СМЕЖНЫЕ ЗАЯВКИ
Данная заявка объявляет приоритет по отношению к предварительной заявке США 61/410 003, поданной 4 ноября 2010 г., и патентной заявке США 13/282 658, поданной 27 октября 2011 г.
ОБЛАСТЬ ПРИМЕНЕНИЯ ИЗОБРЕТЕНИЯ
Данное изобретение относится, по меньшей мере в одном из вариантов осуществления, к способу формирования силиконового гидрогеля, служащего материалом для контактных линз.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Силиконовый гидрогель обычно образуется из силиконовых мономеров или макромеров и гидрофильных мономеров. Во многих случаях смеси желаемых силиконовых мономеров с гидрофильными мономерами не обладают взаимной смешиваемостью. Такие смеси непрозрачны и не могут быть использованы для изготовления материалов, пригодных для применения в качестве контактных линз.
Одно из решений проблемы несмешивающихся смесей подразумевает использование специальных разбавителей, включая вторичные и четвертичные спирты, а также разбавителей со сбалансированными параметрами растворимости и образования водородных связей.
Предыдущие попытки разрешить проблему несмешиваемости опирались на то, чтобы добиться совместимости нерастворимых друг в друге компонентов таким образом, чтобы можно было получить оптически прозрачные линзы. Обычно эти попытки не сказывались благоприятным образом на других характеристиках реакционных смесей, таких как вязкость, скорость отверждения или механические свойства получающихся линз.
ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Изобретение включает в одной из его форм способ формирования силиконовых гидрогелевых материалов для контактных линз путем отверждения реакционной смеси, которая включает по меньшей мере один гидрофильный компонент, один силиконовый компонент и одну боратную добавку. В некоторых случаях использование боратной добавки повышает вязкость смеси или уменьшает время гелеобразования, необходимое для отверждения смеси, при сохранении оптической прозрачности смеси.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Данное изобретение раскрывается со ссылками на сопутствующие чертежи.
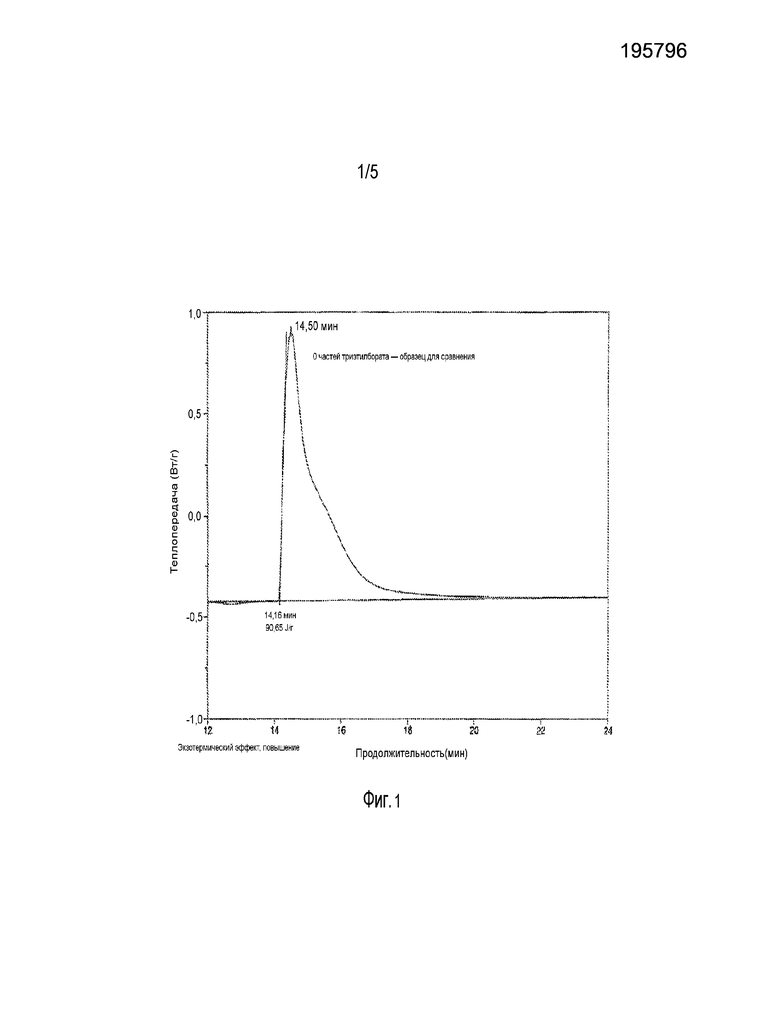
ФИГ. 1 представляет поток тепла при отверждении контрольного образца.
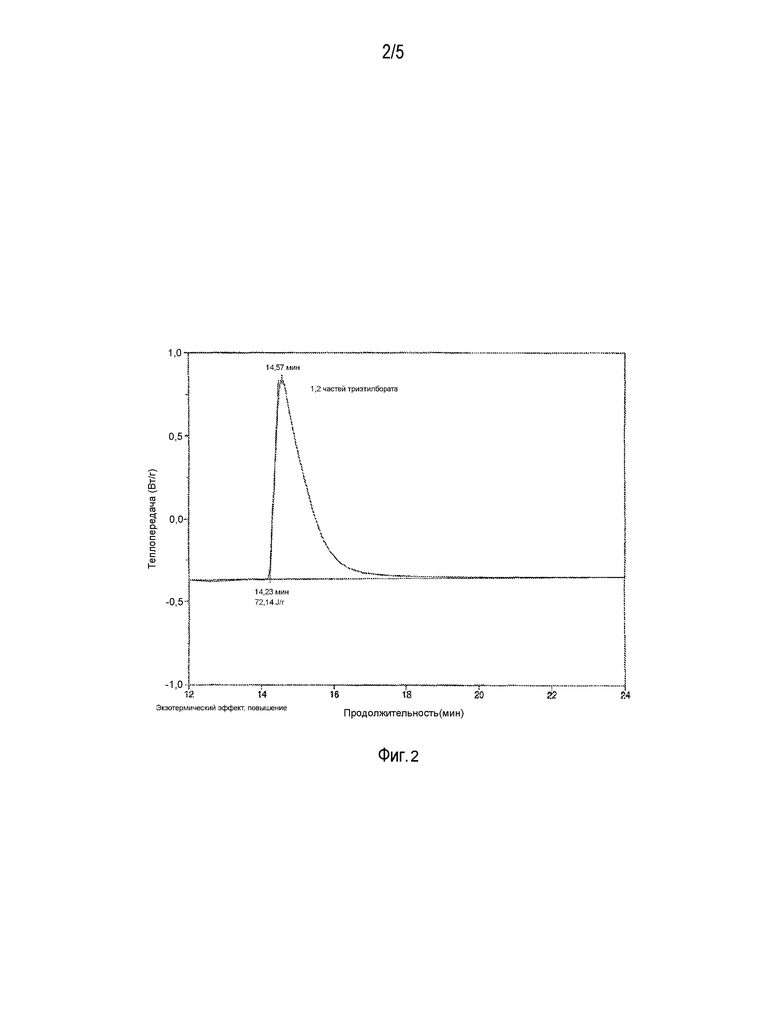
ФИГ. 2 представляет поток тепла при отверждении образца, обработанного боратом.
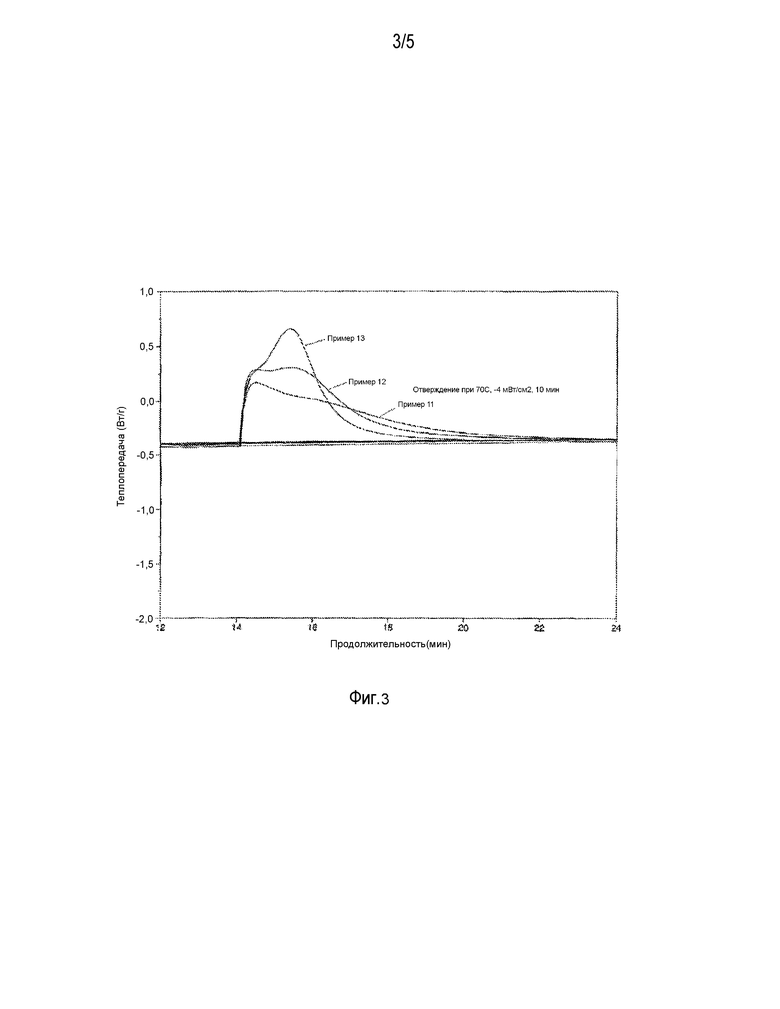
ФИГ. 3 представляет поток тепла для примеров с 11 по 13.
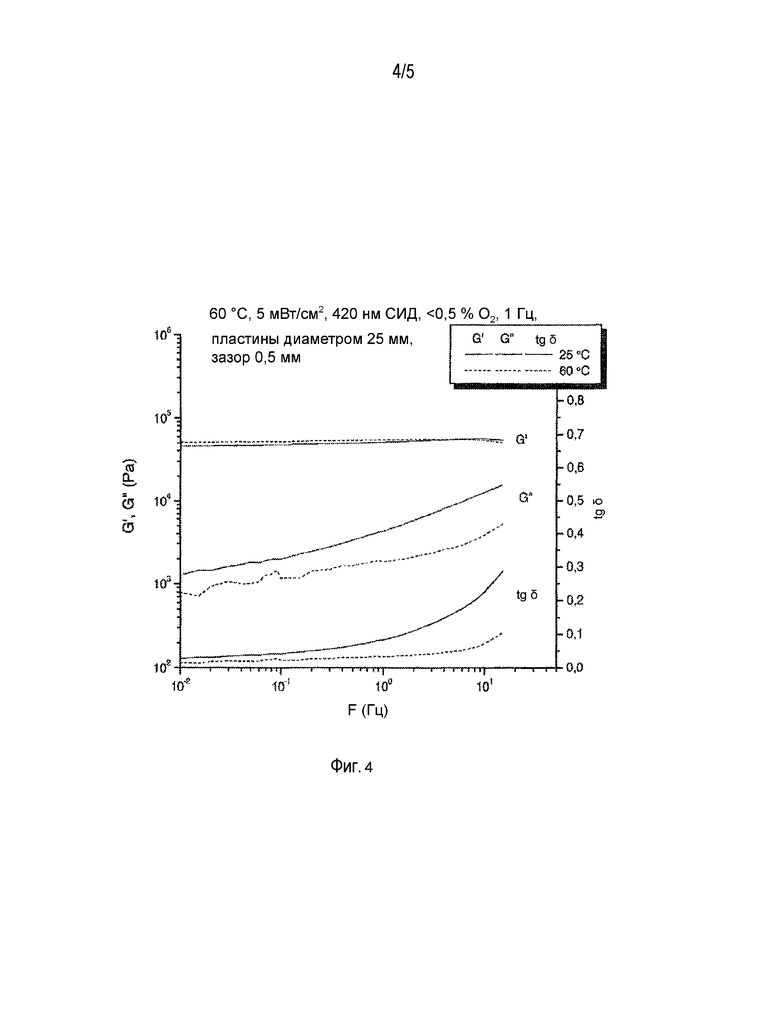
ФИГ. 4 - динамический механический анализ смеси 17.
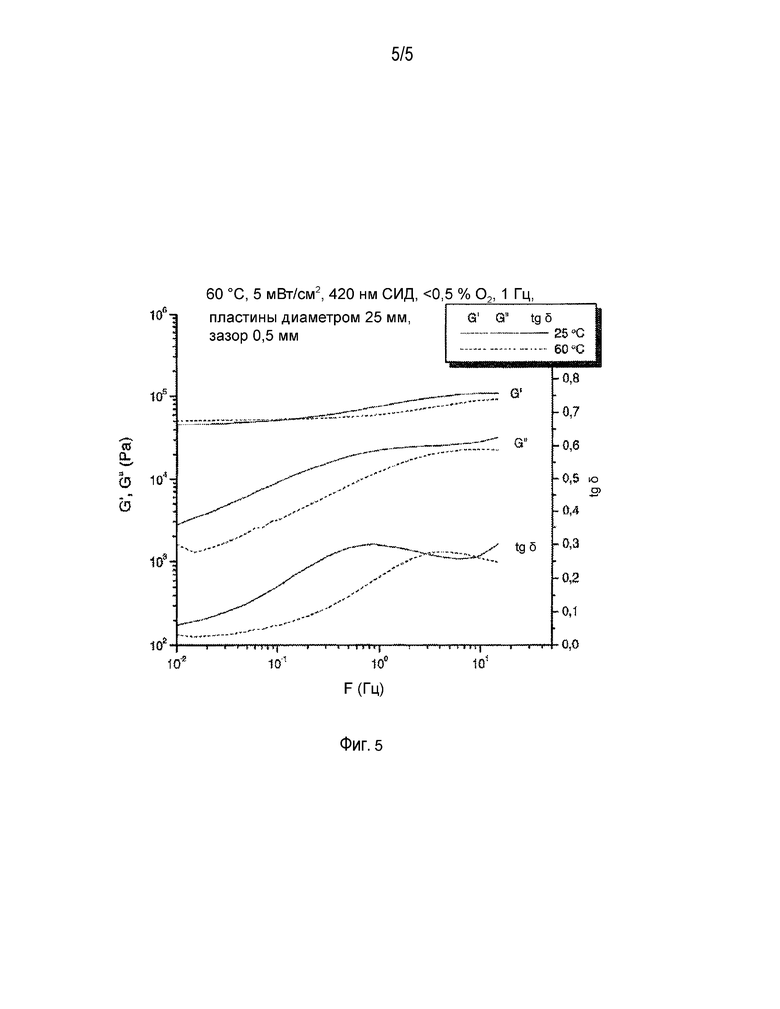
ФИГ. 5 - динамический механический анализ смеси 20.
Для указания аналогичных элементов на разных изображениях используются аналогичные цифровые обозначения. Приведенные примеры иллюстрируют несколько вариантов осуществления данного изобретения, но не должны никоим образом рассматриваться как ограничивающие область изобретения.
ПОДРОБНОЕ ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНОГО ВАРИАНТА ОСУЩЕСТВЛЕНИЯ
Использование боратосодержащих разбавителей было описано при образовании обычных гидрогелей, не содержащих силикона. Например, см. патенты США № 4889664 и 5039459. Однако было неожиданно обнаружено, что бораты можно использовать в смесях, служащих исходными материалами для получения силиконовых гидрогелей, и что такие составы благотворным образом меняют некоторые свойства исходных смесей (вязкость и скорость отверждения) и положительно сказываются на свойствах получающихся линз, таких как оптическая прозрачность.
Изобретение в целом относится к способу формирования силиконового гидрогелевого материала, который включает полимеризующийся гидрофильный компонент, полимеризующийся силиконовый компонент и добавку, обладающую свойствами сложного борного эфира. По меньшей мере один из компонентов включает гидроксильную группу, способную формировать борноэфирную группу.
Используемый в настоящем документе термин «реакционная смесь» означает смесь компонентов, включая реакционно-способные компоненты (такие как мономеры и макромеры), разбавитель, инициаторы, сшивающие агенты и добавки, которые при помещении в условия полимеризации образуют полимер. Реакционно-способными являются те компоненты реакционной смеси, которые в процессе полимеризации становятся неотъемлемой частью полимера либо путем образования химических связей, либо путем механической фиксации в полимерной матрице. Например, реакционно-способные компоненты становятся частью полимера посредством полимеризации, тогда как внутренние смачивающие вещества без полимеризующихся групп, такие как ПВП (поливинилпирролидон), становятся частью полимера путем включения в физическую структуру. Разбавитель и любые иные дополнительные технологические добавки не встраиваются в структуру полимера и не являются частью реакционно-способных компонентов. Такие компоненты удаляются в ходе технологического процесса. Применение таких материалов для производства силиконовых гидрогелей подразумевает использование при изготовлении контактных линз, линз-повязок, интраокулярных линз, а также различных медицинских устройств.
Гидрогель представляет собой гидрированную, поперечно сшитую полимерную систему, которая содержит воду в равновесном состоянии. Когда гидрогели, описанные в данном изобретении, используются для образования контактных линз, они поглощают около 10% весовых воды.
Под «биомедицинским устройством» или «медицинским устройством» в настоящем документе подразумевается любое изделие, предназначенное для использования внутри и/или на поверхности ткани или жидкости организма млекопитающих. Примеры подобных устройств, дополнительно, включают: раневые повязки, катетеры, имплантаты, стенты и офтальмологические устройства, такие как интраокулярные линзы и контактные линзы. В одном из вариантов осуществления биомедицинские устройства представляют собой офтальмологические устройства, в частности контактные линзы, чаще всего контактные линзы, сформированные из силиконового гидрогеля.
Под «контакными линзами» подразумевают офтальмологические устройства, расположенные в глазу или на его поверхности. Подобные устройства могут обеспечивать оптическую коррекцию, применяться в косметических целях, использоваться для блокирования УФ-излучения, ослабления видимого света и защиты от бликов, давать терапевтический эффект, включая залечивание ран, доставку лекарственных препаратов и биологически активных веществ, использоваться в диагностических целях или для контроля текущего состояния пациента, а также обеспечивать перечисленные выше функции в различных сочетаниях. Термин «линза» включает, помимо прочего, мягкие контактные линзы, жесткие контактные линзы, интраокулярные линзы, накладные линзы, офтальмологические вкладыши и оптические вкладыши.
Выражение «полимеризуемые группы» относится к группам, способным к реакциям свободнорадикальной и (или) катионной полимеризации. Характерные, но не ограничивающие примеры свободнорадикальных реакционно-способных групп включают (мет)акрилаты, стирилы, винилы, виниловые эфиры, C1-6алкил(мет)акрилаты, (мет)акриламиды, C1-6алкил(мет)акриламиды, N-виниллактамы, N-виниламиды, C2-12алкенилы, C2-12алкенилфенилы, C2-12алкенилнафтилы, C2-6алкенилфенил-C1-6алкилы, O-винилкарбаматы и O-винилкарбонаты. Характерные, но не ограничивающие примеры катионных реакционно-способных групп включают винилэфирные или эпоксидные группы, а также их смеси. В одном из вариантов осуществления свободнорадикальные реакционно-способные группы включают (мет)акрилаты, акрилоксигруппы, (мет)акриламиды, а также их смеси.
В данных технических условиях термин «(мет)» означает возможность замещения метильным радикалом. Так, термин «(мет)акрилат» относится одновременно и к метакриловому и к акриловому радикалам.
В данных технических условиях термин «борат» относится к сложному эфиру борной кислоты и спирта и включает структуру -В-O-С-. Борат не становится постоянной частью полимерной матрицы (например, хотя он может реагировать с гидроксилсодержащими составляющими реакционной смеси, эти связи подвижны и легко гидролизуются, когда полимер контактирует с водой). Напротив, боратная группа является частью системы разбавления и не становится постоянной частью готовой линзы. Бораты, как правило, удаляются из полимеризованного материала до начала коммерческого использования. Примеры боратов включают сложные эфиры борной кислоты, такие как триметилборат, триэтилборат, три-н-пропилборат, триизопропилборат, трибутилборат, тритербутилборат и триГЭМАборат (три-(2-гидроксиэтилметакрилат)борат). Сложные эфиры борной кислоты обычно отвечают формуле:
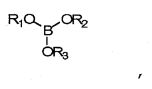
где R1, R2 и R3 - углеводородные остатки. В одном из вариантов осуществления углеводородные остатки - это одновалентные алкильные или арильные группы. В другом варианте осуществления две из этих групп являются двухвалентными и образуют друг с другом ковалентную связь, образуя тем самым циклический борный эфир. В одном из вариантов осуществления три три углеводородные группы идентичны. В другом варианте три углеводородные группы независимо определяются и, таким образом, не являются идентичными. В еще одном варианте исполнения по меньшей мере одна из углеводородных групп представляет собой полимеризующийся компонент, например если R-группа сложного эфира борной кислоты происходит из гидроксильно-функционального реакционно-способного мономера, такого как ГЭМА (2-гидроксиэтилметакрилат), образуется моно-, ди- или триГЭМАборат. В другом варианте R1, R2 и R3 имеют независимое происхождение и являются остатками монофункциональных спиртов.
В другом варианте осуществления по меньшей мере один из углеводородов может представлять собой полимеризующийся компонент, используемый при образовании силиконового гидрогеля. После того как информация, раскрытая в данных технических условиях, будет использована на благо пользователей, открывается возможность использования других источников боратов, которые можно рассматривать для использования в данном изобретении. В некоторых вариантах желательно добавлять бораты в таких концентрациях, которые не приводят к появлению свободных боратов в реакционной смеси. Таким образом, в некоторых вариантах осуществления подходящие количества боратов могут быть определены на основании молярных соотношений путем вычисления мольной доли (в процентах) гидроксильных групп, в которых водород был временно замещен боратом, по отношению к общему количеству гидроксильных групп и групп с боратными заместителями и включают примерно от 5 мольных % ОН (мольная процентная доля функциональных групп -В-О-С- по отношению к сочетанию групп -С-O-H и -B-O-C-) вплоть до почти 100 мольных % OH, исходя из всех гидроксильных ионов в реакционной смеси и разбавителе, а в других вариантах осуществления эта доля составляет от 10 до 80% ОН, а еще в других вариантах осуществления она составляет от 15 до 70%. Мольный % OH, необходимый для реакции, меняется от системы к системе. Мольный % OH не должен быть настолько велик, чтобы реакционная смесь превратилась в гель до полимеризации. Например, в системах, содержащих макромеры полигидроксила, такие как HFM, с добавлением компонентов, содержащих малое количество моногидроксильных компонентов или вообще их не содержащих (таких как ГЭМА), % OH около 25% вызывает нежелательное гелеобразование. Мольный % OH в этих системах соответственно ниже 25% OH, и в некоторых системах мольная доля ОН составляет от 3 до 20% OH. В системах, содержащих значительное количество моногидроксильного компонента, мольная доля (%) OH может достигать 100%. Пример расчета включен в параграф [0081] данного документа.
В данном документе запись -C-OH или -C-O-H относится к гидроксильной группе, присоединенной к атому углерода в молекуле, следовательно, при этом исключаются группы -ОН, входящие в состав воды. Подобным же образом -B-O-C- обозначает группу сложного эфира борной кислоты, содержащуюся в молекуле. В целом рассчитывается мольная концентрация функциональных групп -B-O-C- на грамм смеси. Подобным же образом рассчитывается количество функциональных групп -C-O-H (включая миллимоли -B-O-C-) на грамм смеси. Разделив количество миллимолей -B-O- на общее количество миллимолей O-H и -B-O-C- («ммоль OH») и умножая полученное значение на 100, получаем мольный % OH.
В данных технических условиях фраза «гидрофильный компонент» относится к мономеру или макромеру, который, будучи полимеризован со сшивающим компонентом, содержит не меньше 10% воды. К примерам дополнительных гидрофильных мономеров относятся: ГЭМА (2-гидроксиэтилметакрилат), ДМА (N,N-диметилакриламид), ГМА (глицеринмонометакрилат), 2-гидроксиэтилметакриламид, полиэтиленгликольмонометакрилат, метакриловая кислота, акриловая кислота, N-винилпирролидон, N-винил-N-метилацетамид, N-винил-N-этилацетамид, N-винил-N-этилформамид, N-винилформамид, их сочетания и тому подобные соединения. Другие примеры гидрофильных компонентов приводятся в патенте США № 6822016, содержание которого считается включенным в данный документ посредством упоминания. После того как специалисты ознакомятся с данными техническими условиями, они смогут выявить другие гидрофильные компоненты и использовать их при реализации данного изобретения. Приемлемые количества гидрофильного компонента составляют от 5 до 80%.
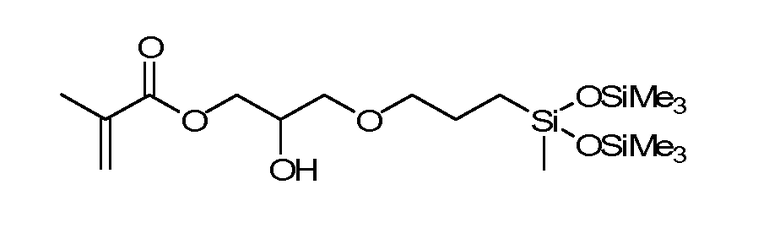
В данных технических условиях фраза «силиконовый компонент» относится к мономеру или макромеру, содержащему силоксан. Примеры включают реакционно-способные полидиалкилсилоксаны, такие как монометакрилоксипропил- и моно-C1-C5 алкилполидиметилсилоксан. К числу подходящих примеров относятся: полидиметилсилоксан с концевыми монометакрилооксипропильными и монометильными группами, полидиметилсилоксан с концевыми монометакрилооксипропильными и моно-н-бутильными группами с молекулярной массой 800-1000 или OH-mPDMS - полиметилдисилоксан с концевыми моно-(3-метаклилокси-2-гидроксипропилокси)пропильными и монобутильными группами.
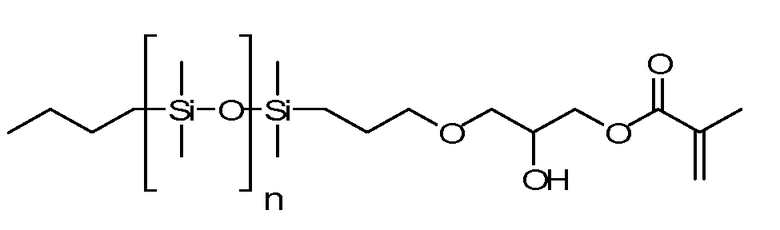
n равно от 1 до 200.
Другие примеры силиконов включают
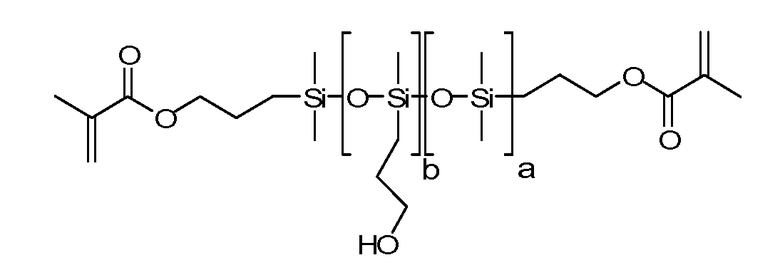
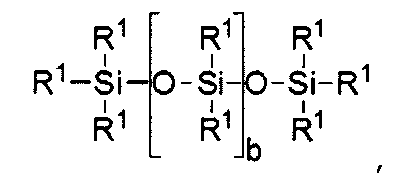
где а равно 10-500 и b равно 1-150.
Другие примеры силиконовых компонентов включают в себя 2-акриловую кислоту, 2-метил-, 2-гидрокси-3-[3-[1,3,3,3-тетраметил-1-[(триметилсилил)окси]дисилоксанил]пропокси]пропиловый эфир (SiGMA).
Прочие, пригодные для целей настоящего изобретения силиконовые компоненты включают соединения, отвечающие формуле I
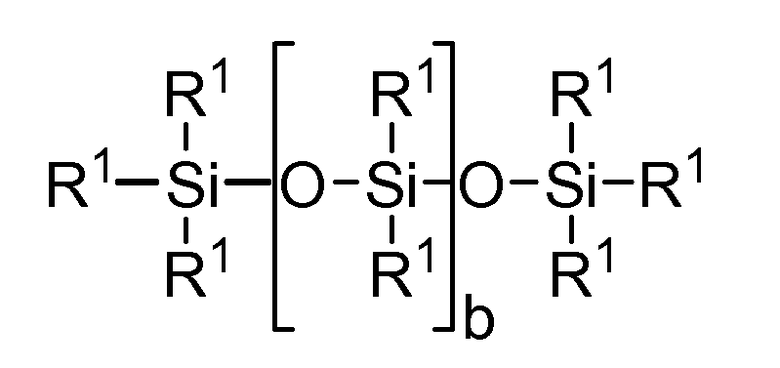
где
R1 независимо выбирают из группы, включающей одновалентные реакционно-способные группы, одновалентные алкильные группы или одновалентные арильные группы, причем каждая из перечисленных химических групп может далее иметь в своем составе функциональные группы, выбираемые из следующего ряда: гидрокси, амино, окса, карбокси, алкилкарбокси, алкокси, амидо, карбамат, карбонат, галоген, а также их различные комбинации. Одновалентные силоксановые цепи имеют в своем составе 1-100 повторяющихся Si-O блоков и могут далее иметь в своем составе функциональные группы, выбираемые из следующего ряда: алкил, гидрокси, амино, окса, карбокси, алкилкарбокси, алкокси, амидо, карбамат, галоген, а также их различные комбинации,
где b = от 0 до 500, причем подразумевается, что если b отлично от нуля 0, то по b имеется распределение с модой, равной указанному значению;
причем по крайней мере один фрагмент R1 представляет собой одновалентную реакционно-способную группу, а в некоторых реализациях настоящего изобретения от одного до трех фрагментов R1 представляют собой одновалентные реакционно-способные группы.
Используемый в настоящей заявке термин «одновалентные реакционно-способные группы» относится к группам, способным к реакциям свободнорадикальной и/или катионной полимеризации. Характерные, но не ограничивающие примеры свободнорадикальных реакционно-способных групп включают: (мет)акрилаты, стирилы, винилы, виниловые эфиры, C1-6алкил(мет)акрилаты, (мет)акриламиды, C1-6алкил(мет)акриламиды, N-виниллактамы, N-виниламиды, C2-12алкенилы, C2-12алкенилфенилы, C2-12алкенилнафтилы, C2-6алкенилфенил-C1-6алкилы, O-винилкарбаматы и O-винилкарбонаты. Характерные, но не ограничивающие примеры катионных реакционно-способных групп включают винилэфирные или эпоксидные группы, а также их смеси. В одном из вариантов осуществления свободнорадикальные реакционно-способные группы включают (мет)акрилаты, акрилоксигруппы, (мет)акриламиды, а также их смеси.
Соответствующие целям настоящего изобретения одновалентные алкильные и арильные группы включают незамещенные одновалентные C1-C16 алкильные группы, C6-C14 арильные группы, такие как замещенные и незамещенные метил, этил, пропил, бутил, 2-гидроксипропил, пропоксипропил, полиэтиленоксипропил, а также их различные комбинации и т.д.
В одной реализации настоящего изобретения b равно нулю, один фрагмент R1 представляет собой одновалентную реакционно-способную группу, по крайней мере три фрагмента R1 выбраны из одновалентных алкильных групп, содержащих от одного до 16 атомов углерода, в другой реализации - из одновалентных алкильных групп, содержащих от одного до 6 атомов углерода. Другие примеры силиконовых компонентов, упомянутых в данном варианте осуществления, включают в себя в том числе 2-метил-, 2-гидрокси-3-[3-[1,3,3,3-тетраметил-1-[(триметилсилил)окси]дисилоксанил]пропокси]пропиловый эфир (SiGMA), 2-гидрокси-3-метакрилоксипропилоксипропил-трис(триметилсилокси)силан, 3-метакрилоксипропилтрис(триметилсилокси) силан (TRIS), 3-метакрилоксипропилбис(триметилсилокси)метилсилан и 3-метакрилоксипропилпентаметилдисилоксан.
В других вариантах осуществления b составляет от 2 до 20, от 3 до 15 или в некоторых вариантах осуществления от 3 до 10. По меньшей мере один концевой фрагмент R1 представляет собой одновалентную реакционно-способную группу, а остальные группы R1 выбраны из одновалентных алкильных групп, содержащих от 1 до 16 атомов углерода, а в другом варианте осуществления - из одновалентных алкильных групп, содержащих от 1 до 6 атомов углерода. В еще одной реализации настоящего изобретения b находится в диапазоне от 3 до 15, один концевой фрагмент R1 представляет собой одновалентную реакционно-способную группу, другой концевой фрагмент R1 представляет собой одновалентную алкильную группу, содержащую от одного до 6 атомов углерода, а остальные фрагменты R1 представляет собой одновалентные алкильные группы, содержащие от 1 до 3 атомов углерода. Характерные, но не ограничивающие примеры содержащих силикон компонентов такой реализации настоящего изобретения включают (полидиметилсилоксан (МВ 400-1000) с концевой моно-(2-гидрокси-3-метакрилоксипропил)-пропил эфирной группой) (OH-mPDMS), (полидиметилсилоксаны (МВ 800-1000) с концевыми моно-н-бутильными и концевыми монометакрилоксипропильными группами) (mPDMS).
В другой реализации настоящего изобретения b находится в диапазоне от 5 до 400 или от 10 до 300, оба концевых фрагмента R1 представляют собой одновалентные реакционно-способные группы, а остальные фрагменты R1 независимо выбираются из одновалентных алкильных групп, содержащих от одного до 18 атомов углерода, которые могут иметь эфирные мостиковые группы между атомами углерода и могут также включать атомы галогенов.
В другой реализации настоящего изобретения от одного до четырех фрагментов R1 представляют собой винилкарбонат или карбамат со следующей формулой:
Формула II
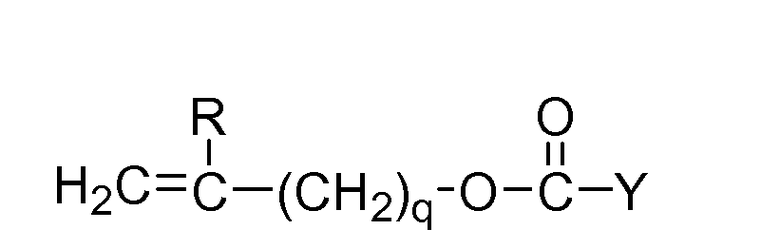
где Y означает O-, S- или NH-;
R означает водород или метил; и q равен 0 или 1.
К числу содержащих силикон-винилкарбонатных или винилкарбаматных мономеров относятся: 1,3-бис[4-(винилоксикарбонилокси)бут-1-ил]тетраметилдисилоксан; 3-(винилкарбонилтио) пропил-[трис (триметилсилокси)силан]; 3-[трис(триметилсилокси)силил] пропил аллилкарбамат; 3-[трис(триметилсилокси)силил] пропилвинилкарбамат; триметилсилилэтилвинилкарбонат; триметилсилилметилвинилкарбамат. Формула III:
Если необходимы биомедицинские устройства с модулем упругости менее 200, только один из фрагментов R1 должен представлять собой одновалентную реакционно-способную группу и не более двух из остальных фрагментов R1 должны представлять собой одновалентные силоксановые группы.
В одном из вариантов реализации изобретения, в котором необходимы силикон-гидрогелевые линзы, линза в соответствии с настоящим изобретением будет изготовлена из реакционной смеси, содержащей по крайней мере 20% по массе, а в некоторых вариантах - приблизительно от 20 до 70% по массе содержащих силикон компонентов в пересчете на общую массу реакционно-способных компонентов, из которых выполнен полимер. Другой класс содержащих силикон компонентов включает в себя полиуретановые макромеры со следующими формулами:
Формулы IV-VI
(*D*A*D*G)a *D*D*E1;
E(*D*G*D*A)a *D*G*D*E1 или
E(*D*A*D*G)a *D*A*D*E1,
где:
D обозначает алкильный бирадикал, алкилциклоалкильный бирадикал, циклоалкильный бирадикал, арильный бирадикал или алкиларильный бирадикал, содержащий от 6 до 30 атомов углерода;
G обозначает алкильный бирадикал, циклоалкильный бирадикал, алкилциклоалкильный бирадикал, арильный бирадикал или алкиларильный бирадикал, содержащий от 1 до 40 атомов углерода, который может иметь в основной цепи эфирные, тиоэфирные или аминовые мостиковые группы;
* обозначает уретановую или уреидо-мостиковую группу;
a равен по крайней мере 1;
A обозначает дивалентный полимерный радикал со следующей формулой:
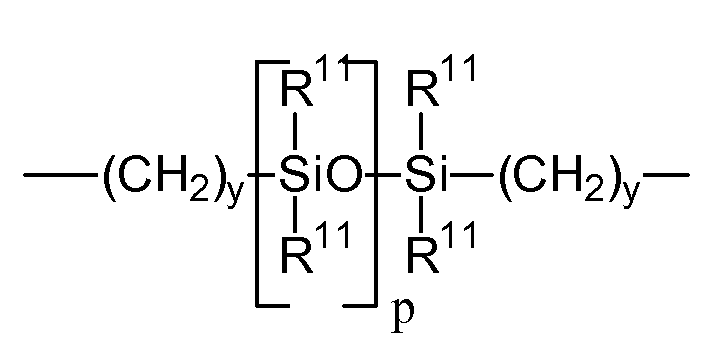
Формула VII
R11 независимо обозначает алкильную или фтор-замещенную алкильную группу, содержащую от 1 до 10 атомов углерода, которая может иметь эфирные мостиковые группы между атомами углерода; y равен по меньшей мере 1, а p означает функциональную группу с молекулярной массой от 400 до 10 000; каждый символ E и E1 независимо означает полимеризующийся ненасыщенный органический радикал, представленный следующей формулой:
Формула VIII
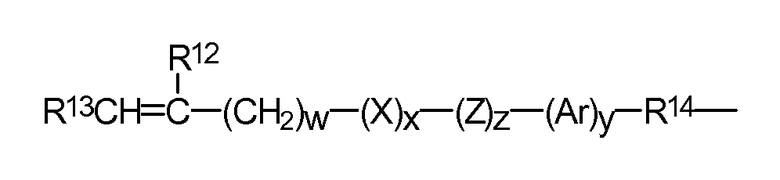
где R12 представляет собой водород или метил; R13 представляет собой водород, алкильный радикал, содержащий от 1 до 6 атомов углерода, или радикал -CO-Y-R15, где Y представляет собой -O-,Y-S- или -NH-; R14 означает двухвалентный радикал с количеством атомов углерода от 1 до 12; X обозначает -CO- или -OCO-; Z обозначает -O- или -NH-; Ar обозначает ароматический радикал с числом атомов углерода от 6 до 30; w - число от 0 до 6; x равно 0 или 1; y равно 0 или 1; z равно 0 или 1.
В одной реализации настоящего изобретения содержащий силикон компонент представляет собой полиуретановый макромер, представленный следующей формулой:
Формула IX
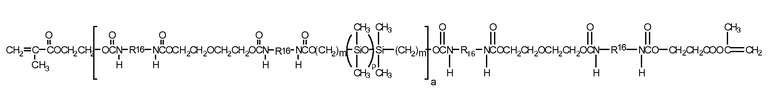
где R16 представляет собой бирадикал диизоцианата после удаления собственно изоцианатной группы, например бирадикал изофоронизоцианата. Другим содержащим силикон макромером, соответствующим целям настоящего изобретения, является соединение по формуле X (где x + y представляет собой число в диапазоне от 10 до 30), получаемое при реакции фторэфира, полидиметилсилоксана с концевой гидроксильной группой, изофоронизоцианата и изоцианатоэтилметакрилата.
Формула X
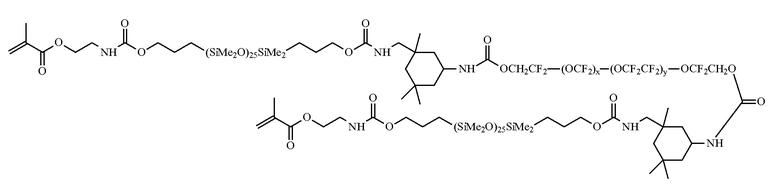
Иные содержащие силикон компоненты, соответствующие целям настоящего изобретения, включают компоненты, описанные в заявке на патент WO 96/31792, такие как макромеры, содержащие полисилоксановые, полиалкиленэфирные, диизоцианатные, полифторуглеводородные, полифторэфирные и полисахаридные группы. Другой класс содержащих силикон компонентов, соответствующих целям настоящего изобретения, включает содержащие силикон макромеры, полученные способом полимеризации с переносом группы, например макромеры, описанные в заявках на патент США № 5314960, 5331067, 5244981, 5371147 и 6367929. Патенты США № 5321108, 5387662 и 5539016 описывают силоксаны с полярной фторированной привитой или боковой группой, имеющей атом водорода при терминальном дифторзамещенном атоме водорода. В заявке на патент США № 2002/0016383 описаны гидрофильные силоксанилметакрилаты, содержащие эфирные и силоксанильные мостиковые группы, и пригодные для поперечной сшивки мономеры, содержащие полиэфирные и полисилоксанильные группы. Любой из перечисленных выше полисилоксанов также может быть использован в качестве содержащего силикон компонента в рамках настоящего изобретения.
После ознакомления с содержанием данных технических условий опытные специалисты смогут выявить другие силиконовые компоненты и использовать их в рамках данного изобретения.
В смеси гидрофильных и силиконовых компонентов по меньшей мере один из компонентов имеет одну или более свободных гидроксильных групп. В одном из вариантов по меньшей мере один гидрофильный компонент имеет по меньшей мере одну гидроксильную группу, которая может быть превращена в группу сложного борного эфира. В другом варианте исполнения по меньшей мере один силиконовый компонент имеет одну свободную гидроксильную группу. В другом варианте исполнения оба компонента (и силиконовый и гидрофильный) имеют по меньшей мере одну свободную гидроксильную группу. Благодаря наличию по меньшей мере одной свободной гидроксильной группы в компонентах получающийся полимер оказывается полиолом, который содержат несколько свободных гидроксильных групп.
Реакционная смесь может также содержать дополнительные компоненты, в том числе такие, которые поглощают ультрафиолетовое излучение, реакционно-способные красители, фотохромные соединения, вещества, облегчающие вынимание отливки из формы, смачиватели, нутрицевтики, фармацевтические соединения, их сочетания и тому подобное. Сшиватели - это соединения с одной или несколькими полимеризуемыми функциональными группами. Примерами сшивателей, используемых в данном изобретении, могут служить TEGDMA (тетраэтиленгликольдиметакрилат), TrEGDMA (триэтиленгликольдиметилакрилат), EGDMA (этиленгликольдиметилакрилат), acPDMS, их сочетания и тому подобное.
Реакционные смеси данного изобретения могут, дополнительно, содержать по меньшей мере один инициатор. Инициаторы включают такие соединения, как лаурилпероксид, перекись бензоила, изопропилперкарбонат, азобисизобутиронитрил и т. п., которые образуют свободные радикалы при умеренно высоких температурах и фотоиницитаорные системы, такие как ароматический альфагидроксикетон и четвертичный амин плюс дикетон. Иллюстративными примерами фотоинициаторных систем являются: 1-гидроксилциклогексилфенилкетон, 2-гидрокси-2-метил-1-енилпропан-1-он, бис(2,6-диметоксибензоил)-2,4-4-триметилфенолфосфиноксид (DMBAPO) и сочетание камфорохинона и этил-4-(N,N-диметиламино)бензоата. Инициатор используется в реакционной смеси в каталитически эффективных количествах, например от 0,1 до 2 весовых частей на 100 частей реакционно-активного компонента. Полимеризацию реакционной смеси можно инициировать с помощью надлежащего выбора нагрева, видимого или УФ-света или других способов в зависимости от используемого инициатора полимеризации. Инициацию можно проводить без фотоинициатора с помощью, например, низковольтного электронного луча. Тем не менее, когда используется фотоинициатор, предпочтительным вариантом является сочетание 1-гидроксициклогексилфенилкетона и бис(2,6-диметоксибензоил)-2,4-4-триметилпентилфосфиноксида (DMBAPO), а предпочтительным способом полимеризации является облучение видимым светом.
Разбавители, подходящие для изготовления изделий данного изобретения, включают в себя простые эфиры, сложные эфиры, алканы, алкилгалиды, силаны и спирты. В одном из вариантов осуществления в состав разбавителей входят спирты, а в другом - четвертичные спирты. Примеры простых эфиров, подходящих для данного изобретения в качестве разбавителей, включают в себя тетрагидрофуран. Примеры сложных эфиров, подходящих для данного изобретения, включают в себя этилацетат. К примерам алкилгалидов, используемых в данном изобретении в качестве разбавителей, относится метиленхлорид. Примеры силоксанов, подходящих для данного изобретения в качестве разбавителей, включают в себя октаметилциклотетрасилоксан. Примеры спиртов, подходящих для данного изобретения в качестве разбавителей, включают в себя гексанол, гептанол, октанол, нонанол, деканол, трет-бутиловый спирт, 3-метил-3-пентанол, изопропанол трет-амиловый спирт и 3,7-диметил-3-октанол. Дополнительные разбавители, подходящие для данного изобретения, описаны в патенте США № 6020445, содержание которого включено в настоящий документ путем ссылки.
Не желая связывать себя какой-либо конкретной теорией, заявители полагают, что, когда боратный разбавитель добавляется к смеси, которая содержит по меньшей мере один вид других гидроксильных соединений, происходит быстрое установление равновесия в реакции переэтерификации борного эфира, при этом некоторая доля этих прочих гидроксильных соединений образует временные сложные борные эфиры. Это повышает плотность сшивания полимеров всех форм, сохраняя при этом оптическую прозрачность раствора. При этом повышается вязкость смеси. Повышенная плотность сшивания также приводит к тому, что время гелеобразования сокращается, что приводит к повышению скорости полимеризации (эффект Тромсдорфа). Уменьшение времени гелеобразования сокращает время отверждения. Время отверждения может быть измерено путем дифференциальной фотокалориметрии с помощью оборудования и методик, приведенной в примере 9 в данном документе. В одном из вариантов исполнения время до достижения 90% отверждения составляет менее 3 минут, а в другом варианте время достижения 95% отверждения было менее 3,6 минуты. В другом варианте осуществления время до достижения 90 или 95% отверждения уменьшалось по меньшей мере на 10% по сравнению с линзами, образованными без участия по меньшей мере одного бората. В другом варианте осуществления время отверждения уменьшилось по меньшей мере на 20%.
Благоприятные эффекты от применения боратов могут быть реализованы различными способами. В одном из вариантов сложный эфир борной кислоты добавляется в реакционную смесь. В другом варианте осуществления борная кислота добавляется в реакционную смесь. В еще одном из вариантов добавляется борный ангидрид. Предполагается, что и борная кислота, и борный ангидрид образуют сложные эфиры борной кислоты in situ, вступая в реакцию конденсации со свободными гидроксильными группами компонентов. В некоторых вариантах осуществления может быть полезным способствовать образованию боратов in situ путем нагревания смеси и/или приложения вакуума для удаления побочного продукта конденсации (например, воды). Давление, способствующее образованию борного эфира, колеблется в широком диапазоне, а температурный диапазон составляет от 25 до 100°C.
В еще одном варианте исполнения по меньшей мере часть гидроксилсодержащих компонентов может быть «предварительно заряжена» путем реагирования их свободных гидроксильных групп с боратами с образованием борного сложного эфира. Когда такие предварительно прореагировавшие компоненты взаимодействуют с другими, содержащими гидроксильную группу компонентами реакционной смеси, реакция переэтерификации борного эфира идет дальше.
В практике известны различные способы количественного определения вязкости жидкостей. Например, при помощи вискозиметра из стеклянной трубки (вискозимер Оствальда или Уббеледе). В качестве альтернативы может использоваться ротационный вискозиметр, например марки MERLIN™ или «Брукфилд». В одном из вариантов осуществления бораты вызывают повышение вязкости не менее чем на 10% по сравнению с идентичной смесью, не содержащей боратов. В другом варианте осуществления бораты повышают вязкость как минимум на 20%. Еще в одном из вариантов осуществления добавление боратов вызывает по меньшей мере 30%-ное увеличение вязкости. Еще в одном из вариантов исполнения наблюдалось по меньшей мере 50%-ное увеличение вязкости.
В одном из вариантов осуществления изобретение относится к промежуточному материалу контактных линз, который включает отвержденный, оптически прозрачный материал для контактных линз. Материал представляет собой продукт полимеризации по меньшей мере одного гидрофильного компонента и одного силиконового компонента, по меньшей мере один из которых содержит гидроксильную функциональную группу, которая может быть частью борного сложного эфира или может существовать в форме -C-O-H. Также в качестве остаточного продукта в отвержденном материале присутствует сложный эфир борной кислоты, о котором шла речь в других разделах данного документа. Этот борат не остается навсегда в составе полимерного материала и обычно удаляется из него посредством гидролиза или экстракции. Способы удаления остаточных соединений из отвержденного материала для контактных линз известны в практике. Например, бораты могут быть извлечены посредством промывки материала растворителями, включающими по меньшей мере гидроксильную группу (такими как спирты, вода и их смеси). В одном из вариантов осуществления, в котором используется по меньшей мере один спирт, этот спирт растворим в воде. Примеры подходящих растворителей включают воду и низшие спирты, такие как метанол, этанол, н-пропанол и изопропанол.
Линзы, изготовленные вышеуказанным способом, отличаются по своим физическим свойствам от тех линз, которые были изготовлены без применения боратов. Например, эти линзы могут быть оптически более прозрачны. В некоторых вариантах % дымки менее 100% по сравнению с линзами CSI, а в других вариантах осуществления - менее 40%.
Хотя вышеуказанное осуждение сосредоточивается на использовании материала в области контактных линз, существуют варианты использования данного изобретения, не связанные с контактными линзами. В качестве иллюстрации (но не ограничения) другие виды применения включают опоры для биологических материалов, впитывающие изделия, частицы для доставки лекарственных препаратов, полимеры и изделия.
Приведенные примеры не ограничивают изобретение. Они предназначены только для предложения способа практического использования изобретения. Специалисты в области контактных линз, а также в других областях смогут найти и другие способы практического использования изобретения. Тем не менее, эти способы будут считаться подпадающими под действие настоящего изобретения.
Дымка измеряется путем помещения увлажненной исследуемой линзы в буферизированный солевой боратный раствор в стеклянной ячейке размером 20×40×10 мм на черном фоне и подсвечивания ее снизу волоконно-оптическим источником света (изготовитель Titan Tool Supply Co., волоконный источник света - световод диаметром 1,3 см (0,5”) мощностью 4-5,4) под углом 66° в направлении, перпендикулярном линзе, которую при этом снимают сверху в направлении, перпендикулярном ее положению, видеокамерой (камера DVC 1300C:19130 RGB с объективом с переменным фокусным расстоянием Navitar TV Zoom 7000), установленной на расстоянии 14 мм от платформы с линзой. Фоновое рассеяние вычитается из рассеяния линзы при помощи вычитания из ее изображения, полученного на пустой ячейке; для этого используется ПО EPIX XCAP V 1.0. Полученное вычитанием изображение в рассеянном свете количественно анализируется путем интегрирования по центрально части линзы размером 10 мм и в сравнении результата с результатом для линзы CSI Thin Lens® силой -1,0 диоптрии, которой произвольно присваивается значение дымки, равное 100, при этом нулевое значение дымки не присваивается какой-либо из линз. Анализируется пять линз, после чего рассчитывают среднее значение, выражая значение дымки в процентах относительно стандартной линзы CSI.
ПРИМЕРЫ
Примеры с 1 по 7, подробно рассмотренные в данных технических условиях, показывают рост вязкости раствора по мере добавления дополнительного количества боратов.
Примеры с 8 по 11 показывают уменьшение времени отверждения компонентной смеси силиконового гидрогеля по мере того, как концентрация боратов возрастает.
Примеры с 12 по 14 также показывают уменьшение времени отверждения различных смесей для получения силиконового гидрогеля по мере роста концентрации бората.
Примеры 15 и 16 показывают, что в некоторых случаях добавка бората улучшает оптические свойства.
Пример 17 показывает рост смешиваемости компонентов смеси, вызванный добавлением боратов. Это приводит к улучшению оптических свойств линзы.
Примеры 18 и 19 показывают количественное уменьшение времени отверждения вследствие добавления боратов в определенные смеси.
Термин «ммоль ОН» относится к общему количеству гидроксильных групп в молях в составе данной реакционной смеси, включая свободные гидроксильные группы и те, что входят в состав борного эфира. Поэтому в указанных выше расчетах ммоль Он может рассчитываться следующим образом. Сначала рассчитывается количество молей ОН в силиконе, разбавителе и гидрофильном компоненте. Силиконовый компонент - это макромер, образовавшийся в примере 1. Молекулярный вес макромера из примера 1 равен 13 550 г/моль. 1 моль этого вещества содержит 30 ОН-групп, следовательно:
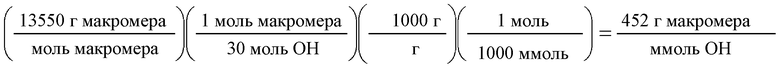
Смесь (5 г в примерах 2-7), не содержащая разбавителя, перед добавкой бората содержит 60% макромера, следовательно:

Однако дополнительные гидроксильные группы (в форме сложного эфира борной кислоты) добавляются с B(OEt)3. Дополнительное количество ОН равно количеству (в ммоль) групп BO, показанных в данных примерах. Значение в столбце миллимолей ОН является суммой этих двух источников гидроксильных групп. Дополнительные подробности приведены согласно примеру.
В следующих примерах используются определенные сокращения.
Пример 1. Приготовление макромера HFM 130:30
Пробу октаметилциклотетраполисилоксана весом 50 г смешали с 9,36 г 1,3,5,7-тетраметилциклотетрасилоксана, 1,96 г 1,3-бис(3-метакрилоксипропил)тетраметилдисилоксана, 70 г дихлорметана и 0,5 г трифторметансульфоновой кислоты в круглодонной колбе. Смесь перемешивали под азотной подушкой при комнатной температуре в течение 24 часов. Смесь промыли 75 мл насыщенного водного раствора карбоната натрия. Нижний слой промыли 75 мл воды и высушили над сульфатом натрия. Смесь пропустили через целитовый фильтр и отогнали растворитель под разрежением, получив промежуточный продукт в виде прозрачного силоксанового «масла».
К 98 г этого промежуточного продукта добавили 0,02 г 2,6-дитербутил-4-метилфенола (BHT), 0,04 г ацетата натрия, 100 мг 10%-го (вес.) раствора хлороплатиновой кислоты в изопропаноле, 18,1 г аллилового спирта и около 100 г изопропанола. Смесь нагрели под азотной подушкой до 50°C и выдержали в течение трех часов. Смесь охладили и профильтровали через целит. Растворитель отогнали под разрежением около 8 торр (отгонка длилась около 3 часов) до получения на выходе продукта (HFM 130:30) в виде прозрачного «масла».
Пример 2. Контроль - 0 M%OH бората
Была приготовлена смесь из 60 весовых частей HFM 130:30 (см. пример 1) и 40 частей N,N-диметилакриламида (DMA) с 0,23 части CGI 819 (бис(2,4,6-триметилбензоил)-фенилфосфиноксид). Часть смеси весом 5 г была перемешана, и ее вязкость была определена визуально. Смесь производила впечатление оптически прозрачной и маловязкой.
Пример 3. 6 M%OH бората
5 г смеси приготовили согласно примеру 2. К этой смеси добавили 25 мкл триэтилбората (0,15 ммоль, 0,02145 г). Получившая смесь оказалась значительно более вязкой, чем контроль из примера 2. В смеси при добавлении бората образовался белый осадок, который, однако, постепенно растворился при перемешивании.
Триэтилборат вносит 20,5 ммоль ОН-групп на грамм.
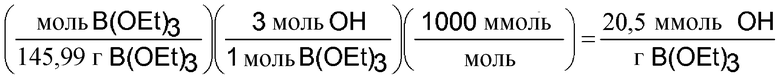
Подобные же расчеты для смесей с использованием HFM (130:30) из примера 1 показывают, что смесь обеспечивает 2,21 ммоль ОН на грамм. Аналогичные способы показывают, что триэтилборат может обеспечить 20,5 боратной функциональной группы (связей B-O) на грамм.
Можно определить количество ммоль OH, которые компонент привносит в грамм смеси. В следующем расчете определяется количество OH, возникающее за счет триэтилбората, но для HFM 130:30 тоже можно провести подобный расчет.
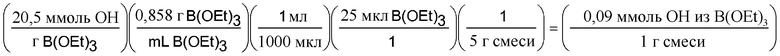
В итоге полученная смесь имеет 6% мольных OH, измеренных относительно гидроксильной функциональной группы. Результаты приведены в таблице 1.
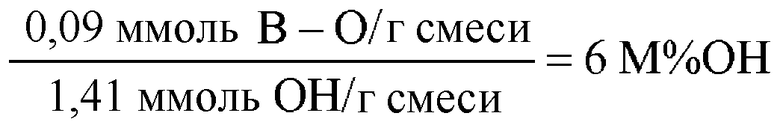
Состав смеси из примера 3
(6,1 M%OH)
Пример 4. 12 M % OH борат
5 г смеси приготовили согласно примеру 2. К этой смеси добавили 50 мкл триэтилбората (0,30 моль). Получившая смесь оказалась значительно более вязкой, чем контроль из примера 2. В смеси при добавлении бората образовался белый осадок, который, однако, постепенно растворился при перемешивании.
Состав смеси из примера 4
Пример 5. 21 M%OH бората
5 г смеси приготовили согласно примеру 2. К смеси было добавлено 100 мкл триэтилбората (0,60 ммоль). Получившая смесь оказалась значительно более вязкой, чем контроль из примера 2. В смеси при добавлении бората образовался белый осадок, который, однако, постепенно растворился при перемешивании.
Состав примера 5
Пример 6. 34 M%OH бората
5 г смеси приготовили согласно примеру 2. К этой смеси добавили 200 мкл триэтилбората (1,20 ммоль). Получившая смесь оказалась значительно более вязкой, чем контроль из примера 2. В смеси при добавлении бората образовался белый осадок, который, однако, постепенно растворился при перемешивании.
Состав смеси из примера 6
Пример 7. 50 M%OH бората
5 г смеси приготовили согласно примеру 2. К этой смеси добавили 375 мкл триэтилбората (2,20 ммоль). Получившая смесь оказалась значительно более вязкой, чем контроль из примера 2. В смеси при добавлении бората образовался белый осадок, количество которого, однако, постепенно уменьшилось при перемешивании Смесь осталась слегка мутной.
Состав смеси из примера 6
Пример 8. Приготовление смеси
Была приготовлена смесь, как показано в таблице 6.
К этой смеси был добавлен разбавитель PAGMBE в таком количестве, что соотношение между компонентами и разбавителем составило 90:10 по весу. К этой смеси добавили 0,039 весовой части CGI-819 на 100 частей смеси. Эта смесь была использована в примерах 9 и 10.
Пример 9. Контроль - 0 мольных % OH бората
Часть смеси из примера 8 была оставлена без добавления боратов для сравнения. Кинетику отверждения под действием света изучали при помощи термоаналитического дифференциального сканирующего калориметра (ДСК) марки TA Instruments 2920, оснащенного ртутной лампой с интенсивностью излучения (после прохождения через сине-фиолетовый стеклянный светофильтр и стеклянный фильтр, непрозрачный для красного излучения, оба марки Melles Griot) около 2 мВт/см2 в видимом и УФ-диапазоне.
Около 10 мг образца было помещено в алюминиевую кювету, которую затем поместили в ДСК. Камеру прибора продували азотом в течение пяти минут. Температуру образца подняли до 70°C. После пятиминутной выдержки образец облучали, и выделяющееся тепло регистрировалось в зависимости от времени. Результаты показаны на фиг. 1.
Пример 10
1,2 части триэтилбората были добавлены к 100 частям смеси, приготовленной в примере 8. При добавлении образовался белый осадок. Получившуюся смесь перемешали и оставили на ночь до тех пор, пока она не стала оптически прозрачной. Вязкость полученной смеси была выше, чем у образца из примера 9.
Кинетику отверждения под действием света изучали, как описано в примере 9. Результаты на фиг. 2 показывают, что у второго графика короче «хвост», т.е. в этом случае отверждение наступало быстрее, чем в смеси, не содержащей боратов.
Пример 11. Подготовка смеси
Смесь была приготовлена, как в таблице 7.
К 55 частям смеси вышеуказанных компонентов добавили 45 частей разбавителя (TPME). Смесь обладала следующими свойствами (0 M%OH бората).
Пример 12. 22,6 мольного % ОН
Используя смесь из примера 11, 55 частей ее смешали с 45 частями разбавителя, состоящего на 91% (вес.) из TPME и на 8,9% из триэтилбората. Полученную смесь дегазировали в течение 15 минут под вакуумом, составлявшим 5,3 кПа (40 мм рт. ст.). Линзы формировались путем облучения в течение 15 минут светом с интенсивностью 1,3 мВт/см2, в качестве излучателя использовались флуоресцентные лампочки Philips TL 20 Вт/0,3 T; температура эксперимента составляла 60°C, использовались пластиковые матрицы с продувкой азотом. Кинетика отверждения под действием света изучалась в соответствии с процедурой, описанной в примере 9, за исключением того, что облучение имело интенсивность 4 мВ/см2. На фиг. 3 видно, что добавление бората уменьшило время полного отверждения по сравнению с той же самой смесью без применения боратов. В таблице 9 показаны расчеты состава в граммах компонента/грамм смеси, ммоль ОН/г компонента, ммоль В-О на грамм компонента, ммоль ОН/г смеси и ммоль ВО на грамм смеси. Мольный % OH составляет 22,6%, и этот показатель был рассчитан делением общего количества ммоль -B-O-C- из последнего столбца на общее количество ммоль ОН в предпоследнем столбце.
Пример 13. 47,7 M%OH бората
Пример 14 практически совпадал с примером 12, за исключением того, что разбавитель состоял из 76% (вес.) TPME и 24% (вес.) триэтилбората.
Кинетика отверждения под действием света изучалась в соответствии с процедурой, описанной в примере 9, за исключением того, что облучение имело интенсивность 4 мВ/см2.
На фиг. 3 видно, что добавление бората уменьшило время полного отверждения по сравнению с той же самой смесью без применения боратов. В таблице 10 показаны расчеты состава в граммах компонента/грамм смеси, ммоль ОН/г компонента, ммоль В-О на грамм компонента, ммоль ОН/г смеси и ммоль ВО на грамм смеси. Мольный % OH составляет 47,7%.
Примеры 15 и 16. GMA-борат - улучшенная смешиваемость компонентов
Как показано в таблице 11, были приготовлены две смеси: одна с применением GMA (пример 15), а вторая с использованием GMA-бората (пример 16). GMA-борат был приготовлен сочетанием 292 весовых частей триэтилбората (Aldrich) и 380 частей GMA (сольное соотношение 2:3 соответственно) и удалением образующегося этанола во вращательном испарителе при 50°C в течение часа при высоком вакууме или до постоянной массы с образованием нового соединения, GMA-бората. Смесь с использованием GMA-бората была профильтрована через стекловату для удаления наблюдаемого незначительного количества осадка, образовавшегося при получении соединения.
GMA/GMA-боратные смеси
Через каждую смесь пропускали азот в течение 5 минут, после чего образцам давали осесть в открытом состоянии в гермобоксе с атмосферой азота в течение 2 часов. Пластиковые матрицы наполнялись в гермобоксе и помещались на расстоянии около 7,6 см (3 дюйма) под лампочками Philips TL09 20 Вт. Линзы отверждались при комнатной температуре в атмосфере азота на протяжении 30 минут. Затем линзы промывали - сначала смесью 50%-ного изопропанола: 50%-ного борного буферного раствора на протяжении 30 минут, затем трижды 100%-ным изопропанолом на протяжении 30 минут каждый раз, а затем смесью 50%-ного изопропанола: 50%-ного борного буферного раствора и, наконец, трижды по 30 минут 100%-ным борным буферным раствором. Линзы из примера 15 (без бората) были белыми, а линзы из примера 16 (с боратной добавкой) были прозрачны. В таблицах 12 и 13 показаны расчеты состава в граммах компонента/грамм смеси, ммоль ОН/г компонента, ммоль В-О на грамм компонента, ммоль ОН/г смеси и ммоль ВО на грамм смеси. Мольный % OH равен 0% для примера 15 и 38,4% для примера 16.
Пример 15
Пример 16
Примеры 17-25. HEAA-борат - улучшенная смешиваемость за счет сокращения количества разбавителя, необходимого для получения прозрачной линзы.
HEAA-борат получали сочетанием 3 молей HEAA с одним молем триэтилбората. Этанол удалялся в роторном испарителе при 50°C под высоким вакуумом в течение 1 часа или до постоянной массы с образованием нового продукта - HEAA-бората. Были приготовлены различные смеси, как показано в таблице 14.
Смеси на основе HEAA и HEAA-бората
(% вес.)
(% вес.)
(% вес.)
(% вес.)
(% вес.)
(% вес.)
(% вес.)
(% вес.)
(% вес.)
Смеси на основе HEAA и HEAA-бората дегазировались в вакуумном эксикаторе при давлении 4 кПа (40 мбар) на протяжении приблизительно 30 минут. Пластиковые матрицы наполнялись в гермобоксе и помещались на расстояние около 7,6 см (3 дюйма) под лампочки Philips TL09 20 Вт. Линзы отверждались при комнатной температуре в атмосфере азота на протяжении 30 минут. Затем линзы промывали - сначала смесью 50%-ного изопропанола: 50%-ного борного буферного раствора на протяжении 30 минут, затем трижды 100%-ным изопропанолом на протяжении 30 минут каждый раз, а затем смесью 50%-ного изопропанола: 50%-ного борного буферного раствора и, наконец, трижды по 30 минут 100%-ным борным буферным раствором. Прозрачность смесей и готовых линз указана в таблице 14. Пример 17 (без бората) оказался несмешивающейся системой, поэтому линзы из смеси номер 17 не делались. Соответствующая смесь с добавкой бората (пример 25) была однородной. Из смеси, приготовленной для примера 18 (без бората), получилась мутная белая линза, тогда как соответствующая смесь с добавкой бората позволила получить прозрачную линзу (пример 23). Расчеты состава в граммах компонента/грамм смеси, ммоль ОН/г компонента, ммоль В-О на грамм компонента, ммоль ОН/г смеси и ммоль ВО на грамм смеси. Мольный % OH для примеров 17-20 равен 0. Мольный % OH для примеров 21-25 составляет 70%, 62,2%, 62,2%, 43,8% и 34,6% соответственно.
Пример 17
Пример 18
Пример 19
Пример 20
Пример 21
Пример 22
Пример 23
Пример 24
Пример 25
Примеры 26-31. Количественные соотношения между временем отверждения и концентрацией бората
A. Рецептура А с боратным разбавителем
Разнообразные смеси показаны в таблице 24. D3O-борат образуется соединением 3 молей D3O с одним молем триметилбората. Метанол удаляется во вращающемся испарителе при 50°C под высоким вакуумом в течение 1 часа или до достижения постоянной массы, образуя новое соединение - D3O-борат. Процентная доля боратного разбавителя меняется в зависимости от того, какую часть составляет борат от общего количества разбавителя. Компоненты смеси были добавлены во флакон и перемешивались мешалкой до тех пор, пока все твердые вещества полностью не перешли в раствор.
Расчеты состава в граммах компонента/грамм смеси, ммоль ОН/г компонента, ммоль В-О на грамм компонента, ммоль ОН/г смеси и ммоль ВО на грамм смеси.
Пример 27
Линзы из примера 27 имели мольный % OH, равный 61,7%.
Пример 28
Линзы из примера 28 имели мольный % OH, равный 46,1%.
Пример 29
Линзы из примера 29 имели мольный % OH, равный 30,6%.
Пример 30
Линзы из примера 30 имели мольный % OH, равный 15,4%.
Пример 31
Линзы из примера 31 имели мольный % OH, равный 0%.
Примеры 32-33. Рецептуры с боратными компонентами и разбавителями
Была приготовлена смесь из 141 весовой части триметилбората, 402 частей D3O, 81 части HEMA и 376 частей SiGMA, мольное соотношение составило 10:19:6:5 соответственно Метанол удалялся во вращающемся испарителе при 50°C под высоким вакуумом в течение 1 часа или до достижения постоянной массы с получением нового соединения - SHD-бората. Две смеси рецептуры А с использованием SHD-бората были подготовлены, как указано в таблице 30. Компоненты смеси были добавлены во флакон и перемешивались мешалкой до тех пор, пока все твердые вещества полностью не перешли в раствор.
Смеси с боратным компонентом/разбавителем
Примеры 34-39.
Смеси с рецептурой из примеров 34-39 были помещены с открытыми крышками флаконов в бокс с азотной атмосферой на срок не менее 1 часа. Пластиковые матрицы заполнялись в гермобоксе и помещались на расстоянии около 7,6 см (три дюйма) под лампочками Philips TL03 20 Вт на 30 минут. Линзы отверждались при комнатной температуре в атмосфере азота на протяжении 30 минут. Затем линзы промывали - сначала смесью 50%-ного изопропанола: 50%-ного борного буферного раствора на протяжении 30 минут, затем трижды 100%-ным изопропанолом на протяжении 30 минут каждый раз, а затем смесью 50%-ного изопропанола: 50%-ного борного буферного раствора и, наконец, трижды по 30 минут 100%-ным борным буферным раствором. Линзы из смесей 10-15 были прозрачными, а линзы из смеси 16 - мутными.
Характеристики отверждения смесей рецептуры А, описанных в таблицах 7 и 8, были изучены с помощью приборов марки TA Instruments модели Q100 (фото-ДКС) с универсальным светодиодным модулем производства Digital Light Labs модели ULM-1-420. Образцы помещались на пластину, продуваемую азотом, и выдерживались в течение 5 минут при 25°C, затем в течение 5 минут при 70°C, а затем инициировалось отверждение путем облучения смесей светом с интенсивностью 4 мВт/см2. Базовые кривые строились с применением сигмоидальной коррекции. Время отверждения рассчитывалось с помощью программного обеспечения TA Universal Analysis 2000. Каждая смесь проверялась несколько раз, и значения в таблице представляют собой средние значения из 2 или 4 реплик. В таблице 9 показаны энтальпия, время выхода на пиковое значение экзотермического эффекта и время, прошедшее до 25, 50, 75, 90 и 95-процентного отверждения.
Время, прошедшее до различной степени отверждения, рассчитывалось как площадь под кривой фото-ДСК. Когда отверждение закончено, кривая возвращается к той же базовой линии, которая регистрировалась до момента включения источника света. Иногда это конечное значение базовой линии может быть чуть больше исходного. В таком случае к сигмовидной базовой линии следует применить коррекцию. Сигмовидной базовой линией называют s-образную линию, которая меняет уровень и/или угол наклона до или после пика. Базовая линия корректируется по зависимости прореагировавшей доли (альфа) продукта от времени. Сигмовидная базовая линия первоначально рассчитывается как прямая линия от начала пика до его окончания. Затем ее пересчитывают для каждой точки между предельными значениями пика как средневзвешенную величину проекций горизонтальных или касательных базовых линий в начале и в конце пика. Для каждой точки коэффициент взвешивания составляет: (1) единица минус альфа умножить на начальную базовую линию и (2) альфа умножить на начальную базовую линию. Площадь затем пересчитывается на новую базовую линию. Если новая площадь отличается от предыдущей более чем на один процент, то ее пересчитывают и сигмовидную кривую сдвигают снова и снова до тех пор, пока разница между двумя последовательными величинами площади не будет отличаться менее чем на один процент. Программное обеспечение, использованное для получения данных, представленных ниже, - Universal Analysis 2000 для Windows 2000/XP/Vista, Version 4.5A, сборка 4.5.0.5, но ДСК от разных производителей обычно имеют собственные средства коррекции сигмовидной базовой линии.
Результаты анализа рецептуры А с боратом в фото-ДСК
отверждения
значения
(мин)
25%-ного отверждения (мин)
50%-ного отверждения
(мин)
75%-ного отверждения
(мин)
90%-ного отверждения
(мин)
95%-ного отверждения
(мин)
Результаты показывают, что по мере повышения доли D3O-боратного разбавителя время отверждения уменьшается. Эксперимент также показывает, что добавление боратов одновременно в качестве компонента и разбавителя (пример 39, 50% SHD-борат) еще сильнее ускоряет отверждение. Сравнивая значения при 95% отверждения, можно видеть, что 50% SHD-бората (пример 39) уменьшают время отверждения чуть больше чем на 50 процентов, а 100% добавки боратного разбавителя (пример 38) уменьшают время отверждения примерно на 38%.
Полученные линзы проверялись на кислородную проницаемость, динамический контактный угол, содержание влаги, а также проверяли их механические свойства, чтобы определить, не оказывает ли добавка бората какого-либо отрицательного влияния на свойства линзы. Результаты, приведенные в таблице 32, показывают, что бораты не оказывают воздействия (или оказывают очень малое воздействие) на эти свойства линз.
Характеристики линз, полученных по рецептуре А, с боратным разбавителем
растворителя
(105±10)
(104±11)
(98±2)
(105±9)
(93±7)
(96±7)
(106±26)
(129±22)
(116±30)
(112±29)
(114±34)
(130±39)
Добавление бората к смесям рецептуры А снижает время отверждения более чем на 50%, не сказываясь отрицательно на свойствах линзы.
Примеры 40-43. Рецептура B. Борат
A. Рецептура В с боратным разбавителем.
TPME-борат образуется сочетанием трех молей TPME с одним молем триметилбората. Метанол удаляли в роторном испарителе при 50°C под высоким вакуумом в течение 1 часа или до достижения постоянной массы с образованием нового соединения, TPME-бората. Различного состава смеси были приготовлены, как показано в таблице 11. Как показано в таблице 11, с использованием TPME-бората было приготовлено несколько смесей рецептуры В. Деканоевую кислоту поставила компания KIC Chemicals Inc. Показатель процентного содержания боратного разбавителя относится к общей процентной доле гидроксильных функциональных групп, внедренных в смесь в виде бората. Компоненты смеси добавили во флакон и встряхивали до полного растворения всех твердых веществ.
Смеси рецептуры В с боратным разбавителем
Смеси с рецептурой из примеров 40-43 были помещены с открытыми крышками флаконов в бокс с азотной атмосферой на срок не менее 1 часа. Пластиковые матрицы заполнялись в гермобоксе и помещались на расстоянии около 7,6 см (три дюйма) под лампочками Philips TL03 20 Вт на 30 минут. Линзы отверждались при комнатной температуре в атмосфере азота на протяжении 30 минут. Затем линзы промывали - сначала смесью 50%-ного изопропанола: 50%-ного борного буферного раствора и затем трижды по 30 минут 100%-ным борным буферным раствором. Линзы, полученные из этих смесей, были прозрачными.
Характеристики отверждения для смесей рецептуры В, описанных в таблице 34, изучались на аппаратуре TA Instruments (фото-ДСК модели Q-100, как в примере 3). В таблице 35 показаны энтальпия, время выхода на пиковое значение экзотермического эффекта и время, прошедшее до 25, 50, 75, 90 и 95-процентного отверждения. Каждая смесь проверялась несколько раз, и значения в таблице представляют собой средние значения из 2 или 4 реплик.
Результаты анализа рецептуры В с боратом в фото-ДСК
Результаты показывают, что по мере повышения доли ТРМЕ-боратного разбавителя время отверждения уменьшается. При сравнении значений, соответствующих 95%-му отверждению, видно, что в примере 43 (100% TPME-борат) время отверждения уменьшилось чуть более чем на 25 процентов по сравнению с показателем из примера 40.
Полученные линзы проверялись на кислородную проницаемость, динамический контактный угол, содержание влаги, а также проверяли их механические свойства, чтобы определить, не оказывает ли добавка бората какого-либо отрицательного влияния на свойства линзы. Результаты, приведенные в таблице 35, показывают, что бораты не оказывают воздействия (или оказывают очень малое воздействие) на эти свойства линз.
Характеристики линз, полученных по рецептуре В, с боратным разбавителем
(103±7)
(117±9)
(110±9)
(97±6)
(104±20)
(115±25)
(113±30)
(99±34)
Путем добавления боратов мы смогли достичь 25%-ного уменьшения времени отверждения без отрицательного воздействия на свойства линзы.
Пример 44-47. Рецептура В с боратными компонентами и без разбавителя
Была приготовлена смесь из 77 весовых частей триметилбората, 805 частей OH-mPDMS и 118 частей HEMA (мольное соотношение составило 10:18:12 соответственно). Метанол удалялся в роторном испарителе при 50°C под высоким вакуумом в течение 1 часа или до достижения постоянной массы с получением нового соединения - ОНН-бората. Несколько смесей рецептуры В без разбавителя с использованием OHH-бората было приготовлено, как описано в таблице 36. Компоненты смеси добавили во флакон и встряхивали до полного растворения всех твердых веществ.
Смеси рецептуры В с боратными компонентами
Смеси с рецептурой из примеров 44-47 были помещены с открытыми крышками флаконов в бокс с азотной атмосферой на срок не менее 1 часа. Пластиковые матрицы заполнялись в гермобоксе и помещались на расстоянии около 7,6 см (три дюйма) под лампочками Philips TL03 20 Вт на 30 минут. Линзы отверждались при комнатной температуре в атмосфере азота на протяжении 30 минут. Затем линзы промывали - сначала смесью 50%-ного изопропанола: 50%-ного борного буферного раствора и затем трижды по 30 минут 100%-ным борным буферным раствором. Линзы, полученные из этих смесей, были прозрачными.
Характеристики отверждения для смесей рецептуры В, описанных в таблице 36, изучались на аппаратуре TA Instruments (фото-ДСК модели Q-100, как в примере 3). В таблице 37 ниже показаны энтальпия, время выхода на пиковое значение экзотермического эффекта и время, прошедшее до 25, 50, 75, 90 и 95-процентного отверждения. Каждая смесь проверялась несколько раз, и значения в таблице представляют собой средние значения из 2 или 4 реплик.
Результаты на фото-ДСК для рецептур группы В без боратных разбавителей
Результаты показывают, что добавление боратов снижает время отверждения.
Полученные линзы проверялись на кислородную проницаемость, динамический контактный угол, содержание влаги, а также проверяли их механические свойства, чтобы определить, не оказывает ли добавка бората какого-либо отрицательного влияния на свойства линзы. Результаты, приведенные в таблице 16, показывают, что бораты не оказывают воздействия (или оказывают очень малое воздействие) на эти свойства линз.
Характеристики линз, сформированных из боратных смесей рецептуры В без разбавителей
(304±27)
(263±31)
(304±30)
(248±33)
(145±31)
(136±28)
(154±37)
(141±32)
Добавление бората к не содержащим разбавителя смесям рецептуры В уменьшило время отверждения без отрицательного воздействия на свойства линзы.
Динамический механический анализ
Смесь 17 (контрольный образец без бората) и смесь 20 (с боратом) были протестированы с использованием динамического механического анализа. Результаты на фиг. 4 и 5 показывают, что эластичность материала, полученного отверждением смесей, содержащих бораты, выше, чем эластичность материала, полученного из тех же смесей, но без содержания бората. Ниже приведены подробности и протоколы испытаний.
Реакция отслеживалась при помощи реометра ATS StressTech [ATS RheoSystems, 52 Georgetown Road, Bordentown, NJ 08505], оснащенного устройством для светового отверждения, которое состояло из термостатируемой ячейки с кварцевой нижней пластиной и алюминиевой верхней пластиной, а также из светодиодной матрицы с длиной волны испускаемого света 420 нм [EXFO Photonic Solutions Inc., 2260 Argentia Rd., Mississauga, ON L5N 6H7, КАНАДА], расположенной под кварцевой пластиной. Интенсивность излучения, измерявшегося на поверхности кварцевой пластины радиометром IL1400A и датчиком XRL140A [International Light, Inc., 17 Graf Road, Newburyport, MA 01950], регулировалась в пределах 5,0±0,1 мВт/см2 при помощи электронного контроллера [Andover Corporation, 4 Commercial Drive, Salem, NH 03079-2800, США]. Температура регулировалась в пределах 60,0±0,1°C.
После того как приблизительно 0,25 мл реакционной смеси было помещено на нижнюю пластину реометра, верхнюю пластину диаметром 25 мм опускали на расстояние 0,500±0,001 мм над поверхностью смеси и оставляли там до тех пор, пока реакция не достигнет стадии гелеобразования. Образец выдерживался в ячейке до достижения состояния температурного равновесия (приблизительно 5 минут, определялось по выравниванию вязкости образца при постоянном усилии сдвига по мере его прогрева), после чего включалась светодиодная матрица и начиналось отверждение. На протяжении периода достижения температурного равновесия камеру с образцом продували азотом с расходом 400 станд. см3/мин. После этой первичной продувки содержание кислорода в камере выдерживалось на уровне 0,2±0,1% при помощи датчика CheckPoint O2 [PBI Dansensor, изготовитель компания Topac, 101 Derby St., #203 Hingham, MA 02043]. Во время реакции при помощи реометра постоянно измерялось напряжение, вызываемое прилагаемой динамической нагрузкой (в быстром осциллирующем режиме), при этом для измерения напряжения и приложенной синусоидальной нагрузки (частота 1 Гц) использовались временные сегменты длительностью меньше одного полного цикла. Динамический модуль сдвига (G'), податливость (G”) и высота зазора определялись как функция от времени воздействия света. По мере протекания реакции модуль сдвига вырос с <1 Па до >0,1 МПа и тангенс δ (=G”/G') снизился от почти бесконечно большой величины до менее чем 1. Для многих реакционно-способных сшивающихся систем точка гелеобразования определяется как δ=1 (точка перехода, в которой G'=G”). К тому моменту, когда G' достигло 100 Па (вскоре после точки гелеобразования), ограничения на высоту зазора между смесью и верхней пластиной были сняты (режим автоматической регулировки напряжения: напряжение=0), т.е. зазор между верхней и нижней пластиной мог изменяться по мере сокращения объема реакционной смеси во время отверждения, и напряжение от этой усадки держалось на минимальном уровне. Измерение величины зазора позволяет оценить, насколько в результате полимеризации уменьшается объем полимера.
После 10-минутного облучения светодиодную матрицу выключили (отверждение было прервано), и был проведен замер изменения температуры между параллельными пластинами реометра. Температура была снижена с 60 до 25°C (1°C/мин) при приложенном напряжении сдвига 3000 Па с частотой 1,0 Гц для определения температуры остекловывания (Tg), после чего материал был нагрет от 25 до 120°C и охлажден от 120 до 25°C с целью определить, возникли ли какие-либо изменения в материале при нагревании до температуры свыше точки отверждения; все температурные замеры проводились при приложении переменного напряжения сдвига 3000 Па с частотой 1,0 Гц и скоростью изменения температуры 1°C/мин.
Для получения спектра механических свойств отвержденных пленок частота изменения напряжения сдвига (при 1000 Па) изменялась от 0,01 до 40 Гц при температуре 60 и 25°C.
Изобретение относится к способам формирования силиконового гидрогеля, служащего материалом для контактных линз. Предложен способ формирования силиконового гидрогелевого материала, включающий этапы: получения смеси полимеризуемых компонентов, содержащей по меньшей мере один гидрофильный компонент и по меньшей мере один силиконовый компонент, где по меньшей мере один полимеризуемый компонент содержит по меньшей мере одну гидроксильную группу, причем дополнительно смесь включает борат в количестве, достаточном для уменьшения времени отверждения по сравнению с идентичной смесью, не содержащей боратов; отверждения смеси для получения отвержденного силиконового гидрогелевого материала. Предложены также варианты указанного способа, способы получения оптически прозрачного материала и промежуточного материала контактных линз, варианты контактных линз и материал для медицинских устройств на основе получаемого гидрогелевого материала. Технический результат - получение силиконового гидрогелевого материала, обладающего хорошей вязкостью и скоростью отверждения и позволяющего формировать контактные линзы с улучшенными механическими свойствами. 9 н. и 27 з.п. ф-лы, 5 ил., 38 табл., 47 пр.
1. Способ формирования силиконового гидрогелевого материала, включающий этапы:
получения смеси полимеризуемых компонентов, которая включает по меньшей мере один гидрофильный компонент и один силиконовый компонент и в которой по меньшей мере один из полимеризуемых компонентов содержит по меньшей мере одну гидроксильную группу, дополнительно такая смесь включает количество по меньшей мере одного бората, достаточное для уменьшения времени отверждения по сравнению с идентичной смесью, не содержащей боратов;
отверждения смеси для получения отвержденного силиконового гидрогелевого материала;
при этом борат выбирают из
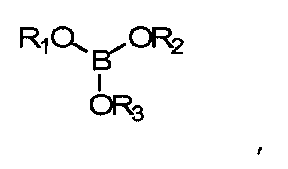
где R1, R2 и R3 независимо выбирают из групп, включающих водород; алкильные группы С1-С16; алкильные группы С1-С16 с замещением атомов углерода атомами О, N; полимеризуемые группы; эфирные группы, амидные группы, сложноэфирные группы;
силиконовый компонент выбирают из
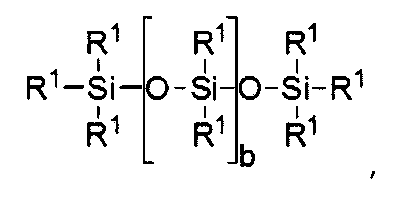
где b находится в диапазоне от 3 до 15, один концевой фрагмент R1 представляет собой одновалентную реакционноспособную группу, другой концевой фрагмент R1 представляет собой одновалентную алкильную группу, содержащую от одного до 6 атомов углерода, а остальные фрагменты R1 представляет собой одновалентные алкильные группы, содержащие от 1 до 3 атомов углерода; или
b равно нулю, один фрагмент R1 представляет собой одновалентную реакционноспособную группу, по крайней мере три фрагмента R1 выбраны из одновалентных алкильных групп, содержащих от одного до 6 атомов углерода.
2. Способ по п. 1, в котором силиконовый компонент содержит мономеры, выбранные из: полидиметилсилоксана (MB 400-1000) с концевой моно-(2-гидрокси-3-метакрилоксипропил)пропил эфирной группой (ОН-mPDMS), полидиметилсилоксанов (MB 800-1000) с концевыми моно-н-бутильными и концевыми монометакрилоксипропильными группами (mPDMS) и 2-метил-, 2-гидрокси-3-[3-[1,3,3,3-тетраметил-1-[(триметилсилил)окси]дисилоксанил]пропокси]пропилового эфира (SiGMA).
3. Способ по п. 1, в котором под временем отверждения подразумевают время, необходимое для достижения 95% отверждения.
4. Способ формирования силиконового гидрогелевого материала, включающий этапы:
получения смеси полимеризуемых компонентов, которая включает по меньшей мере один гидрофильный компонент и по меньшей мере один силиконовый компонент и в которой по меньшей мере один из полимеризуемых компонентов содержит гидроксильную группу, дополнительно такая смесь включает количество бората, достаточное для уменьшения времени отверждения по сравнению с идентичной смесью, не содержащей боратов, при этом бораты выбираются либо в виде борного ангидрида, либо в виде боратов, представленных формулой
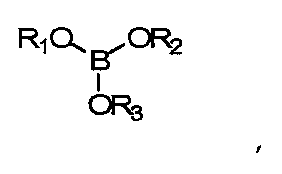
где R1, R2 и R3 независимо выбираются из групп, включающих водород; алкильные группы С1-С16; алкильные группы С1-С16 с замещением атомов углерода атомами О, N; полимеризуемые группы; эфирные группы, амидные группы, сложноэфирные группы, силиконовые компоненты;
отверждения смеси для получения отвержденного материала;
при этом силиконовый компонент выбирают из
где b находится в диапазоне от 3 до 15, один концевой фрагмент R1 представляет собой одновалентную реакционноспособную группу, другой концевой фрагмент R1 представляет собой одновалентную алкильную группу, содержащую от одного до 6 атомов углерода, а остальные фрагменты R1 представляет собой одновалентные алкильные группы, содержащие от 1 до 3 атомов углерода; или
b равно нулю, один фрагмент R1 представляет собой одновалентную реакционноспособную группу, по крайней мере три фрагмента R1 выбраны из одновалентных алкильных групп, содержащих от одного до 6 атомов углерода.
5. Способ по п. 4, где R1, R2 и R3 одновалентны.
6. Способ по п. 4, где по меньшей мере две из групп R1, R2 и R3 двухвалентны и совместно образуют циклический борный эфир.
7. Способ по п. 4, в котором по меньшей мере одна из групп R1, R2 и R3 представляет собой полимеризуемую группу.
8. Способ по п. 4, где R1, R2 и R3 одинаковы.
9. Способ по п. 8, где R1, R2 и R3 одновалентны.
10. Способ по п. 4, в котором борат выбирают из группы, представленной борным эфиром метанола, этанола, пропанола, трет-амилового спирта, 3-метил-3-пентанола, 3,7-диметил-3-октанола, 2-гидроксиметилметакрилата, трипропиленгликольметилового эфира, 2-акриловой кислоты, 2-метил-, 2-гидрокси-3-[3-[1,3,3,3-тетраметил-1-[(триметилсилил)окси]дисилоксанил]пропокси]пропилового эфира (SiGMA), полиметилдисилоксана с моно-(3-метакрилокси-2-гидропропилокси)пропильными и монобутильными концевыми группами (OHmPDMS) и их сочетаниями.
11. Способ по п. 8, в котором R1, R2 и R3 выбирают из группы, включающей н-пропил, изопропил, бутил и трет-бутил.
12. Способ по п. 4, в котором бораты выбирают из группы, состоящей из триметилбората и триэтилбората.
13. Способ по п. 4, в котором в качестве бората используют борную кислоту.
14. Способ по п. 4, в котором на этапе отверждения смесь содержит от 2 до 50 М%ОН бората.
15. Способ по п. 4, в котором смесь имеет вязкость по меньшей мере на 10% большую, чем вязкость идентичной смеси, не содержащей боратов.
16. Способ по п. 4, который дополнительно включает этап удаления по меньшей мере половины боратов из отвержденного материала для контактных линз.
17. Способ по п. 4, в котором время отверждения уменьшается по крайней мере на 10% по сравнению с идентичной смесью, не содержащей боратов.
18. Способ по п. 4, в котором время отверждения уменьшается по крайней мере на 25% по сравнению с идентичной смесью, не содержащей боратов.
19. Способ по п. 4, в котором время отверждения уменьшается по крайней мере на 50% по сравнению с идентичной смесью, не содержащей боратов.
20. Контактная линза, сформированная из силиконового гидрогелевого материала в соответствии со способом по п. 1.
21. Материал, используемый при изготовлении медицинского устройства, включающий силиконовый гидрогелевый материал, изготовленный способом, указанным в п. 1.
22. Материал контактной линзы, сформированный из силиконового гидрогелевого материала в соответствии со способом по п. 4.
23. Способ формирования оптически прозрачного материала, включающий этапы:
получения смеси полимеризуемых компонентов, которая включает по меньшей мере один гидрофильный компонент и по меньшей мере один силиконовый компонент и в которой по меньшей мере один из полимеризуемых компонентов содержит одну гидроксильную группу, дополнительно такая смесь включает достаточное количество боратов для получения более оптически прозрачного материала по сравнению с идентичной смесью, не содержащей боратов;
отверждения смеси для получения отвержденного, оптически прозрачного материала;
при этом борат выбирают из
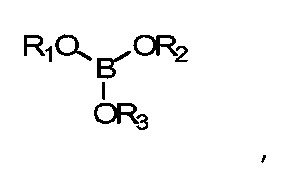
где R1, R2 и R3 независимо выбирают из групп, включающих водород; алкильные группы С1-С16; алкильные группы С1-С16 с замещением атомов углерода атомами О, N; полимеризуемые группы; эфирные группы, амидные группы, сложноэфирные группы;
силиконовый компонент выбирают из
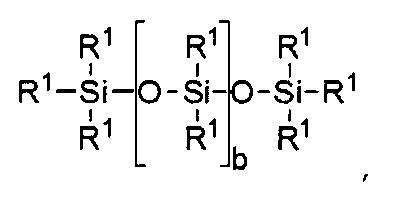
где b находится в диапазоне от 3 до 15, один концевой фрагмент R1 представляет собой одновалентную реакционноспособную группу, другой концевой фрагмент R1 представляет собой одновалентную алкильную группу, содержащую от одного до 6 атомов углерода, а остальные фрагменты R1 представляет собой одновалентные алкильные группы, содержащие от 1 до 3 атомов углерода; или
b равно нулю, один фрагмент R1 представляет собой одновалентную реакционноспособную группу, по крайней мере три фрагмента R1 выбраны из одновалентных алкильных групп, содержащих от одного до 6 атомов углерода.
24. Отвержденная контактная линза, состоящая из отвержденного, оптически прозрачного полимерного материала, сформированного из смеси, которая включает:
гидрогелевую смесь, содержащую по меньшей мере один полимеризуемый гидрофильный компонент и один полимеризуемый силиконовый компонент, в которой по меньшей мере один из полимеризуемых компонентов содержит гидроксильную группу;
борат, представленный формулой
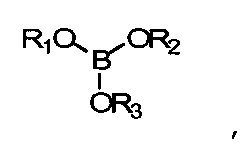
где R1, R2 и R3 независимо выбираются из числа групп, включающих водород; алкильные группы С1-С16; алкильные группы С1-С16 с замещением атомов углерода атомами О, N;
при этом силиконовый компонент выбирают из
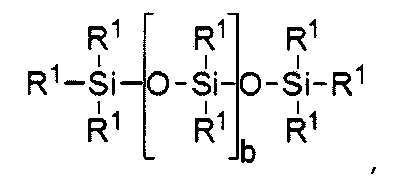
где b находится в диапазоне от 3 до 15, один концевой фрагмент R1 представляет собой одновалентную реакционноспособную группу, другой концевой фрагмент R1 представляет собой одновалентную алкильную группу, содержащую от одного до 6 атомов углерода, а остальные фрагменты R1 представляет собой одновалентные алкильные группы, содержащие от 1 до 3 атомов углерода; или
b равно нулю, один фрагмент R1 представляет собой одновалентную реакционноспособную группу, по меньшей мере три фрагмента R1 выбраны из одновалентных алкильных групп, содержащих от одного до 6 атомов углерода.
25. Контактная линза по п. 24, где бораты выбирают из группы, состоящей из триметилбората и триэтилбората.
26. Контактная линза по п. 24, где по меньшей мере две из групп R1, R2 и R3 двухвалентны и образуют совместно циклический борный эфир.
27. Контактная линза по п. 24, в которых по меньшей мере одна из групп R1, R2 и R3 представляет собой полимеризуемый углеводород.
28. Контактная линза по п. 24, где R1, R2 и R3 одинаковы.
29. Контактная линза по п. 24, где R1, R2 и R3 одновалентны.
30. Контактная линза по п. 24, в которых R1, R2 и R3 выбирают из групп, включающих этил и метил.
31. Контактная линза по п. 24, в которых R1, R2 и R3 выбирают из групп, включающих н-пропил, изопропил, бутил и трет-бутил.
32. Контактная линза по п. 24, в которых борат представлен борной кислотой.
33. Промежуточный материал контактных линз из отвержденного, оптически прозрачного силиконового гидрогеля, включающий:
отвержденный, оптически прозрачный продукт реакции полимеризации по меньшей мере одного гидрофильного компонента и по меньшей мере одного силиконового компонента, в котором по меньшей мере один из полимеризующихся компонентов содержит гидроксильную группу, а реакция полимеризации протекает в присутствии по меньшей мере приблизительно 5 мол.% ОН относительно гидроксильных групп бората, представленного формулой
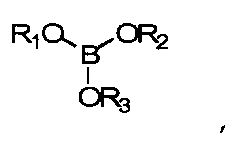
где R1, R2 и R3 независимо выбираются из числа групп, включающих водород и алкильные группы С1-С16; алкильные группы С1-С16 с замещением атомов углерода атомами О, N; полимеризуемые группы; эфирные группы, амидные группы, сложноэфирные группы;
отвержденные, оптически прозрачные контактные линзы, включающие по меньшей мере 2 мол.% бората в качестве остаточного продукта, который не остается навсегда в составе полимерного материала;
при этом силиконовый компонент выбирают из 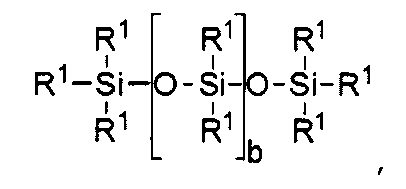
где b находится в диапазоне от 3 до 15, один концевой фрагмент R1 представляет собой одновалентную реакционноспособную группу, другой концевой фрагмент R1 представляет собой одновалентную алкильную группу, содержащую от одного до 6 атомов углерода, а остальные фрагменты R1 представляет собой одновалентные алкильные группы, содержащие от 1 до 3 атомов углерода; или
b равно нулю, один фрагмент R1 представляет собой одновалентную реакционноспособную группу, по меньшей мере три фрагмента R1 выбраны из одновалентных алкильных групп, содержащих от одного до 6 атомов углерода.
34. Способ формирования оптически прозрачного материала, включающий этапы:
получения смеси полимеризуемых компонентов, которая включает по меньшей мере один гидрофильный компонент и по меньшей мере один силиконовый компонент и в которой по меньшей мере один из полимеризуемых компонентов содержит одну гидроксильную группу, дополнительно такая смесь включает достаточное количество борной кислоты для получения более оптически прозрачного материала по сравнению с идентичной смесью, не содержащей борной кислоты;
отверждения смеси для получения отвержденного, оптически прозрачного материала;
при этом борат выбирают из
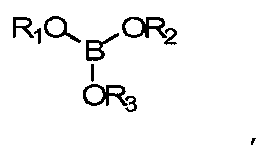
где R1, R2 и R3 независимо выбирают из групп, включающих водород; алкильные группы С1-С16; алкильные группы С1-С16 с замещением атомов углерода атомами О, N; полимеризуемые группы; эфирные группы, амидные группы, сложноэфирные группы;
силиконовый компонент выбирают из
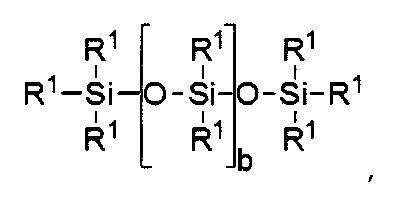
где b находится в диапазоне от 3 до 15, один концевой фрагмент R1 представляет собой одновалентную реакционноспособную группу, другой концевой фрагмент R1 представляет собой одновалентную алкильную группу, содержащую от одного до 6 атомов углерода, а остальные фрагменты R1 представляет собой одновалентные алкильные группы, содержащие от 1 до 3 атомов углерода; или
b равно нулю, один фрагмент R1 представляет собой одновалентную реакционноспособную группу, по крайней мере три фрагмента R1 выбраны из одновалентных алкильных групп, содержащих от одного до 6 атомов углерода.
35. Способ по п. 34, в котором указанную смесь полимеризуемых компонентов нагревают для ускорения реакции конденсации между указанной борной кислотой и гидроксильной группой.
36. Способ по п. 34, в котором указанную смесь полимеризуемых компонентов подвергают воздействию вакуума для удаления продуктов реакции конденсации указанной борной кислоты и указанных гидроксильных групп.
| CN 101526632 A, 09.09.2009 | |||
| US 6367929 B1, 09.04.2002 | |||
| WO 1999060048 A1, 25.11.1999 | |||
| ПОЛИМЕРНАЯ КОМПОЗИЦИЯ ДЛЯ МЯГКИХ КОНТАКТНЫХ ЛИНЗ ПРОДЛЕННОГО НОШЕНИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2007 |
|
RU2334770C1 |

