ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к соединениям, применяемым в медицине, имеет отношение к (S)-(-)-3-(3′-гидрокси)-бутилфталиду и образованному из него и кислоты сложному эфиру, и включает способ получения и применения данных соединений.
УРОВЕНЬ ТЕХНИКИ
Бутилфталид оказывает положительное действие при нарушении функций центральной нервной системы у пациентов с острым ишемическим инсультом, может способствовать восстановлению функций у пациентов, и по большей части превращается в условиях in vivo в два метаболита, 3-3-(3′-гидрокси)бутилфталид и 3-гидрокси-3-бутилфталид:
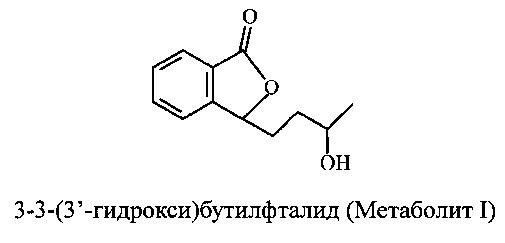

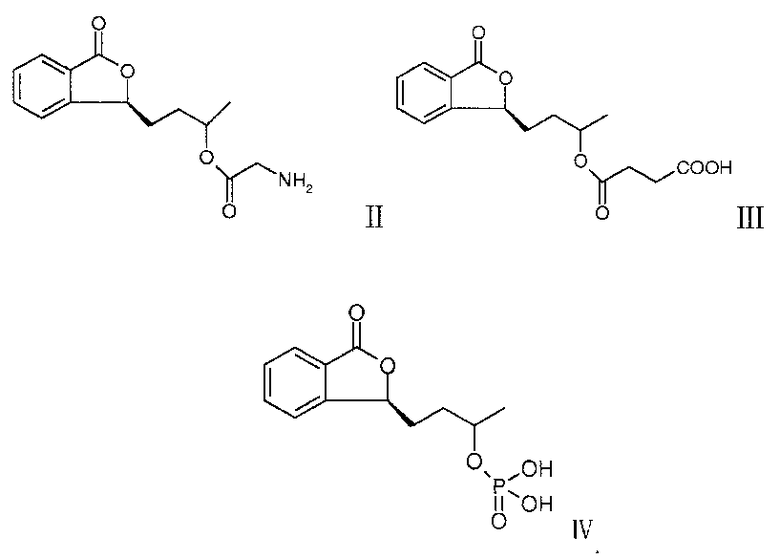
В 2008 году компания «Гуангдонг Жонгке Драг Ресерч энд Девелопмент Ко. Лтд» (Guangdong Zhongke Drug Research & Development Co. Ltd.) и компания «Шандонг Лве Нэчерэл Драг Ресерч энд Девелопмент Ко. Лтд» (Shandong Lvye Natural Drug Research & Development Co. Ltd.) синтезировали сложный эфир янтарной кислоты, сложный эфир глицина и сложный эфир фосфорной кислоты Метаболита I, и продемонстрировали их применение для предотвращения церебральных ишемических заболеваний. В Китайской заявке на патент 200410036628.3 описан новый способ применения гомологов бутилфталида: 3-(3′-гидрокси)бутилфталида и 3-гидрокси-3-бутилфталида. Доказано, что оба эти соединения обладают следующими действиями:
1) выраженное улучшение неврологической симптоматики у крыс, вызванной церебральной ишемией в результате травмы головного мозга;
2) улучшение памяти у крыс при нарушениях, вызванных церебральной ишемией;
3) уменьшение отека мозга у крыс, вызванного церебральной ишемией;
4) снижение частоты апоплексического инсульта у крыс, вызванного церебральной ишемией;
5) улучшение энергетического метаболизма у крыс, нарушения которого вызваны церебральной ишемией;
6) улучшение мозгового кровообращения в пораженном ишемией участке мозга;
7) уменьшение зоны ишемического инсульта у крыс с локальной церебральной ишемией и уменьшение симптомов неврологических расстройств;
8) антитромбоцитарное и антитромбогенное действие;
9) профилактика и лечение деменции.
Исследователи нашей компании обнаружили, что Метаболит I имеет два оптических изомера, и получили соединение 3-3-(3′-гидрокси)бутилфталид (Метаболит I) в обеих, S- и R-конфигурациях, как показано на фигурах ниже, методом асимметрического синтеза:
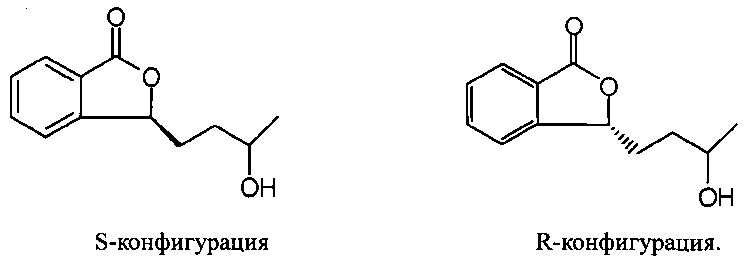
Более того, испытания на животных показали, что R-конфигурация не может влиять на объем церебрального инфаркта у крыс с церебральной ишемией и уменьшать симптомы церебральной ишемии, при этом S-конфигурация приводит к уменьшению симптомов церебральной ишемии и способствует уменьшению объема церебрального инфаркта у крыс с церебральной ишемией. Также обнаружено, что S-конфигурация Метаболита I (соединения по п. 1) улучшает сон.
S-конфигурация Метаболита I (соединения по п. 1) является маслянистой жидкостью и не растворима в воде. Поэтому, чтобы получить ее в дозированной форме для инъекций, мы провели следующие исследования по превращению данного вещества в водорастворимое соединение за счет образования сложного эфира в результате взаимодействия с кислотой с последующим образованием соли для удовлетворения требований к составу.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Задачей данного изобретения является изучение возможности использования производного 3-(3′-гидрокси)бутилфталида для профилактики и лечения ишемического инсульта и разработка способа его получения и применения.
Для решения указанной задачи в данном изобретении использованы следующие технические решения.
Получение соединения формулы 1, как показано на фигуре ниже,
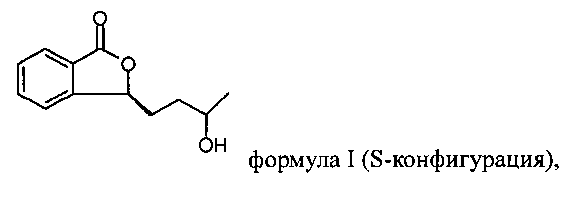
и получение соединения в R-конфигурации, как показано на фигуре ниже,

методом асимметрического синтеза.
Испытания на животных показали, что исследуемое соединение в R-конфигурации не способствует уменьшению объема церебрального инфаркта при церебральной ишемии у крыс, не может уменьшать симптомы церебральной ишемии и не улучшает сон, при этом S-конфигурация оказывает очень положительное действие на уменьшение симптомов церебральной ишемии и может улучшить режим сна у животных.
Соединение формулы 1 является маслянистым веществом, не растворимым в воде. Поэтому мы добавили к нему кислоту для получения сложного эфира, при этом кислота представляет собой фармацевтически приемлемую неорганическую или органическую кислоту. Неорганическая кислота представляет собой азотную кислоту, серную кислоту или фосфорную кислоту. Органическая кислота дополнительно содержит группу одного типа и по меньшей мере одну из амино-, гидрокси- или карбоксил-групп помимо кислотного радикала. Образованная из сложного эфира соль растворима в воде и может быть получена в дозированной форме для инъекций или в форме лиофилизированного порошка для инъекций.
Органическая кислота представляет собой аминокислоту, в частности: глицин, аланин, лизин, аргинин, серии, фенилаланин, пролин, тирозин, аспарагиновую кислоту, глутаминовую кислоту, гистидин, лейцин, метионин, треонин, пироглутаминовую кислоту, триптофан или валин.
При этом предпочтение отдается сложному эфиру, образованному из глицина, показанному на фигуре ниже:
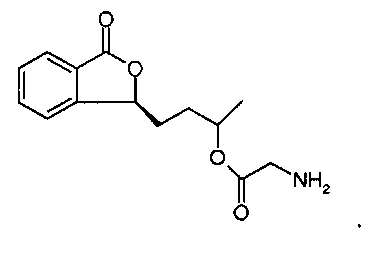
Органическая кислота также может представлять собой и двухосновную карбоновую кислоту, в частности: камфарную кислоту, яблочную кислоту, лимонную кислоту, малеиновую кислоту, янтарную кислоту, щавелевую кислоту, глутаровую кислоту, этандикислоту или малоновую кислоту.
При этом предпочтение отдается сложному эфиру, образованному из янтарной кислоты, показанному на фигуре ниже:
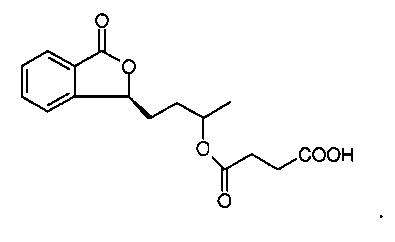
Органическая кислота также может представлять собой: памовую кислоту, гидроксинафтойную кислоту, гентизиновую кислоту, салициловую кислоту, гликолевую кислоту, миндальную кислоту, молочную кислоту, 4-ацетамидобензойную кислоту или никотиновую кислоту.
Соединение формулы I взаимодействует с неорганической кислотой, предпочтительно фосфорной, с образованием сложного эфира, показанного на фигуре ниже:
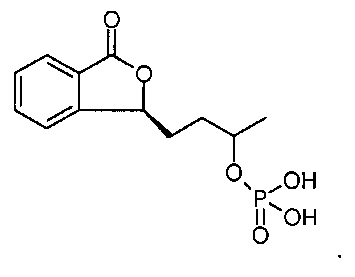
Вышеприведенный сложный эфир далее образует соль, поскольку необходимо получить водорастворимое соединение, чтобы решить проблему растворимости в воде, и в таком виде может быть использован для получения дозируемой формы для инъекций.
Сложный эфир, образованный из соединения формулы I и глицина, как правило, образует гидрохлорид, показанный на фигуре ниже:
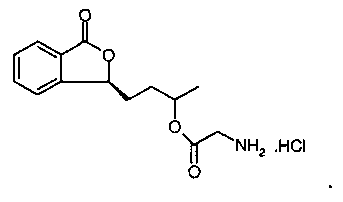
Согласно настоящему изобретению также предложена соль сложных эфиров двухосновных кислот соединения формулы I, образованная при взаимодействии с калием, натрием, магнием или органическим амином. Органический аминорадикал может представлять собой трометамин, диэтаноламин, триэтаноламин, глицин, лизин или аргинин.
При этом предпочтение отдается соли натрия, показанной на фигуре ниже:

Чтобы решить проблему растворимости, из сложного эфира, образованного из соединения I и фосфорной кислоты, далее можно в результате взаимодействия с физиологически приемлемым основанием получить соль, являющуюся солью натрия, солью калия, солью магния или солью органического амина. Органический амин может представлять собой: лизин, глицин, аргинин, трометамин, диэтаноламин или триэтаноламин.
Сложный эфир, полученный из соединения формулы I и фосфорной кислоты, преимущественно образует динатриевую соль, имеющую следующую структурную формулу:
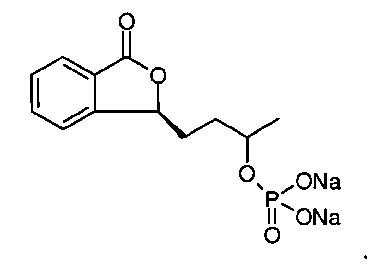
Согласно настоящему изобретению также предложено лекарственное средство для лечения ишемического инсульта, отличающееся тем, что содержит терапевтически эффективное количество соединения общей формулы (1) или его соль и фармацевтически приемлемый носитель. Фармацевтическая композиция может быть в пероральной или инъекционной лекарственной форме.
Исследования раздражения мышечной и сосудистой систем показали, что лекарственное средство вводится инъекционным путем после сложного эфира, полученного из соединения формулы I и кислоты, образующего в дальнейшем соль. Раздражений ни мышечной, ни сосудистой системы не наблюдалось, поэтому лекарственное средство может быть использовано в инъекционной лекарственной форме.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Пример 1: Получение различных соединений
Представленные в данном изобретении соединения очень похожи. Поэтому для того, чтобы подробно, точно и понятно описать способ получения различных соединений, мы представили его одним примером. В следующей цепочке превращения каждое соединение обозначено порядковым номером. Соединения заменены порядковыми номерами для более четкой иллюстрации способа ниже.
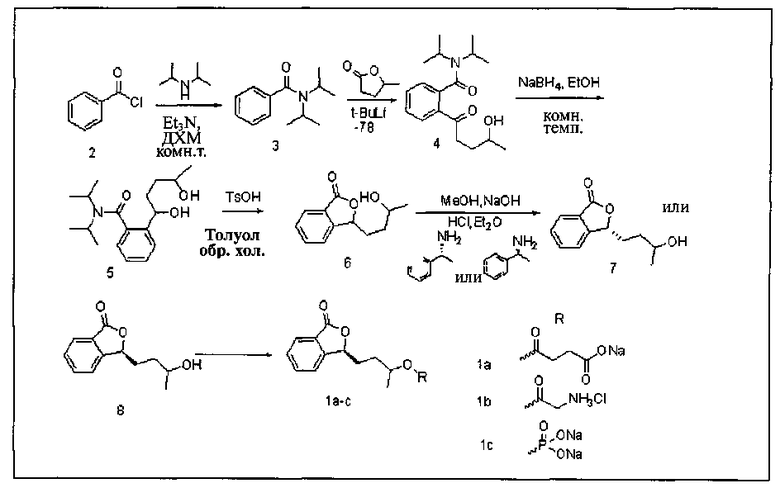
Если на вышеприведенной схеме вместо R подставить различные группы, получаются различные соединения 1a, 1b и 1с.
(1). Получение Соединения 3:
Диизопропиламин (1,3 моль) помещали в круглодонную колбу объемом 2000 мл и добавляли 1000 мл безводного дихлорметана. После растворения добавляли триэтиламин (2,0 моль) и по каплям добавляли Соединение 2 (1,0 моль) на ледяной бане. После этого реакционную смесь нагревали до комнатной температуры и перемешивали в течение ночи. 500 мл дихлорметана добавляли для разведения, и промывали реакционную смесь 5% разбавленной хлороводородной кислотой (500 мл × 1), водой (500 мл × 1) и соляным раствором (500 мл × 1), соответственно, высушивали над безводным сульфатом натрия и концентрировали до получения 202,4 г Соединения 3. Выход неочищенного продукта составлял 99%, МС (m/z): 206,1.
(2). Получение Соединения 4:
Соединение 3 (0,8 моль) растворяли в 500 мл сухого тетрагидрофурана и постепенно по каплям добавляли трет-бутиллитий (1,0 моль) при -78ºС. После этого добавляли тетраметилэтилендиамин (1,2 моль) и перемешивали смесь при -78ºС в течение 30 мин. Лактон (1,0 моль) добавляли по каплям в полученную смесь, постепенно нагревали до комнатной температуры и проводили реакцию в течение 5 ч. Реакцию прерывали добавлением насыщенного раствора хлорида аммония и удаляли органический растворитель перегонкой под уменьшенным давлением. Остаток экстрагировали этилацетатом, концентрировали при пониженном давлении и рекристаллизовывали с получением 222,4 г Соединения 4. Выход составлял 91%.
ЯМР (400 Гц, CDCl3): 8,08-8,06 (м, 1H), 8,04-8,02 (м, 1H), 7,63-7,61 (м, 1Н), 7,60-7,58 (м, 1H), 3,95-3,93 (м, 2Н), 3,40-3,38 (м, 1H), 2,55 (т, J=l,2 Гц, 2Н), 1,64-1,62 (м, 2Н), 1,25(д, J=l,5 Гц, 12Н), 1,21(д, J=l,6 Гц, 3Н); MC (m/z): 306,2.
(3). Получение Соединения 5:
220 г Соединение 4 (0,7 моль) растворяли в 500 мл метанола, борогидрит натрия (1,40 моль) добавляли малыми порциями на ледяной бане и перемешивали в течение ночи при комнатной температуре под слоем газообразного азота. По завершении реакции по каплям добавляли примерно 20 мл концентрированной хлороводородной кислоты, чтобы разложить избыток борогидрита натрия. Метанол удаляли перегонкой под уменьшенным давлением. Остаток экстрагировали дихлорметаном, промывали водой, промывали соляным раствором, высушивали над безводным сульфатом натрия, концентрировали и рекристаллизовывали с получением 204,8 г Соединения 5. Выход составлял 95%. MC (m/z): 308,2.
(4). Получение Соединения 6:
Соединение 5 (0,60 моль) растворяли в 500 мл толуола, добавляли п-толуолсульфоновую кислоту в каталитической концентрации (примерно 1%), смесь немного кипятили и нагревали в колбе с обратным холодильником под слоем газообразного азота в течение 8 дней. Толуол удаляли перегонкой под уменьшенным давлением, дихлорметан добавляли для разбавления смеси, остатки промывали водой, промывали соляным раствором, высушивали над безводным сульфатом магния и пропускали через колонну с получением 106,4 г Соединения 6. Выход составлял 86%. MC (m/z): 207,2.
(5). Получение Соединения 7:
Соединение 6 (0,2 моль) растворяли в 200 мл метанола, при перемешивании добавляли водный раствор гидроксида натрия (16 г/40 мл) в течение 15 мин. Систему нагревали до гомогенного раствора и проводили реакцию при комнатной температуре в течение 2 ч. Реакцию прерывали, и удаляли метанол перегонкой под уменьшенным давлением. В остаточный раствор добавляли дистиллированную воду в нужном для разбавления количестве. Систему охлаждали до -5ºС или ниже, доводили кислотность до рН=3~4 с помощью 5% раствора хлороводородной кислоты, и экстрагировали диэтиловым эфиром (100 × 3). Фракции с диэтиловым эфиром собирали, и охлаждали раствор до -5ºС или ниже. 0,2 моль (R)-(+)-α-фенилэтиламина добавляли медленно по каплям. Температуру системы поддерживали -5ºС или ниже, смесь оставили на 3 ч. Образовалось множество кристаллов, и систему отфильтровывали, чтобы собрать кристаллы. Кристаллы дважды рекристаллизовывали ацетоном или этилацетатом с получением 20,3 г кристаллов, концентрация кристаллов составляла 15 г кристалл/100 мл растворителя. Кристаллы растворяли в 10 объемах дистиллированной воды и добавляли гидроксид натрия для достижения рН=13. (R)-(+)-α-Фенилэтиламин регенерировали экстракцией в диэтиловом эфире, кислотность водной фазы доводили до рН=2 с помощью хлороводородной кислоты, экстрагировали, высушивали и концентрировали с получением неочищенного продукта (R)-(+)-3-(3′-гидрокси)бутилфталида. Неочищенный продукт рекристаллизовывали этанолом с получением 8,7 г Соединения 7. Выход составлял 21%. [α]D=+66,80 (с=1,02, СН3ОН).
ЯМР (400 Гц, CDCl3): 7,91-7,89 (м, 1H), 7,41-7,39 (м, 1H), 7,33-7,31 (м, 1H), 7,30-7,28 (м, 1Н), 5,24 (т, J=1,2 Гц, 1H), 3,40-3,38 (м, 1Н), 2,55 (т, J=1,2 Гц, 2Н), 2,05-2,03 (м, 2Н), 1,45-1,43 (м, 2Н), 1,21(д, J=1,6 Гц, 3Н); MC (m/z): 207,2.
(6). Получение Соединения 8:
Соединение 6 (0.2 моль) растворяли в 200 мл метанола, при перемешивании добавляли водный раствор гидроксида натрия (16 г/40 мл) в течение 15 мин. Систему нагревали до гомогенного раствора, и проводили реакцию при комнатной температуре в течение 2 ч. Реакцию прерывали, и метанол удаляли перегонкой под уменьшенным давлением. В остаточный раствор добавляли дистиллированную воду в нужном для разбавления количестве. Систему охлаждали до -5ºС или ниже и доводили кислотность до рН=3~4 с помощью 5% раствора хлороводородной кислоты, и экстрагировали диэтиловым эфиром (100×3). Фракции с диэтиловым эфиром собирали, и охлаждали раствор до -5ºС или ниже. 0,2 моль (S)-α-фенилэтиламина добавляли медленно по каплям. Температуру системы поддерживали -5ºС или ниже, смесь оставляли на 3 ч. Образовывалось множество кристаллов, и, чтобы собрать кристаллы, систему отфильтровывали. Кристаллы дважды рекристаллизовывали ацетоном или этилацетатом с получением 20,3 г кристаллов, концентрация кристаллов составляла 15 г кристалл/100 мл растворителя. Кристаллы растворяли в 10 объемах дистиллированной воды и добавляли гидроксид натрия для достижения рН=13. (S)-α-Фенилэтиламин регенерировали экстракцией в диэтиловом эфире, кислотность водной фазы доводили до рН=2 с помощью хлороводородной кислоты, водную фазу экстрагировали, высушивали и концентрировали с получением неочищенного продукта (S)-3-(3′-гидрокси)бутилфталида. Неочищенный продукт рекристаллизовывали этанолом с получением 8,7 г Соединения 7. Выход составлял 21%. [α]D=-66,80 (с=1,02, СН3ОН).
ЯМР (400 Гц, CDCl3): 7,91-7,89 (м, 1Н), 7,41-7,39 (м, 1Н), 7,33-7,31 (м, 1H), 7,30-7,28 (м, 1H), 5,24 (т, J=l,2 Гц, 1H), 3,40-3,38 (м, 1Н), 2,55 (т, J=1,2 Гц, 2Н), 2,05-2,03 (м, 2Н), 1,45-1,43 (м, 2Н), 1,21(д, J=l,6 Гц, 3Н); MC (m/z): 207,2.
(7). Получение Соединения 1a:
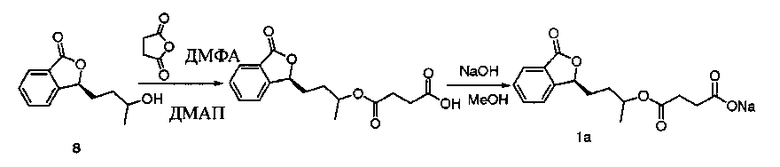
10,0 ммоль Соединения 8, 10,0 ммоль ангидрида бутандикислоты и 10 ммоль ДМАП растворяли в 50 мл ДМФА, нагревали до 85ºС, и проводили реакцию в течение 6 ч. Реакцию прерывали и наливали реакционный раствор в 200 мл ледяной воды, кислотность доводили до рН=2~3 с помощью 0,1 Η хлороводородной кислоты, проводили экстракцию этилацетатом (100 мл × 3). Органическую фазу собирали, трижды промывали соляным раствором, высушивали над безводным сульфатом натрия, перегоняли при пониженном давлении с получением (S)-3-(3′-сукцинат)бутилфталида, который рекристаллизовывали с образованием 2,8 г белого порошка. Выход составлял 91%.
ЯМР (400 Гц, CDCl3): 7,8-7,78 (м, 1H), 7,65-7,63 (м, 1H), 7,49-7,47 (м, 1H), 7,44-7,42 (м, 1H), 5,50-5,46 (м, 1H), 3,82-3,74 (м, 1H), 2,72-2,66 (м, 2Н), 2,62-2,56 (м, 2Н), 2,05-2,03 (м, 2Н), 1,45-1,43 (м, 2Н), 1,21 (д, J=1,6 Гц, 3Н); MC (m/z): 307,3.
Ранее описанный (S)-3-(3′-сукцинат)бутилфталид растворяли в 50 мл метанола и 4 мл 10% раствора гидроксида натрия, нагревали в целях регенерации в течение 2 ч и концентрировали с получением натриевой соли (S)-3-(3′-сукцинат)бутилфталида (Соединения 1a), MC (m/z): 305,3.
(8). Получение Соединения 1b:

10,0 ммоль Соединения 8 растворяли в 50 мл этилацетата и добавляли 1,0 мл пиридина и 10 ммоль ДМАП. Смесь охлаждали до 0~5ºС, при перемешивании добавляли 10,0 ммоль глицина и проводили реакцию в течение 6 ч, температуру поддерживали 0~5ºС. Реакцию прерывали, реакционный раствор выливали в ледяную воду объемом 100 мл, кислотность доводили до рН=7,0 с помощью 0,1 Η хлороводородной кислоты, смесь отстаивали для отделения органического слоя. Водную фазу экстрагировали этилацетатом (50 мл × 3). Органическую фазу собирали, трижды промывали соляным раствором, высушивали над безводным сульфатом натрия, перегоняли при пониженном давлении с получением (S)-3-(3′-глицинат)бутилфталида, который рекристаллизовывали из этанола с получением 2,3 г белого порошка. Выход составлял 87%.
ЯМР (400 Гц, CDCl3): 7,8-7,78 (м, 1H), 7,65-7,63 (м, 1H), 7,49-7,47 (м, 1H), 7,44-7,42 (м, 1H), 5,50-5,46 (м, 1H), 3,82-3,74 (м, 1H), 3,62 (с, 2Н), 2,05-2,03 (м, 2Н), 1,45-1,43 (м, 2Н), 1,21 (д, J=l,6 Гц, 3Н); MC (m/z): 264,3.
Вышеописанный (S)-3-(3′-глицинат)бутилфталида растворяли в 50 мл ацетона и 10 мл диэтилового эфира, и по каплям добавляли диэтиловый раствор гидрохлорида для достижения рН=2. Образовывалось обильное количество белого твердого осадка, который отфильтровывали, высушивали с получением 2,4 г гидрохлорида (S)-3-(3′-глицинат)бутилфталида (Соединения 1b). Выход составлял 80%. MC (m/z): 264,3.
(9). Получение Соединения 1с:
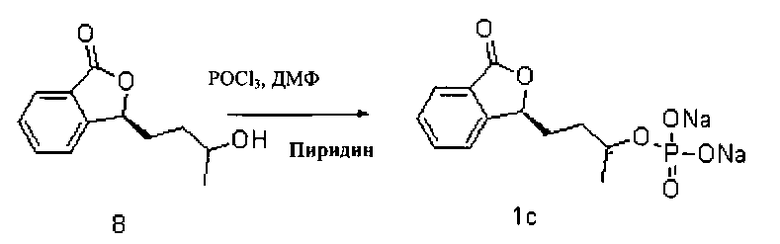
20,0 мл пиридина добавляли к 100 мл ДМФА, охлаждали до -10ºС и при перемешивании добавляли 25,0 мл оксихлорида фосфора. Смесь перемешивали в течение 30 мин и равномерными порциями добавляли 10,0 ммоль Соединения 8. Смесь перемешивали в течение 3 ч, после чего реакцию прерывали. Реакционную смесь выливали в ледяную воду объемом 100 мл, кислотность доводили до рН=2 с помощью 0,1 Η хлороводородной кислоты, и экстрагировали этилацетатом (50 мл × 3). Органическую фазу собирали, трижды промывали соляным раствором и высушивали над безводным сульфатом натрия. Растворитель удаляли перегонкой под уменьшенным давлением с получением (S)-3-(3′-фосфат)бутилфталида. После чего его растворяли в 20 мл этанола, добавляли 10,6 карбоната натрия, реакцию проводили в течение 2 ч при температуре 30ºС. Реакцию прерывали, и добавляли 100 мл ацетона. Смесь оставляли для осаждения при 5ºС, отфильтровывали, высушивали в вакууме с получением 2,8 г динатриевой соли (S)-3-(3′-фосфат)бутилфталида (Соединение 1с), который представлял из себя белый порошок. Выход составлял 85%.
ЯМР (400 Гц, CDCl3): 7,91-7,89 (м, 1H), 7,41-7,39 (м, 1Н), 7,33-7,31 (м, 1H), 7,30-7,28 (м, 1Н), 5,24 (т, J=1,2 Гц, 1Н), 3,40-3,38 (м, 1H), 2,05-2,03 (м, 2Н), 1,65-1,63 (м, 2Н), 1,21 (д, J=1,6 Гц, 3Н); MC (m/z): 331,2.
Пример 2: Получение лиофилизированного порошка для инъекций (S)-(-)-3-(3′-динатрийфосфат)бутилфталида (Соединения 1с в Примере 1)
Взвешивали 10 г полученного (S)-3-(3′-фосфат)бутилфталида натрия (Соединения 1с), добавляли 1000 мл воды для инъекций для растворения, после чего дополнительно добавляли 60 г маннитола. По завершении растворения дополнительно добавляли воду для инъекций до объема 1200 мл. После декарбонизации активированным углем смесь фильтровали через микропористую мембрану, помешали в 7 мл пенициллиновые флаконы по 3 мл раствора, флаконы закупоривали, смесь лиофилизировали и герметично упаковывали с получением лиофилизированного порошка. Спецификация: 20 мг/флакон.
Пример 3: Получение капсул (S)-3-(3′-динатрийфосфат)бутилфталида (Соединение 1с)
Формула: (S)-3-(3′-фосфат)бутилфталид натрия 60 г

Взвешивали точное количество (S)-3-(3′-фосфат)бутилфталид натрия согласно формуле, вещество просеивали через сито в 100 меш, и при равномерном перемешивании добавляли необходимое согласно формуле количество лактозы, высушенной при 80ºС и просеянной через сито в 80 меш. Определяли состав смеси и, если он удовлетворял критериям, смесь засыпали в оболочки капсул №1 с получением капсул.
Пример 4: Получение инъекционного раствора (S)-3-(3′-динатрийфосфат)бутилфталида (Соединения 1с)
Формула: (S)-3-(3′-фосфат)бутилфталид натрия 50 г

Взвешивали точное количество (S)-3-(3′-фосфат)бутилфталид натрия (Соединения 1с) согласно формуле, и добавляли нужное количество воды для инъекций. рН доводили до 6,5-7,2, и добавляли воду для инъекций до 4000 мл. Добавляли 2 г активированного угля для инъекций, смесь кипятили в течение 15 мин, декарбонизировали фильтрованием с отсасыванием. Раствор фильтровали через микропористую мембрану с размером пор 0,22 мкм, помещали в стеклянные ампулы, которые герметично закупоривали, и обрабатывали в автоклаве при 115ºС в течение 30 мин с получением раствора для инъекций.
Пример 5: Влияние на объем церебрального инфаркта у крыс с локальным церебральным инфарктом
(1) Экспериментальные материалы и метод
Крыс Уистара с массой тела 250-280 г выращивали отдельно до и после хирургического вмешательства и содержали при комнатной температуре 23-25ºС. Все крысы имели свободный доступ к еде и воде. Модели транзитной окклюзии средней мозговой артерии, tMCAO, получали согласно методу Лонго и соавт. Крыс анастезировали 10% хлоральгидратом (350 мг/кг, интраперитонеально), температуру тела поддерживали при 37±0,5ºС. Крыс фиксировали на операционном столе в положении «лежа на спине». Кожу разрезали по середине шеи, и аккуратно отделяли общую сонную артерию (ССА), наружную сонную артерию (ЕСА) и внутреннюю сонную артерию (ICA) с правой стороны. ЕСА перевязывали, вырезали, и вытягивали прямо в линию с ICA. На ЕСА делали небольшой разрез и продевали силифицированную нейлоновую нить длиной 4,0 см с закругленным наконечником и диаметром 0,26 мм (покрытую 0,1% полилизином) через это отверстие в ICA примерно на 1,85-2,00 см до передней части мозговой артерии крысы, чтобы заблокировать поступление крови средней мозговой артерии. Через 2 ч ишемии нейлоновую нить осторожно снимали, ЕСА перевязывали и накладывали швы на операционный надрез. Животное помещали обратно в клетку на 24 ч реперфузии.
(2) Экспериментальные группы и введение
Крыс в произвольном порядке делили на 12 групп: модельную контрольную группу, которой вводили воду для инъекций (100 мг/кг); группу Соединения 7 в Примере 1 (25, 50, 100 мг/кг); группу Соединения 8 в Примере 1 (25, 50, 100 мг/кг); группу DL-3-(3′-гидрокси)-бутилфталида (DL сокращенно) (25, 50, 100 мг/кг). Все вещества вводились перорально через 10 мин после ишемии, вызванной окклюзией средней мозговой артерии СМА.
(3) Определение объема церебрального инфаркта
После реперфузионного повреждения у крысы, длившегося 24 ч, крысу обезглавливали и сразу же вынимали мозг. Удаляли обонятельный тракт, мозжечок и нижний отдел ствола головного мозга. Мозг в фронтальной плоскости разрезали на 6 сегментов (с первого по пятый сегмент размер составлял 2 мм/часть, а размер шестого был 4 мм), которые быстро помещали в 5 мл раствора, содержащего 1,5 мл 4% ТТХ и 0,1 мл 1 Μ раствора К2НРО4 для окрашивания (37ºС, защищено от света) на 20~30 мин, при этом их переворачивали один раз в каждые 5 мин. После окрашивания ТТХ нормальная ткань была темной и приобрела красный цвет, а ткань с инфарктным поражением приобрела белый цвет. Каждую группу образцов мозга устанавливали по порядку и фотографировали для сохранности. Рассчитывали размер зоны инфаркта в каждом сегменте и, в конечном итоге, вычисляли объем инфаркта интегрированием площади инфаркта. Объем инфаркта выражали как процент полушария головного мозга, чтобы исключить влияния отека головного мозга.
Объем инфаркта головного мозга (%) = (объем неоперированного полушария - объем неинфарктной части оперированного полушария)/объем неоперированного полушария * 100%
(4) Результаты экспериментов
Через 2 ч после ишемии и 24 ч после реперфузии объем церебрального инфаркта контрольной группы растворителя составлял 33,8%. В группе с имитацией операции не наблюдалось церебрального инфаркта. Результаты по объему церебрального инфаркта в других группах представлены в Таблице 1:

По сравнению с контрольной группой растворителя пероральное введение в группы Соединения 8 и DL-3-(3′-гидрокси)-бутилфталида может значительно уменьшить объем церебрального инфаркта, а в группы Соединения 7 - не приводит к эффективному снижению объема церебрального инфаркта. Группы (-)-3-(3′-гидрокси)-бутилфталида (например, Соединения 8) имели результаты, лучшие, чем группы DL-3-(3′-гидрокси)-бутилфталида, и это свидетельствует о том, что Соединение 8 в S конфигурации является эффективным действующим веществом, тогда как в случае Соединения 7 в R конфигурации не было обнаружено эффекта снижения объема церебрального инфаркта, вызванного церебральной ишемией.
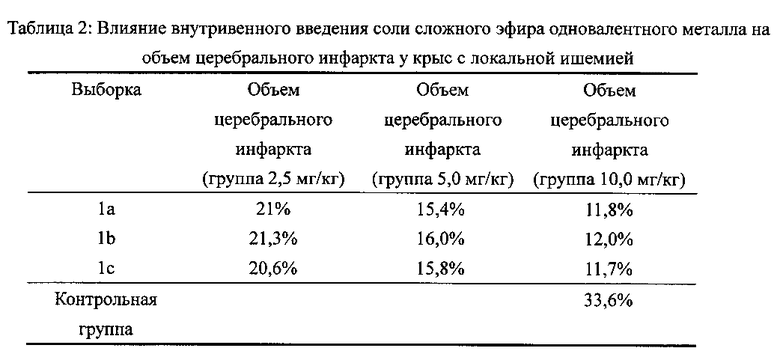
Пример 12: Влияние инъекционного введения 1a, 1b, 1с на объем церебрального инфаркта
(1) Экспериментальные группы и введение
Брали крыс модели фокальной ишемии Примера 11, и в произвольном порядке делили их на 12 групп: группу имитации операции, модельную контрольную группу, (вода для инъекций, 10 мг/кг), 1а группу (2,5 мг/кг, 5,0 мг/кг, 10 мг/кг), lb группу (2,5 мг/кг, 5,0 мг/кг, 10 мг/кг) и 1 с группу (2,5 мг/кг, 5,0 мг/кг, 10 мг/кг). Вещества вводили внутривенно через 10 мин после ишемии, вызванной окклюзией средней мозговой артерии СМА.
(2) Результаты и обсуждение
Определение объема церебрального инфаркта аналогично приведенному в Примере 4. Через 2 ч после ишемии и 24 ч после реперфузии объем церебрального инфаркта контрольной группы растворителя составлял 33,6%. В группе с имитацией операции не наблюдалось церебрального инфаркта. По сравнению с контрольной группой растворителя в каждой из групп может значительно снижаться объем церебрального инфаркта. По сравнению с контрольной группой растворителя во всех выборках может значительно снижаться объем церебрального инфаркта, как показано в Таблице 2.
Пример 13: Исследования на раздражение сосудистой системы соединениями 1a, 1b и 1с
1) Моделирование экспериментов
Взвешивали 1a, 1b и 1с и растворяли в воде для инъекций с получением растворов разных концентраций для двух групп. Группа высокой концентрации: 4,2 мг/мл, группа низкой концентрации: 1,4 мг/мл. Соединения вводили внутривенно в ушные вены кроликов в дозировке 5 мл.
2) Способ применения
Отбирали 8 здоровых новозеландских кроликов и соответственно инъекционно вводили исследуемое лекарственное средство высокой и низкой концентрации в ушную вен левого уха кроликов, тогда как такой же объем инъекционного раствора хлорида натрия инъекционно вводили в ушную вену правого уха кроликов. 8 кроликам успешно вводили исследуемое лекарственное средство высокой и низкой концентрации, после чего им соответственно вводили 0,9% инъекционный раствор хлорида натрия. Такую процедуру проводили раз в день 3 дня подряд. Кроликов соответственно взвешивали перед введением лекарственного средства, через 48 ч и через 14 дней после введения лекарственного средства.
3) Общие наблюдения и подготовка образцов
Каждый день до введения лекарственного средства проводили и записывали наблюдения за поведением животных и реакцией кровеносных сосудов в месте введения инъекции. Через 48 ч после последнего введения лекарственного средства 2 новозеландским кроликам, которым лекарственное средство было введено в высокой и низкой концентрации, соответственно, провели кровопускание. Проводили и записывали визуальные наблюдения за реакцией ткани кровеносных сосудов, затем кроликам ампутировали оба уха у основания (вначале левое ухо, затем правое ухо, и наносили на них метки). Затем часть образца уха кролика отрезали, соответственно, и помещали в 10% нейтральный раствор формальдегида (размер полученного образца составлял 8 см в длину и 1 см в ширину; разрез на дистальном конце находился на расстоянии примерно 0,5 см от первого укола, разрез на проксимальном конце находился на расстоянии примерно 2 см от третьего укола, а проксимальный конец служил концом для подвешивания). 2-х животных, которым вводили исследуемое лекарственное средство высокой и низкой концентрации, соответственно, оставляли для дальнейших наблюдений на 14 дней после введения лекарственного средства и проводили медико-патологическое исследование.
С дистального конца в месте первого укола отрезали один сегмент; и с проксимального конца в месте третьего укол отрезали два сегмента; во время подготовки сегментов кровеносный сосуд разрезали поперек. Сегменты препарировали в обычном парафине, толщина сегмента составляла 4~5 мкм. Перед проведением гистологических исследований образцы окрашивали гематоксилином и эозином.
5) Оценка результатов
Проводили тщательную оценку визуальных наблюдений и патологических исследований. Каждый день до введения лекарственного средства проводили и записывали наблюдения за реакцией стенки кровеносных сосудов в месте введения инъекции. Во время введения лекарственного средства, согласно визуальному осмотру, внутренняя сторона и внешняя сторона васкулярного эпидермиса выглядели красными в месте введения инъекции на левом ухе испытуемых кроликов и контрольном ухе некоторых животных, которым вводили исследуемое лекарственное средство высокой и низкой концентрации, зона покраснения составляла от ОД см × 0,2 см до 0,2 см × 1,0 см. Через 48 ч после последнего введения сосудистый профиль кровеносных сосудов с обеих сторон ушей 4-х кроликов, которым вводили исследуемое лекарственное средство высокой и низкой концентрации, был чистым, толщина ушей кроликов была равномерной, и никаких значительных изменений не наблюдалось. Через 14 дней после последнего введения лекарственного средства проводили некропсию 4-х кроликов, которым вводили исследуемое лекарственное средство высокой и низкой концентрации. Сосудистый профиль кровеносных сосудов с обеих сторон ушей кроликов был относительно чистым, толщина ушей кроликов была равномерной и никаких значительных изменений не наблюдалось.
Некропсию 4-х кроликов, которым вводили исследуемое лекарственное средство высокой и низкой концентрации, проводили через 40 ч после последнего введения препарата, и некропсию других 4-х кроликов, которым вводили исследуемое лекарственное средство высокой и низкой концентрации, проводили в конце второй недели восстановительного периода. Ни одно гистологическое исследование не показало существенных раздражений таких, как дегенерация и некроз тканей сосудов.
Пример 13: Влияние Соединения 7, Соединения 8 и DL-3-(3′-гидрокси)-бутилфталида на сон крыс:
Эксперименты по улучшению сна
Свойства образцов: содержимое капсул с Соединением 7 и Соединением 8 настоящего изобретения представляет собой коричневые частицы.
Поставщик животных: мыши из г. Куньмин, 18~22 г, мужского пола, являлись чистыми животными и поставлялись из лаборатории «Guangdong Medical Lab Animal Center». В помещении, где разводились животные для эксперимента, температура составляла 22±2ºС, относительная влажность была 55~70%, и корм для животных поставляла лаборатория «Guangdong Medical Lab Animal Center».
В эксперименте принимало участие 3 группы: Соединения 7, Соединения 8, DL-3-(3′-гидрокси)-бутилфталида (DL сокращенно), 25 мг/кг, соответственно. И дополнительно проводили эксперимент с дистиллированной водой в контрольной группе.
Подготовка образца: взвешивали 25 мг соответствующего образца и добавляли к нему 20 мл дистиллированной воды с получением гомогенной суспензии, используемой в исследовании.
Способ введения: гаваж
Экспериментальный метод:
Исследование с помощью анестезии пентобарбиталом натрия в надпороговой дозировке:
Отбирали 40 мышей мужского пола с массой тела 18-22 г и в произвольном порядке делили их на 4 группы, по 10 мышей в каждой группе. В течение 30 дней им в последовательном порядке вводили образцы и через 15 мин после введения образцов методом гаважа на 30 день каждой группе животных вводили интраперитонеально инъекции пентобарбитала натрия в дозировке 50 мг/кг массы тела, вводимое путем инъекции количество составляло 0,2 мл/20 г массы тела. Засыпание крыс определяли по исчезновению установочного рефлекса у мышей на 1 мин и более. В течение 60 мин после введения пентобарбитала натрия проводили наблюдения за периодом засыпания и периодом сна каждой группы животных.
Результаты:
Действие образцов в зависимости от массы тела животных
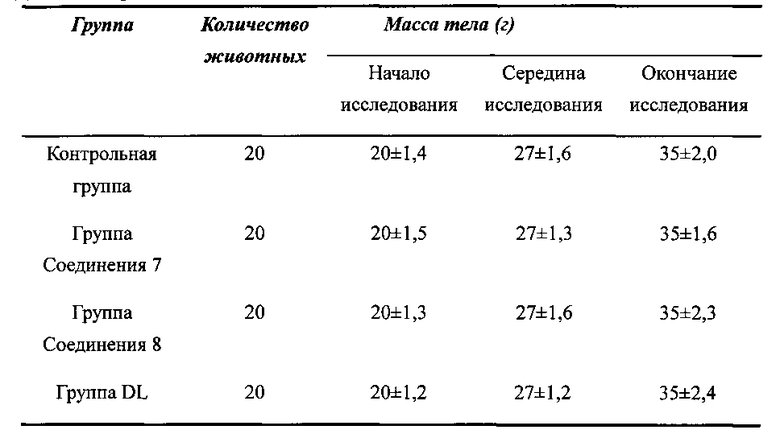
Как показано в таблице выше, масса тела животных каждой группы несильно отличается от массы тела животных контрольной группы.
Влияние на период сна мышей, вызванного действием пентобарбитала натрия в надпороговой дозировке
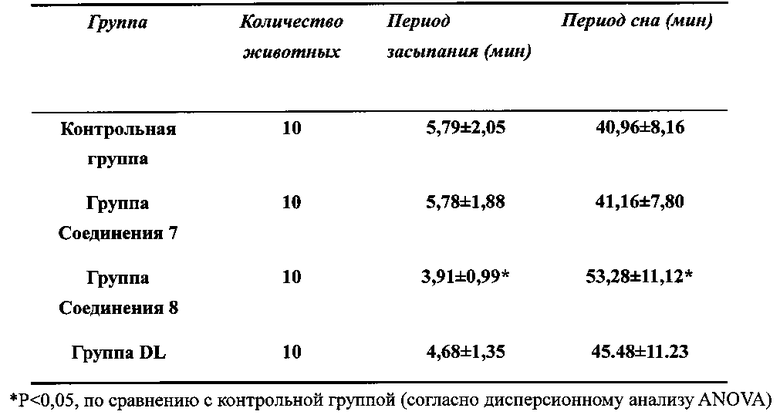
Как показано в таблице выше, период засыпания и период сна животных, вызванного действием пентобарбитала натрия в надпороговой дозировке в группе Соединения 8 ((S)-(-)-3-(3′-гидрокси)-бутилфталида)), значительно отличается от соответствующих показателей контрольной группы.
Исследование анестезией пентобарбиталом натрия в допороговой дозировке:
Отбирали 40 мышей мужского пола с массой тела 18~22 г и в произвольном порядке делили их на 4 группы, по 10 мышей в каждой группе. В течение 28 дней им в последовательном порядке вводили образцы и через 15 мин после введения образцов методом гаважа на 30 день каждой группе животных вводили интраперитонеально инъекции пентобарбитала натрия в дозировке 30 мг/кг массы тела, вводимое путем инъекции количество составляло 0,2 мл/20 г массы тела. Засыпание крыс определяли по исчезновению установочного рефлекса у мышей на 1 мин и более. В течение 25 мин после введения пентобарбитала натрия проводили наблюдения за периодом засыпания и периодом сна каждой группы животных.
Результаты
Влияние на частотность засыпания, вызванного действием пентобарбитала натрия в допороговой дозировке
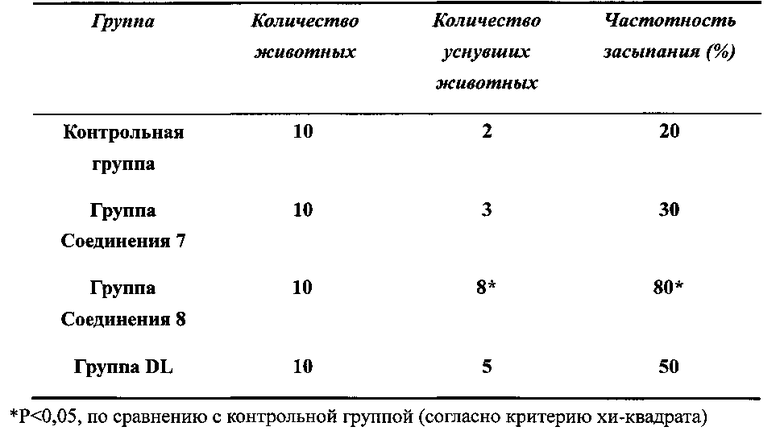
Как показано в таблице выше, количество уснувших животных и частотность засыпания животных, вызванного действием пентобарбитала натрия в допороговой дозировке в группах Соединения 8 ((S)-(-)-3-(3′-гидрокси)-бутилфталида)) и DL, значительно отличается от соответствующих показателей контрольной группы.
Выводы: пероральное введение образцов в течение 30 дней мышам групп Соединения 8 ((S)-(-)-3-(3′-гидрокси)-бутилфталид)) и DL приводят к улучшению сна. Влияние S конфигурации сильнее влияния рацемической DL конфигурации, а влияние DL конфигурации сильнее влияния R конфигурации.
Изобретение относится к новому соединению формулы I, а именно (S)-(-)-3-(3′-гидрокси)-бутилфталиду и сложному эфиру, образованному из соединения формулы I и кислоты, которая представляет собой фармацевтически приемлемую неорганическую или органическую кислоту. Неорганическая кислота относится к: азотной кислоте, серной кислоте или фосфорной кислоте. Помимо кислотного радикала органическая кислота содержит по меньшей мере одну из аминогруппы, гидрокси-группу и карбокси-группу. Ни соединение, представленное формулой I, ни образованный из него сложный эфир не растворимы в воде и могут применяться для лечения и профилактики церебральных ишемических заболеваний и улучшают сон. Сложный эфир, полученный из соединения и кислоты, дополнительно взаимодействует с кислотой или основанием с получением соли, которая растворима в воде и используется для получения инъекционного лекарственного средства. Эксперимент показал, что соль не вызывает раздражения сосудистой системы. Изобретение также относится к фармацевтической композиции на основе полученных соединений. 7 н. и 4 з.п. ф-лы, 5 табл. 13 пр.
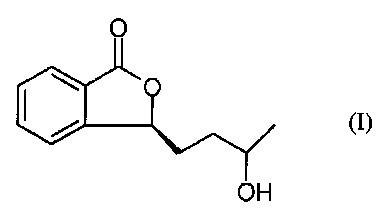
1. Соединение для профилактики и лечения церебрального ишемического заболевания, такое, как показано ниже:
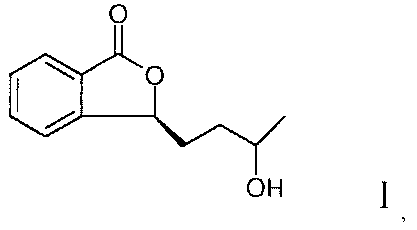
химическое название которого (S)-(-)-3-(3′-гидрокси)-бутилфталид.
2. Сложный эфир, полученный в результате взаимодействия соединения по п. 1 с кислотой, выбранной из фармацевтически приемлемых неорганических кислот или органических кислот.
3. Сложный эфир по п. 2, отличающийся тем, что неорганическая кислота выбрана из группы, состоящей из: азотной кислоты, серной кислоты или фосфорной кислоты; органическая кислота выбрана из группы, состоящей из: глицина, аланина, лизина, аргинина, серина, фенилаланина, пролина, тирозина, аспарагиновой кислоты, глутаминовой кислоты, гистидина, лейцина, метионина, треонина, пироглутаминовой кислоты, триптофана или валина; двухосновная кислота: камфарная кислота, яблочная кислота, лимонная кислота, малеиновая кислота, янтарная кислота, щавелевая кислота, глутаровая кислота, этандикислота, молочная кислота или малоновая кислота; памовая кислота, гидроксинафтойная кислота, гентизиновая кислота, салициловая кислота, гликолевая кислота, миндальная кислота, молочная кислота, 4-ацетамидобензойная кислота или никотиновая кислота.
4. Сложный эфир по п. 2, отличающийся тем, что кислота представляет собой: глицин, янтарную кислоту, фосфорную кислоту; при этом указанный эфир такой, как показано ниже:
5. Сложный эфир по любому из пп. 2-4, имеющий форму соли, полученный в результате взаимодействия соединения формулы I с аминокислотой с образованием сложного эфира, который далее подвергнут взаимодействию с серной кислотой, фосфорной кислотой, сульфокислотой или хлороводородной кислотой; или полученный в результате взаимодействия соединения формулы I с двухосновной кислотой с образованием сложного эфира, который далее подвергнут взаимодействию с натрием, калием, магнием, трометамином, диэтаноламином, триэтаноламином, глицином, лизином или аргинином; или полученный в результате взаимодействия соединения формулы I с фосфорной кислотой с образованием сложного эфира, который далее подвергнут взаимодействию с натрием, калием, магнием, лизином, глицином, аргинином, трометамином, диэтаноламином или триэтаноламином.
6. Сложный эфир в форме соли по п. 5, такой, как показано ниже:


7. Применение соединения по п. 1 для получения лекарственного средства для лечения церебральных ишемических заболеваний.
8. Применение соединения по п. 1 для получения лекарственного средства для улучшения сна.
9. Применение соединения по п. 1 или сложного эфира по пп. 2-6 для получения лекарственного средства для лечения церебральных ишемических заболеваний и улучшения сна.
10. Применение соединения по п. 1 или сложного эфира по пп. 4-6 для получения лекарственного средства для лечения ишемических заболеваний и улучшения сна.
11. Фармацевтическая композиция для лечения ишемических заболеваний и улучшения сна, содержащая эффективное количество соединения по п. 1 или эфира по любому из пп. 2-6 и фармацевтически приемлемый носитель.
| CN 101289438 A 22.10.2008 | |||
| Регистр сдвига | 1984 |
|
SU1257706A1 |
| ПРИМЕНЕНИЕ L-БУТИЛФТАЛИДА ДЛЯ ИЗГОТОВЛЕНИЯ ЛЕКАРСТВЕННОГО СРЕДСТВА ДЛЯ ПРОФИЛАКТИКИ И ЛЕЧЕНИЯ ЦЕРЕБРАЛЬНОГО ИНФАРКТА | 2004 |
|
RU2336870C2 |

