гранулирования, способ гранулирования в барабане, способ гранулирования распылением и сухой способ гранулирования; и устройства, которые используют в указанных способах. В другом варианте осуществления настоящего изобретения может применяться способ гранулирования в псевдоожиженном слое.
В качестве связующего вещества, применяемого при влажном гранулировании, предпочтительным является гидрофильное вещество, и может использоваться, например, полиэтиленгликоль, мальтоза или поливинилпирролидон. Указанные связующие соединения могут использоваться индивидуально или в виде соответствующей комбинации двух или большего количества связующих.
Условия для приготовления используемой при распылении жидкости можно выбрать соответствующим образом, и они специально не ограничиваются.
Полученный гранулированный продукт можно высушить. Способ сушки специально не ограничивается, при условии, что гранулированный продукт можно высушить фармацевтически.
(4) Стадия формования
Устройство и способ, которые используют на данной стадии, специально не ограничиваются, при условии, что фармацевтическую композицию по настоящему изобретению можно формовать.
Примеры способа включают:
способ, при котором лекарственные средства и соответствующие добавки смешивают без гранулирования и сушки; и прессуют в форму, получая таблетки;
способ, при котором проводят стадию гранулирования, к полученному гранулированному продукту добавляют лубрикант и полученную смесь прессуют в форму, получая таблетки;
способ получения двухслойных таблеток путем прокатывания тонких слоев из части с модифицированным высвобождением и части с немедленным высвобождением;
способ получения многослойных таблеток путем прокатывания тонких слоев из множества частей с модифицированным высвобождением и частей с немедленным высвобождением;
способ получения многослойных таблеток путем размещения не содержащего лекарственного средства слоя между частью с модифицированным высвобождением и частью с немедленным высвобождением; и
способ получения таблеток, покрытых оболочкой сухим способом, у которых сердцевину таблетки образует часть с модифицированным высвобождением, а часть с немедленным высвобождением выполняет роль внешнего покрытия. В другом варианте осуществления настоящего изобретения может использовать способ получения двухслойных таблеток.
Примеры таблеточной машины включают многослойную роторную таблеточную машину и масляный пресс.
Условия для таблетирования, такие как давление при таблетировании, специально не ограничиваются, при условии, что могут быть получены двухслойные таблетки и многослойные таблетки. При приготовлении двухслойных таблеток гранулированное вещество для первого слоя и другое гранулированное вещество для второго слоя прокатывают в виде тонких слоев и сжимают в таблеточной машине под давлением от приблизительно 2 кН приблизительно до 20 кН с тем, чтобы получить двухслойные таблетки. В другом варианте осуществления настоящего изобретения гранулированное вещество для первого слоя сжимают в таблеточной машине под давлением от приблизительно 0,1 кН до приблизительно 10 кН, а другое гранулированное вещество для второго слоя наносят на первый слой и сжимают в таблеточной машине под давлением от приблизительно 2 кН до приблизительно 20 кН с тем, чтобы получить двухслойные таблетки. При изготовлении многослойных таблеток давление в таблеточной машине можно, соответственно, регулировать таким образом, чтобы осуществить сжатие.
Твердость полученной таблетки специально не ограничивается, при условии, что таблетка не повреждается в процессе ее изготовления и дальнейшего перемещения. Твердость может составлять, например, от 2 до 20 Н.
(5) Нанесение пленочного покрытия
После таблетирования на полученные таблетки может быть нанесено покрытие.
Способ нанесения пленочного покрытия специально не ограничивается, при условии, что на таблетки можно нанести покрытие фармацевтически. Примеры покрытия включают покрытие, которое наносят в установке барабанного типа, и покрытие, которое наносят методом окунания.
Вещества для нанесения покрытия можно добавлять отдельно или в виде сочетания соответствующего количества двух или нескольких веществ. Степень покрытия специально не ограничивается, при условии, что пленка может быть образована. Степень покрытия составляет, например, от 1% до 10%.
Когда на сердцевину в виде части с модифицированным высвобождением наносят покрытие из части с немедленным высвобождением, с целью получения таблеток с оболочкой, то жидкость для распыления, которую готовят, растворяя или диспергируя компоненты части с немедленным высвобождением в растворителе, таком как вода, можно нанести на сердцевину путем распыления и получить таблетки с покрытием. Степень покрытия специально не ограничивается, при условии, что может быть образована пленка из части с немедленным высвобождением. Степень покрытия составляет, например, от 1% до 20%.
После нанесения пленочного покрытия таблетки с оболочкой могут быть высушены. Способ сушки специально не ограничивается, при условии, что покрытые оболочкой таблетки могут быть фармацевтически высушены. Условия сушки специально не ограничиваются, при условии, что они выбраны соответствующим образом с учетом, например, стабильности препаративной формы. Начальное содержание воды после нанесения пленочного покрытия, предпочтительно, составляет от 0,1% до 2% в соответствии, например, с устойчивостью.
Фармацевтическая композиция для орального введения по настоящему изобретению может использоваться в качестве фармацевтической композиция для лечения симптомов со стороны нижних мочевыводящих путей, связанных с доброкачественной гиперплазией предстательной железы.
Способ изготовления фармацевтической композиции по настоящему изобретению специально не ограничивается, при условии, что требуемая фармацевтическая препаративная форма может быть получена путем соответствующего комбинирования вышеуказанных способов или известных способов.
ПРИМЕРЫ
Настоящее изобретение далее поясняется следующими примерами, сравнительными примерами и экспериментальными примерами, которые ни в коем случае его не ограничивают.
Пример 1
(1) Получение смешанного порошка для части с модифицированным высвобождением
Жидкость для распыления получают, растворяя 1,2 части макроголя 8000 в 4,8 частях воды при перемешивании и суспендируя 0,1 части предварительно измельченного гидрохлорида тамсулозина в указанном растворе. В реактор для гранулирования в псевдоожиженном слое помещают 8,8 частей макроголя 8000 и 50 частей полиэтиленоксида [POLYOX (зарегистрированная торговая марка) WSR-303, производитель - компания Dow] и проводят гранулирование путем распыления используемой при распылении жидкости. Полученный в результате гранулирования продукт сушат и 60,1 части высушенного гранулированного продукта смешивают с 0,3 частями стеарата магния, получая смешанный порошок для части с модифицированным высвобождением.
(2) Получение смешанного порошка для части с немедленным высвобождением
Жидкость для распыления получают, растворяя 1 часть мальтозы в 4 частях воды при перемешивании. Затем смешивают 0,6 частей солифенацина и 2,4 частей маннита и измельчают, а полученную измельченную смесь и 5,9 частей маннита помещают в реактор для гранулирования в псевдоожиженном слое и проводят гранулирование путем распыления используемой при распылении жидкости. Полученный в результате гранулирования продукт сушат и 9,9 части высушенного гранулированного продукта смешивают с 0,1 частями стеарата магния, получая смешанный порошок для части с немедленным высвобождением.
(3) Изготовление таблеток и нанесение покрытия
Полученный смешанный порошок для части с модифицированным высвобождением и полученный смешанный порошок для части с немедленным высвобождением формуют в виде таблеток с помощью многослойной роторной таблеточной машины и получают препаративную форму (двухслойные таблетки) по настоящему изобретению. На полученные двухслойные таблетки наносят покрытие способом распыления с помощью ранее приготовленной жидкости для распыления, которую получают путем растворения и диспегирования 5,04 частей гидроксипропилметилцеллюлозы, 0,95 частей макроголя 6000 и 1,26 части желтого оксида железа(III), и получают фармацевтическую композицию (таблетки с пленочным покрытием) по настоящему изобретению.
Экспериментальный пример 1
Фармацевтическую композицию, полученную в Примере 1, подвергают испытанию на растворимость, которое проводят в соответствии с тестом на растворимость, метод 2 (способ с использованием лопастной мешалки, 50 об/мин), описанным в Фармакопее Японии. Объем используемой при проведении испытания жидкости составляет 900 мл. Проценты растворения солифенацина по прошествии 15 мин, 30 мин и 60 мин с начала испытания приведены в таблице 1.
Пример 2
(1) Получение смешанного порошка для части с модифицированным высвобождением
Жидкость для распыления получают, растворяя 1,2 части макроголя 8000 в 4,8 частях воды при перемешивании и суспендируя 0,1 части предварительно измельченного гидрохлорида тамсулозина в указанном растворе. В реактор для гранулирования в псевдоожиженном слое помещают 8,8 частей макроголя 8000 и 50 частей полиэтиленоксида [POLYOX (зарегистрированная торговая марка) WSR-303, производитель - компания Dow] и проводят гранулирование путем распыления используемой при распылении жидкости. Полученный в результате гранулирования продукт сушат и 60,1 части высушенного гранулированного продукта смешивают с 0,3 частями стеарата магния, получая смешанный порошок для части с модифицированным высвобождением.
(2) Получение смешанного порошка для части с немедленным высвобождением
После того как смешивают и измельчают 0,6 частей солифенацина и 2,4 частей маннита, полученную измельченную смесь, 5,9 частей маннита, 1 часть PEG 8000 и 0,1 часть стеарат магния смешивают и получают смешанный порошок для части с немедленным высвобождением.
(3) Изготовление таблеток
Полученный смешанный порошок для части с модифицированным высвобождением и полученный смешанный порошок для части с немедленным высвобождением формуют в виде таблеток с помощью масляного пресса и получают фармацевтическую композицию (двухслойные таблетки) по настоящему изобретению.
Пример 3
(1) Получение смешанного порошка для части с модифицированным высвобождением
Жидкость для распыления получают, растворяя 1,2 части макроголя 8000 в 4,8 частях воды при перемешивании и суспендируя 0,1 части предварительно измельченного гидрохлорида тамсулозина в указанном растворе. В реактор для гранулирования в псевдоожиженном слое помещают 8,8 частей макроголя 8000 и 50 частей полиэтиленоксида [POLYOX (зарегистрированная торговая марка) WSR-303, производитель - компания Dow] и проводят гранулирование путем распыления используемой при распылении жидкости. Полученный в результате гранулирования продукт сушат и 60,1 части высушенного гранулированного продукта смешивают с 0,3 частями стеарата магния, получая смешанный порошок для части с модифицированным высвобождением.
(2) Получение смешанного порошка для части с немедленным высвобождением
Жидкость для распыления получают, растворяя 0,5 частей поливинилпирролидона (PVP K90) в 4 частях воды при перемешивании. Затем смешивают 0,6 частей солифенацина и 2,4 частей маннита, измельчают и полученную измельченную смесь и 6,4 частей маннита помещают в реактор для гранулирования в псевдоожиженном слое и проводят гранулирование путем распыления используемой при распылении жидкости. Полученный в результате гранулирования продукт сушат и 9,9 частей высушенного гранулированного продукта смешивают с 0,1 частями стеарата магния, получая смешанный порошок для части с немедленным высвобождением.
(3) Изготовление таблеток
Полученный смешанный порошок для части с модифицированным высвобождением и полученный смешанный порошок для части с немедленным высвобождением формуют в виде таблеток с помощью многослойной роторной таблеточной машины и получают фармацевтическую композицию (двухслойные таблетки) по настоящему изобретению.
Сравнительный пример 1
(1) Получение смешанного порошка для части с модифицированным высвобождением
Жидкость для распыления получают, растворяя 1,2 части макроголя 8000 в 4,8 частях воды при перемешивании и суспендируя 0,1 части предварительно измельченного гидрохлорида тамсулозина в указанном растворе. В реактор для гранулирования в псевдоожиженном слое помещают 8,8 частей макроголя 8000 и 50 частей полиэтиленоксида [POLYOX (зарегистрированная торговая марка) WSR-303, производитель - компания Dow] и проводят гранулирование путем распыления используемой при распылении жидкости. Полученный в результате гранулирования продукт сушат и 60,1 части высушенного гранулированного продукта смешивают с 0,3 частями стеарата магния, получая смешанный порошок для части с модифицированным высвобождением.
(2) Получение смешанного порошка для части с немедленным высвобождением
Жидкость для распыления получают, растворяя 204 части гидроксипропилметилцеллюлозы 2919 в 1836 частях воды при перемешивании. Затем смешивают 340 частей сукцината солифенацина и 1360 частей лактозы, измельчают и полученную измельченную смесь, 2125 частей лактозы и 1020 частей кукурузного крахмала помещают в реактор для гранулирования в псевдоожиженном слое и проводят гранулирование путем распыления используемой при распылении жидкости. Полученный в результате гранулирования продукт сушат и 1188 частей высушенного гранулированного продукта смешивают с 12 частями стеарата магния, получая смешанный порошок для части с немедленным высвобождением.
(3) Изготовление таблеток
Полученный смешанный порошок для части с модифицированным высвобождением и полученный смешанный порошок для части с немедленным высвобождением формуют в виде таблеток с помощью масляного пресса и получают фармацевтическую композицию (двухслойные таблетки) для сравнения.
Экспериментальный пример 2
Фармацевтическую композицию, полученную в примерах 1-3 и сравнительном примере 1, подвергают испытанию на растворимость, которое проводят в соответствии с тестом на растворимость, метод 1 (барабанный способ, 100 об/мин), описанным в Фармакопее Японии. Объем используемой при проведении испытания жидкости составляет 900 мл. Степени растворения солифенацина по прошествии 15 мин, 30 мин и 60 мин с начала проведения испытания приведены в таблице 2.
Применимость в промышленности
В настоящем изобретении предлагается фармацевтическая композиция для орального введения, которая включает часть с модифицированным высвобождением, содержащую тамсулозин или его фармацевтически приемлемую соль, и часть с немедленным высвобождением, содержащую солифенацин или его фармацевтически приемлемую соль. Фармацевтическая композиция по настоящему изобретению демонстрирует степень высвобождения, аналогичную степени высвобождения известных препаративных форм, содержащих одно лекарственное средство, и, таким образом, может применяться как способ приготовления препаративной формы, который позволяет получить единую препаративную форму (т.е. объединенную препаративную форму), способную оказывать фармакологическое действие, эквивалентное фармакологическому действию известных препаративных форм, которые содержат одно лекарственное средство.
Как указано выше, настоящее изобретение поясняется со ссылкой на конкретные варианты его осуществления, однако модификации и улучшения, очевидные специалистам из области техники, входят в объем настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ МОДИФИЦИРОВАННОГО ВЫСВОБОЖДЕНИЯ | 2009 |
|
RU2495666C2 |
| СОСТАВ С ДЛИТЕЛЬНЫМ ВЫСВОБОЖДЕНИЕМ ДЛЯ СНИЖЕНИЯ ЧАСТОТЫ МОЧЕИСПУСКАНИЯ И СПОСОБ ЕГО ПРИМЕНЕНИЯ | 2013 |
|
RU2599017C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ КОНТРОЛИРУЕМОГО ВЫСВОБОЖДЕНИЯ АКТИВНЫХ ВЕЩЕСТВ И СПОСОБ ЕЕ ИЗГОТОВЛЕНИЯ | 2003 |
|
RU2291711C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ДИАЦЕРЕИН | 2009 |
|
RU2542461C2 |
| ПРЕПАРАТИВНЫЕ ФОРМЫ ГИДРОКОДОНА С КОНТРОЛИРУЕМЫМ ВЫСВОБОЖДЕНИЕМ | 2000 |
|
RU2230556C2 |
| НОВАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ТАМСУЛОЗИНА И ДУТАСТЕРИДА | 2019 |
|
RU2795928C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ КОНТРОЛИРУЕМОГО ВЫСВОБОЖДЕНИЯ АКТИВНЫХ ВЕЩЕСТВ (ВАРИАНТЫ) И СПОСОБ ЕЕ ПОЛУЧЕНИЯ (ВАРИАНТЫ) | 1998 |
|
RU2179017C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ АКОТИАМИДА И ИНГИБИТОРА ПРОТОННОЙ ПОМПЫ | 2019 |
|
RU2820820C2 |
| КОМПОЗИЦИИ ГИДРОКОДОНА С КОНТРОЛИРУЕМЫМ ВЫСВОБОЖДЕНИЕМ | 2001 |
|
RU2253452C2 |
| ПРЕПАРАТ ДЛИТЕЛЬНОГО ВЫСВОБОЖДЕНИЯ ДЛЯ СНИЖЕНИЯ ЧАСТОТЫ МОЧЕИСПУСКАНИЯ И СПОСОБ ЕГО ПРИМЕНЕНИЯ | 2013 |
|
RU2669565C2 |
Изобретение относится к фармацевтике. Описана двуслойная таблетка для орального применения, включающая тонкий слой с замедленным высвобождением и тонкий слой с немедленным высвобождением. Слой с замедленным высвобождением содержит тамсулозин или его фармацевтически приемлемую соль, полимер, который образует гидрогель, и полиэтиленгликоль с молекулярным весом 8000. Слой с немедленным высвобождением включает солифенацин или его фармацевтически приемлемую соль и гидрофильное вещество. Гидрофильное вещество выбрано из группы, состоящей из D-маннита, мальтозы, полиэтиленгликоля и поливилпирролидона. Указанное гидрофильное вещество используется в качестве связующего вещества и его содержание составляет от 5 до 10 мас.% от массы части с немедленным высвобождением. Изобретение обеспечивает получение фармацевтической композиции в виде единой препаративной формы (т.е. объединенной препаративной формы), обладающей биодоступностью, которая эквивалентна биодоступности известных препаративных форм с одним лекарственным средством. 13 з.п. ф-лы, 3 ил., 3 пр., 2 табл.
1. Двуслойная таблетка для орального введения, содержащая
(1) тонкий слой, включающий часть с замедленным высвобождением, которая содержит тамсулозин или его фармацевтически приемлемую соль, полимер, который образует гидрогель, и полиэтиленгликоль с молекулярным весом 8000, и
(2) тонкий слой, включающий часть с немедленным высвобождением, которая включает солифенацин или его фармацевтически приемлемую соль, и, по меньшей мере, одно гидрофильное вещество, выбранное из группы, состоящей из D-маннита, мальтозы, полиэтиленгликоля и поливилпирролидона, где 85% или более солифенацина растворяется через 15 мин, в соответствии с тестом на растворимость, способом 1 (барабанный способ, скорость вращения 100 об/мин), описанным в Фармакопее Японии, где одно гидрофильное вещество или два или более гидрофильных вещества выбранных из группы, состоящей из полиэтиленгликоля, мальтозы и поливинилпирролидона используются в качестве связующих веществ и содержание связующего составляет от 5 до 10 мас.% от массы части с немедленным высвобождением, где полимером, образующим гидрогель, является полиэтиленоксид.
2. Двуслойная таблетка для орального введения по п. 1, где часть с немедленным высвобождением распадается и/или растворяется прежде, чем часть с замедленным высвобождением образует гель.
3. Двуслойная таблетка для орального введения по п. 1, где 90% или более солифенацина растворяется через 60 мин.
4. Двуслойная таблетка для орального введения по п. 1, где 85% или более солифенацина растворяется через 30 мин и 90% или более солифенацина растворяется через 60 мин.
5. Двуслойная таблетка для орального введения по п. 1, где гидрофильное вещество составляет от 5 мас.% до 99 мас.%.
6. Двуслойная таблетка для орального введения по п. 1, где гидрофильным веществом, используемым в качестве связующего вещества, является мальтоза.
7. Двуслойная таблетка для орального введения по п. 1, где маннит в качестве гидрофильного вещества используют в качестве наполнителя.
8. Двуслойная таблетка для орального введения по п. 1, где средневязкостная молекулярная масса полиэтиленоксида составляет 5000000 или более.
9. Двуслойная таблетка для орального введения по п. 1, где полимер, образующий гидрогель, составляет от 5 мас.% до 95 мас.%.
10. Двуслойная таблетка для орального введения по п. 1, где полиэтиленгликоль в части с замедленным высвобождением представляет собой добавку, которая позволяет воде проникать в препаративную форму.
11. Двуслойная таблетка для орального введения по п. 10, где полиэтиленгликоль имеет такую растворимость, что количество воды, необходимое для растворения 1 г указанной добавки, составляет 5 мл или менее.
12. Двуслойная таблетка для орального введения по п. 11, где количество полиэтиленгликоля составляет от 3 мас.% до 80 мас.%.
13. Двуслойная таблетка для орального введения по п. 1, которая представляет собой таблетку для лечения симптомов со стороны нижних мочевыводящих путей, связанных с доброкачественной гиперплазией предстательной железы.
14. Двуслойная таблетка для орального введения по п. 1, где солифенацин или его фармацевтически приемлемая соль представляет собой солифенацина сукцинат.
| VESICARE (инструкция по применению препарата VESICARE, 2005 Astellas Pharma US, Inc., 01.2008) | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| EA 200401369 A1, 30.06.2005 | |||
| Theo Dekker "Качество лекарственных средств, GMP и биоэквивалентность | |||
| Сравнительные исследования растворения и заявки", ВОЗ, Киев, 3-7 октября 2005 г | |||
| Pharmaceutical Excipients | |||
| London: | |||

